catabolisme du peptide etude de sa clairance par des ...doxa.u-pec.fr/theses/th0231627.pdf · -c-...
TRANSCRIPT

1
École Doctorale Science de la vie et de la santé
THESE
pour obtenir le grade de
Docteur de l’Université PARIS XII
par
Boutaïna EL ABIDA
CATABOLISME DU PEPTIDE β-AMYLOÏDE : Etude de sa clairance par des cellules neuronales
et non neuronales en culture
Soutenue le 16 Décembre 2005 devant le jury constitué par :
Professeur Josette CADUSSEAU Président Professeur Omar BENZAKOUR Rapporteur Professeur Lionel LELIEVRE Rapporteur Docteur Pierre MARCHE Examinateur Docteur Mohamed RHOLAM Examinateur Professeur Patrick VICART Examinateur

2
REMERCIEMENTS
Ce travail a été effectué au sein du laboratoire de Biochimie des Signaux
Régulateurs Cellulaires et Moléculaires dirigé par le Professeur Paul COHEN. Je le
remercie très sincèrement de m’avoir accueillie au sein de son laboratoire durant ma
thèse.
Je remercie Madame Josette CADUSSEAU, de m’avoir fait l’honneur de
présider mon jury de thèse. Je souhaite remercier vivement Monsieur Lionel
LELIEVRE et Monsieur Omar BENZAKOUR d’avoir accepté la lourde tâche de
rapporteur, je remercie également Monsieur Patrick VICART et Monsieur Pierre
MARCHE d’avoir bien voulu être dans mon jury de thèse.
Je tiens à exprimer mes remerciements les plus sincères à Monsieur Mohamed
RHOLAM, mon directeur de thèse, de m’avoir guidée, soutenue et conseillée tout en
me laissant une grande liberté. Je le remercie de la confiance qu’il m’a accordée en
espérant avoir été à la hauteur. Qu’il trouve ici le témoignage de mon respect et de
mon admiration.
J’adresse un grand merci à Madame Christine CLAMAGIRAND pour ses
conseils judicieux et sa disponibilité. Je remercie également Madame Christine
FAHY, pour son soutien et Monsieur Arsène DER GARABEDIAN, pour son aide
et sa serviabilité.
Je tiens à remercier tout particulièrement les membres de l’équipe de
spectroscopie de masse du Muséum National d’Histoires Naturelles de m’avoir permis
d’effectuer les expériences et d’avoir été aussi disponibles. Je leurs suis très
reconnaissante.
Et enfin pour terminer, je voudrais assurer à mes proches, mon affection et ma
reconnaissance pour leur appui inconditionnel et pour toutes les leçons de courage
qu’ils m’ont enseignées. Leur soutien aura grandement contribué au bon avancement
de mon travail.

3
SOMMAIRE LISTE DES ABRÉVIATIONS 5LISTE DES ILLUSTRATIONS 7AVANT-PROPOS 9
INTRODUCTION
VIEILLISSEMENT ET DEMENCES -A- MODIFICATIONS DU CERVEAU LIÉES AU VIEILLISSEMENT. 12
1. Modifications anatomiques. 122. Modifications structurales 12
-B- LE SYNDROME DEMENTIEL. 13
MALADIE D’ALZHEIMER 15
-A- HISTORIQUE DE LA MALADIE. 17 -B- DONNEES STATISTIQUES. 17 -C- LES SYMPTÔMES DE LA MALADIE. 18 -D- LES LÉSIONS HISTOLOGIQUES DE LA MALADIE. 19
1. Les pertes neuronales. 21 2. Les plaques séniles. 21 3. Les dégénérescences neurofibrillaires 22
-E- LES FACTEURS DE RISQUE. 23 1. L’âge. 26 2. Formes familiales de la maladie d’Alzheimer. 26 3. Un gène facteur de risque: le gène de l’ApoE. 26 4. Hyperphosphorylation des protéines tau 27 5. Le stress oxydatif. 28 6. L’inflammation 29
METABOLISME DU PEPTIDE β-AMYLOÏDE 30
-A- LE PEPTIDE β-AMYLOÎDE. 321. Structure du peptide β-amyloïde. 322. Agrégation et toxicité du peptide β-amyloïde. 333. L’hypothèse de la cascade amyloïde. 34
-B- LE PRECURSEUR DU PEPTIDE β-AMYLOÏDE (APP). 361. Le gène de l’APP. 392. Les domaines de l’APP. 393. Le rôle de l’APP. 40
-C- ANABOLISME DU PEPTIDE β-AMYLOÏDE. 401. Protéases. 422. Voies de maturation de l’APP. 43
-D- CATABOLISME DU PEPTIDE β-AMYLOÏDE. 441. Internalisation cellulaire via un récepteur. 472. Dégradation enzymatique. 47
Objectifs. 47

4
MATÉRIELS ET MÉTHODES MATÉRIELS 53
1. Peptides. 542. Lignées cellulaires. 543. Matériel pour le test d’ELISA indirect. 554. Autres matériels. 55
MÉTHODES 561. Entretien des cultures cellulaires. 562. Conditions expérimentales de culture des lignées cellulaires. 573. Test ELISA indirect. 574. RP-HPLC. 595. ESI-Q-TOF-MS. 596. Dichroïsme Circulaire (DC). 607. Clairance des peptides Aβ40. 608. Caractérisation des activités enzymatiques. 61
RESULTATS EXPERIMENTAUX
CLAIRANCE DU PEPTIDE β-AMYLOÏDE NATIF PAR LES CELLULES. 631. Cinétique de disparition du peptide Aβ40 du milieu de culture des
cellules.
632. Mécanisme de disparition du peptide Aβ40 du milieu de culture. 673. Caractérisation des activités enzymatiques impliquées dans la
dégradation du peptide Aβ40.
693.1. Caractérisation de l’activité métalloprotéase. 733.2. Caractérisation de l’activité protéase à sérine. 73
4. Localisation cellulaire des activités enzymatiques. 755. Détermination des sites de clivage du peptide Aβ40. 77
CLAIRANCE DES PEPTIDES AMYLOÏDES MUTÉS. 871. Détermination des profils de clivage protéolytique des peptides. 882. Détermination des sites de clivage des peptides. 93
CONFORMATION DES PEPTIDES β-AMYLOÏDES. 97
DISCUSSION GÉNÉRALE ET PERSPECTIVES 101
RÉFÉRENCES BIBLIOGRAPHIQUES 111
ANNEXES 135

5
LISTE DES ABRÉVIATIONS
ACE Angiotensin-Converting Enzyme ACN Acétonitrile ADAM A Disintegrin and Metalloprotease ADDLs Aβ-Derived Diffusible Ligands ADN Acide Désoxyribonucleique
ADNmt Acide Désoxyribonucleique mitochondrial
AICD APP IntraCellular Domain
AMPc Adenosine MonoPhosphate cyclique
ApoE Apolipoprotéine E
APP Amyloïd Precursor Protein
APPs Soluble Amyloïd Precursor Protein
ARN Acide Ribonucleique
ARNm Acide Ribonucleique messager
ATP Adénosine Tri-Phosphate
AVC Accidents Vasculaires Cérébraux
ASPD Amylospheroid
Aβ β-Amyloïde
Aβ40 β-Amyloïde (1-40)
BACE Beta-site APP Cleaving Enzyme
CAA Cerebral Amyloid Angiopathy
DMEM Dulbecco’s Modified Eagle Medium
DSTA Démence Sénile de Type Alzheimer
ECE Endothelin-Converting Enzyme
EDTA Ethylene Diamine Tetraacetic Acid
ELISA Enzyme Linked ImmunoSorbent Assay
FAD Familial Alzheimer’s Disease
HCDHWA-D Hereditary Cerebral Dominant Hemorrhages With Amyloidosis Dutch type
HtrA High temperature requirements
IDE Insulin-Degrading Enzyme
KPI Kunitz-type Protease Inhibitor
LDL Low-Density Lipoprotein

6
LRP LDL Receptor-related Protein
MAP K Mitogen Activated Protein Kinases
MMPs Matrix MetalloProtéinases
Nct Nicastrine
NEM N-EthylMaleimide
NEP Neutral EndoPeptidase
PBS Phosphate Buffer Saline
PHF Paired Helicoidal Filaments
PS Préséniline
ROS Reactive Oxygen Species
RP-HPLC Reverse-Phase High Performance Liquid Chromatography
RPMI Roswell Park Memorial Institute
SR Scavenger receptor
SVF Sérum de Veau Foetal
TACE TNFα-Converting Enzyme
TGFβ Tumoral Growth Factor β
TOP Thimet OligoPeptidase
TPK I/GSK-3β Tau Protein Kinase I/Glycogen Synthase Kinase-3β
TFA Tri-Fluoroacetic Acid

7
LISTE DES ILLUSTRATIONS
INTRODUCTION.
Figure 1 : Les chemins de la détérioration neuronale.
Figure 2 : Atrophie corticale dans la maladie d’Alzheimer.
Figure 3 : Aspect d’une plaque sénile observée dans la maladie d’Alzheimer à
l’examen microscopique. Figure 4 : Marquage à l’hématine et éosine des dégénérescences neurofibrillaires.
Figure 5 : Localisation des zones affectées par les dégénérescences
neurofibrillaires.
Figure 6 : Schéma de l’activation inflammatoire associée au dépôt amyloïde.
Figure 7 : Représentation structural du peptide Aβ42.
Figure 8 : Représentation schématique du processus pathogène commun aux
maladies conformationnelles
Figure 9 : Evolution des intermédiaires oligomériques dans la formation des fibrilles
du peptide β-amyloïde Figure 10 : Représentation de la cascade amyloïde prenant en compte les données
récentes sur les pathologies Aβ et tau.
Figure 11 : Domaines fonctionnels de l’APP humain et principales mutations
causant les formes familiales de la maladie d’Alzheimer (MA).
Figure 12 : Clivages protéolytiques de l’APP par les secrétases α, β, γ.
RESULTATS :
Figure 13 : Cinétique de disparition du peptide Aβ40 du milieu de culture des cellules
neuronales (A) et non neuronales (B).
Figure 14 : Cinétique de dégradation du peptide β-amyloïde dans les surnageants de
cellules neuronales (A) et non neuronales (B).
Figure 15 : Disparition du peptide [I125]-Aβ40 en la présence des cellules ou des
surnageants cellulaires.
Figure 16 : Inhibition de la dégradation du peptide [I125]-Aβ40.
Figure 17 : Effet du Pefabloc (Pf), de l’ortho-phenanthroline (OP) et du pool
d’inhibiteurs I1 sur la dégradation du peptide [I125]-Aβ40.

8
Figure 18 : Effet du Pefabloc (Pf), de l’ortho-phenanthroline (OP) et du pool
d’inhibiteurs I1 sur la formation des pics P1 et P2.
Figure 19 : Effet des pools d’inhibiteurs I2, I3 et du NEM sur la dégradation du
peptide [I125]-Aβ40
Figure 20 : Mise en évidence de la sécrétion extracellulaire de l’activité sérine-
protéase en la présence de 8-Br-cAMP
Figure 21 : Mise en évidence de la sécrétion extracellulaire de l’activité sérine
protéase.
Figure 22 : Chromatogrammes total d’ionisation (LC-MS) du milieu de culture des
cellules CHO.
Figure 23 : Profil de dégradation du peptide amyloïde radioactif par les cellules CHO
en l’absence ou en la présence du Pefabloc.
Figure 24 : Cinétique de disparition des peptides amyloïdes du milieu de culture de
cellules neuronales.
Figure 25 : Profil de dégradation des peptides amyloïdes natif et «Flemish» par les
cellules CHO.
Figure 26 : Profil de dégradation des peptides amyloïdes murin et «Dutch» par les
cellules CHO.
Figure 27 : Spectres de dichroïsme circulaire des peptides amyloïdes dans le TFE.
Figure 28 : Spectres de dichroïsme circulaire des peptides amyloïdes dans le SDS.
Figure 29 : Schéma représentatif des différentes thérapies.

9
Avant-propos
Les plaques séniles sont des dépôts fibrillaires extracellulaires associés à la
maladie d’Alzheimer et dont le constituant principal est le peptide β-amyloïde
(Aβ). Ce peptide de 4 kDa est produit au cours du métabolisme normal (Haass
1992 ; Shoji,1992) par clivage protéolytique de son précurseur APP (« Amyloid
Precursor Protein »). Comme bon nombre de molécules secrétées, le taux
circulant du peptide Aβ est fonction de l’équilibre entre les voies de sa
biosynthèse à partir de son précurseur et celles de son catabolisme.
L’accumulation du peptide Aβ dans les plaques séniles pourrait donc s’expliquer
par un défaut dans l’une de ces voies, comme par la combinaison des deux.
Les voies de la maturation de l’APP ont fait l’objet de nombreuses études qui
ont été axées principalement sur l’identification et la caractérisation des
protéases (les β- et γ-secrétases en particulier) qui sont impliquées dans la
production du peptide β-amyloïde (Mattson 1997; Rholam et al., Brevets Rhône-
Poulenc Rorer et Université P. M. Curie, 1999; Nunan, 2002). Par contre, ce n’est
que tardivement que la compréhension des mécanismes de régulation du taux du
peptide Aβ a suscité de l’intérêt car il est maintenant admis et démontré que ce
peptide est un produit naturel, secrété dans l’organisme.
Notre travail s’inscrit dans cette optique qui consiste à caractériser tous
les facteurs impliqués dans le catabolisme normal du peptide Aβ. Pour cela, nous
avons abordé ce problème en utilisant différentes techniques d’analyse (ELISA,
RP-HPLC et spectroscopie de masse en mode electrospray) et plusieurs cellules
neuronales et non neuronales (8 lignées). On démontre que :
• le peptide Aβ40 natif présente un profil de dégradation commun à toutes les
cellules neuronales et non neuronales étudiées. Ceci suggère l’existence d’un

10
mécanisme similaire, assez largement distribué dans divers types cellulaires et
qui serait responsable de la protéolyse du peptide.
• les activités enzymatiques responsables de la clairance du peptide Aβ natif
sont de deux types: (i)-une métalloprotéase thiol-dépendante (située
principalement à la surface des cellules) agissant essentiellement sur le peptide
Aβ au niveau des sites Val18-Phe19, Phe19-Phe20 et Lys28-Gly29 et (ii)-une sérine
protéase (secrétée dans le milieu extracellulaire) ayant pour substrats les
fragments peptidiques générés par l’action de la 1ère protéase.
• les profils de clivage protéolytique des peptides amyloïdes, portant les
mutations « Flemish » et « Dutch », et du peptide amyloïde murin révèlent des
variations au niveau du nombre et/ou de l’amplitude des pics générés.
• Les peptides amyloïdes « Dutch » et « Rat » se caractérisent chacun par un
clivage majoritaire et sont de plus clivés par la forme soluble de la
métalloprotéase thiol-dépendante. Par contre, le peptide « Flemish », comme le
peptide amyloïde « natif » ne présente pas de clivage préférentiel.
• L’analyse de la conformation de ces peptides, par dichroïsme circulaire, montre
que ces peptides sont régis par un équilibre conformationnel hélice α ↔
feuillet β. L’effet des solvants (TFE et SDS) révèle des différences
conformationnelles entre les peptides en accord avec les différences
observées au niveau de leur clairance par les différentes cellules.
En conclusion, nous montrons essentiellement que la clairance du peptide β-
amyloïde est fonction de sa conformation. Cette dernière, régie par un équilibre
conformationnel, est très susceptible aux changements subis par son
environnement cellulaire. Par conséquent, lors du vieillissement cellulaire, les
cellules subissent diverses modifications qui peuvent abolir ou amoindrir l’activité
des protéases telles que celles mises en évidence dans ce travail et/ou modifier
la conformation du peptide β-amyloïde. Dans les deux cas, le peptide Aβ pourra
s’accumuler et former des dépôts amyloïdes.

11
INTRODUCTION

12
Chapitre I : VIEILLISSEMENT ET DEMENCES
Au cours du vieillissement, tous les tissus se modifient et perdent leur
élasticité. Ceci est vrai et bien visible pour la peau qui va voir son réseau de
fibres d'élastine et de collagène subir les effets accumulatifs de l'attaque
radicalaire des processus oxydants. De même, les tissus cérébraux subissent les
effets négatifs de l’âge, par exemple, la fluidité des membranes qui entourent les
vésicules de neuromédiateur diminue avec l’âge, il en résulte alors une
perturbation de la conduction de l’influx nerveux. Ce processus de vieillissement
affecte tous les organes ; toutefois leur évolution s’opère sur un mode
différentiel puisqu’il est actuellement reconnu que tous les organes ne vieillissent
pas à la même vitesse.
-A- MODIFICATIONS DU CERVEAU LIÉES AU VIEILLISSEMENT.
Au cours du vieillissement normal, le cerveau subit des modifications
anatomiques et structurales complexes. Ainsi, certains groupes de cellules et
certaines aires cérébrales sont plus sensibles au vieillissement que d'autres. En
outre, la vitesse de vieillissement du cerveau, la nature et l'étendue de ses
modifications chimiques et physiques, ainsi que leurs effets sur les capacités
intellectuelles, varient considérablement selon les individus.
1. Modifications anatomiques. Au cours du vieillissement, le nombre des
neurones diminue de manière différente selon la région cérébrale concernée.
Très peu de neurones meurent dans les aires de l'hypothalamus gouvernant la
sécrétion d'hormones. En revanche, de nombreux neurones dégénèrent dans une
partie du système nerveux reliant la moelle épinière aux hémisphères cérébraux
et dans la substance noire (Mc Geer et al., 1977). Cette dernière est
fonctionnellement très importante car ses neurones jouent un rôle essentiel dans

13
le métabolisme d’un neurotransmetteur impliqué dans le contrôle de la motricité,
la dopamine. Cette dégénérescence entraîne une perte de la masse cérébrale :
une légère diminution (7 %) jusqu’à 50 ans pouvant atteindre 30 % à l’âge de 100
ans. Cependant, le vieillissement peut s'accompagner également de modifications
neuronales apparemment bénéfiques puisqu'elles semblent corriger la perte ou
l'atrophie d’autres neurones (Coleman & Flood, 1987). Ainsi, lors du vieillissement
normal, la longueur moyenne des ramifications dendritiques des neurones de
l’hippocampe augmente entre l’âge mur et 70 ans puis régresse autour de 80 ans.
La croissance de ces dendrites compenserait alors les altérations cérébrales
liées à l’âge. Un autre exemple est celui des cellules gliales telles que les
astrocytes fibreux qui, après la soixantaine, se multiplient et grossissent. Ces
astrocytes, qui sécrètent divers facteurs favorisant la survie des neurones et la
croissance des axones et dendrites, prolifèrent pour compenser la dégradation
progressive du nombre et de la structure des neurones.
2. Modifications structurales. Le cerveau des personnes âgées subit de
nombreux changements au niveau du nombre et/ou de la fonction des
biomolécules vitales. Ainsi, des mutations au niveau de l'ADN peuvent entraîner
une diminution de la quantité ou de la qualité de protéines essentielles et/ou une
augmentation de la quantité ou de l'activité de protéines indésirables (Holliday,
1989). Par exemple, un pigment dit « de vieillissement », la lipofuscine, s’accumule
dans les cellules nerveuses, cardiaques et hépatiques, et diminue ainsi la capacité
fonctionnelle des cellules. L'ADN mitochondrial (ADNmt) a été également
soupçonné de participer à la sénescence du cerveau. En effet, l’accumulation de
nombreuses mutations au niveau de l’ ADNmt, chez les sujets âgés (Lin, 2002), se
traduit par l’inhibition de la synthèse de protéines mitochondriales essentielles
au transport des électrons ou à la synthèse d’ATP (Mandavilli, 2002). Les risques
de dérèglement sont plus grands pour l'ADN mitochondrial que pour l'ADN

14
nucléaire, car les enzymes qui réparent l'ADN sont moins efficaces dans les
mitochondries que dans le noyau. En outre, l'ADN mitochondrial est
probablement davantage soumis à l'attaque des radicaux libres (Wallace, 1992).
L'étude des modifications structurales des protéines, en particulier des
enzymes, au cours du vieillissement, présente de toute évidence un grand intérêt.
Les enzymes qui synthétisent les neuromédiateurs ou les récepteurs de ces
derniers deviennent moins actives avec l'âge ; pour certaines d'entre elles, cette
baisse d'activité est due, en partie, à des modifications ayant lieu après leur
synthèse. Les protéines ainsi modifiées sont généralement affectées dans leur
fonction ou parfois même complètement inactivées et doivent être éliminées de la
cellule, leur élimination est assurée principalement par un grand complexe
protéolytique constitué de plusieurs activités peptidases, le protéasome. Ce
dernier est impliqué non seulement dans l’élimination des protéines altérées
(Grune et al., 1997), notamment par oxydation, mais aussi dans le renouvellement
continu des protéines intracellulaires. Cependant, au cours du vieillissement
cérébral, le protéasome, se trouve lui-même oxydé et donc son activité
protéolytique se trouve altérée (Okada et al., 1999 ; Keller et al., 2000 ; Friguet,
2002). Ceci a pour conséquence une accumulation des protéines oxydées qui
amplifient les dommages oxydatifs.
D'autres molécules que les protéines sont également transformées au cours
du vieillissement. Ainsi, l’oxydation des lipides membranaires (Farooqui &
Horrocks, 1998) perturbe le comportement des membranes qui entourent les
cellules ou les organites intracellulaires et altère les fonctions des enzymes et
des transporteurs membranaires.
Si 5 à 30 % des molécules d'enzymes, de protéines ou d'ARN du cerveau
sont modifiées par rapport à des sujets jeunes, la même proportion de neurones
disparaît des aires cérébrales. Bien qu’une telle disparition de neurones semble
élevée, les capacités intellectuelles sont généralement très peu diminuées.

15
Cependant, lorsqu’il y a atrophie neuronale liée au vieillissement, celle-ci se
produit surtout dans les régions cérébrales intervenant dans l’apprentissage, la
mémoire ou les fonctions intellectuelles complexes. Ce sont ces modifications qui
provoquent des désordres connus sous le nom de démence sénile.
-B- LE SYNDROME DEMENTIEL.
Il s’agit de la transformation la plus grave qui puisse atteindre l'être humain
vieillissant. Le syndrome démentiel se définit par l'apparition d'un déficit
cognitif associant obligatoirement une altération de la mémoire à une autre
atteinte (langage, geste, apprentissage...) et d'intensité suffisante pour
perturber le fonctionnement social ou professionnel et entraîner un déclin
significatif par rapport au fonctionnement antérieur. La démence survient en
l'absence de confusion mentale et d'agitation. On distingue différents types de
démences :
• Les démences dégénératives. Il s'agit d'un groupe d'affections
chroniques, d'étiologie inconnue, de début insidieux et d'évolution lentement
progressive. Elles sont caractérisées par la dégénérescence des cellules
nerveuses vitales et regroupent plusieurs pathologies, notamment, les démences
fronto temporales, la maladie de Parkinson et la démence de type Alzheimer.
Cette dernière représente la forme la plus répandue des démences
dégénératives; 75 % des cas de démence lui sont attribués (Neary et al., 1986).
• Les démences non dégénératives. Cette forme de démence est
caractérisée par une apparition soudaine du syndrome démentiel et par une
régression irrégulière des aptitudes intellectuelles. Les démences vasculaires en
constituent la forme la plus fréquente et sont dues principalement à une anomalie
des vaisseaux du cerveau.
• Les démences mixtes. Dans ces cas, un processus dégénératif est associé
au processus pathogène vasculaire. L'association la plus rencontrée est celle des

16
deux causes les plus fréquentes de démence ; à savoir une maladie d'Alzheimer
et une démence vasculaire. On sait aujourd'hui que les accidents vasculaires
cérébraux (AVC) augmentent le risque de développer une démence de type
Alzheimer (Gainotti et al., 2004).

17
Chapitre II : MALADIE D’ALZHEIMER
Au cours de ces dernières décennies, la maladie d'Alzheimer est devenue un
des problèmes majeurs de santé publique du fait de l'augmentation de la
prévalence des formes séniles en rapport avec l'élévation de l'espérance de vie
et le vieillissement de la population. En effet, cette maladie reste la démence la
plus fréquemment diagnostiquée chez le sujet âgé. Des études américaines ont
montré que la maladie d'Alzheimer représente environ 45% des démences
globales et 75% des démences dégénératives (Neary et al., 1986).
-A- HISTORIQUE DE LA MALADIE.
En 1906, à Francfort, le jeune docteur Aloïs Alzheimer, compétent en
psychiatrie comme en histologie, pratique l’autopsie du cerveau d’une de ses
patientes nommée Auguste D., décédée à 51 ans et qui souffrait d’une
dégradation progressive des facultés cognitives, d’hallucinations, de confusion
mentale et d’une inaptitude psychosociale. Il décrit des types inédits de lésions
cérébrales : des plaques, des enchevêtrements neurofibrillaires et des lésions
d’athérosclérose.
C’est en 1907 que le professeur Emil Kræpelin décrit alors pour la première
fois, dans un traité de psychiatrie, la maladie à laquelle il donne le nom de son
élève Aloïs Alzheimer : "Les malades régressent mentalement, leur mémoire et
leur pensée dépérissent, ils sont égarés et désorientés. Par la suite, ils
développent une certaine agitation. Plusieurs désordres asymboliques
apparaissent, les malades ne comprennent plus les demandes ni les signes, ne
reconnaissent plus les objets ni les images. Les troubles du langage sont
particulièrement profonds, jusqu’au mutisme. Ils sont incapables de manger et de

18
s’occuper d’eux-mêmes. Les malades mettent plusieurs années à atteindre le
stade final et la mort est généralement provoquée par une cause extérieure".
Le nom Alzheimer a été utilisé à l’origine pour qualifier la démence présénile.
Il était donc de tradition de distinguer la maladie d’Alzheimer de la démence
sénile dégénérative qui touche les sujets de plus de 65 ans. Mais, la communauté
des lésions histologiques de ces deux affections a conduit à les réunir dans une
entité unique : la démence sénile de type Alzheimer (DSTA). La définition de la
maladie d’Alzheimer est anatomoclinique. Elle est caractérisée par l’installation
progressive de perturbations cognitives réalisant un tableau de démences associé
à des lésions histologiques caractéristiques du cerveau.
-B- DONNEES STATISTIQUES.
Aujourd’hui, des études statistiques démontrent que les hommes autant que
les femmes, toutes ethnies confondues, sont à risque de développer la maladie
d’Alzheimer. En Europe, la maladie d’Alzheimer est la première cause de démence
et la quatrième cause de décès, précédée par les maladies cardiaques, le cancer
et les accidents vasculaires cérébraux. On estime aujourd'hui que la maladie
d’Alzheimer touche 1 à 5% des sujets de moins de 65 ans et 20 à 40% des
patients de 85 ans et plus (Small, 1997).
En France, on observe que la part des 60 ans et plus, qui représentait 19%
de la population française en 1990, est passée à 20,6% en 2000 (12,5 millions de
personnes). Si la baisse de la mortalité se poursuit au même rythme
qu’aujourd’hui, les plus de 60 ans représenteront 35,1% de la population française
(22,4 millions d’individus) à l’horizon 2050. De plus, la croissance du nombre de
personnes âgées est encore plus spectaculaire quand on se rapproche du haut de
la pyramide des âges. En effet, l’effectif des plus de 75 ans passera de 4,2 à 11,6
millions entre 2000 et 2050 et celui des plus de 85 ans de 1,3 à 4,8 millions. Par

19
ailleurs, l’effectif des 60 ans en 2050 sera le double de celui de 2000, celui des
75 ans le triple et celui des 85 ans le quadruple.
En raison du vieillissement de la population, le nombre de cas et les coûts,
liés aux soins des gens atteints de cette maladie, prendront des proportions
alarmantes dans les années futures.
-C- LES SYMPTÔMES DE LA MALADIE.
Après avoir surmonté de multiples épreuves de la vie, certaines personnes
perdent progressivement les facultés les plus spécifiques de l'Homme : raisonner,
parler et se souvenir. La perte de la mémoire, du jugement, des capacités
intellectuelles et du contrôle émotionnel, que la maladie d'Alzheimer inflige aux
malades, est progressive et inexorable. On désigne souvent les caractéristiques
des symptômes de la maladie sous le nom des quatre A:
• La mémoire est en premier lieu atteinte (Amnésie), avec l’impossibilité pour le
patient d’enregistrer de nouveaux événements. Ces symptômes sont aussi
accompagnés d’une disparition des repères dans le temps et dans l’espace.
• Des troubles du langage apparaissent ensuite (Aphasie) et rendent la
communication difficile. Le patient peut parfois se murer dans le silence.
• La perte de compréhension de l’usage des objets usuels (Apraxie)
s’accompagne de la perte de reconnaissance des objets (Agnosie). Le patient
ne reconnaît plus son entourage en adoptant des attitudes d’indifférence, de
mutisme ou d’agressivité. L’état grabataire est inévitable à terme.
Au niveau cérébral, à mesure que les symptômes apparaissent, la perte des
neurones et de leurs connections s’étend (Touchon, 1999). Cette perte se
décompose schématiquement en 3 phases principales (Figure 1) :

20
• La phase préclinique pendant laquelle les lésions cérébrales s'installent dans le
cortex entorhinal sans traduction clinique. Sa durée est de 10 à 25 ans.
• La phase pré démentielle durant laquelle l'autonomie du patient n'est pas
altérée. L’hippocampe est touché et le sujet souffre de troubles mnésiques, la
mémoire épisodique étant la fonction la plus précocement atteinte. La durée
approximative est de 3 à 5 ans.
• La phase démentielle où les lobes pariétaux et le cortex moteur sont touchés.
Les troubles visuospatiaux s'aggravent menant à une véritable désorientation
temporospatiale. Un syndrome aphaso-apraxo-agnosique des troubles
psychocomportementaux (hallucination, délire, agitation) et des troubles
neurologiques (épilepsie, raideur…) altèrent l'autonomie du patient.
LOBEFRONTAL
LOBEPARIETAL
LOBETEMPORAL
LOBEOCCIPITAL
CORTEX MOTEUR
CERVELET
CORTEX ENTORHINAL
HIPPOCAMPE
LOBEFRONTAL
LOBEPARIETAL
LOBETEMPORAL
LOBEOCCIPITAL
CORTEX MOTEUR
CERVELET
CORTEX ENTORHINAL
HIPPOCAMPE
Figure 1 : Les chemins de la détérioration neuronale. 1 : phase pré-clinique ; 2 : phase pré-démentielle ; 3 et 4 : phase démentielle.

21
-D- LES LÉSIONS HISTOLOGIQUES DE LA MALADIE.
Il n'existe pas de critères diagnostiques absolus pour les démences séniles
de type Alzheimer. La seule certitude est anatomique. En effet, à l'autopsie, les
stigmates de la maladie sont visibles au microscope mais aucune des lésions n'est
complètement spécifique. Cependant, leur réunion signe véritablement la maladie.
1. Les pertes neuronales. Le premier stigmate est la disparition de
certains neurones qui se traduit par un «raccourcissement» du cortex
(Duyckaerts et al., 1985). Bien que d’intensité variable selon les patients, la perte
neuronale dans l’ensemble des régions corticales (cortex, hippocampe, amygdales)
n’excède pas 20% (Figure 2). Cependant, les neuropathologistes n’ont pas pu
déterminer si les pertes neuronales correspondent à une atrophie des cellules
(diminution de leur diamètre) ou à une disparition effective des cellules
neuronales.
A BA B
Figure 2 : Atrophie corticale dans la maladie d’Alzheimer. Coupe cérébrale d’un patient atteint de maladie d’Alzheimer (A) et d’un sujet témoin (B). Hauw J.J., CHU Salpêtrière, Paris.

22
2. Les plaques séniles. Ce sont des dépôts extracellulaires de forme
sphérique (15 à 200 µm de diamètre) dont le constituant principal est un peptide
de 4 KDa, le peptide β-amyloïde. Ces plaques séniles ne sont observées que chez
les sujets âgés normaux ou atteints de la maladie d’Alzheimer et dans la trisomie
21. Elles sont nombreuses au niveau de l’hippocampe et du cortex (Dhenain et al.,
2002). Ces plaques sont constituées de deux composants (Figure 3) :
• une partie centrale très compacte (noyau) contenant des filaments linéaires
de protéines dites amyloïdes et groupés en faisceaux enchevêtrés ou
irrégulièrement dispersés et,
• un amas de terminaisons nerveuses en dégénérescence (couronne), entourant
le noyau, qui sont soit des neurites (axones ou dendrites), soit des
prolongements gliaux (Selkoe, 1991).
Figure 3 : Aspect d’une plaque sénile observée au microscope (objectif х100). Un dépôt dense de substance amyloïde (grande flèche) occupe le centre de la plaque sénile qui est entourée d’une couronne claire (correspondant aux prolongements nerveux) et d’un halo de dépôt diffus (petites flèches).
20 µm

23
L’agencement variable de ces deux constituants permet d’individualiser deux
types de plaques :
• Les plaques diffuses (ou amorphes). Ce sont des dépôts volumineux et mal
limités. On les trouve dans les noyaux gris centraux du cervelet. Ils ne sont
pas vraiment des plaques séniles car ils ne comportent pas les prolongements
nerveux qui forment la couronne, l'aspect des synapses au sein du dépôt ne
semble que rarement modifié et les neurones adjacents paraissent normaux.
On en trouve un grand nombre chez les personnes âgées.
• Les plaques focales. Ce sont des dépôts sphériques, bien limités et plus petits.
Ils sont nombreux dans le cortex cérébral. Le noyau est entouré d'une
couronne de prolongements nerveux qui réalise la véritable plaque sénile.
Le cerveau d’un patient, atteint de la maladie d’Alzheimer, peut contenir ces
deux types de plaques. Par conséquent, une plaque sénile est une structure
complexe évoluant lentement. La maladie d’Alzheimer est également le plus
souvent accompagnée d’une angiopathie amyloïde cérébrovasculaire (CAA pour
« cerebral amyloid angiopathy ») caractérisée par la présence de dépôts
amyloïdes dans les vaisseaux sanguins cérébraux (Glenner, 1984; Miller, 1993;
Roher, 1993).
3. Les dégénérescences neurofibrillaires (NFT pour « neurofibrillary
tangles »). Ce sont des dépôts filamenteux présents dans le cytoplasme des
neurones en dégénérescence (Figure 4). Ces lésions intraneuronales sont
constituées de paires de filaments appariés en hélice (PHFs, pour « paired helical
filaments ») dont le constituant principal est la protéine tau hyperphosphorylée
(Wischik et al., 1988). Cette protéine, phosphorylée normalement, participe à la
stabilisation des microtubules (Brion et al., 1992) qui sont des structures

24
impliquées dans le transport intracellulaire des organites et dans l’organisation
spatiale des neurones.
L’apparition des dégénérescences neurofibrillaires suit une séquence spatio-
temporelle présentant une bonne corrélation avec la progression clinique de la
maladie (Braak, 1991; Duyckaerts, 1998). La dégénérescence neurofibrillaire
commence dans la région hippocampique, s’étend séquentiellement aux régions
temporales puis aux régions associatives polymodales (Figure 5). La présence des
PHFs dans la région hippocampique peut être attribuée au vieillissement cérébral
normal tandis que l’atteinte du cortex temporal est un processus précoce de la
maladie (Delacourte, 1999). Ces observations suggèrent un rôle déterminant des
PHFs dans la neurodégénérescence associée à la maladie d’Alzheimer.
Figure 4 : plusieurs dégénérescences neurofibrillaires sont observées à partir de cerveaux de patients atteins de la maladie d’Alzheimer. L’image est colorée à l’argent.

25
8 8
8
88
8
8
7
77 7
9 9
10
99
9
3
45
6
45
6
1
7
9
10
9
9
8 8
8
88
8
8
7
77 7
9 9
10
99
9
3
45
6
45
6
1
7
9
10
9
9
8 8
8
88
8
8
7
77 7
9 9
10
99
9
3
45
6
45
6
1
7
9
10
9
9
8 8
8
88
8
8
7
77 7
9 9
10
99
9
3
45
6
45
6
1
7
9
10
9
9
Face externe
Face interne
Lobe occipital Lobe frontal
Face externe
Face interne
Lobe occipital Lobe frontal
Figure 5. Localisation des zones affectées par les dégénérescences neurofibrillaires. Les numéros indiqués pour localiser les zones atteintes se rapportent aux stades S1 à S10. Le stade pré clinique de la maladie correspond à une grande quantité de dépôts amyloïdes dans l’ensemble du cortex cérébral, et une pathologie tau qui atteint le cortex temporal antérieur (S4), inférieur (S5) et moyen (S6). Au cours du développement de la maladie, la dégénérescence neurofibrillaire envahit la plupart des régions corticales, en progressant d’une manière hiérarchisée, des régions les plus associatives aux aires primaires motrices et sensitives (stades S7 à S10). Des manifestations cliniques importantes sont toujours observées lorsque les régions associatives multimodales sont très affectées par la pathologie tau
S1. Cortex transentorhinal S2. Cortex entorhinal S3. Hyppocampe S4. Lobe temporal antérieur S5. Gyrus temporal
S6. Gyrus temporal moyen S7. Régions associatives polymodales S8. Aires de Broca, régions associatives unimodales S9. Régions visuelles ou motrices

26
Les NFTs ne sont pas observés uniquement dans la maladie d’Alzheimer et la
trisomie 21, mais aussi dans d’autres affections telles que la démence
pugilistique, le syndrome parkinsonien de l'île de Guam ou la panencéphalite
sclérosante subaiguë (Steele, 1972; Dlouhy et al., 1992; Buee-Scherrer et al.,
1995).
-E- LES FACTEURS DE RISQUE.
A ce jour, les causes exactes de la maladie d’Alzheimer ne sont pas
totalement élucidées. En quête de réponses, les chercheurs ont analysé
différents facteurs pouvant avoir une influence quelconque sur la progression de
la maladie. Ce sont les «facteurs de risque». Certains sont génétiques, mais, en
général, il s’agit de facteurs sporadiques, sans antécédents familiaux et à
étiologie inconnue (Rocchi et al., 2003).
1. L’âge. Il s’agit sans conteste du plus grand facteur de risque de la
maladie d’Alzheimer. En effet, comme nous l’avons vu précédemment (paragraphe
B du chapitre II), la prédominance de la pathologie double tous les 5 ans après
l’âge de 65 ans.
2. Formes familiales de la maladie d’Alzheimer. Un autre grand facteur
de risque de la maladie d’Alzheimer est l’hérédité. En effet, il a été observé que
certains facteurs génétiques influencent le développement de cette maladie.
Dans certains cas, ces facteurs sont directement responsables d'une forme
familiale de la maladie (FAD pour « familial Alzheimer’s disease »). C'est le cas
des mutations des gènes codant pour le précurseur du peptide β-amyloïde (APP),
les préséniline-1 (PS1) et préséniline-2 (PS2) et situés respectivement sur les
chromosomes 21, 14 et 1 (Goate, 1997; Selkoe, 2001). Ainsi, le syndrome de Down
(trisomie 21), associé au développement précoce (vers l’âge de 30 ans) de

27
troubles neuropathologiques semblables à ceux qui caractérisent la maladie
d’Alzheimer (Mann, 1988), résulterait d’une surexpression de l’APP dont le gène
est porté par le chromosome 21 (Goldgaber, 1987; Tanzi, 1987). Par ailleurs, les
mutations du gène PS1 représentent la cause majeure de maladie d’Alzheimer
familiale (Sherrington, 1995) alors que celles du gène PS2 sont plus rares (Levy-
Lahad, 1995; Rogaev, 1995; Sherrington, 1996).D’une manière générale, les
mutations associées aux formes familiales précoces de la maladie d’Alzheimer
conduisent toujours à une surproduction du peptide β-amyloïde et en particulier
de la forme la plus longue Aβ42 (Murayama, 1999).
3. Un gène facteur de risque : le gène de l’ApoE. Dans les cas où les
facteurs génétiques influencent le déroulement de la maladie, on parle de
facteurs de susceptibilité génétique. C'est le cas du gène codant pour
l'apolipoprotéine E (ApoE) située sur le chromosome 19 (Das et al., 1985). L’ApoE
est une protéine du plasma impliquée notamment dans le transport du cholestérol
et dans le métabolisme des lipoprotéines (Mahley, 1988).
L’ApoE humaine a trois principales isoformes (ApoE2, ApoE3 et ApoE4)
correspondant respectivement à trois allèles (ε2, ε3, ε4). L’allèle ε4 représente
un facteur de susceptibilité génétique majeur de la forme tardive de la maladie
d’Alzheimer (Strittmatter, 1993; Wisniewski, 1994). En effet, il est 2 à 4 fois
plus fréquent chez les patients atteints de la maladie d’Alzheimer que dans la
population générale. En revanche, l’allèle ε2 semble avoir un effet protecteur. Ces
isoformes diffèrent par la nature de leurs résidus situés en position 112 et 158 :
Cys112-Cys158 (ApoE2) Cys112-Arg158 (ApoE3) et Arg112-Arg158 (ApoE4). Sachant que
seules les isoformes ApoE2 et ApoE3 forment des dimères par l’intermédiaire de
ponts disulfures, cette différence pourrait être à l’origine des propriétés
critiques de l’isoforme ApoE4 dans la maladie (Evans, 1995; Aleshkov, 1997).

28
In vitro, l’ApoE s’associe au peptide Aβ, via sa région Aβ(13-28)
(Strittmatter, 1993; Bales, 1997; Holtzman, 2000; Munson, 2000). Chez les
personnes saines, le complexe Aβ/ApoE favoriserait la dégradation du peptide Aβ
(Aleshkov, 1997) en inhibant la nucléation d’ oligomérisation du peptide (Evans,
1995). Par contre, chez les sujets atteints de la maladie d’Alzheimer, le
complexe formé serait instable et favoriserait au contraire la formation des
fibrilles amyloïdes (Ma, 1994; Castano, 1995).
L’ApoE peut être détectée également au sein des dégénérescences
neurofibrillaires. Ainsi, une étude sur des neurones en culture a mis en évidence
un clivage protéolytique intracellulaire de l’ApoE qui affecte principalement
l’isoforme E4. Les fragments générés interagissant avec des composants du
cytosquelette et induisant ainsi la formation de filaments (Huang, 2001).
En conclusion, l’ApoE4 interviendrait à la fois dans la formation des lésions
fibrillaires impliquant le peptide Aβ et celle des lésions filamenteuses impliquant
la protéine tau.
4. Hyperphosphorylation des protéines tau. La protéine tau joue un rôle
important au niveau du cytosquelette des neurones en modulant la polymérisation
et la stabilisation des microtubules. L’interaction tau/microtubules est régulée
par la phosphorylation de la protéine tau. Les protéines kinases, impliquées dans
cette phosphorylation, n'ont pas été identifiées de manière certaine, mais il
semblerait que les kinases TPK I/GSK-3β (Tau Protein Kinase I/Glycogen
Synthase Kinase-3β) et MAP-K (Mitogen Activated Protein Kinases) joueraient
un rôle important dans ce processus (Arendt et al., 1995; Sperber et al.,
1995; Takashima et al., 1996). Par ailleurs, il existe un équilibre
phosphorylation/déphosphorylation, régulé par des phosphatases (Trojanowski
1995).

29
Outre la phosphorylation normale de la protéine tau, une hyper-
phosphorylation anormale est observée dans le cas de la maladie d’Alzheimer.
Ainsi, l’affinité de la protéine tau hyperphosphorylée pour les microtubules
diminue et induit leur dépolymérisation, il en résulte une perturbation du
transport axonal qui va entraîner une dégénérescence neuronale (Goedert, 1993 ;
Watanabe et al., 1993). A l'heure actuelle, on ne sait pas exactement l’origine du
mécanisme d’hyperphosphorylation de ces protéines. Il pourrait s’expliquer par :
• Une activité accrue des kinases due à une compartimentalisation cellulaire
différente des MAP-K qui mettent en contact les protéines tau et ces kinases
(Arendt et al., 1995).
• Une activité accrue des kinases due à certains signaux extracellulaires qui
activent d’autres kinases par la voie de signalisation des MAP-K. Par exemple,
la présence du peptide Aβ pourrait induire une hyperphosphorylation de la
protéine tau. En effet, il a été montré que le traitement de culture primaire
de neurones d’hippocampe, par les peptides Aβ40 ou Aβ(25-35), engendre une
phosphorylation des protéines tau due à une activation des kinases TPK
I/GSK-3β par la voie de signalisation des MAP-K (Busciglio et al., 1995;
Takashima et al., 1996).
• Une activité phosphatasique déficiente, dans certaines populations de
neurones ou dans certains compartiments cellulaires, pourrait être l’une des
causes de l’hyperphosphorylation des protéines tau. En effet, il a été
démontré qu’une déphosphorylation des complexes tau-PHF par des
phosphatases in vitro augmente leur capacité à se lier aux microtubules (Gong
et al., 1994 ; Iqbal et al., 1994).
5. Le stress oxydatif. On appelle stress oxydatif, une augmentation
excessive de radicaux libres de l'oxygène qui provoque des dommages
moléculaires irréversibles, tels que des mutations au niveau de l’ADN

30
mitochondrial, la peroxydation des lipides ou la dénaturation des acides aminés.
Tous ces effets entraînent une perte de fonction et d'intégrité cellulaire, voire
la mort cellulaire.
Le stress oxydatif est l’un des facteurs potentialisant l'apparition de la
maladie d'Alzheimer (Sayre, 2001) et le peptide amyloïde apparaît comme un
acteur de premier ordre dans son apparition (Hensley, 1994). En effet, le
peptide amyloïde, sous sa forme oligomérique soluble, s’insèrerait dans la
membrane plasmique des neurones et des cellules gliales (Varadarajan, 2000) et
génèrerait des espèces activées de l’oxygène (ROS pour "reactive oxygen
species"), notamment H2O2, OH et O2- . La formation de ROS est également
induite par l’interaction du peptide Aβ avec certains cations métalliques
présentant des propriétés d’oxydo-réduction, comme Cu2+ ou Fe3+ (Sayre, 2000;
Opazo, 2002).
L’oxydation des protéines et la peroxydation des lipides de la membrane se
traduiraient par une désorganisation de la membrane plasmique conduisant à
différents processus critiques tels que la perte de l’homéostasie du calcium
(Demuro et al., 2005), le dysfonctionnement des récepteurs de certains
neurotransmetteurs, la perte de fonction de certaines protéines de transport
ou encore l’altération de la signalisation cellulaire pouvant conduire à l’activation
de facteurs de transcription ou à des processus d’apoptose.
6. L’inflammation. La présence de microgliocytes et d’astrocytes activés, au
voisinage des plaques séniles (Figure 6), indique qu’une réponse inflammatoire est
associée au dépôt amyloïde (Giulian, 1999). En effet, les microgliocytes libèrent
des cytokines inflammatoires (Rogers & Lue, 2001) dont certaines (Interleukine 1
et TGFβ) augmenteraient la synthèse du précurseur du peptide β-amyloïde. Il en
résulte une formation accrue du peptide amyloïde qui, en favorisant les dépôts
amyloïdes (Lesné et al., 2003), stimule à nouveau la réaction gliale. Les astrocytes

31
contribuent également à la neurodégénérescence en stimulant des processus
apoptotiques (Malchiodi-Albedi et al., 2001).
Par ailleurs, le système du complément est également activé après son
interaction avec le peptide amyloïde sous sa forme fibrillaire (Emmerling, 2000).
En particulier, l’interaction Aβ/C1q (impliquant les 11 premiers résidus du peptide
Aβ) active la voie classique du complément (Rogers, 1992; Velazquez, 1997). Ce
processus induit une cascade d’activations qui conduit à la formation de
complexes lytiques, responsable en partie de la mort neuronale et à une activation
inflammatoire. L’existence de ces processus inflammatoires explique l’effet
préventif apparent des anti-inflammatoires.
Figure 6 : Schéma de l’activation inflammatoire associée au dépôt amyloïde (d'après Abraham 2001; Tenner 2001). Les cadres jaunes, bleus et bruns correspondent respectivement aux étapes de l’activation, à leurs effets positifs et à leurs effets pathogènes.
ACCUMULATION D’Aβ
Activation de la microglie
Activation du complément
Activation des astrocytes
Réparation neuronale
Internalisation d’Aβ, déposition intracellulaire
Génération du complexe lytique, dirigé contre les
neurones
Activation de
protéases
PERTE / ALTERATION
Stress oxydant
Activation de la microglie
Activation du complément
Activation des astrocytes
Réparation neuronale
Internalisation d’Aβ, déposition intracellulaire
Génération du complexe lytique, dirigé contre les
neurones
Activation de
protéases
PERTE/ALTERATION
NEURONALE ET SYNAPTIQUE
Stress oxydant
ACCUMULATION D’Aβ
Activation de la microglie
Activation du complément
Activation des astrocytes
Réparation neuronale
Internalisation d’Aβ, déposition intracellulaire
Génération du complexe lytique, dirigé contre les
neurones
Activation de
protéases
PERTE / ALTERATION
Stress oxydant
Activation de la microglie
Activation du complément
Activation des astrocytes
Réparation neuronale
Internalisation d’Aβ, déposition intracellulaire
Génération du complexe lytique, dirigé contre les
neurones
Activation de
protéases
PERTE/ALTERATION
NEURONALE ET SYNAPTIQUE
Stress oxydant

32
Chapitre III : METABOLISME DU PEPTIDE β-AMYLOÏDE
Depuis les travaux de Glenner et Wong en 1984 (Glenner & Wong, 1984), il a
été démontré que le constituant majeur des plaques séniles est le peptide β-
amyloïde. Bien que son rôle causal dans la maladie n’ait pas été définitivement
établi, diverses observations (Sisodia et al., 1990; Kowall, 1994) suggèrent que le
dépôt de ce peptide joue un rôle capital dans la pathogénèse de la maladie
d’Alzheimer. Cependant, il est à noter que le peptide Aβ est produit au cours du
métabolisme normal et exercerait une activité neurotrophique (Yankner, 1990) et
une activité de rétrocontrôle de l’activité neuronale (Kamenetz, 2003).
Dans le cerveau, le peptide amyloïde peut être détecté sous trois formes :
associé aux membranes, agrégé ou soluble. Chez les sujets sains, la majorité du
peptide Aβ est associé aux membranes tandis que chez les sujets atteints de la
maladie d’Alzheimer, les fractions agrégées (dépôts diffus et plaques amyloïdes)
et solubles augmentent notablement (McLean, 1999; Bush, 2003).
-A- LE PEPTIDE AMYLOÎDE.
Constitué de 39 à 43 résidus, le peptide Aβ coexiste in vivo sous deux
formes principales :
Dans les milieux de culture cellulaire et les fluides biologiques, le peptide
Aβ40 est la forme majeure alors que le peptide Aβ42 ne représente que 10 % de
la totalité du peptide (Seubert et al., 1992; Haass et al., 1992; Shoji et al., 1992;
Golde et al., 1993). Dans la maladie d’Alzheimer, les plaques diffuses sont
composées essentiellement du peptide Aβ42 alors que les plaques séniles sont
composées principalement de la forme Aβ40 (Mori, 1992).
Aβ42:DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA Aβ40:DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVV

33
1. Structure du peptide β-amyloïde. Etant donné la localisation multiple
du peptide amyloïde (au voisinage des membranes, au sein de la cellule ou de
fluides biologiques) sa conformation a été caractérisée par différentes
techniques spectroscopiques dans différents milieux, et notamment en milieu
mimant les membranes phospholipidiques (Soto, 1994; Serpell, 2000; Mandal &
Pettegrew, 2004).
De ces études conformationnelles, il a été déduit une représentation de la
structure secondaire du peptide Aβ42 (Figure 7) qui révèle la présence de deux
coudes β (entre les résidus 6 et 8 et 23 et 27) et deux domaines possédant une
grande probabilité à s’organiser en feuillet β (Aβ(9-21) et Aβ(28-42)). De plus,
les séquences 22-27 et 32-42 du peptide possèdent une fraction importante de
résidus hydrophobes.
La conformation du peptide Aβ lui confère des propriétés d’agrégation qui
sont dépendantes de la concentration, du pH (Burdick et al., 1992; Barrow et al.,
1992) et de la taille du peptide (Jarrett et al., 1993). Par exemple, la faculté
d’agrégation du peptide Aβ42 est nettement supérieure à celle du peptide Aβ40
(Jarrett & Lansbury, 1993).
DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA
~~~~~Coude β
~~~~~~~~~Coude β
1 2 3 4 5 6 7 8 9 10 11 12 13 141516 17181920212223 24 2526 27282930 313233 343536 37 38 3940 4142
Cluster hydrophobe
central
Région très hydrophobe
Conformation αou β
Haute probabilité de conformation en
feuillet β
DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA
~~~~~Coude β
~~~~~~~~~Coude β
1 2 3 4 5 6 7 8 9 10 11 12 13 141516 17181920212223 24 2526 27282930 313233 343536 37 38 3940 4142
Cluster hydrophobe
central
Région très hydrophobe
Conformation αou β
Haute probabilité de conformation en
feuillet β
Figure 7 : Représentation structural du peptide Aβ42 (d’après Soto, 1994; Serpell, 2000a). Les régions présentant une forte tendance à adopter une conformation en feuillet β sont indiquées en bleu. Les résidus formant des clusters hydrophobes sont indiqués en rouge.

34
Il est à noter que le phénomène d’agrégation, observé dans la maladie
d’Alzheimer (Selkoe, 1997; Geula et al., 1998), serait à la base d’autres maladies
telles que les maladies d’Huntington (Perutz, 1996), de Parkinson, des tauopathies
(Goedert et al., 1998) et les maladies à prions (Yankner, 1996; Prusiner, 1997).
Une caractéristique, commune à ces maladies neurodégénératives, est la
conversion des protéines solubles en formes insolubles (Figure 8) qui sont
caractérisées par un taux élevé en structures feuillet β (Carrell & Lomas, 1997;
Kelly, 1998; Rochet & Lansbury, 2000).
2. Agrégation et toxicité du peptide β-amyloïde. Dans les modèles de
cultures cellulaires, la forme monomérique du peptide n’est pas toxique (Lorenzo
& Yankner, 1994). Par contre, dans certaines conditions environnementales, le
peptide Aβ subit des changements conformationnels qui favorisent son
agrégation. Il en résulte des dépôts amyloïdes dans l’espace extracellulaire du
cerveau et des parois des vaisseaux cérébraux sous forme de fibrilles (Capaldi &
Agrégation
Activité toxique Perte de la fonction
biologique
Protéine de conformation pathogène
Changement conformationnel
Protéine normalement repliée
Agrégation
Activité toxique Perte de la fonction
biologique
Figure 8: Représentation schématique du processus pathogène commun aux maladies conformationnelles (cas du fragment 90-231 de la protéine prion) (d’après Cohen, 1999). Le changement conformationnel et l’agrégation d’une protéine sont à l’origine d’une toxicité et d’une perte de

35
Radford, 1998). Un modèle moléculaire de la formation de ces fibrilles a été
proposé (figure 9).
Les peptides, adoptant des structures en feuilletβ, s’associeraient entre eux
pour former un noyau stabilisé en oligomères amyloïdiques (étape 1). La séquence
Lys16-Leu-Val-Phe-Phe20 du peptide Aβ serait impliquée dans la formation du
cœur des fibrilles (Tjernberg et al., 1999). La croissance de ce noyau forme alors
des protofibrilles (étape 2) et c’est l’enlacement de quatre protofibrilles qui
forme la structure hélicoïdale (étape 3) appelée fibrille amyloïde.
Il est à noter que cette organisation fibrillaire du peptide Aβ est favorisée
par son association à certaines protéines telles que les héparane sulfate
protéoglycannes, l’α1-antichymotrypsine et l’apolipoprotéine E (Ma et al., 1994).
Certains métaux tels que l’aluminium, le zinc ou le fer, ont été observés dans les
plaques séniles et semblent également favoriser l’agrégation du peptide in vitro
(Bush et al., 1994 ; Kawahara et al., 1994).
a)-Toxicité des formes fibrillaires du peptide Aβ. De nombreuses
études ont mis en évidence la toxicité des formes fibrillaires du peptide Aβ sur
agrégation du Aβen noyau croissance du noyau
en protofibrille association des protofibrillesen fibrille amyloïde
β-amyloïde en feuillet β
Etape 1 Etape 2 Etape 3
agrégation du Aβen noyau croissance du noyau
en protofibrille association des protofibrillesen fibrille amyloïde
β-amyloïde en feuillet β
Etape 1 Etape 2 Etape 3
Figure 9 : Evolution des intermédiaires oligomériques dans la formation des fibrilles du peptide β-amyloïde (d’après Rochet & Lansbury, 2000).

36
des cultures cellulaires (neurones ou cellules de phéochromocytome de rat)
(Iversen, 1995). Cette toxicité se traduit par une dystrophie des neurites (Pike,
1992) et une perte neuronale (Pike, 1991; Busciglio, 1992) qui sont similaires à
celles observées dans la maladie d’Alzheimer. Le degré de fibrillogenèse du
peptide Aβ, lié à sa conformation en feuillets β, est déterminant dans ces effets
neurotoxiques (Simmons, 1994; Howlett, 1995). Ainsi, in vitro les agrégats
fibrillaires du peptide Aβ42 sont neurotoxiques tandis que les agrégats amorphes
ne présentent aucune toxicité (Lorenzo, 1994). Ces effets ont été confirmés par
plusieurs études in vivo par injection intracérébrale du peptide Aβ (Frautschy,
1991; Kowall, 1991).
b)-Toxicité des oligomères solubles du peptide Aβ. Un résultat plus
surprenant a priori est l’effet neurotoxique important observé pour des
oligomères solubles du peptide Aβ. Ces oligomères neurotoxiques regroupent des
protofibrilles (PFs) (Hartley et al., 1999) et des espèces oligomériques plus
stables, les ADDLs (pour "Aβ-derived diffusible ligands") (Klein, 2001). Ces deux
types d’oligomères présentent une neurotoxicité respectivement sur des cultures
organotypiques du système nerveux central (Lambert, 1998) et sur des neurones
en culture (Hartley et al., 1999). Ces oligomères apparaissent donc comme des
acteurs critiques dans la maladie d’Alzheimer d’autant plus que leur solubilité
permet leur diffusion potentielle dans l’espace cérébral.
Depuis peu, il a été suggéré que des agrégats du peptide amyloïde sous une
forme sphérique (ASPD pour «Amylospheroid ») exerceraient également une
activité neurotoxique sur des cultures primaires de la région septale du cerveau
du rat et que cette toxicité est fortement liée à la taille de ces agrégats (Hoshi,
2003).
3. L’hypothèse de la cascade amyloïde. Cette hypothèse met en évidence
le rôle central du peptide β-amyloïde dans le processus toxique de la maladie

37
d’Alzheimer. Elle est basée sur l’étude des cas de formes familiales de la maladie
qui démontre que la plupart des mutations du gène de l’APP augmentent la
production du peptide Aβ42 (Hardy, 1992; Golde, 2003). Il est à remarquer que
toutes ces mutations se situent à proximité ou dans la région correspondant au
peptide β-amyloïde. Selon cette hypothèse, la fibrillogenèse du peptide amyloïde
est un événement précoce dans l’étiologie de la maladie, à l’origine d’une cascade
de mécanismes pathogènes responsables de la formation des lésions fibrillaires
dues à la protéine tau, de la neurodégénérescence et de la démence (Hardy,
1992; Golde, 2003).
Toutefois, une corrélation entre dépôt amyloïde et neurodégénérescence
n’est pas toujours observée (Cochran, 1991; Irizarry, 1997). En effet, des études
récentes (Klein, 2001) suggèrent que les oligomères solubles d’Aβ peuvent être
considérés comme des acteurs pathogènes importants. De plus, il est apparu que
les lésions fibrillaires de la protéine tau ne peuvent être considérées que comme
des lésions secondaires (Iqbal, 2000). Au vu de ces éléments, l’hypothèse de la
cascade amyloïde a évolué de manière à prendre en compte les autres acteurs
critiques dans la maladie d’Alzheimer (Figure 10).
L’accumulation du peptide amyloïde, qui résulterait d’une surproduction ou
d’un déséquilibre entre production et dégradation, apparaît comme l’événement
déclenchant la maladie. Elle peut être favorisée par :
• des facteurs génétiques (mutations au sein des gènes de l’APP, des PS1 et
PS2, expression de l’allèle ε4 de l’ApoE),
• des prédispositions à la maladie (taux de cholestérol élevé),
• des interactions critiques (cations métalliques, protéines chaperones) ou des
dégradations chimiques du peptide liées au vieillissement protéique
(troncations, isomérisations, racémisations).

38
Cette accumulation conduirait à la formations d’oligomères d’Aβ, de dépôts
diffus et finalement de plaques séniles amyloïdes. Le dépôt amyloïde vasculaire
peut être plus ou moins important selon la prédisposition des sujets
(l’hypertension artérielle et les accidents vasculaires cérébraux sont envisagés
comme des facteurs de risque). Les oligomères solubles d’Aβ présentent des
Production altérée d’Aβ
(syndrome de Down, mutationsde l’APP ou des PS1, PS2, taux de cholestérol élevé)
Métabolisme normal d’Aβ + ApoE4 Métabolisme normal d’Aβ+ vieillissement protéique
ToxicitéActivation de la réponse inflammatoire
Stress oxydantAltération de l’homéostasie du calcium
Démence
Accumulation d’Aβ
- Oligomères solubles- Plaques séniles- Déposition vasculaire
Dépôts diffus, non pathogènes
Altération de la protéine tauHyperphosphorylationFormation des PHFs
Déstabilisation des microtubules
Mort et altération neuronale
Glycosaminoglycanes, protéines chaperones
Cations métalliques, protéines chaperones
Cations métalliques
Production altérée d’Aβ
(syndrome de Down, mutationsde l’APP ou des PS1, PS2, taux de cholestérol élevé)
Métabolisme normal d’Aβ + ApoE4 Métabolisme normal d’Aβ+ vieillissement protéique
ToxicitéActivation de la réponse inflammatoire
Stress oxydantAltération de l’homéostasie du calcium
Démence
Accumulation d’Aβ
- Oligomères solubles- Plaques séniles- Déposition vasculaire
Dépôts diffus, non pathogènes
Accumulation d’Aβ
- Oligomères solubles- Plaques séniles- Déposition vasculaire
Dépôts diffus, non pathogènes
Altération de la protéine tauHyperphosphorylationFormation des PHFs
Déstabilisation des microtubulesAltération de la protéine tau
HyperphosphorylationFormation des PHFs
Déstabilisation des microtubules
Mort et altération neuronale
Glycosaminoglycanes, protéines chaperones
Cations métalliques, protéines chaperones
Cations métalliques
Figure 10 : Représentation de la cascade amyloïde prenant en compte les données récentes sur les pathologies Aβ et tau (d’après Golde, 2003). Le rôle critique de l’apoE4 est placé en amont de l’accumulation du peptide amyloïde mais pourrait également intervenir dans la toxicité liée à Aβ et le dépôt des PHFs. L’accumulation du peptide Aβ pourrait exercer une activité d’activation de la production de l’APP et de l’ApoE, qui n’est pas représentée sur ce schéma.

39
effets pathologiques tandis que les dépôts diffus sont intrinsèquement non
pathogènes et pourraient constituer soit un dépôt lié au vieillissement normal,
soit un stade très précoce de la maladie.
Le dépôt amyloïde apparaît comme un événement précoce, à l’origine de
l’astrocytose, de la gliose, de l’hyperphosphorylation et le dépôt des filaments de
tau. En effet, la toxicité et/ou l’inflammation induite par l’accumulation du
peptide amyloïde seraient responsables de la phosphorylation anormale de tau,
conduisant à une déstabilisation des microtubules, et du dépôt de tau sous forme
de filaments. Cette pathologie tau serait le principal responsable de la perte
neuronale. Enfin, pour les porteurs de l’allèle ε4 de l’apoE, l’apoE4 pourrait
intervenir de façon critique dans le dépôt amyloïde, la toxicité liée au stress
oxydant, la réponse inflammatoire et le dépôt de tau.
-B- LE PRECURSEUR DU PEPTIDE β-AMYLOÏDE (APP).
1. Le gène de l’APP. Le gène, codant pour le précurseur du peptide β-
amyloïde, fut le premier gène isolé (Kang 1987) et associé à une forme familiale
de la maladie d’Alzheimer (Chartier-Harlin et al., 1991; Goate 1997). Il contient
au moins 18 exons différents (Yoshikai et al., 1990). Par épissage alternatif, au
moins 9 ARNm différents sont transcrits à partir de ce gène. Ces différents
ARNm codent pour des protéines dont le contenu en acides aminés varie de 365 à
770 résidus (Sandbrink et al., 1994). Les isoformes APP695, APP751 et APP770
sont les plus fréquentes. Elles se différencient par la présence ou non d’une
séquence KPI (Kunitz-type protease inhibitor) correspondant à un inhibiteur de
protéases à sérine (Ponte et al., 1988). Les neurones humains expriment
principalement l’isoforme APP695 et produisent la majorité de l’APP présent dans
le cerveau, tandis que les isoformes APP751 et APP770 sont rencontrées
principalement dans les cellules gliales.

40
2.Les domaines de l’APP. Cette protéine, d'environ 120-140 kDa, est
composée d’un long domaine extracellulaire, d’un domaine transmembranaire
hydrophobe et d’un court domaine COOH-terminal cytoplasmique (figure 11A) :
• Le domaine extracellulaire commence par un peptide signal, suivi d’une région
riche en cystéine, d’une région riche en résidus aspartate et glutamate, de
plusieurs domaines capables de fixer l’héparine, d’un site de fixation du cuivre,
de même que d’un site de fixation du zinc. Ce domaine contient également un
site de fixation du collagène, une séquence qui serait responsable de l’activité
neurotrophique de la protéine (Ninomiya et al., 1993) et une région possédant
deux sites de glycosylation.
• Le domaine cytoplasmique intracellulaire montre, entre autre, un motif
YENPTY qui fixe les protéines X11 et Fe65. Ces protéines interagissent avec
des facteurs de transcription (Borg et al., 1996). Ce domaine possède aussi
une séquence ayant pour affinité une protéine fixant le GTP (Go).
• Ces deux domaines sont connectés par le peptide Aβ dont la séquence COOH-
terminale est ancrée dans la membrane.
3.Le rôle de l’APP. L’APP est exprimé de façon ubiquitaire dans les tissus
avec des taux différents selon le type cellulaire. Ainsi, l’APP695 est largement
majoritaire dans le système nerveux central alors que l’isoforme APP751 est
essentiellement exprimée dans le tissu périphérique (Weidemann et al., 1989).
Bien que la fonction réelle de l'APP n'ait pas encore été élucidée, des études
suggèrent que cette glycoprotéine pourrait jouer un rôle dans la régulation de la
croissance cellulaire ainsi que dans les interactions d'adhésion dans
l'inflammation, la régénération et la réponse immunitaire (Wallace et al., 1997 ;
Marquez-Sterling et al., 1997). L'APP est une protéine présentant les
caractéristiques d'un récepteur de surface de type I (Kang et al., 1987).

41
Figure 11.(A)-Domaines
fonctionnels de
l’APP humain.
L’APP consiste
en un
large domaine
extracellulaire, un
domaine transmem
branaire hydrophobe et un court domaine C-terminal cytoplasmique. SP: Signal peptide; HBD: Heparinbinding
domain; KPI:Kunitzregion; CBD: Clathrin
binding domain.(B)-Principales mutations de l’APP
causant les formes familiales de la maladie. Certaines mutations sont localisées dans la séquence du peptide β-amyloïde.
1 10 20 30 40
ISEVKMDAEFRHDSGYEVHHQ
KLVFFAEDVGSN
KGAIIGLMVGGVVIA TVIVITLVM
LKKKQ
Swedish 670/671N L
Flemish 692 G
Iowa 694 N
Dutch 693 Italian
693 Arctic
693 QKG
London 717
Australian723
Austrian714
IFrench 715
M
Florida 716V
P
IGLF
NH
2CO
OH
SP
Acidicdom
ainCystein rich
regionKPI
RERMS
Trophio dom
ainAβ
CT26
YENPTY
CollagenCu
X11Fe65
ZnHBD
G0
HBDGlycosylation
CBD
HBD
B BA AFigure 11.(A)-Domaines
fonctionnels de
l’APP humain.
L’APP consiste
en un
large domaine
extracellulaire, un
domaine transmem
branaire hydrophobe et un court domaine C-terminal cytoplasmique. SP: Signal peptide; HBD: Heparinbinding
domain; KPI:Kunitzregion; CBD: Clathrin
binding domain.(B)-Principales mutations de l’APP
causant les formes familiales de la maladie. Certaines mutations sont localisées dans la séquence du peptide β-amyloïde.
1 10 20 30 40
ISEVKMDAEFRHDSGYEVHHQ
KLVFFAEDVGSN
KGAIIGLMVGGVVIA TVIVITLVM
LKKKQ1 10 20
30 401 10 20
30 40ISEVKM
DAEFRHDSGYEVHHQKLVFFAED
VGSNKGAIIGLM
VGGVVIA TVIVITLVMLKKKQ
Swedish 670/671N L
Flemish 692 G
Flemish 692 G
Iowa 694 NIowa 694 N
Dutch 693 Italian
693 Arctic
693 QKG
Dutch 693 Italian
693 Arctic
693 QKG
London 717
Australian723
Austrian714
IAustrian
714I
French 715M
French 715M
Florida 716V
Florida 716V
P
IGLF
NH
2CO
OH
SP
Acidicdom
ainCystein rich
regionKPI
RERMS
Trophio dom
ainAβ
CT26
YENPTY
CollagenCu
X11Fe65
ZnHBD
G0
HBDGlycosylation
CBD
HBD
NH
2CO
OH
SP
Acidicdom
ainCystein rich
regionKPI
RERMS
Trophio dom
ainAβ
CT26
YENPTY
CollagenCollagen
CuCuX11
Fe65
ZnZnHBDHBD
G0
G0
HBDHBD
GlycosylationGlycosylation
CBD
HBD
B BA A

42
Des travaux récents lui associent un rôle dans le transport protéique Kinésine-
dependant (Annaert & De Strooper 2002); la kinésine étant une protéine
impliquée dans le transport axonal de la PS1 (Kamal et al., 2001).
Sur le plan le plus fondamental, l’APP apparaît essentiel à la physiopathologie
de la maladie d’Alzheimer parce que son produit de clivage, le peptide β-amyloïde,
est le constituant majeur des plaques séniles. De plus, le gène de l'APP présente
plusieurs mutations dont certaines se situent dans le domaine correspondant au
peptide Aβ (figure 11B). Si la majorité des mutations engendrent des formes
familiales de la maladie, certaines d’entre elles peuvent provoquer des
pathologies, comme les hémorragies cérébrales, qui diffèrent cliniquement de la
maladie d’Alzheimer. C’est le cas des mutations [G692]-APP (mutation Flemish) et
[Q693]-APP (mutation Dutch) qui sont associées respectivement à une CAA
(cerebral amyloid angiopathy) (Hendriks et al., 1992) et au syndrome HCDHWA-D
(Hereditary Cerebral Dominant Hemorrhages With Amyloidosis of the Dutch
type) (Van Broeckhoven et al., 1990). Ces mutations, en modifiant la conformation
du peptide β-amyloïde, favoriseraient son agrégation dans plusieurs régions
cérébrales (Demeester et al., 2001; Nilsberth et al., 2001; Murakami et al.,
2002).
-C- ANABOLISME DU PEPTIDE β-AMYLOÏDE.
Comme pour un bon nombre de peptides, le peptide β-amyloïde est le produit
de la maturation sélective par protéolyse de son précurseur. Une surproduction
du peptide amyloïde pourrait avec d’autres facteurs favoriser son agrégation, par
conséquent, les voies de son anabolisme ont suscité beaucoup d’intérêts et ont
été largement étudiées.

43
1.Protéases. Sous l’action de différentes protéases de maturation,
appelées secrétases, le précurseur du peptide Aβ peut subir trois clivages
majoritaires (Esch et al., 1990; Sisodia et al., 1990; Koo & Squazzo et al., 1994).
a)-α-secrétase. Cette protéase agit à l'intérieur du peptide Aβ entre
les résidus Lys612 et Leu613 de l'APP695 pour générer un fragment N-terminal
secrété APPsα (APP soluble contenant les 16 premiers acides aminés du peptide
Aβ) ainsi qu'un fragment C-terminal de 83 résidus qui reste ancré à la membrane
(C83). Différentes protéines membranaires, appartenant à la famille des ADAM
(a disintegrin and metalloprotease), ont été identifiées comme ayant une activité
de type α-secrétase : l’ADAM17 (ou TACE) (Buxbaum et al., 1998), l’ADAM10
(Fahrenholz et al., 2000 ; Colciaghi et al., 2002) et l’ADAM9 (Koike et al., 1999).
Des expériences d’hybridation in situ, dans le cerveau humain ou dans les
neurones de cerveau de souris, ont révélé une co-expression de l’APP et de
l’ADAM10, suggérant que cette dernière serait la candidate la plus plausible pour
l’α-secrétase (Marcinkiewicz & Seidah, 2000).
b)-β-secrétase (BACE pour « Beta-site APP Cleaving Enzyme »). Il
s’agit d’une protéase à aspartate de la famille des pepsines, comportant un
domaine N-terminal catalytique contenant deux résidus aspartate impliqués dans
son activité, d’un domaine transmembranaire de 17 résidus et d’une queue
cytoplasmique. Cette protéase clive le précurseur entre les résidus Met596 et
Asp597 pour libérer un fragment N-terminal secrété APPsβ (APP soluble délété
totalement du peptide Aβ) et un fragment C-terminal de 99 acides aminés
contenant le peptide Aβ ancré à la membrane (C99). La BACE peut cliver
également l’APP entre les résidus Tyr606 et Glu607, décrit comme un site de
clivage alternatif de l’APP et dénommé clivage β’-secrétase (Vassar, 1999; Liu,
2002).
Deux enzymes transmembranaires de type aspartyl protéase ont été
identifiées : la BACE1 (Hussain et al., 1999 ; Sinha et al., 1999 ; Vassar et al.,

44
1999 ; Yan et al., 1999 ; Lin et al., 2000) et la BACE2 (Yan et al., 1999). Des
études par knockout, sur des souris BACE1, ont montré que cette enzyme est
majoritaire dans le tissu cérébral (Cai et al., 2001 ; Luo et al., 2001) et qu’elle est
co-exprimée avec l’APP dans plusieurs régions (Marcinkiewicz & Seidah, 2000).
Par contre, l’enzyme BACE2 est peu exprimée dans le cerveau (Bennett et al.,
2000) mais plutôt au niveau du cœur, du rein et du placenta (Farzan et al., 2000).
Ces résultats suggèrent que la BACE1 jouerait un rôle important dans la
biosynthèse du peptide Aβ alors que la BACE2 serait impliquée dans la
vascularisation des tissus systémiques.
c)-γ-secrétase. C’est un complexe multi-protéique constitué de quatre
partenaires : (i)-Un hétérodimère des présénilines (PS1 et PS2) qui joue un rôle
clé dans le clivage et assure une fonction de protéase à aspartate ; (ii)-la
nicastrine (Nct) qui est une glycoprotéine de type I et (iii)-Deux protéines
membranaires, APH-1 et PEN-2. L’arrangement du complexe est encore mal
connu, mais des études récentes ont permis de localiser certains sites
d’interactions entre les protéines partenaires (Morais, 2003; Fraering, 2004).
L’implication des présénilines, dans le clivage par la γ-secrétase, a été mise
en évidence par des mutations du gène codant pour ces protéines, qui induisent
une surproduction du peptide amyloïde Aβ42 (Eckman 1997). Plus récemment,
cette implication a été montrée par l’absence de sécrétion du peptide Aβ dans
des cellules issues de souris doublement invalidées pour PS1 et PS2 (Herreman,
2000). La γ-secrétase agit entre les résidus 636 à 637 du précurseur APP ou des
fragments générés par la secrétase α (fragment C83) et β (fragment C99).
2. Voies de maturation de l’APP. Le précurseur APP peut subir au moins
deux voies de maturation post-traductionnelles (figure 12) :

45
b
a
1 16 17 43
Aβ
p3
DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIAT
APPN C
NSAPPβ C99
C
SécrétionSécrétion
NsAPPα C83
C
α-secrétase β-secrétase
γ-secrétase
p3C
SécrétionDépôts diffus
AβC
SécrétionDépôts amyloïdes
γ-secrétase
CTF CTF
APPN C
NNSAPPβ C99
CC
SécrétionSécrétionSécrétionSécrétion
NNsAPPα C83
CC
α-secrétase β-secrétase
γ-secrétase
p3CC
SécrétionDépôts diffus
AβCC
SécrétionDépôts amyloïdes
γ-secrétase
CTF CTF
b
a
1 16 17 43
Aβ
p3
DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIAT
APPN C
NSAPPβ C99
C
SécrétionSécrétion
NsAPPα C83
C
α-secrétase β-secrétase
γ-secrétase
p3C
SécrétionDépôts diffus
AβC
SécrétionDépôts amyloïdes
γ-secrétase
CTF CTF
APPN C
NNSAPPβ C99
CC
SécrétionSécrétionSécrétionSécrétion
NNsAPPα C83
CC
α-secrétase β-secrétase
γ-secrétase
p3CC
SécrétionDépôts diffus
AβCC
SécrétionDépôts amyloïdes
γ-secrétase
CTF CTF
Figure 12 : Clivages protéolytiques de l’APP par les α, β et γ-secrétases. Les séquences 1-16 et 17-42 d’Aβ sont représentées respectivement en rouge et en bleu. Les domaines cellulaires transmembranaires et cytoplasmiques sont colorés respectivement en jaune et en rose. a-Voies de maturation de l’APP :
• Le clivage par l’α-secrétase conduit à la sécrétion d’un grand fragment N-terminal (sAPPα) et à la formation d’un fragment C-terminal (C83). Le clivage par la γ-secrétase sépare le fragment p3 de la queue cytoplasmique, CTF.
• Le clivage par la β-secrétase conduit à la sécrétion du fragment N-terminal (sAPPβ) et à la formation du fragment C-terminal (C99). Le clivage par la γ-secrétase sépare Aβ de CTF.
• Le clivage par la γ-sécrétase, au sein de la membrane cellulaire, s’effectue en différents sites adjacents et conduit ainsi à une hétérogénéité C-terminale des peptides p3 et Aβ Les peptides p3 et Aβ, sécrétés par la cellule, peuvent donner lieu respectivement à des dépôts diffus et des dépôts amyloïdes, associés à la maladie d’Alzheimer.
b. Séquences des peptides Ab et p3 : • Les barres horizontales de longueurs différentes illustrent
l’hétérogénéité C-terminale des peptides, avec en rouge les peptides majoritairement libérés au cours du clivage.

46
a)-Voie de maturation dite non amyloïdogénique. L'APP, inséré dans la
membrane, subit un clivage entre les résidus Lys16 et Leu17 du peptide Aβ pour
générer les fragments APPsα et C83 (Esch et al., 1990; Sisodia et al., 1990).
L’APPsα, secrété dans la circulation sanguine (Haass & Selkoe, 1993), est un
métabolite qui semble être impliqué dans le processus de coagulation et de
réparation cellulaire (Oltersdorf et al., 1989), dans la prolifération cellulaire
(Pietrzik et al., 1998 ; Ohsawa et al., 1999) et qu’il présente des propriétés
neurotrophiques (Kojro et al., 2001). Le fragment C83 peut ensuite subir une
coupure par la γ-secrétase qui libère le produit nommé p3 dont la fonction reste,
à ce jour, mal connue.
b)-Voie de maturation dite amyloïdogénique. Dans cette voie, l’APP est
clivé au niveau des extrémités du peptide Aβ pour libérer ce peptide intact dans
le milieu extracellulaire. Le premier clivage, par la β-secrétase, génère l’APPsβ et
le fragment C99 contenant la totalité du domaine Aβ. Il est à noter que cette
voie, génératrice du métabolite C99 potentiellement amyloïdogène, existe aussi
chez les sujets sains (Selkoe, 1993). La γ-secrétase clive ensuite le fragment C99
pour générer un fragment C-terminal, appelé AICD (APP intracellular domain) et
plusieurs peptides β-amyloïdes qui diffèrent par leur extrémité C-terminale
(Zhang, 2001). Le fragment AICD, libéré dans le cytosol, a la capacité de se lier à
la protéine Fe65 et à l’histone acétyltransférase Tip60 pour former un complexe
transcriptionnel actif (Cao & Sudhof, 2001). Ce fragment serait donc impliqué
dans la signalisation intracellulaire et la régulation de gènes.
Récemment, il a aussi été montré que ce domaine C-terminal de l’APP était
clivé par différentes caspases, conduisant à la libération d’un court fragment
cytoplasmique C31 qui aurait la capacité d’induire le phénomène d’apoptose des
neurones (Bertrand et al., 2001). Ces résultats suggèrent qu’il existerait un
nouveau mécanisme contribuant à la mort neuronale associée à la maladie
d’Alzheimer.

47
-D- CATABOLISME DU PEPTIDE β-AMYLOÏDE.
Puisque ce peptide est un métabolite continuellement synthétisé dans le
cerveau, il doit exister des mécanismes de clairance qui empêchent son
agrégation. Jusqu’ici, deux mécanismes distincts ont été proposés.
1. Internalisation cellulaire via un récepteur. Des études antérieures, sur
le(s) mécanisme(s) impliqué(s) dans l’internalisation cellulaire du peptide Aβ par
un récepteur, ont eu un certain écho. Ainsi, des travaux in vitro ont montré que le
peptide Aβ s’associe avec l’APOE (Strittmatter et al., 1993) ou avec l’α-2-
macroglobuline (α2M) pour former un complexe qui est ensuite internalisé par le
récepteur de la lipoprotéine LDL (pour « Low-density lipoprotein »), nommé LRP
(pour « LDL receptor-related protein ») (Fagan et al., 1996; Narita et al., 1997;
Hughes et al., 1998). Il a aussi été montré que le peptide Aβ pourrait être
directement internalisé par le récepteur SR (pour « scavenger receptor »)
(Paresce et al., 1996). Ces résultats concordent avec ceux obtenus par les études
impliquant l’α2M et le LRP dans les formes familiales de la maladie d’Alzheimer
(Kang et al., 1997; Blacker et al., 1998).
2. Dégradation enzymatique. A l’heure actuelle, les recherches se sont
orientées vers l’étude du mécanisme de dégradation du peptide Aβ par
différentes protéases. Plusieurs enzymes, susceptibles de contribuer à ce
processus, ont été proposées :
a)-Insulin-degrading enzyme (IDE). L’IDE (EC 3.4.24.56), appelée aussi
insulysine, est une métalloprotéase à zinc, thiol-dépendante, exprimée dans tous
les tissus des mammifères (Kuo et al., 1993). Cette enzyme de 110 kDa est
principalement localisée dans le cytoplasme (Shii et al., 1985) bien qu’il existe
aussi une forme associée à la membrane (Vekrellis et al., 2000). L’IDE clive un

48
très grand nombre de peptides du côté N-terminal des résidus hydrophobes et
basiques (Authier et al., 1996).
En se basant sur la capacité de l’IDE à dégrader des substrats peptidiques,
ayant la capacité de former des fibrilles amyloïdes (l’insuline, le peptide
natriurétique atrial, l’amyline et le peptide Aβ), il a été proposé que cette enzyme
pourrait jouer un rôle protecteur contre les effets toxiques induits par des
molécules amyloïdogènes (Kurochkin, 1998; 2001). En effet, plusieurs études ont
mis en évidence le rôle de l’IDE dans la dégradation du peptide Aβ et ont montré
que l’IDE, purifiée à partir du milieu de culture de cellules microgliales de souris
BV-2, dégradait les peptides Aβ endogènes et synthétiques (Qiu et al., 1998). In
vitro, l’IDE hydrolyse les peptides Aβ40 et Aβ42 entre les résidus His13-His14,
His14-Gln15 et Phe19-Phe20. Ces peptides subissent aussi des clivages secondaires
au niveau des liaisons Lys28-Gly29, Val18-Phe19 et Phe20-Ala21 (Mukherjee et al.,
2000; Chesneau et al., 2000).
La dégradation du peptide Aβ par l’IDE a aussi été mise en évidence dans les
cellules PC12 différenciées et les neurones corticaux (Vekrellis et al., 2000). Les
résultats, portant sur des souris IDE « knockout », ont montré une baisse
significative de la dégradation du peptide Aβ synthétique et une augmentation de
l’accumulation cérébrale du peptide endogène (Farris et al., 2003). Récemment, il
a été montré que l’insulysine dégradait aussi le domaine intracellulaire AICD qui
est un produit de maturation de l’APP par la γ-secrétase. Cette observation
suggère que l’IDE jouerait donc un rôle dans la régulation de l’activité biologique
de ce fragment (Edbauer et al., 2002).
b)-Neutral endopeptidase (NEP). La NEP (EC 3.4.24.11), appelée aussi
néprilysine, est une glycoprotéine membranaire de 90 à 110 kDa appartenant à la
famille des métalloprotéinases neutres à zinc (Kerr & Kenny, 1974). Cette enzyme
est peu exprimée dans le cerveau à l’exception de la région hippocampique
(Barnes, 1995).

49
La néprilysine a la capacité d’inactiver plusieurs substrats tels que le
glucagon, les enképhalines, la substance P, la neurotensine, l’oxytocine, la
bradykinine, le peptide natriurétique atrial et le peptide Aβ en les clivant du côté
N-terminal des résidus hydrophobes (Matsas et al., 1983 ; Roques et al., 1993).
In vitro, la NEP clive les peptides Aβ40 et Aβ42 aux sites Glu3-Phe4, Gly9-Tyr10,
Phe19-Phe20, Ala30-Ile31 et Gly33-Leu34 (Howell et al., 1995).
Plusieurs études ont démontré le rôle important de la NEP dans la
dégradation du peptide Aβ dans le cerveau. Ainsi, l’injection intracérébrale du
peptides Aβ, en la présence ou en absence de différents inhibiteurs de
protéases, a montré que la NEP serait l’enzyme de dégradation majoritaire du
peptide Aβ42 dans le cerveau de rat (Iwata et al., 2000). Par ailleurs, il a été
observé, dans une souris NEP « knockout », une accumulation du peptide Aβ
endogène dans la région hippocampique et une réduction de la dégradation du
peptide Aβ42 exogène (Iwata et al., 2001). Cependant, le catabolisme du peptide,
dans ces conditions, n’a pas été totalement aboli ; suggérant ainsi que d’autres
enzymes peuvent y participer.
c)-Endothelin-converting enzyme (ECE). C’est une métalloprotéase
membranaire à zinc (EC 3.4.24.71) de la même famille que la NEP qui clive divers
peptides dont la bradykinine et la substance P ainsi que les précurseurs des
endothélines (Turner & Murphy, 1996 ; Johnson et al., 1999). Deux enzymes ont
été clonées, l’ECE-1 et l’ECE-2. Il a été montré que l’ECE-1, exprimée dans les
cellules CHO, dégradait le peptide Aβ dans le milieu extracellulaire (Eckman et
al., 2001). De plus, in vitro, l’ECE-1 clive le peptide Aβ40 aux sites Lys16-Leu17,
Leu17-Val18 et Phe19-Phe20 (Eckman et al., 2001). Récemment, d’autres études ont
montré que la quantité endogène des peptides Aβ40 et Aβ42 augmente dans le
cerveau des souris « knockout » ECE-1/ECE-2 (Eckman et al., 2003).
d)-Matrix metalloprotéinases (MMPs). Ces enzymes sont des
endopeptidases à zinc qui sont pour la plupart secrétées. Elles dégradent les

50
composants de la matrice extracellulaire et jouent un rôle essentiel dans la
morphogenèse et la réparation tissulaire (Docherty et al., 1992). Il a été montré
que la MMP-2 (gelatinase A), localisée principalement dans les cellules
microgliales de la substance blanche (Yamada et al., 1995), et la MMP-9
(gelatinase B), exprimée majoritairement dans les neurones pyramidaux de
l’hippocampe, dégradent le peptide Aβ in vitro (Roher et al., 1994 ; Backstrom et
al., 1996).
e)-Thimet oligopeptidase (TOP). Cette enzyme (EC 3.4.24.15)
appartient à la famille des métallopeptidases à zinc incluant la NEP et
l’angiotensin-converting enzyme (ACE) (Pierotti et al., 1990). Cette protéase clive
des neuropeptides tels que la bradykinine, la neurotensine et la somatostatine
(Dahms & Mentlein, 1992). Elle est aussi impliquée dans le catabolisme du peptide
Aβ, mais de manière indirecte. En effet, dans un milieu conditionné de
neuroblastomes transfectés par un gène anti-sens de la protéase TOP, il a été
observé une augmentation de la quantité du peptide Aβ. Par ailleurs, le
prétraitement de ce milieu, avec des inhibiteurs de protéases à sérine, réduit la
dégradation du peptide Aβ. Ces expériences suggèreraient que la protéase TOP
activerait une protéase à sérine capable de dégrader ensuite le peptide Aβ
(Yamin et al., 1999 ; Abraham et al., 2000).
f)-Angiotensin-converting enzyme (ACE). Cette enzyme (EC 3.4.15.1),
plus connue pour catalyser la conversion de l’angiotensine I en angiotensine II, a
été proposée pour sa capacité à inhiber l’accumulation du peptide Aβ40 en le
clivant au niveau de la liaison Asp7-Ser8 (Hu et al., 2001). Par ailleurs, le fragment
peptidique, généré par cette coupure, est dépourvu de toute toxicité. Cependant,
la signification physiologique de ces résultats doit être réexaminée car les
inhibiteurs de ACE n’ont aucun effet sur le catabolisme du peptide Aβ dans
l’hippocampe de rat (Iwata et al., 2000, 2001).

51
g)-Plasmine. Cette protéase à sérine, abondante dans les radeaux
lipidiques, clive in vitro le peptide Aβ40 (Van Nostrand & Porter, 1999) et Aβ42
(Exley & Korchazhkina, 2001) entre les résidus Arg5 et His6. Par ailleurs, il a été
observé une carence en plasmine dans des tissus cérébraux de patients atteints
de la maladie d’Alzheimer (Ledesma, 2000). Par conséquent, la plasmine
constituerait un facteur protecteur envers la maladie (Periz, 2000).
h)-HtrA1 pour « high temparature requirement » Cette enzyme
appartient à une nouvelle classe de sérines protéases oligomériques caractérisée
par la combinaison d’un domaine N-terminal trypsine-like avec un ou plusieurs
domaines C-terminaux PDZ. Ces derniers régulent l’activité protéolytique de
l’enzyme en bloquant l’accès au site actif. Récemment, des études ont montré que
HtrA1 est fortement exprimée dans le cerveau de patients atteints de la maladie
Alzheimer, alors qu’elle s’y trouve en petites quantités chez les individus sains
(Grau et al., 2005). De plus, il a été démontré que cette protéase est capable de
cliver in vitro les peptides Aβ40 et Aβ42 entre les acides aminés Val12 et Gln15.

52
Objectifs
Les plaques séniles sont des dépôts fibrillaires extracellulaires associés à la
maladie d’Alzheimer et dont le constituant principal est le peptide amyloïde (Aβ).
Comme pour tous les peptides biologiquement actifs, le taux circulant du peptide
Aβ dans le cerveau est fonction de l’équilibre entre les voies de sa biosynthèse à
partir de son précurseur biosynthétique (APP) et celles de sa dégradation
enzymatique. Par conséquent, dans le cas d’une maturation normale du précurseur
APP, un défaut de résorption du peptide Aβ par les cellules, pourrait, avec
d’autres facteurs, contribuer à son dépôt extracellulaire. Par exemple, les
mutations «Dutch» ou «Flemish» du peptide Aβ, responsables d’angiopathie
amyloïde cérébrale, induisent des dépôts de peptides amyloïdes dans les
vaisseaux sanguins qui irriguent le cortex cérébral et les méninges.
Pour pouvoir développer des stratégies thérapeutiques adéquates, il est
donc primordial d’étudier le catabolisme normal du peptide β-amyloïde afin de
caractériser tous les mécanismes impliqués dans ce processus. Pour cela, nous
avons développé une approche expérimentale visant à analyser qualitativement et
quantitativement la capacité de plusieurs lignées cellulaires à résorber les
peptides amyloïdes (sauvage et mutés) de leur milieu de culture en espérant
dégager un schéma général de la clairance du peptide β-amyloïde.

53
MATÉRIELS ET MÉTHODES

54
-A- MATÉRIELS
1. Peptides.
Le peptide β-amyloïde (1-40) humain (Aβ40), ainsi que les peptides
hormonaux tels que la bradykinine, le neuropeptide Y ou la somatostatine-28, ont
été commandés chez Sigma (St Quentin Fallavier, France). Les peptides
amyloïdes mutés ([Gly21]-Aβ40, [Gln22]-Aβ40) et le peptide amyloïde murin
([Gly5,Phe10,Arg13]-Aβ40) proviennent de VWR International (Strasbourg,
France). Le peptide Aβ(1-28) provient de Neosystem (Strasbourg, France) et le
peptide radioactif [125I]-Aβ40 (activité spécifique ~2.103 Ci/mmol) a été
commandé chez Amersham Pharmacia Biotech (Orsay, France). Ces différents
peptides ont été aliquotés aux concentrations voulues puis stockés à –20°C
jusqu’à utilisation.
2. Lignées cellulaires.
Les lignées cellulaires U-373, HFF, COS-7, CHO et K-562 proviennent de
l’ATCC (American Type Cell Culture). Les lignées de neuroblastomes humains SK-
N-BE, SH-SY5Y et CHP-100 sont un don du professeur Gerry Melino (Rome,
Leicester). L’ensemble de ces lignées cellulaires, humaines et non humaines, sont
représentatives du Système Nerveux Central (U-373, CHP-100, SH-SY5Y et SK-
N-BE), du Système Immunitaire (K-562) et des tissus périphériques (HFF)
(annexe 1).
Les différents milieux de culture tels que le RPMI 1640, Dulbecco’s
modified Eagle medium (DMEM) et F-12 ainsi que la solution tampon PBS
Dulbecco’s pH 7.4 (1X), la glutamine 200mM (100X), le sérum de veau fœtal et la
solution de trypsine-EDTA (1X) proviennent de Invitrogen (Cergy, France). La
composition des milieux de culture est indiquée dans l’annexe 2. Les plaques de
culture à 6 puits Nunclon sont commandées chez VWR International.

55
3. Matériel pour le test d’ELISA indirect.
Les anticorps anti-Aβ40 L11 sont des anticorps polyclonaux obtenus par
immunisation de six lapins mâles adultes de la race New-Zealand. Ces lapins ont
été immunisés avec le peptide Aβ40 synthétisé par Rhône Poulenc. L’immunisation
a été faite à raison de 8 injections de 100 µg de peptide chacune, avec la
fréquence d’une injection tous les 15 jours. L’immunisation des lapins a été
réalisée dans les locaux de Rhône Poulenc.
Les plaques de micro titration Dynatech 96 puits et les anticorps de chèvre
anti-IgG de lapin couplés à la peroxydase proviennent de VWR International. Le
Tween®20 a été acheté chez Sigma. Le substrat de la peroxydase (TMB Elisa
peroxydase substrate) provient de chez Interchim (Asnières, France).
4. Autres matériels.
Les inhibiteurs bestatine, E64, aprotinine, pefabloc, leupeptine, pepstatine,
antipaïne, benzamidine, chymostatine et α2-macroglobuline proviennent de
Boehringer Mannheim (Mannheim, Allemagne). Le kit MMP Inhibitor Set-I a été
commandé chez Calbiochem (Darmstadt, Allemagne). Les inhibiteurs de
métalloprotéases tels que 1,10-phenanthroline, N-ethylmaleimide,
phosphoramidon, thiorphan et captopril ainsi que le secrétagogue 8Br-cAMP
proviennent de Sigma. L’acétonitrile provient de Acros Organics (Noisy-le-Grand,
France). L’acide trifluoroacétique (TFA) et la colonne SUPELCO C18 (250mm х
4,6mm ; 5µm) ont été achetés chez Sigma Aldrich.

56
-B- MÉTHODES
1. Entretien des cultures cellulaires.
Après décongélation des différentes ampoules, toutes les cellules sont
centrifugées pour éliminer le milieu de congélation. Un comptage sur lame de
Malassez est effectué en utilisant la méthode d’exclusion par le bleu de Trypan.
Ce comptage permet d’ajuster la suspension cellulaire à une concentration de 2 à
3x105 cellules/mL. Les cellules sont ensuite maintenues en culture à 37°C dans un
milieu adéquat et dans une atmosphère humidifiée contenant 5% de CO2.
La lignée cellulaire K-562 est cultivée dans le milieu RPMI 1640. Les lignées
cellulaires U-373, COS-7, CHO, HFF sont cultivées dans le milieu DMEM alors
que les lignées SH-SY5Y, SK-N-BE et CHP-100 le sont dans le milieu F12:DMEM
(v/v). Pour cette première étape de culture, le milieu est supplémenté avec 20 %
de sérum de veau foetal (SVF) et contient de la glutamine 2 mM.
En fonction de leur temps de dédoublement, une dispersion des cellules
s’effectue avec du milieu de culture supplémenté avec 10 % de SVF et contenant
de la glutamine (2 mM) :
• Les cellules, poussant en suspension, sont ramenées à une concentration allant
de 1 à 2x105 cellules/mL tous les 3 à 4 jours. Le nombre de cellules voulu est
obtenu par amplification du volume de culture.
• Dans le cas des lignées cellulaires poussant en monocouche, après élimination
du milieu et rinçage avec du PBS Dulbelcco’s pH 7,4 pour éliminer le sérum, les
cellules sont décollées par une solution de Trypsine-EDTA pendant 5 min à
37°C. La réaction trypsique est arrêtée par addition de milieu enrichi en SVF.
Après leur comptage, les cellules sont remises en culture. Plusieurs passages
d’amplification sont nécessaires pour permettre la prolifération cellulaire.

57
2. Conditions expérimentales de culture des lignées cellulaires.
Lorsque les cellules, poussant en suspension, sont à confluence (70 à 80% de
leur courbe de croissance), le milieu de culture est éliminé par centrifugation
(200хg, Jouan GR412) puis les cellules sont rincées trois fois avec du tampon PBS
Dulbecco’s pH 7,4 et suspendues dans un milieu de culture sans SVF. Les cellules
sont ensuite réparties dans une plaque de culture à 6 puits à raison de 3x105
cellules/mL/puits.
Dans le cas des cellules adhérentes, celles-ci sont décollées par une solution
de trypsine-EDTA (1X) quand le tapis cellulaire atteint 70 à 80 % de confluence.
Après élimination du surnageant, les cellules sont ensuite suspendues dans un
milieu DMEM/SVF (10%) et réparties dans une plaque de culture à 6 puits. Le
nombre de cellules à déposer par puits est calculé en tenant compte de leur
temps de dédoublement et de leur temps d’adhérence sur la plaque (deux jours).
Ainsi, les cellules atteignent le nombre de 3x105 cellules/mL le jour de
l’expérience. Les cellules sont ensuite lavées trois fois au tampon PBS Dulbecco’s
pH 7,4 et le milieu DMEM sans SVF est rajouté à raison de 1mL/puits.
3. Test ELISA indirect.
3.1. Titrage de l’anticorps anti-Aβ40. La détermination de la dilution de
l’anticorps L11 a été effectuée selon le protocole suivant :
• Plusieurs gammes de concentrations décroissantes d’une solution de Aβ40 (5
µg/mL) sont distribuées sur la plaque (9 points : 5; 2,5; 1,25; 0,625; 0,312;
0,157; 0,078; 0,039; 0,019 µg/mL), à raison de 100 µL par puits. La plaque est
ensuite incubée toute une nuit à 4°C.
• Après lavage avec du tampon PBS/Tween 20 (0,1%), les sites de fixation libres
sont saturés pendant 1 heure à 37°C par addition de 100 µL du tampon PBS
contenant du lait écrémé à 5%. Puis 100 µL d’anticorps L11, diluées dans du

58
tampon PBS/Tween 20/lait 5%, sont déposés à différentes dilutions (du
1/1000ème au 1/500ème) et incubés pendant 1 heure à 37°C.
• Après lavage, 100 µL du deuxième anticorps anti-IgG de lapin marqué à la
peroxydase, sont ajoutés.
• La révélation est effectuée par l'addition de 100 µL du kit TMB Elisa
peroxydase substrate (3,3’,5,5’-tetramethylbenzidine et peroxyde
d’hydrogène). Après 10 min d'incubation à l'obscurité, la lecture de la plaque
est effectuée en mesurant la densité optique à 630 nm (Dynatech MR5000).
3.2. Dosage des peptides Aβ40 dans les surnageants. Après avoir déterminé
la dilution optimale de l’anticorps L11, nous avons procédé au dosage des
surnageants contenant les différents peptides amyloïdes par la méthode ELISA
indirect selon le protocole suivant (Voller et al., 1976):
• Après dépôt de 100µL du surnageant par puits, les plaques sont incubées toute
la nuit à 4°C. Après lavage avec une solution de PBS/Tween 20, les puits sont
saturés par une solution de PBS/lait 5% et incubés pendant 1 heure à 37°C.
• Après lavage des plaques, l’anticorps L11 anti-Aβ(1-40), dilué au 1/800ème, dans
du PBS/Tween 20/ lait 5% est déposé sur les plaques.
• Après incubation (1 heure à 37°C) et lavage, le deuxième anticorps anti-IgG de
lapin, dilué au 1/1000ème, est déposé dans les puits.
• Après incubation (1 heure à 37°C) et lavage, le deuxième anticorps anti-IgG de
lapin dilué au 1/1000ème est déposé dans les puits.
• Après incubation (1 heure à 37°C) et lavage, les plaques sont ensuite révélées
par réaction enzymatique en utilisant le kit TMB Elisa peroxydase substrate,
substrat de la peroxydase fixé au deuxième anticorps.
• La lecture des plaques est ensuite réalisée en mesurant l’absorbance à 630 nm
sur un lecteur de plaques (Dynatech MR5000).

59
4. RP-HPLC.
Les différents échantillons sont soumis à une chromatographie liquide à
haute performance en phase inverse (RP-HPLC). Les temps de rétention des
peptides, sur colonne analytique C18, sont déterminés à partir d’un gradient
obtenu par mélange différentiel de la solution A (H2O contenant 0,05% de TFA)
et de la solution B (acétonitrile contenant 0,03% de TFA) selon les conditions
suivantes :
• une élution isocratique avec 10% de la solution B pendant 30 minutes,
• un gradient de 10 à 20% de la solution B en 5 minutes,
• un gradient de 20 à 30% de la solution B en 20 minutes,
• un gradient de 30 à 40% de la solution B en 10 minutes
• un gradient de 40 à 80% de la solution B en 10 min
Le mélange des deux solutions est pompé dans la colonne à un débit constant
de 0,5 mL/min.
5. ESI-Q-TOF-MS.
Les différents échantillons sont soumis à une chromatographie liquide à
haute performance en phase inverse, couplée à un spectromètre de masse Q-TOF
en mode electrospray (Perkin Elmer). Le gradient et les tampons d’élution sont les
mêmes que ceux utilisés pour l’analyse des échantillons par RP-HPLC. Le mélange
des solutions A et B est pompé dans la colonne C18 à un débit constant de 0,5
mL/min.
Les spectres de masse ESI (ElectroSpray Ionization) présentent un
ensemble de pics correspondant aux ions multichargés de type [M+nH]+ où M
correspond à la masse moléculaire du fragment peptidique analysé, et n au
nombre de charges portées par ce peptide ionisé.

60
6. Dichroïsme Circulaire (DC).
Les spectres de dichroïsme circulaire des peptides β-amyloïdes ont été
réalisés sur un appareil de type Jobin-Yvon Mark VI couplé à un PC. Une cellule en
quartz d’un trajet optique de 0,1 cm a été utilisée. Les mesures dans le lointain
UV (185-260 nm) ont été réalisées dans un tampon phosphate ou un mélange
tampon/TFE et tampon/SDS.
Les spectres sont représentés en unités d’ellipticité moyenne par résidu [θ]R
(deg.cm2.dmol-1) qui est reliée au dichroïsme circulaire ∆ε selon l’équation
suivante : [θ]R=3298x∆ε/N, où N est le nombre de résidu.
Les structures secondaires des peptides ont été déduites de leurs spectres
DC par différentes méthodes d’analyse (Venyaminov et Yang, 1996).
7. Clairance des peptides Aβ40.
a)-En présence de cellules. Après transfert des cellules dans un milieu
dépourvu de sérum, 5-10 µg de peptides amyloïdes non radioactifs (1.2-3 µM) ou
2x104 cpm du peptide radioactif [125I]-Aβ40 (4 pM) sont ajoutés aux milieux de
culture. A différents temps d’incubation (1, 2, 4 et 8h), les surnageants
correspondants (S1, S2, S4 et S8) sont prélevés puis analysés. Pour les peptides
non radioactifs, la quantité du peptide restant dans les surnageants S est
mesurée par test d’ELISA indirect ou à partir des chromatogrammes HPLC. Dans
le cas du peptide [125I]-Aβ40, les surnageants sont tout d’abord soumis à un
fractionnement sur RP-HPLC puis la radioactivité des fractions collectées a été
mesurée à l’aide d’un compteur γ (Kontron MR480).
b)-En présence de surnageants. Après transfert des cellules dans un milieu
dépourvu de sérum, les surnageants (S’1, S’2, S’4 et S’8), correspondant à
différents temps d’incubation (1, 2, 4 et 8h), sont prélevés. Ces surnageants,
auxquels on a ajouté 5-10 µg de peptides amyloïdes non radioactifs (1.2-3 µM) ou
2x104 cpm du peptide radioactif [125I]-Aβ40 (4 pM), on été ensuite incubés

61
pendant 1, 2, 4 et 8h, respectivement. Pour les peptides non radioactifs, la
quantité du peptide restant dans les surnageants S est mesurée par test
d’ELISA indirect ou à partir des chromatogrammes HPLC. Dans le cas du peptide
[125I]-Aβ40, les surnageants sont tout d’abord soumis à un fractionnement sur
RP-HPLC puis la radioactivité des fractions collectées a été mesurée à l’aide d’un
compteur γ (Kontron MR480).
8. Caractérisation des activités enzymatiques.
Pour déterminer les familles d’enzymes impliqués dans la clairance des
peptides amyloïdes, différentes expériences ont été réalisées en la présence
d’inhibiteurs (voir annexe 3) :
• Les cellules, transférées dans un milieu dépourvu de sérum, sont d’abord
incubées avec différents inhibiteurs pendant 5 min. Après ajout des peptides
amyloïdes au milieu de culture, les cellules sont ensuite incubées pendant 8h.
• Pour caractériser les protéases extracellulaires, les cellules sont d’abord
incubées pendant 8h. Les surnageants prélevés sont incubés, pendant 5 min,
avec différents inhibiteurs puis pendant 8h après ajout des peptides.
Pour déterminer la localisation des activités enzymatiques, la clairance des
peptides amyloïdes a été analysée en la présence du secrétagogue 8Br-cAMP. Les
cellules sont incubées avec les peptides, en l’absence ou en la présence
d’inhibiteurs, pendant 7h à 37°C. Après ajout du 8Br-cAMP, les cellules sont
ensuite incubées pendant 1h à 37°C.
Tous les surnageants de chaque lignée cellulaire sont soumis à une
séparation par RP-HPLC. Pour le peptide [125I]-Aβ40, la radioactivité de chacune
des fractions collectées est ensuite mesurée à l’aide d’un compteur γ (Kontron
MR480). Pour les peptides non radioactifs, la quantité du peptide restant dans
les surnageants est mesurée à partir des chromatogrammes HPLC.

62
RÉSULTATS EXPÉRIMENTAUX

63
Le peptide β-amyloïde est une molécule continuellement synthétisée dans les
milieux de culture cellulaire et les fluides biologiques (Seubert et al., 1992;
Haass et al., 1992 ; Shoji et al., 1992; Golde et al., 1993). Si les voies de sa
biosynthèse à partir de son précurseur APP ont été largement étudiées (Koo &
Squazzo et al., 1994), les mécanismes de sa dégradation protéolytique sont
encore mal élucidés. Notre stratégie a donc consisté à tenter de caractériser ces
mécanismes en suivant le devenir du peptide synthétique Aβ40 en présence de
lignées cellulaires humaines et non humaines. Celles-ci sont représentatives du
système nerveux central, du système immunitaire et des tissus périphériques.
Nos expériences ont été réalisées dans un milieu dépourvu de SVF car la
présence des protéines du sérum pourrait interférer avec la détection du peptide
β-amyloïde (technique d’ELISA indirect) ou avec la purification des protéines ou
des peptides recherchés (technique d’HPLC).
CLAIRANCE DU PEPTIDE β-AMYLOÏDE NATIF PAR LES CELLULES.
1. Mise en évidence de la disparition du peptide Aβ40 du milieu de
culture des cellules. Afin d’analyser quantitativement la capacité des cellules à
éliminer le peptide β-amyloïde de leur milieu de culture, le peptide Aβ40 a été
incubé en la présence de plusieurs et différentes lignées cellulaires. La
concentration du peptide Aβ40 (1,2 µM) a été choisie de manière à être
largement inférieure à la concentration critique (100 µM) permettant l’initiation
de la fibrillogenèse du peptide Aβ (Lomakin et al., 1996). De plus, l’absence de
polymérisation du peptide a été confirmée par chromatographie d’exclusion (gel
Sephadex G-50).
Après différents temps d’incubation (1, 2, 4 et 8 heures), les surnageants
de chaque lignée cellulaire ont été prélevés, puis la quantité du peptide restant a
été dosée par la technique d’ELISA indirect. Les résultats obtenus sont indiqués
dans la figure 13. On observe, en fonction du temps, une diminution significative

64
0
20
40
60
80
100
0 2 4 6 8 10temps d'incubation (h)
pept
ide
rest
ant (
%)
CHOHFFK-562COS-7
B
0
20
40
60
80
100
0 2 4 6 8 10temps d'incubation (h)
pept
ide
rest
ant (
%)
SH-SY5YSK-N-BEU-373CHP-100
A
Figure 13: Cinétique de disparition du peptide Aβ40 du milieu de culture des cellules neuronales (A) et non neuronales (B). A différents temps d’incubation, la quantité du peptide β-amyloïde restant dans les surnageant (concentration initiale de 5 µg/ml) est mesurée par test ELISA indirect.
0
20
40
60
80
100
0 2 4 6 8 10temps d'incubation (h)
pept
ide
rest
ant (
%)
CHOHFFK-562COS-7
B
0
20
40
60
80
100
0 2 4 6 8 10temps d'incubation (h)
pept
ide
rest
ant (
%)
SH-SY5YSK-N-BEU-373CHP-100
A
0
20
40
60
80
100
0 2 4 6 8 10temps d'incubation (h)
pept
ide
rest
ant (
%)
CHOHFFK-562COS-7
B0
20
40
60
80
100
0 2 4 6 8 10temps d'incubation (h)
pept
ide
rest
ant (
%)
CHOHFFK-562COS-7
B
0
20
40
60
80
100
0 2 4 6 8 10temps d'incubation (h)
pept
ide
rest
ant (
%)
SH-SY5YSK-N-BEU-373CHP-100
A0
20
40
60
80
100
0 2 4 6 8 10temps d'incubation (h)
pept
ide
rest
ant (
%)
SH-SY5YSK-N-BEU-373CHP-100
A
Figure 13: Cinétique de disparition du peptide Aβ40 du milieu de culture des cellules neuronales (A) et non neuronales (B). A différents temps d’incubation, la quantité du peptide β-amyloïde restant dans les surnageant (concentration initiale de 5 µg/ml) est mesurée par test ELISA indirect.

65
de la quantité du peptide Aβ40 du milieu de culture des cellules neuronales
(figure 13A) ou non neuronales (figure 13B). Cependant, on remarque que la
cinétique de disparition du peptide varie selon les lignées cellulaires. Par exemple,
après 4 heures d’incubation, 70% du peptide est éliminé du surnageant des
neuroblastomes humains CHP-100 (figure 13A). En revanche, la quantité du
peptide Aβ40 ne diminue que de 30% en présence des cellules humaines non
neuronales HFF (figure 13B). Pour le même temps d’incubation, les autres lignées
cellulaires sont capables d’éliminer 40 à 50 % du peptide Aβ40 présent dans les
surnageants.
L’ensemble de ces résultats suggère l’existence de mécanismes responsables de la
disparition du peptide Aβ40 du surnageant des cellules; à savoir une dégradation
enzymatique ou une internalisation du peptide par les cellules ou la combinaison des deux.
Comme bon nombre de molécules bioactives, le peptide Aβ peut-être
dégradé par les protéases secrétées par les cellules. Afin de tester cette
hypothèse, nous avons analysé la dégradation du peptide Aβ40 par les
surnageants cellulaires. Pour cela, des surnageants de cellules, incubées pendant
1h (S1), 2h (S2), 4h (S4) et 8h (S8), ont été préparés. Le peptide β-amyloïde a
été ensuite incubé à 37°C en présence de ces surnageants pendant 1, 2, 4 et 8
heures respectivement. Comme il est indiqué dans la figure 14, on n’observe
aucune disparition significative du peptide Aβ40 du milieu extracellulaire. En
effet, quelque soit le temps d’incubation ou le type de surnageant cellulaire testé,
environ 95 % du peptide β-amyloïde a été détecté dans le milieu par ELISA
indirect. Des résultats similaires ont été obtenus pour des temps d’incubation
plus longs (12h ou 24h) ou pour des surnageants cellulaires plus concentrés.
Ces observations démontrent que la disparition du peptide Aβ40 ne peux s’expliquer
par une dégradation extracellulaire .

66
Figure 14 : Cinétique de dégradation du peptide β-amyloïde dans les surnageants de cellules neuronales (A) et non neuronales (B). Les surnageants de chaque type de cellules sont prélevés à 1, 2, 4, 8 heures de culture. 5 µg/ml du peptide Aβ40 est incubé avec ces surnageants pendant 1, 2, 4 et 8 heures à 37°C. La quantité du peptide β-amyloïde restant est mesurée par test ELISA indirect.
85
90
95
100
105
0 2 4 6 8 10Temps d’ incubation time (h)
Pept
ide
rest
ant (
%)
SH-SY5Y SK-N-BE
U-373CHP-100
A
0 2 4 6 8 10Temps d’ incubation time (h)
Pept
ide
rest
ant (
%)
B85
90
95
100
105
K-562COS-7
CHO HFF
85
90
95
100
105
0 2 4 6 8 10Temps d’ incubation time (h)
Pept
ide
rest
ant (
%)
SH-SY5Y SK-N-BE
U-373CHP-100
A85
90
95
100
105
0 2 4 6 8 10Temps d’ incubation time (h)
Pept
ide
rest
ant (
%)
SH-SY5Y SK-N-BE
U-373CHP-100
A85
90
95
100
105
85
90
95
100
105
0 2 4 6 8 100 2 4 6 8 10Temps d’ incubation time (h)
Pept
ide
rest
ant (
%)
SH-SY5Y SK-N-BESH-SY5Y SK-N-BE
U-373CHP-100U-373CHP-100
A
0 2 4 6 8 10Temps d’ incubation time (h)
Pept
ide
rest
ant (
%)
B85
90
95
100
105
K-562COS-7
CHO HFF
0 2 4 6 8 10Temps d’ incubation time (h)
Pept
ide
rest
ant (
%)
B85
90
95
100
105
K-562COS-7
CHO HFF
0 2 4 6 8 100 2 4 6 8 10Temps d’ incubation time (h)
Pept
ide
rest
ant (
%)
B85
90
95
100
105
85
90
95
100
105
K-562COS-7K-562COS-7
CHO HFFCHO HFF

67
2. Mécanisme de la disparition du peptide Aβ40 du milieu de culture. Les
résultats précédents démontrent qu’il existerait un ou plusieurs processus
cellulaire(s) impliqué(s) dans la disparition du peptide Aβ40. Afin de caractériser
ces mécanismes, nous avons utilisé le peptide radioactif [I125]-Aβ40 comme
substrat.
Après incubation du peptide amyloïde avec les différentes lignées cellulaires
pendant 8 heures, les surnageants, prélevés, ont été ensuite soumis à un
fractionnement par RP-HPLC (figure 15). On observe, pour toutes les cellules, une
diminution de la quantité du peptide [I125]-Aβ40 accompagnée par l’apparition
concomittante de deux pics radioactifs nommés P1 et P2. Si ces observations
suggèrent qu’un même processus serait impliqué dans la dégradation protéolytique
du peptide Aβ, les lignées cellulaires se différencient entre elles par le profil des
diagrammes RP-HPLC qui révèlent des différences au niveau des proportions des
pics P1 et P2 (figure 15). Ainsi, pour les cellules neuronales (excepté les cellules
CHP-100), l’aire du pic P1 est supérieure à celle du pic P2. Par contre, toutes les
cellules non neuronalles sont caractérisées par des pics radioactifs ayant des
aires équivalentes.
Le comptage cumulé, de toutes les fractions radioactives récupérées (figure
15), montre qu’il y a conservation du nombre total de cpm (2x103 cpm). Ce
résultat, en accord avec l’absence de radioactivité dans les cellules, démontre
que la disparition du peptide Aβ ne peut pas s’expliquer par une dégradation du
peptide après son internalisation intracellulaire.
Les surnageants de ces cellules ont été aussi testés pour leur éventuelle
capacité à dégrader le peptide Aβ. Les données de la figure 15 montrent que le
peptide [I125]-Aβ40 reste stable pour une période d’incubation de 8 heures, en
accord avec les données obtenues par ELISA indirect (figure 13). Des résultats
similaires ont été obtenus pour des temps d’incubation plus longs (12h ou 24h) ou
pour des surnageants cellulaires plus concentrés ou contenant du SVF.

68
Figure 15 : Disparition du peptide [I
125]-Aβ40 en présence des cellules ou des surnageants cellulaires. Le peptide [I
125]-Aβ40 (2.10
3 cpm)
est incubé avec les cellules ou les surnageants pendant 8 heures. Chaque surnageant est ensuite soumis à une séparation sur colonne C18 par
HPLC puis la radioactivité des fractions collectées est m
esurée par un compteur γ.
CelluleSurnageant
SH-SY5Y
Aβ40
P1
P2
Tem
ps de rétention (min)
CelluleSurnageant
U-373
Aβ40
P1
P2
Temps de rétention (m
in)
CelluleSurnageant
SK-N-BE
Aβ40
P1
P2
c p mCelluleSurnageant
CHP-100
Aβ40
P1P2
c p mCelluleSurnageant
SH-SY5Y
Aβ40
P1
P2
CelluleSurnageant
SH-SY5Y
Aβ40
P1
P2
Tem
ps de rétention (min)
CelluleSurnageant
U-373
Aβ40
P1
P2
Tem
ps de rétention (min)
CelluleSurnageant
U-373
Aβ40
P1
P2
Temps de rétention (m
in)
CelluleSurnageant
SK-N-BE
Aβ40
P1
P2
c p m
Temps de rétention (m
in)
CelluleSurnageant
SK-N-BE
Aβ40
P1
P2
c p mCelluleSurnageant
CHP-100
Aβ40
P1P2
c p mCelluleSurnageant
CHP-100
Aβ40
P1P2
c p mCH
O
c p m
CelluleSurnageant
Aβ40
P1P2
COS-7
CelluleSurnageant
Aβ40
P1P2
Temps de rétention (m
in) K-562CelluleSurnageant
Aβ40
P1P2
Temps de rétention (m
in)
HFF
c p m
Aβ40
P1P2
CelluleSurnageant
CHO
c p m
CelluleSurnageant
Aβ40
P1P2
CHO
c p m
CelluleSurnageant
CelluleSurnageant
Aβ40
P1P2
COS-7
CelluleSurnageant
Aβ40
P1P2
COS-7
CelluleSurnageantCelluleSurnageant
Aβ40
P1P2
Temps de rétention (m
in) K-562CelluleSurnageant
Aβ40
P1P2
Temps de rétention (m
in) K-562CelluleSurnageantCelluleSurnageant
Aβ40
P1P2
Temps de rétention (m
in)
HFF
c p m
Aβ40
P1P2
CelluleSurnageant
Temps de rétention (m
in)
HFF
c p m
Aβ40
P1P2
CelluleSurnageant
CelluleSurnageant
CELLULES N
EURO
NALES
CELLU
LES NON N
EURO
NALES

69
L’nesemble de ces expériences permettent de déduire que la disparition du peptide β-
amyloïde résulterait essentiellement de mécanismes protéolytiques prenant place au niveau de
la surface des cellules.
3. Caractérisation des activités enzymatiques impliquées dans la
dégradation du peptide Aβ. En se basant sur les résultats obtenus
précédemment, nous avons testé l’effet de plusieurs inhibiteurs de protéases
afin d’identifier les types d’activités enzymatiques impliquées dans le mécanisme
de dégradation du peptide Aβ en la présence de cellules. En utilisant des
concentrations permettant une inhibition maximale des protéases connues,
différents inhibiteurs (annexe 3) ont été incubés avec chaque lignée cellulaire
pendant 8 heures à 37°C en la présence du peptide [I125]-Aβ40. Les différents
surnageants ont été ensuite soumis à une séparation par RP-HPLC (figure 16).
L’analyse des données de la figure 17 montre que les inhibiteurs de sérine,
de cystéine, d’aspartate protéases ou d’aminopeptidases (pool I1) n’ont aucun
effet sur le taux de clivage du peptide [I125]-Aβ40. Par contre,
l’orthophénanthroline (1,10-phénanthroline), un chélateur de métaux, inhibe à 85-
95% la dégradation du peptide β-amyloïde en présence des cellules. Cette
inhibition est accompagnée par une diminution totale de la radioactivité des pics
P1 et P2 (figure 16) en faveur de l’augmentation de celle correspondant au pic du
peptide Aβ40.
Par ailleurs, on peut remarquer que les diagrammes RP-HPLC de la
dégradation du peptide [I125]-Aβ40, en la présence de Pefabloc®, ont des profils
différents. En effet, cet inhibiteur de protéases à sérine change le taux de
production des pics radioactifs P1 et P2 au profit du pic P1 (figure 18) sans
produire aucun effet significatif sur le taux de clivage du peptide [I125]-Aβ40
(figure 17).

70
CELLULES N
EURO
NALES
CELLU
LES NON N
EURO
NALES
Figure 16 : Inhibition de la dégradation du peptide [I125]-A
β40. Le peptide [I125]-A
β40 (2.103 cpm
) est incubé pendant 8 heures en présence des inhibiteurs du pool I1, de l’ortho-phenanthroline (O
P) ou du Pefabloc (Pf). Les différents surnageants provenant des cellules neuronales et non neuronales sont soum
is à une séparation sur colonne C18 par RP-HPLC puis la radioactivité des fractions collectées est m
esurée par un compteur γ.
CHP-100
Aβ40
Cellule
Cellule+I1
Cellule+OP
Cellule+Pf
c p m
P1
P2
SH-SY5Y
Aβ40
Cellule
Cellule+I1
Cellule+OP
Cellule+Pf
P1
P2
SK-N-BE
Aβ40
Cellule
Cellule+I1
Cellule+OP
Cellule+PfTemps de rétention (m
in)
c p m
P1
P2
U-373
Aβ40
Cellule
Cellule+I1
Cellule+OP
Cellule+Pf
Temps de rétention (m
in)
P1
P2
CHP-100
Aβ40
Cellule
Cellule+I1
Cellule+OP
Cellule+Pf
c p m
P1
P2
CHP-100
Aβ40
Cellule
Cellule+I1
Cellule+OP
Cellule+Pf
Cellule
Cellule+I1
Cellule+OP
Cellule+Pf
c p m
P1
P2
SH-SY5Y
Aβ40
Cellule
Cellule+I1
Cellule+OP
Cellule+Pf
P1
P2
SH-SY5Y
Aβ40
Cellule
Cellule+I1
Cellule+OP
Cellule+Pf
Cellule
Cellule+I1
Cellule+OP
Cellule+Pf
P1
P2
SK-N-BE
Aβ40
Cellule
Cellule+I1
Cellule+OP
Cellule+PfTemps de rétention (m
in)
c p m
P1
P2
SK-N-BE
Aβ40
Cellule
Cellule+I1
Cellule+OP
Cellule+Pf
Cellule
Cellule+I1
Cellule+OP
Cellule+PfTemps de rétention (m
in)
c p m
P1
P2
U-373
Aβ40
Cellule
Cellule+I1
Cellule+OP
Cellule+Pf
Temps de rétention (m
in)
P1
P2
U-373
Aβ40
Cellule
Cellule+I1
Cellule+OP
Cellule+Pf
Cellule
Cellule+I1
Cellule+OP
Cellule+Pf
Temps de rétention (m
in)
P1
P2
c p m
Aβ40
CHO
Cellule
Cellule+I1
Cellule+OP
Cellule+Pf
P1
P2
Aβ40 CO
S-7
Cellule
Cellule+I1
Cellule+OP
Cellule+Pf
P1P2
Retentiontim
e(m
n)c pm
Aβ40
HFF
Cellule
Cellule+I1
Cellule+OP
Cellule+PfP1
P2
Retentiontim
e(m
n) Aβ40 K-562
CelluleCellule+I1Cellule+O
PCellule+Pf
P1P2
c p m
Aβ40
CHO
Cellule
Cellule+I1
Cellule+OP
Cellule+Pf
P1
P2
c p m
Aβ40
CHO
Cellule
Cellule+I1
Cellule+OP
Cellule+Pf
Cellule
Cellule+I1
Cellule+OP
Cellule+Pf
P1
P2
Aβ40 CO
S-7
Cellule
Cellule+I1
Cellule+OP
Cellule+Pf
P1P2
Aβ40 CO
S-7
Cellule
Cellule+I1
Cellule+OP
Cellule+Pf
Cellule
Cellule+I1
Cellule+OP
Cellule+Pf
P1P2
Retentiontim
e(m
n)c pm
Aβ40
HFF
Cellule
Cellule+I1
Cellule+OP
Cellule+PfP1
P2
Retentiontim
e(m
n)c pm
Aβ40
HFF
Cellule
Cellule+I1
Cellule+OP
Cellule+Pf
Cellule
Cellule+I1
Cellule+OP
Cellule+PfP1
P2
Retentiontim
e(m
n) Aβ40 K-562
CelluleCellule+I1Cellule+O
PCellule+Pf
P1P2
Retentiontim
e(m
n) Aβ40 K-562
CelluleCellule+I1Cellule+O
PCellule+Pf
CelluleCellule+I1Cellule+O
PCellule+Pf
P1P2

71
Figure 17 : Effet du Pefabloc (Pf), de l’ortho-phenanthroline (OP) et du pool d’inhibiteurs I1 sur la dégradation du peptide [I
125]-Aβ40. Les quantités du peptide restant (représentés en pourcentage) ont été estim
ées à partir des diagramm
es HPLC de la figure 16, par
mesure de l’aire du pic attribué à l’espèce peptidique restante en l’absence ou en en présence de divers inhibiteurs présum
és.
125[I]-Aβ restant (%)
CHO
CHP-100
SH-SY5Y
SK-N-BE
U-373
COS-7
HFF
K-562
100806040200
Cellules + I1 +Pf+O
P125[I]-Aβ restant (%)
CHO
CHP-100
SH-SY5Y
SK-N-BE
U-373
COS-7
HFF
K-562CH
OCH
P-100SH
-SY5YSK-N
-BEU-373
COS-7
HFF
K-562CH
OCH
P-100SH
-SY5YSK-N
-BEU-373
COS-7
HFF
K-562
100806040200
100806040200
100806040200
Cellules + I1 +Pf+O
PCellules + I1 +Pf
+OP
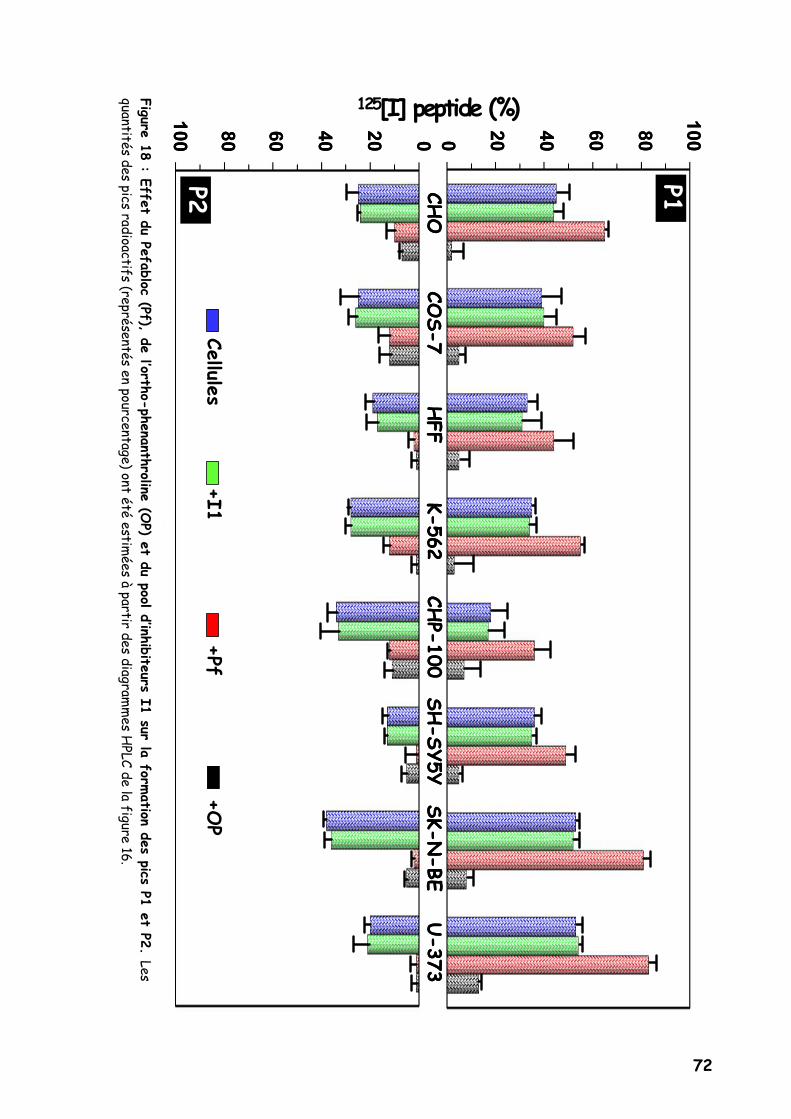
72
Figure 18 : Effet du Pefabloc (Pf), de l’ortho-phenanthroline (OP) et du pool d’inhibiteurs I1 sur la form
ation des pics P1 et P2. Les quantités des pics radioactifs (représentés en pourcentage) ont été estim
ées à partir des diagramm
es HPLC de la figure 16.
CHO
CHP-100
SH-SY5Y
SK-N-BE
U-373
COS-7
HFF
K-562
100806040200080 60 40 20
100
125[I] peptide (%)P1P2
Cellules +I1 +Pf+O
P
CHO
CHP-100
SH-SY5Y
SK-N-BE
U-373
COS-7
HFF
K-562CH
OCH
P-100SH
-SY5YSK-N
-BEU-373
COS-7
HFF
K-562CH
OCH
P-100SH
-SY5YSK-N
-BEU-373
COS-7
HFF
K-562
100806040200
100806040200080 60 40 20
100 080 60 40 20
100
125[I] peptide (%)P1P2
Cellules +I1 +Pf+O
P

73
Ces résultats démontrent que le mécanisme de dégradation du peptide β-amyloïde, en la
présence de cellules neuronales et non neuronales, implique majoritairement deux activités
endoprotéasiques :
• (i)-une activité enzymatique de type métalloprotéase qui cliverait le peptide β-amyloïde
pour libérer le pic P1 et,
• (ii)-une activité enzymatique de type protéase à sérine qui dégrade le pic P1 pour
générer le pic P2.
3.1. Caractérisation de l’activité métalloprotéase. Plusieurs études
portant sur la dégradation du peptide β-amyloïde (Backstrom et al., 1996 ;
Kurochkin, 2001; Song et al., 2001; Iwata et al., 2001; Eckman et al., 2003), ont
montré que celui-ci pouvait être clivé par des métalloprotéases connues telles que
la néprilysine (NEP), l’angiotensin-converting enzyme (ACE), l’endothelin-
converting enzyme (ECE), l’insulin-degrading enzyme (IDE) et certaines
métalloprotéases de matrices (MMP). Des inhibiteurs, connus pour inactiver
spécifiquement ces enzymes, ont été testés pour leur capacité à inhibiter
l’activité enzymatique mise en evidence précédement. Ainsi, la NEP est inhibée
par le thiorphan et le phosphoramidon (Roques et al., 1993) alors que l’ECE-1 l’est
spécifiquement par le phosphoramidon (Rawlings & Barrett, 1995). Le captopril
abolit l’activité de l’ACE (Wei et al., 1992) et les MMP-1, -2, -3, 6 et -9 sont
inhibés par des composés non peptidiques. L’IDE ne possède pas d’inhibiteur
spécifique mais son activité enzymatique est sensible au réactif N-
éthylmaléimide (NEM) connu pour sa capacité à bloquer les groupes –SH (Becker
& Roth, 1995). Ces différents inhibiteurs ont alors été incubés avec le peptide
[I125]-Aβ40 en la présence de toutes les cellules et leurs surnageants récupérés
ont été soumis à une séparation sur RP-HPLC.
Les résultats de la figure 19 montrent que le thiorphan, le phosphoramidon
et le captopril (pool I1) ou les inhibiteurs des MMP (pool I2) n’ont aucun effet
notable sur la dégradation du peptide β-amyloïde. Par contre, on observe une

74
Figure 19 : Effet des pools d’inhibiteurs I1, I2, I3, I4 et du NEM
sur la dégradation du peptide [I125]-A
β40. Les quantités du peptide restant (représentés en pourcentage) ont été estim
ées à partir des diagramm
es HPLC (figure non fournie).
100806040200
120125[I]-Aβ restant (%)
CHO
CHP-100
SH-SY5Y
SK-N-BE
U-373
COS-7
HFF
K-562
Cellules + I2 +I3 +NEM
100806040200
120
100806040200
120125[I]-Aβ restant (%)
CHO
CHP-100
SH-SY5Y
SK-N-BE
U-373
COS-7
HFF
K-562CH
OCH
P-100SH
-SY5YSK-N
-BEU-373
COS-7
HFF
K-562CH
OCH
P-100SH
-SY5YSK-N
-BEU-373
COS-7
HFF
K-562
Cellules + I2 +I3 +NEM
Cellules + I2 +I3 +NEM

75
inhibition de 70-95% (selon les cellules) de l’activité métalloprotéase par le
réactif NEM qui se traduit par la disparition totale des pics radioactifs P1 et P2.
On peut donc en déduire que les protéases NEP, ECE-1, ACE et MMPs ne seraient
pas impliquées dans la dégradation du peptide β-amyloïde par les cellules étudiées. En
revanche, l’activité enzymatique, mise en évidence dans cette étude, est caractérisée par un
profil d’inhibition proche de celui de l’ IDE qui est une métalloprotéase à thiol.
3.2. Caractérisation de l’activité protéase à sérine. La clairance du
peptide [I125]-Aβ40 est accompagnée par l’apparition de deux pics radioactifs
dont les proportions sont variables d’une cellule à l’autre (figure 15). Parmi les
différents inhibiteurs de protéases testés précédemment, les profils des
diagrammes RP-HPLC de la figure 17 révèlent que seul le Pefabloc® a la capacité
d’inhiber la dégradation du pic P1. En effet, les données de la figure 18 montrent
qu’en présence de cet inhibiteur de serpines, l’aire du pic P2 diminue au profit
d’une augmentation de celle du pic P1.
Afin de mieux caractériser cette activité enzymatique, d’autres inhibiteurs
de sérine protéases tels que l’antipaïne, le benzamidine, la chymostatine ou l’α2-
macroglobuline, ont été testés. Après incubation du peptide [I125]-Aβ40 avec les
cellules, en la présence de chacun de ces inhibiteurs pendant 8 heures, les
différents surnageants prélevés ont été soumis à une séparation sur RP-HPLC.
Les résultats obtenus ont révélé que tous ces inhibiteurs (incubés seuls ou
ensemble) n’ont aucun effet notable sur le taux de clivage du pic P1.
Par conséquent, on peut conclure que les protéases telles que la papaïne, la trypsine, la
chymotrypsine, l’élastase, la plasmine ou la thermolysine ne sont pas ou peu impliquées dans
la conversion du pic P1 en pic P2.
4. Localisation cellulaire des activités enzymatiques. Bien que la
métalloprotéase, responsable du clivage du peptide Aβ40, semble être
principalement membranaire, nous n’excluons pas l’existence d’une forme

76
secrétée de cette protéase. La localisation cellulaire de l’activité sérine protéase
reste plus délicate car elle a comme substrat le pic P1 qui est libéré dans les
surnageants après clivage du Aβ40.
Pour préciser ces différents points, nous avons utilisé le secrétagogue 8-
Bromo-cyclo-AMP (8Br-cAMP), un analogue stable de l’AMPc, qui a la capacité
d’activer la voie de sécrétion régulée des cellules pour libérer le contenu des
vésicules de sécrétion. De plus, il est à noter que certaines des cellules étudiées
(U-373, HFF, COS-7, CHO et K-562) sont dépourvues de la voie de sécrétion
régulée. Par conséquent, l’augmentation de la sécrétion d’une protéine
s’expliquerait par une stimulation de la biosynthèse protéique due au 8Br-cAMP.
En effet, l’AMPc peut induire l’activation de promoteurs de certains gènes et
donc l’augmentation de la synthèse des protéines à partir de leurs ARN
messagers (Nikodemova et al., 2003).
Dans une première expérience, les différentes cellules ont été tout d’abord
incubées seules pendant 7 heures puis le 8Br-cAMP (concentration finale de 5
mM) a été ajouté aux milieux. Après 1 heure d’incubation, temps suffisant pour
permettre la libération du contenu des vésicules, les surnageants prélevés ont
été ensuite incubés avec le peptide [I125]-Aβ40 pendant 8 heures puis soumis à
une séparation par RP-HPLC. Les données obtenues ont montré que le peptide β-
amyloïde n’est pas dégradé dans le milieu extracellulaire. On obtient des
résultats similaires pour des surnageants plus concentrés ou pour une incubation
de 24 heures.
Par conséquent, nous pouvons déduire que l’enzyme, responsable de l’activité
métalloprotéase, n’est pas sécrétée par les cellules et/ou le peptide amyloïde natif est résistant
à la protéolyse par les enzymes secrétées.
Dans une deuxième expérience, les cellules ont été incubées avec le peptide
[I125]-Aβ40 pendant 7 heures puis durant 1 heure avec le 8Br-cAMP. Les
surnageants prélevés ont été ensuite soumis à une séparation par RP-HPLC. Les

77
données de la figure 20 montrent une forte augmentation de l’aire du pic P2 au
détriment de celle du pic P1. La même expérience, réalisée en la présence du
Pefabloc® (ajouté en même temps que le secrétagogue), révèle l’effet inverse
(figure 20) : la dégradation du pic P1 est totalement inhibée. Cette conversion
entre les pics P1 et P2 s’expliquerait donc par une augmentation de la sécrétion
de l’activité enzymatique de type protéase à sérine dans le milieu extracellulaire
après stimulation par le 8Br-cAMP. La présence de cette activité enzymatique a
été confirmée par une autre expérience dans laquelle les surnageants, provenant
de l’incubation du peptide [I125]-Aβ40 avec les cellules pendant 8 heures, ont été
ensuite incubés à 37°C pendant 16 heures, en l’absence ou en la présence du
Pefabloc®. Les données de la figure 21 montrent clairement que, durant cette
longue incubation, l’aire du pic radioactif P1 diminue fortement au profit de celle
du pic P2. Inversement, la présence de l’inhibiteur Pefabloc® inhibe fortement la
dégradation du pic P1 qui se traduit par une diminution de l’aire du pic P2 au
profit de celle du pic P1.
L’ensemble de ces résultats confirme, d’une part, que l’activité de type métalloprotéase,
impliquée dans la dégradation du peptide β-amyloïde, est localisée au niveau des membrane
des cellules et démontre, d’autre part, que l’activité de type sérine protéase, impliquée dans la
dégradation du pic P1, est secrétée dans le milieu extracellulaire.
5. Détermination des sites de clivage du peptide β-amyloïde natif. Pour
déterminer les sites de coupure du peptide Aβ40 par les activités enzymatiques
mises en évidence, nous avons soumis les échantillons à une séparation par
chromatographie liquide à haute pression en phase inverse couplée à un
spectromètre de masse Q-TOF en mode electrospray.

78
P1
P2
100806040200080 60 40 20
100
125[I] peptide (%)
CHO
CHP-100
SH-SY5Y
SK-N-BE
U-373
COS-7
HFF
K-562
Cellules Cellules+8Br Cellules+8Br+Pf
P1
P2
100806040200
100806040200080 60 40 20
100 080 60 40 20
100
125[I] peptide (%)
CHO
CHP-100
SH-SY5Y
SK-N-BE
U-373
COS-7
HFF
K-562CH
OCH
P-100SH
-SY5YSK-N
-BEU-373
COS-7
HFF
K-562CH
OCH
P-100SH
-SY5YSK-N
-BEU-373
COS-7
HFF
K-562
Cellules Cellules+8Br Cellules+8Br+Pf
Figure 20 : Mise en évidence de la sécrétion extracellulaire de l’activité sérine-protéase en présence de 8-Br-cA
MP. Les quantités des
pics radioactifs P1 et P2 (représentés en pourcentage) ont été estimées à partir des diagram
mes H
PLC (figure non fournie)

79
Figure 21 : Mise en évidence de la localisation extracellulaire de l’activité sérine-protéase. Les quantités des pics radioactifs
P1 et P2 (représentés en pourcentages) ont été estimées à partir des diagram
mes H
PLC (figure non fournie).
P1CHO
CHP-100
SH-SY5Y
SK-N-BE
U-373
COS-7
HFF
K-562
P2
100806040200080 60 40 20
100
125[I] peptide (%)
Cellules Surnageant 16h Surnageant+Pf16h
P1CHO
CHP-100
SH-SY5Y
SK-N-BE
U-373
COS-7
HFF
K-562CH
OCH
P-100SH
-SY5YSK-N
-BEU-373
COS-7
HFF
K-562
P2
100806040200
100806040200080 60 40 20
100 080 60 40 20
100
125[I] peptide (%)
Cellules Surnageant 16h Surnageant+Pf16h

80
Au vu des résultats précédents, nous avons utilisé les cellules CHO comme
modèle pour identifier les fragments amyloïdes générés après 8 heures
d’incubation de ces cellules avec le peptide Aβ40 en l’absence ou en la présentes
des inhibiteurs identifiés précédemment.
La figure 22 représente les chromatogrammes d’ionisation à partir desquels
ont été déduites les séquences des fragments amyloïdes générés en l’absence ou
en la présence d’inhibiteurs. L’analyse des résultats du tableau 1 révèle la
présence de 14 fragments peptidiques qui sont complémentaires deux à deux.
Cette observation suggère que le peptide β-amyloïde a subis sept clivages
majeurs aux sites suivants: Lys28–Gly29, Asp23–Val24, Phe20–Ala21, Phe19–Phe20,
Val18–Phe19, His14–Gln15 and His13–His14. Sur la figure 22A sont indiqués les
peptides, tronqués en COOH-terminal, et qui sont générés par la protéolyse du
peptide Aβ40 à ces sites. Sachant que tous ces fragments possèdent l’unique
résidu Tyr en position 10, la comparaison des figures 15 et 22A, en se basant sur
les temps de rétention des peptides, a permis de positionner correctement les
différents fragments amyloïdes (figure 23). Sur la base de cette comparaison,
on peut en déduire les conclusions suivantes:
(i)-la contribution du clivage, générant le fragment Aβ(1-13) et son
complémentaire, est insignifiante dans le clivage du peptide Aβ40,
(ii)-le pic P1 contient les fragments peptidiques Aβ(1-18), Aβ(1-19) et Aβ(1-28)
alors que les fragments Aβ(2-14), Aβ(1-20) et Aβ(4-23) sont les composants
du pic P2.
Afin de valider ces résultats, nous avons analysé les fragments amyloïdes
générés après incubation des cellules CHO avec le peptide β-amyloïde en la
présence du Pefabloc® (figure 22B) et des inhibiteurs 1,10-phénanthroline ou
NEM (figure 22C).

81
0.0
1.0
2.0
3.0
0.0
1.0
2.0
3.0
0 10 20 300 10 20 30
Inte
nsité
(104
cps)
Temps d’élution (min)
0.0
0.5
1.0
1.5
0.0
0.5
1.0
1.5
0 10 20 300 10 20 30
Temps d’élution (min)
0 10 20 300 10 20 300.0
1.0
2.0
3.0
0.0
1.0
2.0
3.0
Temps d’élution (min)
A
B
C
Inte
nsité
(104
cps)
Inte
nsité
(104
cps)
2-14
1-19
1-28
1-18
1-13
1-34
1-40
1-20
4-23
2-14
1-19
1-28
1-18
1-13
1-34
1-40
1-20
4-23
1-20
4-23
1-20
4-23
1-40
7-40
4-20 7-
20
1-20
4-23
1-40
7-40
4-20 7-
20
1-20
4-23
1-20
4-23
1-20
4-23
1-40
1-18 1-
28
1-19
5-10
9-25
3-17
1-13
1-40
1-18 1-
28
1-19
5-10
9-25
3-17
1-13
Figure 22 : Chromatogramme total d’ionisation (LC-MS) du milieu de culture des cellules CHO. Le peptide Aβ40 (5 µg/ml) a été incubé avec les cellules CHO pendant 8h (A) et en la présence du Pefabloc® (B) ou du 1,10-phenanthroline (C).

82
Tableau 1 : Détection des fragments du peptide Aβ40 dans le surnageant des
cellules CHO après 8h d’incubation avec le peptide Aβ40 (5µg/ml).
Aβ Séquences des peptides ET 1 10 20 30 40
1-40 DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVV 25,8
1-28 DAEFRHDSGYEVHHQKLVFFAEDVGSNK 14,5
29-40 GAIIGLMVGGVV 26,2
4-23 FRHDSGYEVHHQKLVFFAED 18,8
24-40 VGSNKGAIIGLMVGGVV 25,8
1-20 DAEFRHDSGYEVHHQKLVFF 18,8 21-40 AEDVGSNKGAIIGLMVGGVV 24,2 22-40 EDVGSNKGAIIGLMVGGVV 24,8 23-40 DVGSNKGAIIGLMVGGVV 25,2
21-34 AEDVGSNKGAIIGL 16,2
1-19 DAEFRHDSGYEVHHQKLVF 13,0 20-40 FAEDVGSNKGAIIGLMVGGVV 26,1 20-33 FAEDVGSNKGAIIG 12,0 21-33 AEDVGSNKGAIIG 10,1
34-40 LMVGGVV 15,8
1-18 DAEFRHDSGYEVHHQKLV 10,0
19-40 FFAEDVGSNKGAIIGLMVGGVV 28,0
2-14 AEFRHDSGYEVHH 18,2
15-40 QKLVFFAEDVGSNKGAIIGLMVGGVV 28,5
1-13 DAEFRHDSGYEVH 1,0
14-40 HQKLVFFAEDVGSNKGAIIGLMVGGVV 27,2
15-33 QKLVFFAEDVGSNKGAIIG 20,0 15-28 QKLVFFAEDVGSNK 18,2 18-28 VFFAEDVGSNK 14,0 16-30 KLVFFAEDVGSNKGA 17,8
ET: temps d’ élution (min) des fragments peptidiques déduits des pics de la figure
22A.

83
Figure 23 : Fragments amyloïdes contenant la Tyr10 détectés dans le surnageant après incubation pendant 8h du peptide Aβ40 (5ug/ml) avec les cellules CHO en l’absence (A) ou en la présence (B) du Pefabloc®.
Temps de rétention (min)
cpm
1-13
1-18
1-19
1-28 2-
141-
204-
231-
34
1-40
Temps de rétention (min)
cpm
1-13
1-18
1-19
1-28 2-
141-
204-
231-
34
1-40
Temps de rétention (min)
cpm
1-13
1-18
1-19
1-28
5-10
1-40
3-17
9-25
Temps de rétention (min)
cpm
1-13
1-18
1-19
1-28
5-10
1-40
3-17
9-25
A
B

84
Les données du Tableau 2 confirment l’inhibition de la dégradation des
constituants du pic P1 par l’activité enzymatique de type sérine. En effet, les
composés du pic PII (Aβ(2-14), Aβ(1-20) et Aβ(4-23)) sont absents du
chromatogramme d’ionisation (figure 22B) en total accord avec les données de la
figure 23. Sachant qu’en la présence de cet inhibiteur, le quantité du peptide
Aβ40 reste inchangée, ce résultat démontre que les peptides Aβ(2-14), Aβ(1-20)
et Aβ(4-23) sont les produits de dégradation des fragments Aβ(1-18), Aβ(1-19)
et Aβ(1-28). Cependant, on observe l’apparition de nouveaux peptides (Aβ(3-17),
Aβ(5-10) et Aβ(9-25)) qui ne sont pas présents en l’absence du Pefabloc. Ces
résultats suggèrent l’existence d’autres enzymes capables de cliver les peptides
du pic P1 (Aβ(1-18), Aβ(1-19) et Aβ(1-28)) mais leur action protéolytique est
insignifiante.
Les données du Tableau 3 confirment l’inhibition de la dégradation du
peptide Aβ40 par l’activité enzymatique thiol métalloprotéase, en accord avec les
données obtenues précédemment (figures 17 et 19). Cependant, si l’on observe la
disparition totale des composants des pics P1 et P2, au profit du peptide Aβ40,
de nouveaux peptides sont détectés et plus particulièrement les fragments Aβ(1-
20) et Aβ(4-23). Sachant ces peptides sont aussi les produits de dégradation des
fragments peptidiques du pic PI et qu’ils sont absents en la présence du Pefabloc
(figure 23 et tableau 2), ces résultats suggèrent que les peptides Aβ(1-20) et
Aβ(4-23) peuvent être générés aussi après clivage du peptide Aβ40 par la sérine
protéase secrétée dans le milieu extracellulaire des cellules. Cette observation
est amplement étayée par les données du tableau 4 qui confirment la présence de
ces peptides (Aβ(1-20) et Aβ(4-23)) quant le peptide Aβ40 est incubé avec les
surnageants cellulaires pendant 16h. Cependant, au vu des résultats de la figure
15, ce clivage contribue peu à la dégradation totale du peptide amyloïde.

85
Tableau 2 : Détection des fragments du peptide Aβ40 dans le surnageant des
cellules CHO après 8h d’incubation avec le peptide Aβ40 (5µg/ml) en la présence
de l’inhibiteur Pefabloc®.
Aβ Séquences des peptides ET 1 10 20 30 40
1-40 DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVV 25,8
1-28 DAEFRHDSGYEVHHQKLVFFAEDVGSNK 15,2 29-40 GAIIGLMVGGVV 26,2
1-19 DAEFRHDSGYEVHHQKLVF 13,8 20-40 FAEDVGSNKGAIIGLMVGGVV 26,1 21-40 AEDVGSNKGAIIGLMVGGVV 24,1 22-40 EDVGSNKGAIIGLMVGGVV 24,8 23-40 DVGSNKGAIIGLMVGGVV 25,2
1-18 DAEFRHDSGYEVHHQKLV 10,1 19-40 FFAEDVGSNKGAIIGLMVGGVV 27,5
3-17 EFRHDSGYEVHHQKL 10,2 18-31 VFFAEDVGSNKGAI 19,2 32-40 IGLMVGGVV 22,0
5-10 EFRHDSGY 21,0 11-24 EVHHQKLVFFAEDV 1,0 25-36 GSNKGAIIGLMV 10,3
9-26 GYEVHHQKLVFFAEDVGS 23,8 27-40 NKGAIIGLMVGGVV 24,2
1-13 DAEFRHDSGYEVH 2,0 14-40 HQKLVFFAEDVGSNKGAIIGLMVGGVV 27,2 14-28 HQKLVFFAEDVGSNK 16,2 15-28 QKLVFFAEDVGSNK 18,0 16-28 KLVFFAEDVGSNK 19,2 17-28 LVFFAEDVGSNK 19,3 18-28 VFFAEDVGSNK 14,2 15-40 QKLVFFAEDVGSNKGAIIGLMVGGVV 28,0
15-26 QKLVFFAEDVGS 19,8
20-33 FAEDVGSNKGAIIG 12,2 21-33 AEDVGSNKGAIIG 10,1 21-34 AEDVGSNKGAIIGL 17,1 24-35 VGSNKGAIIGLM 10,2 33-38 GLMVGG 23,8
ET: temps d’ élution (min) des fragments peptidiques déduits des pics de la figure
23B.

86
Tableau 3 : Détection des fragments du peptide Aβ40 dans le surnageant des cellules CHO après 8h d’incubation avec le peptide Aβ40 (5µg/ml) en la présence des inhibiteurs 1,10-phénanthroline et NEM.
Aβ Séquences des peptides ET 1 10 20 30 40
1-40 DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVV 25,8
7-40 DSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVV 26,8
1-20 DAEFRHDSGYEVHHQKLVFF 17,0 4-20 FRHDSGYEVHHQKLVFF 15,5 7-20 DSGYEVHHQKLVFF 18,2 21-40 AEDVGSNKGAIIGLMVGGVV 24,5
4-23 FRHDSGYEVHHQKLVFFAED 17,0 24-40 VGSNKGAIIGLMVGGVV 25,5
Tableau 4 : Détection des fragments du peptide Aβ40 dans le surnageant des cellules CHO de 8h d’incubation qui a subit une incubation de 16h avec le peptide Aβ40 (5µg/ml).
Aβ Séquences des peptides ET 1 10 20 30 40
1-40 DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVV 25,8
1-20 DAEFRHDSGYEVHHQKLVFF 19,0 21-40 AEDVGSNKGAIIGLMVGGVV 24,2 22-40 EDVGSNKGAIIGLMVGGVV 24,7 23-40 DVGSNKGAIIGLMVGGVV 25,1 21-34 AEDVGSNKGAIIGL 18,2 22-34 EDVGSNKGAIIGL 18,2 23-34 DVGSNKGAIIGL 18,2
2-40 AEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVV 27,5 2-23 AEFRHDSGYEVHHQKLVFFAED 18,0 4-23 FRHDSGYEVHHQKLVFFAED 19,0 5-23 RHDSGYEVHHQKLVFFAED 14,2 8-23 SGYEVHHQKLVFFAED 19,0 10-23 YEVHHQKLVFFAED 25,0 24-40 VGSNKGAIIGLMVGGVV 25,5
7-40 DSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVV 27,5 12-40 VHHQKLVFFAEDVGSNKGAIIGLMVGGVV 26,5 14-40 HQKLVFFAEDVGSNKGAIIGLMVGGVV 27,5 15-40 QKLVFFAEDVGSNKGAIIGLMVGGVV 28,5 16-40 KLVFFAEDVGSNKGAIIGLMVGGVV 28,5 12-34 VHHQKLVFFAEDVGSNKGAIIGL 21,8 14-34 HQKLVFFAEDVGSNKGAIIGL 22,2 15-34 QKLVFFAEDVGSNKGAIIGL 23,8 12-38 VHHQKLVFFAEDVGSNKGAIIGLMVGG 27,5

87
Sur la base de ces résultats, nous pouvons par conséquent conclure que la clairance du
peptide β-amyloïde par les cellules se produit selon un mécanisme protéolytique impliquant :
• Une thiol métalloprotéase qui clive le peptide β-amyloïde, au niveau de trois sites
majoritaires Val18-Phe19, Phe19-Phe20 et Lys28-Gly29, pour libérer les fragments Aβ(1-
18), Aβ(1-19) et Aβ(1-28).
• Une sérine protéase qui clive essentiellement les fragments peptidiques Aβ(1-18), Aβ(1-
19) et Aβ(1-28), au niveau du site His14-Glu15, pour libérer le fragment Aβ(2-14). Seul
le clivage du peptide Aβ(1-28), au niveau des sites Phe20-Ala21 et Asp23-Val24, génère
les fragments Aβ(1-20) et Aβ(4-23).
CLAIRANCE DES PEPTIDES AMYLOÏDES MUTÉS
Comme nous l’avons vu précédemment (cf. chapitre III- B-2), le gène de l'APP
présente plusieurs mutations situées au niveau de la séquence correspondant au
peptide β-amyloïde (figure 11). Certaines de ces mutations provoquent des
angiopathies amyloïdes cérébrales; pathologies cliniquement différentes de la
maladie d’Alzheimer (Roher et al., 1993). C’est le cas des mutations de type
« Dutch » et « Flemish » qui engendrent des dépôts amyloïdes dans la paroi des
vaisseaux sanguins irriguant le cortex et les méninges (De Jonghe, 1998 ; Roks et
al., 2000). Par contre, on n’observe aucun dépôt amyloïde chez le rat (Boyd-
Kimball et al.,2004) dont la séquence du peptide Aβ comporte trois substitutions
(R5G, Y10F et H13R) par rapport à la séquence du peptide humain.
Sauvage DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVV
Flemish DAEFRHDSGYEVHHQKLVFFGEDVGSNKGAIIGLMVGGVV
Dutch DAEFRHDSGYEVHHQKLVFFAQDVGSNKGAIIGLMVGGVV
Rat DAEFGHDSGFEVRHQKLVFFAEDVGSNKGAIIGLMVGGVV

88
1. Profils de clivage protéolytique des peptides amyloïdes. Sur la base
des observations précédentes, la dégradation de ces peptides a été analysée afin
de déterminer les conséquences de ces mutations sur leur clairance par les
différentes cellules. Pour cela, ces peptides mutés ont été incubés pendant 8h
avec les différentes lignées cellulaires, puis les surnageants prélevés ont été
ensuite analysés par ELISA indirect ou soumis à une séparation sur RP-HPLC.
Comme pour le peptide natif (figure 13), on observe, en fonction du temps,
une diminution significative de la quantité des peptides «Dutch» et «Flemish» du
milieu de culture des cellules neuronales (figure 24) ou non neuronales. Si la
cinétique de disparition des peptides varie selon les lignées cellulaires, la vitesse
de clairance du peptide «Flemish» est plus rapide que celle du peptide «Dutch».
Par exemple, après 4h d’incubation, 60-80% du peptide «Flemish» est éliminé du
surnageant des cellules neuronales alors que le taux de disparition du peptide
«Dutch» varie entre 10% (CHP-100) et 75% (cellules SY-SH5Y et U-373).
L’ensemble de ces résultats suggère, comme pour le peptide natif, l’existence de
mécanismes responsables de la disparition du peptide Aβ40 du surnageant des cellules.
Les figures 25 et 26 représentent les profils HPLC des peptides amyloïdes
natif et mutés après leur incubation avec les lignées cellulaires CHO pendant 8h.
Tous les peptides amyloïdes sont clivés avec des taux de clivage variant entre
60% à 95%. L’analyse de ces profils révèle des variations au niveau du nombre
et/ou de l’amplitude des pics générés. Ainsi, on observe :
• La présence de pics communs (élués à 61, 63 ou 67 min) pour tous les peptides
mais présentant des amplitudes différentes.
• La présence de pics dominants pour les peptides Aβ « Rat » et «Dutch» élués
respectivement à 56 et 67 min.
• L’absence de clivage préférentiel pour les peptides amyloïdes « natif » et
« Flemish ».

89
0
20
40
60
80
100
0 2 4 6 8 10
pept
ide
rest
ant (
%)
temps d'incubation (h)
SH-SY5YSK-N-BEU-373CHP-100
A
0
20
40
60
80
100
0 2 4 6 8 10
pept
ide
rest
ant (
%)
temps d'incubation (h)
SH-SY5YSK-N-BEU-373CHP-100
B
0
20
40
60
80
100
0 2 4 6 8 10
pept
ide
rest
ant (
%)
temps d'incubation (h)
SH-SY5YSK-N-BEU-373CHP-100
A0
20
40
60
80
100
0
20
40
60
80
100
0 2 4 6 8 100 2 4 6 8 10
pept
ide
rest
ant (
%)
temps d'incubation (h)
SH-SY5YSK-N-BEU-373CHP-100
SH-SY5YSK-N-BEU-373CHP-100
A
0
20
40
60
80
100
0 2 4 6 8 10
pept
ide
rest
ant (
%)
temps d'incubation (h)
SH-SY5YSK-N-BEU-373CHP-100
B0
20
40
60
80
100
0
20
40
60
80
100
0 2 4 6 8 100 2 4 6 8 10
pept
ide
rest
ant (
%)
temps d'incubation (h)
SH-SY5YSK-N-BEU-373CHP-100
SH-SY5YSK-N-BEU-373CHP-100
B
Figure 24. Cinétique de disparition des peptides «Flemish» (A) et «Dutch» (B) du milieu de culture de cellules neuronales. A différents temps d’incubation, la quantité des peptides amyloïdes restant dans les surnageants (concentration initiale de 5 µg/ml) est mesurée par ELISA indirect.

90
Figure 25. Profil de dégradation des peptides natif et «Flemish» après leur incubation avec les cellules CHO pendant 8h. La concentration des peptides est de 5 µg/ml. Les peptides sont élués avec un débit de 0.5 ml/mn sur une colonne C18 selon le gradient défini dans le chapitre Matériels et Méthodes.
0.000
0.005
0.010
0.015Aβ humain
Temps d’élutions(min)
50 55 60 65 70 75 80
Uni
téar
bitr
aire
Uni
téar
bitr
aire
Temps d’élutions(min)
0.000
0.015
0.010
0.015
0.020
0.025
0.030
50 55 60 65 70 75 80
Aβ humain:Modification flemish
Aβ
Aβ
0.000
0.005
0.010
0.015Aβ humain
Temps d’élutions(min)
50 55 60 65 70 75 80
Uni
téar
bitr
aire
Uni
téar
bitr
aire
Temps d’élutions(min)
0.000
0.015
0.010
0.015
0.020
0.025
0.030
50 55 60 65 70 75 80
Aβ humain:Modification flemish
0.000
0.005
0.010
0.015Aβ humain
Temps d’élutions(min)
50 55 60 65 70 75 80
Uni
téar
bitr
aire
0.000
0.005
0.010
0.015
0.000
0.005
0.010
0.015Aβ humain
Temps d’élutions(min)
50 55 60 65 70 75 8050 55 60 65 70 75 80
Uni
téar
bitr
aire
Uni
téar
bitr
aire
Temps d’élutions(min)
0.000
0.015
0.010
0.015
0.020
0.025
0.030
50 55 60 65 70 75 80
Aβ humain:Modification flemish
Uni
téar
bitr
aire
Temps d’élutions(min)
0.000
0.015
0.010
0.015
0.020
0.025
0.030
0.000
0.015
0.010
0.015
0.020
0.025
0.030
50 55 60 65 70 75 8050 55 60 65 70 75 80
Aβ humain:Modification flemish
AβAβ
AβAβ

91
Figure 26. Profil de dégradation des peptides murin et «Dutch» après leur incubation avec les cellules CHO pendant 8h. La concentration des peptides est de 5 µg/ml. Les peptides sont élués avec un débit de 0.5 ml/mn sur une colonne C18 selon le gradient défini dans le chapitre Matériels et Méthodes.
Temps d’élutions(min)
50 55 60 65 70 75 80
Uni
téar
bitr
aire
0.000
0.005
0.010
0.015
0.020Aβ humain:
Modification Dutch
Uni
téar
bitr
aire
Temps d’élutions(min)
0.000
0.020
0.040
0.060
0.080
0.100
0.120
50 55 60 65 70 75 80
Aβ murin
Aβ
Aβ
Temps d’élutions(min)
50 55 60 65 70 75 80
Uni
téar
bitr
aire
0.000
0.005
0.010
0.015
0.020Aβ humain:
Modification Dutch
Uni
téar
bitr
aire
Temps d’élutions(min)
0.000
0.020
0.040
0.060
0.080
0.100
0.120
50 55 60 65 70 75 80
Aβ murin
Temps d’élutions(min)
50 55 60 65 70 75 80
Uni
téar
bitr
aire
0.000
0.005
0.010
0.015
0.020Aβ humain:
Modification Dutch
Temps d’élutions(min)
50 55 60 65 70 75 8050 55 60 65 70 75 80
Uni
téar
bitr
aire
0.000
0.005
0.010
0.015
0.020
0.000
0.005
0.010
0.015
0.020Aβ humain:
Modification Dutch
Uni
téar
bitr
aire
Temps d’élutions(min)
0.000
0.020
0.040
0.060
0.080
0.100
0.120
50 55 60 65 70 75 80
Aβ murinU
nité
arbi
trai
re
Temps d’élutions(min)
0.000
0.020
0.040
0.060
0.080
0.100
0.120
0.000
0.020
0.040
0.060
0.080
0.100
0.120
50 55 60 65 70 75 8050 55 60 65 70 75 80
Aβ murin
AβAβ
AβAβ

92
Parallèlement, nous avons testé la dégradation extracellulaire de ces
peptides dans les surnageants de cellules incubées seules pendant 8h. Les
résultats obtenus ont montré que le peptide amyloïde « Flemish », comme le
peptide amyloïde natif, ne subit aucune dégradation significative par les enzymes
secrétées dans les milieux extracellulaires. Par contre, les peptides amyloïdes
«Rat» et «Dutch» sont clivés par les protéases des surnageants cellulaires avec
des taux de clivage de 20% et 40% respectivement. Notons que le profil de
clivage protéolytique de ces deux peptides est caractérisé par la présence de
produits de clivage retrouvés également en la présence des cellules; l’aire de ces
pics est cependant, moindre dans les surnageants.
Afin de vérifier si les clivages obtenus avec les peptides mutés sont dus à
l’action des mêmes protéases que celles impliquées dans le clivage du peptide
amyloïde natif, nous avons testé les inhibiteurs de protéases utilisés
précédemment. Pour les inhibiteurs 1-10-Phénontroline et NEM, on obtient une
inhibition quasi-totale de la dégradation de tous les peptides mutés. Par ailleurs,
la présence de ces inhibiteurs abolit totalement le clivage des peptides «Rat» et
«Dutch» par les protéases secrétées dans les surnageants cellulaires. Ce résultat
démontre que la métalloprotéase thiol-dépendante existe aussi sous une forme
secrétée.
L’ensemble de ces résultats montre que le peptide amyloïde « natif » et le peptide
amyloïde « Flemish » adopteraient une conformation qui leur confèrent une résistance à
l’action des protéases du surnageant, alors que les peptides « Dutch » et « Rat » seraient plus
accessibles aux clivages par ces protéases.
Afin d’expliquer les différences observées entre les différents profils de
clivage protéolytique obtenu précédemment par HPLC, nous avons soumis nos
échantillons à une analyse par spectroscopie de masse.

93
2. Détermination des sites de clivage des peptides amyloïdes. Afin de
caractériser les sites de coupure des activités enzymatiques responsables du
clivage protéolytique des peptides amyloïdes, nous avons incubé les peptides en la
présence des différentes cellules pendant 8h, les surnageants prélevés ont été
ensuite soumis à une séparation par chromatographie liquide en phase inverse,
couplée à un spectromètre de masse en mode ESI-Q-TOF.
L’ensemble des résultats, indiqué dans les tableaux 5, 6 et 7, met en
évidence plusieurs sites de coupures. Ainsi, les peptides «Flemish», «Rat» et
«Dutch» ont quatre sites de coupures communs avec le peptide natif; il s’agit des
coupures entre les acides aminés Val18-Phe19, Phe19-Phe20, Phe20-Ala21 et Lys28-
Gly29. Ces résultats expliqueraient les pics communs obtenus précédemment au
niveau du profil HPLC des peptides amyloïdes. De plus, on observe la présence de
nouveaux clivages pour les peptides mutés. En effet, le peptide «Flemish» est
clivé au niveau de trois sites supplémentaires Val17-Leu18, Gly21-Glu22 et Glu22-
Asp23, le peptide «Rat» subit deux nouveaux clivages au niveau des sites Arg13-
His14 et leu17-Val18 alors que le peptide «Dutch» possède un seul site de clivage
supplémentaire au niveau des résidus Ala21-Gln22.
Afin de déterminer si certains sites de coupures, obtenus pour les peptides
amyloïdes mutés, pourraient provenir, comme dans le cas du peptide natif, de
l’action de la sérine protéase. Pour cela, nous avons analysé les fragments générés
après une incubation de 8h de chacun de ces peptides en la présence du Pefabloc.
Après analyse des échantillons par spectroscopie de masse, nos résultats
montrent une inhibition des sites de clivage suivants :
• leu17-Val18, Phe20-Ala21, Asp23-Val24 pour le peptide « Rat »
• Leu17-Val18, Phe20-Gly21, Gly21-Glu22, Glu22-Asp23, Asp23-Val24 pour le peptide
«Flemish»
• His14-Gln15, Phe20-Ala21 et Asp23-Val24 pour le peptide « Dutch ».

94
Tableau 5 : Détection des fragments du peptide «Flemish» dans le surnageant
des cellules CHO après 8h d’incubation avec le peptide Aβ40 (5µg/ml).
Aβ(FL) Séquences 1-40 DAEFRHDSGYEVHHQKLVFFGEDVGSNKGAIIGLMVGGVV1-28 DAEFRHDSGYEVHHQKLVFFGEDVGSNK
29-40 GSNKGAIIGLMVGGVV8-22 DAEFRHDSGYEVHHQKLVFFGE
23-40 DVGSNKGAIIGLMVGGVV5-21 RHDSGYEVHHQKLVFFG
22-40 EDVGSNKGAIIGLMVGGVV1-20 DAEFRHDSGYEVHHQKLVFF
21-40 GEDVGSNKGAIIGLMVGGVV1-19 DAEFRHDSGYEVHHQKLVF
20-40 FGEDVGSNKGAIIGLMVGGVV1-18 DAEFRHDSGYEVHHQKLV
19-40 FFGEDVGSNKGAIIGLMVGGVV19-28 FFGEDVGSNK 7-17 DSGYEVHHQKL
18-40 VFFGEDVGSNKGAIIGLMVGGVV18-28 VFFGEDVGSNK 5-13 RHDSGYEVH
14-40 HQKLVFFGEDVGSNKGAIIGLMVGGVV15-40 QKLVFFGEDVGSNKGAIIGLMVGGVV14-28 HQKLVFFGEDVGSNK 15-28 QKLVFFGEDVGSNK

95
Tableau 6 : Détection des fragments du peptide «Dutch» dans le surnageant des
cellules CHO après 8h d’incubation avec le peptide Aβ40 (5µg/ml).
Aβ(DT) Séquences 1-40 DAEFRHDSGYEVHHQKLVFFAQDVGSNKGAIIGLMVGGVV1-28 DAEFRHDSGYEVHHQKLVFFAQDVGSNK
29-40 GAIIGLMVGGVV30-40 AIIGLMVGGVV4-23 FRHDSGYEVHHQKLVFFAQD
24-40 VGSNKGAIIGLMVGGVV3-21 EFRHDSGYEVHHQKLVFFA
22-40 QDVGSNKGAIIGLMVGGVV23-40 DVGSNKGAIIGLMVGGVV1-20 DAEFRHDSGYEVHHQKLVFF
21-40 AQDVGSNKGAIIGLMVGGVV5-19 RHDSGYEVHHQKLVF
20-40 FAQDVGSNKGAIIGLMVGGVV2-14 AEFRHDSGYEVHH
15-40 QKLVFFAQDVGSNKGAIIGLMVGGVV15-28 QKLVFFAQDVGSNK 1-16 DAEFRHDSGYEVHHQK 4-18 FRHDSGYEVHHQKLV
16-30 KLVFFAQDVGSNKGA 19-37 FFAQDVGSNKGAIIGLMVG 34-40 GLMVGGVV

96
Tableau 7 : Détection des fragments du peptide murin dans le surnageant des
cellules CHO après 8h d’incubation avec le peptide Aβ40 (5µg/ml).
Aβ(RT) Séquences 1-40 DAEFGHDSGFEVRHQKLVFFAEDVGSNKGAIIGLMVGGVV1-28 DAEFGHDSGFEVRHQKLVFFAEDVGSNK
29-40 GAIIGLMVGGVV4-23 FGHDSGFEVRHQKLVFFAED
24-40 VGSNKGAIIGLMVGGVV1-20 DAEFGHDSGFEVRHQKLVFF
21-40 AEDVGSNKGAIIGLMVGGVV1-19 DAEFGHDSGFEVRHQKLVF
20-40 FAEDVGSNKGAIIGLMVGGVV1-18 DAEFGHDSGFEVRHQKLV
19-40 FFAEDVGSNKGAIIGLMVGGVV19-28 FFAEDVGSNK 1-17 DAEFGHDSGFEVRHQKL
18-40 VFFAEDVGSNKGAIIGLMVGGVV18-28 VFFAEDVGSNK 1-13 DAEFGHDSGFEVR
14-40 HQKLVFFAEDVGSNKGAIIGLMVGGVV14-28 HQKLVFFAEDVGSNK 15-28 QKLVFFAEDVGSNK 16-28 KLVFFAEDVGSNK 17-28 LFFAEDVGSNK 1-16 DAEFGHDSGFEVRHQK 1-15 DAEFGHDSGFEVRHQ 1-14 DAEFGHDSGFEVRH 1-12 DAEFGHDSGFEV
13-31 RHQKLVFFAEDVGSNKGAI 17-30 LVFFAEDVGSNKGA 31-35 IIGLM

97
Ces résultats montrent que ces sites de coupures sont dus à l’action de la sérine
protéase qui agirait, comme pour le peptide natif, sur les produits de clivage du
peptide Aβ40 par la métalloprotéase thiol-dépendante. Les sites de clivage des
peptides amyloïdes par cette métalloprotéase sont indiqués dans le schéma
suivant :
En conclusion, en la présence des cellules, les peptides amyloïdes mutés et le
peptide amyloïde natif sont tous clivés selon un mécanisme similaire faisant
intervenir principalement deux enzymes qui agissent en tandem. A l’inverse du
peptide natif et du peptide «Flemish», les peptides amyloïdes «Dutch» et «Rat »
sont en revanche clivés par les protéases du surnageant cellulaire. Ce résultat
pourrait s’expliquer par une différence au niveau de la conformation qu’adopte
chaque peptide et qui détermine son accessibilité à l’action des protéases.

98
ANALYSE CONFORMATIONNELLE DES PEPTIDES AMYLOÏDES MUTÉS.
Sur la base des observations précédentes, on peut supposer qu’il existe une
relation fonctionnelle entre la conformation des peptides amyloïdes et leur
clairance par les cellules. Pour cela, une étude conformationnelle des différents
peptides amyloïdes a été menée par dichroïsme circulaire (DC) afin de
déterminer leurs paramètres structuraux.
Pour les peptides ou les protéines modèles (Woody, 1985), leurs spectres DC
présentent, en général, des profils qui sont caractéristiques de leurs structures
secondaires:
• Hélice α : deux minimums négatifs à 208 et 222 nm et un maximum à 190 nm.
• Feuillet β : un minimum négatif à 215 nm et un maximum à 195 nm.
• Coude β : un minimum négatif à 225 nm et un maximum à 205 nm.
• Apériodique ou ″Random Coil″ : un minimum négatif à 198 nm
Différents cosolvants, tels que le TFE et le SDS, ont été utilisés afin de
révéler toutes les différences structurales entre les peptides. Le TFE est un
cosolvant qui a la capacité d’induire les structures secondaires de peptides en
interagissant avec les molécules d’eau (Rizo et al., 1993). Le SDS est par contre
un cosolvant qui mime les micelles (Zhong & Johnson, 1992). C’est une molécule
qui se fixe aux peptides via des interactions hydrophobiques et électrostatiques
(Cardin et al., 1991). Les figures 27 et 28 représentent les spectres obtenus dans
le tampon phosphate et dans des pourcentages croissants de TFE et SDS.
Dans un tampon phosphate (5 mM) à pH 7.5, les spectres de dichroïsme
circulaire des peptides amyloïdes (comme l’ensemble des peptides en général)
présentent essentiellement une bande négative intense autour de 195 nm qui est
une caractéristique typique des polypeptides adoptant une conformation
apériodique (Venyaminov & Yang, 1996). Seul le peptide amyloïde murin se
caractérise par une structure en feuillet β caractérisée par un minimum négatif
autour de 218 nm et un maximum positif proche de 200 nm.

99
∆ε/N (cm-1.M-1)
Longueur d’onde (cm-1)
4020100
-10
-20 30
Aβ(W
t)A
β(Rt)
190200
210220
230240
250
Aβ(F
l)
190200
210220
230240
250
Aβ(D
t)
4020100
-10
-20 30
0% TFE
10% TFE
20% TFE
50% TFE
0% TFE
10% TFE
20% TFE
50% TFE
0% TFE
10% TFE
20% TFE
50% TFE
0% TFE
10% TFE
20% TFE
50% TFE
Figure 27. Spectres de dichroïsm
e circulaire des peptides amyloïdes. Les m
esures sont réalisées à20°C en
présence de 15 µM de
chaque peptide diluédans différents pourcentages de TFE.
∆ε/N (cm-1.M-1)
Longueur d’onde (cm-1)
4020100
-10
-20 30
Aβ(W
t)A
β(Rt)
190200
210220
230240
250
Aβ(F
l)
190200
210220
230240
250
Aβ(D
t)
4020100
-10
-20 30
0% TFE
10% TFE
20% TFE
50% TFE
0% TFE
10% TFE
20% TFE
50% TFE
0% TFE
10% TFE
20% TFE
50% TFE
0% TFE
10% TFE
20% TFE
50% TFE
∆ε/N (cm-1.M-1)
Longueur d’onde (cm-1)
4020100
-10
-20 30 4020100
-10
-20 30
Aβ(W
t)A
β(Rt)
190200
210220
230240
250190
200210
220230
240250
190200
210220
230240
250190
200210
220230
240250
Aβ(F
l)
190200
210220
230240
250190
200210
220230
240250
190200
210220
230240
250190
200210
220230
240250
Aβ(D
t)
4020100
-10
-20 30 4020100
-10
-20 30
0% TFE
10% TFE
20% TFE
50% TFE
0% TFE
10% TFE
20% TFE
50% TFE
0% TFE
10% TFE
20% TFE
50% TFE
0% TFE
10% TFE
20% TFE
50% TFE
Figure 27. Spectres de dichroïsm
e circulaire des peptides amyloïdes. Les m
esures sont réalisées à20°C en
présence de 15 µM de
chaque peptide diluédans différents pourcentages de TFE.

100
Aβ(W
t)
0% SDS
5% SDS
40% SDS
100% SDS
0% SDS
5% SDS
40% SDS
100% SDS
Aβ(R
t)
190200
210220
230240
250
0% SDS
5% SDS
40% SDS
100% SDS
Aβ(F
l)
190200
210220
230240
250
0% SDS
5% SDS
40% SDS
100% SDS
Aβ(D
t)
∆ε/N (cm-1.M-1)
Longueur d’onde (cm-1)
3025151050
-10
-15 20-53025151050
-10
-15 20-5Figure 28. Spectres de dichroïsme circulaire des peptides am
yloïdes. Les mesures sont réalisées à
20°C en présence de 15 µM
dechaque peptide dilué
dans différents pourcentages de SDS.
Aβ(W
t)
0% SDS
5% SDS
40% SDS
100% SDS
0% SDS
5% SDS
40% SDS
100% SDS
Aβ(R
t)
190200
210220
230240
250
0% SDS
5% SDS
40% SDS
100% SDS
Aβ(F
l)
190200
210220
230240
250
0% SDS
5% SDS
40% SDS
100% SDS
Aβ(D
t)
∆ε/N (cm-1.M-1)
Longueur d’onde (cm-1)
3025151050
-10
-15 20-53025151050
-10
-15 20-5
Aβ(W
t)
0% SDS
5% SDS
40% SDS
100% SDS
0% SDS
5% SDS
40% SDS
100% SDS
Aβ(R
t)
190200
210220
230240
250190
200210
220230
240250
190200
210220
230240
250190
200210
220230
240250
190200
210220
230240
250
0% SDS
5% SDS
40% SDS
100% SDS
Aβ(F
l)
190200
210220
230240
250190
200210
220230
240250
190200
210220
230240
250190
200210
220230
240250
190200
210220
230240
250
0% SDS
5% SDS
40% SDS
100% SDS
Aβ(D
t)
∆ε/N (cm-1.M-1)
Longueur d’onde (cm-1)
3025151050
-10
-15 20-5 3025151050
-10
-15 20-53025151050
-10
-15 20-5 3025151050
-10
-15 20-5Figure 28. Spectres de dichroïsme circulaire des peptides am
yloïdes. Les mesures sont réalisées à
20°C en présence de 15 µM
dechaque peptide dilué
dans différents pourcentages de SDS.

101
Pour des pourcentages croissants en TFE et en SDS, les peptides adoptent
des structures fortement hélicoïdales (2 minima négatifs à 207 et 222 nm et un
maximum positif à 192 nm). La comparaison des effets, induits par les deux
cosolvants, montre que le SDS a un effet plus structurant sur les peptides qui
adoptent, déjà à 40 mM, une structure en hélice α. Par contre, le TFE a un effet
différent sur les peptides selon le pourcentage utilisé. Ainsi, pour un pourcentage
de 20% de TFE tous les peptides adoptent une structure hélicoïdale. Par contre,
pour des valeurs inférieures ou supérieures à 20%, la conformation de tous les
peptides est régis par un équilibre conformationnel entre une structure feuillet β
et une structure α.
Cette étude conformationnelle, dans 2 environnements différents, permet
de subdiviser les 4 peptides en 2 groupes; les peptides amyloïdes natif et
«Flemish» formant le 1er groupe et les peptides amyloïdes «Dutch» et «Rat » le
2ème groupe. Cette observation permet d’expliquer les différences observées
précédemment entre les peptides au niveau de leur dégradation par les
protéases.

102
DISCUSSION GÉNÉRALE ET
PERSPECTIVES

103
Bien qu’il soit produit au cours du métabolisme normal, le peptide β-amyloïde
joue un rôle capital dans la pathogenèse de la maladie d’Alzheimer. Jusqu’à
présent, les principales stratégies abordées afin de diminuer le taux circulant du
peptide β-amyloïde dans le cerveau reposent (a)-sur la vaccination contre ce
peptide et (b)-sur l’inhibition ou la modulation des enzymes de maturation de
l’APP (figure 29).
(a)- L’immunisation de souris modèles de la maladie d’Alzheimer induit une
réduction considérable du dépôt amyloïde et des déficits cognitifs (Bard et al.,
2000; Das, 2001). Il apparaît tout de même qu’au-delà d’un stade critique en
dépôt amyloïde, l’effet des immunisations sur la réduction des dépôts amyloïdes
soit limité (Das et al., 2000). Si l’immunisation active par le peptide Aβ42 n’induit
pas de toxicité chez les souris modèles de la maladie d’Alzheimer, les essais
cliniques sur l’homme ont été arrêtés brutalement en phase II, du fait
d’importantes méningo-encéphalites (graves réactions inflammatoires du cerveau)
développées chez 5 % des patients (Check, 2002). Une alternative serait
l'immunisation passive par des anticorps anti-Aβ qui a donné des résultats
encourageants chez des animaux modèles de la maladie (Bard et al., 2000; Das,
2001).
(b)- L’inhibition ou la modulation de la production du peptide amyloïde peut
faire appel soit (i)-au développement d’inhibiteurs des secrétases impliquées dans
le clivage de l’APP, soit (ii)-à la modulation de leur activité.
i)-Inhibition des secrétases. L’inhibition de la β-secrétase ou de la γ-secrétase
diminue la production du peptide Aβ, et apparaît donc comme un moyen
d’inhiber, dès les premiers stades, l’activation de la cascade d’événements
pathogènes responsable de la maladie.
Plusieurs inhibiteurs de la γ-secrétase, qui ciblent les présénilines PS1 et PS2,
ont été décrits (Golde et al., 2001). Le traitement de la maladie chez des
souris modèles par l’un de ces inhibiteurs réduit à la fois le taux du peptide Aβ

104
AP
P
sA
PP
αA
βInactifs
fragments
+-
Voie amyloïdogène
Voie non-amyloïdogène
Dégradation
+
Ac
tiva
tion
de
l’α-s
ec
réta
se
:-agonistes du glutam
ate et de la sérotonine-statines, oestrogènes, testostérone
-activateurs des PKC
Inh
ibitio
n d
e l’β
-se
cré
tas
e:
-inhibiteurs-m
odulateurs de la fonctionIn
hib
ition
de
l’γ-se
cré
tas
e:
-inhibiteurs-Inhibiteurs de la G
SK-3
Activ
atio
n d
e la
NE
P:
-gène thérapie -agonistes de la som
atostatineA
ctiv
atio
n d
e l’ ID
E:
-sélectives activateursA
ctiv
atio
n d
e l’ E
CE
-1
Activ
atio
n d
ela
pla
sm
ine
Neurotoxique
Neuroprotectif
AP
P
sA
PP
αA
βInactifs
fragments
+-
Voie amyloïdogène
Voie non-amyloïdogène
Dégradation
+
Ac
tiva
tion
de
l’α-s
ec
réta
se
:-agonistes du glutam
ate et de la sérotonine-statines, oestrogènes, testostérone
-activateurs des PKC
Inh
ibitio
n d
e l’β
-se
cré
tas
e:
-inhibiteurs-m
odulateurs de la fonctionIn
hib
ition
de
l’γ-se
cré
tas
e:
-inhibiteurs-Inhibiteurs de la G
SK-3
Activ
atio
n d
e la
NE
P:
-gène thérapie -agonistes de la som
atostatineA
ctiv
atio
n d
e l’ ID
E:
-sélectives activateursA
ctiv
atio
n d
e l’ E
CE
-1
Activ
atio
n d
ela
pla
sm
ine
Neurotoxique
Neuroprotectif
Figure 29. Schéma représentatif des différentes voies thérapeutiques

105
et le dépôt amyloïde dans le cerveau (Dovey et al., 2001), et certains
inhibiteurs sont déjà au stade des essais cliniques. Toutefois, les limites de
cette approche sont un risque d’altération du clivage protéolytique d’autres
protéines comme la protéine Notch, dont le clivage par le système
multiprotéique γ-secrétase joue un rôle-clé dans la voie de signalisation de la
protéine, ou bien un risque d’accumulation de fragments transitoires C-
terminaux de l’APP (C99 et C83). Ces fragments ont en effet été détectés
notamment au sein des plaques séniles amyloïdes et des dégénérescences
neurofibrillaires, et présentent comme Aβ un effet neurotoxique (Yankner et
al., 1989) ce qui suggère leur participation à la pathologie (Suh et al., 1997).
L’inhibition du clivage par la β-secrétase, quoique encore peu développée,
apparaît donc comme une alternative moins critique. Les souris invalidées pour
le gène de BACE1 ne produisent pas de peptide Aβ, et ne présentent pas de
phénotype pathologique (Luo et al., 2001). Cependant, la détermination de la
structure tridimensionnelle de BACE1 a montré que le site actif est étendu, et
est donc difficile à cibler par de petites molécules (Hong et al., 2000).
ii)-Modulation du clivage de l’APP : La modulation du clivage de l’APP apparaît
comme une voie intéressante pour limiter la production du peptide amyloïde.
Ainsi, la stimulation de l’α-secrétase diminue la production du peptide Aβ
(Selkoe et al., 2001). Une modulation du clivage de l’APP au détriment de la
production du peptide Aβ peut également être envisagée par des médicaments
modulant la teneur en cholestérol comme les statines (Jick et al., 2000). En
effet, la prévalence de la maladie d’Alzheimer est diminuée pour des
personnes sous traitement par les statines (Jick et al., 2000) et des études
sur cultures cellulaires et in vivo ont montré que l’inhibition de la production
de cholestérol réduit la production d’Aβ, en défavorisant les clivage de l’APP
par les β-et γ-secrétases au profit du clivage par l’α-secrétase (Kojro et al.,
2001). Ce résultat pourrait s’expliquer par le fait que le clivage par les β-et γ-

106
secrétases ont lieu dans des zones membranaires riches en cholestérol, tandis
que le clivage par l’α-secrétase a lieu dans des zones membranaires "fluides", à
faible teneur en cholestérol (Wolozin et al., 2001). Ainsi, une haute teneur en
cholestérol membranaire favoriserait la production d’Aβ tandis qu’une faible
teneur favoriserait la production de sAPPα (Simons et al., 2001). L’avantage de
cette voie thérapeutiques est que les statines sont des médicaments déjà sur
le marché et bien tolérés. Des essais cliniques sont de ce fait déjà en cours.
Une autre piste intéressante, serait l’activation des voies du catabolisme du
peptide β-amyloïde. En effet, comme pour toute protéine (ou tout peptide), il
existe des processus cataboliques responsables de la dégradation du peptide β-
amyloïde circulant (Qiu et al., 1997; 1998). L’altération de ces processus
cataboliques devrait conduire à une augmentation de la concentration du peptide
β-amyloïde qui, compte tenu de ses propriétés physico-chimiques, se polymérise
(Burdick et al., 1992). Au delà de la limite de solubilité de ce peptide, cette
oligomérisation participerait à la formation des premiers dépôts. Ce sont en
effet ces dépôts qui sont à la base de processus donnant naissance aux formes
de β-amyloïde agrégé encore plus résistantes à la protéolyse.
L’étude du catabolisme du β-amyloïde a suscité, ces dernières années,
l’intérêt de plusieurs équipes dont la nôtre. Jusqu’à présent, les travaux des
autres équipes, ont été basés principalement sur le devenir du β-amyloïde en
présence d’une lignée cellulaire particulière (Naidu et al., 1995 ; Knauer et al.,
1992 ; Ida et al., 1996 ; Narita et al., 1997 ; Paresce et al., 1996), et plus
récemment sur la capacité du peptide à être catabolisé par des enzymes
spécifiques connues telle que l’Insulin-degrading enzyme ou IDE (Mukherjee et
al., 2000 ; Chesneau et al., 2000), la néprilysine ou NEP (Howell et al., 1995),
l’Endothelin-converting enzyme ou ECE (Eckman et al., 2001) et l’Angiotensin-
converting enzyme ou ACE (Hu et al., 2001). Compte tenu de cette très disparité entre

107
les différents travaux, nous avons opté pour une approche qui consiste à étudier le devenir du
peptide Aβ40 en la présence de plusieurs lignées cellulaires (neuronales et non neuronales) en
utilisant différentes techniques d’analyse.
A l’inverse de nos résultats, les travaux de certaines équipes (Ida et al.,
1996b ; McDermott et Gibson, 1997; Qiu et al., 1997; 1998; Sinha et Lieberburg,
1999) ont montré que le peptide β-amyloïde natif est dégradé essentiellement
par des protéases secrétées dans les surnageants cellulaires. Ainsi, Naidu et
collaborateurs ont montré que le peptide β-amyloïde était dégradé dans le
surnageant des cellules CHO par divers enzymes de type metallo-, aspartyl- et
thiol-proteases (Naidu et al., 1995). Il a aussi été montré qu’il existait une
enzyme de type métalloprotéase présente dans les surnageants des cellules CHO
et BV-2 et capable de cliver le peptide Aβ40 secrété (Qiu et al., 1997). Par la
suite, cette enzyme a été caractérisée dans le milieu de culture des cellules BV-2
comme étant l’Insulin-degrading enzyme (Qiu et al., 1998). Mentlein et coll. ont
aussi montré l’existence d’une enzyme de type métalloprotéase capable de cliver
le β-amyloïde synthétique dans le surnageant de cellules microgliales de rat
(Mentlein et al., 1998). Par ailleurs, nous n’avons pas observé d’internalisation du
peptide β-amyloïde par les différentes lignées cellulaires étudiées. En revanche,
les travaux des autres équipes montrent une internalisation du peptide par les
cellules fibroblastiques humaines HFF (Knauer et al., 1992) et les neuroblastomes
humains SH-SY5Y (Ida et al., 1996), ou via des récepteur de la lipoprotéine LRP
dans des glioblastomes humains (Narita et al., 1997). Il a aussi été montré une
internalisation de micro-agrégats du peptide β-amyloïde via des récepteurs SR
(scavenger receptor) dans des cellules microgliales de rat (Paresce et al., 1996).
L’ensemble de nos résultats montre que le peptide β-amyloïde est dégradé
via un mécanisme prenant place au niveau de la surface des cellules et que le
profil de dégradation du peptide est le même pour toutes les lignées cellulaires

108
étudiées. Cette observation suggère l’existence d’un mécanisme protéolytique
similaire, assez largement distribué et qui serait impliqué dans la protéolyse du
β-amyloïde. Le profil d’inhibition de ces activités argue en faveur d’un processus
de dégradation impliquant, dans un premier temps, une activité enzymatique de
type métalloprotéase, associée à la membrane des cellules et distincte de
protéases connues pour leur capacité à cataboliser le peptide β-amyloïde telles
que les MMPs (Roher et al., 1994 ; Backstrom et al., 1996), la NEP (Howell et al.,
1995), l’ACE (Hu et al., 2001) et l’ECE-1 (Eckman et al., 2001). Inhibée quasi
totalement par l’ortho-phénanthroline et le N-éthylmaleimide, cette activité
enzymatique de type métalloprotéase à thiol clive essentiellement le peptide β-
amyloïde entre les résidus Val18-Phe19, Phe19-Phe20 et Lys28-Gly29, quel que soit le
temps d’incubation du peptide avec les cellules (4 heures, 8 heures, 16 heures et
24 heures). A ce jour, une seule métalloprotéase à thiol, l’IDE, associée à la
membrane des cellules et capable de dégrader le peptide β-amyloïde, a été
décrite (Vekrellis et al., 2000). Cependant, cette forme membranaire de l’IDE n’a
été observée que dans des neurones différenciés PC12 ; l’enzyme étant
principalement localisée dans le cytoplasme des cellules (Shii K et al., 1985). De
plus, l’identification des sites de clivage du peptide β-amyloïde par une forme
recombinante de l’enzyme IDE révèle des coupures préférentielles (in vitro)
plutôt au niveau des sites His13-His14, His14-Gln15 et Phe19-Phe20 (Mukherjee et al.,
2000 ; Chesneau et al., 2000). Bien que nous ne pouvons pas déduire si cette activité est
l’Insulin degrading-enzyme, ancrée aux membranes, ou une isoforme de cette protéase, nos
résultats constituent la 1ère démonstration révélant le clivage du peptide β-amyloïde au
niveau des membranes de différentes lignées cellulaires neuronales et non neuronales
(Panchal et al., soumis).

109
De plus, notre étude montre, que le processus de dégradation du peptide
amyloïde implique une deuxième activité enzymatique de type sérine protéase.
Sécrétée dans les surnageants cellulaires, cette protéase clive les fragments
peptidiques, générés par l’activité métalloprotéase, au niveau des sites His14-
Glu15, Phe20-Ala21 et Asp23-Val24 pour libérer les peptides Aβ(2-14), Aβ(1-20) et
Aβ(4-23). Cette activité est inhibée par le Pefabloc®, qui agit sur un large
spectre de sérine protéases. En utilisant des inhibiteurs plus ciblés de serpines
tels que l’antipaïne, le benzamidine, la chymostatine ou l’α2-macroglobuline, nous
montrons que les protéases telles la papaïne, la trypsine, la chymotrypsine,
l’élastase, la plasmine et la thermolysine ne sont pas impliquées dans le mécanisme
de dégradation du peptide β-amyloïde. En conclusion, nous montrons que la clairance
du peptide Aβ40 par différentes cellules résulte d’un processus protéolytique qui implique
deux activités qui opèrent en tandem.
Le gène de l'APP présente plusieurs mutations engendrant des formes
familiales de la maladie et connues pour affecter la biosynthèse du peptide β-
amyloïde à partir de son précurseur. En effet, il a été montré que les mutations
de l’APP, situées à l’extérieur de la séquence correspondant au peptide β-
amyloïde induisent une augmentation considérable de sa sécrétion. C’est le cas de
la double mutation «Swedish», située au voisinage du site de clivage de la β-
secrétase, qui augmente considérablement la sécrétion des peptides amyloïdes
Aβ40 et Aβ42 (Citron et al., 1994 ; Haass et al., 1995). De même, les mutations
«London» et «Florida», proches du site de clivage de la γ-secrétase, augmentent
uniquement la sécrétion du peptide Aβ42 (Suzuki et al., 1994 ; Eckman et al.,
1997). D’autres mutations, situées à l’intérieur de la séquence du peptide β-
amyloïde, sont associées à des angiopathies amyloïdes cérébrales et/ou à une
maladie d’Alzheimer précoce. C’est le cas des mutations «Arctic», « Dutch»,
«Flemish», «Iowa» et «Italia » qui sont situées près du site de clivage de l’ α-

110
secrétase. L’effet pathogène de ces mutations découlent de leur capacité à
modifier la conformation du peptide amyloïde qui favorise ainsi sa
protofibrilisation et son agrégation dans plusieurs régions cérébrales (Demeester
et al., 2001 ; Nilsberth et al., 2001 ; Murakami et al., 2002). Des études,
effectuées sur des cellules transfectées par le cDNA de ces peptides mutés, ont
montré que ces mutations engendrent une diminution de la sécrétion des peptides
amyloïdes Aβ40 et Aβ42, liée à l’augmentation de leur capacité à former des
protofibrilles (De Jonghe et al., 1998). Par contre, la mutation «Flemish»
provoque une augmentation de la sécrétion et de la solubilité des peptides Aβ40
et Aβ42 (De Jonghe et al., 1998). Ces observations suggèrent que l’effet
pathogène de ces peptides mutés s’expliquerait par l’augmentation de leur durée
de vie dans le cerveau.
Jusqu’à présent, l’effet de ces mutations sur la dégradation protéolytique
des peptides mutés, n’a fait l’objet que d’un petit nombre d’études (Murakami et
al., 2002; Tsubuki et al., 2003 ; Morelli et al., 2003; 2004). Ainsi, il a été montré
que les peptides «Arctic», «Dutch», «Flemish» et «Italian» sont plus résistants
que le peptide natif à la protéolyse par la NEP (Tsubuki et al., 2003). De même,
l’étude in vitro du clivage des peptides amyloïdes «Flemish» et «Dutch», par l’IDE
purifiée (Morelli et al., 2003; 2004) a révélé sept sites de clivage pour le peptide
«Flemish» (Glu3-Phe4, Phe4-Arg5, His13-His14, His14-Gln15, Val18-Phe19, Phe19-Phe20
et Lys28-Gly29) et six sites de clivage pour le peptide «Dutch» (Glu3-Phe4, Phe4-
Arg5, His13-His14, His14-Gln15, Phe19-Phe20 et Phe20-Ala21). Dans notre travail, nous
avons étudié le clivage protéolytique de ces peptides par les mêmes lignées
cellulaires utilisées pour le peptide natif et déterminé par spectroscopie de
masse les fragments générés. L’analyse des produits de clivage du peptide
amyloïde «Flemish» révèle l’existence de quatre sites de clivage dues à l’action de
la métalloprotéase thiol-dépendante et ayant lieu entre les résidus His13-His14,
Val18-Phe19, Phe19-Phe20 et Lys28-Gly29. Les fragments libérés, par le clivage du

111
peptide à ces sites, serviront de substrats pour la sérine protéase qui va les
cliver au niveau des sites Leu17-Val18, Phe20-Gly21, Gly21-Glu22 et Glu22-Asp23 et
Asp23-Val24. De même, le peptide «Dutch» est clivé en premier lieu par la
métalloprotéase thiol-dépendante au niveau des résidus Val18-Phe19, Phe19-Phe20,
Ala21-Gln22et Lys28-Gly29 pour libérer des fragments qui seront clivés à leur tour
par la sérine protéase au niveau des sites His14-Gln15, Phe20-Ala21 et Asp23-Val24. A
l’inverse du peptide natif et du peptide «Flemish», qui sont résistants à l’action
des protéases du surnageant, le peptide amyloïde «Dutch» est dégradé dans les
surnageants extracellulaires par la même métalloprotéase thiol-dépendante qui
est secrétée dans les milieux de culture. Ces différences pourraient s’expliquer par la
conformation qu’adopterait chaque peptide et qui serait déterminante pour sa résistance ou
son accessibilité à la dégradation protéolytique. L’étude conformationnelle de ces substrats
peptidiques, par dichroïsme circulaire, supporte ces conclusions.
Notre travail a été réalisé sur des lignées cellulaires non transformées dans
le but d’étudier le catabolisme normal du peptide β-amyloïde. De cette étude
émerge le concept que c’est la conformation du peptide amyloïde qui est
essentiellement déterminante dans sa clairance et donc probablement dans la
maladie d’Alzheimer. Par conséquent, il serait intéressant, en utilisant les mêmes
approches (plusieurs lignées cellulaire et différentes méthodes d’analyse),
d'étudier la clairance du peptide amyloïde par des cellules ayant subies diverses
modifications, mimant le vieillissement cellulaire, telles qu’une rupture de
l'homéostasie (ou stress). En effet, lors du vieillissement cellulaire, les enzymes,
responsables de l’élimination du peptide amyloïde, pourraient alors perdre leur
activité. Le peptide pouvant alors s’accumuler et former des dépôts.

112
Une fois les enzymes impliquées dans le catabolisme du peptide amyloïde
seront purifiées, il serait tout à fait possible d’imaginer l’utilisation de ces
protéases comme voie thérapeutique selon trois stratégies :
• La protéase est introduite dans le cerveau par une approche thérapie génique.
• L’activité endogène de la protéase peut être augmentée par un agent
pharmaceutique.
• La transcription du gène codant la protéase peut être sur activée par un
composant pharmaceutique.

113
REFERENCES BIBLIOGRAPHIQUES

114
Abraham, C. R., McGraw, W. T., Slot, F., and Yamin, R. (2000). Alpha 1-antichymotrypsin inhibits A beta degradation in vitro and in vivo. Ann N Y Acad Sci 920, 245-248.
Aleshkov, S., Abraham, C. R., and Zannis, V. I. (1997). Interaction of nascent ApoE2, ApoE3, and ApoE4 isoforms expressed in mammalian cells with amyloid peptide beta (1-40). Relevance to Alzheimer's disease. Biochemistry 36, 10571-10580.
Annaert, W., and De Strooper, B. (2002). A cell biological perspective on Alzheimer's disease. Annu Rev Cell Dev Biol 18, 25-51.
Arendt, T., Holzer, M., Grossmann, A., Zedlick, D., and Bruckner, M. K. (1995). Increased expression and subcellular translocation of the mitogen activated protein kinase kinase and mitogen-activated protein kinase in Alzheimer's disease. Neuroscience 68, 5-18.
Authier, F., Posner, B. I., and Bergeron, J. J. (1996). Insulin-degrading enzyme. Clin Invest Med 19, 149-160.
Bach, R., and Zardini, P. (1992). Long-acting angiotensin-converting enzyme inhibition: once-daily lisinopril versus twice-daily captopril in mild-to-moderate heart failure. Am J Cardiol 70, 70C-77C.
Backstrom, J. R., Lim, G. P., Cullen, M. J., and Tokes, Z. A. (1996). Matrix metalloproteinase-9 (MMP-9) is synthesized in neurons of the human hippocampus and is capable of degrading the amyloid-beta peptide (1-40). J Neurosci 16, 7910-7919.
Bales, K. R., Verina, T., Dodel, R. C., Du, Y., Altstiel, L., Bender, M., Hyslop, P., Johnstone, E. M., Little, S. P., Cummins, D. J., et al. (1997). Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet 17, 263-264.
Bard, F., Cannon, C., Barbour, R., Burke, R. L., Games, D., Grajeda, H., Guido, T., Hu, K., Huang, J., Johnson-Wood, K., et al. (2000). Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 6, 916-919.
Barnes, K., Doherty, S., and Turner, A. J. (1995). Endopeptidase-24.11 is the integral membrane peptidase initiating degradation of somatostatin in the hippocampus. J Neurochem 64, 1826-1832.

115
Barrow, C. J., Yasuda, A., Kenny, P. T., and Zagorski, M. G. (1992). Solution conformations and aggregational properties of synthetic amyloid beta-peptides of Alzheimer's disease. Analysis of circular dichroism spectra. J Mol Biol 225, 1075-1093.
Becker, A. B., and Roth, R. A. (1995). Insulysin and pitrilysin: insulin-degrading enzymes of mammals and bacteria. Methods Enzymol 248, 693-703.
Bennett, B. D., Babu-Khan, S., Loeloff, R., Louis, J. C., Curran, E., Citron, M., and Vassar, R. (2000). Expression analysis of BACE2 in brain and peripheral tissues. J Biol Chem 275, 20647-20651.
Bertrand, E., Brouillet, E., Caille, I., Bouillot, C., Cole, G. M., Prochiantz, A., and Allinquant, B. (2001). A short cytoplasmic domain of the amyloid precursor protein induces apoptosis in vitro and in vivo. Mol Cell Neurosci 18, 503-511.
Blacker, D., Wilcox, M. A., Laird, N. M., Rodes, L., Horvath, S. M., Go, R. C., Perry, R., Watson, B., Jr., Bassett, S. S., McInnis, M. G., et al. (1998). Alpha-2 macroglobulin is genetically associated with Alzheimer disease. Nat Genet 19, 357-360.
Borg, J. P., Ooi, J., Levy, E., and Margolis, B. (1996). The phosphotyrosine interaction domains of X11 and FE65 bind to distinct sites on the YENPTY motif of amyloid precursor protein. Mol Cell Biol 16, 6229-6241.
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 82, 239-259.
Brion, J. P. (1992). [Cellular lesions in Alzheimer's disease: structural and molecular analysis]. Bull Mem Acad R Med Belg 147, 481-489; discussion 490-481.
Buee-Scherrer, V., Buee, L., Hof, P. R., Leveugle, B., Gilles, C., Loerzel, A. J., Perl, D. P., and Delacourte, A. (1995). Neurofibrillary degeneration in amyotrophic lateral sclerosis/parkinsonism-dementia complex of Guam. Immunochemical characterization of tau proteins. Am J Pathol 146, 924-932.
Burdick, D., Soreghan, B., Kwon, M., Kosmoski, J., Knauer, M., Henschen, A., Yates, J., Cotman, C., and Glabe, C. (1992). Assembly and aggregation properties of synthetic Alzheimer's A4/beta amyloid peptide analogs. J Biol Chem 267, 546-554.
Busciglio, J., Lorenzo, A., and Yankner, B. A. (1992). Methodological variables in the assessment of beta amyloid neurotoxicity. Neurobiol Aging 13, 609-612.

116
Busciglio, J., Lorenzo, A., Yeh, J., and Yankner, B. A. (1995). beta-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron 14, 879-888.
Bush, A. I. (2003). The metallobiology of Alzheimer's disease. Trends Neurosci 26, 207-214.
Bush, A. I., Pettingell, W. H., Multhaup, G., d Paradis, M., Vonsattel, J. P., Gusella, J. F., Beyreuther, K., Masters, C. L., and Tanzi, R. E. (1994). Rapid induction of Alzheimer A beta amyloid formation by zinc. Science 265, 1464-1467.
Buxbaum, J. D., Liu, K. N., Luo, Y., Slack, J. L., Stocking, K. L., Peschon, J. J., Johnson, R. S., Castner, B. J., Cerretti, D. P., and Black, R. A. (1998). Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem 273, 27765-27767.
Cai, H., Wang, Y., McCarthy, D., Wen, H., Borchelt, D. R., Price, D. L., and Wong, P. C. (2001). BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci 4, 233-234.
Cao, X., and Sudhof, T. C. (2001). A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 293, 115-120.
Capaldi, A. P., and Radford, S. E. (1998). Kinetic studies of beta-sheet protein folding. Curr Opin Struct Biol 8, 86-92.
Carrell, R. W., and Lomas, D. A. (1997). Conformational disease. Lancet 350, 134-138.
Castano, E. M., Prelli, F., Wisniewski, T., Golabek, A., Kumar, R. A., Soto, C., and Frangione, B. (1995). Fibrillogenesis in Alzheimer's disease of amyloid beta peptides and apolipoprotein E. Biochem J 306 (Pt 2), 599-604.
Chartier-Harlin, M. C., Crawford, F., Houlden, H., Warren, A., Hughes, D., Fidani, L., Goate, A., Rossor, M., Roques, P., Hardy, J., and et al. (1991). Early-onset Alzheimer's disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature 353, 844-846.
Chesneau, V., Vekrellis, K., Rosner, M. R., and Selkoe, D. J. (2000). Purified recombinant insulin-degrading enzyme degrades amyloid beta-protein but does not promote its oligomerization. Biochem J 351 Pt 2, 509-516.

117
Citron, M., Vigo-Pelfrey, C., Teplow, D. B., Miller, C., Schenk, D., Johnston, J., Winblad, B., Venizelos, N., Lannfelt, L., and Selkoe, D. J. (1994). Excessive production of amyloid beta-protein by peripheral cells of symptomatic and presymptomatic patients carrying the Swedish familial Alzheimer disease mutation. Proc Natl Acad Sci U S A 91, 11993-11997.
Cochran, E., Bacci, B., Chen, Y., Patton, A., Gambetti, P., and Autilio-Gambetti, L. (1991). Amyloid precursor protein and ubiquitin immunoreactivity in dystrophic axons is not unique to Alzheimer's disease. Am J Pathol 139, 485-489.
Colciaghi, F., Borroni, B., Pastorino, L., Marcello, E., Zimmermann, M., Cattabeni, F., Padovani, A., and Di Luca, M. (2002). [alpha]-Secretase ADAM10 as well as [alpha]APPs is reduced in platelets and CSF of Alzheimer disease patients. Mol Med 8, 67-74.
Coleman, P. D., and Flood, D. G. (1987). Neuron numbers and dendritic extent in normal aging and Alzheimer's disease. Neurobiol Aging 8, 521-545.
Dahms, P., and Mentlein, R. (1992). Purification of the main somatostatin-degrading proteases from rat and pig brains, their action on other neuropeptides, and their identification as endopeptidases 24.15 and 24.16. Eur J Biochem 208, 145-154.
Das, H. K., McPherson, J., Bruns, G. A., Karathanasis, S. K., and Breslow, J. L. (1985). Isolation, characterization, and mapping to chromosome 19 of the human apolipoprotein E gene. J Biol Chem 260, 6240-6247.
Das, P., Murphy, M. P., Younkin, L. H., Younkin, S. G., and Golde, T. E. (2001). Reduced effectiveness of Abeta1-42 immunization in APP transgenic mice with significant amyloid deposition. Neurobiol Aging 22, 721-727.
De Jonghe, C., Zehr, C., Yager, D., Prada, C. M., Younkin, S., Hendriks, L., Van Broeckhoven, C., and Eckman, C. B. (1998). Flemish and Dutch mutations in amyloid beta precursor protein have different effects on amyloid beta secretion. Neurobiol Dis 5, 281-286.
Delacourte, A., David, J. P., Sergeant, N., Buee, L., Wattez, A., Vermersch, P., Ghozali, F., Fallet-Bianco, C., Pasquier, F., Lebert, F., et al. (1999). The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer's disease. Neurology 52, 1158-1165.
Demeester, N., Mertens, C., Caster, H., Goethals, M., Vandekerckhove, J., Rosseneu, M., and Labeur, C. (2001). Comparison of the aggregation properties,

118
secondary structure and apoptotic effects of wild-type, Flemish and Dutch N-terminally truncated amyloid beta peptides. Eur J Neurosci 13, 2015-2024.
Demuro, A., Mina, E., Kayed, R., Milton, S. C., Parker, I., and Glabe, C. G. (2005). Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem 280, 17294-17300.
Dhenain, M., Privat, N., Duyckaerts, C., and Jacobs, R. E. (2002). Senile plaques do not induce susceptibility effects in T2*-weighted MR microscopic images. NMR Biomed 15, 197-203.
Dlouhy, S. R., Hsiao, K., Farlow, M. R., Foroud, T., Conneally, P. M., Johnson, P., Prusiner, S. B., Hodes, M. E., and Ghetti, B. (1992). Linkage of the Indiana kindred of Gerstmann-Straussler-Scheinker disease to the prion protein gene. Nat Genet 1, 64-67.
Docherty, A. J., O'Connell, J., Crabbe, T., Angal, S., and Murphy, G. (1992). The matrix metalloproteinases and their natural inhibitors: prospects for treating degenerative tissue diseases. Trends Biotechnol 10, 200-207.
Dovey, H. F., John, V., Anderson, J. P., Chen, L. Z., de Saint Andrieu, P., Fang, L. Y., Freedman, S. B., Folmer, B., Goldbach, E., Holsztynska, E. J., et al. (2001). Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J Neurochem 76, 173-181.
Duyckaerts, C., Colle, M. A., Dessi, F., Grignon, Y., Piette, F., and Hauw, J. J. (1998). The progression of the lesions in Alzheimer disease: insights from a prospective clinicopathological study. J Neural Transm Suppl 53, 119-126.
Duyckaerts, C., Hauw, J. J., Piette, F., Rainsard, C., Poulain, V., Berthaux, P., and Escourolle, R. (1985). Cortical atrophy in senile dementia of the Alzheimer type is mainly due to a decrease in cortical length. Acta Neuropathol (Berl) 66, 72-74.
Eckman, C. B., Mehta, N. D., Crook, R., Perez-tur, J., Prihar, G., Pfeiffer, E., Graff-Radford, N., Hinder, P., Yager, D., Zenk, B., et al. (1997). A new pathogenic mutation in the APP gene (I716V) increases the relative proportion of A beta 42(43). Hum Mol Genet 6, 2087-2089.
Eckman, E. A., Reed, D. K., and Eckman, C. B. (2001). Degradation of the Alzheimer's amyloid beta peptide by endothelin-converting enzyme. J Biol Chem 276, 24540-24548.

119
Eckman, E. A., Watson, M., Marlow, L., Sambamurti, K., and Eckman, C. B. (2003). Alzheimer's disease beta-amyloid peptide is increased in mice deficient in endothelin-converting enzyme. J Biol Chem 278, 2081-2084.
Edbauer, D., Winkler, E., Haass, C., and Steiner, H. (2002). Presenilin and nicastrin regulate each other and determine amyloid beta-peptide production via complex formation. Proc Natl Acad Sci U S A 99, 8666-8671.
Emmerling, M. R., Watson, M. D., Raby, C. A., and Spiegel, K. (2000). The role of complement in Alzheimer's disease pathology. Biochim Biophys Acta 1502, 158-171.
Esch, F. S., Keim, P. S., Beattie, E. C., Blacher, R. W., Culwell, A. R., Oltersdorf, T., McClure, D., and Ward, P. J. (1990). Cleavage of amyloid beta peptide during constitutive processing of its precursor. Science 248, 1122-1124.
Evans, K. C., Berger, E. P., Cho, C. G., Weisgraber, K. H., and Lansbury, P. T., Jr. (1995). Apolipoprotein E is a kinetic but not a thermodynamic inhibitor of amyloid formation: implications for the pathogenesis and treatment of Alzheimer disease. Proc Natl Acad Sci U S A 92, 763-767.
Exley, C., and Korchazhkina, O. V. (2001). Plasmin cleaves Abeta42 in vitro and prevents its aggregation into beta-pleated sheet structures. Neuroreport 12, 2967-2970.
Fagan, A. M., Bu, G., Sun, Y., Daugherty, A., and Holtzman, D. M. (1996). Apolipoprotein E-containing high density lipoprotein promotes neurite outgrowth and is a ligand for the low density lipoprotein receptor-related protein. J Biol Chem 271, 30121-30125.
Fahrenholz, F., Gilbert, S., Kojro, E., Lammich, S., and Postina, R. (2000). Alpha-secretase activity of the disintegrin metalloprotease ADAM 10. Influences of domain structure. Ann N Y Acad Sci 920, 215-222.
Farooqui, A. A., and Horrocks, L. A. (1998). Lipid peroxides in the free radical pathophysiology of brain diseases. Cell Mol Neurobiol 18, 599-608.
Farris, W., Mansourian, S., Chang, Y., Lindsley, L., Eckman, E. A., Frosch, M. P., Eckman, C. B., Tanzi, R. E., Selkoe, D. J., and Guenette, S. (2003). Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A 100, 4162-4167.

120
Farzan, M., Schnitzler, C. E., Vasilieva, N., Leung, D., and Choe, H. (2000). BACE2, a beta -secretase homolog, cleaves at the beta site and within the amyloid-beta region of the amyloid-beta precursor protein. Proc Natl Acad Sci U S A 97, 9712-9717.
Fraering, P. C., LaVoie, M. J., Ye, W., Ostaszewski, B. L., Kimberly, W. T., Selkoe, D. J., and Wolfe, M. S. (2004). Detergent-dependent dissociation of active gamma-secretase reveals an interaction between Pen-2 and PS1-NTF and offers a model for subunit organization within the complex. Biochemistry 43, 323-333.
Frautschy, S. A., Baird, A., and Cole, G. M. (1991). Effects of injected Alzheimer beta-amyloid cores in rat brain. Proc Natl Acad Sci U S A 88, 8362-8366.
Friguet, B. (2002). Aging of proteins and the proteasome. Prog Mol Subcell Biol 29, 17-33.
Gainotti, G., Acciarri, A., Bizzarro, A., Marra, C., Masullo, C., Misciagna, S., Tartaglione, T., Valenza, A., and Colosimo, C. (2004). The role of brain infarcts and hippocampal atrophy in subcortical ischaemic vascular dementia. Neurol Sci 25, 192-197.
Geula, C., Wu, C. K., Saroff, D., Lorenzo, A., Yuan, M., and Yankner, B. A. (1998). Aging renders the brain vulnerable to amyloid beta-protein neurotoxicity. Nat Med 4, 827-831.
Giulian, D. (1999). Microglia and the immune pathology of Alzheimer disease. Am J Hum Genet 65, 13-18.
Glenner, G. G., and Wong, C. W. (1984). Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun 122, 1131-1135.
Goate, A. M. (1997). Molecular genetics of Alzheimer's disease. Geriatrics 52 Suppl 2, S9-12.
Goedert, M. (1993). Tau protein and the neurofibrillary pathology of Alzheimer's disease. Trends Neurosci 16, 460-465.
Goedert, M., Spillantini, M. G., and Davies, S. W. (1998). Filamentous nerve cell inclusions in neurodegenerative diseases. Curr Opin Neurobiol 8, 619-632.
Golde, T. E., Cai, X. D., Shoji, M., and Younkin, S. G. (1993). Production of amyloid beta protein from normal amyloid beta-protein precursor (beta APP) and the

121
mutated beta APPS linked to familial Alzheimer's disease. Ann N Y Acad Sci 695, 103-108.
Golde, T. E., and Younkin, S. G. (2001). Presenilins as therapeutic targets for the treatment of Alzheimer's disease. Trends Mol Med 7, 264-269.
Goldgaber, D., Lerman, M. I., McBride, O. W., Saffiotti, U., and Gajdusek, D. C. (1987). Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer's disease. Science 235, 877-880.
Gong, C. X., Grundke-Iqbal, I., and Iqbal, K. (1994). Dephosphorylation of Alzheimer's disease abnormally phosphorylated tau by protein phosphatase-2A. Neuroscience 61, 765-772.
Grune, T., Reinheckel, T., and Davies, K. J. (1997). Degradation of oxidized proteins in mammalian cells. Faseb J 11, 526-534.
Haass, C., Lemere, C. A., Capell, A., Citron, M., Seubert, P., Schenk, D., Lannfelt, L., and Selkoe, D. J. (1995). The Swedish mutation causes early-onset Alzheimer's disease by beta-secretase cleavage within the secretory pathway. Nat Med 1, 1291-1296.
Haass, C., Schlossmacher, M. G., Hung, A. Y., Vigo-Pelfrey, C., Mellon, A., Ostaszewski, B. L., Lieberburg, I., Koo, E. H., Schenk, D., Teplow, D. B., and et al. (1992). Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature 359, 322-325.
Haass, C., and Selkoe, D. J. (1993). Cellular processing of beta-amyloid precursor protein and the genesis of amyloid beta-peptide. Cell 75, 1039-1042.
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer's disease: the amyloid cascade hypothesis. Science 256, 184-185.
Hartley, D. M., Walsh, D. M., Ye, C. P., Diehl, T., Vasquez, S., Vassilev, P. M., Teplow, D. B., and Selkoe, D. J. (1999). Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci 19, 8876-8884.
Hendriks, L., van Duijn, C. M., Cras, P., Cruts, M., Van Hul, W., van Harskamp, F., Warren, A., McInnis, M. G., Antonarakis, S. E., Martin, J. J., and et al. (1992). Presenile dementia and cerebral haemorrhage linked to a mutation at codon 692 of the beta-amyloid precursor protein gene. Nat Genet 1, 218-221.

122
Hensley, K., Carney, J. M., Mattson, M. P., Aksenova, M., Harris, M., Wu, J. F., Floyd, R. A., and Butterfield, D. A. (1994). A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci U S A 91, 3270-3274.
Herreman, A., Serneels, L., Annaert, W., Collen, D., Schoonjans, L., and De Strooper, B. (2000). Total inactivation of gamma-secretase activity in presenilin-deficient embryonic stem cells. Nat Cell Biol 2, 461-462.
Holliday, R. (1989). X-chromosome reactivation and ageing. Nature 337, 311.
Holtzman, D. M., Bales, K. R., Tenkova, T., Fagan, A. M., Parsadanian, M., Sartorius, L. J., Mackey, B., Olney, J., McKeel, D., Wozniak, D., and Paul, S. M. (2000). Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 97, 2892-2897.
Hong, L., Koelsch, G., Lin, X., Wu, S., Terzyan, S., Ghosh, A. K., Zhang, X. C., and Tang, J. (2000). Structure of the protease domain of memapsin 2 (beta-secretase) complexed with inhibitor. Science 290, 150-153.
Hoshi, M., Sato, M., Matsumoto, S., Noguchi, A., Yasutake, K., Yoshida, N., and Sato, K. (2003). Spherical aggregates of beta-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3beta. Proc Natl Acad Sci U S A 100, 6370-6375.
Howell, S., Nalbantoglu, J., and Crine, P. (1995). Neutral endopeptidase can hydrolyze beta-amyloid(1-40) but shows no effect on beta-amyloid precursor protein metabolism. Peptides 16, 647-652.
Howlett, D. R., Jennings, K. H., Lee, D. C., Clark, M. S., Brown, F., Wetzel, R., Wood, S. J., Camilleri, P., and Roberts, G. W. (1995). Aggregation state and neurotoxic properties of Alzheimer beta-amyloid peptide. Neurodegeneration 4, 23-32.
Hu, J., Igarashi, A., Kamata, M., and Nakagawa, H. (2001). Angiotensin-converting enzyme degrades Alzheimer amyloid beta-peptide (A beta); retards A beta aggregation, deposition, fibril formation; and inhibits cytotoxicity. J Biol Chem 276, 47863-47868.
Huang, Y., Liu, X. Q., Wyss-Coray, T., Brecht, W. J., Sanan, D. A., and Mahley, R. W. (2001). Apolipoprotein E fragments present in Alzheimer's disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc Natl Acad Sci U S A 98, 8838-8843.

123
Hughes, S. R., Khorkova, O., Goyal, S., Knaeblein, J., Heroux, J., Riedel, N. G., and Sahasrabudhe, S. (1998). Alpha2-macroglobulin associates with beta-amyloid peptide and prevents fibril formation. Proc Natl Acad Sci U S A 95, 3275-3280.
Hussain, I., Powell, D., Howlett, D. R., Tew, D. G., Meek, T. D., Chapman, C., Gloger, I. S., Murphy, K. E., Southan, C. D., Ryan, D. M., et al. (1999). Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol Cell Neurosci 14, 419-427.
Ida, N., Masters, C. L., and Beyreuther, K. (1996). Rapid cellular uptake of Alzheimer amyloid betaA4 peptide by cultured human neuroblastoma cells. FEBS Lett 394, 174-178.
Iqbal, K., Zaidi, T., Bancher, C., and Grundke-Iqbal, I. (1994). Alzheimer paired helical filaments. Restoration of the biological activity by dephosphorylation. FEBS Lett 349, 104-108.
Irizarry, M. C., Soriano, F., McNamara, M., Page, K. J., Schenk, D., Games, D., and Hyman, B. T. (1997). Abeta deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci 17, 7053-7059.
Iversen, L. L., Mortishire-Smith, R. J., Pollack, S. J., and Shearman, M. S. (1995). The toxicity in vitro of beta-amyloid protein. Biochem J 311 (Pt 1), 1-16.
Iwata, N., Tsubuki, S., Takaki, Y., Shirotani, K., Lu, B., Gerard, N. P., Gerard, C., Hama, E., Lee, H. J., and Saido, T. C. (2001). Metabolic regulation of brain Abeta by neprilysin. Science 292, 1550-1552.
Iwata, N., Tsubuki, S., Takaki, Y., Watanabe, K., Sekiguchi, M., Hosoki, E., Kawashima-Morishima, M., Lee, H. J., Hama, E., Sekine-Aizawa, Y., and Saido, T. C. (2000). Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med 6, 143-150.
Jarrett, J. T., Berger, E. P., and Lansbury, P. T., Jr. (1993). The C-terminus of the beta protein is critical in amyloidogenesis. Ann N Y Acad Sci 695, 144-148.
Jarrett, J. T., and Lansbury, P. T., Jr. (1993). Seeding "one-dimensional crystallization" of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell 73, 1055-1058.
Jick, H., Zornberg, G. L., Jick, S. S., Seshadri, S., and Drachman, D. A. (2000). Statins and the risk of dementia. Lancet 356, 1627-1631.

124
Johnson, G. D., Stevenson, T., and Ahn, K. (1999). Hydrolysis of peptide hormones by endothelin-converting enzyme-1. A comparison with neprilysin. J Biol Chem 274, 4053-4058.
Kamal, A., Almenar-Queralt, A., LeBlanc, J. F., Roberts, E. A., and Goldstein, L. S. (2001). Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature 414, 643-648.
Kamenetz, F., Tomita, T., Hsieh, H., Seabrook, G., Borchelt, D., Iwatsubo, T., Sisodia, S., and Malinow, R. (2003). APP processing and synaptic function. Neuron 37, 925-937.
Kang, D. E., Saitoh, T., Chen, X., Xia, Y., Masliah, E., Hansen, L. A., Thomas, R. G., Thal, L. J., and Katzman, R. (1997). Genetic association of the low-density lipoprotein receptor-related protein gene (LRP), an apolipoprotein E receptor, with late-onset Alzheimer's disease. Neurology 49, 56-61.
Kang, J., Lemaire, H. G., Unterbeck, A., Salbaum, J. M., Masters, C. L., Grzeschik, K. H., Multhaup, G., Beyreuther, K., and Muller-Hill, B. (1987). The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature 325, 733-736.
Kawahara, M., Muramoto, K., Kobayashi, K., Mori, H., and Kuroda, Y. (1994). Aluminum promotes the aggregation of Alzheimer's amyloid beta-protein in vitro. Biochem Biophys Res Commun 198, 531-535.
Keller, J. N., Huang, F. F., and Markesbery, W. R. (2000). Decreased levels of proteasome activity and proteasome expression in aging spinal cord. Neuroscience 98, 149-156.
Kelly, J. W. (1998). The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr Opin Struct Biol 8, 101-106.
Kerr, M. A., and Kenny, A. J. (1974). The purification and specificity of a neutral endopeptidase from rabbit kidney brush border. Biochem J 137, 477-488.
Klein, W. L., Krafft, G. A., and Finch, C. E. (2001). Targeting small Abeta oligomers: the solution to an Alzheimer's disease conundrum? Trends Neurosci 24, 219-224.
Knauer, M. F., Soreghan, B., Burdick, D., Kosmoski, J., and Glabe, C. G. (1992). Intracellular accumulation and resistance to degradation of the Alzheimer amyloid A4/beta protein. Proc Natl Acad Sci U S A 89, 7437-7441.

125
Koike, H., Tomioka, S., Sorimachi, H., Saido, T. C., Maruyama, K., Okuyama, A., Fujisawa-Sehara, A., Ohno, S., Suzuki, K., and Ishiura, S. (1999). Membrane-anchored metalloprotease MDC9 has an alpha-secretase activity responsible for processing the amyloid precursor protein. Biochem J 343 Pt 2, 371-375.
Kojro, E., Gimpl, G., Lammich, S., Marz, W., and Fahrenholz, F. (2001). Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha -secretase ADAM 10. Proc Natl Acad Sci U S A 98, 5815-5820.
Koo, E. H., and Squazzo, S. L. (1994). Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem 269, 17386-17389.
Kowall, N. W. (1994). Beta amyloid neurotoxicity and neuronal degeneration in Alzheimer's disease. Neurobiol Aging 15, 257-258.
Kowall, N. W., Beal, M. F., Busciglio, J., Duffy, L. K., and Yankner, B. A. (1991). An in vivo model for the neurodegenerative effects of beta amyloid and protection by substance P. Proc Natl Acad Sci U S A 88, 7247-7251.
Kumar-Singh, S., Julliams, A., Nuydens, R., Ceuterick, C., Labeur, C., Serneels, S., Vennekens, K., Van Osta, P., Geerts, H., De Strooper, B., and Van Broeckhoven, C. (2002). In vitro studies of Flemish, Dutch, and wild-type beta-amyloid provide evidence for two-staged neurotoxicity. Neurobiol Dis 11, 330-340.
Kuo, W. L., Montag, A. G., and Rosner, M. R. (1993). Insulin-degrading enzyme is differentially expressed and developmentally regulated in various rat tissues. Endocrinology 132, 604-611.
Kurochkin, I. V. (1998). Amyloidogenic determinant as a substrate recognition motif of insulin-degrading enzyme. FEBS Lett 427, 153-156.
Kurochkin, I. V. (2001). Insulin-degrading enzyme: embarking on amyloid destruction. Trends Biochem Sci 26, 421-425.
Lahiri, D. K. (1993). The stability of beta-amyloid precursor protein in nine different cell types. Biochem Mol Biol Int 29, 849-858.
Lambert, M. P., Barlow, A. K., Chromy, B. A., Edwards, C., Freed, R., Liosatos, M., Morgan, T. E., Rozovsky, I., Trommer, B., Viola, K. L., et al. (1998). Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A 95, 6448-6453.

126
Ledesma, M. D., Da Silva, J. S., Crassaerts, K., Delacourte, A., De Strooper, B., and Dotti, C. G. (2000). Brain plasmin enhances APP alpha-cleavage and Abeta degradation and is reduced in Alzheimer's disease brains. EMBO Rep 1, 530-535.
Lesne, S., Docagne, F., Gabriel, C., Liot, G., Lahiri, D. K., Buee, L., Plawinski, L., Delacourte, A., MacKenzie, E. T., Buisson, A., and Vivien, D. (2003). Transforming growth factor-beta 1 potentiates amyloid-beta generation in astrocytes and in transgenic mice. J Biol Chem 278, 18408-18418.
Levy-Lahad, E., Wasco, W., Poorkaj, P., Romano, D. M., Oshima, J., Pettingell, W. H., Yu, C. E., Jondro, P. D., Schmidt, S. D., Wang, K., and et al. (1995). Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science 269, 973-977.
Lin, M. T., Simon, D. K., Ahn, C. H., Kim, L. M., and Beal, M. F. (2002). High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer's disease brain. Hum Mol Genet 11, 133-145.
Lin, X., Koelsch, G., Wu, S., Downs, D., Dashti, A., and Tang, J. (2000). Human aspartic protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor protein. Proc Natl Acad Sci U S A 97, 1456-1460.
Lomakin, A., Chung, D. S., Benedek, G. B., Kirschner, D. A., and Teplow, D. B. (1996). On the nucleation and growth of amyloid beta-protein fibrils: detection of nuclei and quantitation of rate constants. Proc Natl Acad Sci U S A 93, 1125-1129.
Lorenzo, A., and Yankner, B. A. (1994). Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc Natl Acad Sci U S A 91, 12243-12247.
Luo, Y., Bolon, B., Kahn, S., Bennett, B. D., Babu-Khan, S., Denis, P., Fan, W., Kha, H., Zhang, J., Gong, Y., et al. (2001). Mice deficient in BACE1, the Alzheimer's beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat Neurosci 4, 231-232.
Ma, J., Yee, A., Brewer, H. B., Jr., Das, S., and Potter, H. (1994). Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature 372, 92-94.
Mahley, R. W. (1988). Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science 240, 622-630.

127
Malchiodi-Albedi, F., Domenici, M. R., Paradisi, S., Bernardo, A., Ajmone-Cat, M. A., and Minghetti, L. (2001). Astrocytes contribute to neuronal impairment in beta A toxicity increasing apoptosis in rat hippocampal neurons. Glia 34, 68-72.
Mandal, P. K., and Pettegrew, J. W. (2004). Alzheimer's disease: NMR studies of asialo (GM1) and trisialo (GT1b) ganglioside interactions with Abeta(1-40) peptide in a membrane mimic environment. Neurochem Res 29, 447-453.
Mandavilli, B. S., Santos, J. H., and Van Houten, B. (2002). Mitochondrial DNA repair and aging. Mutat Res 509, 127-151.
Mann, D. M. (1988). Alzheimer's disease and Down's syndrome. Histopathology 13, 125-137.
Marcinkiewicz, M., and Seidah, N. G. (2000). Coordinated expression of beta-amyloid precursor protein and the putative beta-secretase BACE and alpha-secretase ADAM10 in mouse and human brain. J Neurochem 75, 2133-2143.
Marquez-Sterling, N. R., Lo, A. C., Sisodia, S. S., and Koo, E. H. (1997). Trafficking of cell-surface beta-amyloid precursor protein: evidence that a sorting intermediate participates in synaptic vesicle recycling. J Neurosci 17, 140-151.
Matsas, R., Fulcher, I. S., Kenny, A. J., and Turner, A. J. (1983). Substance P and [Leu]enkephalin are hydrolyzed by an enzyme in pig caudate synaptic membranes that is identical with the endopeptidase of kidney microvilli. Proc Natl Acad Sci U S A 80, 3111-3115.
Mattson, M. P. (1997). Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev 77, 1081-1132.
McDermott, J. R., and Gibson, A. M. (1997). Degradation of Alzheimer's beta-amyloid protein by human and rat brain peptidases: involvement of insulin-degrading enzyme. Neurochem Res 22, 49-56.
McGeer, P. L., McGeer, E. G., and Suzuki, J. S. (1977). Aging and extrapyramidal function. Arch Neurol 34, 33-35.
McLean, C. A., Cherny, R. A., Fraser, F. W., Fuller, S. J., Smith, M. J., Beyreuther, K., Bush, A. I., and Masters, C. L. (1999). Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol 46, 860-866.

128
Mentlein, R., Ludwig, R., and Martensen, I. (1998). Proteolytic degradation of Alzheimer's disease amyloid beta-peptide by a metalloproteinase from microglia cells. J Neurochem 70, 721-726.
Miller, D. L., Papayannopoulos, I. A., Styles, J., Bobin, S. A., Lin, Y. Y., Biemann, K., and Iqbal, K. (1993). Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer's disease. Arch Biochem Biophys 301, 41-52.
Morais, V. A., Crystal, A. S., Pijak, D. S., Carlin, D., Costa, J., Lee, V. M., and Doms, R. W. (2003). The transmembrane domain region of nicastrin mediates direct interactions with APH-1 and the gamma-secretase complex. J Biol Chem 278, 43284-43291.
Morelli, L., Llovera, R. E., Mathov, I., Lue, L. F., Frangione, B., Ghiso, J., and Castano, E. M. (2004). Insulin-degrading enzyme in brain microvessels: proteolysis of amyloid {beta} vasculotropic variants and reduced activity in cerebral amyloid angiopathy. J Biol Chem 279, 56004-56013.
Mori, H., Takio, K., Ogawara, M., and Selkoe, D. J. (1992). Mass spectrometry of purified amyloid beta protein in Alzheimer's disease. J Biol Chem 267, 17082-17086.
Mukherjee, A., Song, E., Kihiko-Ehmann, M., Goodman, J. P., Jr., Pyrek, J. S., Estus, S., and Hersh, L. B. (2000). Insulysin hydrolyzes amyloid beta peptides to products that are neither neurotoxic nor deposit on amyloid plaques. J Neurosci 20, 8745-8749.
Munson, G. W., Roher, A. E., Kuo, Y. M., Gilligan, S. M., Reardon, C. A., Getz, G. S., and LaDu, M. J. (2000). SDS-stable complex formation between native apolipoprotein E3 and beta-amyloid peptides. Biochemistry 39, 16119-16124.
Murakami, K., Irie, K., Morimoto, A., Ohigashi, H., Shindo, M., Nagao, M., Shimizu, T., and Shirasawa, T. (2002). Synthesis, aggregation, neurotoxicity, and secondary structure of various A beta 1-42 mutants of familial Alzheimer's disease at positions 21-23. Biochem Biophys Res Commun 294, 5-10.
Murayama, O., Tomita, T., Nihonmatsu, N., Murayama, M., Sun, X., Honda, T., Iwatsubo, T., and Takashima, A. (1999). Enhancement of amyloid beta 42 secretion by 28 different presenilin 1 mutations of familial Alzheimer's disease. Neurosci Lett 265, 61-63.

129
Naidu, A., Quon, D., and Cordell, B. (1995). beta-Amyloid peptide produced in vitro is degraded by proteinases released by cultured cells. J Biol Chem 270, 1369-1374.
Narita, M., Holtzman, D. M., Schwartz, A. L., and Bu, G. (1997). Alpha2-macroglobulin complexes with and mediates the endocytosis of beta-amyloid peptide via cell surface low-density lipoprotein receptor-related protein. J Neurochem 69, 1904-1911.
Neary, D., Snowden, J. S., Bowen, D. M., Sims, N. R., Mann, D. M., Benton, J. S., Northen, B., Yates, P. O., and Davison, A. N. (1986). Neuropsychological syndromes in presenile dementia due to cerebral atrophy. J Neurol Neurosurg Psychiatry 49, 163-174.
Nikodemova, M., Kasckow, J., Liu, H., Manganiello, V., and Aguilera, G. (2003). Cyclic adenosine 3',5'-monophosphate regulation of corticotropin-releasing hormone promoter activity in AtT-20 cells and in a transformed hypothalamic cell line. Endocrinology 144, 1292-1300.
Nilsberth, C., Westlind-Danielsson, A., Eckman, C. B., Condron, M. M., Axelman, K., Forsell, C., Stenh, C., Luthman, J., Teplow, D. B., Younkin, S. G., et al. (2001). The 'Arctic' APP mutation (E693G) causes Alzheimer's disease by enhanced Abeta protofibril formation. Nat Neurosci 4, 887-893.
Ninomiya, H., Roch, J. M., Sundsmo, M. P., Otero, D. A., and Saitoh, T. (1993). Amino acid sequence RERMS represents the active domain of amyloid beta/A4 protein precursor that promotes fibroblast growth. J Cell Biol 121, 879-886.
Nunan, J., and Small, D. H. (2002). Proteolytic processing of the amyloid-beta protein precursor of Alzheimer's disease. Essays Biochem 38, 37-49.
Ohsawa, I., Takamura, C., Morimoto, T., Ishiguro, M., and Kohsaka, S. (1999). Amino-terminal region of secreted form of amyloid precursor protein stimulates proliferation of neural stem cells. Eur J Neurosci 11, 1907-1913.
Okada, K., Wangpoengtrakul, C., Osawa, T., Toyokuni, S., Tanaka, K., and Uchida, K. (1999). 4-Hydroxy-2-nonenal-mediated impairment of intracellular proteolysis during oxidative stress. Identification of proteasomes as target molecules. J Biol Chem 274, 23787-23793.
Oltersdorf, T., Fritz, L. C., Schenk, D. B., Lieberburg, I., Johnson-Wood, K. L., Beattie, E. C., Ward, P. J., Blacher, R. W., Dovey, H. F., and Sinha, S. (1989). The secreted form of the Alzheimer's amyloid precursor protein with the Kunitz domain is protease nexin-II. Nature 341, 144-147.

130
Opazo, C., Huang, X., Cherny, R. A., Moir, R. D., Roher, A. E., White, A. R., Cappai, R., Masters, C. L., Tanzi, R. E., Inestrosa, N. C., and Bush, A. I. (2002). Metalloenzyme-like activity of Alzheimer's disease beta-amyloid. Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H(2)O(2). J Biol Chem 277, 40302-40308.
Paresce, D. M., Ghosh, R. N., and Maxfield, F. R. (1996). Microglial cells internalize aggregates of the Alzheimer's disease amyloid beta-protein via a scavenger receptor. Neuron 17, 553-565.
Parvathy, S., Hussain, I., Karran, E. H., Turner, A. J., and Hooper, N. M. (1998). Alzheimer's amyloid precursor protein alpha-secretase is inhibited by hydroxamic acid-based zinc metalloprotease inhibitors: similarities to the angiotensin converting enzyme secretase. Biochemistry 37, 1680-1685.
Periz, G., and Fortini, M. E. (2000). Proteolysis in Alzheimer's disease. Can plasmin tip the balance? EMBO Rep 1, 477-478.
Perutz, M. F. (1996). Glutamine repeats and inherited neurodegenerative diseases: molecular aspects. Curr Opin Struct Biol 6, 848-858.
Pierotti, A., Dong, K. W., Glucksman, M. J., Orlowski, M., and Roberts, J. L. (1990). Molecular cloning and primary structure of rat testes metalloendopeptidase EC 3.4.24.15. Biochemistry 29, 10323-10329.
Pietrzik, C. U., Hoffmann, J., Stober, K., Chen, C. Y., Bauer, C., Otero, D. A., Roch, J. M., and Herzog, V. (1998). From differentiation to proliferation: the secretory amyloid precursor protein as a local mediator of growth in thyroid epithelial cells. Proc Natl Acad Sci U S A 95, 1770-1775.
Pike, C. J., Cummings, B. J., and Cotman, C. W. (1992). beta-Amyloid induces neuritic dystrophy in vitro: similarities with Alzheimer pathology. Neuroreport 3, 769-772.
Pike, C. J., Walencewicz, A. J., Glabe, C. G., and Cotman, C. W. (1991). In vitro aging of beta-amyloid protein causes peptide aggregation and neurotoxicity. Brain Res 563, 311-314.
Ponte, P., Gonzalez-DeWhitt, P., Schilling, J., Miller, J., Hsu, D., Greenberg, B., Davis, K., Wallace, W., Lieberburg, I., and Fuller, F. (1988). A new A4 amyloid mRNA contains a domain homologous to serine proteinase inhibitors. Nature 331, 525-527.
Prusiner, S. B. (1997). Prion diseases and the BSE crisis. Science 278, 245-251.

131
Qiu, W. Q., Walsh, D. M., Ye, Z., Vekrellis, K., Zhang, J., Podlisny, M. B., Rosner, M. R., Safavi, A., Hersh, L. B., and Selkoe, D. J. (1998). Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J Biol Chem 273, 32730-32738.
Qiu, W. Q., Ye, Z., Kholodenko, D., Seubert, P., and Selkoe, D. J. (1997). Degradation of amyloid beta-protein by a metalloprotease secreted by microglia and other neural and non-neural cells. J Biol Chem 272, 6641-6646.
Rawlings, N. D., and Barrett, A. J. (1995). Evolutionary families of metallopeptidases. Methods Enzymol 248, 183-228.
Rocchi, A., Pellegrini, S., Siciliano, G., and Murri, L. (2003). Causative and susceptibility genes for Alzheimer's disease: a review. Brain Res Bull 61, 1-24.
Rochet, J. C., and Lansbury, P. T., Jr. (2000). Amyloid fibrillogenesis: themes and variations. Curr Opin Struct Biol 10, 60-68.
Rogaev, E. I., Sherrington, R., Rogaeva, E. A., Levesque, G., Ikeda, M., Liang, Y., Chi, H., Lin, C., Holman, K., Tsuda, T., and et al. (1995). Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature 376, 775-778.
Rogers, J., Cooper, N. R., Webster, S., Schultz, J., McGeer, P. L., Styren, S. D., Civin, W. H., Brachova, L., Bradt, B., Ward, P., and et al. (1992). Complement activation by beta-amyloid in Alzheimer disease. Proc Natl Acad Sci U S A 89, 10016-10020.
Rogers, J., and Lue, L. F. (2001). Microglial chemotaxis, activation, and phagocytosis of amyloid beta-peptide as linked phenomena in Alzheimer's disease. Neurochem Int 39, 333-340.
Roher, A. E., Kasunic, T. C., Woods, A. S., Cotter, R. J., Ball, M. J., and Fridman, R. (1994). Proteolysis of A beta peptide from Alzheimer disease brain by gelatinase A. Biochem Biophys Res Commun 205, 1755-1761.
Roher, A. E., Lowenson, J. D., Clarke, S., Wolkow, C., Wang, R., Cotter, R. J., Reardon, I. M., Zurcher-Neely, H. A., Heinrikson, R. L., Ball, M. J., and et al. (1993). Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer's disease. J Biol Chem 268, 3072-3083.

132
Roques, B. P., Noble, F., Dauge, V., Fournie-Zaluski, M. C., and Beaumont, A. (1993). Neutral endopeptidase 24.11: structure, inhibition, and experimental and clinical pharmacology. Pharmacol Rev 45, 87-146.
Sandbrink, R., Masters, C. L., and Beyreuther, K. (1994). Similar alternative splicing of a non-homologous domain in beta A4-amyloid protein precursor-like proteins. J Biol Chem 269, 14227-14234.
Sayre, L. M., Perry, G., Harris, P. L., Liu, Y., Schubert, K. A., and Smith, M. A. (2000). In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer's disease: a central role for bound transition metals. J Neurochem 74, 270-279.
Sayre, L. M., Smith, M. A., and Perry, G. (2001). Chemistry and biochemistry of oxidative stress in neurodegenerative disease. Curr Med Chem 8, 721-738.
Selkoe, D. J. (1991). The molecular pathology of Alzheimer's disease. Neuron 6, 487-498.
Selkoe, D. J. (1993). Physiological production of the beta-amyloid protein and the mechanism of Alzheimer's disease. Trends Neurosci 16, 403-409.
Selkoe, D. J. (1997). Alzheimer's disease: genotypes, phenotypes, and treatments. Science 275, 630-631.
Selkoe, D. J. (2001). Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 81, 741-766.
Serpell, L. C. (2000). Alzheimer's amyloid fibrils: structure and assembly. Biochim Biophys Acta 1502, 16-30.
Seubert, P., Vigo-Pelfrey, C., Esch, F., Lee, M., Dovey, H., Davis, D., Sinha, S., Schlossmacher, M., Whaley, J., Swindlehurst, C., and et al. (1992). Isolation and quantification of soluble Alzheimer's beta-peptide from biological fluids. Nature 359, 325-327.
Sherrington, R., Froelich, S., Sorbi, S., Campion, D., Chi, H., Rogaeva, E. A., Levesque, G., Rogaev, E. I., Lin, C., Liang, Y., et al. (1996). Alzheimer's disease associated with mutations in presenilin 2 is rare and variably penetrant. Hum Mol Genet 5, 985-988.
Sherrington, R., Rogaev, E. I., Liang, Y., Rogaeva, E. A., Levesque, G., Ikeda, M., Chi, H., Lin, C., Li, G., Holman, K., and et al. (1995). Cloning of a gene bearing

133
missense mutations in early-onset familial Alzheimer's disease. Nature 375, 754-760.
Shii, K., Baba, S., Yokono, K., and Roth, R. A. (1985). Covalent linkage of 125I-insulin to a cytosolic insulin-degrading enzyme. J Biol Chem 260, 6503-6506.
Shoji, M., Golde, T. E., Ghiso, J., Cheung, T. T., Estus, S., Shaffer, L. M., Cai, X. D., McKay, D. M., Tintner, R., Frangione, B., and et al. (1992). Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science 258, 126-129.
Simmons, L. K., May, P. C., Tomaselli, K. J., Rydel, R. E., Fuson, K. S., Brigham, E. F., Wright, S., Lieberburg, I., Becker, G. W., Brems, D. N., and et al. (1994). Secondary structure of amyloid beta peptide correlates with neurotoxic activity in vitro. Mol Pharmacol 45, 373-379.
Simons, M., Keller, P., Dichgans, J., and Schulz, J. B. (2001). Cholesterol and Alzheimer's disease: is there a link? Neurology 57, 1089-1093.
Sinha, S., Anderson, J. P., Barbour, R., Basi, G. S., Caccavello, R., Davis, D., Doan, M., Dovey, H. F., Frigon, N., Hong, J., et al. (1999). Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 402, 537-540.
Sisodia, S. S., Koo, E. H., Beyreuther, K., Unterbeck, A., and Price, D. L. (1990). Evidence that beta-amyloid protein in Alzheimer's disease is not derived by normal processing. Science 248, 492-495.
Small, G. W., Rabins, P. V., Barry, P. P., Buckholtz, N. S., DeKosky, S. T., Ferris, S. H., Finkel, S. I., Gwyther, L. P., Khachaturian, Z. S., Lebowitz, B. D., et al. (1997). Diagnosis and treatment of Alzheimer disease and related disorders. Consensus statement of the American Association for Geriatric Psychiatry, the Alzheimer's Association, and the American Geriatrics Society. Jama 278, 1363-1371.
Soto, C., Branes, M. C., Alvarez, J., and Inestrosa, N. C. (1994). Structural determinants of the Alzheimer's amyloid beta-peptide. J Neurochem 63, 1191-1198.
Sperber, B. R., Leight, S., Goedert, M., and Lee, V. M. (1995). Glycogen synthase kinase-3 beta phosphorylates tau protein at multiple sites in intact cells. Neurosci Lett 197, 149-153.
Steele, J. C. (1972). Progressive supranuclear palsy. Brain 95, 693-704.

134
Strittmatter, W. J., Weisgraber, K. H., Huang, D. Y., Dong, L. M., Salvesen, G. S., Pericak-Vance, M., Schmechel, D., Saunders, A. M., Goldgaber, D., and Roses, A. D. (1993). Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci U S A 90, 8098-8102.
Suh, Y. H. (1997). An etiological role of amyloidogenic carboxyl-terminal fragments of the beta-amyloid precursor protein in Alzheimer's disease. J Neurochem 68, 1781-1791.
Suzuki, N., Cheung, T. T., Cai, X. D., Odaka, A., Otvos, L., Jr., Eckman, C., Golde, T. E., and Younkin, S. G. (1994). An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science 264, 1336-1340.
Takashima, A., Noguchi, K., Michel, G., Mercken, M., Hoshi, M., Ishiguro, K., and Imahori, K. (1996). Exposure of rat hippocampal neurons to amyloid beta peptide (25-35) induces the inactivation of phosphatidyl inositol-3 kinase and the activation of tau protein kinase I/glycogen synthase kinase-3 beta. Neurosci Lett 203, 33-36.
Tanzi, R. E., Gusella, J. F., Watkins, P. C., Bruns, G. A., St George-Hyslop, P., Van Keuren, M. L., Patterson, D., Pagan, S., Kurnit, D. M., and Neve, R. L. (1987). Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science 235, 880-884.
Tjernberg, L. O., Callaway, D. J., Tjernberg, A., Hahne, S., Lilliehook, C., Terenius, L., Thyberg, J., and Nordstedt, C. (1999). A molecular model of Alzheimer amyloid beta-peptide fibril formation. J Biol Chem 274, 12619-12625.
Touchon, J., and Ritchie, K. (1999). Prodromal cognitive disorder in Alzheimer's disease. Int J Geriatr Psychiatry 14, 556-563.
Trojanowski, J. Q., and Lee, V. M. (1995). Phosphorylation of paired helical filament tau in Alzheimer's disease neurofibrillary lesions: focusing on phosphatases. Faseb J 9, 1570-1576.
Tsubuki, S., Takaki, Y., and Saido, T. C. (2003). Dutch, Flemish, Italian, and Arctic mutations of APP and resistance of Abeta to physiologically relevant proteolytic degradation. Lancet 361, 1957-1958.
Turner, A. J., and Murphy, L. J. (1996). Molecular pharmacology of endothelin converting enzymes. Biochem Pharmacol 51, 91-102.

135
Van Broeckhoven, C., Haan, J., Bakker, E., Hardy, J. A., Van Hul, W., Wehnert, A., Vegter-Van der Vlis, M., and Roos, R. A. (1990). Amyloid beta protein precursor gene and hereditary cerebral hemorrhage with amyloidosis (Dutch). Science 248, 1120-1122.
Van Nostrand, W. E., and Porter, M. (1999). Plasmin cleavage of the amyloid beta-protein: alteration of secondary structure and stimulation of tissue plasminogen activator activity. Biochemistry 38, 11570-11576.
Varadarajan, S., Yatin, S., Aksenova, M., and Butterfield, D. A. (2000). Review: Alzheimer's amyloid beta-peptide-associated free radical oxidative stress and neurotoxicity. J Struct Biol 130, 184-208.
Vassar, R., Bennett, B. D., Babu-Khan, S., Kahn, S., Mendiaz, E. A., Denis, P., Teplow, D. B., Ross, S., Amarante, P., Loeloff, R., et al. (1999). Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735-741.
Vekrellis, K., Ye, Z., Qiu, W. Q., Walsh, D., Hartley, D., Chesneau, V., Rosner, M. R., and Selkoe, D. J. (2000). Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by insulin-degrading enzyme. J Neurosci 20, 1657-1665.
Velazquez, P., Cribbs, D. H., Poulos, T. L., and Tenner, A. J. (1997). Aspartate residue 7 in amyloid beta-protein is critical for classical complement pathway activation: implications for Alzheimer's disease pathogenesis. Nat Med 3, 77-79.
Wallace, D. C. (1992). Mitochondrial genetics: a paradigm for aging and degenerative diseases? Science 256, 628-632.
Wallace, W. C., Akar, C. A., and Lyons, W. E. (1997). Amyloid precursor protein potentiates the neurotrophic activity of NGF. Brain Res Mol Brain Res 52, 201-212.
Watanabe, A., Hasegawa, M., Suzuki, M., Takio, K., Morishima-Kawashima, M., Titani, K., Arai, T., Kosik, K. S., and Ihara, Y. (1993). In vivo phosphorylation sites in fetal and adult rat tau. J Biol Chem 268, 25712-25717.
Weidemann, A., Konig, G., Bunke, D., Fischer, P., Salbaum, J. M., Masters, C. L., and Beyreuther, K. (1989). Identification, biogenesis, and localization of precursors of Alzheimer's disease A4 amyloid protein. Cell 57, 115-126.
Wischik, C. M., Novak, M., Thogersen, H. C., Edwards, P. C., Runswick, M. J., Jakes, R., Walker, J. E., Milstein, C., Roth, M., and Klug, A. (1988). Isolation of a

136
fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A 85, 4506-4510.
Wisniewski, T., Castano, E. M., Golabek, A., Vogel, T., and Frangione, B. (1994). Acceleration of Alzheimer's fibril formation by apolipoprotein E in vitro. Am J Pathol 145, 1030-1035.
Wolozin, B. (2001). A fluid connection: cholesterol and Abeta. Proc Natl Acad Sci U S A 98, 5371-5373.
Yamada, T., Miyazaki, K., Koshikawa, N., Takahashi, M., Akatsu, H., and Yamamoto, T. (1995). Selective localization of gelatinase A, an enzyme degrading beta-amyloid protein, in white matter microglia and in Schwann cells. Acta Neuropathol (Berl) 89, 199-203.
Yamin, R., Malgeri, E. G., Sloane, J. A., McGraw, W. T., and Abraham, C. R. (1999). Metalloendopeptidase EC 3.4.24.15 is necessary for Alzheimer's amyloid-beta peptide degradation. J Biol Chem 274, 18777-18784.
Yan, R., Bienkowski, M. J., Shuck, M. E., Miao, H., Tory, M. C., Pauley, A. M., Brashier, J. R., Stratman, N. C., Mathews, W. R., Buhl, A. E., et al. (1999). Membrane-anchored aspartyl protease with Alzheimer's disease beta-secretase activity. Nature 402, 533-537.
Yankner, B. A. (1996). Mechanisms of neuronal degeneration in Alzheimer's disease. Neuron 16, 921-932.
Yankner, B. A., Dawes, L. R., Fisher, S., Villa-Komaroff, L., Oster-Granite, M. L., and Neve, R. L. (1989). Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer's disease. Science 245, 417-420.
Yoshikai, S., Sasaki, H., Doh-ura, K., Furuya, H., and Sakaki, Y. (1990). Genomic organization of the human amyloid beta-protein precursor gene. Gene 87, 257-263.
Zhang, L., Song, L., Terracina, G., Liu, Y., Pramanik, B., and Parker, E. (2001). Biochemical characterization of the gamma-secretase activity that produces beta-amyloid peptides. Biochemistry 40, 5049-5055.

137
ANNEXES

138
ANNEXE 1
Lignées cellulaires humaines et non humaines représentatives du
Système Nerveux Central, du Système Immunitaire et des tissus périphériques.
CELLULES NEURONALES CELLULES NON NEURONALES
U-373 (glioblastome, astrocytome grade
III)
(ATCC HTB 17)
K-562 (monocyte de leucémie myelogène
chronique)
(ATCC CCL 243)
SH-SY5Y (neuroblastome)
(ICLC HTL 95013)
COS-7 (cellule rénale de singe vert
d’Afrique)
(ATCC CRL 1651)
SK-N-BE (neuroblastome)
(ICLC HTL 96015)
CHO (cellule ovarienne de hamster
chinois)
(ATCC CCL 61)
CHP-100 (neuroblastome) HFF (fibroblaste de peau de prépuce de
nouveau-né) (ATCC CRL 1634)
ATCC American Type Culture Collection ICLC Interlab Cell Line Collection CCL Certified Cell Line Bank CRL Cell Repository Line Bank HTL Human Tumor Line Bank HTB Human Tumor Cell Bank

139
ANNEXE 2
Composition des milieux de culture utilisés
MILIEU RPMI 1640
Amino-acides mg/L Vitamines mg/L Sels et composants mg/L L-arginine HCl 240 D-Biotine 0,200 Ca(NO3)2.4H2O 100 L-asparagine 50 Pantothénate de calcium
D 0,250 KCl 400
Acide L-aspartique 20 Chlorure de choline 3,000 MgSO4.7H2O 100 L-cystine 50 Acide folique 1,000 NaCl 6000
Acide L-glutamique 20 I-inositol 35,000 NaHCO3 2000 L-glutamine* 2mM Nicotinamide 1,000 Na2HPO4 (anhyd.) 800
Glycine 10 acide p-aminobenzoïque 1,000 D-Glucose 2000 L-histidine 15 Pyridoxal HCl 1,000 Glutathion réduit 1
L-hydroxyproline 20 Riboflavine 0,20 Rouge de Phénol - L-isoleucine 50 Thiamine HCl 1,000 HEPES -
L-leucine 50 Vitamine B12 0,005 L-lysine HCl 40 L-méthionine 15
L-phénylalanine 15 L-proline 20 L-serine 30
L-thréonine 20 L-tryptophane 5
L-tyrosine 20 L-valine 20
* rajoutée extemporanément
MILIEU ESSENTIEL MINIMUM DE EAGLE, MODIFICATION DE DULBECCO (DMEM)
Amino-acides mg/L Vitamines mg/L Sels et composants mg/LL-arginine HCl 84 Pantothénate de
calcium D 4,0 CaCl2.2H2O 264
L-cystine 48 chlorure de Choline 4,0 Fe(NO3).9H2O 0,1 L-glutamine* 2mM Acide folique 4,0 KCl 400
Glycine 30 I-inositol 7,2 MgSO4.7H2O 200 L-histidine HCl.H2O
42 Nicotinamide 4,0 NaCl 6400
L-isoleucine 105 Pyridoxine HCl 4,0 NaHCO3 3700 L-leucine 105 Riboflavine 0,4 NaH2PO4.2H2O 141
L-lysine HCl 146 Thiamine 4,0 D-glucose 1000 L-méthionine 30 Rouge de Phénol -
L-phenylalanine 66 HEPES - L-serine 42 Pyruvate de sodium 110
L-thréonine 95 L-tryptophane 16
L-tyrosine 72 L-valine 94
* rajoutée extemporanément

140
MELANGE NUTRITIF F-12 (HAM)
Amino-acides mg/L
Vitamines mg/L Sels et composants mg/L
L-alanine 8,90 Biotine 0,0073 CaCl2.2H2O 44 L-arginine HCl 211 Pantothénate de
calcium D 0,5 CuSO4.5 H2O 0,0024
L-asparagine 13 chlorure de Choline 14,0 FeSO4.7H2O 0,83 acide L-aspartique 13,30 Acide folique 1,3 KCl 223,60
L-cystéine HCl 36 I-inositol 18 MgCl2.6H2O 122 acide L-glutamique 14,70 Nicotinamide 0,036 NaCl 7599
L-glutamine* 2mM Pyridoxine HCl 0,06 NaHCO3 1176 L-alanyl-L-glutamine 217 Riboflavine 0,037 NaH2PO4 (anhyd.) 142
glycine 7,50 Thiamine HCl 0,3 ZnSO4.7H2O 0,86 L-histidine HCl.H2O 21 Vitamine B12 1,40 D-glucose 1802
L-isoleucine 4 Hypoxanthine 4 L-leucine 13 Acide linoléique 0,084
L-lysine HCl 36,50 Acide thioctique DL-68
0,20
L-méthionine 4,50 Rouge de Phénol 1,20 L-phenylalanine 5 Putréscine 2HCl 0,161
L-proline 34,50 Pyruvate de sodium 110 L-serine 10,50 Thymidine 0,70
L-thréonine 12 L-tryptophane 2
L-tyrosine 5,40 L-valine 11,70
* rajoutée extemporanément

141
ANNEXE 3
Concentrations des différents inhibiteurs utilisés.
Inhibiteurs concentration Type de protéases cibles
Aprotinine
ou
E64
ou
Leupeptine Pool I1
ou
Bestatine
ou
Pepstatine
0,3 µM
10 µM
10 µM
40 µM
1 µM
Sérine-protéase
Cystéine-protéase
Sérine- et Cystéine-protéase
Amino-peptidase
Aspartate-protéase
1,10-phénanthroline 1 mM Métalloprotéase
N-ethylmaleimide 1 mM Réactif des groupements thiol
Phosphoramidon
ou
Thiorphan Pool I2ou Captopril
10 µM
1 µM
1 mM
EC 3.4.24.11 (NEP), 24.71 (ECE-1)
NEP
EC 3.4.15.1 (ACE)
MMP Inhibitor Set-I Pool I3 1 µM MMP-1, -2, -3, -8, -9
Pefabloc® 1 mM sérine-protéase
Antipaïne
ou
Benzamidine
ou Pool I4Chymostatine
ou
α2-macroglobuline
100 µM
4 mM
100 µM
100 µM
Papaïne, trypsine
Trypsine-like protéase
Chymotrypsine-like protéase
Elastase, plasmine, thermolysine