cenni di cinetica elettrochimica la grandezza che …studenti.fisica.unifi.it/~aloisi/pdfs/cap ii...
TRANSCRIPT

Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
1
CENNI DI CINETICA ELETTROCHIMICA La grandezza che misura la velocità con cui avviene il processo
all’elettrodo è la densità di corrente, cioè la corrente che fluisce all’elettrodo attraverso l’unità di superficie.
Quando si realizzano le condizioni di equilibrio elettrochimico la corrente è nulla e all’interfase elettrodo/soluzione si stabilisce un potenziale che è quello caratteristico dell’equilibrio. Questo non vuol dire che all’elettrodo non ci sia un processo di trasferimento di carica, ma significa che per un processo
M+ + e → M
La velocità del processo diretto di riduzione uguaglia quella del
processo inverso di ossidazione, per cui il flusso di cariche che esce dall’elettrodo è uguale al flusso di carica che vi arriva. L’equilibrio cioè è un equilibrio dinamico.
Quando la cella è in funzione invece si registra una corrente netta ed il potenziale si discosta dal potenziale di equilibrio di una quantità che prende il nome di sovratensione, ηηηη = E – Eeq.
La dipendenza esponenziale di i da E fu per la prima volta messa in
evidenza da Tafel nella forma logaritmica:
E = a – b ln i
Dove a b sono due parametri sperimentali indipendenti da E. La legge di Tafel è stata verificata in innumerevoli casi e molti sono stati i modelli sviluppati per fornirne una giustificazione teorica.
Ci si riferisce al caso di un processo elettrochimico costituito dal solo stadio di trasferimento di carica. Più in generale, un processo elettrochimico è costituito da più stadi, chimici ed elettrochimici, più uno stadio di trasferimento di massa in soluzione. Quest’ultimo è lo stadio con cui la specie elettroattiva viene rifornita all’elettrodo (diffusione, migrazione o convezione).

Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
2
L’interfase Finché si parla di potenziale di equilibrio si resta nel campo della
termodinamica e non c’è necessità di analizzare in dettaglio la struttura dell’interfase. Quando invece ci si allontana dalle condizioni di equilibrio, come quando si registra un passaggio di corrente, non si possono più applicare le relazioni termodinamiche ed è necessario ricorrere ad un modello di interfase. Occorre ricordare che le relazioni termodinamiche hanno significato statistico in quanto descrivono il comportamento di un numero elevato di particelle, mentre per ricavare la relazione esistente tra corrente e potenziale occorre esaminare una singola particella. Di conseguenza diventa fondamentale la conoscenza dell’interfase.
Modello di Helmholtz Il primo tentativo di spiegare la natura dell’interfase è dovuto ad
Helmholtz (metà del IX secolo).
Nel suo modello l’interfase è vista come un condensatore a piatti piani e paralleli costituito da uno strato di cariche positive o negative su metallo ed uno strato di cariche di segno opposto in soluzione situato alla minima distanza possibile dall’elettrodo. Occorre infatti ricordare che anche in assenza di reazioni di trasferimento elettronico tra elettrodo e soluzione si stabilisce una differenza di potenziale per il solo fatto di mettere a contatto tra di loro due fasi conduttrici. Infatti
uno ione nella massa della soluzione e’ soggetto a forze che sono indipendenti dalla direzione, mentre alla superficie di separazione metallo-soluzione c’e’ una anisotropia delle forze, cioè le particelle cariche possono essere attratte o respinte dalla regione interfasale. Si crea così una separazione di cariche e quindi una differenza di potenziale che per quanto piccola può tradursi in un elevato campo elettrico a causa delle dimensioni ridotte della regione interfasale. Il concetto di due strati di carica di segno opposto e’ l’origine del termine “doppio strato” con in quale si indica la regione interfasale. E’ chiaro comunque che dovrà essere sempre rispettata la condizione di elettroneutralità per cui la carica su metallo dovrà essere uguale ma di segno opposto rispetto a quella sul lato soluzione.
x
φφφφ
-
-
-
-
-
-
-
+
+
+
+
+
+
Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
3
Analogamente a quanto accade per un condensatore elettrico, la caduta di potenziale tra i due piani di carica è lineare.
Un modello simile è estremamente rigido ed ignora l’effetto dirompente dell’agitazione termica che tende a demolire e a disperdere la parete rigida di carica elettrica. Il modello di Helmholtz è tuttavia in grado di spiegare l’esistenza di una capacità all’interfase sebbene non possa spiegarne né la dipendenza dal potenziale né il suo valore realmente misurato ad ogni potenziale. Infatti va sottolineato che l’analogia con un condensatore è limitata dal fatto che la capacità varia con il potenziale, tanto che in realtà si parla di capacità differenziale. In realtà, applicando la formula valida per un condensatore elettrico, C=ε/4πd, in cui ε è la costante dielettrica e d la distanza tra le due armature, attribuendo alla costante dielettrica il valore dell’acqua, ε =80, ed assumendo che la distanza tra le armature sia di circa 2 Å, si ottengono valori di C superiori di un ordine di grandezza superiori a quelli misurati sperimentalmente (tanto per fare un esempio, per Hg la capacità differenziale varia tra circa 10 e 40 µF cm-2, e per Ag ed Au è di poco superiore). Di fatto, non è giusto attribuire all’acqua interfasale lo stesso valore di costante dielettrica dell’acqua massiva in quanto la costante dielettrica è una grandezza termodinamica che come tale esprime il comportamento statistico di un numero notevolmente grande di molecole.
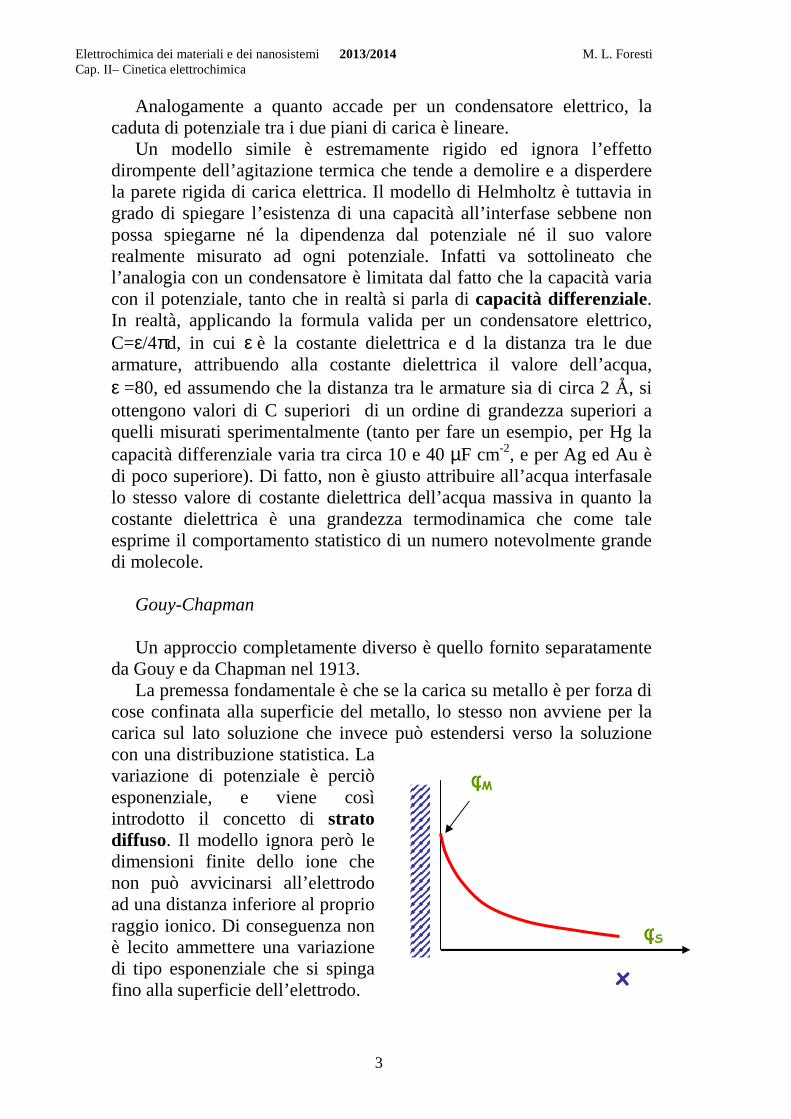
Gouy-Chapman Un approccio completamente diverso è quello fornito separatamente
da Gouy e da Chapman nel 1913. La premessa fondamentale è che se la carica su metallo è per forza di
cose confinata alla superficie del metallo, lo stesso non avviene per la carica sul lato soluzione che invece può estendersi verso la soluzione con una distribuzione statistica. La variazione di potenziale è perciò esponenziale, e viene così introdotto il concetto di strato diffuso. Il modello ignora però le dimensioni finite dello ione che non può avvicinarsi all’elettrodo ad una distanza inferiore al proprio raggio ionico. Di conseguenza non è lecito ammettere una variazione di tipo esponenziale che si spinga fino alla superficie dell’elettrodo.
φφφφS
x
φφφφM
Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
4
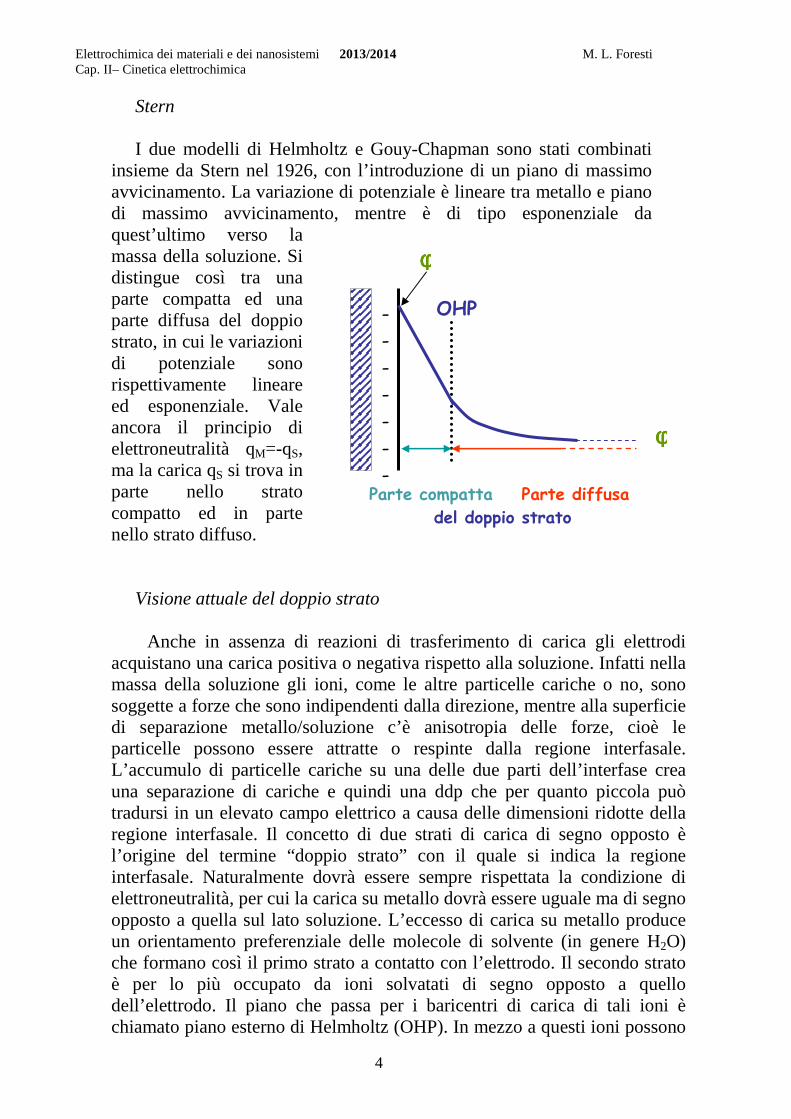
Stern I due modelli di Helmholtz e Gouy-Chapman sono stati combinati
insieme da Stern nel 1926, con l’introduzione di un piano di massimo avvicinamento. La variazione di potenziale è lineare tra metallo e piano di massimo avvicinamento, mentre è di tipo esponenziale da quest’ultimo verso la massa della soluzione. Si distingue così tra una parte compatta ed una parte diffusa del doppio strato, in cui le variazioni di potenziale sono rispettivamente lineare ed esponenziale. Vale ancora il principio di elettroneutralità qM=-qS, ma la carica qS si trova in parte nello strato compatto ed in parte nello strato diffuso.
Visione attuale del doppio strato
Anche in assenza di reazioni di trasferimento di carica gli elettrodi
acquistano una carica positiva o negativa rispetto alla soluzione. Infatti nella massa della soluzione gli ioni, come le altre particelle cariche o no, sono soggette a forze che sono indipendenti dalla direzione, mentre alla superficie di separazione metallo/soluzione c’è anisotropia delle forze, cioè le particelle possono essere attratte o respinte dalla regione interfasale. L’accumulo di particelle cariche su una delle due parti dell’interfase crea una separazione di cariche e quindi una ddp che per quanto piccola può tradursi in un elevato campo elettrico a causa delle dimensioni ridotte della regione interfasale. Il concetto di due strati di carica di segno opposto è l’origine del termine “doppio strato” con il quale si indica la regione interfasale. Naturalmente dovrà essere sempre rispettata la condizione di elettroneutralità, per cui la carica su metallo dovrà essere uguale ma di segno opposto a quella sul lato soluzione. L’eccesso di carica su metallo produce un orientamento preferenziale delle molecole di solvente (in genere H2O) che formano così il primo strato a contatto con l’elettrodo. Il secondo strato è per lo più occupato da ioni solvatati di segno opposto a quello dell’elettrodo. Il piano che passa per i baricentri di carica di tali ioni è chiamato piano esterno di Helmholtz (OHP). In mezzo a questi ioni possono
φφφφ
-
-
-
-
-
-
-
φφφφ
Parte compatta Parte diffusa
del doppio strato
OHP

Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
5
trovarsi altre molecole di solvente più debolmente legate all’elettrodo e quindi orientate in modi diversi.
In vicinanza dell’elettrodo gli ioni solvatati risentono della sua influenza, ma a distanze maggiori i moti termici (disordinanti) cominciano ad avere un’influenza confrontabile con quella (ordinante) esercitata dall’elettrodo. Il risultato è una regione tridimensionale, lo ”strato diffuso” (da non confondere con lo strato di diffusione), nella quale la concentrazione degli ioni che hanno segno opposto all’elettrodo decresce progressivamente fino a raggiungere una distribuzione omogenea nella massa della soluzione. Tanto gli ioni che popolano il piano esterno di Helmholtz quanto quelli che popolano lo strato diffuso sono trattenuti o respinti dalla regione interfasale da forze puramente colombiane e sono perciò considerati come adsorbiti aspecificamente.
L’andamento del potenziale è quello visto in precedenza per il modello di Stern (lineare dalla superficie del metallo all’OHP per poi tendere asintoticamente al valore nella massa della soluzione che per convenzione è di solito posto uguale a zero). Tuttavia la trattazione matematica dello strato diffuso è ancora quella proposta da Gouy-Chapman. (con l’unica modifica riguardante l’introduzione del piano di massimo avvicinamento) basata sull’equazione di Poisson per il potenziale elettrico e sulla legge di distribuzione di Boltzmann per gli ioni in soluzione. Dalla combinazione delle due si ha l’equazione di Poisson-Boltzmann che mette in relazione il potenziale elettrico con la densità di carica e quindi con la concentrazione degli ioni. Secondo l’equazione di Boltzmann il profilo di concentrazione è regolato da un fattore esponenziale il cui argomento è fornito dall'opposto del rapporto tra l'energia potenziale della particella e l'energia di agitazione termica (kT per una singola particella, o RT nel caso di una mole di particelle). In questo caso particolare l'energia potenziale della particella deriva esclusivamente dalla sua carica zi, e corrisponde perciò all'energia potenziale elettrostatica, quale risulta dal prodotto della carica di una mole di particelle (ziF) per il
potenziale elettrico φ(x) alla distanza x alla quale si valuta la concentrazione
-
-
-
-
-
-
- OHP
metallo dipolo H2O
ione positivo

Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
6
ci(x). Perciò, ponendo uguale a zero il potenziale nella massa della soluzione, l’equazione di Boltzmann diventa:
φ−=RT
)x(Fzexpc)x(c i
ii
Il fattore esponenziale di Boltzmann exp[-ziFφ(x)/RT] è minore di 1
quando il suo argomento è negativo, cioè quando la carica zi della particella
ed il potenziale elettrico φ(x) misurato rispetto alla massa della soluzione sono di ugual segno (ziFφ(x)>0); in questo caso infatti le forze elettrostatiche che agiscono all'interfase sono repulsive nei confronti della particella, e tendono quindi a produrne una rarefazione rispetto alla concentrazione massiva (ci(x)<ci).
Naturalmente, quanto più elevata e' la temperatura, tanto maggiore è l'energia di agitazione termica che tende a contrastare l'ordine creato da un potenziale elettrico φ(x) diverso da zero. Infatti il fattore esponenziale di Boltzmann diventa sempre più prossimo all'unità quanto maggiore
diventa l'energia di agitazione termica RT rispetto all'energia potenziale elettrostatica ziFφ(x). Considerazioni analoghe nel caso in cui zi ed il
potenziale elettrico φ(x) hanno segno opposto; in questo caso l'energia potenziale elettrostatica ziFφ(x) è negativa,e le forze che agiscono sulla particella sono attrattive.
La distribuzione di Boltzmann non è tuttavia sufficiente a valutare il profilo di concentrazione della specie i, in quanto il potenziale elettrico φ(x) è a sua volta influenzato dalla presenza di una carica netta non nulla alla distanza x=x; l'alterazione delle concentrazioni delle specie ioniche che popolano l'interfase rispetto ai corrispondenti valori massivi crea infatti una densità di carica ρ(x) diversa da zero. Per densità di carica ad una distanza x dobbiamo intendere la carica totale racchiusa in un elemento di volume dV piccolo rispetto alle dimensioni che cadono sotto la nostra percezione ma sufficientemente grande da contenere un numero elevato (e quindi statisticamente significativo) di specie cariche, divisa per dV. Ciò che si ottiene è una carica riferita all'unità di volume, ma valutata con riferimento ad una posizione spaziale ben precisa, e quindi ad una distanza ben precisa dalla superficie dell'elettrodo.
x
ci ci(x)
ziφ(x) < 0 : attrazione
ziφ(x) > 0 : repulsione

Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
7
Senza addentrarsi nel complesso trattamento matematico, basta riportare che esso porta a collegare tra loro il potenziale al piano esterno di Helmholtz, φd , la densità di carica su metallo, σ , e la concentrazione dell’elettrolita c±.
1 In pratica, dall’equazione di Gouy Chapmann si ricava che per σ=0 anche φd=0, ovvero il potenziale al piano esterno di Helmholtz coincide con il potenziale nella massa della soluzione e non si ha strato diffuso. I potenziali sono ovviamente quelli interni (o Galvani), ben diversi concettualmente dal potenziale applicato nel circuito esterno. Infatti la sola differenza di potenziale significativa da un punto di vista termodinamico è quella ai morsetti della pila costituita da due elettrodi, e non la differenza di potenziale esistente alla singola interfase. Il potenziale applicato dall’esterno per cui la densità di carica σ è nulla viene definito come “potenziale di carica zero”, Ez, ed è un parametro che caratterizza l’interfase. Ez è strettamente collegato al lavoro di estrazione dell’elettrone e può perciò variare anche a seconda dell’orientamento cristallografico del metallo. Ad esempio il potenziale di carica zero di Ag(111) è di circa -0.7 V/SCE, mentre quello di Ag(110) e di circa -1 V/SCE.
In conclusione, l’intera interfase risulta costituita da due condensatori messi in serie:
1C
= 1Ci
+ 1Cd
dove C rappresenta la capacità differenziale dell’intera interfase che è una
grandezza direttamente misurabile, mentre Ci e Cd rappresentano le capacità dello "strato interno" (o "strato compatto") e dello "strato diffuso" rispettivamente. Infatti, ponendo per convenzione uguale a zero il potenziale nella massa della soluzione, il potenziale φd coincide con la differenza di potenziale attraverso lo strato diffuso. Perciò, esprimendo il potenziale elettrico φd in base alla teoria di Gouy-Chapman, la capacità dello strato diffuso Cd coincide con la capacità di Gouy-Chapman, CGC ed ha un andamento simile a quello di una parabola con i rami rivolti verso l'alto. 2
1
φ
πε=σ ±
2
fsinh
cRT2 d2/1
con f≡F/RT
2 =
φ
πε=
φσ= ±
2
fcosh
cRT2
2
f
d
dC d
2/1
d
GC
φ
πε
= ±
2
fcosh
RT2
cF d
2/12

Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
8
Va ricordato che in un sistema di due condensatori posti in serie
predomina quello di capacità minore nel senso che la differenza di potenziale applicata alle estremità del sistema dei due condensatori si accumula per la maggior parte tra le armature di quello di capacità minore. Ora, la capacità dello strato interno Ci è indipendente dalla concentrazione dell'elettrolita mentre la capacità Cd dello strato diffuso dipende in modo
complesso da c± e da σ. Ne segue che per basse concentrazioni di elettrolita e nell'intorno del potenziale Ez di carica zero la capacità di GC, CGC, è inferiore a Ci, per cui predomina su quest'ultima condizionando il valore della capacità sperimentale C.
Per questo motivo C presenta un minimo piuttosto marcato per σ=0, però non cresce con il crescere del valore assoluto di σ così rapidamente come previsto dall’equazione per il semplice fatto che con il crescere di CGC la capacità totale C tende ad essere sempre più controllata da quella, Ci, dello strato interno. Per inciso, la posizione del minimo della capacità C a basse concentrazioni di un elettrolita aspecificamente adsorbito viene comunemente utilizzata per localizzare il potenziale Ez di carica zero.
Come esempio, la figura mostra la forte differenza nella posizione del minimo tra le facce (111) e (110), in accordo con le considerazioni finora fatte.
φφφφd
+ -
2/f de φ−
0
2/f2/f dd ee φ−φ +
2/f de φ
-1.0 -0.5 E / V(SCE)
50
C
µF cm-2
Ag(111)
Ag(110)

Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
9
Va sottolineato che le premesse su cui si basa la teoria di Gouy-Chapman esclude la presenza di adsorbimento specifico. Tuttavia, sebbene il primo strato a contatto con l’elettrodo sia costituito per lo più da molecole di solvente, vi si può trovare anche qualche specie ionica. Gli ioni a contatto con l’elettrodo sono quelli che non hanno sfera di solvatazione o che la hanno parzialmente persa. Per questi ioni si parla di adsorbimento specifico, perché l’adsorbimento non e’tanto legato alla carica dello ione quanto alla sua natura chimica, ed infatti si può avere adsorbimento specifico di ioni che hanno lo stesso segno dell’elettrodo. Il piano che passa per i baricentri di carica di tali ioni costituisce il piano interno di Helmholtz (IHP).
In definitiva la variazione di potenziale nella regione interfasale e’
lineare tra metallo e IHP, ancora lineare tra IHP e OHP ed esponenziale tra OHP e la massa della soluzione.
-
-
-
-
-
-
OHP
metallo dipolo H2O
ione positivo specie adsorbita
IHP

Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
10
Velocità del trasferimento di carica Una giustificazione dell’equazione di Tafel può essere fornita da una
trattazione basata sulla teoria del complesso attivato e sulla separazione formale dei potenziali elettrochimici in un parte chimica ed una parte elettrica.
Nella teoria del complesso attivato la velocità della reazione è proporzionale alla concentrazione di equilibrio del complesso attivato. Tale concentrazione è da considerare trascurabilmente piccola rispetto a quelle dei reagenti e prodotti. La velocità dello stadio di trasferimento di carica è data perciò dalla velocità alla quale i complessi attivati si spezzano per formare molecole del prodotto finale. Questa velocità è espressa da v = c≠/τ , dove τ è il tempo richiesto perché il complesso attivato si spezzi. Poiché il complesso attivato va inteso come una specie altamente instabile, si può ragionevolmente pensare che esso si spezzi alla prima occasione in cui il legame si stira, e cioè alla prima vibrazione. Se ν = 1/τ è la frequenza della vibrazione si ha perciò:
v = c≠ν Considerando poi che in prima approssimazione si può ricavare ν dalla
relazione hν = kT e ponendo per semplicità uguale ad 1 il coefficiente di trasmissione si ha:
≠= ch
kTv (1)
dove k è la costante di Boltzmann, h la costante di Planck, T la
temperatura e c≠ la “concentrazione” del complesso attivato. Il problema è naturalmente quello di determinare c≠ , ma la teoria del complesso attivato si basa su un equilibrio formale tra regente e complesso attivato:
Oz+ + e ↔ ≠
Dove poi naturalmente il complesso attivato evolverà verso i prodotti (in questo caso la forma ridotta R:
≠ → R Allora basta applicare la normale condizione di equilibrio:

Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
11
∆−= ≠≠
RT
Gexp
c
c0
c,
O
(2)
Il suffisso c indica che ci si riferisce al processo di riduzione catodica. Si soprassegna ∆G°≠,c per indicare che si tratta di una variazione di
entalpia libera standard che coinvolge specie cariche. Sappiamo però che:
0
e
0
O
00
ii
0c,G µ−µ−µ=µν=∆ ≠≠ ∑ (3)
dove i vari 0
iµ sono i potenziali elettrochimici standard scomponibili in una parte chimica ed una parte elettrica secondo la relazione:
φ+µ=µ Fzi0i
0
i (4)
0iµ è il potenziale chimico standard della specie i , zi è la sua carica e φ è il
potenziale elettrico della fase in cui si trova la particella i. Ignoriamo per semplicità la parte diffusa del doppio strato, cioè ammettiamo in prima approssimazione che il potenziale al piano esterno di Helmholtz (dove si trova la particella reagente) sia uguale a quello nella massa della soluzione φS, ed ammettiamo che l’elettrone in transito sia delocalizzato (cioè non si trovi totalmente né sulla particella reagente né sul metallo).
La probabilità di trovare l’elettrone sul metallo è 1 prima della reazione, mentre la probabilità di trovarlo sul reagente (che si riduce) è 1 dopo che la reazione è avvenuta. Indichiamo con β la probabilità di trovare l’elettrone sul reagente nello stato di transizione, cioè al momento della formazione del complesso attivato. Naturalmente 0 ≤ β ≤ 1. Se si ammette che la probabilità di trovare l’elettrone a mezza strada tra complesso attivato e metallo sia trascurabile, la
φφφφM
-
-
-
-
-
-
φφφφS
OHP

Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
12
probabilità di trovare l’elettrone sul metallo è il complemento a β, ovvero (1-β). β rappresenta il fattore di simmetria che generalmente è uguale a 0.5.
Sulla base dell’eqn. (4) è possibile valutare i vari 0
iµ che compaiono nell’eqn.( 3):
S0O
0
O zFφ+µ=µ
M0e
0
e Fφ−µ=µ
MS00
F)1(F)z( φβ−−φβ−+µ=µ ≠≠ -(1-β)F rappresenta la frazione di carica su metallo (il segno – è dovuto alla carica dell’elettrone), e (z-β)F rappresenta la frazione di carica sul reagente. Allora:
φ∆β+∆=
=φ+φ−φβ−−φβ−+µ−µ−µ=∆
≠
≠≠
FG
FzFF)1(F)z(G0
c,
MSMS0e
0O
00c,
in cui:
0e
0O
00c,G µ−µ−µ=∆ ≠≠ e SM φ−φ=φ∆
Sostituendo nella relazione di equilibrio (2) si ha:
φ∆β−
∆−=
∆−= ≠≠≠
RT
Fexp
RT
Gexp
RT
Gexp
c
c 0c,
0c,
O
e nell’espressione della velocità (1):
φ∆β−
∆−== ≠
≠ RT
Fexp
RT
Gexpc
h
kTc
h
kTv
0c,
O
cioè :
φ∆β−=RT
Fexpckv O
con:
∆−= ≠
RT
Gexp
h
kTk
0c,

Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
13
Trattandosi di una reazione eterogenea, v rappresenta il numero di moli che reagiscono all’unità di superficie dell’elettrodo nell’unità di tempo, e k rappresenta una costante di proporzionalità. Più precisamente potremo scrivere:
φ∆β−=RT
Fexpckv O
La freccia su v e k sta ad indicare che si tratta del processo diretto (che in
questo caso è quello di riduzione), mentre per il processo inverso si scriverà
v e k . Inoltre si soprassegna cO per indicare che si tratta della concentrazione alla superficie dell’elettrodo, mentre la concentrazione nella massa della soluzione viene indicata con c*O. Infatti la reazione di riduzione cha ha luogo all’elettrodo produce un impoverimento della concentrazione della forma ossidata che si trova in vicinanza dell’elettrodo. Il profilo di concentrazione si ottiene portando in grafico la concentrazione in funzione della distanza dall’elettrodo: a distanze sufficientemente grandi dalla superficie dell’elettrodo la specie non risente del potenziale applicato e la sua concentrazione è uguale a quella nella massa della soluzione, mentre in vicinanza dell’elettrodo essa viene consumata nel processo elettronico e la sua concentrazione è quindi minore di quella massiva.
Trattandosi si un’equazione cinetica, la costante di velocità, kf, di questo
processo diretto di riduzione (f per forward) rappresenta la costante di proporzionalità tra velocità e concentrazione:
Of ckv = quindi:
RT/F
f ekk φ∆β−= (5)
Come è logico aspettarsi, la costante di velocità di un processo di trasferimento di carica dipende dal potenziale, e di conseguenza k coincide con kf solo per ∆φ=0. Dato che poi ∆φ non è direttamente misurabile sarà più conveniente riferirsi al potenziale E che differisce da ∆φ per un termine costante. In tal caso:
x
c
c
c*
O

Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
14
RT/FEf ekk β−=
dove ora:
kk f = per E=0
oppure RT/)EE(F
0f0ekk −β−=
dove k0 rappresenta la costante di velocità standard ovvero la costante di
velocità per E=E0. In modo del tutto analogo è possibile ricavare la velocità del processo
inverso:
R ↔ ≠ → Oz+ + e
In questo caso avremo: 0
R
00a,G µ−µ=∆ ≠≠
dove il potenziale elettrochimico standard del complesso attivato è uguale a quello ricavato in precedenza, mentre per la specie ridotta basta considerare che la carica è (z-1):
MS00
F)1(F)z( φβ−−φβ−+µ=µ ≠≠
S0R
0
R F)1z( φ−+µ=µ
Perciò: φ∆β−−∆=
=φ−−φβ−−φβ−+µ−µ=∆
≠
≠≠
F)1(G
F)1z(F)1(F)z(G0
a,
MSMS0R
00a,
da cui si ricava la velocità:
β−==RT
FE)1(expckckv RRb (6)
con: RT/FE)1(
b ekk β−= oppure
−β−==RT
)EE(F)1(expckckv 0
0b RR (6)
con: RT/)EE(F)1(
0b0ekk −β−=

Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
15
Corrente catodica, anodica e corrente di scambio Per avere la densità di corrente catodica jc basta moltiplicare la velocità del processo diretto di riduzione (che esprime il numero di moli che reagiscono al secondo per unità di superficie dell’elettrodo) per la carica per mole, cioè per il Faraday:
β−−=−=RT
FEexpckFvFj Oc (7)
La natura esponenziale di questa relazione fa sì che piccole variazioni di
campo elettrico all’interfase producano variazioni notevoli della densità di corrente.
In modo del tutto analogo si ricava la densità di corrente anodica:
β−==RT
FE)1(expckFvFj Ra (8)
Nelle espressioni di jc e ja si è adottata la convenzione che attribuisce segno
positivo alla corrente di ossidazione (corrente anodica) e negativo alla corrente di riduzione corrente catodica).
Va notato che una corrente anodica comporta una cessione di elettroni da
parte della specie R che si ossida. Dunque in un processo anodico si avrà un flusso di ioni R verso l’interfase dove tali ioni vengono ossidati. Al contrario il flusso della specie ossidata O formatasi nel processo di ossidazione procederà verso la massa della soluzione. Dunque le correnti anodica e catodica hanno segno contrario.
Ne consegue che la densità di corrente netta è data dalla somma delle
componenti catodica e anodica (equazione di Butler Volmer):
cO
cO
c*O
cR
cR
c*R R O
e e Riduzione:
O + e R Ossidazione:
R – e O
R O

Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
16
]eckeck[Fjjj RT/FE)1(R
RT/FEOac
β−β− +−=+= (9)
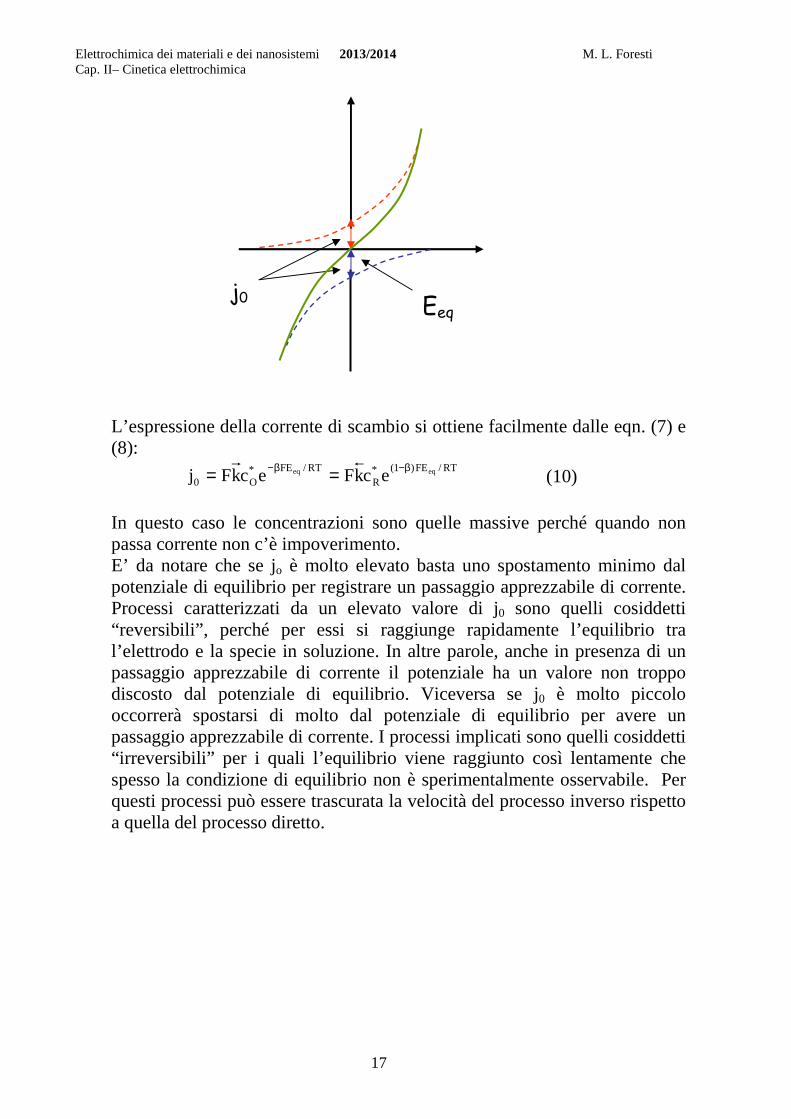
Le curve tratteggiate corrispondono alla reazione diretta e inversa, cioè al processo di riduzione e di ossidazione presi separatamente. Occorre precisare che stiamo parlando del processo che avviene ad un singolo elettrodo, e non del processo elettrochimico che avviene tra due elettrodi.
In assenza di un potenziale imposto dall’esterno, si realizza una situazione di equilibrio tra la reazione diretta di riduzione e quella inversa di ossidazione. Infatti al procedere della reazione diretta di riduzione: O + e → R l’elettrodo dovrebbe assumere una carica sempre più positiva con conseguente aumento del potenziale dell’elettrodo, e dato il segno negativo dell’esponente nell’eqn. (5), la velocità del processo diretto di riduzione dovrebbe progressivamente diminuire. D’altra parte un aumento del potenziale porta ad un aumento della velocità del processo inverso di ossidazione espressa dall’eqn. (6) con la conseguente diminuzione dell’eccesso di carica sul metallo. Ci dovrà perciò essere un valore di potenziale al quale la velocità del processo diretto di riduzione uguaglia quella del processo inverso di ossidazione. Di conseguenza ci dovrà essere un valore di potenziale per cui -jc=ja, e perciò j=0. Questo potenziale è per definizione il potenziale di equilibrio, Eeq, e il valore comune delle densità di corrente diretta e inversa in corrispondenza di esso è la densità di corrente di scambio j0.
ja
E
jc
Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
17
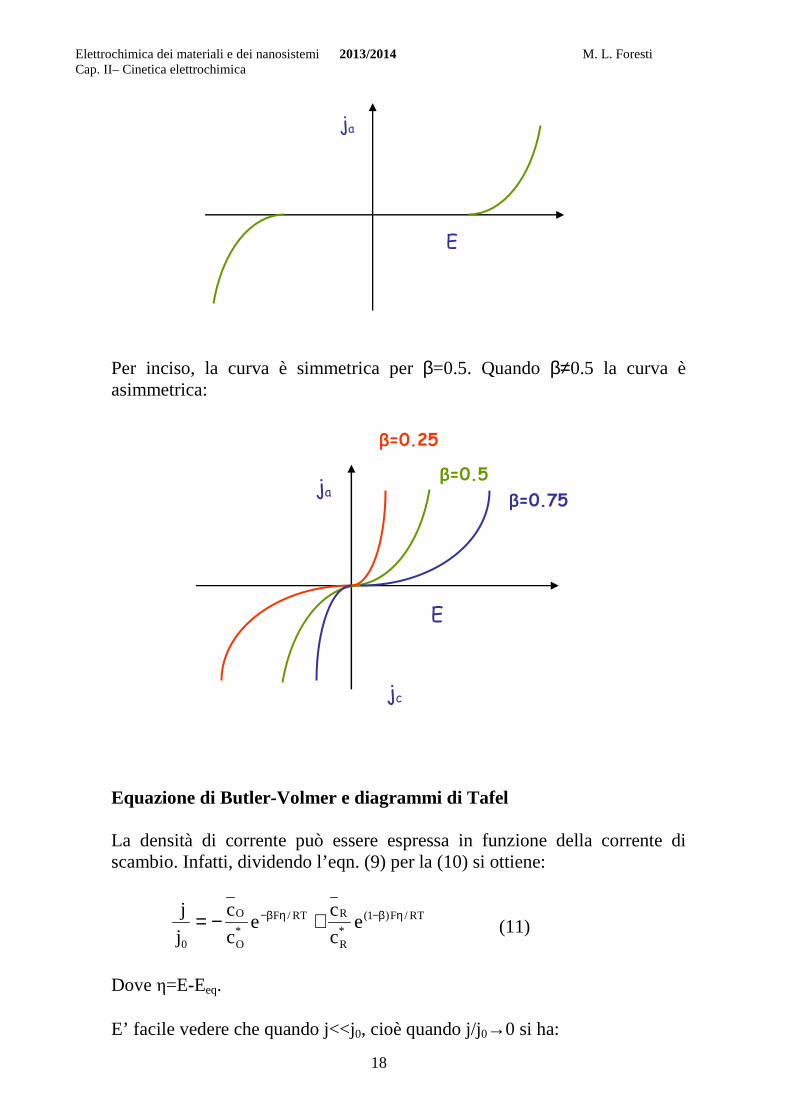
L’espressione della corrente di scambio si ottiene facilmente dalle eqn. (7) e (8):
RT/FE)1(*R
RT/FE*O0
eqeq eckFeckFj β−β− == (10)
In questo caso le concentrazioni sono quelle massive perché quando non passa corrente non c’è impoverimento. E’ da notare che se jo è molto elevato basta uno spostamento minimo dal potenziale di equilibrio per registrare un passaggio apprezzabile di corrente. Processi caratterizzati da un elevato valore di j0 sono quelli cosiddetti “reversibili”, perché per essi si raggiunge rapidamente l’equilibrio tra l’elettrodo e la specie in soluzione. In altre parole, anche in presenza di un passaggio apprezzabile di corrente il potenziale ha un valore non troppo discosto dal potenziale di equilibrio. Viceversa se j0 è molto piccolo occorrerà spostarsi di molto dal potenziale di equilibrio per avere un passaggio apprezzabile di corrente. I processi implicati sono quelli cosiddetti “irreversibili” per i quali l’equilibrio viene raggiunto così lentamente che spesso la condizione di equilibrio non è sperimentalmente osservabile. Per questi processi può essere trascurata la velocità del processo inverso rispetto a quella del processo diretto.
j0
Eeq
Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
18
Per inciso, la curva è simmetrica per β=0.5. Quando β≠0.5 la curva è asimmetrica:
Equazione di Butler-Volmer e diagrammi di Tafel La densità di corrente può essere espressa in funzione della corrente di scambio. Infatti, dividendo l’eqn. (9) per la (10) si ottiene:
RT/F)1(
*R
RRT/F*O
O
0
ec
ce
c
c
j
j ηβ−ηβ− +−= (11)
Dove η=E-Eeq. E’ facile vedere che quando j<<j0, cioè quando j/j0→0 si ha:
ja
E
jc
ja
E
ββββ=0.25 ββββ=0.5
ββββ=0.75

Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
19
RT/)EE(F)1(
*R
RRT/)EE(F
*O
O eqeq ecc
ecc −β−−β− ≅
Riarrangiando i termini e considerando che il rapporto c*O/c*R è espresso dall’equazione di Nernst:
RT/)EE(F
*R
*O 0eqe
cc −=
si ottiene:
RT/)EE(F
R
O0e
c
c −=
Questo significa che il potenziale all’elettrodo e le concentrazioni alla superficie dell’elettrodo sono legati da un’espressione che ha la forma dell’equazione di Nernst, indipendentemente dal fatto che passi corrente. Si ha un passaggio di corrente in quanto le concentrazioni alla superficie dell’elettrodo non sono uguali a quelle massive, però il trasferimento di carica è così veloce che esiste sempre una situazione di equilibrio tra potenziale e concentrazioni superficiali. L’andamento di j espresso dall’eqn (11) è quello già visto, tranne per il fatto che per valori assoluti relativamente elevati di η i fattori esponenziali sono tenuti sotto controllo dai rapporti *
OO c/c e *RR c/c che esprimono
l’impoverimento alla superficie dell’elettrodo e che dipendono appunto da η. In questo caso la corrente raggiunge un valore limite, jlim, in quanto è condizionata dalla velocità con cui ha luogo il trasferimento di massa con cui la specie reagente viene rifornita all’elettrodo (generalmente per diffusione, e la corrente massima è allora la corrente limite di diffusione, id). Finché j si mantiene molto minore del suo valore limite, le concentrazioni alla superficie dell’elettrodo possono essere considerate circa uguali alle corrispondenti concentrazioni massive. In tal caso la (11) diventa:
{ }RT/F)1(RT/F0 eejj ηβ−ηβ− +−= (12)
dove η=E-Eeq rappresenta la sovratensione.
ja
E
jc

Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
20
Questa equazione presenta due forme limite: 1- Se la sovratensione è molto piccola (inferiore a 0.01V) si possono sviluppare gli esponenziali. Infatti per x<<1 si ha: x1ex +≅ Quindi:
RT
Fj
RT
F)1(1
RT
F1jj 0
0
η=
ηβ−++ηβ+−=
Questo significa che per bassi valori di sovratensione l’interfase si comporta come un conduttore ohmico, ovvero la corrente è proporzionale alla ddp. Tenendo conto che un valore è considerato molto minore di un altro quando è inferiore di almeno un ordine di grandezza, si ha che :
per RT
Fηβ<<1 deve essere : |η|<<
F10
RT
β
e dato che in genere β ≅ 0.5 e che RT/F = 25.6 mV a 25°C, questo equivale a dire che
|η|<<5.010
6.25
× ≅ 5 mV
2- Se la sovratensione è grande (>0.1V) si potrà invece trascurare uno dei due esponenziali dell’eqn. (12). Si può pensare che questa approssimazione valga quando uno dei due contributi è inferiore all’1% dell’altro, ad esempio, se:
01.0e
eRT/F
RT/F)1(
≤ηβ−
ηβ−
si ha:
RT/Fe η ≤ 0.01 ovvero :
RT3.2
Fη ≤ -2 da cui: |η|≤ -2
F
RT3.2= -0.118V
Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
21
In particolare, se la sovratensione è molto positiva, cioè η>>0:
RT
F)1(jlnjln 0
ηβ−+=
Se invece la sovratensione è grande ma negativa, η<<0, si ha:
RT
Fjlnjln 0
ηβ+−=
da cui:
RT
Fjlnjln 0
ηβ−=
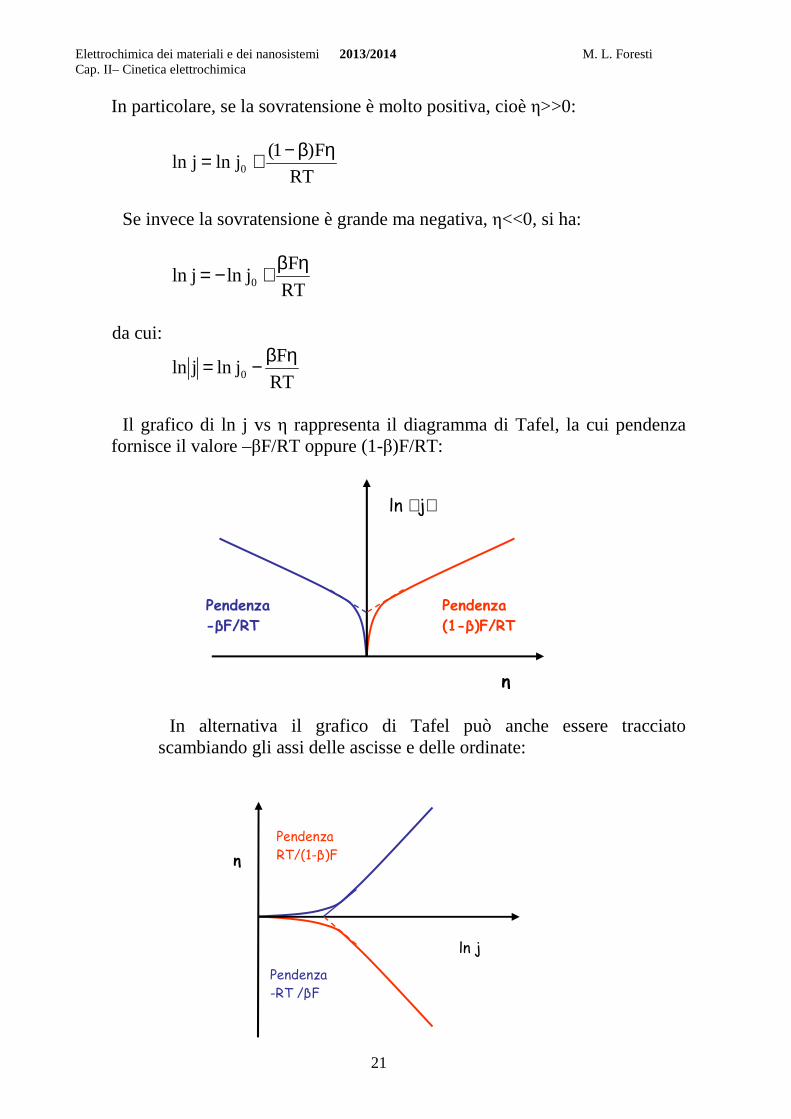
Il grafico di ln j vs η rappresenta il diagramma di Tafel, la cui pendenza
fornisce il valore –βF/RT oppure (1-β)F/RT:
In alternativa il grafico di Tafel può anche essere tracciato
scambiando gli assi delle ascisse e delle ordinate:
η
ln j
Pendenza
RT/(1-β)F
Pendenza
-RT /βF
η
ln j
Pendenza
(1-β)F/RT
Pendenza
-βF/RT

Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
22
Ci siamo riferiti ad un processo elettrochimico corrispondente ad un unico stadio di trasferimento di carica. Più in generale, un processo elettrochimico è costituito da più stadi elementari, di cui almeno uno di trasferimento di carica. La velocità dell’intero processo sarà determinata dallo stadio più “lento” (rds = rate determinino step), che potrà essere sia chimico che elettrochimico. Comunque sia, prima dello stadio lento c’è la possibilità che siano già stati scambiati n elettroni. Nel caso più generale dunque non si parla più di fattore di simmetria, β, bensì di coefficiente di trasferimento di carica, α, che oltre a β include gli eventuali n elettroni scambiati in precedenza. Consideriamo a questo proposito un processo elettrodico globale generico:
A1 + ne ↔ Af
dove A1 è il reagente presente nella massa della soluzione mentre Af
è il prodotto finale. Ammettiamo poi che in generale il processo elettrodico consista di più stadi elementari consecutivi:
A1 ↔ A2 A2 ↔ A3 … A i →A i+1 … Af-1 → Af
di cui lo stadio i-esimo è quello rds che di conseguenza viene indicato con la semplice freccia unidirezionale. In generale non è possibile individuare la natura degli stadi elementari che seguono lo stadio lento, in quanto l'individuazione degli stadi elementari si effettua in base ad uno studio della cinetica del processo, mentre gli stadi successivi a quello lento non hanno nessun effetto su tale cinetica. Questi stadi elementari sono stati scritti in modo schematico, omettendo l'elettrone, nel caso di stadi elettrochimici, od eventuali specie "ancillari" (ad esempio protoni, ossidrilioni o molecole di legante), nel caso di stadi chimici; tali specie ancillari entrano a far parte degli stadi elementari chimici senza peraltro subire una variazione nel loro stato di ossidazione. Usualmente si fa in modo che le concentrazioni massive delle specie ancillari siano di almeno un ordine di grandezza superiori a quella del reagente, in modo che il passaggio di corrente non alteri in modo apprezzabile le concentrazioni di queste specie immediatamente al di fuori del doppio strato rispetto alle corrispondenti concentrazioni

Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
23
massive; quando ciò non sia possibile in modo diretto (per esempio per la concentrazione idrogenionica a pH neutro), si ricorre all'impiego di soluzioni tampone. Operando in questo modo, eventuali stadi chimici che di per sé non sono monomolecolari diventano in pratica "pseudomonomolecolari", in quanto le concentrazioni delle specie ancillari possono essere tranquillamente inglobate in modo formale nelle rispettive costanti di velocità.
La velocità vc dell'intero processo catodico è uguale a quella dello stadio lento i-esimo, che considereremo unidirezionale. Tale velocità sarà data da:
ic ckv = nel caso di stadio lento chimico
fEic eckv β−= ' nel caso di stadio lento elettrochimico
dove k è la costante di velocità dello stadio chimico mentre k' è la
costante di velocità dello stadio elettrochimico per E=0 Per ogni stadio in quasi equilibrio che precede lo stadio i si può
applicare la relazione di equilibrio corrispondente, ovvero la costante dell’equilibrio chimico:.
kk
1kK
c
c =+ se lo stadio è chimico
oppure l’equazione di Nernst:
RT/)EE(F
k
1k 0ke
c
c −−+ = se lo stadio è elettrochimico
Perciò, per calcolare la concentrazione ic basterà applicare la condizione di equilibrio a tutti gli (i-1) stadi in quasi equilibrio che precedono lo stadio lento. Ogni concentrazione può essere perciò espressa in funzione della concentrazione dello stadio precedente, perciò alla fine la concentrazione dello stadio i potrà essere espressa in funzione della sola concentrazione iniziale 1c semplicemente moltiplicando membro a membro le (i-1) equazioni.
In questa moltiplicazione si mette in evidenza che il numero di elettroni scambiati prima dello stadio lento corrisponde alla somma , nc, degli elettroni scambiati in ogni stadio elettrochimico che precede lo stadio lento.
In generale si avrà:

Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
24
( )β+cn se lo stadio rds è elettrochimico
αc
cn se lo stadio rds è chimico
dove cn rappresenta il numero di elettroni scambiati negli stadi che precedono lo stadio lento ed è sicuramente un numero intero, mentre β rappresenta la frazione di elettroni scambiati nello stadio rds ed avrà un valore compreso tra zero e uno (generalmente prossimo a 0.5). Di conseguenza un valore intero di α indica che lo stadio lento è chimico, mentre un valore frazionario indica che lo stadio lento é quello elettrochimico. Si possono perciò stabilire sia la natura dello stadio che determina la velocità, sia il numero di stadi elettrochimici precedenti. Al contrario non si possono ricavare informazioni dirette né sul numero di stadi chimici che precedono lo stadio lento né su tutto ciò che segue lo stadio lento. Ad esempio: � αc= 0.5 : significa che lo stadio lento è elettrochimico e che non ci sono
stadi elettrochimici precedenti. � αc = 1: significa che lo stadio lento è chimico e che prima di esso vi è
già stato uno stadio elettrochimico.
� αc = 1.5 : significa che lo stadio lento è elettrochimico e che prima di esso vi è già stato il trasferimento di 1 elettrone, cioè vi è stato uno stadio elettrochimico.
Con ragionamenti perfettamente analoghi a quelli fatti in precedenza, è
possibile dimostrare che per un processo anodico:
)1( β−+an per uno stadio lento elettrochimico
αa
an per uno stadio lento chimico
In questa equazione an esprime il numero di elettroni che precedono lo stadio lento nel senso anodico e β è ancora il fattore di simmetria di tale stadio lento, se quest'ultimo è uno stadio elettrochimico. Tenendo conto che:
)( ac nn + = n se lo stadio lento è chimico

Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
25
)( ac nn + = n-1 se lo stadio lento è elettrochimico dove n rappresenta il numero totale n di elettroni coinvolti nel processo
elettrodico globale è evidente che in entrambi i casi si ha: αc + αa = n Infatti:
αc + αa = )( ac nn + = n se lo stadio lento è chimico
αc + αa =( )β+cn + )1( β−+an = n se lo stadio lento è elettrochimico Tuttavia occorre notare che ciò è vero solo se lo stadio lento è lo stesso
sia procedendo nel senso catodico che in quello anodico; in caso contrario αc+αa risulterà diverso da n. Quest'ultima circostanza si presenta assai frequentemente, in quanto molto spesso il processo elettrodico nel senso catodico si manifesta a velocità apprezzabile in un campo di potenziali molto più negativo di quello in cui si manifesta il processo anodico, e la notevole separazione tra i due campi di potenziale determina facilmente un cambiamento dello stadio lento.
Occorre sottolineare che mentre β esprime una probabilità ed ha un
significato meccanicistico preciso, α è un parametro sperimentale che esprime la dipendenza lineare di ln j da E. Pertanto α deve essere determinato sperimentalmente proprio da grafici del tipo di Tafel. Nel caso più generale di un generico processo elettrochimico consistente di uno o più stadi, il fattore di simmetria β che compare eqn. (12) dovrà essere sostituito dal coefficiente di trasferimento di carica α.:
]ee[jj RT/FRT/F0
ηαηα− +−= (13)
Va ricordato che esistono un processo diretto ed inverso per entrambe le reazioni che avvengono ai due elettrodi del sistema elettrochimico. Indicando con : • cα : coefficiente di trasferimento di carica per il processo diretto di
riduzione • cα : coefficiente di trasferimento di carica per il processo inverso di
riduzione

Elettrochimica dei materiali e dei nanosistemi 2013/2014 M. L. Foresti Cap. II– Cinetica elettrochimica
26
• aα : coefficiente di trasferimento di carica per il processo diretto di ossidazione
• aα : coefficiente di trasferimento di carica per il processo inverso di ossidazione
le relative pendenze di Tafel risulteranno:
� F
RT
c
c
α=λ per il processo diretto di riduzione
� F
RT
c
c
α=λ per il processo inverso di riduzione
� F
RT
a
a
α=λ per il processo diretto di ossidazione
� F
RT
a
a
α=λ per il processo inverso di ossidazione