clinical pharmacokinetics of the antipurine antifolate (6r...
TRANSCRIPT

Vol. 1. /479-1486. December /995 Clinical Cancer Research 1479
Clinical Pharmacokinetics of the Antipurine Antifolate (6R)-5,1O-
Dideaza-5,6,7,8-tetrahydrofolic Acid (Lometrexol) Administered
with an Oral Folic Acid Supplement1
Stephen R. Wedge, Sudsawat Laohavinij,
Gordon A. Taylor, Alan Boddy,
A. Hilary Calvert, and David R. Newell2Cancer Research Unit. The Medical School, University of Newcastle-
upon-Tyne. Framlington Place. Newcastle-upon-Tyne. NE2 4HH,
United Kingdom
ABSTRACT
(6R)-5,1O-Dideaza-5,6,7,8-tetrahydrofolic acid (lome-
trexol) is an antipurine antifolate which selectively inhibitsglycinamide ribonucleotide formyltransferase. Lometrexol
pharmacokinetics were evaluated in 17 patients (32 courses)
as part of a Phase I study in which folic acid supplementa-
tion was used to improve tolerance to the drug, its clinicalutility being previously limited by severe cumulative toxic-
ity. Lometrexol was administered as an i.v. bolus every 4
weeks at a starting dose of 12 mg/m2, with subsequent
interpatient dose escalation to 16, 30, and 45 mg/m2. p.o.
folic acid (5 mg/day) was given for 7 days before and 7 days
after lometrexol administration. The disposition of total
lometrexol in plasma was best described by a biexponential
model for data acquired up to 12 h after drug administra-tion, although triexponential plasma pharmacokinetics wereoften found to give a more adequate description when datawere available at later time intervals (24 h and greater).
Mean plasma half-lives (± SD) for model-dependent analy-sis were t012a 19 ± 7 mm, t112 �3 256 ± 96 mm, and t112y
(where measurable) 1 170 ± 435 mm. Lometrexol area under
plasma concentration versus time curve was proportional to
the dose administered. Moderate plasma protein binding of
lometrexol was evident (78 ± 3%) with an inverse linear
relationship between fraction of unbound lometrexol and
the concentration of serum albumin. The volume of distri-
bution oflometrexol at steady state was between 4.7 and 158
l/m2. Renal elimination of lometrexol, studied in 19 patients(21 courses), was considerable, accounting for 56 ± 17% of
the total dose administered within 6 h of treatment, and 85
± 16% within 24 h of treatment. These recoveries of Un-changed lometrexol indicate that the drug does not appear
to undergo appreciable systemic metabolism at the range of
concentrations studied.
Received 2/24/95: revised 8/15/95: accepted 8/I 7/95.
I � R. W. and S. L. were supported by Eli Lilly and Company (India-napolis, IN). Financial support was also provided by the North of
England Cancer Research Campaign.
2 To whom requests for reprints should be addressed. Phone: 44-19 1-
222-8233: Fax: 44-191-222-7556.
Lometrexol pharmacokinetics were also examined inseven patients who received 45 or 60 mg/m2 lometrexol as
part of a separate study of the drug given with folinic acid
rescue 5-7 days after treatment. No marked differences
were evident in lometrexol plasma half-lives, plasma clear-
ance, or the extent of plasma protein binding, indicating that
there is not a pronounced pharmacokinetic interaction be-
tween lometrexol and folic acid.
INTRODUCTION
Lometrexol is a folate analogue which selectively inhibits
GAR3 formyltransferase. an enzyme essential for de izos’a purine
biosynthesis (1, 2). This antipurine antifolate exhibits a broad
spectrum of antitumor activity in murine and human xenograft
tumor models. in which the established antifolate methotrexate
demonstrates little or no effect (3).
In early Phase I clinical studies with lometrexol. significant
clinical toxicity was evident, characterized by severe mucositis
and myelotoxicity (thrombocytopenia and leucopenia). which
limited drug administration to only one or two courses (4-7).
These toxicities were unexpected. occurring at drug concentra-
tions which were approximately one hundredth of the 10%
lethal dose in mice (8). A number of clinical responses were
documented, including activity against malignant fibrous histi-
ocytoma (5). non-small cell lung cancer. breast cancer. and
colonic adenocarcinoma (7), which stimulated studies aimed at
the pharmacological amelioration of lometrexol toxicity. Exper-
imentation in mice revealed that the therapeutic index of lome-
trexol was highly dependent on dietary folic acid intake (9. 10),
and suggested that folic acid administration could reduce tox-
icity. without ablating antitumor activity. To enable the devel-
opment of a tolerable and effective schedule for the routine
clinical use of lometrexol, a Phase I study was initiated in which
S mg folic acid/day were given for 7 days before and after
lometrexol administration. After 7 days. this dose of folic acid
resulted in an increase in plasma folate levels from 3 to 64 ng/ml
to 6 to 180 ng/ml in the patients studied.
The Phase I study of lometrexol with ftlate supplementa-
tion provided an opportunity. for the first time. to conduct
detailed clinical pharmacokinetic studies with lometrexol: a
comprehensive pharmacological examination in humans being
previously prohibited by the lack of clinical utility and avail-
ability of a suitable assay. The principal objectives of this
I The abbreviations used are: GAR, glycinamide ribonucleotide: CV.coefficient of variation: AUC. area under the lometrexol plasma con-
centration versus time curve: ClinT. total plasnia clearance: GFR. gb-
merular filtration rate: Vd�, volume of distribution at steady state: FBP,
folate-binding protein.
on July 2, 2018. © 1995 American Association for Cancer Research.clincancerres.aacrjournals.org Downloaded from

1480 Lometrexol with Folic Acid Supplementation
clinical pharmacokinetic study were: (a) to determine the
plasma pharmacokinetics in patients receiving multiple courses
of lometrexol, and thereby define the relationship between
lometrexol AUC and dose, and intra- and interpatient variability
in AUC; (b) to investigate the extent of lometrexol plasma
protein binding; (c) to measure urinary excretion of lometrexol;
and (d) to evaluate the effect of folic acid supplementation on
lometrexol pharmacokinetics to determine whether the im-
proved tolerance of lometrexol produced by folic acid adminis-
tration is a consequence of a pharmacokinetic interaction.
MATERIALS AND METHODS
Patient Eligibility. Patients eligible for this study had a
histiologically documented malignant solid tumor, which was
either refractory to established therapies, or for which no stan-
dard therapy existed. All patients had a predicted life expectancy
of at least 12 weeks, and had recovered from the toxic effects of
previous treatment before entering into the study. i.e., they had
not received any major therapy or investigational drug for at
least 4 weeks (6 weeks if prior therapy included chemotherapy
with a compound known to have delayed toxicity, e.g., a nitro-
sourea). Exclusion criteria included factors which could have
interfered with lometrexol disposition/toxicity or folic acid absorp-
tion and comprised: (a) concomitant medication with probenecid,
trimethoprim, co-trimoxazole, pyrimethamine, prednisolone, anti-
epileptics, or allopurinol; (b) extensive radiotherapy; and (c) in-
flammatory, ulcerative bowel disease or malabsorption syndrome.
All patients were required to have adequate organ function
prior to treatment, with hepatic function characterized by bil-
irubin levels of <25 �i.molIliter, and renal function by a creat-
mine measurement of < 120 �i.molIIiter and a 51Cr-EDTA clear-
ance of >50 ml/min. Informed written consent was given
according to local regulatory requirements.
Study Design. Folic acid (Approved Prescription Ser-
vices Ltd., Leeds, United Kingdom) was given daily as a single
5-mg tablet for 7 days before and 7 days after lometrexol
administration at 4-week intervals. Lometrexol (Lilly Research
Centre, Surrey, United Kingdom) was reconstituted in 0.9%
(v/v) saline and administered as a rapid i.v. bolus over 0.5-1.0
mm at a concentration of 1-10 mg/ml. Patients were admitted to
the Department of Medical Oncology, Newcastle General Hos-
pital, to receive lometrexol and were observed for an additional
24 h after drug administration to ensure that acute toxicity was
not apparent. The performance status of patients was assessed at
least once a week, for a period of 4 weeks, following bometrexol
therapy.
The trial design required three patients, previously un-
treated with lometrexol, to be treated at each dose level. The
first patient entered at each dose level was followed up for 3
weeks before the next patient was entered. At least two patients
per dose level received two courses before dose escalation.
Toxicities were evaluated according to the WHO criteria. If
repeated courses at a given dose level were tolerated without
toxicity greater than WHO grade II, doses were escalated ac-
cording to the clinical judgment of the investigator with ap-
proval of the Medicines Control Agency (London, United King-
dom) and the Local Ethics Committee. The starting dose of
lometrexol was 12 mg/m2, with subsequent escalation to 16, 30,
and 45 mg/m2. Dose escalation increments were determined by
clinical experience at the previous dose level and by data from
a parallel study of lometrexol given with folinic acid ( 1 1 ). No
intrapatient dose escalation occurred.
Pharmacokinetic Studies. Lometrexol pharmacokinet-
ics was determined in 17 patients (32 courses) receiving folic
acid supplementation and in an additional 7 patients (7 courses)
who did not receive folic acid. Plasma samples from patients
receiving bometrexol without folate supplementation were
kindly provided by Drs. C. Sessa and F. Cavalli (Ospedale San
Giovanni, Bellinzona, Switzerland), who were responsible for
an alternative Phase I study that involved folinic acid adminis-
tration ( I 5 mg every 6 h for 1 2 doses), starting 5-7 days after
treatment with lometrexol (I 1).
Blood samples were collected by venipuncture into vacu-
tamer tubes placed on ice and containing the sodium salt of
EDTA as an anticoagulant, and were taken before treatment and
at 5, 15, 30, and 45 mm and at 1, 1.5, 2, 4, 6, 8, 12, and 24 h,
and in some patients at 48. 72, and 96 h, after bometrexol
administration. Samples were immediately centrifuged (1000 X
g, 8 mm, 4#{176}C),and plasma was removed by aspiration with a
Pasteur pipette. Plasma was stored at -20#{176}Cprior to analysis.
The plasma lometrexol concentration was measured by the
HPLC method of Wedge et a!. ( 12), which uses derivitization
and fluorescence detection. Briefly. patient samples were
thawed at room temperature and diluted to I ml with control
human plasma (Red Cross Transfusion Service, Newcastle-
upon-Tyne, United Kingdom) to contain 10-250 ng/ml lome-
trexol. Samples were further diluted ( 1 : 1) with aqueous formic
acid [1% (v/v): pH 3.7] containing 100 ng C’tt-desmethylene
lometrexol (Lilly Research Centre) as an internal standard.
Following rotary mixing and centrifugation, each sample was
subjected to solid-phase extraction using a CS ( I cm) Bondelut
cartridge (Analytichem International, Harbour City. CA). Eluted
samples were evaporated to dryness using a Speedvac concen-
trator (Savant Ltd., Farmingdale, New York) and reconstituted
in 13% (v/v) aqueous formic acid. Oxidation of bometrexol and
the internal standard was achieved by incubation (37#{176}C.90 mm)
with a suspension of manganese dioxide (0.2 mg/mI) in water
and terminated by the addition of a I : 1 mixture of S M NaOH
and 1% (w/v) ammonium carbonate (pH 5) to samples on ice.
Samples were centrifuged (13,000 X g, 10 mm), and 100 p.1 of
the supernatant were analyzed chromatographically. Chromato-
graphic analysis was achieved using an Apex II (Cl 8. 3 p.m; 150
x 4.6 mm) analytical column (Jones Chromatography, Hen-
goed, Glamorgan, South Wales, United Kingdom) and a mobile
phase of I 2% (w/v) acetonitrile in I % (v/v) aqueous acetic acid
(pH 5) containing tetramethylammonium hydrogen sulfate
(0. 1 7 1 g/liter) as an ion pair reagent. Elution was isocratic, at a
flow rate of 1 ml/min, and analyte measurement was by fluo-
rescence detection (E5, 325 nm; EN,, 450 nm).
To assess intraassay variation, each assay was calibrated
using a five-point standard curve of duplicate lometrexol stan-
dards in the range 10-250 ng/ml, prepared in control human
plasma, and extracted/analyzed at the same time as patient
samples. Quantitation was achieved using internal standardiza-
tion by a comparison of peak height ratios. with peak heights
being quantified using Minichrom Software (VG Data Systems
Ltd., Altrincham, Cheshire, United Kingdom). All calibration
on July 2, 2018. © 1995 American Association for Cancer Research.clincancerres.aacrjournals.org Downloaded from

Male 6Female I 1
Median age, yr (range) 49 (30-67)WHO performance status
0 4
I 5
2 8
Primary tumor
Breast 4
Ovarian 3
Malignant melanoma 3
Others 7Prior treatment
Chemotherapy 14
Radiotherapy 9
Clinical Cancer Research 1481
curves were linear (r2 > 0.997), the lower limit of determination
of the assay was 10 ng/ml, and the intraassay CV at nominal
concentrations of 10, 50, and 250 ng/ml was always <8.5%. In
addition, three lometrexol quality assurance samples (10, 50,
and 250 ng/ml), prepared every 2-3 months in bulk, were
assayed in triplicate to assess the interassay CV, which was
always <6%.
Pharmacokinetic Parameters. Plasma lometrexol data
were analyzed using both model-independent and model-depen-
dent analyses. In model-independent analyses. the AUC was
calculated using the log trapezoidal rule (13) with extrapolation
to infinity, using the terminal phase rate constant calculated by
the compartmental analysis. For model-dependent analyses. a
biexponential or triexponential equation was fitted to the con-
centration versus time data using a nonlinear-weighted least-
squares parameter estimation program (ADAPT II, kindly pro-
vided by Dr. S. D. Z. D’Argenio and A. Schumitzky.
Biomedical Simulations Resource, Los Angeles. CA). Data
were weighted as the reciprocal of the estimated variance, where
the SD of the output was assumed to be proportional to the
estimated concentration (constant CV). The model providing the
best fit to each data set was determined using the precision of
the parameter estimates and consideration of the Akaike infor-
mation criterion ( 14). The parameters derived were used to
calculate model-independent (AUC, CITOT, and Vd.,.) or model-
dependent (AUC, CITOT. t112Ot� t112�3, and t111”)’) pharmacokinetic
parameters (13, 15).
Plasma Protein Binding. Lometrexol plasma proteinbinding was determined using [‘4Cllometrexol (specific activ-
ity, 13 �i.Ci/mg). which was kindly provided by Dr. M. D’Incalci
(Istituto Mario Negri, Milano, Italy). This compound was radio-
labeled at the carbonyl group of the benzyl moiety, and had a
radiochemical purity of >88%, as determined by HPLC. Each
patient plasma sample ( 1 ml) was spiked with 3.5 �ig [ ‘4C]lome-
trexol, rotary mixed, and an aliquot (100 p.1) was removed. The
remainder was then subjected to ultrafiltration using an Amicon
Centrifree Micropartition Unit (Amicon Corp., Upper Mill,
Stonehouse, Gloucestershire, United Kingdom) and centrifuga-
tion (1000 X g, 10 mm, 4#{176}C),after which an aliquot (100 p.1) of
ultrafiltrate was removed. [ ‘4C}Lometrexol in the ultrafiltrate
and prefiltered plasma was determined by liquid scintillation
counting, and the ratio was used to calculate the percentage of
unbound lometrexol. Nonspecific binding of [ ‘4Cllometrexol to
the filter was determined to be <4% by ultrafiltering solutions
of f’4Cllometrexol in PBS (0.1 M, pH 7.4; 1 ml) and was
therefore ignored. Samples of plasma from each patient. taken at
< I h and at 12-1 20 h after lometrexol administration were
examined to assess any potential concentration dependency of
lometrexol plasma protein binding, but no significant differ-
ences in binding were found (P = 0.1 1, paired t-test). The mean
of each pair of analyses was therefore used to: (a) relate the
unbound fraction of lometrexol with serum albumin and plasma
protein concentrations and (b) calculate the unbound Vd�� (15).
Urinary Excretion. Urinary excretion was studied in 13
patients (14 courses) for which there was evaluable plasma
pharmacokinetics available, and in an additional 6 patients (7
courses) for which plasma pharmacokinetics was not measured.
Urine samples for lometrexol analysis were collected for 24 h
after drug administration at 6-hour intervals, and 20 ml aliquots
Table I Patient characteristics
Evaluab be patients (courses) 17 (32)
Sex
were stored at -20#{176}Cprior to analysis. Samples were thawed at
room temperature and diluted in control urine to ensure that the
bometrexol concentration would be between 2 and 25 p.g/ml
(i.e. , the dilution factor used was estimated according to the dose
of lometrexol administered and the volume of urine produced in
a 6-h period). The method for sample preparation was as de-
scribed for plasma samples, except C’#{176}-desmethylene bome-
trexol was added at a concentration of I 5 jig/mI, and evaporated
samples were not oxidized, but resuspended in 150 �i.1 of the
mobile phase and 50 p.1 were analyzed chromatographically.
Chromatographic analysis (12) involved a Spherisorb C6 (5 �.am;
150 X 4.6 mm) analytical column (Jones Chromatography) and
isocratic elution with a mobile phase of 13% (w/v) acetonitrile
in aqueous phosphoric acid [1% (v/v), pH 3.2] at a flow rate of
I .5 mI/mm. Each assay included a five-point standard curve
(duplicate samples within the range 0.2-10 pg/ml). and quality
assurance standards at 0.2, 2, and 10 �ig/ml were assayed in
triplicate. The intraassay and interassay CVs for these assays
were found to be <2% and <3%, respectively. Lometrexol
excretion in urine was expressed as a percentage of the admin-
istered dose, and renal clearance was calculated as the ratio of
the total amount excreted and the AUC.
Statistical Methods. All values expressed with a margin
of error represent the mean ± SD. Levels of significance were
calculated using Student’s t test, where P < 0.05 was considered
indicative of a significant difference between groups. The rela-
tionship between lometrexol dose and pharmacokinetic param-
eters was assessed by linear regression analysis and/or a Spear-
man rank correlation.
RESULTSPharmacokinetics of Lometrexol Administered with a
Folic Acid Supplement. The characteristics of the patients
studied are shown in Table I , and plasma lometrexol concen-
tration-time profiles from representative patients receiving 12,
16, 30, or 45 mg/m2 lometrexol are shown in Fig. 1 . Data
collected within the first 12 h of lometrexol administration were
found to be best described by a biexponential equation when
evaluated by compartmental analysis. However. at doses of 30
and 45 mg/m2 lometrexol and where data were available at later
on July 2, 2018. © 1995 American Association for Cancer Research.clincancerres.aacrjournals.org Downloaded from

A‘�. 10E
.�)
C
0
0
C00C00
05<0
0E0
-j
0 150 300 450 600 750
Time (mm)
B=. 100E
.�)
�100
0
� 10C.,C0C., 0.105<0
� 0.01E0
�10.001
Fig. 1 Representative plasma disposition curves for total bometrexol in
patients receiving folic acid supplementation: A, 12 mg/m2 (Li), 16mg/m2 (A); B, 30 mg/m2 (0), 45 mg/m2 (#{149})bometrexol. The lines are
those generated by compartmental analysis.
0 1000 2000 3000
Time (mm)
1482 Lometrexol with Folic Acid Supplementation
0.1
time points (�24 h), plasma elimination could be described
more accurately by a triexponential equation. The most appro-
pnate equations (biexponential or triexponential) fitted to each
data set all had coefficients of determination (ri) of >0.92, i.e.,
0.99 median (range, 0.92-0.99).
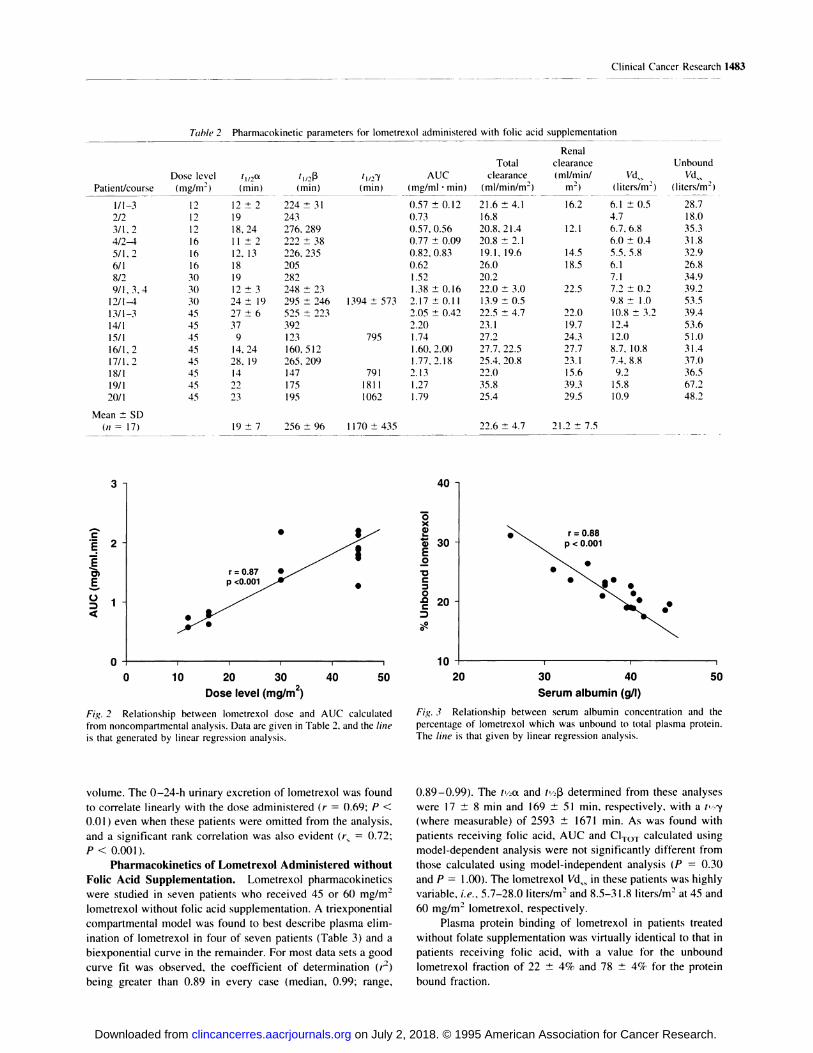
Pharmacokinetic parameters are shown in Table 2 with
parameters for patients receiving more than two courses of
lometrexol being represented as the mean and SD of all courses
studied in that patient. Model-dependent analysis of lometrexol
pharmacokinetics resulted in a t’/2a and t’/213of 19 ± 7 mm and
256 ± 94 mm, respectively, and a twy (where measurable) of
1170 ± 435 mm.
Calculation of both lometrexol AUC and C1TOT by either a
model-independent or model-dependent analysis was not found
to result in any significant difference (P 0.14 and P 0.33,
respectively, paired t test), and there were strong linear relation-
ships for both parameters for both analyses (r > 0.98).
Model-independent analysis indicated that lometrexol
AUC was linearly related to dose (r = 0.88, P < 0.001 ; Fig. 2),
i.e. , plasma clearance of lometrexol was not dose dependent.
However, two patients had a lometrexol AUC and C1TOT which
differed markedly from that observed in other patients treated at
the same dose level. One patient (patient 12) receiving 30
mg/m2 lometrexol had a consistently greater AUC than for
others receiving the same dose, i.e., 2.08-2.28 mg/ml . mm
compared with 1.38 and 1.50 mg/ml . mm (Table 2 and Fig. 2).
Reduced lometrexol clearance in this patient may have been due
to an underlying early left ventricular failure, which could have
decreased cardiac output and thereby reduced tissue perfusion,
combined with a relatively low pretreatment GFR of 75 ml/min,
which may have influenced the renal excretion of lometrexol. In
contrast, at a dose of 45 mg/m2 lometrexol. one patient (patient
19) had a much lower AUC than was measured in others (1.27
mg/mi . mm compared with values of I .60-2.20 mg/mI . mm),with a correspondingly higher C1TOT (35.8 mI/min/m2 compared
to values of 23. 1-27.7 ml/min/m2). This may be attributable to
an unusually large lometrexol Vd�� in this patient of I 5.8 liters!
m2, which in turn could have been caused by the presence of a
bilateral pleural effusion, combined with a high pretreatment
GFR of 165 mI/mm which would have promoted extensive renal
excretion of lometrexol.
No consistent change in the model-independent AUC was
evident following more than one course of lometrexol, with the
possible exception of three patients receiving 45 mg!m2 lome-
trexol every 4 weeks (patients 13, 16, and 17), in whom small
increases in AUC after the second and third courses of bome-
trexol were observed. In patients receiving more than three
courses of bometrexol, no consistent change in clearance could
be found, i.e. , the intrapatient CV of plasma clearance was 14%
(median; range, 3-21%; ii = 5).
4000 5000 The bometrexol Vd�� varied between 4.7 and 15.8 l!m2, and
the unbound lometrexol � calculated from measurements of
lometrexol plasma protein binding (see below), varied between
18.1 and 67. 1 l/m2. The larger Vd.�. of two patients receiving 45
mg/m2 lometrexol may be attributed to the fact that both patients
had pleural effusions. Even if data from these two patients are
excluded, rank correlations are observed between dose level
(mg!m2) and both � (r� = 0.84. P < 0.001) and unbound Vd��
(r� 0.66, P < 0.01).
Plasma Protein Binding of Lometrexol. The percent-
age of unbound lometrexol in plasma was 22 ± 3%. indicating
plasma protein binding of 78 ± 3%. Protein binding determined
at two concentrations of lometrexol (3.5-4.7 and 7.7-2 1 . I p.g!
ml) revealed that binding was not concentration dependent (P
0. 1 1 , paired t test). An inverse linear relationship was apparent
between the lometrexol binding and serum albumin concentra-
tion (r = 0.88; P < 0.001; Fig. 3). The relationship between
unbound lometrexol and total serum protein was not found to be
significant (r 0.38; P > 0.05), when one patient with partic-
ularly low total serum protein (55 g/liter) was removed from the
analysis.
Urinary Excretion of Lometrexol. The 0-24-h cumu-
lative urinary excretion data (Fig. 4) for I 9 patients (2 1 courses)
indicated that the major elimination route for lometrexol was
renal excretion, with 85 ± 16% of the administered dose being
excreted within 24 h of drug administration and 56 ± 17%
within the first 6 h. Urinary excretion studied in two patients
who received two consecutive courses of lometrexol did not
reveal any consistent change in excretion following the second
course of treatment. Two patients had 0-24-h lometrexol un-
nary recoveries of > I 00% of the administered dose, which was
likely to be due to inaccuracies in the measurement of urine
on July 2, 2018. © 1995 American Association for Cancer Research.clincancerres.aacrjournals.org Downloaded from
Patient/course
1/1-3
2/2
3/1, 2
4/2-45/I. 2
6/I
8/2
9/1, 3,4
I 2/1-4
13/1-3
14/1
15/I
16/I , 2
17/1. 2
18/I
19/I
20/I
Mean ± SD(ii = 17)
Renal
Dose level
(mg/m)
12
t1,2a
(mm)
12 ± 2
(mm)
224 ± 31
tiifY(mm)
AUC
(mg/mI . mm)
Total
clearance
(mI/minim2)
clearance
(mllminl
m2)
16.2
Vd�
(liters/ni)
6.1 ± 0.5
Unbound
Vd�
(liters/m)
28.70.57 ± 0.12 21.6 ± 4.1
12 19 243 0.73 16.8 4.7 18.0
12 18, 24 276. 289 0.57, 0.56 20.8. 21.4 12.1 6.7, 6.8 35.3
16 11 ± 2 222 ± 38 0.77 ± 0.09 20.8 ± 2.1 6.0 ± 0.4 31.8
16 12. 13 226. 235 0.82, 0.83 19.1, 19.6 14.5 5.5, 5.8 32.9
16 18 205 0.62 26.0 18.5 6.1 26.8
30 19 282 1.52 20.2 7.1 34.9
30 12 ± 3 248 ± 23 1.38 ± 0.16 22.0 � 3.0 22.5 7.2 ± 0.2 39.2
30 24± 19 295±246 1394±573 2.17±0.11 13.9±0.5 9.8± 1.0 53.5
45 27 ± 6 525 ± 223 2.05 ± 0.42 22.5 ± 4.7 22.0 10.8 ± 3.2 39.4
45 37 392 2.20 23.1 19.7 12.4 53.645 9 123 795 1.74 27.2 24.3 12.0 51.0
45 14, 24 160. 512 1.60, 2.00 27.7. 22.5 27.7 8.7, 10.8 31.4
45 28, 19 265,209 1.77,2.18 25.4,20.8 23.1 7.4,8.8 37.0
45 14 147 791 2.13 22.0 15.6 9.2 36.5
45 22 175 1811 1,27 35.8 39.3 15.8 67.2
45 23 195 1062 1.79 25.4 29.5 10.9 48.2
19±7 256±96 1170±435 22.6 ± 4.7 21.2 ± 7.5
C
E
E
EC)
3.
2
1
0
.
.r = 0.87
p <0.001
.
.
40
05<0
� 30�E0
�0C
0
f 20�
10
r = 0.88p < 0.001
.
..
.
, t I � I 4 4
0 10 20 30 40 50 20 30 40 50
Dose level (mg/m2) Serum albumin (g/l)
Fig. 2 Relationship between bometrexol dose and
from noncompartmental analysis. Data are given in Table
AUC calculated
2. and the line
Fig. 3 Relationship between serum albumin concentration
percentage of lometrexol which was unbound to total plasma
and the
protein.
is that generated by linear regression analysis. The line is that given by linear regression analysis.
Clinical Cancer Research 1483
Table 2 Pharmacokinetic parameters for lometrexol administered with folic acid supplementation
volume. The 0-24-h urinary excretion of bometrexol was found
to correlate linearly with the dose administered (r = 0.69; P <
0.0 1 ) even when these patients were omitted from the analysis,
and a significant rank correlation was also evident (r.� = 0.72;
P < 0.001).
Pharmacokinetics of Lometrexol Administered without
Folic Acid Supplementation. Lometrexol pharmacokinetics
were studied in seven patients who received 45 or 60 mg!m2
lometrexol without folic acid supplementation. A triexponential
compartmental model was found to best describe plasma elim-
ination of lometrexol in four of seven patients (Table 3) and a
biexponential curve in the remainder. For most data sets a good
curve fit was observed, the coefficient of determination (r�)
being greater than 0.89 in every case (median, 0.99; range,
0.89-0.99). The t’ 2a and (‘/43 determined from these analyses
were 17 ± 8 mm and 169 ± 51 mm, respectively. with a t’ .‘y
(where measurable) of 2593 ± 1671 mm. As was found with
patients receiving folic acid, AUC and C1TOT calculated using
model-dependent analysis were not significantly different from
those calculated using model-independent analysis (P = 0.30
and P = 1 .00). The lometrexol Vd�.. in these patients was highly
variable, i.e. , 5.7-28.0 liters!m2 and 8.5-3 1.8 liters!m2 at 45 and
60 mg!m2 bometrexol, respectively.
Plasma protein binding of lometrexol in patients treated
without folate supplementation was virtually identical to that in
patients receiving folic acid, with a value for the unbound
bometrexol fraction of 22 ± 4% and 78 ± 4% for the protein
bound fraction.
on July 2, 2018. © 1995 American Association for Cancer Research.clincancerres.aacrjournals.org Downloaded from

�. 1200
I �I0 60’
I�‘ 20’0C
7////////////////
/////////////////////
/////////////////////
///////////////////////
///////////////////
//////////////////
///////////////////////////
///////////////
III
////////////////////////
///////////////////////
//////////////////////-I
///////////////////////
I7/////
/I
////////////////////////
12 16 30 45
Dose level (mg/m2)
Fig. 4 Relationship between lometrexol dose and the percentage of the
administered dose excreted in urine within 24 h of drug treatment. Each
column represents an individual patient treated with either I 2, 16. 30, or
45 mg/m2 lometrexol.
1484 Lometrexol with Folic Acid Supplementation
0’
DISCUSSION
This study describes the first detailed examination of the
plasma pharmacokinetics of lometrexol, a prototype antipurine
antifolate, administered as an iv. bolus to patients receiving
folate supplementation. The plasma elimination of lometrexol
was adequately described by a biexponential equation when data
are collected up to 12 h after drug administration, although the
inclusion of samples taken at later time points (i.e. , 24 h and
greater) would indicate that a tniphasic model was a more
appropriate description. The a, �3, and “s’ phase half-lives deter-
mined for lometrexol were comparable to those measured for
the classical dihydrofolate reductase inhibitors methotrexate
( 16) and edatrexate ( 10-ethyl- lO-deaza-aminopterin; Ref. I 7),
but not with the thymidylate synthase inhibitor Tomudex, which
differs in having a much longer t#{189}’y of 50-100 h (18). The data
reported here agree with those of Young et a!. (5) who per-
formed a preliminary pharmacokinetic study of lometrexol in
patients not receiving folic acid, using a particle concentration
immunofluorescence assay, and identified biphasic and triphasic
plasma pharmacokinetics. A tertiary elimination phase could
potentially result from release of lometrexol from hepatic stores
or enterohepatic cycling of the drug; two possibilities which
have also been proposed to describe the t�,2’y of methotrexate
(19, 20). The involvement of enterohepatic cycling has also
being implicated in the disposition of dichloromethotrexate in
humans (21) and CB3988 (C2-desamino-C2-methyl-N’#{176}-propar-
gyl-2’-trifluoromethyl-5,8-dideazafolic acid) in the rat (22). The
involvement of such mechanisms in the disposition of lome-
trexol are supported by whole-body autoradiographic studies
using ‘4C-labeled lometrexol in mice maintained on a folate-
deficient diet, which indicate that the liver is the primary organ
for drug accumulation (23). This terminal elimination phase
indicates persistence of lometrexol, which could be related to
the cumulative toxicity encountered in early Phase I studies.
The AUC in individual patients studied after multiple
courses of lometrexol was found to be minimally cumulative in
three patients and noncumulative in five patients. Minimal ac-
cumulation of lometrexol on repeated treatment was also de-
scribed in the earlier Phase I study of Young et a!. (5). Similarly.
in patients receiving repeated Tomudex treatment, no accumu-
lation is evident ( 1 8). Lometrexol AUC was found to increase
linearly with dose, an observation which is in agreement with
data for other classical antifolates such as CB3717 (N’#{176}-propar-
gyl-5,8-dideazafolic acid; Ref. 24), Tomudex (18), and edatrex-
ate (17), but not for methotrexate for which it has been sug-
gested that there is a nonlinear relationship between dose and
AUC following bolus administration (25).
That there was a tendency for the lometrexol Vd�� (and
unbound Vd��) to be larger in patients receiving higher doses of
the drug may be indicative of concentration-dependent protein
binding. Thus, protein binding of lometrexol may be a saturable
phenomenon, with greater drug concentrations resulting in a
larger unbound fraction. Unbound lometrexol would be subject
to rapid cellular uptake and hence an apparently larger �
This possibility is supported by the observation that the fraction
of lometrexol unbound is inversely related to serum albumin
concentration (Fig. 4). A relationship between Vd�� and dose has
also been observed in pharmacokinetic studies with Tomudex in
rats (26).
The Vd�� range (4.7-15.8 liters!m2) from patients treated
with 12-45 mg/m2 lometrexol was, on average, smaller than
that measured in patients who did not receive folate supplemen-
tation, but were treated with 45 and 60 mg/rn2 lometrexol
(5.7-3 1 .8 liters/rn2), and that reported for patients treated in a
previous study (14-32 liters!m2) with doses of 15-60 mg/rn2
(27). This observation may again reflect the possible concentra-
tion-dependent plasma protein binding of lometrexol and the
effect that higher lometrexol concentrations would therefore
have on drug disposition.
Despite these findings, the in vitro protein-binding data did
not indicate that concentration-dependent protein binding of
lometrexol was statistically significant (P > 0.05) between the
concentration ranges of 3.5 and 4.7 and 7.7 and 21 .0 p.g/ml,
although a comparison of binding by a paired Student’s t test
resulted in a P value of 0.1 1, which would indicate a trend
toward reduced lometrexol binding at higher concentrations.
The magnitude of lometrexol protein binding (78%) was
comparable to that of methotrexate (28, 29), but not with the
thymidylate synthase inhibitors CB3717 and Tomudex which
bind more extensively (97% and >90%; Refs. 24 and 26). Since
lometrexol has a high affinity for membrane-bound FBP (30,
31), it should be noted that a component of the protein binding
measured in this study may have involved binding to soluble
FBP. Soluble FBP present in plasma is thought to function as a
folate transport protein (32) and has a Mr 35,000l00,000 (33),
which is above the threshold (30,000) used to determine protein
binding.
Any condition influencing lometrexol protein binding, e.g.,
changes in protein conformation, hyperbilirubinemia, or dis-
placement by concomitant drugs, could increase the cellular
uptake and renal clearance of the drug. The inverse linear
relationship between unbound lometrexol and the concentration
of albumin, which comprises approximately 50% of plasma
on July 2, 2018. © 1995 American Association for Cancer Research.clincancerres.aacrjournals.org Downloaded from

�-�-- Table 3 Pharmacokinetic parameters for bometrexol administered without folic acid supplementation
Patient/course
1/1
2/I
3/I
4/I
5/I
6/I
Dose level t,12a ‘I/2� tI/2� AUC
(mg/m2) (mm) (mm) (mm) (mg/mI ‘ mm)
45 12 136 4020 1.48
45 27 217 1.73
45 22 164 1.18
60 14 128 3885 1.53
60 11 198 1935 3.56
60 6 103 533 2.62
60 27 240 1.70
17 ± 8 169 ± 51 2593 ± 1671
7/I
Mean ± SD(F’ = 7)
Totalclearance
(ml/min/m2)
Vd�
(liters/m2)
28.0
UnboundVd��
(liters/m2)
125.731.2
26.0 5.7 29.5
38.2 6.8 28.5
37.6 31.8 09.0
16.8 13.1 68.2
23.7 9.0 37.9
35.8
30.1 ± 8.1
8.5 46.1
3. Shih. C.. Grindey, G. B.. Houghton, P. J., and Houghton, J. A. in vito
antitumour activity of 5. lO-dideazatetrahydrofolic acid (DDATHF) and
Clinical Cancer Research 1485
proteins. also suggests that albumin concentration may be such
a factor. As a determinant of lometrexol toxicity. however,
plasma protein binding may be of lesser importance if drug
dissociation is induced by high-affinity binding to membrane-
bound FBP or the reduced folate carrier, proteins which facili-
tate the cellular uptake of lometrexol. This could also hold true
for renal excretion, if renal tubular cells contain proteins that are
involved in active secretion of bometrexol.
Renal excretion was found to be the major route of lome-
trexol elimination. with approximately 85% of the administered
dose excreted as unchanged drug within 24 h after bolus iv.
injection. the majority of which (56%) occurred within the first
6 h. The percentage of the lometrexol dose excreted in urine was
found to be comparable to that reported for methotrexate (70-
94%; Ref. 34), but much greater than that for other antifolates in
humans, i.e., edatrexate (13-55%), CB3717 (27%) and tomudex
(12%; Refs. 17, 18, 24), and CB3988 (< 10%) in the rat (22),
which are predominantly eliminated by biliary clearance. Meth-
otrexate is reabsorbed by renal tubules at low concentrations
( 16), but actively secreted when at high concentrations (35),
with reabsorption being attributed to the pteridine ring and
secretion to the p-aminobenzoylglutamate moiety (36). Thus,
bometnexol also has the structural requirements (i.e. , a 5-deaza
pteridine ring and a benzoylglutamate moiety) for bidirectional
transport by renal tubular cells. Indeed, active secretion of
lometrexol was suggested in this study by the finding that the
ratio of unbound renal clearance:GFR (data not shown) was
significantly > I (range, 1 .2 1-2. 15). That a weak linear relation-
ship was observed between the percentage of the lometrexol
dose recovered in the urine and the dose administered could
reflect saturability of lometrexol renal tubular reabsorption. In
comparison. dose-dependent urinary excretion of methotrexatehas also been suggested within a comparatively low dose range
of 25-100 rug (25), but not in high-dose methotrexate treatment
where doses of 50-300 mg/kg are administered (37).
Although systemic metabolism of lometrexol was not stud-
ied, the finding that the majority of the dose was recovered in
urine unchanged and that no metabolite(s) of lometrexol was
detected by HPLC would indicate that the drug does not un-
dergo extensive systemic metabolism at the range of doses
studied. Quinazoline antifolates are also poorly metabolized
(22), while in comparison, the lipophilic nonpolyglutamatable
dihydrofolate reductase inhibitor trimetrexate is found to un-
dergo 95% biotransformation (38).
Folic acid supplementation reduces the toxicity of bome-
trexol, enabling the administration of up to four repeated courses
of treatment (39). The mechanism(s) which underlies modula-
tion of lometrexol toxicity remains to be identified, but the
possibility exists that folic acid may increase the plasma clear-
ance of bometrexol, such that the AUC is lower for a given dose.
However, this would seem unlikely since comparison of plasma
pharmacokinetic parameters for patients receiving 45 mg/m2
lometrexol every 4 weeks, with or without folate supplementa-
tion, showed no marked differences. Unfortunately, samples
were not available to study the urinary excretion and renal
clearance of lometrexol in patients not receiving folic acid.
The disposition of lometrexol administered with a folate
supplement is similar to that of methotrexate in terms of the
kinetics of plasma elimination, extent of protein binding. and
urinary excretion. Plasma elimination of lometrexol was not
markedly different in patients who did not receive a folate
supplement before and during lometrexol administration, and
the improved tolerance of the drug when given with folate
supplementation is therefore unlikely to be due to a pharmaco-
kinetic interaction.
ACKNOWLEDGMENTSWe thank Drs. H. Schmidt (Lilly Research. Brussels, Belgium) and
J. Walling (Lilly Research, Basingstoke, Surrey, United Kingdom) for
supplying bometrexol and C’#{176}-desmethylene bometrexol, and for their
support and encouragement. Thanks are also due to the research nurses,
data managers. and medical staff in the Department of Medical Oncol-
ogy. Newcastle General Hospital. who cared for the patients treated with
bometrexol and obtained samples for the pharmacokinetic study. Finally.the help of Drs. C. Sessa, 0. Pagani, and F. Cavalli in supplying plasma
samples from patients treated without folate supplements is greatly
appreciated.
REFERENCES
I. Boschelli, D. H., Webber, S., Whitley. J. M.. Oronsky. A. L.. andKerwa, S. S. Synthesis and biological properties of 5,10-dideaza-
5,6,7,8-tetrahydrofolic acid. Arch. Biochem. Biophys.. 265: 43-49,
1988.
2. Beardsley. G. P., Moroson, B. A., Taylor, E. C.. and Moran, R. G. A
new folate antimetabolite 5.l0-dideaza-5,6,7.8-tetrahydrofolate is a pa-
tent inhibitorofde nato purine synthesis. J. Biol. Chem.. 264: 328-333,
1989.
on July 2, 2018. © 1995 American Association for Cancer Research.clincancerres.aacrjournals.org Downloaded from

1486 Lometrexol with Folic Acid Supplementation
its diastereomeric isomers. Proc. Am. Assoc. Cancer Res., 29: 283,
1988.
4. Nelson. R., Butler, F., Dugan Jr., W., Davisland C., Stone, M., and
Dyke, R. Phase I clinical trial of LY264618 (dideazatetrahydrofolicacid; DDATHF). Proc. Am. Soc. Clin. Oncol., 9: 76, 1990.
5. Young, C., Curnie, V., Baltzer, L., Trochanowski, B., Eton, 0., Dyke,
R., and Bowsher, R. Phase I and clinical pharmacologic study of
LY264618, 5,10-dideazatetrahydrofolate. Proc. Am. Assoc. Cancer
Res., 31: 177, 1990.
6. Muggia, F., Martin, Y., Ray, M.. Martin, T., Ray. M., Leichman,C. G., Grunberg, S., Gill, I., Moran, R., Dyke, R., and Grindey, G. B.Phase I study of weekly 5, lO-dideazatetrahydrofolate (LY26461 8.
DDATHF-B). Proc. Am. Assoc. Cancer Res., 9: 74, 1990.
7. Ray, M. S., Muggia, F. M., Leichman, C. G., Grunberg. M. G..
Nelson, R. L., Dyke, R. W., and Moran, R. G. Phase I study of(6R)-5,l0-dideazatetrahydrofolate: a folate antimetabolite inhibitory to
de novo punine synthesis. J. NatI. Cancer Inst., 85: 1 154-1 159, 1993.
8. Alati, T., Shih, C., Bewely, J. R., Worzella, J. F., Lewis, S., and
Grindey, G. B. Reversal of the toxicity, but not the antitumour activityof DDATHF by oral folic acid in C3H mice. Lometrexol LY 264618Clinical Investigational Brochure, p. 3.13. Indianapolis: Lilly IndustriesLtd., 1992.
9. Grindey, G. B., Alati, 1., and Shih, C. Reversal of the toxicity but notthe antitumour activity of lometrexol by folic acid. Proc. Am. Assoc.Cancer Res., 32: 324, 1991.
10. Alati, T., Shih, C., Pohland, R. C., Lantz, R. J., and Gnindey, G. B.Evaluation of the mechanism(s) of inhibition of the toxicity, but not theantitumour activity of lometrexol (DDATHF) by folic acid. Proc. Am.Assoc. Cancer Res., 33: 407, 1992.
1 1. Pagani, 0., Sessa, C., Dejong, J., Kern, H., Hatty, S., Schmitt, H.,
and Cavalli, F. Phase I study of lometrexol (DDATHF) given in corn-bination with leucovorin. Proc. Am. Soc. Clin. Oncol., II: 109, 1992.
12. Wedge, S. R., Laohavinij, S., Taylor, G. A., and Newell, D. R.
Measurement of 5, l0-dideaza-5,6,7,8-tetrahydrofolate (lometrexol) in
human plasma and urine by high-performance liquid chromatography. J.Chromatogr. Biomed. Appl., 663: 327-335, 1995.
13. Gibaldi, M., and Pernier, D. Appendix D: estimation of areas.Pharmacokinetics, Ed. 2. pp.445-449. New York: Marcel Dekkcr, Inc.,
1982.
14. Yamaoka, K., Nakagawa, Y., and Uno, T. Application of Akaike’sinformation criterion (AIC) in the evaluation of linear pharmacokinetic
equations. J. Pharmacokinet. Biopharm., 6: 165-175, 1978.
15. Rowland, M., and Tozer, T. N. Clinical Pharmacokinetics: Con-
cepts and Applications. Philadelphia: Lea & Febiger, 1989.
16. Huffman, D. H., Wan, S. H., Azarnoff, D. L., and Hogstraten. B.Pharmacokinetics of methotrexate. Clin. Pharmacol. Then., 14: 572-
579, 1973.
17. Kris. M. G.. Kinahan, J. J., Gralla, R. J., Fanucchi, M. P., Wertheim,
M. S., O’Connell, J. P., Marks, L. D., Williams, L., Farag, F., Young,C. W., and Sirotnak F. M. Phase I trial and clinical pharmacological
evaluation of l0-ethyl-l0-deazaaminopterin in adult patients with ad-vanced cancer. Cancer Res., 48: 5573-5579, 1988.
18. Clarke, S. J., Ward, J., de Boer, M., Planting. A., Verweij. J..Sutcliffe, F., Azab, M., and Judson, I. R. Phase I study of the newthymidylate synthase inhibitor tomudex (ZD1694) in patients with ad-vanced malignancy. Ann. Oncol., 5 (Suppl. 5): 132. 1994.
19. Leme, P. R., Creaven, P. J., Allen, L. M., and Berman, M. Kineticmodel for the disposition and metabolism of moderate and high-dose
methotrexate (NSC-740) in man. Cancer Chemother. Rep., 59: 81 1-817, 1975.
20. Calvert, A. H., Bondy, P. K., and Harrap, K. R. Some observations
on the human pharmacology of methotrexate. Cancer Treat. Rep., 61:
1647-1656, 1977.
21. Hantel, A., Rowinsky, E. K., Noe, D. A., McGuire, W. P. Grochow,
L. B., Vito, B. L., Ettinger, D. S., and Donehower, R. C. Clinical andpharmacologic reappraisal of dichloromethotrexate. J. Nail. Cancer
Inst.,80: 1547-1553, 1988.
22. Newell, D. R., Maxwell, R. J.. Bisset, G. M. F.. Jodrell, D. I.. and
Griffiths, J. R. Pharmacokinetic studies with the antifolate C2-des-amino-C2-methyl-Nt0-propargyl-2’trifluoromethyl-5.8-dideazafolic
acid (CB3988) in mice and rats using in viva ‘9F-NMR spectroscopy.Br. J. Cancer., 62: 766-772, 1990.
23. Grindey, G. B., Alati, T., Lantz, R., Pohland, R., and Shih. C. Role
of dietary folic acid in blocking the toxicity but not the antitumour
activity oflometrexol (DDATHF). Ann. Oncol., 3 (Suppl. I ): 1 13. 1992.
24. Alison, D. L., Newell, D. R., Sessa, C.. Harland, S. J., Hart, L. I.,
Harrap, K. R., and Calvert, A. H. The clinical pharmacokinetics of thenovel antifolate Nt0-propargyl-5.8-dideazafolic acid (CB37 I 7). CancerChemother. Pharmacol., 14: 265-271, 1985.
25. Lawrence, J. R., Steele, W. H., Stuart. J. F. B., McNeilI, C. A..
McVie, J. G., and Whiting, B. Dose dependent methotrexate elimination
following bolus intravenous injection. Eur. J. Clin. Pharmacol., 17:
371-374, 1980.
26. Jodrell, D. I., Newell, D. R., Gibson, W., Hughes, L. R., and
Calvert, A. H. The pharmacokinetics of the quinazoline antifolate ICI
D1694 in mice and rats. Cancer Chemother. Pharmacol.. 28: 331-338,
1991.
27. Taber, L. D., O’Brien, P., Bowsher, R. R., and Sportsman. J. R.
Competitive particle concentration fluorescence immunoassay for mea-
suring 5, lO-dideaza-5,6,7,8-tetrahydrofolic acid (lometrexol) in serum.
Clin. Chem., 37: 254-260, 1991.
28. Henderson, E. S., Adamson. R. H., and Oliverio V. T. The meta-
bolic fate of tritiated methotrexate. II. Absorption and excretion in man.CancerRes., 25: 1018-1024, 1965.
29. Steele, W. H., Lawrence, J. R., Staurt, J. F. B., and McNeilI, C. A.
The protein binding of methotrexate in the serum of patients withneoplastic disease. Cancer Chemother. Pharmacol., 7: 61- 64, 198 1.
30. Wang, X., Shen, F., Freisheim, J. H., Gentry, L. E., and Ratnam, M.
Differential stereospecificities and affinities of folate receptor isoformsfor folate compounds and antifolates. Biochem. Pharmacol., 44: 1898-
1901, 1992.
31. Jansen, G.. Westerhof, G. R., Kathmann, I., Rijksen. G.. and Schor-nagel. J. H. Growth inhibitory effects of 5.10-dideazatetrahydrofolic
acid on variant murine Ll2lO and human CCRF-CEM leukemia cells
with different membrane transport characteristics for (anti)folate com-
pounds. Cancer Chemother. Pharmacol., 28: 1 15-1 17, 1991.
32. Kane, M. A., and Waxman, S. Role of folate binding proteins in
folate metabolism. Lab. Invest.. 60: 737-746, 1989.
33. Waxman, S., and Schreiber, C. Characteristics of folic-acid binding
protein in folate-deficient serum. Blood, 42: 291-301. 1973.
34. Breithaupt, H., and Kuenzlen, E. Pharmacokinetics of methotrexate
and 7-hydroxymethotrexate following infusions of high-dose methotrex-ate. Cancer Treat. Rep., 66: 1733-1741, 1982.
35. Liegler. D. G., Henderson, E. S., Hahn, M. A.. and Oliverio, V. T.
The effect of organic acids on renal clearance of methotrexate in man.Clin. Pharmacol. Ther., 10: 849-857, 1969.
36. Williams, W. M., and Huang. K. C. Renal tubular transport of folicacid and methotrexate in the monkey. Am. J. Physiol.. 242: F484-F490,1982.
37. Isacoff, W. H., Morrison, P. F., Aroesty, J.. Willis, K. L.. Block,
J. B., and Lincoln, T. L. Pharmacokinetics of high-dose methotrexatewith citrovorum factor rescue. Cancer Treat. Rep., 61: 1665-1674,
1977.
38. Marshall, J. L., and DeLap, R. J. Clinical pharmacokinetics and
pharmacology of tnimetrexate. Clin. Pharmacokinet., 26: 190-200.
1994.
39. Wedge, S. R., Laohavinij, S., Taylor. G. A., Newell, D. R., Char-lion, C. J., Proctor, M., Simmons, D., Oakey, A.. Gumbrell. L.. andCalvert, A. H. Modulation of lometrexol toxicity by oral folic acid
administration: a Phase I study. Proc. Am. Assoc. Cancer Res., 34: 274,
1993.
on July 2, 2018. © 1995 American Association for Cancer Research.clincancerres.aacrjournals.org Downloaded from

1995;1:1479-1486. Clin Cancer Res S R Wedge, S Laohavinij, G A Taylor, et al. with an oral folic acid supplement.dideaza-5,6,7,8-tetrahydrofolic acid (Lometrexol) administered Clinical pharmacokinetics of the antipurine antifolate (6R)-5,10-
Updated version
http://clincancerres.aacrjournals.org/content/1/12/1479
Access the most recent version of this article at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://clincancerres.aacrjournals.org/content/1/12/1479To request permission to re-use all or part of this article, use this link
on July 2, 2018. © 1995 American Association for Cancer Research.clincancerres.aacrjournals.org Downloaded from