chapter 4 molecular cloning methods. introduction the significance of gene cloning to elucidate the...
TRANSCRIPT

Chapter 4Chapter 4
Molecular Cloning MethodsMolecular Cloning Methods

Introduction Introduction The significance of gene cloningThe significance of gene cloning
To elucidate the structure and function of genesTo elucidate the structure and function of genes..
i.e.: investigating hGH gene i.e.: investigating hGH gene
hGH gene: <10hGH gene: <10-6 -6 of human genomeof human genome
Problem 1: need kilograms of human Problem 1: need kilograms of human
genome DNA for 1μg hGH genegenome DNA for 1μg hGH gene
Problem 2: how to separate the gene from the Problem 2: how to separate the gene from the
rest DNA rest DNA

4.1 Gene Cloning
The procedure in a gene cloning experiment iThe procedure in a gene cloning experiment iss
1.1. To place a foreign gene into a bacterial cell;To place a foreign gene into a bacterial cell;
2.2. To grow a clone of those modified bacteriaTo grow a clone of those modified bacteria..
The principle factors for gene cloning experiThe principle factors for gene cloning experiment:ment:
Restriction endonucleasesRestriction endonucleases
VectorsVectors
Specific probeSpecific probe

The Role of Restriction Endonucleases Vectors Plasmids as Vectors Phages as Vectors λ Phage Vectors Cosmids M13 phage vectors Phagemids Eukaryotic Vectors Identifying a Specific Clone with a Specific Probe Polynucleotide Probes

4.1.1 The Role of Restriction En4.1.1 The Role of Restriction Endonucleasesdonucleases
RestrictionRestriction : restrict the host range of the : restrict the host range of the virusvirus
Endonucleases :Endonucleases : cut at sites within the fore cut at sites within the foreign DNA ign DNA
How to nameHow to name: the first 3 letters of the Lat: the first 3 letters of the Latin name of the microorganism + the strain in name of the microorganism + the strain designation + Roman numeral designation + Roman numeral


Recognition sequenceRecognition sequence Recognition frequencyRecognition frequency
4 bp4 bp 44 =254 bp
6 bp6 bp 46=4096 bp
8 bp8 bp 4488=65000bp=65000bp Rere cuttersRere cutters

The main advantage of restriction enzyme is there ability to cut a DNA reproducibly in the same place; this is the basis of many techniques used to analyze genes.
Many restriction enzymes make staggered cut in the tMany restriction enzymes make staggered cut in the two DNA strands, leaving a wo DNA strands, leaving a sticky ends, sticky ends, that can basthat can base-pair together briefly.e-pair together briefly.
Enzymes that recognize identical sequences are called Enzymes that recognize identical sequences are called isoschizomers.isoschizomers.

Restriction- modification systemRestriction- modification systemR-M systemR-M system
Almost all restriction nucleases are paireAlmost all restriction nucleases are paired with methylases that recognize and med with methylases that recognize and me
thylate the same DNA sitesthylate the same DNA sites

Figure 4.1 Maintaining restrictioFigure 4.1 Maintaining restriction endonuclease resistance after Dn endonuclease resistance after D
NA replicationNA replication
We begin with an We begin with an EcoEcoRI site that is metRI site that is met
hylated (red) on both strands. After replicathylated (red) on both strands. After replicat
ion, the parental strand of each daughter Dion, the parental strand of each daughter D
NA duplex remains methylated, but the neNA duplex remains methylated, but the ne
wly made strand of each duplex has not bewly made strand of each duplex has not be
en methylated yet. The one methylated straen methylated yet. The one methylated stra
nd in these hemimethylated DNAs is enougnd in these hemimethylated DNAs is enoug
h to protect both strands against cleavage bh to protect both strands against cleavage b
y y EcoEcoRI. Soon, the methylase recognizes thRI. Soon, the methylase recognizes th
e unmethylated strand in each EcoRI site ae unmethylated strand in each EcoRI site a
nd methylates it, regenerating the fully metnd methylates it, regenerating the fully met
hylated DNA.hylated DNA.


Figure 4.2 The first cloning experiment invFigure 4.2 The first cloning experiment involving a recombinant DNA assembled in vitolving a recombinant DNA assembled in vit
ro.ro.
Boyer and Cohen cut two plasmids, pSC101 and RSBoyer and Cohen cut two plasmids, pSC101 and RS
F1010, with the same restriction endonuclease, F1010, with the same restriction endonuclease, EcoEcoRI. RI.
This gave the twolinear DNAs the same stickyends, whThis gave the twolinear DNAs the same stickyends, wh
ich were then linked in vitro using DNA ligase. The inich were then linked in vitro using DNA ligase. The in
vestigators reintroduced the recombinant DNA into vestigators reintroduced the recombinant DNA into E. E.
colicoli cells by transformation and selected clones that we cells by transformation and selected clones that we
re resistant to both tetracycline and streptomycin. Thesre resistant to both tetracycline and streptomycin. Thes
e clones were therefore harboring the recombinant plase clones were therefore harboring the recombinant plas
mid.mid.

Restriction endonucleases recognize specific sequences in DNRestriction endonucleases recognize specific sequences in DN
A molecules and make cuts in both strands. This allows very specifA molecules and make cuts in both strands. This allows very specif
ic cutting of DNAs. Also, because the cuts in the two strands are fric cutting of DNAs. Also, because the cuts in the two strands are fr
equently staggered, restriction enzymes can create sticky ends that equently staggered, restriction enzymes can create sticky ends that
help link together two DNAs to form a recombinant DNA help link together two DNAs to form a recombinant DNA in vitroin vitro..
Summary:Summary:

4.1.2 Vectors4.1.2 VectorsVectors serve as carriers to allow replicaVectors serve as carriers to allow replication of recombinant DNAs.tion of recombinant DNAs.
Origin of replicationOrigin of replication
Multiple cloning site(MCS)Multiple cloning site(MCS)
Selection geneSelection gene PlasmidsPlasmids pBR322 pUC pBR322 pUC
PhagesPhages λphage cosmids M13 λphage cosmids M13
Phagemids Phagemids

Plasmids as VectorsPlasmids as Vectors

Summary:Summary:
The first generations of plasmid cloning vectors were pBR322 and tThe first generations of plasmid cloning vectors were pBR322 and t
he pUC plasmids. The former has two antibiotic resistance genes and a he pUC plasmids. The former has two antibiotic resistance genes and a
variety of unique restriction sites into which one can introduce foreign variety of unique restriction sites into which one can introduce foreign
DNA. Most of these sites interrupt one of the antibiotic resistance geneDNA. Most of these sites interrupt one of the antibiotic resistance gene
s, making screening straightforward. Screening is even easier with the s, making screening straightforward. Screening is even easier with the
pUC plasmids. These have an ampicillin resistance gene and a multiple pUC plasmids. These have an ampicillin resistance gene and a multiple
cloning site that interrupts a partial β-galactosidase gene. One screens fcloning site that interrupts a partial β-galactosidase gene. One screens f
or ampicillin-resistant clones that do not make active β-galactosidase aor ampicillin-resistant clones that do not make active β-galactosidase a
nd therefore do not turn the indicator, X-gal, blue. The multiple cloning nd therefore do not turn the indicator, X-gal, blue. The multiple cloning
site also makes it convenient to carry out directional cloning into two disite also makes it convenient to carry out directional cloning into two di
fferent restriction sites. fferent restriction sites.

Figure 4.3 The plasmid pBR322, showing the locations of 11 unique restriction Figure 4.3 The plasmid pBR322, showing the locations of 11 unique restriction sites that can be used to insert foreign DNAsites that can be used to insert foreign DNA
The locations of the two antibiotic resistance genes (Ampr =ampicillin resistance; TetThe locations of the two antibiotic resistance genes (Ampr =ampicillin resistance; Tet
r =tetracycline resistance) and the origin of replication (ori ) are also shown. Numbers rer =tetracycline resistance) and the origin of replication (ori ) are also shown. Numbers re
fer to kilobase pairs (kb) from the fer to kilobase pairs (kb) from the EcoEcoRI site.RI site.

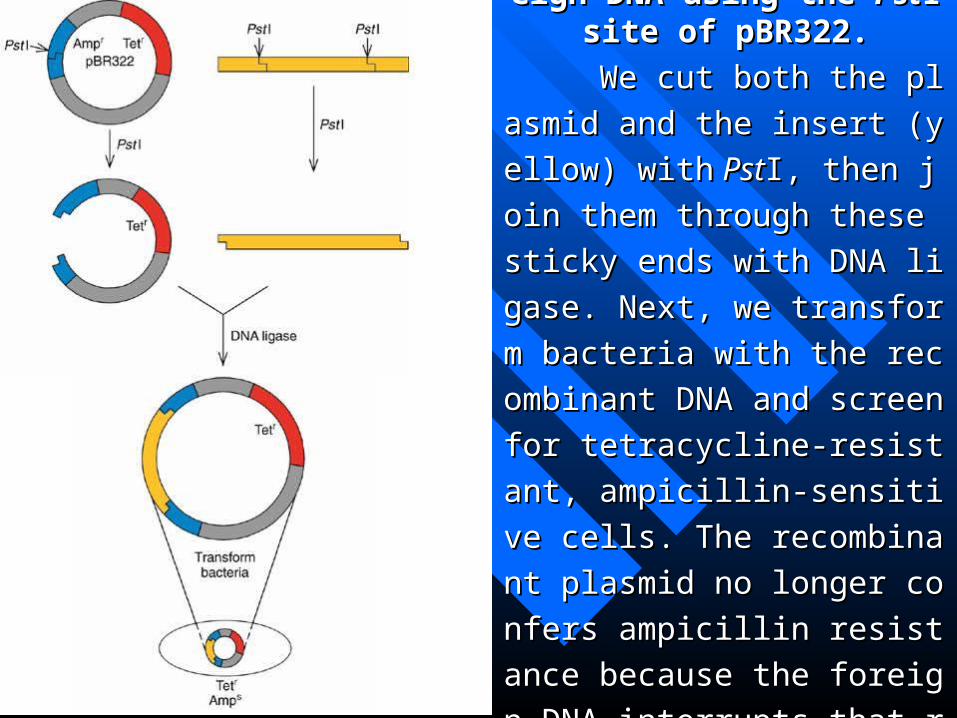
Figure 4.4 Cloning foreign DNFigure 4.4 Cloning foreign DNA using the A using the PstPstI site of pBR322.I site of pBR322.
We cut both the plasmid and thWe cut both the plasmid and th
e insert (yellow) withe insert (yellow) with Pst PstI, then joI, then jo
in them through these sticky ends in them through these sticky ends
with DNA ligase. Next, we transfowith DNA ligase. Next, we transfo
rm bacteria with the recombinant rm bacteria with the recombinant
DNA and screen for tetracycline-rDNA and screen for tetracycline-r
esistant, ampicillin-sensitive cells. esistant, ampicillin-sensitive cells.
The recombinant plasmid no longThe recombinant plasmid no long
er confers ampicillin resistance beer confers ampicillin resistance be
cause the foreign DNA interrupts tcause the foreign DNA interrupts t
hat resistance gene (blue). hat resistance gene (blue).

Figure 4.5 Screening bacteria by replFigure 4.5 Screening bacteria by replica plating. ica plating.
(a)(a) The replica plating process. We touc The replica plating process. We touch a velvet-covered circular tool to the sh a velvet-covered circular tool to the surface of the first dish containing coloniurface of the first dish containing colonies of bacteria. Cells from each of these es of bacteria. Cells from each of these colonies stick to the velvet and can be trcolonies stick to the velvet and can be transferred to the replica plate in the samansferred to the replica plate in the same positions relative to each other. e positions relative to each other. (b)(b) Sc Screening for inserts in the pBR322 ampicreening for inserts in the pBR322 ampicillin resistance gene by replica plating. illin resistance gene by replica plating. The original plate contains tetracycline, The original plate contains tetracycline, so all colonies containing pBR322 will so all colonies containing pBR322 will grow. The replica plate contains ampicilgrow. The replica plate contains ampicillin, so colonies bearing pBR322 with inlin, so colonies bearing pBR322 with inserts in the ampicillin resistance gene wserts in the ampicillin resistance gene will not grow (these colonies are depicted ill not grow (these colonies are depicted by dotted circles). The corresponding cby dotted circles). The corresponding colonies from the original plate can then olonies from the original plate can then be picked.be picked.


pUCpUC

lacZ’ : coding for the amino terminalportion of the enzyme β –galactosidease.
Host E.coli strain carry a gene fragment that codes the carboxyl potion of β –galactosidease;
When X-gal cleaved by β –galactosidease, it releases galactose plus an indigo dye that stains the bacterial colony blue.

Figure 4.7 Joining of vector to insert. (a) Figure 4.7 Joining of vector to insert. (a) Mechanism of DNA ligase. Mechanism of DNA ligase.
Step 1Step 1:: DNA ligase reacts with an AMP do DNA ligase reacts with an AMP donor—either ATP or NAD(nicotinamide adenor—either ATP or NAD(nicotinamide adenine dinucleotide), depending on the type of nine dinucleotide), depending on the type of ligase. This produces an activated enzyme (lligase. This produces an activated enzyme (ligase-AMP). igase-AMP). Step 2Step 2: : The activated enzyme The activated enzyme donates a phosphate to the free 5’-phosphate donates a phosphate to the free 5’-phosphate at the nick in the lower strand of the DNA dat the nick in the lower strand of the DNA duplex, creating a high-energy diphosphate gruplex, creating a high-energy diphosphate group on one side of the nick. oup on one side of the nick. Step 3Step 3: With en: With energy provided by cleavage of the diphosphatergy provided by cleavage of the diphosphate, a new phosphodiester bond is created, seae, a new phosphodiester bond is created, sealing the nick in the DNA. This reaction can ling the nick in the DNA. This reaction can also occur in both DNA strands at once, so talso occur in both DNA strands at once, so two independent DNAs can be joined togethwo independent DNAs can be joined together by DNA ligase. er by DNA ligase.

Figure 4.7 Joining of vector to insert. Figure 4.7 Joining of vector to insert. (b)(b)Alkaline phosphatase prevents vector Alkaline phosphatase prevents vector
re-ligation.re-ligation. Step 1Step 1:: We cut the vector(blue, top left) We cut the vector(blue, top left) withwith Bam BamHI. This produces sticky ends HI. This produces sticky ends with 5’-phosphates(red). with 5’-phosphates(red). Step 2Step 2: : We remWe remove the phosphates with alkaline phosphaove the phosphates with alkaline phosphatase, making it impossible for the vector ttase, making it impossible for the vector to re-ligate with itself. o re-ligate with itself. Step 3Step 3: : We also cuWe also cut the insert(yellow, upper right) with t the insert(yellow, upper right) with BamBamHI, producing sticky ends with phosphatHI, producing sticky ends with phosphates that we do not remove.es that we do not remove. Step Step 44:: Finally, Finally, we ligate the vector and insert together. we ligate the vector and insert together. The phosphates on the insert allow two pThe phosphates on the insert allow two phosphodiester bonds to form(red), but leahosphodiester bonds to form(red), but leave two unformed bonds, or nicks, These ve two unformed bonds, or nicks, These will be completed once the DNA is in the will be completed once the DNA is in the transformed bacterial cell. transformed bacterial cell.

Phages as vectorsPhages as vectors
Natural advantages over plasmid:
They infect cells much more efficiently than plasmids transform cells, so the yield of clones with phage vectors is usually higher.

Summary:Summary: Two kinds of phages have been especially popular as cloning vectors.Two kinds of phages have been especially popular as cloning vectors.
The first of these is λ, from which certain nonessential genes have bee The first of these is λ, from which certain nonessential genes have bee
n removed to make room for inserts. Some of these engineered phages n removed to make room for inserts. Some of these engineered phages
can accommodate inserts up to 20 kb, which makes them useful for builcan accommodate inserts up to 20 kb, which makes them useful for buil
ding genomic libraries, in which it is important to have large pieces of ding genomic libraries, in which it is important to have large pieces of
genomic DNA in each clone. Cosmids can accept even larger inserts—genomic DNA in each clone. Cosmids can accept even larger inserts—
up to 50 kb—making them a favorite choice for genomic libraries. The up to 50 kb—making them a favorite choice for genomic libraries. The
second major class of phage vector is composed of the M13 phages. Thsecond major class of phage vector is composed of the M13 phages. Th
ese vector have the convenience of a multiple cloning site and the furthese vector have the convenience of a multiple cloning site and the furth
er advantage of producing single-stranded recombinant DNA, which caer advantage of producing single-stranded recombinant DNA, which ca
n be used for DNA sequencing and for site-direct mutagenesis. Plasmidn be used for DNA sequencing and for site-direct mutagenesis. Plasmid
s called phagemids have also been engineered to produce single-strandes called phagemids have also been engineered to produce single-strande
d DNA in the presence of helper phages. d DNA in the presence of helper phages.
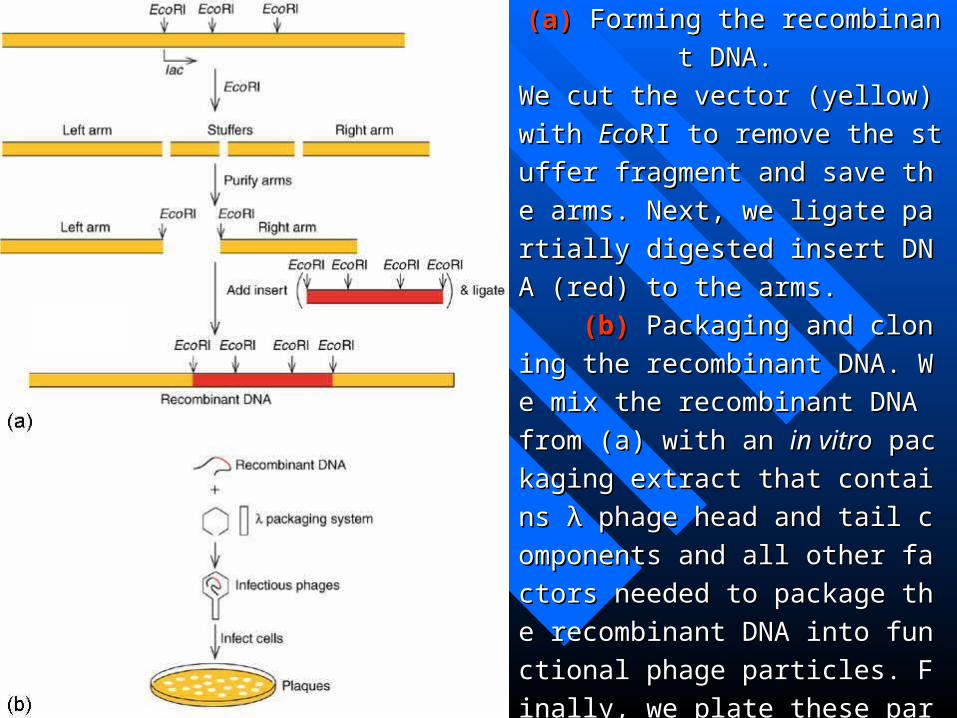
Figure 4.8 Cloning in Charon 4. Figure 4.8 Cloning in Charon 4.
(a)(a) Forming the recombinant DNA. Forming the recombinant DNA.
We cut the vector (yellow) with We cut the vector (yellow) with EcoEcoRI tRI t
o remove the stuffer fragment and save o remove the stuffer fragment and save
the arms. Next, we ligate partially digesthe arms. Next, we ligate partially diges
ted insert DNA (red) to the arms. ted insert DNA (red) to the arms.
(b)(b) Packaging and cloning the recom Packaging and cloning the recom
binant DNA. We mix the recombinant binant DNA. We mix the recombinant
DNA from (a) with an DNA from (a) with an in vitroin vitro packagin packagin
g extract that contains λ phage head and g extract that contains λ phage head and
tail components and all other factors netail components and all other factors ne
eded to package the recombinant DNA eded to package the recombinant DNA
into functional phage particles. Finally, into functional phage particles. Finally,
we plate these particles on we plate these particles on E.coliE.coli and co and co
llect the plaques that form. llect the plaques that form.
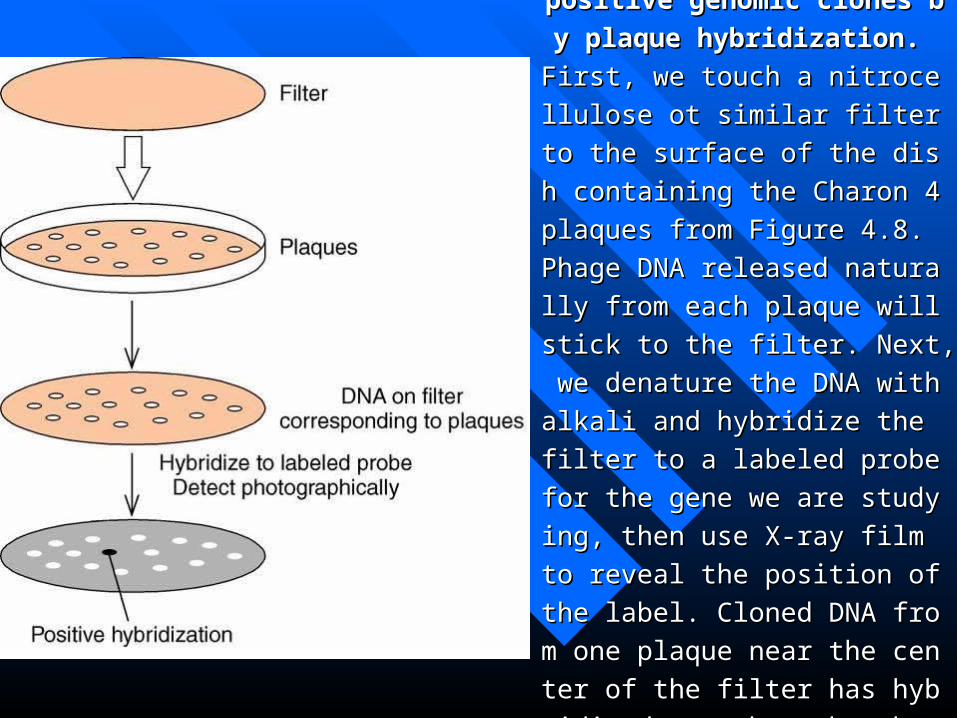
Figure 4.9 Selection of positive genFigure 4.9 Selection of positive gen
omic clones by plaque hybridizatioomic clones by plaque hybridizatio
n. n.
First, we touch a nitrocellulose ot simFirst, we touch a nitrocellulose ot sim
ilar filter to the surface of the dish coilar filter to the surface of the dish co
ntaining the Charon 4 plaques from Fntaining the Charon 4 plaques from F
igure 4.8. Phage DNA released naturaigure 4.8. Phage DNA released natura
lly from each plaque will stick to the lly from each plaque will stick to the
filter. Next, we denature the DNA witfilter. Next, we denature the DNA wit
h alkali and hybridize the filter to a lah alkali and hybridize the filter to a la
beled probe for the gene we are studybeled probe for the gene we are study
ing, then use X-ray film to reveal the ing, then use X-ray film to reveal the
position of the label. Cloned DNA froposition of the label. Cloned DNA fro
m one plaque near the center of the film one plaque near the center of the fil
ter has hybridized, as shown by the dter has hybridized, as shown by the d
ark spot on the film. ark spot on the film.

Cosmids
Behave both as plasmids and as phages;
Contain the cos sites of λ and plasmid origin of replication;
Have room for 40-50 kb inserts.

M13 phage vectors
β –galactosidease gene fragment
pUC family MCS
Single stranded DNA genome

Figure 4.10 Obtaining single-straFigure 4.10 Obtaining single-stranded DNA by cloning in M13 phagnded DNA by cloning in M13 phag
e. e.
Foreign DNA (red), cut with Foreign DNA (red), cut with HinHindIII, is insedIII, is inse
rted into the rted into the HinHindIII site of the double-strandIII site of the double-stran
ded phage DNA. The resulting recombinant ded phage DNA. The resulting recombinant
DNA is used to transform DNA is used to transform E.coliE.coli cells, wher cells, wher
eupon the DNA replicates by a rolling circle eupon the DNA replicates by a rolling circle
mechanism, producing many single-strandemechanism, producing many single-strande
d product DNAs. The product DNAs are cald product DNAs. The product DNAs are cal
led positive (+) strands, by convention. The led positive (+) strands, by convention. The
template DNA is therefore the negative (-) stemplate DNA is therefore the negative (-) s
trand. trand.

PhagemidesPhagemides
Single-stranded;Single-stranded;
Both phage and plasmid characteristics;Both phage and plasmid characteristics;
Help phageHelp phage
Two RNA polymerase promoters (T7and TTwo RNA polymerase promoters (T7and T3)3)


SummarySummary Two kinds of phages have been especially popular as cloning vectors. The Two kinds of phages have been especially popular as cloning vectors. The
first of these is λ, from which certain nonessential genes have been removed tfirst of these is λ, from which certain nonessential genes have been removed t
o make room for inserts. Some of these engineered phages can accommodate o make room for inserts. Some of these engineered phages can accommodate
inserts up to 20 kb, which makes them useful for building genomic libraries, iinserts up to 20 kb, which makes them useful for building genomic libraries, i
n which it is important to have large pieces of genomic DNA in each clone. Cn which it is important to have large pieces of genomic DNA in each clone. C
osmids can accept even larger inserts—up to 50 kb—making them a favorite osmids can accept even larger inserts—up to 50 kb—making them a favorite
choice for genomic libraries. The second major class of phage vector is compchoice for genomic libraries. The second major class of phage vector is comp
osed of the M13 phages. These vector have the convenience of a multiple cloosed of the M13 phages. These vector have the convenience of a multiple clo
ning site and the further advantage of producing single-stranded recombinant ning site and the further advantage of producing single-stranded recombinant
DNA, which can be used for DNA sequencing and for site-direct mutagenesis.DNA, which can be used for DNA sequencing and for site-direct mutagenesis.
Plasmids called phagemids have also been engineered to produce single-stra Plasmids called phagemids have also been engineered to produce single-stra
nded DNA in the presence of helper phages. nded DNA in the presence of helper phages.

4.1.3 Identifying a Specific Clone with 4.1.3 Identifying a Specific Clone with a Specific Probea Specific Probe
Polynucleotide ProbesPolynucleotide ProbesHigh stringencyHigh stringencyLow stringencyLow stringency

SummarySummary Specific clones can be identified using polynSpecific clones can be identified using polyn
ucleotide probes that bind to the gene itself. Knucleotide probes that bind to the gene itself. Kn
owing the amino acid sequence of a gene prodowing the amino acid sequence of a gene prod
uct, one can design a set of oligonucleotides thuct, one can design a set of oligonucleotides th
at encode part of this amino acid sequence. Thiat encode part of this amino acid sequence. Thi
s can be one of the quickest and most accurate s can be one of the quickest and most accurate
means of identifying a particular clone. means of identifying a particular clone.

4.2 The Polymerase Chain Reaction (PCR)4.2 The Polymerase Chain Reaction (PCR)
PCR amplifies a region of DNA between two PCR amplifies a region of DNA between two predetermined sites. Oligo-nucleotides complemepredetermined sites. Oligo-nucleotides complementary to these sites serve as primers for synthesis ontary to these sites serve as primers for synthesis of copies of the DNA between the sites. Each cycle f copies of the DNA between the sites. Each cycle of PCR double the number of copies of the amplifiof PCR double the number of copies of the amplified DNA until a large quantity has been made.ed DNA until a large quantity has been made.



Figure 4.12 Amplifying DNA by the polymerase chain reaction.Figure 4.12 Amplifying DNA by the polymerase chain reaction. Cycle 1:Cycle 1: Start with a DNA duplex (top) and heat it to separate its two strands Start with a DNA duplex (top) and heat it to separate its two strands (red and blue). Then add short, single-stranded DNA primers (purple and yellow) (red and blue). Then add short, single-stranded DNA primers (purple and yellow) complementary to sequences on either side of the region (X) to be amplified. The complementary to sequences on either side of the region (X) to be amplified. The primers hybridize to the appropriate sites on the separated DNA strands; now a primers hybridize to the appropriate sites on the separated DNA strands; now a special heat-stable DNA polymerase uses these primers to start synthesis of special heat-stable DNA polymerase uses these primers to start synthesis of complementary DNA strands. The arrows represent newly made DNA, in which complementary DNA strands. The arrows represent newly made DNA, in which replication has stopped at the tip of the arrowhead. At the end of cycle 1, two DNA replication has stopped at the tip of the arrowhead. At the end of cycle 1, two DNA duplexes are present, including the region to be amplified, whereas we started with duplexes are present, including the region to be amplified, whereas we started with only one. The 5’→3’ polarities of all DNA strands and primers are indicated only one. The 5’→3’ polarities of all DNA strands and primers are indicated throughout cycle 1. The same principles apply in cycle 2. throughout cycle 1. The same principles apply in cycle 2. Cycle 2:Cycle 2: Repeat the Repeat the process, heating to separate DNA strands, cooling to allow annealing with primers, process, heating to separate DNA strands, cooling to allow annealing with primers, and letting the heat-stable DNA polymerase make more DNA. Now each of the four and letting the heat-stable DNA polymerase make more DNA. Now each of the four DNA strands, including the two newly made ones, can serve as templates for DNA strands, including the two newly made ones, can serve as templates for complementary DNA synthesis. The result is four DNA duplexes that have the complementary DNA synthesis. The result is four DNA duplexes that have the region to be amplified. Notice that each cycle doubles the number of molecules of region to be amplified. Notice that each cycle doubles the number of molecules of DNA because the products of each cycle join the parental molecules in serving as DNA because the products of each cycle join the parental molecules in serving as templates for next cycle. This exponential increase yields 8 molecules in the next templates for next cycle. This exponential increase yields 8 molecules in the next cycle and 16 in the cycle after that. This process obviously leads to very high cycle and 16 in the cycle after that. This process obviously leads to very high numbers in only a short time. numbers in only a short time.

4.2.1 cDNA Cloning4.2.1 cDNA Cloning
Nick translationNick translation Reverse transcriptaseReverse transcriptase RNase HRNase H Terminal transferaseTerminal transferase

Figure 4.13 Making a cDNA library. Figure 4.13 Making a cDNA library. This figure focuses on cloning a single cDNA , but This figure focuses on cloning a single cDNA , but the method can be applied to a mixture of mRNAs athe method can be applied to a mixture of mRNAs and produce a library of corresponding cDNAs. nd produce a library of corresponding cDNAs. (a)(a) U Use oligo(dT) as a primer and reverse transcriptase tse oligo(dT) as a primer and reverse transcriptase tocopy the mRNA (blue), producing a cDNA (red) thocopy the mRNA (blue), producing a cDNA (red) that is hybridized to the mRNA template. at is hybridized to the mRNA template. (b)(b) Use RNa Use RNase H to partially digest the mRNA, yielding a set of se H to partially digest the mRNA, yielding a set of RNA primers base-paired to the first-strand cDNA. RNA primers base-paired to the first-strand cDNA. (c)(c) Use Use E.coliE.coli DNA polymerase I under nick translat DNA polymerase I under nick translation conditions to build second-strand cDNAs on the ion conditions to build second-strand cDNAs on the RNA primers. RNA primers. (d)(d) The second-strand cDNA growing The second-strand cDNA growing from the leftmost primer (blue) has been extended afrom the leftmost primer (blue) has been extended all the way to the 3’-end of the oligo(dA) correspondill the way to the 3’-end of the oligo(dA) corresponding to the oligo(dT) primer on the first-strand cDNAng to the oligo(dT) primer on the first-strand cDNA. . (e)(e) To give the double-stranded cDNA sticky ends, a To give the double-stranded cDNA sticky ends, add oligo(dC) with terminal transferase. dd oligo(dC) with terminal transferase. (f)(f) Anneal t Anneal the oligo(dC) ends of the cDNA to complementary olihe oligo(dC) ends of the cDNA to complementary oligo(dG) ends of a suitable vector (black). The recomgo(dG) ends of a suitable vector (black). The recombinant DNA can then be used to transform bacterial binant DNA can then be used to transform bacterial cells. Enzymes in these cells remove remaining nicks cells. Enzymes in these cells remove remaining nicks and replace any remaining RNA with DNA. and replace any remaining RNA with DNA.


Figure 4.15 Using RT-PCR to clone a single cFigure 4.15 Using RT-PCR to clone a single cDNA. DNA.
(a)(a) Use a reverse primer (red) with a Use a reverse primer (red) with a HinHindIII site dIII site
(yellow) at its 5’-end to start first-strand cDNA (yellow) at its 5’-end to start first-strand cDNA
synthesis, with reverse transcriptase to catalyze tsynthesis, with reverse transcriptase to catalyze t
he reaction. he reaction. (b)(b) Denature the mRNA-cDNA hybDenature the mRNA-cDNA hyb
rid and anneal a forward primer (red) with a Barid and anneal a forward primer (red) with a Ba
mHI site (green) at its 5’-endmHI site (green) at its 5’-end. (c). (c) This forward p This forward p
rimer initiates second-strand cDNA synthesis, wrimer initiates second-strand cDNA synthesis, w
ith DNA polymerase catalyzing the reaction. ith DNA polymerase catalyzing the reaction. (d)(d)
Continue PCR with the same two primers to amContinue PCR with the same two primers to am
plify the double-stranded cDNA. plify the double-stranded cDNA. (e)(e) Cut the cD Cut the cD
NA with NA with BamBamHI and HI and HinHindIII to generate sticky dIII to generate sticky
ends. ends. (f)(f) Ligate the cDNA to the Ligate the cDNA to the BamBamHI and HI and HinHin
dIII sites of a suitable vector (purple). Finally, trdIII sites of a suitable vector (purple). Finally, tr
ansform cells with the recombinant cDNA to pransform cells with the recombinant cDNA to pr
oduce a clone. oduce a clone.

Figure 4.16 RACE procedure to fill in the Figure 4.16 RACE procedure to fill in the 5’-end of a cDNA. 5’-end of a cDNA.
(a)(a) Hybridize an incomplete cDNA (red), o Hybridize an incomplete cDNA (red), or an oligonucleotide segment of a cDNA to r an oligonucleotide segment of a cDNA to mRNA (green), and use reverse transcriptmRNA (green), and use reverse transcriptase to extend the cDNA to the 5’-end of tase to extend the cDNA to the 5’-end of the mRNAhe mRNA. (b). (b) Use terminal transferase an Use terminal transferase and dCTP to add C residues to the 3’end of d dCTP to add C residues to the 3’end of the extended cDNA; also, use RNase H to the extended cDNA; also, use RNase H to degrade the mRNA. degrade the mRNA. (c)(c) Use an oligo(dG) p Use an oligo(dG) primer and DNA polymerase to synthesize rimer and DNA polymerase to synthesize a second strand of cDNA (blue). a second strand of cDNA (blue). (d)(d) Perfor Perform PCR with oligo(dG) as the forward primm PCR with oligo(dG) as the forward primer and an oligonucleotide that hybridizes er and an oligonucleotide that hybridizes to the 3’-end of the cDNA as the reverse to the 3’-end of the cDNA as the reverse primer. primer. (e)(e)The product is a cDNA that has The product is a cDNA that has been extended to the 5’-end of the mRNbeen extended to the 5’-end of the mRNA. A similar procedure (3’-RACE) can be A. A similar procedure (3’-RACE) can be used to extend the cDNA in the 3’-directiused to extend the cDNA in the 3’-direction. In that case, there is no need to tail thon. In that case, there is no need to tail the 3’-end of the cDNA with terminal transe 3’-end of the cDNA with terminal transferase because the mRNA already containferase because the mRNA already contains poly(A); thus, the reverse primer would s poly(A); thus, the reverse primer would be oligo(dT). be oligo(dT).

SummarySummary
To make a cDNA library, we can synthesize cDNAs one strand aTo make a cDNA library, we can synthesize cDNAs one strand at a time, using mRNAs from a cell as templates for the first strands ant a time, using mRNAs from a cell as templates for the first strands and these first strands as temletes for the second strands. Reverse trnscrid these first strands as temletes for the second strands. Reverse trnscriptase generates the first strands and ptase generates the first strands and E.coliE.coli DNA polymerase I generat DNA polymerase I generates the second strands. We can endow the double stranded cDNAs wites the second strands. We can endow the double stranded cDNAs with oligonucleotide tails that base-par with complementary tails on a cloh oligonucleotide tails that base-par with complementary tails on a cloning vector. We can then use these recombinant DNAs to transform bning vector. We can then use these recombinant DNAs to transform bacteria. We can use RT-PCR to generate a cDNA from a single type oacteria. We can use RT-PCR to generate a cDNA from a single type of mRNA, but we must know the sequence of the mRNA in order to def mRNA, but we must know the sequence of the mRNA in order to design the primers for the PCR step. If we put restriction sites on the PCsign the primers for the PCR step. If we put restriction sites on the PCR primers, we place these sites at the ends of the cDNA,so it is easy tR primers, we place these sites at the ends of the cDNA,so it is easy to ligate the cDNA into a vector. We can detect particular clones by coo ligate the cDNA into a vector. We can detect particular clones by colony hybridazation with redioactive DNA probes,or with antibodies if lony hybridazation with redioactive DNA probes,or with antibodies if an expression vector such as λgt11 is used.an expression vector such as λgt11 is used.

4.3 Methods of Expressing 4.3 Methods of Expressing Cloned GenesCloned Genes

4.3.1 Expression Vectors4.3.1 Expression Vectors
Expression vectors with strong promotersExpression vectors with strong promoters Inducible Expression VectorsInducible Expression Vectors Expression vectors produce fusion proteinsExpression vectors produce fusion proteins Eukaryotic expression vectors Eukaryotic expression vectors

Figure 4.17
Figure 4.17 Producing a fusion proteFigure 4.17 Producing a fusion prote
in by cloning in a pUC plasmid.in by cloning in a pUC plasmid.
Insert foreign DNA (yellow) into tInsert foreign DNA (yellow) into t
he multiple cloning site (MCS); transcrihe multiple cloning site (MCS); transcri
ption from the ption from the laclac promoter (purple) giv promoter (purple) giv
es a hybrid mRNA beginning with a few es a hybrid mRNA beginning with a few
lacZ’ lacZ’ codons, changing to insert sequenccodons, changing to insert sequenc
e, then back to e, then back to lacZ’ lacZ’ (red). This mRNA i(red). This mRNA i
s translated to a fusion protein containins translated to a fusion protein containin
g a few β-galactosidase amino acids for tg a few β-galactosidase amino acids for t
he remainder ofthe protein. Because the he remainder ofthe protein. Because the
insert contains a translation stop codon, tinsert contains a translation stop codon, t
he remaining he remaining lacZ’ lacZ’ codons are not translcodons are not transl
ated. ated.

Figure 4.18 Using aFigure 4.18 Using a P PBAD BAD vector. vector.
The green fluorescent protein (GFP) gene was cloned into a vector under contrThe green fluorescent protein (GFP) gene was cloned into a vector under contr
ol of theol of the PPBADBAD promoter and promoter activity was induced with increasing concentratipromoter and promoter activity was induced with increasing concentrati
ons of arabinose. GFP production was monitored by electrophoresing extracts from cons of arabinose. GFP production was monitored by electrophoresing extracts from c
ells induced with the arabinose concentrations given at top, blotting the proteins to a ells induced with the arabinose concentrations given at top, blotting the proteins to a
membrane, and detecting GFP with an anti-GFP antibody . membrane, and detecting GFP with an anti-GFP antibody .

Summary:Summary:
Expression vectors are designed to yield the protein Expression vectors are designed to yield the protein
product of a cloned gene, usually in the greatest amount product of a cloned gene, usually in the greatest amount
possible. To optimize expression, these vectors provide possible. To optimize expression, these vectors provide
strong bacterial promoters and bacterial ribosome binding strong bacterial promoters and bacterial ribosome binding
sites that would be missing on cloned eukaryotic genes. sites that would be missing on cloned eukaryotic genes.
Most cloning vectors are inducible, to avoid premature Most cloning vectors are inducible, to avoid premature
overproduction of a foreign product that could poison the overproduction of a foreign product that could poison the
bacterial host cells. bacterial host cells.
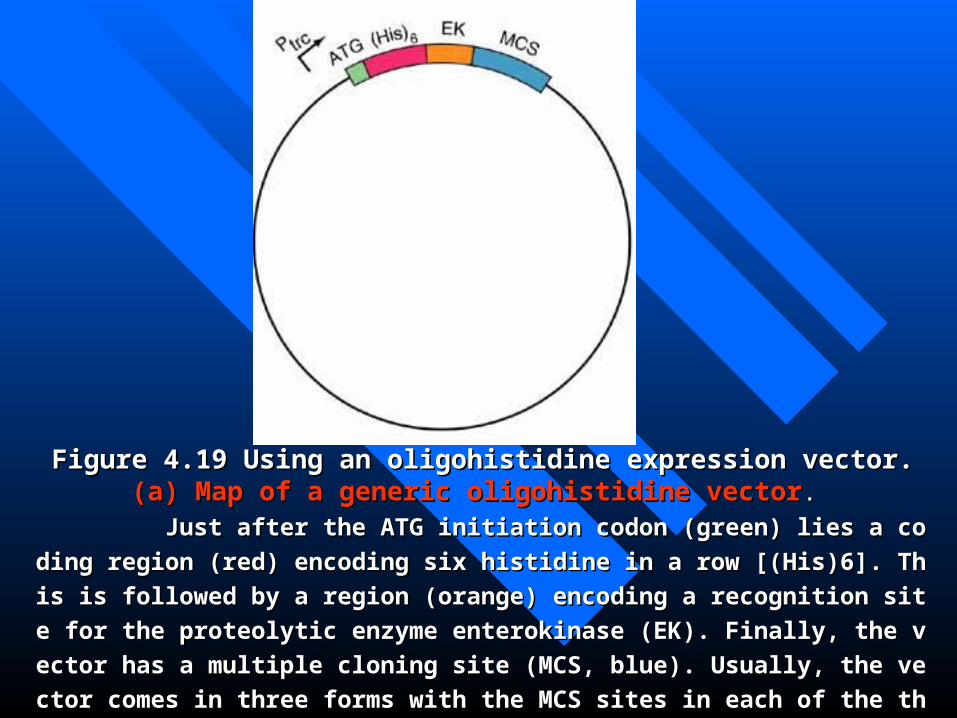
Figure 4.19 Using an oligohistidine expression vector. Figure 4.19 Using an oligohistidine expression vector. (a)(a) Map of a generic oligMap of a generic oligohistidine vectorohistidine vector. .
Just after the ATG initiation codon (green) lies a coding region (red) encoding six Just after the ATG initiation codon (green) lies a coding region (red) encoding six
histidine in a row [(His)6]. This is followed by a region (orange) encoding a recognition histidine in a row [(His)6]. This is followed by a region (orange) encoding a recognition
site for the proteolytic enzyme enterokinase (EK). Finally, the vector has a multiple closite for the proteolytic enzyme enterokinase (EK). Finally, the vector has a multiple clo
ning site (MCS, blue). Usually, the vector comes in three forms with the MCS sites in eaning site (MCS, blue). Usually, the vector comes in three forms with the MCS sites in ea
ch of the three reading frames. One can select the vector that puts the gene in the right ch of the three reading frames. One can select the vector that puts the gene in the right
reading frame relative to the oligohistidine.reading frame relative to the oligohistidine.

Figure 4.19 Using an oligohistidine expressioFigure 4.19 Using an oligohistidine expressio
n vector. n vector. (b)(b) Using the vectorUsing the vector. . 11. Insert the gene of interest (yellow) into the vecto. Insert the gene of interest (yellow) into the vector in frame with the oligohistidine coding region (rer in frame with the oligohistidine coding region (red) and transform bacterial cells with the recombind) and transform bacterial cells with the recombinant vector. The cells produce the fusion protein (reant vector. The cells produce the fusion protein (red and yellow), along with other, bacterial proteins d and yellow), along with other, bacterial proteins (green). (green). 22. Lyse the cells, releasing the mixture of proteins. . Lyse the cells, releasing the mixture of proteins. 33. Pour the cell lysate through a nickel affinity chr. Pour the cell lysate through a nickel affinity chromatography column, which binds the fusion protomatography column, which binds the fusion protein but not the other proteins. ein but not the other proteins. 44. Release the fusion protein from the column with . Release the fusion protein from the column with histidine or with imidazole, a histidine analogue, whistidine or with imidazole, a histidine analogue, which competes with the oligohistidine for binding thich competes with the oligohistidine for binding to the nickel. o the nickel. 5.5. Cleave the fusion protein with enterokinase. Cleave the fusion protein with enterokinase. 66. Pass the cleaved protein through the nickel colu. Pass the cleaved protein through the nickel column once more to separate the oligehistidine from tmn once more to separate the oligehistidine from the desired protein.he desired protein.

SummarySummary::
Expression vectors frequently produce fusion Expression vectors frequently produce fusion
proteins, with one part of the protein coming from coding proteins, with one part of the protein coming from coding
sequences in the vector and the other part from sequences sequences in the vector and the other part from sequences
in the cloned gene itself. Many fusion proteins have the in the cloned gene itself. Many fusion proteins have the
great advantage of being simple to isolate by affinity great advantage of being simple to isolate by affinity
chromatography. The λgt11 vector produces fusion chromatography. The λgt11 vector produces fusion
proteins that can be detected in plaques with a specific proteins that can be detected in plaques with a specific
antiserum. antiserum.

Figure 4.20 Forming a fusion protein Figure 4.20 Forming a fusion protein in λgt11. in λgt11.
The gene to be expressed (green) is iThe gene to be expressed (green) is i
nserted into the nserted into the EcoEcoRI site near the end oRI site near the end o
f the f the lacZlacZ coding region (red) just upstre coding region (red) just upstre
am of the transcription terminator. Thus, am of the transcription terminator. Thus,
upon induction of the upon induction of the lacZlacZ gene by IPTG, gene by IPTG,
a fused mRNA results, containing the in a fused mRNA results, containing the in
serted coding region just downstream of serted coding region just downstream of
that of β-galactosidase. This mRNA is trthat of β-galactosidase. This mRNA is tr
anslated by the host cell to yield a fusion anslated by the host cell to yield a fusion
protein. protein.

Figure 4.21 Detecting positiveλgtFigure 4.21 Detecting positiveλgt11 clones by antibody screening.11 clones by antibody screening. A filter is used to blot proteins A filter is used to blot proteins from phage plaques on a Petri disfrom phage plaques on a Petri dish. One of the clones (red) has proh. One of the clones (red) has produced a plaque containing a fusioduced a plaque containing a fusion protein including β-galactosidan protein including β-galactosidase and a part of the protein of intse and a part of the protein of interest. The filter with its blotted perest. The filter with its blotted proteins is incubated with an antibroteins is incubated with an antibody directed against our protein ody directed against our protein of interest, then with labeled Stapof interest, then with labeled Staphylococcus protein A, which bindhylococcus protein A, which binds specifically to antibodies. It will s specifically to antibodies. It will therefore bind only to the antibodtherefore bind only to the antibody-antigen complexes at the spot cy-antigen complexes at the spot corresponding to our positive clonorresponding to our positive clone. A dark spot on the film placed ie. A dark spot on the film placed in contact with the filter reveals thn contact with the filter reveals the location of our positive clone.e location of our positive clone.

Figure 4.22 Expressing a gene in a baculovirus.

Figure 4.22 Expressing a gene in a baculovirus. Figure 4.22 Expressing a gene in a baculovirus. First, insert the gene to be expressed (red), into a baculovirus transfer vector. IFirst, insert the gene to be expressed (red), into a baculovirus transfer vector. In this case, the vector contains the powerful polyhedrin promoter (Polh), flanked byn this case, the vector contains the powerful polyhedrin promoter (Polh), flanked bythe DNA sequences (yellow) that normally surround the polyhedrin gene, including the DNA sequences (yellow) that normally surround the polyhedrin gene, including a gene (green) that is essential for virus replication, the polyhedrin coding region itsa gene (green) that is essential for virus replication, the polyhedrin coding region itself is missing from this transfer vector. Just downstream of the promoter is a elf is missing from this transfer vector. Just downstream of the promoter is a BamBamHI HI restriction site, which can be used to open up the vector (restriction site, which can be used to open up the vector (step astep a) so it can accept the ) so it can accept the foreign gene (red) by ligation (foreign gene (red) by ligation (step bstep b). In ). In step cstep c, mix the recombinant transfer vecto, mix the recombinant transfer vector with linear viral DNA that has been cut so as to remove the essential gene. Transfer with linear viral DNA that has been cut so as to remove the essential gene. Transfect insect cells with the two DNAs together. This process is known as co-transfection.ct insect cells with the two DNAs together. This process is known as co-transfection. The two DNAs are not drawn to scale, the viral DNA is actually almost 15 times th The two DNAs are not drawn to scale, the viral DNA is actually almost 15 times the size of the vector. Inside the cell, the two DNAs recombine by a double crossover e size of the vector. Inside the cell, the two DNAs recombine by a double crossover that inserts the gene to be expressed, along with the essential gene, into the viral DNthat inserts the gene to be expressed, along with the essential gene, into the viral DNA. The result is a recombinant virus DNA that has the gene of interest under the conA. The result is a recombinant virus DNA that has the gene of interest under the control of the polyhedrin promoter. Next, infect cells with the recombinant virus. Finalltrol of the polyhedrin promoter. Next, infect cells with the recombinant virus. Finally, in y, in step dstep d and and ee, infect cells with the recombinant virus and collect the protein pro, infect cells with the recombinant virus and collect the protein product these cells make. Notice that the original viral DNA is linear and it is missing tduct these cells make. Notice that the original viral DNA is linear and it is missing the essential gene , so it cannot infect cells he essential gene , so it cannot infect cells (f).(f). This lack of infectivity selects automa This lack of infectivity selects automatically for recombinant viruses; they are the only ones that can infect cells. tically for recombinant viruses; they are the only ones that can infect cells.

Summary:Summary:
Foreign genes can be expressed in eukaryotic cells, and these Foreign genes can be expressed in eukaryotic cells, and these
eukaryotic systems have some advantages over their prokaryotic eukaryotic systems have some advantages over their prokaryotic
counterparts for producing eukaryotic proteins. counterparts for producing eukaryotic proteins.
Two of the most important advantages are (1) Eukaryotic proTwo of the most important advantages are (1) Eukaryotic pro
teins made in eukaryotic cells tend to be folded properly, so they teins made in eukaryotic cells tend to be folded properly, so they
are soluble, rather than aggregated into insoluble inclusion bodieare soluble, rather than aggregated into insoluble inclusion bodie
s. (2) Eukaryotic proteins made in eukaryotic cells are modified s. (2) Eukaryotic proteins made in eukaryotic cells are modified
(phosphorylated, glycosylated, etc.) in a eukaryotic manner. (phosphorylated, glycosylated, etc.) in a eukaryotic manner.

4.3.2 Other Eukaryotic Vectors4.3.2 Other Eukaryotic Vectors
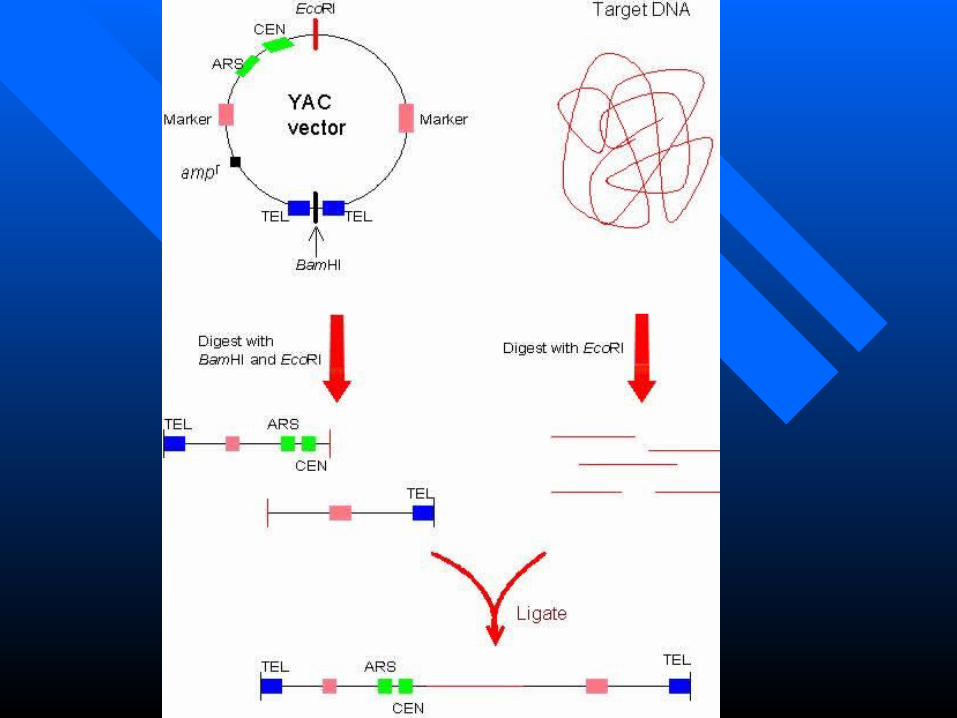
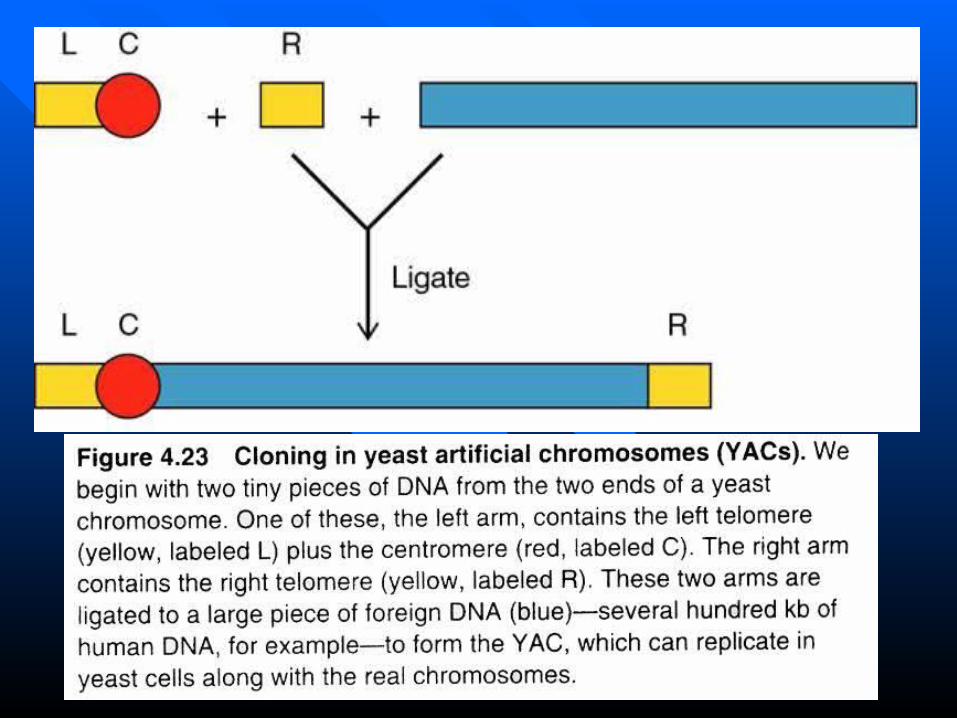
Yeast Artificial chromosomes (YACs)Yeast Artificial chromosomes (YACs) Using the Ti plasmid to transfer genes to plantsUsing the Ti plasmid to transfer genes to plants

Essential components of YAC vectors
•Centromers (CEN), telomeres (TEL) and autonomous replicating sequence (ARS) for proliferation in the host cell.
•ampr for selective amplification and markers such as TRP1 and URA3 for identifying cells containing the YAC vector.
•Recognition sites of restriction enzymes (e.g., EcoRI and BamHI)

BAC vectorsBAC vectors
Bacterial artificial chromosomes are basBacterial artificial chromosomes are based on the F factor of ed on the F factor of E. coliE. coli and can be used and can be used to clone up to 350 kb of genomic DNA in a to clone up to 350 kb of genomic DNA in a conveniently handled conveniently handled E. coliE. coli host. They are host. They are a morre stable and easier to use alternative ta morre stable and easier to use alternative to YAC.o YAC.

Using the Ti Plasmid to Transfer Gees to Using the Ti Plasmid to Transfer Gees to PlantsPlants

Nopaline and octopine Ti plasmids carry a variety o
f genes, including T-regions that have overlapping functi
ons

T-DNA has almost identical repeats of 25 bp at each end in thT-DNA has almost identical repeats of 25 bp at each end in the Ti plasmid. The right repeat is necessary for transfer and integre Ti plasmid. The right repeat is necessary for transfer and integration to a plant genome. T-DNA that is integrated in a plant genation to a plant genome. T-DNA that is integrated in a plant genome has a precise junction that retains 1-2 bp of the right repeat, ome has a precise junction that retains 1-2 bp of the right repeat, but the left junction varies and may be up to 100 bp short of the lbut the left junction varies and may be up to 100 bp short of the left repeat.eft repeat.

Figure 4.24 Crown gall tumors. (a) Formation of a crown gall. 1. Agrobacterium cells enter a wound in the plant, usually at the crown, or the junction of root an stem. 2. The Agrobacterium contains a Ti plasmid in addition to the much larger bacterial chromosome. The Ti plasmid has a segment (the T-DNA, red) that promotes tumor formation in infected plants. 3. The bacterium contributes its Ti plasmid to the plant cell, and the T-DNA from the Ti plasmid into grates into the plant’s chromosomal DNA. 4. The genes in the T-DNA direct the formation of a crown gall, which nourishes the invading bacteria.

Figure 4.24 Crown gall tumors. (b) Photograph of a crown gall tumor g
enetated by cutting off the top of a tobacco plant and inoculating with Agrobac
terium.
This crown gall tumor is a teratoma, which generates normal as wel
l as tumorous tissues springing from the tumor.

Figure 4.25 Using a T-DNA plasmid to intrFigure 4.25 Using a T-DNA plasmid to introduce a gene into tobacco plant. oduce a gene into tobacco plant.
(a)(a) A plasmid is formed with a foreign genA plasmid is formed with a foreign gene (red) under the control of the mannopine syne (red) under the control of the mannopine synthetase promoter (blue). This plasmid is used tthetase promoter (blue). This plasmid is used to transform o transform Agrobacterium Agrobacterium cells. cells. (b)(b) The transformed bacterial cells divide repeThe transformed bacterial cells divide repeatedly. atedly. (c)(c) A disk of tobacco leaf tissue is remA disk of tobacco leaf tissue is removed and incubated in nutrient medium, along oved and incubated in nutrient medium, along with the transformed with the transformed AgrobacteriumAgrobacterium cells. Th cells. These cells infect the tobacco tissue, transferring ese cells infect the tobacco tissue, transferring the plasmid bearing the cloned foreign gene. the plasmid bearing the cloned foreign gene. (d) (d) The disk of tobacco tissue sends out roots The disk of tobacco tissue sends out roots into the surrounding medium. into the surrounding medium. (e)(e) One of these One of these roots is transplanted to another kind of mediuroots is transplanted to another kind of medium, where it forms a shoot. This plantlet grows m, where it forms a shoot. This plantlet grows into a transgenic tobacco plant that can be testinto a transgenic tobacco plant that can be tested for expression of the transplanted gene. ed for expression of the transplanted gene.


Summary:Summary: Molecular biologists can clone hundreds of thousands Molecular biologists can clone hundreds of thousands
of base pairs of DNA at a time in yeast artificial chromosof base pairs of DNA at a time in yeast artificial chromos
omes (YACs). If they wish to transfer cloned genes to plomes (YACs). If they wish to transfer cloned genes to pl
ants, creating transgenic organisms with altered characterants, creating transgenic organisms with altered character
istics, they use a plant vector such as the Ti plasmidistics, they use a plant vector such as the Ti plasmid.