
1
Ataksi og spastisk paraparese uten genetisk
diagnose i en norsk kohort.
Stud. med. Ann-Helen Richvoldsen
Prosjektoppgave ved det medisinske fakultet
Universitet i Oslo
2016
Veileder: Chantal M.E. Tallaksen
Med-veiledere: Iselin Marie Wedding og Siri Rydning

2
Innholdsfortegnelse
Sammendrag ...........................................................................................................................................3
Bakgrunn .................................................................................................................................................5
Innledende ............................................................................................................................................5
Hereditær ataksi ....................................................................................................................................5
Klinisk klassifikasjon .......................................................................................................................5
Diagnose, utredning, typiske funn ....................................................................................................5
Genetisk klassifikasjon .....................................................................................................................6
Patofysiologi.....................................................................................................................................8
Epidemiologi ....................................................................................................................................8
Prognose og behandling ...................................................................................................................8
Hereditær spastisk paraparese ..............................................................................................................9
Klinisk klassifikasjon .......................................................................................................................9
Diagnose, utredning, typiske funn ....................................................................................................9
Genetisk klassifikasjon ...................................................................................................................10
Patofysiologi...................................................................................................................................11
Epidemiologi ..................................................................................................................................11
Prognose og behandling .................................................................................................................11
Mål med oppgaven ...............................................................................................................................12
Metode ...................................................................................................................................................13
Resultater ..............................................................................................................................................14
Hereditær ataksi ..................................................................................................................................14
Gjennomgang av hele pasientmaterialet .........................................................................................14
Sammenligning av trekk ved ulike transmisjonsmåter ...................................................................21
Hereditær spastisk paraparese ...........................................................................................................29
Gjennomgang av hele pasientmaterialet .........................................................................................29
Sammenligning av trekk ved ulike transmisjonsmåter ...................................................................37
Diskusjon ...............................................................................................................................................45
Konklusjon ............................................................................................................................................51
Litteraturliste ........................................................................................................................................52
Vedlegg 1 ...............................................................................................................................................53
Vedlegg 2 ...............................................................................................................................................55
Vedlegg 3 ...............................................................................................................................................56

3
Sammendrag
Bakgrunn: Hereditær ataksi og spastisk paraparese er begge sjeldne, degenerative spinocerebellære
lidelser, som kan nedarves via alle modaliteter for arv. De er kjennetegnet ved progressive
gangvansker, og er klinisk og genetisk svært heterogene tilstander. Det foregår intensivt forskning på
området, og det oppdages stadig nye gener som årsak til sykdommene, men det er fremdeles mange
pasienter som står uten genetisk diagnose.
Det har ved Ullevål Universitetssykehus vært samlet en kohort av disse pasientgruppene siden 2002.
Halvparten av disse pasientene mangler genetisk diagnose. Denne oppgaven er et deskriptivt studie av
disse pasientene, med den hensikt å identifisere særtrekk, mulige fenotyper, trender for debut, klinikk
og progresjon.
Materiale og metode: 230 pasienter med hereditær ataksi og 161 pasienter med hereditær spastisk
paraparese, uten genetisk diagnose har blitt inkludert i denne oppgaven. Disse pasientene var registrert
i databasen via det internasjonale registreringsskjemaet SPATAX, og manglende informasjon i
databasen har blitt innhentet fra tidligere registrerte skjemaer, og ytterligere informasjon innhentet fra
journaler ved hjelp fra veiledere. Alle statistiske analyser har blitt utført ved bruk av IBM SPSS
Statistics versjon 22.
Resultater og konklusjon: 57% av HA og 56% av HSP pasientene var sporadiske. 9% av HA og 6% av
HSP var recessive familier. 26% av HA og 30% av HSP var dominante familier. 65% av HA
pasientene og 51% av pasientene med HSP hadde et komplisert klinisk syndrom, og denne fordelingen
varierte etter transmisjonsmåte, hvor alle recessive hadde en komplisert form, og dominante generelt
hadde en høyere andel rene kliniske tilstander enn øvrige grupper. Gjennomsnitt for debut for HA lå
på 35 år, og for HSP på 27 år, men fåtallet debuterte i denne alderen. Pasientene var fordelt i to
grupper for debut: En tidlig, med topp allerede i løpet av de første leveår, og en sen, med topp i
aldersgruppen 50-60 år. Alder for debut varierte avhengig av transmisjonsmåte, hvor recessive HSP og
HA debuterte generelt tidlig, sporadiske oftere debuterte sent, og dominante HSP oftest debuterte
tidlig, og dominante HA var likt fordelt i tidlig og sen debut-gruppe. Flertallet av både HA og HSP
pasientene hadde bevart gangfunksjon, og pasienter med recessiv arvegang hadde generelt høyest
alvorlighetsgrad. Pasienter som var eldst ved undersøkelse, var hos HA de dominante, med
gjennomsnitt på 58 år, og hos HSP de sporadiske, med gjennomsnitt på 52 år. Recessive var hos begge
pasientgrupper de som har blitt undersøkt i yngst alder (HA 39 år, HSP 37 år), og hadde også levd
med sin sykdom lengst (HA 26 år, HSP 23 år). Sporadiske sykdomstilfeller var de som i gjennomsnitt
hadde levd med sin sykdom i kortest tid (HA 12 år, HSP 18 år). Pasienter med sen debut hadde hos
både HA og HSP en raskere progresjon enn øvrige pasient-grupper, med unntak pasienter med
recessiv arvegang, som både debuterte tidlig og hadde rask progresjon. Sent debuterende, sporadiske
sykdommer hadde raskest progresjon hos både HA og HSP. Det var videre en upåfallende
kjønnsfordeling i alle gruppene. Det var noen få kliniske særtrekk hos noen grupper, slik som en økt
andel med okulomotorisk apraksi hos recessive HA, lett økt andel ekstrapyramidale funn hos
sporadisk HA og økt andel med ataksi og øyemotilitetsforstyrrelser hos recessive HSP. Det er vist stor
klinisk heterogenitet hos pasientene i denne oppgaven, og det er funnet noen få, uspesifikke trender
hos noen grupper i dette pasientmaterialet.

4
Abstract
Background: hereditary ataxia (HA) and spastic paraparesis (HSP) are both rare, degenerative
spinocerebellar disorders, which can be passed down through all forms of inheritance. They are
characterized by progressive difficulty in walking, and are clinically and genetically heterogeneous
conditions. Intensive research is currently being conducted, and new genes are coninuously being
discovered, but there are still many patients who have no genetic diagnosis established.
At Ullevål University Hospital a cohort of patients with HA and HSP has been assembled since 2002.
Half of these patients lack genetic diagnosis. This thesis is a descriptive study of these patients, with
the aim of identifying characteristics, possible phenotypes, trends, onset, clinical features and
progression.
Material and methods: 230 patients with hereditary ataxia and 161 patients with hereditary spastic
paraparesis, without genetic diagnosis have been included in this project. These patients were
registered in the database via the international registration form SPATAX, information missing in the
database has been collected from previously registered forms, additional information was obtained
from medical records with help from supervisors. All statistical analyses have been performed using
IBM SPSS version 22 Statistics.
Results and conclusions: 57% of HA and 56% of HSP patients were sporadic. 9% of HA and 6% of
HSP were from recessive families. 26% of HA and 30% of HSP were from dominant families. 65% of
HA patients and 51% of patients with HSP had a complicated clinical syndrome. This distribution
varied with type of transmission, where all recessive diseases had a complicated form, and dominantly
inherited forms generally had a higher proportion of pure clinical conditions than other groups. The
average age of onset of HA was at age 35, and HSP at age 27, but very few patients actually had a
symptom debut at this age. Instead, the patients were divided into two groups for debut: One early,
with debut already during the first year of life, and one late group, where most had their debut in 50-
60 years of age. Age of onset varied depending on the type inheritance, where recessive HSP and HA
debuted generally early, sporadic often debuted late. Dominant HSP usually debuted early and
dominant HA were distributed equally in one early and one late onset group. The majority of both HA
and HSP patients had preserved walking ability, and patients with recessive inheritance had generally
the highest severity of symptoms. The patients who were the oldest when examined, were in the HA-
group the patients with dominant disease, with an average of 58 years, and in the HSP group the
patients with sporadic disease, with an average of 52 years. Recessive HSP and recessive HA were
both the groups who had been examined at the youngest age (HA 39 years, HSP 37 years) and had
also lived with their illness the longest (HA 26, HSP 23 years). Patients with a sporadic case of the
diseases had on average lived the shortest duration with their illness (HA 12, HSP 18). Patients with
late-onset of their disease, both in HA and HSP, had a faster progression of symptom severity than
other patient groups, except for patients with recessive inheritance, who both debuted early and had
rapid progression. Late-onset, sporadic diseases had the fastest progression in both HA and HSP.
There were further no remarkable differences in gender distribution in all groups. A few clinical
characteristics were found in some groups, such as an increased proportion of patients with
oculomotoric apraxia with recessive HA, slightly increased proportion extrapyramidal findings in
sporadic HA and an increased amount of patients with ataxia and eyemotility disturbances in the group
of patients with recessive HSP. This patient material has shown considerable clinical heterogeneity,
and a few non-specific trends in some groups in this patient sample have been uncovered through this
project.

5
Bakgrunn
Innledende:
Arvelig Ataksi (HA) og arvelig spastisk paraparese (HSP) er begge spinocerebellære
nevrodegenerative lidelser med progressive gangvansker som hovedsymptom, som for mange leder til
permanente funksjonstap. Disse lidelsene har stor heterogenitet, som gjør det vanskelig å skille
mellom ulike former av sykdommene, ettersom forløpene varierer betraktelig mellom individer med
samme type sykdom, selv innen samme familie. Ettersom det er et klinisk overlapp mellom HA og
HSP, undersøkes de gjerne sammen i epidemiologiske og kliniske studier, og en metaanalyse fra 2014
estimerte en kombinert, global prevalens av HSP og HA på 1/10 000 [1].
Tidligere, ble disse sykdommene klassifisert etter klinikk, da delt inn i rene og komplekse tilstander
[2]. Men store framskritt innen genetisk forskning de siste 10 årene har forbedret diagnostisering av
disse tilstandene betraktelig, samt ført til et nytt, genetisk klassifikasjonssystem, hvor det stadig
identifiseres nye genfeil som årsak til tilstandene. Men det er fremdeles mange pasienter som står uten
genetisk diagnose.
Hereditær ataksi
Generelt
Hereditær ataksi er en monogenetisk, heterogen gruppe sykdommer, som er karakterisert av
progredierende inkoordinasjon av gange og andre cerebellære utfall, som inkoordinasjon av hender,
trunkus, tale og øyebevegelser. Den finnes også i episodisk form, hvor symptomene kommer i
perioder på sekunder til dager, avhengig av subtype.
Det presise antall typer av arvelig ataksi er ukjent, men minst 37 autosomale, dominante former er så
langt identifisert, 20 autosomalt recessive, og et mindre antall X-bundene og mitokondrielle former.
[3]
Klinisk klassifikasjon
Britiske nevrolog Harding laget i 1983 et klinisk inndelingssystem, basert på kliniske funn,
sykdomsforløp, assosierte symptomer og nevropatologiske funn. [2] Dette har blitt utbyttet med et mer
dekkende inndelingssystem basert på genetikk.[4] Men klinisk benyttes også begrepene "rene" og
kompliserte/komplekse tilstander, basert på det kliniske bildet til pasienten. De rene tilstandene er
hvor pasienten kun har et cerebellært syndrom, og de komplekse har ulike tilleggssymptomer, slik som
ekstrapyramidale manifestasjoner, spastisitet, muskelsvekkelse, muskelsvinn, kognitiv svikt, perifer
nevropati, epilepsi, oftalmoplegi eller både motoriske og mentale utviklingsforstyrrelser.
Manifestasjoner i andre organsystemer forekommer også, og kan være i form av retinopatier,
cardiomyopati, immunsvikt mm. [4, 5]
Diagnose, utredning, typiske funn
Diagnosen er basert på typisk klinikk med progressive cerebellære utfall, positiv familiehistorikk,
bildediagnostikk, genetisk testing, og utelukkelse av andre, ikke genetiske årsaker til ataksi.
De differensialdiagnoser man må utelukke er ervervede former for ataksi, som konsekvenser ved
alkoholisme, vitamin mangler (B12,E) , multippel sclerose, vaskulær sykdom, primære eller
sekundære tumores, paraneoplastisk syndrom, multisystem atrofi (MSA), infeksjoner (HIV, borrelia,
syfilis mm.) og metabolske sykdommer. [5]

6
Ved klinisk undersøkelse vil man typisk se at pasienten har en bredbaset, ataktisk gange, dårlig
balanse, ataksi i over eller underekstremiterer eller trunkal ataksi. Øyemotilitetet kan være forstyrret,
og man kan se sakkadisk pursuit, nystagmus (horisontal, vertikal og rotatorisk). Noen former for ataksi
har okulomotorisk apraksi, og andre har oftalmoplegi. Dysatri kan ofte observeres, hvor pasienter har
en skanderende tale, og flere pasienter har dysfagi.
Både debut og progresjon varierer mellom de ulike variantene av hereditær ataksi, og innen en og
samme variant og familie.[5] Men man ser oftere en trend til tidlig debut hos de recessive variantene.
Supplerende undersøkelser har funn avhengig av subtype av hereditær ataksi. Ved for eksempel
spinocerebellære ataksier, kan man se tre ulike mønstre for atrofi på MR: Ren, cerebellær atrofi,
olivopontocerebellær atrofi, eller global atrofi av hele hjernen. [6]
Ved nevrografi/EMG kan perifer nevropati iblant avdekkes. Av blodprøver, annet en gen-testing, er
det hovedsakelig ved E-vitamin mangel (som ved AVED) det er mulige, spesifikke funn, og noen
former har økt alfa-fetoprotein (ataksia telengiectasia).[5]
Gen testing utføres strategisk, ettersom det både er dyrt og tidkrevende. Man legger vekt på
transmisjonsmåte, og om mulig, diverse spesifikke funn som kan gi misstanke om ulike typer
hereditær ataksi. Det er vanlig ved dominant hereditær ataksi å teste for SCA1, 2, 3, 6 og 7, ettersom
disse representerer over 50% av de affiserte familiene, og Friedreich ataksi ved recessiv form.[7]
Familiehistorie er viktig å avdekke, ettersom transmisjonsmåte sier mye om hvilke type hereditær
ataksi det dreier seg om. Alle transmisjonsmodaliteter er beskrevet. Ofte kan pasienten mangle
familiehistorie, og ha en tilsynelatende sporadisk form for ataksi, men hvor det misstenkes at pasienten
har en skult familiehistorie. Det er rapportert om høy penetrans av spinocerebellære ataksier, men
alvorlighetsgrad og alder for debut varierer voldsomt, også i familier, som kan gi en falskt negativ
familie anamnese. [4]
Genetisk klassifikasjon
Hereditær ataksi kan arves autosomalt dominant, benevnt som ADCA, autosomal recessivt ,benevnt
ARCA. X-bundet ataksi er sjeldent, men forekommer, og rammer da kun menn. Mitokondriell
transmisjon forekommer også. [4, 5] Rundt 30% av alle cerebellær ataksi pasienter er genetisk
udiagnostisert[3].
ADCA: Denne gruppen kan deles i to typer: den progressive typen, og de episodiske. De progressive
former består av de spinocerebellære ataksier (SCA), som hittil har over 37 ulike varianter, og
dentatorubralpallidoluysisk atrofi (DRPLA). De episodiske ataksiene har 7 ulike varianter.
Spinocerebellære ataksier (SCA) og DRPLA er forårsaket av et stort mangfold av ulike gen-
affeksjoner. De debuterer generelt i voksen alder, og de fleste av tilstandene er sakte progredierende,
hvor man ofte ser atrofi av cerebellum. De enkelte spinocerebellære ataksiene kan ikke skilles fra
hverandre av klinikken eller ved bildestudier alene, da det er et stort overlapp mellom alle tilstandene,
og selv de funn som sees på som typiske for noen SCA, er ikke spesifikke for den enkelte varianten.
Men enkelte kliniske funn kan fungere om en pekepinn[4]. Noen typiske funn som går igjen, er
pyramidale tegn ved SCA1 og 3, kognitiv svikt hos SCA1, 12,14 og 17, chorea hos SCA17 eller
DRPLA, netthinnesvikt hos SCA7 og en relativt ren, sent debuterende ataksi hos SCA6[8].
De vanligste typer av spinocerebellære ataksier, er SCA1, 2, 3, 6 og 7 [9]. Men prevalens varierer med
geografisk lokalisasjon, og man kan for eksempel se en høyere prevalens av DRPLA i Japan, og en
høyere prevalens av SCA3 i Portugal.[5]

7
Episodisk ataksi kommer i anfall med cerebellære utfall, og avhengig av type kan vare i alt fra
sekunder eller dager, og er i all hovedsak uten symptomer mellom anfallene. Men de kan etter mange
år med sykdommen utvikle permanent ataksi. Noen subtyper har myokjimier, hørselstap og vertigo.
Vanligste type er EA2. Disse sykdommene er ionekanalsykdommer. [4, 5]
For videre informasjon om dominante hereditære ataksier, se vedlegg 1.
ARCA: Recessive former for hereditær ataksi er mangfoldige, og de debuterer gjerne tidlig. Disse
tilstandene er oftest komplekse tilstander, og gjerne med flere symptomer fra andre organsystemer i
tillegg til de nevrologiske. De vanligste formene er blant annet Friedrichs ataksi, ataxia
teleangiektasia, ataksi med E-vitamin mangel og ataksi med okulomotorisk apraksi (type 1 og 2).
Friedreich ataksi (FRDA) er den vanligste autosomalt recessive ataksien globalt, men er mindre
representert i Norge. Den debuterer oftest før 25 års alder, og er karakterisert av en sakte
progredierende sensorisk ataksi. 25% har et atypisk forløp, med senere debut og mildere sykdom.
Typiske funn er tap av posisjon og vibrasjonssans, svekkelse og spastisitet i underekstremiteter,
bortfall av reflekser i underekstremiteter, blære dysfunksjon, kardiomyopati og skoliose.[10] Den
kommer i flest tilfeller av triplet repeat ekspansasjoner i FXN, men er ikke assosiert med anticipation.
Ataxia telangiectasia er vanligere i Norge enn FRDA, er karakterisert av progressiv cerebellær ataksi,
har debut i tidlig barndom, og har ofte okulomotorisk apraksi, immunsvikt, choreoathetose,
telangiektasier på konjunktiva, og en høyere risiko for å utvikle kreft, som leukemi og lymfomer. [11]
Ataksi med E-vitamin mangel begynner oftest i sen barndom/tidlig tenår, med dysatri og klønethet og
dårlig balanse grunnet nedsatt propriosepsjon. Fenotypisk kan den ligne på FRDA, men den har oftere
dystoni og sjeldnere kardiomyopati. Diagnosen stilles på grunnlag av FRDA lignende fenotype, og
lave nivåer av E-vitamin, uten fettmalabsorpsjon.[8] Den kan behandles med E-vitamin.
Ataksi med okulomotorisk apraksi (AOA1 og AOA2) er karakterisert av tidlig debut med sent
progredierende cerebellær ataksi, og okulomotorisk apraksi. AOA1 debuterer tidligere enn AOA2, da i
tidlig barndom, og AOA2 i tenår og tidlig voksen alder. De kan ha nevropati, kognitive problemer,
dystoni, og choreoatetose. [5]
Det finnes videre spastiske ataksier, som kan nedarves både recessivt og dominant. Her er spastisiteten
uttalt. Det er 5 typer, SPAX1 til 5. hvor SPAX1 er dominant nedarvet, resterende er recessive
tilstander, hvor man kan finne tilleggsfunn som blant annet optisk atrofi, perifer nevropati og epilepsi,
avhengig av typen SPAX. Autosomalt recessiv spastic ataxia av Charlevoix-Saguenay (ARSACS) er
også en spastisk ataksi, karakterisert av distal nevropati og tidlig debut. [5]
For utfyllende informasjon om recessive ataksier, se vedlegg 2.
X-bunden hereditær ataksi: X-bunden sideroblastisk anemi og ataksi er karakterisert av tidlig debut av
cerebellære symptomer og asymptomatisk, mild anemi. Den kan både være progressiv og non-
progressiv. Noen har pyramidale funn. Hos menn med debut i voksen alder, kan noen ha Fragilt X,
som er et ataksi/tremor syndrom.[8]
Sporadisk ataksi: Et forsøk gjort i 2002 [12], forsøkte å kartlegge sporadiske ataksier hos 112 pasienter
med debut etter 20 års alder, og finne ut hvor mange av pasientene i deres materiale hadde sporadisk
ataksi med en genetisk årsak, og hvor mange som led av multiple system atrofi (MSA-c). De kom
frem til at 29% av deres pasientmateriale hadde en klinisk mulig/ sannsynelig diagnose av MSA-c.

8
4% hadde Friedreichs ataksi, SCA2 ble funnet hos 1%, SCA3 hos 2%, SCA6 6%, og sykdommen
forble uforklarlig hos 58%.
Patofysiologi
Patofysiologi er avhengig av type hereditær ataksi, men hos mange varianter fører det til cerebellær
atrofi og tap av purkinje fibre via ulike mekanismer. [3]
Det er stor heterogenitet i mutasjonstyper som gir ataksi. Mange har en polyglutamin (CAG-
repetisjons ekspansasjoner), slik som sees hos pasienter med SCA1, 2, 3, 6 og,7 og DRPLA, som
dermed også gir anticipasjon. Andre ikke-kodende eksapsasjoner missense mutasjoner, delesjoner,
dupliasjoner, splice og trunkerende mutasjoner forekommer også.[5]
Alle gener hos de dominante cerebellære ataksier virker funksjonelt forskjellig, men har en feil i enten
foldingen av proteiner, aggregering av dem i cellene, de kan ha feil i selve kvalitetskontrolleringen av
proteinene, feilregulert gentranskripsjon, RNA toksisitet, eller endringer i deres synaptiske
transmisjon. [3]
Hos de recessive cerebellære ataksiene, kan man summere opp genmutasjonene til å affisere
mitokontriell funksjon, DNA reparasjon, chaperone aktivitet, synaptisk transmisjon og metabolsk
funksjon.[3]
Epidemiologi
Ved en studie utført i sørøst-Norge i 2009, ble det registrert en prevalens på 6,5/100 000. 4,2/100 000
for autosomal dominant ataksi, 2,3/100 000 for autosomal recessiv, 0,15/100 000 med Friedreich’s
ataxia og 0.4/100 000 for ataxia telangiectasia.[13]
Globale tall utregnet via en metanalyse i 2014 [1] estimerte et tall på 2,7/100 000 dominante ataksier,
hvor SCA3, 2 og 6 var mest vanlig. Recessive ble utregnet til 3,3/100 000, med Friedreich ataksi som
vanligste type. I samme studie fant de at 33-92% av pasienter med dominante hereditære ataksier
manglet diagnose, og 40–46% av pasienter med recessive ataksier.
Prognose og behandling
Prognose er avhengig av hvilke type hereditær ataksi pasienten har. Ved de dominante,
spinocerebellære tilstnadene, kan forventet levetid være forkortet i SCA1, 2,3 og 7, og normal ved
SCA 5, 6 og 14.[8] Ved recessive tilstander er det ofte en alvorligere prognose, som for eksempel ved
Friedreich ataksi, som har gjennomsnitt levealder på 36 år, og rask progresjon av deres symptomer til
de blir rullestolbundet[10].
Med unntak av pasienter med AVED (E-vitamin mangel) og andre mangeltilstander, finnes det kun
symptomatiske behandlinger av hereditær ataksi. Dette går ut på regelmessig fysioterapi,
tilrettelegging med riktige hjelpemidler, hjelp fra logoped, og medisinsk behandling av eventuelle
kompliserende symptomer (ved for eksempel spastisitet eller epilepsi)[4].

9
Hereditær spastisk paraparese
Generelt:
Hereditær spastisk paraparese er en heterogen, arvelig gruppe nevrodegenerative sykdommer, som gir
en lengde-avhengig degenreasjon av corticospinale aksoner, som er klinisk karakterisert av progressiv
spastisitet og svakhet i underekstremitetene. Den ble først beskrevet på 1880-tallet av en tysk
nevrolog, Strumpell, og har lenge vært en sykdom med lite forståelse rundt, fram til de siste 30 år.
Forståelsen har ekspandert sammen men generell kunnskap om genetikk, og det er i dag identifisert
over 50 ulike genfeil som årsak til sykdommen. [14]
Klinisk klassifikasjon:
Britiske nevrolog Harding dannet et klinisk inndelingssystem for sykdommene i 1983, hvor
begrepene "ren" og "kompleks" ble brukt for å beskrive dem, avhengig av tilstedeværelsen av
ledsagende symptomer[2]. Den rene formen gir progressiv spastisitet i nedre ekstremiteter, hvor
autonome forstyrrelser i blære og seksualfunksjon, og lette sensorielle symptomer i
underekstremitetene ikke er uvanelig. Den komplekse formen er i tillegg til karakteristikaene til den
rene formen ledsaget av både nevrologiske og ikke-nevrologiske manifestasjoner. De nevrologiske
tilleggsymptomer kan være ataksi, para-spastisitet i øvre ekstremiteter, neuropati, mental retardasjon,
kognitiv svekkelse, dystoni, ekstrapyramidale tegn (parkinsonisme, chorea, athetose, dyskinesi),
øyemotilitetsforstyrrelser og epilepsi. Ikke nevrologiske manifestasjonen inkluderer retinopati,
makuladegenerasjon, optisk atrofi, katarakt, døvhet, ansiktsdeformasjoner, persisterende oppkast,
hudaffeksjon, gastro-øsofagal refluks og ortopediske abnormaliteter (som for eksempel skoliose). [14]
Diagnose, utredning og funn:
Diagnose er basert på familiehistorie, positiv anamnese på progressive gangvansker, kortikospinale
funn i underekstremitetene, utelukkelse av andre sykdomsforklaringer og genetisk testing.[15]
De differensialdiagnoser man må utelukke, er strukturelle abnormaliteter, andre motor neuron
sykdommer (ALS, PLS), leukodystrofier, multippel sklerose, vitamin B12 mangel, Spinocerebellære
ataksier, friedreich ataksi, metabolske sykdommer, tidlig debuterende Alzheimers grunnet PS1
mutasjoner, dopa-responsiv dystoni, infeksjoner (HIV, syphilis mm.) og mitokondrielle sykdommer.
[14, 15]
Ved klinisk undersøkelse, vil man kunne se varierende grader av spastisitet og svekkelse i
underekstremitetene, hvorav noen individer kun har spastisitet. De muskelgrupper man finner mest
uttalt spastisitet i er hamstrings, quadriceps, adduserende muskulatur og gastrocnemius. Svekkelser er
mest uttalt og oftest funnet i iliopsoas, hamstrings og tibialis anterior. [14] Andre kliniske funn, er
hyperrefleksi i underekstremiterer, og inversjon av plantar refleks. Vibrasjonssans kan ofte være
påvirket helt distalt. Det kan videre finnes andre ledsagende symptomer, men som tidligere beskrevet
omdefinerer dette diagnosen til en kompleks spastisk paraparese.
Debut-alder varierer, selv innen samme familie, og innen samme gen-variant av sykdommer. Noen
genetiske varianter av sykdommen har en generelt tidligere alder for debut, men dette er ikke en fast
regel som kan brukes til å forutsi hvilke gen som er involvert. Det samme gjelder progresjonsrate. Den
kan være relativt statisk, den kan progressivt bli verre, og mange kan få et "funksjonelt platå" av sin
sykdom, med tilsynelatende stabilisering av deres funksjonstap. Ofte kan man se hos pasienter som har
debutert tidlig (eg. før 2års alder) at de har en nonprogressiv sykdom over flere ti-år før det er noen
markant forverring av symptombyrde, mens senere debuterende kan ha en mer stødig, progressiv
variant. [16]

10
MR undersøkelser av CNS er oftest normale. Men det vanligste funnet, om noe finnes, er uttynning av
ryggmarg, spesielt thorakalt. Videre kan man iblant finne abnormaliteter i selve hjernen, som kan være
av verdi for genotypisk strategi. Man kan for eksempel finne gracil corpus callosum, som kan være til
stedet ved blant annet SPG11, SPG4, 7, 15 og så videre. Andre funn kan være spredte hvite substans
lesjoner, atrofi av spesifikke korticale områder og dypere ganglier. Ingen av disse funnene er
spesifikke for kun enkelt genvarianter, men kan fungere som en pekepinn. [17]
Ved nevropatologiske studier, som nevrografi og EMG, er det vanligste funnet ved ren spastisk
paraparese en akson degenerasjon, som er mest uttalt i de distale deler av den kortikospinale banen,
mild tap av anteriore hornceller, eller demyelinisering konsistent med graden av aksonal skade. [14,
16]
Genetisk testing er mer og mer tilgjengelig, og et nyttig verktøy for å bekrefte en eventuell
diagnose[16]. Men ikke alle gener er enda avdekket, og genetisk testing må utføres strategisk, og
oftest rutinemessig over kun noen typer HSP (SPG3A, SPG4, SPG6, SPG7, SPG10, SPG11, SPG17,
SPG31)[18]. Derfor kan ikke genetisk testing alene ekskludere en hereditær spastisk paraparese
diagnose.
Familiehistorien kan ofte være fraværende, som gir inntrykket av å tilsynelatende være en isolert
sykdom, men som egentlig er en sporadisk form hvor familiehistorie ikke har framkommet av ulike
årsaker [15].
Genetisk klassifikasjon:
Det er til nå identifisert over 54 ulike gener som fører til spastisk paraparese, og tallet øker stadig, med
over 71 ulike loci.[18] Den kan videreføres via autosomal dominant (AD) arvegang, autosomal
recessiv (AR), X-bundet (XL) eller mitokondrielt. Sporadiske former kan komme grunnet de novo
mutasjoner, genologisk sensur, non-penetrant AD mutasjoner, eller om kun ett familiemedlem er
påvirket i en liten, og egentlig recessiv familie.[16]
Av autosomalt dominante har flertallet en ren form for spastisk paraparese, da opp til 70-80%, og den
vanligste typen er SPG4 og SPG3A [14]. Noen kliniske trekk ved disse to typene HSP, er en tidlig
debutalder for SPG3A, oftest før 10 års alder, og den kan ha en relativt non progressiv utvikling. Den
er oftest ren HSP, men kan i mer sjeldne tilfeller være assosiert med komplekse trekk, da for eksempel
motorisensoriell nevropati, tynn corpus callosum og kognitiv svikt. SPG4 gir oftest en senere debut,
med et gjennomsnitt på 34 år. Men det er høy variasjon i debutalder, fra fødsel til 80 års alder. SPG4
er i all hovedsak ren, men en liten andel er assosiert med komplekse symptomer, som ataksi, kognitiv
svikt, tynn corpus callosum og muskelsvinn. [14, 19]
Autosomalt recessive former er sjeldne, spesielt i populasjoner med lav forekomst av inngifte foreldre.
Disse er oftest komplekse tilstander, med noen få unntak, henholdsvis SPG5, 24, 28 og 30.Vanligste
type recessiv HSP er SPG11 og SPG15, som begge er klinisk overlappende. De debuterer begge i
tidlig barndom med mental utviklingshemning, og spastisiteten i underekstremitetene tilkommer
senere i livet. [19]
X-bundet HSP har 5 kjente former, vanligste form SPG1, som kun affiserer gutter[14]. Alle typer av
X-bundet HSP er komplekse tilstander. SPG1 er karakterisert av debut i spedbarnsalder med spastisitet
i underekstremitetene og mental utviklingsheming. Den kan videre ha afasi, hydrocephalus og
adduserte tomler. (Også kalt MASA syndrom.[19])

11
For mer informasjon om hereditære spastiske parapareser, se vedlegg 3.
Et viktig poeng vedrørende ulike genotypers særtrekk, er at det er sjeldent faste korrelasjoner mellom
genotype-fenotype, og disse er sjeldent spesifikke nok til å la klinikken gi lys over hvilke genotype
pasienter er affisert med[18]. Dette ettersom det er en voldsom variasjon av fenotype ved de ulike
genetiske feilene, selv innad i familier, hvor noen familiemedlemmer kan ha en ren form, og andre en
kompleks form. Denne variasjonen kan tenktes skyldt i varierende penetrans av genfeilen, og ulike
"modifiserende faktorer" hvor andre gener og miljøfaktorer påvirker sykdommens forløp. [16]
Selve fordelingen av de ulike varianter av SPG i en befolkning, kan man se varierer avhengig av om
det er en klar familiehistorie med spastisk paraparese, eller om det er et sporadisk tilfelle. Det
eksemplifiseres fra data fra et Italiensk nettverk, basert på over 1700 pasienter, hvor en ser en ulik
distribusjon av type HSP mellom sporadiske og familiære tilfeller, hvor det var oftere funnet recessive
sykdommer hos de med tilsynelatende sporadisk forekomst.[18]
Patofysiologi:
Det er mange ulike cellulære prosesser involvert i patofysiologien til spastisk paraparese, avhengig av
hvilket protein deres genfeil affiserer. De ulike mekanismene er feil i membran og aksonal
transportmekanismer, forstyrrelser i endoplasmatic retikulums morfologi, påvirking av mitokodriell
funksjon, feil i DNA reparasjonssystemet, forstyrrelser i vesikkelformasjon, autofagi, abdnormaliteter
i lipid metabolismen og myeliniseringsprosesser, oksydativt stress mm. [16, 18]
Til sammen påvirker disse cellulære prosesser i stor grad de lengste aksonene, da i den kortikospinale
tractus, og i en mindre grad bakstrengens fibre, enten via påvirkning fra innen i nervecellen selv, eller
påvirkning via feil i hjelpeceller.[16]
Epidemiologi:
Det var ved en studie i sørøst Norge estimert en prevalens på 7,4/100 000. ADHSP 5.5/100 000,
ARHSP 0,6/100 000, sporadisk 1,3/100 000. [13] En metaanalyse utført i 2014 regnet en global
prevalens på ca 1,8/100 000 ARHSP og 1,8/100 000ADHSP [1]. I denne samme studien, manglet 45-
67% av dominante HSP genetisk diagnose, og 71-81% av recessive HSP manglet diagnose-
Prognose og behandling:
Prognosen varierer med fenotype. Noen pasienter har en helt normal livslengde, mens andre kan bli
påvirket av kompliserende symptomer eller tidlig invalidisering, og dø tidligere grunnet dette[14].
Det finnes ingen kurativ behandling for spastisk paraparese. All behandling er symptomatisk, og går ut
på fysioterapi og daglig trening, for å opprettholde funksjonsnivå og hindre komplikasjoner som
kontrakturer. Annen behandling går ut på anti-spasmodisk medisiner, slik som Baclofen, eller
injeksjoner med botulinum toksin i spastisk muskulatur, eller oxybutilin for reduksjon av urge
inkontinens. [15]

12
Mål med oppgaven
Det er ved Ullevål sykehus blitt samlet en kohort av pasienter siden 2002, bestående av ca. 550
affiserte individer undersøkt av legene i prosjektet. Målet for prosjektet har vært å identifisere affiserte
pasienter i Norge, kartlegge dem klinisk og registrere blodprøver i en biobank, for å avdekke genetiske
årsaker til de ulike sykdommene, og til slutt å bidra til kartlegging av patofysiologi og fremme
behandlingsstrategier for disse pasientgruppene. Kun ca 50% av pasientene i kohorten har fått en
presis genetisk diagnose. Det foregår flere studier for å finne genetisk årsak i de resterende individer.
Denne oppgaven er et deskriptivt studie, med den hensikt å nøye kartlegge de uavklarte tilfellene av
hereditær spastisk paraparese og ataksi, for å avdekke ulike kliniske karakteristika, finne eventuelle
spesifikke fenotyper, for å mulig orientere genetiske strategier videre.
Alle de kliniske dataene er samlet i forskningsdatabasen, og pasienter uten genetisk diagnose
selekteres for å granske eventuelle likheter med vekt på følgende trekk:
- Debutalder
- Første symptom
- Arvmønster, evt. særtrekk mellom dem
- Assosierte nevrologiske symptomer
- Progresjonsrate
- Alvorlighetsgrad
- Andre assosierte sykdommer
- Andre særtrekk
- Funn (MR, Nevrografi/EMG)

13
Materiale og metoder
Pasientmateriale
Oppgaven er en deskriptivt studie basert på informasjon samlet i en Norsk kohort ved Ullevål sykehus,
startet i 2002, hvor informasjonen er registrert i Excel programvare. Kohorten inneholder pasienter
som har samtykket skriftlig for deltagelse, og består overveiende av voksne individer.
Alle kliniske opplysninger og ytterligere undersøkelser har blitt systematisk registrert etter en
protokoll dannet av SPATAX, den Europeiske og mediterranske nettverk for analyse av
spinocerebellære degenerative lidelser, via to standardiserte kliniske skjemaer.
Pasientene er gradert etter sannsynlighet av affeksjon, fra "sikkert affisert" til "sannsynlig affisert", og
"mulig affisert", gradert etter kliniske tegn, symptomer og familiehistorie. Alle ble inkludert i denne
oppgaven.
Denne preeksisterende informasjonen i databasen har deretter blitt gjennomgått for hånd, alle variabler
som var nedtegnet med ord istedenfor tall har blitt standardisert nummerisk etter skjemaers
spesifikasjon, og de manglende verdier har blitt innhentet fra utfylte skjemaer lagret i permer, og
ytterligere manglende informasjon innhentet fra journaler i samarbeid med veiledere.
Til sammen 230 pasienter med arvelig ataksi og 161 pasienter med hereditær spastisk paraparese er
inkludert i denne oppgaven.
Alle barn og alle med sikker genetisk diagnose har blitt ekskludert fra studien.
Statistisk metode
Data har blitt gjennomgått ved bruk av IBM SPSS Statistics versjon 22. Pasientmaterialet har enten
blitt gjennomgått per individ, eller per proband.
Programmets funksjon "Descriptive Statistics" ble benyttet ved alle statistiske analyser av
pasientmaterialet. "Frequency" ble brukt for alle nominative non-kontinuerlige varibler og alle string
variabler, og "Explore" ved undersøkelse av nominative, kontinuerlige variabler. "Crosstabs" ved
sammenligninger.
Ved undersøkelse av diverse undergrupper, har "Select cases" blitt brukt til å undersøke kun individer
som oppfyller visse krav, og disse undergruppene videre undersøkt på lik linje med hele
pasientmaterialet. Deretter sammenlignet.
Denne informasjon har deretter blitt grafisk framstilt via funksjonen "Graph".
Oppgitte P-verdier har blitt utregnet ved hjelp av GraphPad Software: QuickCalcs, t-test calculator.
Litteratur:
Bakgrunnsinformasjon er basert på et ikke-systematisk søk i McMaster Plus og direkte i Pubmed og
Uptodate,. Det har overveiende blitt selektert oppsummerende litteratur, metaanalyser, og noen få
enkeltstudier.
14
Resultater
Hereditær ataxi:
Generell oversikt
Diagnosesikkerhet:
Alle individer i databasen er klassifisert med diagnosesikkerhet
fra 1-3, hvor 1 er sikker diagnose, 2 er sannsynlig, og 3 er
mulig diagnose. 67,0% har en sikker diagnose. 16,1% har en
sannsynlig diagnose, og 15,2% har en mulig diagnose. 1,7% av
pasientmaterialet var uten oppgitt diagnosesikkerhet.
Av de med usikker diagnose, som da utgjør 35 individer, har
54% kjent familiær arvegang, resterende er sporadiske. Av dem
med kjent arvegang, er 67% dominante. Det er videre et
forholdstall på 1:3 mellom rene og komplekse tilstander.
Kjønnsfordeling:
Det var ingen vesentlig forskjell mellom antall kvinner og menn i pasientmaterialet. Av de med
oppgitt kjønn, er 52,4% av pasientmaterialet menn og 47,6% kvinner. 2,2% har ikke oppgitt kjønn i
databasen.
Stadiet:
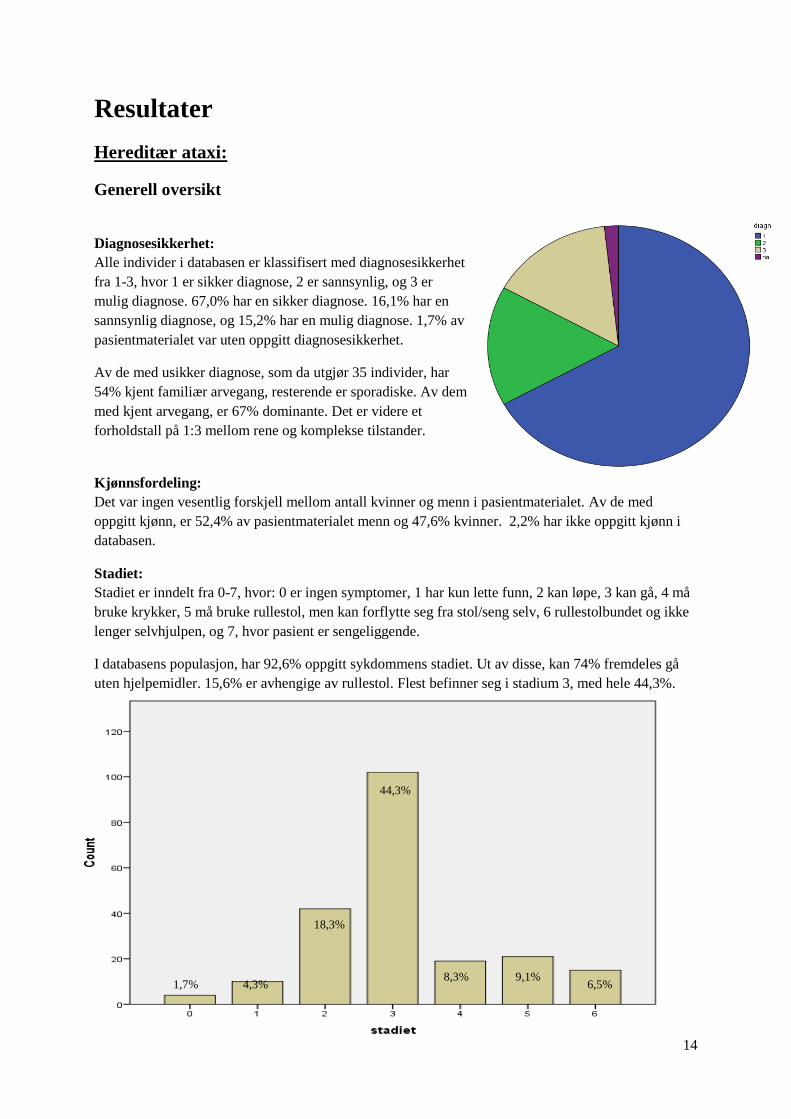
Stadiet er inndelt fra 0-7, hvor: 0 er ingen symptomer, 1 har kun lette funn, 2 kan løpe, 3 kan gå, 4 må
bruke krykker, 5 må bruke rullestol, men kan forflytte seg fra stol/seng selv, 6 rullestolbundet og ikke
lenger selvhjulpen, og 7, hvor pasient er sengeliggende.
I databasens populasjon, har 92,6% oppgitt sykdommens stadiet. Ut av disse, kan 74% fremdeles gå
uten hjelpemidler. 15,6% er avhengige av rullestol. Flest befinner seg i stadium 3, med hele 44,3%.
44,3%
18,3%
4,3% 8,3% 9,1%
6,5% 1,7%

15
Transmisjon:
1,3% har ingen oppgitt informasjon vedrørende transmisjonsmåte.
Det vises her fordeling av transmisjonsmåte mellom antall familier representert i databasen, dvs. per
proband.
Sporadiske tilfeller er pasienter uten familiehistorie, men som det mistenktes å foreligge en ubekreftet
familiær nedarving av sykdommen. Uspesifisert familiær type, er av kjent familiær type, hvorav
arvemønsteret ikke mulig lar seg avgjøre med eksisterende informasjon om pasientens familie.
Det er til sammen 230 individer i databasen, fordelt i 180 familier. Ut av disse familiene, er 56,6%
sporadiske enkeltstående individer. 26,1% er dominante familier, 9,4% er recessive og 6,7% er
uspesifiserte men med kjent familiehistorie.
Dette utgjør til sammen 82 enkeltindivider med dominant arvegang, 27 individer med recessiv
arvegang, 16 individer med uspesifisert familiær arvegang, og 102 sporadiske tilfeller.
Klassifikasjon
Databasen har en overvekt av pasienter med
kompleks sykdomsbilde. Av de med oppgitt
klassifikasjon, er 65,3% komplekse, 31,9%
er rene, og 2,7% er episodiske.
3,5% har ikke oppgitt klassifikasjon.

16
Debut -alder
Gjennomsnittsalder for debut er 35 år (SD 22,3). Men, som tydeliggjort av grafen under, er det et fåtall
av pasientene som debuterer i 30 årene. Isteden, er det to grupper for debut: En tidlig, og en sen.
Hos gruppen med tidlig debut, er det en hovedvekt av debut allerede i de første leveår, som deretter
avtar gradvis jo eldre de blir. 19% av pasientmaterialet har debutert innen de er 10 år.
For å demonstrere pasientgruppen med sen debut, har alle med en alder for debut over eller lik 40 blitt
selektert og fremstilt i grafen over til høyre. Normalfordelingen av denne populasjonen har et
gjennomsnitt på 55,3 år (SD 7,6). 23% av hele pasientmaterialet debuterer i aldersgruppen 50-59.
4,8% av pasientmaterialet har ingen oppgitt debut alder.

17
Debuterende symptom
3% hadde ingen oppgitt debuterende symptom.
77% av dem med oppgitt debut symptom debuterte med ustøhet. 3,5% debuterte med tremor.
Resterende 16,5% debuterte med andre symptomer, herunder dysartri, epilepsi, hukommelsessvikt,
synsproblematikk, svelgvansker, episodiske symptomer, svimmelhet eller svekkelser i ulike muskler.
Alder ved undersøkelse og varighet av sykdom
5,6% av pasientmaterialet har ikke oppgitt varighet av sykdom, og 5,6% hadde ikke oppgitt alder for
undersøkelse.
Gjennomsnittlig alder ved undersøkelse var 53,7 år (SD 17,11). Grafisk er det en topp i aldersklassen
55-65 år.
Gjennomsnittlig varighet var på 18,5 år (SD 15,14). 61% av pasientmaterialet hadde en varighet på
under 20 år.
Symptomer og klinisk bilde
Gjennomgående mangles det fra 8-16% av ulike symptom-opplysninger i pasientmaterialet.
Manglende data er ekskludert fra prosentutregningene.
Cerebellære symtomer:
91,8% av pasientmaterialet hadde gangataksi, og av dem med gangataksi, hadde 1/2 en mild ataksi,
1/3 hadde en moderat ataksi, og 1/6 hadde en alvorlig ataksi. 86,2% av hele pasientmaterialet hadde
ataksi i underekstremitetene, mens 76,8% hadde ataksi i overekstremitetene, begge med tilsvarende
alvorlighetsgrad som gangataksi.
59,8% av pasientmaterialet hadde dysartri, overveiende mild form.
Spastisitet og hyperrefleksi:
17,6% av pasientmaterialet hadde hvilespastisitet, 15,7% hadde ganspastisitet. 16% av

18
pasientmaterialet var kun spastiske i under ekstremitetene, 3% var spastiske både i underekstremiteter
og over ekstremiteter. 5,3% hadde clonus. Hyperrefleksien var overveiende funnet i
underekstremitetene. 7,6% hadde positiv hoffmanns.
Kraft og muskelsvinn:
18% av pasientmaterialet hadde krafstap i underekstremitetene, 16,1 % hadde muskelsvinn. Begge
funn var mest uttalt distalt og hovedsakelig klassifisert som mildt til moderat.
9,5% hadde krafttap i overekstremitetene, 10,5% hadde muskelsvinn, begge igjen mest uttalt distalt,
og hovedsakelig klassifisert som mildt til moderat.
Vibrasjonssans og sensibilitet:
26% av pasientmaterialet hadde uspesifiserte utfall av sensibilitet, alle klassifisert som milde. 61,4%
hadde en nedsatt vibrasjonssans målt ved laterale malleol. 23,9% av hele pasientmaterialet hadde
fullstendig tap av vibrasjonssans.
Tremor:
30% av pasientmaterialet hadde en form for postural, uspesifisert tremor, flertallet en mild utgave.
10,4% av pasientmaterialet hadde hviletremor, overveiende mild form.
Ekstrapyramidale symptomer:
20% av pasientmaterialet har ekstrapyramidale symptomer. 7,2% hadde akinesi, 13,9% hadde amimi,
og 7,3% hadde choreatiske bevegelsesforstyrrelser.
Myokloni:
7,5% hadde myokloni. 3,0% hadde myokymier.
Dysfagi:
23,7% av pasientene hadde mild dysfagi.
Dystoni:
10,3% av pasientene hadde dystoni.
Plantar:
25% av pasientene hadde en bilateral inversjon.
Øyemotilitetsforstyrrelser:
76% hadde en affeksjon av øyemotilitet. 66% av pasientene hadde nystagmus, sakkadisk eller sluggish
pursuit. 4,4% hadde oftalmplegi, og 4,4% hadde okulomotorisk apraksi.
Utviklingshemming:
11,0% av pasientmaterialet hadde en motorisk utviklingshemming. 10,3% hadde en mental
utviklinghemming. 71% av dem med motorisk utviklingshemning, hadde også en mental
utviklingshemning.
Kognitiv svikt:
25,3% av pasientene hadde en kognitiv svikt.
Andre sykdommer/assosierte lidelser:
Det var kun enkelttilfeller av andre assosierte lidelser, der i blant 4 individer med diabetes, 7 med en
historikk med epileptiske anfall, 3 med hjertearytmi, 3 med hypertensjon, 22 med nedsatt hørsel og 5
med hammertær.

19
MR funn
21,3% av pasientmaterialet hadde manglende
informasjon om MR funn.
Av de med oppgitt informasjon, hadde 76,2%
degenerative forandringer infratentorielt og eller
supratentorielt. 9,9% hadde andre funn, herunder
uspesifikke høysignalsforandringer eller vaskulære
funn. 13,8% hadde normale funn.
Progresjon
Hele pasientmaterialet er inkludert. Hver bar er prosentvis inndeling av andel pasienter med ulik
alvorlighetsgrad, innenfor hvert tiår med sykdommen.
Andel som har behov for hjelpemidler for å gå (gruppe 4-6) øker gradvis for hvert tiår med
sykdommen. Hos de som har hatt sykdommen i 20-29 år, har 35% av pasientene behov
forhjelpemiddel for å forflytte seg, 24% bruker rullestol. Til sammenligning har 16% behov for
hjelpemidler ved gange ved varighet under 10 år.

20
Til sammenligning ser man at i gruppen tidligere beskrevet som dem med "sen debut", her valgt
pasienter med debut etter 35 år, er 28% av pasientene med sykdomsvarighet på 10-19 år avhengige av
hjelpemidler for å forflytte seg. Hele 23,1% er avhengige av rullestol. 70% er avhengige av
hjelpemidler ved 20-29 år. Ved varighet under 10 år, er 18,5% avhengige av hjelpemidler ved gange.
Hos gruppen med tidlig debut, her valgt alle med debut før 25 års alder, har kun 23% behov for
hjelpemidler ved 20-29 år med sykdommen. Ingen har behov for hjelpemidler ved varighet under 10
år.
Dette gir inntrykk av at dem med tidlig debut, har en senere progresjon, enn dem med sen debut.
Pasienter med debut før 25 års alder
Pasienter med debut etter 35 års alder
21
Sammenligner av data for recessive, dominante, uspesifisert
familiære arvemønstre og sporadiske tilfeller av ataksi:
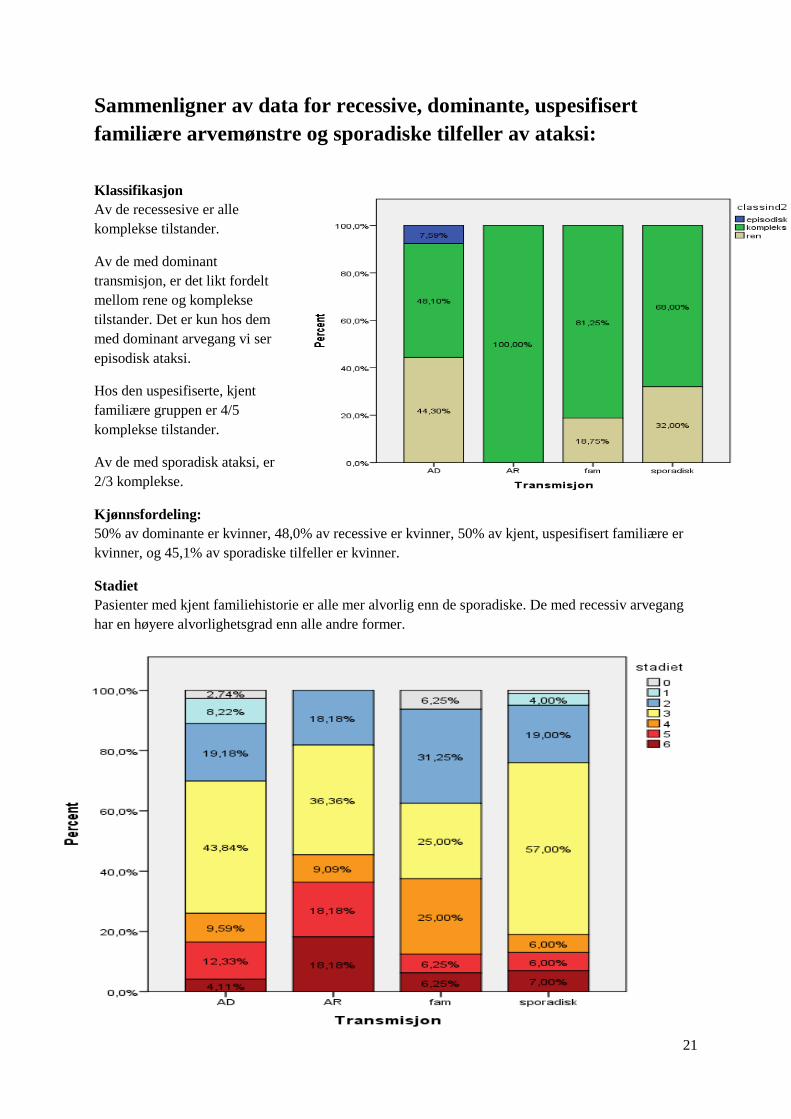
Klassifikasjon
Av de recessesive er alle
komplekse tilstander.
Av de med dominant
transmisjon, er det likt fordelt
mellom rene og komplekse
tilstander. Det er kun hos dem
med dominant arvegang vi ser
episodisk ataksi.
Hos den uspesifiserte, kjent
familiære gruppen er 4/5
komplekse tilstander.
Av de med sporadisk ataksi, er
2/3 komplekse.
Kjønnsfordeling:
50% av dominante er kvinner, 48,0% av recessive er kvinner, 50% av kjent, uspesifisert familiære er
kvinner, og 45,1% av sporadiske tilfeller er kvinner.
Stadiet
Pasienter med kjent familiehistorie er alle mer alvorlig enn de sporadiske. De med recessiv arvegang
har en høyere alvorlighetsgrad enn alle andre former.

22
Debut
Recessive: Dominante:
Uspesifisert familiære: Sporadiske:
P-verdi ved sammenligning av de ulike gjennomsnitt, utregnet via T-test:
AR AD SPOR
AR - <0,0001 -
FAM <0.0001 0.2109 0.8875
SPOR <0,0001 0.0340 -
De med recessiv arvegang har generelt en tidlig debut, gjennomsnitt 13,7 år (SD 15,6). Denne er
signifikant forskjellig fra alle andre grupper, med P-verdi<0,0001.
Dominante har to bølger med debut, en tidlig og en sen, med omtrent like mange individer i begge
gruppene. Gjennomsnitt på 33,7 (SD22,9). Gjennomsnittet er signifikant forskjellig fra sporadiske og
recessive, men ikke fra den uspesifiserte familiære gruppen.
Uspesifisert, kjent familiære har flest debuterende sent, gjennomsnitt på 41, 4 (SD 18,8). Den er
signifikant forskjellig fra recessive, men ikke fra dominante (P=0,21) eller sporadiske (P=0,88)
Sporadiske har flest debuterende i den sene gruppen, gjennomsnitt 40,6 (SD 20,4). Den er signifikant
forskjellig fra recessive og dominante, men ikke mot den uspesifiserte, kjent familiære gruppen.
23
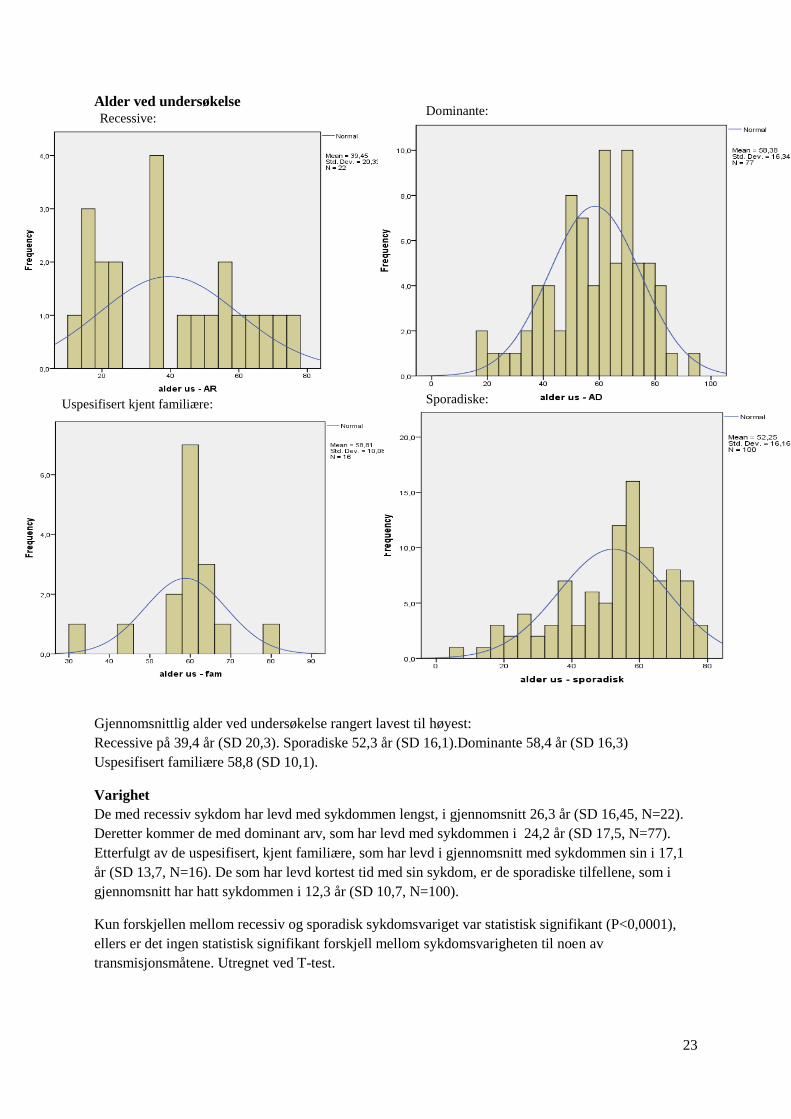
Alder ved undersøkelse
Gjennomsnittlig alder ved undersøkelse rangert lavest til høyest:
Recessive på 39,4 år (SD 20,3). Sporadiske 52,3 år (SD 16,1).Dominante 58,4 år (SD 16,3)
Uspesifisert familiære 58,8 (SD 10,1).
Varighet
De med recessiv sykdom har levd med sykdommen lengst, i gjennomsnitt 26,3 år (SD 16,45, N=22).
Deretter kommer de med dominant arv, som har levd med sykdommen i 24,2 år (SD 17,5, N=77).
Etterfulgt av de uspesifisert, kjent familiære, som har levd i gjennomsnitt med sykdommen sin i 17,1
år (SD 13,7, N=16). De som har levd kortest tid med sin sykdom, er de sporadiske tilfellene, som i
gjennomsnitt har hatt sykdommen i 12,3 år (SD 10,7, N=100).
Kun forskjellen mellom recessiv og sporadisk sykdomsvariget var statistisk signifikant (P<0,0001),
ellers er det ingen statistisk signifikant forskjell mellom sykdomsvarigheten til noen av
transmisjonsmåtene. Utregnet ved T-test.
Recessive: Dominante:
Uspesifisert kjent familiære: Sporadiske:

24
Symptomer og klinisk bilde
De symptomer og funn hvor det fremkommer forskjeller mellom de ulike gruppene og hele
pasientmaterialet, trekkes fram her. Øvrige funn var tilnærmet likt distribuert mellom de ulike
gruppene.
Dem med recessivt arvemønster, manglet gjennomgående mer informasjon enn øvrige grupper, og
hadde gjennomgående en mangel på 20-30% ved alle symptomer/funn, med unntak motorisk
utviklingshemning, hvor kun 7,4% manglet informasjon. Øvrige grupper har gjennomsnittlig mangler
som tidligere spesifisert for hele pasientmaterialet.
Spastisitet
Pasienter med recessivt arvemønster hadde en høyere andel med spastisk gange (48,4%) enn øvrige
pasientgrupper ( ~12%), og en høyere andel med hvilespastisitet (42,9%) enn øvrige grupper (~17%).
Av dem med uspesifisert familiær arvegang hadde ingen spastisitet. Recessive hadde flere tilfeller med
spastisitet i både under ekstremiteter og over ekstremiteter enn øvrige grupper (15,8%, mot ~3%).
Clonus var likt representert i alle pasientgrupper.
Kraft
40% av dem med recessiv arvegang hadde utfall i kraft i underekstremiteter, 30% hadde utfall i
overekstremiteter, mer uttalt distalt. Øvrige grupper hadde ~20% med utfall i kraft i
underekstremiteter, 10% i overekstremiteter.
Plantar
57% av recessive hadde bilateral plantar inversjon, 31% av uspesifisert familiære hadde bilateral
inversjon. Øvrige hadde fra 19-21% tilfeller med bilateral inversjon.
Utviklingshemning:
Pasienter med recessivt arvemønster hadde en høyere andel med motorisk utviklingshemning (36%)
enn sporadiske tilfeller (8,1%) , dominante (6,6%) og uspesifisert familiære (12,5%).
Videre har 40% av dem med recessivt arvemønster en mental utviklingshemning, 4,3% av dem med
dominant arvegang hadde dette, 13,3% av dem med uspesifisert familiært arvemønster, og 7,9% av
sporadiske tilfeller hadde det.
Av recessive med motorisk utviklingshemning, hadde 9/10 også en mental utviklingshemning.
Kognitiv svikt
50% av dem med recessiv arvegang hadde en kognitiv svikt, mens 7,5% av dem med dominant
arvegang hadde dette, 33% av dem med uspesifisert arvemønster, og 31,% av sporadiske tilfeller.
Ekstrapyramidale funn:
Pasienter med kjent familiære former av sykdommen hadde en lavere forekomst av ekstrapyramidale
funn (Uspesifisert kjent familiær: 6,3%, AD:15,3%, AR:18,2%), enn sporadiske tilfeller (27,1%).
Øyemotilitet
Øyemotilitetsfunn er kategorisert som:
0: ingen funn.
1: sakkadisk pursuit og nystagmus.
2:oftalmoplegi.
3 okulomotorisk apraksi/ dyspraksi.
4: Trege sakkader.

25
Den mest uttalte forskjellen, er hvor
overrepresentert okulomotorisk
apraksi/dyspraksi er hos dem med
recessiv arvegang, hvor hele 35% av
gruppen har dette funnet,
sammenlignet med de andre
gruppene, hvor det er kun en liten
andel som har dette funnet.
Andre forskjeller, er hvordan den
sporadiske og den uspesifisert kjent
familiære gruppen har en større andel
enn de to andre gruppene med
oftalmoplegi, med 5,15% og 13,33%
respektivt.
MR funn
Hos dem med recessiv arvegang, var det ingen pasienter uten MR funn. Ellers er funnene relativt likt
distribuert mellom de ulike gruppene.
36% av dominante, 40% av recessive, 0% av uspesifisert kjent familiære, og 10% av sporadiske
manglet informasjon.
0=normale funn
1= degenerative
forandringer
2= andre funn
(HSO, vaskulære)

26
Progresjon
Ettersom det var to grupper for debut for sporadisk og dominante tilfeller, er disse splittet i tre
grupper: en graf over alle individer med sporadisk eller dominante tilfeller, en med kun individer som
debuterte under en alder av 25 år, og en hvor alle har en debut etter alder 40, for å sammenligne
progresjonen i de ulike gruppene. Ettersom det ikke var definerte grupper for debut innenfor recessive
og uspesifisert familiære, og det er få individer i disse gruppene, har disse ikke blitt splittet.
Uspesifisert, kjent familiær arvegang:
Ettersom det er kun 16 individer er her
fordelt på 5 ulike klasser, er det vanskelig
å si noe spesifikt om progresjonen, som
gjenspeiles i grafen til høyre.
Recessiv arvegang
Igjen er det nå kun 22 individer fordelt på 6
klasser, som gjør det vanskelig å si noe
definitivt. Men man får inntrykk av en rask
progresjon til alvorlige sykdomsformer,
ettersom det ikke finnes milde tilfeller av
sykdommen etter en varighet på 20 år.
Verdt å merke seg at det ikke fantes
pasienter med varighet under 10 år.

27
Dominant arvegang
Dem med en tidlig debut, ser ut til å
ha en senere progresjon av deres
sykdom, enn dem med debut etter
fylte 40 år, men fremstillingen av
dataen er påvirket av et lite antall i
gruppen med både tidlig debut og
kort varighet av sykdommen.
Selektert kun dem med debut før alder 25år: Selektert kun dem med debut etter fylte 40 år:

28
Sporadisk ataksi
Dem med en tidlig debut, ser
ut som å ha en senere
progresjon av deres sykdom,
enn dem som debuterer etter
fylte 40 år.
Selektert kun dem med debut før alder 25år: Selektert kun dem med debut etter fylte 40 år

29
Hereditær spastisk paraparese:
Generell oversikt
Diagnosesikkerhet:
78,9% har en sikker diagnose (1 i grafen). 7,5% har en sannsynlig
diagnose (2), og 13,7% har en mulig diagnose(3). Alle pasientene
i databasen hadde oppgitt diagnosesikkerhet.
Av dem med usikker diagnose, som utgjorde til sammen 22
pasienter, var 71% komplekse tilstander, resterende var rene. 36%
var dominant arvelige, 9% var recessive, 4,5% uspesifisert kjent
familiære og 52% var sporadiske.
Kjønnsfordeling:
Det var ingen forskjell mellom antall kvinner eller menn i pasientmaterialet. 49,4% var menn, 50,6%
var kvinner. Kun 0,6% av databasen manglet kjønnsopplysninger.
Stadiet:
Stadiet er inndelt fra 0-7, hvor: 0 er ingen symptomer, 1 har kun lette funn, 2 kan løpe, 3 kan gå, 4 må
bruke krykker, 5 må bruke rullestol, men kan forflytte seg fra stol/seng selv, 6 rullestolbundet og ikke
lenger selvhjulpen, og 7, hvor pasienten er sengeliggende.
1,2% av databasen mangler oppgitt stadiet. Av dem med oppgitt stadiet, kan 71% fremdeles gå uten
hjelpemidler. 18,2% av databasen er avhengige av rullestol. Flest individer befinner seg i stadiet 3,
med 44,7%.

30
Transmisjon:
Alle individer i databasen har oppgitt transmisjonsmåte. Det vises her fordeling av transmisjonsmåte
mellom familiene representert i databasen, dvs. per proband.
Sporadiske tilfeller er pasienter uten familiehistorie, men som det mistenktes å foreligge en ubekreftet
familiær nedarving av sykdommen. Uspesifisert familiær type, er av kjent familiær type, hvorav
arvemønsteret ikke mulig lar seg avgjøre med eksisterende informasjon om pasientens familie.
Det er 161 individer i databasen, fordelt i 120 familier. 30,0% av familiene har dominant arvegang.
5,8% av familiene har en recessiv arvegang. 8,3% har en uspesifisert men kjent familiær arvegang
(abbreviert til fam i alle grafer), og 55,8% er sporadiske sykdomstilfeller.
Dette utgjør totalt 70 enkeltindivider med dominant arvegang, 10 individer med recessiv arvegang, 13
med uspesifisert kjent familiær arvegang, og 68 individer med sporadisk sykdom.
Klassifikasjon
Pasientmaterialet er relativt likt
fordelt mellom komplekse og
rene tilstander.
Av dem med oppgitt
informasjon, har 48,4% en
kompleks sykdomstilstand.
51,5% hadde en ren
sykdomstilstand.
1,2% hadde ingen oppgitt
klassifikasjon.

31
Debut -alder
Gjennomsnitt for debut er 27,14 år (SD 21,68). Men det et fåtall som har debut i denne alderen, og
istedenfor en gruppe som debuterer tidlig, og en sent.
3,1% mangler data for debut.
Hos dem med tidlig debut, er det en topp før en alder av ti år, hvor 27,7% av pasientmaterialet har
debutert. En stor andel av disse har allerede debutert innen 2 års alder.
Hos dem som debuterer sent, her dem etter alder av 40 år, er det et gjennomsnitt på 53 år (SD 8,62).
Grafisk er det fremstilt en topp i alder 50-53, og en topp ved 60-63. 23,2% av pasientmaterialet har
debutert i alder 50-69 år.

32
Debuterende symptom
1,2% hadde ingen oppgitt debuterende symptom.
36% debuterte med ustøhet, 44% debuterte med spastisitet/stivhet, 10% debuterte med uspesifisert
smerte, og 9% debuterte med "annet", herunder krafttap, nummenhet, blæreproblematikk, tremor eller
kramper.
Alder ved undersøkelse og varighet av sykdom
3,1% har ikke oppgitt varighet av sykdom, 0,6% har ikke oppgitt alder ved undersøkelse.
Gjennomsnittlig alder for undersøkelse er 47,7 år (SD 17,59). Toppen i selve grafen ligger nærmere 60
år.
Gjennomsnittlig alder for varighet er 20,8 (SD 16,63). Toppen i grafen ligger nærmere 5 år. 57% av
pasientmaterialet har hatt en varighet under 20 år.
Symptomer og klinisk bilde
Gjennomgående mangles det fra 1,2% til 8,1% av symptomopplysningene.
Spastisitet
85,5% har hvilepastisitet, hvor 2/5 har en mild form, 1/2 har en moderat form, og 1/10 har en alvorlig
form. 88,4% har gangspastisitet, hvor ca. 1/2 har en mild form, ca 2/5 har en moderat form, og 1/5 en
alvorlig form. 80% av pasientmaterialet er spastiske kun i underekstremitetene. 14,2% er spastisk i
både over og underekstremiteter.
39,2% av pasientene har clonus, og hyperrefleksien var hos flest i underekstremitetene, mest uttalt
distalt. 31,8% hadde positiv Hoffmanns.
Cerebellære symptomer
15,6% hadde gangataksi. 10,7% hadde ataksi i underekstremitetene. 16,6% hadde ataksi i
overekstremitene. Ataksien var i all hovedsak klassifisert som mild.

33
Kraft og muskelsvinn
50% av pasientmaterialet hadde krafttap i underekstremitetene, distalt og proksimalt likt påvirket,
flertallet fra mild til moderat affisert. 13,2% hadde muskelsvinn proksimalt i underekstremitetene, og
23,3% hadde muskelsvinn distalt. Muskelsvinnet var hovedsakelig mildt klassifisert, noen moderate.
10% av pasientmaterialet hadde krafttap i overekstremitetene, distalt mer påvirket enn proksimalt, alle
hovedsakelig mildt affisert. 8,1% hadde muskelsvinn distalt i overekstremitetene, hovedsakelig mildt
affisert. Kun et fåtall hadde muskelsvinn proksimalt i overekstremitetene.
Plantar refleks
81,8% hadde bilateral inversjon, 6,3% hadde ensidig inversjon. Kun 8,8% hadde upåfallende plantar
refleks.
Vibrasjonssans og sensibilitet
18,5% av pasientmaterialet hadde fullstendig bortfall av vibrasjonssans ved laterale malleol. 41,7%
hadde upåfallende vibrasjonssans.
25% hadde et uspesifisert, mildt sensibilitetsutfall.
Tremor
2,6% hadde hviletremor, 8,3% hadde postural tremor. Alle klassifisert som milde tilfeller.
Ekstrapyramidale symptomer
5% hadde ekstrapyramidale symptomer. 1,3% hadde akinesi, 4,6% amimi. Ingen pasienter hadde
chorea eller atetose.
Øyemotilitetsforstyrrelser
16,8% hadde øyemotilitetsforstyrrelser, da hovedsakelig i form av nystagmus eller sakkadisk pursuit.
Utviklingsheming
7,9% var mentalt utviklingshemmet. 9,1% hadde en motorisk utviklingshemning. Halvparten av dem
med motorisk utviklingshemning, hadde også mental utviklingshemning.
Kognitiv svikt
12,2% hadde en kognitiv svikt.
Hulfot
18,5% har hulfot. 1/2 er mildt affisert, 3/10 moderat, og 1/5 alvorlig affisert.
Annet
3,9% hadde dysfagi, alle milde. 3,1% hadde dystoni, alle milde. 14,7% hadde dysartri, flertallet milde.
1,3% hadde myokloni. 40% er plaget med uspesifisert smerteproblematikk.
Andre sykdommer/assosierte lidelser:
Det er kun enkelttilfeller av andre lidelser, derav 4 med diabetes, 4 med nyresykdom, 3 med epilepsi, 5
med skoliose, 11 med nedsatt syn, og 3 med psoriasis.

34
MR funn
MR-funn er inndelt etter 0= ingen funn, 1=degenerative
forandringer, 2= andre funn, slik som uspesifikke
høysignalsforandringer og ischemiske funn, og et fåtall med
gracil corpus callosum.
25,5% har ingen informasjon om MR funn.
Av dem med oppgitt informasjon, har 18,3% degenerative
forandringer i cerebellum og/eller cerebrum. 24,2% har
andre funn. 57,5% har normale funn.
EMG/nevrografi
32,9% manglet informasjon om EMG/nevrografi
Av dem med oppgitt informasjon, hadde 63% ingen
funn. 7,4% hadde kun funn i underekstremitetene (1 i
graf), og 29,6% hadde funn både i over-og
underekstremitetene (2 i graf).
Grafene under fremstiller de ulike typer funn i over og
underekstremitetene.
Det er flere med aksonal polynevropati eller både
aksonal og demyeliniserende, enn demyeliniserende alene. Men flertallet av pasienter med funn har en
uspesifisert polynevropati, som gjør forholdstolkningen her upresis. Det er ellers 1 tilfelle med
tynnfiber affeksjon, og 3 tilfeller med denervasjon.
EMG/nevrografi funn, oversikt
1: kun funn u ex
2: funn i både u ex og o ex
0=ingen funn
1=nevrogene forandringer, udefinerte PN
2=aksonal PN
3=demyeliniserende PN
4=aksonal OG demyeliniserende PN
5= tynnfiber
6=denervasjon
35
Progresjon
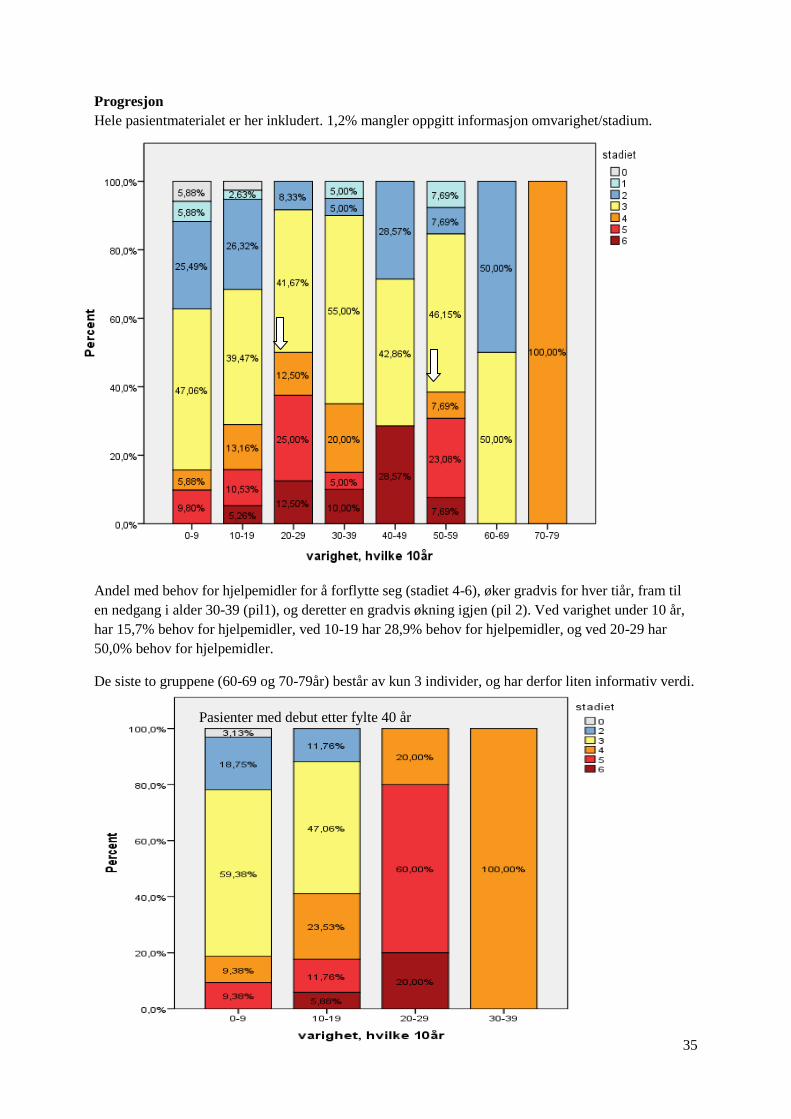
Hele pasientmaterialet er her inkludert. 1,2% mangler oppgitt informasjon omvarighet/stadium.
Andel med behov for hjelpemidler for å forflytte seg (stadiet 4-6), øker gradvis for hver tiår, fram til
en nedgang i alder 30-39 (pil1), og deretter en gradvis økning igjen (pil 2). Ved varighet under 10 år,
har 15,7% behov for hjelpemidler, ved 10-19 har 28,9% behov for hjelpemidler, og ved 20-29 har
50,0% behov for hjelpemidler.
De siste to gruppene (60-69 og 70-79år) består av kun 3 individer, og har derfor liten informativ verdi.
Pasienter med debut etter fylte 40 år
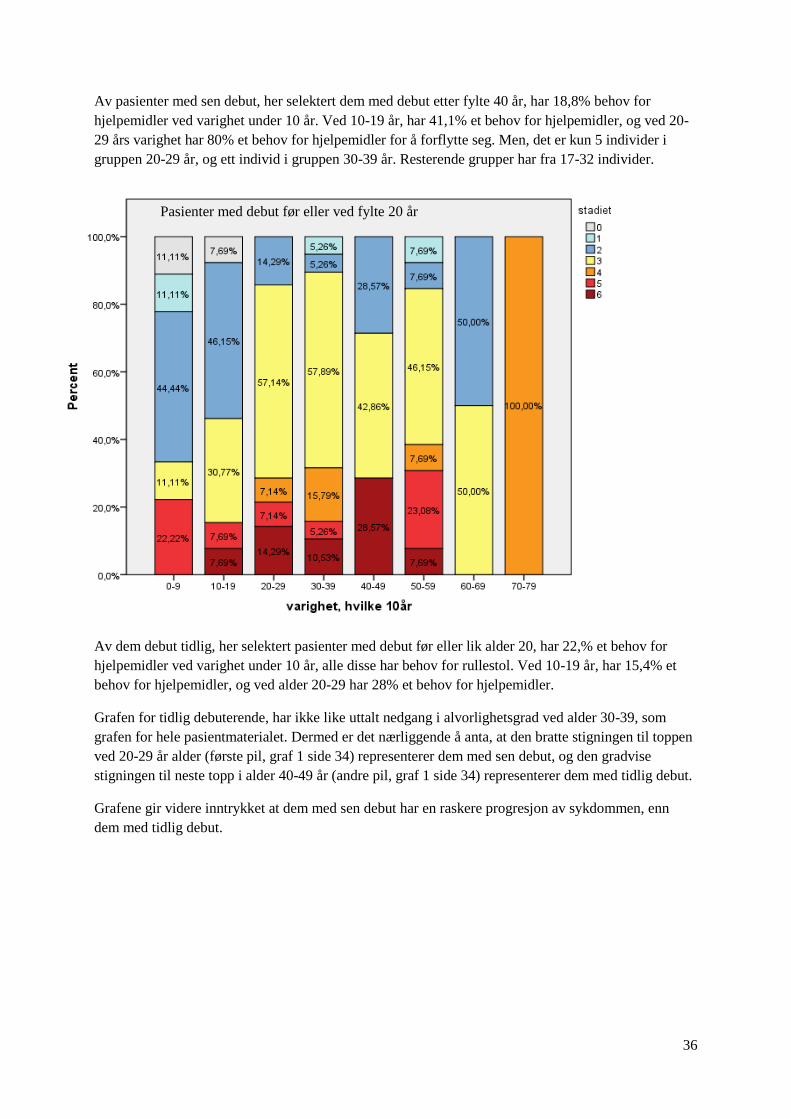
36
Av pasienter med sen debut, her selektert dem med debut etter fylte 40 år, har 18,8% behov for
hjelpemidler ved varighet under 10 år. Ved 10-19 år, har 41,1% et behov for hjelpemidler, og ved 20-
29 års varighet har 80% et behov for hjelpemidler for å forflytte seg. Men, det er kun 5 individer i
gruppen 20-29 år, og ett individ i gruppen 30-39 år. Resterende grupper har fra 17-32 individer.
Av dem debut tidlig, her selektert pasienter med debut før eller lik alder 20, har 22,% et behov for
hjelpemidler ved varighet under 10 år, alle disse har behov for rullestol. Ved 10-19 år, har 15,4% et
behov for hjelpemidler, og ved alder 20-29 har 28% et behov for hjelpemidler.
Grafen for tidlig debuterende, har ikke like uttalt nedgang i alvorlighetsgrad ved alder 30-39, som
grafen for hele pasientmaterialet. Dermed er det nærliggende å anta, at den bratte stigningen til toppen
ved 20-29 år alder (første pil, graf 1 side 34) representerer dem med sen debut, og den gradvise
stigningen til neste topp i alder 40-49 år (andre pil, graf 1 side 34) representerer dem med tidlig debut.
Grafene gir videre inntrykket at dem med sen debut har en raskere progresjon av sykdommen, enn
dem med tidlig debut.
Pasienter med debut før eller ved fylte 20 år

37
Sammenligner av data for recessive, dominante, uspesifisert
familiære arvemønstre og sporadiske tilfeller av HSP:
Klassifikasjon
Av de recessive tilstandene, er alle
komplekse.
Av de dominante sykdommene, er 3/5
rene, resterende komplekse.
Hos den uspesifisert kjent familiære
gruppen, er under 1/10 rene.
Og hos dem klassifisert som
sporadisk, er litt under 3/5 rene.
Kjønnsfordeling:
55,7% av dominante tilfeller er kvinner. 40,0% av recessive er kvinner, 23,1% av kjent, uspesifisert
familiære er kvinner, og 52,2% av sporadiske er kvinner.
Stadiet
Dem med recessiv arvegang har en høyere andel enn andre former som er avhengige av rullestol for å
forflytte seg. Deretter kommer den uspesifisert kjent familiære gruppen, som har flere med behov for
hjelpemidler generelt enn de recessive, men færre bundet til rullestol. Neste gruppe er de sporadiske,
og til slutt har de med dominant arvegang lavest andel med behov for hjelpemidler.
38
Debut
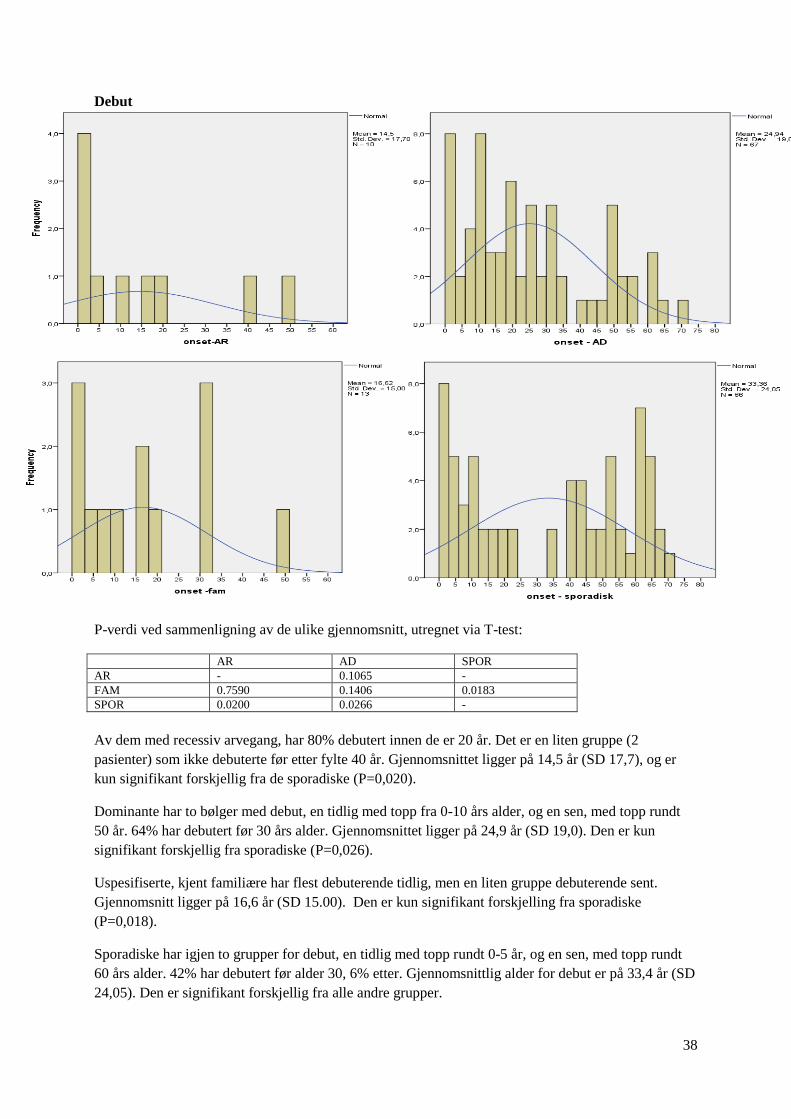
P-verdi ved sammenligning av de ulike gjennomsnitt, utregnet via T-test:
AR AD SPOR
AR - 0.1065 -
FAM 0.7590 0.1406 0.0183
SPOR 0.0200 0.0266 -
Av dem med recessiv arvegang, har 80% debutert innen de er 20 år. Det er en liten gruppe (2
pasienter) som ikke debuterte før etter fylte 40 år. Gjennomsnittet ligger på 14,5 år (SD 17,7), og er
kun signifikant forskjellig fra de sporadiske (P=0,020).
Dominante har to bølger med debut, en tidlig med topp fra 0-10 års alder, og en sen, med topp rundt
50 år. 64% har debutert før 30 års alder. Gjennomsnittet ligger på 24,9 år (SD 19,0). Den er kun
signifikant forskjellig fra sporadiske (P=0,026).
Uspesifiserte, kjent familiære har flest debuterende tidlig, men en liten gruppe debuterende sent.
Gjennomsnitt ligger på 16,6 år (SD 15.00). Den er kun signifikant forskjelling fra sporadiske
(P=0,018).
Sporadiske har igjen to grupper for debut, en tidlig med topp rundt 0-5 år, og en sen, med topp rundt
60 års alder. 42% har debutert før alder 30, 6% etter. Gjennomsnittlig alder for debut er på 33,4 år (SD
24,05). Den er signifikant forskjellig fra alle andre grupper.

39
Alder ved undersøkelse
Gjennomsnittlig alder ved undersøkelse ligger for recessive tilfeller på 37,1 år (SD 12,82 ). For de
dominante, er gjennomsnittlig alder 45,55 år (SD 17,18). Uspesifisert kjent familiære ligger på 44,62
år (SD 12,91), og sporadiske har et gjennomsnitt på 52,1 (SD 18, 43)
Eneste statistisk relevante forskjeller er mellom sporadisk og dominante, og sporadisk og recessive.
Varighet
De med uspesifisert kjent familiære har i gjennomsnitt levd lengst med sin sykdom, da med en
sykdomsvarighet på 28,0 år (SD 15,32). Deretter kommer de med recessiv sykdom, som har i
gjennomsnitt levd med sin sykdom i 22,9 år (SD 14,9). Etter dem er det de dominante, med en
gjennomsnittlig sykdomsvarighet på 21,12 år (SD 15,62), og til slutt sporadiske, med en varighet på
18,82 år (SD 17,96).

40
Symptomer og klinisk bilde
De symptomer og funn hvor det fremkommer forskjeller mellom de ulike gruppene og hele
pasientmaterialet, trekkes fram her. Øvrige funn var tilnærmet likt distribuert.
Spastisitet:
Dem med recessiv arvegang har en høyere andel med affeksjon i både overekstrmeiteter og
underekstremiteter (33%) enn øvrige grupper (~15%). Recessive og uspesifisert familiære hadde
begge en høyere andel med gangspastisitet klassifisert som alvorlige (33% og 38% respektivt), enn
sporadiske og dominante arvelige (10,8% og 16% respektivt). Sporadiske hadde andel med clonus på
33%, dominante på 45%, recessive på 50%, og uspesifisert familiære på 27%.
Ataksi:
Dominante hadde lavest andel med gang ataksi, med kun 7 %. Sporadisk lå på 13%, mens recessive og
uspesifisert familiære lå begge på rundt 45%. Både Uspesifisert familiære og recessive hadde en
høyere andel med ataksi i overekstremitetene (~40%), enn øvrige (~12%)
Kraft og muskelsvinn:
Recessive og uspesifisert kjente familiære har igjen en høyere andel med muskelsvinn (40%) og
muskelsvakhet (70%) enn øvrige grupper (~10% og 50% respektivt), mest uttalt distalt i
underekstremitetene.
Plantar:
Sporadiske tilfeller hadde en høyere andel med bilateral inversjon (88%) enn øvrige grupper, hvor
dominante hadde en andel på 80%, og recessive og uspesifisert kjent familiære lå begge på ~70%.
Vibrasjonssans og overflate sensibilitet:
Uspesifisert familiære og recessive hadde høyest andel med fullstendig bortfall av vibrasjonssans, på
27,3% og 30% respektivt. Deretter kommer sporadiske, med 20,3% fullstendig bortfall, deretter
dominante, med 16,7%.
50% av recessive hadde et uspesifisert sensibilitetsutfall. Dominante og sporadiske på begge rundt 20-
25%, og uspesifisert familiære hadde kun 7,7% sensibilitetsutfall.
Øyebevegelser:
Både recessive og uspesifisert familiære hadde en påvirkning av øyemotilitet, rundt 50%. Øvrige
grupper hadde 10-15% utfall, i all hovedsak sakkadisk pursuit eller nystagmus.
Tremor
Pasienter med dominant arvegang hadde kun 1,5% med postural tremor. Øvrige grupper hadde alle ca
10% med postural tremor. Ingen av gruppene var overrepresentert med hviletremor.
Ekstrapyramidale funn:
20% av recessive (2 pasienter) hadde et ekstrapyramidalt funn. Øvrige pasientgrupper lå på rundt 5 %.
Utviklingshemning:
Hos uspesifisert familiære og recessive hadde begge grupper en andel på ca. 25% mental retardasjon.
Øvrige grupper lå begge på 4,5%.
44% av recessive og 33% uspesifisert kjent familiære hadde en motorisk utviklingshemning. Av
dominante hadde 9% dette, og hos sporadiske hadde 13% en motorisk utviklingshemning.

41
Kognitiv svikt
25% av recessive, 36% av uspesifisert familiære, 13% av dominante og 5% av sporadiske hadde en
kognitiv svikt.
MR funn
Funn ved MR er relativt likt
fordelt mellom de ulike
transmisjonsgruppene, hvor
rundt 60% har normalfunn, med
unntak dem med recessiv
arvegang, hvor 25% er uten
funn, og halvparten har
degenerasjon.
0:Ingen funn
1:degenerative forandringer, atrofi
2: høysignalsforandringer,
ischemiske forandringer.
Nevrografi
Gruppen med høyest andel
EMG/nevrografi-funn, er de
sporadiske, med en andel på
44,2% positive funn. Dernest
kommer uspesifisert kjent
familiære, med 33,3% positive
funn, tett etterfulgt av dem med
dominant arvegang, med 31,6%
positive funn.
Pasientene med lavest andel funn,
er de recessive, med en andel på
16,7%. Videre har alle de
recessive funn både i under
ekstremiteter og i
overekstremiteter, ingen har funn
kun i underekstremiteter alene.
0:Ingen funn
1: funn kun i under ekstremiteter
2: funn både i under og
overekstremiteter.

42
Progresjon
Recessiv arvegang:
Grafen er sterkt preget over at
det kun er 10 pasienter med
recessiv arvegang. Det er kun én
pasient med varighet fra 10-19,
og én pasient med varighet 30-39
år. Dette er et alt for smalt
pasientmateriale til å kunne si
noe definitivt.
Uspesifisert, kjent familiær arvegang:
Det er igjen en meget sparsom
mengde pasienter, og vanskelig
å si noe definitivt ut fra grafen.

43
Dominant arvegang:
Her valgt å fordele pasientene etter
debut, da de med debut under 25 år,
og pasienter med debut etter 40 år.
Grafene gir et inntrykk av at dem
med tidlig debut har en raskere
progresjon, enn dem med sen. Viktig
å notere seg, at det er kun ett individ
med debut over 40 år, med varighet
20-29.
44
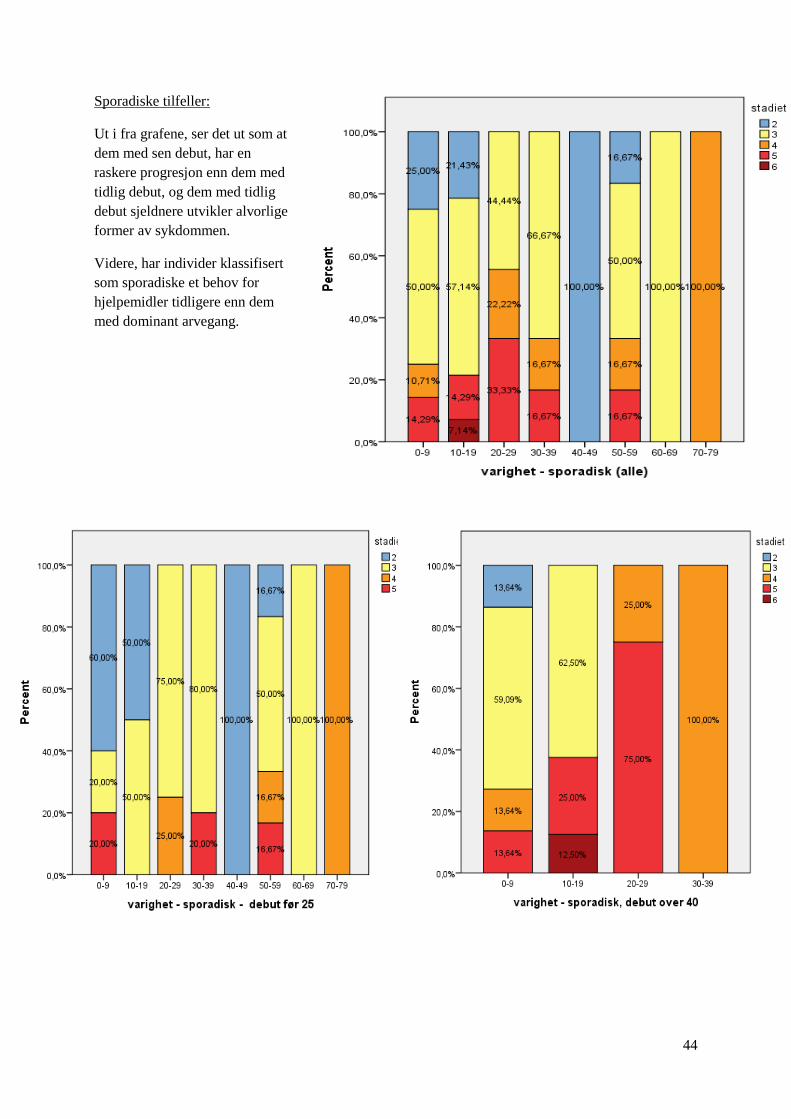
Sporadiske tilfeller:
Ut i fra grafene, ser det ut som at
dem med sen debut, har en
raskere progresjon enn dem med
tidlig debut, og dem med tidlig
debut sjeldnere utvikler alvorlige
former av sykdommen.
Videre, har individer klassifisert
som sporadiske et behov for
hjelpemidler tidligere enn dem
med dominant arvegang.

45
Diskusjon
Hensikten ved denne oppgaven, var å undersøke databasen for å identifisere mulige fenotyper, trender
og mønstre for debut og progresjon hos pasienter som fremdeles står uten genetisk diagnose, for å
mulig identifisere fremtidige genetiske strategier.
Ved gjennomgang av databasen, har det blitt spesielt fokusert på debut-alder, stadium av sykdom,
progresjonsrate, kliniske funn og billeddiagnostikk og nevrografi/EMG. Dette har deretter blitt
sammenlignet mellom ulike transmisjonsmåter.
15% av ataksi pasientene, og 14% av HSP pasientene hadde en usikker/mulig diagnose. Halvparten i
begge gruppene var sporadiske, og halvparten kjent familiære. Det er ukjent hvorfor dem med kjent
familiær historie var usikre diagnoser. Mulig det er pasienter med uspesifikk klinikk i en ellers kjent
affisert familie.
Hereditær ataksi:
Av pasientene undersøkt per proband, var 57% sporadiske. Altså, pasienter uten noen kjent
familiehistorie, men som det antas å foreligge en arvelig form for ataksi. Resterende var kjent
familiære, hvorav 7% ikke hadde klart definert arvegang, 26% hadde dominant arvegang, og 9,4%
hadde en recessiv arvegang.
Kjønnsfordelingen var jevn, og det var ingen overvekt hos verken av transmisjonsmåtene, som ikke er
overraskende, ettersom X-bunden ataksi er sjelden.
Det ble funnet et gjennomsnitt for debutalder på 35 år, men dette er lite illustrativt for hvilke alder
flest debuterte i. Det sees ved grafene for debut (side 15) to topper, en tidlig, hvor flertallet hadde
allerede fått symptomer det første leveåret, og en sen, hvor flertallet debuterte i alder mellom 55-60 år.
Det var kun få pasienter med debut i 30 årene. Når pasientmateriale ble fordelt etter transmisjonsmåte,
kan man se at dominante var fordelt i to grupper, likeledes det totale pasientmaterialet. Hos recessive
derimot, hadde nesten alle debutert før de var 20 år, med unntaket hvor 3 pasienter debuterte rundt 50
års alder. Men detter er ikke utenkelig, ettersom det allerede er beskrevet i litteratur at for eksempel en
prosentandel av Fridreichs ataksi pasienter også har en mildere variant, hvor de debuterer senere i
livet. Sporadiske pasienter i dette pasientmaterialet debuterte generelt senere, men hadde flere grupper
for debut. En gruppe pasienter hadde allerede fått diagnosen ved 1 års alder, deretter var det en liten
topp i tenårene, og så debuterte hovedbulken av pasientene i slutten av 50 årene. Dermed lignet de noe
på de dominante, men hadde en større andel som debuterte sent enn de dominante. De uspesifisert
familiære lignet både på dominante og sporadiske, med to grupper for debut, men en hovedvekt med
sen debut.
Sykdomsstadiet hadde nærmest en normalfordeling, hvor toppen lå over stadium 3, som står for
pasienter som fremdeles kan gå uten hjelpemidler, men ikke lenger kan løpe. 1/4 av pasientene var
avhengig av hjelpemidler for å forflytte seg, flertallet av disse var rullestolavhengige. Mellom de ulike
transmisjonsmåtene, var det hos de recessive hvor det største flertallet hadde behov for hjelpemidler
for å forflytte seg, og de sporadiske hadde minst andel som hadde behov for hjelpemidler.
En konfunderende faktor vedrørende sykdomsstadie, er at det både er ulik alder for debut, alder for
undersøkelse og varighet av sykdom mellom de ulike transmisjonsmåtene. En eldre person kan tenkes
å ha vanskeligheter med gange grunnet høy alder i seg selv, og ikke nødvendigvis kun pga. deres
sykdom. Videre ville en forente en høyere alvorlighetsgrad hos en person som har levd lenge med

46
sykdommen sin, enn dem med kort varighet, som foreksempel kan tenkes hos pasienter med tidlig
debut.
De som da har hatt sykdommen lengst, er de recessive, med et gjennomsnitt på 26 år, som ikke er
overraskende, ettersom disse også er pasientene med generelt tidligst debut. Men dette er delvis
oppveid for ved at de også generelt er undersøkt ved en tidligere alder. Dominante har også et
gjennomsnitt for varighet som ligger nært de recessive, kun 2 år under dem, men undersøkes generelt
senere i livet enn øvrige former av sykdommen. Etter dem kommer den uspesifisert, kjent familiære
gruppen, som har levd med sykdommen i gjennomsnittlig 17 år, og har en sen debut, på 41 år.
Sporadiske har levd kortest tid med sin sykdom, gjennomsnitt på kun 12 år. Men de har også en sen
debutalder (41 år), og har en lavere alder for undersøkelse enn dominante og pasienter med
uspesifisert, kjent familiære sykdommer. Dette kan tenkes grunnet i at pasienter som debuterer med
ataksi, og ikke har en kjent familiehistorie med sykdommen, er raskere til legen og ønsker utredning,
mens pasienter med hvor det er flere kjente familiemedlemmer med en sent progredierende sykdom,
hvor det er kjent det ikke finnes behandling, ikke er like raske å komme til legen for å bli utredet.
Så, på tross av komparativ sykdomsvarighet, har recessive pasienter generelt en høyere
alvorlighetsgrad enn dominante. Sporadiske har lavest alvorlighetsgrad, men har også levd med
sykdommen sin kortest tid, og er noe yngre ved undersøkelse enn pasienter med dominant arvegang.
De uspesifisert familiære er preget av at de kun har totalt 16 pasienter i sin gruppe, og har en stor
spredning av debut over alle aldersklasser.
Progresjonsraten varierte både mellom de ulike transmisjonsmåtene, og mellom hvilke alder man
debuterte i. For hele pasientmaterialet samlet, var det en jevn progresjon av stadium for hver tiår, med
ca 5-10% økning av andel med behov for hjelpemidler ved forflytning for hvert tiår, fram til 30-39 år.
Deretter hadde 40-49års klassen en mindre andel med behov for hjelpemidler, og etter dette var dataen
preget av et lite antall pasienter i varighetsklassene fra 50-79.
Det var videre en synlig forskjell av progresjonsrate mellom alle dem med tidlig debut, og alle med
sen debut. De med sen debut, hadde opp til 20% med behov for hjelpemidler allerede innen 0-9 år med
sykdommen, men det tok 20-29 år før de med tidlig debut hadde den samme andelen med et lignende
behov. Da dette ble videre delt inn etter type transmisjon, og igjen sporadiske pasienter og pasienter
med dominant arvegang ble delt inn etter debut-alder (ettersom det var to klare topper for debut hos
disse gruppene) , så man ulike mønstre innad i de ulike gruppene. Dem med raskest progresjon var
sporadiske individer med sen debut. Ettersom denne gruppen tilsynelatende hadde en raskere
progresjon enn de med sent debuterende dominant ataksi, kan man stille spørsmålstegn om dette er
grunnet i at flere av de sporadiske pasientene har MSA-c, og ikke en hereditær ataksi. Dette er en
sykdom med en rask progresjon av symptomer, som kunne forklart hvorfor sporadiske med sen debut
har da denne raske progresjonen. Ettersom de sporadiske pasientene er undersøkt ved en lavere alder
enn de med dominant arvegang, kan det da ikke forklares med at de har en høy alder. Videre hadde de
sporadiske pasientene den generelt laveste andelen med behov for hjelpemidler, på tross av en meget
rask progresjon hos de med sen debut. Dette kan forklares ved at de hadde den korteste varigheten.
Recessive er dem med nest raskest progresjonsrate, men denne grafen er svært påvirket av et tynt
pasientmateriale. De dominante hadde en svingende inndeling av alvorlighetsgrad avhengig av
varighet, av ukjent årsak. Men det ser ut som at dominante som med en tidlig debut, hadde en tregere
progresjon enn recessive, som i all hovedsak debuterte tidlig, og sporadiske med tidlig debut hadde en
enda tregere progresjon enn de dominante igjen.

47
Av type ataksi, hadde flertallet en komplisert form, med 65% komplekse. Av dem med recessiv
arvegang, hadde alle en kompleks type, som ikke er overraskende. Det var likt fordelt mellom ren og
kompleks hos dem med dominant arvegang, og sporadiske hadde 2/3 med komplekse.
Ved gjennomgang av klinikken, kan noen funn bringes frem. 20% hadde ekstrapyramidale
symptomer, som oftest ble funnet hos pasienter med sporadisk forekommende sykdom. Pasienter med
recessiv arvegang hadde en høyere andel med spastisitet, både ved gange og ved hvile(42-48%), enn
øvrige grupper (12-17%), og de hadde oftere kraftutfall og andre pyramidale funn. De hadde også en
høyere andel med motorisk og mental utviklingshemning (36-40%) og enn øvrige transmisjonsmåter,
som også gjenspeiles i det at absolutt alle av de recessive var kompliserte tilstander. Sporadisk hadde
høyere andel med kognitiv svikt (31%) enn dominante (7%), men recessive hadde igjen høyest andel
(50%) av alle gruppene.
Et interessant klinisk funn, var hvordan så mange av de med recessiv arvegang, hadde en
okulomotorisk apraksi, i forhold til de andre, ettersom det allerede er kjent i litteraturen at det er et
hyppig fenomen ved flere typer recessiv arvet ataksi, som foreksempel ved AOA1 og 2 og ataksia
teleangiektasia. Sporadisk forekommende ataksi og uspesifisert, familiær type hadde derimot høyest
forekomst av oftalmoplegi. Ettersom det hos de sporadiske tilfellene både ble funnet flest
ekstrapyramidale funn, og flere av pasientene hadde oftalmoplegi, kan man igjen stille spørsmålstegn
om dette skyldes en andel med MSA-c.
Vedrørende billeddigagnostiskk, var funn relativt likt distribuert mellom de ulike formene. Av
recessive var det ingen uten MR funn, eller var det ingen slående forskjeller.
Til slutt kan vi se på: hvem er de uspesifisert familiære? Recessive former, eller dominante?
Debutmessig, alvorlighetsgradmessig og klassifikasjonsmessig, lignet de mest på dominante. Men
flere av de kliniske funnene hadde sammenlignbare prosentandeler som hos recessive. Men
overveiende, lignet de mest på dominante, så det kan tenktes at mulig en overvekt av dem er en
dominant form, men det er vanskelig å si noe, ettersom pasientmaterialet er nok så smalt, og det videre
er en meget liten gruppe pasienter, og det er kjent at selv like genotyper, kan ha meget varierende
fenotyper.
Hereditær spastisk paraparese:
Av pasientene undersøkt per proband, var 56% av pasientene sporadiske forekommende HSP. 30% av
familiene hadde dominant arvegang, 6% hadde recessiv, og 8% hadde en uspesifisert kjent arvegang.
Disse tallene er ikke overraskende, ettersom recessiv sykdom er, som tidligere nevnt i oppgaven,
meget sjeldent i samfunn hvor det er få som får barn når de er i nært slektskap med hverandre. Dette
påvirker også alle sammenligninger i denne oppgaven mellom recessive og øvrige former, ettersom
det var kun totalt 10 pasienter med recessiv arvegang.
Kjønnsfordelingen var helt upåfallende for hele pasientmaterialet, men for uspesifisert, kjent familiære
var det en overvekt med mannlige pasienter, hvor 3/4 var menn. Men, det må presiseres at det kun var
13 pasienter i denne gruppen, og det dermed kan like gjerne forklares i dette lave antallet, og ikke
nødvendigvis peker i retning av mulig X-bundene HSP-former.
Gjennomsnitt for debut var på 27 år, men meget få har faktisk debutert i denne alderen. Flertallet
debuterer tidligere, og på grafen er den største toppen allerede ved 2 års alder. Etter dette var det en

48
gradvis nedgang, fram til rundt slutten av 30 årene, og etter dette trappes det opp til en topp rundt 50
års alder, og en ny, enda noe høyere topp rundt 60 års alder. Når pasientmaterialet var delt inn etter
transmisjonsmåte, var det hos de sporadiske tilfellene to klare grupper med debut, en tidlig, og en sen,
med like høye topper i hver gruppe. De med dominant arvegang debuterte flertallet før en alder av 35
år, og det var en gradvis nedgang fra toppen i tidlig barndom, med en liten topp ved 50 års alder.
Recessive var overveiende tidlig debuterende, da før en alder av 20 år, med kun to sene tilfeller. Det
samme gjaldt de uspesifisert, kjent familiære. Ingenting av dette var avvikende i forhold til tidligere
beskrevet litteratur.
I dette pasientmaterialet, kunne flertallet fremdeles gå uten hjelpemidler (71%), men fordelingen av
alvorlighetsgradene varierte noe mellom de ulike transmisjonsmåtene. Her var de recessive og
uspesifisert, kjent familiære formene oftest avhengige av hjelpemidler for å forflytte seg. Etter dem
kom de sporadiske formene. Til slutt hadde de med dominant sykdom den mildeste sykdommen, med
færrest antall som var avhengige av hjelpemidler, og høyest antall som fremdeles kunne løpe.
At recessive former av HSP er mer alvorlig, er allerede kjent i litteratur. Men, man kan da spørre
hvorfor de sporadiske hadde en lett høyere alvorighetsgrad enn de med dominant arvegang. Man ser
da, at de sporadiske undersøkes i en høyere alder enn de med dominant arvegang, med en mye større
andel som ligger over 65 år, og de har et gjennomsnitt for undersøkelse som er 7 år høyere enn de med
dominant arvegang. Varighet av sykdommen er ganske likt mellom de ulike gruppene (med unntak
den uspesifisert familiære), med en varighet på 19-23 år hos disse tre. Det kan dernest tenkes, at de
med sporadisk arvegang, har da denne høyere alvorlighetsgrad grunnet denne høyere alderen ved
undersøkelse.
Selve progresjonen av alvorlighetsgraden til HSP, var karakterisert av to klare topper, som sees på side
34. Da pasientene ble gruppert etter debut tidspunkt, kunne det da virke som at den første toppen etter
3 tiår med sykdommen, representerte pasienter med sen debut (etter fylte 40 år), og den neste toppen
etter 5 tiår med sykdommen representerte pasienter med en tidlig debut. Dette passer godt med
tidligere beskrevet litteratur i denne oppgaven, hvor det ofte kan sees en relativt non progressiv HSP
hos pasienter med tidlig debut, og det utvikler seg mer senere i livet. Dette er klart illustrert også i
denne pasientgruppen. Raskest progresjon forekommer hos dem med sen debut.
Når pasientene ble videre delt inn etter transmisjonsmåte for å undersøke progresjonsraten, ble det
raskt synlig at pasientmaterialet var for smalt hos recessive og uspesifisert, kjent familiære til å kunne
si noe om progresjonen her. Men hos dominante og sporadiske, kom det fram en mulig forskjell i
progresjonsrate, avhengig av om de debuterte i tidlig eller sen alder. Men igjen, dette er påvirket av et
smalt pasientmateriale.
Sporadiske tilfeller med sen debut, hadde en raskere sykdomsprogresjon, enn pasienter med dominant
arvegang og sen debut. Mens, det virket som det var omvendt hos pasientene med tidlig debut, hvor
det var en noe raskere progresjon hos dominante med tidlig debut, enn sporadiske med tidlig debut.
Men det var få sporadiske pasienter med både kort varighet og tidlig debut, så disse forskjellene kan
forklares i dette. Men hos sporadiske med sen debut, hvorfor har de en raskere progresjon enn dem
med dominant arvegang? Er det som tidligere nevnt grunnet den høyere alderen og dermed høyere
sårbarhet vedrørende deres gangfunksjon, eller har disse pasientene en faktisk raskere progredierende
HSP grunnet for eksempel en annen fordeling av genfeil enn de kjent dominante tilfellene, som da
påvirker progresjonsraten?
Klinisk inndeling av pasientene med HSP, viste at det var tilnærmet lik andel rene og kompliserte
tilstander. Når de ble fordelt i grupper etter transmisjonsmåte, viste det seg at recessive pasienter kun

49
hadde kompliserte tilstander, uspesifisert kjent familiære hadde en klar overvekt av kompliserte
tilstander, og både sporadiske og dominante hadde ca 2/3 med rene tilstander, de sporadiske en litt
høyere andel komplekse enn de dominante. I litteratur, så er det tidligere beskrevet å være en andel på
70-80% med rene tilstander hos dominante. Men i dette materialet kom det da fram en andel på 61%,
som er litt lavere. Kanskje dette kan forklares ved at de vanligste formene for HSP, slik som SPG4 og
SPG3A, som begge oftest er rene tilstander, allerede er diagnostisert genetisk, og dermed selektert ut
av dette pasientmaterialet, som da igjen påvirker fordelingen.
De ulike, kliniske funnene viste generelt at både recessive og uspesifisert familiære pasienter hadde
oftere kompliserende funn, og funn med en høyere alvorlighetsgrad enn øvrige grupper. Rent klinisk,
lignet de sporadiske formene mer på dominante, og de uspesifisert familiære, mer på recessive. Noen
forskjeller som kan nevnes, var en økt andel med recessive som hadde øyemotilitetsforstyrrelser enn
sporadiske og dominante. Det var flere med dominant arvegang enn sporadiske som hadde kognitiv
svikt (13%, mot 5% hos sporadiske), på tross av høyere alder hos sporadiske pasienter.
Ved supplerende undersøkelser, som MR og nevrografi/EMG, kom det fram en nokså lik fordeling av
funn. Recessive hadde en høyere andel med atrofi enn øvrige, og en lavere andel med funn på
nevrografi/EMG. Sporadiske hadde en noe høyere andel med funn på nevrografi enn de andre
gruppene. Ved nevrografi/EMG, hadde det store flertallet aksonal eller blandet påvirkning, og kun få
pasienter hadde ren demyelinisering.
Det har jo som tidligere nevnt i oppgaven, blitt oftere observert recessive sykdommer hos pasienter
med sporadisk forekommende HSP, enn hos pasienter med kjent familiehistorie. Og de vanligste
formene for recessiv HSP, har en mental utviklingshemning. Men kun 4,5% av sporadiske pasienter i
dette materialet hadde en mental utviklingshemning. Overveiende virket det som at de hadde mer til
felles med den dominante gruppen. Mens den uspesifisert, kjent familiære gruppen derimot lignet mer
på den recessive. Men den uspesifisert familiære og den recessive gruppen var kontinuerlig preget av
et meget lite antall i hver gruppe.
Feilkilder:
Ved ulike kliniske funn, slik som øyebevegelser, er det en feilkilde at disse pasientene ikke har
systematisk gått gjennom en nevrooftalmologisk undersøkelse, som gjør disse dataene mindre
nøyaktige. Samme gjelder ved MR bilder, da det ikke har vært en og samme radiolog som har
gjennomgått dette, og ofte var ikke cerebellum engang beskrevet, slik at man enkelt kan stille
spørsmålstegn om dette i det hele tatt ble vurdert. MR og nevrografi var dog heller ikke tatt på samme
tidspunks som øvrige undersøkelser ble tatt, og dermed ikke kan måles mot kliniske funn og
alvorlighetsgrad, og det var generelt en stor andel (20-30%)som ikke hadde gjennomgått supplerende
undersøkelser. Dette er nokså vanlig hos pasienter med arvelige, sent degenerative tilstander, hvor det
ikke er alle som ønsker ytterligere utredning, da dette ikke påvirker behandling videre.
Anamnese var heller ikke systematisk tatt for å fange opp eventuelle andre opplysninger, slik som ved
andre assosierte lidelser og sykdommer pasientene muligens har.
Det kan videre nevnes, at på tross av oppgitt p-verdier i resultat-feltene, er det for mange p-verdier
utregnet til å kunne virkelig si noe om statistisk signifikans, men det ble gjort som en kuriositet.

50
Strategier videre:
Når det kommer til å legge grunnlag for videre genetiske strategier for diagnostikk, er dette vanskelig.
Det er som sagt per dags dato ingen fenotype som har en fast korrelasjon med en spesifikk genotype,
hverken hos hereditær ataksi eller hereditær spastisk paraparese. Dette pasientmaterialet viste stor
heterogenitet i klinikk og progresjon, og selv innad i familiene i denne databasen var det ingen fast
fenotype, slik som det ellers er beskrevet i litteraturen. Noen kliniske funn kan peke i retning av
spesifikke genfeil, slik som noen trender avdekket i dette pasientmaterialet, som for eksempel tidligere
nevnte øyemotilitetsfunn og spastisitet.
Det kunne dermed vært interessant å videre undersøke recessive ataksi pasienter med okulomotorisk
apraksi etter dette, eller gå videre med funnene av spastisitet hos de recessive ataksipasientene, eller
videre undersøke om de pasientene med sporadisk ataksi og sen debut mulig har en overvekt med
MSA-c, og ikke hereditær ataksi.
Evaluering av databasen:
Databasen har utfyllende, verdifull informasjon vedrørende disse pasientgruppene, er systematisk
utført og har en oversiktlig og intuitiv framstilling.
Ved gjennomgang av databasen, var det dog mye informasjon som manglet, og mye arbeid gikk med
på å fylle inn denne informasjonen fra skjemaer, spesielt i ataksi databasen. Det var også mye
informasjon som ikke var nedtegnet med de spesifiserte tallverdiene, og istedenfor var beskrevet med
ord i de ulike boksene i databasen. Det gikk da også med mye tid til å korrigere dette.
På tross av etterfylling av data i databasen, manglet det fra 0-30% data. Oftest manglet det fra 2-16%
data, men symptomopplysningene hos pasienter med recessiv ataksi hadde mangler opp til 30%, og
supplerende undersøkelser slik som MR og nevrografi/EMG manglet generelt fra 20-30%.
Skjemaene var lette å fylle in informasjon fra, men det var et problem at ikke alle skjemaer var
fullstendig utfylt. Dette gjorde det vanskelig for en utenforstående å overføre informasjon, ettersom
det dermed ikke alltid var åpenbart om pasienten ikke hadde et spesifikt funn, eller om dette ikke
hadde blitt evaluert.
Videre savnet jeg en egen kolonne for rigiditet, da dette kun var oppsummert med ord i kolonnen for
ekstrapyramidale funn. Dermed gjorde dette det umulig å si noe om hvor mange av pasientene som
manglet informasjon om rigiditet, og hvor mange som hadde faktisk negative funn. Dette var spesielt
relevant hos ataksi pasientene, ettersom MSA-c er en relevant differensial diagnose.
Derfor ville det vært ønskelig med strengere regler for inntasting av data i databasen for å unngå bruk
av ord istedenfor tall, og eventuelt å istedenfor presisere funn med beskrivelser i andre kolonner, slik
at informasjonen lettere kan fremstilles i programvare som SPSS, som hovedsagelig prosesserer
tallverdier.

51
Konklusjon
Denne oppgaven var en deskriptiv studie over pasienter med hereditær ataksi og spastisk paraparese
uten genetisk diagnose, samlet i en norsk kohort siden 2002. Den gikk ut på å identifisere mulige
fenotyper, diverse særtrekk, generell debut-alder og progresjonsrater.
Av pasienter med hereditær ataksi, hadde 1/3 av pasientmaterialet et rent cerebellært syndrom.
Undersøkt per proband, var 57% av dem sporadiske ataksier, 9% var autosomalt recessive familier, og
26% var autosomalt dominante familier. Alle med recessiv arvegang hadde et kompleks
symptombilde, 1/2 av dominant nedarvede hadde dette, og 2/3 av de sporadiske.
Av pasientene med hereditær spastisk paraparese, hadde halvparten av pasientmaterialet en ren
spastisk paraparese. Undersøkt per proband, var 56% av pasientene en sporadisk form, 6% var
recessive familier, og 30% var dominante familier. Av pasienter med sporadisk definert spastisk
paraparese og dominant spastisk paraparese, hadde pasientene i begge gruppene ca. 3/5 en ren form.
Alle med recessiv arvemåte hadde en komplisert form.
Det har ikke blitt funnet noen form for klare kliniske trender, dog noen kliniske funn, slik som noen
spesifikke øyemotilitetsforstyrrelser (okulomotorisk apraksi) og spastisitet gikk igjen hos recessive
former for ataksi, og en lett økt andel med sporadisk form for ataksi hadde ekstrapyramidale funn. Hos
hereditær spastisk paraparese pasientene var det en økt andel av recessive pasienter med ataksi i
forhold til andre grupper. Det var ingen klare særtrekk ved debuterende symptom, eneste som var å
bemerke var en liten andel av ataksi pasienter som debuterte med tremor. Det var ingen overvekt av
assosierte sykdommer hos noen av gruppene. Kjønnsfordeling var upåfallende.
For både spastisk paraparese og ataksi, var det to bølger for debut: En tidlig, med høyeste topp
allerede i det første leveåret, og en sen, med største topp i alderklassen 50-60 år. Tid for debut varierte
også innad i de ulike transmisjonsmåtene, hvor recessive generelt debuterte tidlig, og øvrige var
fordelt i en tidlig og en sen gruppe, og sporadiske var dem med flest debuterende sent.
Flertallet av pasientmaterialet hadde bevart gangfunksjon, men alvorlighetsgraden av deres symptomer
og behov for hjelpemidler for å forflytte seg varierte med transmisjonsmåte, hvor recessive hos både
ataksi og spastisk paraparese-pasienter hadde høyest alvorlighetsgrad.
Ved både ataksi og spastisk paraparese, hadde pasienter med en sen debut en raskere progresjon av
sine symptomer enn pasienter med tidlig debut, med unntak av recessive pasienter, som debuterte
tidlig, og hadde en rask progresjon. Dem med raskeste progresjon hos ataksi pasientene, var
sporadiske ataksier med sen debut, etterfulgt av recessive nedarvede ataksier, deretter sent debuterende
dominante ataksier, og de med senest progresjon var sporadiske og dominante ataksier med tidlig
debut. Hos pasienter med hereditær spastisk paraparese, var det igjen sporadiske pasienter med sen
debut som hadde raskest progresjon. Deretter kom pasienter med recessiv arvegang, etterfulgt av
pasienter med dominant arvegang og tidlig debut, og de med senest progresjon, var pasienter med
dominant arvegang og sen debut, og sporadiske pasienter med tidlig debut.
Det er per i dag ikke etablert faste korrelasjoner mellom genotyper og fenotyper ved disse
sykdommene. Ulik klinikk kan gi misstanke om genotype, men disse lidelsene er karakterisert av stor
heterogenitet, både innen en genotype, og også innen en og samme familie. Ved gjennomgang av dette
pasientmaterialet, ble det funnet stor fenotypisk heterogenitet, også innen de ulike gruppene definert
og sammenlignet. Deskriptive analyser gjort har ikke gitt inntrykk av klart definerte subgrupper, men
noen få og interessante, uspesifikke trender kan sees hos pasienter gruppert etter arvegang.

52
Litteraturliste
1. Ruano, L., et al., The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology, 2014. 42(3): p. 174-83.
2. Harding, A.E., Classification of the hereditary ataxias and paraplegias. Lancet, 1983. 1(8334): p. 1151-5.
3. Smeets, C.J. and D.S. Verbeek, Cerebellar ataxia and functional genomics: Identifying the routes to cerebellar neurodegeneration. Biochim Biophys Acta, 2014.
4. Tallaksen, C.M., [Hereditary ataxias]. Tidsskr Nor Laegeforen, 2008. 128(17): p. 1977-80. 5. Bird, T.D., Hereditary Ataxia Overview, in GeneReviews(R), R.A. Pagon, et al., Editors. 1993,
University of Washington, Seattle, All rights reserved.: Seattle (WA). 6. Manto, M.U., The wide spectrum of spinocerebellar ataxias (SCAs). Cerebellum, 2005. 4(1): p.
2-6. 7. Nemeth, A.H., et al., Next generation sequencing for molecular diagnosis of neurological
disorders using ataxias as a model. Brain, 2013. 136(Pt 10): p. 3106-18. 8. Jayadev, S. and T.D. Bird, Hereditary ataxias: overview. Genet Med, 2013. 15(9): p. 673-83. 9. Shakkottai, V.G. and B.L. Fogel, Clinical neurogenetics: autosomal dominant spinocerebellar
ataxia. Neurol Clin, 2013. 31(4): p. 987-1007. 10. Bidichandani, S.I. and M.B. Delatycki, Friedreich Ataxia, in GeneReviews(R), R.A. Pagon, et al.,
Editors. 1993, University of Washington, Seattle. All rights reserved.: Seattle (WA). 11. Gatti, R., Ataxia-Telangiectasia, in GeneReviews(R), R.A. Pagon, et al., Editors. 1993,
University of Washington, SeattleAll rights reserved.: Seattle (WA).
12. Abele, M., et al., The aetiology of sporadic adult-onset ataxia. Brain, 2002. 125(Pt 5): p. 961-8.
13. Erichsen, A.K., et al., Prevalence of hereditary ataxia and spastic paraplegia in southeast Norway: a population-based study. Brain, 2009. 132(Pt 6): p. 1577-88.
14. Finsterer, J., et al., Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J Neurol Sci, 2012. 318(1-2): p. 1-18.
15. Fink, J.K., Hereditary Spastic Paraplegia Overview, in GeneReviews(R), R.A. Pagon, et al., Editors. 1993, University of Washington, Seattle. All rights reserved.: Seattle (WA).
16. Fink, J.K., Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol, 2013. 126(3): p. 307-28.
17. Faber, I., et al., Clinical features and management of hereditary spastic paraplegia. Arq Neuropsiquiatr, 2014. 72(3): p. 219-26.
18. Lo Giudice, T., et al., Hereditary spastic paraplegia: Clinical-genetic characteristics and evolving molecular mechanisms. Exp Neurol, 2014.
19. Opal, P. and S. Ajroud-Driss, Hereditary spastic paraplegia, in UpToDate, M.C. Patterson, Editor. 2015: UpToDate, Waltham, MA. (Acessed on February 16th, 2016).

53
Vedlegg 1
Tabell over klinikk hos ulike former for dominant hereditær ataksi, hentet og oversatt fra Suman
Jayadev og Thomas D. Birds "Hereditary ataxias: overview".
Type Gen Debutalder Symptomer/funn
SCA1 ATXN1 3–4 tiår (<10 to
>60)
Pyramidalee tegn, perifer neuropati
SCA2 ATXN2 3-4tiår (<10 to
>60)
Trege sakkader, perifer neuropati, nedsatt DTRs, kognitiv
svikt
SCA3 ATXN3 4 tiår (10–70) Pyramidale og ekstrapyramidale tegn; lid retraction,
nystagmus, trege sakkader; amyotrophy fasikuasjoner,
sensorisk tap
SCA4 16q22.1 4–7tiår (19–72) Sensorisk axonal neuropathy, døvhet
SCA5 SPTBN2 3–4 tiår (10–68) Tidlig onset, sen progresjon
SCA6 CACNA1A 5–6 tiår (19–71) Veldig sen progresjon
SCA7 ATXN7 3–4 tiår (0.5–
60)
Synstap med nevropati
SCA8 ATXN8/ 4 tiår (1–65) Sent progresiv, nedsatt vibrasjonssans; sjeldent kognitivt
svikt
SCA10 ATXN80S 4 tiår (12–48) Occasional seizures
SCA11 ATXN10 30 år (15–70) Mild
SCA12 TTBK2 4 tiår (8–62) Sent progressiv ataksi; aksjonstremor ; hyperrefleksi
SCA13 PPP2R2B Barndom eller
voksen alder
Mild intellectual disability, lav høyde
SCA14 KCNC3 3–4 tiår (3–70) Tidlig aksial myoclonus
SCA15 PRKCG 4 tiår (7–66) Ren ataksi, veldig sen progresjon
SCA16 ITPR1 30 år (20–66) Hodet tremor
SCA17 SCA16 4 tiår (3–55) Mental svikt; tidvis chorea, dystoni, myoclonus, epilepsi;
Purkinje celletap
SCA18 TBP Tenår (12–25) Ataksi med tidlig sensorisk/motorisk neuropati, nystagmus,
dysarthria, nedsatt reflexes, mmuskelsvekkelse,
atrophy,fasiculations, Babinski responses
SCA19/22 KCND3 4 tiår (10–51) Sent progressive, sjelden kognitiv svikt, myoclonus,
hyperreflexia
SCA20 11q12.2–
11q12.3
5 tiår (19–64) Tidlig dysarthria, spasmodic dysphonia, hyperreflexia,
bradykinesia; calcification of the dentate nucleus
SCA21 SCA21 6–30 Mild kognitiv svikt
SCA23 PDYN 5–6 tiår Dysarthria, abnellermal eye movements, reduced vibration
og posisjonssans
SCA25 SCA25 1.5–39 Sensorisk neuropati
SCA26 EEF2 26–60 Dysarthria, uregelmessig pursuit
SCA27 FGF14 11 år (7–20) Tidlig-onset tremor; dyskinesia, cognitive deficits; one
Dutch family
SCA28 AFG3L2 19.5 år (12–36) Nystagmus, ophthalmoparesis, ptosis, økt reflekser
SCA29 3p26 Tidlig barndom Learning deficits
SCA30 4q34.3–
q35.1
(45–76) Hyperreflexia
SCA31 BEAN1 5–6 tiår Nellermal sensation
SCA35 TGM6 43.7 ± 2.9 (40–
48) år
Hyperreflexia, Babinski responses; spasmodic torticollis
SCA36 NOP56 52.8 ± 4.3 år Muscle fasiculations, tungeatrofi, hyperreflexia
DRPLA ATN1 3–4 tiår
(8–20 eller 40–
60s)
Chorea, seizures, kognitiv svikt, myoclonus; ofte forvekslet
med Huntington disease
EA1 KCNA1 1–2 tiår (2–15) Myokymia; anfall varer i sekunder til minutter; startle eller
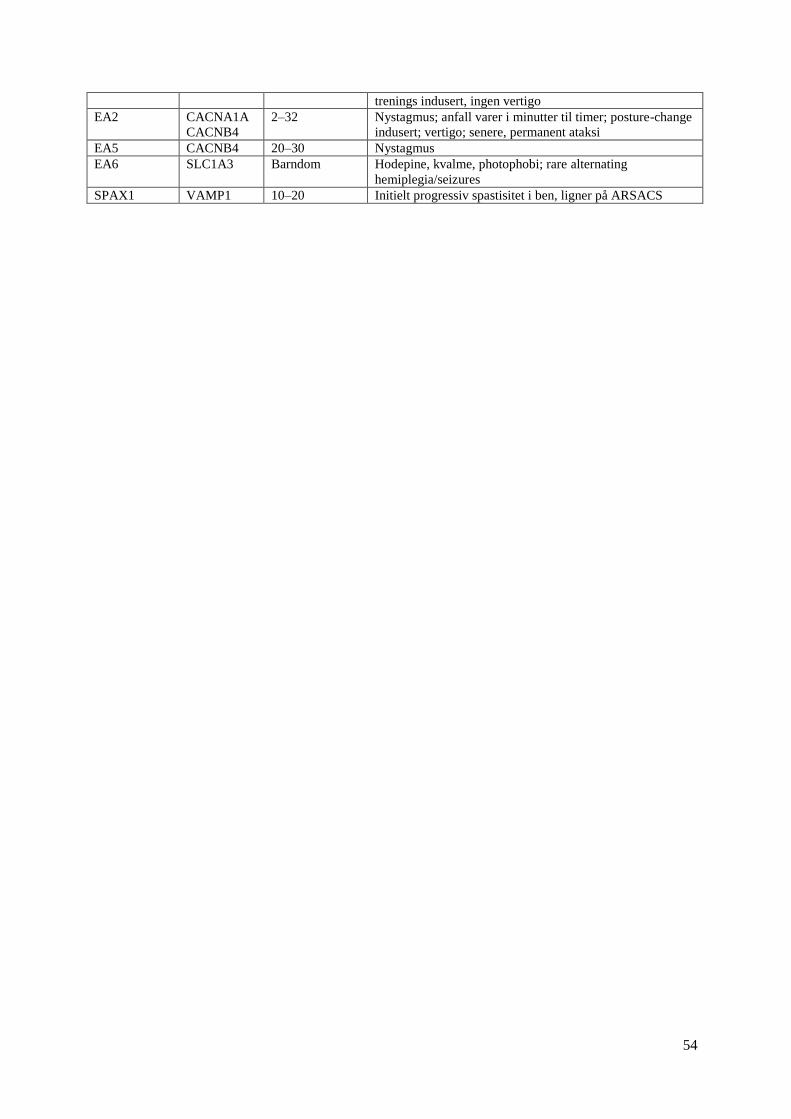
54
trenings indusert, ingen vertigo
EA2 CACNA1A
CACNB4
2–32 Nystagmus; anfall varer i minutter til timer; posture-change
indusert; vertigo; senere, permanent ataksi
EA5 CACNB4 20–30 Nystagmus
EA6 SLC1A3 Barndom Hodepine, kvalme, photophobi; rare alternating
hemiplegia/seizures
SPAX1 VAMP1 10–20 Initielt progressiv spastisitet i ben, ligner på ARSACS

55
Vedlegg 2
Tabell over klinikk hos ulike former for recessiv hereditær ataksi, hentet og oversatt fra Suman
Jayadev og Thomas D. Birds "Hereditary ataxias: overview".
Type Gen Debutalder Symptomer/funn
Friedreich ataksi (FRDA) FXN/frataxin 1–2 tiår
(4–40)
Hyporefleksi, Babinski responses, sensorielt tap
kardiomyopati
Ataksi-telangiectasia ATM 1tiår Telangiectasia, immune deficiency,
kreft, chromosomal instability, økt
α-fetoprotein
Ataksi med vitamin E
deficiency
TTPA 2–52 år, oftest
<20
Ligner på FRDA, head titubation (28%)
Ataksi med oculomotorisk
apraxia type 1
APTX/aprata
xin
Barndom Oculomotorisk apraksi, choreathetosis,
mild intellectual disability, hypoalbuminemia
Ataksi med oculomotorisk
apraxia type 2
SETX 10–22 år Cerebellær atropfi, axonal sensorimotorisk
neuropatti, okulomotorisk apraksi
IOSCA C10ellerf2/t
winkle
Spedbarnsalder Perifer neuropati, athetosis, optic
atrophy, døvhet, ophthalmoplegia,
seizures
Marinesco–Sjögren
syndrom
SIL1 Spedbarnsalder Kognitiv svikt, cataract, hypotoni,
myopathy
Autosomal recessive
spastic ataksi of
Charlevoix-Saguenay
SACS/sacsin Barndom Spastisitet, perifer neuropati, retinal
striation
Refsum disease PHYH;
PEX7
1–6 tiår Neuropathy, døvhet, ichthyosis,
retinopathy
CoQ10 deficiency CABC1;
COQ2;
COQ9;
PDSS1;
PDSS2
Barndom Seizures, kognitiv svikt, pyramidale tegn
MIRAS POLG1 Barndom til ung
voksen alder
Nystagmus, dysarthria, epilepsy,
Cerebellær atrofi på MR
SANDO POLG1 Barndom til ung
voksen alder
Abnormale øye bevegelser, ragged red
fibers, myopathy, dysphagia, neuropathy,
myopathy
Cerebrotendinous
xanthomatosis
CYP27A1 Barndom til ung
voksen alder
Tykke sener, kognitiv svikt, dystoni, white
matter disease, cataract
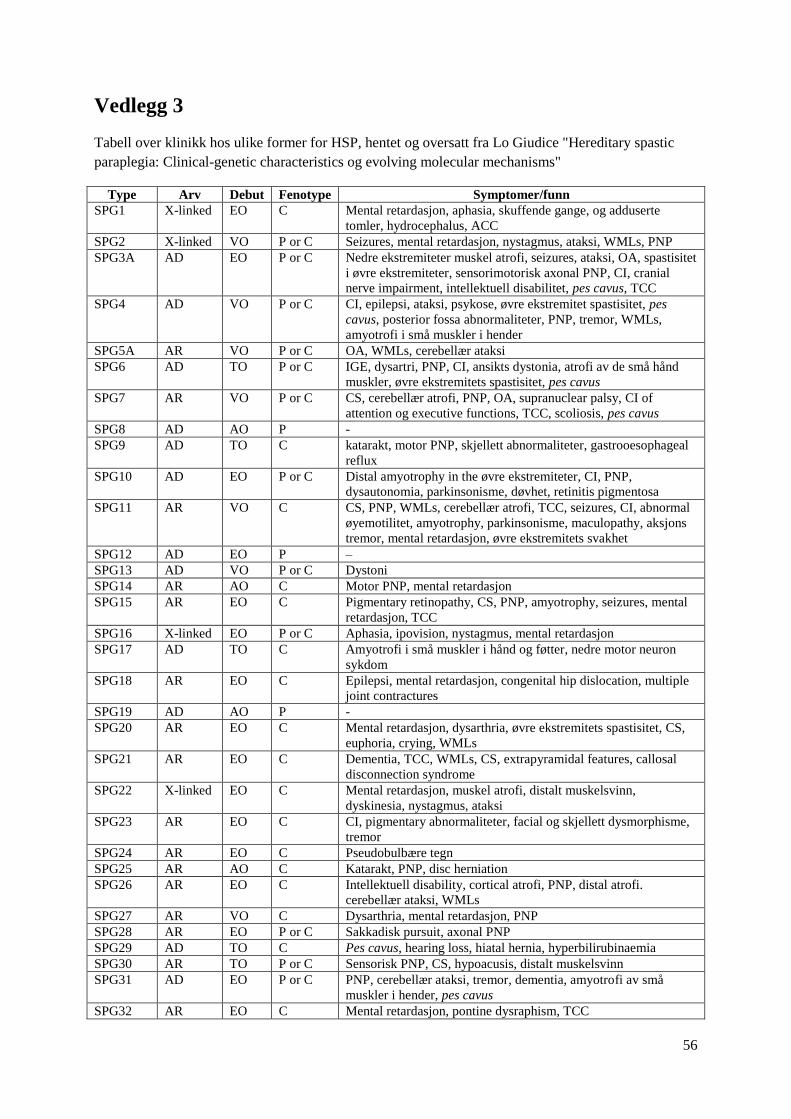
56
Vedlegg 3
Tabell over klinikk hos ulike former for HSP, hentet og oversatt fra Lo Giudice "Hereditary spastic
paraplegia: Clinical-genetic characteristics og evolving molecular mechanisms"
Type Arv Debut Fenotype Symptomer/funn
SPG1 X-linked EO C Mental retardasjon, aphasia, skuffende gange, og adduserte
tomler, hydrocephalus, ACC
SPG2 X-linked VO P or C Seizures, mental retardasjon, nystagmus, ataksi, WMLs, PNP
SPG3A AD EO P or C Nedre ekstremiteter muskel atrofi, seizures, ataksi, OA, spastisitet
i øvre ekstremiteter, sensorimotorisk axonal PNP, CI, cranial
nerve impairment, intellektuell disabilitet, pes cavus, TCC
SPG4 AD VO P or C CI, epilepsi, ataksi, psykose, øvre ekstremitet spastisitet, pes
cavus, posterior fossa abnormaliteter, PNP, tremor, WMLs,
amyotrofi i små muskler i hender
SPG5A AR VO P or C OA, WMLs, cerebellær ataksi
SPG6 AD TO P or C IGE, dysartri, PNP, CI, ansikts dystonia, atrofi av de små hånd
muskler, øvre ekstremitets spastisitet, pes cavus
SPG7 AR VO P or C CS, cerebellær atrofi, PNP, OA, supranuclear palsy, CI of
attention og executive functions, TCC, scoliosis, pes cavus
SPG8 AD AO P -
SPG9 AD TO C katarakt, motor PNP, skjellett abnormaliteter, gastrooesophageal
reflux
SPG10 AD EO P or C Distal amyotrophy in the øvre ekstremiteter, CI, PNP,
dysautonomia, parkinsonisme, døvhet, retinitis pigmentosa
SPG11 AR VO C CS, PNP, WMLs, cerebellær atrofi, TCC, seizures, CI, abnormal
øyemotilitet, amyotrophy, parkinsonisme, maculopathy, aksjons
tremor, mental retardasjon, øvre ekstremitets svakhet
SPG12 AD EO P –
SPG13 AD VO P or C Dystoni
SPG14 AR AO C Motor PNP, mental retardasjon
SPG15 AR EO C Pigmentary retinopathy, CS, PNP, amyotrophy, seizures, mental
retardasjon, TCC
SPG16 X-linked EO P or C Aphasia, ipovision, nystagmus, mental retardasjon
SPG17 AD TO C Amyotrofi i små muskler i hånd og føtter, nedre motor neuron
sykdom
SPG18 AR EO C Epilepsi, mental retardasjon, congenital hip dislocation, multiple
joint contractures
SPG19 AD AO P -
SPG20 AR EO C Mental retardasjon, dysarthria, øvre ekstremitets spastisitet, CS,
euphoria, crying, WMLs
SPG21 AR EO C Dementia, TCC, WMLs, CS, extrapyramidal features, callosal
disconnection syndrome
SPG22 X-linked EO C Mental retardasjon, muskel atrofi, distalt muskelsvinn,
dyskinesia, nystagmus, ataksi
SPG23 AR EO C CI, pigmentary abnormaliteter, facial og skjellett dysmorphisme,
tremor
SPG24 AR EO C Pseudobulbære tegn
SPG25 AR AO C Katarakt, PNP, disc herniation
SPG26 AR EO C Intellektuell disability, cortical atrofi, PNP, distal atrofi.
cerebellær ataksi, WMLs
SPG27 AR VO C Dysarthria, mental retardasjon, PNP
SPG28 AR EO P or C Sakkadisk pursuit, axonal PNP
SPG29 AD TO C Pes cavus, hearing loss, hiatal hernia, hyperbilirubinaemia
SPG30 AR TO P or C Sensorisk PNP, CS, hypoacusis, distalt muskelsvinn
SPG31 AD EO P or C PNP, cerebellær ataksi, tremor, dementia, amyotrofi av små
muskler i hender, pes cavus
SPG32 AR EO C Mental retardasjon, pontine dysraphism, TCC

57
SPG33 AD AO C Pes equinus
SPG34 X-linked VO P –
SPG35 AR EO P or C CI, epilepsi
SPG36 AD VO C Sensory PNP
SPG37 AD VO P -
SPG38 AD VO C amyotrofi av små muskler i hender, PNP
SPG39 AR EO C Distal wasting in øvre ekstremitets, axonal PNP
SPG40 AD AO P or C Hyperrreflexia of øvre ekstremitets, CI
SPG41 AD TO P -
SPG42 AD VO P -
SPG43 AR VO C Amyotrofi av små muskler i hender, bilateral OA, axonal sensory
og motor PNP, brain iron deposits in the globus pallidus
SPG44 AR AO C CI, CS, dysartri, WMLs, pes cavus, TCC, scoliosis, øvre
ekstremiteter er involvert
SPG45 AR EO C Mental retardasjon, pendular nystagmus, OA
SPG46 AR EO C Mental retardasjon, katarakt, cerebellær atrofi, TCC,
hypogonadism in males
SPG47 AR EO C Periventricular WMLs, TCC, microcephaly, epilepsi, waddling
gait, joint hyperlaxity
SPG48 AR AO P or C Spinal cord hyperintensities
SPG49 AR EO C Forsinket psykomotorisk utvikling, mentalt retardasjon, TCC,
cerebral og cerebellær dysfunction, dysmorphic features, sentral
apnea
SPG50 AR EO C Tetraplegic cerebral parese, mental retardasjon, reduction of
cerebral white matter, cerebellær atrofi
SPG51 AR EO C Microcephaly, growth og mental retardasjon
SPG52 AR EO C Delayed speech, stereotypic laughter, growth retardasjon
SPG53 AR EO C Spastisitet in øvre ekstremiteter, forsinkelser i kognisjon og
taleutvikling, kyphosis, pectus carinatum, hypertrichosis
SPG54 AR EO C Mental retardasjon, strabismus, dysarthria, dysphagia, optic-nerve
hypoplasi, short stature, TCC, laterally deviated feet, abnormal
lipid peak on brain spettroscopy, WMLs
SPG55 AR EO C OA, PNP pes equinovarus
SPG56 AR EO P or C TCC, CI, øvre ekstremitets involvement, basal-ganglia
calcification. dystonic postures, WMLs
SPG57 AR EO C OA, PNP
SPG58 AR EO P or C Chorea, myoclonus, ataksi, hypodontia, døvhet, lav høyde, pes
planus, ptosis, forsinket utvikling, WMLs
SPG59 AR EO C Nystagmus, borderline intelligence
SPG60 AR EO C PNP in nedre ekstremiteter, nystagmus
SPG61 AR EO C Loss of terminal digits, acromutilation, PNP
SPG62 AR EO P –
SPG63 AR EO C TCC, WMLs, undervektige og lave
SPG64 AR EO C Pes equinovarus, aggresivititet, forsinket pubertet, microcephaly,
borderline intelligence
SPG65 AR EO P or C TCC, defective myelination, small bilateral cystic occipital
leukomalacia, lærevansker, pes equinovarus
SPG66 AR EO C Corpus callosum og cerebellær hypoplasi, colpocephaly,
borderline intelligence, PNP, pes equinovarus
SPG67 AR EO C Distendert abdomen, borderline intelligence, ACC, vermis
hypoplasi, defective myelination
SPG68 AR EO C Nystagmus, OA, PNP, amyotrophy, dropfot
SPG69 AR EO C Intellektuell disability, døvhet, katarakt
SPG70 AR EO C Scoliosise, bilateral Achilles contracture, borderline intelligence,
nephrotic syndrome
SPG71 AR EO C TCC
SPG72 AD og
AR
EO P -
GAD1 AR EO C Spastisk cerebral parese, mental retardasjon

58
gene
Cct5 gene AR EO C Mutilerende sensorisk neuropati
OPA3
gene
AR EO C OA, chorea, cerebellær ataksi, dementia
BICD2
gene
AR EO P -
MAG
gene
AR EO C Nystagmus
LYST
gene
AR AO C PNP, cerebellær ataksi
ATPase6
gene
Maternal AO C Diabetes mellitus, hypertrofisk kardiomyopati, supraventricular
arytmi, cerebellært syndrom
SPOAN AR EO C OA, PNP, dysartri, rygg deformiteter, fiksasjons nystagmus,
distal amyotrofi, ekstrapyramidale tegn
ACC = agenesis corpus callosum; AO = adult onset; AD = autosomal dominant; AR = autosomal
recessive; C = complicated; CI = cognitive impairment; CS = cerebellær signs; EO = early onset
(infancy); IGE = idiopathic generalized epilepsi; MI = mode of inheritance; NOF = number of families;
OA = optic atrofi; P = pure; PNP = polyneuropathy; TCC = thin corpus callosum; TO = teenage onset;
VO = variable onset (infancy, teenage, or adult); WMLs = white matter lesions.



![0900 Eidi Nafstad [Kompatibilitetsmodus] · 2016-06-06 · Hereditær spastisk paraparese og hereditær ataksi 51 Iktyose (ikke x‐bundet) 40 Mitokondriesykdommer 115 Epidermolysis](https://cdn.vdocuments.net/doc/165x107/5e5a2f32ea4ddd6f8c4cc3d5/0900-eidi-nafstad-kompatibilitetsmodus-2016-06-06-hereditr-spastisk-paraparese.jpg)



