expression and cloning of complementary dna for a human enzyme that repairs o6-methylguanine in dna
TRANSCRIPT

J. Mol. Biol. (1990) 213, 739-747
Expression and Cloning of Complementary D N A for a Human Enzyme that Repairs
O6-Methylguanine in D N A
Hiroshi Hayakawa, George Koike and Mutsuo Sekiguchi Department of Biochemistry, Faculty of Medicine
K y u s h u University, Fukuoka 812, Ja p a n
(Received 20 November 1989; accepted 26 February 1990)
A cell line with an increased resistance to alkylating agents and an extremely high level of 06-methylguanine-DNA methyltransferase activity was isolated after transfection of methyltransferase-deficient Mer- cells with a cDNA library, prepared from methyl- transferase-profieient human Mer + (Rail) cells. Sodium dodecyl sulfate/polyacrylamide gel electrophoresis analysis revealed that a protein, with a molecular weight of approximately 25,000, accepted 3H label from DNA that had been treated with [3H]methylnitrosourea. Since the eDNA for methyltransferase was integrated into the chromosomal DNA, it was recovered by using the polymerase chain reaction. When the eDNA placed in an expression vector p500 was introduced into Mer- cells, the cells acquired an increased resistance to alkylating agents and exhibited a high level of 06-methylguanine-DNA methyltransferase activity. From the transformants the cDNA could be recovered as a part of the auto- nomously replicating plasmid. The nucleotide sequence of the cDNA was determined, and an open reading frame comprising 207 amino acid residues was found. The molecular weight of methyltransferase, calculated from the predicted amino acid sequence, was 21,700. The predicted amino acid sequence of the human methyltransferase exhibits an intensive homology with those of the bacterial counterparts, Ada and Ogt proteins of Escherichia coli and Dat protein of Bacillus subtilis, especially around possible methyl acceptor sites.
1. Introduction
Alkylating agents produce various kinds of alky- lated purine bases and pyrimidine bases in DNA. Among them, 06-methylguanine appears to be most responsible for the induction of mutations and cancers in organisms (Strauss et al., 1975). O6-Methylguanine can pair with thymine to replace cytosine during DNA replication, thus causing a G" C to A ' T transition (Coulondre & Miller, 1977). Such mutations were indeed found in the Ha-ras-1 gene in mammary tumors induced by MNU~f (Sukumar et al., 1983).
Many organisms possess an enzyme, O6-methyl - guanine-DNA methyltransferase, that repairs
t Abbreviations used: MNU, methylnitrosourea; MNNG, N-methyl-N'-nitro-N-nitrosoguanidine; ACNU, 1-(4-amino-2-methyl-5-pyrimidinyl)methyl- 3-(2-chloroethyl)-3-nitrosourea; DME medium, Dulbeceo's Modified Eagle's medium; EB, Epstein-Barr; EBNA, Epstein-Barr virus nuclear antigen; bp, base- pair(s); MGMT, 0~-methylguanine-DNA methyltransferase; RSV, Raus sarcoma virus; LTR, long terminal repeat; MMTV, mouse mammary tumor virus; HB ~, hygromycin B-resistant.
0022-2836/90/120739-09 $03.00/0
06-methylguanine in the DNA. I t transfers methyl groups from 06-methylguanine and/or other methylated moieties of the DNA to its own mol- ecule, thereby repairing DNA lesions in a single-step reaction (for a review, see Lindahl et al., 1988). The structure and function of DNA methyltransferase enzymes in Escherichia coli have been extensively studied. The ada gene, which encodes a 39,000 M r methyltransferase, was cloned (Demple et al., 1985; Margison et al., 1985), and the enzyme was purified to physical homogeneity (Nakabeppu et al., 1985). The enzyme was induced by brief treatment of cells with alkylating agents, and this process was con- trolled by the ada gene itself (Teo et at., 1986; Nakabeppu & Sekiguehi, 1986). In addition to Ada protein, a smaller methyltransferase, encoded by the ogt gene, is present in E. coli (Potter et al., 1987). The dat gene of Bacillus subtilis, coding for a con- stitutive methyttransferase, has also been cloned (Kodama et at., 1989; Morohoshi et al., 1989).
About one-fifth of available human tumor cell lines carry the Mer- phenotype, characterized by the lack of ability to support the growth of MNNG- treated adenovirus, and also by increased sensi- tivity to alkylating agents (Day et al., 1980;
739 ~) 1990 Academic Press Limited

740 H. Hayakawa et al.
Scudiero et al., 1984). The Mer- strains are deficient in the methyltransferase act ivi ty (Yarosh et al., 1983). When the bacterial ada gene, coding for methyltransferase, was introduced into Mer- cells, the cells acquired resistance to alkylating agents (Ishizaki et al., 1986, 1987; Samson et al., 1986; Brennand & Margison, 1986; Ka taoka et al., 1986). I t seems, therefore, tha t 06-methylguanine-DNA methyltransferase plays an impor tan t role in preserving genetic information, hence prevent ing tumor formation.
While a t tempts have been made to purify 06-methylguanine-DNA methyltransferase from mammalian cells, i t is extremely difficult to obtain a homogeneous enzyme preparation, and this has hindered application of the usual procedures for cloning of the cDNA for methyltransferase. Such being the case we used another approach, namely, expression of a cDNA library, prepared from Mer + cells, in Mer- cells and subsequent selection of clones with the Mer + phenotype. We report the cloning of a eDNA for human methyltransferase and determinat ion of the sequence,
2, Mater ia ls and M e t h o d s
(a) Cells and plasmids
HeLa $3 (Mer +) and MR (Mer-) cells (Ishizaki et at., 1986) were provided by Mituo Ikenaga, Kyoto Univer- sity. Vector plasmids pcDEB and p500 were constructed from pHEBol and p220-2, respectively, which were provided by Bill Sugden, University of Wisconsin (Sugden et al., 1985, and personal communication).
(b) Chemicals
ACNU, MNNG and hygromycin B were purchased from Sankyo Co., Tokyo Kasei Co. and Sigma, respectively. [3H]MNU was obtained from New England Nuclear.
(c) Assay of methyltransfera~e activity
Cells from 1 100 mm dish were collected with a rubber policeman, suspended in 1 ml of 20 mM-Tris" HC1 (pH 8-6), 1 mM-EDTA, 1 mM-mercaptoethanol, 5% (v/v) glycerol, and then sonicated. After centrifugation, the supernatant was taken as a crude extract. The extract was incubated at 37°C for 15 min with 3"85/~g of calf thymus DNA (2200 disints/min per/tg) that had been treated with [3H]MNU (1 Ci/mmol) in 20 mM-ammediol'HCi buffer (pH 10) for 15min (Nakabeppu et al., 1985). The reaction was stopped by the addition of triehloroacetie acid followed by heating at 90°C for 15 rain. Acid-insoluble materials were dissolved in 0"1 M-NaOH, neutralized and the radio- activity was counted. In some cases, 0-25 mg of bovine serum albumin (fraction V) was added to the reaction mixture, to ensure the acid precipitation. To analyze proteins labeled with methyl-3H on SDS/polyacrylamide gels, the reaction was stopped by the addition of 0-25 vol. of 0"25 M-Tris. HCI (pH 6-8), 20% (v/v) mercaptoethanol, 9.2% (w/v) SDS, 40% glycerol, followed by heating at 95°C for 90s. The mixtures were then applied to an SDS/12-5°/o polyaerylamide gel for electrophoresis and methyl-SH-labeled proteins were detected by fluorography.
(d) Cell survivals
Cells were seeded at 400 to 10,000 cells]60 mm dish and incubated at 37°C for 1 day. The cells were washed with ssrum-free DME medium and exposed to various concen- trations of ACNU or MNNG in serum-free DME medium for 1 h at 37°C. In the case of MNNG, DME medium was acidified (to pH 6), by C02 to stabilize the chemical. The cells were cultured in 10°/o (v/v) calf serum containing DME medium for 10 to 14 days, and then fixed with 10% (v/v) formaldehyde and stained with 0.1% (v/v) crystal violet. The number of colonies was counted using a magnifying glass.
(e) Preparation of eDNA library
The eDNA library was constructed as described by Okayama & Berg (1983). Briefly, mRNA was prepared from Raft (Mer +) cells by the guanidinium thiocyanate/ CsCl method, followed by oligo(dT)-celluiosc column chromatography. A vector, pcDEB, was digested with KpnI and a poly(dT) tail was added to the termini using the terminal transferase reaction. After removal of one of the poly(dT)-tailed termini by digestion with XbaI, the DNA construct and mRNA were annealed and eDNA was synthesized by reverse transcriptase. After poly(dC)- tailing at the termini, one of the poly(dC)-tailed termini was removed by digestion with HindIII. A poly(dG)- tailed, HindIII-digested linker fragment containing the SP~ promoter (Takebe et al., 1988) was annealed to the eDNA-linked vector, and the DNA was circularized with E. coli ligase. Replacement of the RNA portion by DNA was performed by consecutive reactions with E. coli RNase H, DNA polymerase I and DNA ligasc.
(f ) DNA transfection
Healthy growing cells were trypsinized and washed with electroporation buffer containing 40 mM- Hepes-NaOH (pH 7"05), 167 mM-NaC1, 10 mM-KCI, 1"4 m~-Na2HPO 4, 0-2% (w/v) dextrose. Cells (4x l07) were resuspended in 1 ml of the buffer containing DNA and electroporated at 225 V, 1180 pF in an ice bath (Chu et at., 1987). After 10 min or more in the ice bath, cells were placed in 10% calf serum containing DME medium, and incubated for 3 days at 37°C. Cells (2 x 107) were then subjected to screening for transformants by incubating in a medium containing 300#g of hygromycin B/ml (Blochlinger & Diggelmann, 1984).
(g) Amplification of eDNA
Oligonucleotide primers were synthesized with 381A DNA synthesizer (Applied Biosystems) and purified with SEP-PAK (Millipore). High-molecular-weight DNA was extracted as described by Perucho et al. (1981) and digested with XbaI. The polymerase chain reaction was performed with 1 ~g of each of the primers and 2-5 units of Taq polymerase (Promega), in 10mM-Tris.HC1 (pH 8"8), 50 mM-KCI, 2 mm-MgCI2, 0"01% (v/v) Triton X-100, 4 mg bovine serum albumin/ml (Takara), by using a DNA thermal cycler (Perkin-Elmer Cetus; Saiki et al., 1988). A reaction cycle was 1 rain at 94°C (denaturation), 2 min at 65°C (annealing), and 1 min at 72°C (elongation).
(h) DNA sequencing
DNA was digested with appropriate restriction enzymes and subcloned into multicloning sites of pUC9.

eDNA for Human Methyltransferase 741
SR a promoter or i
SV40 A m ~ ~ 6 S S,J.
L / I VG/C f H, ndlTI , ~
A ~cDNA
pt v
~ ori P HB' An,~
RSV LTR Amp r ~%~llk ~ ~V~rSJ
\ ~ / / or/ P
~ ~ ~ ' ~ J E BN A - 1
(a) (b)
Figure 1. Organizations of vectors used. (a) pcDEB-based eDNA library. (b) p500. p, An and S.J. denote promoter, poly(A) attachment signal and splicing junction, respectively, eDNA, shown by a hatched box, was inserted at a-site indicated. Arrows indicate directions of transcription. Amp r, ampicillin resistance; SV40, simian virus 40; An tk and An sv, herpes simplex virus thymidine kinase and SV40 poly(A) attachment signal, respectively.
The inserts were sequenced by the fluorescent dideoxy method using bacteriophage T7 DNA polymerase TM
(Prober et al., 1987; Sanger et al., 1977).
3. Results
(a) Acquisition of resistance to alkylating agents by eDNA transfection
A eDNA expression library was constructed with mRNA prepared from cultured human Raji (Mer +) cells. As the vector we used pcDEB, which possesses oriP, the replication origin of EB virus (Yates et al., 1984; Sugden et at., 1985; Reisman et al., 1985), and the origin of plasmid pBR322, together with the hygromycin B resistance gene and the ampicillin resistance gene, the former being used for selection in mammalian cells and the latter in bacterial cells (Blochlinger & Diggelmann, 1984). The eDNA was placed downstream from the strong SRa promoter (Takebe et al., 1988) and HeLa MR (Mer-) cells were transfected with the eDNA library. In an at tempt to achieve an autonomous replication of the construct, the p500 plasmid that carries the EBNA-I gene (Yates et at., 1985) was co- transfected. The organizations of the pcDEB-based eDNA library and the helper plasmid p500 are shown in Figure 1.
After transfection with 5/~g of p500 DNA and 500gg of eDNA library, cells (2x10 v) were subjected to screening for transformants by incu- bating in a medium containing 300 #g of hygro- mycin B/ml. When tiny colonies appeared, cells were washed with serum-free DME and treated with 10 #g of ACNU/mt in serum-free DME for one hour at 37 °C. ACNU was used as it selectively kills Mer- cells (Scudiero et at., 1984; Ishizaki et al., 1987). Cells were then incubated in medium containing hygro- mycin B for five to seven days and exposed to 50 #g of ACNU/ml. After repeating this hygromycin B/ACNU (50/~g/ml) selection twice, the number of colonies was counted (Table 1). Fifty colonies showing an increased resistance to both agents were obtained and some of them were cloned. No ACNU- resistant clone was found in the control, to which only the vector DNA had been applied.
The ACNU-resistant clones exhibited an increased resistance to MNNG, but the degree of resistance varied from clone to clone. We chose clone 1H3 with a distinctly high degree of resistance to MNNG, the degree being far greater than that for HeLa $3, the ordinary HeLa (Mer +) strain (Fig. 2(a)). Clone 1H3 showed a relatively high degree of resistance to ACNU, but its degree was less than that for HeLa $3 (Fig. 2(b)). The possible cause for this discrepancy will be discussed below.
Table 1 Screening of hygromycin B/ACN U double-resistant clones following eDNA
transfection
Transformation Number of Ratio of efficiency Number of ACNU'IHB ~ double resistants
DNA (%) HB' clones clones to HB r cells
Vectort 1"8 3"6 x 10 5 0 <2"7 x 10 -6 V e c t o r + e D N A library 1"6 3"2 x l0 s 50 1"6 x 10 -4
t As a negative control, 5/~g of p500 DNA and 500 ~g of t R N A were used for transfection.

742 H. Hayakawa et al.
]i
0-1
0,01
0 " 0 0 1 ~ I . , I , I , I ~ I , 0 I 2 _S 4 ,5 6
MNNG (/~g/ml) (a)
0 50 I00 150
ACNU (Fglml)
(b)
Figure 2. Sensitivity to alkylating agents. (a) Survivals after treatment with MNNG. (b) Survivals after treat- ment with ACNU. (&) HeLa $3 (Mer+); (O) HeLa MR (Mer-); (0) 1H3.
(b) Methyltransferase activity in the transformants
06-Methylguanine-DNA methyltransferase acti- vity was assayed with crude extracts of clone 1H3, as well as of the parental Mer- and the control Mer + strains. As shown in Figure 3(a), clone 1H3 had,
approximately, a 45 times higher level of enzyme activity compared with that of the ordinary Mer + strain, $3. This would explain the exceedingly high degree of resistance to MNNG of this clone.
To determine the molecular size of the methyl- transferase protein, the 1H3 extract was incubated with [SH]MNU-treated DNA and subjected to SDS/ polyacrylamide gel electrophoresis followed by fluorography. As shown in Figure 3(b), we noted a distinct band corresponding to a protein with molecular weight of approximately 25,000. In a control experiment, in which the reaction was per- formed with a purified preparation of bacterial Ada protein carrying methyltransferase activities (Nakabeppu et al., 1985), a band was detected at a position corresponding to the size of this protein (molecular weight 39,000).
(e) Recovery of cDNA for methyltransferase
In an attempt to recover cDNA for methyltrans- ferase, a Hirt extract (Hirt, 1967) was prepared from transformant clone 1H3 and applied to MR (Mer-) cells. No ACNU-resistant cells were obtained from the l0 s hygromycin B-resistant cells exam-
| e 0 •
0"8 t 0"8t r i 2 3 4
~" 6 9 - ~ ! i ~ : : , ' , , ~ ' ! ' : ' - :
u
/ o.~ . tK~'~ ; ~ i ~ ] ~ ~
0 IO0 ZOO ~00 400 500 ( b ) P r o t e | n ( ~ j . g )
(o)
Figure 3.06-Methylguanine-DNA methyltransferase activity of various strains. (a) Transfer of the methyl group from [3H]MNU-treated DNA to hot acid-insoluble fractions of the crude extracts. The enzyme activity is shown by the amount of methyl residues transferred. (A) HeLa $3; (0) HeLa MR; (O) 1H3. (b) Fluorography of the methyl- transferase reaction products. Reaction mixtures containing 50 #g of crude extract protein were subjected to electro- Ph0resis at 25 mA for 3 h. Lane l, MR; lane 2, $3; lane 3, 1H3; lane 4, 0"1 pg of purified Ada protein. [~4C]methylated proteins were used as molecular weight markers.

cDNA for Human Methyltran~ferase 743
ined. High-molecular-weight DNA was then extracted from 1H3 cells and subjected to Southern blot analysis using pcDEB as the probe. Distinct bands corresponding to the vector DNA appeared (data not shown), implying that the cDNA may be integrated into the chromosomal DNA, rather than being present in the plasmid.
The high-molecular-weight DNA was digested with restriction enzymes and used to transfect MR (Met-) cells by electroporation. A large number of hygromycin B-resistant transformants was obtained on transfection with KpnI or XbaI-digested 1H3 DNA, but not with similarly treated MR DNA (data not shown). When methyltransferase activity was assayed, 11 out of 12 independently isolated transformants contained very high levels of the activity and showed an extremely high degree of resistance to MNNG, comparable to that of the original 1H3 cells. Thus, it is evident that the cDNA for methyltransferase is integrated into the chromo- somal DNA and is present in a form closely linked to the hygromycin B resistance gene.
To recover the integrated cDNA from the chromosome, the approach illustrated in Figure 4(a) was followed. Since the vector sequences located on both sides of the cDNA are known, parts of these sequences were synthesized and used for primers to amplify the cDNA sequence by polymerase chain reaction. As shown in Figure 4(b), three fragments were detected in the first transformant 1H3, one of which retained the secondary transformants. Since this band was found in all the secondary trans- formant cells exhibiting high levels of methyl- transferase activity, it seems very likely that it carries cDNA for methyltransferase. From the rela- tive electrophoretic mobility, the size of the DNA was estimated to be 1.1 x l0 a bp, that is, sufficient for coding for a 25,000 M r methyltransferase pro- tein. The cDNA fragment was amplified from the secondary transformant, X2, and inserted into the Sinai site of plasmid pUC9. The resulting plasmid, which can be replicated in E. coli cells, was named pUC9MGMT, for it carries the eDNA for human 06-methylguanine-DNA methyltransferase {MGMT).
(d) Expression of the cDNA on an autonomously replicating vector
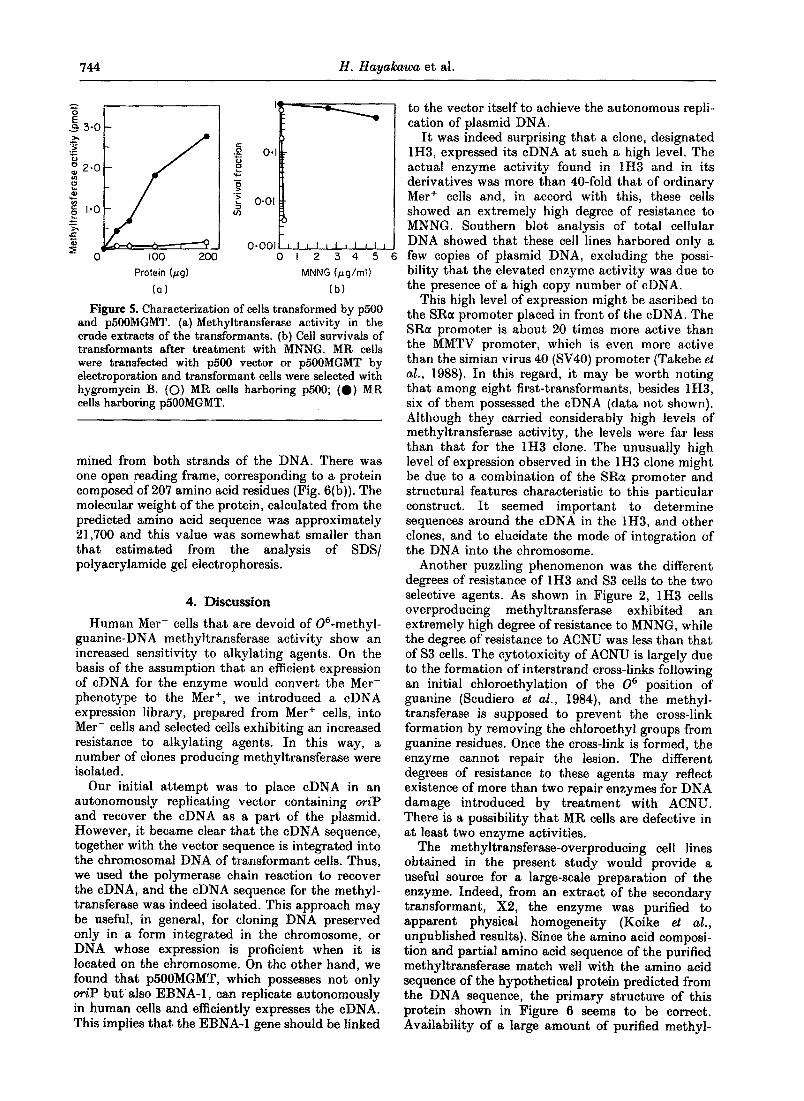
The 1"1 x 10 a bp DNA fragment carrying the cDNA was excised from pUC9MGMT by SalI and HpaI digestion and inserted into the XhoI-KpnI site of the cDNA expression vector, p500. This construct was named p500MGMT, in which the cDNA can be expressed under the control of the RSV LTR promoter. When MR (Mer-) cells were transfected with p500MGMT and selected by hygro- mycin B, a number of transformants exhibiting a distinctly high degree of resistance to MNNG and possessing high levels of methyltransferase activity were obtained (Fig. 5).
Plasmid p500MGMT can be recovered from the Hirt extract {data not shown), thereby indicating
5" GCCT'n'ACITCTAGGCCTGTACGGAAGTG -3' /
SR .[ cDHA An -.4e,,.-
I co
PCR 3'-ATfCGACGTfATI~GTTCAATTGTrGTrG-5'
(a)
1 2 3 4 5 6 7
(b) Figure 4. Amplification of eDNA for 06-methyl -
guanine-DNA methyltransferase. (a)The strategy for amplification by polymerase chain reaction (PCR). SR and An denote the SRa promoter and the SV40 poly(A) attachment site of the vector, respectively. (b) Electro- phoretic analyses of amplified DNA products from the 1st and the secondary transformant DNAs. Lane l, 2 DNA digested with HindIII as markers; lane 2, MR DNA; lane 3, $3 DNA; lane 4, DNA of the 1st transformant clone 1H3; lanes 5 to 7, DNAs of the secondary transformants.
that the plasmid replicates autonomously in human cells once the EBNA-1 is present on the plasmid. This suggests that the EBNA-1 must be linked to the vector DNA to allow the autonomous replication.
(e) Nucleotide sequence of the cDNA for methyltransferase
Nucleotide sequence of the cDNA was determined by using an automatic sequencer. The strategy for sequence analysis is shown in Figure 6(a). All of the sequence corresponding to the cDNA was deter-
744 H. Hayakawa et al.
o E
~ 5 - 0
-~.
~' 2.0
0
f f I00 200
Protein (pg) (a)
8 : = fJ o
( / }
Ii
0"1
0.01
0-001 I t I ~ I ~ I ~ I t
0 I 2 5 4 5
MNNG (pg/ml)
(b)
Figure 5. Characterization of cells transformed by p500 and p500MGMT. (a)Methyltransferase activity in the crude extracts of the transformants. (b) Cell survivals of transformants after treatment with MNNG. MR cells were transfected with p500 vector or p500MGMT by electroporation and transformant cells were selected with hygromycin B. (O) MR cells harboring pS00; (0) MR cells harboring p500MGMT.
mined from both strands of the DNA. There was one open reading frame, corresponding to a protein composed of 207 amino acid residues (Fig. 6(b)). The molecular weight of the protein, calculated from the predicted amino acid sequence was approximately 21,700 and this value was somewhat smaller than that estimated from the analysis of SDS/ polyacrylamide gel electrophoresis.
4. Discussion
Human Mer- cells that are devoid of O~-methyl - guanine-DNA methyltransferase activity show an increased sensitivity to alkylating agents. On the basis of the assumption that an efficient expression of eDNA for the enzyme would convert the Mer- phenotype to the Mer +, we introduced a eDNA expression library, prepared from Met + cells, into Mer- cells and selected cells exhibiting an increased resistance to alkylating agents. In this way, a number of clones producing methyltransferase were isolated.
Our initial attempt was to place eDNA in an autonomously replicating vector containing oriP and recover the eDNA as a part of the plasmid. However, it became clear that the eDNA sequence, together with the vector sequence is integrated into the chromosomal DNA of transformant cells. Thus, we used the polymerase chain reaction to recover the eDNA, and the eDNA sequence for the methyl- transferase was indeed isolated. This approach may be useful, in general, for cloning DNA preserved only in a form integrated in the chromosome, or DNA whose expression is proficient when it is located on the chromosome. On the other hand, we found that p500MGMT, which possesses not only oriP but' also EBNA-1, can replicate autonomously in human cells and efficiently expresses the eDNA. This implies that the EBNA-1 gene should be linked
to the vector itself to achieve the autonomous repli- cation of pla~mid DNA.
I t was indeed surprising that a clone, designated 1H3, expressed its eDNA at such a high level. The actual enzyme activity found in 1H3 and in its derivatives was more than 40-fold that of ordinary Mer + cells and, in accord with this, these cells showed an extremely high degree of resistance to MNNG. Southern blot analysis of total cellular DNA showed that these cell lines harbored only a few copies of plasmid DNA, excluding the possi- bility that the elevated enzyme activity was due to the presence of a high copy number of eDNA.
This high level of expression might be ascribed to the SRa promoter placed in front of the eDNA. The SRa promoter is about 20 times more active than the MMTV promoter, which is even more active than the simian virus 40 (SV40) promoter (Takebe et al., 1988). In this regard, it may be worth noting that among eight first-transformants, besides 1H3, six of them possessed the eDNA (data not shown). Although they carried considerably high levels of methyltransferase activity, the levels were far less than that for the 1H3 clone. The unusually high level of expression observed in the 1H3 clone might be due to a combination of the SRa promoter and structural features characteristic to this particular construct. I t seemed important to determine sequences around the eDNA in the IH3, and other clones, and to elucidate the mode of integration of the DNA into the chromosome.
Another puzzling phenomenon was the different degrees of resistance of 1H3 and $3 cells to the two selective agents. As shown in Figure 2, 1H3 Cells overproducing methyltransferase exhibited an extremely high degree of resistance to MNNG, while the degree of resistance to ACNU was less than that of $3 cells. The cytotoxicity of ACNU is largely due to the formation of interstrand cross-links following an initial chloroethylation of the 06 position of guanine (Scudiero et al., 1984), and the methyl- transferase is supposed to prevent the cross-link formation by removing the chloroethyl groups from guanine residues. Once the cross-link is formed, the enzyme cannot repair the lesion. The different degrees of resistance to these agents may reflect existence of more than two repair enzymes for DNA damage introduced by treatment with ACNU. There is a possibility that MR cells are defective in at least two enzyme activities.
The methyltransferase-overproducing cell lines obtained in the present s tudy would provide a useful source for a large-scale preparation of the enzyme. Indeed, from an extract of the secondary transformant, X2, the enzyme was purified to apparent physical homogeneity (Koike et al., unpublished results). Since the amino acid composi- tion and partial amino acid sequence of the purified methyltransferase match well with the amino acid sequence of the hypothetical protein predicted from the DNA sequence, the primary structure of this protein shown in :Figure 6 seems to be correct. Availability of a large amount of purified methyl-

Aatl(S/ut) SocI 8omHI ~
(o)
(A).
CCCCCCCCGCCCCCCCCGCCGCCCCTTGGTACTTGGAAAA 40
41 ATG GAC AAG GAT TGT GAA ATG AAA CGC ACC ACA CTG GAC AGC CCT 85 1 Met Asp Lys Asp Cys Glu Met Lys Arg Thr Thr Leu Asp Ser Pro 15
8~ TTG GGG AAG CTG GAG CTG TCT GGT TGT GAG CAG GGT CTG CAC GAA 130 16 Leu Gly Lys Leu Glu Leu Ser Gly Cys Glu Gln Gly Leu His Glu 30
131 ATA AAG CTC CTG GGC AAG GGG ACG TCT GCA GCT GAT GCC GTG GAG 175 31 Ile Lys Leu Leu Gly Lys Gly Thr Ser Ala Ala Asp Ala Val Glu 45
176 GTC CCA GCC CCC GCT GCG GTT CTC GGA GGT CCG GAG CCC CTG ATG 220 46 Val Pro Ala Pro Ala Ala Val Leu Gly Gly Pro Glu Pro Leu Met 60
221 CAG TGC ACA GCC TGG CTG AAT GCC TAT TTC CAC CAG CCC GAG GCT 265 61 Gln Cys Thr Ala Trp Leu Asn Ala Tyr Phe His Gln Pro Glu Ala 75
266 ATC GAA GAG TTC CCC GTG CCG GCA CTT CAC CAT CCC GTT TTC CAG 310 76 Ile Glu Glu Phe Pro Val Pro Ala Leu His His Pro Val Phe Gln 90
311 CAA GAG TCG TTC ACC AGA CAG GTG TTA TGG AAG CTG CTG AAG GTT 355 91 Gln Glu Ser Phe Thr Arg Gln Val Leu Trp Lys Leu Leu Lys Val 105
356 GTG AAA TTC GGA GAA GTG ATT TCT TAC CAG CAA TTA GCA GCC CTG 400 106 Val Lys Phe Gly Glu Val Ile Ser Tyr Gln Gln Leu Ala Ala Leu 120
401 GCA GGC AAC CCC AAA GCC GCG CGA GCA GTG GGA GGA GCA ATG AGA 445 121 Ala Gly Asn Pro Lys Ala Ala Arg Ala Val Gly Gly Ala Met Arg 135
446 GGC AAT CCT GTC CCC ATC CTC ATC CCG TGC CAC AGA GTG GTC TGC 490 136 Gly Asn Pro Val Pro Ile Leu Ile Pro Cys His Arg Val Val Cys 150
491 AGC AGC GGA GCC GTG GGC AAC TAC TCC GGA GGA CTG GCC GTG AAG 535 151 Ser Ser Gly Ala Val Gly Asn Tyr Ser Gly Gly Leu Ala Val Lys 165
536 GAA TGG CTT CTG GCC CAT GAA GGC CAC CGG TTG GGG AAG CCA GGC 580 166 Glu Trp Leu Leu Ala His Glu Gly His Arg Leu Gly Lys Pro Gly 180
581 TTG GGA GGG AGC TCA GGT CTG GCA GGG GCC TGG CTC AAG GGA GCG 625 181 Leu Gly Gly Ser Ser Gly Leu Ala Gly Ala Trp Leu Lys Gly Ala 195
626 GGA GCT ACC TCG GGC TCC CCG CCT GCT GGC CGA AAC TGA GTATGTG 671 196 Gly Ala Thr Ser,Gly Ser Pro Pro Ala Gly Arg Asn *** 207
672 CAGTAGGATGGATGTTTGAGCGACACACACGTGTAACACTGCATCGGATGCGGGGCGTG 730
731 GAGGCACCGCTGTATTAAAGGAAGTGGCAGTGTCCTGGG~ 789
790 AA 791
(b)
Figure 6. Nueleotide sequence of eDNA for methyltransferase. (a) Organization of the eDNA and strategy for DNA sequencing. Open box indicates vector sequences. The cross-hatched box indicates the coding sequence and stippled boxes indicate the non-translated regions. Horizontal arrows show the directions and the regions of the sequence analyzed. (b) Nucleotide sequence of the cDNA and the deduced amino acid sequence.

746 H. H a y a k a w a et al.
( o )
cr' , (MGMT)
( M) o,,,,°,.,.
C H )
/ \
( b t " \ t
/ \ t \
' .o .
I,t :vo i:ppo,F,Ip,si,l vF<,.,,vi , Kp_~VI~AVGI OIAIN KR INI 0 LP I [ ~ V ~ H R V l l Oat
iS. subtllls Dat protein
Figure 7. Comparison of the predicted amino acid sequences of 4 methyltransferases from human and prokaryotes. The homologous regions are stippled in (a) or boxed in (b). * denotes the methyl accepter site of Ada protein. The data for bacterial methyltransferases were taken from Nakabeppu et al. (1985), Potter et al. (1987) and Kodama et al. (1989).
transferase protein would facilitate characterization of the enzyme as well as production of the antibody.
The methyltransferase enzyme is widely distri- buted in biological systems, and to date three genes coding for the enzyme have been cloned from bacteria. Among them the Ada protein of E. cell, which carries two distinct methyltransferase activi- ties, has been studied most extensively. A specific cysteine residue close to the carboxy terminus of the protein (Cys321) proved to be an accepter site for 06-methylguanine and 04-methylthymine, while a cysteine residue that is present near the amino terminus (Cys69) accepts a methyl group from one of the two stereoisomers of methylphosphotriester in alkylated DNA (Tee et al., 1986; Takano et al., 1988). The other two methyltransferases, Ogt pro- tein of E, coli and Dat protein of B. subtilis, carry only the former activity, and their possible methyl acceptor sites were deduced from their sequence homology with the Ada protein {Potter et al., 1987; Kodama et al., 1989; Morohoshi et al., 1989). Inspec- tion of the predicted amino acid sequence for the human methyltransferase revealed that there is a high homology with the bacterial enzymes. In particular, the five consecutive amino acid residues around the possible methyl accepter sites were conserved, and two additional amino acid residues surrounding this sequence were identical with those of the Ada protein (Fig. 7). I t is most likely that the eysteine residue within this sequence is the accepter site for a methyl group from the 06-methylguanine of methylated DNA.
Many cell lines established from human cancers show the Mer- phenotype (Day et al., 1980; Yarosh et al., 1983; Scudiero et al., 1984). Conversion of Mer + cells to Mer- may occur as a consequence of stress or malignant transformation in culture, but the Met- cells may be derived from the cancer tissues. The molecular basis for the conversion may involve switching off transcription of the methyl- transferase gene. To elucidate the mechanism it will be necessary to compare the gene and its surrounding sequences in Met + and Met- cells. The successful cloning of the cDNA should make feasible such endeavors.
The vectors used were designed and prepared while H.H. was a visiting fellow in the laboratory of Paul Berg at Stanford University, and we are especially grateful to P. Berg. We also thank K. Arai, N. Arai, Y. Takebe, T. Yokota, K. Yokota, M. Ikenaga and M. Ohara for advice. This work was supported by a grant from the Ministry of Education, Science and Culture (61065007).
References
Bloehlinger, K. & Diggelmann, H. (1984). Mol. Cell. Biol. 4, 2929-2931.
Brennand, J. & Margison, G. P. (1986). Prec. Nat. Aead. Sci., U.S.A. 83, 6292-6296.
Chu, G., Hayakawa, H. & Berg, P. (1987). Nuel. Acids Res. 15, 1311-1326.
Coulondre, C. & Miller, J. H. (1977). J. Mol. Biol. 117, 577-606.
Day, R. S., III, Ziolkowski, C. H. J., Scudiero, D. A., Meyer, S. A.,-Lubinieeki, A. S., Girardi, A. J., Galloway, S. M. & Bynum, G. D. (1980). Nature (London), 288, 724-727.
Demple, B., Sedgwick, B., Robins, P., Totty, N., Waterfield, M. D. & Lindahl, T. (1985). Proc. Nat. Acad. Sci., U.S.A. 82, 2688-2692.
Hirt, B. (1967). J. Mol. Biol.. 26, 365-369. Ishizaki, K., Tsujimura, T., Yawata, H., Fujio, C.,
Nakabeppu, Y., Sekiguchi, M. & Ikenaga, M. (1986). Murat. Res. 166, 135-141.
Ishizaki, K., Tsujimura, T., Fujio, C., Yangpei, Z., Yawata, H., Nakabeppu, Y., Sekiguchi, M. & Ikenaga, M. (1987). Murat. Res. 184, 121-128.
Kataoka, H., Hall, J. & Karran, P. (1986). E M B O J. 5, 3195-3200.
Kodama, K., Nakabeppu, Y. & Sekiguchi, M. (1989). Murat Res. 218, 153-163.
Lindahl, T., Sedgwick, B., Sekiguehi, M. & Nakabeppu, Y. (1988). Annu. Rev. Biochem. 57, 133-157.
Margison, G. P., Cooper, D. P. & Brennand, J. (1985). Nucl. Acids Res. 13, 1939-1952.
Morohoshi, F., Hayashi, K. & Munakata, N. (1989). Nuel. Acids Res. 17, 6531-6543.
Nakabeppu, Y. & Sekiguehi, M. (1986). Proc. Nat. Acad. Sci., U.S.A. 83, 6297-6301.
Nakabeppu, Y., Kondo, H., Kawabata, S., Iwanaga, S. & Sekiguchi, M. (1985). J. Biol. Chem. 260, 7281-7288.
Okayama, H. & Berg, P. (1983). Mol. Cell. Biol. 3, 280-289.
Perucho, M., Goldfarb, M., Shimizu, K., Lama, C., Fogh, J. & Wigler, M. (1981). Cell, 27, 467-476.
Potter, P. M., Wilkinson, M. C., Fitton, J., Carr, F. J., Brennand, J., Cooper, D. P. & Margison, G. P. (1987). Nucl. Acids Res. 15, 9177-9193.

cDNA for Human Methyltransferase 747
Prober, J. M., Trainor, G. L., Dam, R. J., Hobbs, F. W., Robertson, C. W., Zagursky, R. J., Coeuzza, A. J., Jensen, M. A. & Baumeister, K. (1987). Science, 238, 336-341.
Reisman, D., Yates, J. & Sugden, B. (1985). Mol. Cell. Biol. 5, 1822-1832.
Saiki, R. K., Gelfand, D. H., Stoffel, S., Seharf, S. J., Higuehi, R., Horn, G. T., Mullis, K. B. & Erlieh, H. A. (1988). Science, 239, 487-491.
Samson, L., Derfier, B. & Waldstein, E. A. (1986). Proc. Nat. Acad. Sci., U.S.A. 83, 5607-5610.
Sanger, F., Nicklen, S. & Coulson, A. R. (1977). Proe. Nat. Acad. Sci., U.S.A. 74, 5463-5467.
Seudiero, D. A., Meyer, S. A., Clatterbuck, B. E., Mattern, M. R., Ziolkowski, C. H. J. & Day, R. S., III (1984). Cancer Res. 44, 2467-2474.
Strauss, B., Scudiero, D. & Henderson, E. (1975). In Molecular Mechanisms for Repair of DNA, part A (Hanawalt, P. C. & Setlow, R,. B., eds), pp. 13-24, Plenum Press, New York.
Sugden, B., Marsh, K. & Yates, J. (1985). Mol. Cell. Biol. 5,410-413.
Sukumar, S., Notario, V., Martin-Zanea, D. & Barbacid, M. (1983). Nature (London), 306, 658-661.
Takano, K., Nakabeppu, Y. & Sekiguehi, M. (1988). J. Mol. Biol. 201,261-271:
Takebe, Y., Seiki, M., Fujisawa, J., Hoy, P., Yokota, K., Arai, K., Yoshida, M. & Arai, N. (1988). Mol. Ceil. Biol. 8, 466-472.
Teo, I., Sedgwick, B., Kilpatriek, M. W., McCarthy, T. V. & Lindahl, T. (1986). Cell, 45, 315-324.
Yarosh, D. B., Foote, R. S., Mitra, S. & Day, R. S., I I I (1983). Carcinoyene,~is, 4, 199-205.
Yates, J., Warren, N., Reisman, D. & Sugden, B. (1984). Proc. Nat. Acad. Sci., U.S.A. 81, 3806-3810.
Yates, J. L., Warren, N. & Sugden, B. (1985). Nature (London), 313,812-815.
Edited by K. Matsubara