c.!titus!brown! [email protected]! -...
TRANSCRIPT

ì Metagenome assembly – part II C. Titus Brown [email protected]

Warnings
This talk contains forward looking statements. These forward-‐looking statements can be iden@fied by terminology such as
“will”, “expects”, and “believes”.
-‐-‐ Safe Harbor provisions of the
U.S. Private Securi@es Li@ga@on Act
“Making predic@ons is difficult, especially if
they’re about the future.”
-‐-‐ AOributed to Niels Bohr

The computational conundrum
More data => beOer.
and
More data => computa@onally more challenging.

Reads vs edges (memory) in de Bruijn graphs
Conway T C , Bromage A J Bioinformatics 2011;27:479-486
© The Author 2011. Published by Oxford University Press. All rights reserved. For Permissions, please email: [email protected]

2. Big data sets require big machines
For even rela@vely small data sets, metagenomic assemblers scale poorly.
Memory usage ~ “real” varia@on + number of errors
Number of errors ~ size of data set
Size of data set == big!!
(Es@mated 6 weeks x 3 TB of RAM to do 300gb soil sample, with a slightly modified conven@onal assembler.)
GCGTCAGGTAGGAGACCGCCGCCATGGCGACGATG
GCGTCAGGTAGGAGACCACCGTCATGGCGACGATG
GCGTCAGGTAGCAGACCACCGCCATGGCGACGATG
GCGTTAGGTAGGAGACCACCGCCATGGCGACGATG

Soil is full of uncultured microbes
Randy Jackson

SAMPLING LOCATIONS

Great Prairie sampling design
Reference core
Soil cores: 1 inch diameter 4 inches deep (litter and roots removed)
• Spatial samples: 16S rRNA, nifH
• Reference sample sequenced (small unmixed sample)
Reference bulk soil: stored for additional “omics” and metadata
10 M
10 M
1 M
1 M
1 cM
1 cM

0
200
400
600
800
1000
1200
1400
1600
1800
2000
100 600 1100 1600 2100 2600 3100 3600 4100 4600 5100 5600 6100 6600 7100 7600 8100
Iowa Corn
Iowa_Na@ve_Prairie
Kansas Corn
Kansas_Na@ve_Prairie
Wisconsin Corn
Wisconsin Na@ve Prairie
Wisconsin Restored Prairie
Wisconsin Switchgrass
Number of Sequences
Num
ber o
f OTU
s
Soil contains thousands to millions of species (“Collector’s curves” of ~species)

The set of questions for soil -‐-‐ discovery
ì What’s there?
ì Is it really that complex a community?
ì How “deep” do we need to sequence to sample thoroughly and systema@cally?
ì What organisms and gene func@ons are present, including non-‐canonical carbon and nitrogen cycling pathways?
ì What kind of organismal and func@onal overlap is there between different sites? (Total sampling needed?)
ì How is ecological complexity created & maintained?
ì How does ecological complexity respond to perturba@on?

Why are we applying short-‐read sequencing to this problem!?
ì Short-‐read sampling is deep and quan(ta(ve. ì Sta@s@cal argument: your ability to observe rare
organisms – your sensi@vity of measurement – is directly related to the number of independent sequences you take.
ì Longer reads (PacBio, 454, Ion Torrent) are less informa@ve.
ì Majority of metagenome studies going forward will make use of Illumina.
ì BUT this kind of sequence is challenging to analyze.
ì BUT, BUT this kind of sequence is necessary for high complexity environments.

Challenges of short-‐read analysis
ì Low signal for func@onal analysis; no linkage at all.
ì High error rates.
ì Massive volume.
ì Rapidly changing technology.
ì Several approaches but we have seOled on assembly.

Our “Grand Challenge” dataset
0"
100"
200"
300"
400"
500"
600"
Iowa,"Continuous"corn"
Iowa,"Native"Prairie"
Kansas,"Cultivated"corn"
Kansas,"Native"Prairie"
Wisconsin,"Continuous"corn""
Wisconsin,"Native"Prairie"
Wisconsin,"Restored"Prairie"
Wisconsin,"Switchgrass"
Basepairs(of(Sequencing((Gbp)(
GAII" HiSeq"
Rumen"(Hess"et."al,"2011),"268"Gbp
MetaHIT"(Qin"et."al,"2011),"578"Gbp
NCBI"nr"database,""37"Gbp
Total:""1,846"Gbp"soil"metagenome
Rumen"KSmer"Filtered,"111"Gbp

Approach 1: Partitioning
Split reads into “bins” belonging to different source species.
Can do this based almost en@rely on connec@vity of sequences.

Partitioning for scaling
ì Can be done in ~10x less memory than assembly.
ì Par@@on at low k and assemble exactly at any higher k (DBG).
ì Par@@ons can then be assembled independently ì Mul@ple processors -‐> scaling ì Mul@ple k, coverage -‐> improved assembly ì Mul@ple assembly packages (tailored to high varia@on, etc.)
ì Can eliminate small par@@ons/con@gs in the par@@oning phase.
ì An incredibly convenient approach enabling divide & conquer approaches across the board.

Technical challenges met (and defeated)
ì Novel data structure proper@es elucidated via percola@on theory analysis (Pell et al., PNAS, 2012)
ì Exhaus@ve in-‐memory traversal of graphs containing 5-‐15 billion nodes.
ì Sequencing technology introduces false connec@ons in graph (Howe et al., in prep.)
ì Only 20x improvement in assembly scaling L.

Approach 2: Digital normalization
Species A
Species B
Ratio 10:1Unnecessary data
81%
Suppose you have a dilu@on factor of A (10) to B(1). To get 10x of B you need to get 100x of A!
Overkill!!
This 100x will consume disk space and, because of
errors, memory.
(NOVEL)

Digital normalization discards redundant reads prior to assembly.
This removes reads and decreases data size, eliminates errors from removed reads, and normalizes coverage across loci.

Digital normalization algorithm
!
!
for read in dataset:!
!if median_kmer_count(read) < CUTOFF:!
! !update_kmer_counts(read)!
! !save(read)!
!else:!
! !# discard read!
!
! !! Note, single pass; fixed memory.

Downsample based on de Bruijn graph structure (which can be derived online)
A. B.

Shotgun data is often (1) high coverage and (2) biased in coverage.
(MD amplified)

Digital normalization fixes all that.
Normalizes coverage Discards redundancy Eliminates majority of errors Scales assembly drama@cally. Assembly is 98% iden@cal.

Digital normalization retains information, while discarding data and errors

Other key points
ì Virtually iden@cal con9g assembly; scaffolding works but is not yet cookie-‐cuOer.
ì Digital normaliza@on changes the way de Bruijn graph assembly scales from the size of your data set to the size of the source sample.
ì Always lower memory than assembly: we never collect most erroneous k-‐mers.
ì Digital normaliza@on can be done once – and then assembly parameter explora@on can be done.

Quotable quotes.
Comment: “This looks like a great soluHon for people who can’t afford real computers”.
OK, but:
“Buying ever bigger computers is a great soluHon for people who don’t want to think hard.”
To be less snide: both kinds of scaling are needed, of course.

Why use diginorm?
ì Use the cloud to assemble any microbial genomes incl. single-‐cell, many eukaryo@c genomes, most mRNAseq, and many metagenomes.
ì Seems to provide leverage on addressing many biological or sample prep problems (single-‐cell & genome amplifica@on MDA; metagenome; heterozygosity).
ì And, well, the general idea of locus specific graph analysis solves lots of things…

Some interim concluding thoughts
ì Digital normaliza@on-‐like approaches provide a path to solving the majority of assembly scaling problems, and will enable assembly on current cloud compu@ng hardware. ì This is not true for highly diverse metagenome environments… ì For soil, we es@mate that we need 50 Tbp / gram soil. Sigh.
ì Biologists and bioinforma@cians hate: ì Throwing away data ì Caveats in bioinforma@cs papers (which reviewers like, note)
ì Digital normaliza@on also discards abundance informa@on.

Evaluating sensitivity & specificity
Digital normalization+ other filters
Partitioning Velvetk from 19-51
minimus2merge
E. coli @ 10x + soil
98.5% of E. coli

Example
Dethlefsen shotgun data set / Relman lab 251 m reads / 16gb FASTQ gzipped ~ 24 hrs, < 32 gb of RAM for full pipeline -‐-‐ $24 on Amazon EC2
(reads => final assembly + mapping)
Assembly stats: 58,224 con@gs > 1000 bp (average 3kb)
summing to 190 mb genomic ~38 microbial genomes worth of DNA
~65% of reads mapped back to assembly

What do we get for soil?
Total Assembly Total ConHgs % Reads
Assembled
Predicted protein coding
rplb genes
2.5 bill 4.5 mill 19% 5.3 mill
391
3.5 bill
5.9 mill
22% 6.8 mill
466
This es@mates number of species ^
Adina Howe
Pu|ng it in perspec@ve: Total equivalent of ~1200 bacterial genomes Human genome ~3 billion bp

Coverage of Assemblies
Corn Prairie

Nearest reference in NCBI
Most abundant con@gs in Iowa corn metagenome: Unknown; alpha/beta hydrolase (Streptomyces sp. S4); unknown; unknown; hypothe@cal protein HMP (Clostridium clostridioforme) Most abundant con@gs in Iowa prairie metagenome: hypothe@cal protein (Rhodanobacter sp. 2APBS1); hypothe@cal protein (Oryza sa@va Japonica); outer membrane adhesin like proteiin (Solitalea canadensis) ; alcohol dehydrogenase zinc-‐binding domain protein (Ktedonobacter racemifer); alcohol dehydrogenase GroES domain protein (Ktedonobacter racemifer)

(Done with MEGAN)

How many soil samples do we need to sequence??
(Cumula@ve) Adina Howe
Overlap between Iowa prairie & Iowa corn is significant!

Extracting whole genomes?
So far, we have only assembled con9gs, but not whole genomes.
Can en@re genomes be assembled from metagenomic data? Iverson et al. (2012), from
the Armbrust lab, contains a technique for scaffolding metagenome con@gs into ~whole genomes. YES.

Perspective: the coming infopocalypse
ì Assembling about $20k worth of data, we can generate approximately 700 microbial genomes worth of data. (This is only going to go up in yield/$$, note.)
ì Most of these assembled genomic con@gs
(and genes) do not belong to studied
organisms.
ì What the heck do they do??

More thoughts on assembly
ì Illumina is the only game in town for sequencing complex microbial popula@ons, but dealing with the data (volume, errors) is tricky. This problem is being solved, by us and others.
ì We’re working to make it as close to push buOon as possible, with objec@vely argued parameters and tools, and methods for evalua@ng new tools and sequencing types.
ì The community is working on dealing with data downstream of sequencing & assembly. ì Most pipelines were built around 454 data – long reads, and
rela@vely few of them. ì With Illumina, we can get both long con9gs and quan9ta9ve
informa9on about their abundance. This necessitates changes to pipelines like MG-‐RAST and HUMANn.

The interpretation challenge
ì For soil, we have generated approximately 1200 bacterial genomes worth of assembled genomic DNA from two soil samples.
ì The vast majority of this genomic DNA contains unknown genes with largely unknown func@on.
ì Most annota@ons of gene func@on & interac@on are from a few phylogene@cally limited model organisms ì Est 98% of annota@ons are computa@onally inferred: transferred
from model organisms to genomic sequence, using homology. ì Can these annota@ons be transferred? (Probably not.)
This will be the biggest sequence analysis challenge of the next 50 years.

Concluding thoughts on “assembly”
ì We can handle all the data (modulo another year or so of engineering.) Bring it on!
ì Our approaches let us (& you) assemble preOy much anything, much more easily than before. (Single cell, microbial genomes, transcriptomes, eukaryo@c genomes, metagenomes, BAC sequencing…)
ì Seriously. No more problemo. Done. Finished. Kaput.
ì So now what? ì Valida@on. ì Interpreta@on and building general tools. ì Interpreta@on relies on annota9on… (Uh oh.)

What are future needs?
ì High-‐quality, medium+ throughput annota@on of genomes? ì Extrapola@ng from model organisms is both immensely
important and yet lacking. ì Strong phylogene@c sampling bias in exis@ng annota@ons.
ì Synthe@c biology for inves@ga@ng non-‐model organisms?
(Cleverness in experimental biology doesn’t scale L)
ì Integra@on of microbiology, community ecology/evolu@on modeling, and data analysis.

Replication fu
ì In December 2011, I met Wes McKinney on a train and he convinced me that I should look at IPython Notebook.
ì This is an interac9ve Web notebook for data analysis…
ì Hey, neat! We can use this for replica@on! ì All of our figures can be regenerated from scratch, on an EC2
instance, using a Makefile (data pipeline) and IPython Notebook (figure genera@on).
ì Everything is version controlled. ì Honestly not much work, and will be less the next @me.


So… how’d that go?
ì People who already cared thought it was ni}y.
hOp://ivory.idyll.org/blog/replica@on-‐i.html
ì Almost nobody else cares ;( ì Presub enquiry to editor: “Be sure that your paper can be reproduced.” Uh,
please read my leOer to the end? ì “Could you improve your Makefile? I want to reimplement diginorm in another
language and reuse your pipeline, but your Makefile is a mess.”
ì Incredibly useful, nonetheless. Already part of undergraduate and graduate training in my lab; helping us and others with next parpes; etc. etc. etc.
Life is way too short to waste on unnecessarily replicaHng your own workflows, much less other people’s.

Acknowledgements
Lab members involved Collaborators
ì Adina Howe (w/Tiedje) ì Jason Pell ì Arend Hintze ì Rosangela Canino-‐Koning ì Qingpeng Zhang ì Elijah Lowe ì Likit Preeyanon ì Jiarong Guo ì Tim Brom ì Kanchan Pavangadkar ì Eric McDonald
ì Jim Tiedje, MSU
ì Billie Swalla, UW
ì Janet Jansson, LBNL
ì Susannah Tringe, JGI
Funding
USDA NIFA; NSF IOS; BEACON.


ì Current research in my lab Solving the rest of your problems J
Preliminary func@onal analysis

Search SSU rRNA gene in Illumina data
1. Randomly sequencing about 100bp long DNA in microbial genomes;
2. Everything is sequenced;
3. Not limited by primers or PCR bias;
4. Data mining is the challenge;
10^3
10^6 10^7 10^4
Reads #
SSU rRNA Gene length
Expected SSU RNA gene fragments
Genome length

Classification: Pyrotag vs shotgun
RDP-‐pyrotag-‐SSU
silva-‐pyrotag-‐SSU
silva-‐shotgun-‐SSU

Primers used in 454 Titanium sequencing of SSU rRNA gene, using E.coli as an example. Consensus sequences of the primer region from Illumina reads suggest 1) searching method is good and 2)primer bias is minimal at the current E-‐value cutoff.
Start:907 End:1402
Forward
Reverse
1542 bp
Sequence logo of short reads at forward primer region:
AAACTYAAAKGAATTGACGG Current forward primer
GYACACACCGCCCGT Current reverse primer (reverse complement)
Sequence logo of short reads at reverse primer region:

CowRumen – JGI 16s primer mismatches
postion! A T C G Total 1G! 0.001 0.001 0.002 0.996 12154 2T! 0.002 0.983 0.003 0.012 12169 3G! 0.001 0.001 0.002 0.995 12166 4C! 0.001 0.001 0.996 0.002 12143 5C! 0.003 0.001 0.994 0.002 12183 6A! 0.986 0 0.008 0.005 12209 7G! 0.001 0.001 0.002 0.996 12189 8C! 0.001 0.001 0.996 0.002 12198 9A! 0.978 0.001 0.017 0.004 12230
10G! 0.001 0 0.002 0.997 12231 11C! 0.001 0.001 0.996 0.002 12198 12C! 0.002 0.001 0.994 0.003 12185 13G! 0 0 0.002 0.997 12190 14C! 0.001 0.001 0.995 0.003 12195 15G! 0.001 0.001 0 0.998 12213 16G! 0.001 0.001 0 0.998 12206 17T! 0.002 0.974 0.003 0.021 12171 18A! 0.99 0.001 0.006 0.003 12150 19A! 0.995 0.001 0.002 0.002 12106

Running HMMs over de Bruijn graphs (=> cross validation)
ì hmmgs: Assemble based on good-‐scoring HMM paths through the graph.
ì Independent of other assemblers; very sensi@ve, specific.
ì 95% of hmmgs rplB domains are present in our par@@oned assemblies.
Jordan Fish, Qiong Wang, and Jim Cole (RDP)

Streaming error correction.
Does read come from a high-
coverage locus?
Error-correct low-abundance k-mers in
read.
Add read to graph and save for later.
All reads Yes!
No!
First pass
Does read come from a now high-coverage locus?
Error-correct low-abundance k-mers in
read.
Leave unchanged.
Only saved reads
Yes!
No!
Second pass
We can do error trimming of genomic, MDA, transcriptomic, metagenomic data in < 2 passes, fixed memory.
We have just submiOed a proposal to adapt Euler or Quake-‐like error correc@on (e.g. spectral alignment problem) to this
framework.

Side note: error correction is the biggest “data” problem left in
sequencing.
A. B.
XXX
X
XX
X
X
XX
X
C.
XX
X X
XX
Both for mapping & assembly.

End:1402
Consensus of short reads at reverse primer region:
é Figure. Primers used in 454 Titanium sequencing of 16S rRNA gene, using E.coli as an example. Consensus sequences of the primer region from Illumina reads suggest primer bias is minimal at the current E-value cutoff.
Start:907 Forward
1542 bp
Consensus of short reads at forward primer region:
AAACTYAAAKGAATTGACGG Current forward primer

Supplemental: abundance filtering is very lossy.
0.0 20.0 40.0 60.0 80.0 100.0
Total
3.8x par@@on
8.2x par@@on
Largest par@@on
Percentage lost
Percent loss from abundance filtering (all >= 2)
con@gs
bp

11
Velvet Assembler
Coverage of
Unfiltered by
Filtered Assembly
Coverage of
Normalized Filtered by
Unfiltered Assembly
Coverage of
Reference Genes
by Unfiltered
Coverage of
Reference Genes by
Filtered
Unfiltered Assembly Statitics
(No. of Contigs/Assembly
Length(bp)/Max Contig Size (bp)
Normalized Assembly Statitics
(No. of Contigs/Assembly
Length(bp)/Max Contig Size (bp)
Unfiltered Assembly
Requirements (Memory,
GB / Time (hours)
Small Soil 74.5% 98.6% - - 25,470 / 16,269,879 / 118,753 17,636 / 10,578,908 / 13,246 5 / 4Medium Soil 75.4% 98.1% - - 113,613 / 81,660,678 / 57,856 79,654 / 54,424,264 / 23,663 18 / 21Large Soil 50.8% 86.3% - - 554,825 / 306,899,884 / 41,217 290,018 / 159,960,062 / 41,423 33 / 12*Rumen 75.1% 98.3% 17.2% 14.6% 92,044 / 74,813,072 / 182,003 72,705 / 49,518,627 / 34,683 11 / 14Human Gut 79.5% 88.5% - - 543,331 / 234,686,983 / 85,596 203,299 / 181,934,800 / 145,740 76 / 8*Simulated 84.6% 98.3% 4.5% 3.9% 11,204 / 6,506,248 / 5,151 9,859 / 5,463,067 / 6,605 < 1 / < 1
!Meta-IDBA Assembler
Coverage of
Unfiltered by
Filtered Assembly
Coverage of
Normalized Filtered by
Unfiltered Assembly
Coverage of
Reference Genes
by Unfiltered
Coverage of
Reference Genes by
Filtered
Unfiltered Assembly Statitics
(No. of Contigs/Assembly
Length(bp)/Max Contig Size (bp)
Normalized Assembly Statitics
(No. of Contigs/Assembly
Length(bp)/Max Contig Size (bp)
Unfiltered Assembly
Requirements (Memory,
GB / Time (hours)
Small Soil 75.6% 93.9% - - 15,739 / 9,133,564 / 37,738 12,513 / 7,012,036 / 17,048 < 1 / < 1Medium Soil 67.5% 94.5% - - 76,269 / 45,844,975 / 37,738 52,978 / 30,040,031 / 18,882 2 / 2Large Soil N/A N/A - - N/A 395,122 / 228,857,098 / 37,738 > 116 / incompleteRumen 70.4% 94.4% 15.5% 13.0% 60,330 / 47,984,619 / 54,407 48,940 / 33,276,502 / 22,083 12 / 3Human Gut 74.0% 96.5% - - 173,432 / 211,067,996 / 106,503 132,614 / 142,139,101 / 85,539 58 / 15Simulated 86.5% 93.4% 3.8% 3.5% 8,707 / 4,698,575 / 5,113 7,726 / 4,078,947 / 3,845 < 1 / <1
SOAPdenovo Assembler
Coverage of
Unfiltered by
Filtered Assembly
Coverage of
Normalized Filtered by
Unfiltered Assembly
Coverage of
Reference Genes
by Unfiltered
Coverage of
Reference Genes by
Filtered
Unfiltered Assembly Statitics
(No. of Contigs/Assembly
Length(bp)/Max Contig Size (bp)
Normalized Assembly Statitics
(No. of Contigs/Assembly
Length(bp)/Max Contig Size (bp)
Unfiltered Assembly
Requirements (Memory,
GB / Time (hours)
Small Soil 86.6% 95.8% - - 14,275 / 7,100,052 / 37,720 12,801 / 6,343,110 / 13,246 3 / < 1Medium Soil 82.2% 95.7% - - 66,640 / 33,321,411 / 28,695 56,023 / 27,880,293 / 15,721 10 / < 1Large Soil 78.7% 94.2% - - 412,059 / 215,614,765 / 32,514 334,319 / 171,718,154 / 41,423 48 / 11Rumen 84.7% 97.3% 14.7% 13.4% 62,896 / 40,792,029 / 22,875 55,975 / 34,540,861 / 19,044 5 / < 1Human Gut 84.9% 98.5% - - 190,963 / 171,502,574 / 57,803 161,795 / 139,686,630 / 56,034 35 / 5Simulated 93.2% 96.1% 2.5% 2.4% 6,322 / 2,940,509 / 3,786 6,029 / 2,821,631 / 3,764 < 1 / < 1
Table 2. Comparison of unfiltered and filtered assemblies of various metagenome lumps using Velvet,SOAPdenovo, and Meta-IDBA assemblers. Assemblies were aligned to each other, and coverage wasestimated (columns 1-2). Simulated and rumen assemblies were aligned to available referencegenes/genomes (columns 3-4). Total number of contigs, assembly length, and maximum contig size wasestimated for each assembly, as well as memory and time requirements of unfiltered assembly (columnes5-7). Filtered assemblies required less than 2 GB of memory. Velvet assemblies of the unfiltered humangut and large soil datasets (marked as *) could only be completed with K=33 due to computationallimitations. The Meta-IDBA assembly of the large soil metagenome could not be completed in less than100 GB.
Number of Hits to Unique Genes in 112 Reference Genomes
ABC transporter-like protein 306Methyl-accepting chemotaxis sensory transducer 210ABC transporter 173Elongation factor Tu 94Chemotaxis sensory transducer 51ABC transporter ATP-binding protein 44Diguanylate cyclase/phosphodiesterase 36ATPase 36S-adenosyl-L-homocysteine hydrolase 36Adensylhomocysteine and downstream NAD binding 36Ketol-acid reductoisomerase 34S-adenosylmethionine synthetase 34Elongation factor G 34ABC transporter ATPase 33
Table 3. Annotation of highly-connecting sequences from the simulated metagenome with most hits toconserved genes within the 112 reference genomes [8].
Comparing assemblers
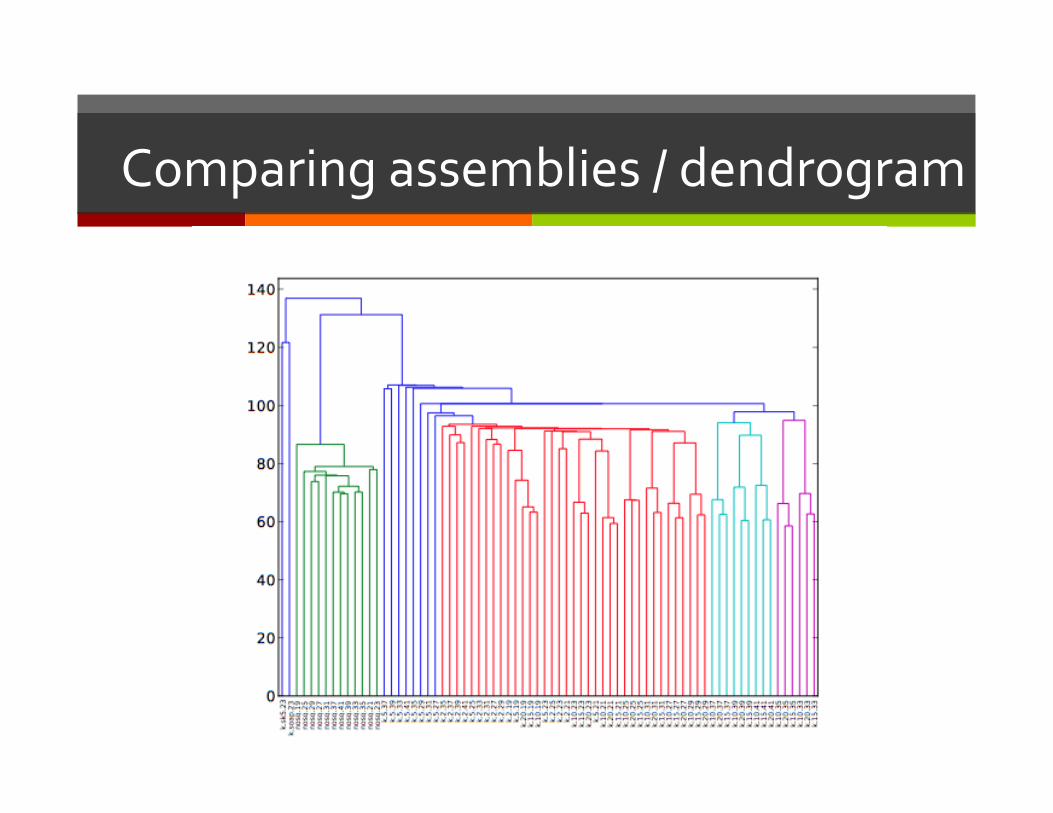
Comparing assemblies / dendrogram

Integrating modeling into data analysis?