composition driven structural phase transitions in the (y1-xlax)2ti2o7 system
DESCRIPTION
Structural characteristics of La-doped pyrochloresTRANSCRIPT

Mat Res Soc Proc 932: 631-638
Composition driven structural phase transitions in the (Y1-xLax)2Ti2O7 system
Elizabeth J. Harvey, Sharon E. Ashbrook, Gregory R. Lumpkin, Simon A.T. Redfern Department of Earth Sciences, University of Cambridge, Downing Street, Cambridge, CB2 3EQ, UK ABSTRACT
The structural characteristics of samples in the (Y1-xLax)2Ti2O7 system have been studied as a function of bulk composition using time-of-flight (TOF) powder neutron diffraction, powder X-ray diffraction (XRD), Electron Microscopy and 89Y (I = !) Magic Angle Spinning Nuclear Magnetic Resonance (MAS NMR). Analysis of the neutron diffraction data suggests the following: For compositions where 0 " x " 0.132, a single cubic phase is present (Fd3# m, Z = 8). Between 0.832 " x " 1, a solid solution with the La2Ti2O7 structure (P21, Z = 4) forms. The two phases coexist between x values of 0.132 and 0.832. XRD suggests that the limits of the two regions of solid solution fall within the ranges of x = 0.1-0.125 and x = 0.875-0.9. The variation in the limits found by the two diffraction methods may be due to stoichiometric errors in the samples. Line-shape analysis of NMR spectra for Y rich compositions suggests that increasing proportions of La are incorporated onto the pyrochlore A site in a statistically random manner, up to the limit of solid solution. The NMR spectra of the monoclinic phase suggest that occupation of the four crystallographically distinct ‘A’ type sites within the structure is not random. At low Y concentrations, two of these sites are preferentially occupied by Y. These sites are suggested to be those found at the edge of the perovskite slabs, which exhibit smaller coordination numbers (based on the number of oxygens within bonding distance of the ‘A’ type cation) than the true perovskite A sites found at the slab centres.
INTRODUCTION
In the evaluation of candidate waste forms for the immobilisation of high level nuclear waste (HLW), crystalline phase assemblages are often thought of as preferable to borosilicate glass based matrices, despite the higher projected costs of fabrication. Multiphase ceramics such as Synroc [1] have been shown to be far more resilient to intrinsic radiation damage and attack by groundwater than glass based waste forms; they do not devitrify over time and the composition of the immobilising medium can be optimised for a particular HLW stream so that the active species present are ‘locked’ into specific atomic positions within long lasting crystalline arrays. The pyrochlore structure (A2B2O7, Fd3# m, Z = 8), is a particularly robust example of a candidate phase for immobilising HLW – natural samples containing U and Th have been found which are in excess of 1 Ga in age [2]. It is therefore considered to be an excellent crystalline species for the immobilisation of actinides, particularly Pu, where the long term (Ma) retention of radionuclides is paramount. In order to critically evaluate the suitability of such a phase, it is necessary to thoroughly understand its susceptibility to changes in composition – these affect its ability to accommodate chemical variations in the waste stream and changes in the degree of waste loading. Here we report the findings of structural studies on the (Y1-xLax)2Ti2O7 system. The Y end member has the pyrochlore structure, whilst a monoclinic structure (P21, Z = 4) is observed for the La rich end member. Both Y and La are useful non-radioactive analogues of the actinides. Here, Y and La are mixed together, nominally on the A sites, in order to facilitate discussion of the effect of altering the average cation size on the structural characteristics of the system, as well as to study aspects of cation partitioning across all the available cation sites in both structures. Figure 1 shows the crystalline structures of a pyrochlore such as Y2Ti2O7 and a La2Ti2O7 type monoclinic phase, illustrating how the arrangement of cation sites within the two structures varies.

Mat Res Soc Proc 932: 631-638
Figure 1 (a) Unit cell of the pyrochlore structure projected down [001]. The large, dark grey spheres are oxygen anions. A3+ cations on the 16d Wyckoff positions (8-fold coordinated) are medium sized and light coloured; B4+ cations on the 16c sites (6-fold coordinated) are smaller and dark grey. The small black unbonded spheres indicate the position of unoccupied 8a sites. (b) Part of the La2Ti2O7 structure showing the perovskite-like ‘slabs’ of TiO6 octahedra and large ‘A’ type cations (black). The geometry of the ‘true perovskite’ (i) and ‘slab edge’ (ii) coordination environments of these cations is also shown.
EXPERIMENTAL
Sample fabrication
Samples with composition (Y1-xLax)2Ti2O7, where x varied from 0 to 1 were synthesized via a mixed-metal-oxide route. Stoichiometric amounts of Y2O3 (99.99%), La2O3 (99.99%) and TiO2 (99.8%) were ball milled in acetone and the resultant slurries were passed through a 38 µm sieve. The dried product was uniaxially pressed at 16 MPa into pellets 20 mm in diameter and ~4 mm high. These were sintered at 1300-1500°C for 48 hours. The pellets were then reground, pressed and resintered under the same conditions. Sample characterization
X-ray diffraction (XRD) was performed on powdered samples using a Bruker D5000 diffractometer fitted with a copper X-ray tube, employing a scintillation counter and a Ni filter. Diffraction patterns were recorded over a 2! range of 10° to 90°, using a step size of 0.02° 2!, counting for 3-4 s per step. It was necessary to employ a Philips 1710 diffractometer, also equipped with a scintillation counter and a Ni filter, operating at a continuous scan speed of 1° min-1 from 10° to 80°, with a paper trace output, because the noise levels observed using the D5000 diffractometer proved to be too high to accurately observe the pyrochlore solid solution limit. Neutron Time-of-flight (TOF) powder diffraction experiments were performed on the high flux POLARIS powder diffractometer at the ISIS pulsed spallation neutron source, Rutherford Appleton Laboratory, UK. Approximately 10 g of sample was loaded into thin walled vanadium sample cans, which were then mounted in an automatic sample changer. Each sample was run at room temperature for a total
(i)
(ii)
(a) (b)
(i)
(ii)
Mat Res Soc Proc 932: 631-638
integrated proton beam current to the ISIS target of 175 µA h (approx 1 hour). Data were collected over a TOF range of 1000-19600 µs, corresponding to d spacings between 0.162 and 3.2 Å. Data were recorded by a bank of 58 3He gas tube detectors positioned at average 2! = 145°. An empty instrument background was subtracted and the data were normalised using an incoherent scattering pattern from a vanadium sample. The diffraction profiles acquired by neutron diffraction were fitted by the Rietveld method [3], using the General Structure Analysis System (GSAS), with the front-end graphical user interface ‘EXPGUI’. Fragments of sintered pellets were examined using a Jeol JSM-820 scanning electron microscope (SEM), equipped with an X-ray energy dispersive unit and operating at an accelerating voltage of 20 kV. The samples were mounted in epoxy-resin, which was polished (using 1 µm diamond grit as final abrasive) before being coated with a conducting layer of carbon approximately 20 nm thick. 89Y (I = !) MAS NMR spectra were acquired using a Varian Infinity Plus spectrometer, equipped with an 11.7 T wide bore magnet, operating at a Larmor frequency of 24.5 MHz for Y. The samples were packed into a 7.5 mm silicon nitride rotor (to minimise any Y background) and rotated at MAS speeds between 6-7 kHz in a conventional Chemagnetics 7.5 mm probe, equipped with a low " tuning attachment. Spectra were acquired either with short flip angle (#/6) pulses and a 100 µs pre acquisition delay (to eliminate probe ringdown) or with a Carr-Purcell-Meiboom-Gill (CPMG) echo train. In both cases, recycle intervals between 1000-3000 seconds were used. Radio frequency field strengths between 20-25 kHz, producing #/2 pulse durations of 10-12 µs were employed. Spectra are referenced to 1 molar YCl
3 (aq).
RESULTS AND DISCUSSION
Solid Solution Limits in the (Y1-xLax)2Ti2O7 system
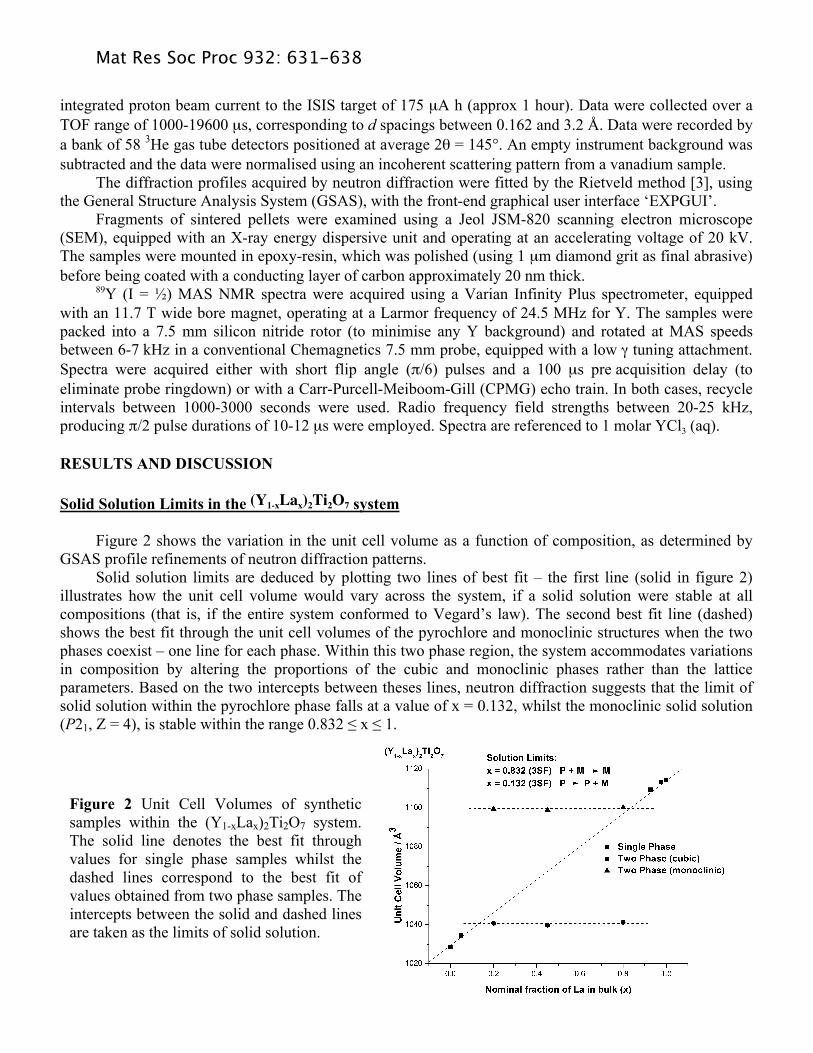
Figure 2 shows the variation in the unit cell volume as a function of composition, as determined by GSAS profile refinements of neutron diffraction patterns. Solid solution limits are deduced by plotting two lines of best fit – the first line (solid in figure 2) illustrates how the unit cell volume would vary across the system, if a solid solution were stable at all compositions (that is, if the entire system conformed to Vegard’s law). The second best fit line (dashed) shows the best fit through the unit cell volumes of the pyrochlore and monoclinic structures when the two phases coexist – one line for each phase. Within this two phase region, the system accommodates variations in composition by altering the proportions of the cubic and monoclinic phases rather than the lattice parameters. Based on the two intercepts between theses lines, neutron diffraction suggests that the limit of solid solution within the pyrochlore phase falls at a value of x = 0.132, whilst the monoclinic solid solution (P21, Z = 4), is stable within the range 0.832 " x " 1.
Figure 2 Unit Cell Volumes of synthetic samples within the (Y1-xLax)2Ti2O7 system. The solid line denotes the best fit through values for single phase samples whilst the dashed lines correspond to the best fit of values obtained from two phase samples. The intercepts between the solid and dashed lines are taken as the limits of solid solution.
Mat Res Soc Proc 932: 631-638
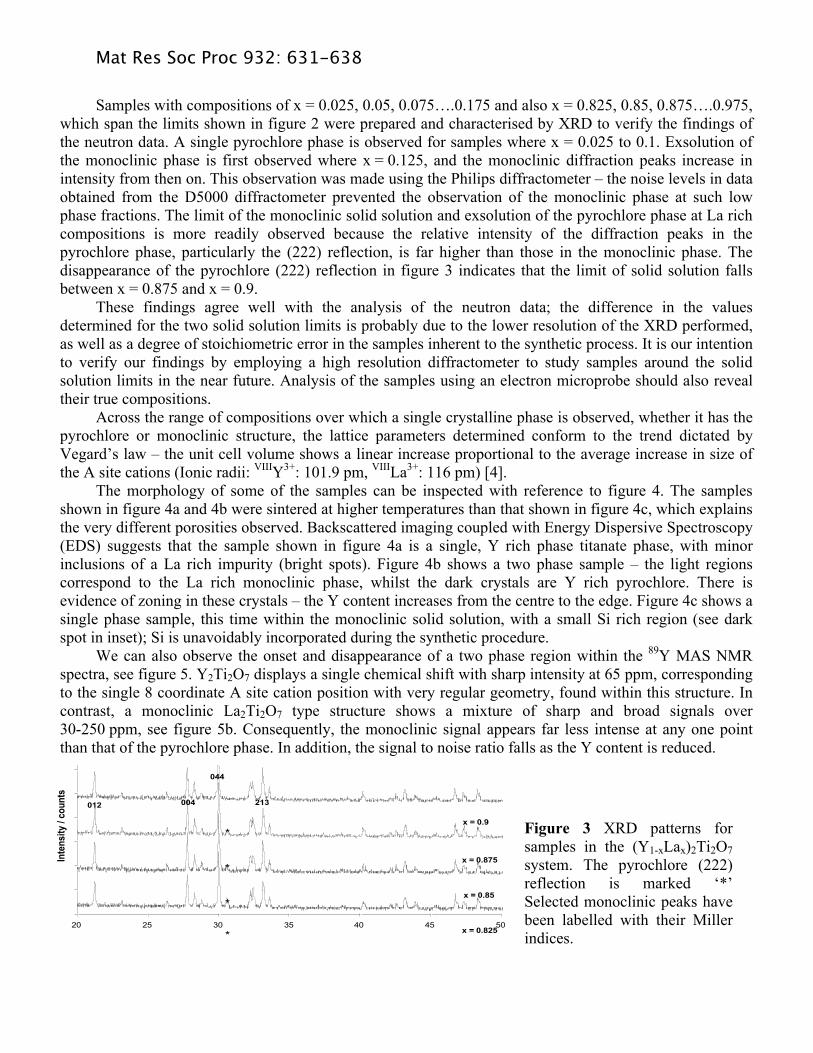
Samples with compositions of x = 0.025, 0.05, 0.075….0.175 and also x = 0.825, 0.85, 0.875….0.975, which span the limits shown in figure 2 were prepared and characterised by XRD to verify the findings of the neutron data. A single pyrochlore phase is observed for samples where x = 0.025 to 0.1. Exsolution of the monoclinic phase is first observed where x = 0.125, and the monoclinic diffraction peaks increase in intensity from then on. This observation was made using the Philips diffractometer – the noise levels in data obtained from the D5000 diffractometer prevented the observation of the monoclinic phase at such low phase fractions. The limit of the monoclinic solid solution and exsolution of the pyrochlore phase at La rich compositions is more readily observed because the relative intensity of the diffraction peaks in the pyrochlore phase, particularly the (222) reflection, is far higher than those in the monoclinic phase. The disappearance of the pyrochlore (222) reflection in figure 3 indicates that the limit of solid solution falls between x = 0.875 and x = 0.9. These findings agree well with the analysis of the neutron data; the difference in the values determined for the two solid solution limits is probably due to the lower resolution of the XRD performed, as well as a degree of stoichiometric error in the samples inherent to the synthetic process. It is our intention to verify our findings by employing a high resolution diffractometer to study samples around the solid solution limits in the near future. Analysis of the samples using an electron microprobe should also reveal their true compositions. Across the range of compositions over which a single crystalline phase is observed, whether it has the pyrochlore or monoclinic structure, the lattice parameters determined conform to the trend dictated by Vegard’s law – the unit cell volume shows a linear increase proportional to the average increase in size of the A site cations (Ionic radii: VIIIY3+: 101.9 pm, VIIILa3+: 116 pm) [4]. The morphology of some of the samples can be inspected with reference to figure 4. The samples shown in figure 4a and 4b were sintered at higher temperatures than that shown in figure 4c, which explains the very different porosities observed. Backscattered imaging coupled with Energy Dispersive Spectroscopy (EDS) suggests that the sample shown in figure 4a is a single, Y rich phase titanate phase, with minor inclusions of a La rich impurity (bright spots). Figure 4b shows a two phase sample – the light regions correspond to the La rich monoclinic phase, whilst the dark crystals are Y rich pyrochlore. There is evidence of zoning in these crystals – the Y content increases from the centre to the edge. Figure 4c shows a single phase sample, this time within the monoclinic solid solution, with a small Si rich region (see dark spot in inset); Si is unavoidably incorporated during the synthetic procedure. We can also observe the onset and disappearance of a two phase region within the 89Y MAS NMR spectra, see figure 5. Y2Ti2O7 displays a single chemical shift with sharp intensity at 65 ppm, corresponding to the single 8 coordinate A site cation position with very regular geometry, found within this structure. In contrast, a monoclinic La2Ti2O7 type structure shows a mixture of sharp and broad signals over 30-250 ppm, see figure 5b. Consequently, the monoclinic signal appears far less intense at any one point than that of the pyrochlore phase. In addition, the signal to noise ratio falls as the Y content is reduced.
20 25 30 35 40 45 50
2_ / degrees
Inte
nsit
y / c
ou
nts
012 004
044
x = 0.825
x = 0.85
x = 0.875
x = 0.9
*
*
*
*
213
20 25 30 35 40 45 50
2_ / degrees
Inte
nsit
y / c
ou
nts
012 004
044
x = 0.825
x = 0.85
x = 0.875
x = 0.9
20 25 30 35 40 45 50
2_ / degrees
Inte
nsit
y / c
ou
nts
012 004
044
x = 0.825
x = 0.85
x = 0.875
x = 0.9
*
*
*
*
213
Figure 3 XRD patterns for samples in the (Y1-xLax)2Ti2O7 system. The pyrochlore (222) reflection is marked ‘*’ Selected monoclinic peaks have been labelled with their Miller indices.

Mat Res Soc Proc 932: 631-638
As a result of these two factors, the transition from a two phase system to a monoclinic solid solution is readily observed by the disappearance of the sharp signal from the pyrochlore phase and also its spinning sidebands - this occurs at a composition of x = 0.9, see figure 5b. In contrast, the limit of the pyrochlore solid solution (and exsolution of the monoclinic phase), is not as easily discerned by this technique. The monoclinic phase is first apparent within the NMR spectra at a composition of x = 0.175, see figure 5a, but given the diffraction results, it is most likely to be present in several previous samples. The NMR spectra of the pyrochlore and monoclinic phases are discussed in more detail within the next section. It is interesting to note the apparent symmetry of the solid solution characteristics of the (Y1-xLax)2Ti2O7 system; the extent of the pyrochlore and monoclinic solid solutions are almost equal – this is very different from the behaviour we have previously observed in the Nd2(Zr1-xTix)2O7 system [5]. In that system the pyrochlore solid solution is far more extensive, whilst there is a very limited range of compositions across which a single monoclinic phase is stable.
Cation ordering in the (Y1-xLax)2Ti2O7 system
In terms of designing a suitable waste form with a crystalline structure, an understanding of how the radioactive nuclei will distribute themselves across the available crystallographic sites is essential. In the pyrochlore structure, there are two types of cation sites – one at (!,!,!), the A site (Wyckoff position 16d) and the other at (0,0,0), the B site (Wyckoff position 16c). There are 16 symmetrically equivalent positions of each cation site within the unit cell (Z = 8). GSAS refinements suggest that both Y3+ and La3+ prefer to occupy the A sites of the pyrochlore phase and that there is minimal disorder with the Ti4+ on the B site. Evaluating the cation disorder in the monoclinic phase from diffraction measurements is more complex, because there are 8 crystallographically distinct cation sites (4 ‘A’ type and 4 ‘B’ type), all in low symmetry positions within the structure. However the four ‘B’ sites in the monoclinic structure are all relatively small, octahedral positions (see figure 1b), whereas the ‘A’ sites are far more irregular and have coordination numbers ranging from 7-12. It therefore seems extremely likely that the small Ti4+ cations will display a strong preference for the ‘B’ sites, whilst the larger Y3+ and La3+ cations favour the ‘A’ sites. In the discussion of NMR data that follows, we assume that Y and La occur only on the A sites in both phases. Were it the case that some disorder of Y across both sites was occurring in pyrochlore, an additional peak
Figure 4 Backscattered SEM micrographs of samples in the (Y1-xLax)2Ti2O7 system. (a) x = 0.025 (b) x = 0.15 (c) x = 0.975. Scale bars of the main images are 20 µm long ($300 magnification). Magnification of inset in (c) is $3300.
1c:
(a) (b) (c
)

Mat Res Soc Proc 932: 631-638
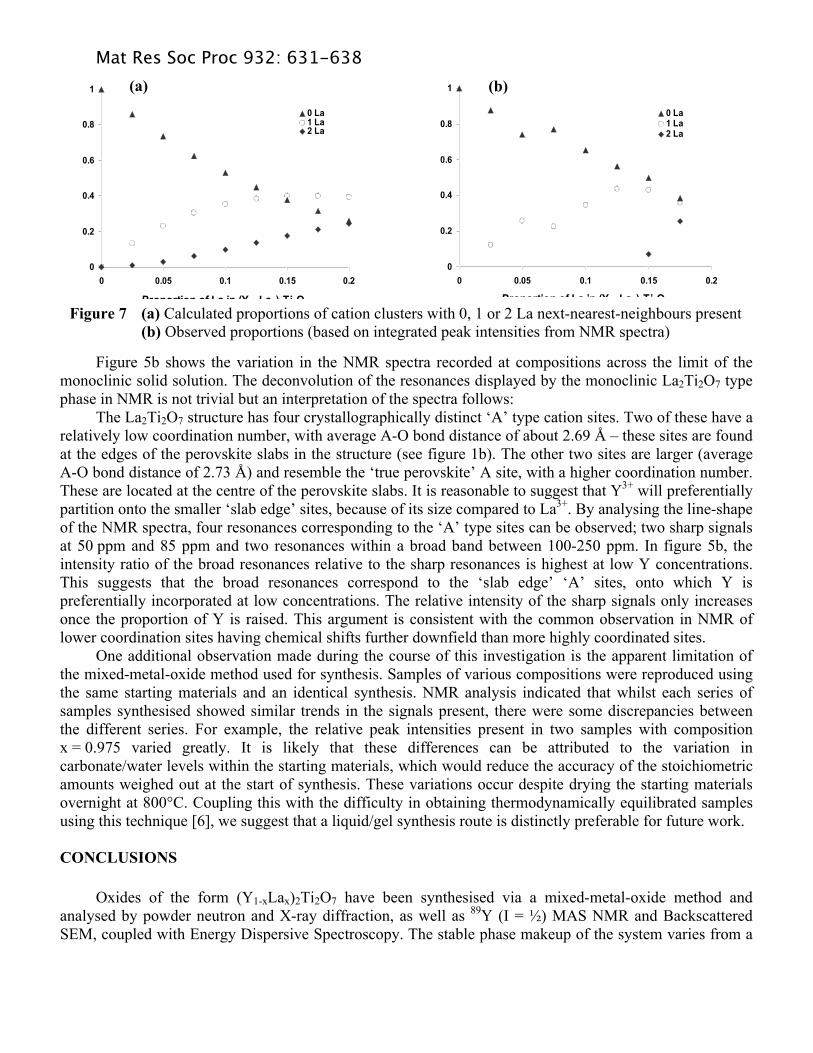
corresponding to resonance of Y in a 6-coordinate environment would also be expected within the spectra, at a value further downfield than that observed for the 8-coordinate peak. This is not observed at any composition measured. Resonances corresponding to the different coordination environments would be expected to be far more separated than the multiple resonances observed in figure 5a upon the introduction of La. Figure 6a illustrates one way of viewing part of the pyrochlore structure, in terms of the nearest cation neighbours surrounding one A site. The Y2Ti2O7 end member shows a single, sharp resonance in the NMR spectra at 65 ppm (figure 5a), indicating that all A sites are equivalent – as would be expected when only one species is distributed across them. Initial additions of La to the system immediately result in the appearance of a second resonance at ~72 ppm. The intensity of this resonance relative to that of the original Y2Ti2O7 signal increases with successive additions of La, and when x reaches 0.125, a shoulder which appears to be a third resonance is observed at ~77 ppm. The intensity of this peak also rises as the La content is further increased. It is likely that these small changes in the chemical shift correspond to the placement of first one and then two La cations within the next-nearest-neighbour environment of a Y cation, see figures 6b and 6c. Figure 7a shows the statistical probability of finding zero, one or two La next-nearest-neighbours within a single ‘cluster’ around a Y atom as a function of composition. The integrated peak ratios measured from the NMR spectra are shown in figure 7b, and suggest that there is no significant deviation from this behaviour. In other words, the placement of a La cation on a specific site neither inhibits, nor encourages the placement of a second La cation within the same cluster. It is not possible to observe any additional resonance that might correspond to 3 La next-nearest-neighbours within a cluster; if it is present, it is masked by thermal noise and the onset of the monoclinic phase.
Figure 5 89Y MAS NMR spectra for samples in the (Y1-xLax)2Ti2O7 system. For ease of inspection, the pyrochlore resonance is sometimes marked ‘*’. (a) samples spanning the limit of the pyrochlore solid solution (b) samples spanning the limit of the monoclinic solid solution.
(a) (b)
Figure 6 Part of the pyrochlore structure showing (a) no La, (b) 1 La and (c) 2 La next-nearest-neighbour A cations around a central Y cation. Large dark spheres are oxygen anions, small light spheres are Ti cations on B sites and medium black spheres are Y cations. La cations have been labelled.
(a) (b) (c)
Mat Res Soc Proc 932: 631-638
Figure 5b shows the variation in the NMR spectra recorded at compositions across the limit of the monoclinic solid solution. The deconvolution of the resonances displayed by the monoclinic La2Ti2O7 type phase in NMR is not trivial but an interpretation of the spectra follows: The La2Ti2O7 structure has four crystallographically distinct ‘A’ type cation sites. Two of these have a relatively low coordination number, with average A-O bond distance of about 2.69 Å – these sites are found at the edges of the perovskite slabs in the structure (see figure 1b). The other two sites are larger (average A-O bond distance of 2.73 Å) and resemble the ‘true perovskite’ A site, with a higher coordination number. These are located at the centre of the perovskite slabs. It is reasonable to suggest that Y3+ will preferentially partition onto the smaller ‘slab edge’ sites, because of its size compared to La3+. By analysing the line-shape of the NMR spectra, four resonances corresponding to the ‘A’ type sites can be observed; two sharp signals at 50 ppm and 85 ppm and two resonances within a broad band between 100-250 ppm. In figure 5b, the intensity ratio of the broad resonances relative to the sharp resonances is highest at low Y concentrations. This suggests that the broad resonances correspond to the ‘slab edge’ ‘A’ sites, onto which Y is preferentially incorporated at low concentrations. The relative intensity of the sharp signals only increases once the proportion of Y is raised. This argument is consistent with the common observation in NMR of lower coordination sites having chemical shifts further downfield than more highly coordinated sites. One additional observation made during the course of this investigation is the apparent limitation of the mixed-metal-oxide method used for synthesis. Samples of various compositions were reproduced using the same starting materials and an identical synthesis. NMR analysis indicated that whilst each series of samples synthesised showed similar trends in the signals present, there were some discrepancies between the different series. For example, the relative peak intensities present in two samples with composition x = 0.975 varied greatly. It is likely that these differences can be attributed to the variation in carbonate/water levels within the starting materials, which would reduce the accuracy of the stoichiometric amounts weighed out at the start of synthesis. These variations occur despite drying the starting materials overnight at 800°C. Coupling this with the difficulty in obtaining thermodynamically equilibrated samples using this technique [6], we suggest that a liquid/gel synthesis route is distinctly preferable for future work. CONCLUSIONS
Oxides of the form (Y1-xLax)2Ti2O7 have been synthesised via a mixed-metal-oxide method and analysed by powder neutron and X-ray diffraction, as well as 89Y (I = !) MAS NMR and Backscattered SEM, coupled with Energy Dispersive Spectroscopy. The stable phase makeup of the system varies from a
0
0.2
0.4
0.6
0.8
1
0 0.05 0.1 0.15 0.2
Proportion of La in (Y1-xLax)2Ti2O7
0 La1 La2 La
0
0.2
0.4
0.6
0.8
1
0 0.05 0.1 0.15 0.2
Proportion of La in (Y1-xLax)2Ti2O7
0 La1 La2 La
Figure 7 (a) Calculated proportions of cation clusters with 0, 1 or 2 La next-nearest-neighbours present (b) Observed proportions (based on integrated peak intensities from NMR spectra)
(a) (b)

Mat Res Soc Proc 932: 631-638
single Y2Ti2O7 pyrochlore type solid solution between x = 0 and x = 0.132 (determined by neutron diffraction), to a single La2Ti2O7 type solid solution with a monoclinic structure between x = 0.832 (neutron diffraction) or x = 0.9 (from XRD and NMR) and x = 1. A two-phase intergrowth of the two solid solutions exists between these limits. The lattice parameters of the single phase samples vary linearly with composition, in accordance with Vegard’s law. NMR spectra around the limit of the Y rich pyrochlore solid solution suggest that incorporation of La onto the pyrochlore A cation site occurs in a statistically random manner, where one La cation has no effect on the positioning of other La cations around it in the structure. The La rich monoclinic solid solution is characterised in the NMR spectra by the presence of four resonances, two sharp signals and two broader signals within a broad band. These have been attributed to the two types of ‘A’ type cation sites present in this structure – the broad resonances to the ‘slab edge’ sites, and the sharp signals to the sites at the centre of perovskite slabs of TiO6 octahedra. The relative intensities of these signals suggest that Y preferentially occupies the ‘slab edge’ sites in the monoclinic structure. The selection of suitable materials for the immobilisation of HLW is a pressing issue. This work expands our understanding of systems containing pyrochlore – a highly robust candidate crystalline phase for the immobilisation of actinide species and builds on our knowledge of the compositional effects on solid solution and cation partitioning within this and related crystalline structures. ACKNOWLEDGEMENTS
The authors gratefully acknowledge support from the Engineering and Physical Sciences Research Council (EPSRC) British Nuclear Fuel Ltd. (BNFL) and the Cambridge Massachusetts Institute (CMI) as well as the Royal Society for the award of a Dorothy Hodgkin Research Fellowship. We are also grateful to the Council for the Central Laboratory of the Research Councils (CCLRC) for the award of neutron beam time at ISIS, Rutherford Appleton Laboratories, and to Ron Smith on POLARIS for invaluable assistance during the collection of neutron data. REFERENCES
1 A. E. Ringwood, Safe disposal of high level nuclear reactor wastes: A new strategy, (Australian National University Press, Canberra), 1978. 2 G. R. Lumpkin, “Alpha-decay damage and aqueous durability of actinide host phases in natural systems,” J. Nucl. Mater., 289, 136 (2001). 3 H. M. Rietveld, “A profile refinement method for nuclear and magnetic structures”, J. Appl.
Crystallogr., 2, 65 (1969). 4 R. D. Shannon, “Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides,” Acta Crystallogr. A 32, 751 (1976). 5 E. J. Harvey, K. R. Whittle, G. R. Lumpkin, R. I. Smith, S. A. T. Redfern, “Solid solubilities of (La,Nd)2(Zr,Ti)2O7 phases deduced by neutron diffraction,” J. Solid State Chem., 178, 800 (2005).
6 Y. Liu, R. L. Withers, L. N. Norén, “The pyrochlore to ‘defect fluorite’ transition in the Y2(ZryTi1-y)2O7 system and its underlying crystal chemistry,” J. Solid State Chem., 177, 4404 (2004).