fce tema 3
TRANSCRIPT

Módulo 1. Módulo 1. SÍNTESIS ORGÁNICA.Tema 1. COMPUESTOS ORGÁNICOS DE NITRÓGENO QUE NO DERIVAN DEL AMONÍACO.-Nitrocompuestos. Estructura y propiedades físicas. Nitroalcanos. Reacciones. Nitroarenos.Preparación y Reacciones.- Isonitrilos. Nitrilos: Preparación y Reacciones.- Azidas. Preparacióny Reacciones.- Diazocompuestos. Reacciones.Tema 2. COMPUESTOS ORGÁNICOS DE AZUFRE.- Introducción.- Tioles y sulfuros.- Sulfóxidos ysulfonas.- Carbaniones estabilizados por azufre.- Sales de sulfonio.- Ácidos sulfónicos yderivados.Tema 3. COMPUESTOS ORGÁNICOS DE FÓSFORO Y SILICIO.- Reacciones de olefinación decompuestos carbonílicos.- Compuestos orgánicos del fósforo.- Compuestos orgánicos del Si.Tema 4. SÍNTESIS ORGÁNICA I.- Introducción a la síntesis orgánica.- Reacciones orgánicascomo instrumento de síntesis.- Interconversión de grupos funcionales.- Reacciones deformación de enlaces C-C.- Adición conjugada a compuestos carbonílicos a,b-insaturados.Tema 5. SÍNTESIS ORGÁNICA II.- Selectividad en Química Orgánica.- Conceptos de gruposprotector, activadores e inductores.- Protección de grupos funcionales.Tema 6. SÍNTESIS ORGÁNICA III.- Introducción.- Síntesis lineal y convergente.- Planteamientodel diseño sintético. Molécula Objetivo. Concepto de sintón y de reactivo equivalente. Análisisretrosintético.- Reglas que han de presidir un buen análisis retrosintético.- Interconversión degrupos funcionales (IGF).- Desconexiones 1,2 y 1,3-difuncionales (1,2-diX y 1,3-diX).-Desconexiones C-C: 1,1 C-C y 1,2 C-C.- Sintones donadores y aceptores.- Reactividad normal(lógica) y reactividad Umpolung (ilógica).- Transposiciones en síntesis.- Ciclación.-Polimerización.
TEMA 3
1

TEMA 3. COMPUESTOS ORGÁ-NICOS DE FÓSFORO Y SILICIO
2
1. Compuestos orgánicos de fósforo
1.1. Fosfinas. Estructura y reactividad
1.2. Reacción de Staudinger
1.3. Reacción de Mitsunobu
1.4. Sales de alquilfosfonio e iluros de fósforo
1.5. Reacción de Wittig
1.6. Ésteres fosfato
1.7. Fosfonatos. Reacción de Horner-Emmons
2. Compuestos orgánicos de silicio
2.1. Estructura y preparación
2.2. Reacciones
2.2.1.Sustitución nucleófila en el silicio
2.2.2. Silil enol éteres
2.2.3. Efecto estabilizante del silicio. Carbocationes. Carbaniones
2.2.4. Rotura electrófila del enlace carbono-silicio
2.2.5. Reacción de Peterson
3. Sumario de reacciones de olefinación de compuestos carbonílicos
Bibliografía:
Warren,
Streitwieser,
Carruthers

3
1. Compuestos orgánicos de fósforo
• Son compuestos orgánicos de P los que contienen enlaces C-P.
• El átomo de P tiene orbitales d accesibles, es más grande, más
polarizable y más electropositivo que el nitrógeno.
1.1. Fosfinas. Estructura y reactividad
• Las fosfinas son los análogos de fósforo de las aminas
• Muchas son comerciales
• Las fosfinas terciarias son las más importantes: a) Son configuracionalmente
estables a temperatura ambiente, b) Son ligandos excelentes.
Estructura
PH2
Etilfosfina
PH
Di-n-butilfosfina
Ph3P
Trifenilfosfina
PH H
H
93º
P
99º
PPh
Ph
103ºÁngulo C-P-C
PhSon más piramidales
que las aminas
Reactividad: Basicidad y Nucleofilia
Las fosfinas son menos básicas que las aminas pero son más nucleófilas
4
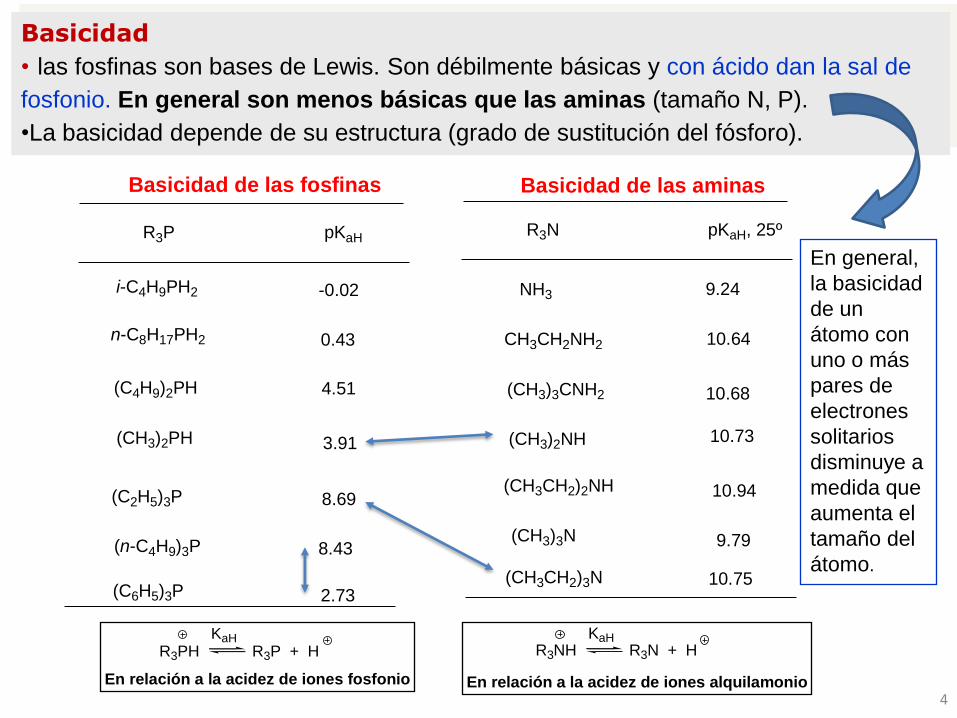
Basicidad
• las fosfinas son bases de Lewis. Son débilmente básicas y con ácido dan la sal de
fosfonio. En general son menos básicas que las aminas (tamaño N, P).
•La basicidad depende de su estructura (grado de sustitución del fósforo).
R3PH R3P + HKaH
En relación a la acidez de iones fosfonio
R3NH R3N + HKaH
En relación a la acidez de iones alquilamonio
Basicidad de las fosfinas
pKaHR3P
(C2H5)3P 8.69
(n-C4H9)3P 8.43
(C4H9)2PH 4.51
(CH3)2PH 3.91
n-C8H17PH2 0.43
(C6H5)3P 2.73
Basicidad de las aminas
pKaH, 25º
NH3 9.24
CH3CH2NH2 10.64
(CH3)3CNH2 10.68
10.73(CH3)2NH
(CH3CH2)2NH 10.94
(CH3)3N 9.79
(CH3CH2)3N 10.75
R3N
-0.02i-C4H9PH2
En general,
la basicidad
de un
átomo con
uno o más
pares de
electrones
solitarios
disminuye a
medida que
aumenta el
tamaño del
átomo.
5
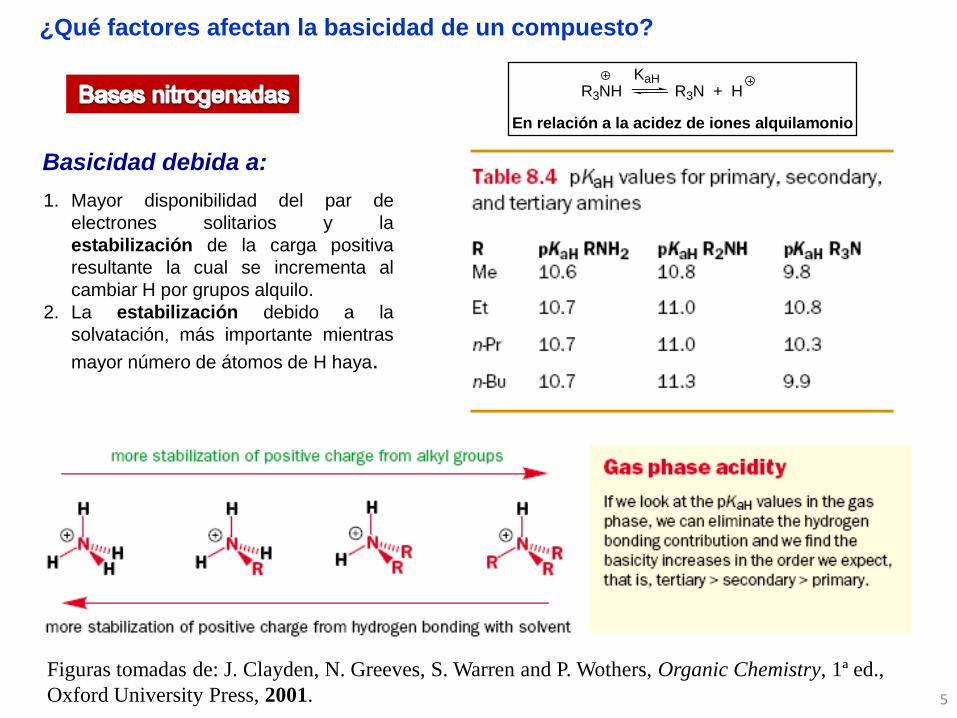
1. Mayor disponibilidad del par de
electrones solitarios y la
estabilización de la carga positiva
resultante la cual se incrementa al
cambiar H por grupos alquilo.
2. La estabilización debido a la
solvatación, más importante mientras
mayor número de átomos de H haya.
Basicidad debida a:
R3NH R3N + HKaH
En relación a la acidez de iones alquilamonio
¿Qué factores afectan la basicidad de un compuesto?
Figuras tomadas de: J. Clayden, N. Greeves, S. Warren and P. Wothers, Organic Chemistry, 1ª ed.,
Oxford University Press, 2001.
6
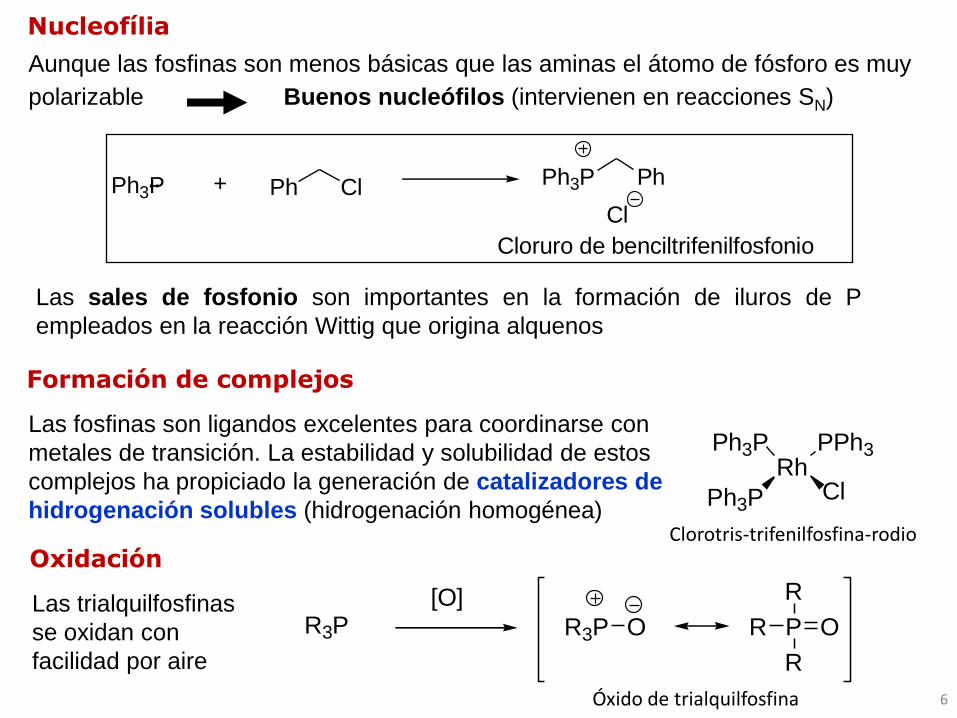
Ph3P + ClPh Ph3P Ph
Cl
Cloruro de benciltrifenilfosfonio
Aunque las fosfinas son menos básicas que las aminas el átomo de fósforo es muy
polarizable Buenos nucleófilos (intervienen en reacciones SN)
Las sales de fosfonio son importantes en la formación de iluros de P
empleados en la reacción Wittig que origina alquenos
¨
Formación de complejos
RhPPh3Ph3P
ClPh3P
R3P[O]
R3P O P O
R
R
R
Oxidación
Nucleofília
Las fosfinas son ligandos excelentes para coordinarse con
metales de transición. La estabilidad y solubilidad de estos
complejos ha propiciado la generación de catalizadores de
hidrogenación solubles (hidrogenación homogénea)Clorotris-trifenilfosfina-rodio
Las trialquilfosfinas
se oxidan con
facilidad por aire
Óxido de trialquilfosfina
7
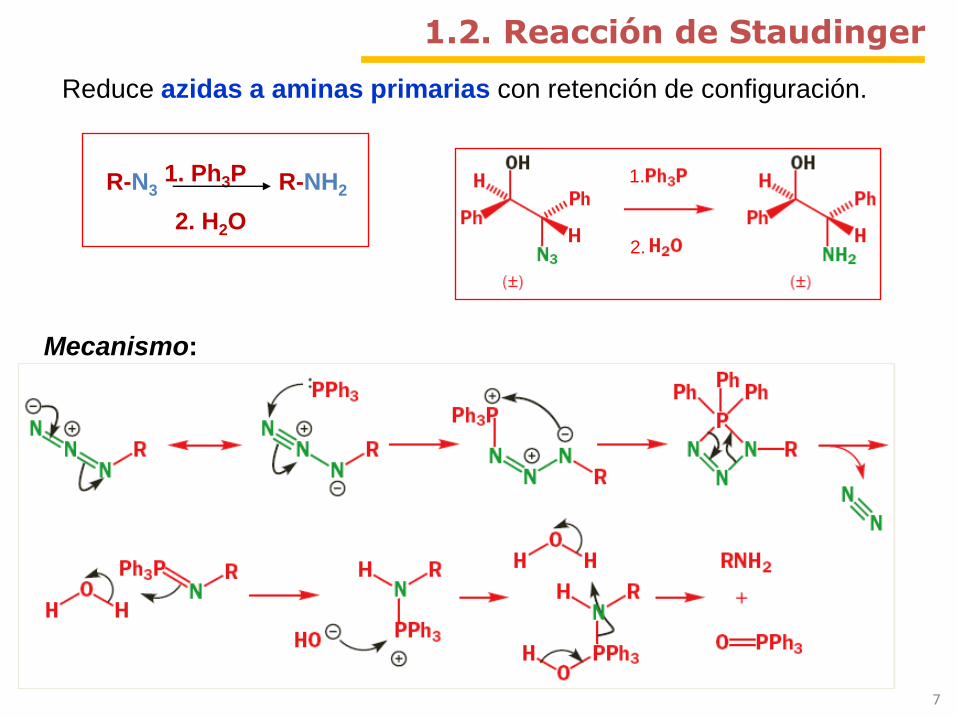
1.
2.
Mecanismo:
R-N3 R-NH21. Ph3P
2. H2O
1.2. Reacción de Staudinger
Reduce azidas a aminas primarias con retención de configuración.
8
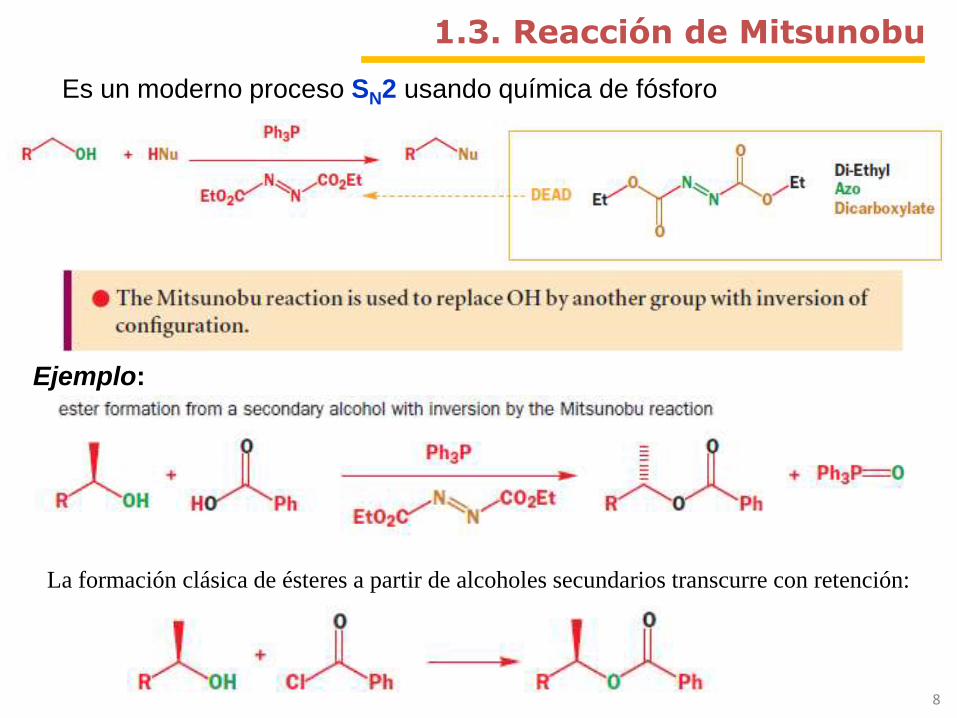
1.3. Reacción de Mitsunobu
Es un moderno proceso SN2 usando química de fósforo
La formación clásica de ésteres a partir de alcoholes secundarios transcurre con retención:
Ejemplo:
9
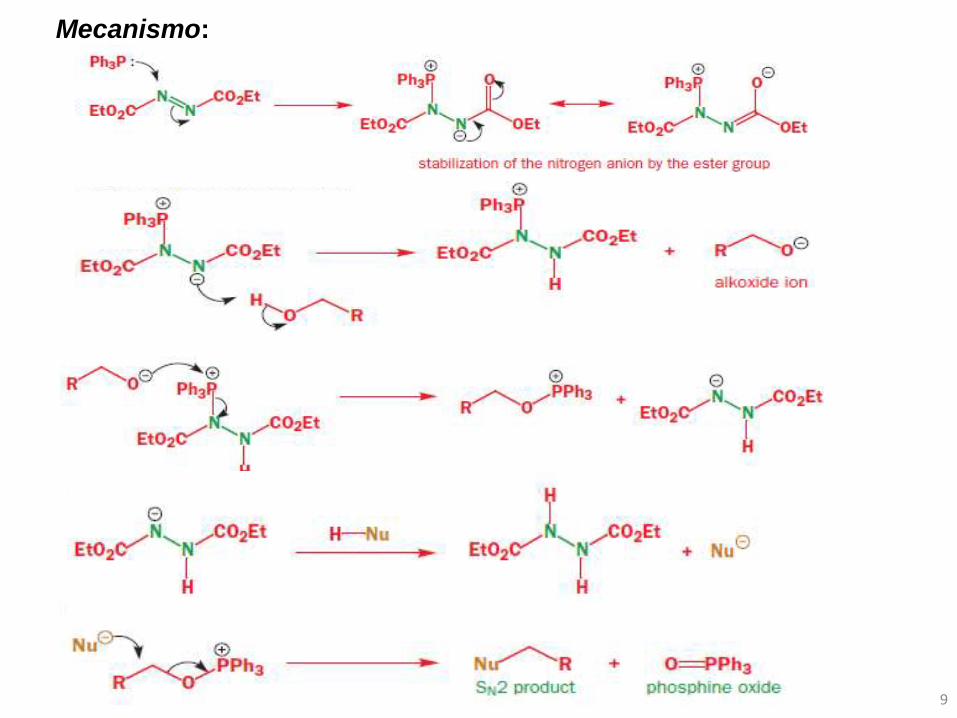
Mecanismo:
10
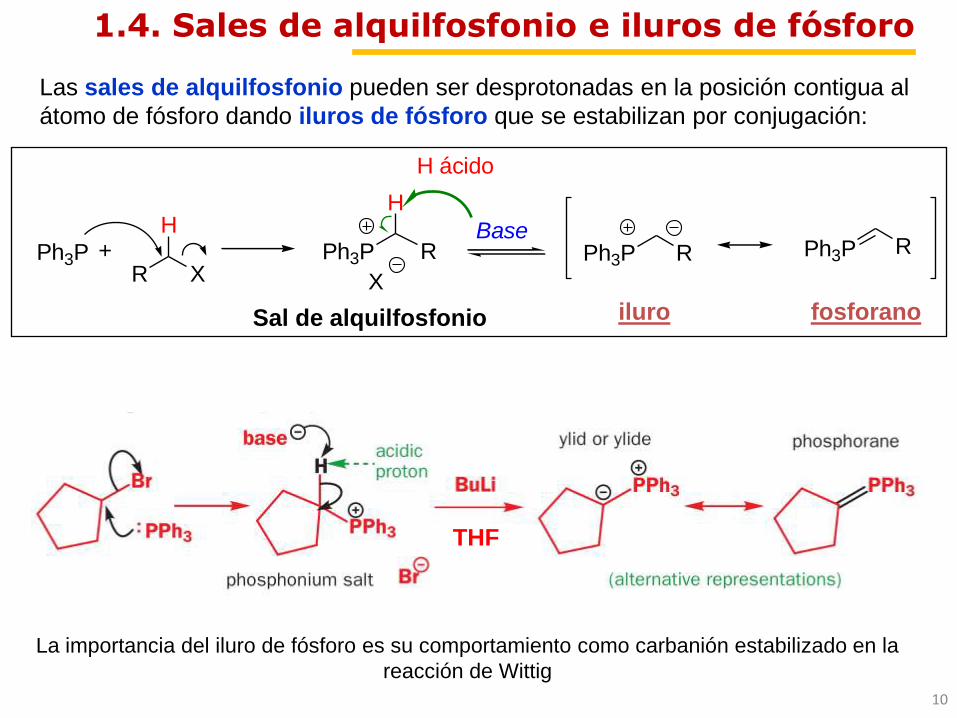
1.4. Sales de alquilfosfonio e iluros de fósforo
iluro fosforanoSal de alquilfosfonio
Ph3P +R X
Ph3P
X
BasePh3P R Ph3P R
HR
H
H ácido
THF
Las sales de alquilfosfonio pueden ser desprotonadas en la posición contigua al
átomo de fósforo dando iluros de fósforo que se estabilizan por conjugación:
La importancia del iluro de fósforo es su comportamiento como carbanión estabilizado en la
reacción de Wittig
11
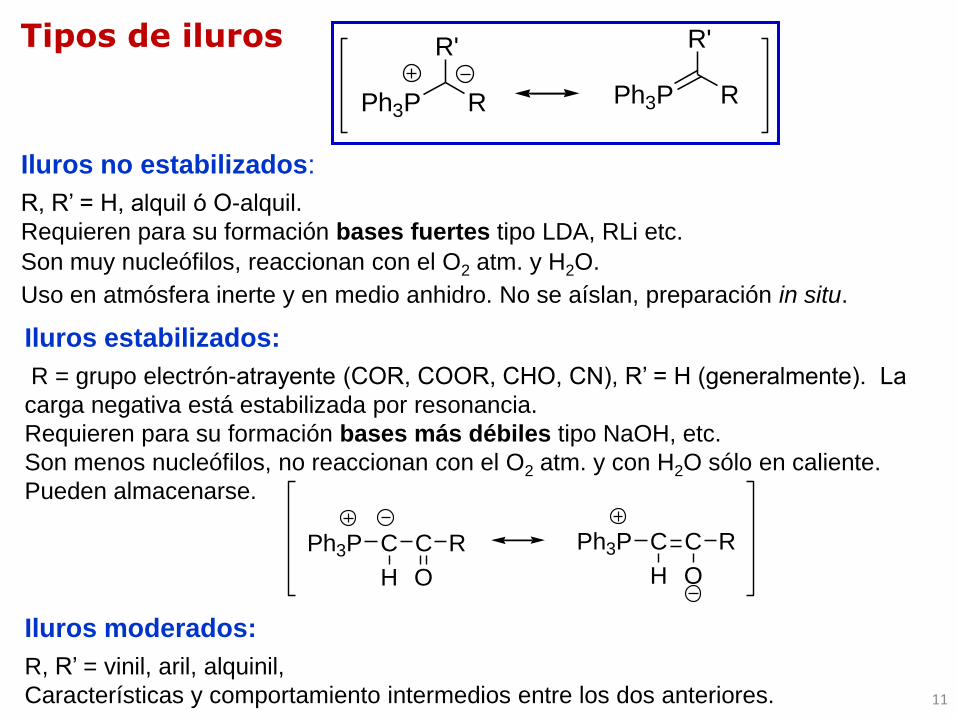
Iluros no estabilizados:
R, R’ = H, alquil ó O-alquil.
Requieren para su formación bases fuertes tipo LDA, RLi etc.
Son muy nucleófilos, reaccionan con el O2 atm. y H2O.
Uso en atmósfera inerte y en medio anhidro. No se aíslan, preparación in situ.
Tipos de iluros
Iluros estabilizados:
R = grupo electrón-atrayente (COR, COOR, CHO, CN), R’ = H (generalmente). La
carga negativa está estabilizada por resonancia.
Requieren para su formación bases más débiles tipo NaOH, etc.
Son menos nucleófilos, no reaccionan con el O2 atm. y con H2O sólo en caliente.
Pueden almacenarse.
Ph3P C C
H O
R Ph3P C C
H
R
O
Iluros moderados:
R, R’ = vinil, aril, alquinil,
Características y comportamiento intermedios entre los dos anteriores.
Ph3P R Ph3P R
R' R'
12
Mecanismo
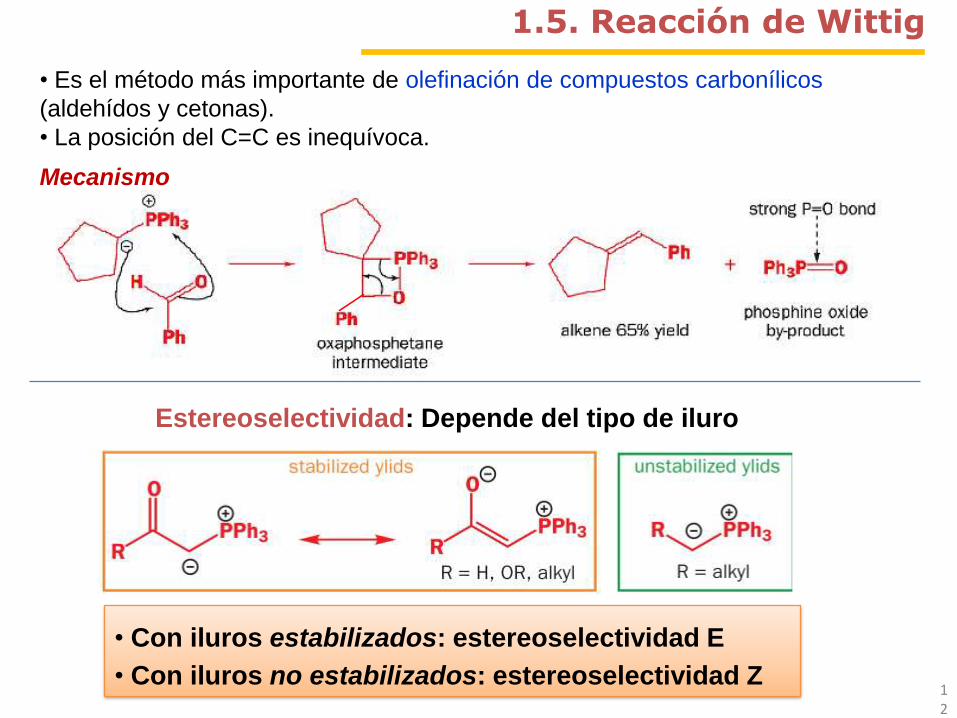
1.5. Reacción de Wittig
• Es el método más importante de olefinación de compuestos carbonílicos
(aldehídos y cetonas).
• La posición del C=C es inequívoca.
Estereoselectividad: Depende del tipo de iluro
• Con iluros estabilizados: estereoselectividad E
• Con iluros no estabilizados: estereoselectividad Z
13
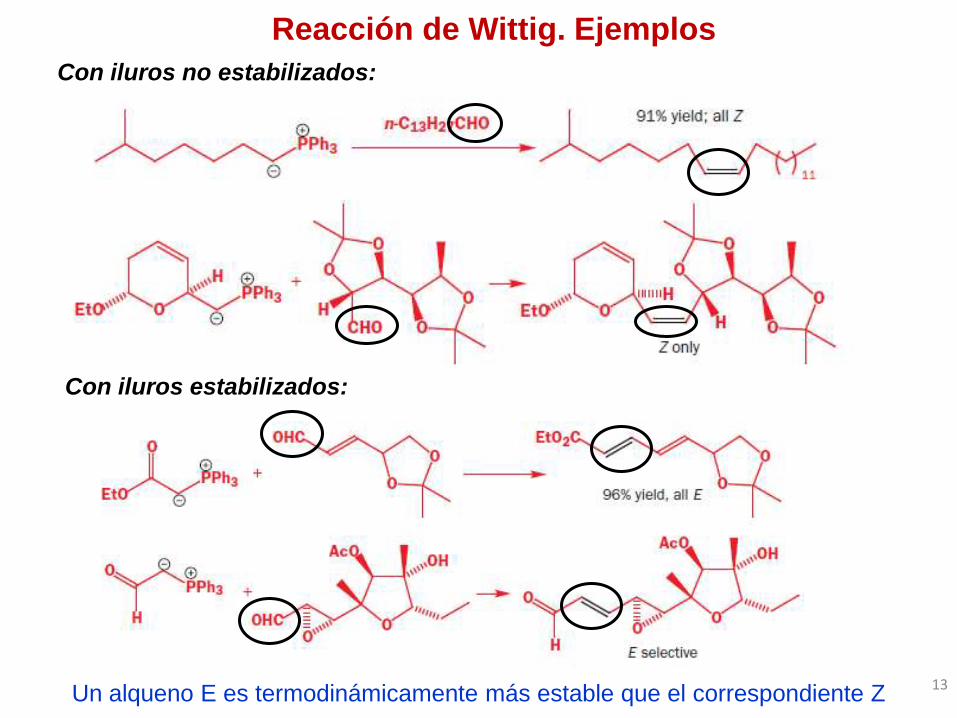
Reacción de Wittig. Ejemplos
Con iluros no estabilizados:
Con iluros estabilizados:
Un alqueno E es termodinámicamente más estable que el correspondiente Z
14
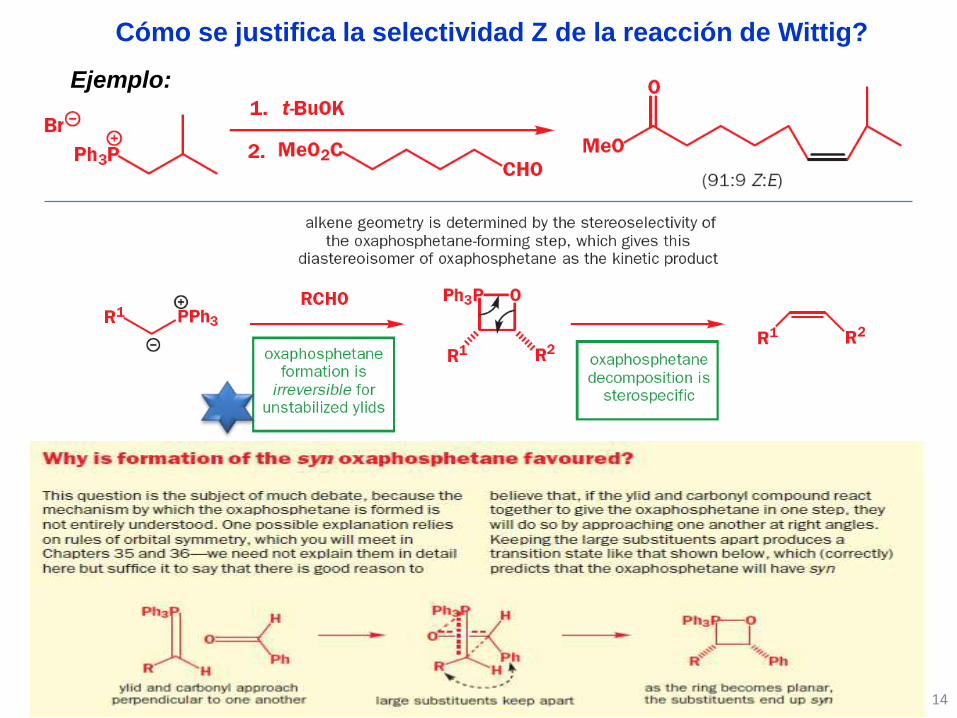
Cómo se justifica la selectividad Z de la reacción de Wittig?
Ejemplo:
15
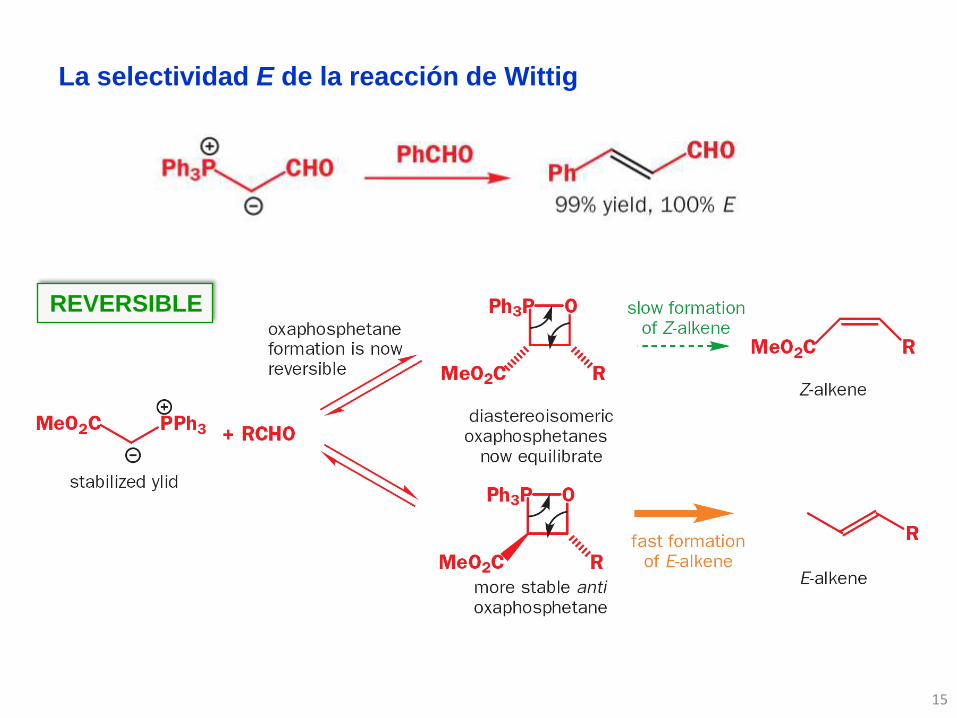
La selectividad E de la reacción de Wittig
REVERSIBLE
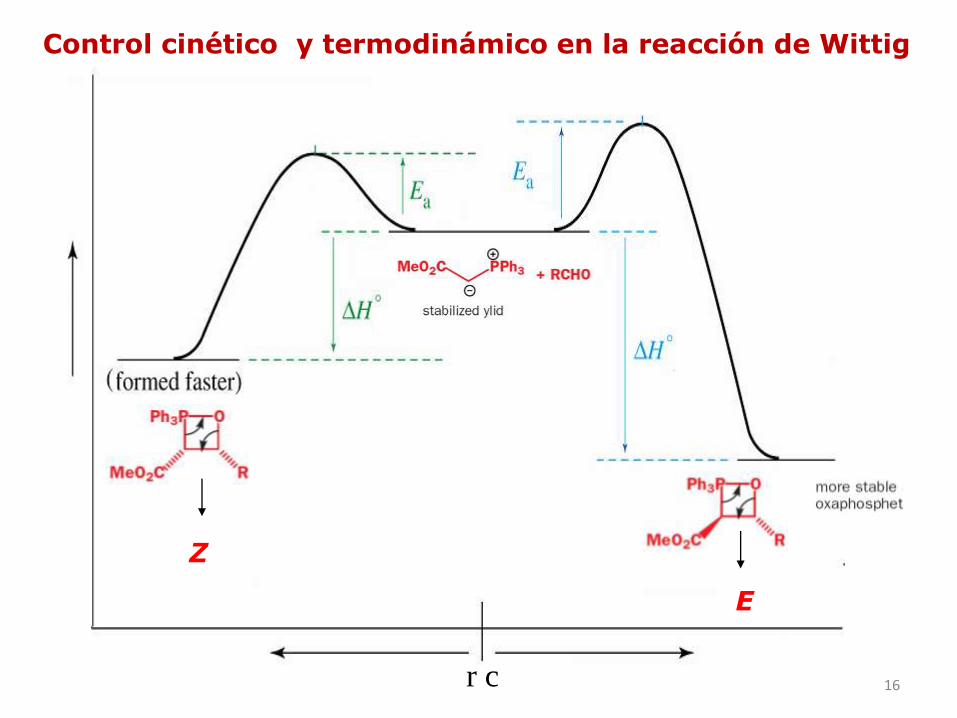
16
Z
E
Control cinético y termodinámico en la reacción de Wittig
r c
17
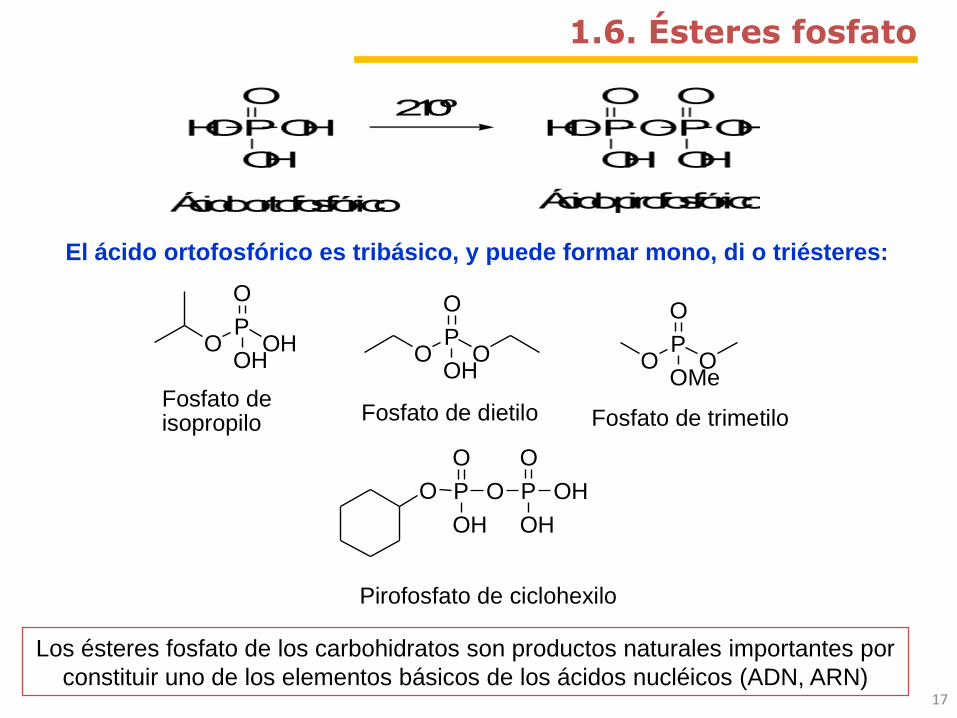
1.6. Ésteres fosfato
El ácido ortofosfórico es tribásico, y puede formar mono, di o triésteres:
OP
OH
O
OH
Fosfato deisopropilo
OP
O
O
OH
Fosfato de dietilo
OP
O
O
OMe
Fosfato de trimetilo
O P
O
O
OH
P
O
OH
OH
Pirofosfato de ciclohexilo
Los ésteres fosfato de los carbohidratos son productos naturales importantes por
constituir uno de los elementos básicos de los ácidos nucléicos (ADN, ARN)
18
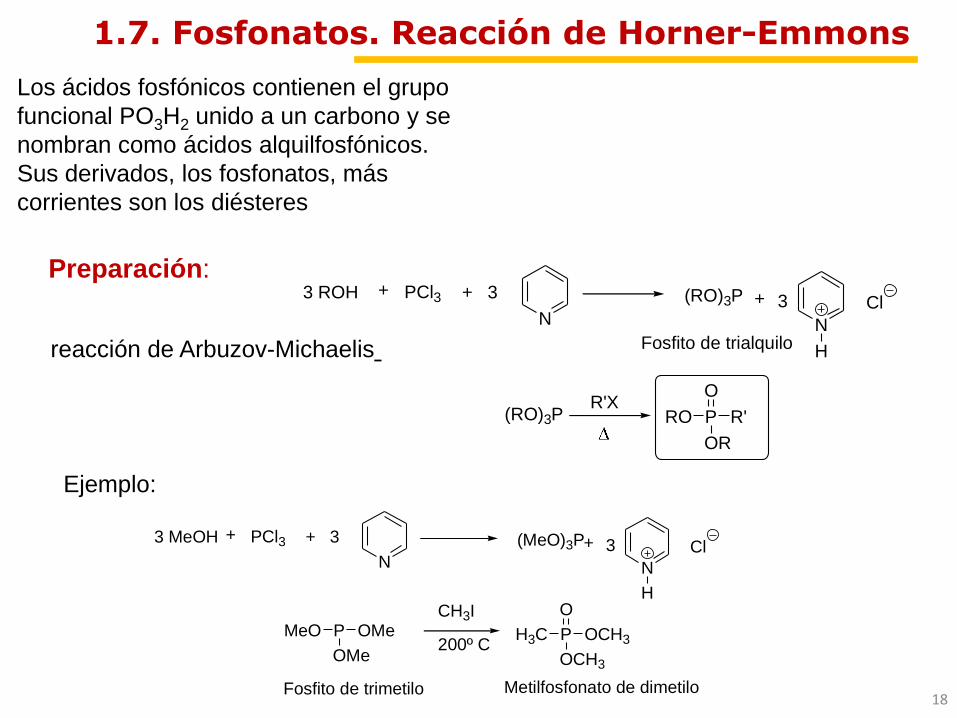
1.7. Fosfonatos. Reacción de Horner-Emmons
Preparación:
Ejemplo:
+ 3
N+
N
H
Cl3
Fosfito de trialquilo
PCl3+3 ROH
R'XP R'
OR
O
RO
(RO)3P
(RO)3P
PMeO OMe
OMe
CH3I
200º CP
O
H3C OCH3
OCH3
Metilfosfonato de dimetiloFosfito de trimetilo
+ 3
N+
N
H
Cl3PCl3+3 MeOH (MeO)3P
Los ácidos fosfónicos contienen el grupo
funcional PO3H2 unido a un carbono y se
nombran como ácidos alquilfosfónicos.
Sus derivados, los fosfonatos, más
corrientes son los diésteres
reacción de Arbuzov-Michaelis
19
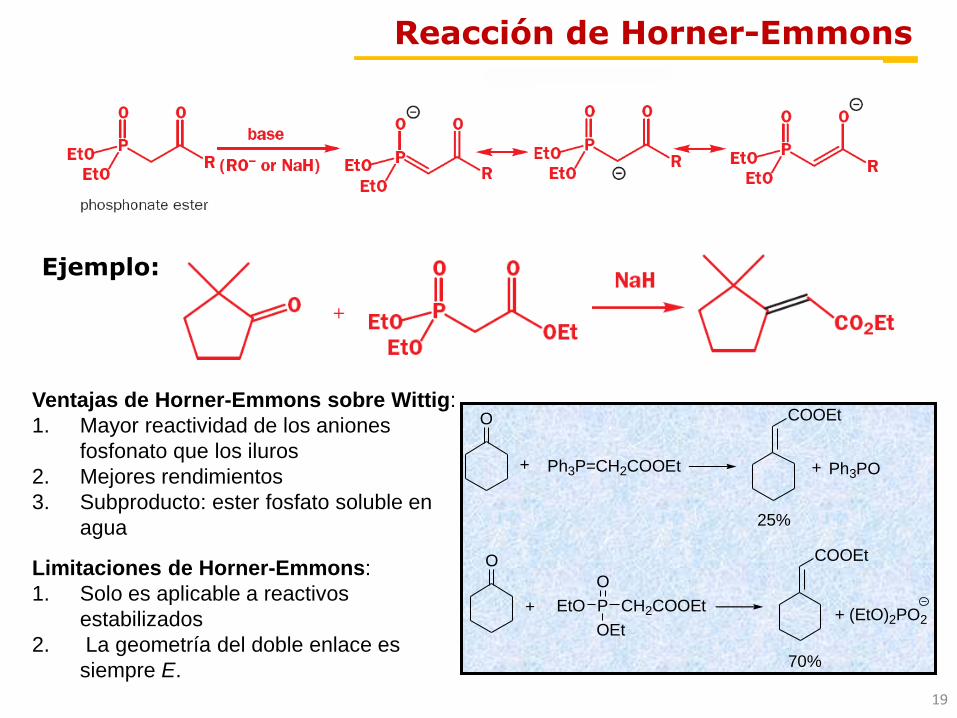
Reacción de Horner-Emmons
Ejemplo:
Ventajas de Horner-Emmons sobre Wittig:
1. Mayor reactividad de los aniones
fosfonato que los iluros
2. Mejores rendimientos
3. Subproducto: ester fosfato soluble en
agua
Limitaciones de Horner-Emmons:
1. Solo es aplicable a reactivos
estabilizados
2. La geometría del doble enlace es
siempre E.
Ph3P=CH2COOEt
O
+
P CH2COOEt
OEt
O
EtO
25%
COOEt
O
+
70%
COOEt
Ph3PO
+ (EtO)2PO2
+
20
2. Compuestos orgánicos de silicio
• Ambos, Si y C, son tetravalents y forman compuestos tetraédricos (sp3).
• El radio atómico del Si es mayor que el del C y su polarizabilidad también.
• C forma compuestos trigonales y lineales muy estables que contienen
enlaces , el Si muy pocos, no participa en enlaces p-p.
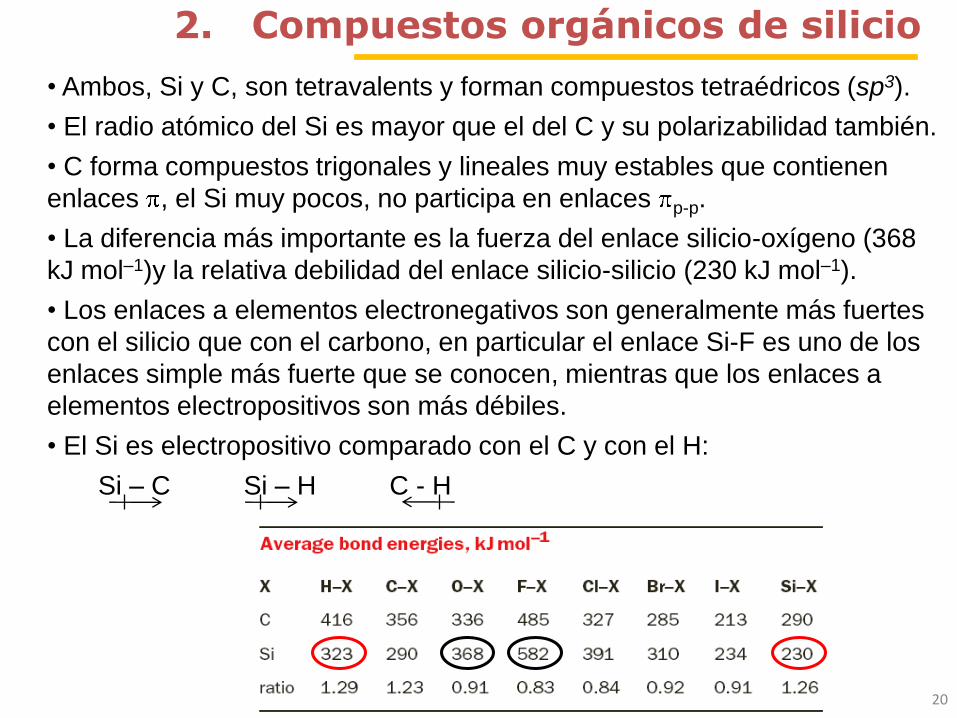
• La diferencia más importante es la fuerza del enlace silicio-oxígeno (368
kJ mol–1)y la relativa debilidad del enlace silicio-silicio (230 kJ mol–1).
• Los enlaces a elementos electronegativos son generalmente más fuertes
con el silicio que con el carbono, en particular el enlace Si-F es uno de los
enlaces simple más fuerte que se conocen, mientras que los enlaces a
elementos electropositivos son más débiles.
• El Si es electropositivo comparado con el C y con el H:
Si – C Si – H C - H
21
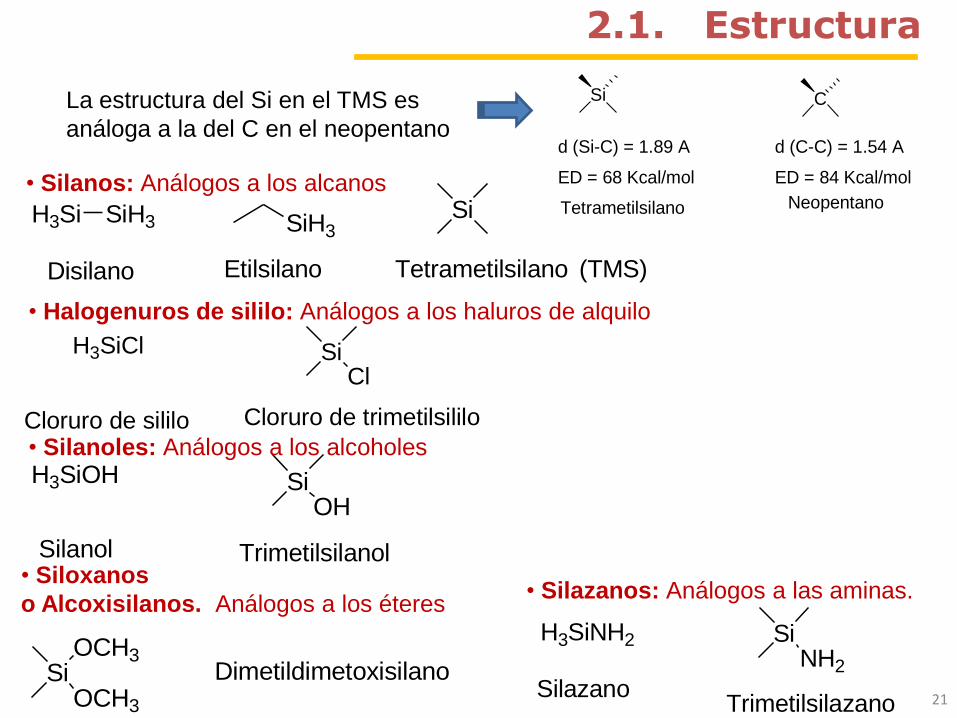
Si
d (Si-C) = 1.89 A
ED = 68 Kcal/mol
C
d (C-C) = 1.54 A
ED = 84 Kcal/mol
Tetrametilsilano NeopentanoH3Si SiH3
Disilano
SiH3
Etilsilano
Si
Tetrametilsilano (TMS)
2.1. Estructura
• Silanos: Análogos a los alcanos
H3SiCl SiCl
Cloruro de sililo Cloruro de trimetilsililo
• Halogenuros de sililo: Análogos a los haluros de alquilo
H3SiOH SiOH
Silanol Trimetilsilanol
• Silanoles: Análogos a los alcoholes
SiOCH3
OCH3
Dimetildimetoxisilano
• Siloxanos
o Alcoxisilanos. Análogos a los éteres
H3SiNH2
Silazano
SiNH2
Trimetilsilazano
• Silazanos: Análogos a las aminas.
La estructura del Si en el TMS es
análoga a la del C en el neopentano
22
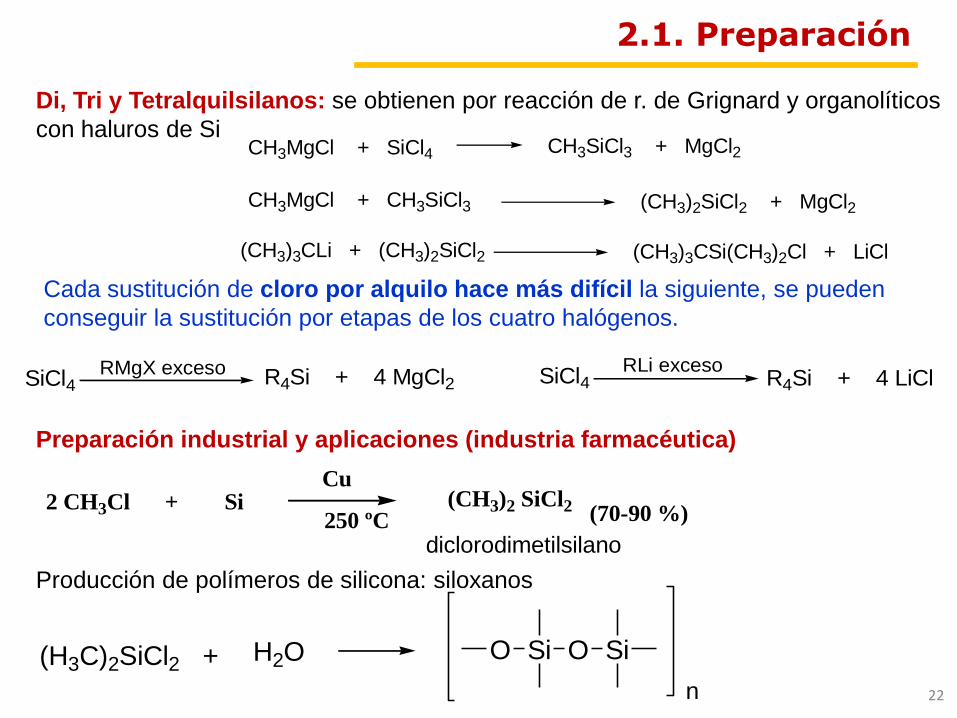
2.1. Preparación
(H3C)2SiCl2 + H2O Si OO Si
n
Producción de polímeros de silicona: siloxanos
Preparación industrial y aplicaciones (industria farmacéutica)
Cada sustitución de cloro por alquilo hace más difícil la siguiente, se pueden
conseguir la sustitución por etapas de los cuatro halógenos.
CH3MgCl + CH3SiCl3
CH3SiCl3 + MgCl2CH3MgCl + SiCl4
(CH3)2SiCl2 + MgCl2
(CH3)3CLi + (CH3)2SiCl2 (CH3)3CSi(CH3)2Cl + LiCl
SiCl4 R4Si + 4 MgCl2 SiCl4 R4Si + 4 LiCl RMgX exceso RLi exceso
Di, Tri y Tetralquilsilanos: se obtienen por reacción de r. de Grignard y organolíticos
con haluros de Si
2 CH3Cl + SiCu
250 ºC(CH3)2 SiCl2 (70-90 %)
diclorodimetilsilano
23
2.2. Reacciones
2.2.1. Sustitución nucleófila en el silicio
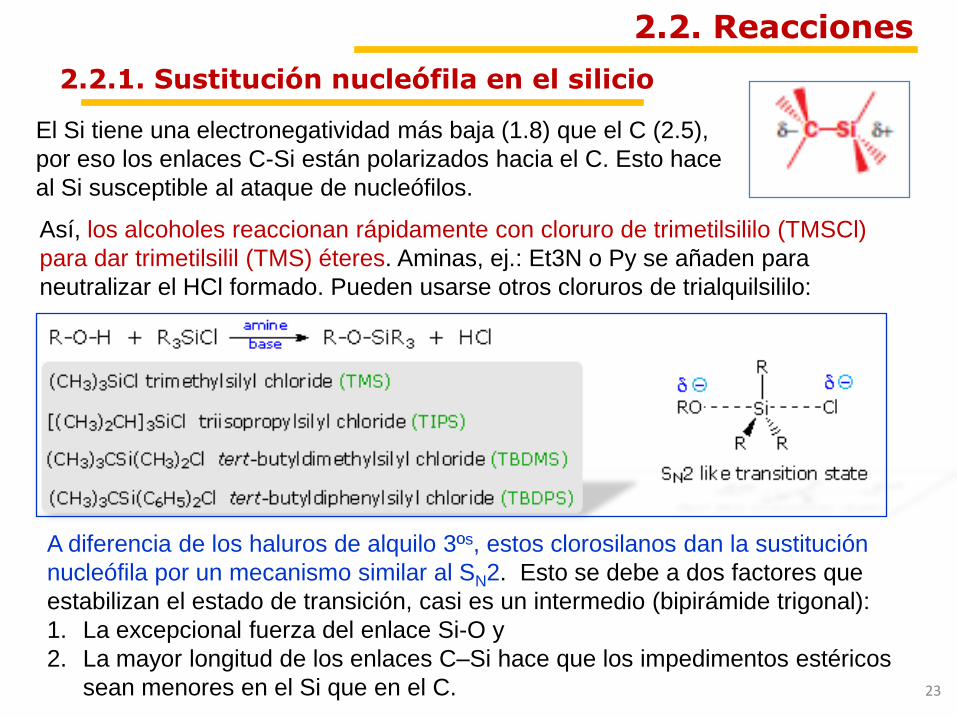
El Si tiene una electronegatividad más baja (1.8) que el C (2.5),
por eso los enlaces C-Si están polarizados hacia el C. Esto hace
al Si susceptible al ataque de nucleófilos.
Así, los alcoholes reaccionan rápidamente con cloruro de trimetilsililo (TMSCl)
para dar trimetilsilil (TMS) éteres. Aminas, ej.: Et3N o Py se añaden para
neutralizar el HCl formado. Pueden usarse otros cloruros de trialquilsililo:
A diferencia de los haluros de alquilo 3ºs, estos clorosilanos dan la sustitución
nucleófila por un mecanismo similar al SN2. Esto se debe a dos factores que
estabilizan el estado de transición, casi es un intermedio (bipirámide trigonal):
1. La excepcional fuerza del enlace Si-O y
2. La mayor longitud de los enlaces C–Si hace que los impedimentos estéricos
sean menores en el Si que en el C.
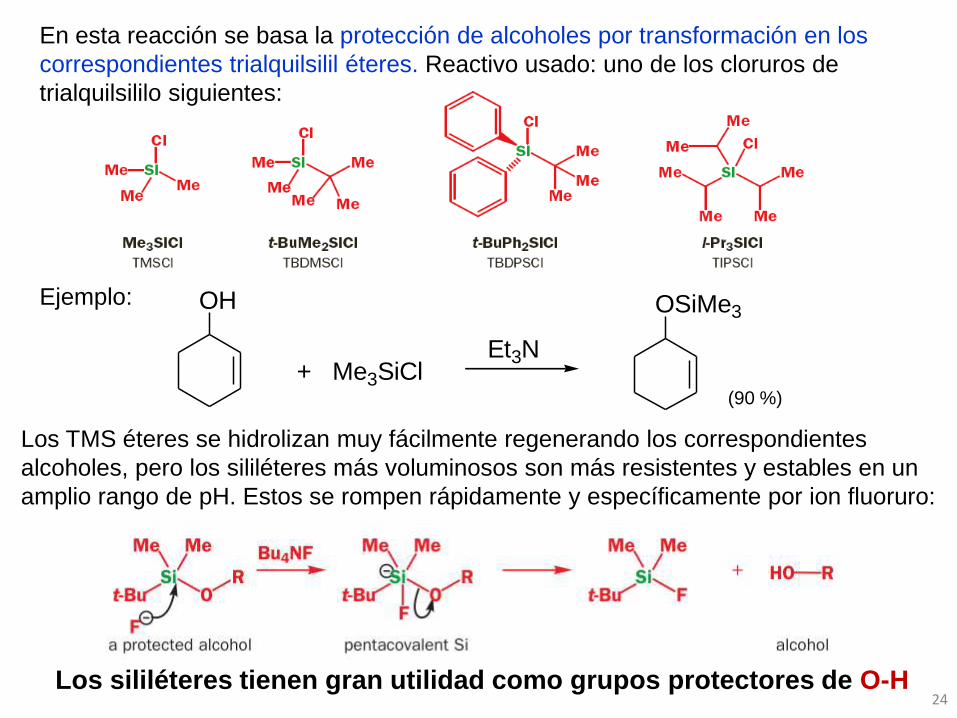
24
OH
+ Me3SiClEt3N
OSiMe3
90%
Los sililéteres tienen gran utilidad como grupos protectores de O-H
Los TMS éteres se hidrolizan muy fácilmente regenerando los correspondientes
alcoholes, pero los sililéteres más voluminosos son más resistentes y estables en un
amplio rango de pH. Estos se rompen rápidamente y específicamente por ion fluoruro:
En esta reacción se basa la protección de alcoholes por transformación en los
correspondientes trialquilsilil éteres. Reactivo usado: uno de los cloruros de
trialquilsililo siguientes:
Ejemplo:
(90 %)
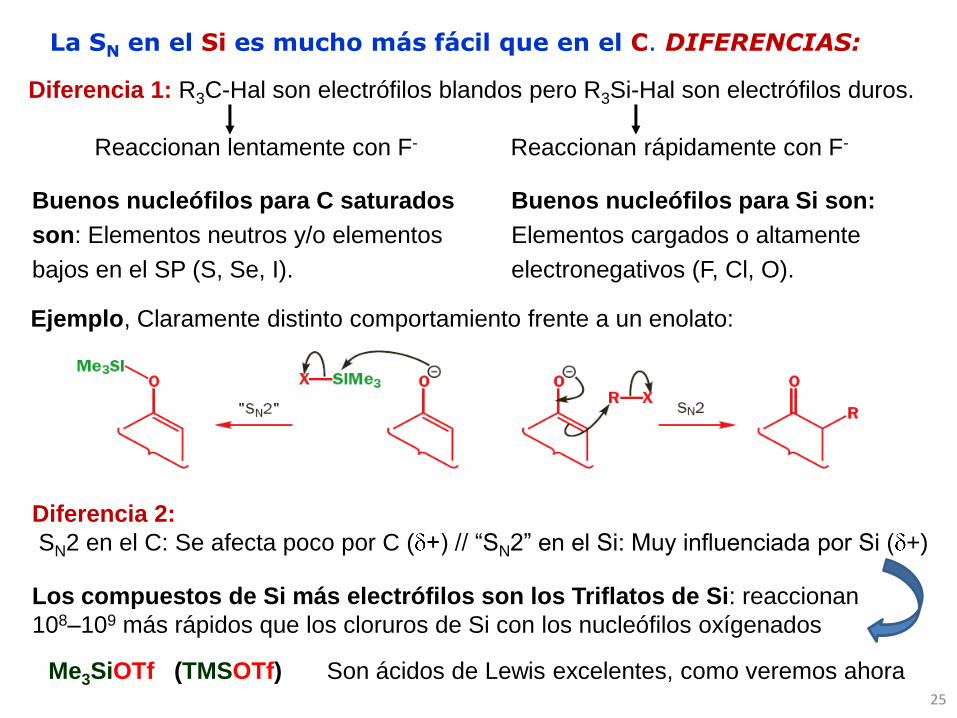
25
Diferencia 1: R3C-Hal son electrófilos blandos pero R3Si-Hal son electrófilos duros.
Reaccionan lentamente con F- Reaccionan rápidamente con F-
Buenos nucleófilos para C saturados
son: Elementos neutros y/o elementos
bajos en el SP (S, Se, I).
Buenos nucleófilos para Si son:
Elementos cargados o altamente
electronegativos (F, Cl, O).
La SN en el Si es mucho más fácil que en el C. DIFERENCIAS:
Ejemplo, Claramente distinto comportamiento frente a un enolato:
Diferencia 2:
SN2 en el C: Se afecta poco por C ( +) // “SN2” en el Si: Muy influenciada por Si ( +)
Los compuestos de Si más electrófilos son los Triflatos de Si: reaccionan
108–109 más rápidos que los cloruros de Si con los nucleófilos oxígenados
Me3SiOTf (TMSOTf) Son ácidos de Lewis excelentes, como veremos ahora
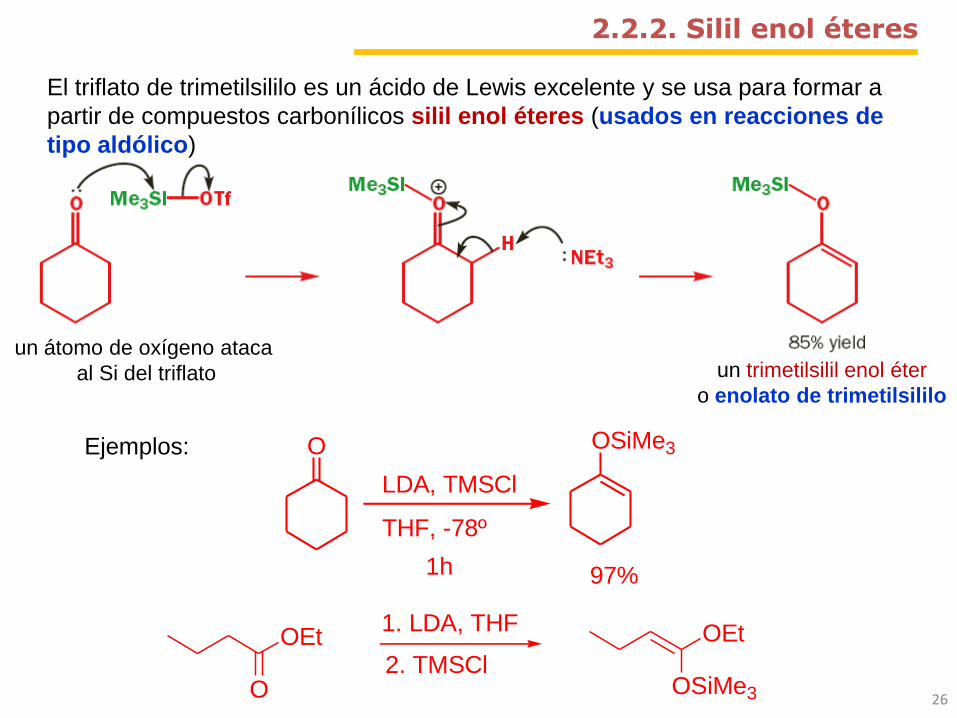
26O
OEt1. LDA, THF
2. TMSClOSiMe3
OEt
O
LDA, TMSCl
THF, -78º
1h
OSiMe3
97%
un átomo de oxígeno ataca
al Si del triflato
2.2.2. Silil enol éteres
El triflato de trimetilsililo es un ácido de Lewis excelente y se usa para formar a
partir de compuestos carbonílicos silil enol éteres (usados en reacciones de
tipo aldólico)
Ejemplos:
un trimetilsilil enol éter
o enolato de trimetilsililo

27
2.2.3. Efecto estabilizante del silicio. Carbocationes. Carbaniones
• El silicio estabiliza una carga + en posición con respecto a él. Así, los
cationes con Si en la posición son importantes intermedios. Esto se
verá ahora en las reacciones de varios tipos de compuestos:
Alquinil silanos / Arilsilanos / Vinilsilanos / Alilsilanos
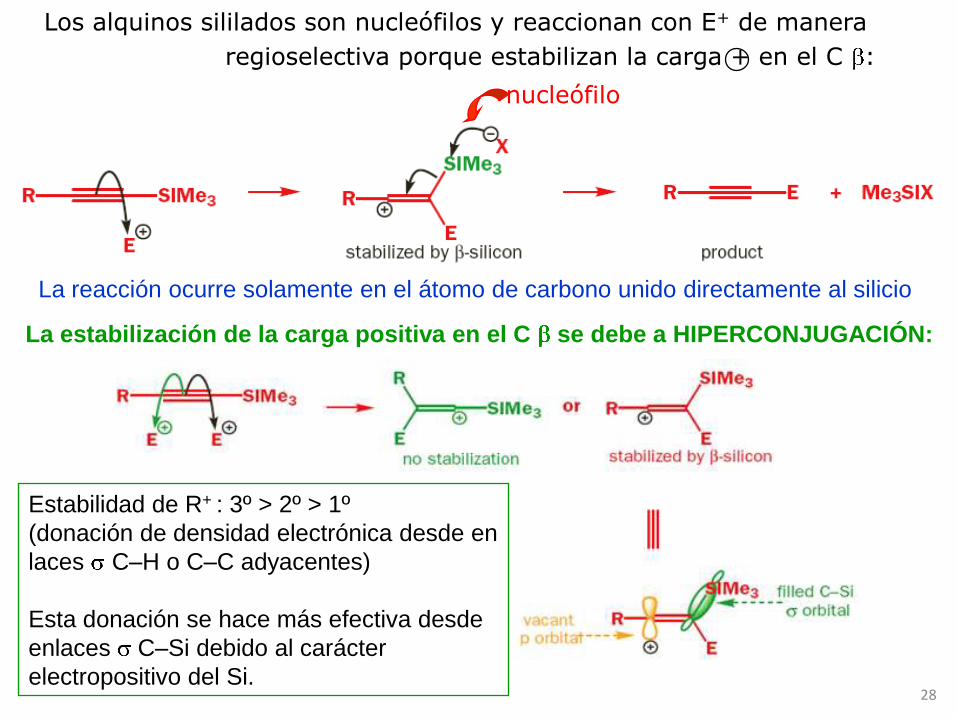
Alquinil silanos
Los alquinil silanos o alquinos sililados se obtienen:
Al igual que los alquinos ordinarios, los alquinos sililados son nucleófilos
frente a electrófilos. Si bien, la presencia del Si controla la regioselectividad
• El silicio estabiliza carbaniones en posición con respecto a él. Iluros:
OLEFINACIÓN DE PETERSON
28
Los alquinos sililados son nucleófilos y reaccionan con E+ de manera
regioselectiva porque estabilizan la carga + en el C :
nucleófilo
La reacción ocurre solamente en el átomo de carbono unido directamente al silicio
Estabilidad de R+ : 3º > 2º > 1º
(donación de densidad electrónica desde en
laces C–H o C–C adyacentes)
Esta donación se hace más efectiva desde
enlaces C–Si debido al carácter
electropositivo del Si.
La estabilización de la carga positiva en el C se debe a HIPERCONJUGACIÓN:
29
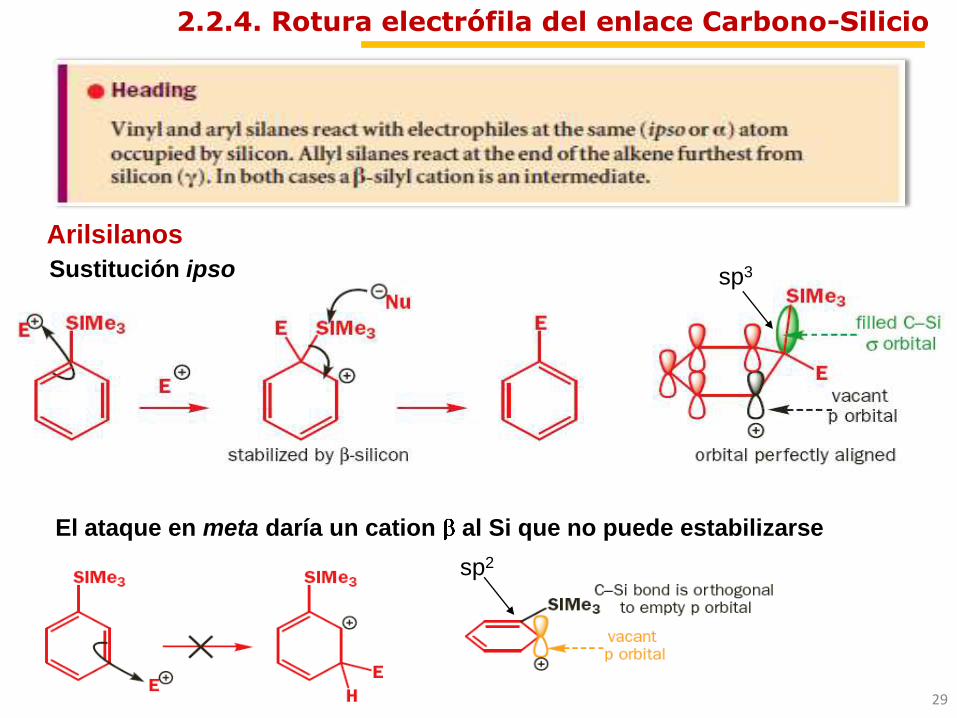
Arilsilanos
Sustitución ipso
El ataque en meta daría un cation al Si que no puede estabilizarse
2.2.4. Rotura electrófila del enlace Carbono-Silicio
sp3
sp2
30
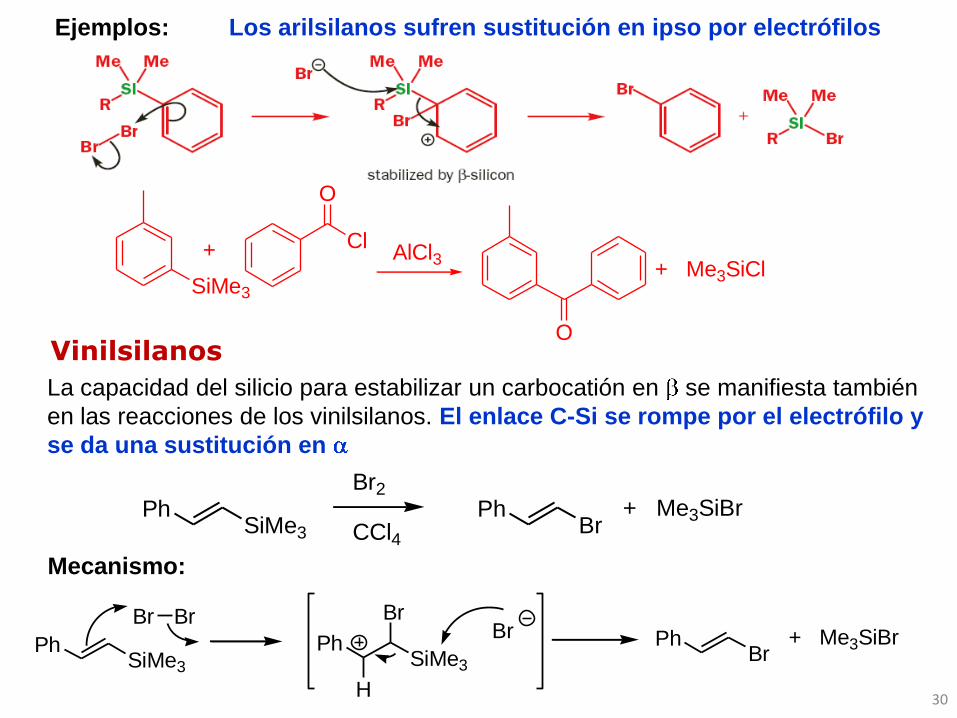
Ejemplos: Los arilsilanos sufren sustitución en ipso por electrófilos
SiMe3
+ Cl
O
AlCl3
O
+ Me3SiCl
Vinilsilanos
La capacidad del silicio para estabilizar un carbocatión en se manifiesta también
en las reacciones de los vinilsilanos. El enlace C-Si se rompe por el electrófilo y
se da una sustitución en
PhSiMe3
Br2
CCl4
PhBr
+ Me3SiBr
PhSiMe3
Br Br
PhSiMe3
H
BrBr Ph
Br+ Me3SiBr
Mecanismo:
31
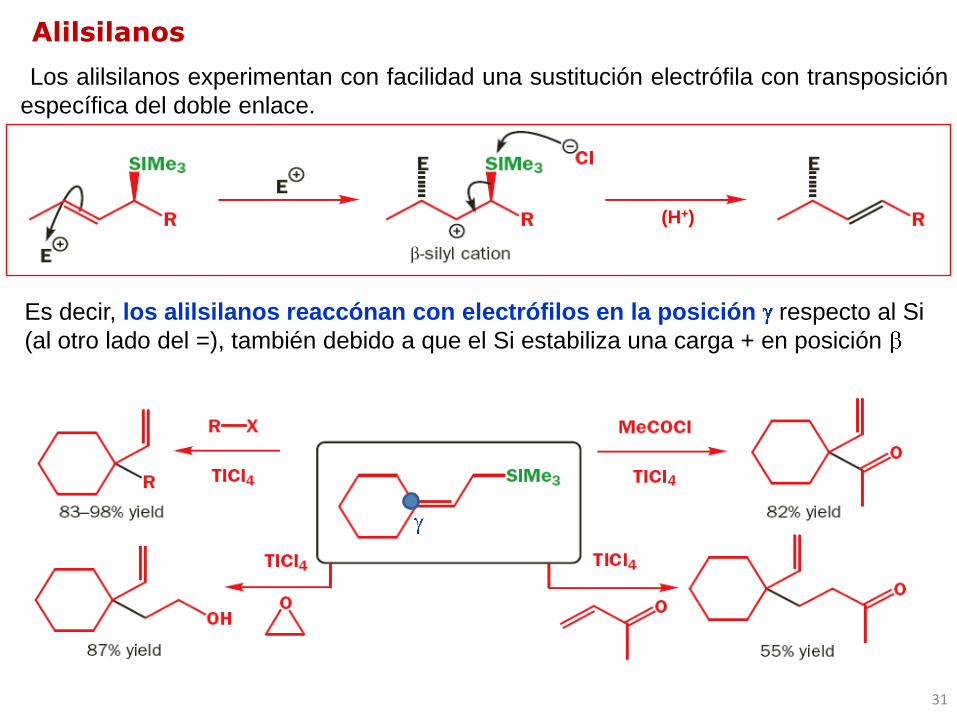
Los alilsilanos experimentan con facilidad una sustitución electrófila con transposición
específica del doble enlace.
Alilsilanos
(H+)
Es decir, los alilsilanos reaccónan con electrófilos en la posición respecto al Si
(al otro lado del =), también debido a que el Si estabiliza una carga + en posición
32
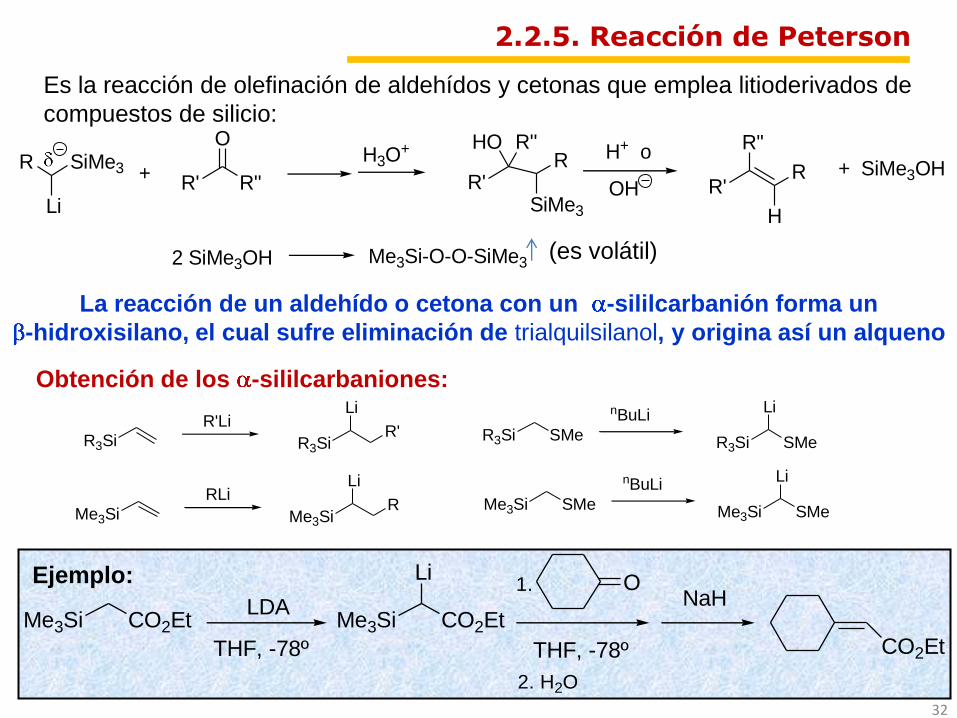
Es la reacción de olefinación de aldehídos y cetonas que emplea litioderivados de
compuestos de silicio:
Obtención de los -sililcarbaniones:
Me3Si
RLi
Me3SiR
Li
R3SiR'Li
R3SiR'
Li
Me3Si SMe
nBuLi
Me3Si SMe
Li
R3Si SMe
nBuLi
R3Si SMe
Li
2.2.5. Reacción de Peterson
Li
SiMe3R+ R' R''
OH3O+
SiMe3
RR'
R''HOH+ o
OH
H
R
R''
R'+ SiMe3OH
2 SiMe3OH Me3Si-O-O-SiMe3
La reacción de un aldehído o cetona con un -sililcarbanión forma un
-hidroxisilano, el cual sufre eliminación de trialquilsilanol, y origina así un alqueno
(es volátil)
LDA
THF, -78º
CO2EtMe3Si CO2EtMe3Si
Li O
THF, -78º CO2Et
1.
2. H2O
NaHEjemplo:
33
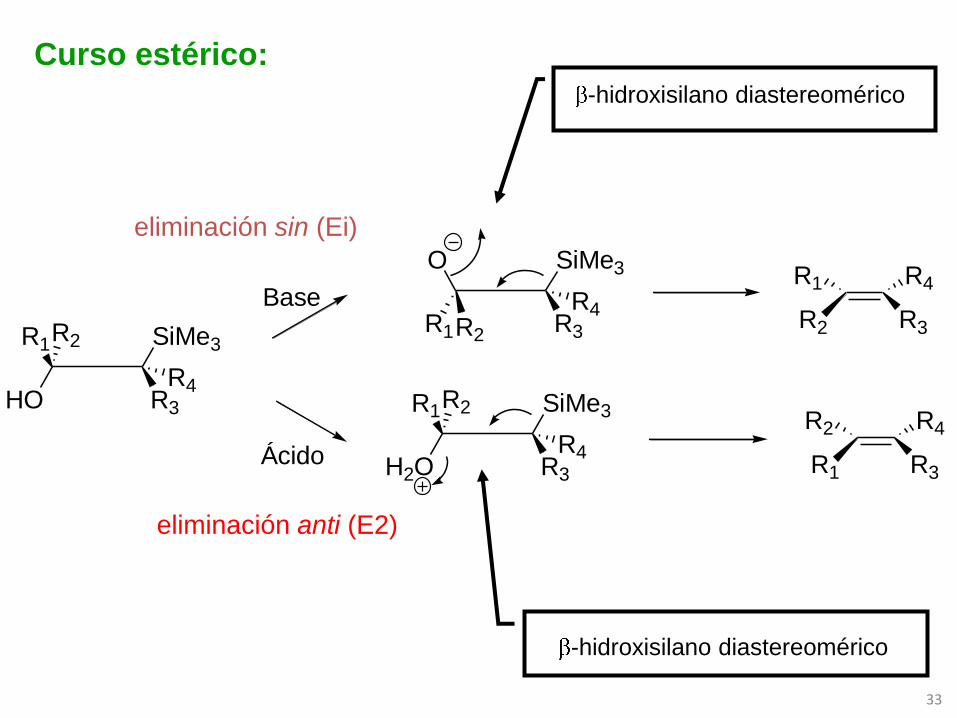
HO
SiMe3R1R2
R3
R4
Base
Ácido H2O
SiMe3R1R2
R3
R4
SiMe3
R3
R4
O
R2R1
R2 R3
R1
R1 R3
R2
R4
R4
Curso estérico:
eliminación anti (E2)
eliminación sin (Ei)
-hidroxisilano diastereomérico
-hidroxisilano diastereomérico
34
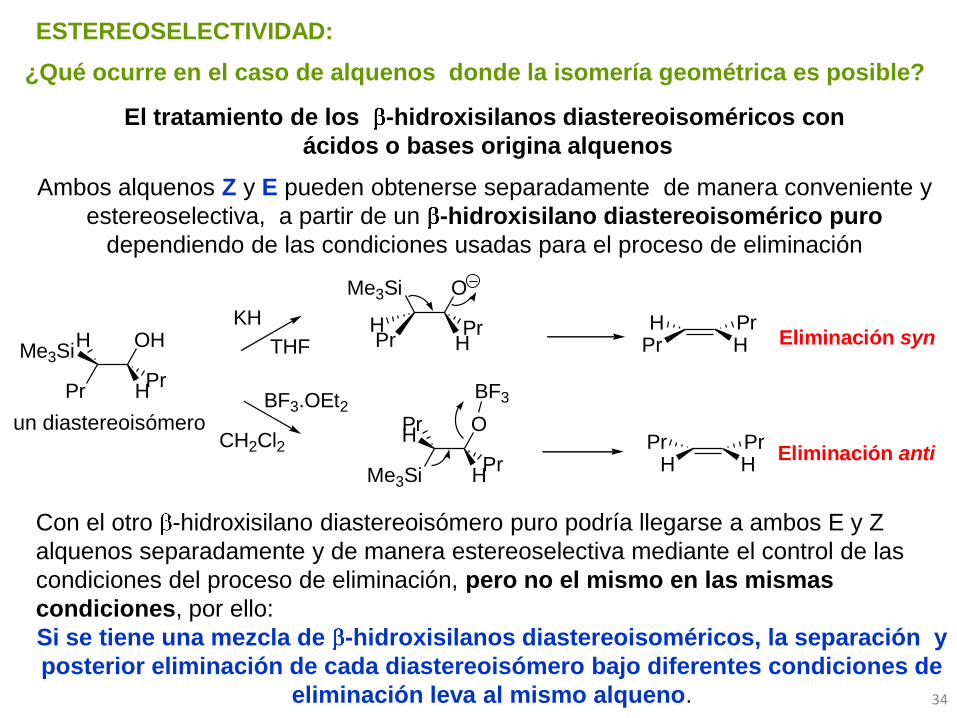
OH
HPr
Pr
HMe3Si
KH
THF
BF3.OEt2
CH2Cl2
O
HPr
O
HPr
Me3Si
PrH
Me3Si
PrH
Pr HPrH
H HPrPr
Eliminación syn
Eliminación anti
BF3
¿Qué ocurre en el caso de alquenos donde la isomería geométrica es posible?
El tratamiento de los -hidroxisilanos diastereoisoméricos con
ácidos o bases origina alquenos
Ambos alquenos Z y E pueden obtenerse separadamente de manera conveniente y
estereoselectiva, a partir de un -hidroxisilano diastereoisomérico puro
dependiendo de las condiciones usadas para el proceso de eliminación
ESTEREOSELECTIVIDAD:
un diastereoisómero
Con el otro -hidroxisilano diastereoisómero puro podría llegarse a ambos E y Z
alquenos separadamente y de manera estereoselectiva mediante el control de las
condiciones del proceso de eliminación, pero no el mismo en las mismas
condiciones, por ello:
Si se tiene una mezcla de -hidroxisilanos diastereoisoméricos, la separación y
posterior eliminación de cada diastereoisómero bajo diferentes condiciones de
eliminación leva al mismo alqueno.
35
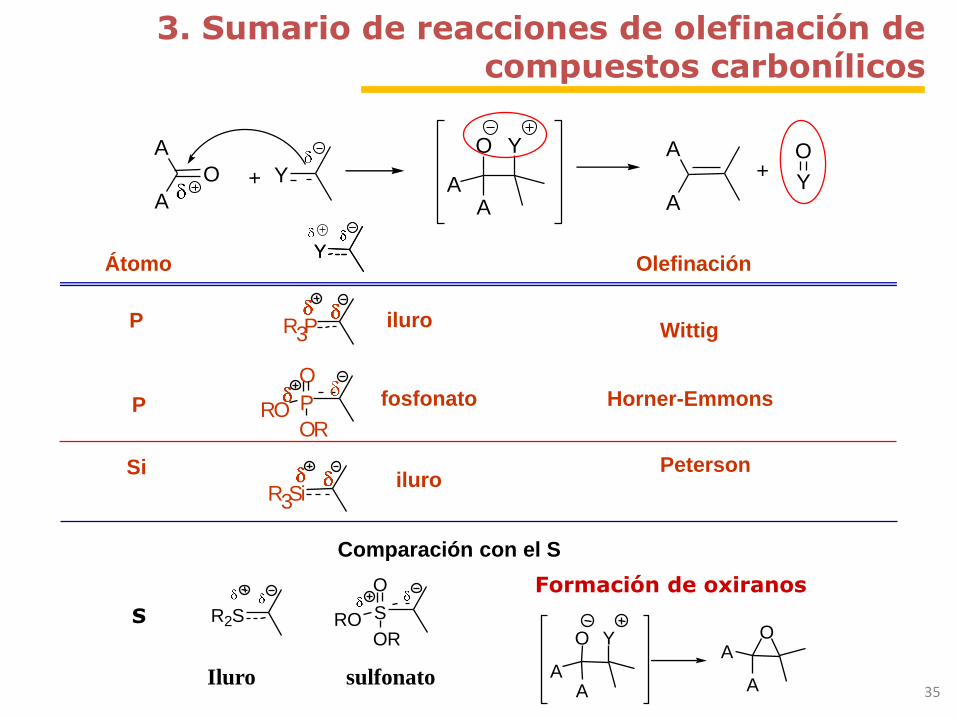
3. Sumario de reacciones de olefinación de compuestos carbonílicos
+
Y
O Y
AA A
A+
Y
OO
A
A
Átomo OlefinaciónYY
P R3Piluro
Wittig
P P
ORRO
Ofosfonato Horner-Emmons
R3Siiluro
PetersonSi
Comparación con el S
S R2S S
ORRO
O
Iluro sulfonato
Formación de oxiranos
O Y
AA
OA
A