development of a single-plasmid-based regulatable gene ... · the plasmid is based on a pbsv2...
TRANSCRIPT

APPLIED AND ENVIRONMENTAL MICROBIOLOGY, Oct. 2009, p. 6553–6558 Vol. 75, No. 200099-2240/09/$08.00�0 doi:10.1128/AEM.02825-08Copyright © 2009, American Society for Microbiology. All Rights Reserved.
Development of a Single-Plasmid-Based Regulatable Gene ExpressionSystem for Borrelia burgdorferi�
Christine R. Whetstine,† Joyce G. Slusser, and Wolfram R. Zuckert*Department of Microbiology, Molecular Genetics and Immunology, University of Kansas Medical Center,
Kansas City, Kansas 66160
Received 12 December 2008/Accepted 14 August 2009
We developed a single-plasmid-based regulatable protein expression system for Borrelia burgdorferi. Expres-sion of a target gene is driven by Post, a hybrid B. burgdorferi ospA-tetO promoter, from a recombinant B.burgdorferi plasmid constitutively expressing TetR. The system was tested using the green fluorescent protein(GFP) as a reporter. Under noninducing conditions, recombinant B. burgdorferi cells were nonfluorescent, noGFP protein was detected, and residual, small amounts of transcript were detectable only by reverse tran-scription-PCR but not by Northern blot hybridization. Upon induction with anhydrotetracycline, increasinglevels of GFP transcript, protein, and fluorescence were observed. This tight and titratable promoter systemwill be invaluable for the study of essential borrelial proteins. Since target protein, operator, and repressor arecarried by a single plasmid, the system’s application is independent of a particular strain background.
Since its first description in 1982 (6), the Lyme diseasespirochete Borrelia burgdorferi has become a model organ-ism for its phylum for studies on bacterial virulence mech-anisms, membrane structure, physiology, and metabolism.An increasing number of molecular tools have been devel-oped (reviewed in reference 23), including fluorescent fu-sion proteins for use in both cytoplasmic and extracytoplas-mic compartments (10, 28).
On the basis of studies determining a minimal bacterialgenome (14, 21), about 400 or more of the 1,813 genes en-coded by the B. burgdorferi type strain B31 (11, 12) can beexpected to be essential for growth on its rich Barbour-Stoen-ner-Kelly II (BSK-II) laboratory medium (2, 37). One cur-rently available but indirect approach to test the indispensabil-ity of a B. burgdorferi gene is to attempt its disruption by anantibiotic resistance cassette; essential genes can be knockedout only if the cell is supplied with an additional functionalcopy in trans on a recombinant plasmid (7, 17). A more directand elegant approach uses a tightly regulatable promoter con-struct either to deplete the wild-type protein or to selectivelyoverexpress a dominant negative mutant. Environmental generegulation by, e.g., temperature or pH is counter-indicated dueto its pleiotropic effects (9, 32). Escherichia coli lac operon-based expression systems responding to induction by isopropyl�-D-1-thiogalactopyranoside have been adapted for use in B.burgdorferi by either optimizing the codon usage of the lacrepressor gene lacI (4) or generating a lac operator-modifiedB. burgdorferi flaB promoter in a specific, engineered strainbackground expressing LacI (13).
Tetracycline-regulated expression is similarly well character-
ized on the molecular level and provides tight control andsensitive induction by compounds that diffuse passively acrossbiological membranes without the requirement of uptake pro-teins. In addition, tetracycline has been used in eukaryotes aswell as in both gram-positive and -negative bacteria (20, 29, 34,36). A typical regulatory setup consists of a constitutively ex-pressed tet repressor, TetR, binding to the tet operator, tetO,within the tet promoter, tetP. Binding of tetracycline to TetRreleases the repressor, leading to expression of the reporterprotein from tetP. Two modifications of this system make itparticularly versatile: (i) the use of the tetracycline analoganhydrotetracycline (ATc), which has a 30-fold higher affinityfor TetR and lower antibiotic activity (15) and thus allows forits use in tetracycline-sensitive bacteria and for transient in-duction of bacterial genes during in vivo experiments (19, 35);and (ii) the additional experimental flexibility provided by theuse of revTetR, a TetR G96E L205S mutant, which binds ATcnot as an inducer but as a corepressor (18). Here, we describethe successful adaptation and evaluation of a tet promotersystem in B. burgdorferi. It allows for a single-plasmid-based,tight, and titratable expression of a specific target gene.
MATERIALS AND METHODS
Strains and culture conditions. E. coli strains TOP10 (Invitrogen) or XL-10Gold (Stratagene) were used for recombinant plasmid construction and main-tenance and were grown in selective Luria-Bertani broth or on solid plates (26).B. burgdorferi B31-e2 (1) and B313 (24) are clones of type strain B31 (ATCC35210). B313 lacks the ospA-harboring 54-kb linear plasmid lp54 (25, 39). B.burgdorferi cells were transformed by electroporation using 10 �g of plasmidDNA following established protocols (27, 33). Transformants were selected insolid BSK-II medium containing 200 �g/ml kanamycin and expanded in selectiveliquid BSK-II medium at 34°C in a 5% CO2 atmosphere (2, 37). For induction ofexpression, anhydrotetracycline hydrochloride ([ATc] IBA GmbH, Gottingen,Germany) was added at final concentrations of 2 ng/ml to 2 �g/ml to liquid B.burgdorferi cultures with a cell density of 1 � 107 cells/ml.
Recombinant plasmids. All plasmids generated and used in this study arederivatives of pBSV2 (33), which replicates autonomously in both E. coli and B.burgdorferi and confers resistance to kanamycin. Plasmids used as target DNA forsequence overlap extension PCR (SOE-PCR) (16) were the following: (i) pASK-IBA14 (IBA GmBH, Gottingen, Germany) with the E. coli tetPO tetracyclinepromoter and operator region, as well as the tetR repressor gene; (ii)
* Corresponding author. Mailing address: Department of Microbi-ology, Molecular Genetics and Immunology, University of KansasMedical Center, Mail Stop 3019, 3025 Wahl Hall West, 3901 RainbowBoulevard, Kansas City, KS 66160-7420. Phone: (913) 588-7061. Fax:(913) 588-7295. E-mail: [email protected].
† Present address: Stowers Institute for Medical Research, 1000 E.50th Street, Kansas City, MO 64110.
� Published ahead of print on 21 August 2009.
6553
on August 13, 2019 by guest
http://aem.asm
.org/D
ownloaded from

pBSV�(ospAp-gfp) (10) with the outer surface protein A (ospA) promoter PospA
driving green fluorescent protein (cycle 3 [c3] GFP) expression; and (iii) pBSV2harboring the flaB promoter PflaB. SOE-PCR was carried out using either Plat-inum Taq or Platinum Pfx polymerase (Invitrogen) using the primers listed inTable 1. Site-directed mutagenesis was carried out using a QuikChange kit(Stratagene).
pCRW50 carries fusions of tetPO to the c3 GFP gene and PflaB to tetR. tetPOwas amplified from pASK-IBA14 using primer pair KpntetPO-fwd and tetPO_gfp-rev. The c3 GFP gene was amplified from pBSVphi(ospAp-gfp) using theprimer pair tetPO_gfp-fwd and gfp_flaBP-rev. The overlapping tetPO and c3GFP gene amplicons were fused using the flanking primer pair KpntetPO-fwdand gfp_flaBP-rev. PflaB was amplified from pBSV2.vsp1 (38) using the primerpair gfp_flaBP-fwd and flaBP_tetR-rev. tetR was amplified from pASK-IBA14using the primer pair flaBP_tetR-fwd and KpntetR-rev. PflaB and tetR were fusedtogether by SOE-PCR using the flanking primers gfp_flaBP-fwd and KpntetR-rev. In a final SOE-PCR, the tetPO-c3 GFP gene-PflaB-tetR cassette was fused by
using the primer pair KpntetPO-fwd and KpntetR-rev and ligated into pCR2.1-TOPO (Invitrogen). The cassette was then excised with KpnI and ligated intopBSV2.
pCRW53 carries a tetO-modified PospA (Post) driving expression of the c3 GFPgene (Fig. 1). The Post promoter fragment was amplified from pBSV�(ospAp-gfp) with the primer pair SacPospA-fwd and ospAtetO-rev. The c3 GFP gene wasamplified from pBSV�(ospAp-gfp) using the primer pair ospAtetO-fwd andSacgfp-rev. The Post-c3 GFP gene fusion was obtained by SOE-PCR with flank-ing primers SacPospA-fwd and Sacgfp-rev. The resulting amplicon was digestedwith SacI and ligated with an SacI-cut pCRW50 plasmid backbone fragment.
Epifluorescence microscopy and flow cytometry. Cultured B. burgdorferi cellswere harvested, washed once, and resuspended in phosphate-buffered salinecontaining 5 mM MgCl2. For an initial qualitative analysis, cells were observedunder epifluorescence using a Nikon Eclipse E600 microscope fitted with a V-2Aand fluorescein isothiocyanate-HYQ filter block and a Retiga EXi camera(QImaging). For a quantitative analysis, 200 �l of cells at approximately 5 � 107
TABLE 1. Oligonucleotides used in this study
Name Target Sequence (5� to 3�)a
KpntetPO-fwd 5� end of tetPO GGTACCATCGAATGGCCAGATGATTAATTCtetPO_gfp-rev 3� end of tetPO CAGAATTGCCCTTTCATTTTTTGCCCTCGTTATCtetPO_gfp-fwd 5� end of gfp GATAACGAGGGCAAAAAATGAAAGGGCAATTCTGgfp_flaBP-rev 3� end of gfp CACAAGAGGCGACAGACATCATTATTTGTAGAGCTCATCgfp_flaBP-fwd 5� end of flaB promoter GAGCTCTACAAATAATGATGTCTGTCGCCTCTTGTGflaBP_tetR-rev 3� end of flaB promoter CTTTTATCTAAACGAGACATCATATGTCATTCCTCCATGflaBP_tetR-fwd 5� end of tetR CATGGAGGAATGACATATGATGTCTCGTTTAGATAAAAGKpntetR-rev 3� end of tetR GGTACCTCATTAAGACCCACTTTCACATTTAAGSacgfp-rev 3� end of gfp TTGTAGAGCTCATCCATGCCATGTGSacPospA-fwd 5� end of ospA promoter GAATTCGAGCTCAAGTCCCAAAACTGGGACospAtetO-rev 3� end of ospA promoter TATATTCTCCTTTTTCTCTATCACTGATAGGGACAAGTATAATT
ATATTATAAGATTAACospAtetO-fwd 5� end of gfp ATAATATAATTATACTTGTCCCTATCAGTGATAGAGAAAAAG
GAGAATATATTATGAAAGgfpRTPCR-fwd 5� internal of gfp TGGCCAACACTTGTCACTACTTTCgfpRTPCR-rev 3� internal of gfp AGCTCATCCATGCCATGTGTAATCpBRori-fwd pBR origin of replication GCGTAATCTGCTGCTTGC AAACpBRori-rev pBR origin of replication AAATCGACGCTCAAGTCAGAGG
a Restriction sites are italicized.
FIG. 1. Plasmid map of pCRW53. The plasmid is based on a pBSV2 plasmid backbone, replicates autonomously in E. coli (pBR origin ofreplication) and B. burgdorferi (paralogous gene families [PF] 57, 50, and 49 flanked by inverted repeats [IR]), and confers resistance to kanamycin(PflgB-Kanr cassette) (33). The hybrid Post promoter consists of the B. burgdorferi ospA promoter PospA and the 19-bp tet operator (tetO; gray box)inserted downstream of its �1 transcriptional start site, replacing 19 nucleotides of the 5� untranslated ospA mRNA shown above tetO. �1, �10,and �35 sequences of PospA are indicated in bold. The formyl-methionyl (fMet) ospA start codon is indicated in bold italics. The tetracyclinerepressor gene tetR is driven by the B. burgdorferi PflaB promoter.
6554 WHETSTINE ET AL. APPL. ENVIRON. MICROBIOL.
on August 13, 2019 by guest
http://aem.asm
.org/D
ownloaded from

cells/ml were subjected to flow cytometry at 100 lb/in2 using a 100-�m nozzle atthe slowest flow rate on a BD LSRII instrument equipped with FACSDiva,version 4.4, software (BD Biosciences). Gating was determined by plotting logforward scatter versus log side scatter, using running buffer alone to determinethe forward scatter threshold. Nonrecombinant B. burgdorferi cells were used todetermine background fluorescence. About 100,000 events were counted foreach sample. The FlowJo program suite, version 7.2.2 (Treestar), was used fordata analysis.
Protein gel electrophoresis and immunoblot analysis. Proteins were separatedby sodium dodecyl sulfate–12% polyacrylamide electrophoresis and visualized byCoomassie blue staining. For immunoblot analysis, proteins were electrophoreti-cally transferred to nitrocellulose membranes (Immobilon-NC; Millipore) usinga Transblot-SD Semi-Dry Transfer Cell (Bio-Rad) as described previously (39).Membranes were blocked and incubated with antibodies in 5% nonfat dry milk,20 mM Tris–500 mM NaCl, and 0.05% Tween 20 as described previously (38).Antibodies used were anti-TetR rabbit polyclonal antiserum (1:500; Abcam),anti-GFP rabbit polyclonal rabbit antiserum (1:3,000; Invitrogen), or monoclonalantibody against B. burgdorferi FlaB (1:10; H9724) (3). Secondary antibodieswere alkaline phosphatase-conjugated goat anti-mouse heavy and light chainimmunoglobulin G or mouse anti-rabbit immunoglobulin G (heavy chain)(Sigma). The alkaline phosphatase substrate for chemiluminescent detection wasCDP-Star (GE Healthcare). A Fujifilm LAS-4000 Luminescent Image Analyzerwas used for data acquisition and analysis.
RNA isolation and Northern blot analysis. Total RNA was isolated from 35-mlcultures using an RNeasy Mini Kit (Qiagen). Prior to isolation, the RNAs werefixed using an RNA Protect Kit (Qiagen). RNA concentrations were measuredusing a NanoDrop 1000 spectrophotometer (Thermo Scientific). Total RNA (1.0�g) was fractionated in a 1.2% formaldehyde-agarose gel and transferred to anImmobilon-NY� membrane (Millipore) by upward capillary transfer (26). RNAladders (0.5 to 10 kb or 0.24 to 9.5 kb; Invitrogen) served as size standards. ADNA probe was generated by PCR using the primer pair ospAtetO-fwd andSacgfp-rev (Table 1) and pBSV�(ospAp-gfp) as a template. Probe labeling andNorthern blot hybridizations were performed using the Gene Images AlkPhosDirect Labeling and Detection System with CDP-Star (GE Healthcare) accord-ing to the manufacturer’s instructions. A Fujifilm LAS-4000 Luminescent ImageAnalyzer was used for data acquisition and analysis.
RT-PCR. Isolated total RNA was treated with DNase I (Invitrogen). Reversetranscriptase PCR (RT-PCR) reaction mixtures consisted of 0.25 �g of totalDNA-free RNA and primers gfpRTPCR-fwd and gfpRTPCR-rev (Table 1)(predicted amplicon of 539 bp) with the GeneAmp EZ rTth RNA PCR kit(Applied Biosystems), according to the manufacturer’s instructions. Reactionmixture incubation conditions were 30 min at 55°C for reverse transcription,followed by 2 min at 94°C, 40 cycles of 1 min at 94°C and 1 min at 55°C, and 7min at 60°C. To check for DNA contamination, Mn(OAc)2 was omitted for thecontrol (without the RT step). To conserve rTth polymerase, alternative controlreactions were carried out with Taq polymerase (New England Biolabs) usingpBRori-fwd and pBRori-rev (Table 1) (predicted amplicon of 533 bp), twoprimers specific for an untranscribed region of pCRW53. Reaction mixtureincubation conditions were identical to those described above. A DNA ladder (1Kb Plus DNA ladder; Invitrogen) served as a size marker.
RESULTS AND DISCUSSION
Given the potential versatility of a tetPO- and TetR-basedexpression system, we decided to adapt it for use in B. burg-dorferi. First, we assessed whether B. burgdorferi was resistantto the ATc concentrations typically used for induction in apreliminary experiment. B. burgdorferi cells did not show anygrowth defect in the presence of the standard ATc concentra-tion for induction (0.2 �g/ml) or at ATc concentrations thatwere 100-fold higher. Next, we tested whether the tet promoteris active and regulatable by ATc in B. burgdorferi. E. coli and B.burgdorferi transformants carrying pCRW50 (tetPO-gfp-PflaB-tetR) were cultured in the presence or absence of ATc. Trans-formed E. coli cells were green fluorescent under both condi-tions, suggesting that the tet promoter was leaky due toinsufficient levels of TetR. Transformed B. burgdorferi cellswere nonfluorescent when grown in BSK-II medium withoutATc. In the presence of 0.2 �g/ml ATc, a very low level of
green fluorescence was observed by epifluorescence micros-copy using a Nikon V-2A filter (data not shown). The level offluorescence remained low in the presence of 2 and 20 �g/mlATc. This indicated that the tet promoter was active at a verylow level in B. burgdorferi and could be repressed by TetR. Thelow activity of nonborrelial promoters had been a major ob-stacle in the initial development of antibiotic markers (5, 31)and was therefore expected.
We hence set out to modify a strong B. burgdorferi promoterthat drives expression of the outer surface protein OspA(PospA). The �35/�10 spacer region in PospA contains 16 nucle-otides (30). It was therefore not entirely surprising that inser-tion of the 19-bp tetO sequence between the promoter’s �10and �35 sequence disrupted activity (data not shown). Replac-ing tetO in the construct with a truncated 16-bp sequence stillcontaining the palindromic sequence interacting with TetR(22) led to a leaky phenotype (data not shown). This suggestedthat the truncated tetO sequence did not bind TetR efficientlyunder noninducing conditions. Thus, we repositioned the op-erator in subsequent constructs.
In pCRW53, we replaced 19 residues downstream of thePospA �1 transcriptional start site with tetO (Fig. 1), resulting inthe hybrid Post promoter driving the expression of gfp. B. burg-dorferi cells carrying pCRW53 derived from both strain B31clones B31-e2 and B313 were brightly green fluorescent in thepresence of ATc yet nonfluorescent in the absence of theinducer. This suggested that the hybrid Post promoter, in com-bination with a constitutively expressed TetR repressor, pro-vided a tight and tetracycline-inducible B. burgdorferi expres-sion system. We therefore decided to evaluate its inductionproperties in more detail.
Northern blotting and RT-PCR were used to assess regula-tion on the transcriptional level. For Northern blot analysis(Fig. 2A and B), total RNA was probed with a PCR-generatedprobe specific for the gfp transcript. A significant signal abovebackground was detected in the RNA from cells grown in thepresence of 75 ng/ml ATc. Three major bands were detected bythe probe, approximately 0.8 kb, 1.2 kb, and 1.8 kb in size. The0.8-kb band corresponds to the predicted size of the gfp tran-script. The detected 1.8-kb band most likely represents a co-transcript of gfp and tetR (1.0 kb in size including the upstreamPflaB sequence) since both operons face the same direction andare not separated by a transcription terminator sequence. Theorigin of the intermediate 1.2-kb band is currently unclear.Interestingly, the gfp transcript levels upon Post induction re-mained significantly lower than those detected in RNA iso-lated from cells harboring pBSV�(ospAp-gfp) (10), where gfpis driven by the unmodified PospA.
Total DNase-treated RNA was used to assay transcriptionlevels by RT-PCR (Fig. 2C and D). Reactions omitting the RTreaction step or using DNA-specific primers did not yield am-plicons, demonstrating that the RNA preparation was free ofdetectable amounts of DNA. As expected from the Northernblot analysis, gfp transcripts were detected in cells grown atATc concentrations above 50 ng/ml. Most importantly, onlyresidual, low levels of transcript were detected under nonin-ducing conditions for both strain backgrounds. Together, thisindicated that the Post promoter is also regulated at the tran-scriptional level and silenced by TetR under noninducing con-ditions.
VOL. 75, 2009 BORRELIA HYBRID Post PROMOTER 6555
on August 13, 2019 by guest
http://aem.asm
.org/D
ownloaded from

Western blot and flow cytometry analyses were performed tomeasure protein expression upon induction. In Western blotswith whole-cell lysates and specific antibodies, no GFP wasdetectable in cells harboring pCRW53 grown at ATc concen-trations of 10 ng/ml and below (Fig. 3A). Weak GFP bandswere observed in lysates from cells grown at 20 ng/ml ATc, withthe band intensities increasing significantly toward higher ATcconcentrations. In line with the above Northern blot data, theTetR signal seemed to increase in intensity as well. Interest-ingly, the above described higher levels of gfp transcript in cellsharboring pBSV�(ospAp-gfp) (10) (Fig. 2A and B) translatedinto only marginally higher protein levels (Fig. 3A). In a West-ern blot analysis of whole-cell lysates obtained over a 72-h timecourse, GFP became detectable within 8 h of induction andreached an apparent plateau at around 40 h postinduction with200 ng/ml ATc (Fig. 3B). Similarly, green fluorescence wasdetected by epifluorescence microscopy in pCRW53-harboringB31-e2 cells at 8 h postinduction (data not shown).
Three independent flow cytometry experiments reproduc-ibly showed an analogous ATc-dependent titration of greenfluorescence. Results from a representative experiment areshown in Fig. 4A, and a cumulative titration response curve isshown in Fig. 4B. The fluorescence of uninduced cells wasindistinguishable from the background fluorescence of nonre-
FIG. 2. Transcriptional analysis of Post. (A and B) Northern blot analysis of B31-e2 (A) and B313 (B) transformants. One microgram of totalRNA was separated in a formaldehyde agarose gel and visualized with ethidium bromide to ensure equal loading (top panel). The blotted RNAwas probed with a PCR-generated gfp fragment (bottom panel). Arrowheads indicate the 0.8-kb and 1.8-kb gfp-tetR (*) transcripts. In panel B, ashorter time exposure of the two rightmost RNA lanes is shown to the right to better visualize the PospA transcripts. bg, background B. burgdorferiB31-e2 or B313 strains; PospA, background strains carrying pBSV�(ospAp-gfp); Post, background strains carrying pCRW53. Numbers below thebracket indicate the ATc concentrations in �g/ml. m, size marker in kb (0.5 to 10 kb in panel A or 0.24 to 9.5 kb in panel B; RNA ladders,Invitrogen). (C and D) RT-PCR analysis of B31-e2 (C) or B313 (D) transformants. A total of 0.25 �g of DNase-treated total RNA was used asa template for gfp-specific oligonucleotide primers. To exclude potential residual DNA in the sample, control reactions either omitting the RTreaction step (�RT control) or using pCRW53 DNA-specific primers (PCR control) were run in parallel. pCRW53, positive PCR control usingpCRW53 plasmid as a template; m, marker in bp (1 kb Plus DNA ladder; Invitrogen). Other sample labeling is identical to that in panels A and B.
FIG. 3. Western blot analysis of Post-driven GFP expression. B.burgdorferi whole-cell lysates were separated by sodium dodecyl sul-fate-polyacrylamide electrophoresis. Samples were normalized for theconstitutively expressed flagellar protein FlaB based on densitometryof a Coomassie blue-stained gel. (A) An equally loaded gel was thenused for Western blot analysis using antibodies against FlaB, TetR,and GFP. bg, background B. burgdorferi B31-e2 (upper three panels) orB313 (lower three panels) strain; PospA, background strains carryingpBSV�(ospAp-gfp); Post, background strains carrying pCRW53. Num-bers below the bracket indicate the ATc concentrations in �g/ml.(B) Time course of GFP expression by B313 carrying pCRW53. Har-vested culture volumes were adjusted to obtain equal cell numbersprior to further densitometry-based loading adjustment as describedabove. GFP was detected by Western immunoblotting. Numbers indi-cate hours postinduction with 0.2 �g/ml ATc.
6556 WHETSTINE ET AL. APPL. ENVIRON. MICROBIOL.
on August 13, 2019 by guest
http://aem.asm
.org/D
ownloaded from
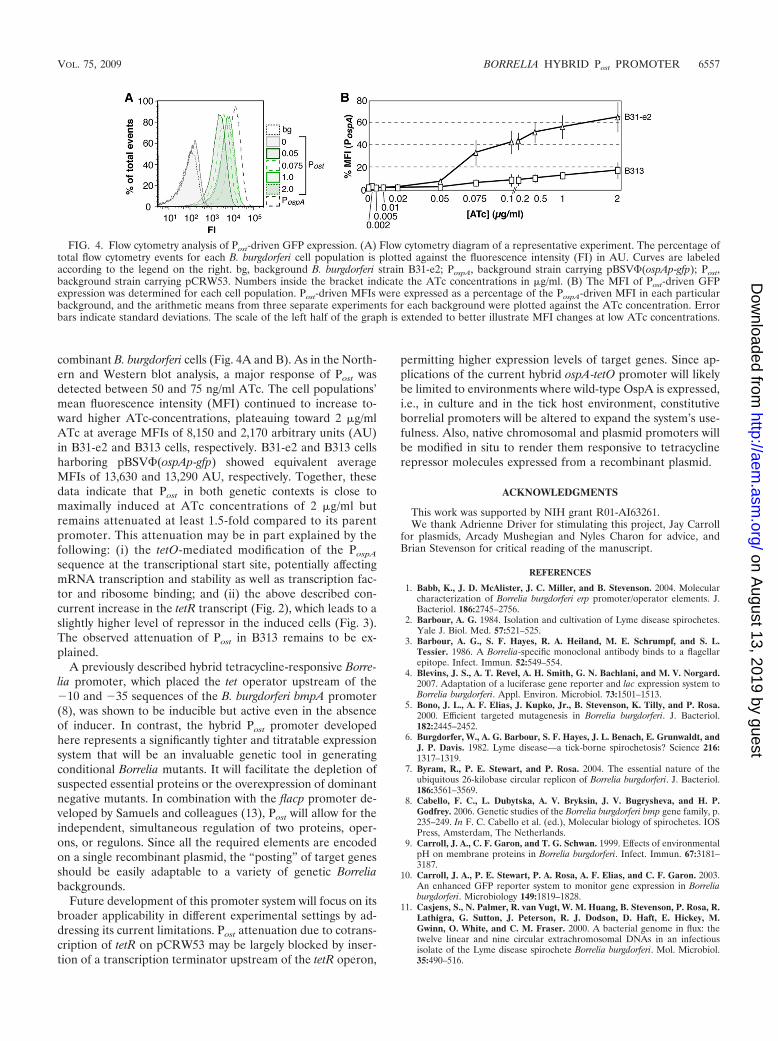
combinant B. burgdorferi cells (Fig. 4A and B). As in the North-ern and Western blot analysis, a major response of Post wasdetected between 50 and 75 ng/ml ATc. The cell populations’mean fluorescence intensity (MFI) continued to increase to-ward higher ATc-concentrations, plateauing toward 2 �g/mlATc at average MFIs of 8,150 and 2,170 arbitrary units (AU)in B31-e2 and B313 cells, respectively. B31-e2 and B313 cellsharboring pBSV�(ospAp-gfp) showed equivalent averageMFIs of 13,630 and 13,290 AU, respectively. Together, thesedata indicate that Post in both genetic contexts is close tomaximally induced at ATc concentrations of 2 �g/ml butremains attenuated at least 1.5-fold compared to its parentpromoter. This attenuation may be in part explained by thefollowing: (i) the tetO-mediated modification of the PospA
sequence at the transcriptional start site, potentially affectingmRNA transcription and stability as well as transcription fac-tor and ribosome binding; and (ii) the above described con-current increase in the tetR transcript (Fig. 2), which leads to aslightly higher level of repressor in the induced cells (Fig. 3).The observed attenuation of Post in B313 remains to be ex-plained.
A previously described hybrid tetracycline-responsive Borre-lia promoter, which placed the tet operator upstream of the�10 and �35 sequences of the B. burgdorferi bmpA promoter(8), was shown to be inducible but active even in the absenceof inducer. In contrast, the hybrid Post promoter developedhere represents a significantly tighter and titratable expressionsystem that will be an invaluable genetic tool in generatingconditional Borrelia mutants. It will facilitate the depletion ofsuspected essential proteins or the overexpression of dominantnegative mutants. In combination with the flacp promoter de-veloped by Samuels and colleagues (13), Post will allow for theindependent, simultaneous regulation of two proteins, oper-ons, or regulons. Since all the required elements are encodedon a single recombinant plasmid, the “posting” of target genesshould be easily adaptable to a variety of genetic Borreliabackgrounds.
Future development of this promoter system will focus on itsbroader applicability in different experimental settings by ad-dressing its current limitations. Post attenuation due to cotrans-cription of tetR on pCRW53 may be largely blocked by inser-tion of a transcription terminator upstream of the tetR operon,
permitting higher expression levels of target genes. Since ap-plications of the current hybrid ospA-tetO promoter will likelybe limited to environments where wild-type OspA is expressed,i.e., in culture and in the tick host environment, constitutiveborrelial promoters will be altered to expand the system’s use-fulness. Also, native chromosomal and plasmid promoters willbe modified in situ to render them responsive to tetracyclinerepressor molecules expressed from a recombinant plasmid.
ACKNOWLEDGMENTS
This work was supported by NIH grant R01-AI63261.We thank Adrienne Driver for stimulating this project, Jay Carroll
for plasmids, Arcady Mushegian and Nyles Charon for advice, andBrian Stevenson for critical reading of the manuscript.
REFERENCES
1. Babb, K., J. D. McAlister, J. C. Miller, and B. Stevenson. 2004. Molecularcharacterization of Borrelia burgdorferi erp promoter/operator elements. J.Bacteriol. 186:2745–2756.
2. Barbour, A. G. 1984. Isolation and cultivation of Lyme disease spirochetes.Yale J. Biol. Med. 57:521–525.
3. Barbour, A. G., S. F. Hayes, R. A. Heiland, M. E. Schrumpf, and S. L.Tessier. 1986. A Borrelia-specific monoclonal antibody binds to a flagellarepitope. Infect. Immun. 52:549–554.
4. Blevins, J. S., A. T. Revel, A. H. Smith, G. N. Bachlani, and M. V. Norgard.2007. Adaptation of a luciferase gene reporter and lac expression system toBorrelia burgdorferi. Appl. Environ. Microbiol. 73:1501–1513.
5. Bono, J. L., A. F. Elias, J. Kupko, Jr., B. Stevenson, K. Tilly, and P. Rosa.2000. Efficient targeted mutagenesis in Borrelia burgdorferi. J. Bacteriol.182:2445–2452.
6. Burgdorfer, W., A. G. Barbour, S. F. Hayes, J. L. Benach, E. Grunwaldt, andJ. P. Davis. 1982. Lyme disease—a tick-borne spirochetosis? Science 216:1317–1319.
7. Byram, R., P. E. Stewart, and P. Rosa. 2004. The essential nature of theubiquitous 26-kilobase circular replicon of Borrelia burgdorferi. J. Bacteriol.186:3561–3569.
8. Cabello, F. C., L. Dubytska, A. V. Bryksin, J. V. Bugrysheva, and H. P.Godfrey. 2006. Genetic studies of the Borrelia burgdorferi bmp gene family, p.235–249. In F. C. Cabello et al. (ed.), Molecular biology of spirochetes. IOSPress, Amsterdam, The Netherlands.
9. Carroll, J. A., C. F. Garon, and T. G. Schwan. 1999. Effects of environmentalpH on membrane proteins in Borrelia burgdorferi. Infect. Immun. 67:3181–3187.
10. Carroll, J. A., P. E. Stewart, P. A. Rosa, A. F. Elias, and C. F. Garon. 2003.An enhanced GFP reporter system to monitor gene expression in Borreliaburgdorferi. Microbiology 149:1819–1828.
11. Casjens, S., N. Palmer, R. van Vugt, W. M. Huang, B. Stevenson, P. Rosa, R.Lathigra, G. Sutton, J. Peterson, R. J. Dodson, D. Haft, E. Hickey, M.Gwinn, O. White, and C. M. Fraser. 2000. A bacterial genome in flux: thetwelve linear and nine circular extrachromosomal DNAs in an infectiousisolate of the Lyme disease spirochete Borrelia burgdorferi. Mol. Microbiol.35:490–516.
FIG. 4. Flow cytometry analysis of Post-driven GFP expression. (A) Flow cytometry diagram of a representative experiment. The percentage oftotal flow cytometry events for each B. burgdorferi cell population is plotted against the fluorescence intensity (FI) in AU. Curves are labeledaccording to the legend on the right. bg, background B. burgdorferi strain B31-e2; PospA, background strain carrying pBSV�(ospAp-gfp); Post,background strain carrying pCRW53. Numbers inside the bracket indicate the ATc concentrations in �g/ml. (B) The MFI of Post-driven GFPexpression was determined for each cell population. Post-driven MFIs were expressed as a percentage of the PospA-driven MFI in each particularbackground, and the arithmetic means from three separate experiments for each background were plotted against the ATc concentration. Errorbars indicate standard deviations. The scale of the left half of the graph is extended to better illustrate MFI changes at low ATc concentrations.
VOL. 75, 2009 BORRELIA HYBRID Post PROMOTER 6557
on August 13, 2019 by guest
http://aem.asm
.org/D
ownloaded from

12. Fraser, C. M., S. Casjens, W. M. Huang, G. G. Sutton, R. Clayton, R.Lathigra, O. White, K. A. Ketchum, R. Dodson, E. K. Hickey, M. Gwinn, B.Dougherty, J. F. Tomb, R. D. Fleischmann, D. Richardson, J. Peterson, A. R.Kerlavage, J. Quackenbush, S. Salzberg, M. Hanson, R. van Vugt, N.Palmer, M. D. Adams, J. Gocayne, J. Weidman, T. Utterback, L. Watthey, L.McDonald, P. Artiach, C. Bowman, S. Garland, C. Fuji, M. D. Cotton, K.Horst, K. Roberts, B. Hatch, H. O. Smith, and J. C. Venter. 1997. Genomicsequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature 390:580–586.
13. Gilbert, M. A., E. A. Morton, S. F. Bundle, and D. S. Samuels. 2007. Artificialregulation of ospC expression in Borrelia burgdorferi. Mol. Microbiol. 63:1259–1273.
14. Glass, J. I., N. Assad-Garcia, N. Alperovich, S. Yooseph, M. R. Lewis, M.Maruf, C. A. Hutchison III, H. O. Smith, and J. C. Venter. 2006. Essentialgenes of a minimal bacterium. Proc. Natl. Acad. Sci. USA 103:425–430.
15. Gossen, M., and H. Bujard. 1993. Anhydrotetracycline, a novel effector fortetracycline controlled gene expression systems in eukaryotic cells. NucleicAcids Res. 21:4411–4412.
16. Ho, S. N., H. D. Hunt, R. M. Horton, J. K. Pullen, and L. R. Pease. 1989.Site-directed mutagenesis by overlap extension using the polymerase chainreaction. Gene 77:51–59.
17. Jewett, M. W., R. Byram, A. Bestor, K. Tilly, K. Lawrence, M. N. Burtnick,F. Gherardini, and P. A. Rosa. 2007. Genetic basis for retention of a criticalvirulence plasmid of Borrelia burgdorferi. Mol. Microbiol. 66:975–990.
18. Kamionka, A., J. Bogdanska-Urbaniak, O. Scholz, and W. Hillen. 2004. Twomutations in the tetracycline repressor change the inducer anhydrotetracy-cline to a corepressor. Nucleic Acids Res. 32:842–847.
19. Lathem, W. W., P. A. Price, V. L. Miller, and W. E. Goldman. 2007. Aplasminogen-activating protease specifically controls the development ofprimary pneumonic plague. Science 315:509–513.
20. Lutz, R., and H. Bujard. 1997. Independent and tight regulation of tran-scriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 25:1203–1210.
21. Mushegian, A. R., and E. V. Koonin. 1996. A minimal gene set for cellularlife derived by comparison of complete bacterial genomes. Proc. Natl. Acad.Sci. USA 93:10268–10273.
22. Orth, P., D. Schnappinger, W. Hillen, W. Saenger, and W. Hinrichs. 2000.Structural basis of gene regulation by the tetracycline inducible Tet repres-sor-operator system. Nat. Struct. Biol. 7:215–219.
23. Rosa, P. A., K. Tilly, and P. E. Stewart. 2005. The burgeoning moleculargenetics of the Lyme disease spirochaete. Nat. Rev. Microbiol. 3:129–143.
24. Sadziene, A., A. G. Barbour, P. A. Rosa, and D. D. Thomas. 1993. An OspBmutant of Borrelia burgdorferi has reduced invasiveness in vitro and reducedinfectivity in vivo. Infect. Immun. 61:3590–3596.
25. Sadziene, A., B. Wilske, M. S. Ferdows, and A. G. Barbour. 1993. The crypticospC gene of Borrelia burgdorferi B31 is located on a circular plasmid. Infect.Immun. 61:2192–2195.
26. Sambrook, J., and D. Russell. 2001. Molecular cloning: a laboratory manual(ed.), Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
27. Samuels, D. S., K. E. Mach, and C. F. Garon. 1994. Genetic transformationof the Lyme disease agent Borrelia burgdorferi with coumarin-resistant gyrB.J. Bacteriol. 176:6045–6049.
28. Schulze, R. J., and W. R. Zuckert. 2006. Borrelia burgdorferi lipoproteins aresecreted to the outer surface by default. Mol. Microbiol. 59:1473–1484.
29. Skerra, A. 1994. Use of the tetracycline promoter for the tightly regulatedproduction of a murine antibody fragment in Escherichia coli. Gene 151:131–135.
30. Sohaskey, C. D., W. R. Zuckert, and A. G. Barbour. 1999. The extendedpromoters for two outer membrane lipoprotein genes of Borrelia spp.uniquely include a T-rich region. Mol. Microbiol. 33:41–51.
31. Stevenson, B., J. L. Bono, A. Elias, K. Tilly, and P. Rosa. 1998. Transfor-mation of the Lyme disease spirochete Borrelia burgdorferi with heterologousDNA. J. Bacteriol. 180:4850–4855.
32. Stevenson, B., T. G. Schwan, and P. A. Rosa. 1995. Temperature-relateddifferential expression of antigens in the Lyme disease spirochete, Borreliaburgdorferi. Infect. Immun. 63:4535–4539.
33. Stewart, P. E., R. Thalken, J. L. Bono, and P. Rosa. 2001. Isolation of acircular plasmid region sufficient for autonomous replication and transfor-mation of infectious Borrelia burgdorferi. Mol. Microbiol. 39:714–721.
34. Stieger, M., B. Wohlgensinger, M. Kamber, R. Lutz, and W. Keck. 1999.Integrational plasmids for the tetracycline-regulated expression of genes inStreptococcus pneumoniae. Gene 226:243–251.
35. Wright, K. J., P. C. Seed, and S. J. Hultgren. 2007. Development of intra-cellular bacterial communities of uropathogenic Escherichia coli depends ontype 1 pili. Cell Microbiol. 9:2230–2241.
36. Zhang, L., F. Fan, L. M. Palmer, M. A. Lonetto, C. Petit, L. L. Voelker, A. St.John, B. Bankosky, M. Rosenberg, and D. McDevitt. 2000. Regulated geneexpression in Staphylococcus aureus for identifying conditional lethal pheno-types and antibiotic mode of action. Gene 255:297–305.
37. Zuckert, W. R. 2007. Laboratory maintenance of Borrelia burgdorferi. Curr.Protoc. Microbiol. Chapter 12:Unit 12C.1.
38. Zuckert, W. R., J. E. Lloyd, P. E. Stewart, P. A. Rosa, and A. G. Barbour.2004. Cross-species surface display of functional spirochetal lipoproteins byrecombinant Borrelia burgdorferi. Infect. Immun. 72:1463–1469.
39. Zuckert, W. R., J. Meyer, and A. G. Barbour. 1999. Comparative analysis andimmunological characterization of the Borrelia Bdr protein family. Infect.Immun. 67:3257–3266.
6558 WHETSTINE ET AL. APPL. ENVIRON. MICROBIOL.
on August 13, 2019 by guest
http://aem.asm
.org/D
ownloaded from