codon bias and heterologous protein expression - …sfi5843/aula2.pdf · codon bias and...
TRANSCRIPT

Codon bias and heterologous protein expression Claes Gustafsson, Sridhar Govindarajan and Jeremy Minshull TRENDS in Biotechnology Vol.22 No.7 July 2004. Komar, A. (2008). Angov E. Codon usage: nature's roadmap to expression and folding of proteins. Biotechnol J. 2011 Jun;6(6):650-9. Review.
from the denatured state [37]. It should be noted, however,that directionality of co-translational folding might haveonly a fine-tuning role in the overall folding processbecause some proteins, for example, circularly permutedproteins and proteins obtained during chemical (Merri-field) synthesis, seem to be correctly folded after thereversal of synthesis directionality [6,38].
A comprehensive understanding of protein foldingrequires the elucidation of the folding mechanism andits pathway under the native conditions, such as thosethat exist in vivo during protein synthesis on the ribosome.In this regard, it is important to note that the appearanceof folding intermediates during co-translational protein
folding is likely to depend on the local translationelongation rates. Obviously, restrictions of chain flexibilityowing to stepwise, rate-dependent folding by defined partsmight be essential for the process [39,40]. Recent in silicomodeling experiments clearly indicate that the rate atwhich various portions of the growing polypeptide chainemerge from the ribosome can be crucial for the foldingprocess and can influence both the folding pathway and thefinal conformation of the protein [40].
What evidence supports the view that an in vivo protein-folding code might be encrypted in the kinetics of mRNAtranslation? First, it should be noted that several lines ofevidence demonstrate that elongation rates are not
Table 1. Representative examples of proteins suggested to fold co-translationally.Protein Domain
organizationStructural class Subunit organization Cellular component Experimental approach
used to suggestco-translational folding
Refs
b-galactosidase(E. coli)
Multidomain Domains belong toall b and a/bstructural classes
Homotetramer Cytoplasm Enzymatic activity,immunological activity
[13–16]
Immunoglobulin(heavy and lightchains);immunoglobulinfragment(s)
Multidomain All b Tetramer (disulphide-linked homodimer ofdisulphide-linkedheterodimers)
Extracellular Disulphide bond formation,subunit association, NMR
[17–19,77]
Serum albumin Multidomain All a Homodimer Extracellular (plasma) Disulphide-bond formation [78]MS2 phage coatprotein
Single domain a and b Dimer Phage capsid Fluorescence anisotropy,immunological activity
[79]
a-globin Single domain All a Monomer (subunit ofheterotetramerichemoglobin)
Cytoplasm Ligand (heme) binding [21]
Bacteriophage P22tailspike protein
Multidomain All b Homotrimer Phage capsid Immunological activity,limited proteolysis
[67,68,80]
Firefly luciferase Multidomain a and b Monomer Peroxisome Enzymatic activity,limited proteolysis
[28,69,81–83]
Bacterial luciferase(b-subunit)
Single-domain a/b protein The active enzymeis a heterodimer
Cytoplasm Enzymatic activity [84]
Reovirus cellattachment protein
Two-domain All b Homotrimer Virion Subunit association [85]
Influenzahemagglutinin
Multidomain All b and coiled coil Homotrimer ofdisulfide-linkedhemagglutininHA1-HA2 chains
Virion membrane Disulphide bond formation,immunological activity
[86–88]
Mammalianrhodanese
Two-domain a/b Monomer Mitochondrial matrix Enzymatic activity [29]
Ricin A-chain Two-domain a and b Ricin is adisulfide-linkedheterodimer ofA and B chains
Cytoplasm Enzymatic activity [89]
Human H-Ras andmouse dihydrofolatereductase (DHFR)
Synthetic fusionof two single-domain proteins,H-Ras andDHFR
a/b H-Ras forms signalingcomplexes with otherproteins;DHFR is a monomer
H-Ras shuttlesbetween theGolgi apparatus andcellular membrane;
Limited proteolysis [90]
DHFR is in thecytoplasm
OmpR (E. coli) Two-domain All a and a/b Momomer andmultimer
Cytoplasm Limited proteolysis [90]
Semliki Forest viruscapsid protein
Two-domain All b Heterodimer Viral capsid Enzymatic activity [72,91]
NF-kB1 Multidomain All b Homodimer orheterodimer
Cytoplasm andnucleus
Subunit association,immunological activity
[92]
Caspase-activatedDNase
Two-domain a and b Heterodimer Cytoplasm andnucleus
Subunit association [93]
Low-densitylipoprotein receptor(LDL-R)
Mutidomain Domains belong toall a, a and b, allb classes
Apparently,monomer
Membrane Disulphide-bond formation,immunological activity
[26]
HIV-1 envelopeglycoprotein
Mutidomain All b and a and b Heterodimer Membrane, virion Disulphide-bond formation [94]
Cystic fibrosistransmembraneconductanceregulator
Mutidomain a/b Can be part ofheteromultimericcomplex
Membrane Limited proteolysis [95]
Review Trends in Biochemical Sciences Vol.34 No.1
18
Profa. Ana Paula - 2015

• Genentech (1977) → 1ª proteína humana (somatostatina) produzida em bactéria Ala-Gly-Cys-Lys-Asn-Phe-Phe-Trp-Lys-Thr-Phe-Thr-Ser-Cys 1 2 3 4 5 6 7 8 9 10 11 12 13 14 ⇓ SÍNTESE do GENE oligonucleotídeos codificando os 14 códons
1ª produção de polipeptídeo funcional a partir de gene sintético

Hoje...
genes clonados de bibliotecas de cDNA ou diretamente do genoma do organismo
original por PCR - genes sintéticos
“Porém, a verdadeira diversão começa depois
que o cDNA é clonado em um vetor de expressão”

proteína não é obtida
Diagnóstico, do ponto de vista prático:
- Estabilidade e estrutura do mRNA - Toxicidade do produto - Solubilidade do produto - Disparidade no “codon bias”

código usado no organismo nativo
≠ Código usado no organismo hospedeiro
"PREFERÊNCIA" DE CÓDONS (Codon Bias)

61 códons → 20 aa e 3 códons de parada
cada aa codificado por 1 (Met ou Trp) a 6 (Arg, Leu, Ser) códons sinônimos
muitas alternativas de sequências de DNA para codificar a mesma
proteína
A frequência com que os ≠ códons são usados variam:
- entre ≠ organismos
- entre proteínas expressas em ↑ ou ↓ níveis no mesmo organismo
Por que ≠s organismos preferem ≠s códons?

Diferenças nas preferências por códons → Forças Evolutivas - conteúdo GC no genoma
- equilíbrio mutação-seleção codon bias + organismo: ↓ diversidade tRNAs isoaceptores ↓ carga metabólica - expressão proteínas heterólogas

Visualização das preferências de códons
Índice de adaptação de códon (CAI): correlação entre a preferência de códons de um gene e seus níveis de expressão
♦ prediz os níveis de expressão de genes endógenos
♦ porém, não avalia a provável compatibilidade entre um gene e um hospedeiro heterólogo.
Análise do componente principal:
mostra as diferenças de preferências de códons entre organismos ≠s
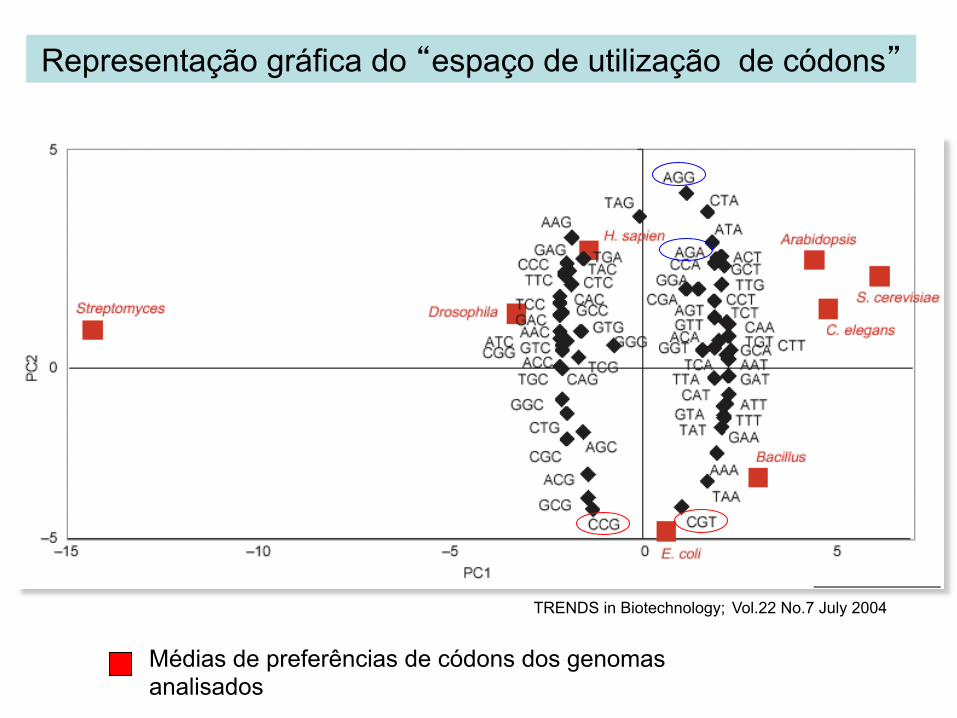
Médias de preferências de códons dos genomas analisados
Representação gráfica do “espaço de utilização de códons”
TRENDS in Biotechnology; Vol.22 No.7 July 2004
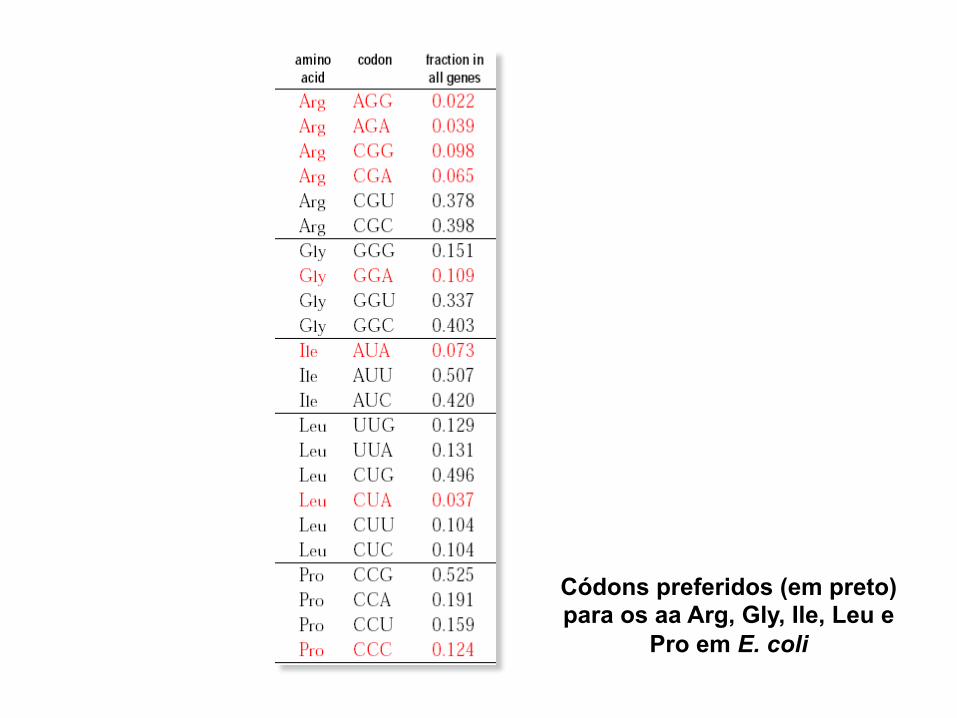
Códons preferidos (em preto) para os aa Arg, Gly, Ile, Leu e
Pro em E. coli

1) Correlação entre códons preferidos e abundância tRNAs
correspondentes disponíveis no hospedeiro Ex: E. coli ↓ AGG e AGA ↓ tRNAArg
4
[códons] ~ [tRNA isoaceptor] → Co-evolução
2) Existência códigos levemente ≠s em organismos ≠s Ex: Ciliados - tRNAs lêem os stop códons TAA e TAG como Glu
Como a preferência de códons afeta a expressão?

Qual o papel dos códons raros na tradução?
• Análises de bioinformática em E. coli mostram que: – Códons > freq. de uso = associados a elementos estruturais
(hélices); – Cluster de codons com < freq. = regiões menos estruturadas – Ou seja: o posicionamento e o agrupamento de códons não é
randômico e se reflete na expressão gênica. Exemplos: EgFABP1 – substituição de códons raros por sinônimos preferenciais em região correspondente a um “linker” impactou a solubilidade do produto recombinante.

• Nem sempre códons raros numa seq. tem efeito negativo na expressão heteróloga;
• Sua posição específica downstream ao códon de iniciação pode modular a expressão.
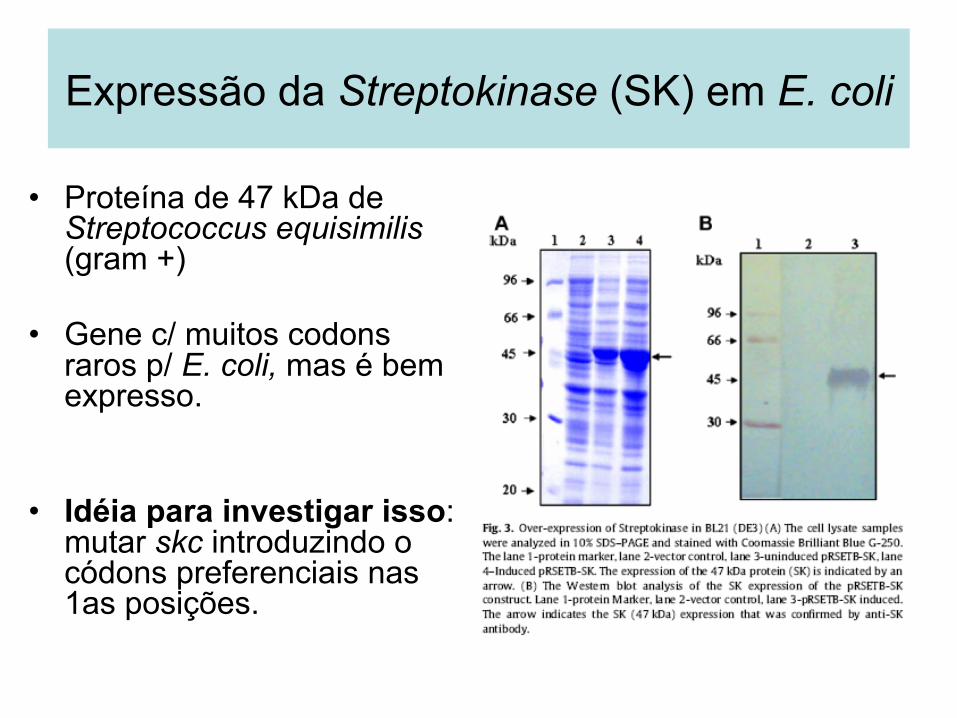
• Proteína de 47 kDa de Streptococcus equisimilis (gram +)
• Gene c/ muitos codons raros p/ E. coli, mas é bem expresso.
• Idéia para investigar isso: mutar skc introduzindo o códons preferenciais nas 1as posições.
Expressão da Streptokinase (SK) em E. coli
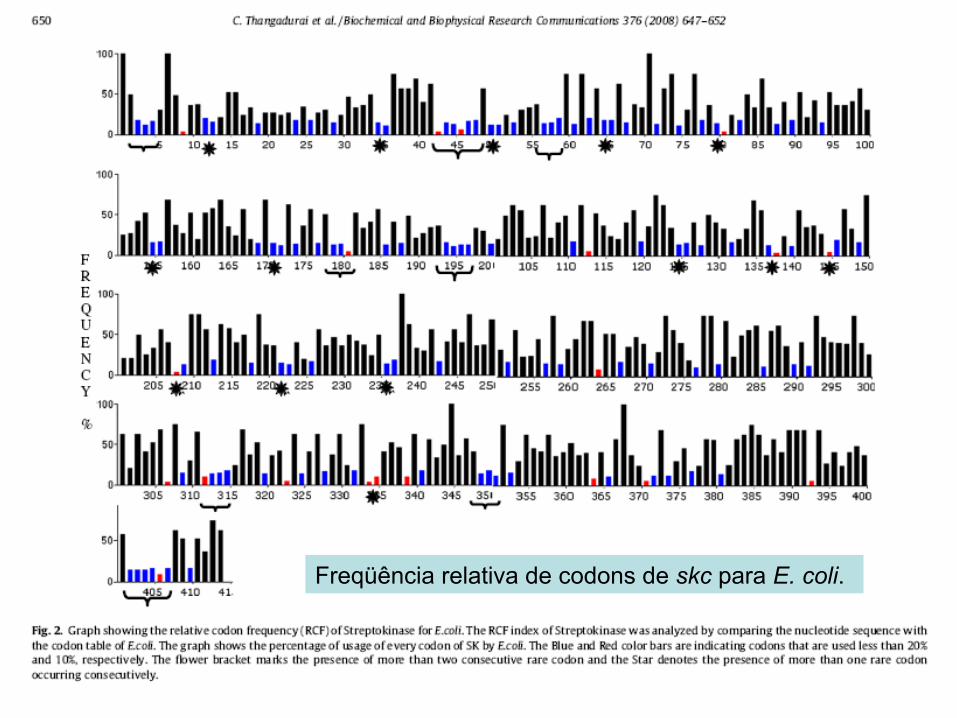
Freqüência relativa de codons de skc para E. coli.
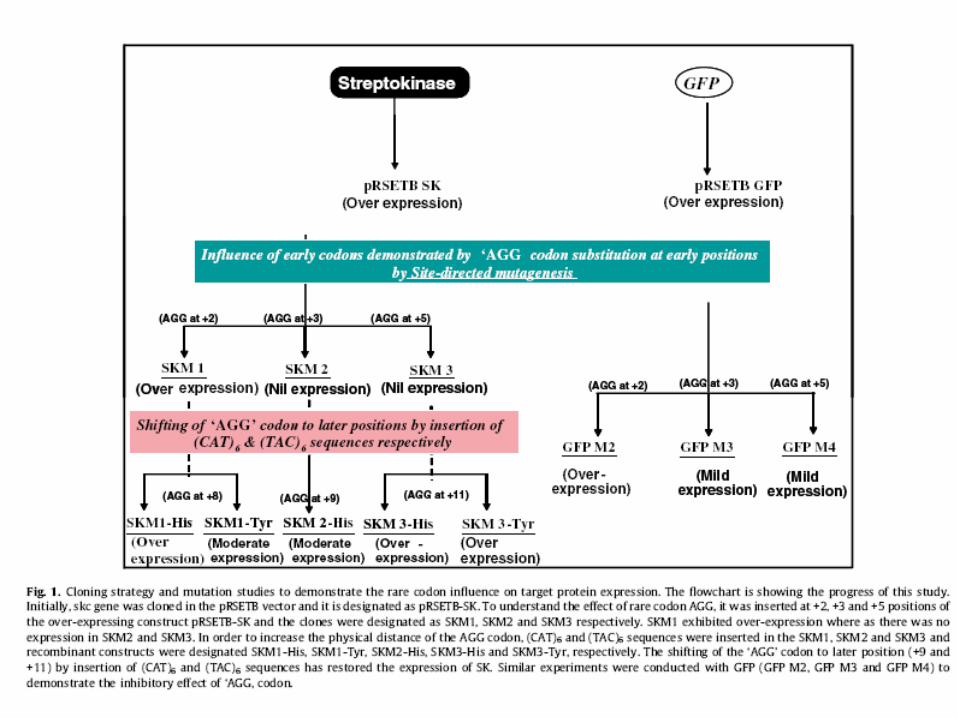
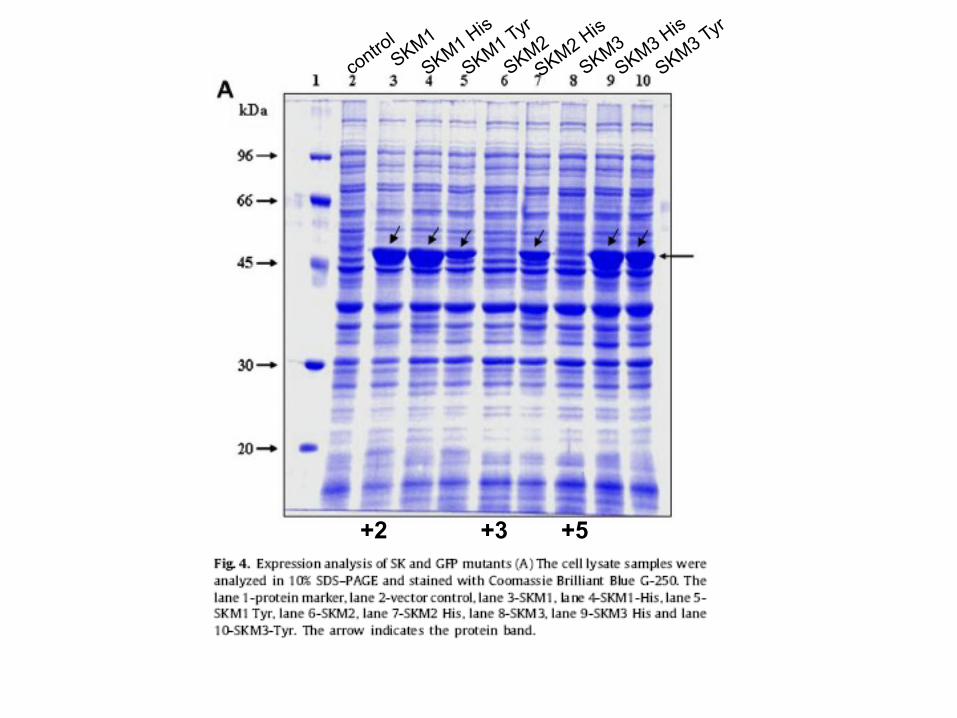
+2 +3 +5

Resumindo: • AGG na posição +3 ou +5 tem um efeito inibitório na
expressão • AGG na posição +2, +9 o +11 leva expressão de skc
Provavelmente devido a estrutura secundária do mRNA... AGG na posição +3 ou +5 causa a formação de uma nova
seqüência SD-like. Nem todos os códons raros afetam a expressão
significativamente...além de depender de sua posição.
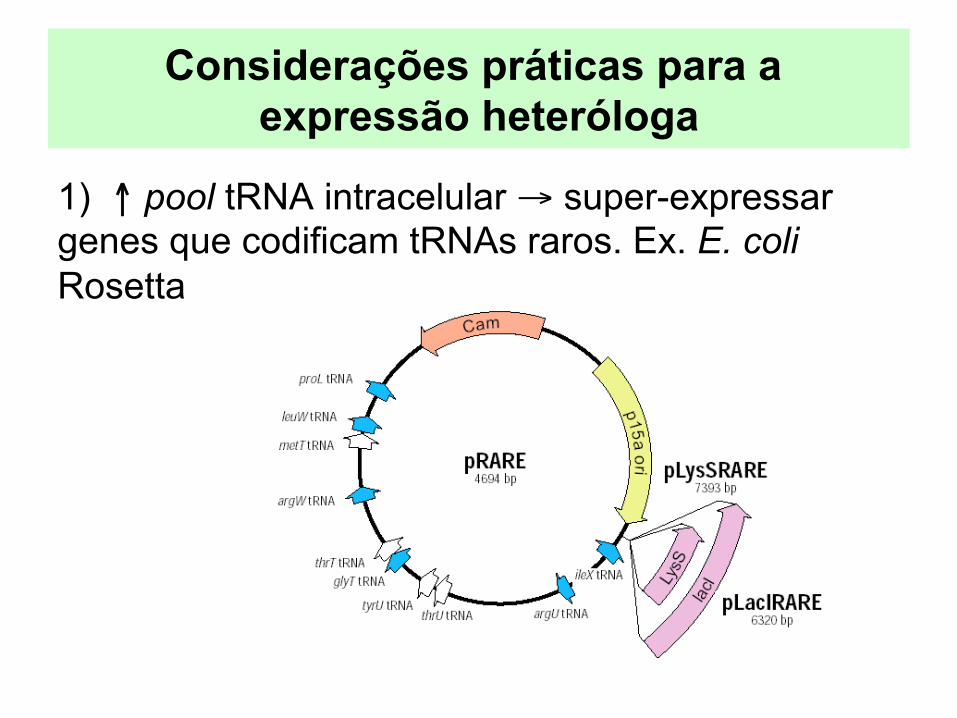
Considerações práticas para a expressão heteróloga
1) ↑ pool tRNA intracelular → super-expressar genes que codificam tRNAs raros. Ex. E. coli Rosetta

Porém... - ≠s tRNAs precisam ser super-expressos para genes de ≠s organismos - estratégia não muito flexível Ex. para vacinas DNA: não dá para modificar o
hospedeiro! - mudanças nas concentrações tRNAs na cel prejudicam seu
o processamento pós-traducional ausência modificações tRNA → ↓ fidelidade tradução - substituição aa - mudança na fase leitura - terminação prematura
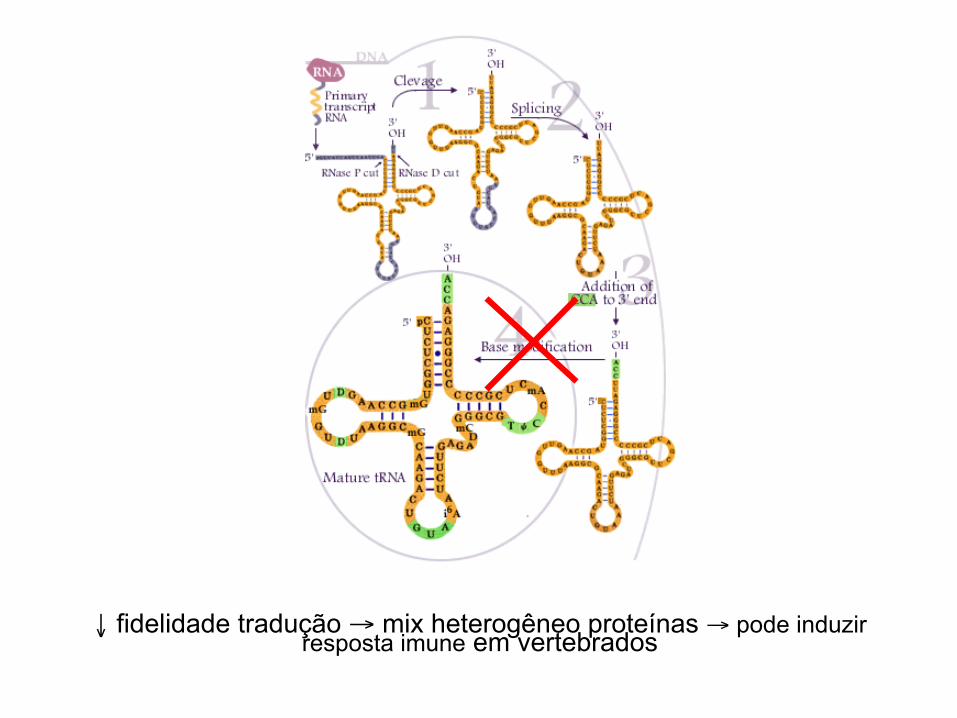
↓ fidelidade tradução → mix heterogêneo proteínas → pode induzir resposta imune em vertebrados

Considerações práticas para a expressão heteróloga
1) ↑ pool tRNA intracelular → super-expressar genes que codificam tRNAs raros 2) Otimizar os códons para o hospedeiro 3) Harmonizar os códons com o hospedeiro

Trocar os códons raros do gene por códons sinônimos preferidos pelo hospedeiro
Mutagênese sítio-dirigidas
Gene sintético
↑ 5-15x expressão proteínas mamíferos em E. coli
Gene sintético → expressão genes de organismos que usam
códigos não comuns Ex: Candida albicans e Tetrahymena (ciliado)
Otimização de códons...

Considerações sobre a síntese gênica 1) cada aa pode ser codificado em média por 3 códons ≠s 3100 (~5x1047) seq nucleotídicas que produziriam a
mesma proteína de 100aa
Quantas seq resultariam em ↑ níveis expressão protéica heteróloga?
Como escolher entre elas?

Desenho do gene: o processo de desenvolvimento no DNA2.0 (https://www.dna20.com/)

© 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 655
Biotechnol. J. 2011, 6, 650–659 www.biotechnology-journal.com
gion; high-frequency codons are translated quick-ly within the protective ribosomal tunnel (Fig. 3B;tunnel is not shown); and as the translocating ribo-some reaches an mRNA segment encoded by low-frequency-usage codons, the rate of translationslows, and allows for the preceding nascent peptide
to gain some helical structure within the tunnel(Fig. 3C).
Over the past decade, several codon adaptationalgorithms have been developed and are availablethrough public website access (Table 1). Needlessto say the success or failure of applying any ap-
Figure 3. Schematic model of co-translational folding on mRNA by ribosomes. (A) Ribosomal complex centered on the translation initiation site, AUG(initiation codon). (B) Nascent polypeptide synthesis within the protective environment of the ribosomal tunnel. (C) Putative translational pause sites inconjunction with co-translational folding occur within the ribosomal tunnel. Differences in codon usage frequency are shown as thick dashed lines witharrowheads for areas representing high-frequency-usage codons, and therefore, translating rapidly (hare) and regions that are double lined represent seg-ments of lower frequency usage codons (i.e., putative pause sites; tortoise) where translation proceeds more slowly to allow nascent polypeptide folding.
Table 1. Codon usage analysis and optimization tools
Algorithm Description Citation
ORFOPT Tunes regional nucleotide composition, codon choice, mRNA secondary structure [43]
Gene Composer Gene and protein engineering using PCR-based gene assembly and PIPE cloning. [100]
Codon Adjusts codon usage by predicting translational pauses and matching codon usage on native gene hosts [62, 63]Harmonization in heterologous hosts
GASCO Codon optimization based on host genome codon bias with the identification of desirable/undesirable motifshttp://miracle.igib.res.in/gasco/ [101]
QPSO Quantum-behaved particle swarm optimization [102]
OPTIMIZER Codons computed based on highly expressed prokaryotic genes, based on CAI [103]http://genomes.urv.es/OPTIMIZER
Gene Designer Synthetic biology workbench using advanced optimization algorithms and an intuitive drag-and-drop [104](DNA 2.0 Inc.) graphic interface
Synthetic Gene Enhanced functionality enabling users to work with nonstandard genetic codes, with user-defined patterns [105]Designer of codon usage, and an expanded range of methods for codon optimization
JCat Codon adaptation with the avoidance of cleavage sites [106]http://ww.prodoric.de/JCat
GeMS Gene design functions, including restriction site prediction, codon optimization for expression, [107]stem-loop determination, and oligonucleotide design
UpGene SIV/HIV coding sequence adaptation for eukaryotic expression [108]http://www.vectorcore.pitt.edu/upgene.htm
© 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 655
Biotechnol. J. 2011, 6, 650–659 www.biotechnology-journal.com
gion; high-frequency codons are translated quick-ly within the protective ribosomal tunnel (Fig. 3B;tunnel is not shown); and as the translocating ribo-some reaches an mRNA segment encoded by low-frequency-usage codons, the rate of translationslows, and allows for the preceding nascent peptide
to gain some helical structure within the tunnel(Fig. 3C).
Over the past decade, several codon adaptationalgorithms have been developed and are availablethrough public website access (Table 1). Needlessto say the success or failure of applying any ap-
Figure 3. Schematic model of co-translational folding on mRNA by ribosomes. (A) Ribosomal complex centered on the translation initiation site, AUG(initiation codon). (B) Nascent polypeptide synthesis within the protective environment of the ribosomal tunnel. (C) Putative translational pause sites inconjunction with co-translational folding occur within the ribosomal tunnel. Differences in codon usage frequency are shown as thick dashed lines witharrowheads for areas representing high-frequency-usage codons, and therefore, translating rapidly (hare) and regions that are double lined represent seg-ments of lower frequency usage codons (i.e., putative pause sites; tortoise) where translation proceeds more slowly to allow nascent polypeptide folding.
Table 1. Codon usage analysis and optimization tools
Algorithm Description Citation
ORFOPT Tunes regional nucleotide composition, codon choice, mRNA secondary structure [43]
Gene Composer Gene and protein engineering using PCR-based gene assembly and PIPE cloning. [100]
Codon Adjusts codon usage by predicting translational pauses and matching codon usage on native gene hosts [62, 63]Harmonization in heterologous hosts
GASCO Codon optimization based on host genome codon bias with the identification of desirable/undesirable motifshttp://miracle.igib.res.in/gasco/ [101]
QPSO Quantum-behaved particle swarm optimization [102]
OPTIMIZER Codons computed based on highly expressed prokaryotic genes, based on CAI [103]http://genomes.urv.es/OPTIMIZER
Gene Designer Synthetic biology workbench using advanced optimization algorithms and an intuitive drag-and-drop [104](DNA 2.0 Inc.) graphic interface
Synthetic Gene Enhanced functionality enabling users to work with nonstandard genetic codes, with user-defined patterns [105]Designer of codon usage, and an expanded range of methods for codon optimization
JCat Codon adaptation with the avoidance of cleavage sites [106]http://ww.prodoric.de/JCat
GeMS Gene design functions, including restriction site prediction, codon optimization for expression, [107]stem-loop determination, and oligonucleotide design
UpGene SIV/HIV coding sequence adaptation for eukaryotic expression [108]http://www.vectorcore.pitt.edu/upgene.htm

Harmonização de códons
• Combina a tendência de uso de códons inerente ao hospedeiro nativo com o mais próximo possível no hospedeiro heterólogo.
• Em termos de enovelamento co-traducional isso pode ser crucial.

A taxa de síntese de proteínas no ribossomo é modulada pelo:
- Uso não-randômico de códons sinônimos;
– Disponibilidade de tRNAs isoaceptores.
Códons + frequentes podem ser traduzidos + rapidamente que aqueles pouco usados (raros);
Códons raros no organismo podem ser
correlacionados a regiões menos estruturadas
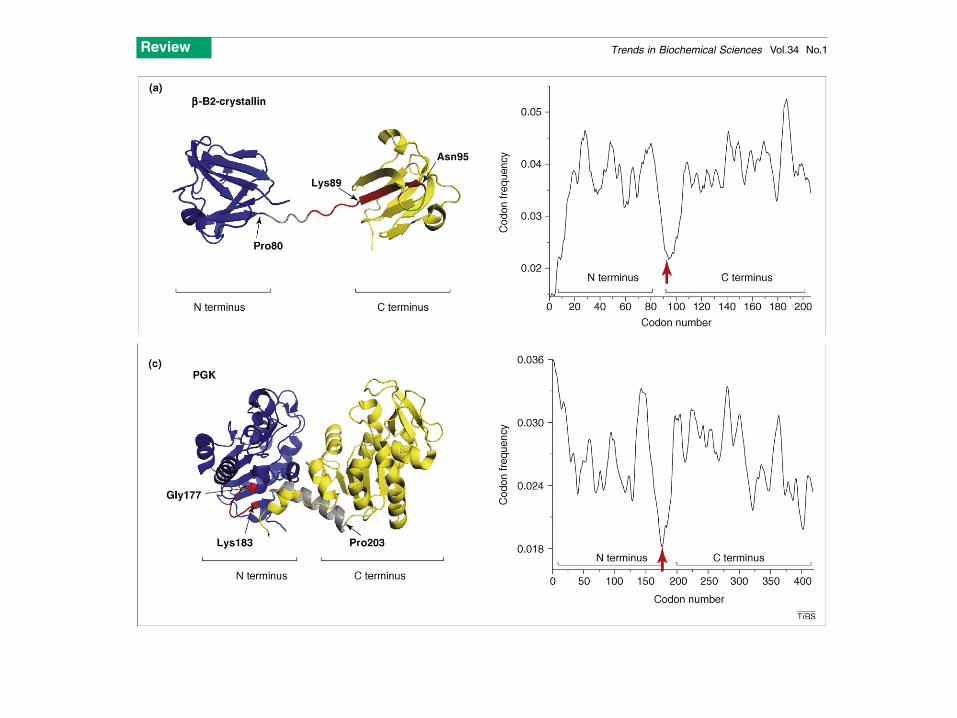
Figure 1. The location of rare codon clusters often indicates domain termini and/or boundaries in multidomain proteins. The left panels show backbone (cartoon)structures of the two-domain proteins whereas the codon frequency profiles are shown on the right (built as described previously in Refs [42,45,52,55,56]). Red arrowsindicate the clusters of rare codons at the domain boundaries. (a) Bovine (Bos taurus) b-B2 crystallin (PDB 2BB2; www.rcsb.org). The N-terminal domain is in blue, the C-terminal domain is in yellow and a portion of the linker connecting the two domains is shown in gray. Residues encoded by the most rarely used codons (cluster) close tothe linker region are in red; Pro80 and Lys89 are at the beginning and the end of the linker peptide connecting the domains; Asn95 marks the end of the first b-structure inthe b-B2 C-terminal domain. (b) Rabbit (Oryctolagus cuniculus) glyceraldehyde-3-phosphate dehydrogenase (GAPDH; PDB 1J0X). The N-terminal domain is in blue, theC-terminal is in yellow and residues encoded by the rarely used codons connecting the two domains are shown in red. The final C-terminal a-helix is shown in green forbetter visualization of the GAPDH structural features. Ser145 and Ser148 are at the beginning and the end of the short linker connecting the two domains. The cluster of
Review Trends in Biochemical Sciences Vol.34 No.1
20
Figure 1. The location of rare codon clusters often indicates domain termini and/or boundaries in multidomain proteins. The left panels show backbone (cartoon)structures of the two-domain proteins whereas the codon frequency profiles are shown on the right (built as described previously in Refs [42,45,52,55,56]). Red arrowsindicate the clusters of rare codons at the domain boundaries. (a) Bovine (Bos taurus) b-B2 crystallin (PDB 2BB2; www.rcsb.org). The N-terminal domain is in blue, the C-terminal domain is in yellow and a portion of the linker connecting the two domains is shown in gray. Residues encoded by the most rarely used codons (cluster) close tothe linker region are in red; Pro80 and Lys89 are at the beginning and the end of the linker peptide connecting the domains; Asn95 marks the end of the first b-structure inthe b-B2 C-terminal domain. (b) Rabbit (Oryctolagus cuniculus) glyceraldehyde-3-phosphate dehydrogenase (GAPDH; PDB 1J0X). The N-terminal domain is in blue, theC-terminal is in yellow and residues encoded by the rarely used codons connecting the two domains are shown in red. The final C-terminal a-helix is shown in green forbetter visualization of the GAPDH structural features. Ser145 and Ser148 are at the beginning and the end of the short linker connecting the two domains. The cluster of
Review Trends in Biochemical Sciences Vol.34 No.1
20

© 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 655
Biotechnol. J. 2011, 6, 650–659 www.biotechnology-journal.com
gion; high-frequency codons are translated quick-ly within the protective ribosomal tunnel (Fig. 3B;tunnel is not shown); and as the translocating ribo-some reaches an mRNA segment encoded by low-frequency-usage codons, the rate of translationslows, and allows for the preceding nascent peptide
to gain some helical structure within the tunnel(Fig. 3C).
Over the past decade, several codon adaptationalgorithms have been developed and are availablethrough public website access (Table 1). Needlessto say the success or failure of applying any ap-
Figure 3. Schematic model of co-translational folding on mRNA by ribosomes. (A) Ribosomal complex centered on the translation initiation site, AUG(initiation codon). (B) Nascent polypeptide synthesis within the protective environment of the ribosomal tunnel. (C) Putative translational pause sites inconjunction with co-translational folding occur within the ribosomal tunnel. Differences in codon usage frequency are shown as thick dashed lines witharrowheads for areas representing high-frequency-usage codons, and therefore, translating rapidly (hare) and regions that are double lined represent seg-ments of lower frequency usage codons (i.e., putative pause sites; tortoise) where translation proceeds more slowly to allow nascent polypeptide folding.
Table 1. Codon usage analysis and optimization tools
Algorithm Description Citation
ORFOPT Tunes regional nucleotide composition, codon choice, mRNA secondary structure [43]
Gene Composer Gene and protein engineering using PCR-based gene assembly and PIPE cloning. [100]
Codon Adjusts codon usage by predicting translational pauses and matching codon usage on native gene hosts [62, 63]Harmonization in heterologous hosts
GASCO Codon optimization based on host genome codon bias with the identification of desirable/undesirable motifshttp://miracle.igib.res.in/gasco/ [101]
QPSO Quantum-behaved particle swarm optimization [102]
OPTIMIZER Codons computed based on highly expressed prokaryotic genes, based on CAI [103]http://genomes.urv.es/OPTIMIZER
Gene Designer Synthetic biology workbench using advanced optimization algorithms and an intuitive drag-and-drop [104](DNA 2.0 Inc.) graphic interface
Synthetic Gene Enhanced functionality enabling users to work with nonstandard genetic codes, with user-defined patterns [105]Designer of codon usage, and an expanded range of methods for codon optimization
JCat Codon adaptation with the avoidance of cleavage sites [106]http://ww.prodoric.de/JCat
GeMS Gene design functions, including restriction site prediction, codon optimization for expression, [107]stem-loop determination, and oligonucleotide design
UpGene SIV/HIV coding sequence adaptation for eukaryotic expression [108]http://www.vectorcore.pitt.edu/upgene.htm
BiotechnologyJournal DOI 10.1002/biot.201000332 Biotechnol. J. 2011, 6, 650–659
650 © 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
1 Introduction
Biomedical and biotechnological research relies onprocesses leading to the successful expression andproduction of key biological products. High-qualityproteins are required for many purposes, includingprotein structural and functional studies. Proteinexpression is the culmination of multistep process-es involving regulation at the level of transcription,mRNA turnover, protein translation, and post-translational modifications leading to the forma-tion of a stable product. Although significantstrides have been achieved over the past decade,advances toward integrating genomic and pro-teomic information are essential, and until suchtime, many target genes and their synthetic poten-tial may not be fully realized.Thus, the focus of this
review is to provide some experimental supportand a brief overview of how codon usage bias hasevolved relative to regulating gene expression lev-els.
Due to their apparent “silent” nature, synony-mous codon substitutions have long been thoughtto be inconsequential. In recent years, this long-held dogma has been refuted by evidence that evena single synonymous codon substitution can havesignificant impact on gene expression levels, pro-tein folding, and protein cellular function [1–4]. It iscertainly conceivable that, by design, nature hasprovided the basic instructions to direct efficientprotein synthesis and folding through the informa-tion encoded at the genetic code level. For most se-quenced genomes, synonymous codons are notused at equal frequencies. Sixty-one codons speci-fy the twenty amino acids found commonly in pro-tein sequences; most of these are specified by morethan one synonymous codon, with the exception ofmethionine and tryptophan.The redundancy in thegenetic code may have evolved as a way to preservestructural information of proteins within the nu-cleotide content [5]. In unicellular organisms, high-frequency-usage codons correlate with abundantcognate isoacceptor tRNA molecules and have
Review
Codon usage: Nature’s roadmap to expression and folding of proteins
Evelina Angov
Division of Malaria Vaccine Development, Walter Reed Army Institute of Research, Silver Spring, MD, USA
Biomedical and biotechnological research relies on processes leading to the successful expressionand production of key biological products. High-quality proteins are required for many purposes,including protein structural and functional studies. Protein expression is the culmination of mul-tistep processes involving regulation at the level of transcription, mRNA turnover, protein trans-lation, and post-translational modifications leading to the formation of a stable product. Althoughsignificant strides have been achieved over the past decade, advances toward integrating genom-ic and proteomic information are essential, and until such time, many target genes and their prod-ucts may not be fully realized. Thus, the focus of this review is to provide some experimental sup-port and a brief overview of how codon usage bias has evolved relative to regulating gene expres-sion levels.
Keywords: Codon usage · Protein folding · Translation
Correspondence: Dr. Evelina Angov, Division of Malaria Vaccine Develop-ment, Walter Reed Army Institute of Research, 503 Robert Grant Avenue,Silver Spring, MD 20910, USAE-mail: [email protected]
Abbreviations: AU, adenine:thymidine; CAI, codon adaptation index; GC,guanidine:cytosine; GFP, green fluorescent protein; ORF, open readingframe; RBS, ribosomal binding site; SD, Shine and Dalgarno
Received 11 January 2011Revised 11 April 2011Accepted 13 April 2011

Utilização dos códons
• A sequência do mRNA, através da distribuição dos códons sinônimos, pode servir como um guia para a cinética de enovelamento co-traducional…
might play a substantial, yet an overall fine-tuning, part inthe co-translational folding process. The impact of trans-lation kinetics on the folding of any given protein would,thus, depend on the stability of the nonproductive inter-mediates forming along the pathway.
Therefore, it should be possible (at least in certain cases)to optimize heterologous protein production and folding bythe appropriate selection of synonymous (both rare andfrequent) codons along the mRNA in a way such that themRNA translation kinetics in the heterologous host wouldmimic its natural translation kinetics in the homologousorganism. Very recent experimental evidence supports thisview; appropriate introduction of (especially) rare synon-ymous codons at selected places in several Plasmodiumfalciparum mRNAs increased both the solubility and the
yield (of up to 1000-fold) of recombinant proteins expressedin E. coli [75].
Concluding remarksSynonymous codon substitutions have, for the most part,been considered to be neutral and silent. Yet, accumulatingevidence indicates that synonymous codons are used inmRNA in a non-randommanner and that the unique codonusage profile of a given mRNA might specify the co-trans-lational folding pathway of the encoded protein. Therefore,codon usage coordinates the active conjunction of thesynthesis and folding of a protein. This idea has attractedan increasing number of proponents and should be con-sidered during optimization of heterologous proteinexpression. However, the effects of non-random synon-
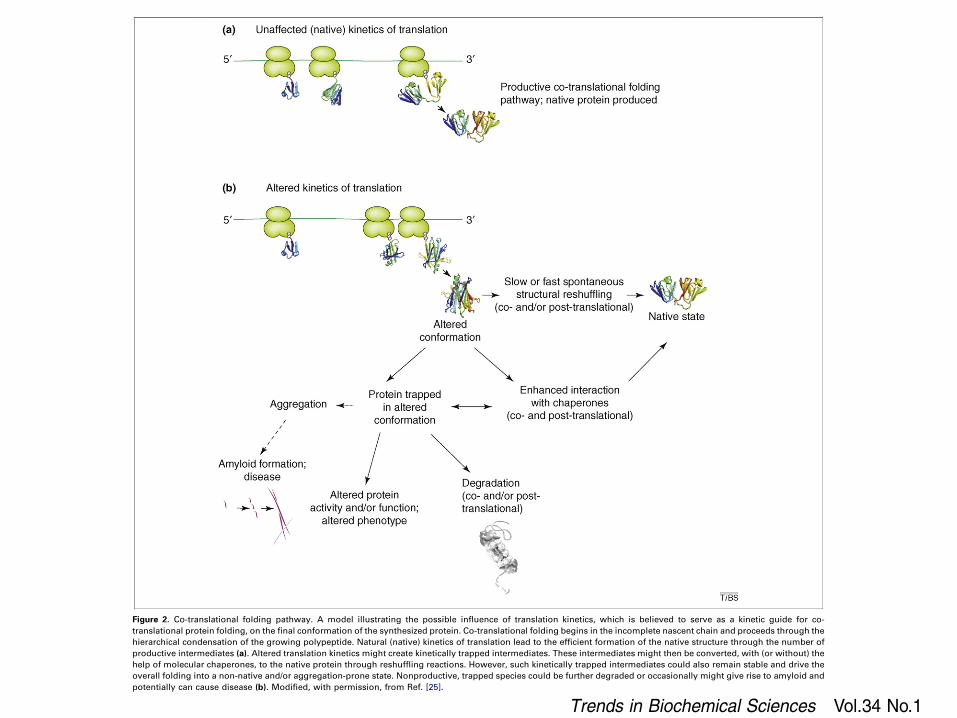
Figure 2. Co-translational folding pathway. A model illustrating the possible influence of translation kinetics, which is believed to serve as a kinetic guide for co-translational protein folding, on the final conformation of the synthesized protein. Co-translational folding begins in the incomplete nascent chain and proceeds through thehierarchical condensation of the growing polypeptide. Natural (native) kinetics of translation lead to the efficient formation of the native structure through the number ofproductive intermediates (a). Altered translation kinetics might create kinetically trapped intermediates. These intermediates might then be converted, with (or without) thehelp of molecular chaperones, to the native protein through reshuffling reactions. However, such kinetically trapped intermediates could also remain stable and drive theoverall folding into a non-native and/or aggregation-prone state. Nonproductive, trapped species could be further degraded or occasionally might give rise to amyloid andpotentially can cause disease (b). Modified, with permission, from Ref. [25].
Review Trends in Biochemical Sciences Vol.34 No.1
22
ymous codon utilization might have a much broaderimpact, which is yet to be uncovered. The nature andspecific features of synonymous codon usage, characteristicfor given genes, cell systems, tissues and organisms, isgenerating renewed interest. Comprehensive databases,containing information on protein structures, folding path-way(s) and features of synonymous codon usage withinencoding mRNAs, would be extremely helpful in addres-sing many aspects of the problem discussed here. Further-more, new approaches to study co-translational foldingsuch as those involving FRET analysis [24,30] or dynamicfluorescence depolarization technique [76] might help toilluminate (in real time) the exact nature of intermediatesforming on the co-translational folding pathway and tobetter address the importance of codon usage and proteintranslation kinetics in in vivo protein folding. These datashould be extremely helpful in further developing a uni-fying concept of protein folding and might have a tremen-dous potential within the biotechnology industry.
AcknowledgementsI apologize to those colleagues whose work I was not able to cite owing tospace limitations. I am grateful to Christopher Hellen, Tatyana Pestovaand Marina Rodnina for their critical reading of the manuscript andmany helpful discussions. I am also grateful to anonymous reviewers forthoughtful and stimulating suggestions. Work in my laboratory is, inpart, supported by the National American Heart Association(www.americanheart.org) grant (0730120N) to A.A.K.
References1 Luheshi, L.M. et al. (2008) Proteinmisfolding and disease: from the test
tube to the organism. Curr. Opin. Chem. Biol. 12, 25–312 Anfinsen, C.B. (1973) Principles that govern the folding of protein
chains. Science 181, 223–2303 Jaenicke, R. and Lilie, H. (2000) Folding and association of oligomeric
and multimeric proteins. Adv. Protein Chem. 53, 329–4014 Onuchic, J.N. andWolynes, P.G. (2004) Theory of protein folding.Curr.
Opin. Struct. Biol. 14, 70–755 Lindberg, M.O. and Oliveberg, M. (2007) Malleability of protein folding
pathways: a simple reason for complex behaviour. Curr. Opin. Struct.Biol. 17, 21–29
6 Jaenicke, R. (1991) Protein folding: local structures, domains, subunits,and assemblies. Biochemistry 30, 3147–3161
7 Chow, M.K. et al. (2006) The REFOLD database: a tool for theoptimization of protein expression and refolding. Nucleic Acids Res.34, D207–D212
8 Ellis, R.J. (1996) Revisiting the Anfinsen cage. Fold. Des. 1, R9–R159 Naylor, D.J. and Hartl, F.U. (2001) Contribution of molecular
chaperones to protein folding in the cytoplasm of prokaryotic andeukaryotic cells. Biochem. Soc. Symp. 68, 45–68
10 Kubelka, J. et al. (2004) The protein folding ‘speed limit’. Curr. Opin.Struct. Biol. 14, 76–88
11 Frydman, J. (2001) Folding of newly translated proteins in vivo: therole of molecular chaperones. Annu. Rev. Biochem. 70, 603–647
12 Bukau, B. et al. (2006) Molecular chaperones and protein qualitycontrol. Cell 125, 443–451
13 Cowie, D.B. et al. (1961) Ribosome-bound b-galactosidase. Proc. Natl.Acad. Sci. U. S. A. 47, 114–122
14 Zipser, D. and Perrin, D. (1963) Complementation on ribosomes. ColdSpring Harb. Symp. Quant. Biol. 28, 533–537
15 Kiho, Y. and Rich, A. (1964) Induced enzyme formed on bacterialpolyribosomes. Proc. Natl. Acad. Sci. U. S. A. 51, 111–118
16 Hamlin, J. and Zabin, I. (1972) b-Galactosidase: immunologicalactivity of ribosome-bound, growing polypeptide chains. Proc. Natl.Acad. Sci. U. S. A. 69, 412–416
17 Bergman, L.W. and Kuehl, W.M. (1979) Formation of intermoleculardisulfide bonds on nascent immunoglobulin polypeptides. J. Biol.Chem. 254, 5690–5694
18 Bergman, L.W. and Kuehl, W.M. (1979) Formation of an intrachaindisulfide bond on nascent immunoglobulin light chains. J. Biol. Chem.254, 8869–8876
19 Bergman, L.W. and Kuehl, W.M. (1979) Co-translational modificationof nascent immunoglobulin heavy and light chains. J. Supramol.Struct. 11, 9–24
20 Fedorov, A.N. and Baldwin, T.O. (1997) Cotranslational proteinfolding. J. Biol. Chem. 272, 32715–32718
21 Komar, A.A. et al. (1997) Cotranslational folding of globin. J. Biol.Chem. 272, 10646–10651
22 Hardesty, B. and Kramer, G. (2001) Folding of a nascent peptide on theribosome. Prog. Nucleic Acid Res. Mol. Biol. 66, 41–66
23 Kolb, V.A. (2001) Cotranslational protein folding.Mol. Biol. (Mosk.) 35,682–690
24 Johnson, A.E. (2005) The co-translational folding and interactions ofnascent protein chains: a new approach using fluorescence resonanceenergy transfer. FEBS Lett. 579, 916–920
25 Komar, A.A. (2008) Protein translation rates and protein misfolding: isthere any link? In Protein Misfolding: New Research (O’Doherty C.B.and Byrne, A.C. eds) Nova Science Publishers (in press)
26 Jansens, A. et al. (2002) Coordinated nonvectorial folding in a newlysynthesized multidomain protein. Science 298, 2401–2403
27 Jenni, S. and Ban, N. (2003) The chemistry of protein synthesis andvoyage through the ribosomal tunnel.Curr. Opin. Struct. Biol. 13, 212–
21928 Makeyev, E.V. et al. (1996) Enzymatic activity of the ribosome-bound
nascent polypeptide. FEBS Lett. 378, 166–17029 Kudlicki, W. et al. (1995) Folding of an enzyme into an active
conformation while bound as peptidyl-tRNA to the ribosome.Biochemistry 34, 14284–14287
30 Woolhead, C.A. et al. (2004) Nascent membrane and secretory proteinsdiffer in FRET-detected folding far inside the ribosome and in theirexposure to ribosomal proteins. Cell 116, 725–736
31 Lu, J. and Deutsch, C. (2005) Folding zones inside the ribosomal exittunnel. Nat. Struct. Mol. Biol. 12, 1123–1129
32 Lim, V.I. and Spirin, A.S. (1986) Stereochemical analysis of ribosomaltranspeptidation. Conformation of nascent peptide. J. Mol. Biol. 188,565–574
33 Ziv, G. et al. (2005) Ribosome exit tunnel can entropically stabilizealpha-helices. Proc. Natl. Acad. Sci. U. S. A. 102, 18956–18961
34 Etchells, S.A. and Hartl, F.U. (2004) The dynamic tunnel. Nat. Struct.Mol. Biol. 11, 391–392
35 Voss, N.R. et al. (2006) The geometry of the ribosomal polypeptide exittunnel. J. Mol. Biol. 360, 893–906
36 Gilbert, R.J. et al. (2004) Three-dimensional structures of translatingribosomes by Cryo-EM. Mol. Cell 14, 57–66
37 Morrissey,M.P. et al. (2004) The role of cotranslation in protein folding:a lattice model study. Polymer 45, 557–571
38 Heinemann, U. and Hahn, M. (1995) Circular permutation ofpolypeptide chains: implications for protein folding and stability.Prog. Biophys. Mol. Biol. 64, 121–143
39 Contreras Martı́nez, L.M. et al. (2006) Protein translocation through atunnel induces changes in folding kinetics: a lattice model study.Biotechnol. Bioeng. 94, 105–117
40 Huard, F.P. et al. (2006) Modelling sequential protein folding underkinetic control. Bioinformatics 22, e203–e210
41 Wolin, S.L. and Walter, P. (1988) Ribosome pausing and stackingduring translation of a eukaryotic mRNA. EMBO J. 7, 3559–3569
42 Krasheninnikov, I.A. et al. (1991) Nonuniform size distribution ofnascent globin peptides, evidence for pause localization sites, and acotranslational protein-folding model. J. Protein Chem. 10, 445–454
43 Komar, A.A. et al. (1999) Synonymous codon substitutions affectribosome traffic and protein folding during in vitro translation.FEBS Lett. 462, 387–391
44 Protzel, A. and Morris, A.J. (1974) Gel chromatographic analysis ofnascent globin chains. Evidence of nonuniform size distribution. J.Biol. Chem. 249, 4594–4600
45 Komar, A.A. and Jaenicke, R. (1995) Kinetics of translation of g Bcrystallin and its circularly permutated variant in an in vitro cell-freesystem: possible relations to codon distribution and protein folding.FEBS Lett. 376, 195–198
46 Hollingsworth, M.J. et al. (1998) Heelprinting analysis of in vivoribosome pause sites. Methods Mol. Biol. 77, 153–165
Review Trends in Biochemical Sciences Vol.34 No.1
23