immuno2008, vol.28, issues 4, intravenous immunoglobulin treatment of immunodeficiency
TRANSCRIPT


Intravenous Immunoglobulin Treatment of Immunodeficiency
Foreword
The Question of When and How
Rafeul Alam, MD, PhD
Consulting Editor
Replacement immunoglobulin therapy saves lives. When a patient is deficient in im-munoglobulins and is unable to produce antibodies in response to pathogens, therationale for immunoglobulin replacement therapy is straightforward. The rationalebecomes more problematic when the immunoglobulin level is only mildly reducedor the antibody response is partial. How do we decide which of these patientswill respond to immunoglobulin therapy and which will not? What are the minimumcriteria for the initiation of immunoglobulin therapy? These are very important issuesthat all practitioners struggle with. Once we decide on immunoglobulin therapy, theimmediate next questions are: which preparation and what route? The issues atstake are the quality of available immunoglobulins, IgA content, the amount ofsalt, sugar, blood-derived nonimmunoglobulin products, stabilizing agents, preser-vatives, and finally the pH and osmolality of the solution. Many of these factors con-tribute to the side-effect profile of the IVIG preparation. The safety and efficacy ofsubcutaneous immunoglobulin have now been well established. So the questionis: which patient is best suited for this treatment as opposed to IVIG? There is a gen-eral consensus on the initial dose of immunoglobulin for replacement therapy. Thedose needs to be adjusted based upon the treatment response. The trough levelof IgG that renders protection from infections and infection-related complicationsmay vary from patient to patient. Third-party payors have their own guidelines forthe trough level of IgG, which may not necessarily be protective against infections.We need a consensus guideline for determining the therapeutically effective IgGtrough level.
Dr. Roifman, a leader in the field, has put together this excellent issue dedicated toIVIG. A group of outstanding experts presents the state of the art on matters that are of
Supported by NIH grants RO1 AI059719 and AI68088, PPG HL 36577, and N01HHSN272200700048C.
Immunol Allergy Clin N Am 28 (2008) xiii–xivdoi:10.1016/j.iac.2008.08.001 immunology.theclinics.com0889-8561/08/$ – see front matter ª 2008 Elsevier Inc. All rights reserved.

Forewordxiv
practical importance to clinicians. The value of this issue to practicing immunologistsand other physicians is enormous.
Rafeul Alam, MD, PhDDivision of Immunology and Allergy
National Jewish HealthUniversity of Colorado Denver Health Sciences Center
1400 Jackson StreetDenver, CO 80206, USA
E-mail address:[email protected]

Intravenous Immunoglobulin Treatment of Immunodeficiency
Preface
Chaim M. Roifman, MD, FRCPC
Guest Editor
Antibody deficiency is the most common clinically significant immunodeficiency. Thisinability to produce specific antibodies against microbial agents can be caused bydefects of B cells, T cells or both arms of the immune system. In addition, selectiveor universal antibody deficiency can be associated with innate immune defects aswell as with a large number of multi-organ syndromes.
Invariably, antibody deficiency ultimately leads to susceptibility to life-threateninginfections and autoimmune manifestations. Equally consistent is the efficiacy of IgGreplacement therapy in preventing infections. Immunoglobulin (Ig) replacement hassaved the lives and dramatically reduced morbidity in numerous patients who haveprimary immunodeficiency, especially in the last 25 years since the introduction of Igthat is suitable for intravenous administration. These products, which were developedby the blood-product pharmaceutical industry, allowed for administration of higherdoses that could build IgG trough levels comparable to normal serum homeostasis.
Appropriate trough levels can be achieved by monthly administration of IVIgor weekly injections of subcutaneous Ig. Each route of infusion has its advantagesand disadvantages, but both are welcomed by patients who can choose which modalitywould best accommodate their lifestyle. To date, approximately ten percent of patientsin North America have chosen the recently introduced subcutaneous route ofIg administration.
This issue provides an important review of all aspects of immunoglobulin therapy,including the definition of patients who need this treatment as well as evolution of prod-ucts and treatment protocols. We also discuss extensively various routes of Ig infusionand their impact on the healthcare system.
Immunol Allergy Clin N Am 28 (2008) xv–xvidoi:10.1016/j.iac.2008.08.002 immunology.theclinics.com0889-8561/08/$ – see front matter ª 2008 Elsevier Inc. All rights reserved.

Prefacexvi
DEDICATION
This issue is dedicated to an exceptional leader, Dr. Fred Rosen, who, through inge-nuity and extraordinary dedication, elevated the field of primary immunodeficiency to itscurrent stature for the benefit of patients.
Chaim M. Roifman, MD, FRCPCDivision of Immunology and Allergy
Department of PediatricsThe Hospital for Sick Children
555 University AvenueToronto, ON M5G 1X8
Canada
E-mail address:[email protected]

Hypogammaglobulinaemia
Patrick F.K.Yong, MRCPa,b, Ronnie Chee, MRCP, FRCPathb,Bodo Grimbacher, MDb,*
KEYWORDS
� Hypogammaglobulinaemia � Primary immunodeficiency� Primary antibody deficiency � Agammaglobulinaemia� Common variable immunodeficiency� Class switch recombination defects
Hypogammaglobulinemia generally can be divided into primary/genetic causes or sec-ondary causes due to other factors, such as sequelae of certain infectious diseases,malignancy, various medications, including immunosuppressants and anticonvulsantsand systemic diseases that result in hypercatabolism or excessive loss of immunoglob-ulin (Ig).1 Depending on the symptoms and severity of the hypogammaglobulinemia,various treatment options are available including replacement Ig therapy, antibiotictreatment or just careful follow-up observation.
The International Union of Immunological Societies (IUIS) has produced regular re-ports on the classification of primary immunodeficiency diseases (PIDs), with the mostrecent update in 2007.2–5 PIDs that resulted in hypogammaglobulinemia were catego-rized within several groups, which included those that caused a combined T and B celldefect, those that resulted in predominantly antibody deficiency, and those that incor-porated other well-defined immunodeficiency syndromes. Other groups of PIDs didnot have significant hypogammaglobulinemia, including diseases of immune dysregu-lation, congenital disorders of phagocyte numbers or function or both, defects ininnate immunity, autoinflammatory disorders, and complement deficiencies.
This article discusses primarily PIDs that result in hypogammaglobulinemia gener-ally following the order in the most recent IUIS classification (Table 1),5 with particularfocus on the more common ones that typically require Ig replacement therapy. Severalof the PIDs classified in this section (ie, X-linked lymphoproliferative syndrome, CD40ligand deficiency, and CD40 deficiency) are also classified elsewhere in the IUISscheme and are discussed in greater depth elsewhere in this issue because they resultfrom T-cell dysfunction or immune dysregulation.
Predominantly antibody deficiency syndromes as a whole make up the greatestproportion of PID diagnoses—up 67% to 77% of all PIDs, as recently published bythe European and Australian registries.6,7 Of the individual antibody deficiency
B. Grimbacher is funded by EU grant MEXT-CT-2006-042316.a Department of Clinical Immunology, Kings College Hospital, London SE5 9RS, UKb Department of Immunology and Molecular Pathology, UCL Immunology Consortium, RoyalFree Hospital and University College London, Pond Street, London NW3 2QG, UK* Corresponding author.E-mail address: [email protected] (B. Grimbacher).
Immunol Allergy Clin N Am 28 (2008) 691–713doi:10.1016/j.iac.2008.06.003 immunology.theclinics.com0889-8561/08/$ – see front matter ª 2008 Elsevier Inc. All rights reserved.

Table 1The International Union of Immunological Societies classification of predominantly antibodydeficiencies
Disease Genetic DefectsSevere reduction in all serum Ig isotypes with profoundly decreased or absent B cells
BTK deficiency BTK/BTK
m Heavy chain deficiency IGHM/m heavy chain
l5 Deficiency CD179B/l5
Iga deficiency CD79A/Iga
Igb deficiency CD79B/Igb
BLNK deficiency BLNK/BLNK
Thymoma with immunodeficiency Unknown
Myelodysplasia Monosomy 7, trisomy 8, and dyskeratosiscongenita have been reported
Severe reduction in serum IgG and IgA with normal, low, or very low numbers of B cells
Common variable immunodeficiencydisorders
TNFRSF13B/TACITNFRSF13C/BAFFRMSH5/Msh5
ICOS deficiency ICOS/ICOS
CD19 deficiency CD19/CD19
X-linked lymphoproliferative syndrome SH2D1A/SAPXIAP/XIAP
Severe reduction in serum IgG and IgA with normal/elevated IgM and normal numbersof B cells
CD40L deficiency TNFSF5/CD154
CD40 deficiency TNFRSF5/CD40
Activation-induced cytidine deaminasedeficiency
AICDA/AID
Uracil-DNA glycosylase deficiency UNG/UNG
Isotype or light chain deficiencies with normal numbers of B cells
Ig heavy chain deletions Deletion at chromosome 14q32
k Chain deficiency Mutation in k constant gene
Isolated IgG subclass deficiency Unknown
IgA deficiency associated with IgGsubclass deficiency
Unknown
Selective IgA deficiency Unknown
Specific antibody deficiency with normal Igconcentrations and normal numbers of B cells
Unknown
Transient hypogammaglobulinemia of infancywith normal numbers of B cells
Unknown
Yong et al692
disorders, common variable immunodeficiency (CVID) made up the largest proportionof entries at 31.6% and 38.4% of all PIDs, respectively.
DISEASES RESULTING IN SEVERE REDUCTION IN ALL SERUM IMMUNOGLOBULIN ISOTYPESWITH PROFOUNDLY DECREASED OR ABSENT B CELLSBTK Deficiency/X-linked Agammaglobulinemia
X-linked agammaglobulinemia (XLA), or Bruton’s agammaglobulinemia, first describedin 1952,8 is the prototypic B-cell immunodeficiency resulting in agammaglobulinemia

Hypogammaglobulinaemia 693
because of a block in B-cell maturation. Almost 40 years later, in 1993, two groupsidentified BTK, the gene responsible for XLA, which encodes a protein tyrosinekinase—Bruton tyrosine kinase (Btk).9,10 The disease is typically characterized bymarked reduction in serum Ig levels (IgG < 2 g/L, IgA and IgM < 0.2 g/L) and circulatingB cells of less than 2%.1 A recent US registry estimated the birth rate for XLA at 1 in379,000 live births, although this number was thought to be an underestimate.11 Otherregistry data have estimated a live birth rate as high as 1 in 100,000 in Norway and aslow as 1 in 20 million in Spain, however.12,13
Btk is encoded over 19 exons spanning 37 kb at Xq2214 and belongs to the Tecfamily of cytoplasmic tyrosine kinases.15 It is present in all stages of B-cell differenti-ation except plasma cells and myeloid cells and platelets but not T cells.16,17 Cross-linking of the B-cell receptor results in phosphorylation and activation of Btk.18 InitiallyBtk is phosphorylated by src family members and then undergoes autophosphoryla-tion.19 Receptor-associated src family members also phosphorylate the immunore-ceptor tyrosine activation motifs on the cytoplasmic tails of Iga and Igb, whichescort the m-heavy chain to the cell surface. Full phosphorylation of the immunorecep-tor tyrosine activation motifs allows Syk, another cytoplasmic tyrosine kinase to dockand be activated via transphosphorylation.20 Syk then phosphorylates downstreamtargets, including B-cell linker protein (BLNK).21 This process allows Btk and PLCg2to bind to BLNK, resulting in phosphorylation of PLCg2 by Btk.22 PLCg2 then gener-ates inositol triphosphate (IP3), a second messenger that binds to receptors on theendoplasmic reticulum leading to calcium release.
Btk mutationsThere is significant variability in Btk mutations, with more than 170 different mutationsidentified and no single mutation accounting for more than 3% of patients in one se-ries.23 The issue as to whether specific mutations are associated with more significantdisease has been difficult to clarify, because in addition to the nature of the mutationand compensating genetic factors, the age of first diagnosis is influenced by environ-mental exposure to infectious organisms, the level of suspicion of the physician, andthe amount of antibiotic use. One study does raise the suggestion that patients withless ‘‘severe’’ mutations (ie, persons with amino acid substitutions or base pair sub-stitutions at sites within the splice consensus site that are conserved, but not invari-ant) are more likely to have a later diagnosis, higher B-cell percentage, and plasmaIgM.24 It should be noted, however, that patients with ‘‘severe’’ mutations (eg, prema-ture stop codons or mutations in the start codon) can have mild disease 25–27 and thatpatients with the same mutation in the same family can have varying degrees ofseverity,25,27,28 which implies that other factors play a role in determining outcomesin XLA.
Clinical featuresXLA is an X-linked recessive disorder that is fully penetrant and manifests in affectedmen. In general, female carriers are asymptomatic, although there are rare exceptionsto the rule, with a recent case report of a daughter of a man with XLA who had all thefeatures of XLA caused by extremely skewed X-inactivation.29
There is marked variability in the clinical course of patients with XLA. In general,most patients become clinically symptomatic by the age of 1 year with nearly all pa-tients manifesting by the age of 5.11,30 It should be noted that 10% to 25% of patientsdevelop symptoms before 3 to 4 months of age, when some degree of maternal anti-body would still be expected to be present.11,30 Most patients had reduced levels of allIg isotypes and markedly reduced circulating B cells, although in most of the series of

Yong et al694
patients, a handful developed symptoms only after the age of 5 and some had Ig levelswithin the normal range, despite confirmed Btk mutations.11,31,32
The mean age at diagnosis was 3.5 years in the Italian series31 and 4 years in theIraninan series.30 The most recent American series showed that patients with a familyhistory were diagnosed at a mean of 2.59 years but patients without a family historywere diagnosed significantly later at a mean age of 5.37 years.11 In general, therewas an inverse correlation between the age at diagnosis and the year of birth, hope-fully indicating greater awareness of the disease.11,30,31 Approximately 25% to 40% ofpatients had a positive family history at the time of birth;11,31,32 however, even in themost recently published series, only approximately a third of patients with a positivefamily history were diagnosed before the onset of clinical symptoms.11 This findingindicates the need for improved genetic counseling in affected families.
Infection is the commonest feature in XLA before diagnosis and during follow-up.The commonest infections involve the respiratory tract (including pneumonia, sinusi-tis, and otitis media) and affect 60% to 80% of patients, most commonly withStreptococcus pneumoniae but also with Haemophilus influenzae, staphylococcus,and pseudomonas species.11,30–32 Diarrhea affects approximately 25% of patients(most commonly with giardia lamblia but also rotavirus, campylobacter, enterovirus,salmonella, and shigella species).11,30–32 A few cases of vaccine-associated paralyticpolio and wild polio have been reported in the various case series.11,31,32 A handful ofcases of Pneumocystis jirovecii infection was reported despite the fact that Btk muta-tions are not known to affect T-cell function.11,33,34 This was attributed to poor nutritionin the children affected.
One case series described a constellation of symptoms in patients who presented ininfancy, including pyoderma gangrenosa, perirectal abscess, cellulitis, or impetigoassociated with pseudomonas or staphylococcal sepsis and neutropenia.32 Inaddition to recurrent infections, patients with XLA also have poorly developedlymphoid tissue, which can be noted clinically in the absence of tonsils and lymphnodes and should alert the clinician to consider the diagnosis.
Patients are prone to long-term complications of chronic lung damage and chronicsinusitis from infection; data have shown that the factors that significantly influencedthe likelihood of chronic lung damage were ‘‘higher mean age at diagnosis’’ and‘‘duration of follow-up’’.31
In addition to conventional pathogens, patients with XLA have been noted to be sus-ceptible to certain more unusual infections. Enteroviral infection (eg, coxsackie andechoviruses) can cause meningitis/encephalitis and, more rarely, hepatitis, pneumonia,and dermatomyositis, resulting in significant morbidity and mortality.35–38 Mycoplasmaarthritis and urethritis were reported in 7 of 52 patients with XLA in one study,39 althoughthe more recent case series did not note this as a frequent complication.11,30,31
The incidence of malignancy in XLA is unclear. Gastric adenocarcinoma, lung can-cer, lymphoproliferative disease, dermatofibrosarcoma protuberans, and colorectalcancer have been reported to occur in patients who have XLA.11,40–44 There was anincrease in colorectal cancer in 3 of 52 patients in one report43 but no other casesin two series of 4445 and 73 patients,31 respectively, and only one case in the largestseries of 201 patients.11 Without formal epidemiologic studies, it is not possible tostate definitively if patients with XLA are more prone to malignancy and if any tumorsoccur more frequently.
Autosomal Recessive Agammaglobulinemia
Approximately 15% of patients with congenital agammaglobulinemia and absentcirculating B cells do not have a mutation in Btk.23 Mutations in the m heavy chain

Hypogammaglobulinaemia 695
(IGHM) are thought to account for approximately 20% to 30% of patients without Btkmutations.46,47 Defects in l5 (CD179B), Iga (CD79A), Igb (CD79B), and BLNK (BLNK)have been identified in a small number of patients with autosomal recessive agamma-globulinemia.48–53 In approximately 5% to 10% of all patients with defects in earlyB-cell development, no clear molecular defect has been identified.
Clinically, patients with the autosomal recessive forms of agammaglobulinemia arenot easily distinguishable from patients with XLA, although there is heterogeneity in theclinical presentation. Patients with m heavy chain deficiency, for example, were notedto generally present at an earlier age with a higher incidence of complications,although two patients in one series had a relatively mild course and were still aliveat age 53 and 49 years despite receiving what would currently be considered subop-timal doses of Ig.46
Thymoma with Immunodeficiency
The association of thymoma with immunodeficiency or Good’s syndrome was origi-nally described in 1955,54 and although there is no formal diagnostic criteria, it is rec-ognized as a separate entity in the IUIS classification.5 Patients generally present intheir 50s,55 but Good’s syndrome can occur in children.56 It occurs with a similarfrequency in men and women. Immunodeficiency can either precede or follow thediagnosis of the thymoma and does not resolve with thymectomy.55
The etiology of Good’s syndrome is not clear, although three possible hypotheseshave been suggested: (1) the possibility that cytokines (eg, limitin in a murine model)can cause B-cell arrest or impair maturation, (2) the loss of the naive or memory T-cellpopulation (and thereby the T-cell help for B cells) in view of the opportunistic infec-tions, and (3) autoimmune destruction of the B cells in view of the studies in thymomapatients showing that T cells or autoantibodies can inhibit erythropoiesis.57
Clinically, these patients are susceptible to recurrent infection with encapsulatedbacteria and diarrhea,55 similar to other patients with agammaglobulinemia. Suscep-tibility to opportunistic infections also suggests a cell-mediated defect. Cytomegalo-virus colitis and retinitis and chronic mucocutaneous candidiasis occur relativelyfrequently;55 infections with P jirovecii pneumonia, human herpesvirus 8, herpessimplex, varicella zoster, and babesiosis have been reported.56–59 Autoimmunephenomena, including myasthenia gravis, pure red cell aplasia, pernicious anemia,diabetes mellitus, polymyositis, and idiopathic thrombocytopenia, also can occur.2,58
Prognosis is generally thought to be worse compared to other antibody deficiencysyndromes. In one series, 10 years after diagnosis only 33% of patients who hadGood’s syndrome were alive compared to 95% of patients with XLA or CVID.45 Theincreased mortality was thought to be caused by disease complications (eg, infection,autoimmune disease, and hematologic complications) rather than the underlyingthymoma, although patient numbers in the largest series were small (7 patients whohad Good’s syndrome vs 240 patients who had CVID).
Apart from the reduced/absent B cells and hypogammaglobulinemia, various otherimmunologic abnormalities have been described, including abnormal CD41/CD81T-cell ratios, CD41 lymphopenia, and reduced T-cell proliferation to mitogens.60
Myelodysplasia
Myelodysplastic syndromes can mimic XLA, and this diagnosis has been included inthe most recent IUIS classification.5,61 A small number of pediatric patients have beenreported in the literature. They can have monosomy 7, trisomy 8, or dyskeratosis con-genita and recurrent infection and low B cells at the onset of disease and the pancy-topenia associated with myelodysplasia.61–63 These patients can have normal specific

Yong et al696
antibody titers and isohemagglutinins titers and low numbers of pro- and pre-B cells inthe bone marrow (unlike patients with XLA who have normal numbers of pro-Bcells).62,63
SEVERE REDUCTION IN SERUM IgG AND IgAWITH NORMAL, LOW, OR VERY LOW NUMBERSOF B CELLSCommon Variable Immunodeficiency
The first description of CVID in 1953 has been credited to Janeway.64 It is currentlyunderstood to be a heterogenous group of predominantly antibody deficiency disor-ders that make up the greatest proportion of patients with symptomatic primary hypo-gammaglobulinemia, with an estimated population prevalence of between 1in 10,000and 1 in 50,000.2 Clinically, it is defined by the presence of recurrent infection, a reduc-tion in IgG (of at least two standard deviations below the mean), and at least one otherIg isotype and a failure to generate a significant specific antibody response after vac-cination or natural infection after other known genetic or acquired causes of hypogam-maglobulinemia have been excluded.1,2
Clinical featuresCVID affects both genders equally, and symptoms can begin at any age, althoughthere are peaks in the first and third decades.65 A significant diagnostic delay of be-tween 4 and 9 years exists in the published case series.6,65,66 In approximately 10%of patients, familial clustering of CVID has been documented, although typically theillness is sporadic.67 IgA deficiency (discussed later) can occur in family members ofpatients with CVID,68 which is consistent with the observation that some patientswith IgA deficiency progress to CVID.69
Recurrent infections (with a similar spectrum to patients with agammaglobulinemia,possibly reflecting antibody deficiency rather than the intrinsic genetic defect) are themost frequent complications in CVID. Recurrent respiratory tract infections occur in upto 98% of patients who have CVID.65 Recurrent sinopulmonary infection can result inchronic sinusitis, hearing loss, and bronchiectasis, which are the principal sources ofmorbidity and (along with lymphoma) mortality in CVID.65 In one cohort, bronchiecta-sis was present in a third of patients at baseline, with a further 12.2% developing it dur-ing follow-up despite appropriate treatment.66 In general, most patients are not moresusceptible to most viral infections and opportunistic infections are rare. Similar to pa-tients with agammaglobulinemia, however, there is also infrequently a predispositionto mycoplasma infection of the joints (11 of 306 patients in one series,39 althoughthese numbers were not replicated in another series of similar size)65 and enteroviralmeningoencephalitis (with no more than 30 patients identified in the literature, mostof whom were on inadequate doses of replacement Ig).37,70
Gastrointestinal disease also occurs frequently in patients who have CVID, affectingup to 20% to 25% of patients.65 The most common infections are with Giardia lamblia,Campylobacter jejuni, and Salmonella species; other prominent findings includenodular lymphoid hyperplasia, inflammatory bowel disease, and nonspecific malab-sorption.65 Severe cytomegalovirus enteritis also has been reported.71
Autoimmune disease complicates CVID in up to 20% to 25% of patients. Autoim-mune cytopenias (particularly autoimmune thrombocytopenia and autoimmunehemolytic anemia) are the most commonly reported conditions, but various other con-ditions, including rheumatoid arthritis, sicca syndrome, pernicious anemia, and sys-temic lupus erythematosus, have been described.65,66 One series reported that theautoimmune thrombocytopenia preceded the hypogammaglobulinemia in 62% ofcases.72

Hypogammaglobulinaemia 697
Nonmalignant lymphoproliferation and granulomatous disease have been de-scribed in patients who have CVID. Up to a third of patients with CVID can developlymphoproliferation, which is reflected as splenomegaly, intestinal lymphoid hyperpla-sia, or lymphadenopathy.65 Lymphoid interstitial pneumonitis also has been re-ported.73 Granulomatous inflammation most commonly affects the lung and hasbeen reported in 8% to 22% of patients with CVID.65,74,75 Multisystemic involvementis not infrequent; granulomatous disease has been described in lymph nodes, spleen,liver, parotid glands, meninges, and bone marrow76 and is often associated witha poor prognosis.74
Patients with CVID are also at greater risk of malignancy, particularly non-Hodgkin’slymphoma and gastric carcinoma, with rates between 18 and 10 to 16 times greaterthan that of healthy individuals, respectively.65,66,77 Other malignancies, such ascolorectal cancer, prostate cancer, breast cancer, ovarian cancer, melanoma, andWaldenstrom’s macroglobulinemia, also have been described, but numbers in thevarious series have been too small to determine if there was a significant increasedrisk.65,66
Immunopathology and classification schemesA host of immunologic abnormalities have been described in the innate and adaptiveimmune systems in patients who have CVID.78–91 It is unclear if these changes arepathogenic or merely represent epiphenomena. In the innate immune system,abnormalities have been described in monocytes,78 monocyte-derived dendriticcells,79–81 and blood myeloid and plasmacytoid dendritic cells.82 Signaling defectsin the TLR9 pathway in plasmacytoid dendritic cells and B cells have been reported.83
There has been a greater focus on the adaptive immune system, and multiple T-cellabnormalities in antigen and mitogen-induced proliferation,65 cytokine production,84
generation of antigen-specific T cells after vaccination,85 cell surface molecule expes-sion (CD40L, attractin),86,88 and T-cell apoptosis87 have been described.
More recent work has shown elevation in serum IL-7 (which plays a role in homeo-static proliferation of lymphocytes) in a subset of patients who have CVID who hadincreased numbers of CD81 T cells with decreased apoptosis and a greater incidenceof splenomegaly and autoimmunity.89 Abnormalities in T-cell receptor signaling affect-ing the cytoplasmic guanine nucleotide exchange factor Vav91 and ZAP-7090 havebeen demonstrated.
Based on the immunologic abnormalities seen in patients who have CVID, variousclassification schemes, mostly based on B-cell phenotype, have been developed tohelp stratify patients for research and prognostic reasons. Bryant and colleagues92
originally divided patients who have CVID based on the ability of lymphocytes toproduce Ig on stimulation in vitro. This method was labor intensive and not widelyadopted; subsequently, two flow cytometric classification systems based on memoryB-cell phenotype were published.93,94 These systems had some differences, and tofurther refine these schemes, the EUROClass scheme was developed after a large trialinvolving 303 patients.95 EUROClass separated patients who had nearly absentB cells (< 1% of lymphocytes), severely reduced switched memory B cells (< 2% oftotal B cells), and expansion of transitional (> 9% of total B cells) or increased CD21low
B cells (> 10% of total B cells). There was a degree of clinical correlation, albeit rela-tively imprecise, with splenomegaly and granulomas more frequently seen in patientswith reduced switched memory B cells and elevated CD21low B cells and lymphade-nopathy more frequently seen in patients with elevated transitional B cells. In additionto the B-cell classification schemes, investigators also classified patients who haveCVID using T cells96 and dendritic cells,97 with a degree of clinical correlation.

Yong et al698
GeneticsThe past few years have seen the discovery of mutations/polymorphisms in five genesthat result/contribute to a CVID phenotype, representing a significant advance in theunderstanding of what was previously poorly understood at a molecular level. Thegenes identified so far affect inducible costimulator ([ICOS] gene: ICOS) on T cells,98
transmembrane activator and calcium-modulator and cyclophilin ligand interactor([TACI] gene: TNFRSF13B),99,100 B-cell activating factor receptor ([BAFF-R] gene:TNFRSF13C),101 CD19102,103 (gene: CD19) on B cells, and MSH5, which is involvedin regulating meiotic homologous recombination and contributes to class switch re-combination (CSR).104
Inducible Costimulator Deficiency
ICOS is expressed on activated T cells and belongs to the CD28 family of costimula-tory surface molecules. It plays a significant role in activating T helper cells andproviding B-cell help by superinduction of IL-10 necessary for terminal B-cell differen-tiation into memory and plasma cells and by binding to ICOS-ligand, which is presenton antigen-presenting cells, including naive B cells.105,106
So far, a total of nine individuals from four families have been identified with thesame homozygous mutation (resulting in a truncated protein) in ICOS since it was firstdescribed in 2003.98,107 All affected patients have the same homozygous haplotype atthe D2S2289 locus near the ICOS gene; all four families are believed to originate froma common founder and are either linked by the House of Habsburg or the RiverDanube.107
Phenotypically, ICOS deficiency results in hypogammaglobulinemia and reducedB-cell numbers, particularly in the IgM memory and switched memory B-cell subsets.This further strengthens the evidence that ICOS plays an important role in late B-celldifferentiation, class switching, and memory B-cell development.98 Patients who haveICOS deficiency were able to generate IgM responses during infection, however.
TACI Deficiency
TACI is a member of the tumor necrosis factor receptor superfamily (TNFRSF) andbelongs to a group of TNFRSF receptors that also includes B-cell maturation antigenand BAFF-R, which play important roles in B-cell survival, development, and antibodyproduction.108 The ligands for TACI and B-cell maturation antigen are BAFF and a pro-liferation-inducing ligand.
Mutations in TACI that result in CVID were first described in 200599,100 and arethought to be present in 8% to 10% of patients with CVID.109 A complicated patternof inheritance with homozygous, heterozygous, and compound heterozygous muta-tions were identified, suggesting that there were autosomal dominant and autosomalrecessive patterns of inheritance. Extracellular (C104R, S144X), transmembrane(A181E), and intracellular (S194X, R202H, Ins204) portions of the molecule were allfound to possess mutations.99,100
The role of heterozygous mutations in CVID is not completely clear because it wassubsequently shown that patients with CVID had unaffected family members with thesame TACI mutation.110 In a large study, the C104R and A181E (but not the R202H)mutations were present in greater frequency in patients who had CVID compared tothe general population.111,112 This finding suggested that TACI mutations (at least inthe heterozygous state) might result in increased disease susceptibility rather than be-ing solely responsible. No clear genotype-phenotype correlation has been shown inpatients with TACI mutations, although there is a suggestion that these patients areat increased risk of autoimmunity and lymphoid hyperplasia.99

Hypogammaglobulinaemia 699
B-cell Activating Factor Receptor Deficiency
BAFF-R deficiency has so far been identified in only one patient—a 60-year-old manwho had a 24 base pair homozygous deletion in exon 2, which codes for the trans-membrane portion of the receptor.101 This deficiency resulted in a block at the transi-tional B-cell stage, leading to low total peripheral B-cell numbers and a percentageincrease in transitional B cells, indicative of the BAFF–BAFF-R role in peripheralB-cell survival. These findings have only been published in abstract form so far; thefull publication is awaited.
CD19 Deficiency
CD19 is a B-cell surface molecule that forms a co-receptor complex with CD21, CD81,and CD225. The co-receptor complex reduces the signaling threshold after antigenbinding to the B-cell receptor,106 and the CD21 component can bind antigen-boundC3d, thus linking recognition of complement to CD19 signaling.113
Five patients with a CVID phenotype from three families have been found to havea total of four different mutations in CD19.102,103Three patients (born to unrelatedColombian parents) had a homozygous two base pair deletion that resulted in a frame-shift and premature stop codon leading to deletion of a large portion of the intracellulardomain. One patient (born to consanguineous Turkish parents) had a single base pairinsertion that resulted in a frameshift and premature stop codon in the proximal regionof the intracellular domain.102 The final patient (born to unrelated Japanese parents)was a compound heterozygote with a splice acceptor site mutation of intron 5 onthe maternal allele, which resulted in skipping of exon 6 and a truncated protein,and a gross deletion on the paternal allele that encompassed the CD19 and at leastthe neighboring ATP2A1 and NFATC2IP genes.103 All patients presented in childhoodwith recurrent infections and hypogammaglobulinemia. One patient was found to havemild thrombocytopenia, which raised the possibility of autoimmunity.103 PeripheralB-cell numbers were normal, but CD51 and memory B cells were reduced. Patientshad normal germinal center formation but poor antibody responses to vaccination.Other publications on patients with CD19 deficiency are underway, which suggeststhat the defect may not be that rare.
MSH5 Mutations
MSH5 is a gene encoded in the major histocompatibility class III region that plays rolein homologous recombination in meiosis but was found to be involved in CSR inmice.104 Subsequently, several nonsynonymous single nucleotide polymorphisms(SNPs) in MSH5 were found in greater frequency in patients who have IgA deficiency(C580G, L85F/P786S, rs3131378) and CVID (Q292H, rs3131378).104 MSH5 was foundto have reduced binding affinity to its heterodimerization partner MSH4 in patientswho had the L85F/P786S allele. Patients who have CVID with heterozygous nonsy-nonymous MSH5 polymorphisms were found to have abnormalities in the Sm-Sa1joints; although controls with heterozygous MSH5 polymorphisms did not have hypo-gammaglobulinemia, there were some subtle differences in the S joint phenotype.Consequently, it is more likely that MSH5 is a disease susceptibility gene ratherthan pathogenic.104
X-linked Lymphoproliferative Syndrome
X-linked lymphoproliferative syndrome was originally described in 1974114 as a rapidlyfatal illness after Epstein Barr virus infection in men with a strong genetic linkage. Theillness can result in fulminant infectious mononucleosis (60% of patients), lymphoma

Yong et al700
(30%), or dysgammaglobulinemia (30%), and patients can present with any or all ofthese features.115 It also has been recognized that Epstein Barr virus infection is notnecessary to trigger the onset of the disease. The mutation responsible for X-linkedlymphoproliferative syndrome was identified in the signaling lymphocyte activationmolecule–associated protein ([SAP] gene: SH2D1A) in 1998.116,117 SAP mutationsonly accounted for approximately 60% of familial X-linked lymphoproliferative syn-drome, however, and defects in the gene encoding the X-linked inhibitor of apoptosis(XIAP) were identified in 2006 in a cohort of some of these patients with no moleculardiagnosis.118 X-linked lymphoproliferative syndrome can mimic CVID, although in oneseries only 1 of 60 patients with CVID had an SH2D1A mutation.119
CLASS SWITCH RECOMBINATION DEFECTS: SEVERE REDUCTION IN SERUM IgG AND IgAWITH NORMAL/ELEVATED IgM AND NORMAL NUMBERS OF B CELLS
Reductions in serum IgG and IgA with a normal or elevated IgM are suggestive of a de-fect in the machinery required for CSR and result in the so-called ‘‘hyper-IgM syn-drome’’ (HIGM), which is somewhat of a misnomer because the IgM can sometimesbe in the normal range. These syndromes can be inherited in an X-linked, autosomalrecessive or autosomal dominant manner.120 Limited epidemiologic data are avail-able, but X-linked HIGM is thought to have an estimated frequency of approximately1 in 500,000 live male births in the United States.121
The first mutation that accounted for these syndromes to be discovered was inCD40 ligand ([CD40L] gene: TNFSF5), which results in the X-linked HIGM syndromethat makes up approximately 30% of patients with CSR defects.122,123 CD40L is pres-ent on the surface of T cells and interacts with CD40 on the surface of B cells (requiredfor Ig class switching) and dendritic cells/monocytes (required for T cell responses).Patients are susceptible to recurrent bacterial infections similar to other patientswith hypogammaglobulinemia but are also prone to infections with opportunisticorganisms, such as P jirovecii, cryptosporidium, toxoplasma, and cytomegalovirus,and neutropenia and autoimmune disease.120,121 A mutation in CD40 (gene:TNFRSF5) resulting in a similar clinical phenotype but inherited in an autosomal reces-sive manner also was described in a small number of patients.124 Another form of anX-linked HIGM syndrome in association with anhidrotic ectodermal dysplasia wasdescribed with mutations in NF-kB essential modulator (NEMO or IKKg), which isrequired for CD40 induced signaling of the transcription factor NF-kB.125,126
CD40L, CD40, and NEMO deficiency all result in a combined antibody and cellularimmune deficit and are discussed elsewhere in this issue. The remaining 70% of pa-tients with class switch defects possess some form of intrinsic B-cell defect that isusually inherited in an autosomal recessive (sometimes autosomal dominant) manner.Mutations in activation-induced cytidine deaminase ([AID] gene: AICDA) and uracil-DNA glycosylase ([UNG] gene: UNG) have been described to account for approxi-mately 40% of these patients, although there remains a substantial group with anas-yet uncharacterized molecular defect.
Activation-Induced Cytidine Deaminase Deficiency
AID deficiency is typically inherited in an autosomal recessive fashion and is charac-terized by defects in CSR and somatic hypermutation (SHM).127 Clinically, thesepatients are prone to recurrent bacterial infections and diarrhea, similar to otherpatients with hypogammaglobulinemia, but they also frequently possess markedenlargement of lymphoid organs, in contrast to patients with XLA who have sparselymphoid tissue. Giant germinal centers (five to ten times the normal size) filled with

Hypogammaglobulinaemia 701
intensely proliferating B cells have been found in lymphoid tissue biopsies. This findingis thought to be caused by continuous antigen stimulation due to lack of SHM second-ary to defective AID.128 Lymphoid hyperplasia has been noted to decrease with Igreplacement.129 IgM-mediated autoimmunity (mostly cytopenias) is seen in approxi-mately one fourth of these patients.127,129 In a case series of 29 patients, the medianage of onset of clinical symptoms was 2 years (range 0.3–12.9) with recognition ofimmunodeficiency at 3.8 years and diagnosis of a HIGM syndrome at 4.9 years.129
IgM levels ranged from 1 to 37 g/L, whereas IgG levels ranged from undetectable to1.5 g/L and IgA ranged from undetectable to 0.2 g/L.129
AID possesses a cytidine deaminase activity domain, an apolipoprotein-B mRNA-editing cytidine deaminase 1 (APOBEC-1)-like domain, and a nuclear localizationsignal and nuclear export signal in the N and C terminal portions of the protein, respec-tively. It is thought to be essential for initiation of the DNA cleavage required for CSRand SHM; 35 different recessive mutations have been identified in 73 patients.128–131
The mechanism by which AID exerts its function is not completely clear, althoughstudies in patients with AID deficiency have helped to shed some light on this. It isthought to act as a DNA-editing enzyme and a docking protein by forming multimericcomplexes.132
Mutations in AID generally result in defective CSR and SHM in keeping with its roleas the inducer of DNA breaks required for these processes, although mutations in theC-terminal portion in a small number of patients have been shown to result only in de-fective CSR but not SHM.133 This information and additional data showing that AIDtruncated for the last ten amino acids was unable to generate CSR but was able togenerate mutations in the Sm region134 was taken to indicate that the C-terminal por-tion of AID played a role in binding a CSR-specific cofactor that helped target AID toSm regions. A particular heterozygous mutation (R109X) in the C-terminal portion hasbeen shown to result in an autosomal dominant form of a HIGM defect.128,135 This isthought to arise because of a dominant negative effect of the mutated allele, which im-plies that a multimeric AID complex is necessary for optimal CSR and SHM.
Uracil-DNA Glycosylase Deficiency
A similar clinical picture to AID deficiency with recurrent infections and lymphadenop-athy was described in three patients with a homozygous defect in UNG, which isa member of a family of glycosylases able to deglycosylate uracil residues onDNA.136 UNG deficiency results in defective CSR but with SHM in normal frequency,albeit with a biased pattern, with almost all mutations at G/C residues being transitionsas opposed to an equal frequency of transitions and transversions at A/T resi-dues.136,137 This finding has been cited as a strong argument for the DNA-editingactivity of AID. AID is thought to deaminate cytosine into uracil. Subsequently, UNGdeglycosylates and removes the uracil residues, which creates an abasic site andallows creation of single-stranded DNA breaks. UNG deficiency interferes with thispathway and results in defective CSR and skewed SHM.136,137
Uncharacterized Molecular Defects that Result in Deficiencies in IsotypeClass Switching
There are still many cases of CSR defects caused by an intrinsic B-cell abnormalitythat are not caused by AID or UNG deficiency.
A CSR defect titled HIGM4 has been described in a group of 15 patients with clinicalfeatures similar to AID deficiency, although slightly milder with some residual IgG pro-duction.138 The specific defect has yet to be identified, although it is thought to be

Yong et al702
downstream to AID and probably due to a selective defect in either a CSR-specificfactor of the DNA machinery or survival signals delivered to B cells.
Another defect thought to be upstream of S region DNA cleavage was described ina group of 16 patients with HIGM and a generally good prognosis, with no autoimmu-nity or lymphoma.128 It was speculated that this condition could be caused by prob-lems with AID targeting (which is poorly understood) to switch regions.
ISOTYPE OR LIGHT CHAIN DEFICIENCIESWITH NORMAL NUMBERS OF B CELLS
Most of the deficiencies in Ig isotypes or light chains occur in otherwise healthyindividuals, and the question of Ig replacement is controversial in persons who aresymptomatic. They are covered here briefly for completeness.
Immunoglobulin Heavy Chain Deletions
Deletions and duplications that affect the heavy chain constant regions in chromo-some 14q32 have been described in 5% to 10% of the healthy white populationwho have no history of recurrent infection.2,139 One or more IgG and IgA subclassesand IgE have been shown to be affected.139 Homozygous individuals lack the relevantsubclasses, and heterozygotes may show diminished levels. Most patients are well,although a few individuals have presented with recurrent infections, which casts doubton the relevance of the immunologic abnormalities.
k Chain Deficiency
k chain deficiency has been reported in two families.2,140 B cells seemed to be normal,although all of them possessed the l light chain. Point mutations in the k chain genewere reported in one family.141
Isolated IgG Subclass Deficiency
Isolated deficiencies in one or more IgG subclasses were first described in 1970 inpatients with recurrent sinopulmonary infections142 and are defined as a reductionin one or more IgG subclasses more than two standard deviations from the mean.By definition, however, approximately 2.3% of the healthy population will have anIgG subclass deficiency and up to 10% to 15% actually do have an IgG4 level belowthe limit of detection.143 Many healthy individuals have been identified with signifi-cantly decreased IgG subclass levels. Consequently, controversy exists as to whetherisolated IgG subclass deficiency does represent a true PID. Some authors haveargued that there is no clinical value in IgG subclass measurement.144–146
Selective IgA Deficiency
Selective IgA deficiency is defined as complete absence of IgA (usually less than thedetection limit of 0.07 g/L in most laboratories) with a normal IgG and IgM in patientsolder than age of 4 and in whom other causes of hypogammaglobulinemia have beenexcluded. It is the commonest Ig deficiency and has a prevalence of 1 in 300 to 1 in700 in whites.2,147–149 Most patients with IgA deficiency are asymptomatic, althoughthere is an increased prevalence of infections, autoimmune disease, atopy, and celiacdisease.150–152 There was a suggestion of increased rates of gastrointestinal malig-nancy and lymphoma in patients who have IgA deficiency,153 but this was not repli-cated in a later study.154 The molecular mechanisms underlying IgA deficiency areunclear, although as noted in some patients, there is a family history of IgA deficiencyand CVID, and some patients can progress to CVID.67–69

Hypogammaglobulinaemia 703
IgA Deficiency Associated with IgG Subclass Deficiency
IgG subclass deficiency has been noted to occur in association with IgA deficiency,and both can present with specific antibody deficiency. The molecular, clinical char-acteristics and frequency of these combined deficiencies in the healthy and patientpopulations remain poorly understood and characterized, although small studies sug-gest that a combination of these defects is more likely to result in clinical disease.155
SPECIFIC ANTIBODY DEFICIENCY WITH NORMAL IMMUNOGLOBULIN CONCENTRATIONSAND NORMAL NUMBERS OF B CELLS
Specific antibody deficiency with normal Ig was first described in 1980156 and is char-acterized by normal levels of IgG, IgA, and IgM but a failure to make antibody re-sponses to vaccination, typically with polysaccharide antigens. Clinically, thesepatients are prone to recurrent sinopulmonary infections; bronchiectasis, diarrhea,and autoimmune disease also have been reported.157 There is no universal definitionas to what constitutes a failure to respond, however. Normal responses are agedependent and not well characterized. Pure polysaccharide antibody responses areunreliable in children younger than age 2 years.158
The components necessary to define what constitutes an adequate response in-clude (1) the increase in antibody titers above baseline, (2) the final antibody concen-tration, and (3) the percentage of serotypes in the vaccine to which the patient hasresponded.
The American practice parameter defines an adequate response to individual sero-types as a postimmunization antibody titer of 1.3 mcg/mL or more or at least fourfoldover baseline.145,158 Patients between 2 and 5 years of age are expected to respond toat least half the vaccinated serotypes; for patients 5 years or older, the consensus rec-ommendation was that there should be a response to 70% of the serotypes, althoughit was acknowledged that there was a degree of controversy regarding this. The IUISclassification is a lot less specific than this.2 The increasing use of conjugated pneu-mococcal vaccines in routine immunization is likely to influence how a diagnosis ofspecific antibody deficiency is made in the future.
A further point about the diagnosis of specific antibody deficiency is whetherpatients who fail to make responses to polysaccharide antigens but not to proteinantigens should be considered a separate group than patients who cannot make re-sponses to polysaccharide and protein antigens.159
Much work still needs to be done with regard to diagnosing and working out themolecular mechanisms underlying specific antibody deficiency. The entity does serveto make the point that in patients with normal Ig levels who have recurrent infections,further detailed evaluation is necessary.
TRANSIENT HYPOGAMMAGLOBULINEMIA OF INFANCY WITH NORMAL NUMBERS OF B CELLS
In 1956, Gitlin and Janeway160 described two infants with temporary hypogammaglo-bulinemia and coined the description ‘‘transient hypogammagloublinemia of infancy’’(THI). This was thought to be caused by prolongation of the nadir in gammaglobulinsnormally seen in infants in the first few months of life after the decline in maternallytransferred Ig. Despite this, until now this group of patients remains poorly character-ized with little understanding of the molecular mechanisms underlying the condition.
The IUIS classification had noted that THI can take up to 36 months to resolve,2
although it has been noted that the period of hypogammaglobulinemia can extendsignificantly beyond infancy. Only approximately half the infants had resolution of

Yong et al704
hypogammaglobulinemia by 24 months in one case series, with resolution seen at upto 14 years of age,161 and a definitive diagnosis of THI can be made only retrospec-tively. Patients with THI were more likely to be male (60%–80%) and generally pre-sented with mild infections, including ear/nose/throat infections, respiratoryinfections, and diarrhea,161–165 although isolated case reports have documentedmore severe infection.166 An increased risk of atopic disease has been noted in pa-tients with THI.164,165
Generally, patients with THI have reductions in IgG and IgA below the lower limit ofnormal and less frequently in IgM; specific antibody production and cell-mediatedimmunity is usually intact.161,165 A few individuals have reduced vaccine responsesthat recover by 3 to 4 years.162 Some recent data indicated that in vitro Ig secretoryresponses were poorer in patients who had THI and that in vitro IgG and IgA (butnot IgM) responses did not normalize at the same time as serum Ig.164 This was inter-preted as possibly caused by some deficiency in class switch mechanisms.
SUMMARY
The predominantly antibody deficiency PIDs that result in hypogammaglobulinemiarepresent an important group of diseases, both clinically and for furthering under-standing of the immune system. Combined, they represent the largest group of PIDdiagnoses that possess a relatively good outcome with Ig replacement therapy. A sig-nificant delay in diagnosis still remains, which can result in significant morbidity andlong-term complications and emphasizes that the need for greater awareness of theseconditions still remains. This heterogenous group of disorders has provided many use-ful insights into our understanding of the immune system, particularly with regard toB-cell development and antibody responses and is likely to continue doing so.
REFERENCES
1. Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunode-ficiencies: representing PAGID (Pan-American Group for Immunodeficiency)and ESID (European Society for Immunodeficiencies). Clin Immunol 1999;93(3):190–7.
2. International Union of Immunological Societies. Primary immunodeficiency dis-eases. Report of an IUIS Scientific Committee. Clin Exp Immunol 1999;118(Suppl 1):1–28.
3. Chapel H, Geha R, Rosen F. Primary immunodeficiency diseases: an update.Clin Exp Immunol 2003;132(1):9–15.
4. Notarangelo L, Casanova JL, Conley ME, et al. Primary immunodeficiencydiseases: an update from the International Union of Immunological Societies-Primary Immunodeficiency Diseases Classification Committee Meeting inBudapest, 2005. J Allergy Clin Immunol 2006;117(4):883–96.
5. Geha RS, Notarangelo LD, Casanova JL, et al. Primary immunodeficiencydiseases: an update from the International Union of Immunological SocietiesPrimary Immunodeficiency Diseases Classification Committee. J Allergy ClinImmunol 2007;120(4):776–94.
6. Eades-Perner AM, Gathmann B, Knerr V, et al. The European Internet-based pa-tient and research database for primary immunodeficiencies: results 2004–06.Clin Exp Immunol 2007;147(2):306–12.
7. Kirkpatrick P, Riminton S. Primary immunodeficiency diseases in Australia andNew Zealand. J Clin Immunol 2007;27(5):517–24.
8. Bruton OC. Agammaglobulinemia. Pediatrics 1952;9(6):722–8.

Hypogammaglobulinaemia 705
9. Vetrie D, Vorechovsky I, Sideras P, et al. The gene involved in X-linked agamma-globulinaemia is a member of the src family of protein-tyrosine kinases. Nature1993;361(6409):226–33.
10. Tsukada S, Saffran DC, Rawlings DJ, et al. Deficient expression of a B cell cyto-plasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell 1993;72(2):279–90.
11. Winkelstein JA, Marino MC, Lederman HM, et al. X-linked agammaglobulinemia:report on a United States registry of 201 patients. Medicine (Baltimore) 2006;85(4):193–202.
12. Matamoros FN, Mila LJ, Espanol BT, et al. Primary immunodeficiency syndromein Spain: first report of the National Registry in children and adults. J Clin Immu-nol 1997;17(4):333–9.
13. Stray-Pedersen A, Abrahamsen TG, Froland SS. Primary immunodeficiency dis-eases in Norway. J Clin Immunol 2000;20(6):477–85.
14. Rohrer J, Parolini O, Belmont JW, et al. The genomic structure of human BTK, thedefective gene in X-linked agammaglobulinemia. Immunogenetics 1994;40(5):319–24.
15. Mano H, Ishikawa F, Nishida J, et al. A novel protein-tyrosine kinase, tec, is pref-erentially expressed in liver. Oncogene 1990;5(12):1781–6.
16. Genevier HC, Hinshelwood S, Gaspar HB, et al. Expression of Bruton’s tyrosinekinase protein within the B cell lineage. Eur J Immunol 1994;24(12):3100–5.
17. Smith CI, Baskin B, Humire-Greiff P, et al. Expression of Bruton’s agammaglob-ulinemia tyrosine kinase gene, BTK, is selectively down-regulated in T lympho-cytes and plasma cells. J Immunol 1994;152(2):557–65.
18. Aoki Y, Isselbacher KJ, Pillai S. Bruton tyrosine kinase is tyrosine phosphorylatedand activated in pre-B lymphocytes and receptor-ligated B cells. Proc Natl AcadSci U S A 1994;91(22):10606–9.
19. Rawlings DJ, Scharenberg AM, Park H, et al. Activation of BTK by a phosphory-lation mechanism initiated by SRC family kinases. Science 1996;271(5250):822–5.
20. Bu JY, Shaw AS, Chan AC. Analysis of the interaction of ZAP-70 and syk protein-tyrosine kinases with the T-cell antigen receptor by plasmon resonance. ProcNatl Acad Sci U S A 1995;92(11):5106–10.
21. Fu C, Turck CW, Kurosaki T, et al. BLNK: a central linker protein in B cell activa-tion. Immunity 1998;9(1):93–103.
22. Humphries LA, Dangelmaier C, Sommer K, et al. Tec kinases mediate sustainedcalcium influx via site-specific tyrosine phosphorylation of the phospholipaseCgamma Src homology 2-Src homology 3 linker. J Biol Chem 2004;279(36):37651–61.
23. Conley ME, Broides A, Hernandez-Trujillo V, et al. Genetic analysis of patientswith defects in early B-cell development. Immunol Rev 2005;203:216–34.
24. Broides A, Yang W, Conley ME. Genotype/phenotype correlations in X-linkedagammaglobulinemia. Clin Immunol 2006;118(2–3):195–200.
25. Kornfeld SJ, Haire RN, Strong SJ, et al. A novel mutation (Cys145/Stop) inBruton’s tyrosine kinase is associated with newly diagnosed X-linked agamma-globulinemia in a 51-year-old male. Mol Med 1996;2(5):619–23.
26. Conley ME, Fitch-Hilgenberg ME, Cleveland JL, et al. Screening of genomicDNA to identify mutations in the gene for Bruton’s tyrosine kinase. Hum MolGenet 1994;3(10):1751–6.
27. Bykowsky MJ, Haire RN, Ohta Y, et al. Discordant phenotype in siblings withX-linked agammaglobulinemia. Am J Hum Genet 1996;58(3):477–83.

Yong et al706
28. Morwood K, Bourne H, Gold M, et al. Phenotypic variability: clinical presentationbetween the 6th year and the 60th year in a family with X-linked agammaglob-ulinemia. J Allergy Clin Immunol 2004;113(4):783–5.
29. Takada H, Kanegane H, Nomura A, et al. Female agammaglobulinemia due tothe Bruton tyrosine kinase deficiency caused by extremely skewed X-chromo-some inactivation. Blood 2004;103(1):185–7.
30. Moin M, Aghamohammadi A, Farhoudi A, et al. X-linked agammaglobulinemia:a survey of 33 Iranian patients. Immunol Invest 2004;33(1):81–93.
31. Plebani A, Soresina A, Rondelli R, et al. Clinical, immunological, and molecularanalysis in a large cohort of patients with X-linked agammaglobulinemia: anItalian multicenter study. Clin Immunol 2002;104(3):221–30.
32. Conley ME, Howard V. Clinical findings leading to the diagnosis of X-linkedagammaglobulinemia. J Pediatr 2002;141(4):566–71.
33. Alibrahim A, Lepore M, Lierl M, et al. Pneumocystis carinii pneumonia in an infantwith X-linked agammaglobulinemia. J Allergy Clin Immunol 1998;101(4 Pt 1):552–3.
34. Dittrich AM, Schulze I, Magdorf K, et al. X-linked agammaglobulinaemia andPneumocystis carinii pneumonia: an unusual coincidence? Eur J Pediatr 2003;162(6):432–3.
35. Misbah SA, Spickett GP, Ryba PC, et al. Chronic enteroviral meningoencephali-tis in agammaglobulinemia: case report and literature review. J Clin Immunol1992;12(4):266–70.
36. Wilfert CM, Buckley RH, Mohanakumar T, et al. Persistent and fatal central-nervous-system ECHOvirus infections in patients with agammaglobulinemia.N Engl J Med 1977;296(26):1485–9.
37. Halliday E, Winkelstein J, Webster AD. Enteroviral infections in primary immuno-deficiency (PID): a survey of morbidity and mortality. J Infect 2003;46(1):1–8.
38. Bardelas JA, Winkelstein JA, Seto DS, et al. Fatal ECHO 24 infection in a patientwith hypogammaglobulinemia: relationship to dermatomyositis-like syndrome.J Pediatr 1977;90(3):396–9.
39. Franz A, Webster AD, Furr PM, et al. Mycoplasmal arthritis in patients with pri-mary immunoglobulin deficiency: clinical features and outcome in 18 patients.Br J Rheumatol 1997;36(6):661–8.
40. Echave-Sustaeta JM, Villena V, Verdugo M, et al. X-linked agammaglobulinae-mia and squamous lung cancer. Eur Respir J 2001;17(3):570–2.
41. Lavilla P, Gil A, Rodriguez MC, et al. X-linked agammaglobulinemia and gastricadenocarcinoma. Cancer 1993;72(5):1528–31.
42. Lederman HM, Winkelstein JA. X-linked agammaglobulinemia: an analysis of96 patients. Medicine (Baltimore) 1985;64(3):145–56.
43. van Der Meer JW, Weening RS, Schellekens PT, et al. Colorectal cancer in pa-tients with X-linked agammaglobulinaemia. Lancet 1993;341(8858):1439–40.
44. Yong PF, Grosse-Kreul D, Maher J, et al. Dermatofibrosarcoma protuberans ina patient with X-linked agammaglobulinaemia. J Clin Pathol 2007;60(10):1162–4.
45. Hermaszewski RA, Webster AD. Primary hypogammaglobulinaemia: a survey ofclinical manifestations and complications. Q J Med 1993;86(1):31–42.
46. Lopez GE, Porpiglia AS, Hogan MB, et al. Clinical and molecular analysis ofpatients with defects in micro heavy chain gene. J Clin Invest 2002;110(7):1029–35.
47. Yel L, Minegishi Y, Coustan-Smith E, et al. Mutations in the mu heavy-chain genein patients with agammaglobulinemia. N Engl J Med 1996;335(20):1486–93.

Hypogammaglobulinaemia 707
48. Ferrari S, Lougaris V, Caraffi S, et al. Mutations of the Igbeta gene cause agam-maglobulinemia in man. J Exp Med 2007;204(9):2047–51.
49. Dobbs AK, Yang T, Farmer D, et al. Cutting edge: a hypomorphic mutationin Igbeta (CD79b) in a patient with immunodeficiency and a leaky defect inB cell development. J Immunol 2007;179(4):2055–9.
50. Wang Y, Kanegane H, Sanal O, et al. Novel Igalpha (CD79a) gene mutation ina Turkish patient with B cell-deficient agammaglobulinemia. Am J Med Genet2002;108(4):333–6.
51. Minegishi Y, Coustan-Smith E, Wang YH, et al. Mutations in the human lambda5/14.1 gene result in B cell deficiency and agammaglobulinemia. J Exp Med 1998;187(1):71–7.
52. Minegishi Y, Coustan-Smith E, Rapalus L, et al. Mutations in Igalpha (CD79a)result in a complete block in B-cell development. J Clin Invest 1999;104(8):1115–21.
53. Minegishi Y, Rohrer J, Coustan-Smith E, et al. An essential role for BLNK inhuman B cell development. Science 1999;286(5446):1954–7.
54. Good RA, Varco RL. A clinical and experimental study of agammaglobulinemia.J Lancet 1955;75(6):245–71.
55. Tarr PE, Sneller MC, Mechanic LJ, et al. Infections in patients with immunodefi-ciency with thymoma (Good syndrome): report of 5 cases and review of theliterature. Medicine (Baltimore) 2001;80(2):123–33.
56. Watts RG, Kelly DR. Fatal varicella infection in a child associated with thymomaand immunodeficiency (Good’s syndrome). Med Pediatr Oncol 1990;18(3):246–51.
57. Agarwal S, Cunningham-Rundles C. Thymoma and immunodeficiency (Goodsyndrome): a report of 2 unusual cases and review of the literature. Ann AllergyAsthma Immunol 2007;98(2):185–90.
58. Souadjian JV, Enriquez P, Silverstein MN, et al. The spectrum of diseases asso-ciated with thymoma: coincidence or syndrome? Arch Intern Med 1974;134(2):374–9.
59. Beck S, Slater D, Harrington CI. Fatal chronic cutaneous herpes simplex asso-ciated with thymoma and hypogammaglobulinaemia. Br J Dermatol 1981;105(4):471–4.
60. Kelleher P, Misbah SA. What is Good’s syndrome? Immunological abnormalitiesin patients with thymoma. J Clin Pathol 2003;56(1):12–6.
61. Conley ME. Early defects in B cell development. Curr Opin Allergy Clin Immunol2002;2(6):517–22.
62. Revy P, Busslinger M, Tashiro K, et al. A syndrome involving intrauterine growthretardation, microcephaly, cerebellar hypoplasia, B lymphocyte deficiency, andprogressive pancytopenia. Pediatrics 2000;105(3):E39.
63. Srivannaboon K, Conley ME, Coustan-Smith E, et al. Hypogammaglobulinemiaand reduced numbers of B-cells in children with myelodysplastic syndrome.J Pediatr Hematol Oncol 2001;23(2):122–5.
64. Janeway C, Apt L, Gitlin D. Agammaglobulinemia. Trans Assoc Am Physicians1953;66:200–2.
65. Cunningham-Rundles C, Bodian C. Common variable immunodeficiency:clinical and immunological features of 248 patients. Clin Immunol 1999;92(1):34–48.
66. Quinti I, Soresina A, Spadaro G, et al. Long-term follow-up and outcome of a largecohort of patients with common variable immunodeficiency. J Clin Immunol 2007;27(3):308–16.

Yong et al708
67. Hammarstrom L, Vorechovsky I, Webster D. Selective IgA deficiency (SIgAD)and common variable immunodeficiency (CVID). Clin Exp Immunol 2000;120(2):225–31.
68. Vorechovsky I, Webster AD, Plebani A, et al. Genetic linkage of IgA deficiency tothe major histocompatibility complex: evidence for allele segregation distortion,parent-of-origin penetrance differences, and the role of anti-IgA antibodies indisease predisposition. Am J Hum Genet 1999;64(4):1096–109.
69. Espanol T, Catala M, Hernandez M, et al. Development of a common variableimmunodeficiency in IgA-deficient patients. Clin Immunol Immunopathol 1996;80(3 Pt 1):333–5.
70. Rotbart HA, Webster AD. Treatment of potentially life-threatening enterovirusinfections with pleconaril. Clin Infect Dis 2001;32(2):228–35.
71. Stack E, Washington K, Avant GR, et al. Cytomegalovirus enteritis in commonvariable immunodeficiency. South Med J 2004;97(1):96–101.
72. Michel M, Chanet V, Galicier L, et al. Autoimmune thrombocytopenic purpuraand common variable immunodeficiency: analysis of 21 cases and review ofthe literature. Medicine (Baltimore) 2004;83(4):254–63.
73. Davies CW, Juniper MC, Gray W, et al. Lymphoid interstitial pneumonitis associ-ated with common variable hypogammaglobulinaemia treated with cyclosporinA. Thorax 2000;55(1):88–90.
74. Bates CA, Ellison MC, Lynch DA, et al. Granulomatous-lymphocytic lung diseaseshortens survival in common variable immunodeficiency. J Allergy Clin Immunol2004;114(2):415–21.
75. Morimoto Y, Routes JM. Granulomatous disease in common variable immunode-ficiency. Curr Allergy Asthma Rep 2005;5(5):370–5.
76. Mechanic LJ, Dikman S, Cunningham-Rundles C. Granulomatous disease incommon variable immunodeficiency. Ann Intern Med 1997;127(8 Pt 1):613–7.
77. Kinlen LJ, Webster AD, Bird AG, et al. Prospective study of cancer in patientswith hypogammaglobulinaemia. Lancet 1985;1(8423):263–6.
78. Cambronero R, Sewell WA, North ME, et al. Up-regulation of IL-12 in monocytes:a fundamental defect in common variable immunodeficiency. J Immunol 2000;164(1):488–94.
79. Bayry J, Lacroix-Desmazes S, Kazatchkine MD, et al. Common variable immu-nodeficiency is associated with defective functions of dendritic cells. Blood2004;104(8):2441–3.
80. Cunningham-Rundles C, Radigan L. Deficient IL-12 and dendritic cell function incommon variable immune deficiency. Clin Immunol 2005;115(2):147–53.
81. Scott-Taylor TH, Green MR, Eren E, et al. Monocyte derived dendritic cell re-sponses in common variable immunodeficiency. Clin Exp Immunol 2004;138(3):484–90.
82. Viallard JF, Camou F, Andre M, et al. Altered dendritic cell distribution inpatients with common variable immunodeficiency. Arthritis Res Ther 2005;7(5):R1052–5.
83. Cunningham-Rundles C, Radigan L, Knight AK, et al. TLR9 activation is defec-tive in common variable immune deficiency. J Immunol 2006;176(3):1978–87.
84. Sneller MC, Strober W. Abnormalities of lymphokine gene expression in patientswith common variable immunodeficiency. J Immunol 1990;144(10):3762–9.
85. Kondratenko I, Amlot PL, Webster AD, et al. Lack of specific antibody responsein common variable immunodeficiency (CVID) associated with failure in produc-tion of antigen-specific memory T cells. MRC Immunodeficiency Group. Clin ExpImmunol 1997;108(1):9–13.

Hypogammaglobulinaemia 709
86. Farrington M, Grosmaire LS, Nonoyama S, et al. CD40 ligand expression isdefective in a subset of patients with common variable immunodeficiency.Proc Natl Acad Sci U S A 1994;91(3):1099–103.
87. Di RM, Serrano D, Zhou Z, et al. Enhanced T cell apoptosis in common variableimmunodeficiency: negative role of the fas/fasligand system and of the Bcl-2family proteins and possible role of TNF-RS. Clin Exp Immunol 2001;125(1):117–22.
88. Pozzi N, Gaetaniello L, Martire B, et al. Defective surface expression of attractinon T cells in patients with common variable immunodeficiency (CVID). Clin ExpImmunol 2001;123(1):99–104.
89. Holm AM, Aukrust P, Damas JK, et al. Abnormal interleukin-7 function in com-mon variable immunodeficiency. Blood 2005;105(7):2887–90.
90. Boncristiano M, Majolini MB, D’Elios MM, et al. Defective recruitment and activa-tion of ZAP-70 in common variable immunodeficiency patients with T cell de-fects. Eur J Immunol 2000;30(9):2632–8.
91. Paccani SR, Boncristiano M, Patrussi L, et al. Defective vav expression andimpaired F-actin reorganization in a subset of patients with common variableimmunodeficiency characterized by T-cell defects. Blood 2005;106(2):626–34.
92. Bryant A, Calver NC, Toubi E, et al. Classification of patients with commonvariable immunodeficiency by B cell secretion of IgM and IgG in response toanti-IgM and interleukin-2. Clin Immunol Immunopathol 1990;56(2):239–48.
93. Warnatz K, Denz A, Drager R, et al. Severe deficiency of switched memoryB cells (CD27(1)IgM(-)IgD(-)) in subgroups of patients with common variableimmunodeficiency: a new approach to classify a heterogeneous disease. Blood2002;99(5):1544–51.
94. Piqueras B, Lavenu-Bombled C, Galicier L, et al. Common variable immunode-ficiency patient classification based on impaired B cell memory differentiationcorrelates with clinical aspects. J Clin Immunol 2003;23(5):385–400.
95. Wehr C, Kivioja T, Schmitt C, et al. The EUROclass trial: defining subgroups incommon variable immunodeficiency. Blood 2008;111(1):77–85.
96. Giovannetti A, Pierdominici M, Mazzetta F, et al. Unravelling the complexity ofT cell abnormalities in common variable immunodeficiency. J Immunol 2007;178(6):3932–43.
97. Yong PF, Workman S, Wahid F, et al. Selective deficits in blood dendritic cell sub-sets in common variable immunodeficiency and X-linked agammaglobulinaemiabut not specific polysaccharide antibody deficiency. Clin Immunol 2008;127(1):34–42.
98. Grimbacher B, Hutloff A, Schlesier M, et al. Homozygous loss of ICOS is asso-ciated with adult-onset common variable immunodeficiency. Nat Immunol 2003;4(3):261–8.
99. Salzer U, Chapel HM, Webster AD, et al. Mutations in TNFRSF13B encodingTACI are associated with common variable immunodeficiency in humans. NatGenet 2005;37(8):820–8.
100. Castigli E, Wilson SA, Garibyan L, et al. TACI is mutant in common variable im-munodeficiency and IgA deficiency. Nat Genet 2005;37(8):829–34.
101. Warnatz K, Salzer U, Gutenberger S. Finally found: human BAFF-R deficiencycauses CVID. XIth Meeting of the European Society for Immunodeficiencies.Versailles (France), October 21–24, 2004.
102. van Zelm MC, Reisli I, van der BM, et al. An antibody-deficiency syndrome dueto mutations in the CD19 gene. N Engl J Med 2006;354(18):1901–12.

Yong et al710
103. Kanegane H, Agematsu K, Futatani T, et al. Novel mutations in a Japanesepatient with CD19 deficiency. Genes Immun 2007;8(8):663–70.
104. Sekine H, Ferreira RC, Pan-Hammarstrom Q, et al. Role for Msh5 in the regula-tion of Ig class switch recombination. Proc Natl Acad Sci U S A 2007;104(17):7193–8.
105. Carreno BM, Collins M. The B7 family of ligands and its receptors: new path-ways for costimulation and inhibition of immune responses. Annu Rev Immunol2002;20:29–53.
106. Carter RH, Fearon DT. CD19: lowering the threshold for antigen receptor stimu-lation of B lymphocytes. Science 1992;256(5053):105–7.
107. Salzer U, Maul-Pavicic A, Cunningham-Rundles C, et al. ICOS deficiency inpatients with common variable immunodeficiency. Clin Immunol 2004;113(3):234–40.
108. Schneider P. The role of APRIL and BAFF in lymphocyte activation. Curr OpinImmunol 2005;17(3):282–9.
109. Bacchelli C, Buckridge S, Thrasher AJ, et al. Translational mini-review series onimmunodeficiency: molecular defects in common variable immunodeficiency.Clin Exp Immunol 2007;149(3):401–9.
110. Zhang L, Radigan L, Salzer U, et al. Transmembrane activator and calcium-modulating cyclophilin ligand interactor mutations in common variable immuno-deficiency: clinical and immunologic outcomes in heterozygotes. J Allergy ClinImmunol 2007;120(5):1178–85.
111. Pan-Hammarstrom Q, Salzer U, Du L, et al. Reexamining the role of TACI codingvariants in common variable immunodeficiency and selective IgA deficiency.Nat Genet 2007;39(4):429–30.
112. Castigli E, Wilson S, Garibyan L, et al. Reexamining the role of TACI codingvariants in common variable immunodeficiency and selective IgA deficiency.Nat Genet 2007;39(4):430–1.
113. Fearon DT, Carroll MC. Regulation of B lymphocyte responses to foreign andself-antigens by the CD19/CD21 complex. Annu Rev Immunol 2000;18:393–422.
114. Purtilo DT, Cassel C, Yang JP. Letter: fatal infectious mononucleosis in familiallymphohistiocytosis. N Engl J Med 1974;291(14):736.
115. Seemayer TA, Gross TG, Egeler RM, et al. X-linked lymphoproliferative disease:twenty-five years after the discovery. Pediatr Res 1995;38(4):471–8.
116. Coffey AJ, Brooksbank RA, Brandau O, et al. Host response to EBV infection inX-linked lymphoproliferative disease results from mutations in an SH2-domainencoding gene. Nat Genet 1998;20(2):129–35.
117. Sayos J, Wu C, Morra M, et al. The X-linked lymphoproliferative-disease geneproduct SAP regulates signals induced through the co-receptor SLAM. Nature1998;395(6701):462–9.
118. Rigaud S, Fondaneche MC, Lambert N, et al. XIAP deficiency in humans causesan X-linked lymphoproliferative syndrome. Nature 2006;444(7115):110–4.
119. Eastwood D, Gilmour KC, Nistala K, et al. Prevalence of SAP gene defects inmale patients diagnosed with common variable immunodeficiency. Clin ExpImmunol 2004;137(3):584–8.
120. Notarangelo LD, Duse M, Ugazio AG. Immunodeficiency with hyper-IgM (HIM).Immunodefic Rev 1992;3(2):101–21.
121. Winkelstein JA, Marino MC, Ochs H, et al. The X-linked hyper-IgM syndrome:clinical and immunologic features of 79 patients. Medicine (Baltimore) 2003;82(6):373–84.

Hypogammaglobulinaemia 711
122. Aruffo A, Farrington M, Hollenbaugh D, et al. The CD40 ligand, gp39, is defec-tive in activated T cells from patients with X-linked hyper-IgM syndrome. Cell1993;72(2):291–300.
123. DiSanto JP, Bonnefoy JY, Gauchat JF, et al. CD40 ligand mutations in x-linkedimmunodeficiency with hyper-IgM. Nature 1993;361(6412):541–3.
124. Ferrari S, Giliani S, Insalaco A, et al. Mutations of CD40 gene cause an autoso-mal recessive form of immunodeficiency with hyper IgM. Proc Natl Acad SciU S A 2001;98(22):12614–9.
125. Doffinger R, Smahi A, Bessia C, et al. X-linked anhidrotic ectodermal dysplasiawith immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet2001;27(3):277–85.
126. Jain A, Ma CA, Liu S, et al. Specific missense mutations in NEMO result in hyper-IgM syndrome with hypohydrotic ectodermal dysplasia. Nat Immunol 2001;2(3):223–8.
127. Revy P, Muto T, Levy Y, et al. Activation-induced cytidine deaminase (AID)deficiency causes the autosomal recessive form of the Hyper-IgM syndrome(HIGM2). Cell 2000;102(5):565–75.
128. Durandy A, Revy P, Imai K, et al. Hyper-immunoglobulin M syndromes causedby intrinsic B-lymphocyte defects. Immunol Rev 2005;203:67–79.
129. Quartier P, Bustamante J, Sanal O, et al. Clinical, immunologic and genetic anal-ysis of 29 patients with autosomal recessive hyper-IgM syndrome due to activa-tion-induced cytidine deaminase deficiency. Clin Immunol 2004;110(1):22–9.
130. Minegishi Y, Lavoie A, Cunningham-Rundles C, et al. Mutations in activation-induced cytidine deaminase in patients with hyper IgM syndrome. Clin Immunol2000;97(3):203–10.
131. Zhu Y, Nonoyama S, Morio T, et al. Type two hyper-IgM syndrome caused bymutation in activation-induced cytidine deaminase. J Med Dent Sci 2003;50(1):41–6.
132. Durandy A, Peron S, Taubenheim N, et al. Activation-induced cytidine deami-nase: structure-function relationship as based on the study of mutants. HumMutat 2006;27(12):1185–91.
133. Ta VT, Nagaoka H, Catalan N, et al. AID mutant analyses indicate requirementfor class-switch-specific cofactors. Nat Immunol 2003;4(9):843–8.
134. Barreto V, Reina-San-Martin B, Ramiro AR, et al. C-terminal deletion of AIDuncouples class switch recombination from somatic hypermutation and geneconversion. Mol Cell 2003;12(2):501–8.
135. Kasahara Y, Kaneko H, Fukao T, et al. Hyper-IgM syndrome with putative dom-inant negative mutation in activation-induced cytidine deaminase. J Allergy ClinImmunol 2003;112(4):755–60.
136. Imai K, Slupphaug G, Lee WI, et al. Human uracil-DNA glycosylase deficiencyassociated with profoundly impaired immunoglobulin class-switch recombina-tion. Nat Immunol 2003;4(10):1023–8.
137. Di NJ, Neuberger MS. Altering the pathway of immunoglobulin hypermutation byinhibiting uracil-DNA glycosylase. Nature 2002;419(6902):43–8.
138. Imai K, Catalan N, Plebani A, et al. Hyper-IgM syndrome type 4 with a B lympho-cyte-intrinsic selective deficiency in Ig class-switch recombination. J Clin Invest2003;112(1):136–42.
139. Brusco A, Cariota U, Bottaro A, et al. Variability of the immunoglobulin heavychain constant region locus: a population study. Hum Genet 1995;95(3):319–26.
140. Zegers BJ, Maertzdorf WJ, Van LE, et al. Kappa-chain deficiency: an immuno-globulin disorder. N Engl J Med 1976;294(19):1026–30.

Yong et al712
141. Stavnezer-Nordgren J, Kekish O, Zegers BJ. Molecular defects in a human im-munoglobulin kappa chain deficiency. Science 1985;230(4724):458–61.
142. Schur PH, Borel H, Gelfand EW, et al. Selective gamma-g globulin deficienciesin patients with recurrent pyogenic infections. N Engl J Med 1970;283(12):631–4.
143. Jefferis R, Kumararatne DS. Selective IgG subclass deficiency: quantificationand clinical relevance. Clin Exp Immunol 1990;81(3):357–67.
144. Maguire GA, Kumararatne DS, Joyce HJ. Are there any clinical indications formeasuring IgG subclasses? Ann Clin Biochem 2002;39(Pt 4):374–7.
145. Bonilla FA, Bernstein IL, Khan DA, et al. Practice parameter for the diagnosisand management of primary immunodeficiency. Ann Allergy Asthma Immunol2005;94(5 Suppl 1):S1–63.
146. Buckley RH. Immunoglobulin G subclass deficiency: fact or fancy? Curr AllergyAsthma Rep 2002;2(5):356–60.
147. Clark JA, Callicoat PA, Brenner NA, et al. Selective IgA deficiency in blooddonors. Am J Clin Pathol 1983;80(2):210–3.
148. Litzman J, Sevcikova I, Stikarovska D, et al. IgA deficiency in Czech healthy in-dividuals and selected patient groups. Int Arch Allergy Immunol 2000;123(2):177–80.
149. Ulfarsson J, Gudmundsson S, Birgisdottir B, et al. Selective serum IgA defi-ciency in Icelanders: frequency, family studies and Ig levels. Acta Med Scand1982;211(6):481–7.
150. Edwards E, Razvi S, Cunningham-Rundles C. IgA deficiency: clinical correlatesand responses to pneumococcal vaccine. Clin Immunol 2004;111(1):93–7.
151. Hanson LA, Bjorkander J, Carlsson B, et al. The heterogeneity of IgA deficiency.J Clin Immunol 1988;8(3):159–62.
152. Alaswad B, Brosnan P. The association of celiac disease, diabetes mellitus type1, hypothyroidism, chronic liver disease, and selective IgA deficiency. Clin Pe-diatr (Phila) 2000;39(4):229–31.
153. Cunningham-Rundles C, Pudifin DJ, Armstrong D, et al. Selective IgA deficiencyand neoplasia. Vox Sang 1980;38(2):61–7.
154. Mellemkjaer L, Hammarstrom L, Andersen V, et al. Cancer risk among patientswith IgA deficiency or common variable immunodeficiency and their relatives:a combined Danish and Swedish study. Clin Exp Immunol 2002;130(3):495–500.
155. Bossuyt X, Moens L, Van HE, et al. Coexistence of (partial) immune defects andrisk of recurrent respiratory infections. Clin Chem 2007;53(1):124–30.
156. Saxon A, Kobayashi RH, Stevens RH, et al. In vitro analysis of humoral immunityin antibody deficiency with normal immunoglobulins. Clin Immunol Immunopa-thol 1980;17(2):235–44.
157. Cheng YK, Decker PA, O’Byrne MM, et al. Clinical and laboratory characteristicsof 75 patients with specific polysaccharide antibody deficiency syndrome. AnnAllergy Asthma Immunol 2006;97(3):306–11.
158. Sorensen RU, Leiva LE, Javier FC III, et al. Influence of age on the response toStreptococcus pneumoniae vaccine in patients with recurrent infections andnormal immunoglobulin concentrations. J Allergy Clin Immunol 1998;102(2):215–21.
159. Kirkpatrick CH. Specific polysaccharide antibody deficiency. Ann AllergyAsthma Immunol 2006;97(3):271.
160. Gitlin D, Janeway C. Agammaglobulinemia, congenital, acquired and transientforms. Prog Hematol 1956;1:318–29.

Hypogammaglobulinaemia 713
161. Whelan MA, Hwan WH, Beausoleil J, et al. Infants presenting with recurrent in-fections and low immunoglobulins: characteristics and analysis of normalization.J Clin Immunol 2006;26(1):7–11.
162. Dalal I, Reid B, Nisbet-Brown E, et al. The outcome of patients with hypogamma-globulinemia in infancy and early childhood. J Pediatr 1998;133(1):144–6.
163. Walker AM, Kemp AS, Hill DJ, et al. Features of transient hypogammaglobuli-naemia in infants screened for immunological abnormalities. Arch Dis Child1994;70(3):183–6.
164. Kidon MI, Handzel ZT, Schwartz R, et al. Symptomatic hypogammaglobulinemiain infancy and childhood - clinical outcome and in vitro immune responses. BMCFam Pract 2004;5:23.
165. Kilic SS, Tezcan I, Sanal O, et al. Transient hypogammaglobulinemia of infancy:clinical and immunologic features of 40 new cases. Pediatr Int 2000;42(6):647–50.
166. Kosnik EF, Johnson JP, Rennels MB, et al. Streptococcal sepsis presenting asacute abdomen in a child with transient hypogammaglobulinemia of infancy.J Pediatr Surg 1986;21(11):975–6.

Genetic SyndromicImmunodefic iencieswith Antibody Defects
Jeffrey E. Ming, MD, PhDa, E. Richard Stiehm, MDb,*
KEYWORDS
� Immunodeficiencies � Genetic syndromes � Immunoglobulin� Antibody � Hypogammaglobulinemia
In this article we review the major syndromic immunodeficiencies with significant an-tibody defects, many of which may require intravenous immunogammaglobulin(IVIG) therapy. We define syndromic immunodeficiency as an illness associatedwith a characteristic group of phenotypic abnormalities or laboratory features thatcomprise a recognizable syndrome. Many are familial with a defined inheritance pat-tern. Immunodeficiency may not be a major part of the illness and may not be pres-ent in all patients; thus, these conditions differ from primary immunodeficiencysyndromes, in which immune abnormalities are a consistent and prominent featureof their disease.
Certain well-recognized primary inmmunodeficiencies, such as Wiskott-Aldrichsyndrome and ataxia-telangiectasia, fit into primary and syndromic immunodeficiencycategories because they have characteristic organ dysfunction or dysmorphology un-related to the immune system and a consistent, well-defined immunodeficiency.1,2
They are not discussed in this article because they have been covered in detail else-where.1–3
The immune defects of syndromic deficiencies may include B-cell (antibody), T-cell(cellular), phagocytic cell, complement, or innate immune defects or a combination ofthereof. All of the illnesses mentioned in this article have antibody deficiency as themain portion or a significant portion of their immune abnormalities. A more completediscussion of all of the syndromic inmmunodeficiencies is available elsewhere.4
The inheritance pattern of each condition and the chromosomal location of the dis-ease-related genes, when known, are indicated in the Tables 1–5. Online mendelianinheritance in man (OMIM)5 numbers are indicated within parentheses in the text.
a Division of Human Genetics, Department of Pediatrics, The Children’s Hospital of Philadelphia,The University of Pennsylvania School of Medicine, 3615 Civic Center Boulevard, Philadelphia,PA 19104, USAb Division of Immunology/ Allergy/ Rheumatology, Mattel Children’s Hospital at UCLA, 10833Le Conte Avenue, 22-387 MDCC, Los Angeles, CA 90095, USA* Corresponding author.E-mail address: [email protected] (E.R. Stiehm).
Immunol Allergy Clin N Am 28 (2008) 715–736doi:10.1016/j.iac.2008.06.007 immunology.theclinics.com0889-8561/08/$ – see front matter ª 2008 Elsevier Inc. All rights reserved.

Table1Syndromic immunodeficiencies associated with growth deficiency
NameInheritance(Chromosome)
AssociatedFeatures Immune Defect Frequency of IDa
Disproportionate short stature
Schimke immunoosseousdysplasia
AR 2q34-q36 Spondyloepiphyseal dysplasia, progressivenephropathy, episodic lymphopenia,pigmentary skin changes
T, B 1111
Short-limb skeletal dysplasiawith immunodeficiency
AR Short-limb skeletal dysplasia, metaphysealdysplasia; may be associated with adenosinedeaminase deficiency or Omenn syndrome;heterogeneous
T, B 1111
Roifman syndrome XL Spondyloepiphyseal dysplasia, retinal dystrophy B 1111
Roifman-Costa syndrome AR Spondylometaphyseal dysplasia, autoimmuneconditions
B, T 1111
Growth hormone pathwaydefects
Various Defects in growth hormone synthesis orsensitivity deficiency, sinopulmonaryinfections
B, T, NK 1
Kabuki syndrome AD Long palpebral fissures, prominent eyelashes,skeletal anomalies, congenital heart disease;increased risk of autoimmune diseases
B 111
CHARGE association ? Coloboma, heart defect, atresia choanae,retarded growth and development, genitalhypoplasia, ear anomalies/deafness
T, B 1
Rubinstein-Taybisyndrome
AD (16p13) Broad thumbs and halluces, prominent nasalseptum below ala nasi, cryptorchidism, mentalretardation
T, B 1
Abbreviations: AD, autosomal dominant; AR, autosomal recessive; B, B-cell defect; ID, immunodeficiency; NK, NK cell defect; Ph, phagocyte defect; T, T-cell defect;XL, X-linked.
a Frequency of ID: 1, < 5% of reported cases with documented ID; 11, 5%–30%; 111, 30%–65%; 1111 , > 65%.
Min
g&
Stieh
m716

Genetic Syndromic Immunodeficiencies 717
SYNDROMES ASSOCIATEDWITH GROWTH DEFICIENCYPrimary Immunodeficiencies Associated with Short Stature
Schimke immune-osseous dysplasiaThis condition (OMIM 242900) is associated with short stature with exaggerated lum-bar lordosis, spondyloepiphyseal dysplasia, defective cellular immunity, and progres-sive renal failure (see Table 1).6,7 Patients may develop glomerulosclerosis andprogress to end-stage renal disease; arteriopathy with cerebral infarcts or ischemiamay be seen. Mutations in the gene encoding the chromatin remodeling proteinSMARCAL1 have been detected in affected patients.8 Patients are prone to viraland bacterial infections and demonstrate decreases in CD4 T-cell numbers, mito-gen-induced proliferation, and delayed cutaneous hypersensitivity responses. Immu-noglobulin levels are often abnormal.7,9
Other Immunodeficiencies Associated with Short Stature
Roifman syndrome (Roifman syndrome 1)Five boys from four families had microcephaly, growth retardation, spondyloepiphy-seal dysplasia, developmental delay, and retinal dystrophy (OMIM 300258).10,11
They had low or absent antibody titers in response to infection, decreased isohemag-glutinins, and decreased mitogenic response to Staphylococcus aureus Cowan A.T-cell numbers and function were normal. They had epiphyseal dysplasia of thehips and long bones and vertebral anomalies. Because all reported patients havebeen male, X-linked recessive inheritance has been suggested.
Roifman-Costa syndrome (Roifman syndrome 2)Four patients, including two siblings of first cousin parents, with spondylometaphysealdysplasia, autoimmune conditions, combined immunodeficiency (low specific anti-body titers, T-cell mitogenic response, and CD4 T-cell count), and recurrent infectionswere described (OMIM 607944).12 A boy born to a consanguineous couple had spon-dylometaphyseal dysplasia, decreased CD4 and CD8 T-cell numbers, recurrent infec-tions, disseminated herpes zoster, and autoimmune disease.13 Similarity in clinicalfeatures with spondyloenchondrodysplasia (OMIM 27155) has been noted.14
Growth hormone pathway defectsPatients with defects in the growth hormone pathway and immunodeficiency havebeen described. In patients with growth hormone deficiency with X-linked agamma-globulinemia (OMIM 307200), individuals have recurrent sinopulmonary infections,short stature, and decreased growth hormone levels without other endocrinologicabnormalities.15 B-cell number and immunoglobulin levels are greatly decreased orabsent, consistent with X-linked agammaglobulinemia. T-cell numbers and functionare normal. Mutations in the gene BTK, the gene associated with isolated X-linkedagammaglobulinemia, have been detected in some—but not all—patients with growthhormone deficiency and X-linked agammaglobulinemia.16–18
Additional immune defects reported in association with isolated growth hormonedeficiency include combined immunodeficiency,19,20 decreased natural killer (NK)cell activity,21 and hypogammaglobulinemia.22 Most children with growth hormonedeficiency do not display an increased susceptibility to infection, however.23,24
Some patients with growth hormone insensitivity were found to have a mutation inthe STAT5B gene. One of the patients who had recurrent skin and respiratory infec-tions had T-cell lymphopenia and low NK and CD4 T-cell numbers.25 Both growth hor-mone and interleukin-2 receptor signaling use Stat5 proteins in their pathways.

Table 2Syndromic immunodeficiencies associated with specific organ dysfunction
Name Inheritance (Chromosome) Associated Features Immune Defect Frequency of IDa
Gastrointestinal
Familial intestinal polyatresia AR Multiple atresias from pylorus torectum
T, B 11
Trichohepatoenteric syndrome AR Severe infantile diarrhea, hepaticcirrhosis, Trichorrhxis nodosa,characteristic facies
B, Ph 1111
Dermatologic
Omenn syndrome AR (11p13) Erythematous dermatitis,eosinophilia, lymphadenopathy,hemophagocytosis; severecombined immunodeficiency
T, B 1111
Griscelli syndrome, type 2 AR (15q21) Partial albinism, frequent pyogenicinfections, lymphohistiocytosis,episodic thrombocytopenia
T, B, NK, Ph 1111
Hypohydrotic/anhidrotic ectodermaldysplasia with immunodeficiency
XL (Xq28) Alopecia, hypo/anhydrosis, toothanomalies,hypogammaglobulinemia
T, B 1111
WHIM syndrome AD Warts, hypogammaglobulinemia,infection, myelokathexis
T, B, Ph 1111
Incontinentia pigmenti XL (Xq28) Erythematous vesiculobullouseruptions, central nervous systeminvolvement, swirling macules ofhyperpigmentation
T, B, Ph 1
Min
g&
Stieh
m718

OLEDAID syndrome XL (Xq28) Anhidrotic ectodermal dysplasia,osteopetrosis, lymphedema
B 1111 (2 cases)
Dyskeratosis congenita XL, AR, AD (Xq28) Atrophy and pigmentation of skin,nail dystrophy, leukoplakia of oralmucosa; risk of cancer of the mouth,anus, skin
T, B, Ph 11
Acrodermatitis enteropathica AR (8q24) Vesiculobullous dermatitis, alopecia,diarrhea; caused by zinc deficiency,may be associated withopportunistic infections
T, B, Ph 11
Netherton syndrome AR (5q32) Trichorrhexis invaginata (bamboohair), ichthyosiform dermatitis,atopic diathesis, skin infections
T, B, Ph 11
Neurologic
Myotonic dystrophy AD (19q13, 3q) Myotonia, muscle wasting, cataract,hypogonadism, cardiac arrhythmia;caused by triplet repeat expansion
B 11
Høyeraal-Hreidarsson syndrome XL (Xq28) Cerebellar hypoplasia, absent corpuscallosum, microcephaly, growthfailure, pancytopenia, fungal sepsis
T, B, Ph 1111
Abbreviations: AD, autosomal dominant; AR, autosomal recessive; B, B-cell defect; ID, immunodeficiency; NK, NK cell defect; Ph, phagocyte defect; T, T-cell defect;XL, X-linked.
a Frequency of immunodeficiency: 1, < 5% of reported cases with documented ID; 11, 5%–30%; 111, 30%–65%; 1111, > 65%.
Gen
etic
Synd
rom
icIm
mu
no
deficie
ncie
s719
Table 3Inborn errors of metabolism associatedwith immunodeficiency
NameInheritance(Chromosome)
AssociatedFeatures
ImmuneDefect
Frequencyof IDa
Congenitaldisordersof glycosylation,Types Ia, Ig, Ik
Various Decreasedglycosylation,hypotonia, poorgrowth, other organsystems may beinvolved dependingon the type
B, Ph 11
Branchedchain aminoacidemias
AR (various) Methylmalonic,propionic, andisovaleric acidemias,acidosis, vomiting,ketosis
T, B, Ph 111
Lysinuric proteinintolerance
AR (14q11) Dibasicaminoaciduria,hepatomegaly,failure to thrive,severe varicella
infection
T, B, Ph, NK 111
Abbreviations: AD, autosomal dominant; AR, autosomal recessive; B, B-cell defect; ID, immunode-ficiency; NK, NK cell defect; Ph, phagocyte defect; T, T-cell defect; XL, X-linked.
a Frequency of ID: 1, < 5% of reported cases with documented ID; 11, 5%–30%; 111, 30%–65%; 1111, > 65%.
Table 4Syndromes associated with chromosomal instability and/or defective DNA repair
NameInheritance(Chromosome)
AssociatedFeatures
ImmuneDefect
Frequencyof IDa
Nijmegenbreakagesyndrome
AR (8q21) Microcephaly, mental retardation,prenatal onset short stature,bird-like facies, malignancy(lymphoma), sinopulmonary andurinary tract infections
T, B 1111
Bloomsyndrome
AR (15q26) Short stature, telangiectatic erythemaof face, sensitivity to sunlight,pneumonia, otitis media, risk forleukemia/lymphoma
T, B, NK 111
ICFsyndrome
AR (20q11) Mental retardation, chromosomalinstability, facial dysmorphism;sinopulmonary, gastrointestinal,cutaneous infections
T, B 1111
Abbreviations: AD, autosomal dominant; AR, autosomal recessive; B, B-cell defect; ICF, immunode-ficiency, centromeric instability, facial anomalies; ID, immunodeficiency; NK, NK cell defect; Ph,phagocyte defect; T, T-cell defect; XL, X-linked.
a Frequency of ID: 1, < 5% of reported cases with documented ID; 11, 5%–30%; 111, 30%–65%; 1111, > 65%.
Ming & Stiehm720

Table 5Syndromes associated with chromosomal abnormalities of number or structure
NameAssociatedFeatures
ImmuneDefect
Frequencyof IDa
Trisomy 21(Down syndrome)
Hypotonia, flat facies, upslantingpalpebral fissures, mental retardation,sinopulmonary infections, risk ofleukemia, autoimmune thyroiditis
T, B, Ph, NK 11
Deletion of shortarm of chromosome 4(4p16) (Wolf-Hirschhornsyndrome)
Growth and developmental deficiency,‘‘Greek helmet’’- like facies,microcephaly, coloboma, respiratoryinfections
B 111
Missing or abnormalX chromosome(Turner syndrome; XO,isoX, ring X)
Short stature, webbed neck, broad chest,ovarian dysgenesis, congenitallymphedema, pulmonary/earinfections, autoimmune disease (eg,thyroid disease, celiac disease,arthritis), gonadoblastoma (if Ychromosome material present)
T, B 11
Abbreviations: AD, autosomal dominant; AR, autosomal recessive; B, B-cell defect; ID, immunode-ficiency; NK, NK cell defect; Ph, phagocyte defect; T, T-cell defect; XL, X-linked.
a Frequency of ID: 1, < 5% of reported cases with documented ID; 11, 5%–30%; 111, 30%–65%; 1111, > 65%.
Genetic Syndromic Immunodeficiencies 721
Kabuki syndromeThis syndrome (OMIM 147920) features short stature, congenital heart disease, devel-opmental delay, skeletal anomalies, and cleft palate.26,27 The distinctive facial featuresinclude long palpebral fissures with eversion of the lower lateral eyelid, prominenteyelashes, and abnormal ears. Frequent infections occur in approximately 60% of pa-tients.28 Hypogammaglobulinemia, including decreased IgG and low IgA, is a commonmanifestation.29,30 Autoimmune conditions, including autoimmune hemolytic anemia,idiopathic thrombocytopenic purpura, and hypothyroidism, also have been reportedand may reflect the underlying immune dysfunction.31,32
CHARGE associationThe abnormalities (OMIM 214800) that comprise the CHARGE association include co-loboma, heart defects, atresia of the choanae, retardation of growth and development,genital hypoplasia, and ear anomalies and/or deafness.33–35 Some patients who haveCHARGE syndrome have been found to have mutations in CHD7.36,37 In this syn-drome, asymmetric facial palsy, esophageal or laryngeal abnormalities, renal malfor-mations, and facial clefts are present. Several patients who have CHARGE associationhave had immune abnormalities, including severe combined immunodeficiency withundetectable thymus tissue,38 decreased T-cell numbers and response to antigen, an-tibody deficiency and impaired T-cell proliferation, and isolated IgG2 deficiency.39 Pa-tients who have CHARGE association who also have the DiGeorge anomaly and didnot have a 22q11 deletion have been described.40 Other affected patients who haveDi George sequence but in whom the 22q11 deletion status was not known havebeen reported.33,41
Rubinstein-Taybi syndromeRubinstein-Taybi syndrome (OMIM 180849) is characterized by broad thumbs andgreat toes, characteristic facial features, short stature, mental retardation, and cardiac

Ming & Stiehm722
abnormalities. Affected individuals have an increased susceptibility to infection. De-creased T-cell numbers, impaired delayed cutaneous hypersensitivity response,42
lymphopenia, thymic hypoplasia,43 poor response to pneumococcal vaccine,44 anda deficit in polysaccharide antibody response45 have been reported. Microdeletionsand truncating mutations in the gene encoding CREB-binding protein have beendetected in several affected patients.46,47 Mutations in the gene EP300, which alsoencodes a transcriptional coactivator, have been detected in three patients.48
Mulvihill-Smith syndromeThis disorder (OMIM 176690) is characterized by pre- and postnatal growth retarda-tion, multiple pigmented nevi, microcephaly, reduced facial fat, genitourinary anoma-lies, and a high-pitched voice.49,50 Infectious complications are common, andimmunodeficiency is often progressive. Impaired T-cell response to mitogen, de-creased CD4 count, and low Ig levels have been described.50–52
SYNDROMES ASSOCIATEDWITH GASTROINTESTINAL DYSFUNCTION
Gastrointestinal abnormalities may lead to malnutrition and secondarily result in an im-munodeficient state. In the syndromes described herein, however, the immunodefi-ciency precedes nutritional deprivation and is likely to be intrinsic to each condition(see Table 2).
Other Syndromic Immunodeficiencies Associated with Gastrointestinal Dysfunction
Familial intestinal polyatresiaMultiple atretic lesions are found throughout the gastrointestinal tract in this condition(OMIM 243150). Severe combined immunodeficiency was described in three affectedbrothers.53 Adenosine deaminase activity was normal. The recurrent infections werenot caused by the intestinal problems because they occurred while the patients stillhad good nutritional status. Several other cases of multiple intestinal atresia associ-ated with immune defects have been described.54–57 Two families with duodenal atre-sia and immunodeficiency were reported.58
Trichohepatoenteric syndromeThis condition (OMIM 222470) is characterized by severe infantile diarrhea, dysmor-phic features (hypertelorism, prominent forehead, flat/broad nose), hepatic cirrhosis,and the hair abnormality of trichorrhexis nodosa. Reported immune defects have in-cluded negative skin tests with absent specific antibody response,59 pancytopenia,60
and hypogammaglobulinemia.61
SYNDROMES ASSOCIATEDWITH CUTANEOUS ABNORMALITIES
Although dermatitis or skin infection often occurs in immunodeficient patients, someimmunodeficiency syndromes present with primarily cutaneous manifestations (seeTable 2). Some of these conditions present with alterations in pigmentation.
Primary Immunodeficiencies Associated with Cutaneous Abnormalities
Griscelli syndromeGriscelli syndrome is an autosomal recessive syndrome of partial albinism, neutrope-nia and thrombocytopenia, and lymphohistiocytosis (OMIM 607624).62–64 Melano-somes accumulate in melanocytes, resulting in large clumps of pigment in hairshafts. Most patients suffer from recurrent and severe fungal, viral, and bacterial infec-tions. T-cell dysfunction, hypogammaglobulinemia, and neutropenia have been re-ported.64 Mutations in the RAB27A gene, which encodes a GTP-binding protein of

Genetic Syndromic Immunodeficiencies 723
the Ras family, were detected in affected individuals.65 A genetically distinct form ofGriscelli syndrome that is not associated with immunodeficiencies has beendescribed.65,66
Omenn syndromeThis autosomal recessive form of familial histiocytic reticulocytosis (OMIM 267700)presents with an erythematous skin rash, eosinophilia, reticulosis, hepatosplenome-galy, protracted diarrhea, alopecia, and lymphadenopathy. A characteristic severecombined immunodeficiency leads to failure to thrive, recurrent infection, and prema-ture death. Mutations in genes that encode any of three proteins that play a role inV(D)J recombination, RAG1, RAG2, or Artemis (DCLRE1C) can cause Omenn syn-drome with SCID.67,68
WHIM syndromeWHIM syndrome (OMIM 193670) is associated with multiple warts, hypogammaglobu-linemia, infection, and myelokathexis (bone marrow retention of neutrophils).69
Neutrophil count is reduced, B-cell numbers and IgG and IgA levels are mildlydecreased, and depressed T-cell numbers and diminished response to mitogen andskin tests have been noted. Mutations in the gene encoding the chemokine receptorCXCR4 were detected.70
Hypohidrotic/anhidrotic ectodermal dysplasiaA subset of patients with this form of ectodermal dysplasia has immune defects(OMIM 300291) and diminished or absent sweat glands, thin and sparse hair, and hy-podontia. The subset with immune defects is genetically distinct from forms withoutimmune defects. The most common immune defect is hypogammaglobulinemia.71,72
The X-linked recessive form is caused by mutations in the IKBKG (also termed NEMO)gene, which is involved in nuclear factor-kB regulation.71,72 An autosomal form causedby mutations in the NFKBIA gene has been described.73
Other Syndromic Immunodeficiencies Associated with Cutaneous Abnormalities
Incontinentia pigmentiLinear erythematous vesiculobullous lesions that evolve into hyperpigmented swirlingmacules on the trunk and proximal extremities are typical findings for this X-linkeddominant neurocutaneous disorder with fetal lethality in most affected male patients(OMIM 308300). Other findings include mental retardation, seizures, alopecia, ocularabnormalities, nail dystrophy, and malformed teeth. In a review of 77 cases, 13%had significant infection, and 4 died of infectious causes.74 No consistent immunologicabnormality has been detected, but decreased neutrophil chemotaxis and impairedproliferative response to phytohemagglutinin have been described.75,76 A girl hadtransient immunodeficiency that resolved, likely because of progressive selectionagainst cells carrying an active mutated X chromosome.77 Mutations in the geneIKBKG, also termed NEMO, cause incontinentia pigmenti.78 The protein is involvedin the regulation of the transcriptional regulator nuclear factor-kB. Mutations in thisgene cause other forms of ectodermal dysplasia associated with immune defects: hy-pohidrotic ectodermal dysplasia and immunodeficiency, a primary immunodeficiency,and OLEDAID syndrome (see following discussion).
OLEDAID syndromeTwo male patients with osteopetrosis, lymphedema, ectodermal dysplasia, anhidrotictype, and immune deficiency (OLEDAID, OMIM 300301), were born from mothers withmild incontinentia pigmenti.72 Both had multiple infections and died from infectious

Ming & Stiehm724
causes. The inflammatory response was poor, and isohemagglutinin titers and titers toPneumococcus (despite documented infection) were decreased. Both patients hada mutation converting a stop codon to a tryptophan in IKBKG.72
Dyskeratosis congenitaDyskeratosis congenita (OMIM 305000) is an X-linked disorder marked by reticulateskin pigmentation, nail dystrophy, leukoplakia of the oral mucosa, aplastic anemia,and an increased risk of malignancy. Progressive bone marrow failure develops inmost patients and is the major cause of early mortality. Neutropenia occurs in mostpatients, and humoral and cellular immune responses may be defective.79,80 Thymicaplasia was reported in two patients.81 The gene that causes dyskeratosis congenita(DKC1) codes for a protein that is predicted to function in ribosome formation.82 Mu-tations in this gene also cause Høyeraal-Hreidarsson syndrome (see later discussion).A less common autosomal dominant form has been described (OMIM 127550) andcan be caused by mutations in TERC,83 TERT,84 or TINF2.85 These gene productsare involved with telomere regulation.
Acrodermatitis enteropathicaAcrodermatitis enteropathica (OMIM 201100), an autosomal recessive disorder char-acterized by diarrhea, dermatitis, and alopecia, is caused by inadequate zinc metab-olism. Severe infection with opportunistic pathogens occurs frequently, and recurrentinfection occurs in 30% of cases.86 Decreased response to phytohemagglutinin andabnormal delayed cutaneous hypersensitivity skin response are typical.87 Hypogam-maglobulinemia and defective chemotaxis of neutrophils and monocytes are variablypresent.86,88 Clinical and immunologic abnormalities resolve after normalization ofserum zinc levels. Mutations in the gene encoding the intestinal zinc transporterSLC39A4 have been detected.89
Netherton syndromeThe triad of trichorrhexis (brittle ‘‘bamboo’’ hair), ichthyosiform erythroderma, andatopic diathesis make up Netherton syndrome (OMIM 256500), an autosomal reces-sive disorder. Recurrent infections occur in 28% of patients, most commonly involvingthe skin.90,91 IgG abnormalities (hypo- and hyper-IgG) are present in 12% to 14% ofpatients. Impairment of delayed cutaneous hypersensitivity response, mitogen re-sponse, and neutrophil phagocytosis can occur. Increased IgE is found in 10%.92 Mu-tations in the gene SPINK5, which encodes a serine protease inhibitor, have beendetected in affected patients.93
Syndromes Associated with Neurologic Dysfunction
Neurologic abnormalities ranging from structural abnormalities to epilepsy or ataxiahave been reported in association with immunodeficiency (see Table 2).
Myotonic Dystrophy
This autosomal dominant condition (OMIM 160900) is a multisystem disorder charac-terized by difficulty in relaxing a contracted muscle. Muscle weakness and wasting,cataracts, hypogonadism, and cardiac conduction defects are also frequent manifes-tations. Cognitive function may deteriorate in adults. In the congenital form, there issevere hypotonia and respiratory insufficiency.
Most cases of myotonic dystrophy are caused by a trinucleotide repeat expansionin the 30 untranslated region of the DMPK gene, which encodes the dystrophia myo-tonica protein kinase.94–96 In general, the size of the expansion correlates with the se-verity of the disease and the age of onset. A large family with features typical of

Genetic Syndromic Immunodeficiencies 725
myotonic dystrophy did not have the repeat expansion in the DMPK gene 97 but had anexpansion in a CCTG repeat in intron one of the ZNF9 gene.98
The most common immunologic abnormality in affected patients is a reduction inIgG level,99 although decreased IgA and IgM levels occasionally have been noted. In-creased repeat length was found to correlate with decreased serum IgG level, de-creased total lymphocyte count, and low T-cell numbers in one study,100 butanother study found no correlation.101 There is generally no increased susceptibilityto infection.102
Høyeraal-Hreidarsson Syndrome
A syndrome of X-linked cerebellar hypoplasia, psychomotor retardation, microceph-aly, growth failure, and progressive pancytopenia has been reported in several af-fected male patients (OMIM 300240). Decreased IgG103 and death from fungalsepsis104,105 have been described. Progressive combined deficiency has been notedin other patients.106,107 This condition is caused by mutations in the DKC1 gene, thesame gene that is mutated in dyskeratosis congenita.106
INBORN ERRORS OFMETABOLISM ASSOCIATEDWITH IMMUNODEFICIENCYCongenital Disorders of Glycosylation, Type I
Congenital disorders of glycosylation (CDG), also known as carbohydrate-deficientglycoprotein syndromes (CDGS), are autosomal recessive disorders characterizedby decreased glycosylation of glycoproteins. In type I CDG, there is a defect in the pro-duction of lipid-linked oligosaccharides or their transfer to nascent proteins. Hypoto-nia and poor growth are present, and other organ system involvement is often present,depending on the type of CDG (see Table 3). Type Ia CDG (OMIM 212065) is causedby a defect in phosphomannomutase 2;108 abnormal fat distribution is characteristic.Severe infections often occur, and decreased IgA or IgG levels, defective response tovaccines, and diminished neutrophil chemotaxis have been observed.109 Type Ig CDG(OMIM 607143) is caused by a defect in the gene that encodes a mannosyltransferase(ALG12).110–112 Microcephaly and male genital hypoplasia are characteristic. Recur-rent infections and decreased IgG levels often occur.110 A short-limb skeletal dyspla-sia was noted in two affected siblings.113 Type Ik CDG (OMIM 608540) is caused bya defect in mannosyltransferase I (ALG1 gene);114–116 refractory seizures, microceph-aly, and early death are characteristic. An affected patient was noted to have de-creased B-cell numbers and absence of IgG.114
Branched Chain Amino Acidurias
Three diseases that affect branched chain amino acid metabolism are associated withleukopenia: methylmalonic acidemia (OMIM 251000), propionic acidemia (OMIM232000), and isovaleric acidemia (OMIM 243500).117–119 The conditions present withmetabolic acidosis, lethargy, failure to thrive, and recurrent vomiting. These individualsare at increased risk for infection, which may precipitate episodes of acidosis. De-creases in B-cell numbers and immunoglobulin levels have been reported.120–122
Lysinuric Protein Intolerance
This condition (OMIM 222700) is marked by defective transport of the dibasic aminoacids lysine, arginine, and ornithine in the intestine and renal tubules, leading to de-creased levels of these substances in the blood, hyperammonemia, protein intoler-ance, and failure to thrive. Decreases in CD4 T-cell numbers,123 lymphopenia,124
IgG subclass deficiency and poor humoral response to vaccination,125 and leukopenia

Ming & Stiehm726
with decreased leukocyte phagocytic activity126 have been reported. Varicella infec-tion may be severe.127
SYNDROMESWITHCHROMOSOME INSTABILITYAND/ORDEFECTIVEDNA REPAIR ASSOCIATEDWITH IMMUNODEFICIENCY
Syndromes associated with chromosome instability often have immune abnormalities,and such patients are often at increased risk for malignancy (see Table 4).
Primary Immunodeficiencies Associated with Chromosome Instabilityand/or Defective DNA Repair
Nijmegen breakage syndromePatients who have Nijmegen breakage syndrome (OMIM 251260) have short stature,microcephaly, and bird-like facies.128 Characteristic facial features include a recedingforehead, prominent midface with a long nose, large ears, and micrognathia. Mentalretardation may occur. There is an increased risk of malignancy, especially lymphoma.Cells from patients who have Nijmegen breakage syndrome are sensitive to ionizingirradiation. Bronchopneumonia and urinary tract infections commonly occur, andthere is an increased risk of otitis media, mastoiditis, and sinusitis. Patients generallyhave abnormal immunoglobulin levels, most commonly including IgG (especially IgG2and IgG4), and may have agammaglobulinemia.129 Reduced CD31 and CD41 cellnumbers with a decreased CD4/CD8 ratio have been noted. A markedly decreasedproliferative response to T-cell mitogens was noted in 94% of patients. Mutations inthe NBS1 gene (also termed Nibrin or p95), which encodes a subunit of the Rad50/Mre11 protein complex involved in double-stranded break repair, were detected inpatients who have Nijmegen breakage syndrome.130,131
Bloom syndromeThis autosomal recessive condition (OMIM 210900) is characterized by growth failure,hypersensitivity to sunlight, and characteristic facial features (malar hypoplasia, mi-crognathia, and prominent ears). Neoplasia, especially leukemia and lymphoma, isgreatly increased and is the most frequent cause of death.132 The diagnosis may beestablished by the finding of an increased number of sister chromatid exchanges incells grown in medium with bromo-deoxyuridine. There is an increased susceptibilityto infection, especially pneumonia and otitis media. Immunologic defects may involvehumoral and cellular responses.133 The product of the BLM gene encodes a RecQDNA helicase that is involved in DNA duplex unwinding and may interact with topoiso-merases or other proteins involved in DNA repair.134
Other Syndromic Immunodeficiencies Associated with Chromosome Instabilityand/or Defective DNA Repair
Immunodeficiency, centromeric instability, and facial anomalies syndromeThis autosomal recessive condition (ICF, OMIM 242860) is comprised of immunode-ficiency, centromeric instability (involving chromosomes 1 and 16, often 9, rarely 2and 10), and facial anomalies (ocular hypertelorism, flat nasal bridge).135,136 Mental re-tardation is frequent. Deletions, breaks, interchanges between homologous and non-homologous chromosomes, and multibranched configurations involving pericentricheterochromatin have been described. The ICF syndrome differs from many otherchromosome instability syndromes in that no hypersensitivity to clastogenic agentshas been demonstrated; hence, it is not a chromosome breakage syndrome.
Severe chronic sinopulmonary, gastrointestinal, and cutaneous infections occur.Generally, at least two immunoglobulin classes are affected in each patient.136,137

Genetic Syndromic Immunodeficiencies 727
Immunoglobulin supplementation can improve the course of the disease;138 T-cellnumbers and lymphoproliferative response to mitogen may be decreased.137,139 Mu-tations in the gene encoding the DNA methyltransferase DNMT3B have been identi-fied;140,141 however, other patients diagnosed with ICF with centromeric instabilityof chromosomes 1 and 16 do not have identified DNMT3B mutations.142,143 Ofnote, the patient reported by Braegger and colleagues144 (OMIM 243340) with intra-uterine growth deficiency, ischiadic hypoplasia, microcephaly, renal dysfunction,cryptorchidism, postaxial polydactyly, and hypogammaglobulinemia was subse-quently diagnosed with ICF.142
SYNDROMES ASSOCIATEDWITH CHROMOSOMAL ABNORMALITIESOF NUMBER OR STRUCTURETrisomy 21
Down syndrome (OMIM 190685) results from trisomy 21 and is associated with mentalretardation, cardiac defects, gastrointestinal abnormalities, leukemia, and early-onsetAlzheimer disease. Affected individuals can experience significant morbidity and mor-tality because of infections, especially respiratory infections (see Table 5).145 Althoughmost individuals do not have clear immune dysfunction, several immunologic abnor-malities have been noted. B lymphocyte counts are often low throughout childhood,and the T lymphocyte count may be low in the first 15 months of life, although thesecounts may normalize with time.146 No relationship between the lymphocyte subpop-ulation sizes and the frequency of infections was detected. Decreased B-cell numbersand low specific antibody response have been reported.145,147 Proliferation in re-sponse to phytohemagglutinin and alloantigens, delayed cutaneous hypersensitivityresponse, and T-cell–mediated killing is variably reduced.145,148 Total NK cell numberis increased but the activity is decreased.148,149 Phagocyte number is normal, but che-motaxis and oxidative metabolism—and hence killing—are impaired.150 There is anincreased incidence of autoimmune conditions.151 Proliferation and interleukin-2 pro-duction in response to phytohemagglutinin were decreased in adult men who hadDown syndrome.152
Partial Deletions of Chromosome 4p
Patients with partial deletions of chromosome 4p or Wolf-Hirschhorn syndrome(OMIM 194190) have prenatal-onset growth deficiency, mental retardation, micro-cephaly, ocular hypertelorism, coloboma of the iris, and seizures.153 The critical regionhas been narrowed to 165 kb on 4p16.3,154 and a second critical region has been pro-posed.155 Patients have frequent episodes of respiratory infections, partly because ofrecurrent aspiration, but antibody deficiencies are also common. Immune defects in-clude common variable immunodeficiency, IgA and IgG2 subclass deficiency, IgA de-ficiency, and impaired polysaccharide responsiveness.156 T-cell immunity is normal.Immunodeficiency does not seem to correlate with deletion size, and all of these pa-tients were deleted for the 4p16.3 critical region. This region likely contains a gene orgenes critical for B-cell function.
Turner Syndrome
Patients with a missing or structurally abnormal X chromosome often present withshort stature, shield chest, congenital lymphedema, and ovarian dysgenesis. The syn-drome is associated with an increased risk for upper respiratory and ear infections, au-toimmunity, and occasional neoplasia. IgG, IgM, and IgA levels may be abnormal.157
Decreased T-cell numbers with poor response to phytohemagglutinin, absent delayedcutaneous hypersensitivity reactions, and common variable immunodeficiency

Ming & Stiehm728
occasionally occur.158–161 The relationship, if any, between the immune defects inTurner syndrome and the X-linked primary immunodeficiencies is unknown.
REFERENCES
1. Geha RS, Notarangelo LD, Casanova JL, et al. Primary immunodeficiencydiseases: an update from the International Union of Immunological SocietiesPrimary Immunodeficiency Diseases Classification Committee. J Allergy ClinImmunol 2007;120:776–94.
2. Mar�odi L, Notarangelo LD. Immunological and genetic bases of new primaryimmunodeficiencies. Nat Rev Immunol 2007;7:851–61.
3. Notarangelo L, Casanova JL, Conley ME, et al. Primary immunodeficiency dis-eases: an update from the International Union of Immunological Societies PrimaryImmunodeficiency Diseases Classification Committee Meeting in Budapest,2005. J Allergy Clin Immunol 2006;117:883–96.
4. Ming JE, Stiehm ER, Graham JM Jr. Syndromic immunodeficiencies: geneticsyndromes associated with immune abnormalities. Crit Rev Clin Lab Sci 2003;40:587–642.
5. OMIM: Online Mendelian Inheritance in Man. OMIM (TM). McKusick-NathansInstitute for Genetic Medicine, Johns Hopkins University (Baltimore, MD) andNational Center for Biotechnology Information, National Library of Medicine(Bethesda, MD), 2006. Available at: http://www.ncbi.nlm.nih.gov/omim/.Accessed May–June, 2008.
6. Saraiva JM, Dinis A, Resende C, et al. Schimke immuno-osseous dysplasia:case report and review of 25 patients. J Med Genet 1999;36:786–9.
7. Boerkoel CF, O’Neill S, Andre JL, et al. Manifestations and treatment of Schimkeimmuno-osseous dysplasia: 14 new cases and a review of the literature. EurJ Pediatr 2000;159:1–7.
8. Boerkoel CF, Takashima H, John J, et al. Mutant chromatin remodeling proteinSMARCAL1 causes Schimke immuno-osseous dysplasia. Nat Genet 2002;30:215–20.
9. Clewing JM, Antalfy BC, Lucke T, et al. Schimke immuno-osseous dysplasia:a clinicopathological correlation. J Med Genet 2007;44:122–30.
10. Roifman CM. Antibody deficiency, growth retardation, spondyloepiphyseal dys-plasia and retinal dystrophy: a novel syndrome. Clin Genet 1999;55:103–9.
11. Robertson SP, Rodda C, Bankier A. Hypogonadotrophic hypogonadism in Roif-man syndrome. Clin Genet 2000;57:435–8.
12. Roifman CM, Melamed I. A novel syndrome of combined immunodeficiency,autoimmunity and spondylometaphyseal dysplasia. Clin Genet 2003;63:522–9.
13. Kulkarni ML, Baskar K, Kulkarni PM. A syndrome of immunodeficiency, autoim-munity, and spondylometaphyseal dysplasia. Am J Med Genet A 2007;143:69–75.
14. Renella R, Schaefer E, LeMerrer M, et al. Spondyloenchondrodysplasia withspasticity, cerebral calcifications, and immune dysregulation: clinical and radio-graphic delineation of a pleiotropic disorder. Am J Med Genet A 2006;140:541–50.
15. Fleisher TA, White RM, Broder S, et al. X-linked hypogammaglobulinemia andisolated growth hormone deficiency. N Engl J Med 1980;302:1429–34.
16. Duriez B, Duquesnoy P, Dastot F, et al. An exon-skipping mutation in the BTKgene of a patient with X-linked agammaglobulinemia and isolated growth hor-mone deficiency. FEBS Lett 1994;346:165–70.

Genetic Syndromic Immunodeficiencies 729
17. Abo K, Nishio H, Lee MJ, et al. A novel single basepair insertion in exon 6 of theBruton’s tyrosine kinase (Btk) gene from a Japanese X-linked agammaglobulin-emia patient with growth hormone insufficiency. Hum Mutat 1998;11:336.
18. Stewart DM, Notarangelo LD, Kurman CC, et al. Molecular genetic analysis ofX-linked hypogammaglobulinemia and isolated growth hormone deficiency.J Immunol 1995;155:2770–4.
19. Tang ML, Kemp AS. Growth hormone deficiency and combined immunodefi-ciency. Arch Dis Child 1993;68:231–2.
20. Maghnie M, Monafo V, Marseglia GL, et al. Immunodeficiency, growth hormone de-ficiency and central nervous system involvement in a girl. Thymus 1992;20:69–76.
21. Kiess W, Malozowski S, Gelato M, et al. Lymphocyte subset distribution and nat-ural killer activity in growth hormone deficiency before and during short-termtreatment with growth hormone releasing hormone. Clin Immunol Immunopathol1988;48:85–94.
22. Ohzeki T, Hanaki K, Motozumi H, et al. Immunodeficiency with increased immu-noglobulin M associated with growth hormone insufficiency. Acta Paediatr 1993;82:620–3.
23. Church JA, Costin G, Brooks J. Immune functions in children treated with bio-synthetic growth hormone. J Pediatr 1989;115:420–3.
24. Spadoni GL, Rossi P, Ragno W, et al. Immune function in growth hormone-defi-cient children treated with biosynthetic growth hormone. Acta Paediatr Scand1991;80:75–9.
25. Bernasconi A, Marino R, Ribas A, et al. Characterization of immunodeficiency ina patient with growth hormone insensitivity secondary to a novel STAT5b genemutation. Pediatrics 2006;118:e1584–92.
26. Niikawa N, Kuroki Y, Kajii T, et al. Kabuki make-up (Niikawa-Kuroki) syndrome:a study of 62 patients. Am J Med Genet 1988;31:565–89.
27. Wilson GN. Thirteen cases of Niikawa-Kuroki syndrome: report and review withemphasis on medical complications and preventive management. Am J MedGenet 1998;79:112–20.
28. Chrzanowska KH, Krajewska-Walasek M, Kus J, et al. Kabuki (Niikawa-Kuroki)syndrome associated with immunodeficiency. Clin Genet 1998;53:308–12.
29. Hostoffer RW, Bay CA, Wagner K, et al. Kabuki make-up syndrome associatedwith an acquired hypogammaglobulinemia and anti-IgA antibodies. Clin Pediatr(Phila) 1996;35:273–6.
30. Hoffman JD, Ciprero KL, Sullivan KE, et al. Immune abnormalities are a frequentmanifestation of Kabuki syndrome. Am J Med Genet A 2005;135:278–81.
31. Kawame H, Hannibal MC, Hudgins L, et al. Phenotypic spectrum and manage-ment issues in Kabuki syndrome. J Pediatr 1999;134:480–5.
32. Ming JE, Russell KL, McDonald-McGinn DM, et al. Autoimmune disorders inKabuki syndrome. Am J Med Genet 2005;32A(3):260–2.
33. Pagon RA, Graham JM, Zonana J, et al. Coloboma, congenital heart disease,and choanal atresia with multiple anomalies: CHARGE association. J Pediatr1981;99:223–7.
34. Tellier AL, Cormier-Daire V, Abadie V, et al. CHARGE syndrome: report of 47cases and review. Am J Med Genet 1998;76:402–9.
35. Graham JM. A recognizable syndrome within CHARGE association: Hall-Hittnersyndrome. Am J Med Genet 2001;99:120–3.
36. Vissers LE, van Ravenswaaij CM, Admiraal R, et al. Mutations in a new memberof the chromodomain gene family cause CHARGE syndrome. Nat Genet 2004;36:955–7.

Ming & Stiehm730
37. Lalani SR, Safiullah AM, Fernbach SD, et al. Spectrum of CHD7 mutations in 110individuals with CHARGE syndrome and genotype-phenotype correlation. AmJ Hum Genet 2006;78:303–14.
38. Boudny P, Kurrer MO, Stamm B, et al. Malakoplakia of the colon in an infant withsevere combined immunodeficiency (SCID) and CHARGE association. PatholRes Pract 2000;196:577–82.
39. Theodoropoulos DS. Immune deficiency in CHARGE association. Clin Med Res2003;1:43–8.
40. de Lonlay-Debeney P, Cormier-Daire V, Amiel J, et al. Features of DiGeorge syn-drome and CHARGE association in five patients. J Med Genet 1997;34:986–9.
41. Wood DJ, David TJ, Chrystie IL, et al. Chronic enteric virus infection in two T-cellimmunodeficient children. J Med Virol 1988;24:435–44.
42. Rivas F, Fragoso R, Ramos-Zepeda R, et al. Deficient cell immunity and mildintermittent hyperaminoacidemia in a patient with the Rubinstein-Taybi syn-drome. Acta Paediatr Scand 1980;69:123–5.
43. Kimura H, Ito Y, Koda Y, et al. Rubinstein-Taybi syndrome with thymic hypopla-sia. Am J Med Genet 1993;46:293–6.
44. Villella A, Bialostocky D, Lori E, et al. Rubinstein-Taybi syndrome with humoraland cellular defects: a case report. Arch Dis Child 2000;83:360–1.
45. Naimi DR, Munoz J, Rubinstein J, et al. Rubinstein-Taybi syndrome: an immune de-ficiency as a cause for recurrent infections. Allergy Asthma Proc 2006;27:281–4.
46. Petrij F, Giles RH, Dauwerse HG, et al. Rubinstein-Taybi syndrome caused bymutations in the transcriptional co-activator CBP. Nature 1995;376:348–51.
47. Petrij F, Dauwerse HG, Blough RI, et al. Diagnostic analysis of the Rubinstein-Taybi syndrome: five cosmids should be used for microdeletion detection andlow number of protein truncating mutations. J Med Genet 2000;37:168–76.
48. Roelfsema JH, White SJ, Ariyurek Y, et al. Genetic heterogeneity in Rubinstein-Taybi syndrome: mutations in both the CBP and EP300 genes cause disease.Am J Hum Genet 2005;76:572–80.
49. Mulvihill JJ, Smith DW. Another disorder with prenatal shortness of stature andpremature aging. Birth Defects Orig Artic Ser 1975;11:368–70.
50. de Silva DC, Wheatley DN, Herriot R, et al. Mulvihill-Smith progeria-like syndrome:a further report with delineation of phenotype, immunologic deficits, and novelobservation of fibroblast abnormalities. Am J Med Genet 1997;69:56–64.
51. Ohashi H, Tsukahara M, Murano I, et al. Premature aging and immunodefi-ciency: Mulvihill-Smith syndrome? Am J Med Genet 1993;45:597–600.
52. Bartsch O, Tympner KD, Schwinger E, et al. Mulvihill-Smith syndrome: casereport and review. J Med Genet 1994;31:707–11.
53. Moreno LA, Gottrand F, Turck D, et al. Severe combined immunodeficiency syn-drome associated with autosomal recessive familial multiple gastrointestinalatresias: study of a family. Am J Med Genet 1990;37:143–6.
54. Walker MW, Lovell MA, Kelly TE, et al. Multiple areas of intestinal atresia associ-ated with immunodeficiency and posttransfusion graft-versus-host disease.J Pediatr 1993;123:93–5.
55. Rothenberg ME, White FV, Chilmonczyk B, et al. A syndrome involving immuno-deficiency and multiple intestinal atresias. Immunodeficiency 1995;5:171–8.
56. Snyder CL, Mancini ML, Kennedy AP, et al. Multiple gastrointestinal atresias withcystic dilatation of the biliary duct. Pediatr Surg Int 2000;16:211–3.
57. Gilroy RK, Coccia PF, Talmadge JE, et al. Donor immune reconstitution afterliver-small bowel transplantation for multiple intestinal atresia with immunodefi-ciency. Blood 2004;103:1171–4.

Genetic Syndromic Immunodeficiencies 731
58. Moore SW, de Jongh G, Bouic P, et al. Immune deficiency in familial duodenalatresia. J Pediatr Surg 1996;31:1733–5.
59. Girault D, Goulet O, Le Deist F, et al. Intractable infant diarrhea associated withphenotypic abnormalities and immunodeficiency. J Pediatr 1994;125:36–42.
60. Landers MC, Schroeder TL. Intractable diarrhea of infancy with facial dysmor-phism, trichorrhexis nodosa, and cirrhosis. Pediatr Dermatol 2003;20:432–5.
61. Fabre A, Andre N, Breton A, et al. Intractable diarrhea with ‘‘phenotypic anom-alies’’ and tricho-hepato-enteric syndrome: two names for the same disorder.Am J Med Genet A 2007;143:584–8.
62. Griscelli C, Durandy A, Guy-Grand D, et al. A syndrome associating partial albi-nism and immunodeficiency. Am J Med 1978;65:691–702.
63. Mancini AJ, Chan LS, Paller AS. Partial albinism with immunodeficiency: Griscellisyndrome. Report of a case and review of the literature. J Am Acad Dermatol1998;38:295–300.
64. Dufourcq-Lagelouse R, Pastural E, Barrat FJ, et al. Genetic basis of hemopha-gocytic lymphohistiocytosis syndrome [review]. Int J Mol Med 1999;4:127–33.
65. Menasche G, Pastural E, Feldmann J, et al. Mutations in RAB27A cause Griscellisyndrome associated with haemophagocytic syndrome. Nat Genet 2000;25:173–6.
66. Pastural E, Barrat FJ, Dufourcq-Lagelouse R, et al. Griscelli disease maps tochromosome 15q21 and is associated with mutations in the myosin-Va gene.Nat Genet 1997;16:289–92.
67. Villa A, Santagata S, Bozzi F, et al. Partial V(D)J recombination activity leads toOmenn syndrome. Cell 1998;93:885–96.
68. Ege M, Ma Y, Manfras B, et al. Omenn syndrome due to ARTEMIS mutations.Blood 2005;105:4179–86.
69. Gorlin RJ, Gelb B, Diaz GA, et al. WHIM syndrome, an autosomal dominant dis-order: clinical, hematological, and molecular studies. Am J Med Genet 2000;91:368–76.
70. Hernandez PA, Gorlin RJ, Lukens JN, et al. Mutations in the chemokine receptorgene CXCR4 are associated with WHIM syndrome, a combined immunodefi-ciency disease. Nat Genet 2003;34:70–4.
71. Zonana J, Elder ME, Schneider LC, et al. A novel X-linked disorder of immunedeficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pig-menti and due to mutations in IKK-gamma (NEMO). Am J Hum Genet 2000;67:1555–62.
72. Doffinger R, Smahi A, Bessia C, et al. X-linked anhidrotic ectodermal dysplasiawith immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet2001;27:277–85.
73. Courtois G, Smahi A, Reichenbach J, et al. A hypermorphic IkappaBalpha mu-tation is associated with autosomal dominant anhidrotic ectodermal dysplasiaand T cell immunodeficiency. J Clin Invest 2003;112:1108–15.
74. Diamantopoulos N, Bergman I, Kaplan S. Actinomycosis meningitis in a girl withincontinentia pigmenti. Clin Pediatr (Phila) 1985;24:651–4.
75. Menni S, Piccinno R, Biolchini A, et al. Immunologic investigations in eightpatients with incontinentia pigmenti. Pediatr Dermatol 1990;7:275–7.
76. Jessen RT, Van Epps DE, Goodwin JS, et al. Incontinentia pigmenti: evidencefor both neutrophil and lymphocyte dysfunction. Arch Dermatol 1978;114:1182–6.
77. Martinez-Pomar N, Munoz-Saa I, Heine-Suner D, et al. A new mutation in exon 7of NEMO gene: late skewed X-chromosome inactivation in an incontinentia pig-menti female patient with immunodeficiency. Hum Genet 2005;118:458–65.

Ming & Stiehm732
78. Smahi A, Courtois G, Vabres P, et al. Genomic rearrangement in NEMO impairsNF-kappaB activation and is a cause of incontinentia pigmenti. The InternationalIncontinentia Pigmenti (IP) Consortium. Nature 2000;405:466–72.
79. Solder B, Weiss M, Jager A, et al. Dyskeratosis congenita: multisystemic disor-der with special consideration of immunologic aspects. A review of the literature.Clin Pediatr (Phila) 1998;37:521–30.
80. Dokal I. Dyskeratosis congenita in all its forms. Br J Haematol 2000;110:768–79.81. Trowbridge AA, Sirinavin C, Linman JW. Dyskeratosis congenita: hematologic
evaluation of a sibship and review of the literature. Am J Hematol 1977;3:143–52.82. Heiss NS, Knight SW, Vulliamy TJ, et al. X-linked dyskeratosis congenita is
caused by mutations in a highly conserved gene with putative nucleolar func-tions. Nat Genet 1998;19:32–8.
83. Vulliamy T, Marrone A, Goldman F, et al. The RNA component of telomerase is mu-tated in autosomal dominant dyskeratosis congenita. Nature 2001;413:432–5.
84. Armanios M, Chen JL, Chang YP, et al. Haploinsufficiency of telomerase reversetranscriptase leads to anticipation in autosomal dominant dyskeratosis congenita.Proc Natl Acad Sci U S A 2005;102:15960–4.
85. Savage SA, Giri N, Baerlocher GM, et al. TINF2, a component of the shelterintelomere protection complex, is mutated in dyskeratosis congenita. Am J HumGenet 2008;82:501–9.
86. Van Wouwe JP. Clinical and laboratory diagnosis of acrodermatitis enteropathica.Eur J Pediatr 1989;149:2–8.
87. Oleske JM, Westphal ML, Shore S, et al. Zinc therapy of depressed cellular im-munity in acrodermatitis enteropathica: its correction. Am J Dis Child 1979;133:915–8.
88. Weston WL, Huff JC, Humbert JR, et al. Zinc correction of defective chemotaxisin acrodermatitis enteropathica. Arch Dermatol 1977;113:422–5.
89. Kury S, Dreno B, Bezieau S, et al. Identification of SLC39A4, a gene involved inacrodermatitis enteropathica. Nat Genet 2002;31:239–40.
90. Greene SL, Muller SA. Netherton’s syndrome: report of a case and review of theliterature. J Am Acad Dermatol 1985;13:329–37.
91. Stryk S, Siegfried EC, Knutsen AP. Selective antibody deficiency to bacterialpolysaccharide antigens in patients with Netherton syndrome. Pediatr Dermatol1999;16:19–22.
92. Smith DL, Smith JG, Wong SW, et al. Netherton’s syndrome: a syndrome of ele-vated IgE and characteristic skin and hair findings. J Allergy Clin Immunol 1995;95:116–23.
93. Chavanas S, Bodemer C, Rochat A, et al. Mutations in SPINK5, encoding a ser-ine protease inhibitor, cause Netherton syndrome. Nat Genet 2000;25:141–2.
94. Brook JD, McCurrach ME, Harley HG, et al. Molecular basis of myotonic dystro-phy: expansion of a trinucleotide (CTG) repeat at the 30 end of a transcriptencoding a protein kinase family member. Cell 1992;68:799–808.
95. Mahadevan M, Tsilfidis C, Sabourin L, et al. Myotonic dystrophy mutation: anunstable CTG repeat in the 30 untranslated region of the gene. Science 1992;255:1253–5.
96. Fu YH, Pizzuti A, Fenwick RG Jr, et al. An unstable triplet repeat in a gene relatedto myotonic muscular dystrophy. Science 1992;255:1256–8.
97. Ranum LP, Rasmussen PF, Benzow KA, et al. Genetic mapping of a secondmyotonic dystrophy locus. Nat Genet 1998;19:196–8.
98. Liquori CL, Ricker K, Moseley ML, et al. Myotonic dystrophy type 2 caused bya CCTG expansion in intron 1 of ZNF9. Science 2001;293:864–7.

Genetic Syndromic Immunodeficiencies 733
99. Wochner RD, Drews G, Strober W, et al. Accelerated breakdown of immunoglob-ulin G (IgG) in myotonic dystrophy: a hereditary error of immunoglobulin catab-olism. J Clin Invest 1966;45:321–9.
100. Nakamura A, Kojo T, Arahata K, et al. Reduction of serum IgG level and periph-eral T-cell counts are correlated with CTG repeat lengths in myotonic dystrophypatients. Neuromuscul Disord 1996;6:203–10.
101. Pan-Hammarstrom Q, Wen S, Ghanaat-Pour H, et al. Lack of correlationbetween the reduction of serum immunoglobulin concentration and the CTGrepeat expansion in patients with type 1 dystrophia correction of Dystrofiamyotonica. J Neuroimmunol 2003;144:100–4.
102. Suzumura A, Yamada H, Matsuoka Y, et al. Immunoglobulin abnormalities inpatients with myotonic dystrophy. Acta Neurol Scand 1986;74:132–9.
103. Hoyeraal HM, Lamvik J, Moe PJ. Congenital hypoplastic thrombocytopenia andcerebral malformations in two brothers. Acta Paediatr Scand 1970;59:185–91.
104. Hreidarsson S, Kristjansson K, Johannesson G, et al. A syndrome of progressivepancytopenia with microcephaly, cerebellar hypoplasia and growth failure. ActaPaediatr Scand 1988;77:773–5.
105. Berthet F, Caduff R, Schaad UB, et al. A syndrome of primary combined immu-nodeficiency with microcephaly, cerebellar hypoplasia, growth failure and pro-gressive pancytopenia. Eur J Pediatr 1994;153:333–8.
106. Knight SW, Heiss NS, Vulliamy TJ, et al. Unexplained aplastic anaemia, immu-nodeficiency, and cerebellar hypoplasia (Hoyeraal-Hreidarsson syndrome)due to mutations in the dyskeratosis congenita gene, DKC1. Br J Haematol1999;107:335–9.
107. Sznajer Y, Baumann C, David A, et al. Further delineation of the congenital formof X-linked dyskeratosis congenita (Hoyeraal-Hreidarsson syndrome). EurJ Pediatr 2003;162:863–7.
108. Matthijs G, Schollen E, Pardon E, et al. Mutations in PMM2, a phosphomannomu-tase gene on chromosome 16p13, in carbohydrate-deficient glycoprotein type Isyndrome (Jaeken syndrome). Nat Genet 1997;16:88–92.
109. Blank C, Smith LA, Hammer DA, et al. Recurrent infections and immunologicaldysfunction in congenital disorder of glycosylation Ia (CDG Ia). J Inherit MetabDis 2006;29:592.
110. Chantret I, Dupre T, Delenda C, et al. Congenital disorders of glycosylation typeIg is defined by a deficiency in dolichyl-P-mannose:Man7GlcNAc2-PP-dolichylmannosyltransferase. J Biol Chem 2002;277:25815–22.
111. Grubenmann CE, Frank CG, Kjaergaard S, et al. ALG12 mannosyltransferase de-fect in congenital disorder of glycosylation type lg. Hum Mol Genet 2002;11:2331–9.
112. Thiel C, Schwarz M, Hasilik M, et al. Deficiency of dolichyl-P-Man:Man7Glc-NAc2-PP-dolichyl mannosyltransferase causes congenital disorder of glycosyl-ation type Ig. Biochem J 2002;367:195–201.
113. Kranz C, Basinger AA, Gucsavas-Calikoglu M, et al. Expanding spectrum ofcongenital disorder of glycosylation Ig (CDG-Ig): Sibs with a unique skeletaldysplasia, hypogammaglobulinemia, cardiomyopathy, genital malformations,and early lethality. Am J Med Genet A 2007;143:1371–8.
114. Kranz C, Denecke J, Lehle L, et al. Congenital disorder of glycosylation type Ik(CDG-Ik): a defect of mannosyltransferase I. Am J Hum Genet 2004;74:545–51.
115. Grubenmann CE, Frank CG, Hulsmeier AJ, et al. Deficiency of the first manno-sylation step in the N-glycosylation pathway causes congenital disorder of gly-cosylation type Ik. Hum Mol Genet 2004;13:535–42.

Ming & Stiehm734
116. Schwarz M, Thiel C, Lubbehusen J, et al. Deficiency of GDP-Man:GlcNAc2-PP-dolichol mannosyltransferase causes congenital disorder of glycosylationtype Ik. Am J Hum Genet 2004;74:472–81.
117. Matsui SM, Mahoney MJ, Rosenberg LE. The natural history of the inheritedmethylmalonic acidemias. N Engl J Med 1983;308:857–61.
118. Muller S, Falkenberg N, Monch E, et al. Propionic acidaemia and immunodefi-ciency. Lancet 1980;1:551–2.
119. Kelleher JF, Yudkoff M, Hutchinson R, et al. The pancytopenia of isovaleric acid-emia. Pediatrics 1980;65:1023–7.
120. Church JA, Koch R, Shaw KN, et al. Immune functions in methylmalonicaciduria.J Inherit Metab Dis 1984;7:12–4.
121. Wong SN, Low LC, Lau YL, et al. Immunodeficiency in methylmalonic acidae-mia. J Paediatr Child Health 1992;28:180–3.
122. Raby RB, Ward JC, Herrod HG. Propionic acidaemia and immunodeficiency.J Inherit Metab Dis 1994;17:250–1.
123. Dionisi-Vici C, De Felice L, el Hachem M, et al. Intravenous immune globulin inlysinuric protein intolerance. J Inherit Metab Dis 1998;21:95–102.
124. Nagata M, Suzuki M, Kawamura G, et al. Immunological abnormalities in apatient with lysinuric protein intolerance. Eur J Pediatr 1987;146:427–8.
125. Lukkarinen M, Parto K, Ruuskanen O, et al. B and T cell immunity in patients withlysinuric protein intolerance. Clin Exp Immunol 1999;116:430–4.
126. Yoshida Y, Machigashira K, Suehara M, et al. Immunological abnormality inpatients with lysinuric protein intolerance. J Neurol Sci 1995;134:178–82.
127. Lukkarinen M, Nanto-Salonen K, Ruuskanen O, et al. Varicella and varicellaimmunity in patients with lysinuric protein intolerance. J Inherit Metab Dis1998;21:103–11.
128. van der Burgt I, Chrzanowska KH, Smeets D, et al. Nijmegen breakage syn-drome. J Med Genet 1996;33:153–6.
129. Group. Nijmegen breakage syndrome: the International Nijmegen BreakageSyndrome Study Group. Arch Dis Child 2000;82:400–6.
130. Matsuura S, Tauchi H, Nakamura A, et al. Positional cloning of the gene forNijmegen breakage syndrome. Nat Genet 1998;19:179–81.
131. Varon R, Vissinga C, Platzer M, et al. Nibrin, a novel DNA double-strand break re-pair protein, is mutated in Nijmegen breakage syndrome. Cell 1998;93:467–76.
132. German J. Bloom’s syndrome: the first 100 cancers. Cancer Genet Cytogenet1997;93:100–6.
133. Kondo N, Motoyoshi F, Mori S, et al. Long-term study of the immunodeficiency ofBloom’s syndrome. Acta Paediatr 1992;81:86–90.
134. Ellis NA, Groden J, Ye TZ, et al. The Bloom’s syndrome gene product is homol-ogous to RecQ helicases. Cell 1995;83:655–66.
135. Tiepolo L, Maraschio P, Gimelli G, et al. Multibranched chromosomes 1, 9, and16 in a patient with combined IgA and IgE deficiency. Hum Genet 1979;51:127–37.
136. Maraschio P, Zuffardi O, Dalla Fior T, et al. Immunodeficiency, centromeric het-erochromatin instability of chromosomes 1, 9, and 16, and facial anomalies: theICF syndrome. J Med Genet 1988;25:173–80.
137. Smeets DF, Moog U, Weemaes CM, et al. ICF syndrome: a new case and reviewof the literature. Hum Genet 1994;94:240–6.
138. Hagleitner MM, Lankester A, Maraschio P, et al. Clinical spectrum of immunode-ficiency, centromeric instability and facial dysmorphism (ICF syndrome). J MedGenet 2008;45:93–9.

Genetic Syndromic Immunodeficiencies 735
139. Fasth A, Forestier E, Holmberg E, et al. Fragility of the centromeric region ofchromosome 1 associated with combined immunodeficiency in siblings. Arecessively inherited entity? Acta Paediatr Scand 1990;79:605–12.
140. Okano M, Bell DW, Haber DA, et al. DNA methyltransferases Dnmt3a andDnmt3b are essential for de novo methylation and mammalian development.Cell 1999;99:247–57.
141. Xu GL, Bestor TH, Bourc’his D, et al. Chromosome instability and immunodefi-ciency syndrome caused by mutations in a DNA methyltransferase gene. Nature1999;402:187–91.
142. Kloeckener-Gruissem B, Betts DR, Zankl A, et al. A new and a reclassified ICFpatient without mutations in DNMT3B and its interacting proteins SUMO-1 andUBC9. Am J Med Genet A 2005;136:31–7.
143. Jiang YL, Rigolet M, Bourc’his D, et al. DNMT3B mutations and DNA methylationdefect define two types of ICF syndrome. Hum Mutat 2005;25:56–63.
144. Braegger C, Bottani A, Halle F, et al. Unknown syndrome: ischiadic hypoplasia,renal dysfunction, immunodeficiency, and a pattern of minor congenital anoma-lies. J Med Genet 1991;28:56–9.
145. Ugazio AG, Maccario R, Notarangelo LD, et al. Immunology of Down syndrome:a review. Am J Med Genet Suppl 1990;7:204–12.
146. de Hingh YC, van der Vossen PW, Gemen EF, et al. Intrinsic abnormalities oflymphocyte counts in children with Down syndrome. J Pediatr 2005;147:744–7.
147. Lockitch G, Singh VK, Puterman ML, et al. Age-related changes in humoral andcell-mediated immunity in Down syndrome children living at home. Pediatr Res1987;22:536–40.
148. Montagna D, Maccario R, Ugazio AG, et al. Cell-mediated cytotoxicity in Downsyndrome: impairment of allogeneic mixed lymphocyte reaction, NK and NK-likeactivities. Eur J Pediatr 1988;148:53–7.
149. Cossarizza A, Monti D, Montagnani G, et al. Precocious aging of the immunesystem in Down syndrome: alteration of B lymphocytes, T-lymphocyte subsets,and cells with natural killer markers. Am J Med Genet Suppl 1990;7:213–8.
150. Barroeta O, Nungaray L, Lopez-Osuna M, et al. Defective monocyte chemotaxisin children with Down’s syndrome. Pediatr Res 1983;17:292–5.
151. Cuadrado E, Barrena MJ. Immune dysfunction in Down’s syndrome: primaryimmune deficiency or early senescence of the immune system? Clin ImmunolImmunopathol 1996;78:209–14.
152. Park E, Alberti J, Mehta P, et al. Partial impairment of immune functions inperipheral blood leukocytes from aged men with Down’s syndrome. Clin Immu-nol 2000;95:62–9.
153. Zollino M, Di Stefano C, Zampino G, et al. Genotype-phenotype correlations andclinical diagnostic criteria in Wolf-Hirschhorn syndrome. Am J Med Genet 2000;94:254–61.
154. Wright TJ, Ricke DO, Denison K, et al. A transcript map of the newly defined 165kb Wolf-Hirschhorn syndrome critical region. Hum Mol Genet 1997;6:317–24.
155. Zollino M, Lecce R, Fischetto R, et al. Mapping the Wolf-Hirschhorn syndromephenotype outside the currently accepted WHS critical region and defininga new critical region, WHSCR-2. Am J Hum Genet 2003;72:590–7.
156. Hanley-Lopez J, Estabrooks LL, Stiehm R. Antibody deficiency in Wolf-Hirsch-horn syndrome. J Pediatr 1998;133:141–3.
157. Lorini R, Ugazio AG, Cammareri V, et al. Immunoglobulin levels, T-cell markers,mitogen responsiveness and thymic hormone activity in Turner’s syndrome.Thymus 1983;5:61–6.

Ming & Stiehm736
158. Cacciari E, Masi M, Fantini MP, et al. Serum immunoglobulins and lymphocytesubpopulations derangement in Turner’s syndrome. J Immunogenet 1981;8:337–44.
159. Donti E, Nicoletti I, Venti G, et al. X-ring Turner’s syndrome with combined immu-nodeficiency and selective gonadotropin defect. J Endocrinol Invest 1989;12:257–63.
160. Robson SC, Potter PC. Common variable immunodeficiency in association withTurner’s syndrome. J Clin Lab Immunol 1990;32:143–6.
161. al-Attas RA, Rahi AH, Ahmed el FE. Common variable immunodeficiency withCD41 T lymphocytopenia and overproduction of soluble IL-2 receptor associ-ated with Turner’s syndrome and dorsal kyphoscoliosis. J Clin Pathol 1997;50:876–9.

History ofImmunoglobulinReplacement
Martha M. Eibl, MD*
KEYWORDS
� Immunoglobulin � IVIg treatment � Primary immunodeficiency� Antibody deficiency � Immune modulation
HISTORYOF IMMUNOGLOBULIN TREATMENT
In 1890, von Behring and Kitasato1 proved that ‘‘bloodserum’’ of rabbits immunizedwith tetanus toxin contained activity against ‘‘tetanus poison,’’ and such blood serumtransferred to rabbits protected these normal (naive) animals against tetanus. In 1901,von Behring was awarded the first Nobel Prize in Medicine or Physiology for his work inthis field. Ehrlich2 demonstrated that protection could be quantitatively correlated tothe amount of antitoxin in the blood.
In 1910, Dr. A. Wolff-Eisner, a 33-year-old physician in Berlin, published ‘‘CurativeSerum Therapy and Experimental Therapy,’’ a handbook for clinic and medical prac-tice.3 His intention—not so far from our own current intentions—was to convey to cli-nicians and practitioners ‘‘the advances of biological science as they relate to therapy.The theoretic [the science] should only be included as far as necessary for the under-standing of treatment.’’ Several infectious diseases and conditions such as allergy andcancer were treated with curative serum. Serum therapy of diphtheria and tetanus wasalready common practice.4
In Germany alone, five companies, including Hochster Farbwerke, Merck-Darm-stadt, and Behring-Hochst, produced curative serum; products from Schering andParke-Davis were also available. There was international cooperation: Calmette5
in Lille, France worked on the treatment of snake venom; Wolff-Eisner in Berlinconcentrated on serum therapy of hay fever and treatment of bacterial diseases.Serum therapy was introduced in the treatment of staphylococcal disease (van deVelde, Holland), streptococcal infection (R. Freund, Berlin), and meningococcal dis-ease (Flexner, New York).6–8 Curative (mainly antitoxic) sera were produced in differentanimal species because serum sickness already was recognized as one of the fearedcomplications of therapy. Convalescent human sera had great advantages compared
Medical University of Vienna, Center for Physiology, Pathophysiology and Immunology, Instituteof Immunology, Borschkegasse 8a, 1090 Vienna, Austria* Immunology Outpatient Clinic, Schwarzpanierstrasse 15/1/9, 1090 Vienna, Austria.E-mail address: [email protected]
Immunol Allergy Clin N Am 28 (2008) 737–764doi:10.1016/j.iac.2008.06.004 immunology.theclinics.com0889-8561/08/$ – see front matter ª 2008 Elsevier Inc. All rights reserved.

Eibl738
with sera of immunized animals, because human serum could be applied without therisk of anaphylaxy and serum sickness.
In 1907, Cenci9 (Italy) applied convalescent human sera for the prevention of mea-sles. McKhann and Chu10 followed in the same indication with material obtained fromthe globulin fraction of placenta in 1933. Treatment with curative sera was performedsuccessfully and saved the lives of many individuals in the first third of the twentiethcentury,5 mainly children with diphtheria and soldiers with tetanus in World War I.Treatment was also widely applied in pneumococcal disease.
Although antibody treatment had been widely used,11 information on serum proteincomposition was incomplete until the 1930s. The proof that antibodies localized tothe immunoglobulin (Ig) compartment of human serum came in the 1930s. Initiallyfractionation was performed by precipitation with different salt concentrations (suchas ammonium sulfate).
In 1938, Karelitz,12 an associate of Schick, published prophylaxis against measleswith the globulin fraction of immune adult serum in The American Journal of Diseasesof Children, but profound understanding on serum proteins became available onlyafter Tiselius and Kabat13 published their pioneering work on electrophoresis ofimmune serum in Science in 1938. Just 2 years later, Cohn and his group14,15 reportedon preparation and properties of serum and plasma proteins, which opened the wayfor Ig prophylaxis and treatment of infectious diseases.
Fractionation of Serum Proteins
Techniques developed by Cohn and his coworkers14,15 in Boston at the beginning ofWorld War II led to the development of the separation of plasma proteins into individ-ual stable fractions with different biologic functions. The basis for Cohn’s fractionationwas to use low concentrations of alcohol by reducing the pH and lowering ionicstrength. The procedure was performed at low temperature, which reduced the likeli-hood of contamination and made large-scale fractionation possible. This method,further refined in cooperation with J.L. Oncley,15 is basically still in use and, withsome additional steps, yields Ig for intramuscular and subcutaneous use. By themid-1940s, however, Cohn14 realized that an Ig product that could be applied intrave-nously was desirable. He recognized that the removal of depressor substances wasnecessary and would require new technology.
In the following years, numerous cooperations were established between Cohn andothers,16 including Elliot Robinson at the Massachusetts Antitoxin and VaccineLaboratory, Charles Janeway17 (Sen.) and John Enders at Harvard, and clinical trialsto prevent viral diseases (eg, measles and hepatitis) were initiated. Because of thelimited availability of blood, Ig products were also prepared from placentas. Duringand after World War II, the supply of gammaglobulin increased as plasma from theAmerican Red Cross became available.
The first Ig products were mainly given to prevent and treat infections: polymyelitis,measles, mumps, pertussis, and hepatitis A.18,19 These Ig disappeared as soon asthe respective diseases could be and were prevented by vaccination. Other Igproducts with defined specificities were developed subsequently and continue tobe used currently (eg, tetanus Ig,20 RHo(D) Ig,21 rabies Ig, hepatitis B Ig,22 and varicellazoster Ig23). Respiratory syncytial virus hyperimmunoglobulin24 was licensed but laterreplaced by a respiratory syncytial virus monoclonal antibody,25,26 one of the fewmonoclonal antibodies that made its way into clinical application in the preventionof an infectious disease—pneumonia caused by respiratory syncytial virus, a danger-ous, potentially fatal disease in premature infants and newborns.

History of Immunoglobulin Replacement 739
Early Treatment of Primary Immunodeficiency with Immunoglobulin
The first patient who had primary immunodeficiency (PID) described by Bruton27 in1952 had recurrent infections and multiple episodes of sepsis. Testing his serum re-vealed the absence of specific antibodies, and serum electrophoresis indicated thatIg were lacking. The boy was treated with subcutaneous gammaglobulin and wasfree of sepsis and severe infections thereafter. The clinical history of the patient indi-cated that in addition to life-threatening bacterial infections, he had mumps four timesbut managed to recover from the disease without problems or complications. Thatfinding, which was an experiment of nature, indicated that antibodies are importantto prevent the disease, but T-cell immunity is sufficient to clear this viral infection. Par-allel to this observation, numerous patients with agammaglobulinemia were identifiedand treated by Janeway and others.17,28 In the early days, however, especially if a prod-uct had not been prepared in the cold, contamination by bacterial toxins occurreddespite the fact that the product passed tests of sterility and pyrogenicity. AlthoughJaneway17 also preferred the subcutaneous route, intramuscular administration of‘‘immune serum globulin’’ became state-of-the-art for the years to come.
Immune serum globulin became standard treatment of patients with antibody defi-ciency syndromes (agammaglobulinemia and hypogammaglobulinemia) in the 1950sand was widely used with this indication for at least the next two decades.29–39 Itsefficacy has been well documented. The products contained mainly IgG traces ofIgM and differing amounts of IgA. Ig treatment substantially improved the prognosisof patients with agammaglobulinemia, hypogammaglobulinemia, and other forms ofsevere antibody deficiencies. The recommended dose was 25 mg/kg/wk. After theefficacy of IgG (for intramuscular use) had been clearly proven in primary immunode-ficiencies in the early 1950s, the British Medical Research Council set up a workingparty to find the optimal dose for the treatment of patients with antibody deficiency40
(hypogammaglobulinemia). Based on the results, a 50 mg/kg/wk dose was regardedas adequate, but it was felt that the previously accepted dose of 100 mg/kg/mo mightbe justified in view of the discomfort the injection caused and with regard to thehigh expense of treatment. In 1966, Janeway17 considered a raise in serum levels of200 mg/dL necessary to prevent invasive bacterial infections and suggested300 mg/kg as an initial dose to be followed by 100 mg/kg/mo.
Products of intramuscular Ig of different manufacturers were similar in compositionand activity as long as the plasma of at least 1000 donors had been pooled. The spec-trum and quality of antibodies were comparable to antibodies in the plasma of thestarting material. Fc-mediated functions were preserved. Antibodies were enrichedapproximately 10- to 20-fold in the 15% to 18% solutions as compared with plasma.Transmission of viral diseases was a major concern because hepatitis B transmissiondid occur when the starting material was contaminated.41–44 If the single donations ofplasma were free of hepatitis B surface antigen and if appropriate fractionation meth-odology was used, however, the final product was proven to be safe with respect tothe transmission of hepatitis B. Serum Ig for intramuscular use also had great disad-vantages. The intramuscular injection was painful, maximum serum levels were notreached before 24 hours and could take several days, and in vivo recovery was usuallyless than 50%.45,46 At higher dosages, the preservative containing mercury causedincreased concern.47,48 These products could not be given intravenously becauseof adverse reactions. These reactions, mainly in children, caused chills and feverbut also could be severe and lead to shock and even fatal reactions.49 Adverse effectsoccurred in 15% to 25% or more of applications and were mainly attributed to aggre-gates in the product. Researchers soon recognized that reactions were more frequent

Eibl740
in agammaglobulinemic patients compared with patients with normal gammaglobulinserum levels, who received these products with the aim of preventing recurrent orsevere infections.
Studies in patients with antibody deficiency demonstrated a reduction of acute andchronic infections by intravenous Ig (IVIg).50–52 Early studies in patients with commonvariable immunodeficiency showed a significant reduction in infection rate as com-pared with before IVIg treatment.53 The superiority of IVIg to intramuscular Ig wasproven by direct comparison.54,55 Ig products that could be applied intravenouslywere clearly desirable, and serum Ig had to be further treated to be tolerated via theIV route.
Different Intravenous Immunoglobulin Products
Several different methods have been tried to arrive at safe and efficacious prepara-tions. Because aggregates were thought to be the main cause of adverse effects,proteolytic treatment was the first choice. Ample evidence from previous timeswhen animal sera treated with pepsin or trypsin were used indicated that this treat-ment reduced reactivity. The first IVIg product from human plasma prepared by pepsintreatment, manufactured by Behringwerke in Germany, has been in clinical use sincethe 1960s. This product contains mainly Fab2 fragments, has no Fc-mediated activity,and has a half-life of 24 hours or less in the circulation. It was principally well tolerated,although it still has some adverse effects in immunodeficient patients. With its shorthalf-life, however, it is clearly unsuitable for the treatment of patients with antibody-deficiency syndromes.
Other subsequently developed IVIg products that could be considered for treatmentof patients with primary immunodeficiency diseases were chemically and enzymati-cally modified preparations. Several IVIgG preparations were created in which the Igwas basically unmodified. Different methods were used to arrive at products thatwere safe when given intravenously and showed wide variation with respect tofunctional integrity (Table 1).56–62
In 1979, a workshop entitled ‘‘Ig: Characteristics and Uses of Intravenous Prepara-tions’’ was organized by J.S. Finlayson and Barbara Alving and sponsored by theBureau of Biologics, the US Food and Drug Administration, and the National Heart,Lung and Blood Institute. The purpose of the workshop was (according to the prefaceby the editors) to review the international use of IVIg preparations in the treatment ofimmunodeficient and immunosuppressed patients, explore potential clinical uses forsuch preparations, discuss the nature of adverse reactions to these products, anddefine, insofar as possible, the ideal characteristics ofIVIg.63 The specialties of themore than 200 attending investigators ranged from basic science to manufacturingto clinical medicine.
The consensus at the 1979 meeting was that IVIg products were indicated for theprovision of antibodies to prevent certain infectious diseases and for the treatmentof patients with antibody deficiency, primary immunodeficiencies with agammaglob-ulinemia and hypogammaglobulinemia, other forms of PID (eg, severe combined im-munodeficiency, combined immunodeficiency), and secondary antibody deficiency.64
At the meeting, individual clinical investigators reported their experience regardingtreatment with the different enzymatically, chemically modified or intact Ig products.In most cases, the number of patients in the individual groups was between 10 and20, and there was agreement that the IV route of Ig treatment was advantageous tointramuscular therapy.65 The adverse reactions observed and reported by the differentinvestigators were virtually identical and are described later in the article.49,66–98 Theconclusion was that IVIg was a promising new option in the treatment of patients

Table 1Intravenous immunoglobulin preparations
Enzymatically treatedProcedure Manufacturer CountryPepsin Behringwerke Germany
Nikon Seiyaku Japan
Kaketsuken Japan
Plasmin Hyland USA
Michigan USA
Green Cross Japan
Merieux France
Chemically treatedProcedure Manufacturer Countryb – propiolactone Biotest Germany
PH4 1 treated pepsin SRC, Sandoz Switzerland
Reduced, alkylated Cutter USA
UnmodifiedProcedure Manufacturer CountrySi O2 QAE sephadex Condie USA
Ethanol DEAE sephadexstabilized with albumin
KABI Switzerland
DEAE sephadex Winnipeg USA
PEG/HES Armour USA
PEG, Plasmin Treatment Green Cross Japan
Salting out, PEG, isoelectric precipation Immuno Austria
Cohn II 1 III 1 PEG 4000 Continental Canada
Ethanol, pH4 Scottish National BloodTransfusion Service
Great Britain
History of Immunoglobulin Replacement 741
with humoral immunodeficiency: impaired antibody production. Adverse events oftenwere seen, but they were manageable. The causes of those adverse events were notcompletely understood.
At the meeting in 1979, a single presentation from Japan69 on 5406 cases of IVIgtreatment for numerous diseases in the specialties of pediatrics, medicine, surgery,and neurosurgery with 82% effectiveness gave one of the early examples of anempiric approach indicating that there might be a much greater scope of treatmentwith IVIg than generally accepted at the time. Researchers soon realized that treat-ment with IgG, the only component of most IVIg products, is necessary and sufficientfor the prevention of infection and the prophylaxis of acute exacerbations of lung dis-ease (pneumonia) and sinusitis,50–52,70 even in patients in whom all isotypes of Ig wereabsent (the agammaglobulinemics). Researchers recognized that antibody quality,especially functions mediated by the Fc portion of the antibody, were important.
The question of antibody diversity was raised on several occasions before anda long time after this workshop in 1979. One of the questions raised was whether itwas necessary to provide the starting material of individual products from the geo-graphic area where the patients are.71 The current view, which is the result ofa long-lasting debate, has been that as long as the number of donations is 15,000to 60,000 per plasma pool for production, the antibody diversity in the product can

Eibl742
be considered adequate.72 Selective antibodies are tested lot-wise to show that anti-toxic, antiviral, and antibacterial qualities are preserved. Extended testing of currentpreparations proves that products have only traces of aggregates, contain 90% ormore monomeric IgG, are free of vasoactive contaminants, and have proven Fab-and Fc-mediated antibody functions.66–68
Fab and Fc Properties
The specific antibody activity, the function of antigen binding, resides in the Fab partof the Ig, and secondary functions of the Ig are carried by the Fc portion of the mol-ecule. The Fc portion determines its isotype (eg, class and subclass). It interacts withreceptors on phagocytic cells (eg, neutrophil granulocytes and macrophages) to fa-cilitate engulfment of antibody-coated cells and pathogens. The Fc portion initiatesactivation of the complement cascade via the classic activation pathway by bindingC1q (an early complement component). It is responsible for the active passage of Igfrom mother to fetus through the placenta and determines the half-life of Ig in the cir-culation. The Fab and Fc regions of the Ig are connected by the hinge region of themolecule. The hinge region is flexible; its function is important for avidity, and it is themost sensitive portion of Ig with respect to chemical and enzyme treatment.
Specific antibody activities are crucial for antigen recognition. Fab fragments aresufficient for toxin neutralization. Antiviral activity depends on recognition throughFab but is much more potent by the intact antibody molecule. The elimination ofbacteria requires the Fab and the Fc moiety of the Ig.
Immunomodulatory Activity
Early observations by Barandun in the 1960s noted that in a patient who had primaryimmunodeficiency disease and hemolytic anemia, signs and symptoms of hemolysisdecreased and Coomb’s test results normalized when treated with IVIg. Parallel tothose observations, a child who had idiopathic thrombocytopenic purpura (ITP) andchicken pox was treated with IVIg, and platelet counts normalized. Subsequent to thoseobservations, other patients with thrombocytopenia were successfully given IVIg.73 Im-bach and colleagues74 followed up on these anecdotes and started clinical studies inchildren with acute and chronic ITP. The dose was 400 mg/kg for 5 days, and almostall patients showed a dramatic response with fast normalization of platelet counts.
After the report by Imbach74 on the effect of IVIg in ITP, numerous studies confirmedthese results,75–82 and other studies have been conducted to investigate the effect ofIVIg in different organ-specific and systemic autoimmune diseases.83–95 These studiesincluded—but were not limited to—investigations in the following areas: hematology(numerous), dermatology (eg, bullous pemphigoid), endocrinology (eg, thyroid dis-ease, diabetes), and neurology (eg, Guillain-Barr�e syndrome, chronic inflammatorydemyelinating polyneuropathy, intractable childhood epilepsy, myasthenia gravis).The effect of IVIg treatment in autoimmune diseases clearly depended on the Fcportion of the Ig. Although chemically modified IVIg with its Fc function mainlypreserved was effective in ITP, pepsin-treated Ig, which contained only the Fab2part of the Ig molecule, had only minimal effect.96,97
Although the mechanism of action of IVIg in different autoimmune diseases was notclear, the following hypotheses have been suggested to explain the results observed:
1. Ig binds activated complement components, solubilizes immune complexes, andinhibits their binding to target cells. Basta and coworkers98 protected guineapigs by Ig in the complement-mediated Forssman shock and emphasized thatthe binding of activated complement components could be the crucial mechanism

History of Immunoglobulin Replacement 743
of the effect of IVIg in hematologic autoimmune diseases.99,100 This hypothesis be-came the basis of a later controlled study that demonstrated the effect of IVIg indermatomyositis.101,102
2. Another hypothesis centered around the inhibition of the binding of antibody-coated target cells to Fc receptors on phagocytic cells: Fc receptor blockade.75,103
Based on this hypothesis, an Ig product with anti RHo(D) specificity was laterlicensed for the treatment of ITP.104–106 An experimental study with an anti-Fcreceptor monoclonal antibody proved the principle but has not been followed upas a routine mode of treatment.107,108
3. The immunomodulatory effect of IVIg was described and thought to be crucial in thedown-modulation of the production of autoantibodies by B cells.109 The importanceof anti-idiotypic antibodies has been emphasized.110 Anti-idiotypic antibodies arepresent in IVIg and antibodies to ab T-cell receptors,111 major histocompatibilitycomplex class I,112 CD45 (Fas),113,114 and other self-determinants115 and werethought to be the effective principle in the treatment of other autoimmune diseases(eg, myasthenia gravis, Guillain-Barr�e syndrome, systemic lupus erythematosus,nephritis).110,116 In addition to their specificities, anti-idiotypic antibodies are leadingto Ig dimerization and the functional importance of the dimers in Fc receptor ligation,and signaling has been demonstrated.110
4. Several studies indicated that IVIg has an immunomodulatory effect, not only onB cells but also on T cells. It down-modulates not only B-cell activation but alsoT- and B-cell interaction and is inhibitory in mixed lymphocyte reaction and mitogenand interleukin-2–induced proliferation of mononuclear cells.117 IVIg products wereshown to contain antibodies against different cytokines, and these antibodies werethought to be important for the immunomodulatory effect.118 Based on the down-regulation of the immune response, IVIg has been studied in bone marrow and solidorgan transplantation.119–123
Modulation of Inflammation by Immunoglobulin
Certain pathophysiologic events (eg, Kawasaki disease, vasculitis) are characterizedby systemic, excessive inflammation. Cells and mechanisms of the innate andadaptive immunity are operational in this process. Phagocytes, B cells, and T cellsare central in mounting inflammatory host response to gram-negative and -positivebacteria and their products, such as lipopolysaccharide and superantigens.124–128 Aderegulated release of inflammatory cytokines results in life-threatening pathologywith fever and organ damage and may lead to shock and fatality. The release of inflam-matory cytokines and their activity can be regulated at different levels: gene expres-sion, translation, release, receptor expression, and neutralization of cytokine activityby antibody or soluble cytokine-binding inhibitors. IVIg has been shown to regulatethe burst of inflammatory cytokines at the level of cytokine receptor interaction withantibodies against cytokines and the induction of receptor antagonists.118,129–133
The effects of monomeric polymeric IgG and immune complexes on Fc receptorshave been studied in several laboratories.75,134–136 Clinical evidence that IVIg treat-ment is safe and effective in a deregulated inflammatory disease has been providedby large, controlled clinical studies of children who have Kawasaki disease, which isa disease with prolonged fever, rash, erythema of palms and soles, and mucosalinflammation with involvement of the oral pharynx and conjunctiva.137 The underlyingpathology is characterized by vasculitis that involves medium and large arteries. Up to25% of children who have this syndrome and received aspirin as the standard treat-ment developed coronary artery lesions with severe sequelae. High-dose IVIg(400 mg/kg on 5 consecutive days or 2 g/kg in one infusion) impressively stopped

Eibl744
the inflammatory symptoms and significantly reduced the prevalence of coronary ar-tery abnormalities to less than 5% when given early in the disease.138,139 Clinical trialswere double-blind, randomized controlled studies that compared the then-standardtreatment of Kawasaki disease with aspirin to aspirin plus IVIg.
Expanding the Scope of Accepted Indications in 1990
The accepted modes of action of IgG, namely provision of antibodies,140–146 inhibitionof Fc receptor binding (on phagocytes), down-modulation of inflammation, and down-modulation of antibody production147–149 (immune response), were the basis of theconsensus in 1990.140 According to the consensus of 1990, IVIg was indicated forthe prevention and treatment of the following disorders:
Primary immunodeficienciesPediatric aidsBone marrow transplantationChronic lymphocytic leukemia with hypogammaglobulinemiaIdiopathic thrombocytopenic purpuraKawasaki syndromeChronic inflammatory demyelinating polyneuropathiesGuillain-Barr�e syndrome
Although a trial to prevent infections in premature infants was one of the firstrandomized trials with intramuscular Ig,150 the question whether treatment with Ig isbeneficial in this vulnerable group of patients is still undecided.
At the 1990 consensus meeting, large clinical trials to answer the question of effi-cacy of Ig in premature infants and neonates were thought to be important. A largemulticenter controlled trial151 could not provide clear results for efficacy, and smallerclinical studies later came to similar conclusions.152,153
Adverse events observed in the course of high-dose treatment were of increasingconcern, as was the transmission of blood-borne pathogens at that time because ofthe theoretic threat of possible HIV transmission by IVIg, which fortunately did nothappen. Transmission of blood-borne pathogens did occur, however, in the 1990s,when hepatitis C was transmitted in clinical trials and with treatment with IVIg of patientswho had PID (see the later section on adverse events). The recently developed test kitsbased on hepatitis C antigen produced by a recombinant technology opened the way toeliminating donations from hepatitis C antibody–positive plasma donors. Industry, in co-operation with the health authorities, reacted in a fast and proper way. By the end of thedecade, all licensed products could be considered free of contaminating viral agents.
In the 1990s, off-label use of IVIg started to widen for numerous autoimmune andinflammatory indications in the treatment of neurologic diseases.154–156 The dosewas usually 2 g/kg either in five applications of 400 mg/kg each or in two applicationsof 1 g/kg each. In the course of expanding high-dose treatment, rare but severeadverse events were noticed, and it became evident that although rare, they mightoccur in patients who had PID and were on regular replacement therapy.
Although during the course of almost two decades [1990–2008] changes in labeledindications for IVIg treatment were minimal, more diseases have been treated off-label.
CONSENSUS STATEMENTS, 1999^2001 (EXPERTMEETINGS, NATIONAL INSTITUTESOF HEALTH, FOODAND DRUG ADMINISTRATION, AND PUBLISHED REPORTS)
The results started to come in from studies initiated to analyze the effect of IVIg in thedifferent areas of autoimmune disease.157,158 Studies of IVIg treatment in 53 different

History of Immunoglobulin Replacement 745
diseases from the areas of hematology, infectious disease, neurology, obstetrics,rheumatology, endocrinology,92–94 and other specialties were investigated. The firststudies, even when based on meta-analysis of several clinical trials, were not largeenough to give clear support for additional indications, even when the results didlook promising. These studies opened the way for further trials, which, almosta decade later, arrived at several indications supported by grade I evidence. Basedon these results, IVIg became a generally accepted standard way of treatment by theend of the twentieth century in numerous neurologic diseases such as Guillain-Barr�esyndrome and chronic inflammatory demyelinating polyneuropathy. By 2003–2004,approximately 60% to 65% of IVIg had already been used for patients with neurologicdisease.72,159
In 2006, a document was prepared by a committee of the American Academy of Al-lergy, Asthma and Immunology on the use of IVIg in human disease.72 This consensusgave a factual review of labeled indications ‘‘as licensed by the FDA’’ and off-labelindications based on (practically all) publications available. A summary of FDA-approved indications for licensed products follows:160
PID or primary humoral immunodeficiencyB-cell chronic lymphocytic leukemia (hypogammaglobulinemia)HIV infection (pediatric)Kawasaki diseaseIdiopathic thrombocytopeniaBone marrow transplantation
Off-label indications increased significantly at that point. More than 80% of the IVIgis used off-label, supported by definitely beneficial and probably beneficial indicationsbased on category Ia-IIb evidence. From the 2006 consensus about the use of IVIg inhuman diseases, the main off-label uses based on category Ia-IIb evidence aresummarized in Table 2.72
The concerns and risks center around adverse events (mainly the rare and serious),or around supply and economic considerations. The concern regarding the
Table 2Off-label uses based on category Ia-IIb evidence
Definitely Beneficial Grade Probably Beneficial GradeGrave’s ophthalmopathy Ib Dermatomyositis and polymyositis IIa
ITP Ia Autoimmune uveitis IIa
Guillain-Barr�e syndrome Ia Lambert-Eaton myasthenic syndrome Ib
Chronic demyelinatingpolyneuropathy
Ia IGM antimyelin paraprotein associatedneuropathy
Ib
Multifocal motor neuropathy Ia Myasthenia gravis Ib-IIa
— — Stiff-man syndrome Ib
— — Neonatal sepsis Ia
— — Rotaviral enterocolitis Ib
— — Toxic epidermal necrolysis and StevensJohnson’s syndrome
IIa
These and additional IVIg products are available in Europe.Data from Orange JS, Hossny EM, Weiler CR, et al. Use of intravenous immunoglobulin in human
disease: a review of evidence by members of the Primary Immunodeficiency Committee of theAmerican Academy of Allergy, Asthma and Immunology. J Allergy Clin Immunol 2006;117:S525–53.

Eibl746
transmission of blood-borne pathogens still exists as a theoretic scenario without anypractical relevance.
Dosage
Numerous publications have dealt with the question of optimal dose as replacementfor patients with primary immunodeficiency disease.72,161–169 After a loading dose,400 mg/kg/mo is the generally accepted standard treatment. Diminished pulmonaryfunction is the major cause of morbidity and mortality in patients with antibodydeficiency syndrome, and it is clear that early diagnosis and prompt initiation of IVIgtreatment significantly reduce these complications. Two large studies analyzed thequestion of dose in patients in whom IVIg treatment was started when impairmentof pulmonary function was already present. In the trial by Roifman and colleagues,167
doses of 200 mg/kg/mo and 600 mg/kg/mo were compared in a cross-over study.This study and the trials conducted by Bernatowska and colleagues,170 proved thatin patients on high-dose treatment, pulmonary function improved. This improvementdid not take place at the lower dose level, which could be attributed to the additionalanti-inflammatory effect of IVIg, which requires treatment at higher dose.
The determination of whether standard doses (eg, 400 mg/kg/mo) are preferable orwhether trough levels should guide IVIg treatment is still undecided. A previous studydemonstrated that trough IgG levels of 500 mg/dL in patients were beneficial,52,167,171
and a recent study indicated that the rate of infection can be further reduced witha trough level of 900 mg/dL.172 This finding might be important for long-term progno-sis.168 Certain conditions in patients who have PID must be treated with a higher dose.Patients with impairment of pulmonary function at presentation might need a total of600 mg/kg/mo in one or two doses, and a few patients with chronic echo virus,meningoencephalitis, a previously fatal disease, may improve and then be maintainedon 1 g/kg per dose, which has to be given repeatedly.173–176
Doses of IVIg required for immunomodulation are much higher. Up to 2 g/kg are rec-ommended and may have to be repeated.138 This usually applies to clinical situationsin which IVIg is given for true immunomodulation and for the treatment of systemicinflammatory conditions (eg, Kawasaki disease).
ADVERSE EVENTSInflammatory Reactions
Reactions during the course of, or immediately after, Ig treatment already wereobserved when Ig was given intramuscularly and have continued to be of concernwith IVIg therapy. These reactions were anaphylactoid in nature.177–183 The followinglist details adverse reactions to IVIg:
FeverChest tightnessChillsDyspneaFacial flushWheezingTachycardiaUrticariaPalpitationRashLowering of blood pressureAnxiety

History of Immunoglobulin Replacement 747
ShockNervousnessLumbar painHeadacheAbdominal painNauseaVomiting
Several investigators gave prophylactic steroids, antihistamines, or anti-inflamma-tory agents to avoid reactions or reduce severity.178,184 In many cases, adverse reac-tions could be avoided just by lowering the rate of infusion. The fact that patients withthe most pronounced impairment of antibody production had the highest likelihood ofadverse reaction made a true anaphylactic reaction most unlikely.177 The question ofwhether these reactions were caused by formation of immune complexes100,180,181 toanticomplementary activity or some other properties of aggregates was a subject ofdiscussion.185,186 The fact that patients with severe adverse reactions often did notshow a drop in complement levels in serum and changes in serum complement levelswere not associated with adverse events indicated that other properties of Ig aggre-gates in addition to complement activation may be operational. IgG aggregates inIVIg products may trigger the release of pharmacologically active mediators by ligationof Fc receptors from macrophages and leucocytes.187–189 Substances such asprostaglandins, platelet-activating factor, and cytokines (eg, tumor necrosis factor-a,interleukin-6) have been described to cause reactions such as fever, bronchospasm,and changes in blood pressure, which are characteristic adverse events of IVIg treat-ment. Another possibility for the release of mediators is that in patients with infections,the antibodies infused react with circulating microbial antigens to form immunecomplexes, which again trigger the release of the respective mediators. Vasoactivecontaminants (eg, enzymes such as prekallikrein activator) that were present inearlier products could have contributed to reactions through the generation ofbradykinin.190,191
The question of whether the frequency and severity of anaphylactoid reactionscould be related to some extent to individual products, their mode of production,and their preparation is still pending. To give definite answers, controlled trials specif-ically designed for comparison of different products are needed, because results ofsuch trials are scarce.172
Because IVIg was infused in large quantities, it was essential that it be free of iso-agglutinins and nonagglutinating antibodies against red cells. Hemolytic reactionshave been described in rare instances.
Transmission of Infectious Agents
Fortunately, because of HIV partitioning during plasma fractionation, Ig products didnot transmit HIV at any time, even in the early 1980s, when the infectious agent wasas yet unknown and testing for antibodies or the virus was not possible. Hepatitis Cvirus transmission did occur, however, with different IVIg products in the late 1980sto mid-1990s.192 In most of the products involved, ion exchange chromatographywas used in purification, but one of the products was prepared by the pH4 pepsintreatment.193
Soon after cases of viral contamination were reported, additional steps and controlsin the course of IVIg manufacturing were implemented. The industry responded withdonor screening, donor testing, inventory management, plasma pooling, and testing.Quality control measures of viral partitioning during fractionation were introduced, and

Eibl748
additional viral inactivation steps were performed. Licensed products currently areconsidered safe with respect to transmission of viral agents, especially HIV and hep-atitis viruses.194 Unknown agents are only of theoretic risk.
Thromboembolic Events
With broad application of IVIg, especially high-dose therapy, previously rare complica-tions have been observed and described more frequently.195 Most of the patientsin whom thromboembolic events occurred in the course of mainly high-dose IVIgtreatment were patients with neurologic diseases or polymorbid patients. Numerousmedical conditions have been recognized as risk factors (eg, hypertension, cardiovas-cular disease, diabetes, kidney disease).196–198 Thromboembolic complications alsowere observed in a few patients with primary immunodeficiency who had been treatedwith a regular dose. Because antibody deficiency syndromes have moved from thepediatric to the adult population and treatment with IVIg of elderly patients whohave PID is not a rarity anymore, underlying morbidity (pulmonary, cardiac, renal)must be taken into consideration in IVIg treatment of patients who have PID.
The mechanisms of action for thromboembolic complications are not well under-stood. Dalakas suggested that increased serum viscosity plays a crucial role.197 Anti-phospholipid antibodies in IVIg also were mentioned as of possible importance.Wolberg and colleagues detected coagulation factor XI as a contaminant in IVIg prod-ucts and showed that IVIg samples that contained factor XI shortened clotting time infactor XI–deficient plasma.183,199
Rapid infusions, which have been suggested because of the inconvenience of theregular lengthy infusion time for patients who had to receive large amounts of IVIg,seemed to be more prone to causing thromboembolic complications. The rate ofadverse events after rapid infusion was higher than reported in usual standard ratesof application. Some reports still favor rapid infusion, mainly because of lowercost.200,201
Renal Complications
Renal complications in the course of IVIg treatment have been described since themid-1980s. Usually, but not exclusively, they occur with high-dose treatment. Thesereactions, although infrequent, resulted in severe morbidity; more than one third ofthe patients required dialysis, and fatalities have been reported.198,202,203 The mech-anisms explaining these adverse effects included sucrose or maltose in the product,because renal lesions caused by sucrose have been known since the 1940s. Riskfactors were similar to those observed in thromboembolic disease, especially oldage (> 60), diabetes, and prior renal disease.204,205
ADVERSE EVENTS CAUSED BY IMMUNOGLOBULIN A
Severe (transfusion) reactions have been described in patients with gastrointestinaldisease who were IgA deficient. After these observations, reactions to IVIg—usuallyanaphylactoid—were contributed to IgA206 in the product and IgA antibodies in thepatient.207–209 In two patients, true anaphylactic reactions were reported; they hadIgE antibodies against IgA.210 During the decades of discussion about anaphylactic/anaphylactoid reactions to IgA, cases in which IgE antibodies could be identified asthe reason for the reaction have been rare. Most patients with anaphylactoid reactionsto IVIg are unable to produce antibodies of any class or specificity, and numerouspatients who had IgA antibodies did not show reaction to IVIg products containing IgA.

History of Immunoglobulin Replacement 749
CURRENT STATUS AND ONGOING DEVELOPMENTS
IVIg treatment is the most commonly applied mode of Ig replacement therapy for pa-tients with antibody deficiency syndromes and the only mode of Ig treatment for allother indications. Intravenous immunoglobulin products available in the United Statescan be seen in Table 3.
Home treatment with immunoglobulin for subcutaneous applicationAlready at the beginning of Ig treatment in the early 1950s, Janeway preferred the sub-cutaneous application of Ig. In 1979, Berger and colleagues described Ig replacementtherapy by slow subcutaneous infusion using small, battery-operated pumps andfound that the ‘‘subcutaneous infusions were tolerated remarkably well with minimallocal reaction and no evidence of adverse systemic effects.’’ Later, subcutaneouslyapplicable Ig preparations without mercury-containing preservatives were made avail-able for clinical studies. During these trials, it became evident that Ig given subcutane-ously is well tolerated.211,212 The most common side effects are local reactions lasting12 to 24 hours after infusion. Systemic reactions seem to occur less frequently. Thedose is usually 100 mg/kg/wk but might be modified slightly according to the situationof the patient. Several papers reported that this way of treatment also makes IgA-deficient individuals tolerant to IgA, so they do not show anaphylactoid reactionseven if treated intravenously afterwards.208 Several licensed products for subcutane-ous application are available in the United States and Europe. This mode of Ig replace-ment opens the way for home treatment. Some patients have a clear preference for IVtherapy once or twice a month, whereas others prefer the subcutaneous way of treat-ment. These latter patients can choose the time at their own convenience and regardthe independence gained by this procedure as a major improvement in their qualityof life.
Monoclonal antibodiesIn the mid-1970s, the epoch-making discovery by Jerne, Kohler, and Milstein (NobelPrize for Medicine or Physiology, 1984) opened the way for the production of
Table 3IVIg products currently available in the United States
Manufacturer Name of Product IndicationsTalecris Gamunex2 Immune globulin intravenous
(human), 10% caprylate/chromatography purified
PID; ITP
Baxter Gammagard liquid immune globulinintravenous (human) 10%(KIOVIG)
PID associated with defectsin humoral immunity
Baxter Gammagard/S/D immune globulinintravenous (human)
PID, ITP, CLL, Kawasaki syndrome
ZLB Behring Carimune NF nanofiltered immuneglobulin intravenous (human)
Immunodeficiency, ITP
Grifols Flebogamma 5% immune globulinintravenous (human)
PID
Octapharma Octagam immune globulinintravenous (human) 5%
PID
Data from Sorensen R. Expert opinion regarding clinical and other outcome considerations in theformulary review of immune globulin. J Manag Care Pharm 2007;13(3):278–83.

Eibl750
monoclonal antibodies in virtually unlimited quantities. The hope that these antibodieswould be used extensively in the prevention and treatment of infectious disease waswidely expressed. Over several decades we learned that monoclonal antibodies witha single specificity recognizing a single epitope on the target antigen cannot substitutefor polyclonal antibodies in prophylaxis and treatment of infectious disease. The task ofhumanization to arrive at products that are not recognized as antigens in the recipientbecame challenging. Monoclonal antibodies are widely used in the treatment of cancerand thromboembolic disease; however, their use in infectious disease is limited andthey still have no place in Ig replacement therapy for antibody-deficient patients.
Increasing understanding in the expression of Fc receptors, ligand binding, and sig-naling led to new hypotheses to explain the numerous immunomodulating propertiesof IVIg.213,214 Most hematopoietic cells, including phagocytes, express high-affinity Fcreceptors that transduce positive signals and low affinity Fc receptors, which down-modulate cell activation to keep the balance. B cells only express the inhibitory Fcreceptor. This FcR has been shown to regulate positive signals coming from theB-cell antigen receptor, which could explain the true immunosuppressive effectobserved in the clinic where IVIg had a long-term effect in patients with autoimmunediseases because of autoantibodies. One is tempted to speculate that this could havebeen the mechanism in one of the first observations by Barandun30 while treatinga patient who had antibody deficiency and hemolytic anemia. The patient turnedCoombs-negative during Ig treatment.
Recently, Ravetch provided experimental evidence that the suppression of inflamma-tion is mediated by sialylation of a defined position in the Ig structure.215–217 Hesuggested that a product containing the Fc part of the IgG with the propersialylation might be 30-fold more effective than an IVIg product and could be pro-duced by recombinant technology. Limited experimental evidence indicates thatsuch a preparation could be developed into a clinical product, but history teachesthat the time course of developments in this area from the first experimentalevidence to successful treatment might span decades. The following list detailsthe chronologic events leading up to the currently available treatment.
1890
Serum of rabbits immunized with tetanus toxin protected normal animalsagainst tetanus1901
Von Behring won the first Nobel Prize in Physiology or Medicine ‘‘for his work onserum therapy, especially its application against diphtheria, by which he hasopened a new road in the domain of medical science and thereby placed inthe hands of the physician a victorious weapon against illness and deaths’’
1890–1910Curative (animal sera) prepared and applied against diseases caused by toxins,
bacterial disease, allergic disease and cancer1907
Convalescent sera in the prevention of measles1910–1930
Human (convalescent) sera preferred to animal sera in prevention and treatmentInitiation of pepsin treatment of animal sera for reduced reactogenicity
1933–1938Prevention of measles with gammaglobulin (placenta)
1940Cold ethanol fractionation

History of Immunoglobulin Replacement 751
1949 until vaccination
Prevention and treatment of poliomyelitis, measles, mumps, pertussis, and hep-atitis A by Ig1952
Treatment of PID with Ig1960
Pepsin-treated IVIg1970
Other chemically, enzymatically modified and unmodified (treated) IVIg products1979
Consensus meeting on IVIg indicated for procurement of antibodiesIVIg has to be safe with unimpaired function
1984C�esar Milstein, with Georges Kohler and Niels K. Jerne, received the Nobel Prize
for Physiology or Medicine for ‘‘theories concerning the specificity in devel-opment and control of the immune system and the discovery of the principlefor production of monoclonal antibodies’’
1990Consensus meeting on IVIg indicated for the provision of antibodies and
immunomodulation.Interference with Fc receptor binding (targets of autoantibodies) ITPAnti-inflammatory effect: Kawasaki diseaseImmunosuppressionGuillain-Barr�e syndromeChronic inflammatory demyelinating polyneuropathy
1995–2001Consensus meetingBasically identical indications to 1990Extensive off-label use
PresentFDA licensed indicationsBasically similar to Consensus 1995–2001Numerous further indications for off-label use based on clinical studies with
clear (CL1-2) evidence
SUMMARY
Soon after the first report by von Behring on toxin neutralization by serum proteins,clinical scientists took the opportunity to apply this principle to a wide range of infec-tious diseases. After important discoveries in the field of biochemistry, it became clearthat antibodies are in the gammaglobulin fraction of serum, that serum can be fraction-ated to obtain gammaglobulins, and that some patients’ serum lacks gammaglobu-lins. The principle: the provision of antibodies with Ig for intramuscular use and laterIVIg to prevent and treat infection, especially in patients with agammaglobulinemia(ie, antibody deficiency, PID), was the governing principle for IVIg use until the1980s, as can be seen in the consensus of 1979. The possibility of modulation ofinflammation, immunomodulation, and immunosuppression with Ig first emerged inthe early 1960s and has expanded since the 1980s. The consensus of 1990 demon-strated examples of clinical use based on immunomodulation. In the last 20 years,the off-label use of Ig exploded mainly for inflammatory and hematologic, neurologic,medical, and dermatologic autoimmune diseases, often based on solid clinical

Eibl752
evidence. The dose is generally 400 mg/kg/mo for the provision of antibodies, up to2 g in single or multiple doses for immunomodulation.
Concerns regarding adverse effects changed during the decades of experience.The early problems of inflammatory reactions are manageable; viral transmissionhas been eliminated by prompt and effective action. Serious adverse events, suchas thromboembolic and renal complications, are rare; however, when they appear,especially in polymorbid patients, they are worrisome. Old and new practical develop-ments move toward home treatment for patients who prefer this mode of therapy. Thequestion of shortage of Ig in the course of extended use and high costs urge us to crit-ically assess available evidence for application of this precious material in treatment.New scientific developments help us to understand the immunomodulatory function ofIg on the cellular and molecular level and may open the way for a different Ig productfor immunomodulation. More than 100 years after initiation of antibody treatment andmore than 50 years after treatment of patients with PID became state-of-the-art, theprovision of antibodies/treatment with Ig (IV or subcutaneous) to patients who havePID remains the life-saving, classic, and first indication for IVIg, and all Ig productscurrently available are licensed for it.
ACKNOWLEDGMENTS
The author would like to thank Barbara Alving, MD, Department of Health andHuman Services, National Institutes of Health, for providing the proceedings of the1979 workshop and Hermann Wolf, MD, for fruitful discussions.
REFERENCES
1. von Behring EA, Kitasato S. Uber das zustandekommen der diphtherie-immuni-tat und der tetanus-immunitat bei tieren. Dtsch Med Wochenschr 1890;16:1113–4.
2. Ehrlich P. Experimentelle untersuchungen uber immunitat. I. Uber Ricin. II. UberAbrin. Dtsch Med Wochenschr 1891;17:976–9, 1218–9.
3. Wolff-Eisner A. Handbuch der serumtherapie und experimentellen therapie: einhandbuch fur klinik und praxis. Munchen: J.F. Lehmanns Verlag; 1910.
4. Verhandlungen der Berliner Medizinischen Gesellschaft 1894.5. Calmette A. Die gifte, die gifttiere und die serumtherapie gegen die tierischen
gifte. Paris: Masson; 1907.6. Flexner S. Experimental cerebro-spinal meningitis in monkeys. J Exp Med 1907;9:
142–67.7. Flexner S. Concerning a serum therapy for experimental infections with Diplo-
coccus intracellularis. J Exp Med 1907;9:168–85.8. Eibl MM, Wedgwood RJ. Intravenous immunglobulin: a review. Immunodefic Rev
1989;1(Suppl):1–42.9. Cenci F. Alcune esperienze di sieroimmunizzaziuone e sieroterapie nel norbillo.
Rivista di Clinica e Pediatrica 1907;5:1017–25.10. McKhann CF, Chu FT. Use of placental extract in prevention and modification of
measles. Am J Dis Child 1933;45:475–9.11. Gibson RB. The concentration of antitoxin for therapeutic use. J Biol Chem 1906;
1:161–70.12. Karelitz S. Prophylaxis against measles with the globulin fraction of immune
adult serum. Am J Dis Child 1938;55:768–75.13. Tiselius A, Kabat EA. Electrophoresis of immune serum. Science 1938;87:416–7.14. Cohn EJ. Blood proteins and their therapeutic value. Science 1945;101:51–6.

History of Immunoglobulin Replacement 753
15. Cohn EJ, Luetscher JA Jr, Oncley JL, et al. Preparation and properties of serumand plasma proteins. J Am Chem Soc 1940;62:3396–400.
16. Cohn EJ. The history of plasma fractionation. In: Merler E, editor. Advances inmilitary medicine, vol. 1. Boston: Little, Brown and Company; 1948. p. 364–443.
17. Janeway CA. The development of clinical uses of immunoglobulins: a review. In:Immunoglobulins. Washington, DC: National Academy of Science; 1970. p.3–14.
18. Stokes J, Maris EP, Gellis SS. Chemical, clinical, and immunological studies onthe products of human plasma fractionation. XI. The use of concentrated normalhuman serum gamma globulin (human immune serum globulin) in the prophy-laxis and treatment of measles. J Clin Invest 1944;23:531–40.
19. Stokes J, Neefe JR. The prevention and attenuation of infectious hepatitis bygamma globulin. JAMA 1945;127:144–5.
20. Nation NS, Pierce NF, Adler SJ, et al. Tetanus; the use of human hyperimmuneglobulin in treatment. Calif Med 1963;98:305–7.
21. Pollack W, Gorman JG, Freda VJ, et al. Results of clinical trials of RhoGAM inwomen. Transfusion 1968;8(3):151–3.
22. Zuckermann AJ. Prophylaxis of hepatitis type B: immunoglobulins and vaccines.Clin Gastroenterol 1980;9(1):65–83.
23. Goebel U, Schwamborn D, Moeller B, et al. Intravenous hyperimmunoglobulin toprevent varicella-zoster infection. Pediatr Infect Dis 1987;6(23):224.
24. Welliver RC. Respiratory syncytial virus immunoglobulin and monoclonal anti-bodies in the prevention and treatment of respiratory syncytial virus infection.Semin Perinatol 1998;22(1):87–95.
25. Storch GA. Humanized monoclonal antibody for prevention of respiratory syncy-tial virus infection. Pediatrics 1998;102:648–51.
26. Centers for Diseases Control and Prevention (CDC). Recommendations of theImmunization Practices Advisory Committee (ACIP) Varicella-Zoster ImmuneGlobulin for the prevention of chickenpox. Morbidity and Mortality Weekly Report(MMWR) 1984;33(7):84–90, 95–100.
27. Bruton OC. Agammaglobulinemia. Pediatrics 1952;9:722–7.28. Good RA, Varco RL. A clinical and experimental study of agammaglobulinemia.
Lancet 1955;75(6):245–71.29. Gitlin D, Janeway DC. Agammaglobulinemia: congenital, acquired and transient
forms. Prog Hematol 1956;1:318–29.30. Barandun S. Die gammaglobulin-therapie: chemische, immunologische und
klinische grundlagen. Bibl Haematol 1964;17:1–138.31. Stiehm ER. Immunodeficieny disorders: general considerations. In: Stiehm ER,
Fulginiti VA, editors. Immunologic disorders in infants and children. Philadelphia:WB Saunders; 1973. p. 145–67.
32. Hitzig WH, Muntener U. Conventional immunoglobulin therapy. In: Gerdsma D,editor. Immunodeficiency in man and animals. Sunderland (MA): Sinauer Asso-ciates; 1975. p. 339–42.
33. Domz CA, Dickson DR. The agammaglobulinemics: relation and implications.Am J Med 1957;23:917–27.
34. Janeway CA, Rosen FS. The gamma globulins. IV. Therapeutic uses of gammaglobulin. N Engl J Med 1966;275:826–31.
35. Report of a WHO Scientific Group. The use of human immunoglobulin. TechnicalReport Series 1966;327.
36. Fudenberg HH, Good RA, Goodman HC, et al. Primary immunodeficiencies:report of a World Health Organization committee. Pediatrics 1971;47:927–46.

Eibl754
37. Cooper MD, Faulk WP, Fudenberg HH, et al. Classification of primary immuno-deficiencies. N Engl J Med 1973;288:966–7.
38. Report of a WHO Scientific Group. Immunodeficiency: technical report series. Doc-ument no. 630. Geneva (Switzerland): World Health Organization; 1978. p. 3–80.
39. Abernathy RS, Strem EL, Good KA. Chronic asthma in childhood: double-blindcontrolled study of treatment with gamma-globulin. Pediatrics 1958;21(6):980–93.
40. MRC Working Party on Hypogammaglobulinaemia. Hypogammaglobulinaemiain the United Kingdom. London: Her Majestys Stationery Office; 1971.
41. Tabor E, Gerety RJ. Transmission of hepatitis B by immuno serum globulin. Lancet1979;2(8155):1293.
42. John TJ, Ninan GT, Rajagopalan HS, et al. Epidemic hepatitis B caused bycommercial human immunoglobulin. Lancet 1979;1(8125):1074.
43. da Silva LC, Sette H, Antonacio F, et al. Commercial gammaglobulin (CGG) asa possible vehicle of transmission of HbsAG in familial clustering. Rev Inst MedTrop Sao Paulo 1977;19:352–4.
44. Petrilli FL, Crovari P, De Flora S. Hepatitis B in subjects treated with a drugcontaining immunoglubulins. J Infect Dis 1977;135:252–8.
45. Morell A, Schnoz M, Barandun S. Build-up and maintenance of IgG serum con-centrations with intravenous immunoglobulin in patients with primary humoralimmunodeficiency. Vox Sang 1982;43:212–9.
46. Smith GN, Griffiths B, Mollison D, et al. Uptake of IgG after intramuscular andsubcutaneous injection. Lancet 1972;1(7762):1208–12.
47. Haeney MR, Carter GF, Yeoman WB, et al. Long-term parenteral exposure tomercury in patients with hypogammaglobulinemia. BMJ 1979;2:12–4.
48. Matheson DS, Clarkson TW, Gelfand EW. Mercury toxicity (acrodynia) inducedby long-term injection of gammaglobulin. J Pediatr 1980;97:153–5.
49. Barandun S, Kistler P, Jeunet F, et al. Intravenous administration of humangammaglobulin. Vox Sang 1962;7:157–74.
50. Ammann AJ, Ashman RF, Buckley RH, et al. Use of intravenous gamma-globulinin antibody immunodeficiency: results of a multicenter controlled trial. ClinImmunol Immunopathol 1982;22:60–7.
51. Lederman HM, Winkelstein JA. X-linked agammaglobulinemia: an analysis of96 patients. Medicine (Baltimore) 1985;64:145–56.
52. Quartier P, Debre M, De Blic J, et al. Early and prolonged intravenous immuno-globulin replacement therapy in childhood agammaglobulinemia: a retrospectivesurvey of 31 patients. J Pediatr 1999;134:589–96.
53. Cunningham-Rundles C, Siegal FP, Smithwick EM, et al. Efficacy of intravenousimmunoglobulin in primary humoral immunodeficiency disease. Ann Intern Med1984;101:435–9.
54. Nolte MT, Pirofsky B, Gerritz GA, et al. Intravenous immunoglobulin therapy forantibody deficiency. Clin Exp Immunol 1979;36:237–43.
55. Roifman CM, Lederman HM, Lavi S, et al. Benefit of intravenous IgG replace-ment in hypogammaglobulinemic patients with chronic sinopulmonary disease.Am J Med 1985;79:171–4.
56. Skvaril F, Probst M, Steinbuch A, et al. Distribution of IgG subclasses incommercial and some experimental g-globulin preparations. Vox Sang 1977;32:335–8.
57. Romer J, Morgenthaler JJ, Scherz R, et al. Characterization of various immuno-globulin preparations for intravenous application. I. Protein composition andantibody content. Vox Sang 1982;42:62–73.

History of Immunoglobulin Replacement 755
58. Morell A, Schurch B, Ryser D, et al. In vivo behaviour of gamma globulin prep-arations. Vox Sang 1980;38:272–83.
59. Ochs HD. Intravenous immunoglobulin therapy of patients with primaryimmunodeficiency syndromes: efficacy and safety of a new immune globulin prep-aration. In: Alving BM, Finlayson JS, editors. Immunglobulins: characteristics anduses of intravenous preparations. Proceedings of a workshop, DHHS Publicationno. (FDA)-80-9005. Washington DC: US Department of Health and HumanServices; 1979. p. 9–14.
60. Pirofsky B, Anderson CJ, Bardana EJ. Therapeutic and detrimental effects of in-travenous immunoglobulin therapy. In: Alving BM, Finlayson JS, editors. Immun-globulins: characteristics and uses of intravenous. Proceedings of a workshop,DHHS Publication no. (FDA)-80-9005. Washington DC: US Department of Healthand Human Services; 1979. p. 15–22.
61. Barandun S, Morell A, Skvaril F. Clinical experiences with immunoglobulin for in-travenous use. In: Alving BM, Finlayson JS, editors. Immunglobulins: character-istics and uses of intravenous preparations. Proceedings of a workshop, DHHSPublication no. (FDA)-80-9005. Washington DC: US Department of Health andHuman Services; 1979. p. 31–5.
62. Mondorf AW, Duswald KH. Intravenous immunoglobulin in high-risk patients: ef-ficacy and tolerance of long-term administration. In: Alving BM, Finlayson, JS,editors. Immunglobulins: characteristics and uses of intravenous preparations.Proceedings of a workshop, DHHS Publication no. (FDA)-80-9005. WashingtonDC: US Department of Health and Human Services; 1979. p. 37–40.
63. Alving BM, Finlayson JS. Preface. In: Alving BM, Finlayson JS, editors. Immun-globulins: characteristics and uses of intravenous preparations. Proceedings ofa workshop, DHHS Publication no. (FDA)-80-9005. Washington DC: US Depart-ment of Health and Human Services; 1979. p. 9–14.
64. Overview of potential uses for immunoglobulin preparations, possible etiologiesof adverse reactions and ideal characteristics of intravenous preparations. In:Alving BM, Finlayson JS, editors. Immunglobulins: characteristics and uses ofintravenous preparations, proceedings of a workshop, DHHS Publication no.(FDA)-80-9005. Washington DC: US Department of Health and Human Services;1979. p. 229–33.
65. Eibl M. Treatment of defects of humoral immunity. Birth Defects Orig Artic Ser1983;19(3):193–200.
66. Kjellman H. Adverse reactions to human immune serum globulin in Sweden(1969–1978). In: Alving BM, Finlayson JS, editors. Immunglobulins: characteris-tics and uses of intravenous preparations. Proceedings of a workshop, DHHSPublication no. (FDA)-80-9005. Washington DC: US Department of Health andHuman Services; 1979. p. 143–50.
67. Hanson LA, Bjorkander J, Ljunggren C, et al. Problems with use of immunoglob-ulins in treatment of immunodeficiency patients. In: Alving BM, Finlayson JS, ed-itors. Immunglobulins: characteristics and uses of intravenous preparations.Proceedings of a workshop, DHHS Publication no. (FDA)-80-9005. WashingtonDC: US Department of Health and Human Services; 1979. p. 150–9.
68. Cassidy JT, Stein LD, Reynolds RT. Intravenous immune globulin: five-yearclinical experience with efficacy and adverse reactions in immunodeficiency.In: Alving BM, Finlayson JS, editors. Immunglobulins: characteristics and usesof intravenous preparations. Proceedings of a workshop, DHHS Publicationno. (FDA)-80-9005. Washington DC: US Department of Health and HumanServices; 1979. p. 161–6.

Eibl756
69. Funakoshi S. Suyama T, Naito R. Use of Intravenous immunoglobulin in Japan.In: Alving BM, Finlayson JS, editors. Immunglobulins: characteristics and usesof intravenous preparations. Proceedings of a workshop, DHHS Publicationno. (FDA)-80-9005. Washington DC: US Department of Health and HumanServices; 1979. p. 219–25.
70. Busse PJ, Razvi S, Cunningham-Rundles C. Efficacy of intravenous immuno-globulin in the prevention of pneumonia in patients with common variableimmunodeficiency. J Allergy Clin Immunol 2002;109(6):1001–4.
71. Brunko P. Community requirements relating to drugs derived from human bloodand plasma. Ann Pharm Fr 1994;52(2):89–98.
72. Orange JS, Hossny EM, Weiler CR, et al. Use of intravenous immunoglobulin inhuman disease: a review of evidence by members of the Primary Immunodefi-ciency Committee of the American Academy of Allergy, Asthma and Immunology.J Allergy Clin Immunol 2006;117:S525–53.
73. Barandun S, Isliker H. Development of immunoglobulin preparations for intrave-nous use. Vox Sang 1986;51:157–60.
74. Imbach P, d’Apuzzo V, Hirt A. High-dose intravenous gammaglobulin for idiopathicthrombocytopenic purpura in childhood. Lancet 1981;1(8232):1228–31.
75. Fehr J, Hofmann V, Kappeler U. Transient reversal of thrombocytopenia inidiopathic thrombocytopenic purpura by high-dose intravenous gamma globulin.N Engl J Med 1982;306:1254–8.
76. Mori PG, Mancuso G, del Principe D, et al. Chronic idiopathic thrombocytopeniatreated with immunoglobulin. Arch Dis Child 1983;58:851–5.
77. Carroll RR, Noyes WD, Rosse WH, et al. Intravenous immunoglobulin administra-tion in the treatment of severe chronic immune thrombocytopenic purpura. AmJ Med 1984;76(Suppl 3a):181–6.
78. Warrier I, Lusher JM. Intravenous gammaglobulin treatment for chronicidiopathic thrombocytopenic purpura in children. Am J Med 1984;76:193–8.
79. Warrier LA, Lusher JM. Intravenous gammaglobulin (Gamimune) for treatment ofchronic idiopathic thrombocytopenic purpura (ITP): a two-year follow-up. AmJ Hematol 1986;23:323–8.
80. Bussel J. Intravenous immune serum globulin in immune thrombocytopenia:clinical results and biochemical evaluation. Vox Sang 1985;49(Suppl 1):44–50.
81. Vos JJ, van Aken WG, Engelffriet CP, et al. Intravenous gammaglobulin therapyin idiopathic thrombocytopenic purpura. Vox Sang 1985;49:92–100.
82. Abe T, Matsuda J, Kawasugi K, et al. Clinical effect of intravenous immuno-globulin on chronic idiopathic thrombocytopenic purpura. Blut 1983;47(2):69–75.
83. Becton DL, Kinney TR, Chaffee S, et al. High-dose intravenous immunoglobulinfor severe platelet alloimmunization. Pediatrics 1984;74:1120–3.
84. Hanada T, Saito K, Nagasawa T, et al. Intravenous gammaglobulin therapy forthromboneutropenic neonates of mothers with systemic lupus erythematosus.Eur J Haematol 1987;38:400–4.
85. Derycke M, Dreyfus M, Ropert JC, et al. Intravenous immunoglobulin for neona-tal isoimmune thrombocytopenia. Arch Dis Child 1985;60:667–9.
86. Berlin G, Selbing A, Ryden G. Rhesus haemolytic disease treated with high-dose intravenous immunoglobulin. [letter]. Lancet 1985;1(8438):1153.
87. Drachman DB, Adams RN, Josifek LF, et al. Functional activities of autoanti-bodies to acetylcholine receptors and the clinical severity of myasthenia gravis.N Engl J Med 1982;307:769–75.

History of Immunoglobulin Replacement 757
88. Morel E, Eymard B, Vernet-der-Garabedian B, et al. Neonatal myasthenia gravis:a new clinical and immunologic appraisal on 30 cases. Neurology 1988;38:138–42.
89. Ruhr G, Kusterer K, Schille M, et al. Treatment of Crohns disease and ulcerativecolitis with 7S-immunoglobulin. [letter]. Lancet 1987;I:170.
90. Pocecco M, de Campo C, Cantoni L, et al. Effect high dose intravenous IgG innewly diagnosed diabetic children. Helv Paediatr Acta 1987;42:289–95.
91. Silverstein J, Maclaren N, Riley W, et al. Immunosuppression with azathioprineand prednisone in recent-onset-dependent diabetes mellitus. N Engl J Med1988;319:599–604.
92. Antonelli A, Navarranne A, Palla R, et al. Pretibial myxedema and high-doseintravenous immunoglobulin treatment. Thyroid 1994;4(4):399–408.
93. Baschieri L, Antonelli A, Nardi S, et al. Intravenous immunoglobulin versus cor-ticosteroid in treatment of Graves ophthalmopathy. Thyroid 1997;7(4):579–85.
94. Kahaly G, Pitz S, Muller-Forell W, et al. Randomized trial of intravenous immuno-globulin versus prednisolone in Graves ophthalmopathy. Clin Exp Immunol1996;106(2):197–202.
95. Czernik A, Bystryn JC. Improvement of intravenous immunoglobulin therapy forbullous pemphigoid by adding immunosuppressive agents: marked improve-ment in depletion of circulating autoantibodies. Arch Dermatol 2008;144(5):658–61.
96. Tovo PA, Miniero R, Fiandino G, et al. Fc-depleted vs intact intravenous immuno-globulin in chronic ITP. J Pediatr 1984;105(4):676–7.
97. Burdach SE, Evers KG, Geursen RG. Treatment of acute idiopathic thrombocy-topenic purpura of childhood with intravenous immunoglobulin G: comparativeefficacy of 7S and 5S preparations. J Pediatr 1986;109(5):770–5.
98. Basta M, Langlois PF, Marques M, et al. High-dose intravenous immunoglobulinmodifies complement-mediated in vivo clearance. Blood 1989;74(1):326–33.
99. Frank MM, Basta M, Fries LF. The effects of intravenous immune globulin oncomplement-dependent immune damage of cells and tissues. Clin ImmunolImmunopathol 1992;62(1 Pt 2):S82–6.
100. Lin RY, Racis SP. In vivo reduction of circulating Clq binding immune complexesby intravenous gammaglobulin administration. Int Arch Allergy Appl Immunol1986;79:286–90.
101. Dalakas MC, Illa I, Dambrosia JM, et al. A controlled trial of high-dose intrave-nous immune globulin infusions as treatment for dermatomyositis. N EnglJ Med 1993;329(27):1993–2000.
102. Basta M, Dalakas MC. High-dose intravenous immunoglobulin exerts its bene-ficial effect in patients with dermatomyositis by blocking endomysial depositionof activated complement fragments. J Clin Invest 1994;94(5):1729–35.
103. Bussel JB. Fc receptor blockade and immune thrombocytopenic purpura.Semin Hematol 2000;37(3):261–6.
104. Bussel JB, Graziano JN, Kimberly RP, et al. Intravenous anti-D treatment ofimmune thrombocytopenic purpura: analysis of efficacy, toxicity, and mechanismof effect. Blood 1991;77(9):1884–93.
105. Becker T, Kuenzlen E, Salama A, et al. Treatment of childhood idiopathic throm-bocytopenic purpura with Rhesus antibodies (anti-D). Eur J Pediatr 1986;45:166–9.
106. Blanchette V, Imbach P, Andrew M, et al. Randomised trial of intravenous immu-noglobulin G, intravenous anti-D, and oral prednisone in childhood acuteimmune thrombocytopenic purpura. Lancet 1994;344(8924):703–7.

Eibl758
107. Clarkson SB, Kimberly RP, Valinsky JE, et al. Blockade of clearance of immunecomplexes by an anti-Fc gamma receptor monoclonal antibody. J Exp Med1986;164(2):474–89.
108. Clarkson SB, Bussel JB, Kimberly RP, et al. Treatment of refactory immunethrombocytopenic purpura with an anti-Fc gamma-receptor antibody. N EnglJ Med 1986;314(19):1236–9.
109. Andemariam B, Bussel J. New therapies for immune thrombocytopenic purpura.Curr Opin Hematol 2007;14:427–31.
110. Kazatchkine MD, Kaveri SV. Immunomodulation of autoimmune and inflamma-tory diseases with intravenous immune globulin. N Engl J Med 2001;345(10):747–55.
111. Marchalonis JJ, Kaymaz H, Dedeoglu F, et al. Human autoantibodies reactivewith synthetic autoantigens from T-cell receptor beta chain. Proc Natl AcadSci U S A 1992;89:3325–9.
112. Kaveri S, Vassilev T, Hurez V. Antibodies to a conserved region of HLA class Imolecules, capable of modulation CD8 T cell-mediated function, are presentin pooled normal immunoglobulin for therapeutic use. J Clin Invest 1996;97:865–9.
113. Prasad NK, Papoff G, Zeuner A, et al. Therapeutic preparations of normal poly-specific IgG (IVIg) induce apoptosis in human lymphocytes and monocytes:a novel mechanism of action of IVIg involving the Fas apoptotic pathway.J Immunol 1998;161:3781–90.
114. Viard I, Wehrli P, Bullani R, et al. Inhibition of toxic epidermal necrolysis byblockade of CD95 with human intravenous immunoglobulin. Science 1998;282:490–3.
115. Dwyer DS, Bradley RJ, Urquhart CK, et al. Naturally occurring anti-idiotypeantibodies in myasthenia gravis patients. Nature 1983;301:611–4.
116. Negi VS, Elluru S, Siberil S, et al. Intravenous immunoglobulin: an update on theclinical use and mechanism of action. J Clin Immunol 2007;27(3):233–45.
117. Andersson J, Skans�en-Saphir U, Sparrelid E, et al. Intravenous immune globulinaffects cytokine production in T lymphocytes and monocytes/macrophages.Clin Exp Immunol 1996;104(Suppl 1):10–20.
118. Okitsu-Negishi S, Furusawa S, Kawa Y, et al. Suppressive effect of intravenousimmunoglobulins on the activity of interleukin-1. Immunol Res 1994;13(1):49–55.
119. Sullivan KM, Kopecky KJ, Jocom J. Immunomodulatory and antimicrobial efficacyof intravenous immunoglobulin in bone marrow transplantation. N Engl J Med1990;323(11):705–12.
120. Winston DJ, Ho WG, Rasmussen LE, et al. Use of intravenous immunoglobulinfor prevention of graft-versus-host disease and infection after allogeneic bonemarrow transplantation. Bone Marrow Transplant 2001;28:187–96.
121. Jordan SC, Vo A, Bunnapradist S, et al. Intravenous immune globulin treatmentinhibits crossmatch positivity and allows for successful transplantation ofincompatible organs in living-donor and cadaver recipients. Transplantation2003;76:631–6.
122. Cordonnier C, Chevret S, Legrand M, et al. Should immunoglobulin therapy beused in allogenic stem-cell transplantation? A randomized, double-blind, doseeffect, placebo-controlled, multicenter trial. Ann Intern Med 2003;139:8–18.
123. Glotz D, Antoine C, Julia P, et al. Desensitization and subsequent kidneytransplantation of patients using intravenous immunoglobulins (IVIg). AmJ Transplant 2002;2:758–60.

History of Immunoglobulin Replacement 759
124. Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N EnglJ Med 2003;348(2):138–50.
125. Munoz C, Carlet J, Fitting C, et al. Dysregulation on in vitro cytokine productionby monocytes during sepsis. J Clin Invest 1991;88:1747–54.
126. Ertel W, Kremer JP, Kenney J, et al. Downregulation of proinflammatory cytokinerelease in whole blood from septic patients. Blood 1995;85(5):1341–7.
127. Faulkner L, Cooper A, Fantino C, et al. The mechanism of superantigen-mediated toxic shock: not a simple Th1 cytokine storm. J Immunol 2005;175(10):6870–7.
128. Sriskandan S, Altmann DM. The immunology of sepsis. J Pathol 2008;214:211–23.
129. Aukrust P, Froland SS, Liabakk NB, et al. Release of cytokines, soluble cytokinereceptors, and interleukin-1 receptor antagonist after intravenous immunoglob-ulin administration in vivo. Blood 1994;84(7):2136–43.
130. Wolf HM, Hauber I, Gulle H, et al. Anti-inflammatory properties of human serumIgA: induction of IL-1 receptor antagonist and Fc alpha R (CD89)-mediateddown-regulation of tumour necrosis factor-alpha (TNF-alpha) and IL-6 in humanmonocytes. Clin Exp Immunol 1996;105(3):537–43.
131. Abe Y, Horiuchi A, Miyake M, et al. Anti-cytokine nature of natural humanimmunoglobulin: one possible mechanism of the clinical effect of intravenousimmunoglobulin therapy. Immunol Rev 1994;139:5–19.
132. Arend WP, Leung DY. IgG induction of IL-1 receptor antagonist production byhuman monocytes. Immunol Rev 1994;139:71–8.
133. Dinarello CA. Is there a role for interleukin-1 blockade in intravenous immuno-globulin therapy? Immunol Rev 1994;139:173–88.
134. Jungi TW, Eiholzer J, Lerch PG, et al. The capacity of various types of immuno-globulin for intravenous use to interact with FC receptors of human monocytesand macrophages. Blut 1986;53(4):321–32.
135. Kimberly RP, Salmon JE, Bussel JB, et al. Modulation of mononuclear phagocytefunction by intravenous gamma-globulin. J Immunol 1984;132:745–50.
136. Mannhalter JW, Ahmad R, Wolf HM, et al. Effect of polymeric IgG on humanmonocyte functions. Int Arch Allergy Appl Immunol 1987;82(2):159–67.
137. Newburger JW, Takahashi M, Burns JC, et al. The treatment of Kawasakisyndrome with intravenous gamma globulin. N Engl J Med 1986;315(6):341–7.
138. Newburger JW, Takahashi M, Beiser AS. A single intravenous infusion of gammaglobulin as compared with four infusions in the treatment of acute Kawasakisyndrome. N Engl J Med 1991;324:1633–9.
139. Newburger JW, Sleeper LA, McCrindle BW, et al. Randomized trial of pulsedcorticosteroid therapy for primary treatment of Kawasaki disease. N EnglJ Med 2007;356(7):663–75.
140. NIH consensus conference. Intravenous immunoglobulin: prevention and treat-ment of disease. JAMA 1990;264(24):3189–93.
141. Intravenous immune globulin for the prevention of bacterial infections in childrenwith symptomatic human immunodeficiency virus infection. The National Insti-tute of Child Health and Human Developments Intravenous ImmunoglobulinStudy Group. N Engl J Med 1991;325(2):73–80.
142. Spector SA, Gelber RD, McGrath N, et al. A controlled trial of intravenousimmune globulin for the prevention of serious bacterial infections in childrenreceiving yidovudine for advanced human immunodeficiency virus infection.Pediatric AIDS Clinical Trials Group. N Engl J Med 1994;331(18):1181–7.

Eibl760
143. Mofenson LM, Moye J Jr, Korelitz J, et al. Crossover of placebo patients to intra-venous immunoglobulin confirms efficacy for prophylaxis of bacterial infectionsand reduction of hospitalizations in human immunodeficiency virus-infectedchildren. The National Institute of Child Health and Human Development Intrave-nous Immunoglobulin Clinical Trial Study Group. Pediatr Infect DisJ 1994;13(6):477–84.
144. Griffiths H, Brennan V, Lea J, et al. Crossover study of immunoglobulin replace-ment therapy in patients with low-grade B-cell tumors. Blood 1989;73(2):366–8.
145. [No authors listed] Intravenous immunoglobulin for the prevention of infectionin chronic lymphocytic leukemia: a randomised, controlled clinical trial. Cooper-ative Group for the Study of Immunoglobulin in Chronic Lymphocytic Leukemia.N Engl J Med 1988;319(14):902–7.
146. Chapel HM, Lee M, Hargreaves R, et al. Randomised trial of intravenous immu-noglobulin as prophylaxis against infection in plateau-phase multiple myeloma.The UK Group for Immunoglobulin Replacement Therapy in Multiple Myeloma.Lancet 1994;343(8905):1059–63.
147. van Doorn PA, Brand A, Strengers PF, et al. High-dose intravenous immunoglob-ulin treatment in chronic inflammatory demyelinating polyneuropathy: a double-blind, placebo-controlled study. Neurology 1990;40(2):209–12.
148. Vermeulen M, van Doorn PA, Brand A. Intravenous immunoglobulin treatment inpatients with chronic inflammatory demyelinating polyneuropathy: a doubleblind, placebo controlled study. J Neurol Neurosurg Psychiatry 1993;56(1):36–9.
149. van der Mech�e FG, Schmitz PI. A randomized trial comparing intravenousimmune globulin and plasma exchange in Guillain-Barr�e syndrome. DutchGuillain-Barr�e Study Group. N Engl J Med 1992;326(17):1123–9.
150. Sweet LK, Howell J, McMurray LG, et al. The use of normal serum gammaglobulin antibodies (human) concentrated (immune serum globulin) in the treat-ment of premature infants. J Pediatr 1946;28:571–3.
151. Fanaroff AA, Korones SB, Wright LL, et al. A controlled trial of intravenousimmune globulin to reduce nosocomial infections in very-low-birth-weightinfants. National Institute of Child Health and Human Development NeonatalResearch. N Engl J Med 1994;330:1107–13.
152. Sandberg K, Fasth A, Berger A, et al. Pretern infants with low immunoglobulinG levels have increased risk of neonatal sepsis but do not benefit from prophy-lactic immunoglobulin G. J Pediatr 2000;137:623–8.
153. Ohlsson A, Lacy J. Intravenous immunoglobulin for suspected or subsequentlyproven infection in neonates. Cochrane Database Syst Rev 2004;CD001239.
154. van der Mech�e FG, van Doorn PA. The current place of high-dose immunoglob-ulins in the treatment of neuromuscular disorders. Muscle Nerve 1997;20(2):136–47.
155. Dalakas MC. Controlled studies with high-dose intravenous immunoglobulin inthe treatment of dermatomyositis, inclusion body myositis, and polymyositis.Neurology 1998;51(6 Suppl 5):S37–45.
156. Gold R, Stangel M, Dalakas MC. Drug insight: the use of intravenous immuno-globulin in neurology: therapeutic considerations and practical issues. NatClin Pract Neurol 2007;3(1):36–44.
157. Ratko TA, Burnett DA, Foulke GE, et al. Recommendations for off-label use ofintravenously administered immunoglobulin preparations. University HospitalConsortium Expert Panel for Off-Label Use of Polyvalent Intravenously Adminis-tered Immunoglobulin Preparations. JAMA 1995;273(23):1865–70.

History of Immunoglobulin Replacement 761
158. Sacher RA, Ivig Advisory Panel. Intravenous immunoglobulin consensus state-ment. J Allergy Clin Immunol 2001;108(4 Suppl):S139–46.
159. Darabi K, Abdel-Wahab O, Dzik WH. Current usage of intravenous immuneglobulin and the rationale behind it: the Massachusetts General Hospital dataand a review of the literature. Transfusion 2006;46(5):741–53.
160. Sorensen R. Expert opinion regarding clinical and other outcome considerationsin the formulary review of immune globulin. J Manag Care Pharm 2007;13(3):278–83.
161. Ochs HD, Fischer SH, Wedgwood RJ, et al. Comparison of high-dose and low-dose intravenous immunoglobulin therapy in patients with primary immunodefi-ciency diseases. Am J Med 1984;76(3A):78–82.
162. Roifman CM, Gelfand EW. Replacement therapy with high dose intravenousgamma-globulin improves chronic sinopulmonary disease in patients with hypo-gammaglobulinemia. Pediatr Infect Dis J 1988;7(5 Suppl):S92–6.
163. Gelfand EW, Reid B, Roifman CM. Intravenous immune serum globulin replace-ment in hypogammagloublinemia: a comparison of high- versus low-dose therapy.Monogr Allergy 1988;23:177–86.
164. Van T, Sussman G, Pruzanski W. Impact of intravenous infusion of low and highdoses of gamma globulins (IVIG) on phagocytic functions in adults with primaryhumoral immunodeficiency. Inflammation 1994;18(4):419–26.
165. Skull S, Kemp A. Treatment of hypogammaglobulinaemia with intravenousimmunoglobulin, 1973–93. Arch Dis Child 1996;74(6):527–30.
166. Liese JG, Wintergerst U, Tympner KD, et al. High- vs low-dose immunoglobulintherapy in the long-term treatment of X-linked agammaglobulinemia. Am J DisChild 1992;146:335–9.
167. Roifman CM, Levison H, Gelfand EW. High-dose versus low-dose intravenousimmunoglobulin in hypogammaglobulinemia and chronic lung disease. Lancet1987;1(8541):1075–7.
168. Eijkhout HW, van Der Meer JW, Kallenberg CG, et al. The effects of two differentdosages of intravenous immunoglobulin on the incidence of recurrent infectionsin patients with primary hypogammaglobulinemia: a randomized, double-blind,multicenter crossover trial. Ann Intern Med 2001;135:165–74.
169. Buckley RH. Long term use of intravenous immune globulin in patients weightprimary immunodeficiency diseases: inadequacy of current dosage practicesand approaches to the problem. J Clin Immunol 1982;2(Suppl):S15–21.
170. Bernatowska E, Madalinski K, Janowicz W, et al. Results of a prospective con-trolled two-dose crossover study with intravenous immunoglobulin and compar-ison (retrospective) with plasma treatment. Clin Immunol Immunopathol 1987;43:153–62.
171. de Gracia J, Vendrell M, Alvarez A, et al. Immunoglobulin therapy to control lungdamage in patients with common variable immunodeficiency. Int Immunophar-macol 2004;47:745–53.
172. Roifman CM, Schroeder H, Berger M, et al. Comparison of the efficacy of IGIV-C,10% (caprylate/chromatography) and IGIV-SD, 10% as replacement therapy inprimary immune deficiency: a randomized double-blind trial. Int Immunophar-macol 2003;3(9):1325–33.
173. Dwyer JM, Erlendsson K. Intraventricular gamma-globulin for the managementof enterovirus encephalitis. Pediatr Infect Dis J 1988;7:530–3.
174. Mease PJ, Ochs HD, Wedgwood RJ. Successful treatment of echovirus menin-goencephalitis and myositis-fasciitis with intravenous immune globulin therapy

Eibl762
in patient with X-linked agammaglobulinemia. N Engl J Med 1981;304:1278–81.
175. Mease PJ, Ochs HD, Corey L, et al. Echovirus encephalitis/myositis in X-linkedagammaglobulinemia. N Engl J Med 1985;313:758.
176. Quartier P, Foray S, Casanova JL, et al. Enteroviral meningoencephalitis inX-linked agammaglobulinemia: intensive immunoglobulin therapy and sequen-tial viral detection in cerebrospinal fluid by polymerase chain reaction. PediatrInfect Dis J 2000;19:1106–8.
177. Part 3. Adverse reactions to immunoglobulins: methods used to produce ‘‘safe’’immunoglobulin preparations. In: Alving BM, Finlayson JS, editors. Immunoglob-ulins: characteristics and uses of intravenous preparations. Proceedings ofa workshop, DHHS Publication no. (FDA)-80-9005. Washington DC: US Depart-ment of Health and Human Services; 1979. p. 137–225.
178. Pirofsky B. Intravenous immune globulin therapy in hypogammaglobulinemia:a review. Am J Med 1984;76(Suppl 3a):53–60.
179. Gislason D, Hanson LA, Kjellman H, et al. Intravenous gamma-globulin infusionsin patients with hypogammaglobulinemia. Vox Sang 1978;34:143–8.
180. Cunningham-Rundles C, Day NK, Wahn V, et al. Reactions to intravenous gam-maglobulin infusions and immune complex formation. In: Nydegger UE, editor.Immunochemotherapy: a guide to immunoglobulin prophylaxis and therapy.London, New York: Academic Press; 1981. p. 447–50.
181. Barandun S, Morell A. Adverse reactions to immunoglobulin preparations. In:Nydegger UE, editor. Immunochemotherapy: a guide to immunoglobulin pro-phylaxis and therapy. London, New York: Academic Press; 1981. p. 223–7.
182. Pierce LR, Jain N. Risks associated with the use of intravenous immunoglobulin.Transfus Med Rev 2003;17:241–51.
183. Wolberg AS, Kon RH, Monroe DM, et al. Coagulation factor XI is a contaminanton intravenous immunoglobulin preparations. Am J Hematol 2000;65(1):30–4.
184. Lederman HM, Roifman CM, Lavi S. Corticosteroids for prevention of adversereactions to intravenous immune serum globulin infusions in hypogammaglobu-linemic patients. Am J Med 1986;81:443–6.
185. Aronson DL, Finlayson JS. Historical and future therapeutic plasma derivatives(epilogue). Semin Thromb Hemost 1980;VI:121–39.
186. Eibl M. Treatment of defects of humoral immunity. In: Wedgwood RJ, Rosen FS,Paul NW, editors, Primary immunodeficiency diseases. Birth defects. New York:Alan R. Liss; 1983. p. 193–200.
187. Goodwin JS, Webb DR. Regulation of the immune response by prostaglandins.Clin Immunol Immunopathol 1980;15:106–22.
188. Passwell JH, Dayer JM, Merler E. Increased prostaglandin production by humanmonocytes after membrane receptor activation. J Immunol 1979;123:115–20.
189. Camussi G, Aglietta M, Coda R, et al. Release of platelet activating factor (PAF)and histamine. II. The cellular origin of human PAF: monocytes, polymorphonu-clear neutrophila and basophils. Immunol 1981;42:191–9.
190. Eibl MM. PKA contamination of immunoglobulin G. [letter]. N Engl J Med 1985;313:581.
191. Alving BM, Hojima Y, Pisano JJ, et al. Hypotension associated with prekallikreinactivator (Hageman-factor fragments) in plasma protein fraction. N Engl J Med1978;299(2):66–70.
192. Rosenfeld EA, Shulman ST, Corydon KE, et al. Comparative safety and efficacy oftwo immune globulin products in Kawasaki disease. J Pediatr 1995;126:1000–3.

History of Immunoglobulin Replacement 763
193. Schiff RI. Transmission of viral infections through intravenous immune globulin.N Engl J Med 1994;331:1649–50.
194. Eibl MM. Intravenous immunoglobulins in neurological disorders: safety issues.Neurol Sci 2003;24:S222–6.
195. Miller JLC, Petteway SR Jr, Lee DC. Ensuring the pathogen safety of intravenousimmunoglobulin and other human plasma-derived therapeutic proteins. J AllergyClin Immunol 2001;108(4):91–4.
196. Klaesson S, Ringd�en O, Ljungman P, et al. Does high-dose intravenous immuneglobulin treatment after bone marrow transplantation increase mortality in veno-occlusive disease of the liver? Transplantation 1995;60(11):1225–30.
197. Dalakas MC. High-dose intravenous immunoglobulin and serum viscosity: riskof precipitating thromboembolic events. Neurology 1994;44(2):223–6.
198. Grillo JA, Gorson KC, Ropper AH, et al. Rapid infusion of intravenous immuneglobulin in patients with neuromuscular disorders. Neurology 2001;57:1699–701.
199. Stangel M, Muller M, Marx P. Adverse events during treatment with high-doseintravenous immunoglobulins for neurological disorders. Eur Neurol 1998;40:173–4.
200. Alving BM, Tankersley DL, Mason BL, et al. Contact-activated factors: contam-inants of immunoglobulins preparations with coagulant and vasoactive proper-ties. J Lab Clin Med 1980;96(2):334–46.
201. Orbach H, Katz U, Sherer Y, et al. Intravenous immunoglobulins: adverse effectsand safe administration. Clin Rev Allergy Immunol 2005;29(3):173–84.
202. Kallenberg CG. A 10% ready-to-use intravenous human immunoglobulin offerspotential economic advantages over a lyophilized product in the treatment ofprimary immunodeficiency. Clin Exp Immunol 2007;150(3):437–41.
203. Brannagan TH III, Nagle KJ, Lange DJ, et al. Complications of intravenousimmune globulin treatment in neurologic disease. Neurology 1996;47:674–7.
204. Gaines A, Varricchio F, Kapit R, et al. Renal insufficiency and failure associatedwith globulin intravenous therapy. MMWR Morb Mortal Weekly Rep 1999;48:518–21.
205. Stewart RR, Winney RJ, Cash JD. Renal toxicity of intravenous immunoglobulin.Vox Sang 1993;65(3):244.
206. Orbach H, Tishler M, Shoenfeld Y. Intravenous immunoglobulin and the kidney:a two-edged sword. Semin Arthritis Rheum 2004;34(3):593–601.
207. Cunningham-Rundles C, Zhou Z, Mankaroius S, et al. Long-term use of IgA-depleted intravenous immunoglobulin in immunodeficient subjects with anti-IgAantibodies. J Clin Immunol 1993;13(4):272–8.
208. Salama A, Schwind P, Schonhage K, et al. Rapid detection of antibodies to im-munoglobulin A molecules by using the particle gel immunoassay. Vox Sang2002;81(1):84–5.
209. Horn J, Thon V, Bartonkova D, et al. Anti-IgA antibodies in common variableimmunodeficieny (CVID): diagnostic workup and therapeutic strategy. ClinImmunol 2007;122:156–62.
210. Salama A, Temmesfeld B, Hippenstiel S, et al. A new strategy for the preventionof IgA anaphylactic transfusion reactions. Transfusion 2004;44(4):509–11.
211. Burks AW, Sampson HA, Buckley RH. Anaphylactic reactions after gammaglobulin administration in patients with hypogammaglobulinemia: detection ofIgE antibodies to IgA. N Engl J Med 1986;314(9):560–4.

Eibl764
212. Chapel HM, Spickett GP, Ericson D, et al. The comparison of the efficacy andsafety of intravenous versus subcutaneous immunoglobulin replacement therapy.J Clin Immunol 2000;20(2):94–100.
213. Wade N. Hybridomas: the making of a revolution. Science 1982;215(4536):1073–5.
214. Baudino L, Nimmerjahn F, Azeredo da Silveira S, et al. Differential contribution ofthree activation IgG Fc receptors (FcgammaRI, FcgammaRIII, and FcgammaRIV)to IgG2a- and IgG2B-induced autoimmune hemolytic anemia in mice. J Immunol2008;180(3):1948–53.
215. Nimmerjahn F, Ravetch JV. Fc-receptors as regulators of immunity. Adv Immunol2007;96:179–204.
216. Anthony RM, Nimmerjahn F, Ashline DJ, et al. Recapitulation of IVIG anti-inflam-matory activity with a recombinant IgG Fc. Science 2008;320(5874):373–6.
217. Nimmerjahn F, Ravetch JV. The antiinflammatory activity of IgG: the intravenousIgG paradox. J Exp Med 2007;204(1):11–5.

IntravenousImmunoglobulins :Evolution ofCommercial IVIGPreparations
John A. Hooper, PhD
KEYWORDS
� Intravenous immunoglobulin � Primary immunodeficiency� Manufacture � Commercial
Human immunoglobulin G (IgG) has been used to treat people with inherited immuno-globulin deficiencies since 1952 when Bruton1 infused a child with undetectable‘‘gamma globulin’’ levels and who suffered from recurrent pneumococcal infections.Subcutaneous infusions of 3.2 g/mo produced measurable gamma globulin levelsand completely eliminated pneumococcal infections. Human IgG soon became thestandard treatment for patients with primary antibody deficiencies who developchronic bacterial infections.
The first human IgG produced on a large scale was known as immune serum glob-ulin or ISG. It was produced by a cold ethanol precipitation process developed in theearly 1940s by E. J. Cohn and his coworkers2,3 in the Department of Physical Chem-istry at Harvard Medical School. ISG was formulated at a protein concentration of165 mg/mL that contained 0.3 Molar glycine, 0.9% (weight/volume) sodium chlorideand 0.1 g/L merthiolate. ISG solutions were adjusted to pH 6.8 � 0.4 and stored at5�C. With time, ISG solutions tended to form particles (aggregates) during storage.Aggregates were generally believed to be the cause of adverse events when ISGwas injected intravenously. Therefore, the first commercial immunoglobulins wererestricted to intramuscular or subcutaneous injections.
INTRAVENOUS IMMUNOGLOBULIN
In 1962, spontaneous complement activation (anticomplement activity) by IgGaggregates was proposed as the principal cause of adverse side reactions whenISG was injected intravenously.4 The desire to eliminate anticomplement activity
BioCatalyst Research LLC, 217 Camelot Drive, Liberty, MO 64068, USAE-mail address: [email protected]
Immunol Allergy Clin N Am 28 (2008) 765–778doi:10.1016/j.iac.2008.06.002 immunology.theclinics.com0889-8561/08/$ – see front matter ª 2008 Elsevier Inc. All rights reserved.

Hooper766
had a significant impact on intravenous immunoglobulin (IVIG) development.5,6
Some manufacturers reduced anticomplement activity by enzymatic digestion orchemical modification. The first IVIG was produced by pepsin digestion and con-tained a principal fragment with two antigen binding sites linked by disulfidebonds.7
The desire to reduce anticomplement activity and produce IVIGs with ‘‘intact’’ IgGas the principal component led some manufacturers to limit pepsin treatments,4 touse the more specific enzyme plasmin,8 and to chemically modify the product. Chem-ically modified IVIGs were produced that were structurally intact, were low in anti-complement activity, and contained no IgG fragments.9–11
Treatments of immunoglobulins with enzymes and chemical modification to sup-press spontaneous complement activation had several unintended consequences.The treatments also reduced important antibody biological activities requiredfor clinical efficacy. For example complement activation by antigen-antibodycomplexes plays an important role in the killing of encapsulated bacteria by leuko-cytes.12 Antibodies that are chemically and physically altered are rapidly removedfrom the circulatory system by the reticuloendothelial system. Thus some anti-bodies in enzyme-digested and chemically modified IVIGs were shown to havereduced bacterial opsonizing activities12–15 and shortened circulating half-lives.16–18
SECOND GENERATION INTRAVENOUS IMMUNOGLOBULIN
All commercial IVIGs are produced from large pools of human plasma by first concen-trating the IgG by cold ethanol fractionation. Although IgG produced by cold ethanolfractionation is relatively pure, it contains trace amounts of highly active contaminantsthat have the potential to cause most of the adverse events previously attributed toaggregates. These contaminants include prekallikrein activator (which initiatesproduction of the potent vasodilator bradykinin), prekallikrein, activated coagulationfactors, complement proteins, and immunoglobulins A and M.6 Other contaminantssuch as plasmin and plasminogen can degrade IgG to form split products and toreduce some antibody activities during ISG storage.19
The desire to produce IVIG that contain native IgG with antibodies that are fullyactive led to development of IVIG using purification with anion exchange (DEAE) chro-matography. The first purified IVIG contained none of the trace contaminants associ-ated with adverse events. Some antibody biological activities such as bacterialopsonization and virus neutralization were higher than in treated products.6 Nowvirtually all commercial IVIGs are produced with an anion exchange chromatographystep and contain relatively low levels of trace contaminants.
Historically, IVIGs were freeze-dried to obtain a preparation that would be stable for2 to 3 years. In 1986, McCue and coworkers20 reported that adjusting the pH to 4.25produced a clear, physically stable IgG solution. Clinical studies demonstrated thatpatients tolerated IgG solutions formulated at a pH significantly lower than thecustomary range of 6.4 to 7.2.21 This product represented a major advance in IVIGproduct formulation.
Table 1 lists commercial IVIG preparations currently (or soon to be) available inNorth America. Of the nine products licensed in the United States, seven are producedby cold ethanol fractionation followed by purification using ion exchange chromatog-raphy. Seven products are formulated as liquids and two are freeze-dried. All areproduced with specific virus inactivation or removal steps incorporated into theirmanufacturing procedures.

Intravenous Immunoglobulins 767
DEVELOPMENT OF VIRUS ELIMINATION PROCEDURES
Transmission of ‘‘homologous serum hepatitis’’ through whole blood, plasma, andserum was a great concern during development of human plasma proteins.1 Yellowfever vaccines stabilized with human serum had produced 23,000 cases of hepatitisin military personnel. Pooled human plasma presented a higher risk of hepatitis trans-mission than whole blood because of the increased probability that pooled plasmawould be contaminated by one or more donors. Human albumin solutions were alsoresponsible for hepatitis transmission.22
Heat can be used to inactivate viruses and proteins. The destruction temperature ofa protein is sharply defined and is different for each protein.23 In the presence of sub-strate, enzymes can be heated to temperatures 10 degrees higher than in the absenceof substrate.23 In 1948, Gellis and coworkers22 reported that hepatitis transmission byalbumin was eliminated by heating it for 10 hours at 60�C. Virus inactivation of albuminby heat treatment was possible because of the discovery that addition of stabilizersincreased the heat resistance of albumin. Human albumin has many binding sitesfor hydrophobic molecules and plays a major role in the transport of fatty acids. Fillingthese sites with the stabilizers acetyltryptophan and caprylic acid allows albumin towithstand heating for 10 hours at 60�C. Since albumin has no measurable biologicalactivity, the full impact of heating albumin is not known.
Unfortunately, other plasma proteins in solution are inactivated by heat and earlyattempts to inactivate viruses in high risk plasma products were unsuccessful. Highrisk plasma products included fibrinogen, Factor VIII concentrate, and Factor IX.24
Heated albumin solutions and immunoglobulins produced by cold ethanol fraction-ation were considered to be low-risk products.24
Factor VIII is rapidly inactivated when heated in solution. However, dried Factor VIIIis relatively heat stable under certain conditions. This observation led to developmentof heat-treated Factor VIII preparations in the 1980s.25,26 Fortunately, HIV was also in-activated in heated Factor VIII but the products had lower biological activities, wererelatively insoluble, and produced a higher incidence of Factor VIII inhibitors.Unfortunately, non-A, non-B hepatitis was not inactivated.25
The perception that immunoglobulins produced by cold ethanol fractionation hada low risk of transmitting virus infections changed in 1983 when Lane reported thatan experimental IVIG produced by cold ethanol fractionation transmitted non-A,non-B hepatitis.27 During this same period, HIV was isolated and shown to be trans-mitted by blood and blood products.28,29 The emergence of HIV and reports of non-A,non-B hepatitis transmission by some IVIG products30,31 caused manufacturers andregulatory agencies to examine existing IVIG manufacturing procedures for theircapacity to eliminate viruses.32–41 Development of dedicated virus inactivation proce-dures for IVIG production was also initiated.42,43
VIRUS INACTIVATION OF INTRAVENOUS IMMUNOGLOBULIN
Studies of IVIG manufacturing procedures revealed that cold ethanol fractionationremoves viruses by two mechanisms: inactivation and partitioning. Several laborato-ries demonstrated that HIV is inactivated by cold ethanol under conditions used in IVIGproduction.33–36,41 However, vesicular stomatitis virus and Sindbis virus, both used asmodels for the hepatitis C virus (HCV), formerly known as non-A, non-B, were stableunder similar conditions.41
Given the success of heat treatment in producing albumin with a low-risk of trans-mitting hepatitis, heat treatments for IVIG were evaluated. One IVIG was stabilized with33% (weight/weight) sorbitol at pH 5.5 and heated at 60�C for 10 hours.43 Several

Table1Production and properties of commercial immunoglobulins
Trade Names Manufacturer Registrations Manufacturing Procedure Composition CommentsGammagard S/D Baxter HealthCare
CorpUnited States, Canada,
European UnionCold ethanol
fractionation, DEAEchromatography, S/D,pH 6.8 � 0.4, freeze-dried
50 mg/mL; 8.5 mg/mLNaCl, 0.3 M glycine,20 mg/mL PEG, 3 mg/mLalbumin, 20 mg/mLglucose
<1 mg/mL IgA
Gammagard Liquid,KIOVIG
Baxter HealthCareCorp
United States,European Union
Cold ethanolfractionation, DEAEchromatography, S/D,nanofiltration, pH 4.85� 0.25, liquid
100 mg/mL; 0.25 Mglycine
—
Intratect Biotest Germany,European Union
Cold ethanolfractionation, octanoicacid/calcium acetatetreatment, S/D, liquid
50 mg/mL;0.3 M glycine
—
Vigam Bio ProductsLaboratory
England — 50 mg/mL; IgG,20 mg/mL humanalbumin, sucrose,glycine, pH 4.8–5.1
In US clinical trials(Gammaplex)
Carimune NF CSL Behring AG United States,European Union
Cold ethanolfractionation, pepsintreatment,nanofiltration, pH 6.6� 0.2, freeze-dried
30. 60, 90 or120 mg/mL;100 mg/mL sucrose,1.2 mg/mL NaCl
—
Sandoglobulin NF liquidCarimune NF liquid
CSL Behring AG Canada Cold ethanolfractionation, pepsintreatment, DEAESephadex batchadsorption,nanofiltration, pH 5.3liquid
120 mg/mL; 100 mML-isoleucine, 120 mML-proline, 80 mMNicotimamide
—
Ho
op
er
768

Privigen CSL Behring AG United States Cold ethanolfractionation,octanoic acidfractionation, anionexchangechromatography,nanofiltration,pH 4.8 � 0.2, liquid
100 mg/mL; 0.25 Mproline
—
Vivaglobin CSL Behring AG United States Cold ethanol, fattyalcohol, DEAEchromatography,activated carbon,heated 10 h @ 60�,pH 6.8 � 0.4, liquid
160 mg/mL; 3 g/L NaCl,0.25 N glycine
Formulated forsubcutaneous injection
Flebogamma 5% Instituto Grifols, SA United States,Spain
Cold ethanol,polyethylene glycolprecipitation, ionexchangechromatography,10 h @ 60�,pH 5.5 � 0.5, liquid
50 mg/mL; 50 mg/mLD-sorbitol, <6 mg/mLpolyethylene glycol
—
Flebogamma 5% DIF Instituto Grifols, SA United States Cold ethanol,polyethylene glycolprecipitation, ionexchangechromatography, pH 4@ 37�, 10 h @ 60�, S/D,nanofiltration, pH 5.5� 0.5, liquid
50 mg/mL; 50 mg/mLD-sorbitol, <3 mg/mLpolyethylene glycol
4 virus elimination steps
(continued on next page)
Intra
ven
ou
sIm
mu
no
glo
bu
lins
769

Table 1(continued)
Trade Names Manufacturer Registrations Manufacturing Procedure Composition CommentsOctagam Octapharma
PharmazeutikaProduktionsges.m.b.H.
United States,European Union
Cold ethanolfractionation, S/D,24 h @ pH 4,pH 5.5 � 0.4, liquid
50 mg/mL; 100 mg/mLmaltose
—
Omr-IgG-am OmrixBiopharmaceuticals Ltd
Israel Cold ethanolfractionation, S/D,24 h @ pH 4,pH 5.5 � 0.4, liquid
50 mg/mL; 100 mg/mLmaltose
In US clinical trials
Gamunex Talecris Biotherapeutics,Inc
United StatesEuropean Union
Cold ethanolfractionation,caprylate precipitation,Q Sepharose-ANXSepharosechromatography,pH 4.25 � 0.25, liquid
100 mg/mL; 0.2 M glycine —
Abbreviations: IgA, immunoglobulin A; NF, nanofiltration; PEG, polyethylene glycol; S/D, solvent-detergent.
Ho
op
er
770

Intravenous Immunoglobulins 771
enveloped viruses and one nonenveloped virus were studied. All viruses were com-pletely inactivated except for HCV. No substantial changes in IgG physicochemicaland biological properties were reported.
In 1988, Horowitz42 reported that a solvent-detergent process, originally developedto inactivate viruses in Factor VIII concentrates, was an effective virus inactivationprocess for IgG solutions. Solvent-detergent virus inactivation was rapidly adoptedby several IVIG manufacturers (Table 2).
Inactivation of hepatitis C and bovine viral diarrhea virus (BVDV, a surrogate forHCV) was reported in liquid IVIG formulated at pH 4.25 and incubated for 21 daysat 21�.38 Pepsin digestion at pH 4 and 37� has also been shown to inactivate severalenveloped viruses.39,40 Recently, incubation of immunoglobulin solutions with caprylicacid has been shown to be an effective procedure for inactivating enveloped viruses.44
VIRUS REMOVAL (NANOFILTRATION)
Manufacturers have long known that clarification filtration of cold ethanol fractionationintermediates in the presence of a filter aid is an effective virus removalprocedure. Somemanufacturers have validated such processes as virus removal steps (see Table 2).
In 1994, Burnouf-Radosevich and colleagues45 reported virus removal from FactorIX and Factor XI solutions by newly developed hollow fiber nanofiltration filters. Thefilters were composed of cellulose layers treated to produce mean pore sizes of15 � 2 and 35 � 2 nanometers (nm). Virus spiking experiments demonstrated thata single dead-end filtration with the 35 nm filter removed >5.7 to 7.8 log10 of HIV-1,BVDV, porcine pseudorabies virus (PRV) reovirus type 3, simian virus 40 (SV 40),and bovine parvovirus, a small (18–25 nm) nonenveloped virus.45
Table 2Dedicated virus inactivation and removal procedures used in IVIG production
Virus Inactivation/Removal Procedure ProductSolvent-detergent inactivation Gammagard S/D
Gammagard Liquid
Flebogamma 5% DIF
Octagam
Omr-IgG-am
Heat inactivation (10 h at 60�C) Vivaglobin
Flebogamma 5%
Flebogamma 5% DIF
Removal by nanofiltration Gammagard Liquid
Carimune NF
Privigen
pH 4 incubation (in process) Flebogamma 5% DIF
Octagam
Omr-IgG-am
Privigen
Low pH incubation in final container (21 d) Gamunex
Low pH incubation at elevated temp in final container Gammagard Liquid
Pepsin treatment Carimune NF
Caprylic acid virus inactivation Gamunex

Hooper772
In studies of immunoglobulin solutions with protein concentrations up to 12 mg/mL,O’Grady and colleagues46 demonstrated that the 35 nm filter removed 6–7 log10
mouse type C retrovirus, SV 40 and PRV, whereas poliovirus was removed by onlya 15 nm filter. Similar results were obtained with 70 mg/mL IgG solutions.47
Omar and Kempf48 studied the effectiveness of nanofiltration to remove small non-enveloped viruses. The viruses studied were bovine enterovirus (BEV,w30 nm),bovine parvovirus (BPV,w18–25 nm) and minute virus of mice (MVM,w18–25 nm).Nanofiltration was performed with filters having nominal pore sizes of 20 and50 nm. Despite their small size, each virus was efficiently removed from 10 mg/mLIgG solutions. The authors demonstrated that removal of viruses smaller in diameterthan the pore sizes of the nanofilter was due to antibodies bound to the viruses.48
Nanofiltration has been adopted by several IVIG manufacturers (see Table 2).
DONOR SCREENING AND PLASMATESTING
Concomitant with development of virus inactivation and removal procedures, scien-tists also recognized the importance of eliminating infected donors and developedmore sensitive tests for blood-borne pathogens. Although people with illnesses arealways excluded from donating blood or plasma, some donors do not feel sick orhave clinical symptoms in the early stages of an infection. During this time (windowperiod), blood or plasma donations may transmit an infection. Thus development ofdonor screening tests involved not only tests for new pathogens but also tests ofever increasing sensitivity to eliminate window period infections.
Gurtler49 has reviewed blood-borne pathogens with respect to their relevance totransfusion. Relevant pathogens are considered to be human pathogens that causechronic, progressive wasting, or lethal diseases; and some infectious agents that arenot prevalent in the transfused population. By these criteria, hepatitis B virus (HBV),HCV, and human immunodeficiency viruses types 1 and 2 (HIV-1 and HIV-2) were char-acterized as relevant. Parvovirus B19, cytomegalovirus, and hepatitis A viruses (HAV)were classified as occasionally relevant. Since this review was published, nonenvel-oped viruses such as Parvovirus B19 and HAV have become more relevant and theSevere Acute Respiratory Syndrome-Corona virus (SARS-CoV) and West Nile virus(WNV) have emerged. Thus relevance of pathogens to transfusion is an evolving concept.
Given the early concern about hepatitis transmission, identification of hepatitisviruses and development of sensitive donor screening tests became a high priority.A sensitive test for HBV was developed in 197250 and was used to eliminate infecteddonors. Unfortunately, the HBV test did not eliminate transfusion-related hepatitis andthe search for one or more non-A, non-B hepatitis viruses was initiated. The AIDSepidemic led to rapid development of a screening test for antibodies to human immu-nodeficiency virus (HIV-1) in 1984.51 In 1989 the genome of a non-A, non-B hepatitisvirus was isolated and used to develop a donor screening test for HCV.52 Today,plasma is screened for antibodies to syphilis, HIV-1, HIV-2, and HCV, and for HBVand HIV antigens. Extremely sensitive tests for HCV, HIV-1, HBV, and parvovirusB19 nucleic acids have recently been introduced and are now being used to furthereliminate window period donations.
PRION REMOVAL
The risk of transmitting prion diseases such as Creutztfedt-Jakob disease (CJD) orVariant Creutzfeldt-Jakob disease (vCJD) by transfusions of human blood or bloodproducts is theoretical at this time. However, the incubation time for development ofCJD disease is so long that it is difficult to quantify the risk.

Intravenous Immunoglobulins 773
There is enough uncertainty about the relationship of vCJD to bovine spongiformencephalopathy (BSE) that regulatory agencies have take steps to reduce the risk.Donors that have spent R6 months in the United Kingdom from 1986 to the presentare not permitted to donate blood or plasma in the United States and Europe. Therecent observation that BSE and scrapie are transmitted from sheep to sheep bytransfusions may support this donor deferral program.
Trejo and colleagues53 studied removal of hamster scrapie protein (PrPsc) duringtwo steps in IVIG production. Western Blot and infectivity tests demonstrated thatPrPsc was removed during two filtration steps. One of the steps was a depth filtrationstep that is common to all IVIG manufacturing procedures.
A similar study was performed by Gregori and colleagues.54 Proteinase K resistantPrP (PrPres) was determined by Western Blot analysis whereas infectivity was mea-sured in hamsters. The authors observed that depth filtration in the presence of filteraids and nanofiltration removed PrPres reactivity and transmissable spongiform en-cephalopathy (TSE) infectivity.
CLINICALTRIALS IN PRIMARY IMMUNODEFICIENCY
In the United States, clinical trials in subjects with primary immunodeficiency havebecome increasingly standardized.55 The Food and Drug Administration (FDA) recom-mends that studies measure the rate of serious bacterial infections during regularinfusions of investigational IVIG for 12 months to avoid seasonal biases. Serious infec-tions are defined as bacteremia/sepsis, bacterial meningitis, osteomyelitis/septicarthritis, bacterial pneumonia, and visceral abscess. Diagnostic criteria are listed. Statis-tical analysis should demonstrate that the upper 99% one-sided confidence interval forthe frequency of acute serious bacterial infections is less than one per subject per year.55
Infusional adverse events are now defined as those that occur up to 72 hours follow-ing an infusion of test product, regardless of other factors that may impact a possiblecausal association with product administration. The target for this safety endpoint isan upper 95% one-sided confidence limit of less than 0.40.55
Pharmacokinetic (PK) data are to be obtained from at least 20 patients. The analysisshould include total IgG and several specific antibodies to derive a plasma concentra-tion-time curve, half-life, area under the curve (AUC0-t; AUC0-infinity), volume ofdistribution, maximum concentration (Cmax), time from start of infusion to Cmax(Tmax), and elimination rate constants. Serum samples for these antibody measure-ments should be taken after a washout period of 3 to 5 estimated half-lives (3–5 infu-sions) investigational IGIV. The FDA also desires that trough IgG and IgG subclasslevels be measured monthly.55
The results of these policies are illustrated in Table 3. The time period for recordinginfusional or temporally associated adverse events has been extended from 30 min-utes to 72 hours postinfusion. Each study has reported the incidence of acute seriousbacterial infections and other bacterial infections. Although not shown in Table 3,pharmacokinetic studies were also performed for each product. The number of PKsubjects ranged from 1462 to 5757.
TRENDS IN IVIGMANUFACTURING
As shown in Table 1, most IVIG products are still produced by cold ethanol fraction-ation but are now further purified with anion exchange chromatography (DEAE anionexchangers or an equivalent). Plasma fractionation by cold ethanol fractionation in-volves precipitating proteins by adjusting pH, salt concentration, temperature, andethanol concentration. Precipitated proteins are removed from proteins still in solution

Tab e 3Re nt clinical trials in patients with primary immunodeficiency disorders
Pro uct
StudyDuration(Months)
PatientsTreated Dose
AcuteSerious BacterialInfect/subj/y
O er BacterialIn ct/subj/y
Related, TemporallyAssociated AEs (%of Infusions) Drug-Related SAEs
Ca mune NFL quid (12%)
6 42 200–800 mg/Kg/21–28 d 0 3 5 21.7%a 0
Fle ogamma 5% 12 51 300–600 mg/Kg/21–28 d 0.061 N 8.2%c 2
Fle ogamma 5% DIF 12 46 300–600 mg/Kg/21–28 d 0.021 1. 6 11.8%c 0
Ga magard liquid 10% 12 61 300–600 mg/Kg/21–28 d 0 0. 7 31.2%c 2 (1 patient)
Ga unex 10% 9 73 100–600 mg/Kg/21–28 d 0.07 0. 8 5.7%a 0
Oc gam 5% 12 46 300–600 mg/Kg/21–28 d 0.1 0 5.5%b 0
Pri igen 10% 12 80 200–888 mg/Kg/21–28 d 0.08 3. 5 18.5%b 5 (1 subject)
Viv globin 16% 15 51 34–352 mg/Kg/wk 0.04 4. Local, 49%;systemic, 5.4%
0
Abb viations: AE, adverse event; infect/subj/y, infections per subject per year NF, nanofiltration; SAE, erious adverse event.a –48 h postinfusion.b –30 min postinfusion.c 72 h postinfusion.Da a from Refs.53–63
Ho
op
er
774
lce
drii
b
b
m
m
ta
v
a
re000–t
thfe.6
R
9
0
1
5
4
s
Table 4United States IVIG distribution data
Year Kg % Increase Liters of Plasmaa
1998 15,000 — 4,285,714
2002 23,000 53% 6,571,429
2003 24,900 8% 7,114,286
2004 26,900 8% 7,685,714
2005 28,200 5% 8,057,143
2006 32,400 15% 9,257,143
2007 34,200 6% 9,771,429
a Assumes 3.5 g IgG obtained per liter of plasma.
Intravenous Immunoglobulins 775
by filtration or centrifugation. The most abundant plasma proteins, IgG and albumin,have vastly different physicochemical properties and are readily separated. However,some IgG and albumin is distributed to other fractions at each precipitation step.
In the classical Cohn-Oncley process, fraction II (IgG) was further purified by at leastthree additional precipitations with IgG losses at each step. Since IgG production isthe driving force behind plasma manufacturing capacity, manufacturers have turnedtheir attention to increasing the amount of purified IgG from plasma. Some manufac-turers limit IgG precipitation from plasma to a single cold ethanol precipitation step toproduce what Cohn referred to as fraction I1II1III. IgG losses are minimized by usingI1II1III (or II1III if fraction I-fibrinogen is precipitated earlier) as the starting material foranion exchange chromatography and the virus inactivation and removal steps thathave been incorporated into the process.
The importance of increasing IgG yield is illustrated in Table 4. Demand for IVIG hasincreased 128% in the past decade. Manufacturers have been able to meet demandby acquiring underutilized facilities, expanding existing facilities, building new facili-ties, and increasing yield. IVIG manufacturing changes have been accompanied byan increase in clinical trials in patients with primary immunodeficiency.
There is also a trend to formulate IVIGs as solutions with a protein concentration of100 mg/mL (10% solutions) and a low pH that favors product stability (pH 4.3 to 5.0.)The increase in IgG concentration from 5% to 10% reduces infusion time, an impor-tant feature for patients with primary immunodeficiency who receive large doses every21 to 28 days all their life. Ten percent IVIGs at low pH are more stable at low ionicstrength and therefore sodium chloride is no longer added. In addition, carbohydratestabilizers are no longer required. Because their tendency to precipitate at increasedionic strength, products of this type may not be diluted with saline or mixed with otherIVIGs that contain sodium chloride.56–58
REFERENCES
1. Bruton OC. Agammaglobulinema. Pediatrics 1952;9:722–7.2. Cohn EJ, Strong LE, Hughes WL Jr, et al. Preparation and properties of serum and
plasma proteins III: a system for the separation into fractions of the protein andlipoprotein components of biological tissues and fluids. J Am Chem Soc 1946;68:459–75.
3. Oncley JL, Melin M, Richert DA, et al. The separation of the antibodies, isoagglu-tinins, prothrombin, plasminogen and beta-lipoprotein into subfractions of humanplasma. J Am Chem Soc 1949;71:541–50.

Hooper776
4. Barandun S, Kistler P, Jeunet F, et al. Intravenous administration of human gammaglobulin. Vox Sang 1962;7:157–74.
5. Aronson DL, Finlayson JS. Historical and future therapeutic plasma derivatives(Epilogue). Semin Thromb Hemost 1980;VI:1231–9.
6. Hooper JA, Alpern M, Mankarious S. Immunoglobulin manufacturing procedures.In: Krijnen HW, Strengers PFW, van Aken WG, editors. Immunoglobulins. Amster-dam: Central Laboratory of the Netherlands Red Cross Blood Transfusion Service;1988. p. 361–80.
7. Schultze HE, Schwick G. Uber neue Moglichkeiten intravenoser gammaglobulin-applikation. Dtsch Med Wochenschr 1962;87:1643–50.
8. Sgouris JT. The preparation of plasmin-treated immune serum globulin for intrave-nous application. Vox Sang 1967;13:71–84.
9. Stephan W. Undegraded human immunoglobulin for intravenous use. Vox Sang1975;28:422–37.
10. Masuho Y, Tomibe K, Matsuzawa K, et al. Development of an intravenousgamma-globulin with Fc activities I: preparation and characteristics ofS-sulfonated human gamma-globulin. Vox Sang 1977;32:175–81.
11. Schroeder DD, Tankersley DL, Lundblad JL. A new preparation of modifiedimmune serum globulin (human) suitable for intravenous administration I: stan-dardization of the reduction and alkylation reaction. Vox Sang 1980;40:373–82.
12. Pollack M. Antibody activity against Pseudomonas aeruginosa in immune globu-lins prepared for intravenous use in humans. J Infect Dis 1983;147:1090–8.
13. Kim KS, Wass CA, Kang JH, et al. Functional activities of various preparations ofhuman intravenous immunoglobulin against type III group B streptococcus.J Infect Dis 1986;153:1092–7.
14. Bender S, Hetherington S. Haemophilus influenzae type b opsonins of intrave-nous imunoglobulins. J Clin Immunol 1987;7:475–80.
15. Steele RW, Steele RW. Functional capacity of immunoglobulin G preparations andthe F(ab0)2 split product. J Clin Microbiol 1989;27:640–64.
16. Janeway CA, Merler E, Rosen FS, et al. Intravenous gamma globulin. Metabolismof gamma globulin fragments in normal and agammaglobulinemic persons.N Engl J Med 1968;278:919–23.
17. Winston DJ, Ho WG, Rasmussen LE, et al. Use of intravenous immune globulin inpatients receiving bone marrow transplants. J Clin Immunol 1982;2(April Supple-ment):42S–7S.
18. Hagenbeek A, Brummelhuis GJ, Donkers A, et al. Rapid clearance of cytomeg-alovirus-specific IgG after repeated intravenous infusions of human immunoglob-ulin into allogeneic bone marrow transplant recipients. J Infect Dis 1987;155:897–902.
19. Tankersley DL, Alving BM, Yi M, et al. Predictive tests for fragmentation ofimmune globulins. In: Alving BM, Finlayson JS, editors. Immunoglobulins, char-acteristics and uses of intravenous preparations. Washington, DC: US Govern-ment Printing Office; 1980. p. 173–7.
20. McCue JP, Hein RH, Tenold R. Three generations of immunoglobulin G prepara-tions for clinical use. Rev Infect Dis 1986;8(Supplement):S374–81.
21. Schiff RI. Intravenous gammaglobulin: pharmacology, clinical uses and mecha-nisms of action. Pediatr Allergy Immunol 1994;5:63–87.
22. Gellis SS, Neefe JR, Stokes J, et al. Chemical, clinical and immunological studieson the products of human plasma fractionation. XXXVI. Inactivation of the virus ofhomologous serum hepatitis in the solutions of normal human serum by means ofheat. J Clin Invest 1948;27:239–44.

Intravenous Immunoglobulins 777
23. Dixon M, Webb EC. Enzyme isolation: methods of purification. In: Dixon M,Webb EC, editors. Enzymes. New York: Academic Press; 1964. p. 36–7.
24. Gerety RJ, Aronson DL. Plasma derivatives and viral hepatitis. Transfusion 1982;22:347–51.
25. Hollinger FB, Dolana G, Thomas W, et al. Reduction in risk of hepatitis transmissionby heat treatment of a human Factor VIII concentrate. J Infect Dis 1984;150:250–62.
26. Colvin BT, Rizza CR, Hill FGH, et al. Effect of dry-heating of coagulation factorconcentrates at 180�C for 72 hours on transmission of non-A, non-B hepatitis.Lancet 1988;ii:814–6.
27. Lane RS. Non-A, non-B hepatitis from intravenous immunoglobulin. Lancet 1983;i:974–5.
28. Barr�e-Sinoussi F, Chermann JC, Rey F, et al. Isolation of a T-lymphotropic retrovi-rus from a patient at risk for AIDS. Science 1983;220:868–71.
29. Centers for Disease Control. Provisional public health service interagency recom-mendations for screening donated blood and plasma for antibody to the viruscausing acquired immunodeficiency syndrome. MMWR Morb Mortal Wkly Rep1985;34:1–5.
30. Ochs HD, Fisher SG, Virant FS, et al. Non-A, non-B hepatitis after intravenousgammaglobulin. Lancet 1985;i:322–3.
31. Bjorkander J, Cunningham-Rundles C, Lundin P, et al. Intravenous immunoglob-ulin prophylaxis causing liver damage in 16 of 77 patients with hypogammglobu-linemia or IgG subclass deficiency. Am J Med 1988;84:107–11.
32. Prince AM, Stephan W, Dichtelmuller H, et al. Inactivation of the Hutchinson strainof non-A, non-B hepatitis virus by combined use of ß-propiolactone and ultravioletirradiation. J Med Virol 1985;16:119–25.
33. Piszkiewicz D, Kingdon H, Apfelsweig R, et al. Inactivation of HTLVIII/LAV duringplasma fractionation. Lancet 1985;ii:1188–9.
34. Wells MA, Wittek AE, Epstein JS, et al. Inactivation and partition of human T-celllymphotrophic virus, type III, during ethanol fractionation of plasma. Transfusion1986;26:210–3.
35. Mitra G, Wong MF, Mozen MM, et al. Elimination of infectious retroviruses duringpreparation of immunoglobulin. Transfusion 1986;26:394–7.
36. H�enin Y, Mar�echal V, Barr�e-Sinoussi F, et al. Inactivation and partition of humanimmunodeficiency virus during Kistler and Nitschmann fractionation of humanblood plasma. Vox Sang 1988;54:78–83.
37. Yei S, Yu MW, Tankersley DL. Partitioning of hepatitis C virus during Cohn-Oncleyfractionation of plasma. Transfusion 1992;32:824–8.
38. Louie RE, Galloway CJ, Dumas ML, et al. Inactivation of hepatitis C virus in lowpH intravenous immunoglobulin. Biologicals 1994;22:13–9.
39. Reid KG, Cuthbertson B, Jones ADL, et al. Potential contribution of mild pepsintreatment at pH 4 to the viral safety of human immunoglobulin products. VoxSang 1988;55:75–80.
40. Kempf C, Jentsch P, Poirier B, et al. Virus inactivation during production of intra-venous immunoglobulin. Transfusion 1991;31:423–7.
41. Hamamoto Y, Harada S, Yamamoto N, et al. Elimination of viruses (human immu-nodeficiency, hepatitis B, vesicular stomatitis and sindbis viruses) from anintravenous immunoglobulin preparation. Vox Sang 1987;53:65–9.
42. Horowitz B. Preparation of virus sterilized immune globulin solutions by treatmentwith organic solvent/detergent mixtures. In: Krijnen HW, Strengers PFW, vanAken WG, editors. Immunoglobulins. Amsterdam: Central Laboratory of theNetherlands Red Cross Blood Transfusion Service; 1988. p. 285–95.

Hooper778
43. Funakoshi S, Uemura Y, Yamamoto N. Virus inactivation and elimination by liquidheat treatment and PEG fractionation in the manufacture of immune globulinintravenous. In: Krijnen HW, Strengers PFW, van Aken WG, editors. Immunoglob-ulins. Amsterdam: Central Laboratory of the Netherlands Red Cross Blood Trans-fusion Service; 1988. p. 313–25.
44. Korneyeva M, Hotta J, Lebing W, et al. Enveloped virus inactivation by Caprylate:a robust alternative to solvent-detergent treatment in plasma derived intermedi-ates. Biologicals 2002;30:153–62.
45. Burnouf-Radosevich M, Appourchaux P, Huart J, et al. Nanofiltration, a newspecific virus elimination method applied to high-purity Factor IX and Factor XIconcentrates. Vox Sang 1994;67:132–8.
46. O’Grady J, Losikoff J, Poily A, et al. Virus removal studies using nanofiltrationmembranes. Dev Biol Stand 1996;88:319–26.
47. Troccoli NM, McIver J, Losikoff A, et al. Removal of viruses from human intrave-nous immune globulin by 35 nm nanofiltration. Biologicals 1998;26:321–9.
48. Omar A, Kempf C. Removal of neutralized model parvoviruses and enterovirusesin human IgG solutions by nanofiltration. Transfusion 2002;42:1005–10.
49. Gurtler L. Blood-borne viral infections. Blood Coagul Fibrinolysis 1994;5:S5–10.50. Alter HJ, Holland PV, Purcell RH, et al. The Ausria test: critical evaluation of sen-
sitivity and specificity. Blood 1973;42:947–57.51. Sarngadharan MG, Popovic M, Bruch L, et al. Antibodies reactive with human
T-lymphotropic retroviruses (HTLV-III) in serum of patients with AIDS. Science1984;224:506–8.
52. Kuo G, Choo Q-L, Alter HJ, et al. An assay for circulating antibodies to a majoretiologic virus of human non-A, non-B hepatitis. Science 1989;244:362–4.
53. Trejo SR, Hotta J, Lebing W, et al. Evaluation of virus and prion reduction in a newintravenous immunoglobulin manufacturing process. Vox Sang 2003;84:176–87.
54. Gregori L, Maring J, MacAuley C, et al. Partitioning of TSE infectivity during eth-anol fractionation of human plasma. Biologicals 2004;32:1–10.
55. Guidance for Industry. Safety, Efficacy, and Pharmacokinetic Studies to SupportMarketing of Immune Globulin Intravenous (Human) as Replacement Therapy forPrimary Humoral Immunodeficiency. Rockville (MD): U.S. Department of Healthand Human Services, Food and Drug Administration. Center for Biologics Evalu-ation and Research; 2005.
56. Prescribing information, Gamunex�, Talecris Biotherapeutics, Inc, November2005.
57. Prescribing information, Gammagard Liquid, Baxter Healthcare Corporation,April 2005.
58. Prescribing information, Privigen�, CSL Behring LLC, July 2007.59. Prescribing information, Vivaglobin�, CSL Behring LLC, April 2007.60. Prescribing information, Flebogamma� 5% DIF, Instituto Grifols. S.A.61. Prescribing information, Octagam�. Octapharma Pharmazeutika Produktionges
m.b.H, March 2007.62. Prescribing information, Carimune� NF, ZLB Behring AG, January 2005.63. Prescribing information, Gammagard S/D, Baxter Healthcare Corporation, March
2007.

SubcutaneousAdministration of IgG
Melvin Berger, MD, PhDa,b,c
KEYWORDS
� IsG � Primary immune deficiency� Subcutaneous � IgG � Efficacy� Pharmacokinetics � Quality of life� Adverse effects
Systemic availability of exogenous antibodies injected subcutaneously (SC) intohumans was recognized more than 100 years ago, when immune animal sera wereused as the major treatment of diphtheria and tetanus.1 Indeed, the characteristicsof adsorption of IgG from SC sites were well understood even then: ‘‘A certain timemust elapse from the injection.until its healing activity in the infected parts of thebody can develop. (Antibody) injected under the skin does not pass straight intoblood vessels but first into the lymphatic vessels. From here it takes several hours be-fore passing gradually into the blood stream and further time still is needed before it isdiffused not only everywhere in the blood stream but also in the extra-vascularfluids.’’1 Despite the slower kinetics of adsorption and distribution of IgG given bythe SC, as opposed to the intravenous (IV), route, SC administration of the exogenousantisera was preferred because of its freedom from the severe and often life-threatening systemic reactions that accompanied IV injections of animal proteins.Decades later, similar freedom from systemic adverse reactions prompted the useof slow SC infusions of human IgG concentrates in patients who had primary immunedeficiency diseases (PIDD) and poor tolerance of intramuscular (IM) injections or thenewly developed IV preparations.2–6 Several studies have shown that subcutaneousimmunoglobulin (SCIG) has similar efficacy to intravenous immunoglobulin (IVIG) inpreventing infections in PIDD patients. The freedom from systemic adverse effectsand the ease with which SC can be administered at home has led to its increasing pop-ularity in recent years, and to documented improvement in quality of life for PIDDpatients.
a Department of Pediatrics, Case Western Reserve University, Cleveland, OH, USAb Department of Pathology, Case Western Reserve University, Cleveland, OH, USAc Jeffrey Modell Center for Primary Immune Deficiencies, Division of Allergy-Immunology,Rainbow Babies and Children’s Hospital, University Hospitals of Cleveland–Case Medical Center,R B & C Room 504, MS 6008B, 11100 Euclid Avenue, Cleveland, OH 44106, USAE-mail address: [email protected]
Immunol Allergy Clin N Am 28 (2008) 779–802doi:10.1016/j.iac.2008.07.002 immunology.theclinics.com0889-8561/08/$ – see front matter ª 2008 Elsevier Inc. All rights reserved.

Berger780
EFFICACY
Although several studies suggest that SCIG treatment is comparable to IV treatment inpreventing infections in PIDD patients, few direct comparisons have been made.Chapel and colleagues7 used a composite score for ‘‘major’’ and ‘‘moderate’’infections in a crossover study comparing the two routes, and reported slight, butnot significantly different, decreases in infection in patients on SC treatment. The num-ber of days lost from work or school due to illness showed no difference in the twogroups.7 This study included 24 subjects who completed 1 year of treatment oneach route, but different IgG preparations were used for the different routes.7 Pacand Bernakowska8 reported a nearly 50% reduction in episodes of infection anddays of antibiotics on 13 months of SC as opposed to the preceding year of IV treat-ment in the same patients. Gardulf and colleagues9 showed a reduction in days in hos-pital per year due to infection in PIDD patients on SC versus those on IV treatment, butthe statistical significance of this difference was not reported. In a recent study in Ger-many in which patients on SCIG were asked to compare retrospectively their previousIVIG with current SCIG treatment regimens, the reported incidence of infection drop-ped from 2.8 � 2.0 days in 6 months on IV to 1.9 � 1.9 days in 6 months on SC(P 5 .021).10 The author and his colleagues recently completed a randomized, cross-over study in the United States, in which 11 patients received the same monthly doseof a single preparation of IgG by the IV and SC routes for 6 months each. Thirteen ep-isodes of infection occurred during the IV phase versus only 9 episodes during the SCphase.11 In one of the largest and most closely monitored studies of SCIG, which in-cluded 51 patients in the United States in a 12-month efficacy period, Ochs and col-leagues12 reported 0.04 serious infections and 4.4 nonserious infections per patientper year. In a 6-month study of 47 patients on the same SC preparation in the Euro-pean Union and Brazil, the annualized incidence of serious infections was also 0.04per patient per year and the incidence of nonserious infections was 4.3 per patientper year.13 These results are comparable to the overall mean of 0.068 serious infec-tions per patient per year and 3.02 nonserious infections per year in the licensing trialsof all IVIG preparations licensed in the United States since 2000. Thus, the resultsindicate that SCIG is equal in efficacy to IVIG in preventing infections in PIDD patients.
Although it may be postulated that elimination of periods with low IgG levels towardthe end of IVIG dosing intervals (see later discussion) might result in better long-termfreedom from morbidity due to chronic sinus or lung infection in PIDD patients, dataare not available that would allow comparison of the long-term effects of SC versusIV IgG administration on the development of bronchiectasis or other changes on CTscans, nor on deterioration of pulmonary function in patients who have PIDD. Similarly,no data are available comparing the efficacy of SC versus IV IgG on the persistence orprogression of chronic sinus disease in PIDD patients with that problem, or on othercomplications of PIDD.
PHARMACOKINETICS
Few published data report traditional pharmacokinetic parameters for IgG adminis-tered by the SC route in immunodeficiency patients. Studies in rabbits have shownthat the peak serum IgG concentration is reached 3.2 days after SC injection of humanIgG and that the bioavailability of IgG given IM or SC is less than that of IgG given IV.14
In another rabbit study, using a preparation of IgG currently marketed in the UnitedStates for IV use, more than 120% of the IV dose was necessary to achieve thesame area under the curve (AUC) of IgG versus time for SC versus IV administration.15

Subcutaneous Administration of IgG 781
The Food and Drug Administration (FDA) criteria for approval of new IgG prepara-tions for IV administration include the requirement that pharmacokinetic parametersfor the new preparation be similar to historical values for previously licensed IV prep-arations.16 The parameters to be reported include the volume of distribution; plasmaconcentration time curve, including the time at which the maximum concentration isreached and the maximum concentration that is achieved; AUC; half-life; and elimina-tion rate constants.16
Partly because of the preclinical data mentioned earlier, partly because of datareceived by the FDA showing that monoclonal antibodies and therapeutic Fc fusionproteins have lower bioavailability when given by the SC or IM as compared with IVroute, and partly because of uncertainty regarding which pharmacokinetic parametersare most relevant for the use of IgG in PIDD patients, the FDA faced a quandary in for-mulating criteria for licensing IgG preparations to be given by the SC route.17 To besure that patients being given SCIG were at least as well protected as those receivingIV preparations, the FDA took the ‘‘conservative’’ position that the AUC for SC IgGmust equal the AUC for the previous IV regimen given to the same patient.17 In con-trast, European regulators require only that the ‘‘trough’’ serum IgG level on SC IgGexceed that achieved on IVIG.18 For the one IgG preparation currently licensed forSC use in the United States, the requirement for equivalence of the AUCs resultedin a ‘‘dosage penalty’’ of 37%.12 That is to say, to achieve the same mean AUC forIgG given SC as compared with IV, an average of 37% more IgG had to be given bythe SC route. It is not clear from published studies that equivalence of the AUC is nec-essary for therapeutic equivalence, and results of a European study in which the samedose was given by the IV and SC routes showed no difference in rates of infection(efficacy) from the North American study in which the SC dose was raised by37%.12,13 In that European study, the mean ‘‘steady-state’’ serum IgG concentrationachieved on SC IgG was 11% higher than the mean trough IgG concentration with thesame total monthly dose being given once every 3 to 4 weeks by the IV route. In con-trast, in the North American study with the 37% dosage increment, the mean steady-state IgG level on SC therapy was 32% higher than the mean IgG trough level on IVtherapy.12 Data from these and other studies comparing IgG levels achieved by theIV and SC routes are presented in Table 1.
The systemic adsorption of IgG from SC sites is considerably slower than that of IV-injected IgG. Sample pharmacokinetic curves for each route of administration areshown in Fig. 1. As seen on the left, an IV infusion of 406 mg/kg of IVIG into a patientwho had X-linked agammaglobulinemia rapidly raised the serum IgG level by morethan 900 mg/dL. The peak IgG typically decreases somewhat over the next 48 hoursas the IgG redistributes into the total extravascular volume, then continues to declinewith first-order kinetics and a half-life of about 22 days.19,20 Thus, patients on typicalregimens of IV infusions every 21 to 28 days are never really at a steady state. Duringthe course of each dosing interval, their serum IgG concentration typically varies bymore than 700 mg/dL around the mean (in this case, 850 mg/dL). In contrast, whenthe IgG dose is fractionated into three or four equal doses given SC at weekly intervals,the slow systemic adsorption from the injection site or sites increases the serum IgGeven as the IgG is diffusing out of the circulation elsewhere in the body. Thus, the peakis markedly truncated. Fractionating the usual 21- or 28-day IV dose into SC dosesgiven weekly, or even more frequently, can result in a true steady state, in which theIgG being adsorbed from SC injection sites essentially replaces that fraction of theprevious dose that has been metabolized. The right side of Fig. 1 shows data forthe same patient maintained on a weekly SC dose equal to 40% of the previous21-day dose; in other words, a monthly dose 20% higher than on IV therapy. The

Table 1Comparison of serum IgG concentrations on intravenous and subcutaneous therapy
Author (Y) (N)IV Dose(mg/kg/mo)
Mean IVTrough(mg/dL)
Mean SCDose (mg/kg/wk)
SC Dose(as % ofIV)
Mean SCtrough(mg/dL)
% D inTrough(SC/IV L100%)a
Chouksey(2003)
5 553 997 138 101.5 1144 14.7
Ochs(2006)
51 465 785 158 136.0 1040 32.0
Gardulf(2008)
47 — 836 89 101.0 922 11.0
Desai(2008)
10 551 1051 138 100.0 1168 11.1
a Mean of individual percent changes.
Berger782
maximum variation around the mean serum IgG concentration of 816 mg/dL is only50 mg/dL.
Fig. 2 shows mean serum IgG levels in 47 patients midway through a 6-month studyin which they received an average weekly dose of 89 mg/kg IgG given SC on days0 and 7 of the time period shown. Again, it is readily apparent that a near-steady stateis achieved, with little variation in the IgG level over the 14-day interval sampled here.21
In a much earlier study of 23 adult CVID patients by Waniewski, and colleagues,22 itwas reported that weekly SC infusions of 100 mg/kg maintained steady-state IgGlevels, with little variation around the mean for more than a year. The time to reachthis steady state was also examined in that study. One group of 6 patients, naıve toIgG therapy, with a mean baseline IgG concentration of 220� 140 mg/dL, was startedon 100 mg/kg IgG SC once a week. After 6 months of weekly infusions, these patients
Tot
al I
gG
400
600
800
1000
1200
1400
1600
0 2 4 6 8 10 12 14 16 18 20 22
Days0 7 14 21
Days
30 gr 5% IVIG (406 mg/kg)Q 3 weeks
12 gr 16% ISG Q 7 days =36 gr in 3 weeks
Fig.1. (Left) Serum IgG levels in a 34-year-old X-linked agammaglobulinemia patient whileon a regimen of 30 g IVIG (406 mg/kg) every 3 weeks. (Right) After a few months on 12 gSCIG once a week.

Fig. 2. Mean steady-state IgG levels in a group of 47 patients receiving weekly SC infusionson days 0 and 7.
Subcutaneous Administration of IgG 783
had a mean serum IgG level of 820 � 180, which was still lower than the apparentsteady state of 990 � 60 mg/dL achieved at 12 months of therapy.22 That concentra-tion then remained fairly constant over 6 months of further treatment with the sameweekly dose of SCIG.22 To obviate the slow and gradual rise in the serum IgG levelobserved in naıve patients given only one infusion a week, Waniewski and col-leagues22 gave another group of patients 100 mg/kg infusions daily for 5 consecutivedays. That group of patients achieved a steady state of approximately 1200 mg/dL bythe end of that 5-day course of treatment, and had mean levels of 1150, 1110, and1120 at 6, 12, and 18 months later, respectively. It may be important to note, however,that the later group of patients had received IM or IV IgG treatment at unspecifiedtimes before beginning on SC treatment, and that they had a mean IgG level of670 � 280 mg/dL just before beginning SC therapy. Nevertheless, many immunolo-gists consider five to seven daily infusions of 90 to 120 mg/kg each satisfactory forquickly bringing up the serum IgG level of a newly diagnosed antibody-deficient pa-tient, so that the patient can then continue on weekly infusions alone.23
In the recent pivotal study of an IgG preparation now licensed by the FDA for SCadministration in the United States, patients who were already on stable IGIV treat-ment regimens were given their initial SC infusion 7 to 10 days after their most recentIV infusion; then, SC infusions were continued on a weekly basis. That schedule resultsin initially higher IgG levels for the next several weeks, which trend downwards overtime to a new steady state. Desai and colleagues11 have also completed a recentstudy using that interval between the patient’s last IV and first SC infusions, followedby weekly SC infusions of one quarter of the previous monthly IVG dose (on a mg/kgbasis). In those patients, the mean steady-state serum IgG concentration on SC infu-sions was 11% higher than the mean trough level had been on IV therapy, and thatsteady state was reached after 10 to 12 weeks of SC treatment.
ADVERSE EFFECTSSystemic Adverse Effects
The difference between the shapes of the pharmacokinetic curves illustrated above forSC versus IV IgG administration underlies one of the most important differencesbetween current IV and SC regimens. Many of the adverse effects associated withIV infusions, such as headaches, fever, and anaphylactoid reactions, occur duringthe infusion or within 48 hours of it, while the serum IgG concentration is rapidlyincreasing or is still near its peak. Elimination of the sudden, rapid increases and tran-sient peaks in the serum IgG concentrations is believed to be responsible for themarked amelioration in systemic adverse effects experienced by patients who transi-tion from IV to SC regimens.24 One of the original rationales for using slow infusions

Berger784
when the IM preparations were given SC was that if large injections of IgG induced therelease of mediators such as prostaglandins or cytokines, which produced chills,fever, and other remote systemic adverse effects, then slow administration mightdecrease the rate at which these putative mediators were released and woulddecrease their concentrations at any point in time,2 which, in turn, would allow com-pensation by normal homeostatic mechanisms. Indeed, many of the systemic adverseeffects associated with IV infusions are considered ‘‘rate related’’ and may be obvi-ated by slowing the rate of the IV infusion.25 With SC administration, a depot is createdin the tissues, from which the IgG reaches the blood stream much more slowly than if itwas infused directly into a vein. Furthermore, the serum IgG level remains nearlyconstant on SC regimens. Hence, rapid release or dramatic systemic effects of anymediators are unlikely to be produced when the exogenous IgG comes into contactwith Fc receptors or antigens.
The freedom from systemic adverse effects with SC administration was apparentwhen the first few reports of slow SC infusions of IM immune serum globulin (ISG) ap-peared in the early 1980s,2–6 and continues to characterize SC administration24 In thereports of Gardulf and colleagues9,26,27 in the early to mid-1990s, systemic adversereactions were reported with less than 1% of SC infusions, as opposed to 46% withthe IV preparations available in that era. Pac and Bernakowska8 reported a series inwhich 15 patients received 780 SCIG infusions over a 13-month period. Systemic ad-verse effects were experienced by only 1 patient, a child who had required steroidsand other premedications before IVIG infusions. Although that patient tolerated theSC infusions for months with no problems, she subsequently suffered systemic reac-tions at two consecutive SC infusions, and was switched back to her previous regimenof IV premedication before standard IVIG doses. None of the other patients had sig-nificant systemic adverse effects and none switched back to IV therapy.8
In only two major studies has the incidence of systemic adverse effects accompa-nying SC infusions been reported as greater than 1%. Chapel and colleagues7
reported that 3.3% of SC infusions were accompanied by systemic adverse effects,as compared with 5% of IV infusions. Ochs and colleagues12 reported that headachesoccurred during, or within 48 hours following, 1.6% of SC infusions, but only 35% ofthese headaches were considered ‘‘severe,’’ requiring prescription medications. Only3 subjects out of 68 withdrew from that year-long study because of infusion-relatedadverse effects; no infusion-related serious systemic adverse reactions occurred.12
Local Adverse Effects
Most patients experience some kind of local reaction, at least transiently, at the sitesof SC infusions. These reactions may include swelling, redness, and a sensationvariously reported as ‘‘itching’’ or ‘‘burning.’’11,12,24,28 The swelling may exceed thatexpected from the volume of fluid injected at the site, but that is unusual. Pruritis,when it occurs, is not usually severe. The patient’s sensation is often more just anawareness of the presence of an area of swelling or redness, rather than true discom-fort. In the trial of Ochs and colleagues,12 these reactions occurred initially in nearly90% of patients, and were mostly considered ‘‘mild’’ or ‘‘moderate’’ (Fig. 3). Only3 patients out of 68 discontinued participation in that study because of local site reac-tions. In almost all cases, local swelling or redness dissipates within hours (Fig. 4).Occasionally a small, pearl-sized nodule or knot may persist for 1 or 2 days. Examina-tion of patients who have infused IgG SC in the same region of the body (ie, anteriorabdominal wall, flanks, or upper thighs) for years has not revealed any long-termchanges such as fibrosis or fat necrosis. In most studies, the incidence of localreactions has been reported to decrease over several months of continuing SC

Fig. 3. Local site reactions in a SC IgG licensing trial. (Top) ‘‘Mild’’ reaction; slight erythemawith minimal swelling. (Bottom) ‘‘Moderate’’ reaction; significant swelling. Swollen area isblanched and surrounded by a ring of erythema few millimeters in width.
Subcutaneous Administration of IgG 785
therapy, as illustrated in the data of Ochs and colleagues (Fig. 5). Most of the data onwhich that and similar curves are based are recorded by the patients in diaries theykeep at home. Therefore, it is likely that a major contribution to the recording of thereactions is subjective. As the patients get accustomed to the local phenomena thataccompany SCIG infusions, they are less likely to report a small degree of local swell-ing as a ‘‘reaction.’’ It is possible that the decreased rate of local reactions associatedwith SC infusions might represent some kind of accommodation of the patient to thespecific product, which might be analogous to the increased rate of reactions associ-ated with switching to a new IV preparation reported in some studies. Biopsy studiesof SC IgG infusion sites at various times after the injections have not been reported.
‘‘Wear-Off’’ or Trough Effects
As opposed to acute adverse effects associated with the high peaks of IgG in the se-rum that accompany bolus IV administration, many patients who receive IVIG infu-sions every 21 to 28 days report general malaise, fatigue, arthralgias or myalgias,and increased susceptibility to infections during the last 7 to 10 days of each dosinginterval. Most patients on IV treatment regimens report that they can feel their IVIG‘‘wearing off’’ before their next infusion is due.29 Often, patients will report that theyhave ‘‘run out of gas’’ or ‘‘feel punk’’ at the end of the IV dosing interval. It is apparentfrom the graphs that these low serum IgG levels are obviated by weekly or more fre-quent SC infusions. Table 1 summarizes the changes in ‘‘trough’’ serum IgG level onSC versus IG therapy reported in several recent studies. As patients achieve stablesteady-state serum IgG levels on SC regimens, they frequently report alleviation of

Fig. 4. Clearing of local site reactions after SC IgG administration. A 14-year-old girl has justfinished infusing approximately 22 mL of a 15% solution of Carimune into each of two ab-dominal sites sequentially. The infusion into the first site (to the patient’s left of umbilicus)was given over about 1 hour and ended 1 hour before the picture was taken. The site of theneedle is still visible, as is some diffuse swelling, but the redness has almost completely dis-sipated. The infusion into the second site (to the patient’s right of umbilicus) was just com-pleted before this picture was taken. Note the mild central swelling surrounding the needlesite, and several centimeters of redness, but no induration per se. The patient was infusedwith 6 g of 15% Carimune solution made by pushing 40 mL of sterile water for injection intoa 6-g bottle of lyophilized IgG. Note that indented scars are from surgical drains or otherprocedures, not from IgG infusions.
Berger786
the symptoms they previously experienced as their serum IgG levels approached theirnadir. This improvement, in turn, may lead to an overall more continuous feeling ofgood health, which likely contributes to the increased quality of life reported bymost PIDD patients who switch from IVIG to SC.27,30–32
QUALITY-OF-LIFE EFFECTS
Several formal studies have been done of the health-related quality of life of PIDDpatients on different modes of therapy, mostly performed by Gardulf and colleaguesin Sweden. European and North American patient populations have been surveyedfor these studies. PIDD patients in the United States have also been surveyed bythe Immune Deficiency Foundation (IDF), but that survey was completed before anySC product was licensed in the United States. Several sources of dissatisfactionexpressed by patients in the 2003 IDF survey are directly related to the sites, timing,or frequency of IV infusions. The specific sources of dissatisfaction with IV regimensresonate with the results of the studies of Gardulf and colleagues, and can be affectedby switching from IV to SC therapy. Most importantly, although 64% of PIDD patientssurveyed by the IDF reported that they were somewhat or very satisfied with the con-venience of the location of their IVIG treatments, satisfaction was reported by 96% ofpatients infused at home, as compared with only 51% of those infused in doctors’offices and 48% of those infused in hospital outpatient clinics. Among those infusedin other locations, only 39% reported that the location was ‘‘very convenient.’’Sixty-nine percent of the patients reported receiving their IVIG infusions on weekdaysbetween 9 AM and 5 PM, and only 64% considered that ‘‘very convenient.’’ Eighty-eightpercent of patients reported receiving IVIG infusions at intervals of 3 weeks or longer,
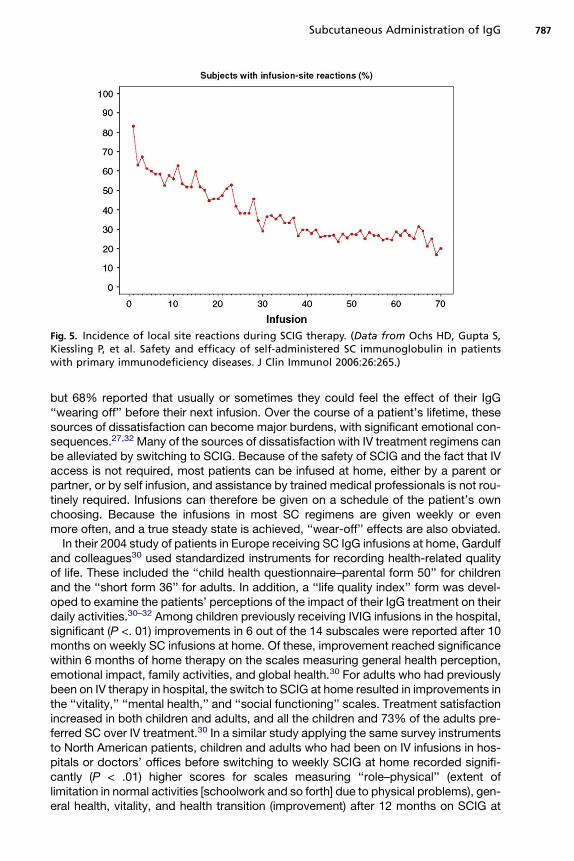
Fig. 5. Incidence of local site reactions during SCIG therapy. (Data from Ochs HD, Gupta S,Kiessling P, et al. Safety and efficacy of self-administered SC immunoglobulin in patientswith primary immunodeficiency diseases. J Clin Immunol 2006:26:265.)
Subcutaneous Administration of IgG 787
but 68% reported that usually or sometimes they could feel the effect of their IgG‘‘wearing off’’ before their next infusion. Over the course of a patient’s lifetime, thesesources of dissatisfaction can become major burdens, with significant emotional con-sequences.27,32 Many of the sources of dissatisfaction with IV treatment regimens canbe alleviated by switching to SCIG. Because of the safety of SCIG and the fact that IVaccess is not required, most patients can be infused at home, either by a parent orpartner, or by self infusion, and assistance by trained medical professionals is not rou-tinely required. Infusions can therefore be given on a schedule of the patient’s ownchoosing. Because the infusions in most SC regimens are given weekly or evenmore often, and a true steady state is achieved, ‘‘wear-off’’ effects are also obviated.
In their 2004 study of patients in Europe receiving SC IgG infusions at home, Gardulfand colleagues30 used standardized instruments for recording health-related qualityof life. These included the ‘‘child health questionnaire–parental form 50’’ for childrenand the ‘‘short form 36’’ for adults. In addition, a ‘‘life quality index’’ form was devel-oped to examine the patients’ perceptions of the impact of their IgG treatment on theirdaily activities.30–32 Among children previously receiving IVIG infusions in the hospital,significant (P <. 01) improvements in 6 out of the 14 subscales were reported after 10months on weekly SC infusions at home. Of these, improvement reached significancewithin 6 months of home therapy on the scales measuring general health perception,emotional impact, family activities, and global health.30 For adults who had previouslybeen on IV therapy in hospital, the switch to SCIG at home resulted in improvements inthe ‘‘vitality,’’ ‘‘mental health,’’ and ‘‘social functioning’’ scales. Treatment satisfactionincreased in both children and adults, and all the children and 73% of the adults pre-ferred SC over IV treatment.30 In a similar study applying the same survey instrumentsto North American patients, children and adults who had been on IV infusions in hos-pitals or doctors’ offices before switching to weekly SCIG at home recorded signifi-cantly (P < .01) higher scores for scales measuring ‘‘role–physical’’ (extent oflimitation in normal activities [schoolwork and so forth] due to physical problems), gen-eral health, vitality, and health transition (improvement) after 12 months on SCIG at

Berger788
home.31 Even more dramatic (P<.001) changes were reported in treatment satisfac-tion, including decreased interference of treatment with other activities, decreasedproblems related to receiving therapy, and increased satisfaction with the treatmentsetting.31 In contrast, among patients who were receiving IV treatment at home beforeentering the study of SCIG, significant improvement in only one scale, ‘‘generalhealth,’’ was reported. These results suggest that major improvements in quality oflife are achieved when PIDD patients are treated at home rather than in hospitals ordoctors’ offices. SC infusions facilitate a transition to home care because of their free-dom from adverse effects and the lack of a requirement for trained medical personnel.Elimination of the low trough levels just before monthly IV treatments, which obviateperiodic increases in susceptibility to infection, is probably responsible for theincrease in general health reported by all patient groups on switching to SCIG, regard-less of the location at which the treatment was administered. These conclusions arefurther supported by the patients’ preferences: 76% of the patients in the North Amer-ican study expressed a preference for SC rather than IV therapy, whereas 91% of thepatients preferred home treatment.31
Despite the apparent advantages of home SCIG treatment for most PIDD patients,not all patients are interested in switching from IV to SC treatment, and not all patientswant to switch from hospital or office to home treatment. In Germany, where home SChas been offered as an alternative to IV treatment since 2003, demographics andattitudes were recorded from 32 adult patients who chose SC therapy and were com-pared with those recorded by 28 adult patients who chose to remain on IV therapy.10
Patients who chose SC treatment were younger (37 � 9 versus 51 � 14 years of age,P < .001) and a higher percentage were employed (69% versus 28%). The predomi-nant reasons patients on IV refused to try SCIG were perceived inconvenience(48%), anxiety about adverse reactions at home (31%), and anxiety over needle sticks(6%). In contrast, the major reasons patients on SC gave for preferring that route wereflexibility (50%), decreased side effects (17%), and constant IgG levels (17%).
Thus, in considering which route and location of therapy might be most appropriatefor any given patient, it is important to consider the individual patient’s life situationand attitudes toward self versus assisted treatment, and the medical advantagesand disadvantages of each route for that individual patient. Although SCIG may bepreferred by many PIDD patients, it is not necessarily the best choice for everyone.
PATIENTSWHOHAVE SPECIAL CONDITIONS
Only a few reports exist of the use of SCIG in PIDD patients presenting unique chal-lenges for IgG therapy, including pregnancy, IgA sensitization, and bleeding disorders.The author and his colleagues’ original motivation for using ambulatory pumps forslow SC delivery of IgG was to facilitate adequate IgG replacement in a pregnant pa-tient who did not tolerate IM injections or plasma transfusions. At times during the thirdtrimester, that patient self administered 140 mL of 16% IgG per week, equaling 22.4 gper week.5 The patient gave birth at term by a spontaneous vaginal delivery, and nei-ther mother nor baby suffered any complications of the therapy or delivery, nor anypostpartum problems. Mother’s and baby’s IgG levels were 620 mg/dL and 880mg/dL, respectively, on day 2 after birth.5 Gardulf and colleagues33 subsequently re-ported a series of 11 pregnancies in nine immunodeficient women who self infused100 mg/kg/wk at home throughout the pregnancies, using a rapid infusion protocol.No complications or serious infections occurred, and in all cases, the cord bloodIgG concentration was equal to, or higher than, the mother’s. No reports have beenpublished describing large series of infants who had antibody deficiency treated

Subcutaneous Administration of IgG 789
with SCIG, but several pediatric centers have been administering IgG in this waybecause the difficulty of obtaining IV access, which is common in babies under 2 yearsof age, is obviated with the SC infusions. The author and his colleagues have treatedseveral patients in this age group with SCIG, and have found weekly doses in therange of 1 mL of 16% IgG (160 mg) per kg, up to 10 mL, are easily tolerated in the thigh.Although some parents may be hesitant to poke their baby, once they see how well thebabies tolerate the small prick of a single 27- or 28-gauge needle, they are quicklyrelieved. Especially if previous attempts at IV therapy have required multiple sticksor the use of scalp-vein infusions, the advantages of the SC route are readily apparent.
Many physicians and patients inquire about the use of the SC route in patients whohave bleeding disorders or anticoagulant therapy. Heparin is commonly self adminis-tered at home by the SC route and, although some local bruising may occur, bleedingfrom the sites is usually not a significant problem. Arora and colleagues34 havereported the use of SCIG in a patient who had von Willebrand’s disease with no specialprecautions or problems, and anecdotal reports exist of the successful use of SCIG inboys who have classic hemophilia.
Another concern that is frequently voiced about IgG treatment of PIDD in general,both IV and SC, is the potential for problems associated with administering IgA-containing IgG preparations to IgA-deficient patients. Although a risk for anaphylaxisexists if preparations containing IgA are given to patients who lack that protein, inpractice this event is extremely rare. Anaphylaxis due to SC injection of IgA-containingpreparations has not been reported, and this procedure is generally regarded as safein PIDD patients, most of whom have some IgA or also lack the capacity to produceIgE (ie, Bruton’s agammaglobulinemia). Sundin and colleagues35 have reported thatadministration of IgA-containing IgG preparations SC was well tolerated by patientswho had detectable anti-IgA antibodies, although the isotypes of the anti-IgA anti-bodies were not reported and none of the patients were specifically tested for IgEagainst IgA. Four of the anti-IgA–positive patients lost their anti-IgA activity afterweeks to months of weekly treatment with an IgG preparation containing up to5 mg/mL of IgA.35 Induction of a state of unresponsiveness to IgA was inferred fromthe absence of IgA-containing complexes in the patient’s sera and the lack of reemer-gent anti-IgA activity, even after switching to low-IgA preparations.35 The presence orabsence of partial IgA deficiency does not seem to correlate with local reactions toSCIG, or with the amelioration of these reactions over time. Because of the theoreticrisk for anaphylaxis, anaphylactoid reactions, and other severe averse effects, it is rec-ommended that the initial doses of SCIG always be given in a clinical setting equippedto treat acute severe reactions in the unlikely event they occur.21
COST
Comparisons of the costs of SC versus IV therapy regimens in the early 1990sreported that substantial savings were achieved by the use of IM preparations bythe SC route.26 Analysis of that data suggests that the savings were mainly due todecreased cost of the stocks of IM ISG compared with the (at that time) new IV prep-arations. In the current era, the major cost of any IgG treatment regimen is the productitself. In turn, the single biggest contribution to the cost of the products is the cost ofthe starting plasma itself. Although marketing and distribution expenses vary, in gen-eral, the costs of SC and IV products supplied by most manufacturers are, and willlikely continue to be, in the same range. Typical regimens for self–SC administrationat home generally use one or more pumps, which may have purchase prices rangingfrom a few hundred to a few thousand US dollars each. Depending on arrangements

Berger790
for purchase or leasing of the pump or pumps, service charges or amortization of thepurchase price may add as much as $50 to $100 a month to the cost of SC regimens.Several popular pumps require special, proprietary syringes or tubing sets, which mayadd $5 to $25 to the cost of each infusion, and elaborate multibranched tubing setswith special SC needles may also drive up the per-infusion costs. Because infusionsare typically given once or twice a week, the cumulative annual costs of some regi-mens may be considerable. In contrast, many IV regimens include facility fees orbed charges, and charges for the nurse or other health professional who starts theIV and monitors the infusion. IV infusions are frequently given using pumps for precisecontrol of the infusion rates, which may add an equipment charge or purchase price tothe cost of the IV regimen. Because IV regimens typically involve infusions that aregiven only once every 3 or 4 weeks, those costs may not accumulate as rapidly asthe cost of the supplies associated with SC regimens. Indirect costs, such as for trans-portation to a hospital or infusion center, parking, and time lost from work, should alsobe considered in comparing the total costs of home SC versus hospital- or office-based IV treatment regimens.
A recent study performed by the Canadian Government Agency for Drugs andTechnologies in Health (CADTH) concluded that home IV treatment would be the leastexpensive program for PIDD patients in that country, with home SC a close second.The estimated direct yearly per-patient costs for treating an ‘‘average’’ adult whohas IVIG in hospital was C$ 21,777; IVIG at home (self administered, without a nurse)was C$19,891; and SC at home was C$ 20,416. When indirect costs, such astransportation and time lost from work were included, the costs were C$ 23,037 forhospital-based IV, C$ 20,302 for home IV (assuming this would be self administered,without a nurse), and C$ 21,033 for self-administered home SCIG. However, when thedecrease in adverse effects and increased quality of life with SC versus IV treatmentwere included in the calculations, the conclusion was reached that SCIG ‘‘dominates’’because of ‘‘greater expected benefits at lower expected costs’’ per quality adjustedlife-year.36 Overall, if the 75% Canadian PIDD population now receiving IgG treatmentswitched from IV to SC therapy, the CADTH estimated that the savings to the govern-ment would be approximately C$ 9,000,000/y.36 This analysis assumed the sameprice per gram of IgG regardless of the route by which it would be administered.
A similar analysis in Germany, using actual data on payments by insurance compa-nies for PIDD patients receiving IgG by both routes, showed that the average price pergram for IgG to be given by the SC route was only 46% of that for IgG to be given bythe IV route. Overall, the budget impact analysis from that study, independent of anyadjustment for improvement in quality of life measurements, showed that yearlysavings for the German statutory health insurance would be 17 to 77 million Eurosannually if 60% of current PIDD patients switched from IV to SCIG.37
PRACTICAL CONSIDERATIONS IN SUBCUTANEOUS IMMUNOGLOBULIN THERAPYFOR PRIMARY IMMUNE DEFICIENCY DISEASESSelection of Infusion Regimens
The author and his colleagues’ initial attempts to give SC IgG infusions were per-formed using 16% ISG preparations in a patient who previously had demonstratedpoor tolerance for IM injections of the same preparations.2 Other early patientswere usually also chronically infected. To avoid local or systemic adverse effects,they administered the ISG slowly, initially at 1 to 2 mL per hour.2,5 Furthermore,because the ISG was generally provided in 10-mL vials, that was the usual volumeinfused into a single site. As other investigators began to use this method in a wider

Subcutaneous Administration of IgG 791
spectrum of patients, it became clear that much faster rates and increased volumesper infusion site could be tolerated. In their 1991 study of 25 PIDD patients routinelyreceiving SCIG infusions for 1.25 to nearly 4 years, Gardulf and colleagues9 reportedthat those patients who had higher body mass indexes were receiving higher doses ateach infusion, and using two pumps with up to four sites in the abdomen or thighs foreach infusion. The mean volume per infusion was 42.8 mL (range 28–60), and themean duration of the infusions was 75.8 minutes (range 45–120 minutes). Eachpatient’s initial infusion was given at a rate of 10 mL/h into each site. After severalinfusions, the rates were increased by 1 mL/site/h every few weeks according toeach patient’s tolerance, until the above rates were acheived.9 Although 36% of thosepatients had previously suffered severe reactions after IM injections, no severe ormoderate reactions were observed after the SC infusions, and only 0.9% of SC infu-sions were accompanied by mild systemic symptoms.9 Subsequently, Gardulf andcolleagues26,38 continued treating patients with these ‘‘rapid’’ infusions at 20 to40 mL/h. Similarly, Gaspar and colleagues39 reported the routine use of 20 mL/h, usu-ally into two sites simultaneously. Hansen and colleagues40 then reported that mostpatients tolerated ‘‘express’’ infusions at rates up to 35 mL/h/site, such that 10 mLcould be delivered per site in 17 minutes. Using multiple syringe drivers to deliverIgG into four sites simultaneously, these investigators reported that 40 mL (6.4 g ofIgG) doses were routinely tolerated in as little as 17 minutes.40
In the only licensing trial of SCIG that has been reported in the United States, limitswere set at 20 mL of IgG per hour with a maximum of 15 mL/site.12 In studies of thistype, conservative regimens are often selected for fear that adverse effects, if they oc-cur, will have a negative impact on the regulatory authorities. It is therefore likely thatsimilar rates and volumes per site will also be selected in future pivotal studies. In con-trast, when IgG preparations marketed for IV or IM use were used ‘‘off label’’ for SCinfusions using drivers that could accommodate syringes as large as 60 mL, a muchbroader range of regimens could be used, and the flexibility of the SC route couldbe exploited more fully. Taking into consideration the patient’s preference as to thenumber of needle sticks (sites) per infusion and the time he/she is willing to spend, itbecomes readily apparent that these two variables have a reciprocal relationship.That is, if more sites are used, the overall time for infusing a given total dose will be de-creased as compared with giving the same dose into fewer sites. Additionally, it be-comes readily apparent that fully grown adults can tolerate greater volumes perindividual site than small children, and that it is likely that patients who have greateramounts of SC tissue (ie, higher body mass index or obese individuals) will alsotolerate more IgG per site than lean individuals. If infusions are given slowly, thenthe infused IgG may be diffusing away even as more is being given, so that morevolume can be put into an individual site without increasing the local discomfort orseverity of any possible reaction. Before any SC preparation was licensed in the UnitedStates, the author and his colleagues reviewed the SC regimens being used by theirpatients, most of whom had been referred because of adverse effects with IVIG. Thesepatients were receiving SC infusions of an IM ISG preparation, or a lyophilized prepa-ration reconstituted to yield a solution with an IgG concentration of approximately15%.41,42 Regimens ranged from 10 mL into one site in less than 1 hour in children,to 40 mL over several hours into one site (usually taken at night while the patientwas sleeping), to 40 to 60 mL given into multiple sites simultaneously in short times.The spectrum of regimens eventually selected by the patients, who ranged in ageand size from preemies initially weighing 5 kg to adults weighing over 130 kg, wasbroad but could be inclusively summarized by calculating the rate in mL of IgG persite per hour per kg body weight. The mean was found to be 0.176 mL/site/kg/h,

Berger792
with a standard deviation of 0.134,42 which is approximately equal to 12 mL/site/h in an‘‘average’’ 70-kg adult, or 3.5 mL/site/h in an ‘‘average’’ 20-kg 6-year-old child.
The flexibility illustrated by the regimens discussed above suggests that the patientshould have a major input into designing his/her own regimen, according to his/herpreferences, tolerance at the local sites, and lifestyle choices. The patient’s prioritiesfor the number of infusions per month, number of needle sticks, and time per infusioncan be easily accommodated if the regimen is selected thoughtfully. A useful algorithmis to determine the total IgG dose to be delivered per month and to ask if the patientalready has firm ideas about the regimen he/she would like to use. If the patient doesnot, a good starting point is to divide the total volume of IgG to be infused during themonth by four and to start with the assumption that one infusion per week will be pre-ferred. The volume to be infused weekly can be rounded off to the nearest multiple ofwhole vials of IgG product, so that the patient can be taught to draw up each dosewithout wasting any of the IgG and without requiring a pharmacist to be involved.The number of pumps that will be available to the patient should then be considered.In most cases in the United States, a patient is likely to have only one pump at home,whereas in the United Kingdom, it is common for the National Health Service (NHS) toprovide two pumps to each patient.43 In Sweden, up to four pumps are frequentlyused for each infusion. Dividing the volume to given in each infusion by the numberof pumps narrows the choices of the size of the syringe that will be used for eachpump, which, in turn, is a major factor in determining which pumps might be usedfor that particular patient.44 The number of sites to be used for each infusion andthe time can then be calculated based on the patient’s preferences. The calculatedvalue given above, 0.18 mL/kg/site/h, can serve as a useful guide and starting point.The product manufacturer’s recommendations in the prescribing information can beused in the same way, but it should be remembered that such information has usuallybeen selected in an arbitrary way to facilitate uniform data collection during clinical tri-als. Once an initial regimen plan is formulated, it should be reviewed carefully with thepatient to be sure it is acceptable and the patient does not foresee any problems incomplying with it. If the time, number of sites, or volume per site in the planned regi-men seems excessive, splitting the dose into two or more infusions per week shouldbe considered. Orders can then be written and the appropriate equipment obtained.As the patient becomes experienced with the infusion regimen, it can be modified.In many cases, the patient’s tolerance of local site reactions increases with repeatedinfusions, and the time for each infusion can be shortened or the volume of IgG infusedper site can be increased.
The paragraph above describes a reiterative process that may be used to arrive atan optimal regimen, individualized for each patient, and is based on the experience inthe author’s clinic with various patients and multiple different IgG products over manyyears. Each of the parameters in Box 1 may be considered independently, and variedto reach a regimen that fits easily into each patient’s individual lifestyle andpreferences. Some investigators have explored options at the extremes of the aboveregimens. For example, Shapiro in Minnesota and Ochs in Seattle have reportedanecdotally that many patients prefer to increase the parameter of the number ofinfusions per month out to 20 to 30, by giving ‘‘daily pushes’’ of 10 mL each, whichis done conveniently by repeatedly pushing 1 to 2 mL from a 10-mL syringe withoutany mechanical pump at all. Daily doses of 10 mL (1.6 g, if a 16% IgG solution isused) are easily given over 5 to 10 minutes in this way, and are well tolerated bymany patients. Infusing 1.6 g every weekday gives a monthly dose of 35.2 g, andthe patient does not have to take infusions on the weekends. In contrast, if the patienttakes a 1.6-g infusion every day, the total monthly dose will be 48.8 g per month.

Box1Establishing an optimal subcutaneous immunoglobulin regimen for any individual patient
1. Predetermination: dose of IgG in mg/kg/mo. Start with estimated dose of 500 mg/kg/mo, usecurrent monthly IV dose, or use current monthly IV dose � correction for bioavailability ifdesired.
2. Parameters that should be selected in accordance with patients preferences. Note that anyindividual parameter in this list can be prioritized and set as the most important variable,which will influence the choices for the other parameters.
Number of infusions per week or month
Volume per infusion
Number of sites per infusion
Volume per site
Total time for each infusion
Decision to use multiple sites simultaneously or sequentially
Number of pumps
Subcutaneous Administration of IgG 793
At another extreme, patients may prefer only a single needle stick per week andmight choose to infuse a large volume into a single site, which may be accomplishedover a long time using a pump that can drive a large (60-mL) syringe, or with a roller-type pump and a reservoir. Examples of this type of regimen include the author and hiscolleagues’ patients who infuse up to 60 mL at rates of 8 to 10 mL per hour while theysleep, and regimens reported by Dr. Charles Kirkpatrick of the University of Colorado,in which a single SC needle with a short ‘‘pigtail’’ tubing that can be closed and cap-ped, is kept in place overnight. IgG is infused into that single site over 2 consecutivedays, which would easily allow doses of IgG as great as 120 mL (19.2 g) to be toleratedby most patients each week. Another variation on a ‘‘standard’’ weekly dosing regi-men was recently reported by Gustafson and colleagues,45 in which doses of200 mg/kg were given SC every other week rather than 100 mg/kg every week. TroughIgG levels on the two regimens were not compared directly, but no increase in adverseeffects (AEs) or infections was reported.45
An alternative approach is to adopt a simplified regimen using a ‘‘rule of twos": "twobottles into two sites over 2 hours.’’ For the average 70-kg adult, an infusion com-posed of two 20-mL vials of 16% IgG given in this way equals 10 mL per site perhour, or 0.143 mL/kg/site/h, well within the range described above. For a 35-kg child,the same rate per site is achieved if two 10-mL vials are used. These regimens provide6.4 g or 3.2 g of IgG per infusion, respectively. The desired total monthly dose can thenbe achieved by varying the number of infusions per month. For example, two 40-mLinfusions per week would provide 51.2 g per month, and two 20-mL infusions perweek would provide 25.6 g per month. Given that any level of activity short of rigorousexercise or total immersion in water is possible while taking these SC infusions, 2 hoursper infusion is easily tolerated once or twice a week by most patients, most of whomspend many more hours than that every day sitting at a desk or in front of a televisionor computer, doing housework, or walking at a moderate pace.
Site Selection and Specific Procedures
Numerous resources describe in detail exact procedures to be used in selecting andpreparing sites and actually administering the infusions.43,46–49 SC injections are most

Berger794
frequently given in the anterior abdominal wall out to the flanks, the anterior or innerthighs, and the backs of the upper arms; these are diagrammed in the cited literature.A useful and easy-to-remember guideline is to be able to ‘‘pinch an inch’’ of SC tissueat any intended site. A key to successful SC infusion is to place the needle sufficientlydeep to avoid intradermal administration. Initially, the author and his colleagues used.75-inch 25-gauge butterfly needles inserted at a 45� to 90� angle to the skin. Now,various needles specifically designed for SC use, with the needle itself mounted at90� to the ‘‘wings’’ or gummed disk and tubing are available.44 Except in the leanestpatients, needles of 11 to 15 mm in length are preferable, to be sure the infusion isgiven into SC fat, and to minimize leakage from the site. When leakage or excessiveredness occurs, it is often because needles that are too short (only 6 mm) havebeen used. Various stainless steel and silastic catheters in the range of 25 to 28 gaugeand 6 to 15 mm in length are now marketed, attached to tubings of various lengths,with as many as four branches attached to one hub,44 for infusing into multiple sitessimultaneously. For most patients, a simple antiseptic wipe with alcohol or chlorhex-idine is all that is needed to prepare the site. Extensive scrubbing is not usually nec-essary, and repeated use of iodine-containing antiseptics such as Betadine mayactually be irritating or cause sensitization. A eutectic mixture of local anesthetics(EMLA) has been successfully used with children and with adults who may have anx-iety about sticking themselves, but it is generally not needed because the needles areso small and precise placement, as with IV catheters, is not necessary. Excessive orirritating tape should be avoided; many patients have more erythema and itching fromthe tape than they do from the SCIG itself. The most important precaution is that thepatient should draw back from the needle with a syringe before starting the pump or‘‘pushing’’ the product, because high-concentration IgG solutions intended for SC orIM use may not necessarily be free from dimers or larger aggregates, which can causeadverse reactions if injected intravascularly. Providers should remember to instructpatients in the proper disposal of ‘‘sharps’’ and other medical waste, and shouldmake sure that proper containers are available and appropriate logistics have beenarranged.
Initiating Subcutaneous Therapy/Teaching the Patient
Various publications and other resources are available for instructing patients to selfadminister SCIG, including illustrated brochures on Ig manufacturer’s Web sites,46,47
a monograph available from the IDF,50 a small pamphlet prepared by the NationalInstitutes of Health Clinical Center nursing staff on how to give a subcutaneous injec-tion,48 and additional information specifically written for patients that is available onthe Web.43,49 Several organizations, including the American Academy of Allergy,Asthma, and Immunology, Case Western Reserve University/University Hospitals ofCleveland, and CSL-Behring, offer courses for physicians, nurses, and pharmacists,to assist with development of expertise within individual practices, institutions, andhome care companies/specialty pharmacies.
Centers in different countries have developed different approaches to actually start-ing SCIG therapy and teaching the patient, parent, or partner to perform the infusionswhen that is appropriate. Several of these are described in the book, ‘‘SubcutaneousIg Training and Education Program.’’23 Frequently, logistics, including the time neces-sary for obtaining approval from the patient’s insurance company and identifying theappropriate home nursing service/specialty pharmacy (when that is necessary), playa major role in determining the approach to be used in any given case. Most patientswho have been referred to the author and his colleagues have already been on IVIG.Many other patients are newly diagnosed but not hospitalized, and it is desirable to get

Subcutaneous Administration of IgG 795
their IgG supplementation going without delay. In these situations, the author and hiscolleagues will submit the necessary paperwork for SC therapy while they start or con-tinue the patient on IV infusions at their center. One or two 10-mL aliquots are removedfrom the total volume of IVIG to be administered, by drawing that amount out with oneor more 10-mL syringes. These aliquots are then infused SC while the main part of thedose is being given IV. Anecdotal and published reports42,51 suggest that most IVpreparations are well tolerated by the SC route. The nurse instructs the patient inhow to administer the SC infusion and demonstrates as he/she starts it. The patientthen experiences the sensation at the infusion site, and is given the opportunity to con-firm that he/she would like to switch to the SC route. Another SC infusion is thenstarted at a different site, and again, the nurse explains each step. That infusionmay be stopped after 3 or 5 mL have gone in, and the patient is invited to starta new infusion at a different site using the IgG remaining in the syringe. In many cases,this step can be done two or three times during the course of a single IV infusion. Afterhe/she has completed an instructional program and observed a nurse starting theinfusions several times, the patient should demonstrate proficiency back to thenurse/physican. A checklist the nurse can initial to document that the patient has suc-cessfully mastered each step can be used to provide documentation that all necessarytechniques were taught correctly. Medicare and Medicaid now have billing codes thatwill allow reimbursement for the time involved in starting and administering SCIGinfusions (obviously, one cannot bill for SC and IV infusions at the same visit), andpreapproval for the SC product and involvement of a specialty pharmacy are notnecessary because the patient is actually receiving an IV product. Teaching theprocedures for SC infusions is continued in the same way at subsequent IV infusionvisits until the patient receives the pump, syringe, and product he/she will actuallyuse at home for self/parent/partner infusions. When that occurs, the author and hiscolleagues ask the patient to bring those materials into their clinic so they can beinstructed with the actual supplies they will use themselves. At that visit, it is not nec-essary to bill for the product, supplies, or pump because that has all been arrangedand provided by the home care company/specialty pharmacy. The next time thepatient is due for an SC infusion, usually 1 week later, the home care nurse is askedto go to the patient’s home and observe/instruct the patient on self infusion. Morethan one visit may be necessary, but that is not usually the case because the patienthas already been instructed by the author’s office staff. It may be helpful if the homecare nurse or someone from the physician’s office confirms that he/she will be acces-sible by telephone during the patient’s first several infusions at home, to provide reas-surance or ‘‘talk the patient through’’ any step at which he/she has questions ordifficulties. Finally, the author and his colleagues ask the patient to return to their clinicafter he/she has taken a few infusions at home without a nurse present. The patientbrings his/her product, pump, and supplies and gives him/herself an infusion underobservation, to be sure all questions are answered, everything is being done in a sterileand safe way, and no problems are evident. After that, the patient continues on homeinfusions, and follow-up visits are scheduled as indicated by the patient’s medicalcondition.
Other approaches include starting naıve patients on IgG replacement by giving fiveto seven daily infusions in the hospital and using each day’s infusion as an opportunityfor instructing the patient. This approach need not be limited to inpatients. Gardulf andcolleagues in Sweden have had good success with conducting a 6- to 8-day course ofinstruction that covers not only the techniques for home SCIG administration but alsobasic information about PIDD, helpful instruction in hygiene for PIDD patients, andother relevant matters. This course is given to small cohorts of newly diagnosed

Berger796
patients who come together at the referral center when the course is given. Thepatients within the cohort have the ability to bond with each other, and then theycontinue as a support group for each other after the formal instruction period iscompleted. In other settings, 24-hour availability of an on-call nurse or pharmacistaccessible by a toll-free phone number, or identification of an ‘‘infusion buddy,’’who either takes his/her own infusion together with another patient or is available byphone, will provide an additional level of comfort and confidence to patients whohave any degree of anxiety or hesitation about proceeding at home.
It is important, particularly for patients who are used to being seen by the physicianat every visit when IV infusions are given in the office setting, to be instructed carefullyabout potential problems with their infusions and complications of their underlyingPIDD that should cause them to contact their physixcian urgently, or when to go toan emergency room or urgent care facility. Although most practices in the UnitedStates will prescribe preloaded epinephrine injectors (such as an Epi-pen) for patientswho infuse IgG at home, in fact, these are rarely needed for patients using the SCroute. The United Kingdom NHS now no longer provides these for home SC patientsbecause they were so rarely actually needed. Finally, patients who are self infusing athome should use special medical waste (biohazard or ‘‘sharps’’) containers andshould discard needles or other used medical equipment cautiously. In addition, pro-visions for collecting and appropriate disposal of the waste containers must bearranged.
Assuring Adherence to Subcutaneous Regimens
In many cases, patients on home SCIG regimens will be seeing the physician or otherhealth care professionals less often than previously, especially if the patient had beenreceiving IGIV in the physician’s office and a ‘‘check-up’’ at each infusion visit. Patientsshould be carefully evaluated for reliability and insight before they are allowed to havethe degree of independence possible with home SCIG regimens. As with all otherusers of blood products, patients should keep a logbook in which the infusion date,lot number, and expiration date of all vials of IgG are recorded. If the patient wantsto keep this log electronically, it should be carefully backed up onto a disc or othermemory device separate from the computer itself, or forwarded to a central memoryservice. Inspection of the logbook or computer record by the physician or nurse at reg-ular intervals should be an important part of each patient’s follow-up. An additionalprecaution may be to ask the patient to mark the label of each vial of IgG with thedate on which it was used, and then to return the empty vials to the physician’s office.
Because the serum IgG concentration tends toward a true steady state if SCIG isgiven weekly or more often, the patient’s serum IgG can be drawn randomly, andshould serve as an indicator of compliance with the prescribed regimen. However,changes in metabolism accompanying different disease states, or protein losses,for example, due to nephropathy or enteropathy, may be the cause if low levels areobserved. In growing children, the dose or regimen may need to be altered everyfew months as the child gains weight.
SUBCUTANEOUS IMMUNOGLOBULIN IN OTHER CONDITIONS
Although the SC route has proved to be useful for delivering the doses of IgG used forreplacement therapy in PIDD, few studies have been carried out of the use of this routein autoimmune and inflammatory diseases for which doses in the range of 1 to 2 g/kgare given over 2 to 4 days once a month. It should be obvious from the data in Fig. 1, inwhich a hypogammaglobulinemic patient sustained an increase in his serum IgG

Subcutaneous Administration of IgG 797
concentration of more than 800 mg/dL when he was given 400 mg/kg of IGIV, thatpatients receiving doses in the range of 1000 to 2000 mg/kg over 1 to 4 days will reachextremely high IgG concentrations. These IgG levels can significantly increase bloodviscosity and may be associated with increased risks for ischemic, thromboembolic,and renal adverse effects.52 Fractionating the same monthly dose into 10, 20, or even30 aliquots, which could be given at 1- to 3-day intervals throughout the month, mightbe expected to decrease the maximum IgG level and the alterations in blood rheologyand might, therefore, be safer. Furthermore, patients who are receiving "high-dose’’IGIV therapy are usually monitored closely during their infusions, so they are frequentlytreated in hospitals or infusion centers rather than at home. It therefore seems likelythat SC infusions would be particularly preferable for many patients in terms of safetyand convenience.
Multiple mechanisms of action have been proposed to account for the efficacy ofhigh-dose IGIV in autoimmune/inflammatory diseases, and it seems likely thatdifferent mechanisms are important in different disease states, depending on theirpathophysiology.53 In any disease state in which an extraordinarily high peak is notrequired, the SC route would seem likely to offer distinct advantages for manypatients. Even in the author’s initial experience with SCIG in the early 1980s, it becameobvious that a motivated patient (a young woman who wanted to safely carry a preg-nancy through to term) could take two 10-mL SC infusions every day, yieldinga monthly dose of nearly 100 g.5 Current regimens for PIDD frequently have patientstaking as much as 60 mL of 16% IgG (9.6 g) over 1 to 2 hours while they are performingnormal activities. Because this regimen is well tolerated, or even preferred over IVinfusions, by most patients, no a priori reason seems to exist as to why multiple infu-sions in that dose range could not be given several days per week. Few studies havebeen carried out of SC dosing in this range for the treatment of autoimmune/inflammatory diseases. In one report of SCIG in patients who had well-documentedchronic idiopathic demyelinating polyneuropathy (CIDP), one patient who could notbe successfully treated with immunosuppressive medications and had only transientresponses to IGIV doses of 800 mg/kg was successfully transitioned to SCIG. Thispatient, a 73-year-old woman weighing 75 kg remained stable with good strengthfor more than 8 months on a regimen of 16 g of IgG per week, given as five separateinfusions of 3.2 g (20 mL) over 2 hours each. The other patient, a 53-year-old man whoalso could not be successfully treated with immunosuppressive and IGIV therapy, hasbeen successfully managed for more than 2 years with only 6.4 g of IgG per week,given as a single infusion of 40 mL of 16% SCIG.54 Neither of these patients hadany side effects from the SC infusions other than local swelling; no systemic adverseeffects were reported at all.54 In the first patient, the cost savings associated with theuse of SCIG rather than IGIV was estimated at 30,000 Euros per year. Cost savingswere not estimated for the other patient.
Koller and colleagues55 have reported on three patients who tried the SC route toreceive IgG for neuromuscular diseases. One patient developed a systemic exanthemafter 9 weeks on SCIG and refused further treatment by that route. Another patient,with biopsy-proven CIDP persisting for more than 2 years, previously suffered multiplerelapses despite corticosteroids and IGIV. He was successfully managed with weeklyinfusions of 100 mg/kg of SCIG and showed marked reduction in disability scores overthe 6 months described in the report. No further relapses occurred and his steroiddose could be reduced. The third patient had multifocal motor neuropathy that hadresponded in the past to IGIV. He was also successfully managed with 100 mg/kgof SCIG per week and did not require any other therapy. It thus seems clear that,although SCIG cannot be widely recommended for use in autoimmune/inflammatory

Berger798
diseases at the present time, reason for optimism exists, and additional studies inthese conditions should be performed.
FUTURE DEVELOPMENTS
In the next few years, we are likely to see continued evolution of SCIG therapy in theUnited States. Multiple IgG products are already marketed for SC use in Europe.Clinical trials are now underway in the United States using a ‘‘next generation’’ 16%solution and with two 10% IgG products currently marketed for IV use. In addition,one manufacturer already has a 20% IgG solution in clinical trials for SC treatmentin PIDD, and another has published a method for generating and reconstituting20% IgG solutions that are easily given through 28-gauge needles.56 Clinical trialsof that type of preparation have not yet been initiated. It is likely that the availabilityof 20% solutions of IgG for SC use will provide further impetus for the use of this routeto deliver high-dose IgG for autoimmune and inflammatory diseases. The availability of20% solutions may also provide advantages for some patients using conventionaldoses of IgG for PIDD, because less volume will be needed to administer the sameamount of IgG. However, studies that show that high-concentration products are tol-erated as well as the alternatives must be completed before they come in clinicalpractice.
Another approach to giving larger volumes of IgG SC is the injection of recombinanthyaluronidase to loosen the SC tissues by temporarily breaking down hyaluronic acidin the extracellular matrix of adipose and connective tissue. Apparently, the matrix isrepaired rapidly, so the tissues are not changed permanently. Recombinant and otherforms of hyaluronidase have been in clinical use in humans for some time for variousindications, including enhancing the effects of local anesthetics, facilitating hydrationby clysis, and in ophthalmic and plastic surgery.57 A recent study has shown that in-tradermal injections of a formulation of a recombinant human hyaluronidase was freefrom allergic reactions in 100 normal subjects.58 Preliminary reports suggest that anSC injection of hyaluronidase just before an infusion of IgG into the same site allowsSC delivery of volumes of 10% IgG similar to those used for monthly IV treatment ofPIDD patients in the same time that would be required for IV administration.59 Obvi-ously, the ability to infuse larger volumes faster by the SC route, without the needfor multiple sites, would be a distinct advantage for many PIDD patients. However,the ability of the weekly SC infusions now used by many patients to raise the ‘‘troughlevel’’ and eliminate ‘‘wear-off’’ effects would not be expected if the whole monthlydose was given at one time. Results of currently ongoing studies are anxiouslyawaited, to assess the safety and tolerance of these regimens and to determinehow hyaluronidase-facilitated SC infusions will affect the treatment of PIDD. Theresults of the current studies are also likely to lead to further studies of SCIG in auto-immune and inflammatory diseases.
SUMMARY
The efficacy of SC administration of antibodies followed by purified IgG was recog-nized and used by von Behring and Bruton. The availability of IgG preparations thatcould be administered safely by the IV route was a long-sought goal that was finallyachieved in the early to mid-1980s. IVIG revolutionized the treatment of PIDD andled to the discovery of the therapeutic value of high-dose IgG in autoimmune and in-flammatory diseases not associated with PIDD. Improved therapy has improved out-comes and expectations, and most PIDD patients can lead fully active and productivelives. Administration of IgG by the SC route is effective and safe, overcomes obstacles

Subcutaneous Administration of IgG 799
to the use of IVIG in some patients, and provides important quality-of-life advantagesto others. The coming years will see increased use of SCIG in PIDD, which will be fa-cilitated by advances leading to higher-concentration IgG products and easier deliv-ery. Additional clinical research and the effects of these advances are likely to leadto increased use of SCIG in autoimmune and inflammatory diseases, in addition tomore widespread use in PIDD.
REFERENCES
1. von Behring E. Serum therapy in therapeutics and medical science. Nobel Lecture1901. Available at: www.nobelprize.org. Accessed January 30, 2008.
2. Berger M, Cupps TR, Fauci AS. Immunoglobulin replacement by slow subcutane-ous infusion. Ann Intern Med 1980;98:55–6.
3. Roord JJ, Van der Meer JW, Kuis M, et al. Home treatment in patients withantibody deficiency by slow subcutaneous infusion of gammaglobulin. Lancet1982;I:689–90.
4. Ugazio AG, Duse M. Re R subcutaneous infusions of gammaglobulin in manage-ment of agammaglobulinemia. Lancet 1982;I:226.
5. Berger M, Cupps TR, Fauci AS. High dose immunoglobulin replacement therapyduring pregnancy. J Am Med Assoc 1982;247:2824–5.
6. Welch MJ, Steihm ER. Slow subcutaneous immunoglobulin therapy in a patientwith reactions to IM immunoglobulin. J Clin Immunol 1983;3:285–6.
7. Chapel HM, Spickett GP, Ericson D, et al. The comparison of the efficacy andsafety of intravenous versus subcutaneous immunoglobulin replacement therapy.J Clin Immunol 2000;20:94–100.
8. Pac M, Bernakowska E. Polish experience with immunoglobulin replacementtreatment by subcutaneous infusion. Central European Journal of Immunology2005;30:78–82.
9. Gardulf A, Hammarstrom L, Smith CIE. Home treatment of hypogammaglobuline-mia with subcutaneous gammaglobulin by rapid infusion. Lancet 1991;338:162–6.
10. Kittner JM, Grimbacher B, Wulff W, et al. Patients’ attitude to subcutaneous immu-noglobulin substitution as home therapy. J Clin Immunol 2006;26(4):400–5.
11. Desai SH, Chouksey A, Poll J, et al. Comparison of efficacy and tolerability ofequal doses of gamunex given IV or SC: a pilot study. J Allergy Clin Immunol2008: submitted for publication.
12. Ochs HD, Gupta S, Kiessling P, et al. Safety and efficacy of self-administeredsubcutaneous immunoglobulin in patients with primary immunodeficiency dis-eases. J Clin Immunol 2006;26:265–73.
13. Gardulf A, Nicolay U, Asensio O, et al. Rapid subcutaneous IgG replacementtherapy is effective and safe in children and adults with primary immunodefi-ciencies - a prospective, multi-national study. J Clin Immunol 2008;26:177–85.
14. ‘‘Pharmacokinetics’’ in Vivaglobin, summary of basis for approval. Available at:www.fda.gov/cber/sba/vivaglobin010906s.pdf. Accessed July 5, 08.
15. Pamarthi M, Taylor G, Scuderi P, et al. Subcutaneous availability of Gamunex inRabbits. J Allergy Clin Immunol 2007;119:s16.
16. US Food and Drug Administration. Guidance for industry: safety, efficacy, andpharmacokinetic studies to support marketing of immune globulin intravenous(human) as replacement therapy for primary humoral immunodeficiency. Avail-able at: http://www.fda.gov/cber/gdlns/igivimmuno.htm. Accessed January 7,2008.

Berger800
17. Aebersold P. Intravenous immunoglobulins in the 21st century: progress andchallenges in efficacy, safety and paths to licensure. FDA workshop 4/13/05.Available at: www.fda.gov/cber/minutes/igiv041305t.htm. Accessed July 5, 2008.
18. Emea. Note for guidance on the investigation of human normal immunoglobulin forsubcutaneous and intramuscular use. CPMP/BPWG/283/00. 2002. Available at:www.emea.europa.eu/pdfs/human/bpwg/028300.en.pdf. Accessed July 5, 2008.
19. Waldmann TA, Strober W, Blaese RM. Metabolism of immunoglobulins. Progressin Immunology 1972;891–903.
20. Mankarious S, Lee M, Fischer S, et al. The half-lives of IgG subclasses andspecific antibodies in patients with primary immunodeficiency who are receivingintravenously administered immunoglobulin. J Lab Clin Med 1988;112(5):634–40.
21. Vivaglobin prescribing information. CSL-Behring. King of Prussia, PA.22. Waniewski J, Gardulf A, Hammarstrom L. Bioavailability of gamma-globulin after
subcutaneous infusions in patients with common variable immunodeficiency.J Clin Immunol 1994;14:90–7.
23. Sewell WAC, editor. Subcutaneous immunoglobulin therapy medical educationprogramme. Oxfordshire (UK): Watermeadow Medical plc; 2006.
24. Berger M. Subcutaneous immunoglobulin replacement in primary immunodefi-ciencies. Clin Immunol 2004;112:1–7.
25. Ballow M. Safety of IGIV therapy and infusion-related adverse events. ImmunolRes 2007;38(1–3):122–32.
26. Gardulf A, Anderson V, Bjorkander J, et al. Subcutaneous immunoglobulinreplacement in patients with primary antibody deficiencies: safety and costs.Lancet 1995;345:365–9.
27. Gardulf A, Bjorvell H, Gustafson R, et al. The life situations of patients with primaryantibody deficiency untreated or treated with subcutaneous gammaglobulin infu-sions. Clin Exp Immunol 1993;92:200–4.
28. Radinsky S, Bonagura V. Subcutaneous immunoglobulin infusion as an alterna-tive to intravenous immunoglobulin. J Allergy Clin Immunol 2003;112:630–3.
29. Immune Deficiency Foundation. Treatment experiences and preferences ofpatients with primary immune deficiency diseases: first national survey. 2003. Avail-able at: www.primaryimmune.org/publications/surveys/Treatment_Experiences_and_preferences_(2003). Accessed July 5, 2008.
30. Gardulf A, Nicolay U, Math D, et al. Children and adults with primary antibodydeficiencies gain quality of life by subcutaneous IgG self-infusions at home.J Allergy Clin Immunol 2004;114:936–42.
31. Nicolay U, Kiessling P, Berger M, et al. Health-related quality of life and treat-ment satisfaction in North American patients with primary immunedeficiencydiseases receiving subcutaneous IgG self-infusions at home. J Clin Immunol2006;26:65–72.
32. Gardulf A, Bjorvell H, Andersen V, et al. Lifelong treatment with gammaglobulinfor primary antibody deficiencies: the patients’ experiences of subcutaneousself-infusions and home therapy. J Adv Nurs 1995;21:917–27.
33. Gardulf A, Andersson E, Lindqvist M, et al. Rapid subcutaneous IgG replacementtherapy at home for pregnant immunodeficient women. J Clin Immunol 2001;21:150–4.
34. Arora R, Newton TC, Nelson MR. Subcutaneous Ig therapy in an 11-year-oldpatient with common variable immunodeficiency and von Willebrand’s disease.Ann Allergy asthma Immunol 2007;99:367–70.

Subcutaneous Administration of IgG 801
35. Sundin U, Neva S, Hammarstrom L. Induction of unresponsiveness against IgA inIgA-deficient patients on subcutaneous immunoglobulin infusion therapy. ClinExp Immunol 1998;112:341–6.
36. Ho C, Membe S, Cimon K, et al. An overview of subcutaneous vs. intravenousimmunoglobulin for primary immunodeficiencies: systematic review and economicanalysis [technology overview #36]. Ottawa (Canada): Canadian Agency for Drugsand Technologies in Health; 2008. Available at: http://www.cadth.ca/index.php/en/publication/785. Accessed August 26, 2008.
37. Hogy B, Keinecke HO, Borte M. Pharmacoeconomic evaluation of immunoglob-ulin treatment in patients with antibody deficiencies from the perspective of theGerman statutory health insurance. Eur J Health Econ 2005;6:24–9 [with Erratumin 6:243, 2005].
38. Gardulf A, Bjorvell H, Gustafson R, et al. Safety of rapid subcutaneous gamma-globulin infusions in patients with primary antibody deficiency. Immunodeficiency1993;4:81–4.
39. Gaspar J, Gerritsen B, Jones A. Immunoglobulin replacement treatment by rapidsubcutaneous infusion. Arch Dis Child 1998;79:48–51.
40. Hansen S, Gustafson R, Smith CIE, et al. Express subcutaneous IgG infusions:decreased time of delivery with maintained safety. Clin Immunol 2002;104:237–41.
41. Berger M, Duff K, Poll J, et al. Immunoglobulin replacement therapy by the sub-cutaneous route using preparations licensed in the US for administration by otherroutes. In: Dalakis M, Spath PJ, editors. Intravenous immunoglobulins in the thirdmillenium. New York: Parthenon; 2004. p. 77–80.
42. Chouksey A, Duff K, Wasserbauer N, et al. Subcutaneous IgG replacement ther-apy with preparations currently available in the US for IV or IM use: reasons andregimens. Allergy Asthma Clin Immunol 2005;1:120–30.
43. Guidelines for subcutaneous IgG therapy. Available at: www.ukpin.org.uk/guidelines3.-01-Administration-SCIG.pdf. Accessed July 5, 2008.
44. Berger M, Duff K, Beal C. Information for patients: pumps and needles used forsubcutaneous IgG. Available at: http://www.rainbowbabies.org/subcu. AccessedJanuary 7, 2008.
45. Gustafson R, Gardulf A, Hansen S, et al. Rapid subcutaneous immunoglobulinadministration every second week: results in high and stable serum IgG levelsin patients with primary antibody deficiencies. Clin Exp Immunol 2008;152:274–9.
46. CSL-Behring, King of Prussia, PA. Vivaglobin patient brochure. Available at: www.vivaglobin.com/PDF/patient_brochure.pdf. Accessed July 5, 2008.
47. Baxter, Vienna, Austria. Subcuvia Patient Brochure. Available at: www.immunediseaseeurope.com/ideu/pdfs/10020_subcuvia_patient_bro_print.pdf.Accessed July 5, 2008.
48. NIH Clinical center nurses: patient information publications. giving a subcutane-ous injection. Available at: www.cc.nih.gov/ccc/patient_education/pepubs/subq.pdf. Accessed January 7, 2008.
49. Berger M, Duff K. ‘‘The story of subcutaneous IgG,’’ and other materials. Availableat: www.rainbowbabies.org/sbcu. Accessed July 5, 2008.
50. Berger M. Subcutaneous IgG therapy in immune deficiency diseases. ImmuneDeficiency Foundation Clinical Focus issue 13, 2008. Available at: www.primaryimmune.org/publications/clinic_focus/cc_feb08.pdf. Accessed July 5,2008.

Berger802
51. Stiehm ER, Casillas AM, Finkelstein JZ, et al. Slow subcutaneous human IGIV inthe treatment of antibody immunodeficiency: use of an old method with a newproduct. J Allergy Clin Immunol 1998;101:848–9.
52. Pierce LR, Jain N. Risks associated with the use of intravenous immunoglobulin.Tansfus Med Rev 2003;17:241–54.
53. Dalakas MC. Mechanism of action of intravenous immunoglobulin andtherapeutic considerations in the treatment of autoimmune neurologic diseases.Neurology 1998;51(Suppl 5):S2–8.
54. Lee DH, Linker RA, Paulus W, et al. Subcutaneous immunoglobulin infusion:a new therapeutic option in chronic inflammatory demyelinating polyneuropathy.Muscle Nerve 2008;37:406–9.
55. Koller H, Schroeter M, Feischen H, et al. Subcutaneous self-infusions of immu-noglbulins as a potential therapeutic regimen in immune-mediated neuropathies.J Neurol 2006;253:1505–6.
56. Dani B, Platz R, Tzannis ST. High concentration formulation feasibility of humanIgG for subcutaneous administration. J Pharm Sci 2007;96:1504–17.
57. Frost GI. Recombinant human hyaluronidase (rHUPH20): an enabling platform forsubcutaneous drug and fluid administration. Expert Opin Drug Deliv 2007;4:427–40.
58. Yocum RC, Kennard D, Heiner LS. Assessment and implication of the allergicsensitivity to a single dose of recombinant human hyaluronidase injection: a dou-ble blind, placebo-controlled clinical trial. J Infus Nurs 2007;30:293–9.

Pharmacokineticsof ImmunoglobulinAdministered viaIntravenous orSubcutaneous Routes
Francisco A. Bonilla, MD, PhDa,b,*
KEYWORDS
� Immunoglobulin � Intravenous � Pharmacokinetics� Subcutaneous
Polyclonal human immunoglobulin G (IgG) therapy is applied in many disease states,including primary and secondary immunodeficiencies, and a wide variety of infectious,autoimmune, and inflammatory disorders.1 The practical use of IgG therapy derives inlarge measure from its peculiar pharmacokinetic properties, which are reviewed in thisarticle. In particular, IgG circulates intact in serum for a period of time that is severalfold longer than almost any other class of serum protein. This permits IgG to be admin-istered intermittently and relatively infrequently (intervals measured in weeks) withpersistent effectiveness between successive administrations.
Although therapeutic replacement of other Ig classes (IgA and IgM) has been stud-ied, we focus our discussion entirely on IgG, because it is the only isotype available forroutine clinical use. IgG may be administered by intramuscular (IM), subcutaneous(SC), or intravenous (IV) routes; IgG administered by each of these routes is abbrevi-ated IMIG, SCIG, and IVIG, respectively. IM injection is no longer considered appro-priate for routine replacement therapy in light of the superior ease of administrationand efficacy via the IV and SC routes. Polyvalent IgG formulated for IM administrationmay be given by the SC route with good results.2 IM injection is still appropriate foradministration of specific Ig for infection prophylaxis (eg, tetanus, rabies, hepatitisB, varicella) because the effectiveness of SC administration of any of these productshas not been determined.
a Division of Immunology, Children’s Hospital Boston, Fegan Building, 6th Floor, 300 LongwoodAvenue, Boston, MA 02115, USAb Department of Pediatrics, 25 Shattuck Street, Harvard Medical School, Boston, MA 02115,USA* Division of Immunology, Children’s Hospital Boston, Fegan Building, 6th Floor, 300 LongwoodAvenue, Boston, MA 02115.E-mail address: [email protected]
Immunol Allergy Clin N Am 28 (2008) 803–819doi:10.1016/j.iac.2008.06.006 immunology.theclinics.com0889-8561/08/$ – see front matter ª 2008 Elsevier Inc. All rights reserved.

Bonilla804
This article focuses mainly on pharmacokinetic studies of polyclonal human IgG viaIV and SC routes administered for replacement and infection prophylaxis in primaryand secondary immunodeficiency states.
PHYSIOLOGYOF IMMUNOGLOBULINS
Important studies of immunoglobulin (Ig) synthesis and catabolism in humans in vivowere conducted in the 1960s. In particular, the work of Waldmann and colleagues3,4
established the fundamental behaviors of various Ig isotypes under a variety of phys-iologic and pathologic conditions. To determine the rate of disappearance of Ig by pro-tein catabolism, these authors injected volunteers with small amounts of radioactive Igof various isotypes and examined the rate of decline of serum radioactivity. Table 1summarizes some of these data. These authors calculated the total body content ofIgG to be 1.1 g/kg with 45% in the intravascular compartment.
These studies quickly determined that IgG catabolism has important differences incomparison to other isotypes. The major contrast is that catabolism of IgG is not onlyslower than all other Ig classes, but it is also proportional to its serum concentration.The fractional catabolic rate of IgG increases as the serum concentration rises andvice versa (Fig. 1), which is not true of any other Ig class. Researchers further observedthat this feature depended entirely on the Fc portion of IgG, because any IgG frag-ments that did not contain Fc were rapidly catabolized (see Table 1), and the ratewas independent of their concentrations. At the time, researchers hypothesized thata saturable protection receptor system for IgG Fc could explain these findings.5
Approximately 30 years later, researchers established that the principal mechanismof regulation of serum IgG level involves the neonatal Fc receptor (FcRn).6 This recep-tor derives its name from its earliest known function—transport of IgG from thenewborn proximal small intestine into the circulation.7 Later, it was found to transportIgG from the maternal bloodstream into the fetus during gestation.8 This receptor isalso expressed on vascular endothelium, where it acts to increase IgG half-life.(The receptor also binds serum albumin and increases its half-life.) More recently,FcRn has been found in various organs and tissues and may play a wider role inIgG physiology and humoral immunity than previously appreciated.9
Table 1Serum half-lives of immunoglobulins in healthy adults
Ig Half-Life (d) FCR Ig Half-Life (d) FCRIgGa 23.0 6.7 IgAa 5.8 25.0
IgG1b 21.0 — IgMa 5.1 18.0
IgG2b 21.0 — IgDa 2.8 37.0
IgG3b 7.1 — IgEa 2.5 89.0
IgG4b 21.0 — — — —
Fca 10-20 — — — —
Faba 0.18 — — — —
L chaina 0.14 — — — —
Abbreviation: FCR, fractional catabolic rate (percent of intravascular pool per day).a Data from Waldmann TA, Strober W, Blaese RM. Variations in the metabolism of immunoglob-
ulins measured by turnover rates. In: Merler E, editor. Immunoglobulins. Washington, DC: NationalAcademy of Sciences; 1970. p. 33–51.
b Data from Morell A, Terry WD, Waldmann TA. Metabolic properties of IgG subclasses in man.J Clin Invest 1970;49:673–80.

0
10
20
30
40
50
60
70
80
90
100
0 20 40 60 80IgG survival t1/2 (days)
Fig.1. Concentration dependence of IgG catabolism. (From Waldmann TA, Strober W, BlaeseRM. Variations in the metabolism of immunoglobulins measured by turnover rates. In:Merler E, editor. Immunoglobulins. Washington, DC: National Academy of Sciences; 1970.p. 38; with permission.)
Pharmacokinetics of Immunoglobulin 805
FcRn is composed of an a chain homologous to human leukocyte antigen classI molecules that also must pair with b2 microglobulin for surface expression and func-tion. The receptor does not bind short peptides, however, and the gene encoding thealpha chain is not located in the human leukocyte antigen gene complex on chromo-some 6 (the FCGRT gene encoding FcRn is on chromosome 19q13.3). The mecha-nism of the concentration dependence of IgG catabolism mediated by FcRn isillustrated in Fig. 2. Promoter polymorphisms of the FCGRT gene lead to variationsin levels of FcRn expression,10 which might underlie some of the variability of IgGlevels between individuals, although this has not been reported.
In 1968, Waldmann and colleagues11 described two siblings with ‘‘familial idiopathichypercatabolic hypoproteinemia’’ who had low serum levels of IgG and albumin. Inthese individuals, the rate of catabolism of IgG was fivefold greater than normals,and serum IgG levels were 130 to 440 mg/dL. Almost 40 years later, these individualswere found to harbor mutations of b2 microglobulin, which reduced expression ofFcRn by more than 80%.12 These siblings are the only two reported patients withthis entity (OMIM #241600).
Understanding the particular pharmacokinetic properties of IgG Fc together withrecombinant DNA technology has led to the development of an entire class of biologictherapeutic agents, the ‘‘IgG fusion proteins.’’ These agents have a non-Ig therapeuticcomponent, linked to an IgG Fc fragment, to ‘‘borrow’’ its ability to confer a prolongedtime in the circulation, enhancing its bioavailability and therapeutic effect.13
IMMUNOGLOBULIN G REPLACEMENT THERAPY
IgG replacement therapy is indicated for primary or secondary immunodeficiencieswith hypogammaglobulinemia or impaired specific antibody formation.1 Purified

Low IgG
IgG
Membrane FcRn
Endocytosis
Recycle to surface
Degradation
in lysosome
High IgG
A
B
Fig. 2. FcRn is expressed on the surface of vascular endothelium (and other cell types thatmay mediate this function, such as monocyte derived cells). Plasma is endocytosed, and acid-ification favors IgG binding to FcRn; an equilibrium is reached with some fraction of IgGbound to FcRn and some free within the endosome. (A) When IgG is relatively low, a smallamount remains unbound after endocytosis and is sent to a degradative pathway, whereasFcRn with bound IgG is shuttled back to the cell surface. Physiologic pH promotes dissocia-tion, and intact IgG is released into the circulation again. In this example, 20% of the IgGthat was taken up was degraded. (B) When the concentration of IgG is relatively high,FcRn becomes saturated, and a greater proportion of circulating IgG remains unboundand is ultimately broken down. In this example, 50% of the IgG taken up was eliminated.
Bonilla806
human IgG preparations have been formulated specifically for IM, SC, and IV admin-istration. Preparations suitable for IM or IV use may be given SC;2 however, prepara-tions intended for IM or SC use may not be given IV because of content of IgGaggregates that may lead to systemic reactions. Dose regimens have been deter-mined empirically in clinical trials to determine the preinfusion (trough) IgG levelsthat are associated with adequate clinical efficacy.1 Several of these studies arealso mentioned here. The minimum trough level consistent with effective protectionin agammaglobulinemic individuals is approximately 500 mg/dL. There is measurableimprovement in outcomes (reduced rate of infections) when trough levels are driven ashigh as 900 to 1000 mg/dL, however. Standard dose regimens for IgG replacement areshown in Table 2.
PHARMACOKINETICS OF INTRAVENOUS IMMUNOGLOBULIN
After administration of relatively large amounts of IVIG (0.1–2 g/kg body mass), the IgGconcentration in serum immediately rises, falls rapidly in the first 1 to 7 days, and thenfalls more slowly thereafter. The initial rapid fall is associated with passage of IgG out

Table 2Common dosing regimens for immunoglobulin G replacement therapy via intramuscular,intravenous, and subcutaneous routes
Dose IntervalConcentration ofInfused IgG (%)
IMIG 0.025 g/kg weekly 16–16.5
IVIG 0.3–0.8 g/kg 2–4 wk 5–12
SCIG 0.05–0.2 g/kg 0.5–2 wk 10–16.5
Low doses are given at shorter intervals; higher doses are given at longer intervals.
Pharmacokinetics of Immunoglobulin 807
of the vasculature into lymph and extracellular fluid compartments. The subsequentdecline is mainly caused by catabolism while IgG in lymph and tissues slowly diffusesback into the circulation. Many studies of healthy individuals model the IgG concen-tration using two compartments: the vascular and extravascular spaces with an equi-librium between the two and with the vascular space being the point of entry and exit(Fig. 3A). The IgG concentration decay curve may be subdivided into two phasescommonly designated as a (early) and b (late) (Fig. 3B). Although this mathematicaldescription is appropriate in many circumstances, it is not universally applied in stud-ies of IVIG pharmacokinetics. Many studies use alternative pharmacologic models(eg, noncompartmental, single compartment).14 Simpler models used to describe lon-ger infusion intervals (weeks) are more likely to deviate from the observed decay curvein the early phase, as opposed to the late phase. The importance of this deviation forpractice is unclear, however. With respect to replacement therapy, primary emphasis
Intravascularcompartment
Extravascularcompartment(s)
IVIGSynthesis
SCIG
CatabolismLoss
IVIG IVIG
Trough IgG level
Early (α) phase
Late (β) phase
Time (days)
Seru
m Ig
G c
once
ntra
tion
0 7 14 2821
A B
Fig. 3. Two-compartment model of IgG pharmacokinetics. (A) IgG is in equilibrium betweenthe vascular space and extravascular areas. IgG is synthesized in the bone marrow anddiffuses into the lymph and then to the blood. SCIG is absorbed from subcutaneous tissues.IVIG enters the vascular space directly. IgG is catabolized in vascular endothelium (and pos-sibly other areas). IgG also may be lost from the vascular space by various mechanisms(eg, protein loss in the intestines or urinary tract). (B) Serum IgG concentration over timeduring IVIG replacement therapy. With IV administration, IgG enters the vascular compart-ment in high concentration, redistributes rapidly into tissue compartments, and then ismore slowly catabolized. The early redistribution phase is sometimes called the a phase,and the later slow decline is the b phase.

Bonilla808
is placed on the trough (immediate preinfusion) IgG level. Many studies—particularlymost studies conducted by IVIG manufacturers—derive an empiric serum or plasmadecay half-life (t1/2) as the principal descriptor of the expected rate of disappearanceof the infused IgG.
Primary Immunodeficiency
Table 3 shows the serum half-lives of several of the IVIG products currently in use.These data were derived mainly from clinical trials of IVIG replacement in patientswith primary immunodeficiency.15–22 Most patients in these trials had either X-linkedagammaglobulinemia or common variable immunodeficiency. The ranges of dosesadministered in these trials vary widely from approximately 0.3 to 0.6 g/kg, and thedose intervals vary. In some cases, dose intervals of 3 or 4 weeks are analyzed
Table 3Elimination half-lives of some intravenous immunoglobulin G products
Productt1/2 (d)Mean � SD
AUC (d*mg/dL)Mean � SD
AUCTimeMean
Dose;Interval n Reference
Carimune NF 38 �15 27,100 � 9400 21 d 0.3–0.8 g/kg; 3 or 4 wk 16 19
ra 16–72 — — — — —
Flebogamma5%
29.9 � 9.5b 29,448 � 9319 28 dc 0.3–0.6 g/kg; 3 wk 10 18
44.6 � 13.4 32,694 � 9848 28 d 0.3–0.6 g/kg; 4 wk 11 —
Flebogamma5% DIF
30 � 9 31,159 � 6572 28 dc 0.3–0.6 g/kg; 3 wk 8 17
r 19–41 r 20,458–40,104 — — — —
32 � 5 32,894 � 3886 28 d 0.3–0.6 g/kg; 4 wk 12
r 25–39 27,650–41,814 — — — —
GAMMAGARD10%
35d 29,139e 28 d 0.3–0.6 g/kg; 3 or 4 wk 57 20
95% CI(31–42)
(95% CI 27,494–30,490)
— — — —
Gamunex 34.9f — — 0.2–0.6 g/kg; 3 or 4 wk 17 16
34.7f — — 0.3–0.5 g/kg; 3 or 4 wk 14 —
Octagam 41 � 17 — — 0.3–0.6 g/kg; 3 or 4 wk 46 21
r 23–84 — — — — —
36 � 11 16,010 � 3460 21 d 0.2–0.42 g/kg; 3 wk 17 15
r 18–56 r 10,380–22,860 — — — —
Privigen 28 � 6 32,820 � 6260 21 d 0.2–0.888 g/kg; 3 wk 3 22
r 22–33 r 28,580–40,010 — — — —
45 � 19 36,390 � 5950 28 d 0.2–0.888 g/kg; 4 wk 22 —
r 21–97 r 19,680–44,340 — — — —
Sandoglobulin 45 � 19 28,400 � 11,700 21 d 0.3–0.8 g/kg; 3 or 4 wk 15 19
r 15–88 — — — — —
Abbreviation: AUC, area under the curve.a Range.b Mean � SE.c Patients on the 3-week interval regimen had one sampling period of 4 weeks for pharmacoki
netic analysis.d Median.e Pharmacokinetic analysis was over 28 days; the reported AUC corresponds to 21 days after
infusion.f Geometric least-square mean; two separate analyses were reported.

Pharmacokinetics of Immunoglobulin 809
together; in others, they are analyzed separately. For cases in which they are analyzedseparately, it is evident that the shorter dose interval (higher cumulative IgG dose overtime) tends to be associated with a shorter half-life. It is also clear that there is tremen-dous individual variation in rates of decay in each study. Sources of variability includedifferent dose regimens, time of sampling, variability in IgG measurement, amount andrate of endogenous IgG production, and actual individual variation in rates of IgGcatabolism. The variability in measured IgG decay rates is so great that it is unlikelythat statistical differences could be established between regimens with one productor between products. Two studies have compared pharmacokinetic parameters fortwo different IVIG preparations.16,19 Both studies concluded that the products werepharmacologically equivalent (no statistical difference in measured parameters).
Replacement dose regimens of IVIG have evolved toward ever higher amounts overthe last few decades. Initial regimens were based on doses that had been adminis-tered IM, and immunodeficient patients were often given a cumulative monthy re-placement dose of 0.1 g/kg (four weekly injections of 0.25 g/kg). One early studycompared pharmacokinetics of IgG in 16 patients who had primary immunodeficiencydiseases (PID) receiving ‘‘low’’ dose (0.1 g/kg monthly) with ‘‘high’’ dose (average 0.35g/kg monthly, titrated to give a trough IgG level of 450 mg/dL).23 The half-lives rangedfrom 22 to 96 days (mean, 43 days) at the lower doses and 20 to 59 days (mean, 33days) at the higher doses. (This product is no longer available and is not included inTable 3.) There was no statistical difference between these values, however. The au-thors also suspected that some level of endogenous IgG production in some patientswas affecting (prolonging) the half-life determinations.
In a more recent study, 41 patients were compared with respect to two differentdose regimens.24 In this study, the ‘‘low’’ dose regimens were 0.3 g/kg and 0.4 g/kgevery 4 weeks in adults or children, respectively; the ‘‘high’’ dose regimens were0.6 g/kg and 0.8 g/kg every 4 weeks in adults and children, respectively. The authorsdid not attempt any pharmacokinetic study other than monitoring the trough IgGlevels, which rose by an average of 45% (from 640 up to 940 mg/dL). This increasewas associated with significant improvements in patients’ clinical courses. Most au-thors emphasize that controlling the trough IgG level, together with monitoring the clin-ical course, is most important for determining the appropriate replacement regimen forindividual patients.
A few recent studies measured half-lives of IgG subclasses after IV administration ofa single product to patients with PID.15,18 These data are summarized in Table 4. Thetotal IgG half-life is dominated by the contributions of IgG1 and IgG2, which haveroughly similar half-lives within studies, and together accounted for 90% to 95% ofthe infused IgG. One study found that IgG3 and IgG4 had relatively shorter half-livesin comparison to IgG1 and IgG2.15 Another study found that IgG3 and IgG4 had similaror longer half-lives in comparison to IgG1 and IgG2, however.18 This study alsocompared pharmacokinetic parameters between patients receiving infusions eitherevery 3 weeks or every 4 weeks and found a trend toward longer half-lives associatedwith patients on the 4-week regimen.
In these studies, the half-life of IgG3 did not seem to be as short as indicated fromradioactive tracer studies (see previous discussion), whereas the half-life ofIgG4 seemed to be either shorter than had been determined by tracer methods15 orpossibly much longer.18 The differences between assessing pharmacokinetics viacatabolism of trace amounts of radiolabeled IgG in healthy individuals in contrast toserum level decay during replacement therapy in immunodeficient patients are sogreat as to severely limit the usefulness of the comparison. Differences between phys-icochemical alterations of IgG subclasses during radioiodination or purification of IgG

Table 4Half-lives of IgG subclasses in intravenous immunoglobulin G replacement therapy for primaryimmunodeficiency disease
Class/Subclass Half-Life (d, Range) AUC (d*mg/dL, Range) AUC Time (d) n ReferenceIgG1 36.3 � 9.2a, r 23–51.5 10,050 � 2470,
5710–15,04021 17 15
26.4 � 8.4b 19,193 � 6074 28 10 18
40.6 � 12.2c 21,657 � 6523 28 11 18
IgG2 37.1 � 13.9, 22.9–62.5a 2990 � 850,1800–4360
21 17 15
33.8 � 10.7b 10,370 � 3282 28 10 18
53.8 � 16.2c 10,776 � 3246 28 11 18
IgG3 28.6 � 10.4, 13–50.2a 664 � 333,359–1739
21 17 15
33.6 � 10.6b 995 � 315 28 10 18
49.9 � 15.0c 1434 � 432 28 11 18
IgG4 15.6 � 14.5, 7.1–24.7a 39 � 21, 22–109 21 17 15
42.2 � 13.4b 489 � 155 28 10 18
75.9 � 22.9c 648 � 195 28 11 18
Abbreviation: AUC, area under the curve.a Data are mean � SD. SeeTable 3 for total IgG data.b Data are mean � SE, 3-week dose regimen. SeeTable 3 for total IgG data.c Data are mean � SE, 4-week dose regimen. SeeTable 3 for total IgG data.
Bonilla810
for IV administration also may affect the pharmacokinetics in vivo. As expected, theareas under the curve for each subclass correlate most closely with the relativeproportions of each IgG subclass in the IVIG preparation.
Some researchers consider that measurement of elimination half-lives by measuringspecific antibody levels may be more accurate than measurement of total IgG. Thetheoretic argument is that there is likely to be less interference via the production ofendogenous specific antibody. One study also measured tetanus specific antibodyafter IVIG infusion.23 The authors found mean half-lives ranging from 27 to 36 daysin the higher dose phases of their study (range of all patients 16–49 days), overall com-parable to rates determined by total IgG (Table 5). Another study in patients who havePID determined IgG decay rates using specific antibody levels (see Table 5).25 Thisstudy also found comparable values of decay rates (mean � SD) for total IgG, 25.9� 18.1; IgG1 29.7 � 18.1; IgG2, 26.9 � 11; and IgG3 15.7 � 5.2 (the value of IgG3tends to be lower than the others in this study; the product is no longer in use andis not included in Tables 3 and 4).
Burn Patients
In some cases, primary or secondary abnormalities of protein catabolism may lead todecreased serum half-life of IgG. Deficiency of b2 microglobulin has been mentionedas one primary cause of increased IgG catabolism (see discussion of bone marrowtransplantation in later section). Burn trauma may lead to a secondary increase inIgG catabolism by an unknown mechanism. In one study of nine patients with largeburns (30%–50% body surface area) who received 0.5 g/kg IVIG once or twice weekly,the half-lives for early (days 0–1) and late (days 1–4) decay were 4.4 and 12.4 days,respectively.26 The ‘‘early’’ period in this study was dominated by redistribution, but

Table 5IgG half-lives determined by specific antibody levels
Organism/Antibody Mean Half-Life (d) ReferenceAdults/children with PID
Tetanus toxoid (3-wk dose interval) 27–36 23
r 16–49, n 5 15 —
Salmonella minnesota lipopolysaccharide 27.2 � 12.4 25
Streptococcus pneumoniae types 1, 3, 6A, 8, 14, 19F 37.6 � 9.8 25
Cytomegalovirus 29.5 � 5.9 25
Infants
Respiratory syncytial virus 21–28 38
0.5–0.75 g/kg monthly n 5 23 —
Neonates
Streptococcus pyogenes group B type III 3.4 28a
Escherichia coli 6.7 28a
Cytomegalovirus 4.7 28a
a Also seeTable 6.
Pharmacokinetics of Immunoglobulin 811
this was also probably occurring to some extent even during the ‘‘late’’ period. In bothphases there may be initial skin loss (although this disappears as the burn heals) andchanges in IgG catabolism induced by systemic effects of the burn trauma. It is alsopossible that the extravascular pool is increased by accumulation of fluid in burnedskin.
Another study looked at 20 patients with large burns (12%–94% surface area) whoreceived 0.5 g/kg IVIG weekly.27 The authors found a half-life of 2 days in the first weekafter injury and a half-life of 6.4 days in the second and third weeks after injury. Thisstudy performed pharmacokinetic analysis over a 96-hour period, still dominated byIgG redistribution. Although these data suggested at least the possibility ofincreased catabolism as a contributing factor, it is not possible to compare theseresults directly with results that measure half-lives in patients receiving replacementevery 3 to 4 weeks.
Neonates
Several studies examined IVIG therapy for prophylaxis and treatment of neonatal sep-sis.28–35 Meta-analyses concluded that adjunct therapy with IVIG reduces mortality inneonatal sepsis36 and is effective as a prophylactic regimen.37 Some of these studieshave included pharmacokinetic analyses. These data are summarized in Table 6.Comparison between these studies is difficult because of the different designs (mainlythe sizes of patients and dose regimens), different pharmacokinetic models, and sam-pling periods. The earliest studies suggested shorter half-lives than those found insubsequent trials. Where included in these reports, data also showed large individualvariations. With the exception of the first study in Table 6, longer sampling periodsseem to be associated with longer half-lives, which suggests that this leads to moreaccurate determination or a systematic bias. Authors emphasize the importance ofmonitoring trough IgG levels during therapy.
For all of the same methodologic reasons noted previously, it is difficult to comparethese studies to those conducted over longer terms in older children and adults.Broadly speaking, studies in neonates indicate somewhat shorter IgG half-lives by

Table 6Intravenous immunoglobulin G half-lives in neonates
BirthWeight (g) Dose (g/kg), Interval
SamplePeriod (d) Half-Life, d (Range) Reference
2140–3,550 0.5, single infusion 42 11.3a � 0.6, n 5 5 33
640–3340 0.5, weekly 7 10.8a, n 5 134 28
750–1000 0.5, single dose 28 22.8a � 5.9 (13.5–30.1), n 5 10 32
0.75, single dose 28 22.6a � 5.6 (11.9–30.1), n 5 10 —
750–1500 0.5, single infusion 28 22.1b (18.1–34.7), n 5 3 31
0.75, single infusion 28 28.7b (19.6–41.5), n 5 4 —
1.0, single infusion 28 19.6b (15.8–26.3), n 5 6 —
< 1000–2000 0.5–1.3, 2–14 d 2–14 days (16–32), n 5 46 30
None (placebo) 10 weeks 33c, n 5 46 —
> 1500 0.25–1.0, single dose 42 24.2a � 7.2, n 5 15 34
< 1000–1500 0.75, 2 wk 14 18.1 (95% CId 15.5–20.6), n 5 35 29
500–2000 0.5, single infusion 56 28.9a � 19.6, n 5 372 35
a Mean, or mean � SD.b Harmonic mean.c Represents maternal IgG.d 95% confidence interval.
Bonilla812
comparison. One study measured the decline in endogenous (maternal-derived) IgG innewborns (see Table 6).30 They determined an elimination half-life of 33 days, compa-rable to studies of patients who have PID (see Table 3). The authors did not accountfor endogenous IgG production, however, which could prolong the measured half-life.The IgG levels in these patients also were much lower than in those receiving ongoingreplacement therapy, leading to a lower catabolic rate (see previous discussion).
At least one study examined IgG elimination by measuring specific antibody inneonates.28 This group found the shortest measured half-lives by this method, rangingfrom 3 to 7 days (see Table 5). The reason for such short half-lives is unclear; this studydid sample over a short period that was likely dominated by the redistribution phase,which may have affected the data based on total IgG as well.
Another study examined the kinetics of antibody to respiratory syncytial virus in in-fants and young children receiving infusions of IVIG with high titer respiratory syncytialvirus antibody (see Table 5).38 Their data showed half-lives comparable to those de-termined by tracer studies in healthy individuals and somewhat lower than determinedby total IgG or other antibodies in older child and adult patients who have PID.
Bone Marrow Transplantation
IVIG has been used as infection prophylaxis for patients undergoing bone marrowtransplantation, and this is one of the US Food and Drug Administration–approvedindications for IVIG therapy.1 Several studies have examined kinetics of IVIG inbone marrow transplantation recipients as determined by total IgG levels and cyto-megalovirus (CMV)-specific antibodies (Table 7).39–43 These studies all tend toshow short IgG half-lives. Comparison among these studies is challenging for the rea-sons noted previously in attempting to compare studies in patients who have PID or inneonates. They are further complicated by variations in the timing of the replacementdosing with respect to the initiation of a myeloablation regimen and the level of

Table 7IVIG elimination in bonemarrow transplantation andmalignancy
Measure Dose (g/kg), Interval Sample Period (d) Mean Half-Life, d ReferenceBone marrow transplantation
Total IgG 0.5, 1–2 wk 7–14 1.3–1.9a, n 5 41 40
Total IgG 0.25–0.5 weekly 7 6.2, n 5 31 42
Total IgG 0.5, weekly 7 5.0 � 6, n 5 9 43
CMV antibody 0.5, weekly 7 2.9–25.5, n 5 27 43
CMV antibody 0.2, 14 d 14 1.3–2.8, n 5 18 41
CMV antibody 0.5, single dose 14 3.5–12.5, n 5 9 39
Chronic lymphocytic leukemia
Total IgG 0.4, 3 wk 21 39.1 � 9.6, n 5 9 44
— — r 24.9–56.5b —
Total IgG 0.1, 3 wk 21 112.7c, n 5 10 45
0.4, 3 wk 21 49.7c, n 5 10 —
0.8, 3 wk 21 44.8c, n 5 10 —
Pneumococcusd 0.1, 3 wk 21 59.5 � 48.3, n 5 10 45
0.4, 3 wk 21 55.3 � 16.8, n 5 10 —
0.8, 3 wk 21 49.7 � 9.1, n 5 10 —
Multiple myeloma
Total IgG 0.1, 3 wk 21 215.6c, n 5 10 45
0.4, 3 wk 21 61.6c, n 5 10 —
0.8, 3 wk 21 42.7c, n 5 10 —
Pneumococcusd 0.1, 3 wk 21 37.1 � 6.3, n 5 10 45
0.4, 3 wk 21 38.5 � 6.3, n 5 10 —
0.8, 3 wk 21 36.4 � 3.5, n 5 10 —
a Where a range is shown in this manner, it represents the range of mean half-lives that werecalculated in various subgroups of patients.b Range of individual patient half-life determinations.c Median.d Twelve serotypes; data are aggregated across serotypes.
Pharmacokinetics of Immunoglobulin 813
pre-existing antibody. All of the studies use short dosing intervals and sampling timesand simple pharmacokinetic models. It is possible that these features again biastoward shorter elimination half-life calculations.
There is great variability between patient subgroups or individual patients. In onestudy43 a large range of half-lives was observed as determined by CMV antibody,and in several cases it exceeded the half-life determined by total IgG level. Thereseemed to be an influence of initial CMV antibody because longer half-lives werefound in patients with higher initial levels. The authors also speculated that theirassumptions regarding stability of ongoing endogenous IgG production during theearly study period may have led to a bias toward a shorter half-life determinationbased on total IgG level.
Another study examined half-lives of IgG subclasses as determined by subclass-specific assays for CMV antibody in two patients.41 In those two patients the totalIgG half-lives were 1.6 and 3.4 days. The CMV-specific antibody half-lives (days)were comparable as follows: IgG1, 1.0 and 2.5; IgG2, 1.0 and 3.0; IgG3, 1.5 and3.4; IgG4, 0.7 and 2.5. As yet, there is no explanation for the apparent increased

Bonilla814
rate of disappearance of IgG from the circulation in patients who receive infusion afterbone marrow transplantation.
Malignancy
A few studies of IgG kinetics in malignancy have been conducted (Table 7), includingchronic lymphocytic leukemia and multiple myeloma.44,45 One study based on totalIgG measurement in replacement therapy for chronic lymphocytic leukemia foundresults comparable to replacement for PID.44 Another study that included chroniclymphocytic leukemia and multiple myeloma found elimination half-lives considerablylonger than any reported previously.45 The data did suggest that higher dose regimensled to shorter half-lives; however, the high IgG levels at baseline in patients withmultiple myeloma appeared possibly to reflect not only increased IgG synthesis butalso reduced catabolism. The prolonged half-lives were also reflected in meas-urements determined by levels of specific (antipneumococcal polysaccharide)antibodies, although the apparent increase was not as marked.
PHARMACOKINETICS OF SUBCUTANEOUS IMMUNOGLOBULIN
Ig administration via the SC route is fundamentally different from IV administration.2
The dose is absorbed slowly and redistributed slowly, whereas concentration-dependent catabolism is ongoing. It is impractical to administer SCIG by a doseregimen comparable to IVIG, because of the relatively limited amount that can beaccommodated subcutaneously (approximately 5–30 mL per site, depending onsize/body mass index), even when multiple sites are used. Although IVIG is usuallyadministered every 3 to 4 weeks, SCIG is dosed weekly, or sometimes twice weekly;the amount of IgG administered over time is generally equivalent. Because the amountadministered at each SCIG infusion is smaller and the interval is shorter, the fluctua-tions in IgG level that are characteristic of IVIG dosing are expected to be muchsmaller. One also would expect these fluctuations to be blunted by the relativelyslow absorption of SCIG from the infusion sites (see Fig. 3A; Fig. 4A).
Few formal pharmacokinetic studies of SCIG have been conducted. One early studycompared IM and SC administration (eight patients each) of 125I-labeled anti-Rho Ig intracer studies similar to those described for IV injection.46 The authors found IM injec-tion to lead to somewhat more rapid uptake, with 70% of maximum plasma levelreached after 1 day, and the maximum level—corresponding to 40% of the injecteddose—occurring after 2 to 4 days. With SC administration, 45% of the maximumplasma level was reached in 1 day, whereas the maximum level—corresponding to33% of the injected dose—was reached in 4 to 6 days. Regardless of the mode ofadministration, plasma levels in the first 24 hours varied erratically, reflecting possiblediscontinuous absorption followed by rapid dilution and early redistribution to othersites. The calculated uptake rates were 0.43 � 0.11 (mean � SD, fraction per day)for IM injection and 0.22 � 0.025 for SC administration. The authors performed twoseparate studies and calculated elimination half-lives independently of IM or SC ad-ministration. In one study of seven patients they found t1/2 5 22 � 4 days; in anotherstudy (n not specified) they reported a range of t1/2 of 22 to 46 days, which was similar(not surprisingly) to results obtained after IV administration.
In an early study of SCIG in 23 patients who had PID, a 16.5% concentration prod-uct formulated for IM use was infused SC at a dose of 0.1 g/kg weekly.47 The authorsfound that weekly dosing required 6 months of therapy to achieve a steady state.Alternatively, if the weekly dose was administered daily for 5 consecutive days (sixpatients), the steady state could be achieved in the first week. Mean steady state
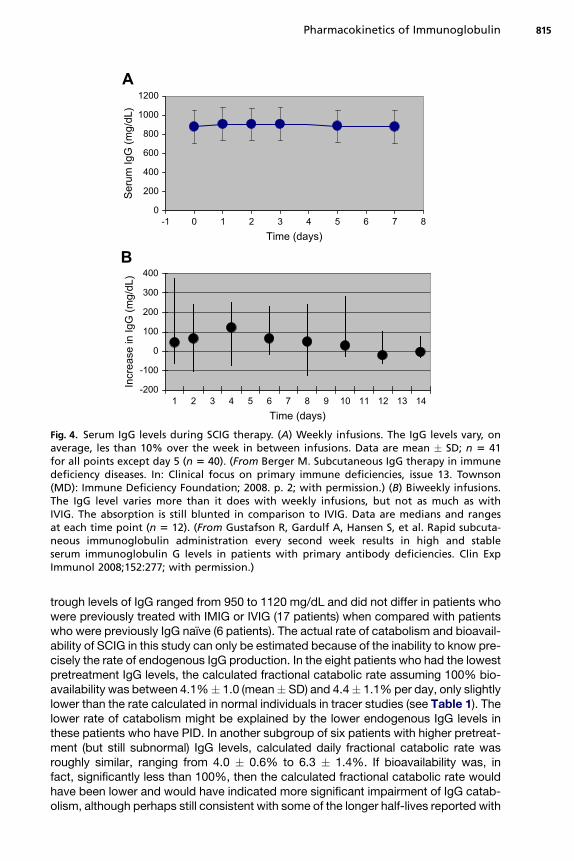
0
200
400
600
800
1000
1200
-1 0 1 2 3 4 5 6 7 8Time (days)
Seru
m Ig
G (m
g/dL
)
-200
-100
0
100
200
300
400
1 2 3 4 5 6 7 8 9 10 11 12 13 14Time (days)
Incr
ease
in Ig
G (m
g/dL
)
A
B
Fig. 4. Serum IgG levels during SCIG therapy. (A) Weekly infusions. The IgG levels vary, onaverage, les than 10% over the week in between infusions. Data are mean � SD; n 5 41for all points except day 5 (n 5 40). (From Berger M. Subcutaneous IgG therapy in immunedeficiency diseases. In: Clinical focus on primary immune deficiencies, issue 13. Townson(MD): Immune Deficiency Foundation; 2008. p. 2; with permission.) (B) Biweekly infusions.The IgG level varies more than it does with weekly infusions, but not as much as withIVIG. The absorption is still blunted in comparison to IVIG. Data are medians and rangesat each time point (n 5 12). (From Gustafson R, Gardulf A, Hansen S, et al. Rapid subcuta-neous immunoglobulin administration every second week results in high and stableserum immunoglobulin G levels in patients with primary antibody deficiencies. Clin ExpImmunol 2008;152:277; with permission.)
Pharmacokinetics of Immunoglobulin 815
trough levels of IgG ranged from 950 to 1120 mg/dL and did not differ in patients whowere previously treated with IMIG or IVIG (17 patients) when compared with patientswho were previously IgG naıve (6 patients). The actual rate of catabolism and bioavail-ability of SCIG in this study can only be estimated because of the inability to know pre-cisely the rate of endogenous IgG production. In the eight patients who had the lowestpretreatment IgG levels, the calculated fractional catabolic rate assuming 100% bio-availability was between 4.1%� 1.0 (mean� SD) and 4.4� 1.1% per day, only slightlylower than the rate calculated in normal individuals in tracer studies (see Table 1). Thelower rate of catabolism might be explained by the lower endogenous IgG levels inthese patients who have PID. In another subgroup of six patients with higher pretreat-ment (but still subnormal) IgG levels, calculated daily fractional catabolic rate wasroughly similar, ranging from 4.0 � 0.6% to 6.3 � 1.4%. If bioavailability was, infact, significantly less than 100%, then the calculated fractional catabolic rate wouldhave been lower and would have indicated more significant impairment of IgG catab-olism, although perhaps still consistent with some of the longer half-lives reported with

Bonilla816
IVIG. The bioavailability of SCIG has not been determined precisely. The authors notedthat IgG subclass levels in patients receiving SCIG differed slightly from the propor-tions in the infused product. Levels of IgG1 and IgG3 were slightly lower, whereaslevels of IgG2 were slightly higher. Levels of IgG4 were low in the product and inpatients. The significance of these subtle differences was unclear.
Only one product is licensed specifically for SC administration in the United States(Vivaglobin, CSL Behring, King of Prussia, Pennsylvania), although any product suitablefor IV administration with concentration of 10% or more or IMIG also may be adminis-tered SC.2 In the licensing studies for Vivaglobin, patients were switched from IVIG toSCIG under a protocol that called for dose adjustment of the SC product to givea time-averaged area under the curve that was equivalent to what had been obtainedpreviously with IVIG. This change required administration of an average of 1.37 times(range 1.02–1.92) the IV dose by the SC route.48 The mean trough IgG levels were768 mg/dL on IVIG, which rose to 1040 mg/dL on SCIG. A rise in the trough IgG levelis expected, because the plasma concentration curve is ‘‘flattened out’’ with SCIG incomparison to IVIG (see Fig. 3B, Fig. 4A). It is also likely, however, because initial phasesof catabolism of IVIG are more rapid as the concentration is supraphysiologic. There isa ‘‘cost’’ of a certain amountof IVIG that is catabolized rapidly to provide an intravascularbolus to fill the extravascular space quickly to create a reservoir for the period betweendoses and prevent the trough level from falling too low. SCIG maintains a more physio-logic balance between the intravascular and extravascular compartments and canmaintain a higher IgG trough level with overall lower amounts of IgG per unit time.
The most recent analysis of SCIG pharmacokinetics included 12 patients (11 com-mon variable immunodeficiency, 1 X-linked agammaglobulinemia) given a 16% con-centration SCIG product at a dose of 0.2 g/kg every 2 weeks.49 These authorscalculated a median half-life of 40.6 days (95% CI 20.1–56.1 days) for total IgG and23.3 days (95% CI 12.7–31.3 days) for tetanus antibody. They speculated (as haveothers) that endogenous IgG production could lead to a slower rate of disappearanceof total IgG, which would not occur in the case of tetanus antibody. The IgG level fluc-tuated somewhat more between infusions, in comparison to what has been observedwith weekly dosing (Fig. 4B).
SUMMARY
IgG equilibrates between intra- and extravascular compartments, and its catabolism iscontrolled by a complex concentration-dependent endocytic mechanism mediated byFcRn. Studies using radioactive labeled IgG in healthy adults indicated an eliminationhalf-life of approximately 23 days, with a lower value for IgG3 of 7 days. A two-compartment pharmacokinetic model fits the data in several studies of bolus IgGreplacement therapy but is not universally applied in studies of IgG decay rates.This difference between studies, together with other methodologic variables (includingdose, interval, sampling schedules and duration, method of measurement of IgG, andpossible actual individual variation in the catabolism of IgG), yields a large range ofcalculated decay rates: 20 to 60 days for total IgG across studies of IVIG. The samevariability is seen when measuring IgG subclasses individually, and the distinct prop-erties (shorter half-life) of IgG3 seen in tracer studies are not as apparent in studies ofIVIG or SCIG replacement pharmacokinetics.
Most studies of IVIG in neonates tend to show somewhat shorter half-lives rangingfrom 10 to 40 days, although the largest study to date yielded results similar to adults.Decay rates determined by specific antibody measurements in adults give resultssimilar to total IgG, whereas in neonates the half-lives are significantly shorter. The

Pharmacokinetics of Immunoglobulin 817
half-life of IVIG seems to be short in patients with severe burns and patients who haveundergone bone marrow transplantation, which is not likely to be entirely explained bymethodologic variables. Alternatively, studies of IVIG in malignancies such as chroniclymphocytic leukemia and multiple myeloma suggest that reduced IgG catabolismmay lead to a longer half-life of IVIG, but this has not been established clearly.
SCIG leads to more physiologic IgG levels because the peaks and nadirs betweeninfusions are blunted by slow absorption and maintenance of closer equilibriumbetween intra- and extravascular compartments. Although SCIG is usually givenweekly (sometimes more often), a recent study suggests that a 2-week interval isalso practical. Because of the individual variability of IgG kinetics and distinct alter-ations in certain disease states, periodic measurement of IgG trough levels is essentialduring replacement therapy, regardless of the route of administration.
REFERENCES
1. Orange JS, Hossny EM, Weiler CR, et al. Use of intravenous immunoglobulin inhuman disease: a review of evidence by members of the primary immunodefi-ciency committee of the American Academy of Allergy, Asthma and Immunology.J Allergy Clin Immunol 2006;117:S525–53.
2. Berger M. Subcutaneous immunoglobulin replacement in primary immunodefi-ciencies. Clin Immunol 2004;112:1–7.
3. Waldmann TA, Strober W. Metabolism of immunoglobulins. Prog Allergy 1969;13:1–110.
4. Waldmann TA, Strober W, Blaese RM. Variations in the metabolism of immuno-globulins measured by turnover rates. In: Merler E, editor. Immunoglobulins.Washington, DC: National Academy of Sciences; 1970. p. 33–51.
5. Brambell FW, Hemmings WA, Morris IG. A theoretical model of gamma-globulincatabolism. Nature 1964;203:1352–4.
6. Junghans RP, Anderson CL. The protection receptor for IgG catabolism is thebeta2-microglobulin-containing neonatal intestinal transport receptor. Proc NatlAcad Sci U S A 1996;93:5512–6.
7. Simister NE, Mostov KE. An Fc receptor structurally related to MHC class I anti-gens. Nature 1989;337:184–7.
8. Leach JL, Sedmak DD, Osborne JM, et al. Isolation from human placenta of theIgG transporter, FcRn, and localization to the syncytiotrophoblast: implications formaternal-fetal antibody transport. J Immunol 1996;157:3317–22.
9. Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat RevImmunol 2007;7:715–25.
10. Sachs UJ, Socher I, Braeunlich CG, et al. A variable number of tandem repeatspolymorphism influences the transcriptional activity of the neonatal Fc receptoralpha-chain promoter. Immunology 2006;119:83–9.
11. Waldmann TA, Terry WD. Familial hypercatabolic hypoproteinemia: a disorder ofendogenous catabolism of albumin and immunoglobulin. J Clin Invest 1990;86:2093–8.
12. Wani MA, Haynes LD, Kim J, et al. Familial hypercatabolic hypoproteinemiacaused by deficiency of the neonatal Fc receptor, FcRn, due to a mutantbeta2-microglobulin gene. Proc Natl Acad Sci U S A 2006;103:5084–9.
13. Zhou H. Clinical pharmacokinetics of etanercept: a fully humanized solublerecombinant tumor necrosis factor receptor fusion protein. J Clin Pharmacol2005;45:490–7.

Bonilla818
14. Koleba T, Ensom MH. Pharmacokinetics of intravenous immunoglobulin:a systematic review. Pharmacotherapy 2006;26:813–27.
15. Alyanakian MA, Bernatowska E, Scherrmann JM, et al. Pharmacokinetics of totalimmunoglobulin G and immunoglobulin G subclasses in patients undergoingreplacement therapy for primary immunodeficiency syndromes. Vox Sang2003;84:188–92.
16. Ballow M, Berger M, Bonilla FA, et al. Pharmacokinetics and tolerability of a newintravenous immunoglobulin preparation, IGIV-C, 10% (Gamunex, 10%). VoxSang 2003;84:202–10.
17. Berger M. A multicenter, prospective, open label, historically controlled clinicaltrial to evaluate efficacy and safety in primary immunodeficiency diseases(PID) patients of Flebogamma 5% DIF, the next generation of Flebogamma.J Clin Immunol 2007;27:628–33.
18. Berger M, Pinciaro PJ. Safety, efficacy, and pharmacokinetics of Flebogamma5% [immune globulin intravenous (human)] for replacement therapy in primaryimmunodeficiency diseases. J Clin Immunol 2004;24:389–96.
19. Borte M, Davies SV, Touraine J-L, et al. Clinical properties of a novel liquidintravenous immunoglobulin: studies of patients with immune thrombocytopenicpurpura and primary immunodeficiencies. Transfus Med Hemother 2004;31:126–34.
20. Church JA, Leibl H, Stein MR, et al. Efficacy, safety and tolerability of a new 10%liquid intravenous immune globulin [IGIV 10%] in patients with primary immuno-deficiency. J Clin Immunol 2006;26:388–95.
21. Ochs HD, Pinciaro PJ. Octagam 5%, an intravenous IgG product, is efficaciousand well tolerated in subjects with primary immunodeficiency diseases. J ClinImmunol 2004;24:309–14.
22. Privigen. Prescribing information. King of Prussia (PA): CSL Behring; 2008.23. Schiff RI, Rudd C. Alterations in the half-life and clearance of IgG during therapy
with intravenous gamma-globulin in 16 patients with severe primary humoralimmunodeficiency. J Clin Immunol 1986;6:256–64.
24. Eijkhout HW, van Der Meer JW, Kallenberg CG, et al. The effect of two differentdosages of intravenous immunoglobulin on the incidence of recurrent infectionsin patients with primary hypogammaglobulinemia: a randomized, double-blind,multicenter crossover trial. Ann Intern Med 2001;135:165–74.
25. Mankarious S, Lee M, Fischer S, et al. The half-lives of IgG subclasses andspecific antibodies in patients with primary immunodeficiency who are receivingintravenously administered immunoglobulin. J Lab Clin Med 1988;112:634–40.
26. Shirani KZ, Vaughan GM, McManus AT, et al. Replacement therapy with modifiedimmunoglobulin G in burn patients: preliminary kinetic studies. Am J Med 1984;76:175–80.
27. Hansbrough JF, Miller LM, Field TO Jr, et al. High dose intravenous immunoglobulintherapy in burn patients: pharmacokinetics and effects on microbial opsonizationand phagocytosis. Pediatr Infect Dis J 1988;7:S49–56.
28. Chirico G, Rondini G, Plebani A, et al. Intravenous gammaglobulin therapy forprophylaxis of infection in high-risk neonates. J Pediatr 1987;110:437–42.
29. Kinney J, Mundorf L, Gleason C, et al. Efficacy and pharmacokinetics of intrave-nous immune globulin administration to high-risk neonates. Am J Dis Child 1991;145:1233–8.
30. Kyllonen KS, Clapp DW, Kliegman RM, et al. Dosage of intravenously adminis-tered immune globulin and dosing interval required to maintain target levels ofimmunoglobulin G in low birth weight infants. J Pediatr 1989;115:1013–6.

Pharmacokinetics of Immunoglobulin 819
31. Noya FJ, Rench MA, Courtney JT, et al. Pharmacokinetics of intravenous immu-noglobulin in very low birth weight neonates. Pediatr Infect Dis J 1989;8:759–63.
32. Noya FJ, Rench MA, Garcia-Prats JA, et al. Disposition of an immunoglobulin intra-venous preparation in very low birth weight neonates. J Pediatr 1988;112:278–83.
33. Weisman LE, Fischer GW, Hemming VG, et al. Pharmacokinetics of intravenous im-munoglobulin (sandoglobulin) in neonates. Pediatr Infect Dis 1986;5:S18–188.
34. Weisman LE, Fischer GW, Marinelli P, et al. Pharmacokinetics of intravenousimmunoglobulin in neonates. Vox Sang 1989;57:243–8.
35. Weisman LE, Stoll BJ, Kueser TJ, et al. Intravenous immune globulin prophylaxisof late-onset sepsis in premature neonates. J Pediatr 1994;125:922–30.
36. Jenson HB, Pollock BH. Meta-analyses of the effectiveness of intravenousimmune globulin for prevention and treatment of neonatal sepsis. Pediatrics1997;99:E2.
37. Ohlsson A, Lacy JB. Intravenous immunoglobulin for preventing infection in pretermand/or low-birth-weight infants. Cochrane Database Syst Rev 2000:CD000361.
38. Groothuis JR, Levin MJ, Rodriguez W, et al. Use of intravenous gamma globulin topassively immunize high-risk children against respiratory syncytial virus: safetyand pharmacokinetics. The RSVIG Study Group. Antimicrob Agents Chemother1991;35:1469–73.
39. Bosi A, De Majo E, Guidi S, et al. Kinetics of anti-CMV antibodies after adminis-tration of intravenous immunoglobulins to bone marrow transplant recipients.Haematologica 1990;75:109–12.
40. Cottler-Fox M, Lynch M, Pickle LW, et al. Some but not all benefits of intravenousimmunoglobulin therapy after marrow transplantation appear to correlate withIgG trough levels. Bone Marrow Transplant 1991;8:27–33.
41. Hagenbeek A, Brummelhuis GJ, Donkers A, et al. Rapid clearance of cytomegalo-virus-specific IgG after repeated intravenous infusions of human immunoglobulininto allogeneic bone marrow transplant recipients. J Infect Dis 1987;155:897–902.
42. Rand KH, Gibbs K, Derendorf H, et al. Pharmacokinetics of intravenous immuno-globulin (Gammagard) in bone marrow transplant patients. J Clin Pharmacol1991;31:1151–4.
43. Rand KH, Houck H, Ganju A, et al. Pharmacokinetics of cytomegalovirus specificIgG antibody following intravenous immunoglobulin in bone marrow transplantpatients. Bone Marrow Transplant 1989;4:679–83.
44. Chapel HM, Hargreaves R, Lee M, et al. Intravenous immunoglobulin therapy inpatients with multiple myeloma. Immunodeficiency 1993;4:77–8.
45. Sklenar I, Schiffman G, Jonsson V, et al. Effect of various doses of intravenous poly-clonal IgG on in vivo levels of 12 pneumococcal antibodies in patients with chroniclymphocytic leukaemia and multiple myeloma. Oncology 1993;50:466–77.
46. Smith GN, Griffiths B, Mollison D, et al. Uptake of IgG after intramuscular andsubcutaneous injection. Lancet 1972;1:1208–12.
47. Waniewski J, Gardulf A, Hammarstrom L. Bioavailability of gamma-globulin aftersubcutaneous infusions in patients with common variable immunodeficiency.J Clin Immunol 1994;14:90–7.
48. Ochs HD, Gupta S, Kiessling P, et al. Safety and efficacy of self-administeredsubcutaneous immunoglobulin in patients with primary immunodeficiencydiseases. J Clin Immunol 2006;26:265–73.
49. Gustafson R, Gardulf A, Hansen S, et al. Rapid subcutaneous immunoglobulinadministration every second week results in high and stable serum immunoglob-ulin G levels in patients with primary antibody deficiencies. Clin Exp Immunol2008;152:274–9.

Self - infusionProgrammes forImmunoglobulinReplacement at Home:Feasibil ity, Safetyand Efficacy
Malini V. Bhole, MSc, MRCPCH, MD, Janet Burton, MSc, RQN,Helen M. Chapel, MA, MP, FRCP, FRCPATH*
KEYWORDS
� IVIg � SCIg � Self-infusion � Home therapy
Since the first paper was written on the use of therapeutic immunoglobulins in patients,1
great progress has been made in immunoglobulin therapy, including the manufactur-ing processes, types of products available, routes of administration, and safety andefficacy of self-infusion at home. Currently, immunoglobulin replacement can beachieved effectively via intravenous (IVIg) or subcutaneous (SCIg) administration.Both options have been shown to be safe and equally efficacious in decreasing theincidence and severity of infections in patients with antibody deficiencies.2 Addition-ally, high-dose immunoglobulin therapy is used in a range of medical conditions,and patients can be trained to self-administer these infusions at home. This article dis-cusses the feasibility and development of successful home therapy programs alongwith safety, efficacy, and cost-effectiveness.
HISTORICAL BACKGROUND
Regular replacement with immunoglobulin infusions is the mainstay of treatment in pa-tients with antibody deficiency. Once preparations of immunoglobulin (IVIg) that weresafe to administer intravenously became available, large doses could be given tomaintain adequate serum IgG levels to prevent infection. In most patients, monthlydoses of 300 to 400 mg/kg body weight were sufficient to achieve immunoglobulin
Department of Clinical Immunology, Nuffield Department of Medicine and Oxford RadcliffeHospitals, Oxford, UK* Corresponding author. Level 4A Academic Street, John Radcliffe Hospital, Headley Way,Oxford OX3 9DU, UK.E-mail address: [email protected] (Helen M. Chapel).
Immunol Allergy Clin N Am 28 (2008) 821–832doi:10.1016/j.iac.2008.06.005 immunology.theclinics.com0889-8561/08/$ – see front matter. Crown Copyright ª 2008 Published by Elsevier Inc. All rights reserved.

Bhole et al822
levels above the lower limit of the normal range,3 although monthly infusions often re-sulted in wide fluctuations between the peak and trough levels. A steadier state wasachievable by administering the same total monthly dose in divided doses at shorterintervals. An increased frequency of infusions resulted directly in increased costs oftreatment, both in terms of hospital resources and patient time (time off school orwork). Home therapy for IVIg administration was first introduced as a measure toachieve a satisfactory balance among adequate treatment, patient convenience,and cost-effectiveness. Based on the vast experience of patients with hemophiliawho were self-administering factor VIII at home for many years,4–8 home therapy pro-grams for IVIg were investigated in the United States in the early 1980s9–12 and werequickly adapted in the United Kingdom.13 Despite the reduction in costs that resultsfrom avoidance of the need for hospital space and staff, home therapy programsdid not become popular outside of the United States and the United Kingdom untilrecently. Some countries banned such programs on safety or moral grounds (patientsshould not be taught to perform venepuncture), whereas other health care systems didnot adapt easily to medical treatment without direct supervision. The UK Departmentof Health was willing to give approval in 1986, provided that there was ongoing datacollection regarding safety and efficacy. Feasibility had already been proven in thehemophilia community, and professional home care companies were providingproducts and consumables for patients to treat themselves with hemophilia products,renal dialysis facilities, or—in the case of HIV patients—intravenous antibioticsat home.
Once immunoglobulin therapy by the subcutaneous route (SCIg) was established inthe early 1990s, self-infusion for SCIg became widespread. Originally, subcutaneousinfusions of immunoglobulin were done in the late 1970s with products designed forintramuscular use and a slow infusion rate.14–22 This method was abandoned as theinfusions were accompanied by multiple reactions, sterile abscesses associatedwith mercury used as a preservative, long infusion times, and also required inefficientbattery-operated pumps. There was renewed interest in the early 1990s after technicalimprovements in the available products, infusion pumps, and protocols,23–26 particu-larly in Sweden,24,27,28 because the use of IVIg was banned there by the government atthat time after an outbreak of transmitted hepatitis C in IVIg. Once new methods ofviral inactivation were introduced and the efficacy of SCIg for infection preventionhad been proven to match that of IVIG in immunodeficient patients,2 studies in Swe-den showed safety and cost-effectiveness. Since then, the use of subcutaneousimmunoglobulin has increased exponentially. Increased interest in the use of thesubcutaneous route for immunoglobulin replacement has led to further developmentof products specifically intended for subcutaneous use. A survey conducted by theEuropean Society for Immunodeficiency in 2002 reported that 7% of Europeanpatients were on regular immunoglobulin replacement by the subcutaneous route,indicating the rapid expansion of the use of SCIg in Europe.29
FEASIBILITYOF SELF-INFUSIONS
Neither intramuscular immunoglobulin injections nor the slow administration of immu-noglobulins subcutaneously was feasible or sufficiently safe to use at home. The intra-venous route was the method of choice for self-administration in the 1980s. Thewidespread use of self-administration for other products and the changing cultureof health care in many countries made this approach feasible for IVIg therapy. Profes-sional medical bodies in the United Kingdom and health care providers in the UnitedStates and United Kingdom recommended that the concept be proven safe and

Self-infusion Programmes for Immunoglobulin 823
required that patients be entered into recognized programs in which they were for-mally trained. Training involved the practice of venepuncture to secure venous access,the availability of a suitable infusion partner in case of a reaction at home, and proof ofunderstanding by the patient and the partner of the potential side effects of intrave-nous administration of IVIg. Using training protocols, this approach proved to be fea-sible, safe, and efficacious in adults and children.9–11,13,30,31 All patients who requiredimmunoglobulin replacement therapy were not suitable for home therapy programs forvarious reasons, including poor venous access, personal circumstances, or lack ofa suitable infusion partner. Introduction of rapid subcutaneous replacement protocolssince 1991 has increased the number of patients in home therapy programs, becauseSCIg obviates the need for venous access and, in some countries, the immediate needfor an infusion partner on site.24,32
The practice of immunoglobulin replacement differs from country to country. In theUnited Kingdom, newly diagnosed patients are loaded rapidly with weekly IVIg and, ifsuitable, are enrolled into a home therapy program as soon as they are stable on im-munoglobulin therapy. The choice of route for immunoglobulin infusions depends onseveral factors, including platelet count, amount of subcutaneous tissue available,dose of immunoglobulin required to prevent infections, and ease of venous access.Practice differs in Sweden, where even newly diagnosed patients are started on sub-cutaneous replacement and all patients are subsequently trained to self-infuse athome. The availability of two equally efficacious and safe routes of immunoglobulin re-placement allows flexibility of choice that is adaptable to the various stages of life.Children may prefer the subcutaneous route but revert to IVIg in their teenage yearsto lessen the infusion frequency. Once independent, however, they often prefer to un-dertake SCIg until they are unable to self-infuse and revert to IVIg in the clinic setting inolder age. Such choice and adaptability improve the overall quality of life.
EFFICACYOF HOME INFUSIONS
The evidence that regular replacement immunoglobulin (in any setting) reduces the in-cidence and severity of infections in patients with antibody deficiency has been pre-viously reported. It was important to show that it applied to those infusing at hometoo. In the first report with home therapy and self-administration of IVIg, Ochs and col-leagues11 reported 1.7 infections per patient per year compared to 2.5 infections/pa-tient/year in the previous year of hospital-based infusions. The reduction in infectionswas probably secondary to shortening the interval between doses (10 to 15 days ver-sus 4 weeks), resulting in a higher IgG trough level and similar differences betweenpeak and trough levels. This improved efficacy with shorter intervals was only feasiblewith the home therapy program at that time because of scarcity of hospital facilitiesand staff. Similar efficacy in infection prevention for immunoglobulin usage at homewas reported by other investigators, and figures for adults and children were similar.2,9
Although equal numbers of days were lost from school or work because of illness,days lost from work by patients or parents for infusion attendances were considerablyreduced.13 Kobayashi and colleagues31 reported successful home administration ofIVIg by parents for 12 children aged 2 to 17, with no difference in the frequency andseverity of infections when compared to the previous phase of IVIg in a clinic setting.
Self-administered SCIg is equal in efficacy to IVIg in terms of trough levels and in-fection rates. A multicenter randomized crossover study of 40 patients who receivedIVIg or SCIg for a year before crossing over to the alternative mode of delivery showedcomparable trough levels in both phases.2 There were no significant differences in thenumbers of infections between the two treatments. Outcome was measured using

Bhole et al824
infection scores, which took into account the frequency and severity of infections.Outcomes for patients who self-infused at home were no different from outcomesfor patients who received their therapies in the clinic. Two other trials that indepen-dently compared retrospective data for IVIg when patients were transferred to SCIg(in Europe, Brazil, and North America) reported the incidence of serious bacterial in-fections as 0.04/patient/year and the overall annual incidence of all minor plus majorinfections as 4.4/patient.26,34 The data from patients in Europe and the United Stateswere slightly different because of the use of an increased dose of SCIg in the UnitedStates (namely, 137% of that used intravenously), whereas European studies used thesame cumulative doses (g/kg/mo) for both routes. Overall, the data from patients onhome therapy with SCIg showed increased protection against serious infections aswell as a reduction in self-reported infections.28,35–38
Efficacy depends on compliance, and subjective and objective monitoring of com-pliance is essential. In the United Kingdom, compliance is evidenced by maintenanceof serum IgG trough levels on blood samples taken by the patient immediately beforethe infusion and returned to the center by mail for measurement of trough IgG levels.IgA and IgM levels are also measured to check for reversal or progression of immuno-deficiency. C-reactive protein gives a measure of ongoing inflammation (usually in thelungs or gastrointestinal tract), and liver function tests are done for early detection ofpossible transmission of hepatitis viruses.
SAFETY
The safety profile of immunoglobulin replacement treatment at home is crucial. Itincludes the absence of infusion-related adverse reactions and reactions caused bypossible transmitted infections, because home therapy programs result in lessfrequent attendance at hospital for medical follow-up.
The first prospective study of adverse reactions in patients self-infusing IVIg athome was a multicenter study in the United Kingdom with 119 patients.30 No seriousreactions were reported. Only 19 moderate adverse reactions were recorded from2031 infusions (0.7%), all of which resolved without medical aid. Further analysisshowed that most were related to pre-existing infections and were avoidable, givinga rate of 0.5% for adverse reactions. This study was further extended to include a totalof 459 patients from centers across the United Kingdom. The rate of adverse reactionsin the larger study remained consistent at 0.7%,39 still without any serious reactions.
The high safety profile of SCIg makes it another appropriate route for self-adminis-tration at home. In addition to ongoing infection, accidental infusion into small bloodvessels has been reported as a possible explanation of moderate systemic reactionin one patient.35 Several studies from many centers across the world have endorsedthe safety of self-administration of SCIg at home.2,23–27,34,35,40,41 SCIg has been usedsuccessfully with minimal adverse reactions in patients with previous severe reactionsto IVIg. It also has been used safely in patients with anti-IgA antibodies,42–44 althoughthe actual significance of anti-IgA antibodies remains unclear. The safety and efficacyof self-administered SCIg and IVIg have been demonstrated during pregnancy,15,24,45
although the site for SCIg infusions likely changes from the abdomen to the upperthigh.
QUALITYOF LIFE
One of the major impacts of self-administered immunoglobulin replacement at homehas been on the quality of life of patients with primary antibody deficiencies. Since theintroduction of home therapy programs, most patients in countries that have adopted

Self-infusion Programmes for Immunoglobulin 825
home therapy have indicated their preference to receive their infusions at home. Thereasons and benefits suggested from oral responses from patients are increased self-confidence, better understanding of their condition, greater independence, improveddisease control, decreased loss of time from school or work, decreased cost, and min-imal disruption of daily activities.10,12 Peer group support has been proposed as an un-expected but important benefit of home training programs.
One of the early studies to evaluate patient responses to home treatment system-atically was a multicenter study based on analysis of completed questionnaires.46
The Life Quality Index, developed by investigators specifically for patients undergoingIVIg treatment, assessed quality of life. A similar questionnaire-based study froma Swedish group showed that weekly self-administered SCIg led to improved qualityof life as measured by better functional status, improved general health, higher troughlevels leading to fewer breakthrough infections, and more patient independence.28,47
Different outcome questionnaires or instruments have been used in published re-ports, including Life Quality Index, Medical Outcomes Study 36-Item Short FormHealth Survey, and the Child Health Questionnaire Parental Form. (For a detailed listof available questionnaires, see reference.48) The most complete studies have usedstandardized survey questionnaires as tools to measure health-related quality of life,which is defined as a subjective perception of the impact of disease and treatmenton daily life, including physical, psychological, and social aspects.49 The other impor-tant measure of patient-reported outcome is treatment satisfaction, as defined by pa-tients’ reports on their treatment experience and satisfaction toward the treatment,encouraging patient compliance.50,51 Studies from Sweden and the United Stateshave demonstrated that immunoglobulin replacement dramatically improved theresults of health-related quality of life, bringing it on par with normal population.47,52
Patients reported fewer infections and decreased anxiety regarding their health.Further improvement in health-related quality of life and treatment satisfaction has
been achieved with the relative ease and safety of self-administered SCIg. Technicalimprovements in available products and infusion pumps have had a favorable impacton the overall quality of life among adults and children with primary antibody defi-ciencies. As with patients on IVIg, patients on SCIg report an increased sense ofself-control, increased flexibility of treatment with minimal disruption of daily life,decreased time off work or school, improved family relations, and greater wellbeing.23,24,28,32,36–38,40,46,47,53
COST-EFFECTIVENESS OF SELF-INFUSIONS AT HOME
In addition to being a safe and efficacious treatment option with significant improve-ment in the quality of life, self-infusions at home may reduce the overall costs of treat-ment for patients and health care providers, depending on the health care system.Early reports of home infusions from the United States estimated savings of between$195 and $355 per infusion.11,31 This calculation was based on savings from cliniccharges, pharmacy expenses, and professional fees and excluded transportationcosts and time lost from work/school. In 1988, Ryan and colleagues54 reported thatwithin the UK National Health Service, the annual cost per patient was £884 for hos-pital therapy compared to £601 for the first year of home therapy and £530 for subse-quent years, although this study did not include the cost of the immunoglobulinproduct. In contrast, Sorensen and colleagues12 included the additional processingcosts for immunoglobulin home delivery and noted no cost savings between hospitalinfusions and home therapy in the United States. A health-economic evaluation of im-munoglobulin therapy done in Sweden reported savings of approximately $10,000 per

Bhole et al826
patient per year on changing from hospital-based IVIg to self-infused SCIg at home,27
and similar savings were reported from other countries.40,55 Because most of thesecalculations depended on a differential in the price of the relevant products and placeof prescribing, however, and as this is no longer the case, these studies needrepeating.
EXTENSION OF HOME THERAPY PROGRAMS
High-dose immunoglobulin is used increasingly for immunomodulation in autoimmunediseases. To maintain a reasonable and functional quality of life, patients require higherdoses of immunoglobulin treatment at regular intervals. The experience gained fromsuccessful home therapy programs for patients with primary antibody deficiencieshas encouraged some centers to offer training in self-infusions to these pa-tients.56–59 As with primary immunodeficiencies, self-administered infusions athome allow optimal disease control and vastly improve quality of life for patientswith potentially debilitating conditions.
REQUIREMENTS FOR A SELF-INFUSION HOME THERAPY PROGRAM
A successful home care program for self-infusion of immunoglobulin depends on sev-eral factors, including general acceptability of home treatments, perception of need bypatients, and an expert and dedicated team to provided the service (Box 1). In partic-ular, the initiation and successful continuation of an efficient home therapy programrequires planning in terms of funding and practicalities. These factors include re-sources and training for staff, criteria for patient selection, protocols for patient trainingand regular monitoring, organization of regular supply of product and consumables,support staff (including specialist nurses and clinical immunologists), and general sup-port from relevant disease-specific patient organizations.
Planning
The best team to provide home therapy service is one that is already involved inimmunoglobulin replacement therapy within the hospital setting. This ensures long-term continuity of care and support for the patients. Self-infusion at home for mostpatients enables more cost-effective use of available resources. Before starting a pa-tient on such a program, it is important to secure funding to ensure ongoing and timelysupply of the immunoglobulin product and the service on a long-term basis. Themechanisms differ from country to country depending on the existing health caresystem, regardless of whether the source of funds is the government, a health careprovider, or medical insurance companies.
Box1Factors that influence a successful home treatment program
� General acceptability of treatments at home
� Acceptance of legalities and liabilities involved by the existing health care system
� High degree of motivation on the part of the patients
� Educational resources for teaching patients all aspects of home treatments
� Availability of resources to deliver consumables and products to homes
� Dedicated and experienced team of doctors and nurses to provide the service

Self-infusion Programmes for Immunoglobulin 827
Staff Resources and Training
Training of medical and nursing staff involved in a home therapy program is directedtoward appropriate assessment of patients for self-infusions, ability to translatemedical principles in simple terms and demonstrate practical techniques in a pa-tient-friendly way, and the acquisition of appropriate skills for patient monitoring, trou-bleshooting, and patient support (Box 2).
Patient Selection
Some of the general criteria required for selection of a patient for the home therapyprogram are listed in Box 3, although practice differs from center to center and countryto country. Home therapy may not be suitable in certain groups of patients, especiallyelderly patients and patients with difficult social or personal circumstances. It is impor-tant to undertake a risk assessment for each patient before enrollment into a self-infusion program. Governments and health care providers have gradually acceptedthis principle, and IVIg and SCIg are self-infused at home in many European and NorthAmerican countries rather than infused at home by a trained nurse.
Patient Training
The key aspects of training programs for selected patients and their infusion partnersare detailed in Box 4. Training may be undertaken either individually or in small groups.The length of the training period varies depending on the protocol and availableresources but must be long enough to ensure proof of competence on the part ofthe attendees. Patient education includes knowledge about the product used, impor-tance of a hygienic environment and aseptic techniques, correct techniques for self-administration, precise documentation of infusion logs, and safe disposal methods.Emphasis is placed on the importance of postponing an infusion if the patient is unwellbecause of increased risk of an adverse event during acute bacterial infection.60
Patients and their partners must be trained to recognize adverse reactions andknow the immediate measures required to treat such a reaction. Documentation of di-rect observation, assessment of competency, and feedback is essential at each stage.
Resources at Home
There are no particular requirements for resources at home other than the timely andregular supply of the immunoglobulin product and other consumables required forhome administration. Inclusion of a method for safe disposal of clinical waste andsharps bins is important, as well as the completion of monitoring and infusion logs. Pa-tients are required to complete infusion logs recording details of infusions and batch
Box 2Training and teaching skills to be acquired by immunology staff
� Ability to assess suitability of patients for home therapy
� Ability to translate basic medical science and understanding to patients and their infusionpartners
� Ability to explain principles and practicalities of self-administration of immunoglobulins,such as aseptic technique, venipuncture, and blood sampling
� Acquisition of knowledge and skills required for patient monitoring
� Ability to troubleshoot and provide patient support

Box 3Criteria for patient selection
� Ability to understand the basic principles and practicalities involved
� High degree of motivation and reliability
� Presence of a suitable infusion partner to understand the basic principles and provideappropriate support for the procedure
� Technical skills of patient and infusion partner
� Ease and availability of IV access for IVIg
� Suitable home environment (eg, general hygiene, area for infusing, pets)
� Availability of appropriate storage space for product and consumables (may needa refrigerator)
� Availability of immediate telephone access in case of emergency
Bhole et al828
numbers of the immunoglobulin used in the case of need for urgent look-back, as fol-lowed the outbreak of hepatitis C in 1994.61 In some countries these logs are com-pleted on Web-based systems, but paper records are equally satisfactory. Patientsmay maintain regular symptom diaries and, in the United Kingdom, are required toorganize regular blood tests every 6 to 12 weeks for trough immunoglobulin levels,liver function tests, and C-reactive protein levels. In addition to regular medical follow-up consultations, reviews by nursing staff are important to check for the maintenanceof practical skills.
Specialist Support
Advice and support are often needed for patients on home therapy programs, espe-cially in the event of practical infusion problems, during intermittent infections, or
Box 4Key aspects of patient training and education
� Knowledge of the nature of product used, current dose/volume required
� Importance of a hygienic environment, including sterility of work surfaces and consumablesthroughout the infusion
� Correct technique of self-administration, including
Noting batch numbers and expiry date
Reconstituting product where required
Priming of giving sets
Showing venipuncture skills
Monitoring drip rates during infusion
� Precise documentation of infusion logs
� Prompt recognition of adverse reactions both minor and major
� Immediate steps to be taken in the event of an adverse reaction
� Safe disposal methods

Self-infusion Programmes for Immunoglobulin 829
during periods of stress or illness. An experienced medical member of the specialistteam must be available at all times.
General Support
Patients are encouraged to join the relevant national patient organization and sharetheir experiences and concerns with others. Peer group support is important for thedevelopment of coping skills and improvement in quality of life.
Protocols around the world have developed on similar lines, and most centers followthese principles despite variations in health care provision in different countries.12,31 Inaddition, guidelines and protocols around the world have been developed on similarlines, and most centers follow these principles despite variations in healthcare provi-sion in different countries.33,53
SUMMARY
This review of the currently available literature from more than two decades of clinicalexperience with self-infusions of immunoglobulin at home provides evidence to sup-port the feasibility, safety, and efficacy in all age groups. Self-infusions at home notonly increase patient confidence and their understanding of immunodeficiency butalso contribute to the improvement of health-related quality of life. Such home therapyprograms should be encouraged, and wherever possible, experienced centers shouldextend their services to include patients who require immunoglobulin therapy for im-munomodulation. Home therapy programs play an important role in the long-termhealth outcome.
ACKNOWLEDGMENTS
Many individuals have contributed to the development of programs throughout theworld for more than 30 years. In our center we owe a special debt of gratitude to SisterVeronica Brennan, who established the first home therapy training center in the UnitedKingdom and subsequently helped other centers here and in Europe to set up similarprograms.
REFERENCES
1. Bruton OC. Agammaglobulinemia. Pediatrics 1952;9:722–8.2. Chapel HM, Spickett GP, Ericson D, et al. The comparison of the efficacy and
safety of intravenous versus subcutaneous immunoglobulin replacement therapy.J Clin Immunol 2000;20:94–100.
3. Morell A, Schnoz M, Barandun S. Build-up maintenance of IgG serum concentra-tions with intravenous immunoglobulin in patients with primary humoral immuno-deficiency. Vox Sang 1982;43:212–9.
4. Evensen SA, Thaule R, Groan K. Self-therapy for haemophilia in Norway: effect ontransfusion frequency and days lost from work. Acta Med Scand 1979;205:395–9.
5. Ingram GI, Dykes SR, Creese AL, et al. Home treatment in haemophilia: clinical,social and economic advantages. Clin Lab Haematol 1979;1:13–27.
6. Kaufert JM. Social and psychological responses to home treatment of haemo-philia. J Epidemiol Community Health 1980;34:194–200.
7. Levine PH. Efficacy of self-therapy in hemophilia: a study of 72 patients withhemophilia A and B. N Engl J Med 1974;291:1381–4.
8. Rizza CR, Spooner RJ. Home treatment of haemophilia and Christmas disease:five years’ experience. Br J Haematol 1977;37:53–66.

Bhole et al830
9. Ashida ER, Saxon A. Home intravenous immunoglobulin therapy by self-adminis-tration. J Clin Immunol 1986;6:306–9.
10. Ochs HD, Fischer SH, Lee ML, et al. Intravenous immunoglobulin home treatmentfor patients with primary immunodeficiency diseases. Lancet 1986;1:610–1.
11. Ochs HD, Lee ML, Fischer SH, et al. Self-infusion of intravenous immunoglobulinby immunodeficient patients at home. J Infect Dis 1987;156:652–4.
12. Sorensen RU, Kallick MD, Berger M. Home treatment of antibody-deficiency syn-dromes with intravenous immune globulin. J Allergy Clin Immunol 1987;80:810–5.
13. Chapel H, Brennan V, Delson E. Immunoglobulin replacement therapy by self-infusion at home. Clin Exp Immunol 1988;73:160–2.
14. Bayston K, Leahy MF, McCreanor JD, et al. Subcutaneous gammaglobulin: effec-tive management of hypogammaglobulinaemia. N Z Med J 1985;98:652.
15. Berger M, Cupps TR, Fauci AS. High-dose immunoglobulin replacement therapyby slow subcutaneous infusion during pregnancy. JAMA 1982;247:2824–5.
16. Berger M, Cupps TR, Fauci AS. Immunoglobulin replacement therapy by slowsubcutaneous infusion. Ann Intern Med 1980;93:55–6.
17. Leahy MF. Subcutaneous immunoglobulin home treatment in hypogammaglobu-linaemia. Lancet 1986;2:48.
18. Roord JJ, van der Meer JW, Kuis W, et al. Home treatment in patients with anti-body deficiency by slow subcutaneous infusion of gammaglobulin. Lancet1982;1:689–90.
19. Stiehm ER, Casillas AM, Finkelstein JZ, et al. Slow subcutaneous human intrave-nous immunoglobulin in the treatment of antibody immunodeficiency: use of anold method with a new product. J Allergy Clin Immunol 1998;101:848–9.
20. Ugazio AG, Duse M, Plebani A, et al. Subcutaneous infusion of gamma globulinsin the management of agammaglobulinemic patients. Birth Defects Orig Artic Ser1983;19:213–5.
21. Ugazio AG, Duse M, Re R, et al. Subcutaneous infusion of gammaglobulins inmanagement of agammaglobulinaemia. Lancet 1982;1:226.
22. Welch MJ, Stiehm ER. Slow subcutaneous immunoglobulin therapy in a patientwith reactions to intramuscular immunoglobulin. J Clin Immunol 1983;3:285–6.
23. Abrahamsen TG, Sandersen H, Bustnes A. Home therapy with subcutaneous im-munoglobulin infusions in children with congenital immunodeficiencies. Pediatrics1996;98:1127–31.
24. Gardulf A, Hammarstrom L, Smith CI. Home treatment of hypogammaglobulinae-mia with subcutaneous gammaglobulin by rapid infusion. Lancet 1991;338:162–6.
25. Thomas MJ, Brennan VM, Chapel HH. Rapid subcutaneous immunoglobulin infu-sions in children. Lancet 1993;342:1432–3.
26. Gardulf A, Nicolay U, Asensio O, et al. Rapid subcutaneous IgG replacementtherapy is effective and safe in children and adults with primary immunodefi-ciencies: a prospective, multi-national study. J Clin Immunol 2006;26:177–85.
27. Gardulf A, Andersen V, Bjorkander J, et al. Subcutaneous immunoglobulin replace-ment in patients with primary antibody deficiencies: safety and costs. Lancet 1995;345:365–9.
28. Gardulf A, Bjorvell H, Andersen V, et al. Lifelong treatment with gammaglobulinfor primary antibody deficiencies: the patients’ experiences of subcutaneousself-infusions and home therapy. J Adv Nurs 1995;21:917–27.
29. Quinti I, Pierdominici M, Marziali M, et al. European surveillance of immunoglob-ulin safety: results of initial survey of 1243 patients with primary immunodefi-ciencies in 16 countries. Clin Immunol 2002;104:231–6.

Self-infusion Programmes for Immunoglobulin 831
30. Brennan VM, Cochrane S, Fletcher C, et al. Surveillance of adverse reactions inpatients self-infusing intravenous immunoglobulin at home. J Clin Immunol 1995;15:116–9.
31. Kobayashi RH, Kobayashi AD, Lee N, et al. Home self-administration of intrave-nous immunoglobulin therapy in children. Pediatrics 1990;85:705–9.
32. Hansen S, Gustafson R, Smith CI, et al. Express subcutaneous IgG infusions: de-creased time of delivery with maintained safety. Clin Immunol 2002;104:237–41.
33. Roifman CM, Levison H, Gelfand EW. High-dose versus low-dose intravenousimmunoglobulin in hypogammaglobulinaemia and chronic lung disease. Lancet1987;1:1075–7.
34. Ochs HD, Gupta S, Kiessling P, et al. Safety and efficacy of self-administeredsubcutaneous immunoglobulin in patients with primary immunodeficiency dis-eases. J Clin Immunol 2006;26:265–73.
35. Gardulf A, Bjorvell H, Gustafson R, et al. Safety of rapid subcutaneous gamma-globulin infusions in patients with primary antibody deficiency. Immunodeficiency1993;4:81–4.
36. Gardulf A, Nicolay U, Math D, et al. Children and adults with primary antibodydeficiencies gain quality of life by subcutaneous IgG self-infusions at home.J Allergy Clin Immunol 2004;114:936–42.
37. Kittner JM, Grimbacher B, Wulff W, et al. Patients’ attitude to subcutaneous immu-noglobulin substitution as home therapy. J Clin Immunol 2006;26:400–5.
38. Nicolay U, Kiessling P, Berger M, et al. Health-related quality of life and treatmentsatisfaction in North American patients with primary immunodeficiency diseasesreceiving subcutaneous IgG self-infusions at home. J Clin Immunol 2006;26:65–72.
39. Brennan VM, Salome-Bentley NJ, Chapel HM. Prospective audit of adverse reac-tions occurring in 459 primary antibody-deficient patients receiving intravenousimmunoglobulin. Clin Exp Immunol 2003;133:247–51.
40. Gaspar J, Gerritsen B, Jones A. Immunoglobulin replacement treatment by rapidsubcutaneous infusion. Arch Dis Child 1998;79:48–51.
41. Radinsky S, Bonagura VR. Subcutaneous immunoglobulin infusion as an alterna-tive to intravenous immunoglobulin. J Allergy Clin Immunol 2003;112:630–3.
42. Eijkhout HW, van den Broek PJ, van der Meer JW. Substitution therapy in immu-nodeficient patients with anti-IgA antibodies or severe adverse reactions to pre-vious immunoglobulin therapy. Neth J Med 2003;61:213–7.
43. Gustafson R, Gardulf A, Granert C, et al. Prophylactic therapy for selective IgAdeficiency. Lancet 1997;350:865.
44. Sundin U, Nava S, Hammarstrom L. Induction of unresponsiveness against IgA inIgA-deficient patients on subcutaneous immunoglobulin infusion therapy. ClinExp Immunol 1998;112:341–6.
45. Gardulf A, Andersson E, Lindqvist M, et al. Rapid subcutaneous IgG replacementtherapy at home for pregnant immunodeficient women. J Clin Immunol 2001;21:150–4.
46. Daly PB, Evans JH, Kobayashi RH, et al. Home-based immunoglobulin infusiontherapy: quality of life and patient health perceptions. Ann Allergy 1991;67:504–10.
47. Gardulf A, Bjorvell H, Gustafson R, et al. The life situations of patients with primaryantibody deficiency untreated or treated with subcutaneous gammaglobulin infu-sions. Clin Exp Immunol 1993;92:200–4.
48. Gardulf A, Nicolay U. Replacement IgG therapy and self-therapy at homeimprove the health-related quality of life in patients with primary antibody defi-ciencies. Curr Opin Allergy Clin Immunol 2006;6:434–42.

Bhole et al832
49. Anderson RT, Aaronson NK, Wilkin D. Critical review of the international assess-ments of health-related quality of life. Qual Life Res 1993;2:369–95.
50. Strasser S, Aharony L, Greenberger D. The patient satisfaction process: movingtoward a comprehensive model. Med Care Rev 1993;50:219–48.
51. Weaver M, Patrick DL, Markson LE, et al. Issues in the measurement of satisfac-tion with treatment. Am J Manag Care 1997;3:579–94.
52. Howard V, Greene JM, Pahwa S, et al. The health status and quality of life ofadults with X-linked agammaglobulinemia. Clin Immunol 2006;118:201–8.
53. Sigstad HM, Stray-Pedersen A, Froland SS. Coping, quality of life, and hope inadults with primary antibody deficiencies. Health Qual Life Outcomes 2005;3:31.
54. Ryan A, Thomson BJ, Webster AD. Home intravenous immunoglobulin therapyfor patients with primary hypogammaglobulinaemia. Lancet 1988;2:793.
55. Hogy B, Keinecke HO, Borte M. Pharmacoeconomic evaluation of immunoglob-ulin treatment in patients with antibody deficiencies from the perspective of theGerman statutory health insurance. Eur J Health Econ 2005;6:24–9.
56. Koller H, Schroeter M, Feischen H, et al. Subcutaneous self-infusions of immuno-globulins as a potential therapeutic regimen in immune-mediated neuropathies.J Neurol 2006;253:1505–6.
57. Lee DH, Linker RA, Paulus W, et al. Subcutaneous immunoglobulin infusion:a new therapeutic option in chronic inflammatory demyelinating polyneuropathy.Muscle Nerve 2008;37:406–9.
58. Schleinitz N, Jean E, Benarous L, et al. Subcutaneous immunoglobulin adminis-tration: an alternative to intravenous infusion as adjuvant treatment for dermato-myositis? Clin Rheumatol 2008;27:1067–8.
59. Sewell WA, Brennan VM, Donaghy M, et al. The use of self infused intravenousimmunoglobulin home therapy in the treatment of acquired chronic demyelinatingneuropathies. J Neurol Neurosurg Psychiatr 1997;63:106–9.
60. Misbah SA, Chapel HM. Adverse effects of intravenous immunoglobulin. DrugSaf 1993;9:254–62.
61. Healey CJ, Sabharwal NK, Daub J, et al. Outbreak of acute hepatitis C followingthe use of anti-hepatitis C virus–screened intravenous immunoglobulin therapy.Gastroenterology 1996;110:1120–6.

ImmunoglobulinReplacement Therapyin Children
Maria Garcia-Lloret, MD, Sean McGhee, MD, Talal A. Chatila, MD*
KEYWORDS
� Intravenous immunoglobulin therapy� Subcutaneous immunoglobulin therapy� Primary immunodeficiency diseases
The benefit of immunoglobulin (IG) replacement in primary antibody deficiencies (AD)is unquestionable. Many of these congenital disorders present early in life and thistherapy is often first implemented in the young. For many of these children, IG infu-sions will remain a requirement for the foreseeable future. No other therapy has dem-onstrated to be as efficacious as IG in reducing the number and severity of infectiouscomplications in pediatric patients with AD. The consensus among pediatric immunol-ogists is that, when combined with close clinical monitoring, timely and appropriate IGreplacement could ultimately extend the life expectancy of these young patients toapproach that of the general population.
The general concerns surrounding IG therapy affect adults and children equally.Issues regarding efficacy in the ever-expanding applications of IG, the predictedshortages of this drug, and the rising costs of therapy have been comprehensivelyaddressed in a number of recent reviews.1–5 This article focuses on the indicationsof IG replacement in children, with an emphasis on the specific diagnostic problemsencountered in this population. Also presented is an overview of the practical aspectsof IG administration in the pediatric setting, including the recognition and managementof adverse reactions. Finally, briefly discussed is the advent of subcutaneous IG,a therapeutic IG modality with the potential to have a great impact in the quality oflife of children with AD and their families.
INTRAVENOUS IMMUNOGLOBULIN FOR ANTIBODY REPLACEMENT THERAPY
Intravenous immunoglobulin (IVIG) is a fractionated blood product made from pooledhuman plasma. Available in the United States since the early 1980s, it rapidly
Division of Immunology, Allergy and Rheumatology, Department of Pediatrics, The DavidGeffen School of Medicine at the University of California at Los Angeles, MDCC 12-430,10833 Le Conte Avenue, Los Angeles, CA 90095–1752, USA* Corresponding author.E-mail address: [email protected] (T.A. Chatila).
Immunol Allergy Clin N Am 28 (2008) 833–849doi:10.1016/j.iac.2008.07.001 immunology.theclinics.com0889-8561/08/$ – see front matter ª 2008 Elsevier Inc. All rights reserved.

Garcia-Lloret et al834
substituted the use of intramuscular preparations as replacement therapy in ADstates. Because it is manufactured from plasma from thousands of individuals, IVIGcontains a mixture of antibodies against a wide spectrum of infectious pathogens.The concentration of antibodies against hepatitis B, diphtheria, measles, tetanus,and polio in the final product must comply with Food and Drug Administration require-ments. Titers against other pathogens, including those that more frequently affect pa-tients with AD, such as Streptococcus pneumonia and Haemophilus influenzaesubtype B, are presently not regulated by the FDA. These titers can vary significantlyamong different products and even from batch to batch.6,7
To comply with World Health Organization and FDA guidelines, more than 90% pro-tein content in commercial IVIG should be monomeric IgG with a distribution of IgGsubclasses close to that in normal plasma.8,9 Traces of IgM and IgA are present inall products, but the content of the latter can vary significantly between manufacturersdepending on the method of IgG purification followed. Other immunomodulatory pro-teins, such as cytokines, soluble CD4 and CD8 and CD40, and HLA molecules are alsopresent in varying amounts.1,10 The risk of transmission of infectious pathogens by thisblood-derived product is minimized by the careful selection of donors, plasma anti-body screening, and various procedures of viral inactivation.
Since the early 1990s the distinction between IVIG products has increased becauseof refinements in manufacturing.11 Most of these products have proved to be effica-cious in the treatment of AD when compared with historical untreated controls orpatients treated with intramuscular IG. The methods of purification, viral inactivation,and the addition of stabilizers vary between different manufacturers and can affect theclinical performance of the different products. Physicians need to be aware of thesedifferences because that could influence their decision in selecting the appropriateproduct for each individual patient. Further, no one IVIG product currently on the mar-ket has approval for all of the FDA-sanctioned indications.
There are notably few studies comparing side by side the efficacy of different IVIGproducts.12 In one such study, patients treated with an IVIG product prepared witha less harsh method of viral inactivation had fewer infections that those who receiveda solvent-detergent treated IVIG.13 Differences in efficacy between IVIG preparationshave also been reported in children with Kawasaki disease.14
Production methods not only can affect efficacy but also tolerability. High sodiumand sucrose-containing products, for instance, may be contraindicated in patientswith marginal cardiac or renal function. This is also an important consideration inneonates and infants. Reduced blood volumes and immature renal function putsthis population particularly at risk of developing electrolytic imbalances or volumeoverload. For these patients, IVIG products with a higher protein concentration,low osmolarity, and neutral pH constitute the best option. IVIG with products withreduced IgA content may be preferred in patients with IgA deficiency who are stillable to produce antibodies of IgE or IgG isotype, because these patients are atrisk of developing anaphylactic-type reactions when they receive IgA containingblood products.15
IMMUNOGLOBULIN REPLACEMENT IN CHILDREN
In general, IG replacement therapy is indicated for patients with primary or secondaryAD only if they have recurrent or severe infections and defective antibody production.The efficacy of IVIG in this setting is primarily related to the well-known attributes ofIgG antibodies to neutralize bacterial toxins, superantigens, and viruses; activatecomplement; and promote phagocytosis and antibody-mediated cytotoxicity.

Immunoglobulin Replacement Therapy in Children 835
Additional benefits are probably drawn from the less well-characterized anti-inflam-matory and immunomodulatory properties of IVIG.1,10
In AD disorders, the host’s ability to mount a protective antibody response againstmicrobial pathogens is markedly impaired. Conceptually, AD can be divided into twogroups: the hypogammaglobulinemias, in which deficits in antibody synthesis result indecreased levels of IGs, and the functional antibody defects in which the serum IGsare within the normal range but where the production of antigen-specific responsesis defective.
HYPOGAMMAGLOBULINEMIA
Because of the substantial physiologic variation in the concentration of serum IGs infirst few years of life, the correct interpretation of IG levels in pediatric patients relieson reference to age-matched controls rather than on absolute values. Evaluation ofpremature babies requires further adjustment according to their gestational age.16
In general terms, a child with serum IgG levels of less than 2 SD below the mean forage is considered to be hypogammaglobulinemic.17 The levels of the other isotypes(IgA and IgM) usually, but not always, correlate with those of IgG, which is the mostabundant serum IG. Although decreased IgG values do not necessarily herald a pri-mary immunodeficiency, the finding of hypogammaglobulinemia in a young patientwarrants further investigation.
Low IGs in children can result from multiple causes, many of which are unrelated toa primary immunodeficiency (Box 1). In a recent retrospective study from the Chil-dren’s Hospital of Philadelphia, about half of the cases of hypogammaglobulinemiawere caused by a pre-existing condition known to be accompanied by decreasedIG.18 In those in which the IG levels were obtained as part of a diagnostic work-up,only 50% were found to have an immunodeficiency.
PRIMARY IMMUNODEFICIENCIES
The first and foremost indication of IG replacement is to decrease the infections in pa-tients with hypogammaglobulinemia and impaired antibody responses. The prototyp-ical diseases in this group are the agammaglobulinemias: X-linked or autosomalrecessive.19 The diagnosis of this condition is usually made in the second or thirdyear of life in a child with a history of recurrent infection, profoundly decreased IGs,and extremely low or absent B cells. In these young patients, early institution of IGtherapy can be life-saving.
Marked hypogammaglobulinemia across the three isotypes with conserved B cellsnumbers suggests the diagnosis of common variable immunodeficiency (CVI),whereas decrease in IgG and IgA with normal to elevated IgM is the hallmark of thehyper-IgM syndrome.20–22 Children with CVI or hyper-IgM syndrome have a severeimpairment in antibody responses and, like agammaglobulinemic patients, usuallysuffer from recurrent sinopulmonary infections that can be ameliorated by the regularIG infusions. IG replacement is also indicated in infants with severe combined immu-nodeficiency awaiting transplant and in those in which B-cell function is not restoredfollowing transplantation.
IgG subclass deficiency rarely results in marked hypogammaglobulinemia. Immu-nologists commonly request this determination in a child with recurrent infectionsand normal levels of total IgG.23 In the absence of impaired antibody responses, thesignificance of a depressed level of any of the IG subclasses is unclear and IG replace-ment is not indicated.

Box1Causes of hypogammalglobulinemia in children
Decreased production
Primary antibody defects
Transient hypogammaglobulinemia of infancyc
X-linked agammaglobulinemiaa
Autosomal-recessive agammaglobulinemiaa
Hyper-IgM syndromea
Common variable immunodeficiencya
Ataxia-telangiectasiaa
Severe combined immunodeficiencya
Prematurityb
Malignancyc
Posttransplant (solid organ, bone marrow transplant)b
Chemotherapyc
Drugs
Increased loss
Congenital heart disease
Nephrotic syndromec
Intestinal lymphangiectasia
Burns
Severe atopic dermatitisc
Increased catabolism
FcRN mutations
Myotonic dystrophy
Sepsis
a IVIG customarily recommended.b Recommended in selected cases.c IVIG rarely indicated.
Garcia-Lloret et al836
Transient hypogammaglobulinemia of infancy is the most common cause of symp-tomatic hypogammaglobulinemia in children under the age of 2.24 This diagnosis canonly be made in retrospect when the child’s IG level reaches age-appropriate levels.Transient hypogammaglobulinemia of infancy follows a benign course, although a fewof the young children originally diagnosed with transient hypogammaglobulinemia ofinfancy develop a more permanent defect.25,26 Most patients with transient hypogam-maglobulinemia of infancy do well with appropriate antibiotic management but a fewmay require short-term IVIG support. The benefits of IVIG in these young patientsshould be balanced against the possibility of interfering with the normal maturationof the immune system, because at least in vitro, IVIG suppresses both T- and B-cellresponses.3,27 For those who go on IVIG, periodic re-evaluation of their immune func-tion is imperative.

Immunoglobulin Replacement Therapy in Children 837
SECONDARY HYPOGAMMAGLOBULINEMIASProtein-Losing Enteropathy
Protein-losing enteropathy is a condition characterized by severe loss of serum pro-tein into the intestine. Hypogammaglobulinemia can occur in this setting, often asso-ciated with severe hypoalbuminemia and edema. A number of conditions have beenassociated with protein-losing enteropathy. In children, gastrointestinal disordersand congenital heart disease are the leading causes.28,29 Protein-losing enteropathyis a known complication of the Fontan circulation and in other cardiac disorders wherean impaired mesenteric circulation results in an ischemic insult of the gastrointestinalmucosa and enteral protein loss.28 Impairment of the lymphatic drainage of the gas-trointestinal tract can also lead to protein-losing enteropathy.30 In addition to hypo-gammaglobulinemia, patients with intestinal lymphangiectasia, also can presentwith T-cell lymphopenia of varying degrees, rising the suspicion of a combined primaryimmunodeficiency diseases (PID). IgG levels in protein-losing enteropathy are usuallymoderately decreased, but they can be very low. Even under these circumstances, IGreplacement is not indicated because there is no evidence that infections in patientswith protein-losing enteropathy occur at a higher rate or are more severe than in com-parable patients with similar comorbidities. It can be argued that, in the face of ongo-ing protein losses, IG administration is futile. Correction of the underlying disorderusually results in normalization of the IG levels.
Nephrotic Syndrome
A low level of serum IgG with normal or increased IgM is a common finding in childrenwith steroid-sensitive nephrotic syndrome in relapse and in remission.31 Originally pre-sumed to be secondary to urinary protein loss, the hypogammaglobulinemia of ste-roid-sensitive nephrotic syndrome is now thought to result from complex immunemechanisms intrinsic to the pathogenesis of this disease. A recent study of 44 childrenwith steroid-sensitive nephrotic syndrome showed that the IgG subclass distributionvaries depending on the stage of the disease, leading to the suggestion that the pref-erential loss of certain IgG subclasses may underlie the unusual susceptibility ofpatients to pneumococcal infections.32 Although functional antibody defects may bea feature of steroid-sensitive nephrotic syndrome, there is no evidence that IGreplacement is useful in this condition and it should not be recommended.
Medications
Several classes of medications can lead to secondary hypogammaglobulinemia.33
These include glucocorticoids and other immunosuppressants, chemotherapeuticagents, and anticonvulsants. In most of these cases, the hypogammaglobulinemiais mild and of no clinical significance. IG replacement is not indicated. Discontinuationor substitution by an alternative drug should be considered in the rare instances whereIG levels are substantially reduced or if the patient develops unusual or recurrentinfections.
Hypogammaglobulinemia Caused by Increased IgG Catabolism
Although generalized hypercatabolic states (eg, infection) are often accompanied byquantitative or qualitative defects in IG production, decreased levels of serum IgGcan also result from primary disorders of IG degradation or turnover. Hypogammaglo-bulinemia is a feature of familial hypercatabolic hypoproteinemia, which is caused bymutations in the b2-microglobulin gene,34 a component of the neonatal Fc receptor(FcRN), which is critically involved in serum IgG homeostasis.35

Garcia-Lloret et al838
Although the mechanism is still unclear, accelerated IgG catabolism is also thoughtto be behind the hypogammaglobulinemia observed in some patients affected withmyotonic dystrophy.36 Interestingly, the gene associated with muscular dystrophy isupstream of the gene encoding the alpha chain of FcRn on chromosome 19, and ithas been suggested that there might be a direct or indirect influence on the expressionof FcRN and consequently in the catabolic rate of IGs.37 Although the levels of IgG inpatients with myotonic dystrophy occasionally are in the range observed in primaryimmunodeficiency, for the most part they do not suffer from recurrent infections andrarely warrant IG support.
FUNCTIONAL ANTIBODY DEFECTSPrimary Specific Antibody Defects
Children who have recurrent sinopulmonary infections with encapsulated bacteria,normal or near normal IgG levels, and impaired antibody responses may pose a diag-nostic challenge. Some of these children have additional features that suggest a CVIphenotype and the presumption is that eventually the total IG levels will fall. Othersnever meet criteria for CVI and their antibody defects remain discrete, leading to thediagnosis of specific antibody deficiency (SAD). The impaired response against poly-saccharide antigens is the best characterized feature of SAD.38 More recently, Alach-kar and colleagues39,40 showed that children and adults with SAD have decreasednumbers of switched memory B cells, which have been argued to play a cardinalrole in the protection against encapsulated bacteria.
The current view is that IG replacement in SAD should only be considered in the faceof recurrent pyogenic infections poorly controlled with antibiotic therapy. In childrenwith SAD who are started on IVIG, the recommendation is to re-evaluate them aftera year. If antibody responses improve and infections do not recur, therapy shouldbe discontinued.
Impaired polysaccharide responses are found in about one third of patients withDiGeorge syndrome, which is primarily considered a T-cell defect. These patients sel-dom warrant IVIG administration. Variable defects in antibody production have beenreported in a number of complex immunodeficiencies, such as the hyperimmunoglo-bulinemia E syndrome, Wiskott-Aldrich syndrome, and X-linked proliferative dis-ease.41–43 The efficacy of IVIG therapy in these rare disorders is mostly anecdotalbut IG supportive therapy is routinely offered to these children in some centers.16
HIV Infection
Profound abnormalities in cellular and humoral immunity are the hallmark of HIV infec-tion. Despite normal or even elevated levels of total serum IGG, children infected withHIV often have impaired antibody responses and suffer from recurrent infections withcommon pyogenic bacteria, such as S pneumonia and H influenzae. This is in contrastto adults, where opportunistic infections are the major concern. IVIG therapy is nowpart of the standard of care of pediatric HIV patients, this being one of the six FDA-approved indications for the drug.4 This indication followed the findings from two largerandomized, placebo-controlled trials conducted in the early 1990s that demonstratedthe benefits of IVIG infusions (400 mg/kg/4weeks) in reducing the number of seriousbacterial infections in HIV-infected children.44,45 The advent of more effective antire-troviral therapies, such as highly active retroviral therapy may, however, change thisprospect. In a recent study of 15 HIV-infected children by Grisaru-Soen and col-leagues46 short-term (<3 months) withdrawal of IVIG was not associated with a signif-icant increase in incidence of infections or a decline in immunologic function.

Immunoglobulin Replacement Therapy in Children 839
Neonatal Sepsis
Neonates are a population at high risk for disseminated infection because of the rela-tive immaturity of their host defense mechanisms.16 In terms of humoral immunity, ma-ternal IgG can offer considerable protection but the infant’s antibody responses tonewly encountered antigens are either delayed (proteins) or absent (polysaccharides).The poor opsonic capacity of neonatal serum leads to inefficient phagocytosis andbacterial killing, These latter abnormalities are even more pronounced in prematurebabies whose IG levels are markedly lower than those in infants born at term. Theseobservations have provided the rationale for the use of IVIG to improve the outcomeof neonatal sepsis.
A number of studies have addressed the efficacy of IVIG in the management of neo-natal infections. An earlier meta-analysis of trials found significant reduction in themortality of neonatal sepsis when IVIG was added to conventional therapies.47 In con-trast, administration of IVIG seems to be only marginally beneficial for preventing neo-natal sepsis and is probably not justified.48
Sepsis in Pediatric Patients
In septic syndromes, the increased demands and the hypercatabolic state often leadto functional antibody deficits that could be partially corrected with IVIG infusions. In-deed, adjuvant therapy with IVIG decreases mortality by more than 30% in adult and inpediatric patients with bacterial sepsis or septic shock as demonstrated by meta-anal-ysis of eight trials involving 492 patients.49 Using slightly different selection parame-ters, another group reported similar conclusions.50 Of note, IVIG products with highIgM content (not available in the United States) seemed to be superior in this setting,likely because of the increased capacity of pentameric IgM to activate complementand to opsonize gram-negative bacteria.51 Despite this promising evidence, at thepresent time IG replacement is by no means customary in the treatment of microbialsepsis. Further studies are required to delineate the precise indications, timing, dos-age, and IVIG in the management of this disorder.
DOSAGE ANDADMINISTRATION
The primary goal of IG replacement is to reduce the incidence and the severity of in-fections in patients with AD. Although the efficacy of IG therapy was apparent from thevery first clinical trials, the optimal dose to achieve this goal is still a matter of investi-gation.52–54 A number of studies established the superiority of higher IVIG doses (ie,400–600 mg/kg versus 100–200 mg/kg every 3–4 weeks) in reducing the rate of infec-tions, decreasing hospitalization and antibiotic usage, and improving pulmonary out-comes in patients with primary hypogammaglobulinemia.55–58
On the basis of these observations, the standard recommended IG replacementdose for children with AD is 100 mg/kg/wk. Doubling this standard dose may furtherdecrease the number of bacterial and viral infections and should be considered in se-lected patients as recently proposed by Eijkhout and colleagues59 In this double-blind,randomized, crossover study of 43 patients with AD, 18 of whom were children, dou-bling the standard dose of IVIG significantly reduced the number and duration of infec-tions. These findings suggest that, in selected patients, higher doses of IVIGassociated with increased trough levels decrease long-term complications, especiallypulmonary ones. For ease and convenience, when the intravenous route is chosen, theinfusions are administered every 3 to 4 weeks. Patients with severe hypogammaglo-bulinemia (<100 mg/dL) may benefit from a total ‘‘loading’’ dose of 800 mg/kg given

Garcia-Lloret et al840
in two separate doses a few days apart, followed by monthly injections of 400 to 500mg/kg.4,16
The average half-life of IgG is 21 days, but IgG metabolism shows significant vari-ations among individuals.60 Active infection, endocrine disorders, and autoimmunityhave all been associated with increased IgG catabolism.61 These comorbidities, whichcould potentially reduce the effective dose of replacement IG, are not unusual in pa-tients with PID. Genetic factors can also play a significant role, as illustrated by thehigher catabolism of IgG in patients with mutations in the b2-microglobulin chain ofthe FcRn.34 It is preferable to assess the adequacy of IG replacement in terms ofthe residual or trough levels of serum IgG rather than on the absolute dose infused.In general, serum IgG troughs of 500 to 600 mg/dL are effective in preventing acutebacterial infections in hypogammaglobulinemic patients. At replacement doses of 500mg/kg/mo, these levels are usually attained after the sixth infusion (or about sixhalf-lives), once redistribution to the tissues is complete and a steady state isreached.60
Residual serum IgG should be monitored every 2 months until steady state isreached and every 6 months thereafter. In children, periodic dose adjustments are re-quired during periods of accelerated growth but excessive monitoring of IG levelsshould be avoided. Higher residual IG levels (>800 mg/dL) may be indicated inselected patients with protracted sinus infections or progressive lung damage.4,57
In children with mild to moderate decreases in serum IG (CVI) or in those with func-tional antibody deficits and normal levels of IGs (SAD), trough levels are more difficultto interpret given that these patients retain some antibody synthetic capabilities.17,62
Some immunologists aim for trough levels of 300 mg/dL higher than the preinfusionlevels, whereas others favor troughs in the midrange of the normal for age. Dosingcan be more complex, but a starting dose of 400 mg/kg/mo is generally acceptable.In some patients, increasing IG doses may be offset by concomitant enhancementIgG catabolism.60 Increasing the dose does not necessarily raise residual levels of IgG.
ADVERSE REACTIONS
Although IG therapy is generally considered safe, adverse reactions associated withIVIG administration are not uncommon (Box 2). Because most adverse reactions oc-cur in the first few infusions, it is advisable to initiate IG therapy in a hospital setting andunder the supervision of a physician experienced in this type of treatment. In that way,adjustments in dosage, type of product, and rate of infusion can be made to ensureoptimal tolerability.
The reported frequency of IVIG-associated adverse reactions ranges between 2%and 25% of all infusions, depending in the particular disease or patient populationstudied.63,64 At replacement doses in patients with AD, this frequency is in the orderof 10% or less. IVIG-associated reactions tend to be mild to moderate in natureand, as a rule, occur during the first few infusions of the product. In this setting, chil-dren are not more likely to experience IVIG-associated adverse reactions than adults.Common symptoms, such as flushing, headaches, and malaise, tend to subside insubsequent administrations. A common practice in many centers is to premedicatepatients with acetaminophen and antihistamines with the aim of minimizing thesetype of reactions. Often, slowing the rate of infusion suffices to abate the symptoms.Because each IVIG product potentially has unique safety and tolerability profiles, it isnot uncommon to find that patients who react to one IVIG product tolerate the infusionwith no problems when switched to another brand.

Box 2Adverse reactions associatedwith IVIG therapy
Mild to moderate
Flushinga
Chillsa
Fevera
Headachea
Back pain
Chest pain
Bronchospasm
Nausea
Myalgiaa
Aseptic meningitis
Transaminitis
Increase creatinine
Severe
Renal failure
Convulsions
Thrombosis and stroke
Pulmonary edema
Hemolysis
Anaphylaxis
a IVIG customarily recommended.
Immunoglobulin Replacement Therapy in Children 841
The pathogenesis of IG-associated adverse reactions is variable and depends onthe type of product, the amount and the rate of infusion, and the clinical characteristicsof the patients. High-dose infusions, for instance, may induce to the formation of IGaggregates or immune complexes that potentially can prompt a generalized inflamma-tory response. A similar mechanism may be in patients with active infection, which isconsidered a relative contraindication for IVIG infusion. Severe adverse reactions,such as strokes, acute lung injury, kidney failure, anaphylaxis, and even death, haveall been reported in association with IVIG therapy.65–68 Fortunately these are veryrare events and tend to occur in patients receiving high-dose, repeated infusions fordisorders other than AD.63,64
IVIG is a human blood–derived product and, as such, its administration carries thepotential risk of transmission of infectious pathogens. Manufacturing techniques nowinclude a multipronged strategy to reduce the risk of potential infections, but in thepast there have been a few instances in which this complication has been docu-mented.69 Notably, in the mid-1990s there were several reports of transmission ofhepatitis C through IVIG infusions. Most of these cases were patients with PID,some were children.70 Infectious lots were traced to a single manufacturer whosestrategy had been to exclude donors positive for hepatitis C antibodies. No episodesof viral transmission caused by IVIG products have been reported after the institutionof dedicated viral inactivation methods.

Garcia-Lloret et al842
SUBCUTANEOUS IMMUNOGLOBULIN
Although IVIG clearly improves the quality of life for children with AD, there remaindrawbacks to its use. Venous access, in particular, is a serious and potentially life-threatening concern for chronically ill children, including those with immune defi-ciencies. IVIG infusion requires newly obtaining venous access on a monthly basisin children. This causes some psychologic distress and pain, and establishment of ve-nous access tends to become more difficult over time, placing the patient and riskshould resuscitation be required, and potentially compromising the ability to deliverimmune globulin. On occasions, indwelling permanent central catheters are recom-mended to facilitate the infusion. Such an intervention places an already immune-deficient child at even higher risk for sepsis, thrombosis, arrhythmias, and emboli.The impact of this on the antibody-deficient child should not be underestimated.
An alternative exists using clysis, an older method of fluid delivery by subcutaneousinfusion. Although clysis is clearly inferior to intravenous infusion for saline resuscita-tion, it is adequate for IG infusion and evades many of the issues that plague intra-venous administration of IG. In a typical subcutaneous infusion of IG, a moreconcentrated preparation of IVIG is delivered by a catheter and small volume infusionpump into the subcutaneous tissue of the abdomen, thigh, or arm. The antibodysolution is gradually absorbed from the subcutaneous tissue by lymphatics and isreturned to the circulation by normal lymphatic pathways. This results in more stablelevels of IgG over time, limits the fluid load imposed on the patient, and avoids therequirement for obtaining venous access.
Although the FDA only recently approved the first formulation for subcutaneousinfusion in the United States (Vivaglobin), subcutaneous infusion of immune globulin(SCIG) has been in use since the 1970s and was in widespread use in Europe formany years before United States approval.71–82 In general, SCIG is at least equivalentto IVIG in reduction of infections and outcomes, with improved quality of life for pa-tients and substantially reduced cost.76,83,84 SCIG may result in slightly higher troughlevels of IgG, probably because of the increased frequency of infusion.79 Although thenumber of infections is generally the same between SCIG and IVIG, some studies haveshown improved outcomes over time and in bone marrow transplant patients that cor-related with higher trough levels, suggesting some benefit to higher steady-state IgGlevels.59,85–87 Other advantages to SCIG is the more limited fluid load and no associ-ation with renal failure, a concern for sucrose-containing intravenous preparations.
There are limitations to this procedure. Principally, the volume of fluid that can bedelivered subcutaneously is limited, requiring the concentration of the IG preparationand potentially requiring infusion in multiple sites. In addition, because of the volumelimitation, the infusion must be given weekly, rather than on a monthly basis. Despitethe volume limitations, subcutaneous infusion has even been used successfully fora dermatomyositis patient who did not tolerate intravenous administration.88 The pri-mary adverse events are local site reactions, including redness, swelling, and pain.Although these are almost universal at the start (91%), as infusions continue these be-come less problematic and seldom require return to intravenous infusion.77
Because subcutaneous access requires limited technical skill, parents and even thechildren themselves can deliver the infusion without the need for nursing services orbeing logistically dependent on an infusion center. Loss of work or school time isalso not an issue for patients receiving SCIG.
Although SCIG has clear advantages over IVIG, for pediatric use the increased num-ber of needle sticks is a concern. Although it is easier to obtain access, the number ofneedle exposures is increased fourfold because of the weekly infusion. This has been

Immunoglobulin Replacement Therapy in Children 843
a problem for some children with severe needle fears. A properly done subcutaneouspuncture causes trivial pain and the principal problem that must be addressed in chil-dren is avoiding the negative stigma associated with the needle. Ideally, this problemshould be managed from the beginning with careful and graded exposure to the infu-sion apparatus. Anesthetic creams should be used for the first few infusions to elim-inate any possible pain associated with puncture.
Other strategies are also being investigated to reduce the number of infusions nec-essary for subcutaneous use of immune globulin. For example, it has already beendemonstrated that similar results can be obtained by infusing once every 2 weeks us-ing twice the dose. This can usually be tolerated, although a greater number of sitesare needed to accommodate the increased volume.89 Furthermore, a version of10% IVIG solution (Gammagard or Kiovig) modified for subcutaneous infusion is cur-rently in phase III trials. This preparation has a version of hyaluronidase included toimprove diffusion through subcutaneous tissue, allowing a greater volume to bedelivered. This permits the infusion to be delivered subcutaneously once per monthwith similar trough levels and infectious outcomes.90,91 It is not clear whether thesame advantage of higher trough levels and more stable steady-state levels can beexpected from these alternative preparations.
Despite the concern by providers and parents about increased exposure to needlesticks, children tolerate the procedure well and most children and families prefer sub-cutaneous infusion to intravenous infusion.92 The savings to the family and society areconsiderable, with SCIG infusions saving $10,100 per patient-year over intravenousinfusion in 1997 in a study in Sweden.76 Similar impacts on quality of life were alsofound in a North American study.93 SCIG also seems to be safe in pregnancy. Althoughit carries a pregnancy class C rating from the FDA, at least 12 pregnant women havereceived SCIG without any evident harm to the pregnancy.94,95 Patients with mildbleeding disorders also can tolerate therapy.96 IgA-deficient patients, who are atrisk for anaphylaxis caused by contaminating IgA in IVIG, can be tolerized to IgA usingsubcutaneous infusion.97,98 Although patients who have had severe reactions to IVIGcan still have severe reactions to SCIG, these reactions are less common and manypatients who do not tolerate IVIG may tolerate SCIG.99,100
REFERENCES
1. Jolles S, Sewell WA, Misbah SA. Clinical uses of intravenous immunoglobulin.Clin Exp Immunol 2005;142:1–11.
2. Mahadevia PJ. The pocketbook: pharmacoeconomic issues related to intrave-nous immunoglobulin therapy. Pharmacotherapy 2005;25:94S–100S.
3. Nimmerjahn F, Ravetch JV. Anti-inflammatory actions of intravenous immuno-globulin. Annu Rev Immunol 2008;26:513–33.
4. Orange JS, Hossny EM, Weiler CR, et al. Use of intravenous immunoglobulin inhuman disease: a review of evidence by members of the Primary Immunodefi-ciency Committee of the American Academy of Allergy, Asthma and Immunol-ogy. J Allergy Clin Immunol 2006;117:S525–53.
5. Clynes R. Protective mechanisms of IVIG. Curr Opin Immunol 2007;19:646–51.6. Givner LB. Human immunoglobulins for intravenous use: comparison of avail-
able preparations for group B streptococcal antibody levels, opsonic activity,and efficacy in animal models. Pediatrics 1990;86:955–62.
7. Lamari F, Anastassiou ED, Tsegenidis T, et al. An enzyme immunoassay to deter-mine the levels of specific antibodies toward bacterial surface antigens in human

Garcia-Lloret et al844
immunoglobulin preparations and blood serum. J Pharm Biomed Anal 1999;20:913–20.
8. Anonymous. Appropriate uses of human immunoglobulin in clinical practice:memorandum from an IUIS/WHO meeting. Bull World Health Organ 1982;60:43–7.
9. Anonymous. Requirements for the collection, processing and quality control ofblood, blood components and plasma derivatives. WHO technical report1989; Series No 786 Annex.
10. Sewell WA, Jolles S. Immunomodulatory action of intravenous immunoglobulin.Immunology 2002;107:387–93.
11. Siegel J. The product: all intravenous immunoglobulins are not equivalent. Phar-macotherapy 2005;25:78S–84S.
12. Schiff RI, Williams LW, Nelson RP, et al. Multicenter crossover comparison of thesafety and efficacy of Intraglobin-F with Gamimune-N, Sandoglobulin, and Gam-magard in patients with primary immunodeficiency diseases. J Clin Immunol1997;17:21–8.
13. Roifman CM, Schroeder H, Berger M, et al. Comparison of the efficacy of IGIV-C,10% (caprylate/chromatography) and IGIV-SD, 10% as replacement therapy inprimary immune deficiency: a randomized double-blind trial. Int Immunophar-macol 2003;3:1325–33.
14. Rosenfeld EA, Shulman ST, Corydon KE, et al. Comparative safety and efficacyof two immune globulin products in Kawasaki disease. J Pediatr 1995;126:1000–3.
15. Cunningham-Rundles C, Zhou Z, Mankarious S, et al. Long-term use of IgA-depleted intravenous immunoglobulin in immunodeficient subjects with anti-IgAantibodies. J Clin Immunol 1993;13:272–8.
16. Stiehm ER. Human intravenous immunoglobulin in primary and secondaryantibody deficiencies. Pediatr Infect Dis J 1997;16:696–707.
17. Agarwal S, Cunningham-Rundles C. Assessment and clinical interpretation ofreduced IgG values. Ann Allergy Asthma Immunol 2007;99:281–3.
18. Onigbanjo MT, Orange JS, Perez EE, et al. Hypogammaglobulinemia in a pedi-atric tertiary care setting. Clin Immunol 2007;125:52–9.
19. Conley ME, Broides A, Hernandez-Trujillo V, et al. Genetic analysis of patientswith defects in early B-cell development. Immunol Rev 2005;203:216–34.
20. Cunningham-Rundles C, Bodian C. Common variable immunodeficiency:clinical and immunological features of 248 patients. Clin Immunol 1999;92:34–48.
21. Bacchelli C, Buckridge S, Thrasher AJ, et al. Translational mini-review series onimmunodeficiency: molecular defects in common variable immunodeficiency.Clin Exp Immunol 2007;149:401–9.
22. Bonilla FA, Geha RS. Primary immunodeficiency diseases. J Allergy Clin Immu-nol 2003;111:S571–81.
23. Buckley RH. Immunoglobulin G subclass deficiency: fact or fancy? Curr AllergyAsthma Rep 2002;2:356–60.
24. Dalal I, Reid B, Nisbet-Brown E, et al. The outcome of patients with hypogamma-globulinemia in infancy and early childhood. J Pediatr 1998;133:144–6.
25. Moschese V, Graziani S, Avanzini MA, et al. A prospective study on children withinitial diagnosis of transient hypogammaglobulinemia of infancy: results from theItalian primary immunodeficiency network. Int J Immunopathol Pharmacol 2008;21:343–52.

Immunoglobulin Replacement Therapy in Children 845
26. Dorsey MJ, Orange JS. Impaired specific antibody response and increasedB-cell population in transient hypogammaglobulinemia of infancy. Ann AllergyAsthma Immunol 2006;97:590–5.
27. Takei S, Arora YK, Walker SM. Intravenous immunoglobulin contains specific an-tibodies inhibitory to activation of T cells by staphylococcal toxin superantigens[see comment]. J Clin Invest 1993;91:602–7.
28. Driscoll DJ. Long-term results of the Fontan operation. Pediatr Cardiol 2007;28:438–42.
29. Chehade M, Magid MS, Mofidi S, et al. Allergic eosinophilic gastroenteritis withprotein-losing enteropathy: intestinal pathology, clinical course, and long-termfollow-up. J Pediatr Gastroenterol Nutr 2006;42:516–21.
30. Radhakrishnan K, Rockson SG. The clinical spectrum of lymphatic disease. AnnN Y Acad Sci 2008;1131:155–84.
31. Schnaper HW. The immune system in minimal change nephrotic syndrome.Pediatr Nephrol 1989;3:101–10.
32. Kemper MJ, Altrogge H, Ganschow R, et al. Serum levels of immunoglobulinsand IgG subclasses in steroid sensitive nephrotic syndrome. Pediatr Nephrol2002;17:413–7.
33. Jaffe E, Lejtenyi C, Noya F, et al. Secondary Hypogammaglobulinemia. ImmunolAllergy Clin North Am 2001;21:141–63.
34. Wani MA, Haynes LD, Kim J, et al. Familial hypercatabolic hypoproteinemiacaused by deficiency of the neonatal Fc receptor, FcRn, due to a mutantbeta2-microglobulin gene. Proc Natl Acad Sci U S A 2006;103:5084–9.
35. Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat RevImmunol 2007;7:715–25.
36. Larsen B, Johnson G, van Loghem E, et al. Immunoglobulin concentration andGm allotypes in a family with thirty-three cases of myotonic dystrophy. ClinGenet 1980;18:13–9.
37. Pan Q, Hammarstrom L. Molecular basis of IgG subclass deficiency. ImmunolRev 2000;178:99–110.
38. Paris K, Sorensen RU. Assessment and clinical interpretation of polysaccharideantibody responses. Ann Allergy Asthma Immunol 2007;99:462–4.
39. Alachkar H, Taubenheim N, Haeney MR, et al. Memory switched B cell percent-age and not serum immunoglobulin concentration is associated with clinicalcomplications in children and adults with specific antibody deficiency and com-mon variable immunodeficiency. Clin Immunol 2006;120:310–8.
40. Carsetti R, Rosado MM, Wardmann H. Peripheral development of B cells inmouse and man. Immunol Rev 2004;197:179–91.
41. Holland SM, DeLeo FR, Elloumi HZ, et al. STAT3 mutations in the hyper-IgEsyndrome. N Engl J Med 2007;357:1608–19.
42. Nichols KE, Ma CS, Cannons JL, et al. Molecular and cellular pathogenesis ofX-linked lymphoproliferative disease. Immunol Rev 2005;203:180–99.
43. Ochs HD, Slichter SJ, Harker LA, et al. The Wiskott-Aldrich syndrome: studies oflymphocytes, granulocytes, and platelets. Blood 1980;55:243–52.
44. Spector SA, Gelber RD, McGrath N, et al. A controlled trial of intravenousimmune globulin for the prevention of serious bacterial infections in childrenreceiving zidovudine for advanced human immunodeficiency virus infection.Pediatric AIDS Clinical Trials Group. N Engl J Med 1994;331:1181–7.
45. Intravenous immune globulin for the prevention of bacterial infections in childrenwith symptomatic human immunodeficiency virus infection. The National

Garcia-Lloret et al846
Institute of Child Health and Human Developments Intravenous ImmunoglobulinStudy Group. N Engl J Med 1991;325:73–80.
46. Grisaru-Soen G, Lau W, Arneson C, et al. Randomized controlled trial of short-term withdrawal of I.V. immunoglobulin therapy for selected children with humanimmunodeficiency virus infection. Pediatr Int 2007;49:972–7.
47. Ohlsson A, Lacy JB. Intravenous immunoglobulin for suspected or subse-quently proven infection in neonates. Cochrane Database Syst Rev 2004:CD001239.
48. Ohlsson A, Lacy JB. Intravenous immunoglobulin for preventing infection in pre-term and/or low-birth-weight infants. Cochrane Database Syst Rev 2004:CD000361.
49. Kreymann KG, de Heer G, Nierhaus A, et al. Use of polyclonal immunoglobu-lins as adjunctive therapy for sepsis or septic shock. Crit Care Med 2007;35:2677–85.
50. Laupland KB, Kirkpatrick AW, Delaney A. Polyclonal intravenous immunoglobu-lin for the treatment of severe sepsis and septic shock in critically ill adults: a sys-tematic review and meta-analysis. Crit Care Med 2007;35:2686–92.
51. Trautmann M, Held TK, Susa M, et al. Bacterial lipopolysaccharide (LPS)-specific antibodies in commercial human immunoglobulin preparations:superior antibody content of an IgM-enriched product. Clin Exp Immunol1998;111:81–90.
52. Cunningham-Rundles C, Siegal FP, Smithwick EM, et al. Efficacy of intravenousimmunoglobulin in primary humoral immunodeficiency disease. Ann Intern Med1984;101:435–9.
53. Eibl MM, Cairns L, Rosen FS. Safety and efficacy of a monomeric, functionallyintact intravenous IgG preparation in patients with primary immunodeficiencysyndromes. Clin Immunol Immunopathol 1984;31:151–60.
54. Nolte MT, Pirofsky B, Gerritz GA, et al. Intravenous immunoglobulin therapy forantibody deficiency. Clin Exp Immunol 1979;36:237–43.
55. Liese JG, Wintergerst U, Tympner KD, et al. High- vs low-dose immunoglobulintherapy in the long-term treatment of X-linked agammaglobulinemia. Am J DisChild 1992;146:335–9.
56. Quartier P, Debre M, De Blic J, et al. Early and prolonged intravenous immuno-globulin replacement therapy in childhood agammaglobulinemia: a retrospectivesurvey of 31 patients. J Pediatr 1999;134:589–96.
57. Roifman CM, Gelfand EW. Replacement therapy with high dose intravenousgamma-globulin improves chronic sinopulmonary disease in patients with hypo-gammaglobulinemia. Pediatr Infect Dis J 1988;7:S92–6.
58. Roifman CM, Levison H, Gelfand EW. High-dose versus low-dose intravenousimmunoglobulin in hypogammaglobulinaemia and chronic lung disease. Lancet1987;1:1075–7.
59. Eijkhout HW, van Der Meer JW, Kallenberg CG, et al. The effect of two differentdosages of intravenous immunoglobulin on the incidence of recurrent infectionsin patients with primary hypogammaglobulinemia: a randomized, double-blind,multicenter crossover trial. Ann Intern Med 2001;135:165–74.
60. Schiff RI, Rudd C. Alterations in the half-life and clearance of IgG during therapywith intravenous gamma-globulin in 16 patients with severe primary humoralimmunodeficiency. J Clin Immunol 1986;6:256–64.
61. Blaese RM, Strober W, Levy AL, et al. Hypercatabolism of IgG, IgA, IgM, andalbumin in the Wiskott-Aldrich syndrome: a unique disorder of serum proteinmetabolism. J Clin Invest 1971;50:2331–8.

Immunoglobulin Replacement Therapy in Children 847
62. Busse PJ, Razvi S, Cunningham-Rundles C. Efficacy of intravenous immuno-globulin in the prevention of pneumonia in patients with common variable immu-nodeficiency. J Allergy Clin Immunol 2002;109:1001–4.
63. Ballow M. Safety of IGIV therapy and infusion-related adverse events. ImmunolRes 2007;38:122–32.
64. Pierce LR, Jain N. Risks associated with the use of intravenous immunoglobulin.Transfus Med Rev 2003;17:241–51.
65. Ahsan N. Intravenous immunoglobulin induced-nephropathy: a complication ofIVIG therapy. J Nephrol 1998;11:157–61.
66. Burks AW, Sampson HA, Buckley RH. Anaphylactic reactions after gamma glob-ulin administration in patients with hypogammaglobulinemia: detection of IgEantibodies to IgA. N Engl J Med 1986;314:560–4.
67. Hamrock DJ. Adverse events associated with intravenous immunoglobulin ther-apy. Int Immunopharmacol 2006;6:535–42.
68. Sekul EA, Cupler EJ, Dalakas MC. Aseptic meningitis associated with high-doseintravenous immunoglobulin therapy: frequency and risk factors. Ann Intern Med1994;121:259–62.
69. Schleis TG. The process: new methods of purification and viral safety. Pharma-cotherapy 2005;25:73S–7S.
70. Bjoro K, Froland SS, Yun Z, et al. Hepatitis C infection in patients with primaryhypogammaglobulinemia after treatment with contaminated immune globulin.N Engl J Med 1994;331:1607–11.
71. Abrahamsen TG, Sandersen H, Bustnes A. Home therapy with subcutaneousimmunoglobulin infusions in children with congenital immunodeficiencies. Pedi-atrics 1996;98:1127–31.
72. Alyanakian MA, Bernatowska E, Scherrmann JM, et al. Pharmacokinetics of total im-munoglobulin G and immunoglobulin G subclasses in patients undergoing replace-ment therapy forprimary immunodeficiency syndromes. VoxSang 2003;84:188–92.
73. Berger M, Cupps TR, Fauci AS. Immunoglobulin replacement therapy by slowsubcutaneous infusion. Ann Intern Med 1980;93:55–6.
74. Bjorkander J, Wadsworth C, Hanson LA. 1040 prophylactic infusions withan unmodified intravenous immunoglobulin product causing few side-effects in patients with antibody deficiency syndromes. Infection 1985;13:102–10.
75. Chapel HM, Spickett GP, Ericson D, et al. The comparison of the efficacy andsafety of intravenous versus subcutaneous immunoglobulin replacement ther-apy. J Clin Immunol 2000;20:94–100.
76. Gardulf A, Andersen V, Bjorkander J, et al. Subcutaneous immunoglobulinreplacement in patients with primary antibody deficiencies: safety and costs.Lancet 1995;345:365–9.
77. Gardulf A, Nicolay U, Asensio O, et al. Rapid subcutaneous IgG replace-ment therapy is effective and safe in children and adults with primary immu-nodeficiencies: a prospective, multi-national study. J Clin Immunol 2006;26:177–85.
78. Gaspar J, Gerritsen B, Jones A. Immunoglobulin replacement treatment byrapid subcutaneous infusion. Arch Dis Child 1998;79:48.
79. Ochs HD, Gupta S, Kiessling P, et al. Safety and efficacy of self-administeredsubcutaneous immunoglobulin in patients with primary immunodeficiency dis-eases. J Clin Immunol 2006;26:265–73.
80. Remvig L, Andersen V, Hansen NE, et al. Prophylactic effect of self-administeredpump-driven subcutaneous IgG infusion in patients with antibody deficiency: a

Garcia-Lloret et al848
triple-blind cross-over study comparing P-IgG levels of 3 g l-1 versus 6 g l-1.J Intern Med 1991;229:73–7.
81. Ugazio AG, Duse M, Re R, et al. Subcutaneous infusion of gammaglobulins inmanagement of agammaglobulinaemia. Lancet 1982;1:226.
82. Waniewski J, Gardulf A, Hammarstrom L. Bioavailability of gamma-globulin aftersubcutaneous infusions in patients with common variable immunodeficiency.J Clin Immunol 1994;14:90–7.
83. Gardulf A, Borte M, Ochs HD, et al. Prognostic factors for health-related qualityof life in adults and children with primary antibody deficiencies receiving SCIGhome therapy. Clin Immunol 2008;126:81–8.
84. Gardulf A, Nicolay U. Replacement IgG therapy and self-therapy at home im-prove the health-related quality of life in patients with primary antibody defi-ciencies. Curr Opin Allergy Clin Immunol 2006;6:434–42.
85. Cottler-Fox M, Lynch M, Pickle LW, et al. Some but not all benefits of intravenousimmunoglobulin therapy after marrow transplantation appear to correlate withIgG trough levels. Bone Marrow Transplant 1991;8:27–33.
86. Leen CL, Yap PL, McClelland DB. Increase of serum immunoglobulin level intothe normal range in primary hypogammaglobulinaemia by dosage individualiza-tion of intravenous immunoglobulin. Vox Sang 1986;51:278–86.
87. Ochs HD, Fischer SH, Wedgwood RJ, et al. Comparison of high-dose and low-dose intravenous immunoglobulin therapy in patients with primary immunodefi-ciency diseases. Am J Med 1984;76:78–82.
88. Schleinitz N, Jean E, Benarous L, et al. Subcutaneous immunoglobulin adminis-tration: an alternative to intravenous infusion as adjuvant treatment for dermato-myositis? Clin Rheumatol 2008;27:1067–8.
89. Gustafson R, Gardulf A, Hansen S, et al. Rapid subcutaneous immunoglobulinadministration every second week results in high and stable serum immunoglob-ulin G levels in patients with primary antibody deficiencies. Clin Exp Immunol2008;152:274–9.
90. Bjorkander J, Nikoskelainen J, Leibl H, et al. Prospective open-label study ofpharmacokinetics, efficacy and safety of a new 10% liquid intravenous immu-noglobulin in patients with hypo- or agammaglobulinemia. Vox Sang 2006;90:286–93.
91. Church JA, Leibl H, Stein MR, et al. Efficacy, safety and tolerability of a new 10%liquid intravenous immune globulin [IGIV 10%] in patients with primary immuno-deficiency. J Clin Immunol 2006;26:388–95.
92. Fasth A, Nystrom J. Safety and efficacy of subcutaneous human immunoglobu-lin in children with primary immunodeficiency. Acta Paediatr 2007;96:1474–8.
93. Nicolay U, Kiessling P, Berger M, et al. Health-related quality of life and treat-ment satisfaction in North American patients with primary immunodeficiency dis-eases receiving subcutaneous IgG self-infusions at home. J Clin Immunol 2006;26:65–72.
94. Berger M, Cupps TR, Fauci AS. High-dose immunoglobulin replacementtherapy by slow subcutaneous infusion during pregnancy. JAMA 1982;247:2824–5.
95. Gardulf A, Andersson E, Lindqvist M, et al. Rapid subcutaneous IgG replace-ment therapy at home for pregnant immunodeficient women. J Clin Immunol2001;21:150–4.
96. Arora R, Newton TC, Nelson MR. Subcutaneous immunoglobulin therapy in an11-year-old patient with common variable immunodeficiency and von Willebranddisease. Ann Allergy Asthma Immunol 2007;99:367–70.

Immunoglobulin Replacement Therapy in Children 849
97. de Albuquerque Campos R, Sato MN, da Silva Duarte AJ. IgG anti-IgAsubclasses in common variable immunodeficiency and association with severeadverse reactions to intravenous immunoglobulin therapy. J Clin Immunol 2000;20:77–82.
98. Sundin U, Nava S, Hammarstrom L. Induction of unresponsiveness against IgAin IgA-deficient patients on subcutaneous immunoglobulin infusion therapy. ClinExp Immunol 1998;112:341–6.
99. Quinti I, Soresina A, Agostini C, et al. Prospective study on CVID patients withadverse reactions to intravenous or subcutaneous IgG administration. J ClinImmunol 2008;28:263–7.
100. Stiehm ER, Casillas AM, Finkelstein JZ, et al. Slow subcutaneous human intra-venous immunoglobulin in the treatment of antibody immunodeficiency: use ofan old method with a new product. J Allergy Clin Immunol 1998;101:848–9.

IntravenousGammaglobulinTreatment in HIV-1Infection
Avi Deener, MDa, Ami Mehra, MDa, Larry Bernstein, MDa,Jenny Shliozberg, MDa, Arye Rubinstein, MD, PhDa,b,*
KEYWORDS
� Immunodeficiency � B cells � T cells� Highly active antiretroviral therapy (HAART) � HIV-1 � AIDS� Thrombocytopenia � Opportunistic infections
HISTORICAL BACKGROUND OF GAMMAGLOBULIN REPLACEMENT IN HIV/AIDS
In 1978–1979, children with an unusual clinical and immunologic profile were observedat Albert Einstein College of Medicine.1–3 These children presented with recurrent bac-terial infections; several had multiple septic episodes with the same organism thatfailed to respond to specific antibodies.1,4 Their clinical presentation appeared typicalfor a B-cell deficiency such as Bruton’s agammaglobulinemia. In contrast to parents ofchildren with congenital B-cell deficiencies, parents of these children also presentedwith immune aberrations, including cell-mediated immunity and B-cell immunity,which suggested that a transmittable infectious agent may be involved. Researcherssoon recognized that the children’s B-cell deficiency was unique in its immunologicand histopathologic profile. They had a prominent generalized lymphadenopathy,and their lymph node biopsies revealed a marked proliferation of B cells. They alsoexhibited a pan-hypergammaglobulinemia that encompassed all three immunoglobu-lin classes: IgA, IgM, and IgG.1–3 Despite the hypergammaglobulinemia, they failed tomount specific antibody responses to various antigens.5 Most prominent was theirinability to mount specific antibodies to polysaccharide antigens.
This work was supported by Grant No.AI051519 from the National Institutes of Health.a Department of Pediatrics, Division of Allergy and Immunology, Albert Einstein College ofMedicine and Montefiore Hospital Medical Center, 1525 Blondell Avenue, Bronx, NY 10461, USAb Department of Microbiology and Immunology, Albert Einstein College of Medicine andMontefiore Hospital Medical Center, 1525 Blondell Avenue, Bronx, NY 10461, USA* Corresponding author. Department of Pediatrics, Division of Allergy and Immunology, AlbertEinstein College of Medicine and Montefiore Hospital Medical Center, 1525 Blondell Avenue,Bronx, NY 10461.E-mail address: [email protected] (A. Rubinstein).
Immunol Allergy Clin N Am 28 (2008) 851–859doi:10.1016/j.iac.2008.06.001 immunology.theclinics.com0889-8561/08/$ – see front matter ª 2008 Elsevier Inc. All rights reserved.

Deener et al852
Treatment of these children, who were later identified as being infected with HIV-1, withintravenous immunoglobulin (IVIG) not only abolished the recurrent bacterial infections 6–8
but also induced previously unexpected immunomodulatory effects.8–14 The introductionof antiretroviral therapieshadan effect on immune functions inpediatricand adult patientsinfected with HIV-1, in most instances deviating the need for IVIG.
USE OF INTRAVENOUS GAMMAGLOBULIN IN THE PRE-ANTIRETROVIRAL ERA
The B-cell immune system in children infected with HIV-1 is more compromised thanthat of adults who have HIV-1. Maturation of the B-cell compartment is physiologicallyincomplete at birth, and protection against common pathogens is provided bypassive transplacental transfer of maternal IgG. Even babies with Bruton’sagammaglobulinemia usually enjoy an infection-free interval of 6 to 9 months throughpassive immunity. This is not always the case in infants who are infected with HIV-1.Because humoral (B-cell) and cellular (T-cell) immune defects are noted in pregnantwomen who are infected with HIV-1, the protection of their infants through passiveimmunity is incomplete. This finding explains the often earlier onset of bacterial andseptic infections in individuals who are infected with HIV-1. The maturation of B cellsin infants who are infected with HIV-1 is further compromised by the inadequate inter-action provided by dysfunctional T cells and by abnormal thymic epithelium function.Decreased levels of circulating thymulin, a marker of thymic dysplasia, have beennoted in infants infected with HIV-1.15
In 1979, our rationale for the introduction of IVIG in children infected with HIV-1 wasbased on documents of in vivo defective primary and secondary antibody responsesto a T-cell–dependent bacteriophage neoantigen (4X174), tetanus toxoid, anda B-cell–dependent pneumococcal polysaccharide antigen.5,16 The poor secondaryresponses to these antigens included abnormal class switches from IgM to IgG.16
Further in vitro evidence of B-cell dysfunction was the poor lymphocyte proliferativeresponse to Pokeweed mitogen and Staphylococcal aureus, a T-cell–independentB-cell mitogen.1–3 Pokeweed mitogen also failed to induce in vitro B-cell secretionof IgG in the presence of normal T lymphocytes.12
The IVIG regimen used between 1979 and 1982 was the same as that used forpatients who had agammaglobulinemia (200–300 mg/kg monthly). In 1982, evidenceemerged for an associated multitude of immune aberrations, including markedly ele-vated circulating immune complexes,9,13 elevated serum neopterin levels,10 elevatedserum tumor necrosis factor a,11 elevated serum b2-microglobulin,14 isomorphicelevation of serum lactate dehydrogenase,17 and T-cell dysregulation of humoralimmunity.12 The IVIG regimen was subsequently modified to address the additionalfactors. The dose was increased to 300 mg/kg every 2 weeks (instead of every4 weeks). The following account details the clinical and immune alterations by IVIGin a 10-year follow-up of 112 children aged 9 months to 6 years who were treatedby the latter intensified protocol.
Infectious Complications
Only mild upper respiratory infections occurred. No additional episodes of sepsis werenoted at 300 mg/kg biweekly as compared with 13 serious bacterial infections inchildren who received 200 to 300 mg/kg monthly.
Immunomodulatory Effects
Serum IgG levels did not increase over baseline in IVIG-treated patients, whereasa progressive increase in serum IgG was noted in most untreated infants. After IVIG

Intravenous Gammaglobulin 853
treatment there was a statistically significant decrease in b2-microglobulin and lactatedehydrogenase levels. As markers of immune activation, neopterin10 and tumornecrosis factor a also decreased or normalized.11 Circulating immune complexeslevels decreased in 77% of patients on the intensified IVIG protocol but in none of24 untreated patients and in 10 patients who received the monthly IVIG dose of200 mg/kg.9,13
In children infected with HIV-1, the CD81 T-cell compartment is markedly increased,but these cells are of poor function, as documented by the failure of CD81 T cells tosuppress in vitro Pokeweed mitogen–driven IgG secretion.12 The reduced Pokeweedmitogen–driven IgG secretion normalized in 80% of studied subjects after IVIG treat-ment. The absent in vitro Concanavalin A generation of suppressor T cells was restoredin IVIG-treated patients.12 Statistically significant increases in percentage and absoluteT-cell counts were observed in 43% of treated patients. This trend persisted in mostpatients for up to 10 years until highly active antiretroviral therapy (HAART) was intro-duced.6–8 In vitro lymphocyte mitogenic responses to phytohemagglutinin increasedtransiently in 71% of patients who received IVIG.6
A large National Institute of Child Health and Human Development (NICHD) random-ized double-blind placebo controlled multicenter IVIG study on children infected withHIV-1 was conducted 10 years later. This study corroborated most of the earlier find-ings, although the benefits achieved were not as robust. The IVIG regimen used wasdifferent in that 400 mg/kg were administered every 4 weeks.18–21 In this trial, childrenwho had a baseline CD41T-cell count of more than 200/mL remained free from seri-ous laboratory-proven and clinical diagnosed bacterial infections.18–20 In contrast toour studies with the higher IVIG dose, no statistically significant benefit was noted inchildren with fewer than 200 CD41T-cells/mL. This difference is probably due tothe less intensive IVIG regimen. The NICHD study confirmed the immunomodulatoryeffect on CD41 T cells with a statistically significant slower decline in CD41 T-cellcounts.21
IVIG also was used to treat several autoimmune manifestations, most prominentlythrombocytopenia, which is a frequent complication of several conditions, includingpediatric HIV-1 infection,22 sepsis,23 and conditions that involve antiplatelet IgG.24
The incidence of thrombocytopenia in children infected with HIV-1 who were onprophylactic IVIG was lower than in untreated patients. Thrombocytopenia, includingseptic thrombocytopenia, responded promptly with increased platelet counts afterhigh-dose IVIG at 1 g/kg.22–24 The numeric platelet responses were not alwaysassociated with a favorable clinical outcome, however. In contrast to adults withHIV-1–associated thrombocytopenia, children may have antiplatelet antibodies andautoantibody-mediated vasculitis or clotting factor abnormalities.25 As a result, theyhave more frequent episodes of bleeding and a poorer prognosis.
Intravenous Gammaglobulin in Adults Who Have HIV-1 Infection
Questions have been raised as to whether IVIG plays a role in adults who are infectedwith HIV-1.26 HIV-1 infected adults may retain their full complement of B cells, includ-ing anamnestic antibody responses, for a while because they were formed before theirHIV-1 infection. As a result, adults who have HIV-1 infection are less susceptible to in-fections with common pathogens. HIV-1 can, however, profoundly affect humoral im-munity in a subset of infected adults. Polsky and colleagues27 and Selwyn andcolleagues28 reported a marked increase in the number of bacterial pneumonias,mainly caused by Staphylococcus pneumoniae and Haemophilus influenza, in adultswho have AIDS, most of whom were substance abusers. Janoff and colleagues29

Deener et al854
estimated that the rate of pneumococcal bacteremia among patients who have AIDSwas 100-fold higher than in age-matched controls.
These findings formed the rationale for use of IVIG in adults. Several uncontrolledclinical trials have not shown conclusive benefits to routine IVIG prophylaxis. Therewas also no evidence that IVIG delayed progression of HIV-1–related diseaseor had immunomodulatory effects as noted in children who were infected withHIV-1. In a small randomized IVIG trial, Schrappe-Bacher and colleagues30 showedno changes in CD41 cell counts or other clinical and immunologic parameters.These studies should not exclude the use of IVIG for adults who are infectedwith HIV-1 with recurrent bacterial infections that are not well controlled with anti-biotics. In this subset of patients, the use of bacterial vaccines was first studied.Because initial uncontrolled studies suggested an acceptable immunogenicity ofpolysaccharide vaccines, pneumococcal vaccines were recommended as a stan-dard of care for adults who are infected with HIV-1 in North America and the UnitedKingdom. Unfortunately, a meta-analysis of prospective randomized pneumococcalvaccine trials did not show efficacy among the subgroup of North American adultsinfected with HIV-1.31
A second clinical trial with a pneumococcal vaccine was evaluated in Africa, whereinvasive pneumococcal disease was particularly prevalent in adults who are HIV-1 in-fected. The study showed not only lack of efficacy but also evidence of vaccineharm,32 probably caused by immune activation. We have shown in an animal modelthat HIV-1 was enhanced by the capsular polysaccharide of Cryptococcus neofor-mans.33 Brichachek and colleagues34 noted an increased plasma HIV-1 burden afterchallenge with a pneumococcal vaccine. We reported transient up-regulation of HIV-1transcription after a phage FX174 vaccine16 and pneumococcal immunizations. Afterbacteriophage immunization, a transient decline in CD41 cells was noted. It was rec-ommended that vaccination should preferably be administered only in tandem withantiretroviral therapy.
Because it is unclear what level of antibodies is protective in HIV-1 infected patientsand the finding that bacterial vaccines may be ineffective or harmful, prophylacticIVIG may be an option in patients with documented recurrent bacterial infectionsand absent or marginal bacterial vaccine responses. There is evidence for theuse of IVIG in adults with autoimmune thrombocytopenia. The incidence of throm-bocytopenia in adult patients who have HIV-1 is estimated at 65 per 10,000persons, whereas the incidence of thrombocytopenia in HIV-1–negative patientsis estimated at 1.5 per 10,000 individuals.35,36
The introduction of HAART has been effective in mitigating thrombocytopenias, butno studies are available on the incidence of autoimmune thrombocytopenia for patientstreated with HAART. The two most common treatments, steroids and splenectomy,remain undesirable for patients who have thrombocytopenia and HIV-1, regardlessof whether they are on HAART. In a study by Cai and colleagues,37 the use ofsteroids was associated with an increased risk of developing Kaposi’s sarcoma.Splenectomy was associated with an increased susceptibility to infections withencapsulated organisms.
In a recent study by Gyongyossy-Issa and colleagues,38 the thrombopoieticresponses to IVIG treatment were evaluated in patients who were uninfectedwith HIV-1 and had immune thrombocytopenic purpura and patients who hadHIV-1–associated autoimmune thrombocytopenia. The initial response to IVIG inHIV-1 autoimmune patients who had thrombocytopenia was similar to that seen inpatients who were not infected with HIV-1. Often-repeated doses were required tomaintain an adequate response, however.39 As a result, the question was raised as

Intravenous Gammaglobulin 855
to whether low-dose IVIG is effective. In 13 patients treated with low-dose IVIG(400 mg/kg/wk for 5 weeks), Majluf-Cruz and colleagues40 demonstrated a sustainedresponse 3 months after the end of treatment.
Anti-D therapy has been studied in the treatment of HIV-1–associatedthrombocytopenia as an alternative to IVIG. In a prospective study, 14 Rh1 patientswho had HIV-1 received 12 to 25 mg/kg of anti-D IgG intravenously on 2 consecutivedays. Nine patients had a rapid and significant increase in their platelet counts.These data suggested that anti-Rh IgG is effective and safe in HIV-1–relatedthrombocytopenic purpura.40 A small, prospective, randomized study by Scaradavouand colleagues41 compared anti-D therapy with IVIG. Nine patients who were Rh1and had HIV-1 were treated with IVIG or anti-D for 3 months. Patients who weretreated with anti-D demonstrated higher mean peak platelet counts and longerduration of response. Anti-D resulted in a mean peak platelet count of 77,000/mLcompared with only 29,000/mL after IVIG (P 5 .07). The mean duration of responsewas significantly longer in patients treated with anti-D (41 days) compared with IVIG(19 days; P 5 .01). This study may support the use of anti-D as first-line therapyin patients who are Rh1 and have HIV-1 and need urgent treatment forthrombocytopenia.
In a retrospective study, Lesprit and colleagues42 compared the cost of anti-D ther-apy to high-dose IVIG. The cost of IVIG per treated episode was $4269, comparedwith $2716 for anti-D therapy. Overall, based on the evaluation of incidence, treatmentpatterns, and hospital care required, anti-D therapy was associated with significantlylower costs. Because lower doses of IVIG also were found to be effective, however,40
it remains more cost effective than anti-D therapy.Rheumatic manifestations may develop at any time during the clinical spectrum of
HIV-1 infection, but they are usually seen more often in late stages. Various disordersmay be seen, particularly Reiter’s syndrome and undifferentiated spondyloarthrop-athy. Most patients do well with conventional anti-inflammatory therapy, but somepatients require immunosuppressive-cytotoxic therapy. IVIG has not been studied inthese situations, except for in cases of polymyositis, in which patients who receivedhigh-dose IVIG showed dramatic improvement.43
USE OF INTRAVENOUS GAMMAGLOBULIN IN THE ERAOF HIGHLYACTIVEANTIRETROVIRALTHERAPIES
Recent advances with HAART have dramatically reduced mortality and morbidity andprolonged life expectancy. Immune restoration is an important component of HAART.There is an initial increase in CD41 and CD81 T cells and B cells followed by improvedin vitro lymphocyte proliferative responses and the return of delayed-type skin hyper-sensitivity reactions.44 Not all patients demonstrate a substantial T-cell reconstitution,and no controlled studies evaluated the restoration of the functional integrity of thehumoral immune responses. Most studies on the restoration of B-cell function orthe lack thereof were performed in children with HIV-1 infection and may not reflectthe situation in adults.
Questions that were raised discussed whether HAART restores the humoralimmune system in children who are infected with HIV-1 to function in a relativelynormal way; that is, to recognize an antigen, produce antibodies to that antigen,and create and sustain memory B cells for quick production of protective antibodiesin the event of re-exposure. One of the first studies that examined this issue waspublished in 1996. In the face of a measles epidemic in New York City, Arpadiand colleagues45 studied measles antibody titers among children aged 9 to 168

Deener et al856
months who had HIV-1. They correlated measles titers with CD41 cell counts alongwith other parameters, including age of first measles immunization and the numberof immunizations. They found that CD41 T-cell counts had a positive correlationwith one’s ability to maintain measles antibodies. Among children who had noT-cell immunosuppression, 82% maintained immunity, whereas among the mostseverely compromised (CD41 T-cell counts % 600/mL) only 53% had positivemeasles antibody titers. Overall, 72% of previously vaccinated children maintainedimmunity compared with 95% of healthy children. Treatment with zidovudine at thetime of vaccination was significantly associated with the children testing seroposi-tive to measles.
Moir and colleagues46 studied the mechanisms by which uncontrolled HIV-1infection may compromise B-cell function. They showed that in viremic patients(patients who were either on HAART-resistant regimens or were noncompliant withHAART) compared with aviremic patients who had HIV-1, B cells were less responsiveto CD41 T cells. This finding was still true when controlling for CD41 T-cell numbers.Their studies showed that this B-cell dysfunction was likely related to B cells’ inabilityto up-regulate CD25 (interleukin-2 receptor) in response to appropriate CD41 T-cellsignaling in the presence of HIV-1 viremia. Thus, HIV-1 viremia may cause an inherentB-cell dysfunction independent of T-cell function.
Bekker and colleagues47 documented that even after immune reconstitution witheffective HAART, B-cell memory was not restored to vaccination (MMR) and naturallyoccurring viral pathogens (VZV, CMV, EBV). The most imperative of B-cell functions—providing specific antibodies to previously exposed antigens—was not restored.Overall, 40% of the patients lost immunity to measles, 38% lost immunity to mumps,and 11% lost immunity to rubella. Only 43% maintained immunity to all three patho-gens, and 20% lost varicella zoster virus titers after being followed longitudinally onHAART.
B-cell reconstitution by HAART was also studied by Rosenblatt and colleagues48 inchildren aged 4 months to 17 years who were HIV-1 infected and lost their titers to tet-anus and then underwent HAART therapy. These children had only mild to moderateimpairment in their immune status before enrolling in this study; their percentage ofCD41 cells was more than 15% with a median absolute count of 976 cells. After re-immunization with tetanus, most (74%) had a good response 4 weeks later; however,the percentage of patients with a positive titer dropped to 67% at 8 weeks, 53% at 18weeks, and 38% at 32 weeks.
In a study that compared patients who had common variable immune deficiency,patients who underwent splenectomy, and patients who had HIV-1 infection, Hartand colleagues49 reported a decreased response to Pneumovax among individualswho had HIV-1 infection and were on HAART. They showed that this poor responsewas caused by a relative deficiency in IgM memory B cells. This finding provided anexplanation for the increased risk of invasive pneumococcal disease among peoplewho have HIV-1 infection. These data suggested that a segment of patients whohave HIV-1 who exhibit poor B-cell functions, respond poorly to bacterial vaccines,and have recurrent bacterial infections may be candidates for prophylactic IVIGtreatment.
No controlled studies have documented the immunomodulatory effect of IVIG onpatients who are infected with HIV-1 and are on HAART; nor have studies exam-ined the effect that IVIG may have on reducing morbidity and mortality. The use ofIVIG for autoimmune manifestations on HAART remains controversial and anunchartered field. It plays a role in autoimmune thrombocytopenia for the pre-HAART era.

Intravenous Gammaglobulin 857
REFERENCES
1. Rubinstein A, Sicklick M, Gupta A, et al. Acquired immunodeficiency withreversed T4/T8 ratios in infants born to promiscuous and drug-addicted mothers.JAMA 1983;249:2345–9.
2. Rubinstein A. Acquired immunodeficiency syndrome in infants [editorial]. AmJ Dis Child 1983;137:825–6.
3. Rubinstein A, Bernstein L, Small C. Autoantibodies to AIDS-T cells. Ann N Y AcadSci 1985;437:508–9.
4. Bernstein L, Krieger BZ, Novick B, et al. Bacterial infection in the acquiredimmunodeficiency syndrome of children. Pediatr Infect Dis 1985;4:472–5.
5. Bernstein L, Ochs H, Wedgewood R, et al. Defective humoral immunity in childrenwith AIDS. J Pediatr 1985;107:352–7.
6. Rubinstein A, Calvelli T, Rubinstein R. Intravenous gammaglobulin for pediatricHIV-1 infection: effects on infectious complications, circulating immunecomplexes and CD4 cell decline. Ann N Y Acad Sci 1993;693:151–7.
7. Calvelli T, Rubinstein A. Immunodeficiency: pediatric AIDS. In: Rosen FS, editor.Clinical aspects of immunology. 5th edition. Oxford, England: Blackwell ScientificPublication, Ltd; 1993. p. 1341–68.
8. Calvelli TA, Rubinstein A. Intravenous gamma-globulin in infant acquiredimmunodeficiency syndrome. Pediatr Infect Dis 1986;5:S207–8.
9. Ellaurie M, Calvelli T, Rubinstein A. Human immunodeficiency virus (HIV)circulating immune complexes in infected children. AIDS Res Hum Retroviruses1990;6:1437–44.
10. Ellaurie M, Calvelli T, Rubinstein A. Neopterin levels in pediatric HIV infection aspredictor of disease activity. Pediatr Infect Dis J 1992;11:286–9.
11. Ellaurie M, Rubinstein A. Tumor necrosis factor in pediatric HIV-1 infection. AIDS1992;6:1265–8.
12. Gupta A, Novick ME, Rubinstein A. Restoration of suppressor T-cell functions inchildren with AIDS following intravenous gamma globulin treatment. Am J DisChild 1986;140:143–6.
13. Ellaurie M, Calvelli T, Rubinstein A. Immune complexes in pediatric HIV infection.Am J Dis Child 1990;144:1207–9.
14. Ellaurie M, Rubinstein A. Beta-2-microglobulin concentrations in pediatric humanimmunodeficiency virus infection. Pediatr Infect Dis 1990;9:807–9.
15. Rubinstein A, Novick BE, Sicklick MJ, et al. Circulating thymulin and thymosin a1
activity in pediatric acquired immunodeficiency ayndrome (in vivo and in vitrostudies). J Pediatr 1986;109:422–7.
16. Rubinstein A, Mizrachi Y, Bernstein L, et al. Progressive specific immune attritionafter primary, secondary and tertiary immunizations with bacteriophage 4X174 inasymptomatic HIV-1 infected patients. AIDS 2000;14:F55–62.
17. Silverman B, Rubinstein A. Serum lactate dehydrogenase in adults and childrenwith AIDS and ARC: possible indicator of B cell proliferation and disease activity.Am J Med 1985;78:728–36.
18. The NCHD Intravenous Immunoglobulin Study Group. Intravenous immuneglobulin for the prevention of bacterial infections in children with symptomaticHIV infection. N Engl J Med 1991;325:73–80.
19. Mofenson LM, Moye J Jr, Bethel J, et al. Prophylactic intravenous gammaglobulin inHIV-infected children with CD41 counts of 0.20/L or more. JAMA 1992;268:483–8.
20. Mofenson LM, Moye J Jr, Korelitz J, et al. Crossover of placebo patients to intra-venous immunoglobulin confirms efficacy for prophylaxis of bacterial infections

Deener et al858
and reduction of hospitalizations in HIV-infected children. Pediatr Infect Dis J1994;13:477–84.
21. Mofenson LM, Bethel J, Moye J Jr, et al. Effect of intravenous immunoglobulin(IVIG) on CD41 lymphocyte decline in HIV-infected children in a clinical trial ofIVIG infection prophylaxis. J Acquir Immune Defic Syndr 1993;6:1103–13.
22. Ellaurie MD, Shah K, Bernstein LJ, et al. Thrombocytopenia in pediatric AIDS.Pediatrics 1988;82:905–8.
23. Burns ER, Lee V, Rubinstein A. Treatment of septic thrombocytopenia withimmune globulin. J Clin Immunol 1991;11:363–8.
24. Ellaurie M, Burns ER, Rubinstein A. Platelet associated IgG in pediatric HIV infec-tion. Pediatr Hematol Oncol 1991;8:179–85.
25. Burns ER, Krieger BZ, Bernstein L, et al. Acquired circulating anticoagulants inpediatric AIDS. Pediatrics 1988;82:763–5.
26. Yap PL. Does intravenous immune globulin have a role in HIV-infected patients?Clin Exp Immunol 1994;97(Suppl 1):59–67.
27. Polsky B, Gold JWM, Whimbey E, et al. Bacterial pneumonias in patients withAIDS. Ann Intern Med 1986;104:38–41.
28. Selwyn PA, Feingold AR, Hartel D, et al. Increased risk of bacterial pneumonias inHIV-infected intravenous drug users without AIDS. AIDS 1988;2:267–72.
29. Janoff EN, Breiman RF, Daley CL, et al. Pneumococcal disease during HIV infec-tion: epidemiologic, clinical and immunologic perspectives. Ann Intern Med1992;117:314–24.
30. Schrappe-Bacher M, Rasokat H, Bauer P, et al. High dose intravenous gamma-globulin in HIV-infected adults with AIDS-related complex and Walter Reed 5.Vox Sang 1990;59(Suppl 1):3–14.
31. Fine MJ, Smith MA, Carson CA, et al. Efficacy of pneumococcal vaccination:a meta-analysis of randomized controlled trials. Arch Intern Med 1994;154:2666–77.
32. French N, Nakiyingi J, Carpenter LM, et al. 23 Valent pneumococcal polysaccha-ride vaccine in HIV-1 infected Ugandan adults: double-blind, randomized andplacebo controlled trial. Lancet 2000;355:2106–11.
33. Pettoello-Mantovani M, Casadevall A, Kollmann TR, et al. Enhancement of HIV-1infection by the capsular polysaccharide of Cryptococcus neoformans. Lancet1992;339(8784):21(1)–3.
34. Brichachek B, Swindells S, Janoff EN, et al. Increased plasma HIV-1 burden fol-lowing antigenic challenge with pneumococcal vaccine. J Infect Dis 1996;174:1191–9.
35. Morris L, Distenfeld A, Amorosi E, et al. Autoimmune thrombocytopenic purpurain homosexual men. Ann Intern Med 1982;96:714–7.
36. Simpson KN, Coughlin CM, Eron J, et al. Idiopathic thrombocytopenia purpura:treatment patterns and an analysis of cost associated with intravenous immuno-globulin and anti-D therapy. Semin Hematol 1998;35(1):58–64.
37. Cai J, Zheng T, Lotz M, et al. Glucocorticoids induce Kaposi’s sarcoma cell pro-liferation through the regulation of transforming growth factor. Blood 1997;89:1491–7.
38. Gyongyossy-Issa MI, Bussel JB, Carter CJ, et al. Comparison of thrombopoiesisduring ITP and HIV-ITP and response to intravenous gammaglobulin treatment.Platelets 2003;14(5):267–76.
39. Oksenhendler E, Bierling P, Brossard Y, et al. Anti-RH immunoglobulin therapy forhuman immunodeficiency virus–related immune thrombocytopenic purpura.Blood 1988;71(5):1499–502.

Intravenous Gammaglobulin 859
40. Majluf-Cruz A, Luna-Castanos G, Huitr�on S, et al. Usefulness of a low-doseintravenous immunoglobulin regimen for the treatment of thrombocytopeniaassociated with AIDS. Blood 1993;82(5):1415–21.
41. Scaradavou A, Cunningham-Rundles S, Ho JL, et al. Superior effect ofintravenous anti-D compared with IV gammaglobulin in the treatment of HIV-thrombocytopenia: results of a small, randomized prospective comparison. AmJ Hematol 2007;82(5):335–41.
42. Lesprit P, P�edrono G, Molina JM, et al. ANRS 114-Pneumovac Study Group. Im-munological efficacy of a prime-boost pneumococcal vaccination in HIV-infectedadults. AIDS 2007;21(18):2425–34.
43. Viard JP, Vittecog D, Lacroix C, et al. Response of HIV-1 associated polymyositisto intravenous gammaglobulin. Am J Med 1992;92:580–1.
44. Smith KY, Valdez H, Landay A, et al. Thymic size and lymphocyte restoration inHIV infected patients following 48 weeks of therapy with zidovudine, lamivudineand ritonavir. J infect Dis 2000;181:141–7.
45. Arpadi SM, Markowitz LE, Baughman AL, et al. Measles antibody in vaccinatedhuman immunodeficiency virus type 1-infected children. Pediatrics 1996;97(5):653–7.
46. Moir S, Ogwaro KM, Malaspina A, et al. Perturbations in B cell responsiveness toCD41 T cell help in HIV-infected individuals. Proc Natl Acad Sci USA 2003;10:6057–62.
47. Bekker V, Scherpbier H, Pajkrt D, et al. Persistent humoral immune defect inhighly active antiretroviral therapy-treated children with HIV-1 infection: loss ofspecific antibodies against attenuated vaccine strains and natural viral infection.Pediatrics 2006;118(2):e315–22.
48. Rosenblatt HM, Song LY, Nachman SA, et al. Tetanus immunity after diphtheria,tetanus toxoids, and acellular pertussis vaccination in children with clinically sta-ble HIV infection. J Allergy Clin Immunol 2005;116:698–703.
49. Hart M, Steel A, Clark SA, et al. Loss of discrete memory B cell subsets is asso-ciated with impaired immunization responses in HIV-1 infection and may be a riskfactor for invasive pneumococcal disease. J Immunol 2007;178:8212–20.

Economic Assessmentof Different Modalitiesof ImmunoglobulinReplacement Therapy
Stephen K. Membe, MDEa, Chuong Ho, MDa, Karen Cimon, MLTa,Andra Morrison, BSca, Amin Kanani, MD, FRCP(C)b,ChaimM. Roifman, MD, FRCPCc,*
KEYWORDS
� Subcutaneous � Intravenous � Cost-effectiveness� Cost-minimization � Immunoglobulin� Immunodeficiency
The most common significant inherited immunodeficiency is characterized by an inad-equate production of antibodies. This feature is shared by a variety of genetically andphenotypically distinct disorders such as x-linked agammaglobulinemia, commonvariable immunodeficiency, or hyper-immunoglobulin M (IgM) syndrome.1 The func-tional defect in these disorders might be limited to the B cell or T cell compartmentsor involve both domains of the immune system. The lack of antibodies and thus thefailure to respond to vaccinations predisposes these patients to invasive and poten-tially life-threatening infections. Unable to actively immunize these patients, attemptswere made to replace antibodies with those derived from blood of immunocompetentindividuals.2,3 Initially only limited quantities of immunoglobulins (Ig) could be deliveredthrough plasma infusions or intramuscular injections.4,5 This treatment was painful andquantitatively inadequate. A major advancement in this field was the introduction ofimmunoglobulins suitable for intravenous use (IVIg).6 In the early 1980s, IVIg contain-ing reduced IgG aggregates and low concentrations of IgA was introduced.6 IVIgcould be administered in large doses which could achieve physiologic serum IgGthrough levels.7–9 This in turn dramatically reduced the frequency of infections andimproved already existing lung disease.7–9
a Canadian Agency for Drugs and Technologies in Health (CADTH), 600-865 Carling Avenue,Ottawa, ON, Canada K1S 5S8b St. Paul’s Hospital and the University of British Columbia, 1081 Burrard Street, Vancouver, BC,Canada V6Z 1Y6c The Hospital for Sick Children, Division of Immunology and Allergy, 555 University Avenue,Toronto, ON, Canada M5G 1X8* Corresponding author.E-mail address: [email protected] (C.M. Roifman).
Immunol Allergy Clin N Am 28 (2008) 861–874doi:10.1016/j.iac.2008.06.008 immunology.theclinics.com0889-8561/08/$ – see front matter ª 2008 Elsevier Inc. All rights reserved.

Membe et al862
The intravenous (IV) route is presently the most common route of administration forIg.10–12 In general, IVIg has been successfully used in many patients and is consideredto be safe and well tolerated.7,9–12 Adverse reactions to IVIg range in severity from mildheadaches to rare episodes of stroke.7,13 Individual products vary in their propensityto cause reactions. Also, infusion rates and concentrations of solutions are factors inpatient tolerability. Poor venous access in some patients (eg, infants) may result inmultiple attempts at venipuncture for each infusion. In some patients indwelling centralvenous devices are required, with the associated risks of infection and thromboem-bolic complications.
The subcutaneous route of administration of immunoglobulins (SCIg) was initiallyexclusively used in Sweden.14,15 It has been gradually introduced to the rest of Europeand more recently to North America. Advantages of SCIg include the lack of require-ment for vascular access and increased patient autonomy due to self-administration.Disadvantages include frequent injections due to volume limitations of the amount thatcan be administered at one time and slow buildup of serum Ig trough levels. Lastly, theuse of SCIg is contraindicated in patients with bleeding tendencies and in those withvarious skin conditions. In addition, doses beyond 400–600 mg/kg/mo, which arecommonly used in autoimmune disorders, cannot be practically administered throughthe subcutaneous (SC) route.
The available Ig products on the market are prepared using the classic Cohn coldalcohol fractionation process, first developed more than 50 years ago.2,3 In this pro-cess, plasma from large pools of donors yields a fractionated serum of 95% to 99%IgG and traces of other immunoglobulins.6 The final product must be as free from bac-terial and viral contamination as possible. There are several methods for ensuring this,including: donor screening, molecular testing to determine viral load, virus partitioningusing Cohn-Oncley fractionation, Kistler-Nitschmann fractionation, chromatographyor filtration, virus inactivation using pH 4 incubation, solvent-detergent, heat, caprylateor low pH formulation, and antibody-mediated virus neutralization.16
IVIg and SCIg are available in liquid or lyophilized form. The liquid form takes lesspreparation before use. Some products require refrigeration, and are available onlyin one or two concentrations.17 Standard IVIg treatment usually consists of infusionsonce a month.8,12 The usual maintenance dose for primary immunodeficiency (PID)is 400 mg/kg to 600 mg/kg infused every three to four weeks.8,12 Infusions usuallytake one to four hours9 and use standard IV administration equipment. Infusions aremost commonly administered in a hospital or outpatient setting, but can be adminis-tered at home.
Pharmacokinetic studies show a rapid increase in serum IgG levels immediatelyafter the infusion (peak) and a subsequent gradual decline (trough). The role of IgGpeaks in combating infection remains unknown, but trough levels above 5 g/L appearsuperior to lower serum IgG levels in preventing infections and improving lungdisease.8
In comparison, SCIg therapy consists of weekly or biweekly administration, and theusual maintenance dose is 100 mg/kg, resulting in an accumulated monthly dose sim-ilar to that of IVIg therapy.18 The dose is self-administered or parentally administeredat home, and the required equipment is tubing, needles or catheters, syringes, and aninfusion pump.18,19 The initial infusion rate starts at 10 mL/h, and can be increased un-til the maximum rate of 22 mL/h is reached.9 Infusions typically take one to four hours.9
SCIg infusions do not result in a peak in serum Ig levels and trough levels rise graduallyover a period of up to six months. Consequently, beginning treatment with IVIg andsubsequent switching to SCIg is common practice. In preparation for home infusions,patients or parents are typically required to complete four to six educational and

Economic Assessment of Immunoglobulin Replacement Therapy 863
training sessions at a hospital. Medical and nursing checkups are usually done everyfourth week during the training period, including self-infusions under supervision ata clinic.
Economic assessment of these modalities of treatment is important and reflects onthe health care system evaluated. In this article we have reviewed here studies per-formed to date including a recent extensive Canadian cost-minimization analysis(CMA).
REVIEWOF ECONOMIC STUDIES
Economic studies were conducted from the perspectives of the health care systems inSweden, Germany, the UK, and France.20–22,24 The characteristics of the four reportsare shown in Table 1. The cost parameters and assumptions in each study were re-viewed to determine the extent to which the results can be applied to Canadian set-tings. The results and cost information from these studies appear in Table 2.
Several studies have described economic assessments which compare IVIg andSCIg. The studies appear similar in terms of clinical end points, cost items thatwere included in the analysis, results of the cost-effectiveness analysis, assumptionabout treatment settings for SCIg and IVIg patients, and assumption about the com-parative effectiveness of SCIg and IVIg. The difference between the studies is thedegree to which SCIg is cost-effective, owing to differences in study perspectivesand differences in the per gram prices of immunoglobulin preparations (whichaccounts for >78% of the total costs of treatment) across countries. The major as-sumption in all the studies was equal benefit and harm of IVIg and SCIg.
In a cost analysis (CA) study, Gardulf and colleagues21 compared the yearly costs ofhome- and hospital-based SCIg to those of hospital-based IVIg from the perspectiveof the Swedish health care system. The per-patient yearly costs associated with IgGpreparations, materials, personnel, rooms, and administrative overhead for each Igtherapy were computed. The original values in Swedish kronor were converted toUnited States dollars (one US$ equals 7.8 kronor), using 1993 prices. The authorsused yearly costs to compare hospital-based IVIg with hospital- and home-basedSCIg. The yearly cost of hospital-administered SCIg was US$4,656 with more thanhalf (51%) of the cost attributed to IgG preparations. In comparison, the yearly costof hospital-administered IVIg was US$14,124, and 93% of the cost was attributedto IgG preparations. The yearly cost of SCIg administered at home was US$3,096,with 78% of the cost attributed to IgG preparations. The authors concluded that byreplacing hospital-based IVIg with home-based SCIg, the per-patient yearly costwould be reduced by more than US$10,000.21
We identified two issues with the Gardulf and colleagues21 study. First, the implicitassumption in the CA that there was equal effectiveness between the two routes ofadministration could not be derived from the clinical data presented in the study.Second, the actual cost of therapy by either route of administration may have beenunderestimated because the study only accounted for costs accrued to the Swedishhealth care system. The costs associated with transportation of patients, manage-ment of adverse events (AE), and time spent by caregivers were not considered inthe analysis.
Hogy and colleagues20 performed a CMA based on Chapel and colleagues’crossover clinical study,23 which found no differences in efficacy or adverse reactionrates between Ig therapy given subcutaneously or intravenously. The CMA took theperspective of the German statutory health insurance, including only those coststhat could be reimbursed (eg, costs due to Ig, premedication for IVIg patients,

Table1Characteristics of economic studies
Author and CountryStudy Design, Perspective,Interventions Study Population Clinical Outcomesa Cost Considered
Gardulf et al,21
SwedenCASwedish health care systemHome SCIg versus hospital
SCIg and hospital IVIg
165 PID patients aged13 years to 76 years
Frequency of adverse systemicreactions, occurrence andintensity of tissue reactionsand serum IgG changes
Ig preparation, materials,personnel, rooms,administrative overhead
Hogy et al,20
GermanyCMAGerman statutory health
insuranceHome SCIg versus hospital
IVIg
Subgroup analysis:adults (75 kg) andchildren (40 kg)
Used result of previous study,involving 30 PID patientsthat showed no significantdifferences in infection andAE rates between SCIg andIVIg
Ig, pre-medications, infusionpump, physicians, diagnosticprocedures, sick leave forchildren’s caregivers
Liu et al,24 UK CMAUK health systemApplication of appropriate
UK assumptions to Hogyet al37 cost calculations
Home SCIg versus homeIVIg and hospital IVIg
Hogy et al subgroupanalysis: adults (75 kg)and children (40 kg)
As in Hogy et al; no significantdifferences in infection andAE rates between SCIg andIVIg
As in Hogy et al; Ig,pre-medications, infusionpump, infusion materials,physicians, diagnosticprocedures, sick leave forchildren’s caregivers
Haddad et al,22
FranceCAPublic payer (France)Home SCIg versus hospital
SCIg and hospital IVIg
Not stated No clinical data provided;authors assume equaleffectiveness between SCIgand IVIg
Hospital admission,transportation to and fromhospital, Ig acquisition cost(pre-tax), homecare nursing,rental cost of administrationpumps and perfusion kits
Abbreviations: AEs, adverse events; CA, cost analysis.a Primary outcome: infection rate; secondary outcome: AE rate, serum Ig levels.
Mem
be
et
al
864

Table 2Results of reviewed economic studies
Author (Funding Source)SensitivityAnalysis on CostParameters Study End Point Study Results
Gardulf et al21 (SwedishMedical ResearchCouncil)
Not performed Total yearly cost of SCIgtreatment (home andhospital) versus IVIgtreatment (hospital)
Hospital-based IVIg 5 $14,124/yHospital-based SCIg 5 $4,656/yearHome-based SCIg 5 $3,096/year
(costs in US$ at 1993 prices)
Hogy et al20 (funding sourceunstated)
Patients’ weight; monthly Igdose; price/g of IVIg andSCIg; costs of pump,treatment procedures, andpre-medications; and sickleave
Total yearly cost of SCIgtreatment versus IVIgtreatment for eachsubgroup
Adult: IVIg 5 V31,027;SCIg 5 V14,893
Children: IVIg 5 V17,329;SCIg 5 V8,659(costs in 2003 prices)
Liu et al24 (funding sourceunstated)
Not performed Total yearly cost of SCIgtreatment versus IVIgtreatment for each subgroup(costs in 2005 £)
Adult: IVIg at home 5 £11,580Adult: IVIg at hospital 5 £18,600Adult: SCIg at home 5 £11,760Children: IVIg at home 5 £6,540Children : SCIg at home 5 £6,720
Haddad et al22 (funding sourceunstated)
Not performed Monthly treatment costof SCIg and IVIg
Hospital IVIg (20 g/mo) 5 V1,192.19Home IVIg (20 g/mo) 5 V1,033Home IVIg (40 g/mo) 5 V2,034.50Hospital SCIg (20 g/mo) 5 V2,908.76Home SCIg (20 g/mo) 5 V1,518Home SCIg (40 g/mo) 5 V2,507–V2,729
Eco
no
mic
Asse
ssmen
to
fIm
mu
no
glo
bu
linR
ep
lace
men
tTh
era
py
865

Table 3Base-case costs and assumptions
Item SCIgIVIg
Assumpt ns SourceHome HospitalAdult’s weight NA — — 70 kg Hogy et al,20 Liu et al,24 PID
Survey27
Child’s weight — — — 40 kg
Monthly Ig dose, adult — 28 g — Average onthly dose/kgof bod weight 0.4 g foreither oute
Liu et al24
Monthly Ig dose, child — 16 g —
Dosing intervals Weekly — Monthly — CBS (Mathias Haun,personal communication,2007) and ZLB (DavidBarnes, personalcommunication, 2007)
Ig price/g C$57.75 — C$57.75
Monthly treatmentor diagnostic cost
NA — C$87.00 4 h of n ses’ time plusphysic ns’ fees, costs aresame r home- andhospit l-based IVIg
National cost list,26,32,a
Monthly hospital charges NA NA C$110.72 Same ch ges for adult andchild; st covers infusionmater ls, administrativesuppo , datamanag ment, andfollow ps
Calculated using expertopinion and Nationalcost list26
Infusion pump cost/y C$525.12 NA — Similar p mp prices forchild a d adult patients,2 pum s required/patien 1 pump lasts5 year
Berger,18 Hogy et al20
Liu et al24 http://www.marcalmedical.com
Mem
be
et
al
866
io
myr
uriafoa
arcoiarte-u
unpt,s

Infusion material cost /y forhome IVIg and SCIg
C$377.43 C$377.43 NA Same costs of infusionm terials for SCIg andh me-based IVIg; forh spital-based IVIg, costi luded in-hospitalc arges
Berger,18 Liu et al,24 NorfolkMedical30
Monitoring costs every6 mo
C$55.00 C$55.00 NA On every 6 mo SCIg, andh me-based IVIg patientsr uire monitoring byn rse or physician
z
Transportation costs/mo NA NA C$10.50 Pat nt chooses to travel byp blic transit or taxi;a erage travelingd tance to clinics orh spitals 15 km inO tario
Ontario Maternity ExpertPanel25
Cost of time lost fortreatment/h
C$8.56 C$8.56 C$18.92 Op rtunity cost of timet en to administerh me-IVIg or SCI andh spital IVIg is forgonev lue of unpaid work andp id work respectively
Statistics Canada;28 PublicHealth Agency ofCanada;29 PID Survey27
Abbreviations: CBS, Canadian Blood Services; IMIg, intramuscular immunoglobulin; NA, not app able. Liu et al.24
a Expert opinion.
Eco
no
mic
Asse
ssmen
to
fIm
mu
no
glo
bu
linR
ep
lace
men
tTh
era
py
867
aoo
nch
ceoequ
ieuvison
poakooaa
lic

Membe et al868
infusion pumps and materials used with SCIg, treatment and diagnostic procedures,and sick leave for children’s caregivers). The costs of managing AEs and time lostfrom work for adults were considered to be identical for SCIg and IVIg and thuswere excluded.
The yearly costs of SCIg and IVIg therapy were derived from the monthly costs foradults and children, calculated as a function of their respective average monthly Igdose (ie, determined by average body weight and market price per gram of Ig). Thequantity of health care resources (treatment and diagnostic procedures) used wasobtained from a survey of 18 German PID-treatment centers. All costs were expressedin 2003 euros (V).
The base case results show that the yearly costs to treat an adult patient wereV31,027 (IVIg) and V14,893 (SCIg). The yearly costs for a child were V17,329 (IVIg)and V8,659 (SCIg). For both subgroups, Ig acquisition costs were the main cost driver,accounting for >80% of the total yearly cost for each treatment arm. One-way sensi-tivity analyses were conducted to factor in the uncertainties and variations in resourceuse, average body weight, monthly dose, and prices. In adult patients, the incrementalcosts between IVIg and SCIg were more sensitive to changes in the price of Ig and lessso to the changes in body weight. In children, the incremental costs were most re-sponsive to changes in body weight.
The study by Hogy and colleagues20 showed that compared to IVIg, SCIg was costsaving because its price was 50% cheaper. However, this may not hold in the long-runas market forces will likely eliminate such per-unit cost differences. The study did notitemize the resources included in the calculations. As a result, it was unclear whetheroverhead costs and hospital charges were accounted for. Moreover, the ability to gen-eralize the results is questionable, because the authors did not describe the methodsused for the survey that was the basis for most of the cost calculations.
More recent work by Liu and colleagues24 was instrumental in examining the costdrivers associated with IVIg and SCIg. The authors did not develop a new analysis,but investigated whether Hogy and colleagues’20 costing assumptions applied inthe United Kingdom (UK). This involved an examination of the cost parameters usedin Hogy and colleagues’20 study and their underlying assumptions, followed by appli-cation of the appropriate UK costs parallel to the original German costs. An average2003 monthly exchange rate of one pound (£) for V1.453 and an annual inflationrate of 2.9% were used to calculate the equivalent UK costs in £ for 2005.
Regarding the costing assumptions between the two countries, Liu and col-leagues24 established that IVIg and SCIg are administered at home and in the hospitalin the UK. In adults, the yearly price and cost of treatment or diagnostic procedures forIVIg were lower in the UK than in Germany (ie, £10,800 versus £22,194 and £180 ver-sus £407 respectively). The yearly cost of a SCIg pump was higher in the UK (£180versus £113). The costs associated with sick leave for caregivers of children and se-nior/disabled adult patients receiving IVIg and SCIg did not apply in the UK, and thecosts due to infusion materials and hospital charges for IVIg (originally excluded inHogy and colleagues’ study) were applied in the UK costing assumptions. The remain-ing cost parameters (ie, SCIg price, premedication for IVIg patients, and materialsused for SCIg) were applicable and within the reasonable cost range in the UK.
The results of Liu and colleagues’24 study showed that the yearly costs for home-based IVIg and SCIg therapy were similar: £11,580 (IVIg) and £11,760 (SCIg) peryear for adults and £6,540 (IVIg) and £6,720 (SCIg) per year for children. However,the cost of hospital-based IVIg was significantly higher at £18,600 per year. The differ-ences in cost were attributed to hospital services, infusion materials, and treatmentand diagnostic procedures for IVIg patients.

Economic Assessment of Immunoglobulin Replacement Therapy 869
Haddad and colleagues22 compared the monthly costs of treatment between IVIgand SCIg from the perspective of the public payer in France. The comparison involvedsix treatment alternatives: hospital 20 g IVIg, hospital 20 g SCIg, home 20 g IVIg, home20 g SCIg, home 40 g IVIg, and home 40 g SCIg. The costs due to hospital admission,transportation to and from hospital, pre-tax Ig acquisition, homecare nursing, andrental of administration pumps and perfusion kits were included. The treatment inter-vals were set at once per month for IVIg and four times per month for SCIg patients.
The base-case monthly costs of treatment were:
� IVIg 20 g 5 V1,192.19 (hospital) and V1,033 (home)� SCIg 20 g 5 V2,908.76 (hospital) and V1,518 (home)� IVIg 40 g 5 V2,034.50 (home)� SCIg 40 g 5 V2,507–V2,729 (home)
The acquisition and hospital costs for either route of administration were major costdrivers. For example, with IVIg 20 g, approximately 57% of the total treatment cost isdue to the cost of Ig, and 39% is due to hospital charges. For SCIg, approximately30% of the total treatment cost is due to the cost of Ig, and 64% is due to hospitalcharges. When Ig was administered at home (by IV or SC), the IVIg patient had lowercosts associated with the rental of administration pumps and perfusion kits, and IVIgacquisition.
PRIMARY ECONOMIC ASSESSMENT IN CANADA
In the present study, SCIg and IVIg were assumed to yield identical clinical outcomes,as per Chapel and colleagues.23
The base-case results from the CMA for an adult and a child by treatment appear inTables 4 and 5 respectively. These results show that for each treatment arm, Ig pricesaccount for >85% of the total cost of therapy. The results show a cost differencebetween SCIg and hospital-based IVIg therapies of about $2,000 Canadian dollars(C$) each year per patient, when indirect costs are included; and a cost advantagefor home-based IVIg compared with SCIg. The difference is about C$1,400 per yearper patient when indirect costs are excluded.
Table 4Results of adult yearly cost by treatment in Canada
Cost Items SCIgIVIg
Home Based Hospital BasedImmunoglobulin C$19,404.00 C$19,404.00 C$19,404.00
Treatment by physician and nurse NA NA C$1,044.00
Hospital NA NA C$1,328.64
Infusion pump C$525.12 NA NA
Infusion materials C$377.43 C$377.43 NA
Monitoring C$110.00 C$110.00 NA
Total direct treatment C$20,416.55 C$19,891.43 C$21,776.64Transportation NA NA C$126.00
Time lost because of treatment C$616.32 C$410.88 C$1,135.20
Total direct and indirect C$21,032.87 C$20,302.31 C$23,037.84
Abbreviation: NA, not applicable.

Table 5Results of child yearly cost by treatment in Canada
Cost Items SCIgIVIg
Home Based Hospital BasedImmunoglobulin C$11,088 C$11,088 C$11,088
Treatment by physician and nurse NA NA C$1,044.00
Hospital NA NA C$1,328.64
Infusion pump C$525.12 NA NA
Infusion materials C$377.43 C$377.43 NA
Monitoring C$110.72 C$110.72 NA
Total direct treatment C$12,101.27 C$11,576.15 C$13,460.64Transportation NA NA C$126
Time lost because of treatment C$616.32 C$410.88 C$1,135.20
Total direct and indirect C$12,717.59 C$11,987.03 C$14,721.84
Abbreviation: NA, not applicable.
Table 6Base-case parameters, ranges of sensitivity analysis, and rationale
ItemBase-CaseParameters
SensitivityAnalysisRange
Rationalefor Range
Adult’s weight 70 kg 45–95 kg � 25 kg
Child’s weight 40 kg 2–70 kg Infant to adult
Ig monthly dose/kg bodyweight
0.4 g 0.15 g–0.60 g Range reportedfrom PID survey27
IVIg price/g C$57.75 C$50.00–70.00 Current range (CBS)
SCIg price/g C$57.75 C$50.00–70.00 CBS range
Infusion pump (SCIg) cost C$525.12/y C$262.56–787.68 � 50%
Infusion materials cost C$377.43/y C$188.71–566.14 � 50%
Treatment costs (hospitalIVIg)
C$1,044.00/y C$522.00–1,566.00 � 50%
Transportation cost(hospital IVIg)
C$10.50/mo C$5.00–20.00 Bus fare to taxi fare
Monitoring cost (SCIgand home IVIg)
C$55.00/6 mo C$0.00–220.00 0–4/y
Time lost because oftreatment (SCIg)
C$8.56/h(unpaid work)
C$4.28–25.68 0.5–3 h/infusion(once a month)
Hospital charges (IVIg) C$1,328.64/y C$664.32–$1992.96 �50%
Time lost because oftreatment (home IVIg)
C$8.56/h(unpaid work)
C$8.56–51.36 1–6 h/infusion(once a month)
Time lost because oftreatment (hospital IVIg)
C$18.92/h(paid work)
C$18.92–151.36 2–6 hours/infusion
Abbreviation: CBS, Canadian Blood Services (Mathias Haun, personal communication, 2007).
Membe et al870

Yearly Cost difference (SCIg - Hospital IVIg) - adult
-3000 -2050 -2000 -1500 -1000 -500 0Adult body weightPer gram SCIg pricePer gram IVIg priceTime lost for treatmentTransportation costMonitoringInfusion materialsInfusion pumpTreatment/diagnosticHospital charges
Fig.1. Adults (SCIg and hospital IVIg). IVIg, intravenous immunoglobulin; SCIg, subcutaneousimmunoglobulin.
Economic Assessment of Immunoglobulin Replacement Therapy 871
Two factors explain the small cost difference between the two routes. First, the Igacquisition cost that constitutes most of the total costs for each treatment alternativeis similar. Second, for SCIg therapy, the high prices of the pump, monitoring, andinfusion materials offset approximately half the gains from averted hospital chargesand treatment or diagnostic costs.
In the sensitivity analysis, we varied each cost driver to account for uncertainty in theuse of resources for each subgroup. We recorded the resulting incremental cost dif-ferences for each parameter to determine their respective magnitude of influence tothe base-case incremental cost differences. The sensitivity ranges for each parame-ter, with the justification, are tabulated against base-case parameters in Table 6.
The results of the sensitivity analysis are shown in tornado diagrams, depicting theinfluence of each parameter on incremental cost. Tornado diagrams for children arereplicas of those for adults, because the dollar value for each variable (except theper gram price of Ig that is the same for each intervention) is applied to adults andchildren. Except for the first comparison, we display only the adult tornado diagrams.For adults (Fig. 1) and children (Fig. 2), the results of the sensitivity analysis show thatthe yearly incremental cost differences between SCIg and hospital-based IVIg aremore responsive to hospital charges, treatment costs, costs of infusion pumps, andcosts of infusion materials. Overall, the cost differences are not driven by the pergram price of IVIg and SCIg, because they are assumed to be in the same range.
Yearly Cost difference (SCIg - Hospital IVIg) - Child
-3000 -2500 -2000 -1500 -1000 -500 0
Child body weight
Per gram SCIg price
Per gram IVIg price
Time lost for treatment
Transportation cost
Monitoring
Infusion materials
Infusion pump
Treatment/diagnostic
Hospital charges
Fig. 2. Children (SCIg and hospital IVIg). IVIg, intravenous immunoglobulin; SCIg, subcutane-ous immunoglobulin.

Yearly cost difference (home IVIg - hospital IVIg)
-4000 -3500 -3000 -2500 -2000 -1500 -1000 -500 0
Adult body weight
IVIg price
Treatment/diagnostic
Transportation cost
Monitoring
Infusion materials
Time lost for treatment
Hospital charges
Yearly cost difference (home IVIg -SCIg)
Bodyweight
-1200 -1000 -800 -600 -400 -200 0
Infusion materials
Monitoring
Ig price
Infusion pump
Time lost for treatment
A
B
Fig. 3. (A) Yearly incremental cost difference (home IVIg–hospital IVIg). (B) Yearly cost differ-ence (home IVIg–SCIg).
Membe et al872
Hospital charges, time lost because of treatment, and infusion materials are majordrivers of the cost differences between home-based IVIg and hospital-based IVIg(Fig. 3A). The yearly incremental cost differences between SCIg and home-basedIVIg are responsive to the cost of infusion pumps and time lost because of treatment.The per gram prices of SCIg and IVIg, infusion materials, body weight, and monitoringwere assumed to be in the same sensitivity range between the two interventions.Therefore, they do not influence the cost differences (Fig. 3B).
We have shown here that differences in cost effectiveness between hospital-basedIVIg and home based IVIg or SCIg are modest. Home IVIg appeared to be the poten-tially most cost saving Ig route of administration. Home SCIg, although to a lesser de-gree, could also save costs when compared with hospital IVIg. Future studies shoulddetermine the safety and long term efficacy of home treatment.
SUMMARY
In spite of major differences in the health care systems in various countries, somecommon themes emerged. First, because the cost of Ig constitutes the lion’s shareof this therapy, lower costs of SCIg in some countries influenced cost analysis studiesin favor of SCIg when compared with IVIg. Second, if SCIg and IVIg costs are compa-rable, then home-based treatment appears more cost effective than hospital-basedtherapy. The extent of this advantage varies among countries.

Economic Assessment of Immunoglobulin Replacement Therapy 873
In Canada the CMA show only minimal cost differences between the two treat-ments. This is attributed to the parity pricing per gram of SCIg and IVIg in this country,and the fact that the acquisition cost of Ig made up the largest proportion of total costsof either treatment. In our analysis, the cost differences resulted from differences be-tween the sums of transportation costs, hospital charges, and treatment or diagnosticcharges for hospital-based IVIg and infusion pumps, infusion materials, and monitor-ing for SCIg. As a result, we found the cost difference between treatments to be small,because the costs due to the materials required for SCIg infusion offset most of thegains from the avoidance of hospital and treatment or diagnostic charges. As a result,home-based IVIg shows a larger net gain from the avoidance of hospital charges.
REFERENCES
1. Cooper MD, Schroeder HW. Primary immune deficiency diseases. In: FD,Kasper DL, Fauci SA, editors. Harrison’s principles of internal medicine. 16th edi-tion. New York: McGraw-Hill; 2005. p. 1939–47.
2. Cohn EJ, Strong LE, Hughes WL, et al. Preparation and properties of serum andplasma proteins. IV: a system for the separation into fractions of protein and lipo-protein components of biological tissues and fluids. J Am Chem Soc 1946;68:459–75.
3. Cohn E. The separation of blood into fractions for therapeutic value. Ann InternMed 1947;26:341–52.
4. MacLennan S, Barbara JA. Risks and side effects of therapy with plasma andplasma fractions. Best Pract Res Clin Haematol 2006;19(1):169–89.
5. Roifman CM, Lederman HM, Lavi S, et al. Benefit of intravenous IgG replacementin hypogammaglobulinemic patients with chronic sinopulmonary disease. AmJ Med 1985;79(2):171–4.
6. Rousell RH, Pennington JE. An historical overview of immunoglobulin therapy. In:Yap PL, editor. Clinical applications of intravenous immunoglobulin therapy. NewYork: Churchhill Livingstone; 1992. p. 1–15.
7. Nowak-Wegrzyn A, Lederman HM. Supply, use, and abuse of intravenous immu-noglobulin. Curr Opin Pediatr 1999;11(6):533–9.
8. Roifman CM, Levison H, Gelfand EW. High-dose versus low-dose intravenous im-munoglobulin in hypogammaglobulinaemia and chronic lung disease. Lancet1987;1(8541):1075–7.
9. Stiehm ER. Appropriate therapeutic use of immunoglobulin. Transfus Med Rev1996;10(3):203–21.
10. Roifman CM, Balter M, Blanchette V, et al. The Consensus Working Group. Pres-ent and future uses of intravenous immune globulin (IVIG): a Canadian multidis-ciplinary consensus-building initiative. Toronto: Maclean Hunter Healthcare/Sante; 1997.
11. Hanna K, et al. Intravenous immune globulin use in Canada. Can J Clin Pharma-col 2003;10(1):11–6.
12. Lemieux R, Bazin R, Neron S. Therapeutic intravenous immunoglobulins. MolImmunol 2005;42(7):839–48.
13. Lederman HM, Roifman CM, Lavi S, et al. Corticosteroids for prevention ofadverse reactions to intravenous immune serum globulin infusions in hypogam-maglobulinemic patients. Am J Med 1986;81(3):443–6.
14. Gardulf A, Hammarstrom L, Smith CI. Home treatment of hypogammaglobulinaemiawith subcutaneous gammaglobulin by rapid infusion. Lancet 1991;338(8760):162–6.

Membe et al874
15. Gardulf A, Nicolay U, Asensio O, et al. Rapid subcutaneous IgG replacementtherapy is effective and safe in children and adults with primary immunodefi-ciencies—a prospective, multinational study. J Clin Immunol 2006;26(2):177–85.
16. Schleis T. SJ Formulary considerations for IGIV products. East Rutherford (NJ):U.S. Pharmacist; 2005.
17. Schulz KF, Chalmers I, Hayes RJ, et al. Empirical evidence of bias. Dimensions ofmethodological quality associated with estimates of treatment effects in con-trolled trials. JAMA 1995;273(5):408–12.
18. Berger M. Subcutaneous immunoglobulin replacement in primary immunodefi-ciencies. Clin Immunol 2004;112(1):1–7.
19. Helbert M, Farragher A. Subcutaneous immunoglobulin for patients with antibodydeficiency. Br J Hosp Med (Lond) 2007;68(4):206–10.
20. Hogy B, Keinecke HO, Borte M. Pharmacoeconomic evaluation of immunoglob-ulin treatment in patients with antibody deficiencies from the perspective of theGerman statutory health insurance. Eur J Health Econ 2005;6(1):24–9.
21. Gardulf A, Andersen V, Bjorkander J, et al. Subcutaneous immunoglobulinreplacement in patients with primary antibody deficiencies: safety and costs.Lancet 1995;345(8946):365–9.
22. Haddad L, Perrinet M, Parent D, et al. �Etude comparative du cout du traitement �adomicile des immunoglobulines intraveineuses ou sous-cutan�ees a vis�ee substi-tutive. Rev Med Interne 2006;27(12):924–6.
23. Chapel HM, Spickett GP, Ericson D, et al. The comparison of the efficacy andsafety of intravenous versus subcutaneous immunoglobulin replacement therapy.J Clin Immunol 2000;20(2):94–100.
24. Liu Z, Albon E, Hyde C. The effectiveness and cost effectiveness of immunoglob-ulin replacement therapy for primary immunodeficiency and chronic lymphocyticleukaemia: a systematic review and economic evaluation. Birmingham (UK):Department of Public Health and Epidemiology, University of Birmingham;2005.
25. Ontario College of Family Physicians. Babies can’t wait - Ontario College ofFamily Physicians project summary. Available at: http://meds.queensu.ca/prn/downloads/OMCEP%20App_H_to_I.pdf. Accessed May 30, 2007.
26. Jacobs P, Assiff L, Bachynsky J, et al. A national list of provincial costs forhealth care: Canada 1997/8. Version 1.0. Edmonton (Canada): Institute of HealthEconomics; 2000.
27. Treatment experiences and preferences of patients with primary immune defi-ciency diseases: first national study. Available at: http://www.primaryimmune.org/pubs/IDF_survey_complete.pdf. Accessed August 2, 2007.
28. Statistics Canada. Payroll employment, earnings and hours. The Daily. December21, 2006. Available at: http://www.statcan.ca/Daily/English/061221/d061221c.htm. Accessed June 7, 2007.
29. Stephens T, Joubert N. The economic burden of mental health problems inCanada. Chronic Dis Can 2001;22(1):18–23.
30. Norfolk Medical [price catalog]. Skokie (IL): Norfolk Medical Products, Inc.; 2005.

Management ofPrimary AntibodyDefic iency withReplacement Therapy :Summary of Guidelines
ChaimM. Roifman, MD, FRCPCa,*, Melvin Berger, MD, PhDb,Luigi D. Notarangelo, MDc
KEYWORDS
� Primary antibody deficiency � IgG replacement� Intravenous immunoglobulin
In managing primary antibody deficiency with replacement therapy, the followingguidelines should be observed:
1. All patients should be referred to an immunologist for diagnosis and initiation oftreatment. Long-term management can be shared with family physicians andpediatricians.
2. The following investigations should be performed as appropriate in individualcases:
a DivHospb JefRainbc Har* CorE-ma
Immudoi:10889
Blood cell count and differentialSerum immunoglobulinsAntibody titers to immunizations with protein antigens, as well as polysaccharide
antigens, (at age greater than 24 months)Polymerase chain reaction assay for HIV and other chronic viral infections
(hepatitis B and C)Lymphocyte surface marker analysisLymphocyte function tests such as mitogen and antigen proliferationLiver and renal function testsAssessment of end-organ damage may be required includingLung function
ision of Immunology and Allergy, Department of Paediatrics, The Research Institute ofital for Sick Children, 555 University Avenue, Toronto, Ontario, Canada M5G 1X8frey Modell Center for Primary Immune Deficiencies, Division of Allergy-Immunology,ow Babies and Children’s Hospital, University Hospitals of Cleveland, Cleveland, OH, USAvard Medical School, Division of Immunology, Children’s Hospital, Boston, MA, USAresponding author.il address: [email protected] (C.M. Roifman).
nol Allergy Clin N Am 28 (2008) 875–8760.1016/j.iac.2008.07.003 immunology.theclinics.com-8561/08/$ – see front matter ª 2008 Elsevier Inc. All rights reserved.

Roifman et al876
CT scans of lung and sinusesGastrointestinal endoscopyBone marrow and/or liver biopsyDetailed mutation and additional molecular analyses of the immunologic defect
should be considered when appropriate.
3. Patients who have significant antibody deficiency require doses of intravenous im-munoglobulin (IVIG) between 0.4 and 0.8 g/kg every 3 to 4 weeks (or the equivalentgiven in divided doses once or twice a week subcutaneously) to achieve trough IgGserum levels of at least 5 g/L (ideally, 6.5–10 g/L). If IgG half-life is shorter than3 weeks and/or if treatment effects are not satisfactory, the frequency of infusionsmay be increased to every 2 weeks and/or the dose may be increased.
4. In patients who have significant antibody deficiency, IgG replacement should begiven regularly and should not be interrupted. IVIG should not be given intermit-tently because effective serum IgG trough levels may drop to nonprotective levelson cessation of treatment.
5. The first three infusions ideally should be given in a qualified center for monitoringof severe adverse reactions. Subsequent infusions can be administered by familyphysicians in community hospitals or at home by homecare nurses.
6. After replacement therapy has been established, pretreatment serum IgG troughlevels should be obtained monthly and should be followed by an immunology spe-cialist at least every 6 months to monitor treatment effectiveness and complica-tions. Individual patients may require more frequent visits with the immunologistand/or other specialists.
7. If a patient experiences severe adverse reactions to a licensed IVIG product,a different brand of IVIG or subcutaneous IgG should be tried. If adverse reactionspersist, premedication with corticosteroids, antihistamines, and/or antipyreticsfrequently is effective. Slowing the infusion rate should alleviate minor reactions.If it has been established that a patient tolerates only certain brands, the brand ofIgG given to that patient should not be changed without obtaining permission fromthe responsible immunologist.
8. Subcutaneous IgG should be offered as substitute for IVIG for patients who havepoor intravenous access. Because subcutaneous immunoglobulin can be self-ad-ministered safely at home, it has been offered as an alternative for selected patients.
9. Patients who have developed chronic lung disease (bronchiectasis) should re-ceive special attention. The minimal monitoring regimen is yearly assessment in-cluding a pulmonary function test and biannual CT scans. Aggressive antibiotictreatment is needed during acute exacerbations of lung disease. Prolonged orcontinuous antibiotic treatment may be helpful in chronically infected patients,and these patients often need adjuvant therapies such as physiotherapy, bron-chodilators/inhaled corticosteroids, and percussion devices to promote pulmo-nary toilet. If bronchiectasis is localized and cannot be controlled adequatelywith IVIG and antibiotics, a lobectomy should be considered.
10. Indications for IgG replacement include common variable immunodeficiency,X-linked agammaglobulinemia, autosomal-recessive agammaglobulinemia, hyper-IgM syndromes, dysgammaglobulinemia, or antibody deficiencies associated withsyndromes such as Wiskott Aldrich syndrome, ataxia telangiectasia, and others.
It is recommended that analysis and interpretation be performed by immunologistsin immunology service laboratories who have the experience, knowledge, and skill toperform DNA sequencing, protein immunoblotting, and, ideally, analysis of the func-tional pathway of the target gene.