dynamic aspects of phosphorus in four- and five ... · four-and five-coordinated compounds dna as...
TRANSCRIPT

Dynamic aspects of phosphorus in four- and five-coordinatedcompounds : DNA as an exampleCitation for published version (APA):van Lier, J. J. C. (1983). Dynamic aspects of phosphorus in four- and five-coordinated compounds : DNA as anexample. Eindhoven: Technische Hogeschool Eindhoven. https://doi.org/10.6100/IR38658
DOI:10.6100/IR38658
Document status and date:Published: 01/01/1983
Document Version:Publisher’s PDF, also known as Version of Record (includes final page, issue and volume numbers)
Please check the document version of this publication:
• A submitted manuscript is the version of the article upon submission and before peer-review. There can beimportant differences between the submitted version and the official published version of record. Peopleinterested in the research are advised to contact the author for the final version of the publication, or visit theDOI to the publisher's website.• The final author version and the galley proof are versions of the publication after peer review.• The final published version features the final layout of the paper including the volume, issue and pagenumbers.Link to publication
General rightsCopyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright ownersand it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights.
• Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal.
If the publication is distributed under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license above, pleasefollow below link for the End User Agreement:www.tue.nl/taverne
Take down policyIf you believe that this document breaches copyright please contact us at:[email protected] details and we will investigate your claim.
Download date: 06. Apr. 2020

DYNAMIC ASPECTSOF PHOSPHORUS IN FOOI·"AND FIVE-COORDINATED COMPOUNDS
DNA as an example
. J.J.C. VAN LIER

DISSERTATIE DRUKKERIJ .... b .... HELMOND
TELEFOON 04920-23981

DYNAMIC ASPECI'S OF PHOSPHORUS IN FOUR· AND FIVE.COORDINATED COMPOUNDS

DYNAMIC ASPECfS OF PHOSPHORUS IN FOUR- AND FIVE-COORDINATED COMPOUNDS
DNA as an example
PROEFSCHRIFT
TER VERKRIJGING VAN DE GRAAD VAN DOCrOR IN DE TECHNISCHE WETENSCHAPPEN AAN DE TECHNISCHE HOGESCHOOL EINDHOVEN, OP GEZAG VAN DE RECfOR MAGNIFICUS, PROF. DR. S. T. M. ACI<El{MANS, VOOR EEN COMMISSIE AANGEWEZEN DOOR HET COLLEGE VAN, DEKANEN IN HET OPENBAAR TE VERDEDIGEN OP
DINSDAG 25 OKTOBER 1983 TE 16.00 UUR
DOOR
JOHANNES JACOBUS CORNELIA VAN LIER
GEBOREN TE 's-GRAVENHAGE

DIT PROEFSCHRIFT IS GOEDGEKEURD DOOR
DE PROMOTOREN
PROF. DR. H.M. BUCK
EN
PROF. DR. J.B.F.N. ENGSERTS


"Wijs is hij/zij~ die van aZZen ?Jeet te tel"en"
- De Talmud -

Chapter I
Chapter 11
CONTENTS
General introduetion
1.1 Recent devetopments in organo
phosphorus chemietry
1.2 Generat properties of penta-aoordinated phosphorus aompounde
1.3 Different aonfigurations of DNA
1.4 Eiologiaal properties of DNA
1.5 Scope of this thesis
Referencee and Notes
Lithium halide and lithium perchlorate binding to phosphates. A multi-nuclear magnetic resonance spectroscopie study
11.1 Introduetion
11.2 The reaction of a five-membered
cyclic P(IV) compound ~ith alco
hol in the presence of lithium
haZides
11.3 Solvent- and salt-induced diffe
rential shielding effects in a
five-membered cyclia phosphonate
11.4 Lo~-temperature 31 P NMR inveeti
gation on a five-membered cyclic
phosphate in the presence of
lithium fluoride
11.5 7Li-4 36cl- and 81 Br NMR investi
gations on salt/phosphate aggre
gates in acetone
11.6 Conalusions
11.7 E~perimental
Beferences and Notes
8
25

Chapter 111
Chapter IV
Chapter V
Dynamics of penta-coordinated phosphorus 56 in the backbene of DNA III.l Introduetion
III.Z Model desaription of the B-.z
transition in DNA
111,3 CND0-2 and MNDO quantum-ahemiaaZ
calculations on model systems
representative for DNA
III.4 Discussion
Appendi~: Theory of the CND0-2
and MNDO quantum-ahemiaal methode
Beferences and Notes
B-Z transition in methylated DNA. A quantum-chemical study of model systems
IV.l Introduetion
IV.2 MNDO aalaulations
IV.3 Methylation of cytosine
IV.4 Methylation of guanine IV.S Discussion
Beferences and Notes
80
Molecular aspects of methylated adenine 97 in DNA. A quantum-chemical study of model systems
V. 1 Introduetion
V. 2 s-.z transition in aZternating
d(A-T)n potymers
V.3 MNDO caleuZations
V. 4 Computational data and stereo
model studies
V. 5 Discussion
Beferences and Notes

Chapter VI
Appendix
SuiD.ID.ary
SaiD.envatting
Tetraoxaspirophosphoranes with a six- 116 -membered ring as model compounds for_ the hydrolysis of ribonucleic acids by RNase A VI.1 Introduation
VI.2 NMR speatrosaopia study of the
formation of tetrao~aspirophos
phoranes aontaining a si~
-membered ring and a P-H bond
VI.3 Base-aataLyzed ring aLosure as
a modeL foP the aation of RNase A
VI.4 Disaussion
VI.S E~perimentaL
Referenaes and Notes
143
147
150
CurriculuiD. vitae 153
Dank'\l\Joord 154

CHAPTER I
General introduetion
I.1 Reaent developments in organophosphorus chemistry.
The research i~ phosphorus chemistry has developed rapidly inthelast decade 1
, A number of systems reveal the unique possibilities of phospporus for coordination in different valenee states 2 • Recent progress has stimulated research o~ the structural and dynamic stereochemistry of organophosphorus compounds, particularly in the field of biomolecules. In this respect, the discovery of stabie penta-coordinated (P(V)) organophosphorus derivatives 3 has been of paramount importance. Westheimer's~- 6 studies on the hydrolysis of five-membered cyclic phosph(on)ates have greatly advanced the understanding of the mechanistic aspects of phosphorylation
reactions (i.e. substitution at phosphorus). The enormous rate enhancement of the phosphorylation, as observed.for these compounds, was elegantly explained by assuming P(V) trigonal bipyramidal (TBP) intermediates with the capability of ligand reorganizations (pseudorotation, vide infra).
Interestingly, group transfer reactions (i.e. substitution at a phosphorus ligand) may also proceed via P(V) TBP intermediates. Voncken, Castelijns, van Aken and Buck 1 - 9 clearly established that, in this case, the selective step inheres a nucleÓphilic attack on one of the pseudo-equatorial positions in the TBP configuration.
P(V) TBP intermediate structures certainly play an important role in bicmolecules containing phosphorus. A well-known example of a biochemica! process in which this P(V) intermediacy has been accepted, is the hydrolysis of ribonucleic acids, catalyzed by bovine pancreatie ribonuclease
8

-
WHis 119
Fig. I.l Catalytia meahanism of RNase A.
ROH = ~·· 0
' \

(RNase A). This hydrolysis occurs via anchimeric assistance of the 2'-0H group of the ribose ring 10 - 12 , which is in close proximity to histidine 12 and the 3'-phosphate group. In the first transphosphorylation step, histidine 12 abstracts the 2'-0H proton thus facilitating apical attack on the phosphate group and formation·of a P(V) TBP intermediate (Fig. 1,1), This intermediate is stabilized by hydrogen bonding between protonated lysine 41 and the equatorial anionic oxygen ligands. Activation of the leaving 5'-nucleotide by protonated histidine 119 generates the 2',3'-cyclic phosphate 13
'14
, which, aft~r a similarly catalyzed hydrolysis, gives rise to the products (see Fig. 1.1).
Like RNase A, staphyZococcal nuclease hydrolyzes nucleic acids but: in contrast, operates on both RNA and DNA substrates15. Of the many known nucleases this enzyme is perhaps. the best characterized. The crystal structure has been resolved for the native enzyme 16 as wellas for the nuclease-thymidine-3',5'-diphosphate (pdTp)-calcium ion complex 17
(resolution, 1.5 ~). The X-ray data 16 • 17 and additional NMR studies 18 • 19 implicate two tyrosines (85 and 113) at the active site in direct interaction with the nucleotide inhibitor, pdTp 17 • Moreover, in this complex 17 the Ca 2+ ion is coordinated by asparagine 21, asparagine 40 and glutamine 43 and located at 4.7 ± 0.2 ~ distance from the phosphorus atom. The X-ray data 17 suggest that an intervening water or hydroxyl ligand can be accommodated as a ligand of the ca2+
ion. Subsequent nucleophilic attack of this ligand at phosphorus generates a P(V) TBP intermediate 20
•21 (Fig. !.2).
This intermediate structure is stabilized by hydrogen-bonding from the protonated arginines 35 and 87 to the anionic oxygen ligands. Proton transfer from arginine 87 to the 0(5') atom leads tp apical departure of the 5'-nucleotide. Interestingly, hydrolysis of the model substrate deoxythymidine-3'-phosphate-5'-p-nitrophenylphosphate, catalyzed by this enzyme, gave exclusive formation of p-nitrophenyl phosphate, whereas nonenzymatic base-catalyzed hydrolysis of the same substrate causes displacement of nitrophenoxide, a much better leaving group than the 5'-oxyanion of deoxythymidine 22 • This is a
10

0 I
" I
, _,b -go -;)>o. ___ r,~, Tyr'
113 5 --- • :.. - - - H + Arg --~
Co o '" s1
)p~3~0 + -#\ 0 Arg H ----0 b 35 0-H
_, Asp;-1- ---1
... *'+ ...... '·Ca' /' - '-GI u~-3----r Asp'
40
Fig. I.2 Proposed cataZytic mechanism for the action of
staphyZoaoccaZ nucZease on DNA substrates.

strong argument in favour of the proposed mechanism.
!.2 GeneraZ properties of penta-aoordinated phosphorus
aompounds.
An important factor governing the stereochemistry of P(V) compounds, in contrast to P(IV) compounds, is that the distribution of the ligands around the central phosphorus atom cannot be spherically symmetrical, i.e., the ligands in P(V) compounds are not equivalent. 23 As shown by X-ray analysis 24 - 26 , usually two types of structures of very similar energy23 are considered, the TBP and the square pyramid (SP). The TBP and SP geometries are characterized by nonequivalent bonding. In the TBP there are three equatorial and two apical substituents, whereas in the SP one apical and four basal ligands are encountered (Fig. I.3). In the
a a apical ligand e, I b~. I .. b
a:
'p-e b_....:p~b b: bas al ligand
e"J e: equator i al ligand a
TBP SP
Fig. I.J Different geometrie a of P(V) aompounds.
TBP configu;ation, the apical honds are langer and weaker than the equatorial bonds 27
• Apical sites are preferred by electron-withdrawing ligands, whereas electron-donating substituents !end to occupy equatoral positions 28
• This 'polarity rule is basedon experimental data 29 • 30 and is supported by semi-empirica! calculations 31 ' 32 • The polarity rule can be explained on the basis of a hybridization of the Pz and dz2 orbitals in the TBP to account for the apical honds, combined with three sp 2 orbitals in the equatorial plane 33
• Although this classica! description of the phosphorus hybridization in a TBP structure offers a good explanation for the observed selectivity, recent ESR studies of phosphoranyl radicals performed by Hamerlinck~ Schipper and Buak 3 ~- 36 reveal a high content of s-character in the apical
12

honds. This means that the discriminatien between the equatorial and apical ligands is determined by a smal! overbalance of s-character in the equatorial position, thus still supporting the physical organic properties of TBP phosphoranes. Furthermore, it has been found that smal! rings are easily accommodated if they.span an apical and an equatorial position. This strain rute 6 is a result of the 90° angle between apical and equatorial honds in the TBP and is demonstrated by the fact that in most of the stabie phosphoranes the phosphorus atom is part of a cyclic system. The diffuse dzz orbital accounts for the greater apical bond length. Apical sites are preferentially occupied by electron-withdrawing substituents, whereas the equatorial sp2 orbitals demand for electrens from the corresponding ligands. In addition, equatorial ligands are more capable to form d~-pn bonds to phosphorus (backdonation) 21
•
One of the consequences of the differences in bond strength in a TBP is that leaving groups preferentially depart from an apical position 6
-31
• Conversely, as required by the principle of microscopie reversibility 6
, nucleophilic attack on tetra~coordinated phosphorus forms TBP intermediates and/or transition states in which the extra ligand occupies an apical position. Interestingly, the TBP configuration is stereochemically non-rigid23
, This was established first for PF 5 by 19F-NMR showing one fluorine resonance 37 ,
while other investigations 38 indicate that the fluorines exchange their positions fast on the NMR time scale. A mechanism which accounts for this permutational isomerization is the Berry pseudorotation (BPR) 39 • In this process two equatorial and both apical ligands change place via an intermediate SP configuration, the remaining ligand being the pivot (Fig. 1.4). The energy harrier for BPR is considerably increased upon introduetion of ligands with different electron-withdrawing character or in case of small rings 31 •
Therefore, an alternative process which accounts for the same permutation is the Turnstite rotation (TR) which may
13

TBP SP TBP
Fig. I.4 The BePry pseudorotation proaess.
be favoured in (bi)cyclic phosphoranes 40 ' 41 (Fig. I.S). From X-ray diffraction studies it has been established that in the solid state the structure of P(V) compounds is distorted more or less from anideal TBP toward an SP geometry 42 ,
and that these distortions closely follow the local c2v eenstraint of the Berry intramolecular exchange coordinate. These data suggest that opening of an equatorial angle in the TBP is associated with an approximately equal degree of closing of the axial angle 43
•
top pair
bottam
Fig. I.& Turnstile rotation.
14

1.3 Different configurations of DNA.
The detailed knowledge with respect to the structural and dynamic stereochemistry of organophosphorus compounds (vide supra) can also be applied to the phosphate groups in the helix backbene of the DNA molecule. It is well known that the genetic information is linearly encrypted in the sequence of bases of nucleic acids which are the genetic material of all living organisms. The means by which the information is contained and transmitted in these molecules is a subject of extensive research which has been greatly advanced by the application of recombinant DNA techniques 44 •
Another major souree of information is the elucidation of the three-dimensional structures of nucleic acids by means of high-resalution single crystal X-ray diffraction studies 45 •
The main structural elements of nucleic acids are i) a five-membered sugar ring which is a ribose for RNA
and a 2'-deoxyribose for DNA. ii) the heterocyclic bases adenine (A). guanine (G).
cytosine (C), thymine (T) and. in case of RNA, uracil (U) replacing T. The bases are bound to C(l') of the sugar ring in the ~-configuration.
iii) the 3'-5'-phosphodiester linkage joining the individual nucZeosides (i.e. the combination of sugar and base).
Double-stranded nucleic acids are formed by Watson-Crick 46
hydrogen bonding between the complementary G and C or A and T(U) bases in both strands of the structure. The complementary strands are anti-paraZZeZ, i.e. have opposite S'-.3' direction. The numbering scheme 47 for the various structural units which bas been used in this work is given in Fig. !.6. DNA usually crystallizes as a right-handed double helix with anti conformation of the base residues (B-DNA) 48
• Other right-handed helices have been classified and designated as A-DNA 49 ' 50 , C-DNA 51 and D-DNA52 ' 53 •
Double-stranded DNA eligomers and polymers containing a strictly alternating CG sequence display unusual conformational properties. Using circulair dichroism (CD) studies, PohZ and Jovin 54 demonstrated that poly d(G-C) undergoes a
15

thymine
cytosine
Fig. I.6 Structure of a DNA fragment containing the four common bases in DNA~ tagether with the numbering
echemes ueed for the pyrimidine (C~ T) and purine (A~ G) bases and the 2'-deo~yriboses. In the cor
reeponding RNA oligomer# the 2'-deo~yriboee wiZZ be replaced by ribose and T wiZZ be substituted
by u.
salt-induced conformational change which is characterized by a speetral inversion in high-salt solution. This transition appeared to be reversible, Interestingly, 1H- and 31 P-NMR studies revealed that the d(G-C) 8 duplex in high-salt solution contained two different types of nucleotide and phosphate group conformations including glycosidic dihedral angles which are different from B-DNA 5 5
'56
• Recently, Wang et al, 51 reported a novel Zeft-handed Z-DNA helix with the sequence d(C-G) 3 . The dG residues in the structure have an C(3')-endo (3E) pucker and a syn conformation in contrast
16

to the C(2')-endo ( 2E) pucker and anti conformation which is observed for B-DNA (Fig. I.7). A similar left-handed duplex structure bas been observed in d(G-C) 3
58 and d(G-m5c) 3
59 single crystals. In solution, the Raman spectra
\ 0
0
J o-o~P~
\ 0
NHz
er Z-DNA
Fig. I.? Geometrie differenaes between B-DNA and Z-DNA for
a 5'-dpG fragment.
of the high- and low-salt forms of poly d(G-C) differ from each other 60 • Recent Raman studies show that the spectrum of crystalline d(G-C) 3 is essentially the same as that of poly d(G-C) in high-salt solution61
• Thus the conformations are identical. This means that the salt-induced conversion as observed for the poly d(G-C) duplex can be interpreted as a transition from a right-handed helix into its left-handed isomer (B-.z transition). Furthermore, X-ray diffraction data on Z-DNA single crystals 57
-59 revealed the unusual
y- and yt conformations around the C(4')-C(S') bond. A yt conformation bas also been established for model organophosphorus substrates in the active site of RNase A10 - 12
and of staphylococcal nuclease 17• Interestingly, for RNase A,
intermediate P(V) TBP structures are known to be involved in the hydrolysis of ribonucleic acids, whereas for staphylococcal nuclease similar intermediates have been proposed (c.f. Chapter I.l). These results seem to parallel the calculated decreased preferenee of the y+ conformer in favour of the y- and yt conformers upon P(IV)-P(V) TBP activatien of the phosphate groups in the helix backbone of the DNA structure (c.f. Chapter III).
Characteristic differences between right-handed and left-handed helices are given in Table I.l. Additional studies revealed that the B-.z transition is generally observed
17

Table I.1 St~ucturaZ pa~amete~s in DNA.
Helix sense Right-handed Left-handed Designation A B c D z Sugar pucker 3Ea ZE ZE ZE ZE (dC); 3E (dG) Glycosyl angle anti anti anti anti anti (dC); syn (dG) Twist per base 32.7° 36° 38.6° 45° -60°/2b
pair Bases per turn 11 10 9.33 8 12
Rise per base 2.6 3.4 3.3 3.0 3.7 pair, Ä Base tilt0 19° -60 -80 -16° -70
a 3E, C(3')-endo; 2E, C(2')-endo. bAs a result of the alternating sugar pucker the repetitive unit is a dinucleotide. 0 Deviation from a plane perpendicular to the helix axis.
in DNA structures with an alternating purine-pyrimidine sequence such as poly d(A-C). poly d(G-T) 62 and poly d(G-m5c). poly d(G-m5C) 63
• Moreover, Z-DNA structures have been detected in vivo 6 ~' 65 • Very recently, Singleton et aZ. 66 showed that a Z-DNA is induced by supercoiling (i.e. higher order structure of DNA) under physiological conditions. Therefore, the mechanistic aspects of the B-.Z transition deserve attention.
I.4 Biologiaal p~ope~tiea of DNA.
DNA has two major and discrete functions. One is to carry the genetic information that brings about the specific phenotype of the cel!. The other function of DNA is its own replication67 • In DNA ~epliaation, a comple,mentary copy of each strand is first made. Since each new strand remains bound to one of the old strands the net result is two double helices, each identical with the one originally present. Thus, for duplicating the genotype of the cell, DNA serves as a template for converting one chromosome into two identical chromosomes. This 'semi-conservative 1 mode of replicatien
18

of DNA was originally predicted by Watson and Crick~ 6 •
DNA, whicJl resides in the nucleus of the cell, does not determine the amino acid sequence of a protein directly. Instead, the base sequence of DNA serves as a template for the synthesis of a single strand of messenger RNA (mRNA). This transcription process produces mRNA with a base sequence complementary to the transcribed DNA strand, with uracil replacing thymine. Very recently, Wang et al. 68 reported the detailed three-dimensional structure of a short DNA-RNA hybrid helix joined to double helical DNA and showed that this fragment adopts a helix close to 11-fold DNA (A-DNA). Once mRNA has been sythesized, it passes out of the nucleus into the cytoplasm to the ribesomes where translation of the base sequence into an amino acid sequence of a protein is accomplished.
I.S Scope of this thesis.
As evidence in favour of the important dynamic role of phosphorus in the DNA molecule is increasing 10 , it is useful to apply the knowledge of phosphorane intermediates to the phosphate groups in the helix backbone of DNA in order to obtain a better understanding of the salt-induced B-.z transition. Chapter II describes a study of the interaction of model organophosph(on)ates with lithium salts in solution using multi-nuclear NMR. The 35c1- and 81 Br NMR results can be explained by assuming fast equilibria between lithium balides in the complexed (with phosph(on)ates) and the free form. The 7Li NMR chemica! shift and line-broadening data reveal complexation of the phosphoryl oxygen atom by the Li+ ion, preferentially in 1:1 mol ratio salt/phosphate. Furthermore, kinetic studies on the combined phosphorylation and group transfer (de-alkylation) in the salt/phosphate/ methanol system reveal that the phosphorylation reaction is retarded in the presence of lithium halides. The magnitude of the effect is related to the ionic radius of the halide anion as well as to the solvation properties of this anion.
19

The data therefore provide evidence for the proximity of the halide anion to the phosphorus atom. Low-temperature 31 P NMR measurements combined with CND0-2 calculations reveal that the close-ion pair structure is the NMR observed configuration, whereas short-lived P(V) structures are encountered as intermediates in the reactions. Based on these data and on three-dimensional structural features of B and Z forms of DNA, a detailed model description of the salt-induced B-.z transition in DNA with alternating purine-pyrimidine sequences is given in Chapter III. The performed CND0-2 and MNDO quantum-cnemical calculations support the suggested selective role of P(V) TBP intermediates within 5'-dpC structural units in DNA as an initial inducer of the B-.Z transition. Moreover, the calculations reveal a significant difference in selectivity between purine and pyrimidine bases in the structure which offers a good rationalization for the observed features in the process.
Chapter IV provides evidence for the considerable impact of specific methylation of C.G base pairs on the relative stability of B and Z isomers of DNA based on calculations using methylated tetrahydrofuryl model systems. The obtained data are related to known experimental details. The calculations reveal that an important stabilization of the Z conformer is obtained upon specific methylation. The possible biologica! significanee of methylated cytosine residues in the genomic (supercoiled) DNA is discussed in relation to the obtained computational results and known experimental data.
The molecular aspects of methylated adenine in DNA,
specifically focused on an altered anti-syn equilibrium due to selective methylation, are discussed in Chapter V, based on calculations on tetrahydrofuryl model structures and experimental data.
In Chapter VI, model compounds derived from tetraoxaspirophosphoranes are used to study the base-catalyzed ring opening and ring ciosure process. This behaviour mimics the possible role of the enzymatic sites in the active cleft of RNase A during hydrolysis of ribonucleic acids.
20

HefePenoes and Notes.
1. For up-to-date reviews on the subject, see the series "Organophosphorus Chemistry" (Specialist Periodical Reports), S. Trippett, ed., The Chemica! Society, London.
2. The following abbreviations are used: P(IV) tetra-coordinated phosphorus; P(V) penta-coordinated phosphorus; TBP trigonal bipyramid(al); SP square pyramid(al); BPR Berry pseudorotation; TR Turnstile rotation.
3. F. Ramirez, R.B. Mitra and N.B. Dessai, J. Am. Chem. Soc., 1960, 82, 2651.
4. J. Kumamoto, J.R. Cox and F.H. Westheimer, J. Am. Chem. Soc., 1956, ?8, 4858,
5. A. Eberhard and F.H. Westheimer, J. Am. Chem. Soc., 1965, 8?, 253.
6. F.H. Westheimer, Acc. Chem. Res., 1968, 1, 70. 7. W.G. Voncken, Ph. D. Thesis, Eindhoven University of
Technology, 1976. 8. A.M.C.F. Castelijns, Ph. D. Thesis, Eindhoven University
of Technology, 1979. 9. D. van Aken, Ph. D. Thesis, Eindhoven University of
Technology, 1981. 10. R.R. Holmes, "Penta-coordinated Phosphorus" (ACS Monograph
176), Vol.II, American Chemica! Society, Washington D.C., 1980, 180.
11. F.M. Richards and H.W. Wyckoff, "The Enzymes", Vol.IV, P.D. Boyer, ed., Academie Press, New York, 1971.
12. D.G. Gorenstein, A.M. Wyrwycz and J, Bode, J. Am. Chem. Soc., 1976, 98, 2308.
13. R.R. Holmes, J.A. Deiters and J.C. Galluci, J. Am. Chem. Soc., 1978, 100, 7393.
14. C.A. Deakyne and L.C. Allen, J. Am. Chem. Soc., 1979, 101, 3951.
15. C.B. Anfinsen, P. Cuatrecasas and H. Taniuchi, "The Enzymes", P.D. Boyer, Ed., 3rd ed., Vol. IV, Academie Press, New York, 1971, 177.
16, F.A. Cotton and E.E. Hazen, Jr., "The Enzymesn, P.D. Boyer, Ed., 3rd ed., Vol. IV, Academie Press, New York,
21

1971, 153. 17. F.A. Cotton, E.E. Hazen, Jr. and M.J. Legg, Proc. Natl,
Acad. Sci. u.s.A., 1979, 76, 2551 and raferences cited therein.
18. J.L. Markley and 0. Jardetzky, J. Mol. Biol., 1970, 50,
223. 19. G.C.K. Roberts and 0. Jardetzky, Adv. Prot. Chem., 1970,
24, 44 7. 20. A.S. Mildvan, Annu. Rev. Biochem., 1974, 43, 357. 21. A.S. Mildvan and C.M. Grisham, Struct. Bonding(Berlin),
1974, 20, 1. 22. B.M. Dunn, C. DiBello and C.B. Anfinsen, J. Biol. Chem.,
1973, 248, 4769. 23. R. Luckenbach, "Dynamic Stereochemistry of Penta-coordi
nated Phosphorus and Related Elements", G. Thieme, Stuttgart, 1973.
24. E.L. Muetterties and R.A. Schunn, Quart. Rev. Chem. Soc., 1966, 20, 245.
25. T.E. Clark, R.O, Day and R.R. Holmes, Inorg. Chem., 1979, 18, 1653.
26, T.E. Clark, R.O. Day and R.R. Holmes, Inorg. Chem., 1979, 18, 1668.
27. F. Ramirez and I. Ugi, "Advances in Physical Organic Chemistry", Vol. 9, V. Gold, ed., Academie Press, London, 19 71 •
28. P. Gillespie, P. Hoffmann, H. Klusacek, D. Marguarding, S. Pfohl, F. Ramirez, E.A. Tsolis and I. Ugi, Angew. Chem., 1971,83,691.
29. E.L. Muetterties, W, Mahler and R. Schmutzler, Inorg. Chem., 1963, 2, 613,
30. E.L. Muetterties, K.J. Packer and R. Schmutzler, Inorg. Chem., 1964, 3, 1298.
31. D. Marguarding, F. Ramirez, I. Ugi and P. Gillespie, Angew. Chem., 1973, 85, 99.
32. F. Keil and W. Kutzelnigg, J. Am. Chem. Soc., 1975, 97,
3623. 33. R.F. Hudson and M. Green, Angew. Chem., 1963, 75, 47. 34. J.H.H. Hamerlinck, Ph. D. Thesis, Eindhoven University
22

of Technology, 1982. 35. J.H.H. Hamerlinck, P. Schipper and H.M. Buck, J. Org.
ehem., 1983, 48, 306. I
36. J.H.H. Hamerlinck, P. Schipper and H.M. Buck, J. Am. ehem. Soc., 1983, 105, 385.
37. H.S. Gutowski, D.M. Meeall and e.P. Slichter, J. Chem. Phys., 1953, 21, 279.
38. H.S. Gutowski and A.D. Liehr, J. ehem. Phys., 1953, 20,
1652. 39. R.S. Berry, J. ehem. Phys., 1960, 32, 933. 40. F. Ramirez, S. Pfohl, E.A. Tsolis, J.F. Pilot, e.P. Smith,
I. Ugi, D. Marguarding, P. Gillespie and P. Hoffmann, Phosphorus, 1971, 1, 1.
41. I. Ugi, D, Marguarding, H. Klusacek, P. Gillespie and F. Ramirez, Acc. ehem. Res., 1971, 4, 288.
42. R.R. Holmes, Acc. ehem. Res. 1979, 12, 257. 43. H.-B. Bürgi and J.D. Dunitz, Acc. ehem. Res., 1983, 16,
153. 44. M. Singer, "Genetic Engineering", A. Hollaender and J.
Setlow, Eds., Vol. I, Plenum Press, New York, 1979. 45. N.e. Seeman, "Nucleic Acid Geometry and Dynamics", R.H.
Sarma, ed., Pergamon Press, New York, 1980, 47. 46. J.D. Watson and F.H.e. erick, Nature(Lond.), 1953, 171,
737. 47. Abbreviations and symbols follow IUPAC-IUB Recommenda
tions, see Eur. J. Biochem., 1983, 131, 9. 48. H.R. Drew, R,M, Wing, T. Takano, C. Broka, s. Tanaka,
K. Itakura and R.E. Dickerson, Proc. Natl. Acad. Sci. u.s.A., 1981, 78, 2179.
49. B.N. Conner, T. Takano, S. Tanaka, K. Itakura and R.E. Dickerson, Nature(Lond.), 1982, 295, 294.
50. A.H.-J. Wang, S. Fujii, J.H. van Boom and A. Rich, Proc. Natl. Acad. Sci. U.S.A., 1982, 79, 3968,
51. s. Arnott, R. Chandrasekaran, D.W.J. Hukins, R.s.c. Smith and L. Watts, J. Mol. Biol., 1974, 88, 523,
52. S. Arnott and E. Selsing, J. Mol. Biol., 1975, 98, 265. 53. A. Mahendrasingam, N.J. Rhodes, D.C. Goodwin, e. Nave,
W.J. Pigram, W. Fuller, J. Brahms and J. Vergne,
23

Nature(Lond.), 1983, SOl, 535, 54. F.M. Pohl and T.M. Jovin, J. Mol. Biol. 1972, 67, 375. 55. D.J. Patel, L.L. Canuel and F.M. Pohl, Proc. Natl. Acad.
Sci. u.s.A., 1979, 76, 2508. 56. D.J. Patel, "Stereodynamics of Molecular Systems", R.H.
Sarma, ed., Pergamon Press, New York, 1979, 397. 57. A.H.-J. Wang, G.J. Quigley, F.J. Kolpak, J.L. Crawford,
J.H. van Boom, G. van derMareland A. Rich, Nature(Lond,), 1979, 282, 680.
58. H. Drew, T. Takano, S. Tanaka, K. Itakura and R. Dickerson, Nature(Lond.), 1980, 286, 567.
59. A.H.-J. Wang, S. Fujii, J.H. van Boom and A. Rich, Cold Spring Harhor Symp. Quant. Biol., 1982, 47, 5.
60. F.M. Pohl, A. Renade and M. Stockburger, Biochim. Biophys. Acta, 1973, SS6, 85.
61, T.J. Thamann, R.C. Lord, A.H.-J. Wang and A. Rich, Nucleic Acids Res., 1981, 9, 5443.
62, s. Arnott, R. Chandrasekaran, D.L. Birdsall, A.G.W. Leslie and R.L. Ratliff, Nature(Lond.), 1980, 28S, 743.
63. J. Nickol, M. Behe and G. Felsenfeld, Proc. Natl. Acad. Sci. u.s.A., 1982, ?9, 1771.
64, A. Nordheim, M.L. Pardue, E. M. Lafer, A. Möller, B.D. Stollar and A. Rich, Nature(Lond.), 1981, 294, 417.
65. H. Hamada and T. Kakunaga, Nature(Lond.), 1982, 298, 396. 66. C.K. Singleton, J. Klysik, S.M. ~tirdivant and R.D.
Wells, Nature(Lond.), 1982, 299, 312. 67. A. Kornberg, "DNA Replication", A.C. Bartlett and P.
Brewer, eds., W.H. Preeman and Company, San Francisco, 1980.
68. A.H.-J. Wang, s. Fujii, J.H. van Boom, G.A. van der Marel, S.A.A. van Boeckel and A. Rich, Nature(Lond.), 1982, 299, 601.
24

CHAPTER 11
LlthiuiD halide and lithluiD perchlorate
binding to phosphates. A IDulti -nuclear
IDagnetic resonance spectroscopie study
11.1 Introduation.
Growing recognition of the importance of biologically relevant metal ion interactions with nucleic acids and nucleotides has stimulated research focused on the chemistry of the complexes formed 1 - 4 • lnteractions between alkaline earth metal species and nucleosides (without phosphate group) are weak and can be explained by means of only a few specified binding criteria 5 , 6 • In nucleic acids, the phosphate-to-metal bonding dominates. Recent ab initia
d . . . f L.+ N + B 2+ dM 2+ . h stu 1es on 1nteract1ons o 1 , a , e an g Wlt H2Po4- reveal significant electron transfer for all complexes, except those invalving Na+ 7 • This implies that these interactions are not purely ionic. Ion pairs in organic solvents have been studied extensively by combinations of multi-nuclear magnetic resonance techniques 8 • 9 •
Recently a thorough study has been performed by Caatelijns 10
' 11 on the reactivity, the stereochemistry and the kinetics of de-alkylation reactions of phosphates and phosphonates with LiCl and LiBr in solvents of various polarities. The NMR- and kinetic data provided strong evidence for the involvement of a penta-coordinated (P(V)) intermediate with only a moderate charge separation in the rate-determining step of the de-alkylation reaction (Fig. 11.1). Although no conclusion could be drawn concerning the amount of charge separation in the P(V) intermediate, it was clearly demonstrated that its reactivity obeyed the stability rules of pentavalent trigonal bipyramidal (TBP)
25

0 0 . 11 •
R, /P, /R' LiX "o·;t 'o' . ~
R" oR-o, 1
' + 'p -o-: .... u
R-0 I' Li ... _ , e - R'-X+ ;p~
R'/ 0 X :F,Cl,Br ~! 0 0
'\ .. Fig. II.1 P(V) intermediate in the de-alkylation reaation.
phosphorus compounds. These results are in contrast with previous work which argues in favour of an "Arbusov"-type mechanism in which reactions proceed via a nucleophilic attack on a saturated carbon, linked through oxygen with the tetravalent phosphonium ion (A) 12 - 1 '*. Even in reactions in which the initial formation of a P(V) compound was established, the de-alkylation was supposed to occur via an intramolecular SNZ displacement from the ion-pair, although a a2s + crza- thermal pericyclic disproportionation reaction from the covalent P(V) compound (B) was not ruled
A
OR
RO .. ,, I C?u~~-OR
I x R
• B .
out 15 - 20 • With this in mind, it was tempting to investigate the nature of the interaction of lithium balides and lithium perchlorate with model phosphates and a model phosphonate in salution by means of multi~nuclear NMR. Therefore 1H NMR spectra of 2-isopropoxy-2-oxo-5-methyl-1,2-oxaphosphol-4-ene (compound 1, Fig. II.2) were recorded in a number of organic solvents. Also 7Li-, 35crand 81 Br NMR spectra of the salt/phosphate complexes in acetone were recorded to yield chemica! shift data for the 7Li nuclei and line-width data for the 35c1- and 81 Br nuclei. Moreover, the solvent- and salt-dependent nonequivalence of the methylene protons in 1 was ex~ined. Besides the study of aggregates by physical means, it also appeared
26

worthwhile to investigate the effect of lithium halides on the rate of phosphorylation of 2-methoxy-2-oxo-4,5-dimethyl-1,3,2-dioxaphosphol-4-ene (compound 2, Fig. 11.2). Bromide anions cause a remarkable decrease in the rate of phosphorylation toward methanol in different cyclic phosph(on)ates 11
• In order to establish whether this retardation is a more general property of halide anions in organic solvents, detailed kinetic data of the system LiX(X=F, Cl, Br)/2/methanol in deuteriochloroform were
determined. Finally, low-temperature 31 P spectra for the combination 2/LiF in tetrahydrofuran were investigated to examine the possibility of the formation of a covalent P-F bond. Some of the conclusions are corroborated by the results of CND0-2 quantum-chemical calculations.
II.2 The Peaation of a five-membePed ayatic P(IV) compound
with alcohol in the pPeaence of lithium halidea.
Addition of LiX (X=F, Cl, Br, Cl04) to solutions of compounds 1-4 (Fig. II.Z) in acetone reveals deshielding
0'\, YO . CH3 0'\_p/OJCH3
CH3 /p I / \ ~0 , H H3C-O 0 CH3
~-~~·'7 ', CHJ H H 1 2
0 11 rQ1
,.-P..\:'o~
if: ~ 4
Fig. II.2 Model aompounda 1-4.
of the proton resonances in the 1H NMR most likely as a consequence of the complexation of the phosphoryl oxygen atom with the lithium cation. This results also in a
27

deshielding of the phosphorus atom. These results are in accord with former workon salt/phosphate aggregates 11 •
21-
24•
The above-mentioned adducts all disproportionate on prolonged standing to the corresponding de-alkylated products in case of LiF, LiCl and LiBr. The weakly nucleophilic CI04 ion is not capable of inducing the de-alkylation process.
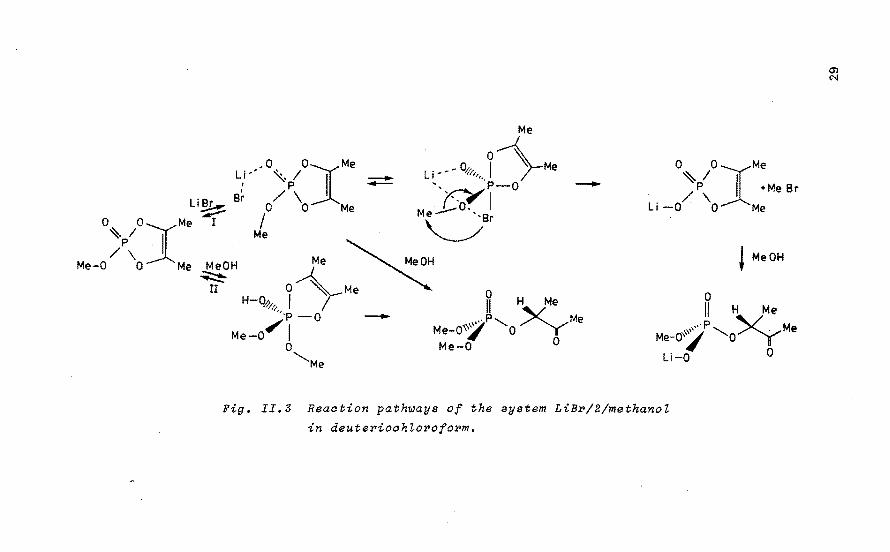
Addition of one equivalent of methanol to a 0.5 M salution of 2 in tetrahydrofuran (THF) results in a fast phosphorylation reaction. However, in the presence of one equivalent of LiBr, almost complete de-alkylation is observed 11 • In order to elucidate the observed decreasein the rate of phosphorylation, the model system LiX(X=F, Cl, Br)/2/methanol was studied in CDC1 3 in equimolar ratio of the components (0.3 M). To dissect the reaction rates of the combined phosphorylation (Path II, Fig. II.3) and de-alkylation (Path I) in ~he system, kinetic measurements in CDC1 3 were performed on 2/methanol (equimolar ratio, 0.3 M) and 2/LiBr (equimolar ratio, 0.3 M). In view of the poor solubility of LiBr in CDCI 3 at 298 K (8.10- 3 mol/L), the effective LiBr concentratien in salution during the de-alkylation process can be regarded as constant and the reaction rate becomes pseudo-first order in phosphate with -d!Pl/dt = k2.!PJ and k2 = k1.!BJ in which !Pl is the concentratien of phosphate (mol/L) a~d (BJ is the concentratien LiBr (mol/1). This gives ln(IPJt/[PI 0) = -k1.!Bl.t with an average k1 = 3.41 10-3 L/mol s. [BI was estimated at 10-Z mol/1 of LiBr. Earlier kinetic
measurements of the de-alkylation (Path I) in the more polar acetone-d6 revealed that the reaction rate was first order in phosphate and first order in LiBr 10
• In case of phosphorylation (Path II), kinetic data on the system 2/methanol in deuteriochloroform revealed that the reaction was first order in phosphate and of zero order in methanol
-3 -1 with an average rate constant of 2.15 10 s • As the phosphorylation proceeds approximately 24 times faster than the de-alkylation, it is reasanabie to neglect the latter path in case of the phosphate/salt/alcohol system. Therefore,
28
Me
• 0 OJMe o 01 ~ Me Li • • '\. 1
1
_ L j -- - w/,,,.. r I p ._. ', P-O
L'B B~ / \ ~I ~~ o o Me M L'o~ ~ e- ,
0 0 Me I / ~Br
Me-O:>~OlMe MeOH Me Me ......... MeOH
~ 0~ Me ~
-
0 H-Q~,~. I I l,,·p-o -
Me-0,.- I 11 H Me
, .. P'-.. y Me Me-o''l o' Y Me-0 ° 0
'-...Me
0 OJMe ~I P I +MeBr
/ \ Li-0 0 Me
~ MeOH
0 11 H Me
··P XyM "'''' -......... . e Me-0\"/ 0
Li-0 O
Fig. II.3 Reaation path~aya of the ayatem LiBP/2/methanoZ
in deutePioahZoPofoPm.

this system can be approximated by a first order reaction in phosphate. The kinetic first order plots of the phosphorylation (Fig. !!.4) reveal that the reaction rate decreases
0
In [ Pl t
I 5.0
6.0
0
Fig. II.4
T=298 K
Li Br
LiF
sa I t-f ree
600 1200 1800 2400 3000 3600
---- t Is I Least squares plot of Zn[P]t vs. t for the system
LiX(X=F$ CZ$ Br)/2/methanoZ in CDCZ 3•
The initiaZ amounts of the different compounds are equimoZar (O,S M).
in the order salt-free>fluoride>chloride>bromide (Table II.1). This result is particularly striking because complexation of the phosphoryl oxygen by the lithium cation increases the phosphorylating ability of this compound toward alcohols. The measured rate constant for the phosphorylation is somewhat lower than the real value because a slight amount of methanol is located in the inner solvation shell of the lithium cation. As a consequence, the amount of free methanol is slightly reduced.
The results show a relation between the reaction rate of the phosphorylation and the ionic radius of the different anions studied. The observed relation is not linear because of the different solvent reorientations induced by the halide ions involved 9 • This creates unique solvation spheres for the various anions. As a result of their ordered solvation spheres, the halide ions, which appear as close-ion pairs 9
30

Table. II.1 Kinetia data of the system LiX(X=F~ CZ~ Br)/2/
methanol in CDCZ 3 at T=298 K.a
salt . b t 1, m1n. r,a Ä 10 3k, s -1 d krel
e
- 5.57 - 2.15 1.00
LiF 11 • 38 1.33 1.02 0.48
LiCl 13.17 1.81 0.88 0.40
LiBr 31 • 81 1.96 0.36 0.17
aThe initia! concentrations of the various compounds are equimolar (0.3 M).
bobtained via the equation t!=ln2/60k. aAnionic radius without solvation spheres.
'
dDetermined via the slope of the straight line in Fig. 11.4. e -3 krel=k/(2,15 10 ).
in combination with the metal ion, are capable of shielding the phosphorus atom against a nucleophilic attack of methanol (Fig. II.5). Consequently, the phosphorylation of methanol is retarded in comparison with the salt-free experiment. Most likely, attack of methanol proceeds by a displacement of the halogen atom followed by a fast intramolecular nucleophilic attack at the phosphorus atom. These kinetic data therefore provide evidence for the proximity of the halide ion to the phosphorus atom.
0 . ; ' ir---·uu--~-0
0 .. •' i
J~~---~----.--e Fig. II.5 Stereosaopia ORTEP drawing of the LiBr/2 aggre
gate. The alosest P-Br distanae in the aompZe~
was aaZaulated to be 4.0 i. The different radii
used for saaZing do not repreaent aotvation.
31

11,3 Solvent-and salt-induced diffe~ential shielding effects
in a five-membe~ed cyalic phosphonate.
In order to improve the knowledge about the location of the Li+ ion in the salt/phosphate complexes studied here, the paramagnatie Eu3+ ion was used in combination with compound 1, Addition of Eu(fod) 3
25 to a solution of 1 in CDC1 3 reveals deshielding for all the proton resonances in the NMR spectrum due to phosphoryl oxygen complexation by the Eu3+ ion (Fig. II.6). The shift of the tertiairy isopropoxy proton (4) shows a high concentratien dependence. Obviously, the Eu3+ ion is located in the proximity of this proton. This confirms the previously observed complexation of the phosphoryl oxygen atom by the Li+ ion. Moreover, the asymmetrie ring in the structure causes, via preferred orientation of the Eu(fod) 3 complex, increased shift differences of the isopropoxy methyl gróups at higher Eu(fod) 3
~ppm
l
0 0 Me
14 ; >çr ~0 H
4 12 Me : 5
1 Me H. H ! 3' 3
10
8
oL-----~o.~2------~o.~4----~o~.a~----~o~.8~----~1~~~--eq Eu lfodl3
Fig. II.6 60 MHz 1H NMR data of compound 1 in CDCZ 3 ~ith diffe~ent concent~ations of Eu(fodJ 3 at P=298 K.
ö in ppm vs. TMS,
concentrations (viz. two resonances 1). The asymmetrie location of the Eu3+ ion with respect to the ring methylene
32
protons (3 and 3 1) causes a different deshielding for both
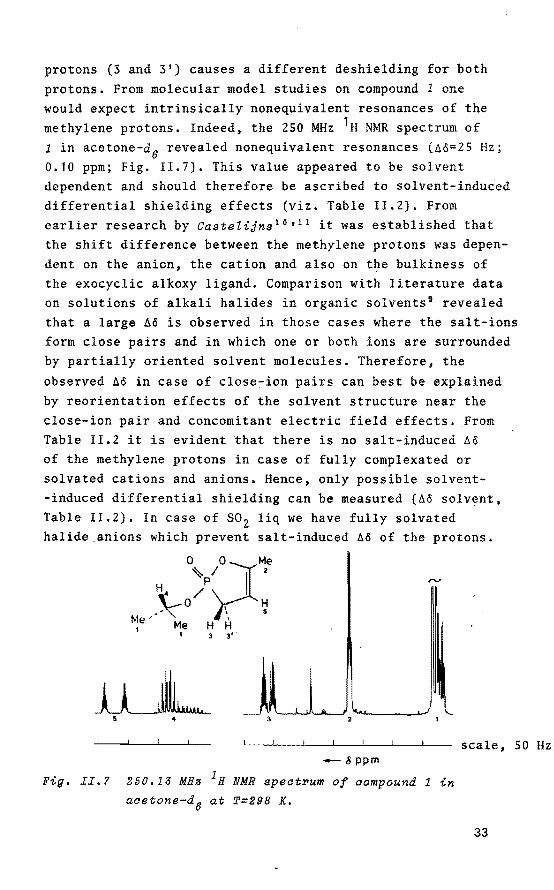
protons. From molecular model studies on compound 1 one would expect intrinsically nonequivalent resonances of the methylene protons. Indeed, the 250 MHz 1H NMR spectrum of 1 in acetone-d6 revealed nonequivalent resonances (Ao=25 Hz; 0.10 ppm; Fig. 11.7). This value appeared to be solvent dependent and should therefore be ascribed to solvent-induced differential shielding effects (viz. Table !1.2). From earlierresearch by CasteZijns 10 • 11 it was established that the shift difference between the methylene protons was dependent on the anion, the cation and also on the bulkiness of the exocyclic alkoxy ligand. Comparison with literature data on solutions of alkali halides in organic solvents 9 revealed that a large Aö is observed in these cases where the salt-ions form close pairs and in which one or both ions are surrounded by partially oriented solvent molecules. Therefore, the
observed Ao in case of close-ion pairs can best be explained by reorientation effects of the solvent structure near the close-ion pair and concomitant electric field effects. From Table 11.2 it is evident that there is no salt-induced Aê of the methylene protons in case of fully complexated or solvated cations and anions. Hence, only possible solvent-induced differential shielding can be measured (Aê solvent, Table II.2). In case of so2 liq we have fully solvated halide_anions which prevent salt-induced Aö of the protons.
o~Pçro I t.;e H4 / .\-a \ ~
Me· Me H H I I 3 3'
-IJ ppm
Fig. II,? 250.13 MHa 1H NMR spect~um of compound 1 in
acetone-d6 at T=298 K.
scale, 50 Hz
33

Table II.2 Solventand salt effeats on the ahemiaal shift
differenae äö of the methylene protons of 1. 4
clóse;-ion salt solvent solventa saltd . b pa1r M ppm llO ppm
- - (CD 3) 2SO 0 -- - co2c1 2 0.048 -- -. CDC1 3 0.100 -
yes LiC104 CD3No2 0 0.048 yes LiCl(satd) CD3No 2 0 0.060 yes LiCl(sàtd) (CD3) 2co 0.01 0.050 yes LiBr(1 equiv) (CD3) 2Co 0.01 0.286 yes LiBr(satd) C6D6 0. 1768 0.504 no LiCl04 SOz liq 0 0 no LiBr(1 equiv) so2 liq 0 0 no LiBr (CD3) 2so 0 0
no A1Cl 3 (CD3) 2CO 0.010 0 no LiBr/kryptofix (CD3) 2CO 0.010 0
(1: 1)f
a250.13 MHz 1H NMR (T=298 K). bAccording to Popov 6 and Weingärtner 9
• aSolvent-induced differentlal shielding. d~o(salt): measured total effect, containing contributions from both solvent and ion pairs. eThis large effect can be explained by aromatic-solvent-induced• shift (ASIS) which is known for phenyl fragments. fKryptofix 221/LiBr in 1:1 mol ratio.
In case of A1C1 3 we have a Lewis aaid ions (X=F, Cl, Br) in the presence of 7Li NMR measurements of a 1:2 mixture
which.forms A1C1 3xhalide anions 11
, 26
of kryptofix 221 and LiBr in acetone revealed two resonances of equal intensity at o( 7Li) 2.43 ppm and o( 7Li) -0.11 ppm which can be attributed to the LiBr in salution and the fully complexed kryptofix 221.Li+, respectively. In a 1:1 mixture there is no free LiBr in solution. As a result there is no salt-induced ~ö.
34

Formation of a covalent bond between the Li+ ion and the phosphoryl oxygen atom would lead to a more positively charged phosphorus atom. In order to investigate a possible change in net atomie charge on the phosphorus atom upon addition of lithium salt, the accurate values of the 2JHH geminal coupling constants 27 had to be determined via computer simulation of the 250 MHz 1H NMR spectra of 1
in acetone-d6, in the presence of öne equivalent of LiBr in acetone-d6 and in benzene-de (Fig. II.8-II.10). The results are summarized in Table 11.3. From this table it is evident, that the difference in 2JHH between acetone-de and acetone-d6 with one equivalent of LiBr is 0.34 Hz. This implies a negligible change in the net atomie charge on the phosphorus atom. Moreover, this conclusion is consistent with the observed small (approximately 1 ppm) 31 P chemical shift differences which occur upon addition of salt.
al ~~--l-~~~~--'---'--- scale, 10Hz
Fig. II.B RecoPded (a) and computeP-eimulated (bJ 2S0.1J
MHs 1n NMR epectPum of the methylene pPotone of compound 1 in acetone-d6• Notice the Pesemblance of the fine structuPe in the lo~-field domain (nearly forbidden traneitione).
35

Ao __ -..
··~~···~-~· scale 10 Hz o.so _ ~ppm '
Fig. II.9 Reaorded (a) and aomputer-simulated (b) 250.13
MHs 1H NMR speatrum of the methylene protons of
aompound 1 in the presenae of one equivalent
LiBr in aaetone-d6 ,
-~ppm
Fig. II.10 Reaorded (a) and aomputer-simulated (b) 250,13
MHz 1H NMR speatrum of the methylene protons
of aompound 1 in bensene-d6•
36

Tab~e II.3 Indireat aoup~ing aonstants {J) and chemiaal
shift values (W) via computer-simulated
260.13 MHz 1H NMR spectra of 1. 0 0 Me
~p))' s Me /' .\-o '. ~ Me' H H H
2 J
W a '
ppm J, Hz
Acetone-d 6 , Rms error=0.042 W(1) J 12 12.64 3 24 2.75 W(2) 0.538 3 13 15.74 3 25 2.55 W(3) 0.548 3 14 33.61 334 3.08 W(4) 3.201 3 15 0.80 335 2.28 W(S) 0 3 23 -18.67 345 1.44
Benzene-d6 , Rms error=0.116 W(1) 3 12 13.46 3 24 2. 72 W(Z) 0,681 313 15.24 3 25 2.49 W(3) 0.503 J14 33.54 3 34 3.03 W(4) 2.917 315 0,85 3 35 2.30 W(S) 0 3 23 -18.41 345 1. 45
Acetone-d0/LiBr 1:1 mol ratio, Rms error=0.167 W(1) J12 14.28 3 24 2.69 W(Z) 0.551 313 15.03 3 25 2.48 W(3) 0.656 3 14 34.58 3 34 2.74 W(4) 3.297 J 15 0.74 335 2.36 W(5) 0 3 23 -19.01 345 1.43
aW(2) and W(3) may be interchanged •.
37

31 . . t. f. 11.4 Lo~-temperature P NMR ~nvest~ga ~on on a ~ve-
membered ayctic phosphate in the presenae of
lithium fluoride,
Earlier work concerning the kinetics of de-alkylation reactions in organophosphorus compounds strongly points to the involvement of penta-coordinated intermediates 10 '
11•
In this. respect it is interesting to investigate possible intermediate structures by means of low-temperature 31 P NMR techniques. Recentworkof Granoth et at. 28 showed structures intermediate between halophosphoranes and phosphonium halides with covalent phosphorus-halogen honds, which have a very large degree of ionic character. 31 P chemica! shifts were measured near ö( 31 P) +40 ppm which points to phosphonium structures. Granoth took recourse to electric field induced contributions in order to explain the deshielding of one of the aromatic protons H
0 (Fig. !1.11), Over such small dis-
,_-'fr~ ~~-x b
Ho
Fig. II.11
tances relatively large uncertainties exist in the use of the formalism, both in order of magnitude and direction29 ,
Moreover, an ion pair suitably oriented with respect to the C-H
0 bond, could very well result in the same effect, i.e.
a covalent or ionic P-X bond (X = halogen) is not necessary in order to rationalize deshielding of the H
0 proton. The
orientation of the halide ion with respect to the phosphorus atom is prQbably determined largely by steric factors.
For the salt/phosphate aggregates studied here it is interesting to determine whether a stabie penta-coordinated structure can be trapped at low temperature. In case of the bromide anion, compound 2 revealed the highest rate of de-alkylation and thus a concomitant low activatien energy (Ba::::: 12.5 kJ/mol) to form the intermedia te state 11
• Therefore, low-temperature 101 MHz 31 P NMR spectra of 2 in THF-d8 were
38

recorded. The low dielectric constant of THF suppresses the ion pair dissociation (e=7.4, T=298 K) thus supporting the generation of a penta-coordinated intermediate with moderate charge separation. Covalent character of the phosphorus-fluoride interaction is accurately measurable due to the large 1JPF z1000 Hz 30 , Addition of one equivalent LiF at' 213 K to a salution of 2 (0,3 M) in THF-d 8 resulted in a line broadening of 1.75 Hz and an upfield shift of 0.40 ppm. No doublet in the 31 P resonance could be resolved in the temperature region 213-298 K. A similar result was obtained in case of NaF.
Recently Richman et a~. 31 publisbed the 31 P- and 19F NMR results of cyclenfluorophosphoranes and demonstrated. a clear distinction between covalent and ionic structures. In the ionic structures, no 1JPF was observed whereas in the penta-coordinated cornpounds couplings of 800-900 Hz were rneasured. Based on these observations and our 31 P NMR data it can therefore be concluded that for the salt aggregates studied here, NMR techniques reveal a change in solvation but no penta-caordinated intermediate structure with a life-time short on the NMR time scale 32 • In addition, different interrnediate geometries of compound 2 with one equivalent LiBr were calculated using the CND0-2 rnethod 33 •
(Por the theory of the CND0-2 method, see Appendix Chapter III). Optimization of the different structures a, b and a toward lowest energy (Fig. II.12) revealed a decreasing stability in the order a > b > c. The kinetic data on de-alkylation reactions point to penta-coordinate interrnediate structures 10 •
Me
Me-0,_ ~~Me 'P-O
Li-O,...j Br
c
Fig. II.12 DilfePent geometPiee of the LiBP/2 comple~.
a) c~oee-ion paiP stPuatuPe.
b) phosphonium etPuctuPe.
c) penta-cooPdinate etPuctuPe,
39

Combined with the CND0-2 calculations this implies the formation of a short-lived structure of type a as a result of
an interaction of the d-orbitals of the phosphorus atom with the unpaired electron pairs of the bromide anion. Structure a seems the most stable configuration observable using NMR spectroscopy.
II.S 7Li-~ 35ct- and 81 Br NMR investigations on
salt/phosphate aggregates in aaetone.
The behaviour of the Li+ ions in the salt/phosphate aggregates in acetone was studied by means of 7Li NMR. With respect to the behaviour of the anion in the salt/phosphate complexes, 35c1- and 81 Br NMR techniques were applied. The data of the 7Li, 35c1 and 81 Br studies are shown in Fig. II.13 - II.16 and in Tables II.4- II.S.
The properties of 7Li nuclei are quite favourable for NMR studies. The resonance lines of the Li+ ion in solutions are exceptionally narrow cw1 ~z Hz)
3 ~ and chemica! shifts (vs. 4.0 M aqueous LiCl04 solution) can be measured with considerable accuracy. Literature data available for chlorineand bromine NMR in non-aqueous solvents are limited. Halide ion quadrupale relaxation rates have·been reported for methanol 9
'35
' 36 , dimethylsulfoxide 9 ' 35 , nitromethane 37 ,
formic acid 9 , N-methylformamide 9 , dimethylformamide 9 ,
acetonitrile 9 , acetone 9 and for mixtures of acetonitrjle 38 ,
methanol 36 ' 39 and acetone 39 with water. From Table II.4 it is evident that the 7Li chemica!
shifts of lithium bromide are concentratien dependent. This concentratien dependenee can be attributed to the formation of contact-ion pairs, i.e. to cases where the anion directly replaces a solvent molecule or molecules in the inner solvation shell of the cation8
, It has been previously observed40 ' 41 that the contact-ion pair equilibrium strongly depends on the donor ability of the solvent molecule as well as on the bulk dielectric constant e of
40
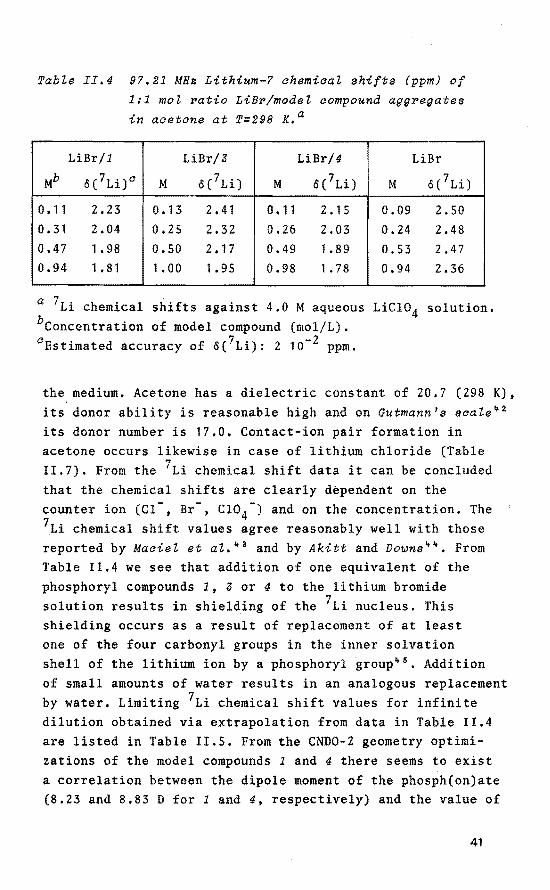
Tabte II.4 97.21 MHs Lithium-? ahemiaat shifts (ppm) of
1:1 mol ~atio LiB~/modet compound agg~egates
in acetone at T=298 K,a
LiBr/1 LiBr/3 LiBr/4 LiBr
Mb ö( 7Li) 0 M ó( 7Li) M o{ 7Li) M o( 7Li)
0.11 2.23 0. 13 2.41 0.11 2.15 0.09 2. 50 0.31 2.04 0,25 2.32 0.26 2.03 0.24 2.48 0.47 1.98 0.50 2.17 0.49 1.89 0.53 2.47 0,94 1. 81 1. 00 1. 95 0.98 1. 78 0.94 2.36
a 7Li chemica! shifts against 4.0 M aqueous LiCio4 solution. bconcentration of model compound (mol/L). 0 Estimated accuracy of ö{ 7Li): 2 10- 2 ppm.
the medium. Acetone has a dielectric constant of 20,7 (298 K), its donor ability is reasonable high and on Gutmann's scale~ 2
its donor number is 17.0. Contact-ion pair formation in acetone occurs likewise in case of lithium chloride (Table 11.7). From the 7Li chemica! shift data it can be concluded that the chemica! shifts are clearly dèpendent on the counter ion (Cl-, Br-, CI04-) and on the concentration. The 7Li chemica! shift values agree reasonably well with those reported by Maciel et al. 43 and by Akitt and Downs 44 • From Table 11.4 we see that addition of one equivalent of the phosphoryl compounds 1, 3 or 4 to the lithium bromide solution results inshielding of the 7Li nucleus. This shielding occurs as a result of replacement of at least one of the four carbonyl groups in the inner solvation shell of the lithium ion by a phosphoryl group 45
• Addition of small amounts of water results in an analogous replacement by water. Limiting 7Li chemica! shift values for infinite dilution obtained via extrapolation from data in Table 11.4 are listed in Table 11.5. From the CND0-2 geometry optimizations of the model compounds 1 and 4 there seems to exist a correlation between the dipole moment of the phosph(on)ate (8.23 and 8.83 D for 1 and 4, respectively) and the value of
41

Tabte II.5 Limiting vatues at T=298 K foF infinite ditution
of tithium-7 chemiaat shifts (ppm) and bFomine-81 line widths (kHz) of 1:1 mol Fatio comple~es
of model aompound/salt in aoetone.
limiting va lues
compound o( 7Li)a W (81Br)b !
LiBr sec 2.53 6.3 1/LiBr 2.36 8.5 3/LiBr 2.53 6.6 4/LiBr 2.26 7.2
a -2 Estimated accuracy: 2 10 ppm; chemical shift in ppm vs. 4.0 M aqueous LiC104 solution. bEstimated accuracy: 0.5 kHz.
the limiting 7Li chemical shift. A larger dipale moment seems to result in a larger shielding of the 7Li nucleus. Compound 3 reveals a different behaviour (dipole moment, 13.05 D). The results of the 81 Br line width measurements of the 1:1 LiBrimodel compound complexes are plotted in Fig. II.13. There is considerable broadening of the 81 Br resonance with increasing concentration of the complexes in acetone. The observed concentration-dependent broadening of the 81 Br resonance is indicative of contact-ion pair formation because of asymmetrical soivation of the bromide anion, caused by a contribution of the (Li-0-P) fragment, thus enhancing the electrical anisotropy around the 81 Br nucleus. This results into an increase in line width with increasing salt concentration. The contribution of the (Li-0-P) fragment to the electrical anisotropy of the ellipsoidal 81 Br nucleus can also account for the difference in limiting values of the 81 Br line widths in the 1:1 complexes salt/phosphate and in LiBr (viz. Table II.S). The limiting 81 Br line widths were obtained via extrapolation from data in Fig. 11.13 to infinite dilution. Table II.4 and Fig. II.13 reveal, that no distinction can be made between the cyclic and the acyclic model compounds. The different behaviour of 3 (Fig. 11.13) might be explained
42

14
T • 298 K
4 12 LiBr sec
WV..kH~
0 o.o o.e o.a 1.0
- (1:1) LiBrimodel compound mol/ltr
Fig. II.13 67.55 MHs Bromine-81 line ~idths (W~) of
LiBrimodel compound aggregates in equimolar
ratio in acetone.
by a larger distartion of the solvent structure due to a relatively higb molecular weight (M, 326 versus 176 and 182 for 1 and 4 respectively) and by the relatively high molecular dipole of this compound (CND0-2 calculation, 13.05 D). The distartion of the solvent structure in case of 3 is reflected in an extra line width of the 81 Br nucleus with increasing phosphate concentration.
Tables II.6 - II.S show the influence of increasing model compound concentratien on the 7Li chemical shift with constant concentratien of LiBr, LiCl and LiCl04 , respectively. These tables reveal, that the 7Li chemica! shift is dependent on the kind of phosphate molecule involved but independent on the nature of the anion (Cl- vs. Br-) 46 • The 7Li measurements reveal a fast equilibrium reaction in case of the salt/phosphate aggregates. Addition of extra lithium salt and dilution to a previously measured concentratien results in exactly the same 7Li chemical shift. The concentratien dependenee of the 7Li chemical shift increases with increasing salt concentration.
43

M"
0 0.09 0.26 0.65 1.16 1.66
Table II.6 9?.21 MHa Lithium-? chemical shifts (ppm) of salt/phosphate agg~egates in acetone at T=298 K.a
1 3 4 6('Li) MC o('Li)- jfd li('Li) .. ö('Li) Mf ö('Li) Mil li('Li)
2.51 0 2.42 0 2.50 0 2.46 0 2.50 0 2.44 l!-23 0.12 2.30 0.13 2.89 0.14 2.86 0.16 1.78 0.16 2.20 1.84 0.44 2.01 0.27 2.28 0.89 2.21 0.54 1.20 0.49 1.78 1.38 0.86 1.66 0.57 2.09 0.54 2.18 0.94 0.93 0.85 1.43 0.98 1.50 1.25 0.94 1.91 0.81 2.00 1.14 0.86 1.17 1.20 0.75 1.25 1.78 1.14 1.86 1.35 0.77 1.86 1.13
1.47 1.78
aLiBr concentration constant and model compound concentration variable; 7Li chemica! shifts vs. 4.0 M aqueous LiC104 solution; M is the concentration of the model compound (mol/L); estimated accuracy of ö( 7Li): 2 10-2 ppm. bO.lO M LiBr. 0 0.58 M LiBr. d0.13 M LiBr. 6 0.40 M LiBr. fo.11 M LiBr. g0.45 M LiBr.
Table II.? 9?.21 MHs Lithium-? chemical shifts (ppm) of salt/phosphate agg~egates in acetone at T=298 K.a
1 3 4 M" o('Li)" M 6('Li) M 6('Li)
0 2.27 0 2.27 0 2.27 0.19 1.82 0.08 2.21 0.08 2.07 0.34 1.59 0.42 2.07 0.21 1.85 0.68 1.24 0.85 1.93 0.49 1.54 1.01 1.02 un 1.81 0.87 1.80 1.33 0.85
. aLiCl concentration saturated (0.08 mol/L) and model compound concentration variable; 7Li chemica! shift vs. 4.0 M aqueous LiC104 solution. bM is the concentration of the model compound (mol/L). 0 Estimated accuracy of ö(7Li): 2 10- 2 ppm.
Addition of water results in shielding of the 7Li nucleus analogous to the results of Popov 8 • This shielding increases with decreasing salt concentration and is independent of the anion (Cl~ vs. Br-). Recent ab initio studies suggest that in the Li+/H2Po4- complex. hydration of the Li+ ion does
not significantly alter the extend of covalency of the metal-phosphate bond, although it weakens the direct complex
44

formation by decreasing the net atomie charge of the cation7 •
The concentration-dependent chemica! shift differences in case of the 7Li nucleus are in the same range as those observed for 1:1 complexes (viz. Table II.4). Therefore, in spite of
Tabte II.B 97.21 MHz Lithium-? ahemiaat shifts (ppm) of
satt/phosphate aggregates in aaetone at T=298 K.a
1 s 4 Mb 6 ('Li) MC Md ö('Li} M• ó( .) Mil
0 2.25 0 0 2.27 0 2.08 0 2.26 0 2.09 0.22 1.71 0.19 0.19 2.07 0.13 1.98 0.21 1.72 0.21 1.87 0.54 1.22 0.36 0.40 1.87 0.31 1.85 0.46 1.26 0.37 0.85 0.96 0.70 0.69 1.72 0.64 1.68 0.83 0.81 0.62 1.00 0.85 1.17 0.89 1.52 0.88 1.50 1.17 0.66 0.88 1.18 0.76 1.09 1.26 1.32 1.35 1.28 1.88 0.61 1.34
aLiCl04 concentratien constant and model compound concentratien variable; 7Li chemica! shifts vs. 4.0 M aqueous LiCl04 solution; M is the concentratien of the model compound (mol/L); estimated accuracy of o( 7Li): 2 10- 2 ppm. b0.13 M LiCl04 • a0.49 M LiCl04 • d0.11 M LiCl04 • 8 0.57 M LiCl04• fo.12 M LiCl04• g0.52 M LiCl04 •
1.73 1.46 1.15 0.90
For variable LiC104 concentratien (mol/L) in ácetone without model compound the following 7Li chemica! shifts were measured: 0.05 M, 2.37 ppm; 0.10 M, 2.30 ppm; 0.20 M, 2.23 ppm; 0.40 M, 2.15 ppm; and 0.81 M, 2.05 ppm.
the large excess of model compound in comparison with lithium salt, it can be concluded that there is a high preferenee to replace onty one of the four carbonyl groups in the inner solvation shell of the lithium cation by a phosphoryl fragment and exclusively 1:1 salt/phosphate complexes have to be considered. In addition, CND0-2 calculations performed on the 1:1 salt/phosphate aggregates, reveal a concomitant decrease in the net atomie charge of the 7Li nucleus (0.3 e)
in comparison with the uncomplexed lithium salt which behaves as a close-ion pair. This large decrease in net atomie charge and the increased sterical bindrance predicted in case of 2:1, 3:1, and 4:1 mol ratio phosphate/salt suggest that the latter structures become highly unlikely.
45
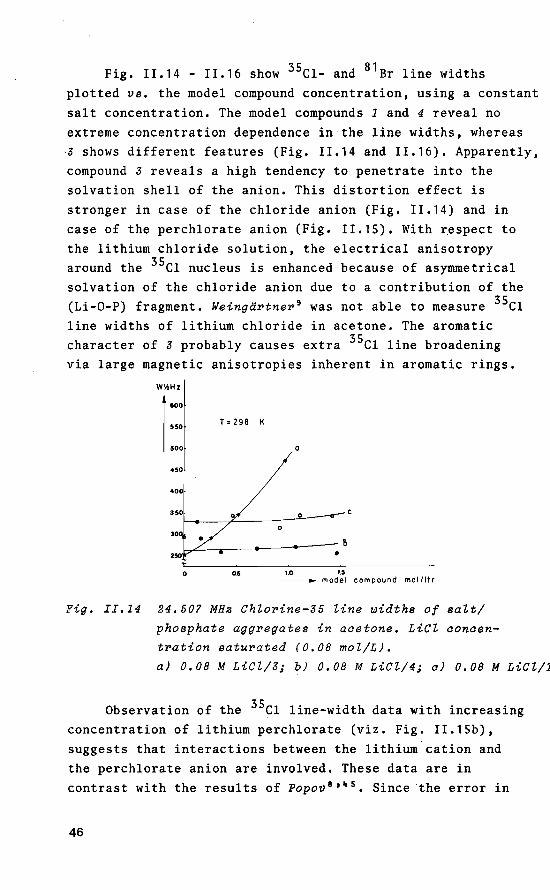
Fig. 11.14 - 11.16 show 35c1- and 81 Br line widths plotted vs. the model compound concentration, using a constant salt concentration. The model compounds 1 and 4 reveal no extreme concentratien dependenee in the line widths, whereas ,3 shows different features (Fig. 11.14 and 11.16). Apparently, compound 3 reveals a high tendency to penetrate into the solvation shell of the anion. This distortien effect is strenger in case of the chloride anion (Fig. 11.14) and in case of the perchlorate anion (Fig. 1!.15). With respect to the lithium chloride solution, the electrical anisotropy around the 35c1 nucleus is enhanced because of asymmetrical solvation of the chloride anion due to a contribution of the (Li-0-P) fragment. Weingärtner 9 was not able to measure 35c1 line widths of lithium chloride in acetone. The aromatic character of 3 probably causes extra 35c1 line broadening via large magnetic anisotropies inherent in aromatic rings.
WY.Hz
800
550 T = 298 K
500 a
450
400
0
0
0 05 1.0 1.5 -model compound mollltr
Fig. II.14 24.507 MHz Chlorine-35 Zine widths of salt/
phosphate aggregates in aaetone. LiCZ aonaen
tration saturated (0,08 mol/L).
a) 0.08 M LiCZ/3; b) 0.08 M LiCZ/4; a) 0,08 M LiCZ/1
Observation of the 35c1 line-width data with increasing concentratien of lithium perchlorate (viz. Fig. 11.15b), suggests that interactions between the lithium cation and the perchlorate anion are involved. These data are in contrast with the results of Popov 8 '~ 5 • Since the error in
46
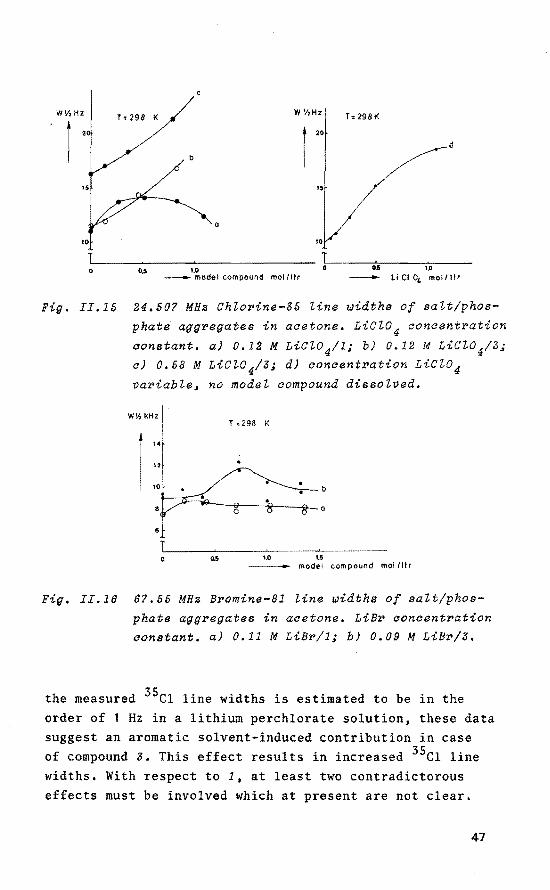
W'/,HZ
) 2
0
Fig. II.15
c
T = 298 K W 'hHz T: 298 K
l 20
d
IS
0
0.5 1.0 Cl5 1.0
- model compound mollltr - li Cl 04 mol/l!r
24.507 MHz Ch~orine-35 ~ine widths of sa~t/phos-
phati aggregates in aaetone. Lic~o 4 aonaentration
constant. a) 0.12 M LiC~0 4/1i b) 0.12 M LiC~04/3; a) 0.58 M LiC~04/3; d) aonaentration LiCl04 variable, no mode~ compound disso~ved.
W\7 kHz I T :298 K
::1 tor
1.0 1.5 model compound mollllr
Fig. II.18 87,55 MHz Bromine-81 ~ine widths of salt/phos
phate aggregates in acetone. LiBr aoncentration constant. a) 0.11 M LiBr/1; b) 0.09 M LiBr/3.
the measured 35c1 line widths is estimated to be in the order of 1 Hz in a lithium perchlorate solution, these data suggest an aromatic solvent-induced contribution in case of compound 3. This effect results in increased 35c1 line widths. With respect to 1, at least two contradictarous effects must be involved which at present are not clear.
47

11.6 ConaZusions.
Kinetic experiments on the system 2/methanol in CDC1 3 revealed that the phosphorylation reaction is retarded in the presence of lithium halides. The magnitude of the effect is related to the ionic radius of the halide anion as well as to the solvation properties of this anion. Larger solvation shells result in more efficient shielding of the phosphorus atom toward methanol. These data therefore provide evidence for the proximity of the halide anion to the phosphorus atom.
1H NMR spectra of Eu(fod) 3 complexes of 1 in CDC1 3 confirm the intrinsic magnetic nonequivalence of the ring methylene protons. Addition of lithium balides to solutions of 1 causes effects which are superficially analogous but which are related to solvent reorientation in the vicinity of the ring methylene protons.
Accurate values of 2JHH determined via c~mputer simulation of 1H NMR spectra of 1 in different environments (acetone-d6 , benzene-d6 , acetone-d6/lithium bromide) revealed a negligible change in the net atomie charge on the phosphorus atom. This conclusion seems consistent with the small (approximately 1 ppm) 31 P chemical shift differences which occur upon addition of salt to the phosphates. The low-temperature 31 P NMR results, in whieh 2 and lithium fluoride or sodium fluoride were dissolved in THF-d8 , showed no 1JPF in the temperature range 213-298 K. This indicates that no covalent P-F bonds are present. Thus, the NMR data can be readily reconciled with changes in solvation but provide no evidence for penta-coordinated intermediate structures with a life-time short on the NMR time scale. In addition, different types of intermediate geometries of 2/LiBr in equimolar ratio (close-ion pair/phosphate complex a, phosphonium spècies b and penta-coordinate structure a) were calculated by means of the CND0-2 method. Optimization of the various parameters toward lowest total energy revealed a decreasing stability in the order a > b
> a. This implies a short-living P(V) structure as a result
48

of the interaction of the d-orbitals of phosphorus with the unpaired electron pairs of the bromine. The close-ion pair structure a seems the most stable, NMR observed configuration.
The 7Li NMR spectra of several lithium halides in acetone are consistent with workof Popov 8 and Weing~PtneP 9 •
The same is true for the 35c1- and 81 Br NMR measurements of the anions with the exception of the 35c1 NMR results of lithium chloride in acetone. 7Li NMR chemica! shift and line-broadening data reveal complexation of the phosphoryl oxygen atom by the Li+ ion. All four phosphorus compounds investigated here show a preferenee for 1:1 salt/phosphate complexes. The 35c1- and 81 Br,NMR results can be explained assuming fast equilibria between lithium halides in the complexed (with phosphates) and the free form.
II.7 E~perimental.
Apparatus.
NMR spectra were recorded using a Bruker WM-250 spectrometer eperating at a fieldstrengthof 5.7 T. For 1H spectra (250.13 MHz), chemical shift data were measured against tetramethylsilane (TMS). 1H NMR data on compound 2 in the presence of Eu(fod) 3 were obtained using a Varian EM-360 A spectrometer eperating at a fieldstrengthof 1.37 T (60 MHz). For 7Li (97.21 MHz), chemica! shift data were measured against a 4.0 M aqueous LiCl04 solution. Line widths, measured with an accuracy of 0.2 Hz, were of the order of 2 Hz. 35c1 spectra were obtained at a frequency of 24.507 MHz. Line-broadening functions in the ranges 3, 10 and 50 Hz were used, depending on the line width to be observed. 81 Br spectra were obtained at a frequency of 67.55 MHz. Line~broadening parameters were set at 100 Hz. Line widths of the 35c1 and 81 Br resonances were determined with an estimated accuracy of 10% as an average of two to four
49

measurements (each of 100-10000 pulses). All spectra, except the low-temperature 31 P, were recorded at 298 K. 31 P spectra (101 .27 MHz) were obtained with a resolution of 0.07 Hz. Chemica! shifts are reported relative to 85\ external H3Po4 ; negative values refer to shielding. Some of the 250 MHz proton spectra were simulated (32 K data points) in order to establish accurate values for the coupling constants and chemical shift parameters.
Preparations.
- 2-Isopropo~y-2-o~o-5-methyZ-1~2-o~aphosphoZ-4-ene 1
This compound was prepared from the corresponding chlorooxaphospholene10. B.p. 50 °C/0.01 mm; yield 60\. 1H NMR (CDC1 3) o 1.37(d,6H, 3JHH=6 Hz,CH3); 1.99(m,3H,ring CH3); 2.40(d of m,2H, 2JPH=14 Hz,ring CH 2); 4.80(sept,1H, 3JHH=6 Hz,isopropoxy H); 4.97(d of m,1H, 3JPH=34 Hz,ring H).
- 2-Metho~y-2-o~o-4~5-dimethyl-1~3~2-dio~aphosphot-4-ene 2
This compound was prepared from the trimethyl phosphite biacetyl adduct with acetyl bromide .in acetonitrile according to the procedure given by,Ramirez 47 •
B.p. 75-77 °C/0.8 mm; yield 85\, 1H NMR (CDC1 3) o 1.93(s,6H,CH3); 3.83td,3H, 3JPH=12 Hz,OCH3). 31 P NMR (CDC1 3) o 11.9
Reagents and model aompounds.
Lithium perchlorate, lithium bromide, lithium chloride, lithium fluoride and potassium fluoride (Merck AG, Darmstadt) were azeotropically refluxed (benzene solution) to remove the water with a special adapter. Evaporation of the anhydrous salution in vacuo under dry nitrogen yielded the dry salt. After drying, all the lithium salts were stored under a dry nitrogen atmosphere. Triphenyl phosphate (3~
Aldrich) was azeotropically dried in a similar way as the
50

lithium salts. This compound was dried just before use. Triethyl phosphate (4, Aldrich) and the cyclic organophosphorus compounds 1 and 2 were freshly distilled under reduced pressure just before use.
Solvents.
Acetone (Merck AG) was distilled over Drierite and further dried over molecular sieves. Methanol (Merck AG) was first fractionally distilled from calcium hydride under a nitrogen atmosphere and storedover molecular sieves (3A). Tetrahydrofuran (Merck AG) was fractionally distilled from calcium hydride under a nitrogen atmosphere. All deuterated solvents used were stored over molecular sieves.
Solutions.
In view of the hygroscopicity of solvents and of lithium s~lts, all solutions were freshly prepared and the NMR sample tubes were filled under a nitrogen atmosphere. NMR spectra of salt/phosphate complexes were obtained immediately after sample preparation. In this way, the influence of de-alkylation reactions on chemica! shifts and line-width data could be neglected.
Kinetics of the phosphoPylation Peaation of compound 2 with
methanol in the pPesenae of lithium halide.
Reaction rates were determined using a 60 MHz Varian EM-360 A spectrometer. In an NMR sample tube, 0.3 M solutions of methanol and lithium halide were prepared in CDC1 3• After addition of an equimolar amount of 2 (0.3 M) and quick stirring, the sample tube was immediately transferred to the NMR spectrometer. The temperature of the probe was kept at 298 K. The reactions were foliowed by determining the relatively amounts of the starting compound 2 (by integration of the corresponding methyl singlet) and the acyclic phosphate at different time intervals. For the acyclic
51

phosphate, the methyl doublet (JHH=7 Hz) at 6 1.48 was monitored. In order to correct for a slight amount of de-alkylated product, the ring methyl singlet at 6 1.77 was used. In the case of LiBr, the methyl bromide singlet (6 2.61) was also monitored. This was not possible for methyl chloride (6 3.10) and methyl fluoride in the analogous series due to their low boiling point. In order to exclude the influence of moisture, the reactions were per
formed in anhydrous CDC1 3• The lithium halides were thoroughly dried by azeotropic removal of water. Each reaction was carried out at least three times and the obtained rate constants appeared to be in reasonable agreement (10%) with one another. Pseudo-first-order kinetics was performed over 10 t 1 values for each experiment.
2
52

Heferences and Notes.
1. L.G. Marzilli, Prog. Inorg. Chem., 1977, 23, 255, 2. D.G. Hodgson, Prog. Inorg. Chem., 1977,23,211. 3. V, Swaminatban and M. Sundaralingam, Crit. Rev. Biochem,,
1979, 6, 245. 4. R.B. Martin, "Metal !ons in Biologica! Systems", H. Sigel
and M. Dekker, eds., Pergamon Press, New York, 1979, Vol.8.
5. L.G. Marzilli, B. de Castro, J.P. Caradonna, R.C. Stewart and C.P. Van Vuuren, J. Am. Chem. Soc., 1980, 102, 916.
6. U.P. Strauss, C. Helfgott and H. Pink, J. Phys. Chem., 1967, ?1, 2550.
7. P. Liebmann, G. Loew, A.D. McLean and G.R. Pack, J. Am. Chem. Soc., 1982,104,691.
8. Y.M. Cahen, P.R. Handy, E.T. Roach and A.I. Popov, J. Phys. Chem., 1975, ?9, 80,
9. H. Weingärtner and H.G. Hertz, Bet. Bunsenges. Phys. Chem., 1977, 81, 1204.
10. A.M.C.F. Castelijns, D. van Aken, P. Schipper, J.J.C. van Lier and H.M. Buck, Reel. Trav. Chim. Pays-Bas, 1980, 99, 380.
11. A.M.C.F, Castelijns, Ph.D. Thesis, Eindhoven University of Technology,1979.
12. B.A. Arbusov, Pure Appl. Chem., 1964, 9, 307. 13. G. Asknes and D. Asknes, Act. Chem. Scand., 1964, 18, 38. 14. R.G. Harvey and E.R. de Sombre, 11Topics in Phosphorus
Chemistry", Wiley Interscience, New York, 1964, Vol.1, 57. 15. A. Skowrónska, J. Mikolajczak and J. Michalski, J. Chem.
Soc., Chem. Comm., 1975, 791. 16. J. Michalski, J. Mikolajczak, M. Rapulski and A.
Skowrónska, Phosphorus Sulfur, 1978, 4, 233. 17. R.G. Weiss and E.I. Snyder, J. Org. Chem., 1971, 36, 403. 18. R. Aneja, A.P. Davies and J.A. Knaggs, J. Chem. Soc.,
Chem. Comm., 1973, 110. 19. R. Aneja, A.P. Davies and J.A. Knaggs, Tetrahedron
Lett., 1974, 1, 67. 20. L.A. Jones, C.E. Sumner,Jr., B. Franzus, T.T.S. Huang
and E.I. Snyder, J. Org. Chem., 1978, 43, 2821.
53

21. N.G. Osipenko, E.S. Petrov, Yu.I. Ranneva, E.N. Tsvetkov and A.I. Shatenstein, Zh. Obshch. Khim., 1976, 47, 2172.
22. N.G. Osipenko, E.S. Petrov, E.N. Tsvetkov, Yu.I. Ranneva and A.I. Shatenstein, Zh. Obshch. Khim., 1976, 46, 2647.
23. A. Hong, J. Lee, J.G. Verkade, J. Am. Chem. Soc., 1976, 98, 6547.
24. E. Breuer, D.M. Bannet, Tetrahedron, 1978, 24, 997. 25. Eu(fod) 3 : Tris (6,6,7,7,8,8,8-heptafluoro-2,2-dimethyl-
3,5-octanedionato) europium. 26. Kryptofix 221: 4,7,13,16,21-Pentaoxa-1,10-diazabicyclo
!8.8.5.Jtricosane. 27. Substitution of an electronegative atom in a S-position
leads to a negati ve change in 2 JHH' See: "Proton and Carbon-13 NMR Spectroscopy. An Integrated Approach.", R.J. Abraham and P. Loftus, eds., Heyden & Son Ltd., London, 1978, 47.
28. I. Granoth and J.C. Martin, J. Am. Chem. Soc., 1981, 103, 2711.
29. M.E. van Dommelen, J.W. de Haan and H.M. Buck, Org. Magn. Reson., 1980, 14, 497.
30. M.M. Crutchfield, C.H. Dungan, J.H. Letcher, V. Mark and J.R. Van Wazer, "Topics in Phosphorus Chemistry", M. Grayson and E.J. Griffith, eds., Interscience Publishers, New York, 1967, Vo1.5, 280.
31. J.E. Richman and R.B. Flay, J. Am. Chem. Soc., 1981, 103, 5265.
32. In an attempt to extend the life-time of the intermediate structure, 2-(1,1,1,3,3,3-hexafluoro)isopropoxy-2-oxo-4,5-dimethyl-1,3,2-dioxaphosphol-4-ene was prepared. Upon addition of one equivalent LiX(X=F, Cl, Br), this compound showed no de-alkylation because of the strongly reduced tendency for P=O bond formation from this ligand.
33. D. Rinaldi, Comput. Chem., 1976, 1, 109. Program 290, Quanturn Chemica! Program Exchange, Indiana University.
34, 35.
W1: Width of a resonance line at half height (Hz). ï
B. Lindman, S. Forsén and E. Forslind, J. Phys. Chem., 1968, 72, 2805.
36, C. Hall, G.L. Hallerand R.E. Richards, Mol. Phys.,
54

1969, 16, 377. 37. R.E. Gentzler, T.R. Stengle and C.H. Langford, J. Chem.
Soc., Chem. Comm., 1970, 1257. 38. T.R. Stengle, Y.-C.E. Pan and C.H. Langford, J. Am. Chem.
Soc., 1972, 94, 9037. 39. R.E. Richards and B.A. Yorke, Mol. Phys., 1963, 6, 289. 40. M.S. Greenberg, R.L. Bodner and A.I. Popov, J. Phys.
Chem., 1973, 77, 2449 and references listed therein. 41. U. Mayer and V. Gutmann, Struct. Bonding (Berlin), 1972,
12, 113. 42. V. Gutmann, "Coordination Chemistry in Non-aqueous
Solvents", Springer Verlag, Vienna, 1968, 19. 43. G.E. Maciel, J.K. Hancock, L.F. Lafferty, P.A. Mueller
and W.K~ Musker, Inorg. Chem., 1966, 5, 554. 44. J.W. Akitt and A.J. Downs, "The Alkali Metals Symposium",
The Chemical Society, Londen, 1967, 199. 45. R.G. Baum and A.I. Popov, J. Sol. Chem., 1975, 4, 441. 46. J.J.C. van Lier, L.J.M. van de Ven, J.W. de Haan and
H.M. Buck, J. Phys. Chem., 1983, in press. 47. F. Ramirez, J.F. Mareeek and J. Ugi, J. Am. Chem. Soc.,
1975, 97, 3809.
55

CHAPTER 111
DynaiDics of penta- coordinated
phosphorus in the hackhone of DNA
III.1 Introduation.
DNA usually crystallizes as a right-handed double helix with an anti conformation of the base residues (B-DNA) 1 (a.f. Chapter I.3). However, synthetic double-stranded DNA polymers and eligomers containing a strictly alternating (G-C)n sequence display unusual conformational properties. In this respect, PohZ and Jovin 2 were the first to show that there is a salt-induced cooperative conformational change of a synthetic poly d(G-C) duplex. With the aid of circular dichroism (CD) techniques and Raman spectroscopy they showed that increasing concentratien of either NaCl, NaCl04 or MgC1 2 resulted in a conformational change from one double-helical form of the molecule to another 3 '~. The conformational change can also be initiated by ethanol 5 ' 6 , This transition has recently been establi&hed as a conversion of a right-handed helix into a left-handed high-salt structure. X-ray crystallographic studies on d(G-c) 2 and d(G-C) 3 duplexes, crystallized in high-salt solution, revealed structures which differ from the B-DNA duplex in the left-handed helix which has a helical periodicity of 12 base pairs, in the deoxyribose conformations which are CJ3')-endo (N) for dG and C(2')-endo (S) for dC, and in the G bases which are in a syn conformation7 - 10 (Fig. III.l). As the sugar-phosphate backbene in these structures fellows a zig-zag course, they are designated as Z-DNA. Furthermore, the "sugar residues have an alternating orientation in such a way that the dG residues have the deoxyribose 0(1') pointing up while the dC residues have the deoxyribose 0(1')
56

pointing down. Because of this alternation, the repeating
unit in the helix is not a mononucleotide as in B-DNA, but a dinucleotide 7 • CD and diffraction patterns consistent with the Z form have also been obtained for fibersof poly d(G-C). poly d(G-C) and poly d(A-C).poly d(G-T) 2 ' 11 • In addition, analysis of the conformational behaviour of an alternating AT sequence in a polymer duplex in CsF solution also indicates the distinct dynamics of a partial B_.z transition12 •
----C2'
B-DNA Z-ONA
Fig. III.1 Ittustvation of the most apparent geometria
differenaes between B-DNA and Z-DNA for a
5'-dpG fragment.
On the basis of these studies it was suggested that, in genera!, any DNA with a repetitive purine-pyrimidine sequence can adopt a Z-DNA structure in high-salt solutions or in nucleophilic solvents 7 ' 11 • One of the questions which remains is concerned with the mechanism of these isomerie interconversions. The conformation of the Z-DNA fragment illustrates how salt effects are transmitted into a specific orientation of the base pairs and the organization of the sugar-phosphate chains. From the experiments of compounds 1-4 with lithium balides in acetone (a,f, Chapter II), it is clear that phosphorus is able to extend its coordination from four to five by the close proximity of the halide anion 13 while most of the time preserving its close-ion pair/phosphate structure. Furthermore, the kinetic experiments on 2 in CDC13 in the presence of methanol and lithium halide clearly showed that the halide anion is capable of shielding the phosphorus atom to some extend toward a nucleophilic attack of methanol. However, the observed phosphorylation reaction still proceeds much faster than the de-alkylation reaction. As pentavalent
57

phosphorus intermediates are the reactive species in phosphorylation reactions, this implies the introduetion of selectivity in the system due to the structural characteristics of the trigonal bipyramidal configuration (c.f. Chapter !.2). A comparable situation might be encountered in the phosphate residues in the helix backbene of the double-stranded DNA molecule in aqueous high-salt environment (i.e. a concentrated aqueous salt solution). Due to the decrease in dielectric constant of this solution, as a result of the high salt concentration, the dissociation of the salt will be suppressed and the aalt/DNA-phosphate/water system can be compared with the salt/2/methanol system. Therefore, it can be speculated that the fifth coordination ligand for phosphorus is affered by water, promoted by the cations which take care of the shielding of the anionic oxygens. The conformational change induced by ethanol (vide supPa) can also be considered as a change in coordination of phosphorus. For the acyclic phosphate groups in DNA, the phosphorylation reaction with water can be excluded. Moreover, in this system there will be no de-alkylation reaction (i.e. breaking of the internucleotide P-0-C bond) because of the strongly reduced tendency for extra P=O bond formation in dialkylphosphates and because of the concomitant high activation energy for de-alkylation in acyclic phosphates in comparison with these of their cycli~ analogues 13 • The suggestion made for the occurrence of the Z-DNA fragment, which is based on the fact that the phosphate groups are closer to each other on two sides of the minor groeve than in B-DNA as a result of decreased oxygen repulsions in high-salt solutions, is oversimplified. Local hydratien introduces an effective shielding of the anionic oxygens by hydragen bridging and/or complexation of water with P(IV), which results in a partial positive charge on the oxygen coordinated with phosphorus, thus reducing the overall charge of the phosphate moiety. The latter approach can be used as a more generalized model for·conformational induction by nucleophilic solvents like ethanol or spermine 7 , used during the crystallization of the Z-DNA fragment. Hence, a change
58

in the coordination of phosphorus from a P(IV) into a P(V) trigonal bipyramid (TBP) activates the phosphate groups in the DNA backbone, which in turn initiates conformational transformations leading to Z-DNA. On the basis of CND0-2 and MNDO quantum-chemical calculations a mechanism for the B-.z transition in ds DNA is given.
III .2 Model descz>iption of the B-Z transition in DNA.
Recently, two structures of Z-DNA were established and designated as z1 and z11
10 (Fig. III.2). On the basis of X-ray analysis it was suggested that the z1 structure is stabilized by a water molecule, located in the 5 1 -dGp fragment between one of the oxygens of 3'-phosphate and the exocyclic NH 2 group of G. The z11 structure is stabilized by virtue of a hydrated Mg 2
+ ion between 0- (phosphate) and N(7) of the G base. A remarkable feature of the Z structures is that only G adopts a syn conformation. Upon B-Z transition, the dihedral angle C(8)-N(9)-C(l')-C(2') changes by 170° in the dG residues, whereas the dihedral angle C(6)-N(1)-C(1')-
'\05'
ZII-DNA
Fig. III.2 ZI and ZII structures in detail. In the ZI structure# the conformation around the C(5')-0(5 1 )
bond is 6- tJhereas in the ZII structure this conformation is 6+.
59

-C(2') changes by 50° ~ithin the anti domain of the dC residues (Fig. III.B-9, vide infPa). In order to explain the salt induced B_.z isomerization in the d(G-C) 3 duplex, Wang et at. 7 put forward a mechanism in which a cascade of internal motions is suggested. Initiatien of the isomerization by breaking of the Watson-CPick base pairs is foliowed by rotation of G and dC residues, which, after rejoining of the hydrogen honds, results in the Z-DNA structure.
Combined with the proposed P(IV)~P(V) TBP activatien of the phosphate residues in the helix backbone, the sequence of these various steps was simulated by means of semi-empirica! quantum-chemical calculations, specifically focused on the rotations around the C(4')-C(5') bond and the glycosidic C(l')-N honds. Simple model systems have been used which are believed to be reasonably representative of the complexity of the B-.z isomerization. The initiatien of the isomerization most likely inheres a change in coordination of phosphorus from P(IV) into P{V) TBP. This primary activatien of the phosphate groups in the helix backbone is enhanced by complexation with solvated roetal ions, which make the phosphorus atom more susceptible to attack by a fifth ligand (e.g. water, nucleophilic solvents) 1 ~. The P(V) TBP which is then formed has the shielded oxygen anions in an equatorial position and the extra ligand in an apical position 15 • The other apical site is·linked to the deoxyribose unit via 0(5') or 0(3') (Fig. III.3), CND0-2 calculations, performed on both isoroers I and II, show that apical location of 0(5') is energetically favoured by 21 kJ/mol as was calculated for water as the fifth ligand. As a consequence of its apical location in the P(V) TBP, the electron density on 0(5') will increase, thus propagating the rotation of the deoxyribose ring around the C(4')-C{5') bond as aresult of the repulsion between the net electron density on 0(5') and 0(1') of the deoxyribose ring. Deprotonation of, for example, the water ligand (vide infPa) leads to a further increase in the electron density on 0(5'}, thus causing a further rotatien of the deoxyribose ring. The rotatien of the deoxyribose ring around the C(4')-C(S') bond is reflected
60

H H ""'-+/
0 -o ', 1 /d- ribose
'p -03' -a' I OS'
""- d-ri bose
0(5') atom as an apical ligand.
Isomer I
0(5') atom as an equatorial ligand.
Isomer II
Fig. III.S Both poeeibte geomet:riee of the P{V) TBP inte:r
mediate# fo:rmed upon apicat attack of a wate:r motecu te.
in the dihedral angle O(S')-C(S')-C(4')-0(1'). The rotation of the deoxyribose ring around the C(4')-C(S') bond will also be accompanied by a rotation around the glycosidic C(1')-N bond. Propagation of these rotations is effected by a synchronous rotation of the equatorial oxygen ligands around the apical axis of the TBP through which they become more accessible for shielding by the metal ion. Hence the rotation around the C(4'}-C(S') bond induced by P(IV)-P(V) activatien and the rotation around the C(1 ')-N bond represent a coupled and complex process. Note that after ligand rearrangement (Turnstile pseudorotation) in the TBP, the energetically less favoured apical location of the 0(3') atom (Fig. III.3, Isomer II) leads to an increased repulsion between the net electron density on 0(3') and 0(1') of the deoxyribose ring. This effect cannot result in a rotation of the deoxyribose ring and implicates that, of the two possible isomerie P(V) structures, onty the t:rigonat bipy:ramid
with apical Zocation of 0(5 1 )# i.e. a 5 1-Zinked phoephate
g:roup# :resutts in a :rotation of the deo~y:ribose :ring.
Stereo model studies, performed on a double-stranded DNA structure, indicate only minor steric hindrance in the C(3')-0(3')-P.part of the helix backbone during the s-.z transition. Studies on pseudorotational motions in furanose rings are in agreement with these observations 16
• 17 • In a
61

double-helical DNA structure with an alternating CG sequence, generation of P(V) TBP intermediates can lead to activated dpC or dpG structures. From the P(V) TBP dynamics and the results of Wang et al. 7
, it is apparent that the B--Z transition can only be induced by symmetrical generation of P(V) TBP intermediates in 5'-dpC fragments. This is the result of a restricted rotation around the C(1')-N bond in the case of pyrimidine bases, which is a consequence of the steric bindrance of the carbonyl C(Z) fragment and. the deoxyribose ring. In contrast, purine bases are relatively free to rotate around the glycosidic bond 18
• The high rotational harrier of C around the C(1 1 )-N bond requires that C remains anti with respect to the deoxyribose ring throughout the entire B--Z transition. In this way, a rotation of the deoxyribose ring forces C to rotate out of the C.G base plane with concomitant separation of the base pair (Fig. III.4). When the entire dC residue rotates, rejoining of the hydrog~n bridges becomes possible after a restrictionless anti--syn conformational change of the complementary G base, Furthermore, the induced rotation of dC fragments in one strand of the structure, farces the adjacent 5'-dpG phosphate groups toadopt the yt conformation of the C(4')-C(5') bond (see Fig. 1!1.6). The rotations around the C(4')-C(5 1
) bond and the C(1')-N bond within s•-dpG residues are accommodated by ft pucker change from C(Z 1 )-endo (S) into C(3 1 )-endo (N). The other possibility, activatien of the 5 1 -dpG phosphate groups does not lead to disruption of the Watson-C~ick base pairs, since the G base accommodates the rotation of the deoxyribose ring on going from the anti to the syn conformation without a significant activatien energy. The left-handed helical fragment thus formed has opposite polarity with respect to the Z-DNA structure but shows most of the conformational characteristics of a Z-DNA structure 19 • However, a left-handed structure of this kind has not yet been observed. A B--Z transition in DNA fragments with nonalternating purine-pyrimidine sequence requires both anti and syn conformations of identical base residues. Considering the unique conformational preferenee
62

,W
'W
Fig. III.4 Rotations of the G and dC fragments in a 5'-dpGpC
structure afte~ the P(V) TBP intermediate has
been formed. Adoption of the Z aonfiguration is
further assoaiated with aomplexation of the 2+ equatorial oxygen atoms to metal ions (e.g. Mg ),
of the base residues, induced by the environment, this kind of B--Z transition becomes highly unlikely. In conclusion, consirlering the B-.z transition in DNA with an alternating
GC sequence, it seems that only symmetrical generation of P(V).TBP intermediates within dpC fragments, in which 0(5') is situated in an apical position of the TBP, leads to a Z-DNA structure. Thus, the concept of selective P(IV)-P(V) activation in the helix backbone and the restricted rotation of pyrimidine bases around the glycosidic bond accounts for the dynamics of the B--Z isomerization.
Consirlering the dynamic motions of the G and dC fragments in one strand of the DNA structure during the B--Z transition it is apparent that the rotating dC residue imposes sterical constraints on the anti--syn rotational movement of both adjacent G bases. Therefore, the G residue, in going from the anti to the syn position, preferentially passes through a transition state in which C(8) and C(2') are in eclipsed conformation (see Fig. III.S).
63

III.3 CND0-2 and MNDO quantum-ahemicat aatcutations
on model systems ~ep~esentative fo~ DNA.
The calculations were performed using the CND0-2 and MNDO method. The CND0-2 metbod is based on the complete neglect of differential overlap which introduces electron-electron repulsion integrals in the simplest possible way 20
• 21 • The basic approximations of the MNDO (modified neglect of differential overlap) include a semi-empirica! model for the two-centre repulsion integrals 22
'23 (see also Appendix,
Chapter III). The tetrahydrofurfuryl phosphate mono anion was used as a simplified model system for studying the role of the active P(V) TBP sites in the dynamics of the B_.z isomerization process. This model system probes the influence of penta-coordination of phosphorus in the DNA backbone upon the dihedral angle ![0(5')-C(5')-C(4')-0(1']. Special attention was focused on a possible shift in the energy minimum on the potential surface (which is reflected in a change of the dihedral angle !) as a result of P(IV)-P(V) coordination change in the structure. The results of the CND0-2 calculations are given in Table III.1 for various apical ligands (ROH, RSH, RNH 2; R•H, Me) 2
-. The geometries were basedon the P(V) TBP having 0(5') in the apical position, since this conformation is 21.0 kJ/mol more stabie than that having 0(3') in apical position as wa~ calculated with water as the fifth ligand. One of the equatorial oxygen anions is shielded by a proton in order to simulate the complexation with metal ions. The calculations indicate that in the optimized system the dihedral angle ! increases in a P(V) TBP activated system as compared to the P(IV) structure i.e. the energy minimum is shifted toward larger values of ! (A! is given in Table III.1 and in Fig. III.S). As the optimized value of ! in the P(V) TBP model system reflects a time-averaged contribution of the rotamer populations y+, y- and yt this suggests that the P(IV)-P(V) activation results in a decreased preferenee of the y+ conformer in favour of the y
and yt conformers of the C(4')-C(S') bond. The latter conformations are observed in Z-DNA single crystals 7 • Deprotonation
64

Table III.l Influence of pPotonation and depPotonation of the intPoduced ligand (L) on the dihedral angle V[O(S')-C(S 1 )-C(4 1 )-0(1')]; CND0-2 optimized
results.a +L-H
/o .. I H • p-o-,-o/1
HSb, os·
HSo~ H4' A
-- +H•
I Net atomie charge (e)
Lè wo 61f1 1 0( 1 '} 0{5') p L
d 54.4 0.0 -0.267 -0.289 0.370 --HNMe A 57.5 3.2 -0.274 -0.316 0.340 -0.031
B 65.5 11. 1 -0.291 -0.348 0.409 -0.320
NH 2 A 57.7 3.3 -0.274 -0.316 0. 336 -0.076
B 67.4 13.0 -0.294 -0.350 0.410 -0.365
OH A 57.4 3.0 -0.274 -0.318 0. 348 -0. 133
B 66.9 12.5 -0.296 -0.355 0.451 -0.436
aEnergy minimalization as a function of the dihedral angles v.~ and 0: ~=~[P-0(5')-C(5')-C(4')1 and 0=0(0(3')-P-0(5')-C(S')l. bsimilar results were obtained with L=SMe, SH, OMe or OC(O)H. 0~V=V-54.4°. dResults for model compound 1n P(IV) configuration with y+ conformation of the C(4')-C(5') bond.
of the fifth ligand (e.g. water) leads to a further increase in ~V (11-13°). Variatiens in the ÁV values were found to be relatively small, considering the various apical ligands 25
•
The electronic distribution15 in the P(V) TBP intermediate, abstracted from the CNDO-Z calculations, supports the qualita~ tive arguments, vide infra. In the P(V) TBP, the net electron density on 0(5') is increased compared to that in the P(IV) configuration (difference 0.03 e), Deprotonation of the fifth ligand results in an additional increase of 0.03 e
(see Table III.1). The accommodation of the fifth ligand also affects the electron density at 0(1 ') of the deoxyribose ring. Thus~ there is theoretical evidence to support the role
65
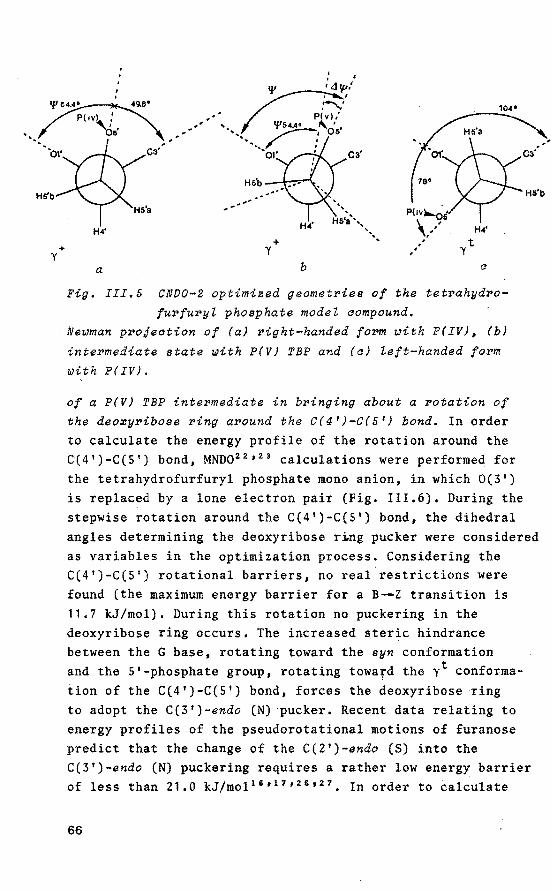
+ y
104°
+ y
a b a
Fig. III.& CND0-2 optimised geometries of the tetrahydro-
furfuryZ phosphate model aompound. Ne~man projection of (a} right-handed form ~ith P(IV)~ (b)
intermediate state ~ith P(V) TBP and (a) Zeft-handed form ~ith P( IV).
of a P(V) TBP intermediate in bringing about a rotation of the deo~yribose ring around the C(4 1 )-C(& 1 ) bond. In order to calculate the energy profile of the rotatien around the C(4')-C(S') bond, MND0 22 ' 23 calculations were performed for the tetrahydrofurfuryl phosphate mono anion, in which 0(3') is replaced by a lone electron pair (Fig. 111.6). During the stepwise rotation around the C(4')-C(S') bond, the dihedral angles determining the deoxyribose r~ng pucker were considered as variables in the optimization process. Considering the C(4')-C(S') rotational harriers, no real restrictions were found (the maximum energy harrier for a B_.z transition is 11.7 kJ/mol). During this rotation no puckeringin the deoxyribose ring occurs. The increased steric bindrance between the G base, rotating toward the syn conformation and the 5'-phosphate group, rotating toward the yt conformation of the C(4')-C(S') bond, forces the deoxyribose ring to adopt the C(3')-endo (N) ·pucker. Recent data relating to energy profiles of the pseudorotational motions of furanose predict that the change of the C(2')-endo (S) into the C(3')-endo (N) puckering requires a rather low energy harrier of less than 21.0 kJ/mol 16 ' 17 ' 26 ' 27 , In order to calculate
66
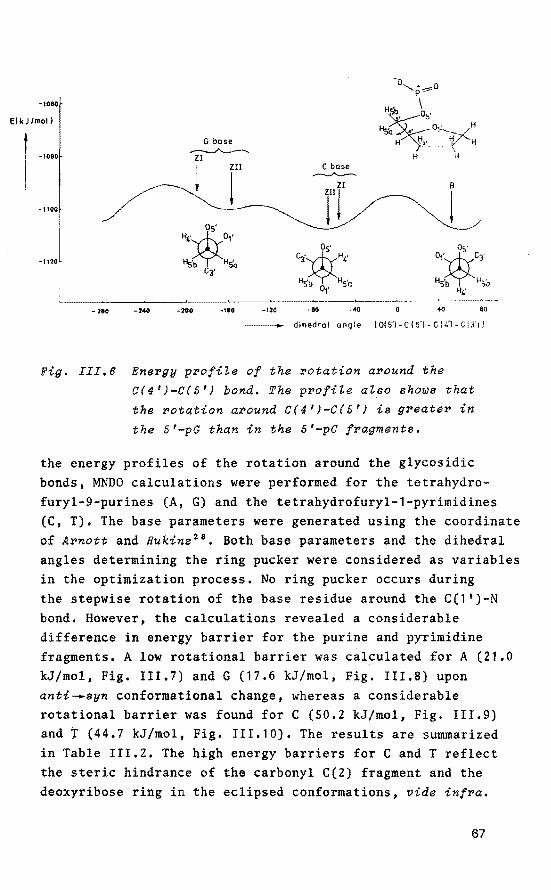
·1060
El kJ/mol)
l ... -1100
·1120,
280 -240
G base ~
Zl
~ Zll
!
·200 ·••o
C base
rr B
!
-120 -ao -•o 40 80
dihedral angle [0(5'1- C I 5')- C (4'! Cl3ï J
Fig. III.6 Energy profite of the rotation around the
C(4'}-C(5'} bond. The profile also shows that
the rotation around C(4'}-C(5'} is greater in
the 5'-pG than in the 5'-pC fragments.
the energy profiles of the rotatien around the glycosidic bonds, MNDO calculations were performed for the tetrahydrofuryl-9-purines (A, G) and the tetrahydrofuryl-1-pyrimidines (C, T). The base parameters were generated using the coordinate of Arnott and Hukins 28 • Both base parameters and the dihedral angles determining the ring pucker were considered as variables in the optimization process. No ring pucker occurs during the stepwise rotatien of the base residue around the C(1')-N bond. However, the calculations revealed a considerable difference in energy harrier for the purine and pyrimidine fragments. A low rotational harrier was calculated forA (21.0 kJ/mol, Fig. III.7) and G (17.6 kJ/mol, Fig. III.8) upon anti-.syn conformational change, whereas a considerable rotational harrier was found for C (50.2 kJ/mol, Fig. III.9) and t (44.7 kJ/mol, Fig. III.10). The results are summarized in Table III.2. The high energy barriers for C and T reflect the steric bindrance of the carbonyl C(2) fragment and the deoxyribose ring in the eclipsed conformations, vide infra.
67

B·Z transition
~ B
! Ct o1• c8=:vc4 z
Hf
Of C2•
C8~C4 Ca~C4 H,· H1' C2· 01'
-1&0 -120 -•o -40 0 40 ~ 120 180 200
___ ,.. dihedral angle [Cf8l- Nf9 J- Cf fl- Cf 2'1 I
Fig. III.7 Ene~gy p~ofile of the ~otation a~ound the C(l')-N
bond~ aalaulated using the tet~ahyd~ofu~yl-9-
adenine aystem.
B-Z trans i !ion
~-B
~. !
-180 -120 -80 -40 80 120 160 200
di hed ral angle !CISJ- Nf91-Cf1'1- C 12'11
Fig. III.B Ene~gy p~ofile of the ~otation a~ound the C(l'J-N
bond~ aalaulated using the tet~ahyd~ofu~yl-9-
guanine system.
68

E (kJ/mol!
400
380 B
! rr -2&0 -220 -140 -100 -20 20 60 100
dihedral angle [Ctó)-NI11-CI0 -C12'll
Fig. III.9 Energy profiLe of the rotation around the C(1')-N
bond~ aaZautated using the tetrahydrofuryt-1-aytosine system.
E( kJ /mol)
1
60
B
40
~ 20
-260 •220 -180 •140 -100 ·10 -20 20 100
- dihedral angle !C!6)-NI1)-CI1'1-Ciill
Fig. III.10 Energy profile of the rotation around the C(1')-N bond~ aaZauZated using the tetrahydrofuryt-1-thymine syetem.
69

In spite of the simplified model systems used, the calculated minima of the energy profiles reflect reasonably well the known X-ray data of the Z-DNA structures with alternating GC sequence 7 - 10 • This is shown in Fig. III.8-9 for the values of the glycosidic dihedral angles in the z1 and z11 form of DNA which reside in the minimum of the energy profile. For B-DNA, the 'single-point' represents the value of the glycosidic dihedral angle in an 'idealized' B configuration (c.J. Chapter 1.3). Most likely, in this case, the geometry of the B-DNA structure prevents adaptation of the pyrimidine glycosidic dihedral angle toward the energy minimum of the calculated profile. The difference in the shape of the energy harriers in the case of T with respect to C can be explained on the basis of the difference between the two structures. InT, the N(3) allows for a slight adjustment in the eclipsed conformations via an out-of-plane bending of the C(2) carbonyl group, as revealed by the MNDO calculations. This conformational adjustment is impossible for N(3) in C.
Table III.2 Energy harrier upon anti-syn conformational change and energy gain during the B_.z tran
sition for the bases A# T# C and G in the
tetrahydrofuryl fragment,a
Residue E for . anti-syn change B-Z transition
kJ/mol kJ/mol
A 21.7 -4.2 T 44.7 -8.4
c 50.2 -12.5
G 17.6 -2.1
aThe data are abstracted from the energy profiles shown in Fig. III.7 - 10. In these figures, the formation energy is plottedas a function of the dihedral angles C(8)-N(9)-C(1')-C(2') for G and A and C(6)-N(1)-C(1')~C(2 1 ) for C and T. The formation energy is linearly correlated with the total energy of the system.
70

111.4 Discuseion.
The CND0-2 computational results on the tetrahydrofurfuryl phosphate mono anion clearly show that the dihedral angle V!O(S')-C(S')-C(4 1 )-0(1 ')1 increases upon a P(1V)-P(V) TBP coordination change in the structure, vide sup~a. This suggests that the P(1V)-P(V) TBP activatien results in a decreased
+ preferenee for the y conformer of the C(4')-C(S') bond in favour of the y- and yt conformers which are required to adopt a Z-DNA structure. However, these computations concern the initiating step in the B-.Z transition and reveal no information with regard to the total reaction coordinate of the rotation around the C(4')~C(S') bond upon P(1V)-P(V) TBP activatien in the system. Therefore, the potential surface of the tetrahydrofurfuryl phosphate mono anion, in which 0(3') is replaced by a lone electron pair, was calculated by the MNDO method from fixed combinations of the dihedral angle V. Analysis of the ground state energy levels of the y+, y- and yt conformers (see Fig. 111.6) leads to the conclusion that the y+ and y- conformers are favoured over the yt conformer in case of a P(IV) model compound. However, the possible altered conformational preferenee of the C(4')-C(S') bond in P(V) TBP model compounds could not be computed, as the MNDO program does not include a parametrization for penta-coordinate structures.
In the case of the d(G-C) 3 duplex, the CND0-2 and MNDO calculations show a high selectivity for direct P(1V)P(V) activatien of the phosphorus atoms within 5'-d(G-C) units in both strands of DNA. Symmetrical generation of the relatively long-lived P(V) TBP intermediates results in the B--Z transition via a selective pathway. A detailed mechanism is suggested, incorporating the role of the P(V) TBP intermediates in the initia! step of a selective rotation of the dC units. Analysis of ground state energy levels and energy harriers in the calculated rotation profiles of the four base fragments revealed a difference in the selectivity between purines and pyrimidines. G and A bases have no restrictions upon anti-syn conformational change, whereas
71

high energy harriers are calculated for C and T bases. Thus, C and T bases prefer to adjust within the anti domain upon B-.z isomerization. The energy gain, upon adjustment, influences the final stage of the B-.z transition. The calculated minima in the energy profiles reflect with reasonable accuracy the known X-ray data of the Z-DNA structures which possess a repetitive GC sequence, vide supra. The higher preferenee for G bases to adopt the syn conformation18 (Z-DNA) can be explained on the basis of increased solvation of the exposed NH2 group. In contrast, the NH2 group in A bases is exposed to the medium, both in the anti and syn position. In the MNDO calculations, no solvation properties of structures in B and Z conformations were taken into account. This may offer a rationalization for the computed higher stability of A bases in the syn position with respect to G bases (viz. Table 111.2, entry 2). Generalizing the quantum-chemical results, only 5'-d(G-C), 5'-d(G-T), 5'-d(A-C) and 5 1 -d(A-T) fragments are selected. Consrquently, the cooperative B_.z transition can only preeeed in DNA helices with repetitive GC, GT, AC or AT sequences. This is consis-tent with experimental data publisbed on Z-DNA structures 7 • 10 - 12
72

Appendix
Theory of the CND0-2 and MNDO quantum-chemicaZ methode.
The self-consistent field (SCF) method has been originally developed by Hartree and Fock 2 !' 30 for calculations on atoms. When the number of electrens is even the wave function of the ground state of the system can in most cases be approximated by a closed-shell configuration:
or
(III.l)
For closed-shell molecules each of the space orbitals ~, ••• ~nis occupied by two electrons, one with a spin c~,)
and the other withBspin (f1). In the case of molecules, the ene-electron wave function ~i extends over the whole molecule. The molecular orbitals can further be written as linear combinations of atomie orbitals (LCAO):
(III.2)
~~ is anatomie orbital on atom ~. The coefficients ei~' which measure the contribution of each atomie orbital in the molecular orbitals, are parameters determined by a variational procedure, i.e. chosen so as to minimize the expression:
(III .3)
The variatien theerem requires for each molecular orbital i, that the coefficients ei~ satisfy the following sets of simultaneous equations:
1 , ••• , n) (III .4)
73

where F~v = ~~~(1)F(l)~v(l)dT 1
and S~v = ~~~(1)~v(l)dT 1 •
By using the expansion in atomie orbitals and by neglectihg S~v for p;v, the matrix elements can be written as:
+ IIP 0 (<~vl lpcr> - l<PPI lvcr>) pcr P
where H~v ~~ (1)Hcore(1)~ (1)dT IJ V 1
(III.S)
After iteration to selfconsistency, the total electronic energy E of a closed-shell molecule is given by 31 :
E =HP (He +HIP (<11vi!Pcr>- i<llPIIvO'>)I (III.6) J,JV J,JV pV pa pa
In the Complete Neglect of Differential Overlap (CNDO) metbod only valenee electrans are treated explicitly, the inner shells being treated as part of a rigid core 32 • 33 •
~p's are treated as if they form an orthonormal set: Spv = ÖJJV (Kronecker delta),
All two-electron integrals which depend on the overlap of charge densities of different orbitals are neglected.
This means that <uvl lpcr>. ö ö <llJJIIPP> = ö ö y JJV p~ pv pcr AB
with p on atom A and p on atom B. Thus yJJP is set equal to yAB' measuring an average repulsion between an electron in a valenee orbital on A and another in a valenee orbital on B. HPJJ is given by
HPJJ = <~1- ~! V2
- VAip> - B~A<piVBiu> = UJJJJ- B~AVAB (III.7)
with UPP = -l(IJJ + AJJ} - (ZA - !)YAA
in which 111 is the valenee state ionization potential and AJJ the electron affinity of the orbital u on atom A.
74

ZA represents the effective nuclear charge of atom A. H =0 for Jl!V. Gore matrix elem~nts HllP' where t is on JlV Jl atom A and t is on atom B, are given by
P HllP = aABsllP in which aAB is the bonding parameter. aAB can be approximated by
in which aA and B~ are adjustable empirically determined parameters. Onder these approximations 20 , the matrix elements of the Fock Hamiltonian reduce to:
(III .9)
The expression (III.9) applies even if Jl and v are on the same atom. Then SJlV=O and the potential integral yAB is replaced by y AA. Initial es·timates of the LCAO coefficients are obtained via
F(o) = -1(1 +A) Jlll 2 Jl Jl
and
Although the CND0-2 method certainly inheres some deficiencies 34 ' 35 , it seems a suitable method to be used for relatively complex structures. The quantum-chemical computations on the tetrahydrofurfuryl phosphate mono anion have been per!ormed wi th the semi-empiric.al CND0-2 methad using the GEOMO program21
• This program performs LCAO calculations and allows direct minimization of energy with respect to any geometrie parameter. The metbod for the minimization of the energy is the classica! conjugate gradient method with the variabie metric, developed by Murtagh and Sargent 36
• The SCF iteration procedure is executed until the energy converges within 9.6 10-7 kJ and optimization is stopped when the minimum relative quadratic difference allowed for two consecutive values of atomie coordinates is less than 1.7 10-9 Ä2•
The quantum-chemical calculations on the nucleoside model systems have been performed with the semi-empirica!
75

MNDO (Modified Neglect of Differential Overlap) method22
which includes a semi-empirica! model for the two-centre rep~lsion integrals 2 s,s 7 ' 38 • The two-centre-one-electron
core resonance integrals ePP' where ~P is on atom A and ~P is on atom B, are considered proportional to the overlap ·
integral SPP
(III.10)
with fl(RAB) = Icet + e!) in which et and a: are characteristic parameters of the atomie orbitals $P and $P. Por the calculation of the two-centre-one-electron terms Vpv,B between an electron in the distribution f~fv of atom A and the core of atom B, the empirica! relation
(III.ll)
is used in which ZB represents the effective nuclear charge of atom B. In this equation the effect of the core of atom B is simulated by the charge distribution s 8s 8 • In the MNDO procedure, Vpv,B is calculated together with the core-core repulsions according to:
(III.12)
the atomie parameter a is used in the iteration procedure to obtain the lowest total energy of the system. Normally, the contribution of the core-core repulsion f 2(RAB) in equation (III.11) is neglected. Molecular geometries, reaction enthalpies, ionization potentials and dipole moments, calculated by the MNDO method, show a good eerrelation with experimental datafora number of molecules 39 •
The SCP iteration procedure in the computations is executed until the energy converges within 2.1 10-4 kJ.
The calculations have been performed on the Burroughs 7700 Computer at the Computing Centre, Eindhoven University of Technology.
76

Referencee and Notee.
1. R. Wing, H. Drew, T. Takano, C. Broka, S. Tanaka, K. Itakura and E. Dickerson, Nature(Lond.), 1980, 287,
755. 2. P. M. Pohl and T.M. Jovin, J. Mol. Biol, 1972, 67, 375. 3. D.J. Patel, L.L. Canuel and F.M. Pohl, Proc. Natl. Acad.
Sci. u.s.A., 1979, 76, 2508. 4. P.M. Pohl, A. Renade and M. Stockburger, Biochem,
Biophys. Acta, 1973, 335, 85. 5. F.M. Johl, Nature(Lond.}, 1976, 260 1 365. 6. D.M. Gray, S.P. Edmondson, D. Lang, M. Vaughan and
c. Nave, Nucleic Acids Res., 1979, 6, 2089. 1. A.H.-J. Wang, G.J. Quigley, F.J. Kolpak, J.L. Crawford,
J.H. van Boom, G. van derMareland A. Rich, Nature(Lond.), 1979, 282, 680.
8. J.L. Crawford, P.J. Kolpak, A.H.-J. Wang, G.J. Quigley, J.H. van Boom, G. van der Mare! and A. Rich, Proc. Natl. Acad. Sci. U.S.A., 1980, 77, 4106.
9. H. Drew, T. Takano, S. Tanaka, K. Itakura and R.E. Dickerson, Nature(Lond.), 1980, 286, 567.
10. A.H.-J. Wang, G.J. Quigley, P.J. Kolpak, G. van der Marel, J.H. van Boom and A. Rich, Science(Wash. D.C.), 1981, 211, 171.
11. S. Arnott, R. Chandrasekaran, D.L. Birdsall, A.G.W. Leslie and R.L. Ratliff, Nature(Lond.), 1980, 283, 743.
12. M. Vorlicková, J, Kypr, V. Kleinwhichter and E. Palecek, Nucleic Acids Res., 1980, 8 1 3965.
13. A.M.C.P. Castelijns, D. van Aken, P. Schipper, J.J.C. van Lier and H.M. Buck, Reel. Trav. Chim. Pays-Bas, 1980, 99. 380.
14. J.J.C. van Lier, L.J.M. van de Ven, J.W. de Haan and H.M. Buck, J. Phys. Chem., 1983, in press.
15. R. Luckenbach in "Dynamic Stereochemistry of Pentacoordinated Phosphorus and Related Elements", Georg Thieme, Publishers, Stuttgart, 1973.
16. W.K. Olson, J. Am. Chem. Soc., 1982, 104, 278. 17. J.W. Keepers and T.L. James, J. Am. Chem. Soc., 1982,
77

104, 929. 18, J.M. Neumann, W. Guschlbauer and S. Tran-Dinh, Eur. J.
Biochem., 1979, 100, 141. 19. A.H.-J. Wang, personal communications, Discussion Meeting
on "Structural Dynamics in DNA", April 27-29, 1982, Veldhoven, The Netherlands.
20. J.A. Pople and D.L. Beveridge, "Approximate Molecular Orbital Theory", McGraw-Hill, New York, 1970.
21. D. Rinaldi, Comput. and Chem., 1976, 1, 109; Program 290, Quanturn Chemistry Program Exchange, Indiana University.
22. W. Thiel, 1978, Program 353, Quanturn Chemistry Program Exchange 11, Indiana University.
23. M.J.S. Dewar and W. Thiel, J. Am. Chem. Soc., 1977, 99,
4899. 24. CND0-2 and MIND0-3 calculations gave similar results. 25. J.J.C. van Lier, L.H. Koole and H.M. Buck, Reel. Trav.
Chim. Pays-Bas, 1983, 102, 148. 26. M. Levitt and A. Warshell, J. Am •. Chem. Soc., 1978, 100,
2607. 27. W.K. Olson and J.L. Sussman, J. Am. Chem. Soc., 1982,
104, 210.
28. S. Arnott and D.W.L. Hukins, Biochem. Biophys. Res. Comm., 1972, 47, 1504.
29. J.E. Lennard-Jones, Trans Faraday Soc., 1929, 2S, 668. 30. R.S. Mulliken, J. Chem. Phys., 1935, 3, 375. 31. c.c.J. Roothaan, Rev. Mod. Phys., 1951, 23, 69. 32. J.C. Slater, Phys. Rev., 1930, 3S, 509. 33. J.C. Slater, Phys. Rev., 1959, 34, 1293. 34. J.N. Murrell and A.J. Harget, "Semi-empirica! Self
-consistent Field Molecular Orbital Theory of Molecules", Wiley Interscience, London, 1972.
35. A.R. Gregory and M.N. Paddon-Row, J. Am. Chem. Soc., 1976, 98, 7521.
36. B.A. Murtagh and R,W,H, Sargent, Comput. J., 1970, 13,
185. 37. M.J.S. Dewar, "The Molecular Orbital Theory of Organic
Chemistry", McGraw-Hill, New York, 1970.
78

38, M.J.S. Dewar, M.L. McKee and H.S. Rzepa, J. Am. Chem. Soc., 1978, 100, 3607.
39. M.J.S. Dewar and W. Thiel, J. Am. Chem. Soc., 1977, 99,
4907.
79

CHAPTERIV
B-Z transition in methylated DNA.
A qu.antniD- chemical study o:f model systems
IV.l Introduation.
In Chapter III a model description of the B-.z transition in DNA was given in which the initiating step inheres a P(IV)-P(V) activatien of the phosphate groups in the helix backbone. On the basis of CND0-2 and MNDO quantum-chemical calculations a good rationalization of the observed selectivity could be obtained. Generally, Z-DNA structures are found with alternating purine-pyrimidine sequences 1 • Recently, circular dichroism (CD) and 31 P NMR studies on synthetic oligonucleotides of d(G-C)n units, inserted within the plasmid pRW751, revealed that these fragments undergo a cooperative transition to a left-handed Z-DNA structure, thereby influencing the supercoil of the plasmid 2 • Addition of Z-DNA-specific antibodies to active DroaophiZa chromosarnes revealed the ~xistence in vivo of local Z-DNA fragments 3
• Furthermore, a possible d(G-T) 25 . d(A-C) 25 z site within one of the introns of a human aardiaa muacZe aatin gene was reported 4
• A biologica! role for left-handed sites in vivo remains to be elucidated 5
'6
• In biologically active DNA sequences, occasionally nature has ·prese~ved alternating sequences of guanine and cytosine residues. Por example, there is a highly conserved segment in radent parvoviruses near the origin of replicatien which contains a total of 20 alternating G and C residues in two segments 7 • Furthermore, a segment of the histidine D gene of SaZmoneZZa has a mÜtational 'hotspot' which occurs at a segment of eight alternating GC residues 8 • Probàbly these sequences are
80

associated with Z-DNA structures. Chemical agents can modify DNA and alter its stability.
There are many agents which are specific for guanine alkylation in the 0(6), N(7) or C(8) position. Several of these are highly active carcinogens, such as N-acetoxy-N-acetyl-2-aminofluorene (AcAFln), which alkylates in the C(B) position, and nitrosamines, nitrogen mustards, nitrosoureas and aflatoxin which alkylate the N(7) position 9 •
It is possible that the low level of alkylation which is seen in these carcinogens may be related to the formation in vivo of Z-DNA structures. Recent studies on double-stranded polymers with the alternating sequence d(G-m5C) 10 • 11
and d(m7G-C) 12 strongly point to a Z-DNA structure under physiological conditions. Apparently, methylation of C.G base pairs in DNA considerably increases the stability of the Z-DNA isomer. In addition, AcAFln modification of dG residues on the C(8) position in the case of an alternating dG-dC sequen~e, destabilizes the B-DNA form in favour of the Z-DNA isomer 13 - 15 • X-ray data on the d(G-m5c) 3 mini-duplex, crystallized in low-salt solution, revealed the conformational features of the Z-DNA structure 16 •
The C.G base pair forms part of the outer surface of Z-DNA. Therefore, the C(S) position of cytosine is likely to be more accessible there to an enzyme than in B-DNA. GpC sequences are relatively infrequent in eukaryotic DNA, but they are often highly methylated on cytosine C(S), especially in inactive genes. It has been speculated that the signal for methylation may be physical in nature 17 and an enzyme recognizing a GpC Z-DNA segment may be the basis for this modification. Structural analysis of the d(G-C) 3 Z-DNA fragment led Wang et at. 18 to a description of the B-.Z isomerization process. From these results and the performed CND0-2 and MNDO calculations 19 on model systems representative for DNA, it seems that the B-Z transition in DNA with alternating dG-dC sequence exclusively proceeds via symmetrical generation of P(V) TBP intermediates within dpC fragments in the helix backbone in which 0(5') is situated in an apical position of the
81

trigonal bipyramid. X-ray data on the d(G-m5c) 3 Z-DNA fragment reveal analogous conformational characteristics 16
•
Therefore the B-.Z transition in DNA fragments with any alternating (methylated) dG-dC sequence proceeds also via the indicated pathway20 (Fig. IV.1).
~~o,.-H
·- pu \
·· o3 o-H
0 -V-~·0----IMn• )oq
o/ . '
__ ~ ~
A
r-IJ /0,~ -~
hydroxyfosforc c
hydroxyfosfor c'
~ 0{ / o·.-- (Mn• )oq
·o-P / "· OS' ,..O-H
'-I H
.
"' a'
Fig. IV.l Generation of the intermediate P(V) TBP structures
Band B' by nucleophilic attack of a water molecule
(A) which represents the initiating step. EquatoriaZ skietding (Mn+; etahilizes the intermediate
structure. The Zess stabZe isomers (B' and C') are con
verted to B and C respectiveZy by the proaess of pseudo
rotation (PR). The C and C' structures are hydrozyphospho
ranes which result upon proton toss in the eztra ligand,
ApicaZ loaation of 0(5') in the TBP is the energeticaZZy
favoured intermediate~ sinae this conformation is 21 kJ/mol
more stabZe than for O(ö') in apical position (B')~ as was.
caZculated for water as the fifth ligand. The most important
dynamic motions are indicated in B. The pyrimidines (pyr)
are cytosine or 5-methyZcytosine; the purines (pu) are
guanine or 6-~ 7- or 8-methytguanine.
82

With the aid of the semi-empirica! quantum-chemical MNDO method, the impact of methylation on the B--Z transition was obtained from calculations on a set of representative model systems.
IV.Z MNDO aalaulations.
By means of the MNDO method 21 ' 22 the energy profile of the stepwise rotation around the glycosidic C(l')-N bond was calculated for the various methylated residues in the tetrahydrofuryl model system. The results are compared with those for the unmethylated bases (c,f, Chapter III). The base parameters and the dihedral angles determining the ring pucker were considered as variables in the optimization process. No ring pucker occurs during the rotation of the base residue around the glycosidic bond. This indicates that a pucker change from C(Z')-endo (S) into C(3')-endo (N) within dpG fragments, as observed in the B-.Z transition, is solely due to an increased steric bindrance between the phosphate moiety and the base residue.
In accord with these observations, calculations on a dpG fragment with eyn conformation of the guanine base revealed the preferenee fora C(3')-endo (N) pucker 19 •
The results of the calculations on the various model systems are presented in Fig. IV.3- 4 and in Table IV.1. The substitution sites in B-DNA are indicated in Fig. IV.Z. The calculated energy profiles support the observed selectivity in the B--Z transition. The anti-eyn conformational change is unfavourable for pyrimidine residues (Fig. IV.3). However, energy release occurs upon adjustment of the pyrimidinebase within the anti domain (Table IV.l). In contrast, purine residues show a low rotational energy harrier upon anti-eyn rotation of the base (Fig. IV.4).
83
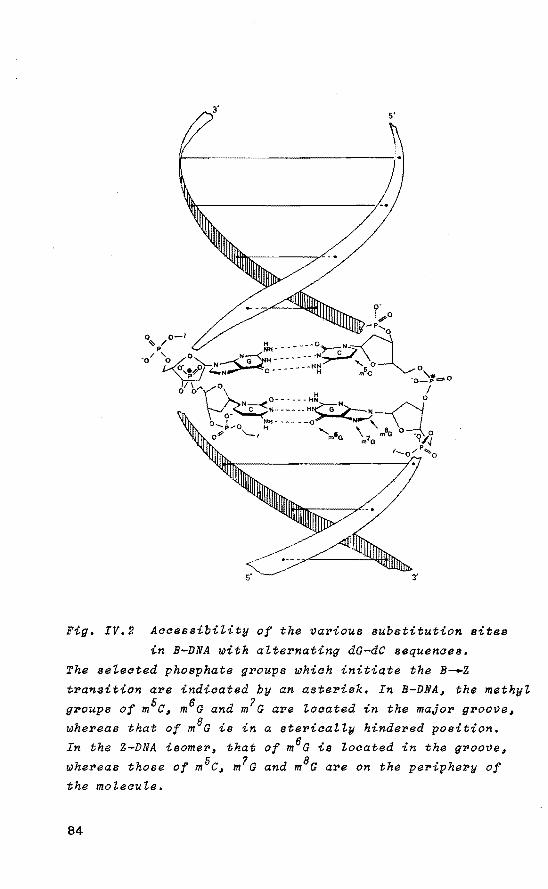
Fig. IV.2 Accessibi"lity of the vavious substitution sites
in B-DNA with a"ltevnating dG-dC sequences.
The setected phosphate gvoups which initiate the B-.Z
transition ave indicated by an asterisk, In B-DNAi the methyt
gvoups of m5ci m6G and m7G ave Zocated in the major gvoovei
wheveas that of m8G is in a stevicaZZy hindered position.
In the Z-DNA isomev, that of m6G is Zocated in the gvoove,
wheveas those of m5c, m7G and m8G are on the periphevy of
the mo"lecu"le.
84

TabZe IV.l Enevgy bavrier upon anti-syn con[ormationaZ
change of cytosine or guanine and energy gain during the B--Z transition for these bases in the tetrahydrofuryZ fragment.a
Resîdue E for
dm'C dC dm•o dm'G dm8G dG
anti-syn change
kJ/mol
89.3 50.2 24.7 22.5 40.5 17.6
B-Z transition
kJ/mol
-14.5 -12.5 -10.8 -12.5 -39.4 - 2.1
aThe data are abstracted from the energy curves in Fig. III.3 - III.4. In those figures the formation energy is plotted as a function of the dihedral angles C(8)-N(9)-C(l')-C(2') for purine residues and C(6)-N(1)-C(1')-C(2') for pyrimidine residues. The formation energy is linearly correlated with the total energy of the system.
IV.3 MethyZation of cytosine.
Methylation of dC residues on the C(5) position is performed after DNA synthesis 23 • In the tetrahydrofuryl-1-(5-methylcytosine) fragment (Fig. IV.3B) an unusually high anti-syn energy barrier was calculated (89.3 kJ/mol) with respect to the same transition in the tetrahydrofuryl-1-cytosine system (50.2 kJ/mol, Fig. IV.3A). The higher barrier in case of the dm5c residue can be rationalized by an electronic effect on the carbonyl C(Z) group and the C(Z') methylene fragment in the tetrahydrofuryl ring due to the para-substituted methyl group. For the ciosest C(Z') hydrogen atom, the net electron density in the eclipsed conformation is +0.025 e in the case of cytosine and +0,069 e
in the case of 5-methylcytosine. The net electron densities on 0(2) and C(2') during steric bindrance between the carbonyl C(Z) group and the C(Z') fragment in the eclipsed conformation are -0.347 e, -0.009 e, respectively in case
85
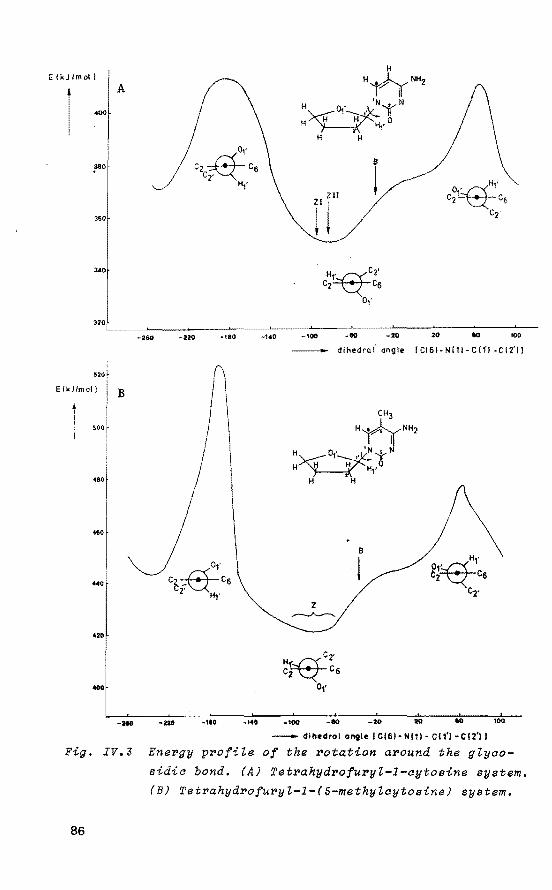
Ei kJ /moll A
400
360
340
-260 -220
520
E [kJ/mail B
500
480
460
.. 0
420
400
-210 ·220
•180
r ' \
'
-110 -140
-140
H
H~NHz 'N N
H~o,·~ 'r W~'Hfo
H H
rr r
dihedral· angle ICI6l-NI1l-Cffi-Cf2'1l
-20 100
- dlhedral antle I Cl61- Nl11· CI1'1·CI2'11
Fig. IV.Z Energy profite of the rotation around the gtyco-
86
sidia bond. (A) Tetrahydrofuryt-1-cytosine system.
(B) Tetrahydrofuryt-1-(S-methyZcytosine) system.

of cytosine and -0.360 e, -0.030 e, respectively in case of 5-methylcytosine. The more powerful repulsion induced by the dm5C residue thus considerably increases the anti-syn activatien energy. From incorporation of dm 5c residues in a Z-DNA model a possible hydrophobic interaction (i.e. attractive van der Waals interaction) between the 5-methyl group in C
and the C(2') hydragen atoms of the. adjoining dG residue is apparent. In B-DNA the 5-methyl group is located in the major groeve, exposed to the solvent, whereas in Z-DNA the methyl group is situated in a more shielded environment. Both the steric and electronic factors account for the observed enhanced stability of the Z conformation upon methylation of the C residues.
Bivalent and trivalent cations have a powerful effect on the double-stranded poly d(G-m5C) transition 10 • The effects of these cations on the B--Z transition can be rationalized in terms of stahilizing interactions with the equatorial oxygen anions of the P(V) TBP intermediate structure.
IV.4 Methylation of guanine.
Alkylating agents react with guanine in DNA to form a variety of products 2 ~. Methylation of the tautomerie enoZ form of guanine on the 0(6) position results in the carcinogenic 6-methylguanine structure 25 ' 26 , Since the latter may hydrogen-bond to thymine as well as to cytosine, it can be misreadas a dA residue 27
• In the C.G base pair, 0(6) of guanine is directly hydrogen-bonded with a NH 2 hydrogen atom of cytosine. Therefore, methylation of dG on the 0(6) position diminishes the total H-bond interaction within the C.m6G base pair.
A moderate anti-syn activatien energy of 24.7 kJ/mol in the tetrahydrofuryl-9-(6-methylguanine) fragment was calculated whereas a considerable stabilization of the syn conformation occurs (-10~8 kJ/mol with respect to the anti conformation, Fig. IV.4B). Therefore it seems that 6-methyla-
87

A
2o.o' B E (kJ /mol)
1
.
0.0
-20.0
- 40:
-60.0
-1110
B
!
-120 -80
B
!
-120 -80
B-Z transition
0
NyÁN-H H~ J.L A
H~· No N NH2 1 ,. H H'
H , Hr H H
0 40 80 1:10 180 200 - dihedrol angle ICIBI- Nt91-CI1'l-CI2'll
B-Z transition
Q
0 40 80 12{) 160 200 - dihedrol ongle I CIS)- N\91- C tfl- C!2'l I
Fig. IV.4 Ene~gy p~ofile of the ~otation a~ound the glyco
sidic bond. (A) Tet~ahyd~ofu~yl-9-guanine system. (B) Tetvahydvofu~yl-9-(6-methylguanine) system.
88

ted dG residues incorporated in a double-stranded DNA structure with alternating dG-dC sequence promote the B--Z isomerization process. However, no data are available to support this suggestion.
Methylation of dG residues on the N(7) position is enhanced by many mutagens and carcinogens. To respond to this damage a glycosylase for dm7G has been found in several cell types 28 • The calculations on the tetrahydrofuryl-9-(7-methylguanine) model system (Fig. IV.4C) reveal that the methyl group introduces a positive charge which is delocalized mainly on H(1') (+0,25 e), C(8) (+0,15 e) and the methyl carbon atom (+0.20 e). An anti-syn activatien energy of 22.5 kJ/mol was calculated. In converting from B-DNA to Z-DNA the average distance between phosphate groups in the helix diminishes. Because of the positively charged imidazole fragment a considerable enhancement of the B--Z transition is reached due to a neutralizing effect on adjacent phosphate groups.
As a model for the N-acetoxy-N-acetyl-2-aminofluorene (AcAFln) reacting with the C(8) position of the dG residue, the tetrahydrofuryl-9-(8-methylguanine) system (Fig. IV.4D) was selected. Studies on oligonucleotides have shown that, on reaction with AcAFln the guanine structures are forced into the syn conformation29
'30
• In Z-DNA, the C(8) position of guanine is locàted readily accessible on the outer surface. In the isomerie B form however, the C(8) hydragen atom is in close contact with the helix backbene and reaction with the large AcAFln molecule can only preeeed with considerable distartion of the backbone, forcing the guanine derivative into the syn conformation. This accounts for the observed stability. In B-DNA there is a high preferenee for insertion of the fluorene ring into the double helix while the dG residue will be outside the helix. Pu~~man et aZ. 31 calculated a higher reactivity for B-DNA, most likely because they underestimated the important geometrical differences between B-DNA and Z-DNA. The calculated high anti-syn energy harrier in case of m8G (40.5 kJ/mol) results from the steric hindrance of the 8-methyl group and the closest C(Z')
89
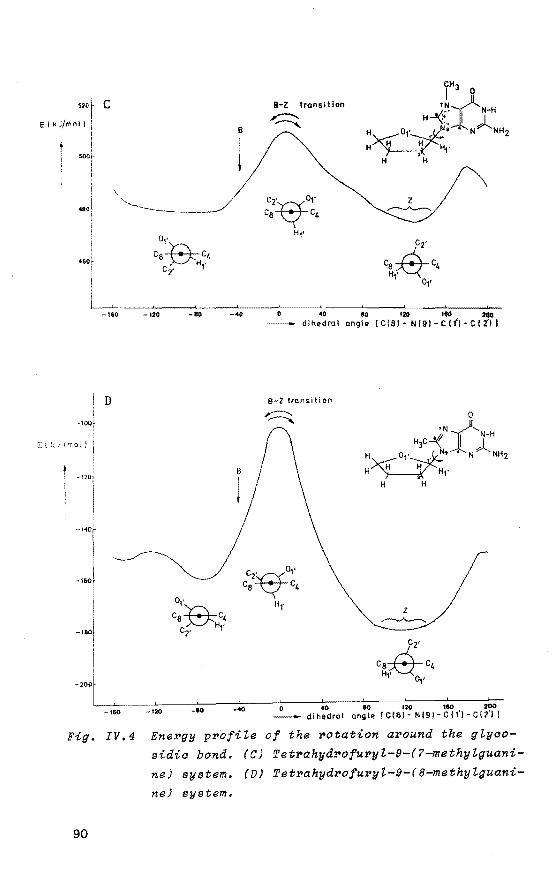
520t c ElkJ/moll i e
50{)/-1 1
.. ai \
a,. Cs~C4
Hr Cz•
-160 -120 _...,
D
-•sof
I
CH3 I o 8-Z transition
'Ni: 8 ~-· I N-H
,~;. """""' H H H H1'
2 H H
c2~1' z ca c4 ~
Ht• C2'
eal2i-c4 Ht'
Oj•
BQ 120 200 dlhedral angle [ C 181 - N l9l - C (I') - C I 2'1 l
a-z trans i ti on
0
•N/-N H
~c~ -! .. i H~01·~• N.., NHz
H~'Ht• H H
-200L . .L--------'----__,____..___.._._ 0 40 80 120 IlO 200
-160 -120 -80 -40 - dihedral angle [CIS!- Nl9l·C!t'l-CI21l
Fig. IV.4 Energy profiLe of the rotation around the gtycosidic bond. (C) Tetrahydrofuryt-9-(7-methytguani
ne) syatem. (D) Tetrahydrofuryt-9-(8-methylguani
ne) syatem.
90

hydragen atom in the eclipsed conformation. Rotation of this base residue toward a syn position diminishes the steric interaction as is reflected in the high energy gain during the B-.Z transition (-39.4 kJ/mol).
IV.S Discussion.
In spite of the simplified model syste~s used to describe the complex dynamics of C.G base pairs with different substituted methyl groups during the B-.z transition, the calculated minima of the energy profiles are in accord with the reported X-ray data 16 • 18 '
32• Driven by the impetus
of methylation, an extra stabilization of the Z isomer is obtained. The energy gain (~E) during the B-.z transition in double-stranded dinucleotide fragments with different locations of the methyl group was calculated. Differences in stacking energy and solvation properties of B and Z structures were not taken into account. These effects may alter the order of relative stahilities as reflected in the 8E value. The results are given in Table IV.Z. Literature data on the B--Z transition in double-stranded DNA fragments with (methylated) alternating dG-dC sequences 10 • 12 •la,aa
show a close correlation between the observed critica! salt concentrations (i.e. the concentratien at which 50% of the DNA is converted to the Z-DNA form) for the various methylated structures reported so far and the calculated ~E values. A smaller contribution of stahilizing cations and anions is needed if the Z-DNA conformation confers a high intrinsic stability. In addition, low critical salt ~oncentrations to induce the B--Z transition correlate with a high 8E value. In the case of the dm8G derivative the results are compared with data on modification of guanine by AcAFln 3 ~• 35 ,
The reverse z~B transition can be induced by de-methylation of the structure and by decreasing the salt concentration. Both effects result in a relaxation toward the isomerie B-DNA form.
91

Table IV.2 Energy gain (áE) during B_.z transition in 100%
methylated dinualeotide duplex fragments ~ith
different substitutions of the methyl group.a
Structural unit AE Methylation Critica! eonen of
Na Cl MgCI2
kJ/mol % M mM
d(G-C) -29.2 0 2.5 700 d(m6G-C) -46.6 d(m7G-C) -50.0 90 0.35 d(m8G-C) -103.8 20-25 0.06 d(G-m'C) -33.2 100 0.7 0.6 d(m6G-m'C) -50.6 d(m7G-m5C) -54.0
~5, 100b d(m8G-m'C) -107.8 0.2
a~E is calculated as the sum of the difference in ground
-state energy levels between the B and Z conformation for the various duplex structures. Data are abstracted from Fig. IV.3 - 4, the experimental results from ref. 10, 12, 13, 33 and 35. bdG 3.5% reacted with N-acetoxy-N-acetyl-2-aminofluorene,
dC 100% methylated at 5 position.
Special attention was focused on the possible biologica! role of d(G-m5C) sites in the genome. In the introduetion several suggestions have been made 5 ' 6 , One further possibility seems quite interesting. Although m5c residues have a high probability of undergoing mutation t~ T by a de-amination reaction 36 , the d(G-m5C) sequence occurs rather frequently in eukaryotic DNA 37
• Recentworkof Hamada et al. 4 showed that Z-DNA sequences are highly repeated in the human genome. Methylated C is also present in higher plant DNA, all of which are part of the basic trinucleotide CXG (X=purine or pyrimidine) 38
, The m5C.G base pairs inhere a strenger H-bond interaction than the corresponding C.G base pairs 39
•
Therefore it seems unrealistic to assume that activatien of d(G-m 5C)n (n=1, 2 ,,) sites in the genome results in local opening of this region. Instead, activatien of the
5'-dpm5c phosphate groups in these structures may lead to a B_.z isomerization process via a selective pathway. Recent work by Stirdivant et al. 40 on supercoiled recombinant
92

plasmids containing alternating d(G-C)n sites has shown that the salt-induced B-.z t~ansition ~esults in a synch~o
nous ~eta~ation of the supe~coiled st~uctu~e while the reverse process, z-.B ~elaxation ~esults in an ove~winding
of the supercailed st~ucture. lnterestingly, studies have indicated that DNA supercoiling can modulate transcription activity. Furthermore, G~uenbaum et al. 38 affered additional support that, in genera!, gene expression is correlated with a relative undermethylation at (G-m5C) sites in the gene while inactive genes are normally methylated at C(S) of cytosine in (G-C) sites. From the recently reported experimental details on recombinant plasmids containing an alternating (G-m5C) sequence~ 1 it is clear that undermethylation of cytosine destabilizes the Z-DNA form under physiotogical conditions in favour of the isomerie B-DNA structure. Considering these results it can be envisaged that a similar situation is encountered in genomic DNA. Undermethylation of specific (G-m5C) Z sites within the genomic material then results in a Z--B refolding process. This process results in a synchronous overwinding of the flanking supercailed region, thereby increasing the total number of supercoils present in the system. As a consequence, protein binding sites within this region (e.g. TATA box) 42
undergo a relative shift in the supercailed structure, probably allowing specific protein-DNA interactions leading to gene activity. Methylation of the G residues within these Z sites could easily perturb this mechanism by promoting an irreversible Z-DNA site. However, studies on the human
yöS-globin gene region suggest that a low level of DNA methylation may be a necessary, but not a sufficient condition for gene expression in higher eukaryotes 43
•
93

Refe~enaes and Notes.
1. S. Arnott, R. Chandrasekaran, D.L. Birdsall, A.G.W. Leslie and R.L. Ratliff, Nature(Lond.), 1980, 283, 743.
2. J. Klysik, S.M. Stirdivant, J.E. Larson, P.A. Hart and R.D. Wells, Nature(Lond.), 1981, 290, 672.
3. A. Nordheim, M.L. Pardue, E.M. Lafer, A. Möller, B.D. Stollar and A. Rich, Nature(Lond.), 1981, 294, 417.
4. H. Hamada and T. Kakunaga, Nature(Lond.), 1982, 298, 396. 5. A. Rich, Science(Wash. D.C.), 1981, 214, 1108. 6. C.R. Cantor, Cell, 1981, 2S, 293. 7. C.R. Astell, M. Smith, M.B. Chow and D.C. Ward, Cell,
1979, 1'1, 691. 8. K. Isono and J. Yourno, Proc. Natl. Acad. Sci. U.S.A.,
1974, '11, 1612. 9. C.E. Searle, "Chemica! Carcinogens", American Chemical
Society, Wash. D.C., 1978. 10. M. Behe and G. Felsenfeld, Proc. Natl. Acad. Sci. U.S.A.,
1981, '18, 1619. 11. Abbreyiations for nucleotides follow IUB/CEJB Recommen
dations, see Eur. J. Biochem., 1970, 1S, 203; e.g. dC, 2'-deoxycytidine; d(G-C)n, structure with n alternating dG-dC fragments;d(mxG-m5C), dinucleotide structure with x-methylguanine and 5~methylcytosine incorporated, in which the phosphorus is bound.to 0(5') of the cytidine residue.
12. A. Möller, A. Nordheim, S.R. Nichols and A. Rich, Proc. Natl. Acad. Sci. u.s.A., 1981, 78, 4777,
13. E. Sage and M. Leng, Proc. Natl. Acad. Sci. U.S.A., 1980, '17' 4597.
14. R.M. Santella, D. Grunberger, I.B. Weinstein and A. Rich, Proc. Natl. Acad. Sci. U.S.A., 1981, 78, 1451.
15. E. Sage and M. Leng, Nucleic Acids Res., 1981, 9, 1241. 16, A.H.-J. Wang, S. Fujii, J.H. van Boom and A. Rich, Cold
Spring Harhor Symp. Quant, Biol., 1982, 4'1, 5. 17. A.P. Bird, M.H. Taggart and B.A. Smith, Cell, 1979,
1'1, 889. 18. A.H.-J. Wang, G.J. Quigley, F.J. Kolpak, J.L. Crawford,
94

J.H. van Boom, G. van der Marel and A. Rich, Nature (Lond.), 1979, 282, 680.
19. J.J.C. van Lier, L.H. Koole and H.M. Buck, Reel. Trav. Chim. Pays-Bas, 1983, 102, 148.
20. J.J.C. van Lier, M.T. Smits and H.M. Buck, Eur. J. Biochem., 1983, 132, ss.
21. M.J.S. Dewar and W. Thiel, J. Am. Chem. Soc., 1977, 99,
4899, 22. W. Thiel, Program 353, Quantum Chemistry Program
Exchange 11, Indiana University, 1978. 23. F. Kalousek and N.R. Norris, J. Biol. Chem., 1969, 244,
1157. 24. G.P. Margison and P.J. O'Connor, "Chemical Carcinogens
and DNA", P.L. Glover, ed., CRC Press, West Palm Beach, FL, 1979, 111.
2S. K.W. Fowler, G. Büchi and J.M. Essigmann, J. Am. Chem. Soc., 1982, 104, 10SO.
26. R. Goth and M.F. Rajewski, Z. Krebsforsch., 1974, 37. 27, P. Lawley and c. Martin, Biochem. J., 1975, 145, 85. 28. J, Laval, J. Pierre and F. Laval, Proc. Natl. Acad.
Sci. u.s.A., 1981, 78, 852. 29. J.F. Lefevre, R.P.P. Fuchs and M.P. Daune, Biochemistry,
1978, 17, 2561. 30. D. Grunberger, S.H. Blobstein and I.B. Weinstein, J.
Mol. Biol., 1974, 82, 459, 31. A. Pullman and B. Pullman, Nucleic Acids Res., 1980,
B, 3917. 32. H. Drew, T. Takano, s. Tanaka, K. Itakura and R.E.
Dickerson, Nature(Lond.), 1980, 286, 567, 33, F.M. Pohl and T.M. Jovin, J. Mol. Biol., 1972, 67, 375, 34. R.P.P. Fuchs and M.P. Daune, FEBS Lett., 1971, 92, 207. 3S. R.M. Santella, D. Grunberger, A. Nordheim and A. Rich,
Biochem. Biophys. Res. Commun., 1982, 106, 1226. 36. M. Ehrlich and R.Y.-H. Wang, Science(Wash. D.C.), 1981,
212, 1350, 37. A. Razin and A.D. Riggs, Science(Wash. D.C.), 1980,
210, 604. 38. Y. Gruenbaum, T. Naveh-Many, H. Cedar and A. Razin,
95

Nature(Lond.), 1981, 292, 860. 39. M. Ehrlich, K. Ehrlich and J.A. Mayo, Biochim. Biophys.
Acta, 1975, 395, 109. 40. S.M. Stirdivant, J. Klysik and R.D. Wells, J. Biol.
Chem., 1982, 257, 10159. 41. C.K. Singleton, J. Klysik, S,M, Stirdivant and R.D,
Wells, Nature(Lond.), 1982, 299, 312. 42. H.B. Gamper and J.E. Hearst, Cell, 1982, 29, 81. 43. L.H.T. van der Ploeg and R.A. Flavell, Cell, 1980, 19,
947.
96

CHAPTER V
rolec-ular aspects o:t IDethylated adenine in DNA.
. A quantuiD-cheiDical study o:t IDodel systeiDs
V.1 Introduation.
Alkylating agents react with nucleic acids to form a variety of products, dependent upon the nature of the base residue, electrophilicity of the agent and presence of activating enzymes 1
• Strong electrophiles such as N-methyl-N-nitrosourea and N-methyl-N•-nitro-N-nitrosoguanidine are believed to react preferentially with oxygen donors via the highly reactive diazonium ion. Weakly electrophilic agents such as dimethyl sulfate and methyl methane sulfonate tend to show high specificity for ring nitrogen methylation. Much of the damage to DNA by chemica! carcinogens occurs through alkylation on the N(3) and N(7) positions of the purine bases 2 , which enhances the rate of depurination of that base by breakage of the glycosidic C(1')-N bond 3 ' 4 •
The resulting apurinic site in DNA is relatively stabie and leads to an increased mutagenesis since a DNA polymerase was shown to copy past apurinic sites on natura!, biologically active DNA 5 • For adenine, the substantial influence of pH on its alkylation pattern is well established. BeasZey et aZ. 6 revealed that N(3) alkylation of the adenine residue is dominant under mildly acidic or neutral conditions. TopaZ et aZ. 7 studied the introduetion of alkylated residues into double-stranded DNA.and showed that the dm3ATP precursor incorporates in vitro at T template sites but not at A, G or C sites. Both m3A and dm3A structures were found to inhibit intracellular protein degradation8 and recently, the partial purification of a human m3A-DNA glycosylase was reported 9 • Methylation
97

of the N(6) position in adenine results in a structure which is involved in the restriction-modification system in bacteria 10 and possibly in control of gene expression and differentiation in eukaryotic cells 11
'12
• E.aoZi contains m6A mainly in GATC sequences 13 and it seems that these residues are involved in strand discriminatien during mismatch correction in DNA 14 • BodeZZ et al. 15 compared the difference in reactivity of N-ethylnitrosourea toward the hydrogen-bonded 6-NH2 group of the adenine base in
ds DNA and the same site in single-stranded (ss) DNA. They found the reactivity to be temperature independent due to the N(6) atom having an electron pair not involved in hydrogen-bonding and, thus, available for reaction. Alkylation of dA residues on N(7) and C(8) is enhanced by many electrophilic agents 2 • The dm7A structure is highly susceptible to hydrolytic cleavage of the glycosidic bond 3
'4
• Studies on mononucleotides have shown that substitution of H(8) in adenine by a bulky substituent restricts the base to the syn range 16 • DNA structures usually crystallize as a B-DNA helix 17 • However, as discussed in Chapter III, strictly alternating d(G-C)n sequences can undergo a cooperative transition into a Z-DNA structure 18 • 19 ,
Alkylating agents can modify DNA and alter its stability. In Chapter IV it was shown that the impetus of methylation of C.G base pairs in DNA confers to .the intrinsic stability of the Z-DNA isomer 20
• It is interesting to investigate whether a similar situation is encountered in DNA structures with alternating d(A-T)n sequences. By means of the semi-empirica! MNDO quantum-chemical method, the effect of methylation of the tetrahydrofuryl-9-adenine model system on the anti-syn equilibrium was examined. The possibility for an alternating methylated d(A-T)n21 sequence to adopt a left-handed duplex is discussed. Furthermore, special attention is focused on the unique properties of the dm6A system toward restriction-modification enzymes.
98

v.z B_.z t~ansition in atte~nating d(A-T)n potyme~s.
X-ray diffraction studies of d(G-C) 2, d(G-C) 3 and d(G-m5c) 3 single crystals showed that these mini-duplexes are in the Z-DNA conformation22 - 26 • Substitution of a C.G base pair by an A.T base pair generally destabilizes the Z-DNA helix. For example, circular dichroism (CD) spectroscopy on the ds alternating polymer d(T-G)n.d(C-A)n reveals that, in comparison with alternating d(G-C)n structures, more stringent conditions (concentrated NaCl solution and partial modification by N-acetoxy-N-acetyl-2-aminofluorene) are required to promote formation of the left-handed helix27
•
The polymer d(A-T)n shows the characteristics of a partial a-.z transition28 ' 29 • However, x-ray data on the dp(A-T)z mini-duplex reveal a B-DNA structure, in which the deoxyribose pucker is C(3')-endo (N) for dA fragments and C(Z')-endo (S) for dT fragments 30
, whereas the values of the glycosidic dihedral angles are almost at the limits of the anti domain 31
• This 'alternating B-DNA' structure, first proposed by Ktug et at. 32
, provides an elegant explanation for the occurrence of 'kinks' in natural DNA, where the helical axes meet at an angle with a concomitant negative change in the duplex winding 33
-35
• The pucker change from C(Z')-endo (S) into C(3')-endo (N) within dpA fragments, as observed in the dp(A-T) 2 structure_, can be explained on the basis of an increased steric bindrance between the phosphate group and the N(9)-C(8)-N(7) moiety of the adenine base which is in a nearly eclipsed conformation with the C(1')-0(1') bond.
From comparison of the structural features of the dp(A-T) 2 duplex with Z-DNA, the similarity in deoxyribose pucker is striking. However, the syn conformation of dA residues, required to adopt a Z-DNA structure, is absent in dp(A-T) 2 due to a lack of stahilizing solvation. With rèspect to the syn conformation of deoxypurines, Wang et al. 22 pointed out that water hydrogen-bonding to the C(2)-NH2 group of the guanine base is an important stahilizing factor, which is absent in adenosine.
99

From the computational results presented in Table V.l (vide infPa) it is evident that the syn conformation of adenine can be promoted by selective methylation. It is not unlikely that under certain conditions the lack of stahilizing solvation in these fragments will be more than compensated by the high energy gain as a result of an anti-syn transition. In Chapter III , the selective role of P(V) TBP intermediates in the initiating step of the s-.z transition was discussed 3
'. In a double-helical DNA fragment with alternating d(A-T)n sequence, generation of P(V) intermediates can lead to activated dpA or dpT structures. In analogy with alternating d(G-C)n sequences it is clear that the B~Z transition exclusively infers symmetrical generation of activated dpT fragments in both strands of the DNA structure. This is based on the calculated high anti-syn activatien energy in case of dT residues, whereas a low anti-syn energy harrier was found for dA residues 36 • Considering the proposed distinct mechanism of the B-.z transition in the alternating d(A-T)n sequence, this implies that the T base within the rotating dT residue remains in anti position throughout the entire process, whereas the complementary A base adopts the syn
conformation in order to rejoin the hydrogen-bonds in the Z-DNA structure.
V.3 MNDO eaZauZations.
In order to rationalize the impact of specific methylation of the adenine base on the anti-syn equilibrium, the energy profile of the stepwise rotatien around the glycosidic C(1 1 )-N bond was calculated for several methylated structures (c.f. Chapter III, Appendix). The adenine derivatives were selected on the basis of their possible biologica! significance. The modified neglect of differentlal overlap (MNDO) method 37
' 38 was used and the base parameters and dihedral angles determining the ring pucker were considered as variables in the optimization process. No ring pucker
100

was observed during the rotation of the base residue around the glycosidic bond in the tetrahydrofuryl system. The results of the computations on the various model system. The are presented in Fig. V.2 - 7 and in Table V.1 (see V.5~
Discussion). Table V.1 gives energy parameters which are of importance in the B-.z isomerization process. The energy profiles for the tetrahydrofuryl-1-thymine and tetrahydrofuryl-9-adenine systems are given in Chapter III.3.
3' 5'
~.
Fig. V.l AccessibiLity of the various substitution sites
in B-DNA with aLternating dA-dT sequences.
The seLected phosphate groups which initiate the B-.z
transition are indicated by an asterisk. In B-DNA~ the methyL
groups of m6A and m7A are Loaated in the major groove~ whereas that of m8A is in a stericaLLy hindered position.
The methyL group of m3A is Located in the minor groove in
B-DNA or in a shieLded environment in oase of Z~DNA.
In the isomerie Z-DNA structure~ that of m6A is situated
.in the groove~ whereas those of m7A and m8A are on the
periphery of the moLecuLe.
101
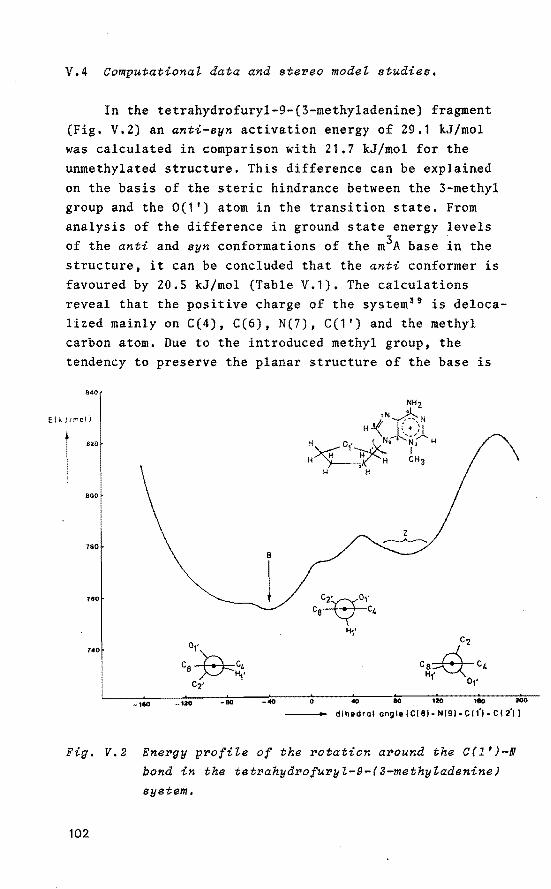
V.4 Computational data and stereo model studies.
In the tetrahydrofuryl-9-(3-methyladenine) fragment (Fig. V.2) an anti-syn activation energy of 29.1 kJ/mol was calculated in comparison with 21.7 kJ/mol for the unmethylated structure. This difference can be explained on the basis of the steric bindrance between the 3-methyl group and the 0(1') atom in the transition state. From analysis of the difference in ground state energy levels of the anti and syn conformations of the m3A base in the structure, it can be concluded that the anti conformer is favoured by 20.5 kJ/mol (Table V.1). The calculations reveal that the positive charge of the system 39 is delocalized mainly on C(4), C(6), N(7), C(1') and the methyl carbon atom. Due to the introduced methyl group, the tendency to preserve the planar structure of the base is
840
ElkJI!roli
820
NHz
rN~N H -'< _{ ;,,)_
H~· N,• N3 H 1 • I H H H H CH ,. 3
H H
800
160 B
760 l 740 o,·
Ca~c4 Hf C2'
-160 -120 -80 -40 0 40 80 120 1eG 200
di he drol ongle! Cl8l- Nl91- C 11'1- C I 2'11
Fig. V.2 Energy profile of the rotation around the C(l')-N bond in the tetrahydrofuryl-9-(3-methyladenine)
eyetem.
102

lost. An out-of-plane bending of the CH3-N fragment was calculated in order to diminish the strong steric interaction between the 3-methyl group of the base in the syn position and the tetrahydrofuryl ring. From incorporation of m3A residues in a double-stranded B-DNA model a strong steric interaction was noticed between the 3-methyl group, located in the minor groove, and the adjacent H(l') atom within the dm3A structure. This interaction can only be diminished upon adjustment of the m3A base within the anti domain with concomitant distortien of the helix backbone. In addition, Cerutti 39 has suggested that the presence of 3-alkylpurines in DNA would induce large deviations from the B-DNA structure. The calculations show that the syn conformation of the m3A base, required to adopt the Z-DNA structure, is highly disfavoured. Therefore, it seems that dm3A residues incorporated in a double-stranded B-DNA helix with alternating d(A-T)n sequence cause local distortions in the helix backbene and preclude isomerization into its left-handed Z-DNA conformer.
In the tetrahydrofuryl-9-(N6-methyladenine) system (Fig. V.3) an anti-syn activatien energy of 22.4 kJ/mol was calculated. In addition, a higher stability of the syn
conformer with respect to the anti conformer was computed (-10.2 kJ/mol). Despite the calculated preferenee for the m6A base to adopt the syn conformation, NMR spectroscopie data on N(6)-methylated ApA structures in aqueous solution reveal an anti conformation of the base residues 35
• Since specific solvation effects were not taken into account in the calculations, the observed discrepancy may only be an apparent one. Specific cationic complexation of N(7) of the adenine base, which would considerably stabilize the syn
conformer in high-salt solution22 , is eliminated due to the sterical hindrance imposed by the adjacent N(6)-methyl group. Therefore, it seems that a low probability exists for an alternating d(m6A-T)n sequence to adopt a Z-DNA duplex in high-salt solution, From incorporation of m6A structures in a double-stranded B-DNA model it is evident that the hydrogen atom not involved in interaction with the compie-
103

mentary thymine base will be substituted by a methyl group, which is then situated in the major groove of the molecule. Stereo model studies suggest a hydrophobic interaction between the methyl group and the adjacent base residue X (X=purine or pyrimidine) within a 3'-(Xpm6A) fragment in the helix. Evaluation 'of the thermodynamic results of OZsthoorn et aZ, 40 indeed shows that the introduetion of methyl in the N(6) position of adenine promotes intramalecular stacking by a relative decrease of the entropy of unstacking. N(6)-methylation drastically increases the hydrophobicity of the major groove which, in turn, ought to affect regulatory protein binding. The restrietion enzymes Dpnii and Mboi seem to be inhibited by the methylation of adenine on N(6), whereas cleavage by Dpni requires the presence of m6A in GATC sites 41 • Cleavage by the enzymes Sau JA~ Fnu EI and Pfai was found to be insensitive to the presence of m6A in GATC recognition sites 42 • As a result of the steric constraints, imposed by the adjacent
100
E!kJ/mol)
B
60 1
40
20
OL---~~--~~---=-----~------~----~---~=----~~---~~--~ •180 -120 -80 -40 0 40 80 120 180 200 - dihedral angle [CI81-NI9J-C(fl-C(2') I
Fig. V.J Energy profile of the rotation around the C(1 1 )-N
bond in the tetrahydrofuvyl-9-(N6-methyZadenine)
system.
104

base pairs in the stacked structure, the rotation of the exocyclic 6-CH3NH group around the C(6)-N(6) bond in dm6A is restricted. Therefore, the energy profile of the rotatien around the C(6)-N(6) bond was calculated as a function of the dihedral angle C(*)-N(6)-C(6)-C(S) in the tetrahydrofuryl-9-(N6-methyladenine) system. The base residue was fixed in the anti position. The allowed domain with respect to the B-DNA structure is indicated in Fig. V.4 and, according to the calculations, reflects an essential sp 2-like hybridi~ation of N(6) with C(*)-N(6)-C(6) angle of 126.8° and H-N(6)-C(6) angle of 116.8°. The N(6) hydrogen atom is located in the base plane at 2.49 Ä from N(1), while the methyl group is free to bend out-of-plane (maximum angle of deviation with respect to B-DNA structures is 45°), since only negligible change in the total energy of the system was computed during this process. This sp 2-like
160
E!kJimol)
140
120
100
80
-120 -•o -40 0 40 80 120 180 200
--- dihedrol angle ICI•l· N!6l-CI6l·C!Sll
Fig. V.4 Ene~gy p~ofile of the ~otation a~ound the C(6)-N(6)
bond in the tet~ahyd~ofu~yl-9-(N6-methyladenine) st~uctu~e.
105

hybridization is considerably stabilized by n-bond interaction between the lone electron pair on N(6) and the aromatic ring system. Illustrative of the high contribution of resonance structures with C(6)-N(6) double-bond character is the calculated decrease in C(6)-N(6) bond length from 1.375 Ä to 1.357 Ä upon rotation of the exocyclic 6-CH3NH group from ± 45° into the base plane (0°). The computational results imply that the methyl group, located in the major groove of B-DNA, can easily adopt the required specific orientation in order to be recognized by m6A sensitive restrietion enzymes. During this process the position of the hydrogen atom, involved in H-bond interaction with the 0(4) atom of thymine, is not changed.
For isolated dm6A fragments, the rotation around the C(6)-N(6) bond is not restricted and may result in inversion of the N(6) atom. The energy profile of the rotation around the C(6)-N(6) bond reveals two maxima, each corresponding with a tetrabedral configuration of N(6) with C(6)-N(6) bondlengthof 1.408 Ä. Apparently, sp 2-like hybridization of N(6) is favoured by 24.2 kJ/mol over sp3-like hybridization due to stahilizing n-bond interaction. The transition state results from enlargement of the dihedral angle C(*)-N(6)-C(6)-C(5) above 45° (B-DNA domain) which forces the N(6)-hydrogen atom out of the base plane, simultaneously reducing the C(6)-N(6) double-bond character. These unique properties inherent in the dm6A system were investigated in detail by selecting the 4-N-methylaminopyrimidine system which considerably reduces the computation time. The iso-total-energy contour map was calculated from fixed combinations of the dihedral angles C(*)-N(4)C(4)-C(5) and H(•)-N(4)-C(4)-C(5). The other parameters determining the geometry were kept variabie in the optimization process. Out of two possible inversions, only the one relevant with respect to Watson-Cviak type base pairing was studied (Fig. V,5), The energy plot reveals two minima (180°,~45°), each corresponding with a tetrabedral configuration of N(4), separated by a planar transition state (175°,0°) only 2.1 kJ/mol higher in energy. As this energy
106

80
;;:; u $0 . u . .:! 40 z . u - 20
"' c 0
. ö 0
" .. .s:: ."
-20
-40
150 170 180 210 230
di he d r o I a ng Ie l H t•l -NI 41 - C (4 I- ( 5ll
Fig. V.S Iso-totaZ-enevgy contouv map of the inversion in
the 4-N-methyZaminopyrimidine system. AZZ para
meteva e~aept both dihedvaZ angZes ~ere considered as vaviables in the optimization process.
difference is small in comparison with the accuracy of the MNDO method 37 it seems reasonable to assume that the inversion of the N(4) atom requires a low activatien energy due to stahilizing n-bond interaction in the system. The pathway leading to inversion of N(4) is represented by the continuous curve in the plot. (Note that for the 4-aminopyrimidine system an activatien energy of 8.1 kJ/mol was calculated for pyramidal inversion of N(4). Apparently, in this system, the absence of the electron-donating methyl
107

group results in diminished N(4)-C(4) ~-bond interaction in the planar transition state). The dashed curve in Fig. V.S represents the reaction pathway for inversion, relevant to Watson-Criak type base pairing in the dm6A system, and strongly stresses the similarity between both systems. The presence of the imidazole fragment in the dm6A structure results in a 0.03 Ä shorter C(6)-N(6) bond length in comparison with the pyrimidine derivative due to increased ~-bond interaction. Recent 1H dynamic NMR spectroscopie studies on simple acyclic trialkylamines show clear evidence for pyramidal inversion at nitrogen 43 ,
Calculations on the tetrahydrofuryl-9-(7-methyladenine) model system (Fig. V.6) reveal that the positive charge 39
is delocalized mainly on C(1 '), C(S), N(7), N(9) and the methyl carbon atom. An anti-syn activatien energy of 25.3 kJ/mol was calculated, whereas a stabilization of the syn
conformer occurs (-13.0 kJ/mol, Table V.1). Pullman et al. 44
calculated the pot~ntials on the surface envelopes of a poly dA.poly dT B-DNA structure and showed that the most negative potential, implicating the highest reactivity toward electrophiles, is located in the minor groove of the molecule. Their results concerning the states of the principal reactive sites on the adenine base, in relation to its potential and steric accessibility, suggest that the accessibility decreases in the order N(7)>C(8)>N(3)> N(6), while the potential decreases in the order N(3)>N(7)> C(8)>N(6). Hence, the N(7) atom in adenine is highly susceptible to alkylation.
Incorporation of m7A base residues in a B-DNA model revealed participation in normal Watson-Criak type base pairing and no local distartion of the DNA duplex structure, since the methyl group is located in the major groove. Model studies suggest a hydrophobic interaction between the methyl group and the adjacent X base (X=purine or pyrimidine) within a 3'-(m7ApX) fragment in the helix. Free rotation of the methyl group will be severely restricted, due to van der Waals interactions with the adjacent 6-NH2 group. In a Z-DNA structure, the phosphate groups are closer to
108

(kJ/mol)
800
780
760
740
720
~oL_ ____ ~,n=-----~,2~o-----~"~---~~=---~o~--~~~---. •• ~--~.2~o--~.~H.---,200~-----dihedral angle ICI8l-NI9l-CI1'l-CI2'll
Fig. V.6 EnePgy pPofile of the Potation aPound the C(l')-N
bond in the tetPahydPo[uPyl-9-(7-methyZadenine)
system.
each other on two sides of the minor groove than in B-DNA. There
fore, the average distance between phosphate groups in a B-DNA structure with alternating d(m7A-T)n sequence diminishes during conversion into Z-DNA. Because of the positively charged dm7A structures, an enhancement of the B-.Z transition would be expected due to a neutralizing effect on adjacent phosphate groups. However, no data are available to support this suggestion.
As a model for C(8) alkylation the tetrahydrofuryl-9-(8-methyladenine) system was selected. The calculated high anti-syn energy harrier in the system (46.5 kJ/mol, Fig. V.7) results from steric bindrance between the 8-methyl group and the ciosest C(2') hydragen atom in the eclipsed conformation. Rotation of the m8A base residue around the glycosidic bond toward a syn position diminishes the steric interaction as is reflected in the calculated high energy gain of -39.5 kJ/mol. In accord with this result, studies
109

120
NHz
H~C.~N~~ NJ.l .. A H~O~·~• N, H
H'~'H H 'H
, .. EIKJimol}
100
80
60
dihedral angle [CI81-NI9) -ctfl· Cl2'1 J
Fig. V.7 Energy profile of the rotation around the C(l')-N
bond in the tetrahydrofuryl-9-(8-methyladenine) system,
on mononucleotides have shown that substitution of H(S) by a bulky substituent restricts the adenine base to the syn range 16 • The calculations and the available experimental data suggest that m8A base residues show a high preferenee for the syn conformation. As a consequence, a double-stranded alternating d(m8A-T)n polymer has the ability to adopt a Z-DNA structure. The fact that the double-stranded alternating polymer d(T-G)n.d(C-A)n adopts a left-handed helix upon partial modification of C(8) 27 supports this conclusion. In a Z-DNA model it can be shown that the C(S) position of adenine is located in a readily accessible position on the periphery of the molecule. In the isomerie B form however, the C(S) hydrogen atom is in close contact with the helix backbone. Within a 3'-(m8ApX) fragment (X=purine or pyrimidine), introduetion of the methyl group results in strong
110
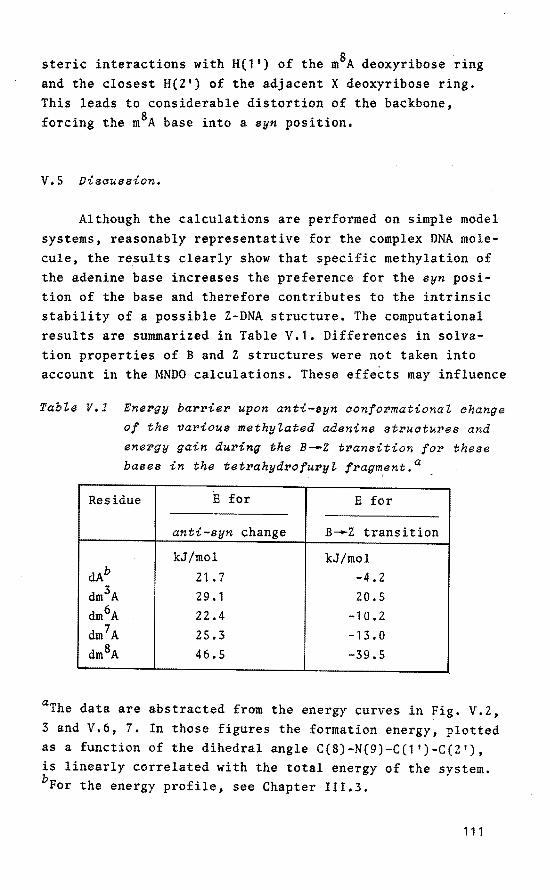
steric interactions with H(l') of the m8A deoxyribose ring and the ciosest H(2') of the adjacent X deoxyribose ring. This leads to considerable distartion of the backbone, forcing the m8A base into a syn position,
V.S Discussion.
Although the calculations are performed on simple model systems, reasonably representative for the complex DNA molecule, the results clearly show that specific methylation of the adenine base increases the preferenee for the syn position of the base and therefore contributes to the intrinsic stability of a possible Z-DNA structure. The computational results are summarized in Table V.1. Differences in solvation properties of B and Z structures were not taken into account in the MNDO calculations. These effeèts may influence
TabZe V,l Ene~gy ba~~ie~ upon anti-syn confo~mationaZ change
of the vavious methyZated adenine st~uctuves and
enevgy gain duving the B-.z t~ansition fo~ these bases in the tetvahyd~ofu~yZ f~agment.a
Residue E for E for
anti-syn change B-.z transition
kJ/mol kJ/mol dAb 21.7 -4.2 dm3A 29.1 20.5
dm6A 22.4 -10.2 dm7A 25.3 -13.0 dm8A 46.5 -39.5
aThe data are abstracted from the energy curves in Fig. V.Z, 3 and V.6, 7. In those figures the formation energy, plotted as a function of the dihedral angle C(8)-N(9)-C(1')-C(2'), is linearly correlated with the total energy of the system. bFor the energy profile, see Chapter III.3.
111

the calculated energy parameters during the B~Z transition for the various methylated structures. Out of the methylated structures stuclied here, it is likely that the alternating d(m8A-T)n duplex can undergo a cooperative transition into a Z-DNA isomer. Potential sites for alkylation in the ds helix are relatively inaccessible to direct alkylation in
vivo. Deoxyribonucleoside residues in the cellular DNA precursor pool are generally more susceptible to methylation. Moreover, methylation of unpaired DNA residues may offer a direct explanation for the chromosome replicatien point specificity of alkylating agents in bacteria~ 5 '~ 6 •
Due to altered conformational preferenee around the glycosidic bond this process may lead to incorrect base pairing and eventually to mutagenesis.
112

Raferences and Notès.
1. P.N. Magee and J.M. Barnes, Advance in Cancer Res., 1968, 10, 3453.
2. B. Singer, M. Kroger and M. Carrano, Biochemistry, 1978, 17, 1246.
3. G.P. Margison and P.J. O'Connor, Biochim. Biophys. Acta, 1973, 331, 349.
4. D.M. Kirtikar and D.A. Goldtwait, Proc. Natl. Acad. Sci. u.s.A., 1974, 71, 2022.
5. T.A. Kunkel, C.W. Shearman and L.A. Loeb, Nature(Lond.), 1981, 291, 349.
6. A.E. Beasley and M. Rasmussen, Aust. J. Chem., 1981, 34, 1107.
7. M.D. Topal, C.A. Hutchison and M.S. Baker, Nature(Lond.), 1982, 298, 863.
8. P.O. Seglen and P.B. Gordon, Proc. Natl. Acad. Sci. u.s.A., 1982, 79, 1889.
9. T.P. Brent, Biochemistry, 1979, 18, 911. 10. M. Meselson, R. Juan and J. Heywood, Annu. Rev. Biochem.,
1972, 41, 447. 11. R. Holliday and J.R. Pugh, Science(Wash. D.C.), 1975,
187, 226. 12. R. Sagerand R. Kitchin, Science(Wash. D.C.), 1975,
189. 426.
13. S. Lacks and B. Greenberg, J. Mol. Biol., 1977, 114, 153. 14. B. Glickman, P. van den Elsen and M. Radman, Mol. Gen.
Genet., 1978, 183, 307, 15. W.J. Bodeli and B. Singer, Biochemistry, 1979, 18, 2860. 16. s.s. Tavale and H.M. Sobell, J. Mol. Biol, 1970, 48, 109. 17. R. Wing, H. Drew, T. Takano, C. Broka, S. Tanaka,
K. Itakura and R.E. Dickerson, Nature(Lond.), 1980, 287, 755.
18. R.D. Wells, T.C. Goodman, W. Hillen, G.T. Horn, R.D. Klein, J.E. Larson, U.R. MUller, S.K. Neuendorf, N. Panayotatos and S.M. Stirdivant, Prog. Nucleic Acids Res. Mol. Biol., 1980, 24, 167.
113

19. W. Zacharias, J.E. Larson, J. Klysik, S.M. Stirdivant, and R.D. Wells, J. Biol. Chem., 1982, 2S7, 2775,
20. J.J.C. van Lier, M.T. Smits and H.M. Buck, Eur. J. Biochem., 1983, 132, 55.
21. Abbreviations for nucleotides follow IUB/CEBJ recommendations, see Eur. J. Biochem., 1970, lS, 203; e.g. dA, 2'-deoxyadenosine; dm3ATP, 2'-deoxy-3-methyladenosinetriphosphate; d(mxA-T), dinucleotide structure with x-methyladenine incorporated, in which the phosphorus is bound to 0(5') of the thymidine residue,
22. A.H.-J. Wang, G.J. Quigley, P.J. Kolpak, J.L. Crawford, J.H. van Boom, G. van derMareland A. Rich, Nature(London), 1979, 282, 680.
23. J.L. Crawford, F.J. Kolpak, A.H.-J. Wang, G.J. Quigley, J.H. van Boom, G. van der Marel and A. Rich, Proc. Natl. Acad. Sci. U.S.A., 1980,77,4106.
24. H. Drew, T. Takano, S. Tanaka, K. Itakura and R.E. Dickerson, Nature(Lond.), 1980, 286, 567.
25. A.H.-J. Wang, G.J. Quigley, P.J. Kolpak, G. van der Mare!, J.H. van Boom and A. Rich, Science(Wash. D.C.), 1981, 211, 171.
26. A.H.-J. Wang, S. Fujii, H. van Boom and A. Rich, Cold Spring Harbor Symp. Quant. Biol., 1982, 47, 5,
27. R.D. Wells, J.J, Miglietta, J. Klysik, J.E. Larson, S.M. Stirdivant and W. Zacharia~, J. Biol. Chem, 1982, 2f57, 10166.
28. H. Shindo, R.T. Simpsen and J.S. Cohen, J. Biel. Chem., 1979, 2f54, 8125.
29. J. Kypr, M. Vorlicková, M. Budesinsky and v. Sklenar, Biochim. Biophys. Res. Commun., 1981, 99, 1257.
30. M.A. Viswamitra, z. Shakked, P.G. Jones, G.M. Sheldrick, S.A. Salisbury and 0. Kennard, Biopolymers, 1982, 21,
513. 31. M. Sundaralingam, Biopolymers, 1969, 7, 821. 32. A. Klug, A. Jack, M. Viswamitra, 0. Kennard, Z. Shakked
and T. Steitz, J. Mol. Biol., 1979, 131, 669, 33. F.H.C. Crick and A. Klug, Nature(Lond.), 1975, 2SS, 530, 34. K.J. Millerand J.F. Pycior, Biopolymers, 1979, 18, 2683.
114

35. C. Altona, Reel. Trav. Chim. Pays-Bas, 1982, 101, 413. 36. J.J.C. van Lier, L.H. Koole and H.M. Buck, Reel. Trav.
Chim. Pays-Bas, 1983, 102, 148. 37. M.J.S. Dewar and W.J. Thiel, J. Am Chem. Soc., 1977,
99, 4899. 38. W. Thiel, Program 353, Quantum Chemistry Program
Exchange 11, Indiana University, 1978. 39, P.A. Cerutti, "Molecular Mechanisms for Repair of DNA",
P.C. Hanawalt and R.B. Setlow, eds., Plenum Press, New York, 1975, 3.
40. C.S.M. Olsthoorn, C.A.G. Haasnoot and C. Altona, Eur. J. Biochem., 1980,106,85,
41. G.T. Vovis and S. Lacks, J. Mol. Biol., 1977, 115, 525. 42. R.E. Streeck, Gene, 1980, 12, 267. 43. C.H. Bushweller, S.H. Fleischman, G.L.Grady, P. McGoff,
C.D. Rithner, M.R. Whalon, J.G. Brennan, R.P. Marcantonio and R.P. Domingue, J. Am. Chem. Soc., 1982, 104,
6224. 44. B. Pullman, R. Lavery and A. Pullman, Eur. J. Biochem,
1982, 124, 229.
45. E. Cerda-Olmeda, P.C. Hanawait and N. Guerola, J. Molec. Biol., 1968, SJ, 705.
46. T.A. Hince and S. Neale, Mutat. Res., 1974, 24, 383.
115

CHAPTER VI
Te"traoxaspirophosphoranes -wi"th a six
IDeiDbered ring as IDodel coiDpounds for "the
hydrolysis of ribonucleic acids by RNase A
VI.l Introduation.
In the previous Chapters III-V, a model description was offered for the B -z transition in (methylated) DNA with alternating d(purine-pyrimidine) sequences. The initiating step in the isomerization process was supposed to involve a P(IV)-P(V) TBP activatien of the 5'-dp(pyrimidine) residues. The observed selectivity in the transition could be explained on the basis of the well-known features of P(V) TBP structures (c.f, Chapter I) and on the difference in anti-syn conformational adjustment of the various (methylated) base residues. Although, for DNA structures, the occurrence of P(V) TBP intermediates is still a point of controversy, recent high-resolution 1H NMR experiments of Kooteet at. 1
, performed on DNA representative P(V) organophosphorus compounds, point to·the predicted selectivity.
A classica! example of a biochemica! process, in which the role of P(V) TBP intermediate structures has been accepted, is the hydralysis of ribonucleic acids by RNase A. Here, the proximity of the 2'-0H group on the ribose with regard to the phosphate group, produces a substantial rate acceleration of the first step in the reaction. This step is known 2 to involve a nucleophilic attack by the 2'-0H group of the ribose ring on the vicinal phosphate group, leading to a 2' ,3'-cyclic phosphate via a penta-coordinated intermediate (a.f. Chapter I). The formation of the transition state is considerably facilitated largely due to the decrease in translational entropy of the reacting groups 3 •
116

GasteZijne et aZ.~,s studied the reaetion of several stable eyelie penta-eoordinated oxyphosphoranes with FS03H in
1 13 31 cn 2c1 2 by means of low-temperature H, C and P NMR teehniques and observed dynamie equilibria between penta-eoordinated oxyphosphoranes and their isomerie tetra-eoordinated enol phosphonium ions. From the observed large negative entropy of a~tivation inherent in the equilibria it was concluded that the rigidity introdueed by a five-membered ring faeilitates ring elosure of the phosphonium ion. In addition, van Aken et aZ. 6 '
7 performed similar experiments, suggesting that intramoleeular phosphorylation of the 2'-0H group in RNA is strongly faeilitated by the presenee of the ribose ring. In this Chapter attention will be foeused on the eatalytie role of the base in the aetive site of RNase A. The experiments were carried out with model eompounds. For this purpose tetraoxaspirophosphoranes were seleeted in order to obtain experimental evidenee for a base-eatalyzed ring opening respeetively ring elosure proeess. The most remarkable property of tetraoxaspirophosphoranes with a P-H bond and two five-membered rings is their ability to give rise to a tautomerie equilibrium between the tri- and penta-eoordinated form. The first example of this P(III)-P(V) equilibrium was offered by Burgada et aZ. 8 (Fig. VI.1, a). Laterit was demonstrated 9
-13 that the phenomenon was quite general and
could be represented by Fig. VI.1, b. The equilibrium
--
-- {
0'-. R' XH R' P-o/X/ b
x/
X=O, NH, NR; R'=stabiZizing factors (eubetituente, unsaturation)
Fig. VI.l
117

is controlled by electronic factors, steric factors, symmetry properties of the tri- and penta-coordinated molecules or external factors such as temperature and basicity of the solvent 1 ~. An example of symmetry properties and electronic factors is given in Fig. VI.2. Upon heating the asymmetrical tetraoxaspirophosphorane 1, a mixture of the symmetrical compounds 2 and J is formed (symbiotic effect) 13 , whereas, due to the electron-donating character of the pinacol ring in 1 (i.e. weak solvation of the OH group after ring opening), exclusive generation of 4 proceeds via ring opening of
H
+ Co,~,.........oJ o/ 'o
J
--4
[
OH 2 +
OH
Fig. VI.2
the less stabie glycol fragment. Addition of free 2,3-dimethylbutanediol-2,3 (pinacol) to 1 results in conversion to the more stabie compound 2 with simultaneous release of glycol. This process, proceeding via the reactive isomer 4, can be regarded as a transesterification. Spirophosphoranes with a P-H bond are normally obtained by the reaction of a-hydroxyacids or alkyl tartrates with tri-coordinated phosphorus compounds 11
• Oxidation of spirophosphoranes with a P-H bond leads to hydroxyphosphoranes which are in equilibrium with their corresponding phosphates. The first stabie hydroxy-
118

©r"'r~~M~ OH
HCl ©(+o:g o1 'o CH2Cl2 0 I \0
15
~2 u 8
©r"'P:©J ©CvoJQJ o1 'o 0 I \0
8 7 OH Fig. VI.3
phosphorane 8 was prepared by Rami~ez et al. 15 by the action of dry hydrogen chloride on the silyloxyphosphorane 15 (Fig.
VI.3). In solution, 8 is in equilibrium with the phosphate 7.
This process is slow on the NMR time scale below 10 °C in acetonitrile. With diazomethane, 8 gives the methoxyphosphorane B. Similar hydroxyphosphoranes have been obtained from POC1 3 and hydroxyacids in the presence of pyridine or from the
PH-phosphorane by oxidation with DMSO in DMF 16 ~ The latter structures were isolated as the salts with DMF or with triethylamine.
Introduetion of a six-membered ring in tetraoxaspirophosphoranes with a P-H bond results in a considerable decrease
in stability. Only one example of this kind of structure has been reported and confirmed by 31 P NMR spectroscopy (Fig. VI.4). However, despite the stahilizing carbonyl group and
""·J~~h P-O
0/1
\_l Fig. VI.4
both phenyl substituents, this compound could not be isolated17. In contrast, spirophosphoranes of the sametype but with a P-OR bond are relatively stable 18 - 20 and show a high
preferenee for an apical-equatorial alignment of the six-mem-
119

bered ring 21• Por tetraoxaspirophosphoranes with a six
membered ring it is interesting to investigate a possible P(III)-P(V) equilibrium. Therefore, the ring-ciosure and ring-opening behaviour of the compounds depicted in Fig. VI.S was stuclied with the aid of NMR spectroscopie techniques. The obtained data are rationalized on basis of Pearson's
HASB theory 22 (hard acid soft base) and ring orientation
Fig. VI.6
effects 23 • In the formation of compound 9 a base-aatalyzed
proton transfer from the hydroxyl oxygen to the phosphorus atom was observed during ring closure. This process mimics the possible role of the base in the active site of RNase A in selective hydralysis of ribonucleic acids 2 ~- 26 •
VI.Z NMR speatrosaopia study of the formation of tetra
o~aspirophosphoranes aontainin~ a si~-membered ring
and a P-H bond.
In order toprepare 5-hydrido-2,3-benzo-1,4,6,10-tetraoxa-5-phosphaspiro[4,5)dec-2-ene (compound 9) the synthetic routes A and B were used (Fig. VI.6). By reacting equimolar amounts of 1,2-dihydroxybenzene 12 and 2-chloro-1,3,2-dioxaphosphorinane 13 in the presence of a base, the corresponding P(V) precursor 14 is obtained, while analogous reaction of propanediol-1 ,3 1'1 with 2-chloro-4,5-benzo-1,3,2--dioxaphospholane 18 leads to the P(V) precursor 16. The advantage of this procedure is that, upon generation of the isomerie precursors 14 and 15, ciosure of the five-respectively six-membered ring can be determined independently with
120
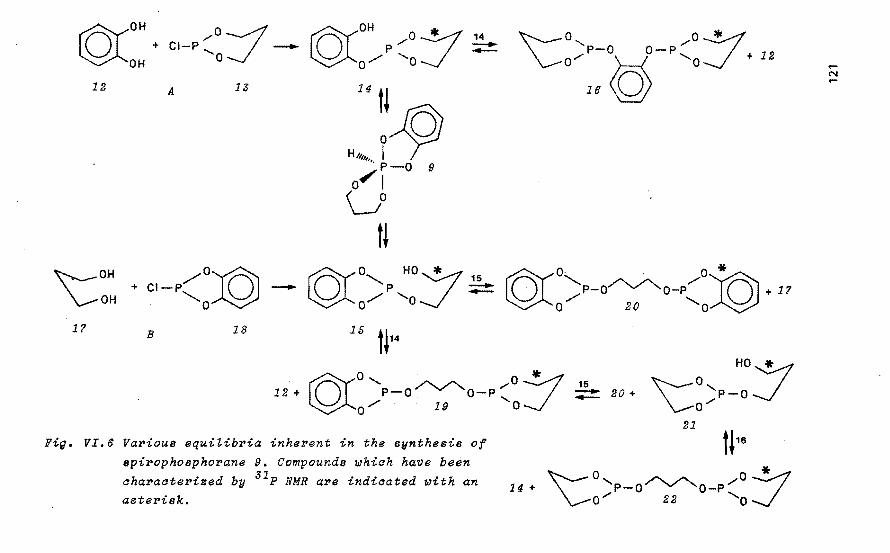
rAYOH +
~OH 12
~OH
\..-oH 1? B
,.....oJ CI-P -'a
A
18
rAYOH p,.....O~ ~o/ 'o-/
14 --
Fig. VI.6 Various equilibria inherent in the synthesis of spirophosphorane 9. Compounds whiah have been aharaaterized by 31 P NMR are indicated with an asterisk.
14 +
15 --
-N
20 +

the aid of 31 P NMR spectroscopy at various temperatures. Surprisingly both synthetic methods gave rise to products with identical 31 P NMR spectra and nearly similar product distribution, indicative of a common intermediate structure. A typical 31 P NMR spectrum, obtained via route A, is given in Fig. VI.7. Besides eight resonances in the P(III) domain (ö 130.0; 129.9; 129.7; 127.8; 127.5, double; 124.0 and 123.7), no phosphates could be found. Moreover, a broad multiplet (ö 31 P=-21.8 ppm) was detected with extremely low intensity, which can be attributed to the symmetrical 5-hydrido-2,3,7,8-benzo-1,4,6,9-tetraoxa-5-phosphaspiro[4,4]non-2,7-diene (28, Table VI.l). In the absence of spirophosphorane 9, bimolecular reaction of 14 (route A) can only lead to formation of the diphosphite 16, while bimolecular reaction of 15 (route B) results in ex~lusive generation of diphosphite 20 (Fig. VI.6). Consiclering the obtained product distribution in both synthetic routes, it can therefore be concluded that the spirophosphorane 9 must be involved as an intermediate in the isomerization 14~15.
22
19 21
19
20 15 16
14
100Hz /cm
Fi~. VI.? 36.43 MHz 31 P NMR speatrum obtained via route A.
122
The aorrespondin~ struatures are ~iven in TabZe
VI.1 and in Fi~. VI.6,

By studying the behaviour of 5-hydrido-2,3-benzo-1,4,6,9--tetraoxa-5-phosphaspiro[4,4l-non-2-ene, Munos et al. 21
established the formation of several diphosphites due to subsequent reactions. However, not all the proposed structures in their reaction scheme were characterized. In order to investigate the possible formation of diphosphites, the model compounds 25-27 were prepared and characterized by means of 1H, 13e and 31 p NMR spectroscopy (Table VI.1), The close resemblance in 31 P chemica! shifts of compounds 25-27 to those observed for the reaction mixture confirmed that these structural units were present in the products. Therefore compounds 16 and 19-24 were synthesized. The corresponding NMR parameters are listed in Table VI.l. Subsequent addition of these various compounds to the reaction mixture established the presence of compounds 16 and 19-22 by comparative 31 P NMR measurements. To elucidate the two remaining resonances in the 31 P NMR spectrum, 0.5 mmol of 12 was dissolved in 0,4 ml of ene13 in an NMR sample tube. The salution was cooled at -20 °e while equimolar amounts of base and 13 were quickly added. This resulted in instantaneous formation of two products (ö 123.7 main signa!; 127.5) which can be ascribed to the equilibrium 14::.;!!::15. This equilibrium is in fact an example of a fast intramolecular transesterification process. eooling of the salution to -50 °e resulted in a slight shift of the equilibrium in favour of compound 14, but even at this temperature no 31 P resonance indicative of the intermediate 9
could be detected. The preferential formation of 14 at low temperature can be rationalized on basis of its more rigid structure in comparison with compound 15. Addition of a slight excess of base to the reaction mixture immediately results in enhanced conversion o'f 14 into 15. This observation leads to the conclusion that the interconversion 14~15, presumably proceeding via the intermediate 9, is a baae-aatalysed process. Warming of the solution to room temperature leads to gradual appearance of the products as depicted in Fig. VI.6. Once the more stable diphosphites 16, 19, 20 and 22 are formed, the original 14~15 equilibrium condition cannot be obtained any more by cooling of the reaction mixture. The
123
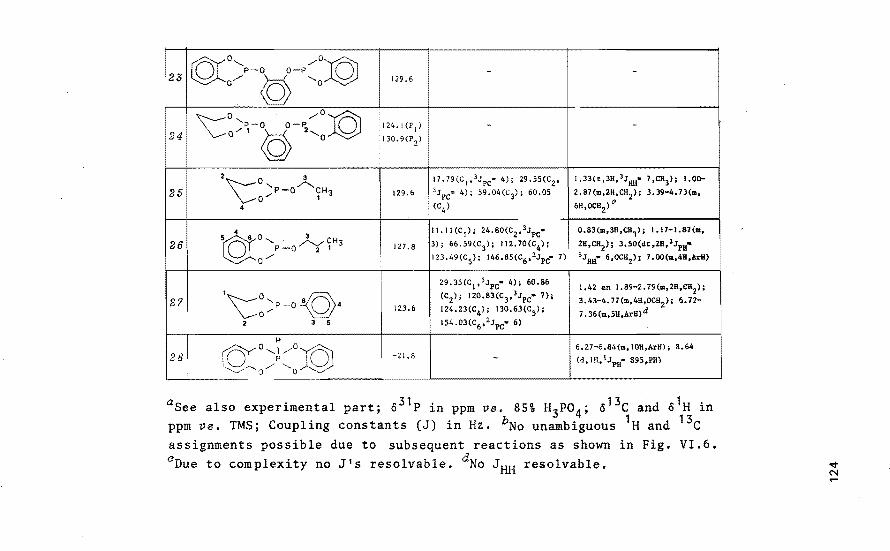
123 ~o, /o~ P-O O-P 0 - -c/ ~ 'o 129.6
'\::0 /0:© ,~,-~-~, 0 124, 1 (P 1) -24 0 0
130.9(P2)
2\::0 3 17.79(C1,l.;pc= 4); 29.55(C2 , 1.33(t,3H,3JHH• 7,CH3); I.Oo-
2S 'P-0./'--.CH 129.6 lJpc= 4); S9.04(c3); 60,05 2,87(m,2~,cH2 ); 3.39-4.73(m, 0_... I 3
4 (C4) 6H,OCH2)
4 0 3
li,II(C 1); 24.80(C2 ,3Jpc= 0.83(m,3H,CH1); 1.17-1,81{m,
26 5©! '- ~CH3 127.8 3); 66.59(C3); 112,70(C
4); 2H,CH2); 3,50(dt,2H, 3JPH• 0 P-O 2 1
0/ 123,49(C5); 146,85(C6 , 2Jpc• 7) i 3Jau· 6,oca2); 7.00(m,4H,ArH)
29.35(C1,l.;pc• 4); 60.86 1,42 en 1.89-2.79(m,2H,CH2);
27 1
'\:::~P-O \94 (C2); 120.83(c3,3.;pc• 7); 3,43-4,77(m,4H,OCH2); 6.72-
123.6 124.23(C4); 130.63(C
5);
7 ,36(m,5H,ArH)d 2 3 5 154.03(c6 , 2JPc· 6)
H
~o,l/oif)i 6.Z7-6.84(m,IOH,ArH); 8,64
28 /",~J· -21.8 - (d, IH, 1JPH= 895,PH)
') 0 ..//
aSee also experimental part; ö31 P in ppm va. 85% H3Po4 ; ö13c and ö1H in ppm vs. TMS; Coupling constants (J) in Hz. bNo unambiguous 1H and 13c assignments possible due to subsequent reactions as shown in Fig. VI.6. aDue to complexity no J's resolvable. dNo JHH resolvable.

liO
14
15
16
I
19
20
21
22
TabZe VI.l NMR speatPoscopia data (CDCZ 3, 2? °CJ of the
compounds shown in Fig. VI.6 and reZated model compounds.a
compound 63! p ël3c &lH
©(OH __.0
o/P'oJ 123.7 b -
©rO HOJ o:P 'o 127.5 b -
1\::0, ,oJ 124.0 29.25(C 1,2Jpç• 3}i 60.93(C2); 0,93-2.84(m,4H,CH2
) i 3.26-4.93 o".P-g-P'o
2 3 0 112.70(C3); 124.70(C
4); (m,SH,OCH2 l i 6.68-7.12(m,4H,
14S.IJ(c5l ArH) 0
5
©lo, ~ o-:J -P-O O-P"' 127.8(P
1)
b o/ 1 2 'o 129.9(P
2)
4 3
5 o, 2 /0~ 32.35(C 1); 60.80(C
2); 112.94 1.58(m,2H, 3JHH• 6,CH
2); 3.53
©t P-o"'Y'o-P 0 127 .s (C
3); 123.62(C
4); 146.68(c
5, (dt,4H, 3JPH•3JHH• 6,0CH2);
2J = 9) 6.71-7 .17(m,8H,Arl!) o/ 'o PC
'\:o,P_::; b -129.7
o'
I 0 4 0-:/ 1
29.5Hc 1,JJPc· 6); 33.76(c2l; l.l0-2.83(m,6H,CH2); 3.46-
'\: 0~P-o~o-P~0 130.0 60.19(C
3); 60.66(C4 ) 4.80(m,l2H,OCH
2) c
3
1.0 N

high percentage of compound 22 (which contains P-0-alkyl bonds) in the reaction mixture can be rationalized on basis of the higher stability of the P-0-alkyl bond in comparison with the P-0-aryl bond. Formation of a smal! quantity of compound 28 due to the equilibrium 12 + 20~28 + 15 was confirmed by 31 P NMR measurements but is not presented in Fig. VI.6. The 31 P chemica! shift of compound 14 was also determined via addition of 12 to a salution of compound 18, This leads to gradual appearance of 14 due to the equilibrium 12 + 18~14 + 14 and eventually results in formation of the other products, vide supra. In order to investigate the ring ciosure of compound 15, 0.5 mmol of 1? was dissolved in 0.4 ml of ene13 in an NMR sample tube. The salution was caoled at -20 °e and, after addition of equimolar amounts of base and 18, two different 31 P chemica! shifts appeared (ö 127,5 main signa!, 123.7) which can be ascribed to the equilibrium 15~14. At -20 °e, a slow conversion of 15 into 14 was observed, which could be accelerated by addition of excess base. Hence, it can be concluded that ciosure of the six-membered ring in 15 is also a base-catalyzed process but proceeds more slowly than ciosure of the five-membered ring in the isomerie structure 14. Apparently, the rigid character of the five-membered ring fragment in 14 results in orientation of the free hydroxyl group in close proximity to the phosphorus atom while this effect is less pronounced in compound 15,
due toa gain in translational entropy23 • Addition of 1? to a salution of compound 20 gives rise to formation of 15 with subsequent conversion into 14 and 19. Purification of the asymmetrical diphosphite 24 (Table VI.l) was not possible due to its low intrinsic stability. Therefore, the presence of this compound during the reaction course could not be detected. Despite the complexity of this system, the overall reaction scheme presented in Fig. VI.6 offers a good explanation for the various obtained products. A 31 P chemica! shift indicative of spirophosphorane 9 could not be detected, even at a temperature of -50 °C. Apparently, 9 is a high energy species in comparison with 14 and 15. Upon replacement of the 1,2-dihydroxybenzene fragment in compound 9 by glycol, one
126

OH oJeMe
Cl-P::oJ -Me
f OH o-y Me Me
Me\:0'- /0-:/'Me Me4{ +
OH
29
Me
.Me~OH \._.oH
A 80
B 35
M .r Me e4 P
o/ 'o
81 ,.fr
32
31 --
32 --Fig. VI.B VaPious equitibPia inhePent in the synthesis of
spiPophosphoPane 11. Compounds whioh have been
ohaPaotePised by 31 P NMR aPe indioated with an
astePisk.
P-O O-P o.r '-t--1 'o + 29 Me4
33
36

obtains the spirophosphorane 10 which is depicted in Fig. VI.S (vide supl'a), Due to the absence of an aromatic ring fragment, this spirophosphorane is less stable than compound 9 as aresult of diminished delocalization10
• Application of the synthetic routes A and B in a similar way as was described for compound 9 gave rise to identical 31 P NMR spectra. Elucidation of the various 31 P chemica! shifts pointed to complete analogy of this system with the one presented in Fig. VI.6. Therefore, the experimental details are nat given here. Introduetion of a pinacol ring instead of a glycol fragment results in a contribution to the intrinsic stability of the corresponding spi~ophosphorane 11 which is depicted in Fig. VI.S. On basis of the known high stability of closed pinacol rings 13 , the isomerization 32~31 may be restricted. Moreover, starting from the P(V) precursor 32, generation of 5-hydrido-2,2,3,3,8,8-hexamethyl-J ,4,6,10-tetraoxa-S-phosphaspiro[4,5)decane 11 by nucleophilic attack of the free hydroxyl oxygen atom will he retarded as a result of increased basicity of the phosphorus atom. For the reversed isomerization 31~32, orientation effects as well as the high energy content of the intermediate compound 11 are of crucial significance. With respect to the pinacol fragment in 31,
the steric bindrance between the four methyl groups in the eclipsed conformation results in a more favourable trans orientation of the free hydroxyl group thus diminishing the possibility of ring closure. In accordance with these predictions, reaction of equimolar amounts pinacol 29 and 2-chloro-5,5-dimethyl-1,3,2-dioxaphosphorinane 30 in the presence of one equivalent base (route A) gave rise to two resonances in the P(III) domain (o 121.9 main signa!; 121.7) which can be attributed to compounds 31 and ss. No resonances indicative of S7 or 38 could be detected (viz. Experimental). Moreover, smal! amounts of products with ö
31 P -21/+14 ppm
and 1JPH =690Hz were detected, indicative of phosphonates 28 ,
No products with closed five-membered rings were found which excludes generation of the spirophosphorane 11 during the reaction course. The 31 P chemica! shift value of compound 31
was also confirmed via addition of excess 29 to a salution
128

Table VI.2 31 P NMR spectroscopie data (CDCt 3~ 2? °CJ of
the compounds depicted in Fig. VI.B.a
NO compound 03lp
Me
31 -fOH Q::/Me 121. 7 Me4
p /
o/ 'o
Me
32 {O' HO :/'Me 147.7 Me, P, o/ o
Me Me
33 Me\:: a, "0::/Me 121.9 P-0 0 P, o" '-f-1 0
Me,
{o' ..-o} 36 Me, P-0 ~O-P Me, 147,5
0 / Me Me 'o
asee also experimental part.
of compounds 29, 31 and 33. This leads to more intense appearance of compound 31 in the 31 P NMR spectrum due to the equilibrium 29 + 33~31 + 31, Preparatien of 31 at -20 °e in an NMR sample tube leads only to a minor amount of 33. Addition of excess base and variable temperature conditions in the range +30 °e /-50 °e failed to give the isomerie structure 32. Application of route B by reacting equimolar amounts of 2,2-dimethylpropanediol-1,3 34 and 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane JS with one equivalent base resulted in exclusive formation of 32 (main product) and 36.
The corresponding 31 p NMR parameters are listed in Table VI.Z. Therefore, ciosure of the six-membered ring in compound 32 must he ruled out which was also confirmed by lew-temperature 31 P NMR measurements. Addition of excess 34 to a solu-. tion of 32, 34 and 36 resulted in more intense appearance of 32 in the 31 P NMR spectrum due to the equilibrium 34 + 36~ 32 + 32. In conclusion, the synthetic routes A and B are
129
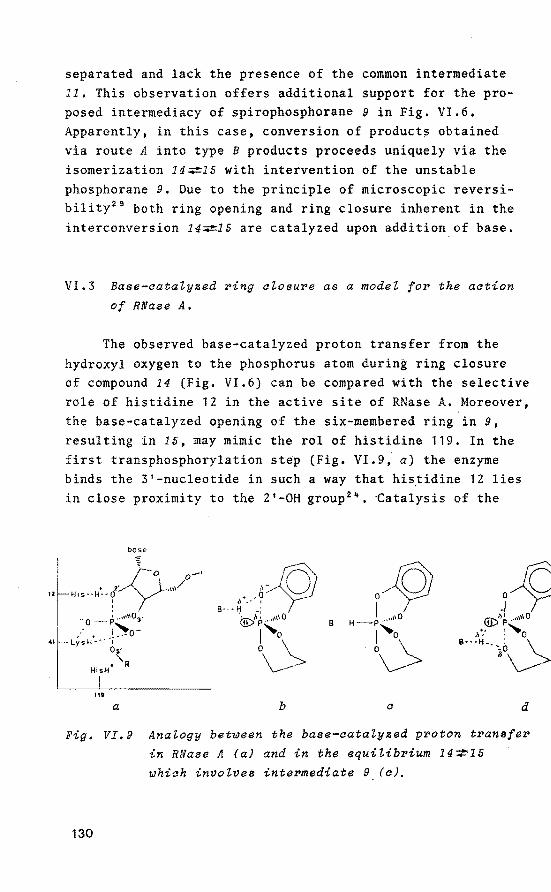
separated and lack the presence of the common intermediate 11, This observation offers additional support for the proposed intermediacy of spirophosphorane 9 in Fig. VI.6. Apparently, in this case, conversion of product~ obtained via route A into type B products proceeds uniquely via the isomerization 14~1E with intervention of the unstable phosphorane 9. Due to the principle of microscopie reversibility29 both ring opening and ring closure inherent in the interconversion 14~1E are catalyzed upon addition of base.
VI.3 Baee-aatalyzed ~ing alosu~e as a model fo~ the action
of RNaee A.
The observed base-catalyzed proton transfer from the hydroxyl oxygen to the phosphorus atom during ring closure of compound 14 (Fig. VI.6) can be compared with the selective role of histidine 12 in the active site of RNase A. Moreover, the base-catalyzed opening of the six-membered ring in 9,
resulting in IE, may mimic the rol of histidine 119. In the first transphosphorylation step {Fig. VI.9, a) the enzyme binds the 3'-nucleotide in such a way that histidine 12 lies in close proximity to the 2'-0H group 24 • ~atalysis of the
B
l!t
a b c d
Fig. VI.9 Analogy bet~een the baee-catalyzed proton t~anefe~
in RNase A (a) and in the equilibrium 14~15
~hiah involves inte~mediate 9 (c).
130

abstraction of the 2'-0H proton by histidine facilitates intramolecular nucleophilic attack on the phosphorus atom and generates the penta-coordinate intermediate. Stahilizatien of the intermediate structure is accomplished by hydrogen bonding from protonated lysine 41 to the anionic oxygen ligands. Subsequent activatien of the leaving group by proton transfer from histidine 119 to the 5'-nucleotide generates the 2',3'-cyclic nucleotide. In the comparative situation (b), the rigid character of the 1,2-dihydroxybenzene fragment, which results in a favoured orientation of the free hydroxyl group toward the phosphorus atom 5 , nicely accounts for the fixed orientation of the 2'-0H group in the 3'-nucleotide unit. The lone pair in b replaces one of the equatorial anionic oxygen ligands while the apically located oxygen in the six-membered ring 21 simulates the departing 5 1 -nucleotide unit 30 , The excess base in solution mimics the catalytic role of histidine 12 and histidine 119 in the active site of the enzyme. Hence, base-catalyzed proton transfer from the oxygen to the phosphorus atom results in the intermediate spirophosphorane 9 (a). In the absence of base, Burgada et al. 1 ~
proposed an analogous intermediate state for the proton transfer. The presence of the six-membered ring reduces the required activatien energy to generate the intermediate c
and therefore has a stahilizing influence. Subsequent base-catalyzed proton transfer from phosphorus to the apically located oxygen atom of the six-membered ring results in structure d.
VI.4 Discussion
The combined NMR data point to spirophosphoranes with a P-H bond and a six-membered ring although, due to their instability, these compounds could not be detected with the aid of low-temperature 31 P NMR techniques. Application of both route A and B in order to prepare the spirophosphorane offers a possibility to establish the formation of a common intermediate. Strong support in favour of the spirophosphorane
131

intermediates comes from comparison of Fig. VI.6 and Fig. VI.8. Due to the absence of the intermediate 11 in Fig. VI.8, product• obtained via route A cannot be converted into phosphites obtained via route B and vice ve~sa, as was established with 31 P NMR techniques at various temperatures. The formation of the intermediate structure 9 (Fig. VI.6) readily
accounts for the observed product distribution. This implies that possible other routes which might explain the interconversion of type A products into type B products can be ruled out. Closure of the six-membered ring in 1& was found to praeeed more slowly than closure of the five-membered ring in 14. This can be explained on the basis of the decrease in translational entropy in five-membered ring fragments if compared with their six-membered analogues. Introduetion of a six-membered ring in spirophosphoranes results in considerable destabilization if compared to analogous structures consisting of two five-membered rings. The latter structures have been detected as stable intermediates in the aleoholysis of phosphoramidites 31
'32 and in transesterification reactions 33
•
The rate of the interconversion 14~1& is enhanced upon addition of excess base and leads to various products through a series of equilibria. However, interconversion of both isomerie P(V) precursors 31 and 32 was not detected which can be rationalized on the basis of a high concomitant activation energy, electronic factors (vide sup~a) and orientation of the free hydroxyl group.
VI.5 E~pe~imental.
Appa~atus.
1H NMR spectra were recorded on a Varian EM-360 A spectrometer. The 13c (22.63 MHz) and 31 P (36.43 MHz) spectra were obtained using a Bruker HX90-R spectrometer equipped with a Digilab-FT-NMR-3 computer. Standards were tetramethylsilane (TMS) for 13c and 1H NMR, and 85% H3Po4 for 31 P NMR. 31 P chemica! shifts downfield from 85% H3Po4 are positive.
132

Melting points were measured with a Mettler FP1.
Reagents and so~vents.
1,2-Dihydroxybenzene 12 was purchased from Merck and recrystallized after azeotropic removal of the water in benzene solution. Propanediol-1,3 17 was purchased from Fluka, dried over K2eo3 and fractionally distilled under reduced pressure before use. 2,3-Dimethylbutanediol-2,3 (pinacol, 29) and 2,2-dimethylpropanediol-1,3 34 were purchased from Aldrich and recrystallized from dry benzene before use. Diethylaniline (BDH Laberatory Reagents), triethylamine (Fluka)and pyridine (Merck) were kept over KOH pellets and fractionally distilled before use. Special care was taken in handling the phosphorus compounds because of their moisture sensitivity. Therefore, all preparations were carried out in dry nitrogen atmosphere. After purification all compounds were stored at -20 °e under nitrogen. enei3 , used for NMR samples, was stored over molsieves (4A). Reduced pressures are given in mm Hg.
Prepara ti ons.
Route A (Fig. VI.6)
To a cooled (0 °e) and stirred solution of 2.76 g (25 mmol) of 12 and 3.76 g (25 mmol) of diethylaniline in 40 ml of dry diethylether, 3.51 g (25 mmol) of 13 dissolved in 10 ml of dry ether was added dropwise over 30 min. The reaction mixture was then stirred for 15 min. and warmed to room temperature (rt) over 30 min. The diethylanilinium salt was filtered off and washed twice with 10 ml portions of dry ether. The clear solution, together with the combined washings, was evaporated in vacuo at rt. The resulting light oil was stored at -20 °e under dry nitrogen. For NMR experiments an aliquot (0.2 ml) was dissolved in 0.3 ml of ene1 3 and transferred to an NMR sample tube. For low-temperature 31 P measurements (-20 °e), SS mg (0.5 mmol) of 12 was dissolved in 0.4 ml of ene1 3 in an NMR sample tube. The solution was
133

kept at -20 °C while 75 mg (0.5 mmol) of diethylanilfne and 70 mg (0.5 mmol) of 13 were quickly added. 31 P NMR {CDC1 3) o 123.7 (main signa!) indicative of 14.
Route B (Fig. VI.6)
To a cooled (0 °C) and stirred solution of 3.81 g (50 mmol) of 17 and 7.52 g (50 mmol) of diethylaniline in 40 ml of dry ether, 8,73 g (50 mmol) of 18 dissolved in 15 ml of dry ether was added dropwise over 45 min. The reaction mixture was then stirred for 15 min. and warmed to rt over 30 min. The salt was filtered off and washed twice with 10 ml portions of dry ether. The clear solution, together with the combined washings, was evaporated in vacuo at rt. The resulting light oil was stored at -20 °C under dry nitrogen. For NMR experiments an aliquot (0.2 ml) was dissolved in 0.3 ml of CDC1 3 and transferred to an NMR sample tube. For low-temperature 31 P NMR measurements (-20 °C), 38 mg (0.5 mmol) of 17 is dissolved in 0.4 ml of CDC1 3 in an NMR sample tube. The solution was kept at -20 °C while 75 mg (0.5 mmol) of diethylaniline and 87 mg (0.5 mmol) of 18 were quickly added. 31 P NMR (CDC1 3) o 127.5 (main signal) indicative of 15.
Prepared according to the procedure described by Lucas et al. 3 ~.
B.p. 64-65 °C/15 mm; yield 54%. 1H NMR (CDC1 3) ö 1.19-3,00 (m, 2H, CH 2); 3.45-5,00(m, 4H, OCH 2), 31 P NMR (CDC1 3) ö 154.0.
~ 2,2'-(o-Phenyldiorey)-1,3,2-dioreaphoephorinane-1',3',2'
-dioreaphoephorinane 18
To a cooled (0 °C) and stirred solution of 2.57 g (23.5 mmol) of 12 and 6.96 g (47 mmol) of diethylaniline in 20 ml; of dry ether, 6.55 g (47 mmol) of 13 dissolved in 30 ml of dry ether was added dropwise over 45 min. The reaction mixture was then stirred for 15 min. and maintained at 4 °C for 16 h. The salt was filtered off and washed twice with 10 ml portions
134

of dry ether. The clear solution, tagether with the combined washings, was evaporated in vacuo at rt foliowed by fractional distillatÎon. B.p. 153-160 °C/0,003 mm; yield 52\. The product contains 10\ of 19. For NMR data, see Table VI.1.
Prepared according to the procedure described in Houben-Weyt 35•
B.p. 76 °C/8,2 mm; yield 85\, 1H NMR (CDC1 3) ö 7,12(m, 5H, ArH). 31 P NMR (CDC1 3) ö 173,8,
-2~2'-(1~3-Propanedio~y)-4~5-benzo-1~3~2-dio~aphoephotane-
-1'~3'~2'-dio~aphoephorinane 19
To a cooled (0 °C) and stirred salution of 3.60 g (20 mmol) of crude 21 and 2.98 g (20 mmol) of diethylaniline in 15 ml of dry ether, 3,49 g (20 mmol) of 18 dissolved in 15 ml of dry ether was added dropwise over 30 min. The reaction mixture was then stirred for 15 min., the saltwas filtered off and wasbed twice with 10 ml portions of dry ether. The clear solution, tagether with the combined washings, was evaporated in vacuo at rt. Several attempts to distill the crude product failed. For NMR data, see Table VI.1.
-2~2'-(1 1 3-Propanediorey)-4 1 5-benzo-1,3,2-dio~aphoephotane-
-41151-benzo-11131,21-dio~aphoepholane 20
Toa caoled (0 °C) and vigorously stirred salution of 1.80 g (25 mmol) of 17 and 7.52 g (50 mmol) of diethylaniline in 40 ml of dry ether, 8.73 g (50 mmol) of 18 dissolved in 15 ml of dry ether was added dropwise over 45 min. The reaction mixture was stirred foranother 15 min. and maintained at 4 °C for 16 h. The saltwas filtered off and washed twice with 10 ml portions of dry ether. The clear solution, tagether with the combined washings, was evaporated in vacuo at rt and fractionally distilled, B.p. 174-176 °C/0.04 mm; yield 79\. For NMR data, see Table VI.l.
135

To a cooled (10 °C) and stirred salution of 6.06 g (50 mmol) of diethylaniline and 3.18 g (50 mmol) of 17 in 40 ml of dry ether, 7,03 g (50 mmol) of 13 dissolved in 15 ml of dry ether was added dropwise over 1 h. The reaction mixture
wasthen cooled to 4 °C for 16 h., the saltwas filtered off and washed twice with 10 ml portions of dry ether. The clear solution, tagether with the combined washings, was evaporated in vacuo at rt. Yield 8,6 g of crude product which consisted of equimolar amounts of 21 and 22. Several
attempts to distill the crude product failed due to the equilibrium 21 + 21~22 + 17. For NMR data, see Table VI.1.
- 2~2 1 -(1~3-Propanediozy)-1~3~2-diozaphosphorinane-1 1 ~3'$2 1 -
-dioxaphosphorinane 22
Preparatien analogous to the one described for 20. B.p. 132-136 °C/0.005 mm; yield 79%. For NMR data, see Table VI.1.
- 2~2'-(o-Phenyldiozy)-4 1 ~-benzo-1 1 3$2-diozaphospholane-4' 1 ~ 1 -
-benzo-11131$2'-diozaphospholane 23
Toa stirred salution (rt) of 1.75 g (10 mmol) of 18 dissolved in 30 ml of dry ether, 0.55 g (5 mmol) of 12 and 1.01 g (10 mmol) of triethylamine dissolved in 20 ml of dry ether was added dropwise over 30 min. The reaction mixture was then stirred for 15 min., the saltwas filtered off and washed twice with 5 ml portions of dry ether. The clear solution, together with the combined washings was evaporated in vacuo at rt. 31 P NMR (CDCI 3) ó 129.6 and ó -21.8 (5% 28).
-2~2'-(o-Phenyldiozy)-4~~-benzo-1 1 3 1 2-diozaphospholane-
-1'~31121-diozaphosphorinane 24
Prepared on NMR scale due to its instability. For 31 P NMR measurements 50 mg (0,2 mmol) of 28 was dissolved in 0.4 ml of CDCI 3 in an NMR sample tube at rt. Quick addition of
136

20 mg (0.2 mmol) of triethylamine and 28 mg (0.2 mmol) of 13 resulted in formation of 24 (main product), 20 and 16.
, 31 P NMR (CDC13) ö 124.1 and ö 130.9 (equal intensity) indicative of 24.
Toa cooled (10 °C) and stirred salution of 4.60 g (0.1 mol) of dry ethanol and 10.12 g (0.1 mol) of triethylamine in 40 ml of dry ether, 14.05 g (0.1 mol) of 2 dissolved in 10 ml of dry ether was added dropwise over 1 h. The reaction, mixture was warmed to rt and stirred for 30 min. The salt was filtered off and wasbed twice with 20 ml portions of dry ether. The clear solution, together with the combined washings, was evaporated in vacuo at rt, foliowed by fractional distillation. B.p. 78 °C/25 mm; yield 50%. For NMR data, see Table VI.l.
Prepared by a metbod analogous to that described by Crofts
et aZ. 36• B.p. 108-109 °C/10 mm; yield 84%. For NMR data,
see Table VI.l.
-2-Pheno~y-1~3~2-dio~aphosphorinane 27
Prepared in essentially the same way as 25, B.p. 90-92 °C/ 0.75 mm; yield 83%. For NMR data, see Table VI.l.
-s-Hydrido-2~3~7~8-benzo-1~4~6~9-tetrao~a-5-phosphaspiro
[4~4]non-2~7-diene 28
Prepared according to the metbod described by Munoz et aZ. 12 •
Yield 82%. 31 P NMR (CDC1 3) ó -21.8.
Route A and B (Fig. VI.S).
For these routes the same procedure was foliowed as described
137

for route A and B, Fig. VI.6 (vide supra). The relevant 31 P NMR data of the various compounds are listed in Table VI.2.
Prepared according to the procedure described in Houben-WeyZ 31•
B.p. 83-84 °C/25 mm; yield 75%. 1H NMR (CDC1 3) 6 0,84 and 1.26 (s, 6H, CH3); 3.30-3.78 and 4.08-4.49(m, 4H, OCH2). 31 P NMR (CDC1 3) ö 146.7.
-2~2'-(1~1~2,2-TetramethyZ-1~2-ethanediozy)-5~5-dimethyl-
-1~3,2-diozaphosphorinane-5',5'-dimethyZ-11,31,21-diozaphos-
phorinane 35
Toa cooled (-78 °C) and stirred solution of 1.18 g (10 mmol) of 29 and 2.02 g (20 mmol) of triethylamine in 30 ml of dry ether, 3.37 g (20 mmol) of 30 dissolved in 20 ml of dry ether was added dropwise over 20 min. The reaction mixture was then stirred for 15 min., warmed up and maintained at 4 °C for 16 h. The salt was filtered off and wasbed twice with 10 ml portions of dry ether. The clear solution, tagether with the combined washings, was evaporated in vacuo at rt. Yield 3.8 g of crude product. 31 P NMR (CDC1 3) ö 121.9 (main product, 33); 121.7 (31) and resonances between ö +14 and -21 (phosphonates) 21
•
Prepared according to the procedure described in Houben-Weyzao.
B.p. 69.5 °C/6.5 mm; yield 54%. 1H NMR (CDC1 3) ó 1,32 and 31 1.50(m, 12H, CH3). P NMR (CDCI 3) ö 175.5.
•2,2 1 -(2~2-DimethyZ-1,3-propanediozy)-4,4,5,5-tetramethyZ
-1,5~2-diozaphospholane-4'~41,51,51-tetramethyZ-11131121-
-dioroaphospholane 36
Preparatien analogous to that described for 33. Nearly quantitative yield of crude 36. 31 P NMR (CDC1 3) ö 147.7 (32), 147.5 (main product, 36) and resonances with ö +13 (phosphonates
138

-2~2'-(2~2-Dimethyl-1~3-propanedio=yJ-5$5-dimethyZ-1~3~2-
dio=aphosphorinane-5'~5'-dimethyZ-1'~3'121-dio=aphospho
rinane 37
Preparation analogous to that described for JJ. Nearly quantitative yield of 37 resulted after evaporation of the dry ether in vacuo at rt. M.p. 44-46 °C. 1H NMR (CDC1 3) ö 0.74 (s), 0.99(s) and l.ZS(s) (18H, CH~); 3.03-4.38(m, 12H, OCH2). 13 . ~ 4 C NMR (CDC1 3) ö 22.37(s, r1ng CH3); 23.51(d, JPC = 6Hz, CH3); 33.59(d, 3JPC = 4Hz, ring C); 37,81(t, 3JPC = 6Hz, C);
2 ' • 68.38(d, Jpc = 16.7 Hz, CH 2); 69.58(s, r1ng CH 2). 3lp NMR (CDC1 3) ö 121.9.
-5-Hydrido-21 21 31 31 71 71 8,8-oatamethyZ-1,4,6 1 9-tetrao=a-5-
phosphaspiro[4,4]nonane 38
Prepared according to the procedure described by Sanchez et
aZ. 38• The crude product 38 was crystallized from petroleum
ether (60-80). Yield 90%, 1H NMR (CDC1 3) ö 1.20(m, 24H, CH?); 1 31 ·' 7.14(d, 1H, JPH =799Hz, P-H). P NMR (CDC1 3) ö -40.6.
139

Referenaea and Notea,
1. L.H. Koole, E.J. Lanters, J.J.C. van Lier, L.J.M. van de Ven and H.M. Buck, J. Am. Chem. Soc., submitted.
2. F.M. Richards and H.W. Wyckoff, "The Enzymes", P.D. Boyer, ed., Academie Press, New York, 1971, Vol. IV, Chapter 24.
3.a R.D. Gandour, "Transition States of Biochemica! Processes", Chapter 14, R.D. Gandour and R.L. Schowen, eds., Plenum Press, New York, 1978, 539, and references cited;
3.b S.J. Benkovic and K.J. Schray, "Transition States of Biochemica! Processes", Chapter 13, R.D. Gandour and R.L. Schowen, eds., Plenum Press, New York, 1978.
4. A.M.C.F. Castelijns, P. Schipper, D. van Aken and H.M. Buck, J. Org. Chem., 1981,46,47.
5. A.M.C.F, Castelijns, Ph. D. Thesis, Eindhoven University of Technology, 1979.
6. D. van Aken, L.M.C. Paulissen and H.M. Buck, J. Org. Chem., 1981, 46, 3189,
7. D. van Aken, Ph.D. Thesis, Eindhoven University of Technology, 1981.
8. R, Burgada, D. Houalla and R. Wolf, C.R. Acad. Sci. Paris C., 1976, 264, 356.
9. D. Bernard, C. Laurenco and R. Burgada, J. Organmetal. Chem., 1973, 47, 113.
10. R. Burgada, Phosphorus Sulfur, 1979, 6, 435, 11. M. Koenig, A; Munoz, B. Garrigues and R. Wolf,
Phosphorus Sulfur, 1976, 2, 237. 12. A. Munoz, M. Sanchez, M. Koenig and R. Wolf, Bull. Soc.
Chim. Fr., 1974, ,B-10, 2193. 13. R. Burgada, Bull. Soc. Chim. Fr., 1975, 1-2, 407. 14. R. Burgada and C. Laurenco, J. Organmetal. Chem., 1974,
86, 255. 15. F. Ramirez, M. Nowakowski and J.F. Marecek, J. Am. Chem.
Soc., 1977, 99, 4515, 16. A. Munoz, B. Garrigues and M. Koenig, J. Chem. Soc.,
Chem. Comm., 1978, 219. 17. B. Garrigues, A. Munoz, M. Koenig, M. Sanchez and
140

R. Wolf, Tetrahedron, 1977, 33, 635. 18. B.C. Chang, W.E. Conrad, D.B. Denney, D.Z. Denney, R.
Edelman, R.L. Powell and D.W. White, J. Am. Chem. Soc., 1971, 93, 4004.
19, B.A. Arbusov, Y.M. Marreev, U.S. Vinogradova and Y.Y. Samitov, Dokl. Akad. Nauk. S.S.S.R., 1972, 205, 843.
20. S.A. Bone and S. Tripett, J. Chem. Soc., Chem. Comm., 1975, 1583.
21. P.J.J.M. van Ool and H.M. Buck, Reel. Trav. Chim. Pays-Bas, 1983, 102, 215.
22. A. Munoz. Bull. Soc. Chim. Fr., 1977, ?-8, 728. 23. See for the references 4-7 and 18-21 cited in this
Chapter. 24. K.G. Scrimgeour, "Chemistry and Control of Enzyme
Reactions", Academie Press, London, 1977, 185 and references cited therein.
25. D.G. Gorenstein, A.M. Wyrwicz and J. Bode, J. Am. Chem. Soc., 1976, 98, 2308.
26. D.A. Usher, D.I. Richardson, Jr. and D.G. Oakenfull, J. Am. Chem. Soc., 1970, 9 1 4699.
27. A. Munoz, M.-T. Boisdon, J.-F. Brazierand R. Wolf, Bull. Soc. Chim. Fr., 1971,4, 1423.
28. R. Burgada, H. Germa, M. Willson and F. Mathis, Tetrahedron, 1971, 2?, 5833.
29, F.H. Westheimer, Acc. Chem. Res., 1968, 1, 70. 30. D. Marguarding, F. Ramirez, I. Ugi and P. Gillespie,
Angew, Chem., 1973, 85 1 99. 31. M.T. Boisdon, C. Malavaud, F. Matbis and J. Barrans,
Tetrahedron Lett., 1977, 3501. 32. L. Lafaille, F. Matbis and J. Barrans, Compt. rend.,
1977, 285, c, 575. 33. M.A. Pudovik, S.A. Terent'eva, Yu.B. Mikhailov and A.N.
Pudovik, J. Gen. Chem., 1982, 52, 1144 •. 34. H.J. Lucas, F.W. Mitchell, Jr. and C.N. Scully, J. Am.
Chem. Soc., 1950, 72, 5491. 35. Houben-Weyl, "Methoden der Organischen Chemie", Phos
phorverbindungen, II, 1964, 49. 36. P.C. Crofts, J.H.H. Markes and H.N. Rydon, J. Chem. Soc.,
141

1958, 4250. 37. Houben-Weyl, "Methoden der Organischen Chemie". Phos
phorverbindungen, II, 1964, 248. 38. M. Sanchez, R. Wolf, R. Burgada and F. Mathis, Bull.
Soc. Chim. Fr., 1968, 2, 773.
142

Appendix
Energiea (E~ kJ/mol) of the MNDO optimized geometriea for the (methyZated) tetrahydrofuryZ-9-purinea (dPu~ A and G) and (methylated) tetrahydrofuryZ-1-pyrimidines (dPyr~ C and T)~ aaZauZated as a funation of the gZyaosidia dihedral angle Xin the atruature. For the (methyZated) dPu struatures the dihedraZ angZe X is defined as X!C(8)-N(9)-C(l')-C(2'Jl
whereas for the (methyZated) dPyr struatures this angle is X[C ( 6) -N ( 1) -C (1 ') -C ( 2 ')] ,
Chapter liL
dA model system xo E xo E xo E
195 100.576 65 102.062 -60 85.941
180 98.950 50 . 106.870 -75 85.108
165 93.389 35 109.080 -90 84.764
150 88.845 10 105.283 -105 85.305 135 86,654 -5 99.834 -120 86.665 110 88,266 -20 95.258 -135 89.817
95 91.926 -35 90.402 -150 94.719 80 97,054 -50 87.244
dG model system xo E xo E xo E
195 -146.830 75 -153.451 -45 -161.155 18() -148.654 60 -149.152 -60 -163.111 175 -150,413 45 -145.140 -75 -163.504 160 -156.231 30 -143,642 -90 -163.657 135 -162.322 15 -145.218 -105 -162.766 120 -162.928 0 -149,728 -120 -161.555 105 -160.657 -15 -153.580 -135 -158.198
90 -157.760 -30 -158.236 -150 -152.596
143

dC model system xo E xo E xo E
100 372.698 -20 373.009 -140 374.815 80 384.919 -40 364.106 -160 398.731 60 411.665 -60 355.153 -180 415.059 40 391.106 -80 351.136 -200 411.583 20 379.756 -100 343.333 -220 390.446 0 376.150 -120 359.638 -240 372.706
dT model system xo E xo E xo E 95 36.257 -25 34.641 -14S 36.432 80 4S.153 -40 28.963 -160 47.886 65 74.461 -ss 23.524 -175 73.805 50 58. 17 4 -70 20.519 -190 73.818 35 46.076 -85 19.784 -20S 63.827 20 40.229 -100 24.051 -220 48.171
5 37.988 -115 27.178 -235 28.571 -10 36.911 -130 30.601 -250 31.332
Chapter IV ~
dm5c model system xo E xo E xo E
100 451.004 20 448.875 -10S 426.242 95 454.436 5 446.563 -120 428,433 80 465.064 -10 444.909 -135 432.311 70 470.281 -25 440.982 -150 439.857 65 471.991 -40 433.959 -175 523.293 60 479.003 -ss 426.975 -190 484.385 55 473.457 -70 422.200 -215 459.223 50 468.136 -85 421.051 -230 443.252 35 455.320 -100 424.547 -245 444.006
144

dm6G model system xo E xo E xo E
19S -29.332 7S -37.971 -4S -40.S72
180 -21.413 60 -31.870 -60 -43.849
16S -38.834 4S -26.69S -7S -43.9S1
1SO -4S.811 30 -21.188 -90 -42.S86
13S -49.149 1S -1S.S99 -10S -40.S7S
120 -49.397 0 -16.S71 -120 -39.086
10S -47.281 -1S -24.870 -13S -37.734
90 -43.320 -30 -33.963 -1SO -34.929
dm7G model system xo E xo E xo E
19S 490.897 70 487.247 -ss 480.606
180 49S.S63 so 493.262 -70 478.284
16S 48S.926 3S 498.843 -8S 478.318
1SO 479.117 20 S06.103 -90 478.431
13S 47S.933 s S09.772 -10S 479.343
11S 476.097 -10 S02.981 -120 480.038
100 478.712 -2S 494.633 -13S 480.689
9S 479.9S1 -40 486.3S4 -1SO 483.S94
80 484.310
dm 8G model system xo E xo E xo E
190 -1S0.674 70 -167.249 -so -1S2.832
17S -162.436 ss -161.1SO -6S -1S9.S86
160 -173.172· 40 -147.243 -80 -160.1SO
14S -179.S41 2S -129.131 -9S -1SS.992 13S -174.221 1 0 -108.090 -11 0 -1S0.770
120 -17S.149 -s -102.762 -12S -149.111 10S -181.S7S -20 -121.990 -140 -1S1.146
90 -178.648 -3S -140.111 -1SS -1S2.S13 8S -177.119
145

Chapter V.
dm3A dm6A dm7A dm8A xo E E E E
185 820.915 101 .662 761.957 75.110 170 820.466 90.665 752.595 63.653 155 811.719 83.717 746.224 56.136 140 797.723 80.401 743.248 52.632 125 784.870 79.862 743.189 51.892 11 0 778.008 81.535 745.279 52.889
95 775.718 84.930 748.751 55.478 80 776.721 89,648 753.121 60.223 65 779.681 95.219 757.957 68.174 50 783.714 100,509 763.068 80.088 35 778.229 105.504 769.352 96.072
"ZO 773.768 110.766 777.317 115.689 5 772.772 112.132 781.202 136.839
-10 763.385 106.063 775.099 129.647 -25 757.259 97.451 765.353 108.960 -40 755.240 90.073 755.965 91.289 -55 757.294 85.792 749.565 79.687 -70 757.630 84.582 746.623 74.561 -85 758.636 85,451 746.091 75.752
-100 761.926 87.046 746.664 80.650 -115 767.593 88.425 7 4 7 '• 118 84.874 -130 776,523 89.332 747.276 84.135 -145 791.322 91.014 748.486 80.396 -160 812.021 95,015 753.227 78.471
146

SuJDJDary
This thesis describes investigations concerning the molecular-mechanistic aspects of phosphorus in four- and five-coordinated compounds. In particular, attention has been focused on the role of the phosphate groups in the helix backbone of the double-stranded DNA molecule. It is known that DNA structures with strictly alternating purine-pyrimidine base sequences can be converted from a right--handed B-DNA helix into their left-handed Z-DNA isomers. This B-.z transition involves rotatien of deoxypyrimidine residues and anti-.syn conformational change of the purine bases. Moreover, this transition proceeds cooperatively and is induced by nucleophilic solvents and/or a high-salt concentration. In order to elucidate a possible role of the phosphate groups in the helix backbone, the nature of the interaction of (cyclic) model organophosph(on)ates with lithium salts in aprotic solvents was studied using multi-nuclear NMR techniques. The 35c1- and 81 Br NMR data can be explained by assuming fast equilibria between lithium balides in the complexed (with phosph(on)ates) and the uncomplexed form. The 7Li NMR results reveal complexation of the phosphoryl oxygen atom by the Li+ ion, preferentially in 1:1 mol ratio salt/phosph(on)ate. Additional 31 P NMR studies combined with quantum-chemical calculations reveal that a close-ion pair structure of the salt/phosphate aggregates is the NMR observed configuration, whereas short-lived pentavalent (P(V)) structures are encountered as intermediatas in the phosphorylation and de-alkylation (group transfer) reactions. Furthermore, kinetic studies of cyclic phosphate in.the presence of methanol and lithium halide clearly show
147

that the phosphorylation reaction is retarded due to the shielding capacity of the halide anion, which is in close proximity to the phosphorus atom. Based on these results and on semi-empirica! quantum-chemical calculations on model systems, representative for the DNA structure, a mechanism of the B-.Z transition is given. The primary step in the · transition is supposed to involve an activatien of the pbospbate groups in the helix backbene from a tetrabedral (P(IV)) into a trigonal bipyramidal (TBP) P(V) intermediate structure. This activation can be accomplished by nucleophilic attack of a fifth ligand at phosphorus (e.g. water), promoted by the metal ions which take care of the shielding of the equatorial oxygen anions. The selectivity is introduced by the difference in orientation of the apical and equatorial ligands with respect to the phosphorus atom. The calculations reveal t~at a P(V) TBP structure in which 0(5') accupies an apical ?Osition is the energetically favoured intermediate. As a result of the electronic distribution in the TBP, the net electron density on 0(5') and 0(1 ') increases which results in a selective rotatien of the deoxyribose ring in the structure in such a way that the average 0(5')-0(1') distance is increased. The rotation of the deoxyribose ring around the C(4')-C(S') bond is also accompanied by a rotation around the glycosidic C(1 ')-N bond. Additional calculations on nucleoside model systems reveal a remarkable difference between purine and pyrimidine bases. Purine bases are relatively free to rotate around the glycosidic bond, whereas a considerable anti-.syn activatien energy was calculated for pyrimidine residues. This explains the observed selectivity in the B-.Z transition. In this way, a rotatien of the deoxyribose ring, induced by a 5'-linked P(V) TBP intermediate, forces pyrimidines to rotate out of the purine-pyrimidine base plane with concomitant separation of the base pair. When the entire deoxypyrimidine residue rotates (restricted rotation of the base), rejoining of the hydrogen-bonds becomes possible after a restrictionless anti-syn
conformational change of the complementary purine base. Special attention bas also been focused on the impact
148

of specific methylation of C.G and A.T base pairs in the DNA structure with respect to the B-Z transition. The calculations are performed on nucleoside model systems and reveal a considerable stabilization of the Z-DNA conformer, in comparison with the B-DNA conformer, upon selective methylation. The quantum-chemical results are combined with stereo model studies and are consistent with known experimental data. As Z-ONA structures have been detected in vivo, a possible biologica! function of the B_.z transition in genomic material is suggested.
Besides, the role of phosphorus in a TBP has been further investigated with tetraoxaspirophosphorane model compounds with a six-membered ring under base-catalyzed conditions. Ciosure of the six-membered ring is found to praeeed more slowly than ciosure of the five-membered ring which can be explained on the basis of an increased translational entropy in open six-membered ring fragments. Por one of these compounds, the rate of ring ciosure respectively ring opening could he considerably enhanced upon addition of base. These reactions show a great correspondence with the base-catalyzed hydralysis of RNA.
149

Sa:rnenvatting
In dit proefsihrift wordt een onderzoek beschreven naar de moleculair-mechanistische aspecten van fosfor in vier-en vijf-gecoördineerde verbindingen. In het bijzonder is aandacht besteed aan de rol van de fosfaatgroepen in de ruggegraat van dubbelstrengs DNA. Het is vastgesteld dat DNA st'ructuren, welke strikt alternerende purine-pyrimidine basesequenties bezitten, kunnen isomeriseren van een rechtshandige B-DNA helix naar een linkshandige Z-DNA structuur, Deze B--Z overgang gaat gepaard met een rotatie van desoxypyrimidine fragmenten en een anti-eyn conformatieverandering van de purine basen. Dit coöperatieve proces wordt geïnduceerd door nucleofiele oplosmiddelen en/of een hoge zoutconcentratie. Teneinde inzicht te verkrijgen in een mogelijke functie van de fosfaten in DNA voor deze B-Z overgang, is de complexering bestudeerd van (cyclische) model organofosf(on)aten met lithium bromide en lithium chloride in aprotische oplosmiddelen met behulp van multi-kernspinresonantie (NMR) technieken. De 35c1- en 81 Br NMR resultaten kunnen worden verklaard door snelle evenwichten te veronderstellen tussen met fosf(on)aten gecomplexeerde lithium halogeniden en niet gecomplexeerde lithium halogeniden. De 7Li NMR resultaten tonen aan dat het fosforyl zuurstofatoom wordt gecomplexeerd door het lithium kation met een hoge voorkeur voor een 1:1 molverhouding zout/fosf(on)aat. Uit 31 P NMR studies aan zout/fosf(on)aat aggregaten, gecombineerd met quanturnchemische berekeningen, kan worden geconcludeerd dat met behulp van NMR technieken een ionpaar wordt waargenomen, terwijl kortlevende pentavalente (P(V)) structuren optreden als intermediair in de fosforylerings- en de-alkyleringsreacties.
150

Kinetische studies aan een cyclisch fosfaat in aanwezigheid van methanol en lithiumhalogeniden tonen duidelijk aan dat de fosforyleringsreactie wordt vertraagd door de afscherming van het fosforatoom door het halogenide anion. Op basis van bovenstaande resultaten en aanvullende semi-empirische quantumchemische berekeningen aan modelsystemen, welke representatief zijn voor de DNA structuur, is een gedetailleerd mechanisme voor de B--Z overgang opgesteld. De primaire stap in dit proces bestaat uit een activering van de fosfaatgroep, waarbij de fosfor van een tetraedrische omringing overgaat in een kortlevende vijf-omringde trigonale bipyramide (TBP). Deze coördinatieverhoging vindt plaats door additie van een nucleofiel agens op P(IV). De aanwezige metaalionen stabiliseren de gevormde TBP door complexatie met de equatoriale zuurstofanionen. De selectiviteit wordt geïntroduceerd door het verschil in ori~ntatie van de apicale en equatoriale liganden met betrekking tot het fosforatoom. De berekeningen tonen aan dat een P(V) TBP structuur, waarin 0(5') een apicale positie inneemt, energetisch het meest stabiele intermediair is. Als gevolg van de electronische verdeling in de TBP neemt de electronendichtheid toe op 0(5') en 0(1 ').Dit resulteert in een selectieve rotatie van de desoxyribosering in de structuur en wel zodanig dat de gemiddelde 0(5')-0(1') afstand wordt vergroot. De rotatie van de desoxyribosering rond de C(4')-C(5') binding is gekoppeld aan een rotatie rond de glycoside C(1')-N binding. Berekeningen aan nucleoside modelsystemen tonen een opmerkelijk verschil aan tussen purine en pyrimidine basen. Purine basen kunnen vrij roteren rond de glycoside binding, terwijl deze rotatie is gehinderd in het geval van pyrimidine basen. Dit verklaart de waargenomen selectiviteit bij de B-.z overgang. Een rotatie van de desoxyribosering, geïnduceerd door een 5'-gebonden P(V) TBP intermediair, dwingt pyrimidines om uit het purine-pyrimidine basevlak te roteren onder gelijktijdige verbreking van de waterstofbruginteractie. Na rotatie van het gehele desoxypyrimidine fragment (gehinderde rotatie van de base) worden de waterstofbruggen weer hersteld doordat de complementaire purine base een anti--ayn conformatieverandering ondergaat.
151

Speciale aandacht is tevens besteed aan het effect van specifieke methylering van C.G en A.T basenparen met betrekking tot de B~z overgang. De quanturnchemische berekeningen zijn uitgevoerd aan nucleoside modelsystemen en tonen aan dat gemethyleerde structuren een grotere stabiliteit bezitten in de Z conformatie dan de corresponderende B conformatie. De berekeningen zijn gecombineerd met stereomodelstudies en zijn consistent met bekende experimentele gegevens. Aangezien Z-DNA structuren in vivo zijn aangetoond~ is tevens ingegaan op de mogelijke biologische betekenis van een B-.Z overgang in genetisch materiaal •.
Bovendien is de rol van fosfor in een TBP nader onderzocht aan de hand van tetraoxaspirofosforaan modelverbindingen met een zesring onder base-gekatalyseerde omstandigheden. Het sluiten van de zesring blijkt langzamer te verlopen dan het sluiten van de vijfring, wat kan worden verklaard op basis van een toename in de translatie-entropie in open zesring fragmenten. Voor een van deze modelstoffen blijkt het ringopenen respectievelijk ringsluiten aanzienlijk versneld te worden in aanwezigheid van base. Deze reacties vertonen grote analogie met de base-gekatalyseerde hydrolyse van RNA.
152

CurriculuJD. vitae
De schrijver van dit proefschrift werd geboren op 28 februari 1955 te 's-Gravenhage. Na het behalen van het diploma atheneum B aan het Sint Miehiellyceum te Geleen in 1973, werd in hetzelfde jaar begonnen met de studie op d~ afdeling der Scheikundige Technologie aan de Technische Hogeschool te Eindhoven. In september 1976 werd hem de Chevron Chemie-prijs toegekend. Het afstudeerwerk werd verricht bij de vakgroep Organische Chemie o.l.v. prof. dr. H.M. Buck en dr. ir. ·D. van Aken. In juni 1979 werd het ingenieursexamen met lof afgelegd.
Vanaf 1 juli 1979 tot 1 september 1979 was hij als wetenschappelijk assistent in dienst van de Technische Hogeschool. Vanaf 1 september 1979 tot 1 september 1983 werd, in dienst van de Nederlandse Stichting voor Zuiver Wetenschappelijk Onderzoek, het onderzoek, beschreven in dit proefschrift, uitgevoerd onder leiding van prof. dr. H.M. Buck,
Per 24 oktober 1983 vervult hij de militaire dienstplicht bij de Koninklijke Nederlandse Marine, waar hij als assistent eliniseh-chemicus gedetacheerd zal worden aan het Marine Hospitaal te Overveen.
153

Dankwoord
Velen hebben aan de totstandkoming van dit proefschrift bijgedragen. Mijn promotor prof. dr. H.M. Buck dank ik voor de waardevolle suggesties, die de loop van het onder~oek mede hebben bepaald. In het bijzonder wil ik tevens danken ir. W.J.A. Reinders, ir. L.H. Koole, ir. M.T. Smits, ir. J.H.M. Stroucken en ir. R.J.M. Hermans voor het werk dat zij tijdens hun afstudeerperiode hebben verricht. Het samenwerken met deze mensen is voor mij een voortdurende bron van inspiratie geweest.
Dr. ir. J.W. de Haan en ing. L.J.M. van de Ven ben ik zeer erkentelijk voor hun bijdrage aan het tot stand komen van Hoofdstuk 11 en dr. G.J. Visser van het Rekencentrum van de TH-Eindhoven voor de aanpassing van het MNDO programma aan de Burroughs computer.
Voor de uitvoering van het proefschrift ben ik veel dank verschuldigd aan mevr. P. Meyer-Timan voor de korrecties op het gebruik van de Engelse taal en de hr. H. Eding voor de voortvarende wijze waarop de tekeningen en de lay-out zijn verzorgd.
The work described in this thesis was supported by the Netherlands Foundation for Chemica! Research (S.O.N.) with financial aid from the Netherlands Organization for the Advancement of Pure Research (Z.w.o.)
154

Stellingen
1. Bij de bepaling van de critische vrije energie van supercoiling, welke benodigd is om een locale B-.z overgang in een recombinant plasmide te induceren, wordt geen rekening gehouden met de invloed van intrinsiek aanwezige concurrerende factoren zoals palindroom sequenties en (A+T}-rijke locaties.
D.B. Haniford en D.E. Pulleyblank, Nature (Lond.), 1983, 302, 632; C.J. Benham, J. Mol. Biol., 1981, 150, 43.
2. Het gevonden verschil in conformatie tussen de C(4')-C(5') binding in adenosine-5'-monofosfaat en het modelnucleotide 5'-deoxy-5'-adenosine azijnzuur moet worden toegeschreven aan sterische hindering tussen het ribosefragment en de 6'-methyleengroep.
T. Ishida, T. Miyazaki, M. Inoue, A. Ota, T. Kurihara en M. Sugiura, J, Chem. Soc. Perkin Trans. I, 1983, 1325.
3. Bij de studie van de aggregatie van n-alkylhalogeniden in een apolair milieu wordt ten onrechte met de mogelijke vorming van inverse micellen geen rekening gehouden.
G.W. Brady, Acc. Chem. Res., 1974, ?, 174 en referenties daarin.

4. De bewering van Turro dat de ~-interactie in een geêxciteerde dubbele band wordt verbroken door rotatie, berust op een onjuiste interpretatie van de volgorde der gebeurtenissen.
N.J. Turro, "Modern Molecular Photochemistry", B. Cummings, Menlo Park, 1978.
5. De 13c chemica! shift veranderingen van n-nonyltrimethylammoniumbromide in water bij toenemende amfifielconcentratie kunnen worden verklaard door grotere bijdragen van anti conformeren.
B.O. Persson, T. Drakenberg en B. Lindman, J. Phys. Chem., 1979 1 23, 3011; R.J.E.M. de Weerd, proefschrift TH Eindhoven, 1983.
6. De verklaring die gegeven wordt voor het verdwijnen van het ESR signaal bij foto-geinduceerde rèductie van v4
+
in Ca2NaMg 2v3o12 houdt geen rekening met de mogelijke vorming van triplet toestanden.
G. Oversluizen, proefschrift TH Eindhoven, 1983.
7. Aangezien de concentratieverhouding van ozon en stikstofoxiden in verontreinigde lucht wordt bepaald door chemische evenwichten, is het gewenst om voor deze componenten gecombineerde grensadvieswaarden in te voeren.
w.-c. Wang en N.D. Sze, Nature (Lond.), 1980,288,589.

8. Het gegeven dat voor planten de schadelijke invloed van zwaveldioxide aerosolen onder winterse temperaturen die van chemische bestrijdingsmiddelen overtreft, is een aanbeveling voor het invoeren van klimatologisch afhankelijke grensadvieswaarden voor deze component.
C.K. Baker, M.H. Unsworth en P. Greenwood, Nature (Lond.), 1982, 299, 149; S.A.W. Gerstl en A. Zardecki, Nature (Lond.), 1982, 300, 436.
9. Bij de discussie over de risico's van het recombinant DNA onderzoek dreigt de betekenis van het fundamentele DNA onderzoek op de achtergrond te raken.
Eindrapport "Brede DNA Commissie", Staatsuitgeverij, 's-Gravenhage, augustus 1983.