fshd disease mechanisms and models - rochester, … · fshd disease mechanisms and models . an...
TRANSCRIPT

Silvère M. van der Maarel, Ph.D. Leiden University Medical Center
FSHD Disease Mechanisms and Models

An Integrative Approach
Patient Organizations
Regulatory Bodies
Funding Agencies
Basic & Medical
Scientists
Industry
FSHD families
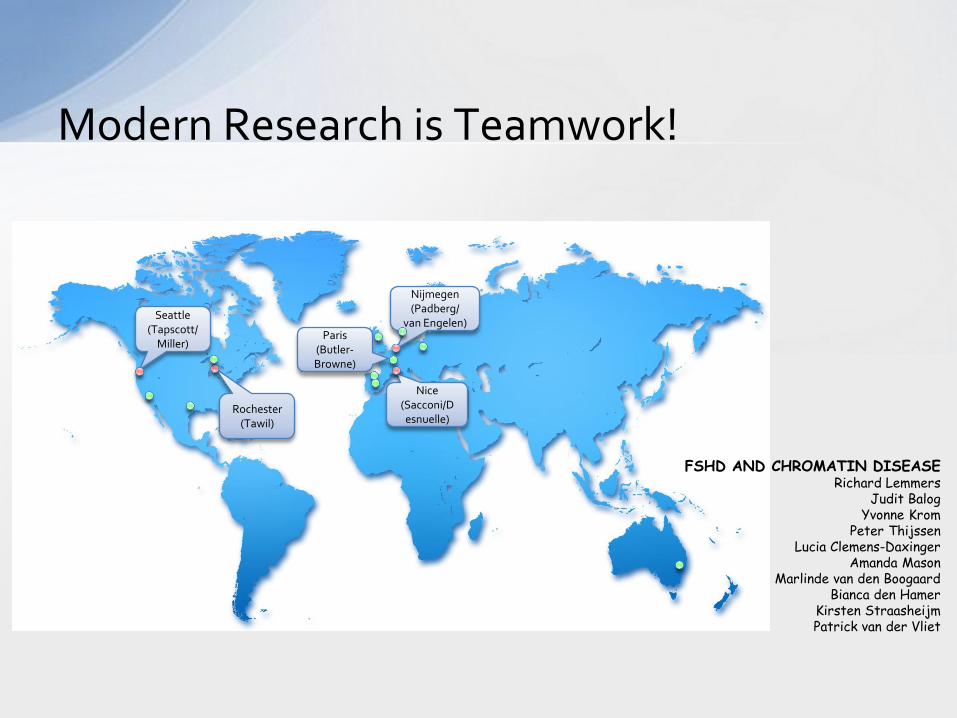
Modern Research is Teamwork!
Seattle (Tapscott/
Miller)
Rochester (Tawil)
Nice (Sacconi/Desnuelle)
Nijmegen (Padberg/
van Engelen) Paris
(Butler-Browne)
FSHD AND CHROMATIN DISEASE Richard Lemmers
Judit Balog Yvonne Krom
Peter Thijssen Lucia Clemens-Daxinger
Amanda Mason Marlinde van den Boogaard
Bianca den Hamer Kirsten Straasheijm Patrick van der Vliet

Modern Research is Teamwork!
Geraldi Norton Foundation
& the Eklund Family
George & Jack Shaw
& the Shaw Family Foundation

• Fields Center was established in 2007: – Strategic Alliance to create a clinical/scientific network
between Rochester-Leiden-Seattle-Nijmegen-Nice – Expedite Research and Therapy Development – Non-exclusive
– Protocols freely available – Sharing resources – Standards for Registries – Standards of care, diagnosis – 50+ publications
– www.urmc.rochester.edu/fields-center/
FSHD and the Fields Center

• Long tradition of: – Genetic research – Molecular and cellular biology
– DNA diagnosis – Assistance in diagnosis
FSHD at the LUMC
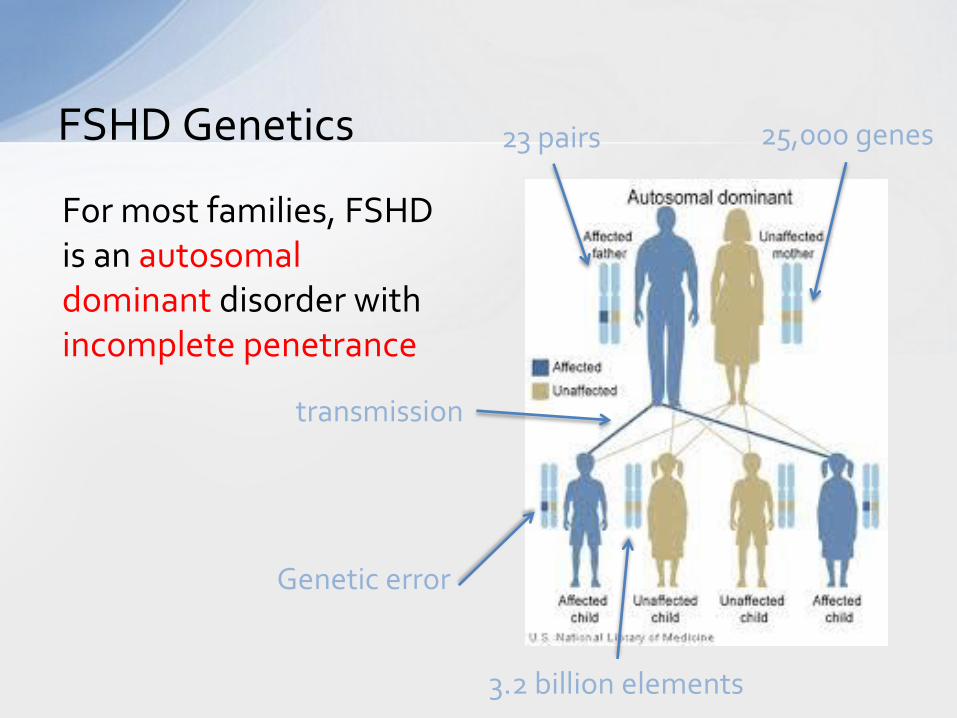
FSHD Genetics
For most families, FSHD is an autosomal dominant disorder with incomplete penetrance
23 pairs 25,000 genes
transmission
Genetic error
3.2 billion elements
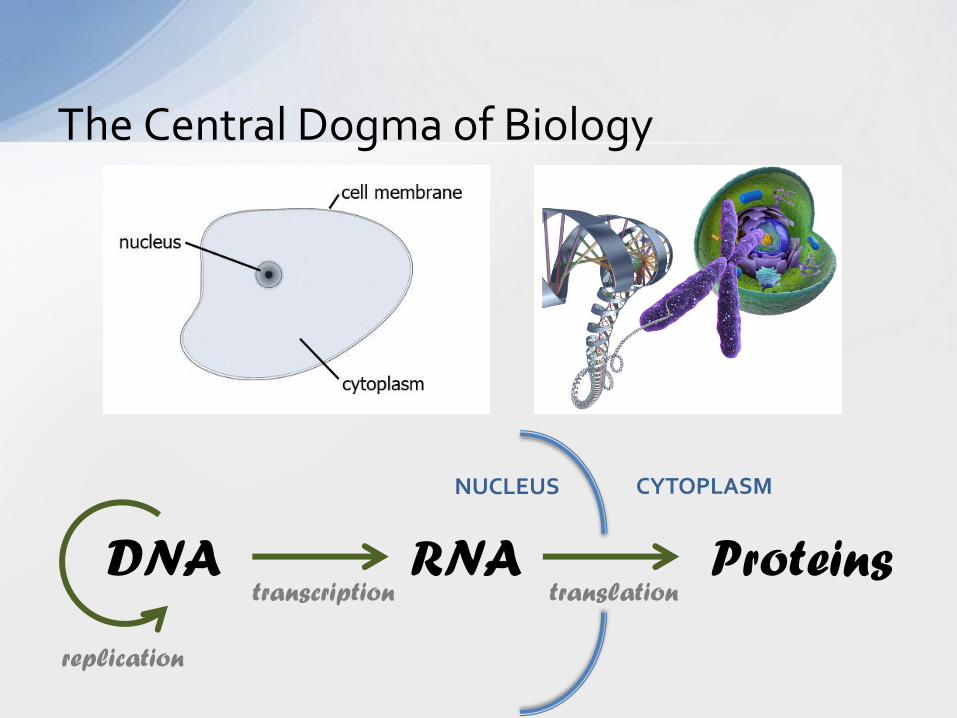
The Central Dogma of Biology
DNA RNA Proteins
replication
transcription translation
NUCLEUS CYTOPLASM
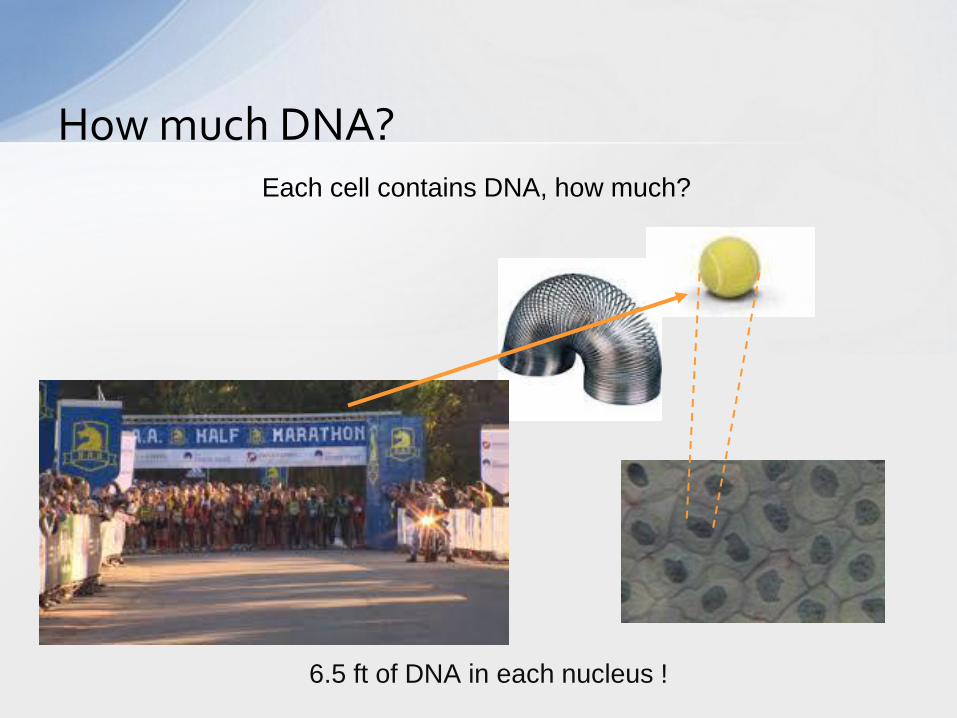
How much DNA?
6.5 ft of DNA in each nucleus !
Each cell contains DNA, how much?
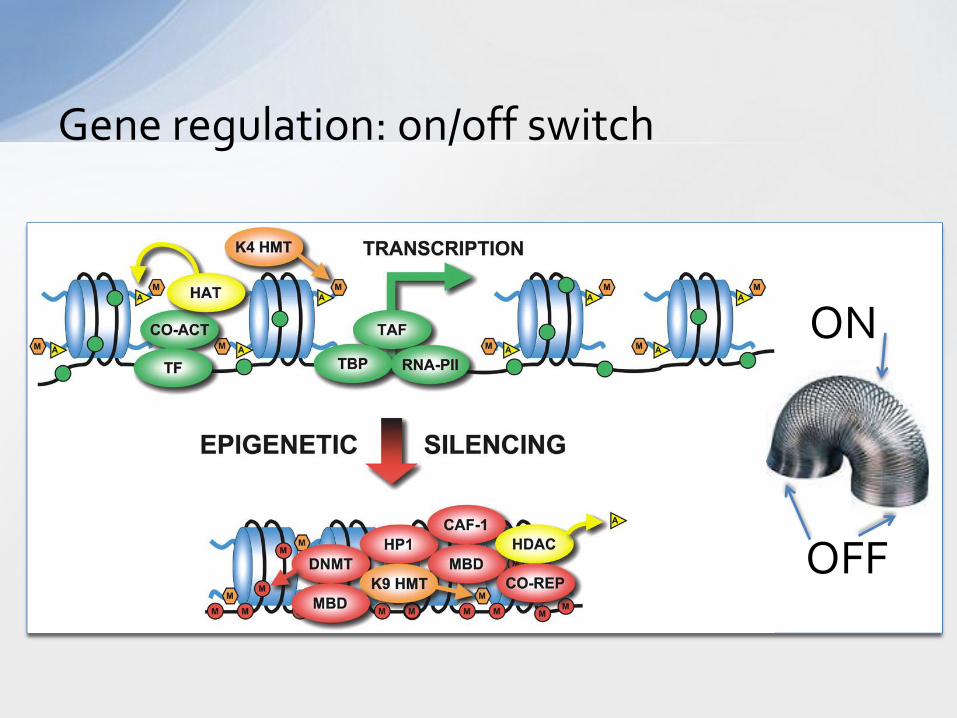
Gene regulation: on/off switch
ON
OFF

• FSHD is caused by the inappropriate production of the DUX4 protein in muscle of FSHD individuals (Lemmers et al., Science 2010)
Breakthrough in 2010
…“If we were thinking of a collection of the genome’s greatest
hits, this would go on the list,” said Dr.Francis Collins, a human
geneticist and director of the National Institutes of Health.
THE FRONT PAGE
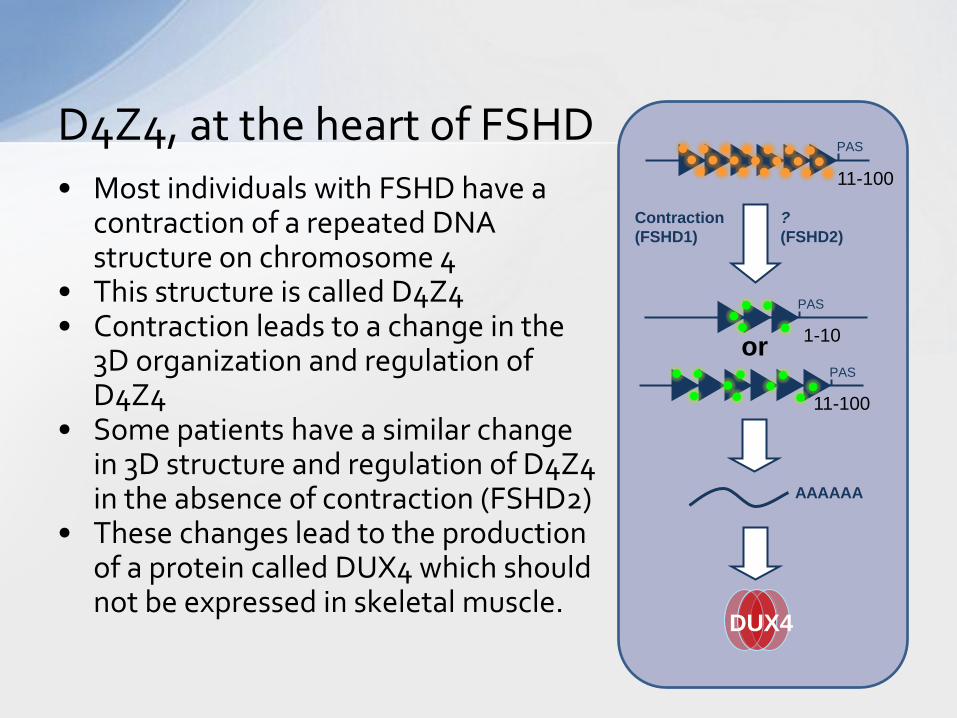
• Most individuals with FSHD have a contraction of a repeated DNA structure on chromosome 4
• This structure is called D4Z4 • Contraction leads to a change in the
3D organization and regulation of D4Z4
• Some patients have a similar change in 3D structure and regulation of D4Z4 in the absence of contraction (FSHD2)
• These changes lead to the production of a protein called DUX4 which should not be expressed in skeletal muscle.
D4Z4, at the heart of FSHD
AAAAAA
DUX4
11-100
1-10
11-100
Contraction
(FSHD1)
?
(FSHD2)
or
PAS
PAS
PAS
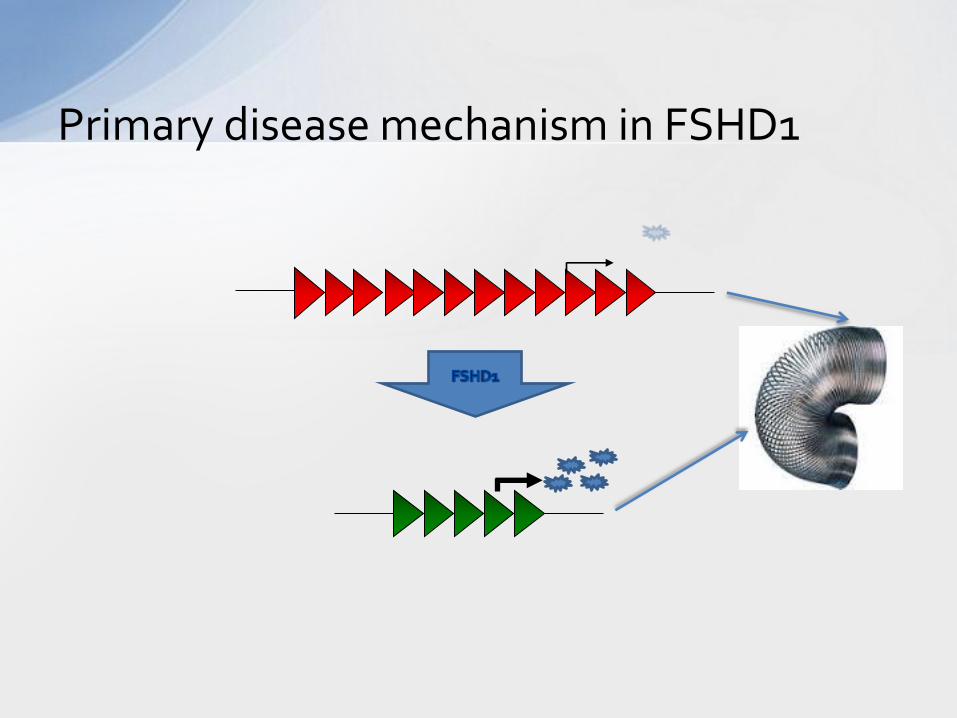
Primary disease mechanism in FSHD1
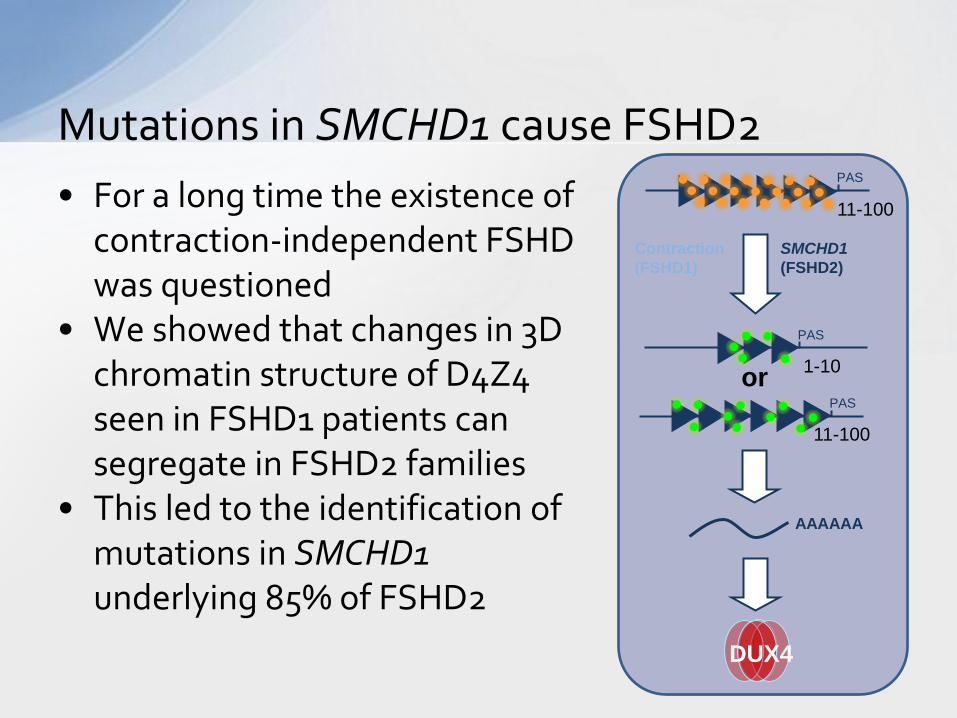
• For a long time the existence of contraction-independent FSHD was questioned
• We showed that changes in 3D chromatin structure of D4Z4 seen in FSHD1 patients can segregate in FSHD2 families
• This led to the identification of mutations in SMCHD1 underlying 85% of FSHD2
Mutations in SMCHD1 cause FSHD2
AAAAAA
DUX4
11-100
1-10
11-100
Contraction
(FSHD1)
SMCHD1
(FSHD2)
or
PAS
PAS
PAS
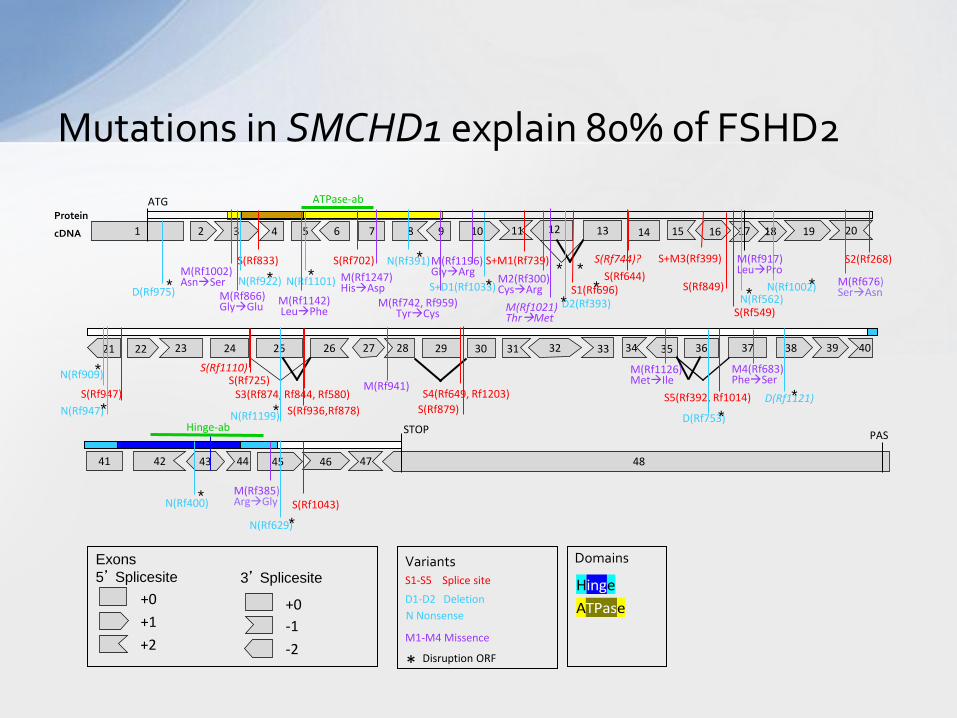
Mutations in SMCHD1 explain 80% of FSHD2
M(Rf1196) GlyArg
M1-M4 Missence
+0
+1
+2
S1-S5 Splice site
D1-D2 Deletion
Exons
5’ Splicesite Variants Domains
Hinge
ATPase +0
-2
-1
3’ Splicesite
2 1 5 7 6 8 9 10 3
ATG ATPase-ab
Protein
cDNA 4
S+D1(Rf1033)
D2(Rf393)
S+M1(Rf739)
M2(Rf300) CysArg
13 20 17 18
S1(Rf696)
15 19 14
S2(Rf268)
43 45 41 42 47 48 44
PAS Hinge-ab STOP
36 37 32 39 34 40
S5(Rf392, Rf1014)
31 33
M4(Rf683) PheSer
26 29 23 27 24 25
S3(Rf874, Rf844, Rf580) S4(Rf649, Rf1203)
30 35 22 21
11 16
46
38
12
28
* * *
* *
Disruption ORF *
S+M3(Rf399)
N Nonsense
S(Rf833) S(Rf702)
N(Rf909)
S(Rf879)
S(Rf1043)
S(Rf947)
S(Rf549)
S(Rf849)
S(Rf1110)
S(Rf744)?
D(Rf1121)
N(Rf400)
N(Rf947)
N(Rf629)
N(Rf1101) N(Rf922)
S(Rf936,Rf878)
N(Rf1002)
M(Rf742, Rf959) TyrCys
M(Rf1021) ThrMet
M(Rf1126) MetIle
M(Rf385) ArgGly
M(Rf941)
* * * M(Rf676)
SerAsn
* *
*
N(Rf562) * D(Rf975) M(Rf866) GlyGlu
M(Rf1002) AsnSer M(Rf1247)
HisAsp
N(Rf391) * S(Rf644)
M(Rf917) LeuPro
S(Rf725)
N(Rf1199) * D(Rf753)
M(Rf1142) LeuPhe
*
*
*
*
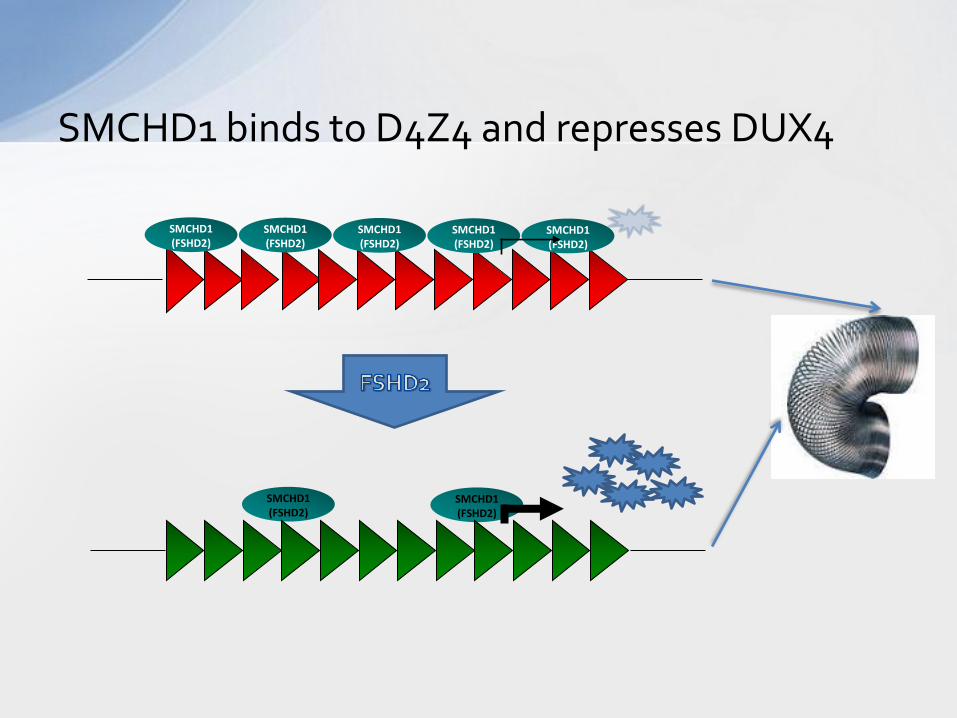
SMCHD1 binds to D4Z4 and represses DUX4
SMCHD1 (FSHD2)
SMCHD1 (FSHD2)
SMCHD1 (FSHD2)
SMCHD1 (FSHD2)
SMCHD1 (FSHD2)
SMCHD1 (FSHD2)
SMCHD1 (FSHD2)
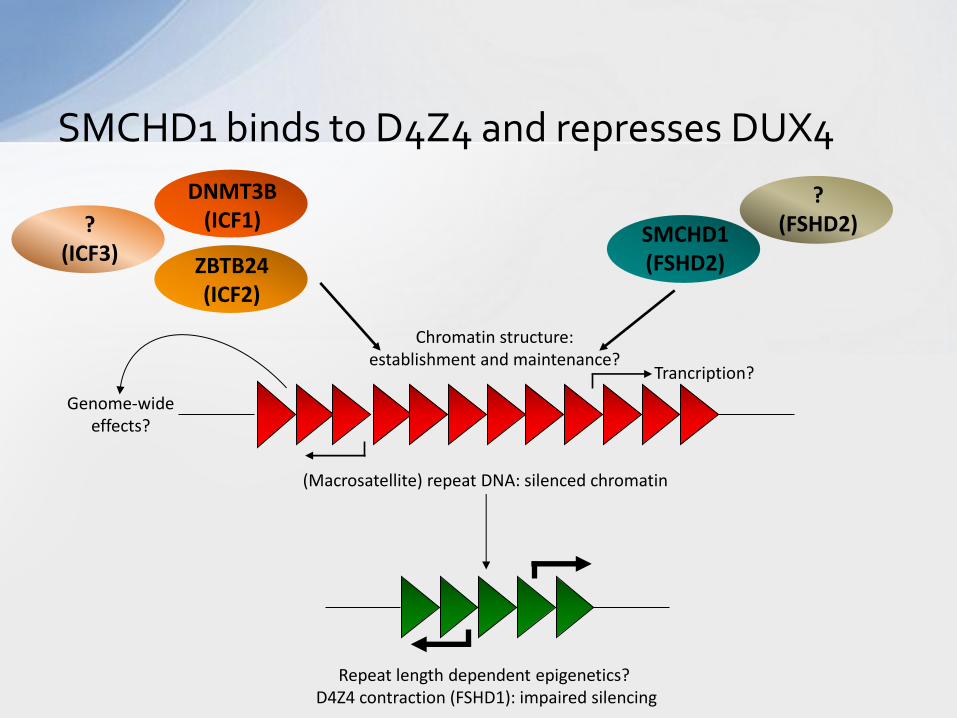
SMCHD1 binds to D4Z4 and represses DUX4
Repeat length dependent epigenetics? D4Z4 contraction (FSHD1): impaired silencing
(Macrosatellite) repeat DNA: silenced chromatin
SMCHD1 (FSHD2) ZBTB24
(ICF2)
Chromatin structure: establishment and maintenance?
Genome-wide effects?
Trancription?
DNMT3B (ICF1)
? (FSHD2) ?
(ICF3)
Clinical Variability
• Large variability in onset, progression and
severity;
• Between families and within families;
• What protects gene-carriers from becoming
affected?;
• Environmental factors?
• Genetic modifiers of D4Z4?
• Role for SMCHD1?
Lan
do
uzy
an
d D
ejer
ine
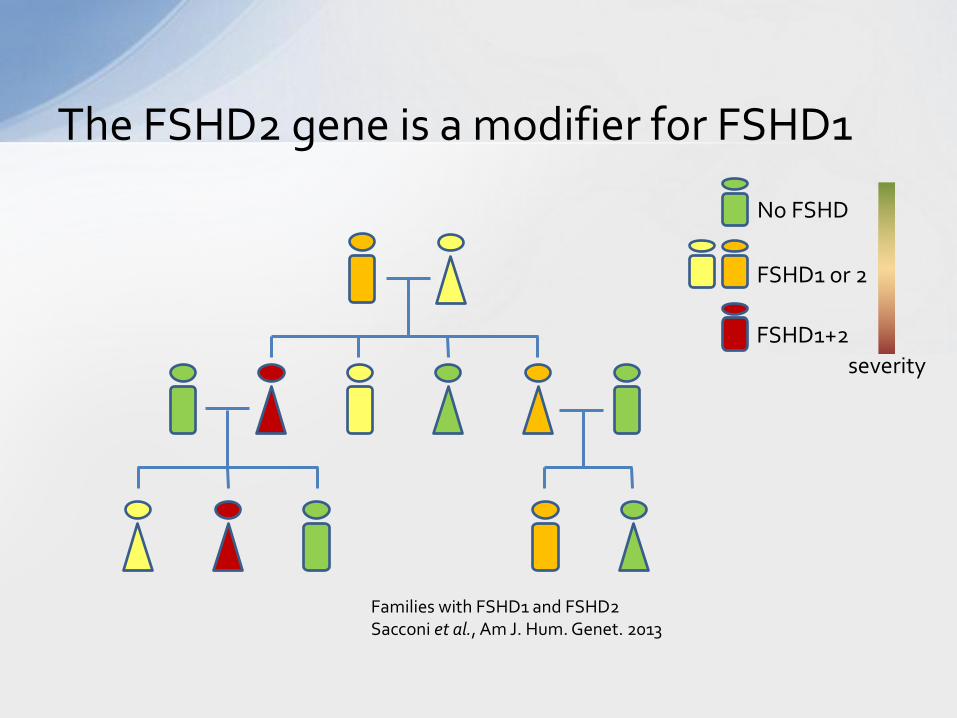
The FSHD2 gene is a modifier for FSHD1
No FSHD
FSHD1 or 2
FSHD1+2
severity
Families with FSHD1 and FSHD2 Sacconi et al., Am J. Hum. Genet. 2013
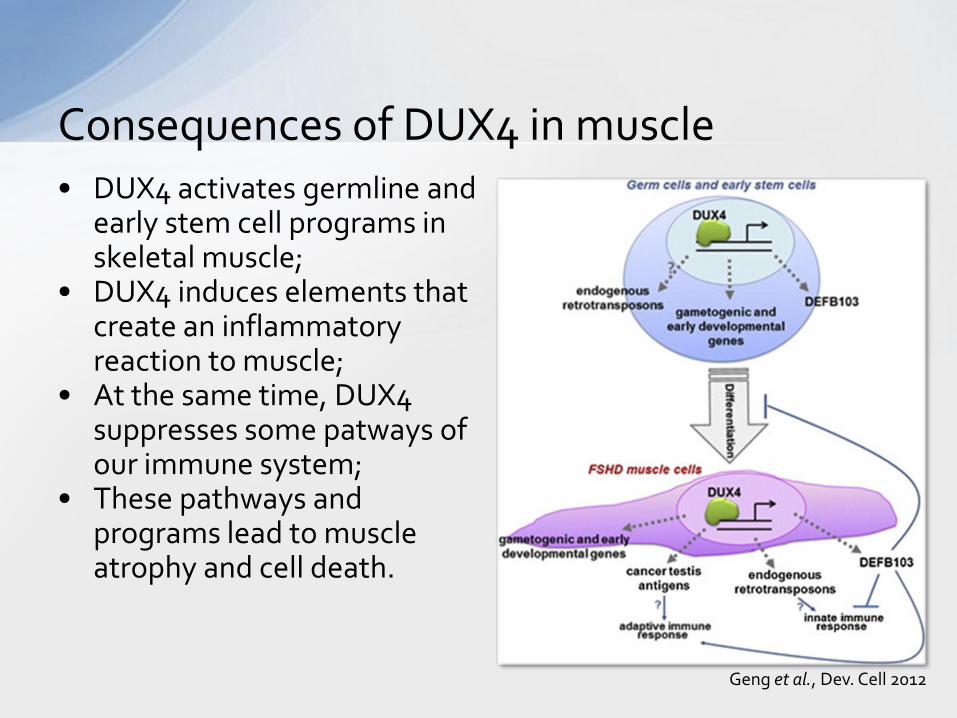
• DUX4 activates germline and early stem cell programs in skeletal muscle;
• DUX4 induces elements that create an inflammatory reaction to muscle;
• At the same time, DUX4 suppresses some patways of our immune system;
• These pathways and programs lead to muscle atrophy and cell death.
Consequences of DUX4 in muscle
Geng et al., Dev. Cell 2012

• Translational research: – Increase our understanding of disease
mechanism; – Translate our findings to models that
allow validation of the mechanism; – Identify potential targets for therapy; – Apply disease models for drug
screens; – Validate hits from drug screens; – Clinical trials
What is next?
Mouse models
Muscle cell culture models
Yeast models

Disease Models for FSHD
Any disease model for FSHD should take into account the bursts of expression pattern of DUX4
Tapscott lab, Seattle
DUX4 at the tipping point
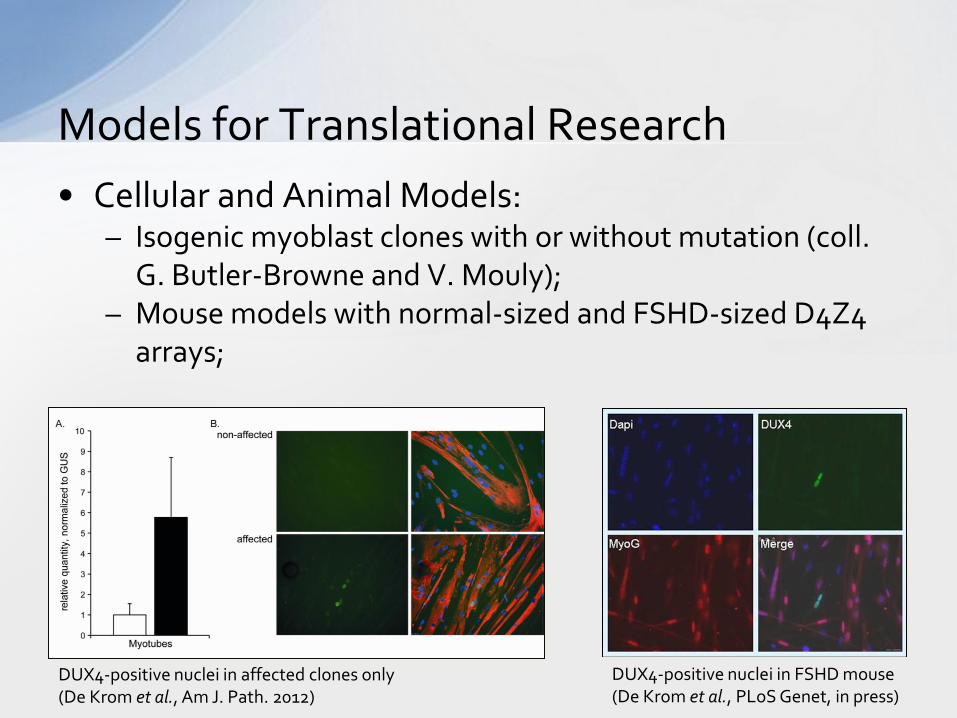
• Cellular and Animal Models: – Isogenic myoblast clones with or without mutation (coll.
G. Butler-Browne and V. Mouly); – Mouse models with normal-sized and FSHD-sized D4Z4
arrays;
Models for Translational Research
DUX4-positive nuclei in FSHD mouse (De Krom et al., PLoS Genet, in press)
DUX4-positive nuclei in affected clones only (De Krom et al., Am J. Path. 2012)
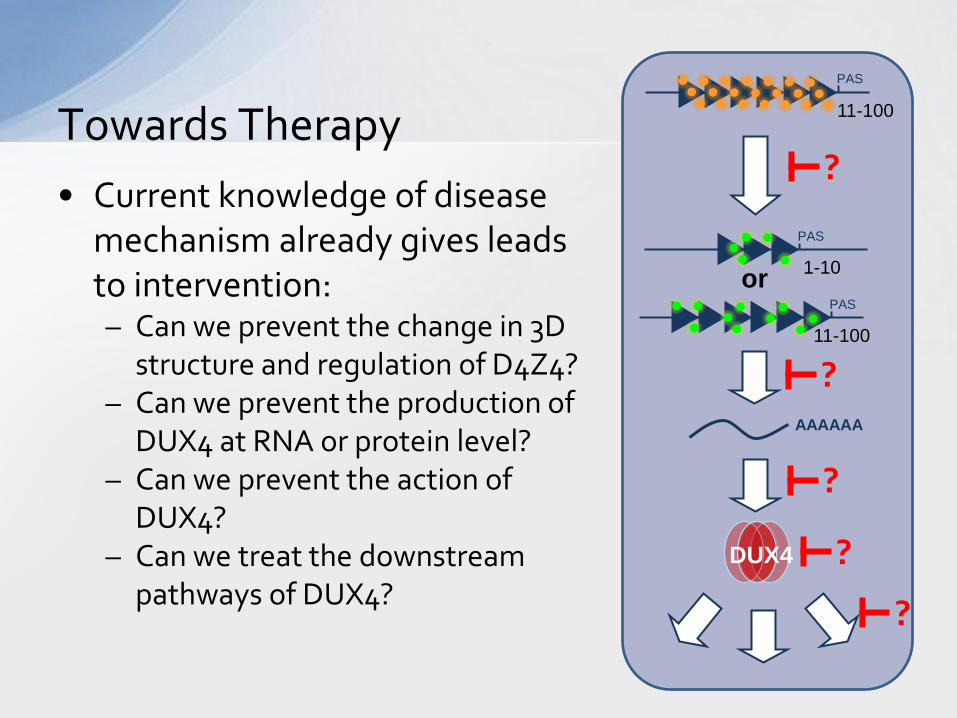
• Current knowledge of disease mechanism already gives leads to intervention: – Can we prevent the change in 3D
structure and regulation of D4Z4? – Can we prevent the production of
DUX4 at RNA or protein level? – Can we prevent the action of
DUX4? – Can we treat the downstream
pathways of DUX4?
Towards Therapy
AAAAAA
DUX4
11-100
1-10
11-100
or
PAS
PAS
PAS
?
?
?
?
?

• There are at least two genetic forms of FSHD – The common form FSHD1 (1-10 D4Z4 units) – The rare form FSHD2 (mostly mutations in SMCHD1)
• Both forms can be genetically confirmed with great accuracy
• Both forms have an identical disease mechanism – Expression of DUX4 in skeletal muscle
• Some individuals have FSHD1 and FSHD2 – Individuals have more variable disease severity
• We have uncovered the mechanistic basis of FSHD
Take home messages

• Not possible to predict, but we have the essentials: – We have a plausible disease mechanism – We know the target – We have (animal) models to test the therapeutic
molecules
• The DMD gene was identified in 1987 and only now there is some hope, but: – We have learned from the past: translational research – In the meantime the life expectancy for DMD has
dramatically increased: quality of care
How much longer?