h.s.p. &i. t. p dr:fatma ali altamimi. pediatric rheumatology consultant
TRANSCRIPT

H .S.P. &I. T. P
Dr:Fatma Ali AlTamimi .
Pediatric rheumatology consultant .

Objectives1 -Outline an appropriate evaluation for apatient
with thrombocytopenia.
2-Explain the pathogenesis, clinical features and treatment of ITP and H S P.
3-Identify the indications for platelet transfusion for I TP .

Introduction
Estimated overall annual incidence of new cases of vasculitis is 53.3 per 100,000 children under 17 years of age . The most common vasculitides are Henoch-Schönlein purpura (HSP) , with estimated annual incidence of 20 per 100,000 in children less than 17 years of age. Reported geographical variations in vasculitis may reflect an environmental influence. A number of factors have been reported to be associated with the development of vasculitis, including infections, drugs, allergy and vaccination.

CONT..…
Purpura may result from disruption in vascular integrity [trauma ,infection,vasculitis ,collagen disorders] or due to abnormalities in primary or secondary hemostasis[thrombocytopenia , abnormal platelet function ,or clotting factor defeciency].
Asystematic approach to the evaluation of achild with purpura helps guide the work up and identify the appropriate treatment. [algorithm].



Definition:
Vasculitides are disorders defined by the presence of inflammation in a blood vessel wall (vasculitis).
The inflammation may occur as a primary process or secondary to an underlying
disease .

Clinical symptoms depending upon
the types and location of the vessels involved.
the extent of inflammation.
and subsequent vessel wall damage with associated hemodynamic changes.

Blood vessel damageBlood vessel damage
Thickening of vessel wall Attenuation of vessel wallThickening of vessel wall Attenuation of vessel wall
Aneurysm formation orAneurysm formation orDisruption of the vessel wallDisruption of the vessel wallwith hemorrhage into tissuewith hemorrhage into tissue
Tissue or organ ischemiaTissue or organ ischemia
Luminal narrowing Luminal narrowing or occlusionor occlusion Vessel wall thinningVessel wall thinning
Vasculitis = Inflammation of the Blood VesselVasculitis = Inflammation of the Blood Vessel

Vasculitis: Histological and Clinical CorrelationVasculitis: Histological and Clinical Correlation
Palpable PurpuraPalpable PurpuraDisruption of the vessel wall with Disruption of the vessel wall with
red blood cell extravasation into tissuered blood cell extravasation into tissue
Copyright © The McGraw-Hill Companies, Inc. All rights reserved.
Courtesy of Carol A. Langford

Chapel Hill Consensus Conference on the Nomenclature of Systemic Vasculitis
Small vessel vasculitisWegener’s granulomatosisChurg-Strauss syndromeMicroscopic polyangiitisHenoch-Schonlein purpuraEssential cryoglobulinemic vasculitisCutaneous leukocytoclastic angiitis
Medium-sized vessel vasculitis
Polyarteritis nodosaKawasaki disease
Large-vessel vasculitis
Giant cell (temporal) arteritis
Takayasu arteritis


Henoch-Schonlein Purpura
Most common systemic vasculitis in children affecting between 10 and 20 per hundred
thousand children every year..Immune mediatedDeposition of IgA immune complexes.
Occurs more often in fall, winter, and spring.Rare in the summer.
About 50% of cases are preceded by URI’s.Streptococcus is often implicated .
Vaccines, insect bites, viruses have also been reported as triggers.

PATHOGENESIS
Immunological; IgA vasculitis associated with [IgA] deposition ,C3, and fibrin depsition.
Immunologic, genetic, and environmental factors play arole.
The characterstic finding of HSP is leukocytoclastic vasculitis accompanied by IgA immune complexes within affected organ.Tpredominant cell typeswithin the inflammatory infiltrate are
neutrophils and monocytes .



Clinical Presentation
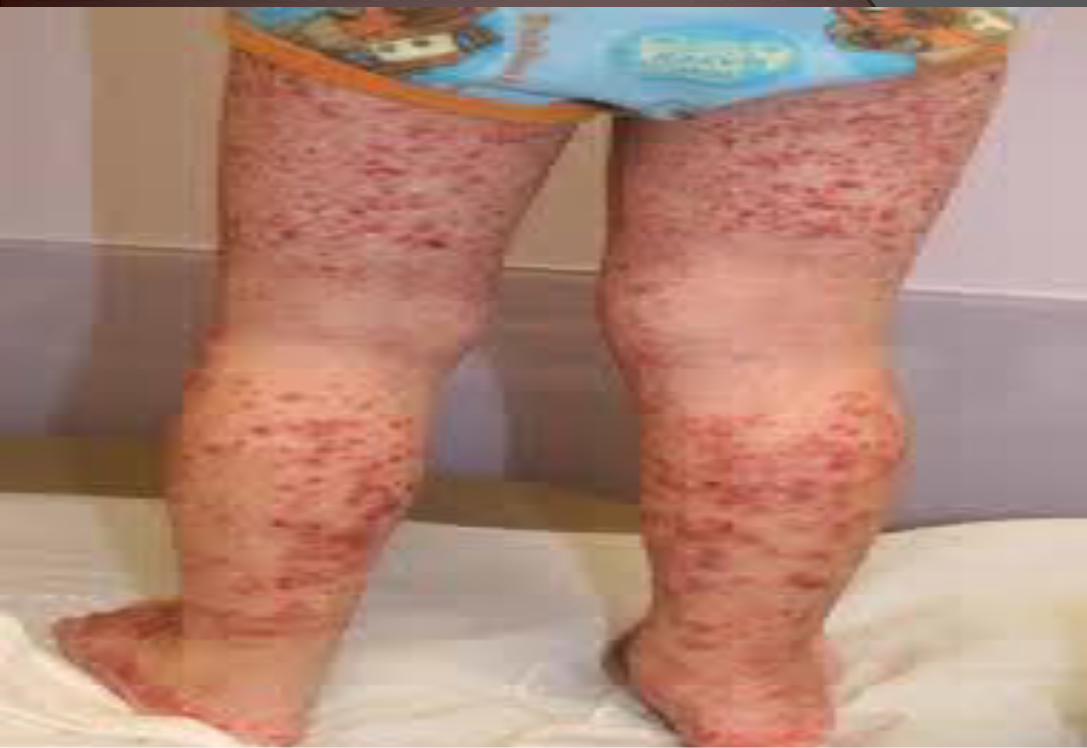
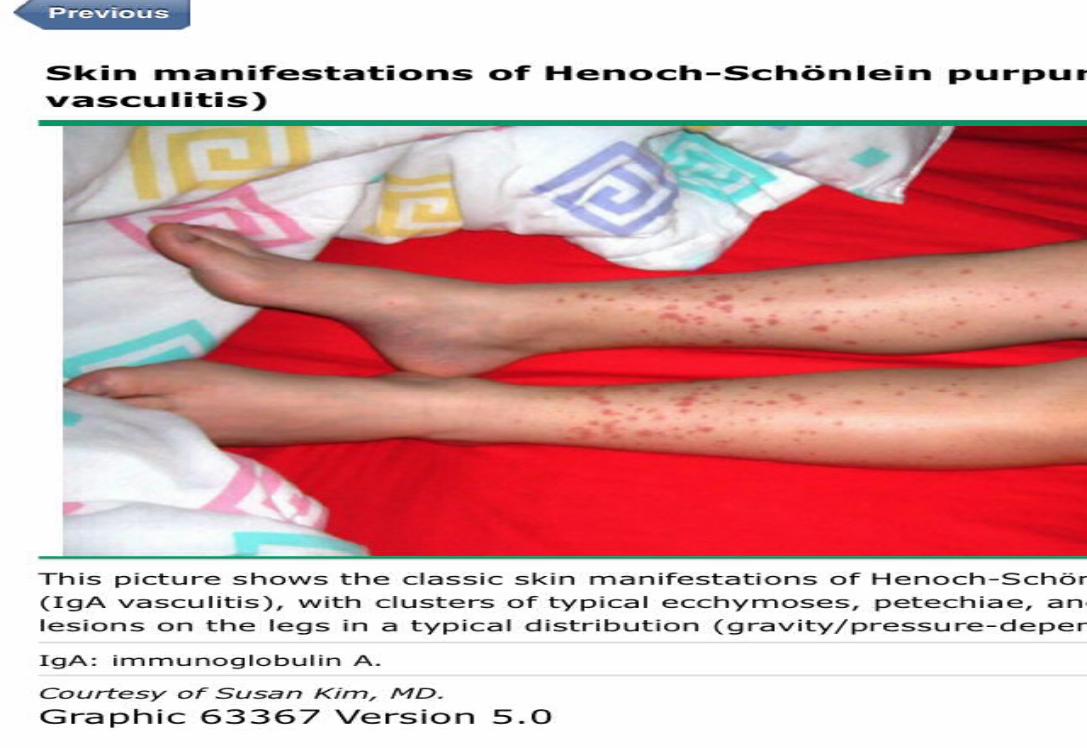
*50% of cases present before the age of five. Classic tetradPalpable purpura (100%)In absence of thrombocytopenia or coagulopathy
Arthritis or arthralgia (75%)
Abdominal pain (50%)
Renal disease (21-50%)


Gastrointestinal symptoms
Occur in 50% of HSP patients.Range from mild ;nausea ,vomiting, abdominal pain , transient paralytic ileus to more significant findings;gastrointestinal hemorrage,bowel ischemia and necrosis, intussusception,and bowel perforation.
Intussusception is the most common gastrointestinal complication 2.3-3.5 %;small bowel can be detected by ultrasonography of abdomen.

Renal disease
20 -54% of HSP patients.
The most common presentation is hematuria with or without red cell casts and no or mild proteinuria.
The finding in renal biopsy are identical to IgAnephropathy.
Urine analysis should be done in all patients.

The A C R CRITERIA OF H S P
Palpable purpura
Age at onset < 20 years
Acute abdominal pain
Biopsy showing granulocytes in the walls of small arterioles and or venules.

Laboratory studies
.Nonspecific - Complete blood count
- Erythrocyte sedimentation rate (ESR) - C-reactive protein.Organ involvement
-Creatinine -Urinalysis – Liver enzymes
-Electrocardiogram - Echocardiogram -Creatinine phosphokinase
-Chest roentgenogram -Sinus roentgenograms
-Electromyography/nerve conduction studiesAngiographyBiopsy

Treatment: Often a self-limited disease. simple medicines like
paracetamol and or ibuprofen are helpful for relief from fever, joint and abdominal pain. .
Those with severe abdominal pain, once the clinician is happy that there is not a severe complication called intussusception, may benefit from a short course of prednisolone.

Supportive care;
Adequate hydration
Rest
Pain killer.

HOspitalization
Inability to maintain adequate hydration.
Sever abdominal pain.
Significant G I bleeding
Changes in mental status.
Sever joint involvement with limitation of movement.
Renal insufficiency , hypertention ,nephrotic syndrome.

Pitfalls in the diagnosis and management of HSP
The trick is identifying which children have severe renal involvement, so that more aggressive treatment can be given earlier , younger children tend to have a better
outcome than older children and adults. . Another common pitfall is to diagnose "atypical" HSP
when in fact much more aggressive treatment is required. . Clinicians should be alert to the possibility of more aggressive forms of vasculitis in children with unusually severe presentation of what initially looks like HSP.

I T P

Decreased productionIncreased destructionSequestrationPseudothrombocytopenia

Artifactually low platelet count due to in vitro clumping of plateletsUsually caused by antibodies that bind platelets only in presence of chelating agent (EDTA)Seen in healthy individuals and in a variety of disease statesDiagnosis:
Marked fluctuations in platelet count without apparent causeThrombocytopenia disproprotionate to symptomsClumped platelets on blood smear
"Platelet satellitism" - platelets stuck to WBCAbnormal platelet/leukocyte histogramsPlatelet count varies with different anticoagulants

PSEUDOTHROMBOCYTOPENIA
Platelet clumping in EDTA No clumping in heparin

PSEUDOTHROMBOCYTOPENIA
Platelet “satellitism”Blood 2012;119:4100

Childhood (acute) ITP
Adult (chronic) ITP

Marrow failure (pancytopenia)aplastic anemia, chemotherapy, toxinsB-12, folate or (rarely) iron deficiencyViral infectionDrugs that can selectively reduce platelet productionAlcoholEstrogensThiazidesChlorpropamideInterferonAmegakaryocytic thrombocytopeniamyelodysplasia (pre-leukemia)immune? (related to aplastic anemia)Cyclic thrombocytopenia (rare)Inherited thrombocytopenia

Immune destructionIntravascular coagulation (DIC or localized)MicroangiopathyDamage by bacterial enzymes, etc

Autoimmune (ITP)ChildhoodAdultDrug-inducedHeparinQuinine, othersImmune complex (infection, etc)AlloimmunePost-transfusion purpuraNeonatal purpura

Immune complex-mediated platelet destructionChildhood ITPBacterial sepsisHepatitis C, other viral infectionsActivation of coagulation cascadeSepsis with DICVascular/endothelial cell damageViral hemorrhagic feversRocky Mountain Spotted FeverDamage to platelet membrane components by bacterial enzymes (eg, S pneumoniae sialidase)Decreased platelet productionViral infections (EBV, measles)Mixed production defect/immune consumptionHIV infection

DRUGS MOST LIKELY TO CAUSE DRUGS MOST LIKELY TO CAUSE THROMBOCYTOPENIATHROMBOCYTOPENIA
Hematology 2009;153
**
*
*
*
*

Childhood form (most < 10 yrs old)May follow viral infection, vaccinationPeak incidence in fall & winter
~50% receive some treatment≥75% in remission within 6 mo
Adult formNo prodromeChronic, recurrences commonSpontaneous remission rate about 5%

DEFINITION;
Isolated immune mediated thrombocytopenia.
Peripheral blood platelet count< 100,000/microl.
It is an aquired benign disorder.
Primary I .T .P .

pathogenesis
Autoantibodies usually Ig G directed against platelet membrane antigens; glycoprotein I
Ib/IIIa complex. The antibody coated platelets have shortened half life because of accelerated clearance by tissue macrophages in the spleen.
The same antibodies may inhibit platelet production.Tcell mediated cytotoxicity.

EPEDMIOLOGY
Annual incidence between 1 and 6.4 cases per 100,000 children.
Peak incidence between 2 and 5 years.
Peak in the spring and early summer.

CLINICAL MANIFESTATIONS
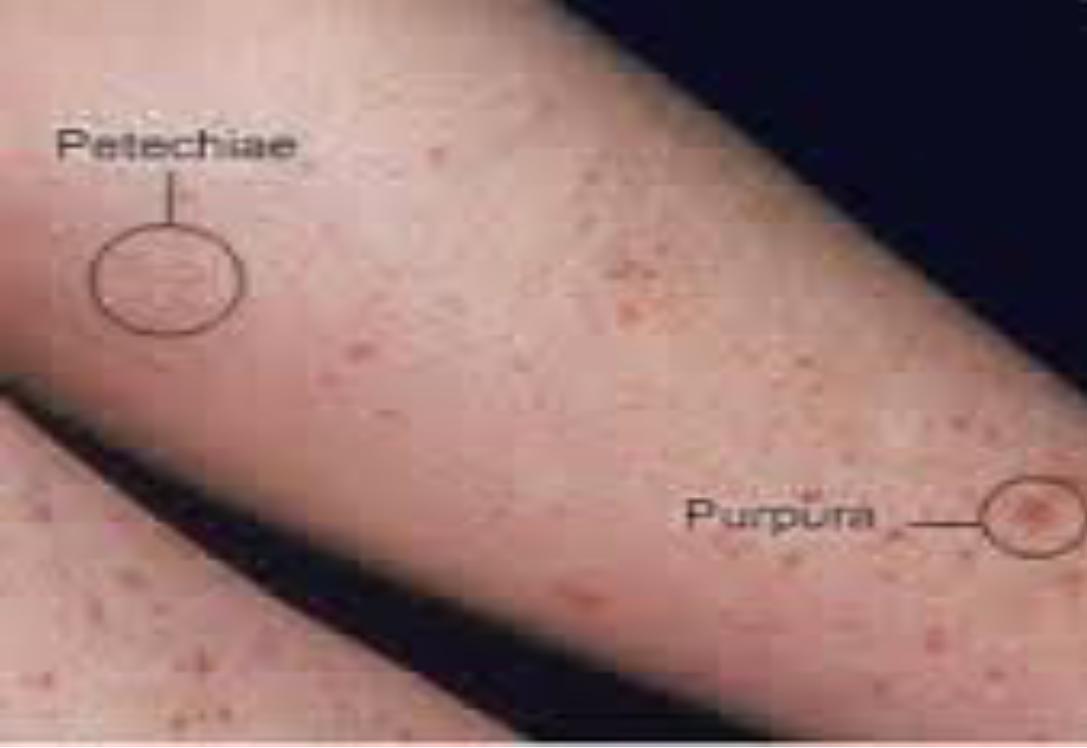
Sudden appearance of petechial rash, bruising and or bleeding in otherwise healthy child.
HISTORY;
60 ./History of prior infection within the past month.
6WKs post MMR vaccine 2.6per 100,000 doses.
If other symptoms present another cause of thrombocytopenia should be considered.

PHYSICAL FINDING;
Cutaneous bleeding[ dry purpura ]; petechiae , purpura, and ecchymoses ;60%.
Mucosal bleeding[ wet purpura]; 40%.
Serious hemorrhage;3%
Intracranial hemorrhage; 0.1-0.8% ;25% mortality.

LABORATORY FINDINGS:
PLT count< 100,000/microl.
Peripheral blood smear to rule out morphological abnormalities in the RBC s or
WBCs .
PLTs size are normal.

DISEASE COURSE
Newly diagnosed I T P ; 3 M
Persistent I T P ;3- 12M
Chronic I T P; > 12 M

DIAGNOSIS
It is adiagnosis of exclusion characterized by isolated thrombocytopenia.
INITIAL EVALUATION;
CBC , Retic ,Direct coombs test, Immunoglobulins level.

DIAGNOSTIC CRITERIA;
P L T COUNT <100,000 microl.
Otherwise normal CBC Retics
Normal peripheral blood smear.
No clinically apparent associated conditions that may cause thrombocytopenia.

MANAGEMENT;
Supportive care;
Restrict physical activities with risk of trauma.
Avoid medication with antiplatelet activity; Aspirin ,ibuprofen, N S AI D Ss, anticoagulants.
Monitor the disease course until full recovery is assured.

Indication for pharmacological interventionThe guidelines of the American Society of Heamatology[A S H];
No pharmacologic intervention for children with no bleeding regardless of platelet count.
Pharmacologic intervention for any child with sever bleeding ;.I V IG; 1 g/ kg, Glucocorticoids, Anti –D immunoglobulin.

Platelets > 20-30 K, no bleeding: no Rx30-70% recover within 3 weeks
Platelets < 10K, or < 20K with significant bleeding: IVIg or corticosteroidsprednisone, 1-2 mg/kg/daysingle dose IVIg 0.8-1g/kg as effective as repeated dosing Splenectomy reserved for chronic ITP (> 12 mo) or refractory disease with life-threatening bleedingpre-immunize with pneumococcal, H. influenzae and meningococcal vaccines
Management

Possible mechanisms of action:Slowed platelet consumption by Fc receptor blockadeAccelerated autoantibody catabolismReduced autoantibody productionDose: 0.4 g/kg/d x 5 days (alternative: 1 g/kg/d x 2 days)About 75% response rate, usually within a few days to a weekOver 75% of responders return to pre-treatment levels within a month
Advantages: rapid acting, low toxicityDisadvantages: high cost, short duration of benefit, high relapse rateIndications: Lifethreatening bleeding; pre-operative correction of platelet count, steroids contraindicated or ineffective
Intravenous immunoglobulin therapy


Life threatening bleeding;
Rare in children.
I C H in 0.5%
Associated with head trauma and P L T count<10,000.
Management by; P L T transfusion and IVIG and methylprednisolone together.
