hematology rivas2009lecture2
TRANSCRIPT

RBC DisordersANEMIAS
Antonio Rivas PA-C
Lecture 2. 2009

Objectives
Discuss the etiology, pathogenesis, clinical features, laboratory evaluation, and management of:
Aplastic anemia Iron deficiency anemia Megaloblastic anemia Anemia of chronic disease Hemolytic anemia Sickle cell disease Thalassemias Sideroblastic anemia

Disruption of the normal stem cell development can result in:- Underproduction of mature cells •Aplastic anemia
- Overproduction of mature cells • Myeloproliferative disease
- Failed differentiation with with production of increased immature cells
• Myelodisplasia • Acute leukemia
Disorders of Hematopoiesis
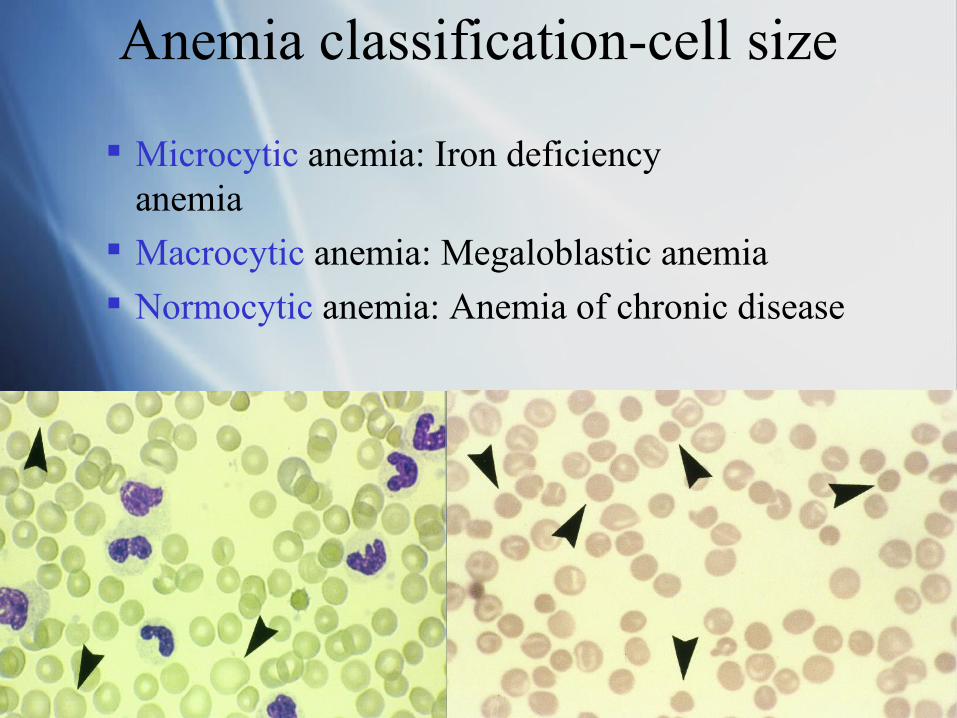
Anemia classification-cell size
Microcytic anemia: Iron deficiency anemia
Macrocytic anemia: Megaloblastic anemia
Normocytic anemia: Anemia of chronic disease

Differential Dx.Anemias - MCV
Low MCV
Thalassemia
Iron def.
Lead Pois.
Sideroblastic
Normal MCV
Acute B. loss
Aplastic A.
Chronic Dis.
Hemoglobinop-athies
Hemolysis
Iron def.
High MCVAlcohol abuseAplastic A.B12 def.Folate defHemolysisHypothyroidismLiver DsMyelodysplastic s

Reticulocyte count vs Retic index Reticulocyte: young circulating Rbc that exhibits
basophilia
Retic count: characterize the bone marrow attempt to compensate, if at all, for the anemia present
The Retic. Index: more useful, corrects for the hematocrit abnormalities

Retic index
Less than 2 found:
Hypoproliferative anemias
Dis. Of heme or globin synthesis Iron deficiency and other metabolic anemias
B12 or folate deficiency
Chronic dis.
Lead poisoning
Thalassemias

Retic index Greater than 2 found:
Hyperproliferative anemias Acute blood loss
Nutrient replacement(B12,folate,Iron) before resolution of the anemia
Hereditary or acquired hemolysis
Polycythemia

Pathophysiology Immune hemolytic anemia
Warm hemolytic anemia(IgG) Cold hemolytic anemia(IgM)
Hemolysis causes extrinsic to RBCs Microangiopathic hemolytic anemia infection
Hemolysis due to disorders RBC membrane Inherited membrane abn.(HS,HE) Acquired membrane abn.(PNH,Spur cell anemia)
Hemolysis due to disorders in RBC enzymes G6PD deficiency
Hemoglobinopathies Sickle cell disease Thalassemia(alpha,beta)

Anemia Reduction of RBC mass or HGB
concentration
Symptoms reflect the rapidity of onset
Pat. with acute hemorrhage/massive hemolysis may exhibit symptoms of hypovolemic shock
Slowly established anemia produce few symptoms

Anemia Symptoms
Fatigue
Decreased exercise tolerance
Dyspnea
Palpitations

Anemia, physical exam
Major sign is pallor of skin, and mucous membranes
May develop tachycardia
Possible audible flow murmurs
Patients with hemolysis could present with jaundice and splenomegaly

Laboratory evaluation Reticulocyte count differentiates between
failure of RBC production (low retic) and increased RBC destruction (increased retic)
Peripheral blood smear provide clues to the cause of the anemia: Spherocytes in immune hemolysis Schistocytes in microangiopathic hemolysis

Laboratory eval. Cont.
Sickle and target cell in hemoglobinopathies Tear drop cells and nucleated RBCs in
myelofibrosis and marrow infiltration Intracellular parasites in Malaria and Babesiosis Pencil shape cells in severe iron deficiency Hypersegmented neutrophils and large plts. In
megaloblastic anemia Immature blasts in leukemias

Laboratory eval.cont. MCV is important in anemia with low retic count, to
differentiate according to cell size Bone marrow analysis helpful in patients with low
retic count, provides information about causes of anemia
If retic count is increased no need for bone marrow analysis, need to determine if decreased RBC mass is due to bleeding or hemolysis
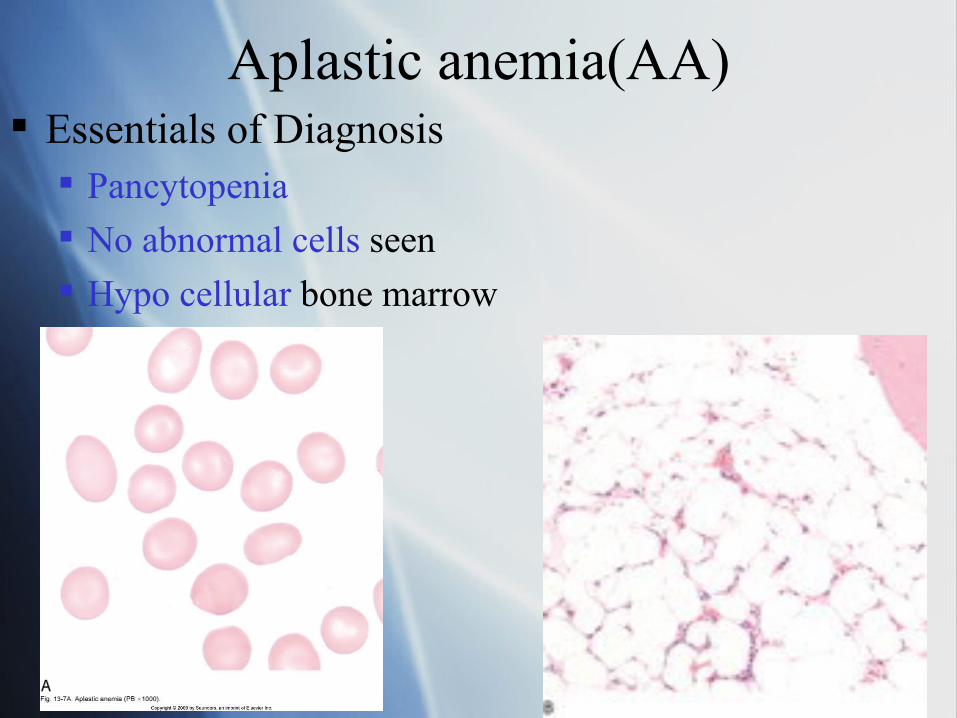
Aplastic anemia(AA) Essentials of Diagnosis
Pancytopenia
No abnormal cells seen
Hypo cellular bone marrow

AA - General considerations Hematopoietic stem cell failure Rare disorder Incidence 1-5 cases per million Affects young adults(20-25yo) and older adults
(60-65yo) Three fold higher incidence in developing
countries (Thailand and China) Most common cause: suspected autoimmune
suppression of hematopoiesis by T-cell mediated cellular mechanism

AA. Acquired causes Drugs : dose related, chemotherapeutic agents,
antibiotics (chloramphenicol , trimethoprim, sulfamethoxazole)
Idiosyncratic(unproven): chloramphenicol, quinacrine, NSAIDS, cimetidine ,penicillamine
Toxins: benzene/hydrocarbons,insecticides Viral infections: hepatitis,EBV,HIV Immune mediated: GVHD ,immuno-
deficiency/SLE, paroxysmal nocturnal hemoglobinuria(PNH) Radiation Pregnancy

AA-Congenital causes
Small proportion of the cases Defective genetic material (DNA ,gene mutations,
etc.) Fanconi’s anemia(most common)
Schwachmann-Diamond syndrome
Dyskeratosis congenita

AA- signs/symptoms
Clinical onset can be insidious or abrupt Early in the disease not all cell lines are
always affected Weakness-fatigue due to anemia Vulnerability to infections if leukopenia is
present Mucosal/skin bleeding associated to
thrombocytopenia

AA-Laboratory findings
Pancytopenia
Anemia may be severe, always associated to decreased reticulocytes
RBC morphology is unremarkable
Hypo cellular marrow analysis
No abnormal cells seen

AA-Differential Diagnosis
From other causes of pancytopenias: Myelodisplasia/acute leukemia: exhibit
morphologic abnormalities in the cells and increased blasts, or abn. Cytogenetics in bone marrow
Hairy cell leukemia: presence of splenomegaly and abnormal Lymphoid cells in bone marrow
SLE, disseminated infection , or hypersplenism: normocellular bone marrow

AA-treatment Current approaches geared to:
Replacing defective marrow: Stem cell transplant
or
Controlling an overactive immune response:
using antithymocyte globulin (ATG) and other
immunesuppresors such as Cyclosporine; Cyclophosphamide

AA-treatment Based on severity of disease:
• mild cytopenias - monitor patient
• severe cases as indicated by blood count results showing:
- anemia, with retic count < 1%
- neutrophil count <500cells/ul
- Plt. count <20,000./ul
- bone marrow cellularity <25%
have poor survival, 2-6 months, without treatment

AA-treatment Supportive care with broad-spectrum antibiotics,
antifungals, and antivirals is warranted in advanced neutropenia
RBC and Plt. Transfusions are helpful for very symptomatic patients
Allogeneic bone marrow transplant offers better outcome (75-90% survival rate) on patients 30 years of age and younger
Immunosuppressive therapy achieve restoration of marrow function in 70-80% of the patients, with a 90% survival rate in 5 years (patients older than 40 years of age)

Hypoproliferative anemias
Microcytic anemia: Iron deficiency anemia
Macrocytic anemia:Megaloblastic anemia
Normocytic anemia:Anemia of chronic disease
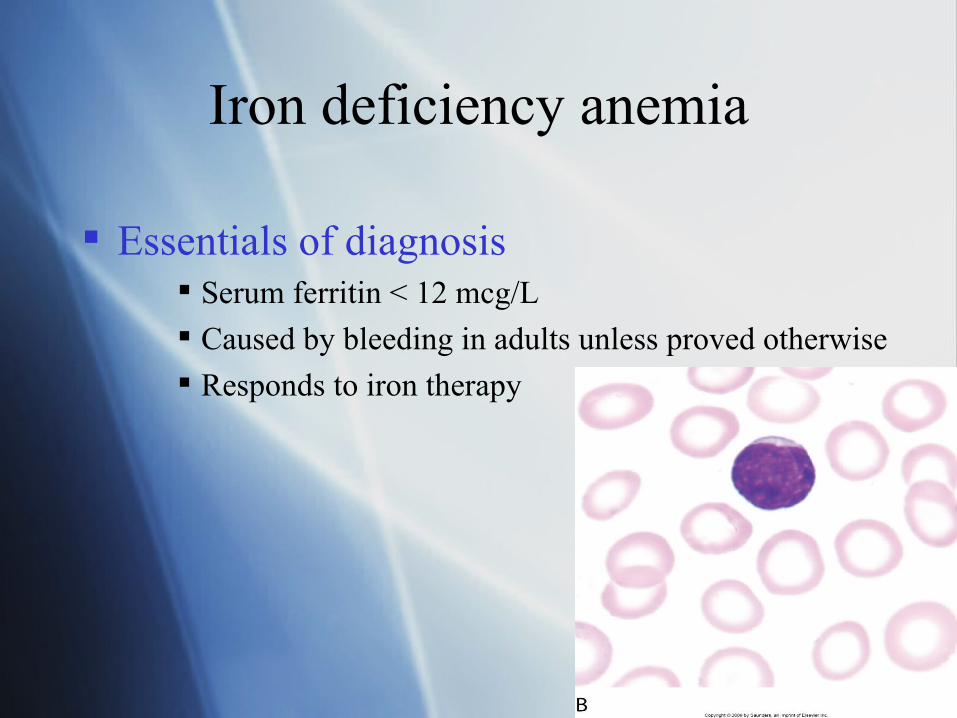
Iron deficiency anemia
Essentials of diagnosis Serum ferritin < 12 mcg/L
Caused by bleeding in adults unless proved otherwise
Responds to iron therapy

Iron deficiency anemia
Most common cause of anemia worldwide
Average american diet contains 10-15mg of iron per day (10% is absorbed)
Iron Absorption in proximal small intestine
Balanced daily iron absorption and loss at 1mg/day

Iron deficiency anemia
Iron Bound to transferrin in plasma Serum iron: men 50mg/kg, women 40 mg/kg 60-75% found in Hgb Iron Stored as ferritin in the liver, spleen, BM,and
muscle Pre-menopausal women could loose15mg of iron
per month, and 900mg/pregnancy Investigate GI blood loss in iron deficient men and
postmenopausal women

Iron def.anemia, causes
Most frequent cause of iron def. is blood loss
Decreased iron absorption is rare, occasional after gastric surgery
Hemoglobinuria in traumatic hemolysis due to prosthetic heart valve
Frequent blood donors

Iron deficiency -symptoms
In severe cases, skin and mucosal changes: Smooth tongue
Brittle nails
Cheilosis
Dysphagia-esophageal webs (Plummer-Vinson syndrome
Pica (ice,dirt)

Iron def.anemia laboratory Dx.
Early Iron storage depletion stage - no changes on RBC morphology
Decreased total serum iron concentration Increased total iron binding capacity (TIBC) Decreased ferritin levels Advanced iron deficiency affects RBC
morphology: microcytic - hypochromic cells, target cells, pencil shaped cells

Differential diagnosis
Anemia chronic disease - NL or increased iron stores in bone marrow, NL or elevated ferritin level, low serum iron level, TIBC NL or low.
Thalassemia - greater degree of microcytosis , changes on the RBC morphology occur earlier

Treatment of Iron deficiency Oral Ferrous sulfate (325mg TID)
Improve compliance, gradually escalating the dose, taken with food
Improvement in 3 weeks, return to baseline in 2 months, continue for 3-6 months to replenish stores
Parenteral Iron in case of oral intolerance, possible anaphylactic reaction

Megaloblastic anemia-general Results from block of synthesis of nucleotide
precursors of DNA Maturation of cell nucleus is arrested, maturation
of cytoplasm continues
Most common causes: Vitamin B12 (Cobalamin) deficiency, more
common Folate deficiency Medications that inhibit DNA synthesis / block
folate metabolism Myelodysplasia
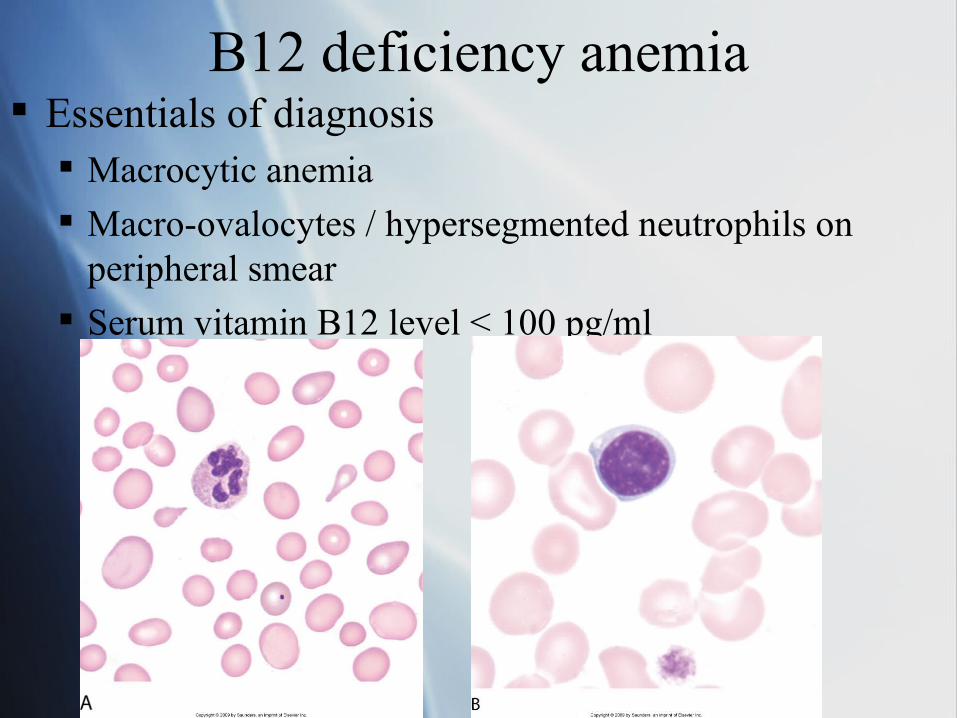
B12 deficiency anemia Essentials of diagnosis
Macrocytic anemia
Macro-ovalocytes / hypersegmented neutrophils on peripheral smear
Serum vitamin B12 level < 100 pg/ml

B12 deficiency anemia-general
B12 absorbed from animal protein in the diet
Absorbed in the terminal ileum bound to intrinsic factor, a protein secreted by gastric
parietal cells
B12 stored in the liver 3 years supply
Daily losses 3-5 mcg, daily absorption 5mcg
Dietary B12 deficiency is rare Vegans diet

B12 deficiency anemia- causes
Pernicious anemia: Autoimmune atrophy of gastric parietal cells ( most common) Gastrectomy
Pancreatic insufficiency failure to inactivate competing binding
protein

B12 deficiency anemia- causes
Bacterial overgrowth in the intestine
Inflammatory bowel disease (crohn’s disease)
Tapeworm infection
Congenital Intrinsic Factor or Transcobalamin II deficiency

Megaloblastic anemia symptoms Often severe anemia at presentation
Insidious onset
Yellowish of skin pallor and jaundice
Mucosal changes glossitis, anorexia, diarrhea, cheilosis
B12 deficiency leads to complex neurologic syndrome:
paresthesias - early difficulty with balance - late

Megaloblastic anemia cont.
Pernicious anemia patients present with atrophic gastritis - achlorhydria
associated with increased risk for gastric carcinoma
association with other autoimmune conditions such as: IgA deficiency and endocrine insufficiency

Laboratory findings Variable severity anemia
possible pancytopenia MCV: 110-140 fl
Peripheral smear large ovalocytes, hypersegmented neutrophils, and large platelets
Hyper cellular bone marrow abnormally large precursors
Increased bilirubin and lactate dehydrogenase enzyme (LDH)
intramedullary destruction of RBCs

Laboratory findings Schilling test :
Radioactive B12 given orally
with a large parenteral dose of unlabeled B12.
Oral absorption determine by: measuring radioactivity in the urine;
B12+ IF + different isotope given at the same time,
selective absorption of the B12+IF suggest pernicious anemia

Schilling test cont. If neither isotope is absorbed,
possible bacterial overgrowth, treat with antibiotics and repeat test
Supply pancreatic enzymes and repeat test to rule out pancreatic insufficiency
New antibody tests for Anti-parietal cell AB , and Anti-IF AB now available

Differential diagnosis
Folic Acid deficiency where: RBC folate is low
Vitamin B12 levels in blood are normal

Treatment Intramuscular B12-
100mcg/dose daily for 7days, then weekly for a month, and monthly for life
Accompanied with Folic acid 1-5 mg /day, oral Folate alone will not correct neurological
symptoms of B12 deficiency

Prognosis Rapid response to therapy,
peak within 7-10 days
Increased reticulocyte count 2 days after therapy
Monitor patient Hypo-kalemia,
Hyper-urecemia,
Hypo-phosphatemia
Neurologic manifestations improve slowly,
some are irreversible

Anemia of chronic disease Occur in patients with:
chronic inflammatory conditions (RA) Infections malignant (cancer) autoimmune diseases (SLE)
Usually normocytic-normochromic occasionally microcytic
Common causes: EPO deficiency (chronic renal failure) Direct inhibition of erythropoiesis Poor iron incorporation to developing Rbcs Shortened erythrocyte survival

Symptoms Related to the underlying disease
SLE: increased fatigue, susceptibility to infections
RA: arthritis, Fatigue / decreased endurance
HIV: Fatigue weight loss lymphadenopathy

Laboratory findings Low serum iron level
TIBC is also reduced
Transferrin saturation >10%
Ferritin level normal or increased
MCV normal or slightly reduced
RBC morphology- non diagnostic

Treatment Mainstay
EPO replacement in the case of chronic renal failure
Anemia will improve if the underlying chronic condition is treated
Some other conditions also respond to EPO treatment, good results in
M.Myeloma, R.Arthritis, and HIV infection

EPO-Treatment Purified recombinant
erythropoietin (epo alfa) 30,000 units once weekly, SQ
Very expensive, used when quality of life clearly improve by hematologic response

Hemolytic anemiasGeneral
Premature loss of RBCs by hemolysis either: extrinsic (reticuloendothelial system)
intrinsic ( blood vessels)
Reticulocytosis as a compensatory response of a normal
bone marrow
Only other condition causing reticulocytosis in anemia is blood loss
Classification Hemolytic Anemia Immune hemolytic anemia
IgG mediated - warm AIHA IgM mediated - cold
Causes extrinsic to erythrocytes Micro-angiopathic (DIC,TTP,drugs,eclampsia, valvular
hemolysis)
Erythrocyte membrane disorders Inherited - H.spherocytosis/elliptocytosis Acquired - PNH, Spur cell anemia
Enzymopathies G6PD deficiencyPK
Hemoglobinopathies Sickle cell disease Thalassemia
Sickle cell anemia essential Dx. Sickled cells on blood smear
Positive family history and lifelong history of hemolytic anemia
Recurrent painful episodes
Hemoglobin S, major Hgb
present on Hgb
electrophoresis

Sickle cell disease - general Belongs to the hemoglobinopathies group
Point mutation in DNA that results in substitution of valine for
glutamine in the sixth position on the beta-globin chain of hemoglobin (beta-sickle chain)
Homozygous genotype(SS)
Resulting Hgb is called Hgb S (sickle)
2 alpha chain
2 beta sickle chain

Sickle cell disease - general Found in African and Mediterranean population
One birth out of 400 in american blacks will produce a child with sickle cell anemia
Onset during the first year of life when fetal Hgb is replaced by adult Hgb
Hgb S is less soluble susceptible to polymerization and precipitation
Trigger factors include Dehydration
Hypoxia,
acidosis,
and high altitudes

Sickle cell anemia symptoms Anemia symptoms
Jaundice
Hepatomegaly
Splenomegaly in children
Enlarged heart, systolic murmurs
Poorly healing ulcers over the lower tibia
Increased susceptibility to encapsulated bacteria (strep. pneumoniae infections)
Delayed puberty

Sickle cell anemia - symptoms Acute complications - vaso-occlusion
Painful crises - ischemia (extremities,chest, abdomen, back)
Acute chest syndrome - life threatening Priapism Cerebrovascular events
Thrombotic/hemorrhagic strokes
Aplastic crisis Splenic sequestration osteomyelitis

Sickle cell anemia - symptoms Chronic manifestations
Chronic renal disease
Chronic pulmonary disease
Sickle hepatopathy
Retinopathy
Avascular necrosis
Skin ulcers

Sickle cell anemia- Lab.findings Hematocrit
20-30%
Increased WBC count (12-15000/mcl)
Thrombocytosis Reticulocytosis
(10-25%)
High indirect bilirubin level Peripheral smear:
Sickled cells Nucleated RBCs Howell - Jolly bodies Target cells

Sickle cell anemia Dx. tests Screening tests
Confirmatory test- hemoglobin electrophoresis showing
85-98% Hgb S present
No Hgb A present in homozygous
Variable amount of Hgb F present
Prenatal testing available(fetal cells), counseling should be offered

Sickle cell anemia-treatment None available for primary disease Folic acid supplementation Blood transfusions for aplastic and hemolytic
crisis Pneumococcal vaccine Antibiotics in case of infections In acute painful episodes,
identify trigger and treat if found. hydrate patient oxygen supplementation as needed analgesics
Sickle cell anemia-treatmentExchange blood transfusion indicated to decreased Hgb S to 30-40% in :
Chest syndrome
Bone marrow necrosis
Priapism

New treatments
Hydroxyurea : increases Hgb F concentration, less susceptible to polymerization.
Survival advantage
Decreased in WBC count

Sickle cell trait Patients with heterozygous genotype (AS)
Clinically normal
Acute painful episodes under extreme conditions only
Hematologically normal, no anemia, normal RBCs
May have inability to concentrate urine, and experience episodes of hematuria
Screening test-positive, Hgb electrophoresis will show 40% Hgb S
No treatment is necessary. Genetic counseling

Hgb C disease
From substitution of glutamic acid to lysine in the sixth position of the beta globin chain
Homozygous Hgb C causes very mild disease, almost silent, mild anemia, mild jaundice, and pigment
gallstones
RBC morph.target cells, Hgb C crystals in a few cells (rectangular)
Compound heterozygous for HgbS/HgbC are more symptomatic, possible retinopathy and splenic sequestration crisis

Thalassemia Hemoglobinopathies
Hereditary reduction in the synthesis of globin chain
alpha or beta
Hypochromic -microcytic anemia because of defective hemoglobinization of RBCs
Often confused with iron deficiency, decreased MCV

Thalassemia Most common inherited hemolytic disorder Defective Hgb caused by decreased production of
at least one globin chain Common in persons of Mediterranean, African
and Southeastern ancestry Alpha thalassemia due to:
gene deletion causing decreased alpha globin chain common in persons from Asia and China, less in blacks
Beta thalassemia caused by: point mutations, leading to reduced or absent beta-globin
chain synthesis primarily affects persons of Mediterranean origin, Chinese and
black to a lesser extent

Essentials of diagnosis
Microcytosis out of proportion to the degree of anemia
Positive family Hx or lifelong personal Hx of microcytic anemia
Microcytes, acanthocytes, and target cells in blood smear
In beta-thalassemia elevated levels of Hgb A2 or F

Alpha thalassemia
Three alpha globin gene present-silent carrier Two alpha globin gene present-trait
only mild microcytic anemia
Only one alpha globin chain present, Hgb H disease(thalassemia minor or intermedia) Pallor and splenomegaly present. infections can cause hemolytic crisis
All four alpha globin gene deletion- affected fetus is stillborn (hydrops fetalis)

B-Thalassemia
B-Thalassemia minor clinically
asymptomatic microcytosis, mild anemia
B-thalassemia Intermediate produce moderate hemolysis
severe anemia
main complication iron overload (non transfusion dependent

Signs and Symptoms B-Thalassemia major (Cooley’s anemia)
homozygous normal at birth after 6 months develop severe
anemia(need transfusion) growth failure bony deformities pathologic fractures and hepatosplenomegaly

B-Thalassemia major
Improved with blood transfusion,
but iron overload develops(hemosiderosis), deposits in heart(heart failure),
liver(cirrhosis),
and endocrine glands (endocrinopathies)

Laboratory findings
Alpha thalassemia trait: mild anemia,MCV 60-75fl, RBC count normal, microcytes, hypochromia, occasional target cells and acanthocytes
Hgb H disease: more marked hemolytic anemia, MCV 60-70 fl, RBC morphology includes all trait changes and poikilocytosis, increased retic count

Laboratory findings
Beta thalassemia minor : modest anemia, MCV 55-75 fl , RBC count normal or increased, peripheral smear similar to alpha thalassemia, basophilic stippling present
Beta thalassemia major: severe anemia, hematocrit of 10% without transfusion, bizarre peripheral smear with microcytes, poikilocytosis , hypochromia, basophilic stippling, target cells and nucleated RBCs

Differential Dx
Mild thalassemias similar to Iron deficiency anemia, but with lower MCV and a more normal RBC count. Peripheral blood smear is more striking in thalassemias, iron studies are normal in thalassemias
Thalassemia major is confused with other hemoglobinopathies, Dx by Hgb electrophoresis

Treatment
B-thalassemia minor require no treatment Hgb H disease should take folate supp. B-thalassemia major patients receive transfusions
as needed Splenectomy may help if hypersplenism Deferoxamine given as an iron-chelating agent Allogeneic bone marrow transplantation has been
successful, treatment of choice for beta-thalassemia

G6PD deficiency X-linked recessive disorder in american black
men Episodic hemolysis in response to oxidant
drugs or infection Reduced levels of glucose-6-phosphate
dehydrogenase between hemolytic episodes Commonly found in people of African,
Mediterranean, Sephardic Jews and Chinese descents

G6PD deficiency
Most female carriers are asymptomatic Partially protects the patient from malaria Clinical problems arise only when the affected
individual is exposed to oxidative stress (infections, drugs )
Oxidized Hgb denatures and precipitates forming Heinz-bodies, that damage RBC membrane, cells are removed by spleen

Signs and Symptoms
Patients usually healthy,
episodes of hemolysis self limiting, because damage red cells are removed from circulation and replaced by functionally normal RBCs
Mediterranean variants may have hemolysis with fava beans exposure

Laboratory Test
During hemolytic episodes there is reticulocytosis, and increased indirect bilirubin
Bite-cells in smear: indicates pitting of Hgb aggregates by the spleen

Treatment
Usually self limiting No specific treatment necessary If hemoglobinuria develops maintain
adequate urine output Have patient avoid Fava beans,
antimalarials, sulfonamides, nitrofurantoin, analgesics, vitamin K

Sideroblastic Anemia
Reduced Hgb synthesis because of failure to incorporate heme into protoporphyrin
Iron accumulates in the mitochondria Prussian blue stain reveals ringed sideroblasts in
bone marrow-cells with iron deposits around the nucleus
Acquired disorder, seen in alcoholism, and lead poisoning
Stage in evolution of a bone marrow disorder that terminates in acute leukemia

Sideroblastic Anemia

Signs and Symptoms/ Laboratory
Moderate anemia, Hematocrit 20-30%, transfusion occasionally required
MCV is normal or increased Dimorphic population of RBC in smear one
normal other hypochromic Lead poisoning shows basophilic stippling Bone marrow iron stores are increased with ringed
sideroblasts present Increased serum iron and transferrin