clearance (cl) describes the efficiency of irreversible elimination of a drug from the body by...
TRANSCRIPT


CLEARANCE (CL) describes the efficiency of
irreversible elimination of a drug from the body by
� excretion of unchanged drug. � Metabolic conversion of the drug. Clea r an ce is d e fi n ed as “ t h e v o lum e o f b lood c lea r ed o f d r u g p e r u n it t im e ” Clearance are additive
t o t a l c lea r an ce = (CL ) + (CL )
H R
CLINICAL PHARMACOKINETICS
CLINICAL PHARMACOKINETICS

Clearance determines the maintenance dose
rate (MD) required to achieve a desired serum drug
concentration (SDC) at steady state.
MD (mg/hr) = desired SDC (mg/L) x CL (L/hr)
or = Css X CL
Why is CL important ?Why is CL important ?
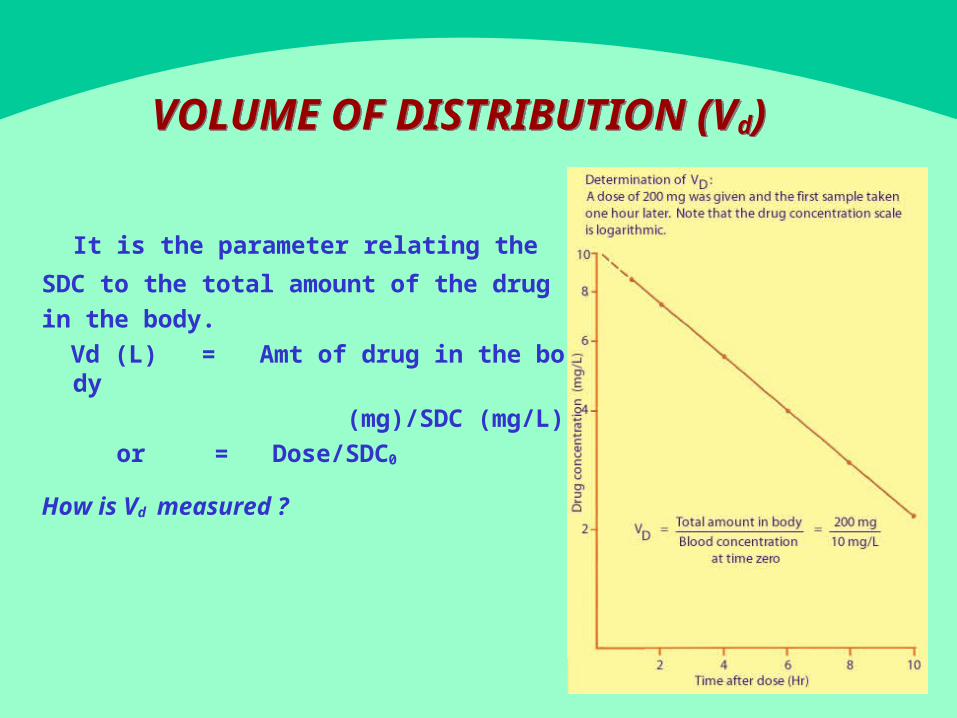
VOLUME OF DISTRIBUTION (Vd
) VOLUME OF DISTRIBUTION (Vd
)
It is the parameter relating the
SDC to the total amount of the drug in the body.
Vd (L) = Amt of drug in the body (mg)/SDC (mg/L)
or = Dose/SDC0
How is Vd measured ?
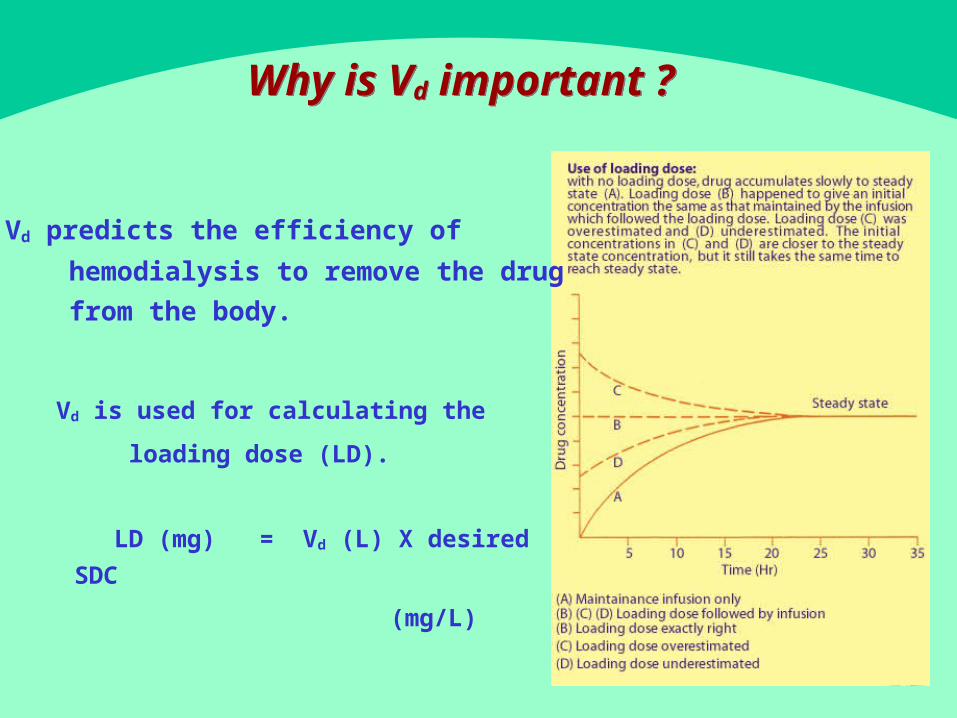
� Vd is used for calculating the
loading dose (LD).
LD (mg) = Vd (L) X desired SDC
(mg/L)
Why is Vd important ? Why is Vd important ?
� Vd predicts the efficiency of
hemodialysis to remove the drug
from the body.
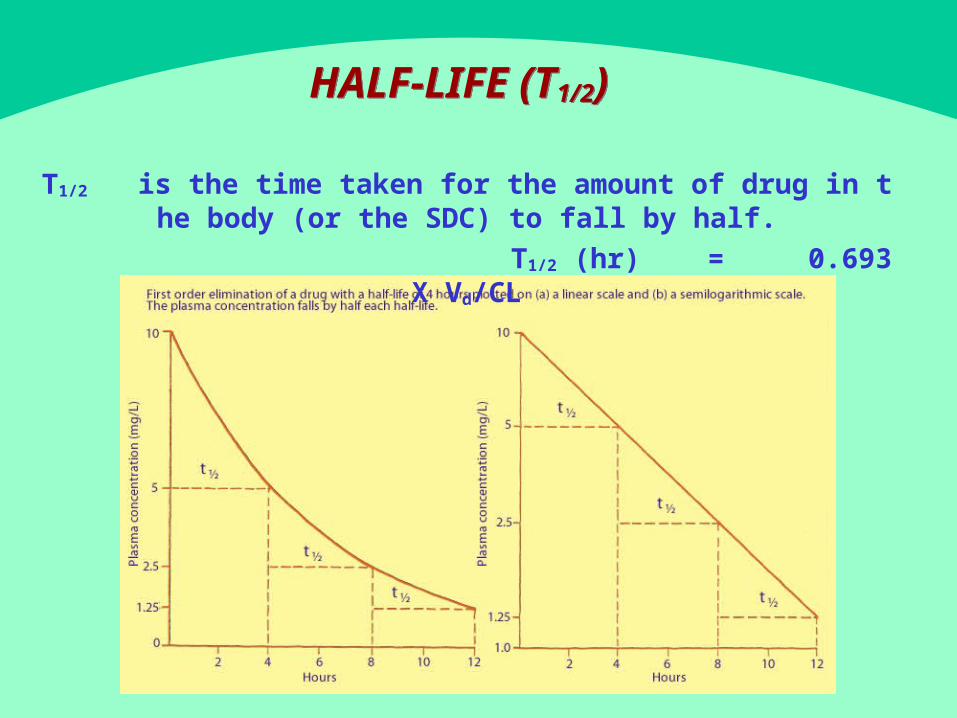
- HALF LIFE (T12/ )- HALF LIFE (T12/ )
T12/ is t h e t im e t ak en fo r t h e am ou n t o f d r u g in t h e b o d y ( o r t h e SDC) t o fa l l b y h a l f .
T12/ (hr) = 0.693 X Vd/CL
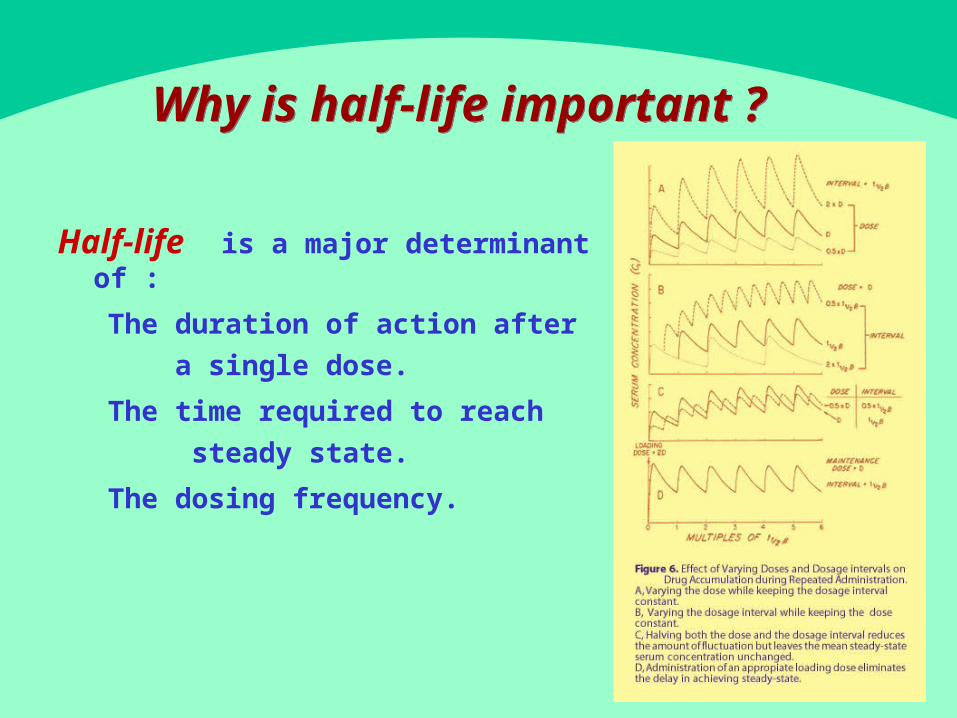
Why is - half lifeimportant ?
Why is - half lifeimportant ?
-Half life is a m a jo r d e t e rm in an t o f :
� The duration of action after
a sin g le d ose .
� The time required to reach
st ead y st a t e .
� The dosing frequency.

BIOAVAILABILITY AND FIRST PASS CLEARANCE
BIOAVAILABILITY AND FIRST PASS CLEARANCE
C.Intestinal motility•Dissolution of slowly soluble drugs (digoxin, sustained release formulations)•Chemical degradation or metabolism by microflora•Affected by : drugs (opiates, anticholinergics, metoclopramide); disease states (gastroenteritis)D.Drug interactions in the gut lumen•Complexation (tetracyclines with divalent metal ions)•Adsorption (anion exchange resins)•Food interactions (many antibiotics)E.Passage through the gut wall•Physico-chemical characteristics of the drug (quaternary ammonium compounds)•Metabolism by enzymes in the intestinal endothelium
Absorption is t h e ex t en t t o w h ich in t act d r u g is absor bed
f r om t h e g u t lum en in t o p o r t a l c i r cu la t io n .
A.Dissolution•Physico-chemical properties of drug•Crystal size and form•Excipients•Special dosage forms (sustained release, enteric coated)•pH (stomach and small intestine)B.Gastric emptying rate•Stability of drug at acid pH•Solution or solid dosage forms (liquids and small particles empty more quickly)•Affected by: food; antacids; drugs (opiates, anticholinergics, metoclopramide); disease (autonomic neuropathy)
Table 1 Determinants of drug absorption from the gut

First pass clearance is the extent to which a drug is removed by t he liver during its first passage in the portal blood through the liver t
o the systemic circulation.

Hepatic enzyme % extracted BioavailabilityActivity first pass (% escaping first pass extraction)A.Low extraction ratio drugNormal 2 98Doubled (induced) 4 96Halved (inhibited) 1 99B.High extraction ratio drugNormal 90 10Doubled (induced) 95 5Halved (inhibited) 83 17All the dose is assumed to be absorbed intact from the gut lumen. The low extraction ratio example is characteristic of theophylline which has an intrinsic clearance (see previous article Aust Prescr 1990; 13:88 – 9) of about 15 mL/min. The high extraction ratio example is characteristic of verapamil which has an intrinsic clearance of about 15 000 mL/min. Liver blood flow is normally around 1500 mL/min.
Table 2Effects of increase or decrease in hepatic drug metabolising enzyme activity on bioavailability
Why is first pass clearance important ?
Why is first pass clearance important ?
� Variability in drug response.� Relationship between oral and intravenous or parenter
al doses.
� Alternative route of administration.� Drug interactions.� Liver disease.

Bioavailability is the fraction of the dose which reache s the systemic circulation as intact drug. It depends both on
how well the drug is absorbed and how much escapes being r emoved by the liver before reaching the systemic circulation
.

Absolute Bioavailability (F) is a m easu r e o f
b ioav a i lab i l i t y o f a d r u g g iv en b y an y r ou t es ag a in st an
in t r a v en ou s r ou t e .
F = AUC0/AUCi
I f d i f fe r en t d oses a r e u sed , t h en F = AUC0 x D ose i/AUCi X Dose0
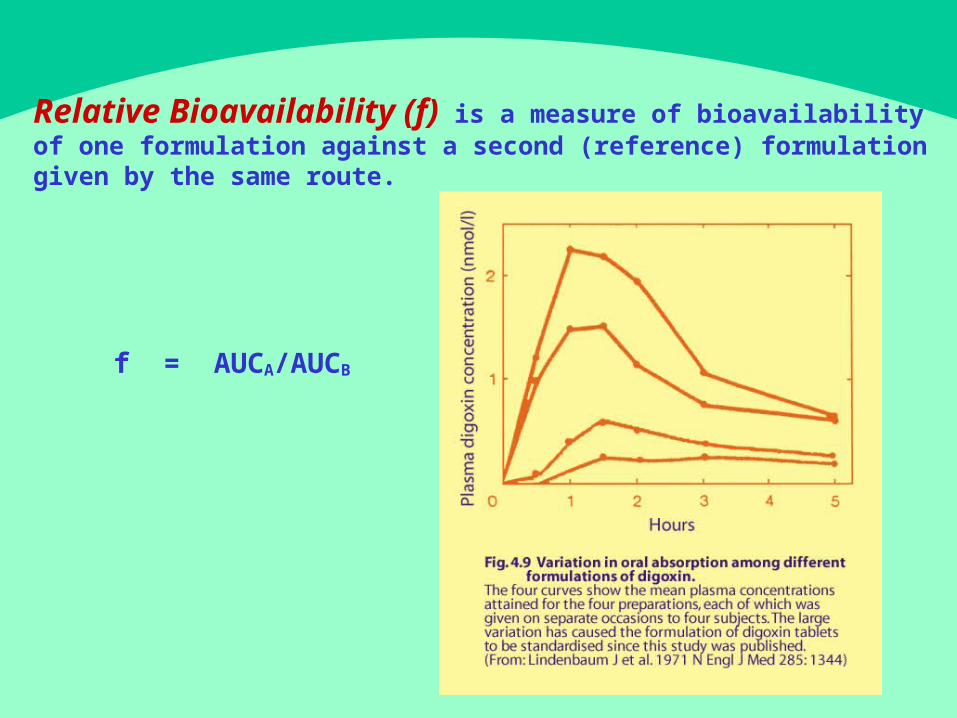
Relative Bioavailability (f) is a measure of bioavailability of one formulation against a second (reference) formulation
given by the same route.
f = A UCA/AUCB
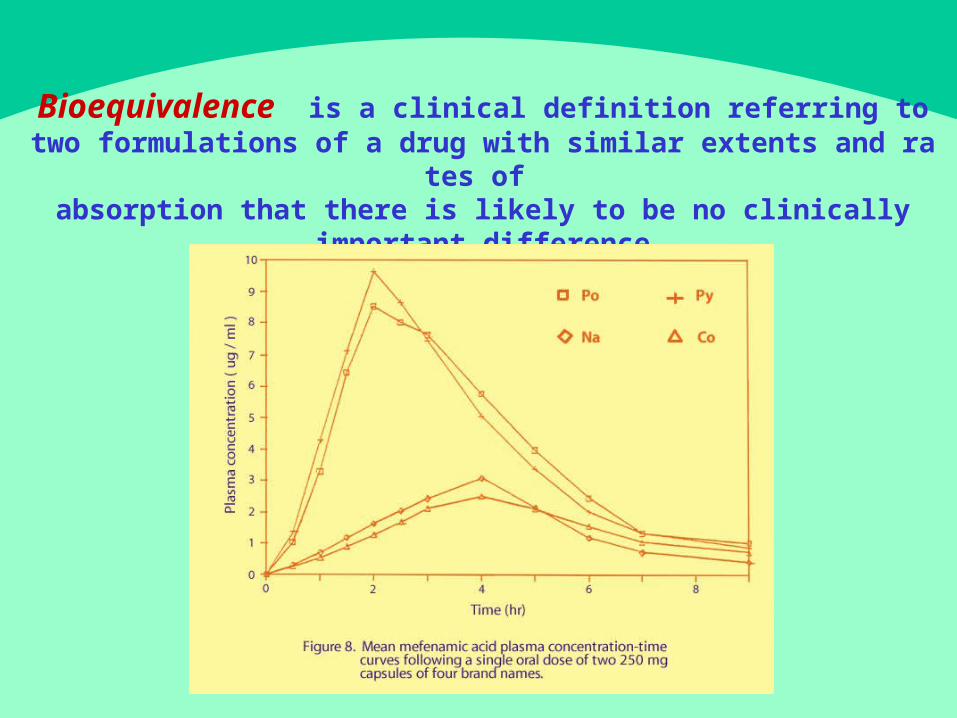
Bioequivalence is a c l in ica l d e fi n i t io n r e fe r r in g t o t w o fo rm u la t io n s o f a d r u g w it h sim i la r e x t en t s an d r a t es o
f absorption that there is likely to be no clinically
important difference.

Therapeutic Equivalence. If two formulations are
bioequivalence, t hey ar e usual l y as hav i ng t her apeut i c
equiv alence prov ided that
� no impurity or pyrogen
� h av e t h e sam e am ou n t s o f st e r eo isom e r ic
activei ngr edi ent s.

Why is bioavailabilit y important ?
Why is bioavailabilit y important ?
Bioavailability is the major parameter that indicate s how
m u ch d r u g r each sy st em ic c i r cu la t io n . B ioav a i lab i l i t y st u d y
is n eed ed fo r d r u g s w h ich h av e on e o r m o r e o f t h e f o l low in g
characteristics : 1 . Narrow therapeutic index : digoxin, theophylline,
antiepileptic drugs, antibiotics, 2 . Lack o f t h e r ap eu t ic e f fe ct s is d an g e r ou s : d ig o x in ,
antiepileptic drugs, antibiotics, 3. Poorabsorption : digoxin, phenytoin, carbamazepine - 4. Non linear elimination kinetics : phenytoin 5 . High first pass clearance : verapamil

FDA approves the drug formulations that have -b ioav a i lab i l i t y in t h e r an g e o f 8 0 1 2
5% of the original orinnovator’sbr and, t hi s may not be appr opr i at e i n
ce r t a in si t u a t io n s, e .g ., Case A. Changing from a formulation with bioavailability
o f 80% t o a fo rm u la t io n w it h 125% b ioav a i lab i l i t y . Since F x MD = Css x CL
Css = F x MD/CL If a formulation with 80% availability is used
Css1 = 0.8 x MD/CL If switching to a formulation with 125% availability
2Css = 1 25. x MD/CL2 1Css /Css = 1 250 8 1 56. / . = .
The SDC increases about 5 0 % from initial levels

Case B. Ch an g in g f r om a fo rm u la t io n w it h b ioav a i lab i l i t y
o f 125% t o a fo rm u la t io n w it h 80% b ioav a i lab i l i t y .
2 1 08125 064Css /Css = . / . = .
The SDC decreases about 3 5 % from initial levels
Con sid e r t h e case o f am in og ly cosid es, i f p a t ien t s sw it ch
t o fo rm u la t io n s w it h v e r y h ig h b ioav a i lab i l i t y t h e y m ay
suffer toxic effects at high peak and high trough levels.

Predicting how drugs will behave pharmacokinetically.
Predicting how drugs will behave pharmacokinetically.
Known parameters 80 3 7Totalclearance(L/hr) dddddddddddd ddd () 500 25 420
dddddddd dddddddd ddddddddd 01 01 08
dddddd (/) 90 90 90
Predicted Renal clearance (L/hr) (fraction 8 excreted unchanged X total Cl) Hepatic clerance (L/hr) 72 Hepatic extraction ratio (hepatic Cl/liver blood flow) 0.8 20Maximumoralbi oavai l abi l i t y ( %) dd dddddddddddddddddddd dd ddddd ddddd dd / when gi ven or al l y Yes
Drug A Drug B Drug C
Affected by liver blood flow when given intravenously YesDecrease dose in liver disease Yesdddddddd dddd dd ddddd ddddddd dd- 0693 4Halflife(hr) = . XVd/Cl d(/)6 ddddd dddddd ddddd dddd() 20

Questions
Comments
Questions
Comments&