entendiendo la farmacocinética
TRANSCRIPT

Entendiendo PK
Hernán Darío Aguirre HResidente de Medicina Interna
Universidad de La Sabana.Departamento de Farmacología Clínica y Terapéutica

PK
La concentración de un fármaco que se alcanza en su lugar de acción es la consecuencia de los siguientes procesos:
• Absorción
• Distribución
• Eliminación-Metabolismo-Excreción

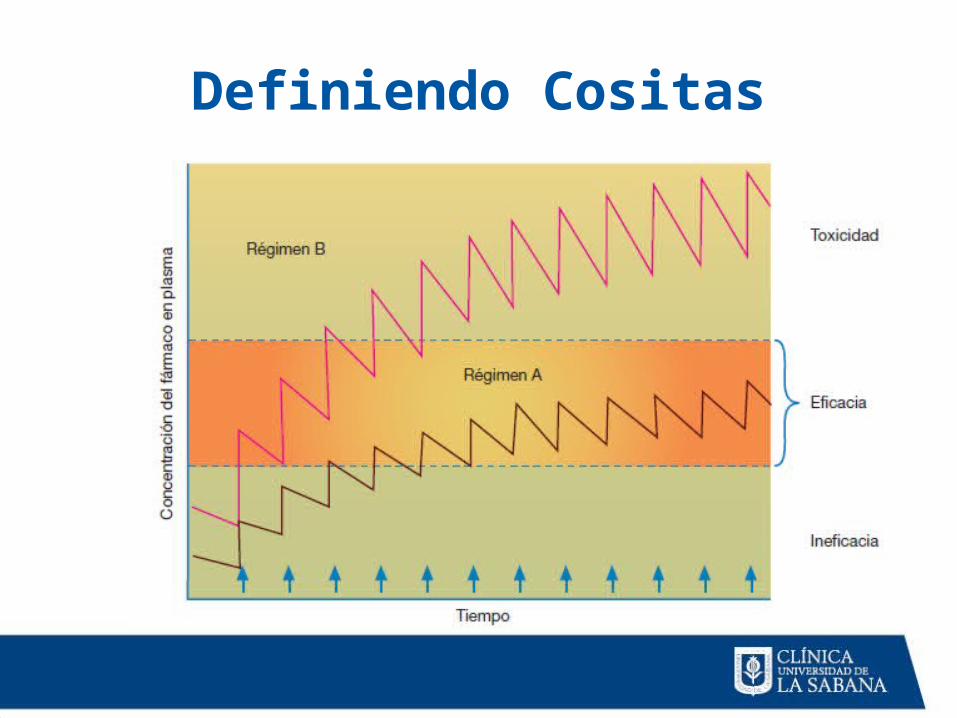
Definiendo Cositas
• Concentración mínima eficaz (CME).
• Concentración mínima tóxica (CMT).
• Índice terapéutico (IT).
• Período de latencia (PL).
• Intensidad del efecto (IE).
• Duración de la acción (TE).

Definiendo Cositas

Definiendo Cositas
Definiendo Cositas

Variabilidad individual
• Factores genéticos y ambientales
• Factores fisiológicos.
• Factores patológicos.
• Factores iatrógenos.

Absorción
• La absorción de un fármaco puede ser entendida como el paso de una sustancia atreves de una membrana que separa el exterior del interior del organismo.
• Depende de la permeabilidad de la membrana (P), de la superficie de absorción (Sa) y del gradiente de concentración del fármaco a ambo lados de la membrana (C1-C2):
Absorción = P · Sa · (C1 – C2)

¿Absorción = P · Sa · (C1 – C2)Y listo?
• Ionizada/no Ionizada
• Difusión pasiva
• Proteínas en la membrana
• Proteínas Trasportadoras de la Membrana

Absorción vs Biodisponibilidad
• Desde el punto de vista del efecto de los fármacos, es más importante la biodisponibilidad entendida como la velocidad y cantidad del fármaco que llega a la circulación sistémica.
• La absorción propiamente dicha no es más que un elemento de esa biodisponibilidad que depende también de la eliminación presistémica.

Eliminación Presistemica
• Eliminación por las heces antes de que se complete su absorción (fL).
• Expulsados de nuevo a la luz intestinal por la glucoproteína P o metabolizarse en el epitelio intestinal (fI).
f = fL · fI · fH
• Inhibición por primer paso hepático o en los pulmones, antes de llegar a la circulación sistémica (fH)
• La fracción del fármaco administrado que llega a la circulación sistémica, será el producto de las tres fracciones (luminal, intestinal y hepática):
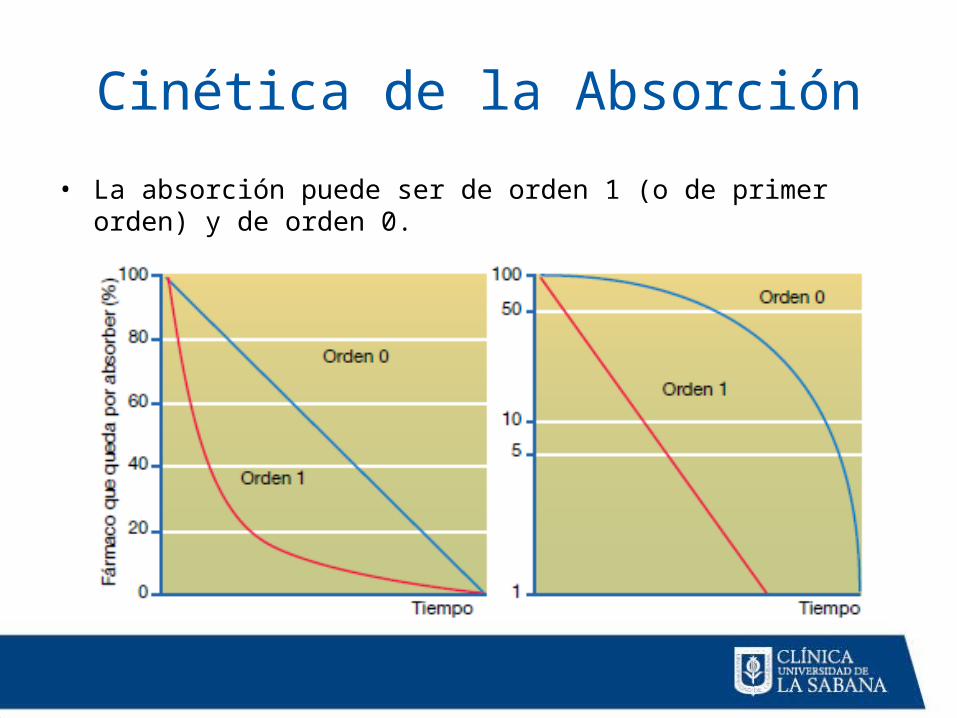
Cinética de la Absorción
• La absorción puede ser de orden 1 (o de primer orden) y de orden 0.

Biodisponibilidad
• Indica la velocidad y la cantidad de la forma inalterada de un fármaco que llega a la circulación sistémica y, por lo tanto, está disponible para acceder a los tejidos y producir un efecto.
• La cantidad absorbida suele valorarse mediante el AUC de concentraciones plasmáticas, la Fracción de absorción biodisponible (f), y la velocidad de absorción por la forma de esa curva expresada por la concentración máxima (Cmáx) y el tiempo en que se alcanza (tmáx).

Variación en la Biodisponibilidad

Bioequivalencia
• La equivalencia farmacocinética entre dos preparados farmacéuticos, es decir, que tengan una velocidad de absorción y una fracción de absorción lo suficientemente similares como para que pueda asumirse que tendrán la misma eficacia y seguridad.

Bioequivalencia
Las diferencias en la cantidad absorbida producen diferencias en el AUC y la Cmáx, mientras que las diferencias en la velocidad de absorción producen diferencias en la Cmáx y el tmáx.

Distribución
• La distribución de los fármacos permite su acceso a los órganos en los que debe actuar y a los órganos que los van a eliminar, y condiciona las concentraciones que alcanzan en cada tejido.

Modificadores de la Distribución
• Transporte en la sangre y unión a proteínas plasmáticas
• Distribución en los tejidos- Distribución regional- Distribución en áreas especiales
• Cinética de la Distribución.
• Volumen aparente de la Distribución.

Unión a proteínas plasmáticas
• La fijación a la albúmina es la más frecuente.
• Aunque la carga de la albúmina a pH de 7,4 es negativa, fija tanto fármacos ácidos como bases mediante enlaces iónicos y, ocasionalmente, enlaces covalentes.
• La cantidad de fármaco unido a proteínas depende de:
La concentración de fármaco libre.
La constante de asociación (K1/K2).
El número de sitios de fijación libres.
La concentración de proteína.

Distribución RegionalEl fármaco disuelto en la sangre pasa de los capilares a los
tejidos a favor del gradiente de concentración.
• Características del fármaco:
Tamaño de la molécula.LiposolubilidadGrado de ionización Unión a las proteínas plasmáticasFlujo sanguíneo del órgano,Características del endotelio capilar.
• Características del paciente:
InflamaciónPHTejido graso.

Compartimentos Farmacocinéticos
• El compartimento central incluye
El agua plasmáticaEl agua intersticial El agua intracelular
• Fácilmente accesible.
• Tejidos bien irrigados, como el corazón, el pulmón, el hígado, el riñón, las glándulas endocrinas y el SNC (si el fármaco atraviesa bien la BHE).

Compartimentos Farmacocinéticos
• El compartimento periférico superficial:
• Tejidos menos irrigados• Piel• Grasa• Músculo • Médula ósea, así como • Depósitos celulares (proteínas y
lípidos) a los que los fármacos se unen laxamente.
• El compartimento periférico profundo incluye:
• Los depósitos tisulares a los que el fármaco se une con mayor fuerza y de los que, por lo tanto, se libera con mayor lentitud

Compartimentos Farmacocinéticos
• Modelo Monocompartimental:
a) Antes de la administración.
b) después de la administración la distribución es rápida y uniforme.

Compartimentos Farmacocinéticos
• Modelo Bicompartimental:
a) Antes de la administración
b) Inmediatamente después el fármaco difunde a los órganos bien irrigados.
c) Se equilibra con el resto del
organismo.

Compartimentos Farmacocinéticos
• Modelo Tricompartimental:
a) Antes de la administración
b) Inmediatamente después, el fármaco difunde a los órganos bien irrigados
c) Se equilibra con el resto del organismo.
d) Acumulación continúa en los órganos a los que el fármaco se fija fuertemente.

Volumen aparente de distribución
• (Vd) no es un volumen real, sino un volumen aparente que relaciona la cantidad total del fármaco que hay en el organismo en un determinado momento con la concentración plasmática.
Cantidad del fármacoVD: ------------------------------ Concentración Sérica

Volumen aparente de distribución
• Duda:
El volumen aparente de distribución de un fármaco es el volumen en el que tendría que haberse disuelto la dosis administrada de un fármaco para alcanzar la concentración plasmática observada.

Vcentral y Vbeta
• El volumen central se utiliza para calcular la dosis inicial en los fármacos que actúan en el compartimento central.
• El volumen en equilibrio se utiliza para calcular la dosis inicial de los fármacos que actúan en el compartimento periférico (p. ej., digoxina, fenitoína o teofilina).

Eliminación
• La concentración activa del fármaco en el organismo humano disminuye como consecuencia de dos mecanismos:
Metabolismo.Excreción.
• Los fármacos liposolubles, aunque se filtren por el riñón, se reabsorben y deben metabolizarse (principalmente en el hígado) a metabolitos más polares.
• Estos metabolitos, junto con los fármacos hidrosolubles, se excretan principalmente por el riñón y la bilis.

Metabolismo

Metabolismo

Metabolismo

Excreción
• Los fármacos se excretan, por orden decreciente de importancia por:
• Vía urinaria• Vía biliar-entérica. • El sudor• La saliva• la leche • Los epitelios descamados.

Excreción Renal
• Filtración Glomerular
• Secreción TubularCationes Vs Aniones
• Reabsorción tubularLiposolubilidad + PH

Excreción Biliar
• En la membrana apical o canalicular del hepatocito hay:
Glucoproteína P, que elimina cationes orgánicosProteínas MRP2 que eliminan aniones orgánicos a la bilis
• En la membrana basal hay Proteínas MRP1 que eliminan aniones orgánicos del hepatocito a la sangre.

Y todo esto para que?
• Mucha cosita y poca utilidad…

Cuidado Critico
Alvarez F. Drugs 2012; 72 (4)

Cuidado Critico
Alvarez F. Drugs 2012; 72 (4)

Cuidado Critico
Roberts, J. Lancet Infect Dis 2014;14: 498–509

Cuidado Critico
Alvarez F. Drugs 2012; 72 (4)

¿Qué deben hacer los médicos para mejorar la forma en que dosifican los antibióticos?
Poner 1 gramito se siente bien, pero poner 2 se siente mejor.
¿Sera este un adecuado raciocinio?
Gracias.
Should IV Antibiotics Be Administered by Prolonged Infusion? Can J Hosp Pharm. 2010 May;63(3):246-9