disolución / desilución semestre 2018-1depa.fquim.unam.mx/amyd/archivero/presentaciondiso... · 1...
TRANSCRIPT


Back to few years….

GENERALIDADES DE DISOLUCIÓN

DISOLUCIÓN
Es el proceso por medio del cual unasustancia sólida (soluto), se dispersa en eldisolvente para dar una solución(dispersión molecular homogénea)

PRUEBA DE DISOLUCIÓN
Procedimiento por medio del cual se determina la cantidad de activo disuelto en un tiempo determinado bajo ciertas condiciones.

OBJETIVO
El objetivo de la prueba de disolución esusarla como una herramienta en eldesarrollo y control de calidad de losmedicamento.

DISOLUCIÓN - OBJETIVO• Proporciona información en áreas clave:
- Detectar cambios en la propiedadesfisicoquímicas del producto terminadoque puedan afectar la velocidad dedisolución del activo y por lo tanto sudesempeño in vivo.
- Diferenciar productos que han sidomanufacturados usando diferentesprocesos y/o formulaciones durante lafase de desarrollo

DISOLUCIÓN
• La disolución ha probado ser una pruebasimple, con un costo – eficiencia razonable.
• Es una determinación rigurosa para evaluarlas características de liberación de unaforma farmacéutica.

DISOLUCIÓN
La disolución no debe ser demasiado sensibleque detecte diferencias entre lotes, cuando nohay diferencias in vivo.

DISOLUCIÓN
• Procedimiento debe ser lo suficientementerobusto para dar resultados reproducible día adía.
• Procedimiento debe poderse transferir de unlaboratorio a otro

ALERTA
• Si un lote difiere significativamente en suscaracterísticas de disolución y muestra unatendencia hacia arriba o hacia abajo, es unaAlerta segura de que algún factor del material,formulación, o proceso esta fuera de control.

Aparato 1 - Canastilla
• Desventajas:
▫ Algunas sustancias quedan adheridas a la malla.▫ Extremadamente sensible a gases
disueltos.▫ Flujo inadecuado cuando las partículas
dejan la canastilla y flotan en el medio.

13
Aparato 1 - Canastilla

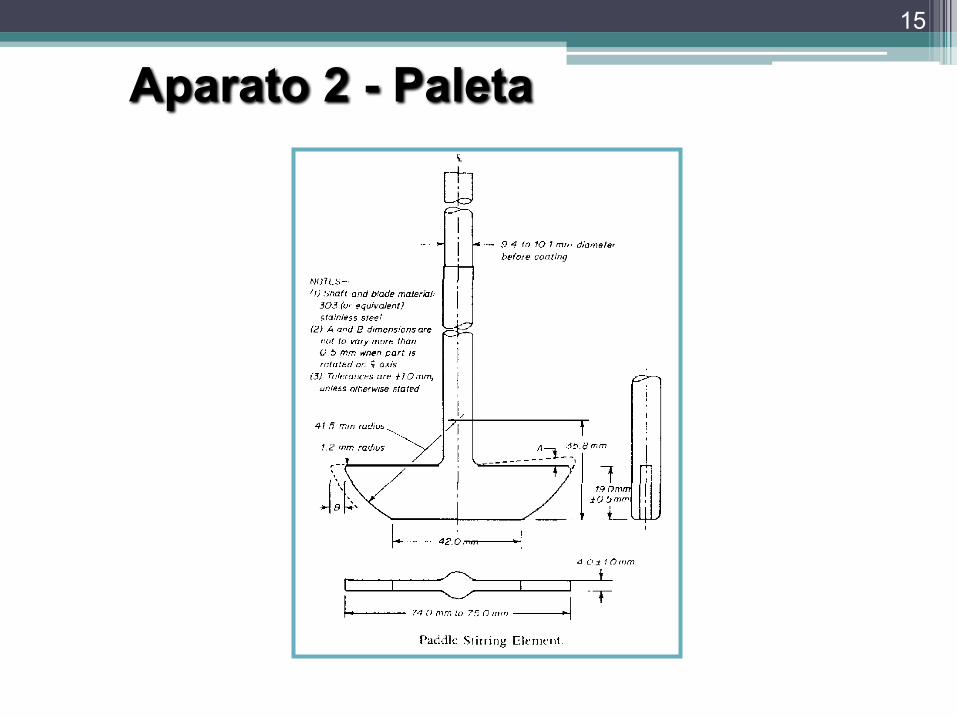
Aparato 2 - Paleta
• Desventajas:
▫ Importante la geometría de la paleta y del vaso.▫ Le afecta el más mínimo desajuste en la
orientación de la paleta.
15
Aparato 2 - Paleta

Capítulos de la USP relacionados disolución
ü701. Desintegración.ü711. Disolución.ü724. Drug release.ü1087. Disolución intrínseca.ü1088 Evaluación In vivo – In vitro de formas
farmacéuticas.ü1090. Guías de Bioequivalencia In vivo.ü1092. Desarrollo y Validación de procedimientos de
disolución.ü1225. Validación de métodos compendiales.

Criterios de aceptación
• Q, se define como la cantidad de ingredienteactivo (fármaco) disuelto expresado comocomo un porcentaje del contenido declarado• No se requieren valores de Q que excedan el 80%,
debido a que se necesitan hacer ajustes para lavaloración y los rangos de uniformidad de contenido
• Los valores típicos de Q, están en el rango del 70 al80%.

Criterios farmacopeicos
• Stage 1 (Etapa 1): Cada unidad al menos Q + 5%• Stage 2 (Etapa 2): Promedio de las 12 unidades
igual o mayor a Q y ninguna unidad menor a Q–15 %.• Stage 3 (Etapa 3): Promedio de las 24 unidades
igual o mayor a Q, no más de 2 unidades menores a Q – 15 % y ninguna unidad menor a Q- 25 %.

CRITERIOS FARMACOPEICOS
EtapaNúmero de tabletas a ensayar
Número de tabletas
para evaluar con los criterios
1 6 6
2 6 12
3 12 24

Determinaciones “Cualitativas”
En etapas de desarrollo de la disolución.
La observación visual detectará problemas con la formulación sin necesidad de cuantificar la prueba

Determinaciones cualitativas
• Tiempo en el que la cápsula o el recubrimiento de latableta se rompe.• Tiempo requerido para la disgregación completa.• Comportamiento de la cápsula con un determinado
“sinker”.• Efectividad del mezclado en el vaso.• Efectividad del procedimiento de degasificación.

Procedimiento de disolución Etapas
• 1. Preparación de la muestra.Incluye hasta la toma de muestra.
• 2. Preparación para su análisis.
Puede ser leída o inyectada después defiltrarla o diluida para llegar a laconcentración final.

Medio de disolución – Gases disueltos
• Los líquidos están en equilibrio con el gas que se encuentra en su interfase.
• La cantidad de gases disueltos depende de la temperatura
• ¿ La cantidad de gases disueltos aumenta o disminuye con la temperatura?

Gases disueltos
• Al liberarse los gases distorsionan el valor de los porcentajes disueltos.
• ¿ Cómo se detectan los gases disueltos?
• 1. Al observarse el medio con un color “plata opaco” – presencia de partículas microscópicas de gas.• 2. Cuando se aprecian burbujas

¿ Como afectan las burbujas ?• 1. Modificando la dinámica de los fluidos
• 2. Modificando el área contacto líquido-sólido
• Por ejemplo las burbujas se pueden “Adherir a la malla de la canastilla” o el producto disgregado.• En disoluciones automatizadas las burbujas se
pueden acumular en las celda del espectro y afectar la lecturas de absorbancia

¿ Como afectan las burbujas ?
• Pueden ocasionar que las partículas se adhieran al aparato y a las paredes del vaso.
• Pueden ocasionar que las partículas floten incrementando la velocidad de disolución
• Pueden afectar el área disponible disminuyendo la velocidad de disolución.
• Incrementan la variación en los resultados, por ejemplo en prednisona hasta un 12 %.
• Según el tipo de aparato y forma farmacéutica se puede observar un incremento o decremento.

¿ Cómo eliminar los gases disuletos?
• Degasificar el medio para mantener lasconcentración de gases por debajo del valor dela saturación.

Vaciado del medio de disolución

pH
• Medios de disolución no amortiguados pueden variar su pH.
• El pH del agua puede variar desde 6 a 7.2.
• Durante el desarrollo se realizan perfiles “pH VS solubilidad”
• Durante el desarrollo de la prueba se debeverificar el valor del pH al inicio y al final de laprueba.

pH
• La absorbancia puede variar al cambiar el pH
• Si la absorbancia cambia con el pH, por ejemploen disoluciones de liberación prolongada, sedebe verificar el pH de los estándares, sicambian significativamente con respecto al pHde las muestras, deben prepararse variosestándares

Volumen del medio
• Es necesario que el volumen del medio se mantenga constante.
• Pérdida en el volumen por muestreo puede corregirse en los cálculos si el factor de corrección es menor al 10 %.
• Si se excede el 10 %, los volúmenes extraídos deben ser reemplazados.

¿ Cómo se puede determinar si el medio se evapora?
• Se puede pesar el vaso con el medio de disolución antes y después de la disolución.
• ¿ En qué medio ambientes hay mayor evaporación?

¿ Cómo evitar la evaporación del medio?
• Una práctica común es medir el medio, colocar en el equipo de disolución y esperar a que el medio alcance los 37ºC – Tapar los vasos.

Altura de las canastillas y paletas
• A 25 mm +/- 2 mm con respecto al fondo

Temperatura
• ¿ Por qué hay que controlar la temperatura en la prueba de disolución?
• Durante el desarrollo hay que realizar perfiles de temperatura VS solubilidad.
• La temperatura debe monitorearse durante la prueba.
• Especificaciones: 37 +/- 0.5ºC

Temperatura
• Vasos de plástico VS Vasos de vidrio

Revoluciones por minuto
• Velocidad especificada +/- 4 %.
• ¿ Calcule el límite de rpm para una disolución que se lleva a cabo a 75 rpm?

Punto de muestreo
A no menos de 1 cm de la pared del vaso

Tiempo de muestreo
• Especificación: Tiempo de muestreo +/- 2 %.
• Utilizar un cronómetro calibrado.

Enzimas
• El uso de enzimas en los fluidos simulados gástrico e intestinal dependerá del producto y se debe justificar.
• Por ejemplo para cápsulas de gelatina se usan enzimas (pepsina para el gástrico y pancreatinapara el intestinal) para disolver la película que puede formarse e impide la disolución del fármaco.

SINKERS
• Debe detallarse las características del sinker.
• Debe emplearse el mismo sinker cuando setransfiere un método analítico, a deben ser lomás semejante posible.

NOM – 177 SSA1 – 2013
Establece las pruebas y procedimientos para demostrar que un medicamento es intercambiable.
Requisitos a que deben sujetarse los terceros autorizados que realicen las pruebas
42

NOM – 177 SSA1 – 2013
• 7.3 Validación del método analítico
• 7.3.1. El método analítico que se utilice pararealizar el perfil de disolución debe estardebidamente validado.
43

Sistema de clasificación biofarmacéutico
Clase I: AS, APVelocidad de disolución > tiempo de vaciado gástrico
Clase II: BS, APDisolución es el paso limitante
Clase III: AS, BPAbsorción es el paso limitante
Clase IV: BS, BPFármacos problemáticos.
1 10 100 1000 10000 100000 mL
Volumen acuoso requerido (pH 1 - 7.5)
0.001
0.0001
0.00001
0.000001

PERFIL DE DISOLUCIÓN
• Utilizar una curva de calibración de la sustanciade referencia para calcular por interpolación laconcentración del fármaco disuelto.

Fórmula para comparar los % disueltos del producto innovador con nuestro producto

PRUEBAS FARMACOPEICAS CON MULTIPLES ETAPAS, EJEMPLO