cinetica di reazione - zanasi.chem.unisa.itzanasi.chem.unisa.it/download/cinetica.pdf · cinetica...
TRANSCRIPT

CINETICA DI REAZIONE
La cinetica chimica, anche chiamata cinetica di reazionee lo studio delle velocita e dei meccanismi di reazione.
Un sistema reagente non e all’equilibrio, cosı la cineticadi reazione non e parte della termodinamica ma e unabranca della cinetica.
Gli aspetti cinetici di una reazione chimica sono impor-tanti quanto quelli termodinamici: la costante termodi-namica di equilibrio ci informa sulla resa di reazione perogni data P e T , ma se la velocita e troppo bassa lareazione non sara piu economica.
Le applicazioni della cinetica di reazione abbondano:industria, praparativa organica, inquinamento atomos-ferico, motori, ossidazione, organismi viventi, catalisi ecatalisi enzimatica.
Reazione omogenea: avviene in un’unica fase.Reazione eterogenea: coivolge specie presenti in dueo piu fasi.Le reazioni omogenee vengono divise in reazioni in fasegassosa e reazioni in soluzione.
1

Velocita di reazione
Consideriamo la reazione omogenea in un sistema chiuso
aA + bB + . . . → eE + fF + . . .
La velocita alla quale ogni reagente e consumato e pro-porzionale al suo coefficiente stechiometrico
dnA/dt
dnB/dt=
a
b
1
a
dnA
dt=
1
b
dnB
dt
dove nA ecc. e il numero di moli di A presenti al tempot.
Si definisce velocita di conversione J di una reazioneomogenea come
J ≡ −1
a
dnA
dt= −1
b
dnB
dt= . . . =
1
e
dnE
dt=
1
f
dnF
dt= . . .
Poiche A scompare dnA/dt e negativo e J e positivo.All’equilibrio J = 0.
Effettivamente, la relazione −a−1dnA/dt = e−1dnE/dt none detto debba essere soddisfatta se la reazione avvienein piu steps: in questo caso A viene prima convertitoin un intermedio e la relazione istantanea tra dnA/dt ednE/dt puo essere complicata. Se, come e frequente-mente vero, le concentrazioni di tutti gli intermedi direazione sono molto piccole in tutta la reazione, allorai loro effetti possono essere trascurati.
2

La velocita di conversione J e una quantita estensi-va. La velocita di conversione per unita di volume,J/V , e una quantita intensiva ed e chiamata velocitadi reazione r
r ≡ J
V=
1
V
(−1
a
dnA
dt
)e dipende da T , P e le concentrazioni delle specie nel sis-tema omogeneo. In molti casi (non tutti) V e costante ocambia in modo trascurabile, cosı che (1/V )(dnA/dt) =(dnA/V )/dt = d[A]/dt. Allora a V costante si ha
r = −1
a
d[A]
dt= −1
b
d[B]
dt= . . . =
1
e
d[E]
dt=
1
f
d[F]
dt= . . .
Unita di misura comuni di r sono mol dm−3 s−1.
3

Leggi cinetiche
In molti casi (non tutti) si trova sperimentalmente che
r = k[A]α[B]β . . . [L]λ
dove usualmente α, β, . . . , λ sono interi o semiinteri.
k e chiamata costante di velocita o coefficiente direazione ed e funzione di T e P .
La reazione e detta essere di ordine α rispetto ad A, diordine β rispetto a B ecc.. Gli esponenti α, β, . . . sonoanche chiamati ordini parziali. La somma α+β + . . .+λ ≡ n e l’ordine totale (o semplicemente ordine) direazione.
Le unita di misura di k sono concentrazione1−n tempo−1.
La costante di velocita di una reazione di primo ordine(n = 1) ha unita s−1 e non dipende dalle concentrazioni.
L’espressione di r come funzione delle concentrazioni echiamata legge cinetica.
4

Alcune leggi cinetiche osservate per sistemi omogeneisono
(1) H2 + Br2 → 2HBr r = k[H2][Br2]1/2
1+j[HBr]/[Br2]
(2) 2N2O5 → 4NO2 + O2 r = k[N2O5]
(3) H2 + I2 → 2HI r = k[H2][I2]
(4) 2NO + O2 → 2NO2 r = k[NO]2[O2]
(5) CH3CHO → CH4 + CO r = k[CH3CHO]3/2
(6) 2SO2 + O2NO−→ 2SO3 r = k[O2][NO]2
(7) H2O2 + 2I− + 2H+ → 2H2O + I2 r = k1[H2O2][I−]+k2[H2O2][I−][H+]
(8) Hg2+2 + Tl3+ → 2Hg2+ + Tl+ r = k
[Hg2+2 ][Tl3+][Hg2+]
dove k dipende fortemente dalla T ed e diverso in ognireazione.
5

Nella reazione (1) j e una costante.
Le reazioni da (1) a (6) sono in fase gassosa; le reazioni(7) ed (8) sono in soluzione acquosa.
Per la reazione (1) il concetto di ordine non e applicabile.
Nella reazione (7) entrambi i termini hanno un ordine,ma la velocita di reazione totale non ha un ordine.
La reazione (5) ha ordine 3/2.
Nella reazione (6) la specie NO aumenta la velocita direazione ma non appare nella equazione chimica, per-tanto e un catalizzatore.
Nella reazione (8) l’ordine rispetto a Hg2+ e −1.
Notare che gli esponenti nelle leggi cinetiche di reazionenon sono i coefficienti stechiometrici: l’espressione delleleggi cinetiche di reazione non puo essere dedotta dallastechimetria di reazione.
L’uso delle concentrazione nelle leggi cinetiche e corret-to solo nei casi ideali.
6

Meccanismi di reazione
Di norma una equazione chimica indica la stechiometriacompleta della reazione, ma non ci dice nulla circa ilprocesso, o meccanismo che effettivamente ha luogo.Ad esempio per la reazione
2SO2 + O2NO−→ 2SO3
e stato proposto il seguente processo a due steps
O2 + 2NO → 2NO2
NO2 + SO2 → NO + SO3
Per ottenere l’equazione stechiometrica occore moltipli-care per due il secondo step e sommarlo al primo.La specie NO2, che si forma nel primo step ed e con-sumata nel secondo, e detta intermedio di reazione.
Ci sono buone prove per credere che la reazione
2N2O5 → 4NO2 + O2
proceda secondo il meccanismo
N2O5 ⇀↽ NO2 + NO3
NO2 + NO3 → NO + O2 + NO2
NO + NO3 → 2NO
In questo caso ci sono due intermedi di reazione NO3 eNO.
7

Gli steps del meccanismo proposto devono sommarsiper dare la reazione stechiometrica completa. Nel casoprecedente moltiplicando per 2,1,1 rispettivamente glisteps a,b,c e sommando si ottiene la reazione stechio-metrica.
Il numero di volte per il quale un dato step deve esseremoltiplicato e detto numero stechiometrico, da nonconfondersi con i coefficienti stechiometrici.
Ogni step di un meccanismo di reazione e chiamatoreazione elementare. Una reazione semplice consistedi un sigolo step elementare. Una reazione complessao composita consiste di due o piu steps elementari.
La reazione di decomposizione di N2O5 e una reazionecomplessa. La reazione di Diels-Alder etilene + buta-diene → cicloesene e ritenuta essere una reazione sem-plice.
La forma della legge cinetica di reazione e unaconseguenza del meccanismo di reazione.
Alle volte la legge cambia al variare di T , indicando cheil meccanismo cambia con T .Altre volte si trova, ad esempio, che r = k[A]1.38, in-dicando che due o piu meccanismi procedono proba-bilmente contemporaneamente, un migliore descrizionepotrebbe essere data da r = k′[A]k′′[A]2.
8

Pseudo ordine
Per l’idrolisi del saccarosio
C12H22O11+H2O → C6H12O6(glucosio)+C6H12O6(fruttosio)
si trova che r = k[C12H22O11]. Poiche il solvente parte-cipa alla reazione, si dovrebbe avere r = k′[C12H22O11]w[H2O]v.Poiche l’acqua e sempre presente in grande eccesso, sipuo ritenere la sua concentrazione costante ed inglo-bare il termine [H2O]v nella costante di velocita, cioek = k′[H2O]v.
Questa reazione e detta essere di pseudo (ordine appar-ente) primo ordine.
Un catalizzatore aumenta la velocita di reazione senzaessere consumato. Reazioni catalizzate mostrano unordine apparente (pseudo ordine).
9

Misura della velocita di reazione
Per misurare r occorre seguire la concentrazione deireagenti o dei prodotti nel tempo.
Mediante il metodo chimico, ad intervalli di tempo, siprelevano campioni della miscela di reazione, si rallentao si blocca la reazione (raffreddamento, togliere il catal-izzatore, diluizione, aggiungere una specie che reagiscerapidamente con uno dei reagenti) e si analizza chimi-camente la mistura.
I metodi fisici sono di norma piu accurati e meno te-diosi: si misura una proprieta fisica del sistema comefunzione del tempo. In questo modo la reazione vineseguita continuamente.Pressione (gas, ∆n, !), volume (liquidi, dilatometro,polimerizzazioni), intensita di una banda di assorbimen-to, attivita ottica, conducibilita (soluzioni ioniche), in-dice di rifrazione (liquidi).
Metodo statico, metodo a flusso.
I metodi sopra sono utlizzabili quando il tempo di dimez-zamento e maggiore del secondo. Molte reazioni im-portanti hanno t1/2 che vanno da 100 − 10−11 (reazioniveloci).
10

Integrazione delle leggi cinetiche
Molti dei metodi utilizzati per dedurre la legge cineticadai dati cinetici, si basano sul confronto tra le predi-zioni che si ottengono dalle leggi possibili con i datisperimentali.
Per ottenere le concentrazioni in funzione del tempooccorre integrare le leggi cinetiche.
Reazioni di primo ordineSupponiamo che aA → prodotti sia una reazione di primoordine con r = k[A], la legge cinetica e
r = −1
a
d[A]
dt= k[A]
Definendo kA ≡ ak si ha
d[A]
dt= −kA[A]
Per integrare questa equazione differenziale, separiamole variabili [A] e t
d[A]
[A]= −kAdt
Integrando si ottiene
ln[A]2[A]1
= −kA(t2 − t1)
11

Se lo stato 1 e lo stato iniziale quando [A] = [A]0 et = 0, allora
ln[A]
[A]0= −kAt
dove [A] e la concentrazione al tempo t
[A] = [A]0e−kAt.
Il grafico di ln [A]0[A]
contro t da una retta di pendenza kA.
Per una reazione di primo ordine [A] diminuisce espo-nenzialmente nel tempo
La velocita di reazione sara
r = k[A]0e−kAt
Ponendo [A] = 12[A]0 si ottiene
kAt1/2 = − ln1
2= 0.693
12

Reazioni di secondo ordineLe forme piu comuni delle leggi cinetiche di secondoordine sono r = k[A]2 e r = k[A][B].
Forma 1. Supponiamo che la reazione aA → prodottisia di secondo ordine con r = k[A]2,allora
r = −1
a
d[A]
dt= k[A]2.
Definendo kA ≡ ak si ha∫ 2
1
d[A]
[A]2= −kA
∫ 2
1dt
1
[A]1− 1
[A]2= −kA(t2 − t1)
1
[A]− 1
[A]0= kAt [A] =
[A]01 + kAt[A]0
13

Inoltre, si puo notare che il grafico di 1/[A] contro t euna retta di pendenza kA.
Il tempo di dimezzamento si trova ponendo [A] = 12[A]0
e t = t1/2 e si trova
t1/2 =1
[A]0kA
tutto cio sempre per una reazione di secondo ordinecon r = k[A]2.
In questo caso il tempo di dimezzamento dipende da[A]0, in contrasto con le reazioni di primo ordine.
Prima di prendere in considerazione la forma 2, apriamouna piccola parentesi riguardante la decomposizione difrazioni razionali in elementi semplici. In particolareesamineremo il caso:
1
f(x)dove f(x) = (x − a)α(x − b)β . . .
Il teorema generale di decomposizione di frazioni razion-ali (vedi un testo di calcolo differenziale ed integrale)porta al seguente risultato
1
f(x)=
A0
(x − a)α+
A1
(x − a)α−1+ . . . +
Aα−1
(x − a)
+B0
(x − b)β+
B1
(x − b)β−1+ . . . +
Bβ−1
(x − b)+ . . .
14

dove i coefficienti A0, A1, . . . , B0, B1, . . . si determinanoponendo le frazioni allo stesso denominatore comune eduguagliano i numeratori.Esempio. Decomporre la frazione 1
(x+1)3 (x−2)in elemen-
ti semplici. Applicando il teorema si ottiene
1
(x + 1)3 (x − 2)=
A0
(x + 1)3+
A1
(x + 1)2+
A2
(x + 1)+
B0
(x − 2)
Mettendo a denominatore comune (che e la f(x)) eduguagliando i numeratori si ha
1 = A0(x−2)+A1(x+1)(x−2)+A2(x+1)2(x−2)+B0(x+1)3
Espandendo si ha
1 = −2A0 − 2A1 − 2A2 + B0 + (A0 − A1 − 3A2 + 3B0)x
+(A1 + 3B0)x2 + (A2 + B0)x3
da cui si ottiene il sistema di equazioni
1 = −2A0 − 2A1 − 2A2 + B0
0 = A0 − A1 − 3A2 + 3B0
0 = A1 + 3B0
0 = A2 + B0
che puo essere semplificato notando che per x = 2 siottiene immediatamente B0 = 1
27e per x = −1 si ottiene
A0 = −13, per cui A2 = −B0 = − 1
27e A1 = −3B0 = −1
9.
La decomposizione cercata sara
− 1
3(x + 1)3− 1
9(x + 1)2− 1
27(x + 1)+
1
27(x − 2).
15

Forma 2. Consideriamo ora la reazione aA + bB →prodotti con legge cinetica r = k[A][B]. Si ha
1
a
d[A]
dt= −k[A][B]
Questa equazione ha tre variabili [A], [B] e t ed occorreeliminarne una (non t ovviamente).Le quantita di A e B che reagiscono sono proporzionaliai loro coefficienti stechiometrici pertanto
∆nB
∆nA=
b
a
e a volume costante
b
a=
∆[B]
∆[A]=
[B] − [B]0[A] − [A]0
da cui si ricava [B]
[B] = [B]0 − b
a[A]0 +
b
a[A].
Sostituendo nell’equazione differenziale, separanto le vari-abili ed integrando si ha∫ 2
1
d[A]
[A]([B]0 − ba−1[A]0 + ba−1[A])= −
∫ 2
1akdt
che possiamo riscrive come∫ 2
1
d[A]
[A](ab−1[B]0 − [A]0 + [A])= −
∫ 2
1bkdt
16

Ponendo x = [A] e r = ab−1[B]0−[A]0, riscriviamo l’argo-mento dell’integrale del lato sinistro come 1
x(x+r)e, ricor-
dando la discussione dulla decomposizione della frazionirazionali fatta precedentemente, si ha
1
x(x + r)=
1
rx− 1
r(x + r)(r �= 0) (∗)
che si integra facilmente per dare (r �= 0)∫dx
x(x + r)=
lnx
r− ln(x + r)
r+ C =
1
rln
x
x + r+ C
Allora ∫ 2
1
d[A]
[A](ab−1[B]0 − [A]0 + [A])
=1
ab−1[B]0 − [A]0ln
[A]
ab−1[B]0 − [A]0 + [A]
∣∣∣∣2
1
=1
ab−1[B]0 − [A]0ln
[A]
ab−1[B]
∣∣∣∣2
1
=1
ab−1[B]0 − [A]0ln
[A]2/[A]1[B]2/[B]1
= −bk(t201/F302336.963 1 Tf0.4865 0 TD(t)Tj/F1 1 Tf118616.5280 11.9552 4 201.705 Tm0 Tc(1)Tj496.528 0 0 16.528[
[B]0∞◦/F3336.963b36.96 2′3.865 T4574[A]0
l
n
1
/
5
3
k
/
[B]1

Il grafico del lato sinistro dell’equazione (**) contro iltempo e una retta di pendenza k.In questo caso il concetto di t1/2 non si applica, poiche
quando [B] = 12[B]0 si ha che [A] �= 1
2[A]0, tranne quando
A e B sono fatti reagire in proporzioni stechiometriche.
Nel caso speciale in cui i reagenti sono mischiati in pro-porzioni stechiometriche si ha che [B]0/[A]0 = b/a, per-tanto a[B]0− b[A]0 = 0 e l’equazione (**) non si applica(vedi anche condizione r �= 0 nella decomposizione (*)).Riconsiderando la realzione
[B] = [B]0 − b
a[A]0 +
b
a[A]
si vede che se a[B]0 − b[A]0 = 0 allora vale anche a[B]−b[A] = 0, a indicazione del fatto che la proporzionestechiometrica si mantiene lungo tutto il corso dellareazione, e la legge cinetica diventa
−1
a
d[A]
[A][B]= −1
b
d[A]
[A]2= kdt
che si integra in modo simile alla forma 1, per dare
1
[A]− 1
[A]0= kBt kB ≡ bk
18

Reazioni di terzo ordineLe forme piu comuni sono r = k[A]3, r = k[A]2[B] er = k[A][B][C].
Forma 1. Invece di delineare l’integrazione della leggecinetica di terzo ordine d[A]/dt = −kA[A]3, consideriamoil caso generale
d[A]/dt = −kA[A]n
Integrando si ha∫ 2
1[A]−nd[A] = −kA
∫ 2
1dt
[A]−n+1 − [A]−n+10
−n + 1= −kAt (n �= 1)
[A]1−n = [A]1−n0 + (n − 1)kAt (n �= 1)
Ponendo [A] = 12[A]0 e t = t1/2 si ottiene il tempo di
dimezzamento
t1/2 =2n−1 − 1
(n − 1)[A]n−10 kA
(n �= 1)
Notiamo che le equazioni sopra si applicano per tuttii valori di n tranne 1. In particolare queste equazionivalgono per n = 0, n = 1/2 e n = 3/2.
19

Forma 2. Consideriamo la reazione aA+bB → prodotti,con legge cinetica r = k[A]2[B]. Si ha
1
a
d[A]
dt= −k[A]2[B]
Analogamente al caso di secondo ordine, si potra scri-vere la relazione
a[B] = a[B]0 − b[A]0 + b[A]
ed integrando si ha∫ 2
1
d[A]
[A]2(ab−1[B]0 − [A]0 + [A])= −
∫ 2
1bkdt
Ponendo x = [A] e r = ab−1[B]0 − [A]0, riscriviamo l’ar-gomento dell’integrale del lato sinistro come 1
x2(x+r), che
puo essere decomposto come
1
x2(x + r)=
A0
x2+
A1
x+
B0
x + r
dove
1 = A0(x + r) + A1x(x + r) + B0x2
da cui si ricava facilmente che A0 = 1/r, A1 = −1/r2 eB0 = 1/r2. Allora
1
x2(x + r)=
1
rx2− 1
r2x+
1
r2(x + r)(r �= 0)
20

che si integra facilmente per dare (r �= 0)∫1
x2(x + r)= − 1
rx−ln x
r2+
ln(x + r)
r2+C = − 1
rx+
1
r2ln
x + r
x+C
Allora∫ 2
1
d[A]
[A]2(ab−1[B]0 − [A]0 + [A])= − 1
ab−1[B]0 − [A]0
1
[A]
∣∣∣∣2
1
+1
(ab−1[B]0 − [A]0)2ln
(ab−1[B]0 − [A]0 + [A])
[A]
∣∣∣∣2
1
= −bk(t2 − t1)
Ponendo δab = a[B]0 − b[A]0 si ottiene
b
δab
(− 1
[A]
∣∣∣∣2
1
+b
δab
lnab−1[B]
[A]
∣∣∣∣2
1
)
=b
δab
(1
[A]1− 1
[A]2+
b
δab
ln[B]2/[B]1[A]2/[A]1
)= −bk(t2 − t1)
Pertanto
1
δab
(1
[A]0− 1
[A]+
b
δabln
[B]/[B]0[A]/[A]0
)= −kt
Analogamente al caso di secondo ordine, l’equazionenon si applica quando i reagenti sono mischiati in pro-porzioni stechiometriche, in quanto δab = 0 (vedi anchecondizione r �= 0 nella decomposizione). In questo casola legge cinetica diventa simile alla forma 1:
1
a
d[A]
[A]2[B]=
1
b
d[A]
[A]3= −kdt
21

Forma 3. Consideriamo la reazione aA + bB + cC →prodotti, con legge cinetica r = k[A][B][C]. Si ha
1
a
d[A]
dt= −k[A][B][C]
e, poiche i reagenti vengono consumati proporzional-mente ai loro coefficienti stechiometrici, valgono le re-lazioni
a[B] = a[B]0 − b[A]0 + b[A] = δab + b[A]
a[C] = a[C]0 − c[A]0 + c[A] = δac + c[A]
ed integrando si ha∫ 2
1
d[A]
[A](b−1δab + [A])(c−1δac + [A])= −
∫ 2
1
bc
akdt
Ponendo x = [A], r = b−1δab e s = c−1δac, riscriviamo l’ar-gomento dell’integrale del lato sinistro come 1
x(x+r)(x+s),
che puo essere decomposto come (r, s �= 0)
1
x(x + r)(x + s)=
1
rsx+
1
r(r − s)(x + r)+
1
s(s − r)(x + s)
Integrando si ottiene (r, s �= 0)∫1
x(x + r)(x + s)=
ln x
rs+
ln(x + r)
r(r − s)+
ln(x + s)
s(s − r)+ C
22

Pertanto∫ 2
1
d[A]
[A](b−1δab + [A])(c−1δac + [A])=
ln[A]
b−1δabc−1δac
∣∣∣∣2
1
+ln([A] + b−1δab)
b−1δab(b−1δab − c−1δac)
∣∣∣∣2
1
+ln([A] + c−1δac)
c−1δac(c−1δac − b−1δab)
∣∣∣∣2
1
= −∫ 2
1
bc
akdt
da cui si ottiene
1
b−1δabc−1δacln
[A]2[A]1
− 1
b−1δab(c−1δac − b−1δab)ln
[B]2[B]1
+1
c−1δac(c−1δac − b−1δab)ln
[C]2[C]1
= −bc
ak(t2 − t1)
considerando che c−1δac−b−1δab = a(bc)−1(b[C]0−c[B]0) =a(bc)−1δbc si ha
a
δabδacln
[A]
[A]0− b
δabδbc
ln[B]
[B]0+
c
δacδbc
ln[C]
[C]0= −kt
L’equazione non vale se i reagenti sono mischiati in pro-porzioni stechiometriche, in quanto δab = δac = 0 (vedianche condizione r, s �= 0 sopra). In questo caso lareazione si riduce alla forma 1. Nel caso solo [A] e [C]siano in rapporto stechiometrico vale la forma 2.
23

Reazioni di primo ordine reversibiliTutto cio che abbiamo visto fino ad ora e strettamentevalido solo se la costante di equilibrio e infinita.Questo non significa che i risultati ottenuti siano inutili,anzi si trova che il tutto funziona abbastanza bene apatto che la reazione sia lontana dall’equilibrio.
Sia la reazione chimica reversibile A⇀↽C di primo ordinein entrambe le direzioni: forward (f) e back (b), cosıche sia rf = kf [A] e rb = kb[C]. Pertanto
−(
d[A]
dt
)f
= rf = kf [A]
(d[A]
dt
)b
= rb = kb[C]
(notare i segni!) Assumendo la presenza di intermedi inconcentrazione trascurabile, si ha la velocita totale datada
d[A]
dt=
(d[A]
dt
)f
+
(d[A]
dt
)b
= −kf [A] + kb[C]
Ora ∆[C] = −∆[A], ovvero
[C] − [C]0 = −([A] − [A]0)
[C] = [C]0 + [A]0 − [A]
Sostituendo si ottiene
d[A]
dt= kb[C]0 + kb[A]0 − (kf + kb)[A]
24

Prima di integrare notiamo che: (i) quanto t → ∞ ilsistema raggiunge l’equilibrio, ovvero le velocita dellereazioni diretta ed inversa diventano uguali; (ii) all’e-quilibrio le concentrazioni delle specie sono costanti, inparticolare d[A]
dt= 0. Quindi
kb[C]0 + kb[A]0 = (kf + kb)[A]eq (∗)Allora
d[A]
dt= (kf + kb)([A]eq − [A])
Separando le variabili si ha
d[A]
[A]− [A]eq= −(kf + kb)dt
ed integrando si ottiene
ln[A]− [A]eq[A]0 − [A]eq
= −(kf + kb)t
[A]− [A]eq = ([A]0 − [A]eq)e−(kf+kb)t (∗∗)
dove [A]eq si ottiene dalla (*). Notare la stretta somiglian-za tra la (**) e la legge cinetica di primo ordine
[A] = [A]0e−kAt
che e un caso particolare della (**) con [A]eq = 0 ekb = 0 (notare che il coefficiente stechiometrico a estato assunto, per semplicita, uguale ad 1).
25

Il grafico di [A] contro t che si ottiene dalla (**) pre-senta un decadimento esponenziale simile a quello dellereazioni di primo ordine, tranne che [A] → [A]eq quandot → ∞.
Reazioni di primo ordine consecutiveFrequentemente un prodotto di una reazione diventa unreagente in una reazione successiva, come, ad esempio,in un meccanismo di reazione a piu passi.Consideriamo il caso di due reazioni consecutive irre-versibili di primo ordine
Ak1−→ B
k2−→ C
(i coefficienti stechiometrici sono assunti 1 per sem-plicita). La velocita delle reazioni sono r1 = k1[A] er2 = k2[B]. Le velocita di variazione di [B] dovute allaprima e alla seconda reazione sono (d[B]/dt)1 = k1[A] e(d[B]/dt)2 = −k2[B] rispettivamente. Cosı
d[B]/dt = (d[B]/dt)1 + (d[B]/dt)2 = k1[A] − k2[B]
26

In totale abbiamo tre equazioni differenziali accoppiate:
d[A]/dt = −k1[A] d[B]/dt = k1[A]−k2[B] d[C]/dt = k2[B]
Assumiamo che solo A sia presente nel sistema a t = 0:
[A]0 �= 0 [B]0 = 0 [C]0 = 0
La prima equazione e di primo ordine e si integra perdare
[A] = [A]0e−k1t
Sostituendo questo risultato nella seconda si ha
d[B]/dt = k1[A]0e−k1t − k2[B]
Si tratta di una equazione lineare di primo ordine
dy
dx+ P (x)y = Q(x)
che ha soluzione
y = v(x)
[∫Q(x)
v(x)dx + C
]dove
v(x) = e−∫
P (x)dx
Riscriviamo l’equazione come
d[B]/dt + k2[B] = k1[A]0e−k1t
e calcoliamo
v(t) = e−∫
k2dt = e−k2t
27

Pertanto la soluzione e
[B] = e−k2t
[∫k1[A]0e−k1t
e−k2tdt + C
]=
k1[A]0k2 − k1
e−k1t + Ce−k2t
Ora a t = 0, [B] = [B]0 = 0, da cui si ricava
C = − k1[A]0k2 − k1
Allora
[B] =k1[A]0k2 − k1
(e−k1t − e−k2t)
Invece di sostituire e risolvere la terza equazione, [C] puoessere ricavato dalla conservazione della materia: il nu-mero di moli presenti e costante nel tempo (attenzioneai coefficienti stechiometrici), ovvero [A]+[B]+[C]=[A]0
[C] = [A]0 − [A]0e−k1t − k1[A]0
k2 − k1(e−k1t − e−k2t)
da cui si ottiene
[C] = [A]0
(1 − k2
k2 − k1e−k1t +
k1
k2 − k1e−k2t
)
28

Grafico delle concentrazioni [A], [B] e [C] contro t.
k2 = 6k1
k2 =1
6k1
Notare il massimo per l’intermedio B.
29

Reazioni competitive di primo ordineFrequentemente una specie puo reagire in modi diversiper dare vari prodotti. Ad esempio, la nitrazione deltoluene puo avvenire nelle tre posizioni orto, meta opara.
Consideriamo il caso piu semplice di due reazioni com-petitive di primo ordine irreversibili con coefficienti ste-chiometrici uguali ad 1:
Ak1−→ C
Ak2−→ D
Si ha d[A]/dt = −k1[A] − k2[A] = −(k1 + k2)[A], checoincide con quella di primo ordine con kA = k1 + k2,quindi
[A] = [A]0e−(k1+k2)t
Per quanto riguarda [C] si ha
d[C]/dt = k1[A] = k1[A]0e−(k1+k2)t
Assumendo [C]0 = 0 per t = 0 si ottiene
[C] = − k1[A]0k1 + k2
e−(k1+k2)t
∣∣∣∣t
0
= − k1[A]0k1 + k2
(e−(k1+k2)t − 1
)
[C] =k1[A]0k1 + k2
(1 − e−(k1+k2)t
)(∗)
30

Similarmente dall’integrazione di d[D]/dt = k2[A] si ot-tiene
[D] =k2[A]0k1 + k2
(1 − e−(k1+k2)t
)(∗∗)
Da notare che la somma delle costanti di velocita k1+k2
appare in entrambe le espressioni che danno [C] e [D].
Dividendo la (*) per la (**) si ottiene
[C]/[D] = k1/k2
valida a qualsiasi istante durante la reazione. In altreparole, in base alle ipotesi fatte, le quantita di C e Dottenute dipendono dal rapporto tra le velocita delle duereazioni in competizione.In generale, pero, si deve tener conto anche delle reazionicontrarie
Ck−1−→ A D
k−2−→ A
e, alle volte, anche della reazione di interconversioneC ⇀↽ D.
31

Se si aspetta un tempo infinito il rapporto [C]/[D] saradeterminato dal rapporto Kc1/Kc2 tra le costanti di equi-librio (sistema ideale), infatti
Kc1
Kc2=
[C]eq/[A]eq[D]eq/[A]eq
=[C]eq[D]eq
In questo caso si ha il controllo termodinamico dei prodot-ti: la specie con il ∆G0 piu negativo sara favorito.
Durante le fasi iniziali, quando le velocita delle reazioniinverse o di interscambio sono trascurabili, vale [C]/[D] =k1/k2 e si ha il controllo cinetico dei prodotti.
Se k−1, k−2 e kinter � k1 e k2 i prodotti sono cineticamentecontrollati anche quando [A]≈ 0.
Se k1/k2 1 e Kc1/Kc2 � 1 si ha che C e favoritocineticamente e D e favorito termodinamicamnete; inquesto caso la resa relativa dei prodotti dipendera dalfatto se c’e controllo cinetico (reazioni inverse lente e/otempi brevi) o termodinamico (reazioni inverse velocie/o tempi lunghi).
32

Integrazione numericaLa legge cinetica ci da d[A]/dt e, dopo integrazione, cipermette di calcolare [A] a qualsiasi tempo t a partireda [A]0 a t = 0.
L’integrazione puo essere eseguita numericamente in-vece che analiticamente. Il metodo numerico e moltoimportante nel caso di sistemi caratterizzati da moltereazioni simultanee indipendenti, che spesso non hannosoluzioni analitiche.
Per illustrare il metodo, consideriamo un caso semplice:d[A]/dt = −k[A]. Per un intervallo di tempo moltopiccolo possiamo fare l’approssimazione
d[A]/dt ≈ ∆A/∆t ∆A ≈ −k[A]∆t
Partendo da [A]0 a t = 0 calcoliamo
[A]1 ≈ [A]0 − k[A]0(t1 − 0)[A]2 ≈ [A]1 − k[A]1(t2 − t1)
...
Questa approssimazione diventa sempre piu accuratadiminuendo ∆t.
33

Determinazione delle leggi cinetiche
Restringeremo la nostra attenzione alla forma
r = k[A]α[B]β . . . [L]λ
Vedremo come e possibile risalire dai dati sperimentaliprima ad α, β, . . . poi a k.Discuteremo 3 metodi.
1. Metodo del tempo di dimezzamento. Si applicaquando r = k[A]n. Nel caso n = 1 si ha che t1/2 nondipende da [A]0: t1/2 = 0.693/kA. Quando n �= 1 si ha
t1/2 =2n−1 − 1
(n − 1)[A](n−1)0 kA
che puo essere trasformata in
log t1/2 = log2n−1 − 1
(n − 1)kA+ (1 − n) log[A]0
Il grafico di log t1/2 contro log[A]0 da una retta di pen-denza 1 − n. Notare che questo vale anche quandon = 1.
In pratica si procede nel seguente modo: (1) si grafi-ca [A] contro t; (2) si sceglie un valore [A]’ e si trova1/2[A]’, l’intervallo di tempo corrispondera a t1/2 per[A]’; (3) si sceglie un secondo valore [A]” e si trova1/2[A]”, il ∆t sara t1/2 per [A]”; (4) si ripete varie voltefino a che i dati sperimentali lo consentono; (5) si graficalog t1/2 contro log[A] e si trova la pendenza.
34

Questo metodo ha lo svantaggio che la reazione deveessere seguita per un tempo abbastanza lungo. Unmiglioramento consiste nell’usare il tempo frazionale tα,definito come il tempo richiesto affinche [A]0 scenda aα[A]0.
(α[A]0)1−n = [A]1−n
0 + (n − 1)kAtα (n �= 1)
tα =α1−n − 1
(n − 1)[A]n−10 kA
(n �= 1)
log tα = logα1−n − 1
(n − 1)kA+ (1 − n) log[A]0 (n �= 1)
tα = −(logα)/kA (n = 1)
Il grafico di log tα contro log[A]0 e una retta di pendenza1 − n. Un valore conveniente e α = 0.75
Esempio. I dati per la dimerizzazione 2A → A2 di uncerto composto sono:
[A] 68.0 50.2 40.3 33.1 28.4 22.3 18.7 14.5t 0 40 80 120 160 240 300 420
dove [A] e in mmol dm−3 ed il tempo in minuti, trovarel’ordine di reazione mediante il metodo del tempo didimezzamento.
35

Portiamo in grafico [A] contro t (vedi figura a sinistra)e ricaviamo per interpolazione i seguenti valori di [A]0(mmol/L) contro t1/2 (min)
[A]0 68 60 50 40 30t1/2 114 129 159 195 251log[A]0 1.833 1.778 1.699 1.602 1.477log t1/2 2.057 2.111 2.201 2.290 2.400
Graficando log t1/2 contro log[A]0 (vedi grafico a destra)si ottiene una retta di pendenza −0.97 = 1−n, pertantola reazione e di secondo ordine.
36

2. Metodo della velocita iniziale. Si misura la veloc-ita iniziale r0 variando la concentrazione iniziale di unreagente alla volta.Supponiamo di aver misurato r0 per [A]0,1 e [A]0,2, man-tenendo costanti [B]0, [C]0, . . . .Il rapporto
r0,2
r0,1=
k[A]α0,2[B]β0[C]γ0 . . .
k[A]α0,1[B]β0[C]γ0 . . .=
[A]α0,2
[A]α0,1
da cui si ricava facilmente α.
Una procedura piu attendibile si trova facendo vari testsvariando [A]0 in un intervallo piu ampio e, poiche ln r0 =ln k+α ln[A]0+β ln[B]0+. . ., il grafico di r0 contro ln[A]0,mantenendo [B]0+ . . . costanti, e una retta di pendenzaα.
La velocita iniziale r0 si puo determinare graficando [A]contro t e trovando la tangente a t = 0.
Gli ordini β, γ, . . . si ricavano in modo analogo.
37

3. Metodo dell’isolamento. Si opera in modo che laconcentrazione iniziale di uno dei reagenti sia molto piupiccola delle concentrazioni iniziali degli altri reagenti,ad esempio [A]0 � [B]0, [A]0 � [C]0 etc.Quindi le concentrazioni di tutti i reagenti, tranne A,rimarranno praticamente costanti nel tempo.La legge cinetica diventa
r = k[A]α[B]β . . . [L]λ = j[A]α
dove
j = k[B]β . . . [L]λ
rimane essenzialmente costante.Sotto queste condizioni la reazione ha pseudo ordine α,ricavabile, ad esempio, mediante il primo metodo visto.Per trovare β, . . . si procede analogamente, rendendodi volta in volta la concentrazione iniziale di uno deireagenti molto piu piccola delle altre.
Attenzione: molti testi suggeriscono di ottenere l’or-dine di reazione “by trial and error”. Ad esempio, sela legge cinetica e del tipo r = k[A]n, si grafica ln[A]verso t, [A]−1 verso t, [A]−2 verso t ecc., il miglior adat-tamento rettilineo dara l’ordine di reazione n = 1,2, . . . ,rispettivamente. Questo metodo e pericoloso perchespesso non e facile decidere quale di questi grafici e ilpiu rettilineo, specialmente se la reazione non e stataseguita a lungo. Notare che nel caso di primo ordine[A]0/[A] = ekt ≈ 1 + kt, per piccoli valori di kt.Inoltre, se l’ordine di reazione e semi-intero si puo giun-gere facilmente ad una conclusione sbagliata.
38

Determinazione di k. Dopo aver trovato l’ordine rispet-to ad ogni specie, si puo procedere alla determinazionedi k attraverso la pendenza del grafico piu appropriato.Ad esempio, se si e trovato che la legge cinetica e deltipo r = k[A][B] allora il grafico di ln([B]/[A]) verso t euna retta di pendenza k(a[B]0 − b[A]0).Il metodo dei minimi quadrati e l’uso di pesi statisticipermette di trattare i dati in modo appropriato.
Esempio. Trovare la costante cinetica della reazione3ArSO2H → ArSO2SAr + ArSO3H + H2O per la qualesono stati misurati i seguenti dati cinetici:
[A] 100 84.3 72.2 64.0 56.8 38.7 29.7 19.6t 0 15 30 45 60 120 180 300
dove [A] e in mmol/L ed il tempo in minuti.
Prima di tutto determiniamo l’ordine di reazione medi-ante il metodo del tempo di dimezzamento. Il graficodi [A] verso t e
39

Per interpolazione si ricavano le coppie [A]0 (mmol/L),t1/2 (min)
[A]0 100 90 80 70 60 50t1/2 77 83 93 108 124 162log[A]0 2.000 1.954 1.903 1.845 1.778 1.699log t1/2 1.886 1.919 1.968 2.033 2.093 2.210
Il grafico di log t1/2 verso log[A]0, a sinistra, e una rettadi pendenza 1 − n = −1.06323, da cui si trova che lareazione e di secondo ordine.Allora, portando in grafico 1/[A] (L/mol) contro t (min)si ha, a destra, una reatta di pendenza kA = 0.13647 Lmol−1 min−1, da cui si ottiene k = 0.000758 L mol−1
sec−1.
40

Leggi cinetiche e costanti di equilibrio per reazionielementari
Una reazione complessiva e data, in generale, da unaserie di reazioni elementari che costituiscono il meccan-ismo di reazione.Il numero di molecole che reagiscono in uno step ele-mentare e detto molecolarita (non esiste per reazionitotali).
A → P unimolecolare2A → P
A + B → P
}bimolecolari
3A → P2A + B → P
3A + B + C → P
}trimolecolari
Step elementari con molecolarita > 3 non sono noti, acausa della bassissima probabilita di urto a n corpi conn > 3. Molte reazioni elementari sono unimolecolari obimolecolari, le trimolecolari sono poco comuni a causadella loro bassa probabilita.
Consideriamo la reazione B+C→prodotti: anche se nontutti gli urti tra B e C conducono ai prodotti, la velocitadi reazione r = J/V e proporzionale alla frequenza dellecollisioni B-C per unita di volume. Per un gas ideale(vedi teoria cinetica dei gas) si trova che la frequenzadelle collisioni per unita di volume e data da
ZBC = π(rB + rC)2
[8RT
π
(1
MB+
1
MC
)]1/2(NB
V
)(NC
V
)
41

In altre parole, per una reazione elementare bimolecolarein un gas ideale si ottiene
r = k
(NB
V
)(NC
V
)r = k[B][C]
Allo stesso modo, per una reazione elementare trimole-colare in un gas ideale, la velocita di reazione e pro-porzionale alla frequenza delle collisioni a 3 corpi perunita di volume e, pertanto, r = k[A][B][C].
Per le reazioni unimolecolari in un gas ideale, c’e unaprobabilita fissata che ogni particolare molecola B→Prodotti(decomposizione, isomerizzazione) nell’unita di tempo.Pertanto, il numero di molecole che reagiscono nell’u-nita di tempo e proporzionale al loro numero NB:
J = kNB r = J/V = kNB/V r = k[B]
Considerazioni simili si applicano anche a reazioni ele-mentari in una soluzione ideale o idealmente diliuita.
In conclusione, per un sistema ideale, la legge cineticaper una reazione elementare aA+ bB+ cC →Prodotti e
r = k[A]a[B]b[C]c
42

Consideriamo la reazione elementare reversibile in unsistema ideale
aA + bBkf⇀↽kb
cCdD
Le leggi cinetiche per la reazione diretta (f) ed inversa(b) sono rf = kf [A]a[B]b e rb = kb[C]c[D]d rispettiva-mente.
Durante il procedere della reazione queste due velocitatendono allo stesso valore e all’equilibrio rf,eq = rb,eq,cioe
kf [A]aeq[B]beq = kb[C]ceq[D]deqIn altre parole
kf
kb
=[A]aeq[B]beq[C]ceq[D]deq
= Kc
Pertanto, per una reazione elementare, si ha che
Kc =kf
kb
Quindi, se kf kb, allora Kc 1 e la posizione diequilibrio favorisce i prodotti.
43

Meccanismi di reazione
La legge cinetica osservata fornisce informazioni sul mec-canismo di reazione, in quanto ogni meccanismo propos-to deve rendere conto di tale legge.
Di norma, l’esatta deduzione della legge cinetica dallediverse equazioni differenziali associate ad un meccan-ismo a molti passi elementari non e possibile, a causadelle difficolta matematiche.
Pertanto, si ricorre a metodi approssimati quali:
• approssimazione dello stadio cineticamente deter-minante;
• approssimazione dello stato stazionario.
Approssimazione dello stadio cineticamente deter-minante.In questo caso il meccanismo di reazione si assume chesia composto da in sequenza:
• una o piu reazioni reversibili che restano vicine al-l’equilibrio durante la reazione;
• uno stadio relativamente lento, cineticamente de-terminante;
• una o piu reazioni rapide.
44

Consideriamo, ad esempio, il seguente meccanismo com-posto da reazioni unimolecolari
Ak1⇀↽k−1
Bk2⇀↽k−2
Ck3⇀↽k−3
D
nel quale il passo centrale sia cineticamente determi-nante.
Cio si verifica quando:
k−1 k2B → C lentoB → A veloce
}passo 1 ≈ equilibrio;
k3 k2
k3 k−2
}in questo modo il passo 2 e
effettivamente il collo di bottiglia.
La velocita complessiva e quindi determinata dal secon-do passo che non e mai all’equilibrio. Inoltre [C] sarasempre piccola e potremo trascurare la reazione inversadello step 2 (non reversibile).
In tali circostanze, e irrilevante che i passi successivisiano reversibili o meno.La legge cinetica osservata dipendera dalla natura degliequilibri che precedono lo stadio lento.La grandezza relativa di k1 confrontata con k2 e irrile-vante ai fini della validita dell’approssimazione.Per quanto riguarda la reazione inversa totale, lo sta-dio determinante rimane sempre il secondo, come si puodesumere dalle condizioni sopracitate.
45

Esempio. La legge cinetica della reazione (in faseacquosa)
H+ + HNO2 + C6H5NH2Br−−→ C6H5N
+2 + 2H2O
osservata e r = k[H+][HNO2][Br−]. Il seguente mecca-nismo e stato proposto:
H+ + HNO2
k1
⇀↽k−1
H2NO+2 rapido
H2NO+2 + Br− k2→ ONBr + H2O lento
ONBr + C6H5NH2k3→ C6H5N
+2 + H2O + Br− veloce
dove il secondo step e quello cineticamente determi-nante. La velocita di reazione e r = d[C6H5N
+2 ]/dt e,
poiche lo step 3 e molto piu veloce dello step 2, si puoritenere che d[C6H5N
+2 ]/dt = d[ONBr]/dt, da cui segue
che la velocita di reazione e quella dello step 2:
r = k2[H2NO+2 ][Br−]
La specie H2NO+2 e un intermedio di reazione ed r deve
essere espressa in termini di reagenti e prodotti. Per farcio si ricorre all’equilibrio 1:
Kc,1 =k1
k−1=
[H2NO+2 ]
[H+][HNO2]e [H2NO+
2 ] =k1
k−1[H+][HNO2]
Sostituendo si ha r =k1k2
k−1[H+][HNO2][Br−]
In accordo con la legge osservata e con k = k1k2/k−1.
46

Esercizio. Per la reazione in soluzione acquosa acida
H2O2 + 2H+ + 2I− → I2 + H2O
la legge cinetica osservata e
r = k1[H2O2][I−] + k2[H2O2][I
−][H+]
indicante che la reazione procede mediante due mecca-nismi simultanei. Supponendo che uno dei due mecca-nismi sia
H+ + I− ⇀↽ HI rapidoHI + H2O2 → H2O + HOI lento
HOI + I− → I2 + OH− veloceOH− + H+ → H2O veloce
trovare la legge cinetica.
Lo step 2 e lo stadio cineticamente determinante, allorasi ha che la velocita di reazione puo essere approssimatacome r = k2[HI][H2O2].
Inoltre, essendo [HI] = Kc,1[H+][I−], sostituendo si ot-tiene
r = k2Kc,1[H+][I−][H2O2]
In accordo con una parte della legge osservata.
47

Approssimazione dello stato stazionarioFrequentemente gli intermedi di reazione, che appaiononei meccanismi proposti, sono specie molto reattive enon si accumulano significativamente durante il corsodella reazione stessa. Ad esempio, data la reazione R →I → P, dove I (intermedio) e una specie molto reattiva,si ha che [I]�[R] e [I]�[P].
Dopo un periodo di induzione, durante il quale [I] crescerapidamente, la pendenza della curva I diventa moltopiccola. Pertanto, si puo frequentemente assumere conbuona approssimazione che d[I]/dt = 0.Questa e chiamata approssimazione dello stato stazionario,in quanto, dopo il periodo di induzione, la velocita diformazione della specie I uguaglia la sua velocita discomparsa, cosı che [I] rimane pressocche costante.
48

Esempio. Riconsideriamo il meccanismo
H+ + HNO2 ⇀↽ H2NO+2
H2NO+2 + Br− → ONBr + H2O
ONBr + C6H5NH2 → C6H5N+2 + H2O + Br−
senza fare ipotesi circa le velocita relative dei singolipassi. Si ha
r = d[C6H5N+2 ]/dt = k3[ONBr][C6H5NH2]
La specie ONBr si forma nello step 2 con velocita
(d[ONBr]/dt)2 = k2[H2NO+2 ][Br−]
ed e consumata nello step 3 con velocota
(d[ONBr]/dt)3 = −k3[ONBr][C6H5NH2]
Applicando l’approssimazione dello stato stazionario allaspecie ONBr si trova
d[ONBr]/dt = (d[ONBr]/dt)2 + (d[ONBr]/dt)3 = 0
k2[H2NO+2 ][Br−] = k3[ONBr][C6H5NH2]
[ONBr] =k2
k3
[H2NO+2 ][Br−]
[C6H5NH2]
Sostituendo si ottiene
r = k2[H2NO+2 ][Br−]
49

Applichiamo ora l’approssimazione dello stato stazionarioalla specie H2NO+
2
d[H2NO+2 ]
dt= k1[H
+][HNO2]−k−1[H2NO+2 ]−k2[H2NO+
2 ][Br−] = 0
da cui si ricava che
[H2NO+2 ] =
k1[H+][HNO2]
k−1 + k2[Br−]
Sostituendo nell’espressione della velocita si ottiene
r =k1k2[H+][HNO2][Br−]
k−1 + k2[Br−]
Mediante l’approssimazione dello stato stazionario si ot-tengono, in generale, espressioni piu complicate di quelleche si ricavano applicando l’approssimazione dello sta-dio cineticamente determinante. Sotto certe condizioni,pero, si possono ricondurre una all’altra.
Ad esempio, nel caso precedente, se si fa l’ipotesi chek−1 k2[Br−] si ottiene lo stesso identico risultato. In-fatti, questo significa che k−1[H2NO+
2 ] k2[Br−][H2NO+2 ],
ovvero che la velocita dello step 2 e piccola rispetto al-la velocita dello step 1 inverso, condizione necessariaaffinche lo step 2 sia cineticamente determinante.
50

Riepilogando, per applicare l’approssimazione dello sta-dio cineticamente determinante occorre:
• porre r = alla velocita dello stadio lento;
• eliminare ogni riferimento a specie intermedie me-diante le Kc degli equilibri precedenti.
Per applicare l’approssimazione dello stato stazionariooccorre:
• porre r = alla velocita di formazione dei prodotti,ultimo step;
• eliminare [I] utilizzando d[I]/dt = 0;
• ripetere il punto precedente se ci sono altri inter-medi fino al primo step.
Di norma, l’approssimazione dello stato stazionario for-nisce espressioni piu complicate di quelle che si otten-gono applicando l’approssimazione dello stadio cinetica-mente determinante.
51

Dalla legge cinetica al meccanismoEsaminiamo ora, brevemente, come sia possibile, datauna legge cinetica osservata sperimentalmente, immag-inare possibili meccanismi che siano consistenti con lalegge cinetica stessa.
Regola 1a. Se la legge cinetica e del tipo r = [A]α[B]β . . . [L]λ,con α, β, . . . , λ interi positivi, la composizione totale diatomi e di cariche reagenti nello step cineticamente de-terminante e data da αA + βB + . . ..Esempio. La reazione in fase gassosa 2NO + O2 →2NO2 ha legge cinetica r = k[NO]2[O2]. Cercandodi immaginare un meccanismo contenente uno stadiocineticamente determinante, la regola precedente for-nisce la composizione totale di atomi reagenti uguale aN2O4. Alcuni possibili step cineticamente determinanticon composizione totale di atomi reagenti N2O4 sono:
(a) N2O2 + O2 →prodotti;
(b) NO3 + NO →prodotti;
(c) 2NO + O2 →prodotti;
(d) N2 + 2O2 →prodotti.
Ogni meccanismo avente come step cineticamente de-terminante uno degli step sopra, conduce alla correttalegge cinetica.
52

Infatti si ha:
(a) in questo caso lo stadio cineticamente determinantedeve essere preceduto da uno step di formazione diN2O2 a partire da due molecole di NO; un mecca-nismo plausibile e
2NO ⇀↽ N2O2 veloceN2O2 + O2 → 2NO2 lento
applicando l’approssimazione dello stadio cinetica-mente determinante si ha
r = k2[N2O2][O2][N2O2] = Kc,1[NO]2
r = k2Kc,1[NO]2[O2]
(b) in questo caso un possibile meccanismo e
NO + O2 ⇀↽ NO3 veloceNO3 + NO → 2NO2 lento
da cui segue che
r = k2[NO3][NO][NO3] = Kc,1[NO][O2]
r = k2Kc,1[NO]2[O2]
53

(c) in questo caso il meccanismo e costituito da ununico step trimolecolare
2NO + O2 → 2NO2
al quale segue immediatamente che
r = k[NO]2[O2]
(d) in questo caso occorre immaginare, a livello pu-ramente teorico, un meccanismo che porti inizial-mente alla formazione di N2 e O2 a partire da NO,ad esempio
2NO ⇀↽ N2 + O2 veloceN2 + 2O2 → 2NO2 lento
dal quale si ottiene
r = k2[N2][O2]2
[N2] = Kc,1[NO]2[O2]−1
r = k2Kc,1[NO]2[O2]
A parte quest’ultimo caso, non e noto quale dei treprecedenti sia quello corretto. D’altra parte, i mecca-nismi (a-c) pottrebbero avvenire simultaneamente e lalegge cinetica osservata sarebbe la medesima.
54

Regola 1b. Se la legge cinetica e del tipor = [A]α[B]β . . . [L]λ/[M]µ[N]ν . . . [R]ρ,
con α, β, . . . , λ, µ, ν, . . . , ρ interi positivi, la composizionetotale di atomi e di cariche reagenti nello step cinetica-mente determinante e dato da
αA + βB + . . . + λL − µM − νN − . . . − ρR;inoltre le specie µM, νN, . . . , ρR appaiono come prodottinell’equilibrio che precede lo stadio cineticamente deter-minante e non sono reagenti in quest’ultimo.Esempio. La reazione in soluzione acquosa
Hg2+2 + Tl3+ → 2Hg2+ + Tl+
procede con la legge cinetica
r = k[Hg2+
2 ][Tl3+]
[Hg2+]
Un meccanismo contenente uno stadio cineticamentedeterminante puo essere dedotto applicando la regolaprecedente: la composizione totale di atomi e di carichereagenti nello step lento sara Hg2+
2 + Tl3+ − Hg2+ =HgTl3+; inoltre la specie Hg2+ risulta nei prodotti del-l’equilibrio precedente lo step lento e non appare neireagenti di quest’ultimo. Allora, un possibile meccanis-mo e
Hg2+2
⇀↽ Hg2+ + Hg veloce
Hg + Tl3+ → Hg2+ + Tl+ lento
55

Infatti, applicando l’approssimazione dello stadio cineti-camnte determinante si ottiene
r = k2[Hg][Tl3+]
[Hg] = Kc,1[Hg2+2 ][Hg2+]−1
r = k2Kc,1[Tl3+][Hg2+2 ][Hg2+]−1
Notare che apparentemente r = ∞ all’inizio della reazionequando [Hg2+] = 0. In realta la legge cinetica non e val-ida negli istanti iniziali, ma solo dopo il raggiungimentodell’equilibrio nel primo step. Poiche tale equilibrio e ve-loce in confronto allo step cineticamente determinante,cio non produce effetti significativi sulla legge osservata.
Regola 2. Se la legge cinetica contiene un fattore[B]1/2, probabilmente il meccanismo contiene uno stepdurante il quale le molecole di B si dividono in due specieprima dello step lento.Esempio. Consideriamo una reazione catalizzata daH+, prodotti dalla dissociazione di un acido debole, se-guita da uno step lento che consuma gli H+, seguito dauno step veloce che rigenera gli H+:
CH3COOH ⇀↽ CH3COO− + H+ veloce
H+ + A → . . . lento
. . . → H+ + . . . veloce
Lo step lento determina la velocita r = k[H+][A]. Poicheil catalizzatore H+ e rigenerato velocemente la sua con-centrazione rimane costante e [H+] = [CH3COO−].
56


L’aggiunta di specie particolari puo aiutare a confermareo meno un meccanismo proposto. Ad esempio, l’idrolisidi certi alogenuri alchilici RCl+H2O → ROH+H++Cl−ha legge cinetica r = k[RCl]. Il meccanismo propostoconsiste in uno step lento, cineticamente determinanteRCl → R++Cl− seguito da uno step veloce R++H2O →ROH+H+ La formazione del carbocatione viene eviden-ziata dal fatto che in presenza di N−
3 la legge cineticanon cambia e, contemporaneamente, si forma una certaquantita di RN3.
Specie sostituite isotopicamente possono aiutare a chiarireun meccanismo. Ad esempio, traccianti isotopici mostra-no che per la reazione di esterificazione di un alcol pri-mario o secondario con un acido carbossilico, l’ossigenodella molecola d’acqua che si forma proviede, di norma,dall’acido: R18OH+R′CO16OH → R′CO18OR+H16OH.
Riassumendo, per studiare la cinetica di una reazioneoccorre: (a) stabilire reagenti, prodotti e stechiometriadella reazione complessiva; (b) seguire la variazione delleconcentrazioni nel tempo, per molte volte cambiando leconcentrazioni iniziali; (c) analizzare i dati ottenuti in(b) per trovare la legge cinetica; (d) proporre plausibilimeccanismi consistenti con la legge trovata; (e) dareevidenza ad un meccanismo tentando di determinare gliintermedi di reazione.
58

Dipendenza dalla temperatura delle costanti cinetiche
Le costanti cinetiche dipendono fortemente da T : ap-prossimativamente, per molte reazioni a T ambiente, kraddoppia o triplica per ∆T ≈ 10.
Nel 1889 Arrhenius noto che i dati k(T) di molte reazionipotevano essere adattati all’equazione
k = Ae−Ea/RT
dove A (fattore pre-esponenziale) ed Ea (energia diattivazione) sono costanti caratteristiche della reazione.Le unita di misura di A sono le medesime di k; quelle diEa sono uguali a quelle di RT , ossia kJ/mol o kcal/mol.
Prendendo i log si ha
ln k = lnA − Ea
RTo log10 k = log10 A − Ea
2.303RT
Attenzione, e lecito prendere il log solo di quantita senzadimensioni; pertanto le equazioni sopra non hanno unreale senso fisico.Comunque, se la legge di Arrhenius vale, il grafico dilog10 k verso 1/T e una retta di pendenza −Ea/2.303Red intercetta log10 A. Questo permette di determinareEa ed A, tipicamente con un errore di 1 kcal/mol Ea eun fattore 3 per quanto riguarda A.
59

Esempio. Per la reazione C2H5I+OH− → C2H5OH+I−in etanolo, sono stati misurati i seguenti dati k(T) (vedigrafico a sinistra)
104k/(Lmol−1s−1) 0.503 3.68 67.1 1190T/K 289.0 305.2 332.9 363.8
Determinare Ea edA.
Calcoliamo la tabella contenente log10 k contro 1/T (ve-di grafico a destra)
log10[k/(Lmol−1s−1)] -4.2984 -3.4342 -2.1733 -0.92451/T/(K−1) 0.00346 0.00328 0.00300 0.00275
La retta ha una pendenza di −4730K = −Ea/2.303R, dacui si ricava Ea = 4730×2.303×1.987 ≈ 21.6 kcal/mol.L’intercetta e 12.06 = log10 A, da cui si ha A ≈ 1.2 ×1012.
60

L’equazione di Arrhenius vale per quasi tutte le reazionielementari omogenee e per molte reazioni complesse.Interpretazione semplice: due molecole collidenti hannobisogno di una certa quantita minima di energia cineticadi moto relativo per iniziare la rottura di certi legami epermettere la formazione di nuovi composti.Per una reazione unimolecolare una certa energia min-ima e richiesta per isomerizzare o decomporre la mole-cola; tale energia proviene da collisioni.La legge di distribuzione di Maxwell-Boltzmann contieneun fattore e−ε/kT e si trova che la frazione di collisionicon energia cinetica relativa delle molecole lungo la lineadi collisione > εa e pari a
e−εa/kT = e−Ea/RT
dove Ea = NAεa e l’energia cinetica molecolare espressasulla base di una mole.
Per una tipica energia di attivazione di 20 kcal/mol ≈80 kJ/mol, solo una minuscola frazione delle collisioniha energia cinetica > Ea.
61

Notare che una bassa energia di attivazione compor-ta una reazione veloce, al contrario, un’alta energia diattivazione comporta ad una reazione lenta.
Il rapido aumento di k con T e dovuto principalmenteal maggior numero di collisioni aventi energia superioreall’energia di attivazione.
Nell’equazione di Arrhenius sia A che Ea sono costan-ti. In realta, si dimostra, sulla base di sofisticate teoriedi reazione, che entrambi i parametri dipendono da T .Quando Ea RT (che e vero per molte reazioni chimiche)la dipendenza dalla temperatura di Ea ed A e troppo pic-cola per essere determinate sulla base di dati cinetici chesono, di norma, poco accurati.
La definizione generale di energia di attivazione e
Ea ≡ RT 2d ln k
dt
Se Ea non dipende da T , l’equazione sopra si integra perdare l’equazione di Arrhenius, dove anche A non dipendeda T .La definizione generale di A e
A ≡ keEa/RT
dove sia A che Ea possono essere funzioni di T .
62

I valori osservati di energia di attivazione per moltereazioni chimiche elementari variano tra 0 e 80 kcal/mole tendono ad essere minori per le reazioni bimolecolari.Per reazioni unimolecolari A va tipicamente da 1012 a1015 s−1. Per reazioni bimolecolari valori tipici di Acadono da 108 a 1012 L mol−1 s−1.
La ricombinazione di due radicali liberi per formare unamolecola stabile poliatomica non richiede la rottura dilegami e, pertanto, non ha energia di attivazione.
Esempio. Calcolare Ea per una reazione la cui costantecinetica a T ambiente raddoppia aumentando T di 10gradi. Ripetere il calcolo nel caso in cui triplichi.
k(T2)
k(T1)=
Ae−Ea/RT2
Ae−Ea/RT2= exp
(Ea
R
T2 − T1
T1T2
)Prendendo i log
Ea = RT1T2(∆T)−1 ln[k(T2)/k(T1)]
Ea = 1.987 × 298 × 308 × 0.1 × ln(2o3)
Ovvero Ea ≈ 13 kcal/mol per il raddoppio; Ea ≈ 20kcal/mol se triplica.
63

Siano kf e kb le costanti cinetiche di una reazione ele-mentare e della sua inversa e siano Ea,f e Ea,b le cor-rispondenti energie di attivazione.Ricordando che kf/kb = Kc, si ha
ln kf − ln kb = lnKc
Differenziando rispetto a T si ottiene
d ln kf/dt − d ln kb/dt = d lnKc/dt
Utilizzando la definizione di Ea si ha
Ea,f/RT 2 − Ea,b/RT 2 = d lnKc/dt
Ora, considerando che (van’t Hoff a V costante) d lnKc/dt =∆U◦/RT 2, si ottiene
Ea,f − Ea,b = ∆U◦
valida per una reazione elementare fra gas ideali e,comunque, sufficientemente accurata anche per reazioniin soluzione (ideali o idealmente diluite).
64

Consideriamo ora una reazione complessa composta davari step elementari. Nel caso in cui sia possibile appli-care l’approssimazione dello stadio cineticamente deter-minante, k ha tipicamente la forma k1k2/k−1, dove k1 ek−1 sono le costanti cinetiche delle reazioni diretta ed in-versa dell’equilibrio che precede lo step 2 cineticamentedeterminante. Applicando l’equazione di Arrhenius si ha
k =k1k2
k−1=
A1A2
A−1e−(Ea,1−Ea,−1+Ea,2)/RT
Riconducibile ancora alla forma k = Ae−Ea/RT con ener-gia di attivazione complessiva Ea = Ea,1 − Ea,−1 + Ea,2.Notare la possibilila di avere Ea < 0.
Se la reazione procede attraverso due maccanismi incompetizione, la costante cinetica globale non obbediscealla legge di Arrhenius. In questo caso la legge cinetica ela somma delle leggi cinetiche dei rispettivi meccanismi.Anche nel piu semplice dei casi e del tipo r = −(k1 +k2)[A] e la costante cinetica complessiva risulta essere
k = k1 + k2 = A1e−Ea,1/RT + A1e
−Ea,1/RT
che non ha la forma dell’equazione di Arrhenius.
Il fatto che una reazione complessa segua la legge di Ar-rhenius, dipende dalla forma della legge cinetica globale.
65

Leggi cinetiche per sistemi non ideali
Per una reazione elementare in un sistema ideale, levelocita delle reazioni diretta ed inversa contengono leconcentrazioni delle specie reagenti, cosı come la costantedi equilibrio Kc.Per un sistema non ideale la costante di equilibrio perla reazione elementare
aA + bB ⇀↽ cC + dD
e
K◦ =acC,eqa
dD,eq
aaA,eqa
bB,eq
Pertanto, sembra ragionevole che le leggi cinetiche direazioni elementari in un sistema non ideale debbanoessere:
rf?= kfaa
AabB e rb
?= kba
cCad
D (∗)Infatti, ponendo rf = rb si ottiene K◦ = kf/kb.
Per molto tempo si e ritenuto che cio fosse corretto.Comunque, se si scrive
rf=kfY aaAab
B e rb=kbY acCad
D (∗∗)dove Y e una qualche funzione di T, P e concentrazioni,ponendo rf = rb all’equilibrio si riottiene K◦ = kf/kb.
I dati cinetici per reazioni ioniche in soluzione acquosachiaramente mostrano che la (*) e sbagliata e che laforma corretta delle leggi cinetiche e la (**)
66

Cosı per una soluzione non ideale, la legge cinetica perla reazione elementare aA+bB→ prodotti e
r=k∞Y (γA[A])a(γB[B])b ≡ kapp[A]a[B]b
dove i γ sono i coefficienti di attivita sulla scala delleconcentrazioni e Y e un parametro che dipende da T, Pe dalle concentrazioni.La costante cinetica apparente e definita come kapp ≡k∞Y γa
AγbB. Al limite di diluizione infinita si raggiunge il
comportamento ideale, i γ → 1, cosı come Y → 1.
La vera costante cinetica k∞ puo essere ottenuta mis-urando kapp a varie concentrazioni ed estrapolando adiluiziuone infinita.
Quando k∞ e nota Y puo essere determinato per ognicomposizione del sistema come
Y =kapp
k∞γaAγb
B
67

Reazioni unimolecolari
La maggior parte delle reazioni elementari sono bimole-colari o unimolecolari. Reazioni unimolecolari possonoessere di isomerizzazione, ad esempio cis-CHCl=CHCl→ trans-CHCl=CHCl, o di decomposizione, ad esempioCH3CH2I→ CH2=CH2+HI.
Le reazioni elementari bimolecolari avvengono quandol’energia cinetica relativa delle due molecole collidenti Ae B supera Ea, la collisione porta alla rottura di legamie alla formazioni di nuovi legami.
Per quale motivo una molecola dovrebbe spontanea-mente dividersi o isomerizzare?
Sembra ragionevole supporre che una molecola A ac-quisti l’energia di attivazione necessaria urtando un’al-tra molecola. In questo caso si dovrebbe osservareuna legge cinetica di secondo ordine, in contrasto conl’osservazione sperimentale di una cinetica di primo or-dine per le reazioni unimolecolari.
La risposta a questo problrma venne data da Lindemannnel 1922.
68

Lindemann propose il seguente dettagliato meccanismoper spiegare la reazione unimolecolare A→B+(C):
A + Mk1
⇀↽k−1
A∗ + M
A∗ k2−→ B(+C)
In questo schema A∗ e una molecola energizzata dellaspecie A (non e un complesso attivato), che ha abbas-tanza energia vibrazionale per decomporsi o isomeriz-zare: la sua energia vibrazionale supera Ea del secondostep.Durante la collisione l’energia cinetica di M e trasfor-mata in energia vibrazionale di A. Qualsiasi molecola Mpuo portare A ad un alto livello vibrazionale; cosı M puoessere un’altra molecola A, una molecola dei prodotti oqualsiasi altra molecola presente nella fase gassosa o insoluzione che non appare nella reazione globale.
La velocita di reazione e r = d[B]/dt = k2[A∗] ed appli-cando l’approssimazione dello stato stazionario si ha
d[A∗]/dt = k1[A][M]− k−1[A∗][M] − k2[A
∗] = 0
[A∗] =k1[A][M]
k−1[M] + k2
Sostituendo si ottiene
r =k1k2[A][M]
k−1[M] + k2
La legge cinetica non ha un ordine definito.
69

Ci sono due casi limite:
k−1[M] k2 r = (k1k2/k−1)[A] (1)k2 k−1[M] r = k1[A][M] (2)
In fase gassosa, la (1) e chiamata limite di alta pressione,poiche il grande valore di [M] porta a k−1[M] k2, valeanche per soluzioni liquide; La (2) e invece chiamatalimite di bassa pressione.La legge cinetica ad alta pressione e di primo ordine,quella a bassa pressione e di secondo ordine (pseudoprimo ordine). Allo stesso medesimo risultato si giungeanche applicando l’approssimazione della stadio cineti-camente determinate: ad alta pressione lo step lento eil secondo mentre il primo e all’equilibrio, per cui si ot-tiene la (1); a bassa pressione lo step lento e il primo,per cui vale la (2).
Il fatto essenziale nel meccanismo di Lindemann e chetrascorre un certa quantita di tempo tra l’energizzazionedi A ad A∗ e la decomposizione di A∗ a prodotti (tempodi vita di A∗). Questo intervallo di tempo consente allostep -1 di avvenire, ed il quasi equilibrio degli steps 1 e-1 produce la cinetica di primo ordine. Notare che untempo di vita nullo, ed anche k2 → ∞, comporta unacinetica di secondo ordine.La specie vibrazionalmente eccitata A∗ ha un tempo divita non nullo perche la molecola ha molti legami, ed oc-corre tempo affinche l’energia vibrazionale si possa accu-mulare su quel particolare legame che si rompe durantela reazione. Conseguentemente una molecola biatomicanon si puo decomporre con una cinetica di primo ordine(vedi anche dopo trimolecolari).
70

La costante cinetica unimolecolare sperimentale kuni edefinita da r = kuni[A], dove r e la velocita osservata.Secondo il meccanismo di Lindemann si ha che
kuni =k1k2[M]
k−1[M] + k2=
k1k2
k−1 + k2/[M]
Nel limite ad alta pressione kuni,P=∞ = k1k2/k−1. Dimin-uendo la pressione iniziale P0, kuni diminuisce lentamente.A pressioni iniziali molto basse kuni = k1[M] e kuni diminuiscelinearmente con P0. Questo comportamento e statoconfermato sperimentalmente per reazioni unimoleco-lari in fase gassosa (vedi grafico per l’isomerizzazioneCH3NC → CH3CN a 230 ◦C).
L’espressione di Lindemann puo essere riscritta come1/kuni = k−1/k1k2 + 1/k1[M], dove si nota un previsionedi andamento lineare di k−1
uni contro P−10 .
71

Sperimentalmente si osservano deviazioni sensibili dal-la linearita (vedi grafico sempre per la reazione CH3NC→CH3CN): il meccanismo di Lindemann considera k2 costanteper tutte le molecole A∗; invece e l’energia vibrazionalee maggiore diventa la probabilita che A∗ isomerizzi o sidecomponga.
Reazioni trimolecolariSono rare. Forse il miglior esempio di una reazione tri-molecolare in fase gas e la ricombinazione di due atomi:l’energia rilasciata nel formare il legame chimico diventaenergia vibrazionale e, se non e presente un terzo corpoper asportare l’energia in eccesso, la molecola dissociadurante la sua prima vibrazione. Ad esempio
I + I + M → I2 + M r = k[I]2[M]
dove M e un qualsiasi altro atomo o molecola.
72

La ricombinazione
CH3 + CH3 → C2H6
non richiede un terzo corpo perche l’energia in eccessopuo essere distribuita su molti legami.In pochi casi speciali la ricombinazione di due atomiavviene in assenza di un terzo corpo M, ovvero quandol’energia in eccesso puo essere emessa come luce da unostato eccitato della molecola.
Durante la ricombinazione di due atomi nessun legamechimico viene rotto, pertanto la Ea dovrebbe essere nul-la. La costante cinetica della ricombinazione di due ato-mi di I e stata misurata in funzione della temperaturamediante esperimenti di flash-photolysis. Si e trovatoche k diminuisce aumentando T , ovvero Ea < 0. Ciosi spiega considerando che se da una parte la frequen-za degli urti a tre corpi aumenta con T , dall’altra laprobabilita che l’energia in eccesso venga assorbita daM diminuisce (in maggior misura). Valori tipici di Ea perla ricombinazione di atomi A+B+M→AB+M vanno da0 a −4 kcal/mol.
73

La decomposizione di I2, come di qualsiasi altra molecolabiatomica, procede come reazione inversa della ricom-binazione
I2 + M → I + I + M
(tempo di vita di I∗2 ≈ nullo). Pertanto la Ea di decom-posizione risulta leggermente minore di ∆U◦. Nel casodi
C2H6 → 2CH3
si ha che Ea = ∆U◦.
La ricombinazione di una molecola triatomica ha spessobisogno di un terzo corpo M, altrimenti l’extra energiavibrazionale puo rapidamente concentrarsi su un legamee dissociare la molecola. Ad esempio
O2 + O + M → O3 + M
La reazione inversa di questa reazione elementare mostrache la decomposizione dell’ozono procede come proces-so bimolecolare.
Le reazioni in fase gas tra NO e Cl2,Br2,O2, ad esempio2NO+Cl2 → 2NOCl, sono cineticamente di terzo ordine,ma non e ancora chiaro se esse procedono come ununico step trimolecolare o come due steps bimolecolario entrambi.
74

Reazioni a catena
Una reazione a catena e costituita da una serie di passielementari attraverso i quali un intermedio e consuma-to, i regenti sono convertiti in prodotti e l’intermedioe rigenerato. Questo ciclo e ripetuto moltissime volte,cosı che piccole quantita di intermedio producono gran-di quantita di prodotti.La maggior parte delle combustioni, esplosioni e polimer-izzazioni sono reazioni a catena e, molto spesso, coin-volgono radicali liberi come intermedi.
Una delle reazioni a catena meglio conosciute e
H2 + Br2 → 2HBr
La legge cinetica osservata per questa reazione in fasegassosa nel range di temperature da 500 a 1500 K e
r =1
2
d[HBr]
dt=
k[H2][Br2]1/2
1 + j[HBr]/[Br2]
dove j e una costante che dipende molto poco dalla tem-peratura e risulta ≈ 0.12. Poiche aumentando [HBr] lavelocita r diminuisce, si dice che il prodotto HBr inibiscela reazione.
L’esponente 1/2 suggerisce la decomposizione del Br2.Un atomo di Br puo reagire con H2 per dare HBr e H.Ogni atomo di H prodotto puo reagire con Br2 per dareHBr e Br, rigenerando l’intermedio Br.
75

Pertanto, si crede che il meccanismo di reazione sia
Br2 + Mk1
⇀↽k−1
2Br + M
Br + H2
k2
⇀↽k−2
HBr + H
H + Br2k3−→ HBr + Br
Lo step 1 innesca il meccanismo, lo step -1 lo termi-na. Gli steps 2 e 3 formano la catena e sono detti dipropagazione. Lo step -2 inibisce la reazione.
Per ogni atomo di Br prodotto nello step 1 si hannomolte ripetizioni degli step 2 e 3. Lo step -3 e troppolento per contribuire al meccanismo.
Allora, dato il meccanismo sopra, la velocita di for-mazione del prodotto e
d[HBr]
dt= r2 − r−2 + r3 (= r3 + r−2 − r−2 + r3 = 2r3)
= k2[Br][H2] − k−2[HBr][H] + k3[H][Br2]
che contiene la concentrazione dei radicali Br e H. Ap-plicando l’approssimazione dello stato stazionario si ha
d[H]
dt= r2 − r−2 − r3 = 0 (r2 = r3 + r−2)
d[Br]
dt= 2r1 − 2r−1 − r2 + r−2 − r3 = 0
76

Sommando le due equazioni si ottiene 2r1 − 2r−1 = 0,cosı che r1 − r−1, ovvero la velocita di innesco uguagliala velocita di terminazione:
k1[Br2][M] = k−1[Br]2[M]
da cui si ricava
[Br] = (k1/k−1)1/2 [Br]1/2
Per ottenere [H] usiamo la prima delle condizioni di statostazionario
k2[Br][H2] − k−2[HBr][H] − k3[H][Br2] = 0
Sostituendo [Br] si ha
[H](k−2[HBr] + k3[Br2]) = k2(k1/k−1)1/2[Br2]
1/2[H2]
da cui
[H] =k2(k1/k−1)1/2[Br2]1/2[H2]
k−2[HBr] + k3[Br2]
Allora
r =1
2
d[HBr]
dt= r3 = k3[H][Br2]
r =k2(k1/k−1)1/2[Br2]1/2[H2]
(k−2[HBr] + k3[Br2])/k3[Br2]
r =k2(k1/k−1)1/2[H2][Br2]1/2
1 + (k−2/k3)[HBr]/[Br2]
che, ponendo k = k2(k1/k−1)1/2 e j = k−2/k3, cor-risponde alla legge cinetica osservata.
77

Il meccanismo della reazione a catena puo essere in-
nescato termicamente, fotochimicamente (Br2hν−→ 2Br)
o anche chimicamente (vapori di Na+Br2 → NaBr+Br).
Poiche gli atomi o le molecole che propagano la catena(carriers) producono molte molecole di prodotto, anchepiccole quantita di specie che li eliminano portano a fortirallentamenti della reazione (inibitori).
Nel meccanismo H2+Br2, per ogni propagatore di cate-na consumato se ne produce un altro (1:1). In altricasi si ha che per ogni propagatore consumato se neproducono piu di uno (1:n). In tale circostanza di han-no le reazioni a catena ramificata, per le quali la ve-locita di reazione puo aumentare enormemente dandoluogo in molti casi ad esplosioni. Ad esempio, per lareazione 2H2 + O2 → 2H2O, la ramificazione includeH + O2 → OH + O e O + H2 → OH + H. (Reazionialtamente esotermiche non a catena possono risultareesplosive in quanto il rapido aumento della temperaturaprovoca un rapido aumento di k.)
Un ulteriore esempio di reazione a catena ramificata edato dalla combustione degli idrocarburi. In questo casosi hanno meccanismi molto complessi, si pensi che per lacombustioni del CH4 si hanno 22 steps e 12 intermedi,tra cui le specie CH3, CH3O, CH2O, HCO, H, O, OH,OOH.
78

Polimerizzazione madiante radicali liberi
Consideriamo la cinetica della polimerizzazione causa-ta da radicali liberi in fase liquida (in un solvente onel monomero puro). Siano I ed M l’iniziatore e ilmonomero. Il meccanismo di reazione e
Iki−→ 2R· iniziazione
R · +Mka−→ RM· addizione
RM · +Mkp1−→ RM2· propagazione
RM2 · +Mkp2−→ RM3· propagazione
... ...
RMm · +RMn· kt,mn−→ RMm+nR terminazione
L’iniziazione puo avvenire per decomposizione termicadi I per formare una piccola quantita di radicali R·, adesempio (C6H5COO)2 → 2C6H5COO·.In alcuni casi lo step di terminazione comporta il trasferi-mento di un atomo di H tra le due catene (disproporzion-amento).
Per semplificare le cose, si assume che la reattivita deiradicali negli step di propagazione sia indipendente dallaloro dimensione, ossia
kp1 = kp2 = . . . ≡ kp
Allo stesso modo si assume che la dimensione dei rad-icali non modifichi la costante cinetica dello step diterminazione.
79

Comunque, occorre considerare che le velocitad[RMm+nR]/dt e d[RM2nR]/dt delle reazioni di termi-nazione elementari RMm ·+RMn· → RMm+nR e 2RMn· →RM2nR sono proporzionali alla frequenza con la quale iradicali reagenti si incontrano per unita di volume disoluzione, e che la frequenza d’urto fra radicali ugualicontiene un fattore 1/2 rispetto a quella relativa all’in-contro di radicali diversi (vedi teoria delle collisioni).Quindi la costante cinetica di terminazione fra radicaliuguali e 1/2 qualla di terminazione fra radicali diversi:
kt ≡ kt,nn =1
2kt,mn m �= n
La velocita di consumo del monomero e
rM = −d[M]
dt= ka[R·][M]+kp[RM·][M]+kp[RM2·][M]+. . .
−d[M]
dt≈ kp[M]
∞∑n=0
[RMn·] ≡ kp[M][Rtot·]
dove [Rtot·] rappresenta la concentrazione totale di tuttii radicali. Poiche vi sono centinaia o migliaia di ter-mini di grandezza significativa nella sommatoria, l’avercambiato ka in kp nel primo termine non ha alcuna con-seguenza.
80

Per determinare [Rtot·] applichiamo l’approssimazionedello stato stazionario ad ogni radicale:
d[R·]/dt = 0, d[RM·]/dt = 0, d[RM2·]/dt = 0, . . .
che sommate equivalgono a d[Rtot·]/dt = 0.
Durante lo step di addizione e durante quelli di propagazioneun radicale e consumato ed uno e generato: questi stepsnon modificano [Rtot·]. Pertanto solo gli steps di innescoe di terminazione producono effetti su d[Rtot·]/dt.Per quanto riguarda lo step di innesco, occorre tenere inconsiderazione che non tutti i radicali [R]· iniziano unacatena polimerica, alcuni si ricombinano per dare I, acausa dell’effetto “gabbia” del solvente, altri si perdonoreagendo con il solvente stesso. Pertanto si ha
(d[Rtot·]/dt)i = (d[R·]/dt)i = 2fki[I]
dove f e la frazione di radicali che reagiscono con M(tipicamente 0.2 < f < 0.8).Il contributo dato dalla terminazione dei radicali conte-nenti n monomeri e
(d[RMn·]/dt)t = −2kt,nn[RMn·]2 − kt,mn[RMn·]∑m �=n
[RMm·]
(d[RMn·]/dt)t = −2kt[RMn·]∞∑
m=0
[RMm·] = −2kt[RMn·][Rtot·]
81

La velocita totale di consumo dei radicali negli steps diterminazione si trova sommando su tutti i possibili valoridi n
(d[Rtot·]/dt)t =∞∑
n=0
(d[RMn·]/dt)t = −2kt[Rtot·]∞∑n
[RMn·]
(d[Rtot·]/dt)t = −2kt[Rtot·]2
Tornando alla condizione di stato stazionario si ha
d[R·]/dt = (d[Rtot·]/dt)i+(d[Rtot·]/dt)t = 2fki[I]−2kt[Rtot·]2 = 0
da cui si ricava
[Rtot·] = (fki/kt)1/2[I]1/2
Pertanto la velocita di consumo del monomero diventa
rM = −d[M]
dt= kp
(fki
kt
)1/2
[M][I]1/2
La reazione e di primo ordine rispetto al monomero edi ordine 1/2 rispetto all’iniziatore.
82

Il grado di polimerizzazione DP di una molecola polimer-ica e il numero di monomeri nel polimero. Ora, se inun breve intervallo di tempo dt durante la reazione dipolimerizzazione, M molecole di monomero sono consu-mate e P molecole di polimero di varia lunghezza sonoprodotte, per la legge di conservazione della massa siha che le P molecole di polimero devono contenere untotale di M unita monomeriche. Il grado di polimeriz-zazione medio e < DP >= M/P .A volume costante si puo definire DP come rapporto divariazioni di concentrazione:
< DP >≡ − d[M]
d[Ptot]= − d[M]/dt
d[Ptot]/dt
dove [Ptot] e la concentrazione totale di molecole dipolimero.Poiche una molecola di polimero si forma quando dueradicali si combinano si ha che
d[Ptot]/dt = −1
2(d[Rtot·]/dt)t = kt[Rtot·]2 = fki[I]
Per cui
< DP >=kp
(fki
kt
)1/2[M][I]1/2
fki[I]=
kp[M]
(fkikt)1/2 [I]1/2
Una bassa concentrazione di iniziatore rispetto alla con-centrazione del monomero favorisce un alto < DP >.Se il modo dominante di terminazione e il disproporzion-amento < DP > e la meta.
83

Reazioni velociLe costanti cinetiche variano in un intervallo enorme divalori.In soluzione acquosa, la piu veloce reazione elementaredi secondo ordine conosciuta e H3O+(aq)+OH−(aq) →2H2O, per la quale k = 1.4× 1011 L mol−1 s−1 a 25 ◦C.In fase gassosa, per la reazione B(g)+C(g)→ prodot-ti, un limite superiore e stabilito dalla frequenza dellecollisioni:
ZBC = π(rB + rC)2
[8RT
π
(1
MB+
1
MC
)]1/2(NB
V
)(NC
V
)Se la reazione avviene ad ogni collisione (questo non evero per la maggior parte delle reazioni) si trova che lavelocita di reazione e
rmax = −d[B]
dt=
ZBC
NA=
ZBC
NA[B][C][B][C]
kmax =ZBC
NA[B][C]= πNA(rB+rC)2
[8RT
π
(1
MB+
1
MC
)]1/2
Ora, a 300 K e per valori tipici MB = 30 g/mol, MC = 50g/mol, rB + rC = 4 A, si trova
kmax = π(6.022×1023)(4×10−10)2
[8 × 8.3145 × 300
π× 160
3
]1/2
ovvero kmax = 1.76 × 108 m3 mol−1 s−1 ≈ 1011 dm3
mol−1 s−1. La ricombinazione di radicali in fase gas (adesempio 2ClC· → C2Cl6) hanno spesso valori di questoordine di grandezza.
84

La velocita di reazioni estremamente lente puo esseredeterminata marcando un reagente con un isotopo ra-dioattivo. Dopo alcune setimane si separa un prodottoe se ne misura la radioattivita. In questo modo sonostate determinate costanti cinetiche di secondo ordinedell’ordine di 10−12 dm3 mol−1 s−1 e reazioni di primoordine con tempi di dimezzamento di 105 anni.
Molte reazioni sono troppo veloci per poter essere se-guite con i metodi classici gia accennati. Si ricorre atecniche diverse. Mediante il metodo a flusso continuo
Il miscelamento avviene in circa 1/2–1 ms nel puntoM. Nel punto P si determina la concentrazione di unaspecie, ad esempio spettroscopicamente. Variando ladistanza del punto P da M e la velocita del flusso siottengono le concentrazioni dei reagenti a vari tempi. Ilmetodo si usa per reazioni in fase liquida ed anche infase gas, sostituendo le siringhe e le soluzioni con bulbicontenenti i gas.
85

Il metodo a flusso interrotto
sfrutta il rapido miscelamento dei reagenti e si osser-vano le concentrazioni delle specie nel punto P moltovicino ad M, dopo aver interrotto il flusso all’interno deltubo di osservazione.Entrambi i metodi sono applicabili a reazioni con tem-pi di dimezzamento nell’intervallo che va da 10 a 10−3 s.
Il limite principale dei metodi a flusso e stabilito dal tem-po di miscelamento. Una delle tecniche piu importantiper lo studio di reazioni aventi velocita ancora piu ele-vate e il metodo del rilassamento. Si considera un sis-tema all’equilibrio e molto rapidamente si cambia unadelle variabili che determinano la posizione di equilib-rio. Seguendo l’avvicinamento del sistema al suo nuovoequilibrio, si puo determinare la costante cinetica.
86

(rilassa1)
87

(rilassa2)
88

(rilassa3)
89

(rilassa4)
90

(rilassa5)
91

(rfl1)
92

(rfl2)
93

(cat1)
94

(cat2)
95

(cat3)
96

(cat4)
97

(cat4a)
98

(ce1)
99

(ce2)
100

(ce3)
101

(ce4)
102

(ce5)
103

(ce6)
104

(ce7)
105

(ce8)
106

(ac1)
107

(ac2)
108

(ac3)
109

(ac4)
110


Adsorbimento di gas su solidiL’attivita catalitica, molto importante dal punto di vistaindustriale, di vari solidi quali Pt, Pd, Ni, ecc., e laconseguenza dell’adsorbimento di gas.
Il solido sulla cui superficie avviene l’adsorbimento edetto adsorbente o substrato. Il gas adsorbito e dettoadsorbato.
L’adsorbimento avviene nella regione di interfase solido-gas e deve essere distinto dall’assorbimento, che com-porta la penetrazione del gas all’interno della fase solida.
Si distingue l’adsorbimento fisico dall’adsorbimento chim-ico, quest’ultimo detto anche chemiadsorbimento. Nel-l’adsorbimento fisico le molecole del gas sono trattenutesulla superficie del solido da forze intermolecolari relati-vamente deboli di van der Waals. Nel chemiadsorbimen-to sulla superficie del solido avviene una reazione chimi-ca ed il gas e trattenuto da legami chimici relativamenteforti.
L’adsorbimento fisico non e specifico; il chemiadsorbi-mento lo e. Ad esempio, se la temperatura e sufficien-temente bassa, l’N2 e fisicamente adsorbito su qualsiasisolido; a temperatura ambiente l’N2 e chemiadsorbitosu Fe, W, Ca e Ti, ma non su Ni, Ag, Cu o Pb. L’Ausolido chemiadsorbe O2, C2H2 e CO ma non adsorbeH2, CO2 o N2.
112

La variazione di entalpia di chemiadsorbimento e sostanzial-mente maggiore in valore assoluto di quella dell’adsor-bimento fisico. Tipicamente il ∆H di chemiadsorbi-mento va da −40 a −800 kJ/mol, mentre il ∆H diadsorbimento fisico va da −4 a −40 kJ/mol.
Durante il chemiadsorbimento possono rompersi legamichimici cosı come si formano (ad esempio, H2 e chemi-adsorbito su metalli come atomi di H), pertanto sarannopossibili ∆H di chemiadsorbimento sia negativi che pos-itivi.Comunque, c’e da aspettarsi che il ∆S di chemiadsor-bimento di un gas su di un solido sia abbondantementenegativo, cosı che per avere una significativa quantitadi chemiadsorbimento il ∆H deve essere significativa-mente negativo.Un eccezione e rappresentata dal chemiadsorbimento diH2(g) su vetro. In questo caso le due moli di H che siformano da una mole di H2 hanno una grande mobilitasulla superficie del solido e, conseguentemente, hannouna entropia maggiore che consente un ∆H leggermentepositivo.
Solo uno strato di gas puo essere chimicamente ad-sorbito sulla superficie di un solido. Altri strati di gaspossono formarsi in virtu dell’adsorbimento fisico.L’adsorbimento fisico avviene in quantita significativesolo a temperature vicine al punto di ebollizione del gas.
113
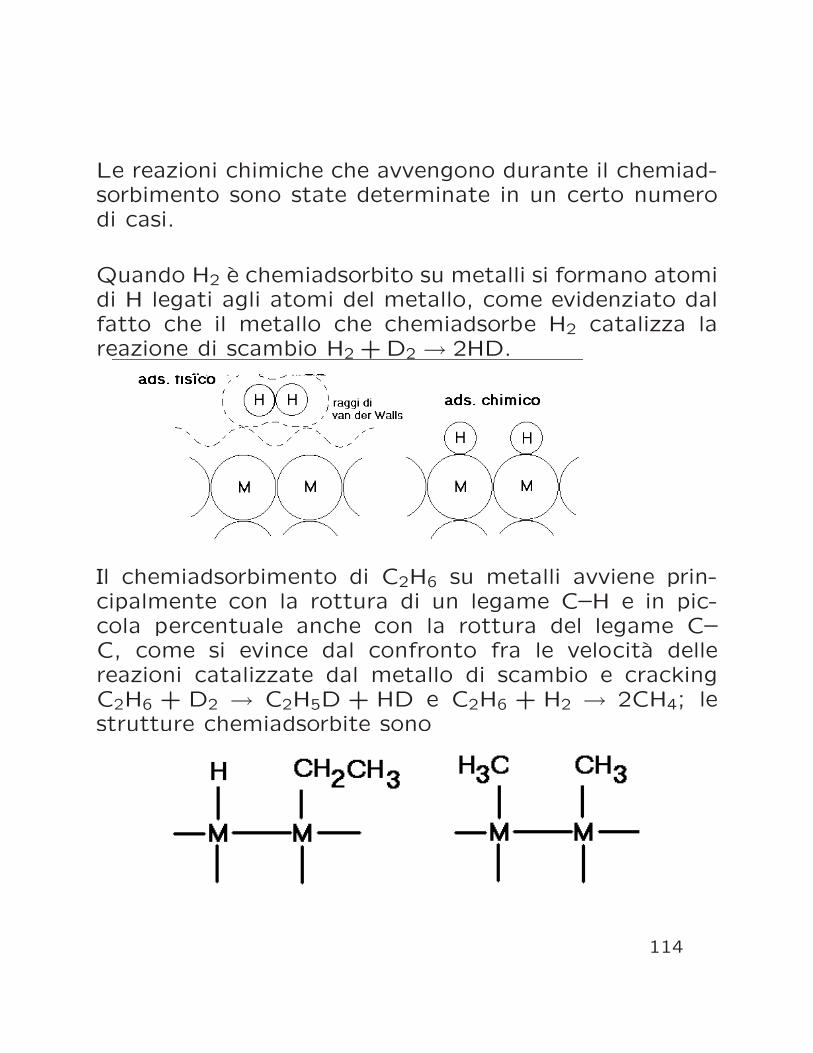
Le reazioni chimiche che avvengono durante il chemiad-sorbimento sono state determinate in un certo numerodi casi.
Quando H2 e chemiadsorbito su metalli si formano atomidi H legati agli atomi del metallo, come evidenziato dalfatto che il metallo che chemiadsorbe H2 catalizza lareazione di scambio H2 + D2 → 2HD.
Il chemiadsorbimento di C2H6 su metalli avviene prin-cipalmente con la rottura di un legame C–H e in pic-cola percentuale anche con la rottura del legame C–C, come si evince dal confronto fra le velocita dellereazioni catalizzate dal metallo di scambio e crackingC2H6 + D2 → C2H5D + HD e C2H6 + H2 → 2CH4; lestrutture chemiadsorbite sono
114
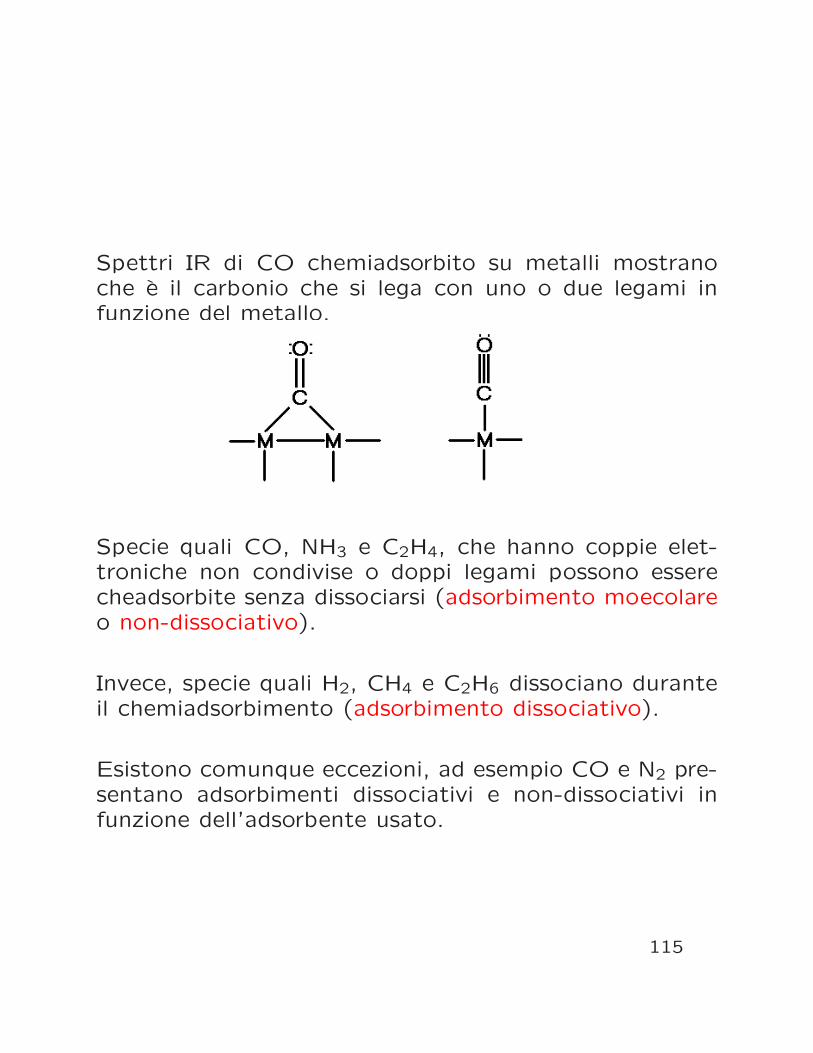
Spettri IR di CO chemiadsorbito su metalli mostranoche e il carbonio che si lega con uno o due legami infunzione del metallo.
Specie quali CO, NH3 e C2H4, che hanno coppie elet-troniche non condivise o doppi legami possono esserecheadsorbite senza dissociarsi (adsorbimento moecolareo non-dissociativo).
Invece, specie quali H2, CH4 e C2H6 dissociano duranteil chemiadsorbimento (adsorbimento dissociativo).
Esistono comunque eccezioni, ad esempio CO e N2 pre-sentano adsorbimenti dissociativi e non-dissociativi infunzione dell’adsorbente usato.
115

(ia1)
116

(ia2)
117

(ia3)
118

Isoterma di LangmuirLa frazione θ di siti attivi e
θ =bP
1 + bP
Essa corrisponde anche al rapporto v/vmon, dove v e ilvolume di gas adsorbito a P e vmon e il volume limite digas adsorbito ad alta pressione, quando un monostratodi gas ricopre l’intera superfice. Pertanto
v =vmonbP
1 + bP(∗)
Per verificare se l’isoterma di Langmuir si adatta ad unset di dati sperimentali, si prende il reciproco della (*)
1
v=
1
vmonbP+
1
vmon
Il grafico di 1/v contro 1/P fornisce una retta se i datisperimentali obbediscono l’isoterma di Langmuir.
Se due gas A e B sono adsorbiti senza dissociarsi sullastessa superfice, le ipotesi di Langmuir danno
θA =bAPA
1 + bAPA + bBPB
v
vmon=
bAPA + bBPB
1 + bAPA + bBPB
119

Il meccanismo della catalisi eterogenea e noto soloper alcune reazioni, tra le quali la reazione di ossidazionedel CO catalizzata da Pt o Pd (una delle tante impor-tanti reazioni che avvengono nelle marmitte catalitichee tra le piu conosciute). Indicando con un asterisco ilsito di adsorbimento si ha
CO + ∗ ⇀↽CO|∗
e O2 + 2∗ → 2O|∗
CO|∗
+O|∗
→ CO2 + 2∗
La maggior parte dei catalizzatori eterogenei sono:
• metalli di transizione con orbitali d parzialmente va-canti che possono essere usati per legare le speciechemiadsorbite, ad esempio Fe, Co, Ni, Pd, Pt, Cr,Mn, W, Ag e Cu;
• ossidi metallici quali Al2O3, Cr2O3, V2O5, ZnO,NiO e Fe2O3,
• acidi come H3PO4 e H2SO4.
Un buon catalizzatore deve avere un ∆H di adsorbimen-to moderato: valori bassi comportano poco adsorbimen-to e basse velocita di reazione; valori alti comportanoun forte legame di adsorbimento dei reagenti e, quindi,scarsa tendenza a reagire fra loro.
120

Un altro fattore importante e l’area della superfice diesposizione. Per questo motivo il catalizzatore e spessodistribuito su supporti porosi, quali silica gel, allumina,carbonio (charcoal) e terra di diatomee. Il supporto puoessere inerte o contribuire all’attivita catalitica.L’attivita ed il tempo di vita di un catalizzatore possonoessere aumentati aggiungendo sostanze particolari chia-mate promotori. Ad esempio il Fe finemente suddivisousato nella sintesi dell’NH3 contiene piccole quantita diossidi, in particolare di Al, che ostacolano la formazionedi cristalli di Fe piu grandi (sintering) che ridurrebbe lasuperficie e, quindi, l’attivita catalitica.
Piccole quntita di certe sostanze che si legano forte-mente al catalizzatore lo possono inattivare o avve-lenare. Questi veleni possono essere presenti nei reagen-ti come impurita o possono formarsi come prodotti sec-ondari di reazione. Veleni catalitici includono compostidello S, N e P contenenti lone pairs di elettroni, ad es-empio H2S, CS2, HCN, PH3, CO, e certi metalli comeHg, Pb, As. Poiche il piombo avvelena i catalizzatori,deve essere eliminato dalla benzina che alimenta le au-tomobili equipaggiate con marmitte catalitiche.La quantita di veleno sufficiente ad eliminare l’attivitacatalitica e di norma molto minore di quella richiestaper coprire completamente la superfice del catalizzatore.Questo indica che che l’attivita catalitica e confinata aa una piccola frazione dei siti superficiali, detti siti attivio centri attivi. La superfice di un solido non e liscia euniforme su scala atomica.
121

Per una reazione in fase fluida catalizzata da un solidosi hanno i seguenti steps:
1. diffusione dei reagenti verso la superfice del solido;
2. chemiadsorbimento di almeno uno dei reagenti;
3. reazione chimica tra molecole adsorbite su siti adi-acenti (meccanismo di Langmuir-Hinshelwood, piucomune) o tra una molecola adsorbita e molecolenella fase fluida collidenti con la superfice (mecca-nismo di Rideal-Eley, meno comune);
4. deadsorbimento dei prodotti dalla superfice;
5. diffusione dei prodotti verso la fase fluida.
Nel caso di reazione tra due molecole adsorbite, la mi-grazione di queste sulla superfice puo avvenire fra glistep 2 e 3.
Un trattamento generale coinvolge la velocita di tutti ecinque gli steps ed e complicato. In molti casi uno diquesti step, ad esempio il terzo, vedi di seguito, e piulento degli altri e solo questo puo essere considerato.
Lo step 3 puo essere costituito da piu reazioni chimicheelementari, che non sono di norma conosciute. Perquesto motivo si assume lo step 3 come un unico stepunimolecolare o bimolecolare, oppure come contenenteuno stadio elementare cineticamente determinate segui-to da uno o piu step rapidi.
122

Poiche stiamo assumendo che l’adsorbimento (2) e ildeadsorbimento (4) siano molto piu veloci dello step 3,l’equilibrio tra adsorbimento e deadsorbimento e man-tenuto per ogni specie durante la reazione. Pertantouseremo l’isoterma di Langmuir, tenendo a mente chesi tratta di una ulteriore semplificazione del trattamento,che si sovrappone alle altre gia fatte.
La velocita di conversione J di una reazione eterogenea-mente catalizzata e definita in modo analogo a quelladi una reazione normale
J =1
νB
dnB
dt
dove νB e il coefficiente stechiometrico di qualsiasi specieB che compare nella reazione complessiva.
In questo caso, pero, la reazione avviene sulla superficedel catalizzatore e J sara proporzionale all’area A dellasuperfice. Si definisce rs come la velocita di conversioneper unita di area di superfice
rs ≡ J
A =1
A1
νB
dnB
dt
123

Ora, supponiamo che la reazione elementare sulla su-perfice sia lo step unimolecolare A → C + D. In questocaso rs e proporzionale al numero di molecole di A ad-sorbite per unita di area nA/A, a sua volta proporzionalea θA. Pertanto si ha che rs = kθA, dove k e una costantecinetica avente dimensioni mol cm−2 s−1.
Poiche le specie C e D competono con A per i siti diadsorbimento si ha che
θA =bAPA
1 + bAPA + bCPC + bDPB
rs = kbAPA
1 + bAPA + bCPC + bDPB
Se i prodotti C e D sono adsorbiti molto debolmente1 + bAPA bCPC + bDPB, quindi
rs = kbAPA
1 + bAPA
Pertanto
rs = kbAPA a bassa pressione, primo ordiners = k ad alta pressione, zero ordine
Ad alta pressione la superfice e completamente copertacon A ed un ulteriore aumento della pressione non hainfluenza.Ad esempio la decomposizione di PH3 su W segue ques-ta legge cinetica, essendo di primo ordine sotto 10−2 torre di ordine zero sopra 1 torr.
124

Supponiamo ora che la reazione elementare che avvienesulla superfice del catalizzatore sia bimolecolare A+B →C+D e che antrambi i reagenti siano adsorbiti sulla su-perfice.In questo caso la velocita di reazione e proporzionale alprodotto delle concentrazioni di superfice (nA/A)(nB/A),che a sua volta e proporzionale a θAθB, cioe rs = kθAθB.Usando l’isoterma di Langmuir si ottiene
rs = kbAbBPAPB
(1 + bAPA + bBPB + bCPC + bDPD)2
Nel caso in cui una specie, ad esempio B, e adsorbitamolto piu tenacemente delle altre bBPB 1 + bAPA +bCPC + bDPD, quindi
rs = kbAPA
bBPB
ed il reagente B inibisce la reazione. Questa situazioneapparentemente paradossale puo essere capita realizzan-do che quando B e piu fortemente adsorbito di A lafrazione si superfice occupata da A va a zero, cosı comela velocita di reazione. La velocita massima di reazionesi ha quando A e B sono egualmente adsorbiti.Un esempio e dato da 2CO+O2 → 2CO2 su Pt, dove lavelocita e inversamente proporsionale alla pressioine delCO. Il CO si lega piu fortemente del O2 anche all’atomodi Fe nell’emoglobina diventando un veleno fisiologico.
125

Nel caso di meccanismo bimolecolare di Rideal-Eley
A(ads) + B(g) → prodotti
la velocita risulta proporzionale a θAPB ed usando l’isoter-ma di Langmuir per θA si ottiene una legge cineticadiversa da quella vista in precedenza.
Quando lo step cineticamente determinante e l’adsor-bimento, come nel caso della sintesi dell’NH3 (adsor-bimento di N2), o il deadsorbimento, la legge cineticarisultante non segue una equazione del tipo di Langmuir.
In questi casi occorre prendere in considerazione la ci-netica dell’adsorbimento e/o del deadsorbimento.
In altri casi ancora, la cinetica della migrazione dellespecie reagenti sulla superfice del solido risulta determi-nante.
126