gammopatie e mieloma multiplo - unità funzionale di ... · mieloma multiplo: complicanze...
TRANSCRIPT

GAMMOPATIE E MIELOMA MULTIPLO
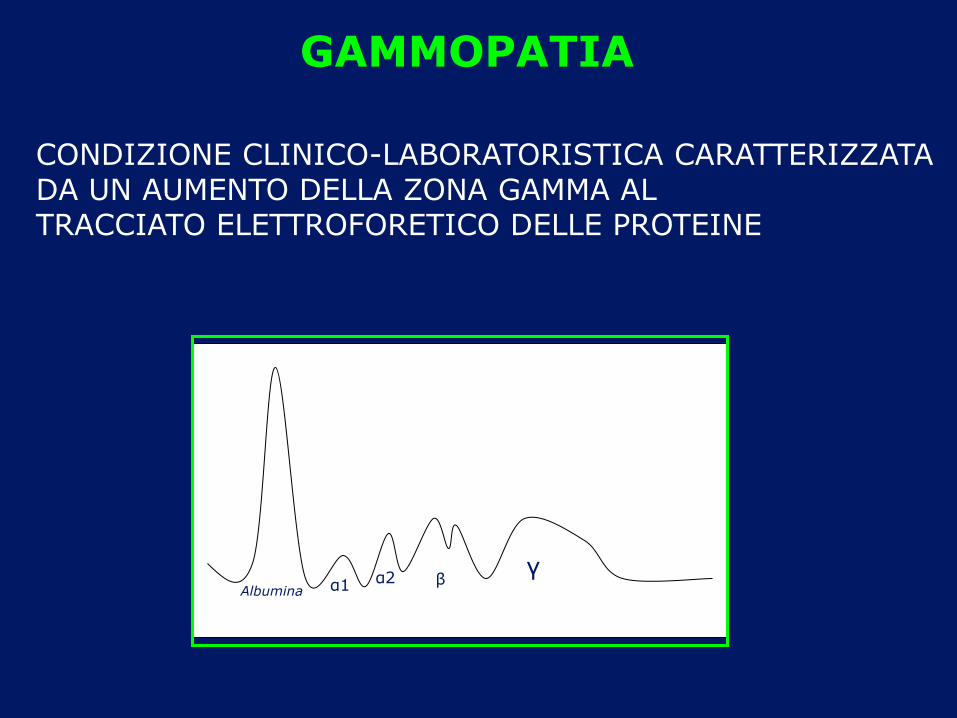
GAMMOPATIA
CONDIZIONE CLINICO-LABORATORISTICA CARATTERIZZATA DA UN AUMENTO DELLA ZONA GAMMA AL TRACCIATO ELETTROFORETICO DELLE PROTEINE
Albumina α1 α2 β γ
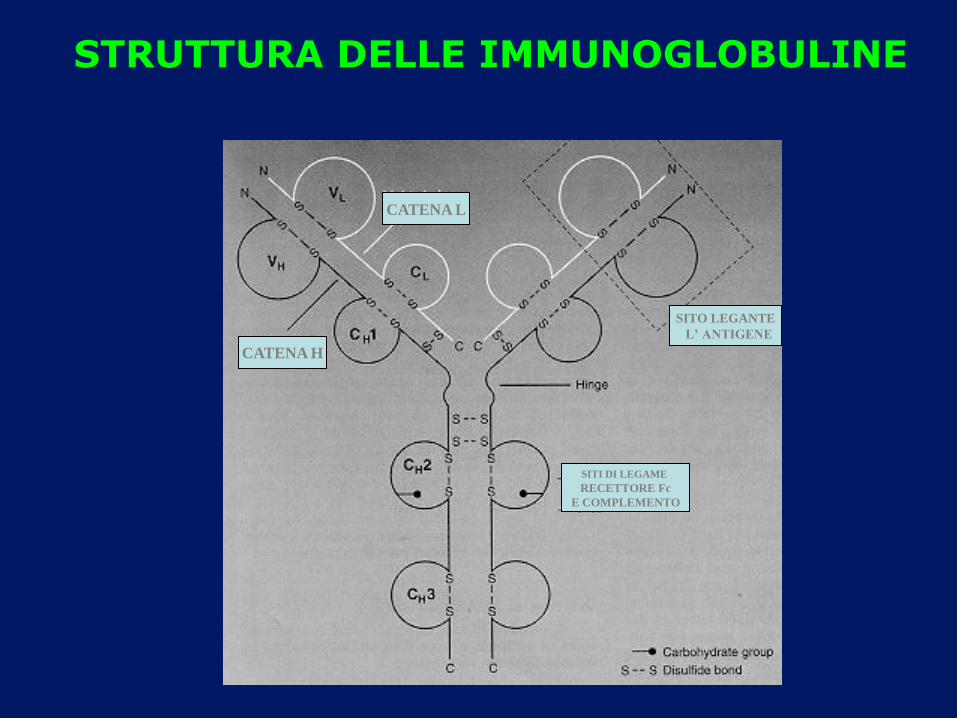
CATENA L
CATENA H
SITO LEGANTE
L’ ANTIGENE
SITI DI LEGAME
RECETTORE Fc
E COMPLEMENTO
STRUTTURA DELLE IMMUNOGLOBULINE

IgG IgA IgM IgD IgE
cat. H
cat. L K o K o K o K o K o
mg/dL 1000-1.400
200-300
40-150 3 <1
Emivita 21 g 6 g 5 g 3 g 2 g
Secrezioni esterne
+ +++ + ? ?
Passaggio placentare
+ - - - -
PROPRIETA’ DELLE IMMUNOGLOBULINE

IgG1 IgE
IgA
IgM
STRUTTURA DELLE IMMUNOGLOBULINE

ANTIGENICITA’ DELLE IMMUNOGLOBULINE
• ISOTIPO
• IDIOTIPO
• ALLOTIPO
• Classi anticorpali: IgG, IgA,
IgM, IgD, IgE
• Determinanti antigenici sulla
porzione ipervariabile delle Ig
• Polimorfismi della sequenza
Ia nelle regioni costanti H o L

LINFONODO
PLASMACELLULA
A VITA LUNGA CELL. Pre-B
PLASMACELLULA
A VITA BREVE
LINFOPLASMOCITA
CENTRO GERMINATIVO
PLASMABLASTO
LINFOBLASTO
LINFOCITA B VERGINE
MIDOLLO OSSEO
}
}
antigene
IND
IPE
ND
EN
TE
D
IPE
ND
EN
TE

GAMMOPATIA POLICLONALE
Stimolazione di più cloni di PC con incremento di una o diverse classi di Ig che esprimono entrambe le catene leggere
Condizioni cliniche: - epatopatie
- mal collagene (LES, AR, Sjogren) - infezioni (TBC, EBV, toxoplasmosi, endocarditi) - sarcoidosi - fibrosi cistica

GAMMOPATIA MONOCLONALE
Stimolazione di un SINGOLO clone di PC con incremento di un SOLO tipo di catena pesante e di catena leggera
ASSOCIATA A:
MGUS (56%) Mieloma multiplo (18%) Amiloidosi (10%) Linfomi non HDG (5%) LLC (2%) Macrogl.Waldestrom (2%)

Collagenopatie e malattie autoimmuni: AR, Crohn,
polimialgia reumatica, LES, sclerodermia, Sjogren, artrite psoriasica, miastenia gravis
Malattie cutanee: pioderma gangrenosus, psoriasi,
orticaria, scleromixedema
Malattie endocrine: iperparatiroidismo
Malattie epatiche: epatite, cirrosi
Malattie infettive: micobatteri, CMV, endocardite batterica, AIDS
Neoplasie: carcinomi colon, polmone, prostata,
GAMMOPATIA MONOCLONALE
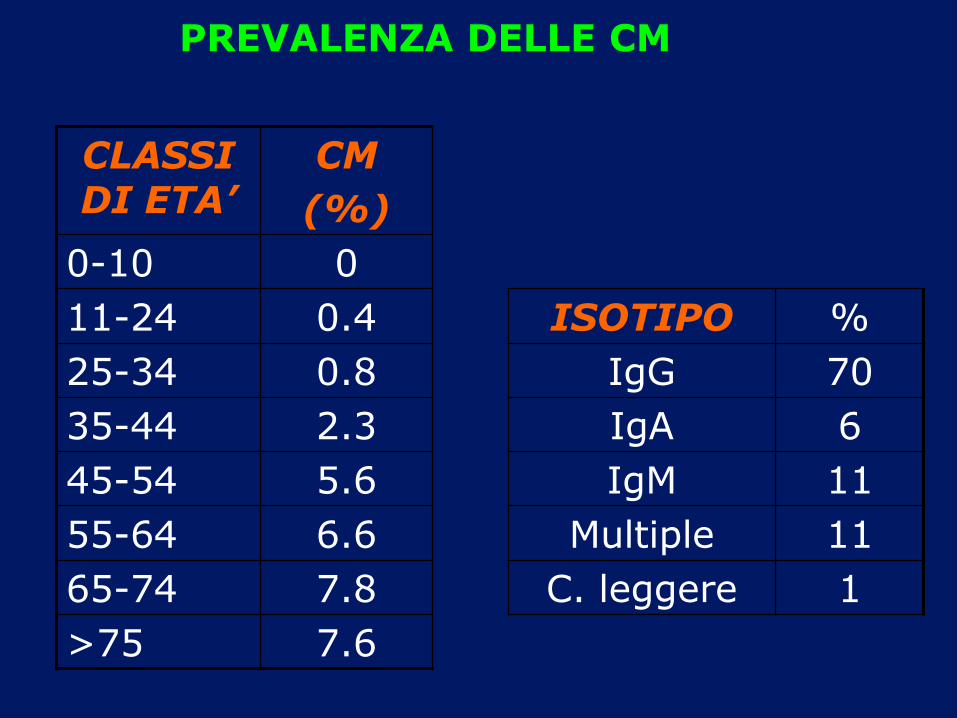
CLASSI DI ETA’
CM
(%)
0-10 0
11-24 0.4 ISOTIPO %
25-34 0.8 IgG 70
35-44 2.3 IgA 6
45-54 5.6 IgM 11
55-64 6.6 Multiple 11
65-74 7.8 C. leggere 1
>75 7.6
PREVALENZA DELLE CM

MGUS
INCIDENZA: 1% popolazione >50 anni; 3% popolazione >70 anni
DIAGNOSI: CM sierica <3 gr/L
<5% PC midollari assenza di lesioni osteolitiche, anemia, ipercalcemia funzione renale nella norma
EVOLUZIONE: 25% sviluppa patologia linfoproliferativa
55% presenta malattia stabile 15% decesso per altre cause 5% picco scompare
FOLLOW-UP: esami ematici periodici semestrali (proteine tot,
elettroforesi e dosaggio immunoglobuline)
TERAPIA: nessuna
CLINICA: assenza di sintomi
riscontro laboratoristico occasionale

• Il rischio attuale di trasformazione di MGUS in MM è del:
• 6.1% a 5 anni
• 15.4% a 10 anni
• 31.3% a 20 anni
EVOLUZIONE DELLE MGUS
FATTORI DI RISCHIO
Elevati livelli iniziali di CM Forme IgA e IgM Catene leggere vs pesanti ( 70% vs 30%)

E’ POSSIBILE
PREVEDERE L’EVOLUZIONE DI UNA
MGUS A MM?
NON CON CERTEZZA (ma il Ratio / può essere indicativo)
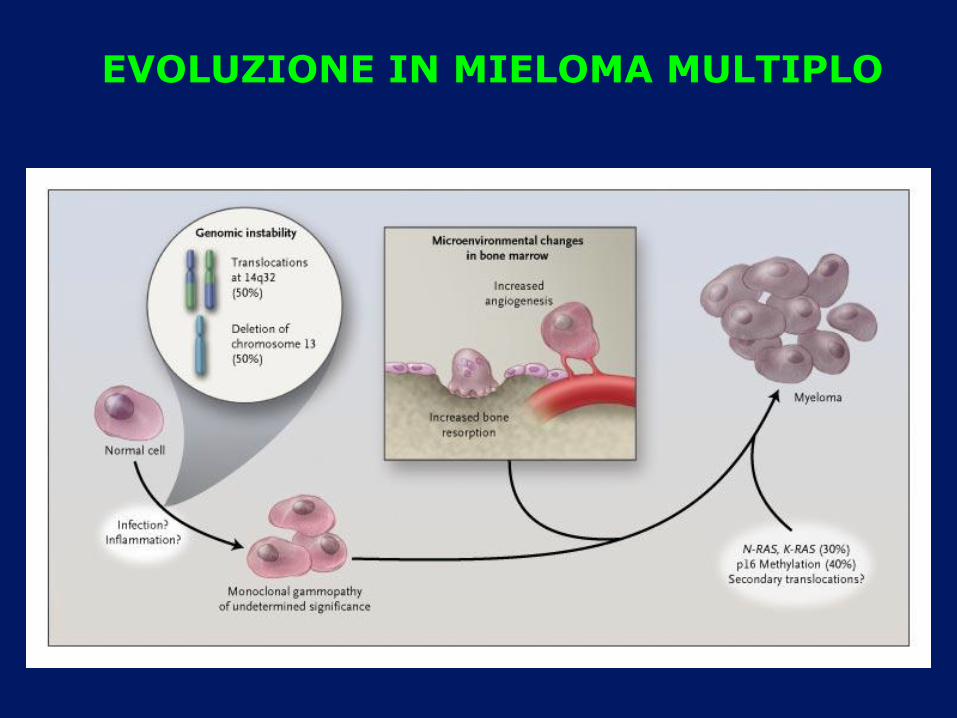
EVOLUZIONE IN MIELOMA MULTIPLO

ESISTE UNA STRETTA CORRELAZIONE TRA MGUS E
MIELOMA MULTIPLO
Un sostegno alla teoria dei “two-hits”
I evento oncogeno comparsa della CM
(“MGUS”)
II evento oncogeno Mieloma multiplo

MIELOMA MULTIPLO
PATOLOGIA CARATTERIZZATA DALLA PROLIFERAZIONE NEOPLASTICA DI UN SINGOLO CLONE DI PLASMACELLULE AD ELETTIVA LOCALIZZAZIONE MIDOLLARE
IN GRADO DI PRODURRE ELEVATE QUANTITA’ DI IMMUNOGLOBULINE
MONOCLONALI (CM)
DEFINIZIONE

EPIDEMIOLOGIA
EZIOLOGIA
MIELOMA MULTIPLO
1% DI TUTTE LE NEOPLASIE 10% DELLE NEOPLASIE EMATOLOGICHE INCIDENZA ANNUALE: 3-4 /100.000/anno POPOLAZIONE NEGRA/BIANCA: 2:1 ETA’ MEDIA DIAGNOSI: 68 ANNI 4% ETA’ INFERIORE AI 40 ANNI M:F=1:1
CAUSA ANCORA SCONOSCIUTA ESPOSIZIONE A RADIAZIONI ESPOSIZIONE A METALLI PESANTI, ASBESTO, BENZE, FITOFARMACI • SUSCETTIBILITA’ GENETICA • FLOGOSI CRONICA (continua stimolazione Ag) • HHV8

• UN’INDAGINE PRELIMINARE INDICA 100 NUOVI CASI DI MM/ANNO NELLE PROVINCIE DI FIRENZE E PRATO
• LA SOPRAVVIVENZA MEDIA DEL MM DALLA
DIAGNOSI È DI 3 ANNI, CON UN RANGE DA POCHI MESI A DIVERSI ANNI.
EPIDEMIOLOGIA E SOPRAVVIVENZA

MM:PATOGENESI
Il mieloma non è patologia neoplastica della PC, ma del sistema linfoide B, di una cellula B che è antecedente alla maturazione in plasmacellula.
Infatti: linfociti B circolanti del soggetto presentano Ig di superficie con le stesse caratteristiche isotipiche ed idiotipiche della CM
IPERMUTAZIONE SOMATICA
SELEZIONE ANTIGENICA
RICOMBINAZIONE IGH SWITCH

STROMA
IL-6
IL-6
IL-6
IL-6R
IL-1
TNF-
M-CSF
HGF
IL-6 IL-6
Molecole
adesione IL-1
TNF-
M-CSF
HGF
NEOANGIOGENESI
Fas
MM:PATOGENESI

MM: TIPO
• SECERNENTE
• BENCE JONES
• NON SECERNENTE
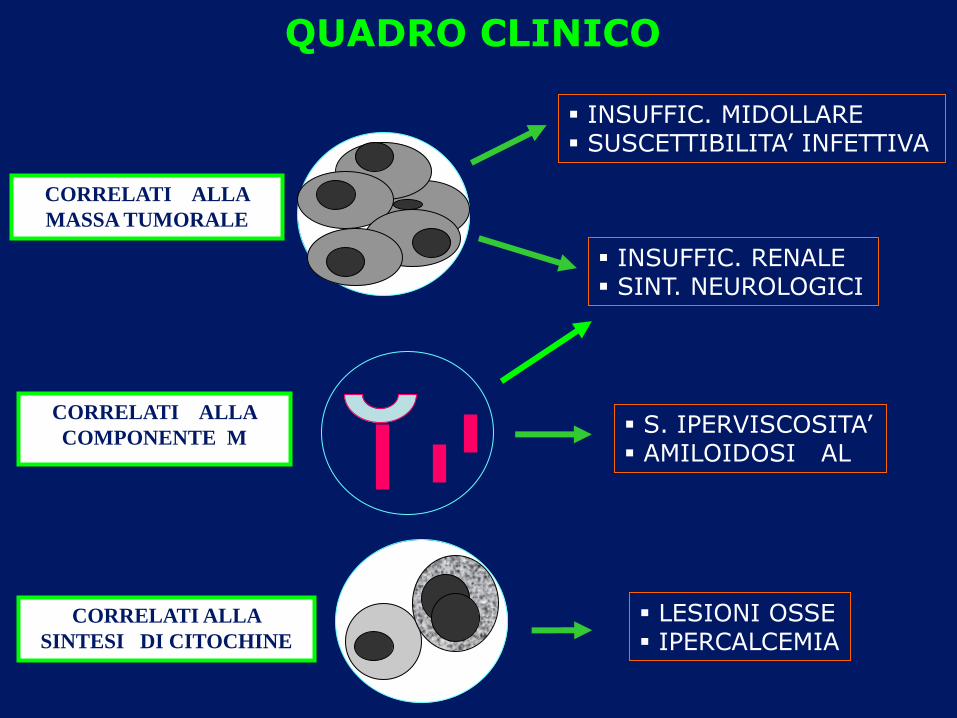
CORRELATI ALLA
COMPONENTE M
CORRELATI ALLA
SINTESI DI CITOCHINE
S. IPERVISCOSITA’ AMILOIDOSI AL
INSUFFIC. RENALE SINT. NEUROLOGICI
LESIONI OSSE IPERCALCEMIA
INSUFFIC. MIDOLLARE SUSCETTIBILITA’ INFETTIVA
CORRELATI ALLA
MASSA TUMORALE
QUADRO CLINICO

MM E DOLORI OSSEI
SEDI: rachide, bacino, cranio, coste, ossa lunghe (omero-femore)
TIPO LESIONE: lisi a stampo, osteoporosi diffusa, fratture
EZIOPATOGENESI: citochine (OAF: Osteoclast Activing Factors) IL6, TNF, IL1 che creano aumento dell’attività degli osteoclasti
DIAGNOSI: Rx scheletro (RMN e scintigrafia)

MM E DOLORI OSSEI
LESIONI DELLE DIPLOE DELLA TECA
CRANICA IN REGIONE FRONTALE E
MASSE ESPANSIVE
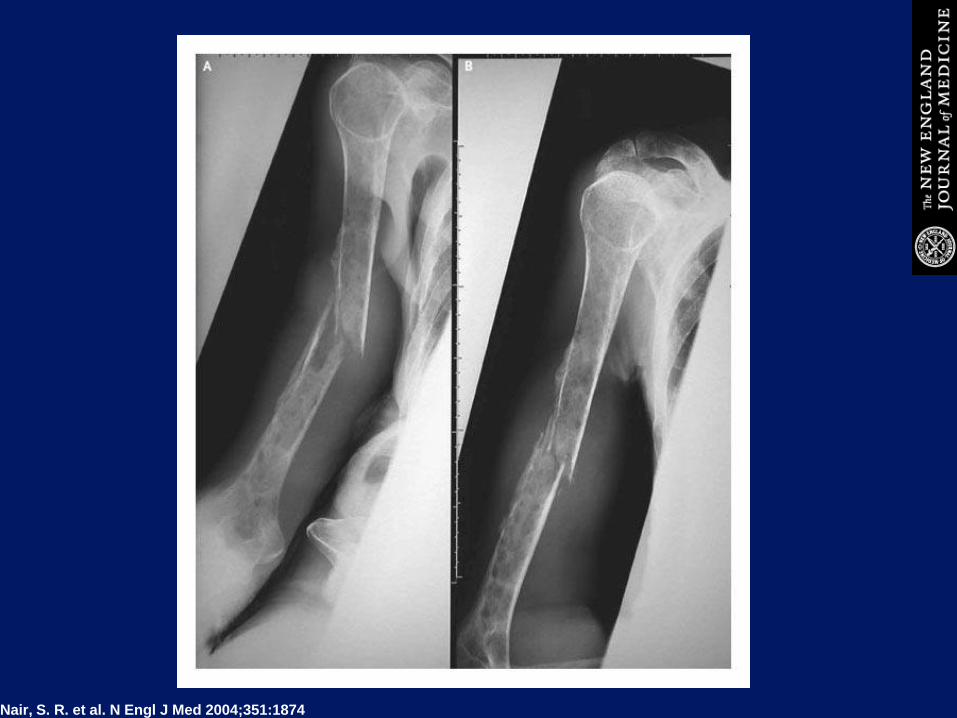
Nair, S. R. et al. N Engl J Med 2004;351:1874
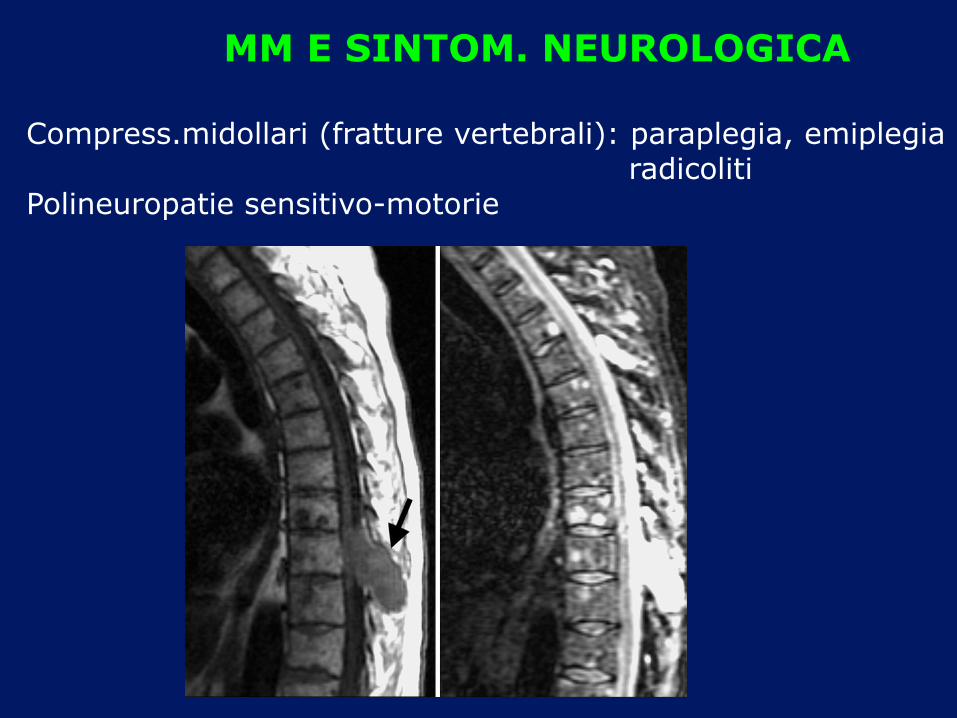
MM E SINTOM. NEUROLOGICA
Compress.midollari (fratture vertebrali): paraplegia, emiplegia radicoliti Polineuropatie sensitivo-motorie
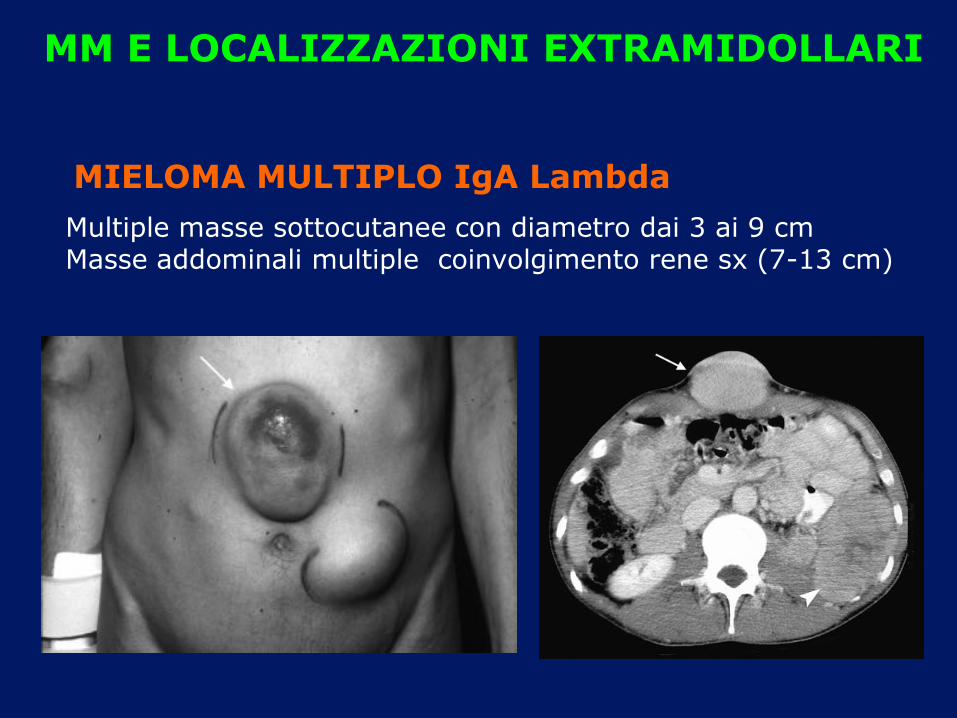
MM E LOCALIZZAZIONI EXTRAMIDOLLARI
Multiple masse sottocutanee con diametro dai 3 ai 9 cm Masse addominali multiple coinvolgimento rene sx (7-13 cm)
MIELOMA MULTIPLO IgA Lambda

MM E LOCALIZZAZIONI SNC
LIQUOR

MM E ALTERAZ. LABORATORIO
• Anemia (normocromica-normocitica)
• Piastrinopenia e leucopenia (grado infiltrazione midollare)
• VES e PRC
• Ipercalcemia
• Iperuricemia
• Creatinina e azotemia
• Proteine totali
• Albumina
• Altre Ig (non CM)
• Elettroforesi: 80% picco monoclonale
10% ipogammaglobulinemia

Test diagnostici di base
• Emocromo completo, formula leucocitaria
• Creatinina e creatinina clearance
• Azotemia
• Calcemia, calciuria
• Uricoemia
• LDH
• Fosfatasi alcalina
• Proteine totali e frazionate
• Dosaggio quant. Ig
• Immunoelettroforesi
• 2-microglobulina
• PCR
• Bence Jones quant.
• Rx torace
• Eco addome
• Rx scheletro
• (RMN)
• BOM

Test diagnostici con significato
prognostico
• 2 –microglobulina
• PCR
• LDH
• Massa tumorale
• Funzione renale
• Correla sopravvivenza
• Correla livelli IL-6
• Correla sopravivenza
• Indipendente da 2-
microglobulina
• Aumenta in quadri infiammatori, …….

• Indice di marcatura
delle plasmacellule
• Citogenetica
• Correla con attività proliferativa plasmacellulare
• Correla con sopravvivenza: MS 56 mesi se LI <3% vs 19 se LI >3%
• Anomalie presenti nell’80-90% casi (traslocazioni, trisomie)
• Cariotipo sfavorevole (11q,
-13)
Test diagnostici di livello n

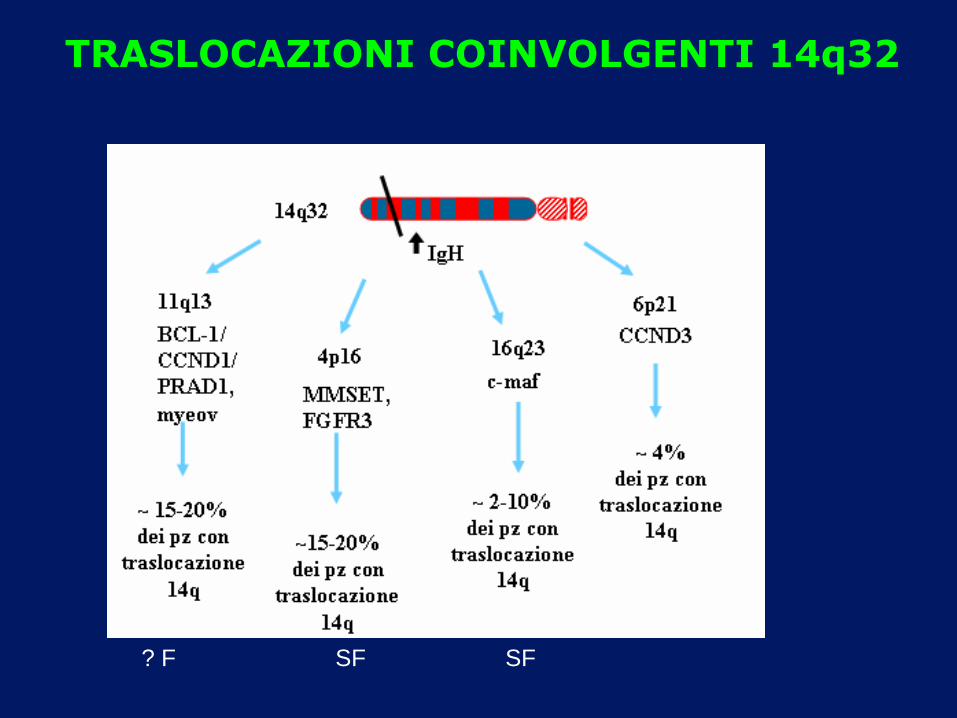
TRASLOCAZIONI COINVOLGENTI 14q32
? F SF SF

FISH: MONOSOMIA 13q
FREQUENZA 30-50% FATTORE PROGNOSTICO SFAVOREVOLE

Rx CONVENZIONALE e RMN
• La Rx convenzionale dello scheletro in toto fornisce informazioni sulle entità e sede delle lesioni osteolitiche
• La RMN della colonna può evidenziare positività in >50% dei pazienti con mieloma indolente (“mieloma asintomatico con 4 lesioni ossee e assenza di insufficienza renale”)
• La Scintigrafia ossea può mettere in evidenza aree di rimaneggiamento osseo, ma queste non sono significative per mieloma

MM: STRISCIO PERIFERICO
emazie a rouleaux

MM: ASPIRATO MIDOLLARE

MAGGIORI:
• Plasmocitoma alla biopsia del tessuto
• Plasmacellule midollari 30%
• CM:
– IgG >3.5 g/dL
– IgA >2 g/dL
– BJ 1g/24h
MINORI:
• Plasmacellule midollari 10-29%
• CM < ai livelli maggiori
• Lesioni osse litiche
• Diminuzione delle Ig normali
– IgG <600 mg/dL
– IgA <100 mg(dL
– IgM <50 mg/dL
DIAGNOSI: 1 criterio maggiore + 1 minore , oppure 3 criteri minori
CRITERI DIAGNOSTICI

CLASSIFICAZIONE DELLE GAMMAPATIE
MONOCLONALI
• MGUS: presenza di una CM sierica o urinaria, ma non sono rispettati i criteri diagnostici per il Mieloma
• MIELOMA SOLITARIO OSSEO: osteolisi unica o regionale, qualora non siano soddisfatti i criteri diagnostici per il mieloma multiplo
• PLASMOCITOMA EXTRAOSSEO: massa mielomatosa localizzata in sede extraossea, qualora non siano soddisfatti i criteri diagnostici per il mieloma multiplo
• LEUCEMIA PLASMACELLULARE: plasmacellule circolanti superiori al 20% o comunque > 20.000/L
• SMOULDERING MIELOMA (MIELOMA INDOLENTE): mieloma asintomatico e senza caratteri di progressività
• MIELOMA MULTIPLO
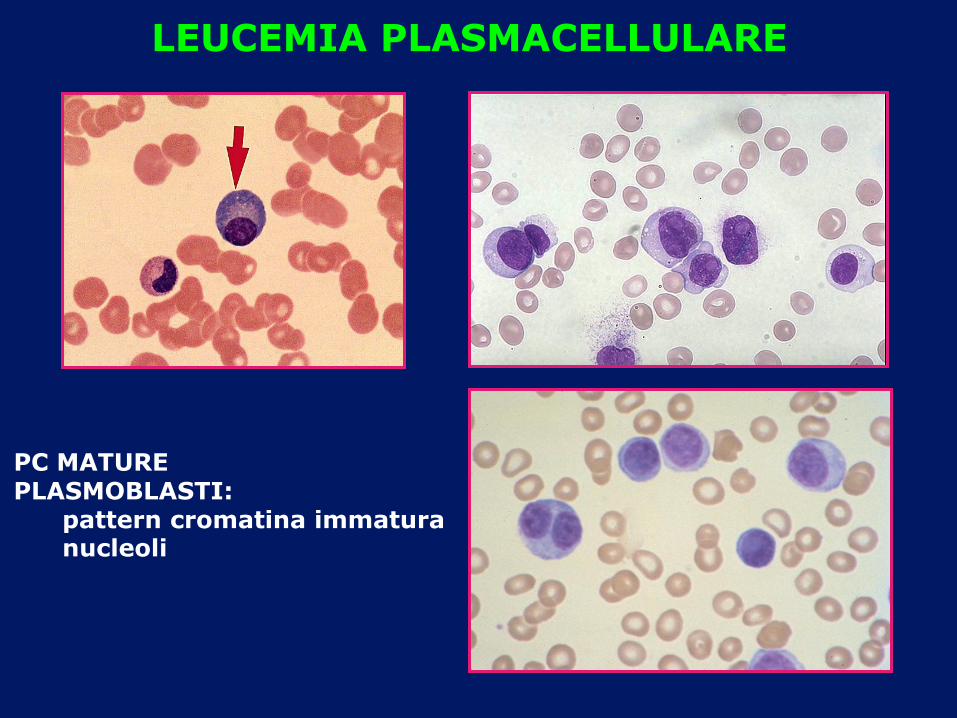
LEUCEMIA PLASMACELLULARE
PC MATURE PLASMOBLASTI: pattern cromatina immatura nucleoli

AMILOIDOSI
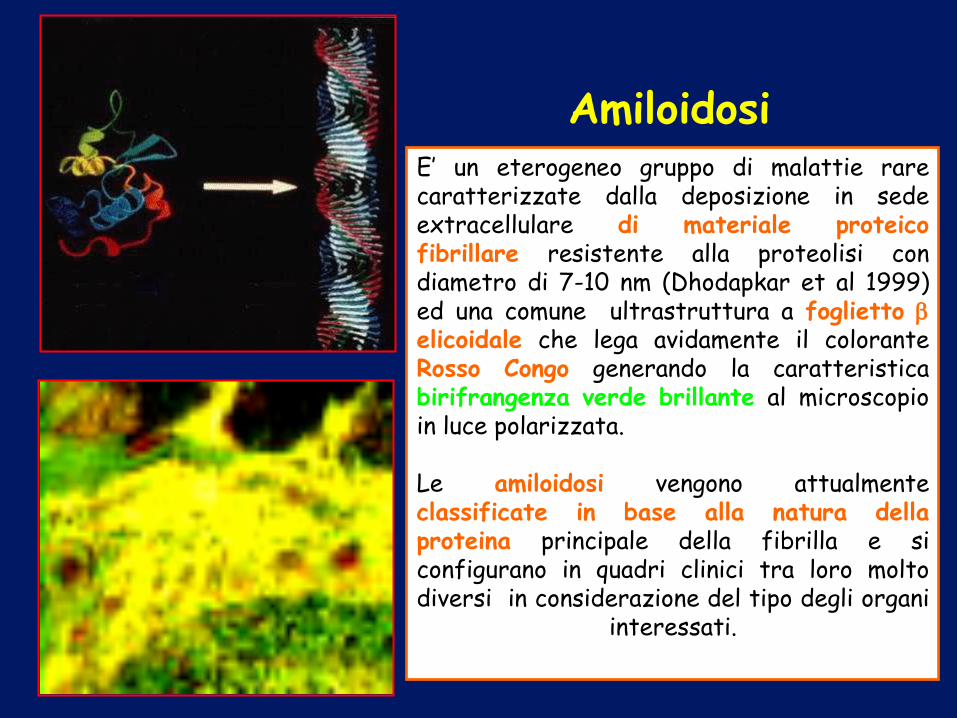
Amiloidosi E’ un eterogeneo gruppo di malattie rare caratterizzate dalla deposizione in sede extracellulare di materiale proteico fibrillare resistente alla proteolisi con diametro di 7-10 nm (Dhodapkar et al 1999) ed una comune ultrastruttura a foglietto elicoidale che lega avidamente il colorante Rosso Congo generando la caratteristica birifrangenza verde brillante al microscopio in luce polarizzata. Le amiloidosi vengono attualmente classificate in base alla natura della proteina principale della fibrilla e si configurano in quadri clinici tra loro molto diversi in considerazione del tipo degli organi
interessati.

Classificazione
• Amiloidosi primitiva (AL), (AH)
• Amiloidosi secondarie (SAA)
• Amiloidosi familiari (TTR,AFib,APO I, ApoII)
• Amilodosi localizzate (cerebrale quali la m. di Alzheimer e alcune malattie da prioni)
SISTEMICHE

Amiloidosi AL
• L'amiloidosi AL è la forma sistemica più comune nel mondo occidentale
• La sua incidenza negli USA è 9 casi per milione/anno
• Circa il 15% dei pazienti con mieloma multiplo presenta depositi di amiloide.
• Fibrille di catene leggere di Ig monoclonali prodotte da un clone plasmacellulare midollare
• Prevalenza delle catene lambda

Sintomi di esordio
• Edemi
• Astenia
• Perdita di peso
• Dispnea
• Ipotensione ortostatica
• Disturbi dell’alvo
• Sindrome del tunnel carpale (20%)
• Porpora periorbitale
• Macroglossia (10%)

Coinvolgimento d’organo
Rene (30%) → proteinuria, s.nefrosica, IR
Cuore (25%) →cardiomiopatia restrittiva con disfunzione diastolica, difetti di conduzione e le tachiaritmie atriali o ventricolari
Fegato (20-25%) → epatomegalia, fosfatasi alcalina e transaminasi elevate in un terzo dei pazienti; iperbilirubinemia infrequente e prognosticamente sfavorevole
SNP → (33%) neuropatia periferica sensitivo-motoria, tipicamente distale, simmetrica, progressiva soprattutto alle estremità inferiori; (15%) sistema nervoso autonomo con ipotensione ortostatica, sincope, vertigine, alterazioni dell'alvo, disfunzione vescicale o impotenza.

INQUADRAMENTO DIAGNOSTICO
•Immunofissazione sierica ed urinaria diagnostiche nel 90-95% dei casi •Catene leggere libere sieriche: RATIO •Immunoistochimica per clonalità plasmacellulare
DETERMINAZIONE DELLA CM
DIMOSTRAZIONE ISTOLOGICA DEI DEPOSITI DI AMILOIDE con Rosso Congo e con immunoistochimica
• biopsia GPO (pos 70-80%); salivari minori, retto • biopsia midollo osseo • biopsia renale, epatica, cardiaca: grave rischio emorragico

INQUADRAMENTO DIAGNOSTICO
PROTEINURIA, CREAT.CLEARENCE
TROPONINA SIERICA, NT-ProBNP
•un aumentato spessore delle pareti ventricolari, e del SIV •dilatazione atriale sinistra e disfunzione diastolica •iperecogenicità del miocardio con il caratteristico aspetto granulare
ECOCARDIO/DOPPLER, ECG: valutazione SIV,FE
ECOGRAFIA ADDOMINALE, FOSFATASI ALCALINA GGT, TRANSAMINASI
EMG, POTENZIALI EVOCATI
FATTORE X STUDIO GENETICO PER TTR, aFIB, LISOZIMA ,APO-AI, APO-AII

INDIRIZZI TERAPEUTICI
ATTUALI NEL
MIELOMA MULTIPLO

MIELOMA MULTIPLO: COMPLICANZE
• IPERCALCEMIA: anoressia, nausea, vomito, sete intensa, poliuria, stipsi, debolezza, alterato stato coscienza
• LESIONI OSSEE E FRATTURE
• ANEMIA TRASFUSIONE DIPENDENTE
• INFEZIONI
• INSUFFICIENZA RENALE
• NEUROLOGICHE

INDIRIZZI TERAPEUTICI
• Chemioterapia convenzionale – I
• Chemioterapia convenzionale – II
• Chemioterapia ad alte dosi – TMO autologo
• Chemioterapia ad alte dosi - TMO allogenico
• Chemioterapia ad alte dosi – TMO “mini”allo
• Interferone-
• Talidomide
• Vaccino anti-idiotipo
• Anticorpi monoclonali anti-IL6

TERAPIA DI SUPPORTO E
DELLE COMPLICANZE
• Bifosfonati (dolore e fratture)
• Eritropoietina (anemia)
• Fisioterapia
• Pronto trattamento antibiotico largo spettro per infezioni /antimicotico
• Vaccinazioni per influenza e pneumococco
• IgG ad alte dose e.v.
• Tearapia della IR, fino alla dialisi
• RT locale, specie sulla colonna, e fino a decompressione chirurgica
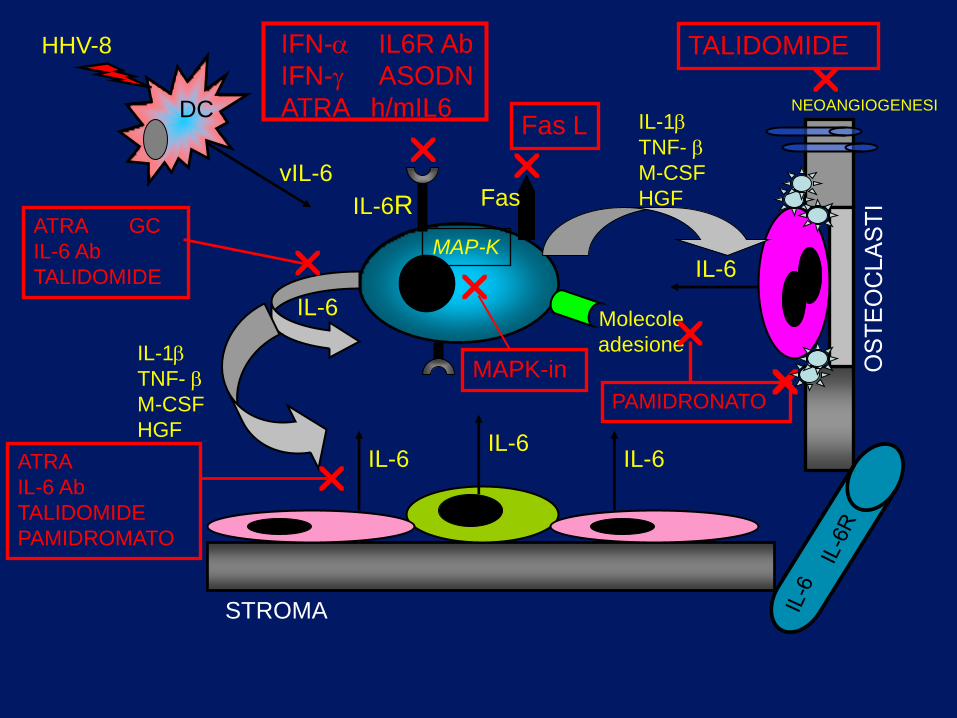
STROMA
IL-6
IL-6
IL-6
DC
HHV-8
vIL-6
IL-6R
IL-1
TNF-
M-CSF
HGF
MAP-K
IL-6 IL-6
Molecole
adesione IL-1
TNF-
M-CSF
HGF
NEOANGIOGENESI
Fas
IFN- IL6R Ab
IFN- ASODN
ATRA h/mIL6
MAPK-in
Fas L
ATRA
IL-6 Ab
TALIDOMIDE
PAMIDROMATO
TALIDOMIDE
ATRA GC
IL-6 Ab
TALIDOMIDE
PAMIDRONATO