inactivation of sodium channel scn8a (na 1.6) in purkinje
TRANSCRIPT

Inactivation of Sodium Channel Scn8A (Nav1.6) in Purkinje NeuronsImpairs Learning in Morris Water Maze and Delay but Not Trace Eyeblink
Classical Conditioning
Diana S. Woodruff-PakTemple University and Albert Einstein Healthcare Network
John T. GreenUniversity of Vermont
Stephen I. Levin and Miriam H. MeislerUniversity of Michigan School of Medicine
To examine the isolated effects of altered currents in cerebellar Purkinje neurons, the authors usedScn8aflox/flox, Purkinje cell protein-CRE (Pcp-CRE) mice in which Exon 1 of Scn8a is deleted only inPurkinje neurons. Twenty male Purkinje Scn8a knockout (PKJ Scn8a KO) mice and 20 male littermateswere tested on the Morris water maze (MWM). Subsequently, half were tested in 500-ms delay and halfwere tested in 500-ms trace eyeblink conditioning. PKJ Scn8a KO mice were impaired in delayconditioning and MWM but not in trace conditioning. These results provide additional support for thenecessary participation of cerebellar cortex in normal acquisition of delay eyeblink conditioning andMWM and raise questions about the role, if any, of cerebellar cortex in trace eyeblink conditioning.
Keywords: place learning, cerebellar cortex, interpositus nucleus, cerebellum-dependent learning,hippocampus-dependent learning
The cerebellar cortex and hippocampus are normally activatedin delay eyeblink classical conditioning (reviewed in Thompson’s,2005, study), but hippocampal lesions do not prevent acquisitionor retention in the delay paradigm (Schmaltz & Theios, 1972).Hippocampal lesions block acquisition and retention in trace eye-blink conditioning (Kim, Clark, & Thompson, 1995; Moyer, Deyo,& Disterhoft, 1990; Solomon, Vander Schaaf, Thompson, &Weisz, 1986). Cerebellar cortical aspirations caused impairment intrace conditioning that was overcome with postlesion presentationsof additional trials (Woodruff-Pak, Lavond, & Thompson, 1985).These results with physical lesions led us to conclude that cere-bellar cortex supported the normal rate of acquisition and retentionin trace eyeblink conditioning. Thus, the perspective has been thatthe hippocampus plays different roles in delay and trace eyeblink
conditioning, but the cerebellar cortex supports the normal rate ofacquisition in both paradigms (Green & Woodruff-Pak, 2000).Recent studies in mice suggest that the cerebellar cortex, like thehippocampus, plays different roles in delay and trace eyeblinkconditioning (Kishimoto, Hirono, et al., 2001; Kishimoto, Kawa-hara, et al., 2001). The cerebellar cortex is critical for normalacquisition in delay, but cerebellar cortical involvement in traceeyeblink conditioning is less well delineated. Spatial learning inthe Morris water maze also requires coordinated action of severalbrain regions, including the cerebellar cortex and hippocampus. Inthis study, we tested mice created with gene targeting to haveselective disruption of sodium channels in cerebellar cortical Pur-kinje neurons to examine the contribution of cerebellar cortex indelay and trace eyeblink classical conditioning and in the Morriswater maze.
Delay and Trace Eyeblink Classical Conditioning andAssociated Circuitry
In the delay classical conditioning procedure, a neutral stimulusserves as the conditioned stimulus (CS). The CS is presented forabout half a second before the onset of a reflex-eliciting stimuluscalled the unconditioned stimulus (US). In eyeblink conditioning,the US is a corneal air puff or stimulation to the obicularis oculimuscles that causes an eyeblink. Organisms learn to blink to theCS before the onset of the US, and the learned response is calledthe conditioned response (CR). In the trace classical conditioningprocedure, the CS does not overlap the US. The CS onsets, then itis turned off, and a blank “trace” period ensues before the onset ofthe US.
The dorsolateral interpositus nucleus ipsilateral to the condi-tioned eye is the essential site for acquisition and retention of CRs
Diana S. Woodruff-Pak, Department of Psychology, Temple University,and Albert Einstein Healthcare Network, Philadelphia; John T. Green,Department of Psychology, University of Vermont; Stephen I. Levin,Department of Human Genetics and Unit for Laboratory Animal Medicine,University of Michigan School of Medicine; Miriam H. Meisler, Depart-ment of Human Genetics, University of Michigan School of Medicine.
This research was supported by National Institutes of Health GrantsAG21925 (awarded to Diana S. Woodruff-Pak) and NS34509 (awarded toMiriam H. Meisler) as well as a grant from the Albert Einstein Society(awarded to Diana S. Woodruff-Pak). Stephen I. Levin acknowledgessupport from University of Michigan Training Program for Veterinariansin Biomedical Research Grant NIH T32 RR07008. We are grateful to MaxChae for testing the mice.
Correspondence concerning this article should be addressed to Diana S.Woodruff-Pak, Department of Psychology, Temple University, 1701 North13th Street, Philadelphia, PA 19122. E-mail: [email protected]
Behavioral Neuroscience Copyright 2006 by the American Psychological Association2006, Vol. 120, No. 2, 229–240 0735-7044/06/$12.00 DOI: 10.1037/0735-7044.120.2.229
229

in both delay and trace eyeblink classical conditioning (Bracha,2004; Christian & Thompson, 2003; Medina, Nores, Ohymama, &Mauk, 2000; Steinmetz, 2000; Thompson, 1986; Woodruff-Pak etal., 1985). Electrophysiological recording of single- and multiple-unit activity in the cerebellum indicated that Purkinje neurons incerebellar cortex and principle cells in interpositus nucleus indiscrete regions of the cerebellum undergo learning-inducedchanges during delay eyeblink conditioning (Green & Steinmetz,2005; McCormick & Thompson, 1984; Thompson, 1990). It hasbeen demonstrated for delay and assumed to hold true for trace thatthe cerebellar cortex as well as the interpositus nucleus use infor-mation received about the CS, conveyed by the mossy fiber systememanating from the pontine nucleus, and information about theUS, relayed by the climbing fiber system originating from theinferior olive. Converging CS and US signals relayed to Purkinjeneurons in cerebellar cortex and principle cells in interpositusnucleus support normal acquisition.
Acquisition of trace eyeblink conditioning requires a coordi-nated network of brain substrates and neurotransmitter systems(e.g., Harvey, Quinn, Liu, Aloyo, & Romano, 2003; Mamounas,Thompson, & Madden, 1987). Extensive evidence supports anessential role for the hippocampus in trace eyeblink conditioningin several species (Clark & Squire, 1998; Kim et al., 1995; Moyeret al., 1990; Solomon et al., 1986; Weiss, Bouwmeester, Power, &Disterhoft, 1999), including mice (Sakamoto et al., 2005; Tseng,Guan, Disterhoft, & Weiss, 2004). There is also some evidencethat medial prefrontal cortex plays a role in acquisition and reten-tion (Kronforst-Collins & Disterhoft, 1998; Powell, Skaggs,Churchwell, & McLaughlin, 2001; Weible, McEchron, & Dister-hoft, 2000). However, these structures play an essential role atdifferent points of time after acquisition (Takehara, Kawahara, &Kirino, 2003). The medial prefrontal cortex, hippocampus, andcerebellum are essential in the mediation of initial acquisition, butas days and weeks elapse after acquisition, the neural circuitry isreorganized to use primarily the medial prefrontal cortex andcerebellar interpositus nucleus (Kim et al., 1995; Takehara et al.,2003).
Morris Water Maze and Associated Circuitry
The Morris water maze task was devised as a means to assessspatial learning and memory in rodents (Morris, Garrud, Rawlins,& O’Keefe, 1982). Animals are introduced into a pool of opaquewater from various positions and are trained to escape the coolwater by finding a hidden (submerged) platform. Reference todistal extramaze cues enables the animals to learn to escape byfinding and crawling onto the platform. A cued condition in whichthe hidden platform is made visible serves as a control for sensoryor motor deficits. Whereas the hippocampus is essential for acqui-sition in the Morris water maze, place learning in the water mazecan be accomplished without corticohippocampal circuitry, includ-ing the perirhinal, postrhinal, and entorhinal cortices (Burwell,Saddoris, Bucci, & Wiig, 2004). However, lesions in distinct brainregions outside the hippocampus—such as the striatum, basalforebrain, cerebellum (D’Hooge & De Deyn, 2001), and pontineand inferior olive cerebellar afferents (Gasbarri, Pompili, Pacitti, &Cicirata, 2003)—impair place learning in this task.
Impairment in the hidden platform condition with normal per-formance on the cued platform condition has been observed in
rodents with genetic and nongenetic lesions of the cerebellum.Cerebellar mutant mice (e.g., Lurcher, Purkinje cell degeneration[pcd], nervous), rats with lesions of either the lateral cerebellarcortex or dentate nucleus, and rats with selective Purkinje celllesions caused by intracerebroventricular injections of OX-7-saporin are impaired in place but not in cued learning (reviewed inLalonde & Strazielle’s, 1999, study).
Genetic Lesions in Mice
Physically introducing a lesion into the cerebellum throughaspiration, electrolytic, or chemical means normally introducessome collateral brain damage, and it is difficult to emulate themagnitude of the lesion with control mice. In addition, physicallesions are challenging to make in an identical fashion to eachanimal. Brain lesions in mutant mice are selective, but they arerarely “pure.” The mutants typically have additional central ner-vous system impairments that could affect performance on learn-ing and memory tasks.
Gene targeting in mice introduces a means to achieve greaterprecision in brain lesions. In the case of a conditional knockout,ablation of gene function can be restricted to certain tissue or celltypes and specific developmental stages. One means to alter brainfunction is to target neuronal firing patterns via ion channels. Afamily of 10 sodium channel genes encodes voltage-gated sodiumchannels that are responsible for generating the rising phase ofneuronal action potentials (Meisler, Kearney, Escayg, MacDonald,& Sprunger, 2001). The sodium channel protein Nav1.6 is encodedby Scn8a, a sodium channel gene that is widely expressed inneurons throughout the central and peripheral nervous system(Burgess et al., 1995; Schaller, Krzemien, Yarowsky, Krueger, &Caldwell, 1995). Nav1.6 is also the primary sodium channel at thenodes of Ranvier in myelinated axons (Caldwell, Schaller, Lasher,Peles, & Levinson, 2000).
To examine the isolated effects of altered currents in cerebellarcortical Purkinje neurons, we used the recently described floxedallele of Scn8a (Levin & Meisler, 2004b). In Scn8aflox/flox, Pur-kinje cell protein-CRE (Pcp-CRE) mice, Exon 1 of Scn8a isdeleted specifically in Purkinje neurons (Levin, 2004). These miceare called Purkinje Scn8a knockout (PKJ Scn8a KO) mice becausethe Scn8a gene that codes for the sodium channel protein Nav1.6is knocked out selectively in Purkinje neurons. The elimination ofthe sodium channel protein Nav1.6 in Purkinje neurons results in aselective impairment of subthreshold sodium currents, 10-foldreduction in spontaneous firing, and halved maximal firing rates ofPurkinje neurons (Levin et al., 2005). The lesion is restricted to theScn8a sodium channel in Purkinje neurons and does not affectother cerebellar or noncerebellar neurons.
Purkinje neurons provide the only cerebellar cortical input todeep cerebellar nuclei, including the interpositus nucleus. Our aimin the current study was to determine whether reduced Purkinjeneuron firing would affect acquisition in delay and trace eyeblinkclassical conditioning and in the Morris water maze. The hip-pocampus is known to be essential in the Morris water maze, andthere are data from several species demonstrating a role for cere-bellar cortex and Purkinje neurons. The hippocampus is alsoessential in trace eyeblink classical conditioning. The limited datathat are available have been interpreted to indicate that the role ofcerebellar cortex is similar in trace and delay eyeblink condition-
230 WOODRUFF-PAK, GREEN, LEVIN, AND MEISLER

ing. The specific question to be addressed is the role of cerebellarcortex in delay versus trace eyeblink classical conditioning andMorris water maze using mice with a genetic lesion that selectivelyimpairs sodium channels in Purkinje neurons.
Method
Subjects
A total of 40 male mice were tested: 20 were PKJ Scn8a KO mice, and20 were wildtype littermates. All mice were generated, genotyped, andmaintained at the University of Michigan School of Medicine (Levin,2004; Levin & Meisler, 2004a, 2004b). At the age of 4 months, they wereshipped to Philadelphia and maintained in the Central Animal Facility atAlbert Einstein Medical Center. PKJ Scn8a KO mice were mildly ataxic,but the deficit was so subtle that animal testers at Einstein could notreliably discriminate the mice by phenotype. Thus, the animal testers wereblind to the genotype of the mice. The colony room was temperature andhumidity controlled and ventilated with a dedicated system. Room lightingwas timed for a 12:12-hr light–dark cycle. Housing consisted of a poly-carbonate microisolator filtered-top cage. All mice had ad libitum access tosterile food (PMI autoclavable rodent lab diet RHI5010) and water. Thisresearch was approved by Albert Einstein Medical Center’s InstitutionalAnimal Care and Use Committee.
Training Sequence
All mice were trained and tested over the same sequence during a periodof 5 weeks. Ten wildtype and 10 PKJ Scn8a KO mice were in tested in thefirst 5-week series, and the sequence was repeated a second time with 10additional wildtype and 10 additional PKJ Scn8a KO mice. All mice weretrained first on the Morris water maze. Next, mice received surgery toimplant stimulating and recording electrodes for eyeblink classical condi-tioning. After recovery from this surgery, half of the wildtype and half ofthe PKJ Scn8a KO mice in the first group were tested for 10 daily sessionsin the 500-ms delay procedure, and the other half were tested in the 500-mstrace procedure. Five additional days of extinction training followed. Thisseries was repeated a second time so that 40 mice (20 wildtype, 20 PKJScn8a KO) were trained in the Morris water maze, and 20 mice (10wildtype, 10 PKJ Scn8a KO) were trained in either 500-ms delay or500-ms trace eyeblink conditioning followed by extinction training.
Morris Water Maze
Apparatus. The training apparatus was a circular pool that had adiameter of 100 cm and was 60 cm deep. The pool was located in alaboratory room that contained camera and computer equipment, a portablepartition to reduce the viewing area, and various visual cues. The interiorof the pool was painted white. The water temperature was maintainedbetween 20 °C and 26 °C, and the depth was 16 cm. White nontoxic paintwas used to make the water opaque. The hidden platform was an 11-cmsquare white tile platform positioned 1 cm below the surface of the water.The same platform was used for the visible platform, and it was marked bya white flag (10 cm � 7 cm) suspended 15 cm above it on a wooden stick.
General procedures. Training in the Morris water maze took place onDays 1–5. Around the test room in close proximity to the pool weremultiple cues such as artificial plants and flowers and graphic prints.Computer and camera equipment used to record the session were alsovisible to the mouse. Each trial was initiated by placing the mouse in thewater at the edge of the pool in a quadrant either opposite or adjacent to thequadrant containing the platform. The start locations were varied amongthe three quadrants not containing the platform, with three different startlocations being used in each block of four trials. The platform remained inthe same location on every trial during the hidden platform task and varied
across the four quadrants in the visible platform task. Each trial lasted 120 sor until the subject located the platform. Subjects that did not find theplatform were guided to it, placed on it, and given a latency score of 120 s.Whether the platform was located or not, each mouse was required to spend15 s on the platform at the end of each trial. Between blocks of four trials,the mice were placed in individually heated, terry cloth lined plasticholding cages for at least 30 min.
Hidden platform training. Each subject was given three blocks of fourtrials each (12 trials/day/mouse) for 3 consecutive days of training. Micewere returned to the holding cage between blocks. On the 4th training day,the subjects were given a probe trial, in which the platform was removedfrom the pool. After swimming for 120 s, the mouse was removed from thepool and returned to its holding cage. Time in each quadrant of the poolwas scored. We videotaped and recorded the training trials and the probetrial using the Spontaneous Motor Activity Recording and Tracking pro-gram manufactured by Panlab (Barcelona, Spain).
Time to reach the platform (latency to escape) was recorded for eachtrial and averaged for each block of four trials. The probe trial wasanalyzed to measure the amount of time spent in each quadrant and thenumber of crossings made over the platform location in the trained quad-rant and the equivalent area in the untrained quadrants.
Visible platform training. On the 5th day of training, all mice weregiven the visible platform task. A flag attached to the platform made itvisible. Training was the same as in the hidden platform version except thelocation of the platform and the start position were varied across trials. Thelatency to escape was recorded.
Eyeblink Classical Conditioning
Surgery. Twenty PKJ Scn8a KO and 20 wildtype littermate micereceived surgery to implant recording and stimulating electrodes for eye-blink classical conditioning. Surgery occurred during the week after micecompleted training in the Morris water maze. For anesthesia, a nonre-breathing Isoflurane administration system was used. Anesthesia was in-duced in a chamber, and two surgical platforms were used so that multiplesurgeries could be performed simultaneously. Anesthesia was induced withO2 � 3% Isoflurane at a flow rate of 1 L per minute. The mouse wasintroduced to the induction chamber and was in a surgical plane ofanesthesia within 1 min. The Isoflurane was then reduced to 2.5% as themouse was placed on a surgical platform and fitted with a nose cone foranesthesia maintenance throughout the procedure. Opthalmic ointment wasapplied to each eye to prevent drying, and mice were covered with gauzestrips to maintain normal thermoregulation. Four Teflon-coated stainlesssteel wires (0.003-in bare, 0.0045-in coated; A-M Systems, Everet, Wash-ington), soldered to a four-pin male header (Jameco Electronics, Belmont,California), were implanted intramuscularly in the obicularis oculi of theleft upper eyelid. Wires were stripped of Teflon and carefully placed suchthat only the muscle-embedded wire was bare. To ensure that the wires didnot move or recede back into the periorbital cavity, we glued the wires tothe skull. The two wires most rostral were used to record differentialelectromyography (EMG) activity, and the two most caudal were used todeliver the eyeblink-eliciting stimulus. When all wires were placed, thefour-pin header (headstage) was cemented to the skull and the incision wasclosed. Following surgery, mice were given Baytril antibiotic (85 mg/kgsc) to prevent infection and Buprenex (0.075 mg/kg sc) for analgesia.Recovery from surgery took place for a minimum of 5 days.
Eyeblink conditioning procedure. The conditioned eyeblink trainingapparatus consisted of four sound- and light- attenuating chambers (MedAssociates, St. Albans, Vermont). Each chamber contained a beaker inwhich the mouse was placed, a copper Faraday cage covering the beaker,a ventilation fan, and a wall-mounted speaker. A shielded four-conductorwire was attached to the mouse’s headstage and was used to deliver ablink-eliciting stimulus to the obicularis oculi and to record EMG activity.EMG activity was passed through a 300–5,000 Hz filter and amplified by10 K. The signal was then integrated and digitized before being read into
231LEARNING IN PURKINJE SODIUM CHANNEL KNOCKOUT MICE

a system compatible with IBM (White Plains, New York) described by G.Chen and Steinmetz (1998) for processing. Data were collected in RAMand saved to a hard drive for offline analyses.
Delay and trace eyeblink classical conditioning took place on Days11–20 and extinction in eyeblink conditioning took place on Days 21–25.The stimulus onset asynchrony between the CS and US was identical forthe delay and trace paradigms and was 500 ms. The white noise CS andobicularis oculi stimulation US were the same and generated by the sameequipment for delay and trace conditioning. The only difference betweenthe two paradigms was the duration of the CS that was 600 ms in delay and250 ms in trace. Each training session was controlled by a program writtenin C�� language (G. Chen & Steinmetz, 1998) and run on an IBM-PCcompatible 386 computer. The intertrial interval was random, ranging from15 s to 30 s at 1-s intervals. Mice were tested in groups of four. Eachsession lasted approximately 1 hr. Mice were allowed to move freelyaround the cage during testing. The ventilation fan remained on andgenerated a 70-dB background noise. There were 100 trials presented inblocks of 10. For the 10 daily acquisition sessions, each block consisted of9 paired trials and 1 CS-alone test trial. For delay eyeblink conditioning,paired trials included a 600-ms, 85-dB, 1-kHz tone CS, followed 500 msafter its onset by a 100-ms 0.5 mA stimulation US. For trace eyeblinkconditioning, paired trials included a 250-ms, 85-dB, 1-kHz tone CS,followed 500 ms after its onset (and 250 ms after its offset) by a 100-ms0.5 mA stimulation US. The trace interval was 250 ms—a trace intervaldemonstrated to be hippocampus dependent (Tseng et al., 2004). It wasdetermined by observation that a 0.5 mA stimulus was sufficient to causea blink/head jerk in all mice. For the five daily extinction trials, only the600-ms delay CS or the 250-ms trace CS was presented. Thus, for delaythere were 100 trials of a 600-ms, 85-dB, 1-kHz tone CS with no USpresentation, and for trace there were 100 trials of a 250-ms, 85-dB, 1-kHztone CS with no US presentation.
Each session was computer-scored with a macro written in Visual Basic,which analyzed each trial individually for responses. Whenever EMGactivity in the obicularis oculi exceeded five standard deviations abovebaseline, a response was considered to have occurred. If a response tookplace in the first 100 ms prior to the CS onset, then the trial was excludedas a bad trial. For each session, several variables were observed. A startlewas scored if the response occurred in the first 60 ms after the CS onset.A CR was scored if a response occurred after the 60 ms startle period andbefore the US onset 500 ms after CS onset. An unconditoned response wasscored if no response occurred prior to the US onset.
Results
Delay Eyeblink Classical Conditioning
In comparison with wildtype littermates, PKJ Scn8a KO miceproduced fewer CRs and exhibited lower CR amplitude. Afterseven acquisition sessions, a mouse with midrange performanceamong PKJ Scn8a KO mice had 16.7% CRs with a CR amplitudeof 0.40 units/bin (see Figure 1A), whereas a mouse with midrangeperformance among wildtype littermate mice had 75.6% CRs witha CR amplitude of 4.01 units/bin (see Figure 1B). Comparisons ofmean group performance demonstrated that acquisition of CRsdiffered in wildtype and PKJ Scn8a KO mice (see Figure 2A). A2 (Genotype) � 10 (Training Sessions) repeated measures analysisof variance (ANOVA) with percentage of CRs as the dependentvariable indicated a nonsignificant main effect of genotype, asignificant main effect of training sessions, F(9, 162) � 7.53, p �.0001, and a significant interaction between genotype and trainingsessions, F(9, 162) � 2.05, p � .037. Examination of the signif-icant Genotype � Training Sessions interaction with t tests tocompare the individual session means indicated that wildtype
littermate mice produced significantly more CRs in Sessions 3, 7,8, 9, and 10.
Using CR amplitude as the dependent variable, we found similarresults for training sessions, F(9, 162) � 5.57, p � .0001, andinteraction, F(9, 162) � 2.02, p � .040, as well as a nonsignificant
Figure 1. Delay eyeblink classical conditioning in Purkinje Scn8a knock-out (PKJ Scn8a KO) mice. A and B: Electromyography recorded from eyemuscles (obicularis oculi) of the upper left eyelid averaged by blocks ofnine paired tone conditioned stimulus (CS)–unconditioned stimulus (US)trials for 10 blocks (90 trials) in Session 7 of 500-ms delay eyeblinkclassical conditioning. Each point called a “bin” represents 3 ms, andnumbers are shown in bins rather than in milliseconds. Total trial lengthwas 1,350 ms. Lines are drawn to indicate the onset of the CS and US.There were 249 ms in the pre-CS period before CS onset. CS onset ismarked, and then there were 500 ms between CS onset and US onset(marked). There were 601 ms in the post-US period. A response was scoredif it exceeded mean pre-CS activity by five standard deviations. Perfor-mance is shown for a PKJ Scn8a KO mouse (A) and a wildtype littermatemouse (B) that scored in the middle of their respective groups. For bothmice, all 90 trials were good trials and were scored. For the PKJ Scn8a KOmice, there were 16.7% conditioned responses (CRs). Short-latency alpharesponses (0–60 ms) occurred in 6.7% of the trials. For the wildtypelittermate mouse there were 75.6% CRs. Short-latency alpha responsesoccurred in 14.4% of the trials.
232 WOODRUFF-PAK, GREEN, LEVIN, AND MEISLER
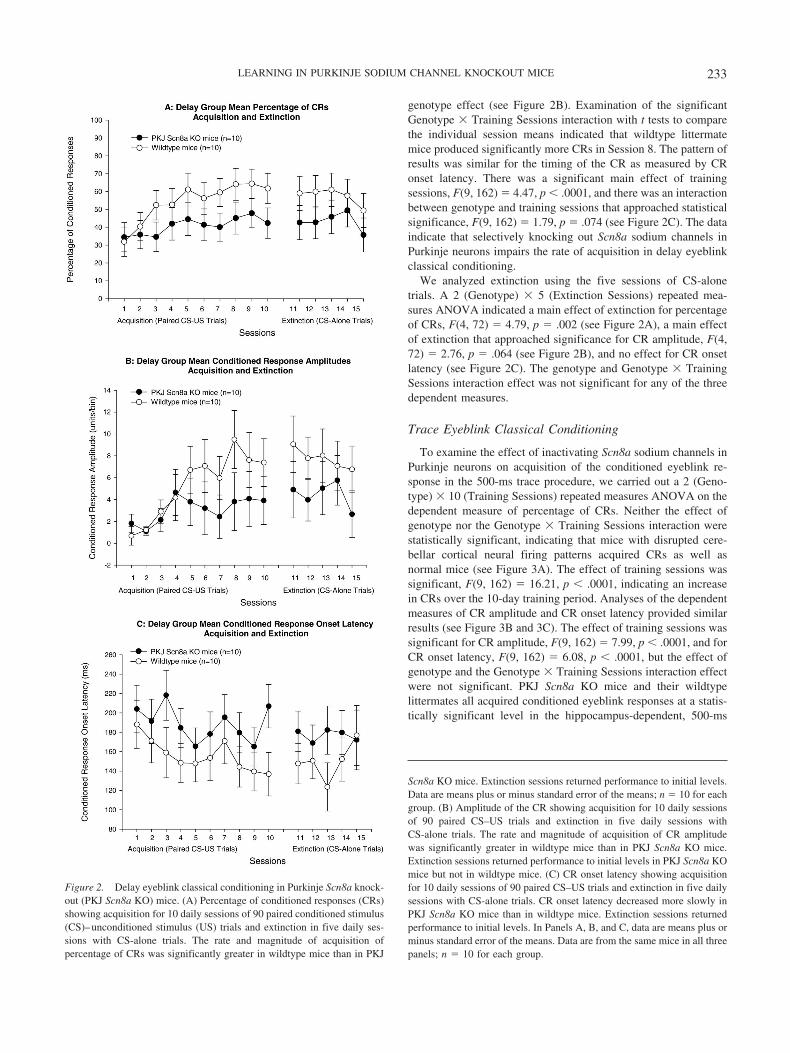
genotype effect (see Figure 2B). Examination of the significantGenotype � Training Sessions interaction with t tests to comparethe individual session means indicated that wildtype littermatemice produced significantly more CRs in Session 8. The pattern ofresults was similar for the timing of the CR as measured by CRonset latency. There was a significant main effect of trainingsessions, F(9, 162) � 4.47, p � .0001, and there was an interactionbetween genotype and training sessions that approached statisticalsignificance, F(9, 162) � 1.79, p � .074 (see Figure 2C). The dataindicate that selectively knocking out Scn8a sodium channels inPurkinje neurons impairs the rate of acquisition in delay eyeblinkclassical conditioning.
We analyzed extinction using the five sessions of CS-alonetrials. A 2 (Genotype) � 5 (Extinction Sessions) repeated mea-sures ANOVA indicated a main effect of extinction for percentageof CRs, F(4, 72) � 4.79, p � .002 (see Figure 2A), a main effectof extinction that approached significance for CR amplitude, F(4,72) � 2.76, p � .064 (see Figure 2B), and no effect for CR onsetlatency (see Figure 2C). The genotype and Genotype � TrainingSessions interaction effect was not significant for any of the threedependent measures.
Trace Eyeblink Classical Conditioning
To examine the effect of inactivating Scn8a sodium channels inPurkinje neurons on acquisition of the conditioned eyeblink re-sponse in the 500-ms trace procedure, we carried out a 2 (Geno-type) � 10 (Training Sessions) repeated measures ANOVA on thedependent measure of percentage of CRs. Neither the effect ofgenotype nor the Genotype � Training Sessions interaction werestatistically significant, indicating that mice with disrupted cere-bellar cortical neural firing patterns acquired CRs as well asnormal mice (see Figure 3A). The effect of training sessions wassignificant, F(9, 162) � 16.21, p � .0001, indicating an increasein CRs over the 10-day training period. Analyses of the dependentmeasures of CR amplitude and CR onset latency provided similarresults (see Figure 3B and 3C). The effect of training sessions wassignificant for CR amplitude, F(9, 162) � 7.99, p � .0001, and forCR onset latency, F(9, 162) � 6.08, p � .0001, but the effect ofgenotype and the Genotype � Training Sessions interaction effectwere not significant. PKJ Scn8a KO mice and their wildtypelittermates all acquired conditioned eyeblink responses at a statis-tically significant level in the hippocampus-dependent, 500-ms
Figure 2. Delay eyeblink classical conditioning in Purkinje Scn8a knock-out (PKJ Scn8a KO) mice. (A) Percentage of conditioned responses (CRs)showing acquisition for 10 daily sessions of 90 paired conditioned stimulus(CS)–unconditioned stimulus (US) trials and extinction in five daily ses-sions with CS-alone trials. The rate and magnitude of acquisition ofpercentage of CRs was significantly greater in wildtype mice than in PKJ
Scn8a KO mice. Extinction sessions returned performance to initial levels.Data are means plus or minus standard error of the means; n � 10 for eachgroup. (B) Amplitude of the CR showing acquisition for 10 daily sessionsof 90 paired CS–US trials and extinction in five daily sessions withCS-alone trials. The rate and magnitude of acquisition of CR amplitudewas significantly greater in wildtype mice than in PKJ Scn8a KO mice.Extinction sessions returned performance to initial levels in PKJ Scn8a KOmice but not in wildtype mice. (C) CR onset latency showing acquisitionfor 10 daily sessions of 90 paired CS–US trials and extinction in five dailysessions with CS-alone trials. CR onset latency decreased more slowly inPKJ Scn8a KO mice than in wildtype mice. Extinction sessions returnedperformance to initial levels. In Panels A, B, and C, data are means plus orminus standard error of the means. Data are from the same mice in all threepanels; n � 10 for each group.
233LEARNING IN PURKINJE SODIUM CHANNEL KNOCKOUT MICE

trace procedure. Inactivation of Scn8a sodium channels in Purkinjeneurons did not impair learning in trace eyeblink conditioning.
We examined extinction by comparing the two groups of micein a 2 (Genotype) � 5 (Training Sessions) repeated measuresANOVA using responses in the CS-alone condition. The effects ofgenotype, training sessions, and the Genotype � Training Sessionsinteraction were not statistically significant for any of the threedependent measures (see Figure 3).
Morris Water Maze
During hidden platform training, PKJ Scn8a KO mice spentlonger searching for the platform (see Figure 4A). To examinegenotype differences in the hidden platform condition of the Mor-ris water maze, we carried out a 2 (Genotype) � 9 (TrainingSessions) repeated measures ANOVA on the latency to escape.The effect of genotype was statistically significant, F(1, 39) �4.72, p � .036 (see Figure 4B). PKJ Scn8a KO mice took signif-icantly longer to reach the hidden platform and escape than didwildtype mice. To determine whether the mild ataxia of the PKJScn8a KO mice affected their swimming speed and hence themotor capacity to reach the platform, we carried out a 2 (Geno-type) � 3 (Training Sessions) repeated measures ANOVA forlatency to escape in the visible platform condition. Performance ofthe two groups of mice was similar when the platform was visible(see Figure 4C).
The fourth training session in the Morris water maze followed 3days of hidden platform training when the hidden escape platformwas removed from the pool. Probe trials were used to measureretention of place learning. One measure taken from probe trialswas the number of times the mouse crossed over the formerplatform area. This measure was taken from each quadrant, andmice that learned the position of the hidden platform crossed overthe area in the trained quadrant more often than the same area inthe other three untrained quadrants. A 2 (Genotype) � 4 (Quad-rant) repeated measures ANOVA comparing the number of cross-ings demonstrated that all mice made significantly more crossingsin the trained quadrant, F(3, 117) � 18.01, p � .001. However,PKJ Scn8a KO mice made significantly fewer crossings in thetraining quadrant than did wildtype mice, F(3, 117) � 5.32, p �.002 (see Figure 5A).
Figure 3. Trace eyeblink classical conditioning in Purkinje Scn8a knock-out (PKJ Scn8a KO) mice. (A) Percentage of conditioned responses (CRs)showing acquisition for 10 daily sessions of 90 paired conditioned stimulus
(CS)–unconditioned stimulus (US) trials and extinction in five daily ses-sions with CS-alone trials. The rate and magnitude of acquisition ofpercentage of CRs was similar in wildtype and PKJ Scn8a KO mice. Intrace conditioning, the extinction sessions did not return performance toinitial levels. (B) Amplitude of CRs showing acquisition for 10 dailysessions of 90 paired CS–US trials and extinction in five daily sessionswith CS-alone trials. The CR amplitude was similar in wildtype mice thanin PKJ Scn8a KO mice. Extinction sessions did not return performance toinitial levels. (C) CR onset latency showing acquisition for 10 dailysessions of 90 paired CS–US trials and extinction in five daily sessionswith CS-alone trials. CR onset latency was similar in PKJ Scn8a KO miceand wildtype mice. Extinction sessions did not return performance to initiallevels. In Panels A, B, and C, data are means plus or minus standard errorof the means. Data are from the same mice in all three panels; n � 10 foreach group.
234 WOODRUFF-PAK, GREEN, LEVIN, AND MEISLER

Another measure of retention of place learning made during theprobe trial session was the time spent swimming in each quadrant,regardless of whether mice passed over the platform area. Groupmeans indicated that wildtype mice demonstrated the normal pat-tern by spending the longest period swimming in the trainingquadrant (see Figure 5B). However, PKJ Scn8a KO mice spent thelongest period swimming in a quadrant adjacent to the trainingquadrant. A 2 (Genotype) � 4 (Quadrant) repeated measuresANOVA revealed a significant effect of quadrant, F(3, 117) �5.45, p � .002. Both groups of mice spent more time in the trainedquadrant and one of the adjacent quadrants than in the other twoquadrants. The difference between the two genotype groups ap-proached statistical significance, F(1, 39) � 3.19, p � .082, andthe Genotype � Quadrant interaction effect was not significant.
Discussion
To examine the contribution of the cerebellar cortex in delayand trace eyeblink conditioning and in the Morris water maze, wetested PKJ Scn8a KO mice that had Exon 1 of Scn8a sodiumchannels deleted specifically in Purkinje neurons. In comparisonwith wildtype littermates, PKJ Scn8a KO mice were impaired inthe acquisition of CRs in 500-ms delay eyeblink classical condi-tioning and in place learning in the hidden platform condition ofthe Morris water maze. PKJ Scn8a KO mice were not impaired in500-ms trace eyeblink classical conditioning or in the visibleplatform condition of the Morris water maze. These results indi-cate that subthreshold sodium currents and normal firing patternsin Purkinje neurons in cerebellar cortex are essential for normalacquisition in delay eyeblink conditioning and place learning in theMorris water maze. The results challenge our previously heldconception of the role of cerebellar cortex in trace conditioning,but the data are consistent with several previous studies of traceconditioning in mutant mice with abnormal cerebellar corticalfunction.
Characteristics of PKJ Scn8a KO Mice
In mice with global loss of Scn8a expression (Dick, Boakes,Candy, Harris, & Cullen, 1986; Sprunger, Escayg, Tallaksen-Greene, Albin, & Meisler, 1999), cerebellar morphology is notaffected. Consistent with this observation, brains from 8-week-oldPKJ Scn8a KO mice had no cerebellar malformations. Foliation,layering, and cellular organization appeared grossly normal, sug-gesting that postnatal knockout of Scn8a did not interfere signif-icantly with structural development of the cerebellum (Levin,2004). Purkinje neurons of 2–3-week-old global Scn8a null micehave specific disruptions in sodium current kinetics (Raman,Sprunger, Meisler, & Bean, 1997) that were similar in PKJ Scn8aKO mice (Aman et al., 2004). There is a selective impairment insubthreshold sodium currents that affects the capacity of Purkinjeneurons to fire repetitively. PKJ Scn8a KO mice have a 10-foldreduction in spontaneous firing of Purkinje neurons. The maximalfiring rate of Purkinje neurons in PKJ Scn8a KO mice is 50% ofthe maximal firing rate in wildtype littermates (Levin et al., 2005).
Motor coordination and motor learning were quantitatively as-sessed in PKJ Scn8a KO mice with the accelerating rotorod test(Levin & Meisler, 2004a). Whereas wildtype mice improvedmarkedly during four daily training sessions, PKJ Scn8a KO mice
initially performed more poorly than wildtype mice and showedminimal improvement over training sessions. Muscle weakness didnot contribute to impaired performance on the rotorod as PKJScn8a KO mice performed as well as wildtype mice on thehanging wire assay (Levin & Meisler, 2004a). The lack of muscleweakness in PKJ Scn8a KO mice distinguishes them from micewith global Scn8a mutations (Kearney et al., 2002; Sprunger et al.,1999).
Reduced Purkinje Neuron Firing and the Morris WaterMaze
The Morris water maze is typically associated with the hip-pocampus. However, this task engages a number of neural sys-tems, the impairment of which can affect acquisition. Goodlett,Hamre, and West (1992) carried out a classic study of Purkinjeneuron involvement in the Morris water maze in pcd mutant mice.Groups of pcd mutant mice and wildtype littermates were tested at30 days of age when Purkinje neuron loss had just occurred, andat 50 and 110 days when the mice were more mature. At all ages,pcd mutant mice had severe deficits in spatial navigation as did thePKJ Scn8a KO mice in the current study. Numerous investigatorshave reported cerebellar cortical involvement in the Morris watermaze in mutant mice and lesioned rats (reviewed in Lalonde &Strazielle’s, 1999, study). The contribution of the current study isto demonstrate that abnormal firing patterns in morphologicallyintact Purkinje neurons impair acquisition and retention in thistask. The fact that PJK Scn8a KO mice acquired CRs normally inthe hippocampus-dependent trace eyeblink classical conditioningprocedure underscores the fact that their hippocampus is normal.The genetic lesion is specific to electrophysiological properties ofPurkinje neurons in cerebellar cortex, and this very specific lesionstill impaired place learning. The mild ataxia in PKJ Scn8a KOmice did not impair their ability to swim rapidly. When the hiddenplatform was made visible, the latency of PKJ Scn8a KO mice toswim to it was similar to the swimming latency of wildtypelittermates.
Reduced Purkinje Neuron Firing and Delay EyeblinkClassical Conditioning
Cerebellar cortical Purkinje neurons affect the rate of acquisi-tion of conditioned eyeblink responses when the CS and USoverlap in the delay paradigm. Purkinje neuron number is highlycorrelated with rate of acquisition in delay eyeblink classicalconditioning in New Zealand white rabbits (Woodruff-Pak &Trojanowski, 1996) and C57BL/6J mice (Woodruff-Pak, in press).Mice homozygous for the pcd mutation that are totally devoid ofPurkinje neurons acquired CRs in the delay eyeblink classicalconditioning procedure at a dramatically slowed rate and to alower maximal level than did their wildtype littermates (L. Chen,Bao, Lockard, Kim, & Thompson, 1996). Histological examina-tion of interpositus nucleus in pcd mutant mice revealed no qual-itative differences from wildtype mice (L. Chen et al., 1996).However, retrograde degeneration of inferior olive neurons(Ghetti, Norton, & Triarhou, 1987) and altered dopamine receptorand transporter levels (Delis, Mitsacos, & Giompres, 2004) havebeen observed in pcd mutant mice. This pathology may haveaffected acquisition of CRs. In the case of PKJ Scn8a KO mice,
235LEARNING IN PURKINJE SODIUM CHANNEL KNOCKOUT MICE

236 WOODRUFF-PAK, GREEN, LEVIN, AND MEISLER

cerebellar morphology, including Purkinje neuron morphology, isintact. Impaired acquisition of CRs in delay eyeblink conditioningis exclusively associated with reduced firing in Purkinje neurons.
Other lines of evidence indicate that whereas the cerebellarcortex and Purkinje neurons are normally engaged in acquisition ofCRs, it is plasticity in the interpositus nucleus that is the basicsubstrate for acquisition and retention (e.g., Kleim et al., 2002;Krupa, Thompson, & Thompson, 1993). Purkinje neurons are theonly efferents to cerebellar deep nuclei. Firing patterns of Purkinjeneurons unquestionably affect function in neurons in the interposi-tus nucleus. It is possible that our results with impaired delayeyeblink conditioning in PKJ Scn8a KO mice represent disruptionof interpositus nucleus input from mossy fiber and climbing fiberpathways essential to acquisition of CRs. Future studies will de-termine whether alterations in Purkinje neuron firing patterns playa direct or more indirect role in affecting acquisition of CRs indelay eyeblink classical conditioning.
A cellular model system proposed as a mechanism for informa-tion storage in the cerebellum is long-term depression (LTD). Inthis model, coactivation of climbing fiber and parallel fiber inputs
to a Purkinje cell induces a persistent, input-specific depression ofthe parallel fiber-Purkinje cell synapse (for a review, see Linden &Connor, 1995). Mouse models used to explore relationships be-tween LTD and acquisition in delay eyeblink conditioning havedemonstrated a consistent correlation between impaired cerebellarcortical LTD and impaired delay eyeblink conditioning (C. Chen etal., 1995; Miyata et al., 2001; Shibuki et al., 1996). The alteredcurrents in Purkinje neurons in PKJ Scn8a KO mice may impedethe persistent, input-specific depression of the parallel fiber-Purkinje neuron synapse normally resulting from coactivation ofparallel fiber and climbing fiber inputs. Scn8a is essential forcomplex spiking in Purkinje cells (Raman et al., 1997), a phenom-enon resulting from combined parallel fiber and climbing fiberinput (Thach, 1967). LTD is likely impaired in PKJ Scn8a KOmice.
Reduced Purkinje Neuron Firing and Trace Conditioning
There were differences between delay and trace conditioning inPKJ Scn8a KO mice in both acquisition and extinction. Acquisi-
Figure 5. Morris water maze in Purkinje Scn8a knockout (PKJ Scn8a KO) mice. (A) Number of platformcrossings in the four quadrants of the pool. PKJ Scn8a KO mice crossed over the quadrant where the hiddenplatform had been significantly fewer times. (B) Time spent in the four quadrants of the pool. PKJ Scn8a KOmice spent more time in the quadrant where the hidden platform had been, although there was a trend for PKJScn8a KO mice to spend more time in an adjacent quadrant as compared with wildtype mice. In Panels A andB, data are means plus or minus standard error of the means. Data are from the same mice in both panels; n �20 for each group.
Figure 4 (opposite). Morris water maze in Purkinje Scn8a knockout (PKJ Scn8a KO) mice. (A) Video trackingfor one 120-s trial in Session 7 for a PKJ Scn8a KO mouse (left) and for a wildtype mouse (right), indicatingthe long search period for the PKJ Scn8a KO mouse and the relatively direct pathway to the hidden platformtaken by the wildtype mouse. (B) Latency to the hidden platform in PKJ Scn8a KO and wildtype mice. PKJScn8a KO mice took significantly longer to find the hidden platform in all sessions. (C) Latency to the visibleplatform in PKJ Scn8a KO and wildtype mice. PKJ Scn8a KO mice were able to swim to the visible platformas rapidly as wildtype mice, demonstrating that mild ataxia did not affect their swimming speed. In Panels B andC, data are means plus or minus standard error of the means. Data are from the same mice in all three panels;n � 20 for each group.
237LEARNING IN PURKINJE SODIUM CHANNEL KNOCKOUT MICE

tion in PKJ Scn8a KO mice was impaired in delay but not traceconditioning, and extinction was significant in both PKJ Scn8a KOmice and wildtype littermates in delay but not in trace eyeblinkconditioning. Kishimoto, Hirono, et al. (2001) reported normalextinction in trace eyeblink conditioning in mice with impairedLTD in rostral cerebellar cortex that acquired trace conditioningnormally. Extinction was not different between PKJ Scn8a KOmice and wildtype littermates in delay or trace conditioning. Wehave no explanation for why both groups of mice failed to showextinction in trace but not delay conditioning.
The role of cerebellar cortex has been investigated much morethoroughly in delay than in trace eyeblink classical conditioning.In the relative absence of data on cerebellar cortical involvement intrace, our 20-year-old study of cerebellar cortical lesions and traceconditioning in rabbits (Woodruff-Pak et al., 1985) continues to becited as support for cerebellar cortical involvement in trace. Aftertraining rabbits in a trace eyeblink conditioning paradigm with a250-ms CS and 500-ms trace, we aspirated cerebellar cortex ipsi-lateral to the trained eye. At retest, rabbits initially produced fewCRs and required several sessions of additional training to produceCRs. On the basis of these data on trace conditioning and manystudies on delay conditioning, we made the assumption that thecerebellar cortex is required for acquisition at a normal rate forboth delay and trace eyeblink conditioning. Data on eyeblinkconditioning in mutant mice with cerebellar cortical impairmentdemonstrating poor delay but normal trace conditioning (Kishi-moto, Hirono, et al., 2001; Kishimoto, Kawahara, et al., 2001)were inconsistent with our conceptions of the role of cerebellarcortex in trace eyeblink conditioning.
A great deal is known about the ontogeny of associative learningin mammals, particularly in rodents (Stanton, 2000). Studies haveexamined relationships between the development of eyeblink con-ditioning and the physiological maturation of the cerebellum inwildtype rodents (Freeman & Nicholson, 2001; Nicholson & Free-man, 2004). However, there is limited knowledge about the pat-terns of cerebellar development in mice with altered genes. At thepresent time, the studies demonstrating normal trace conditioningin organisms with impaired cerebellar cortices are limited to mu-tant (Kishimoto, Hirono, et al., 2001; Kishimoto, Kawahara, et al.,2001) and transgenic mice in the current study. It is possible thatthese mice are unique, having developed alternate brain pathwaysto compensate for abnormal cerebellar cortical function. Addi-tional studies of cerebellar cortex in other mammals will addperspective on this issue.
In this study, we used delay and trace paradigms that werematched as closely as possible. We used identical white noise CSsand obicularis oculi stimulation USs with identical 500-ms inter-vals between the onset of CS and US. The sensory input pathwaysand stimulus onset asynchrony were the same. The only differencebetween the paradigms was that the CS in trace turned off after 250ms, leaving a 250-ms gap between its offset and the US onset. InC57BL/6 mice, this 250-ms gap makes the hippocampus essentialfor acquisition of CRs (Tseng et al., 2004). To be hippocampusdependent, the length of the trace must exceed 300 ms in rabbits(Moyer et al., 1990) and 1,000 ms in humans (Clark & Squire,1998). When the time period of the trace interval exceeds somespecies-specific length, the cerebellum can no longer form anassociation between the CS and US. Additional brain circuitryfrom higher cortical regions is required. It is well documented that
the hippocampus is essential when the critical trace time interval isexceeded, and evidence supporting a role for prefrontal corticalareas is increasing. Results with trace conditioning and PKJ Scn8aKO mice in this study, coupled with results with trace conditioningin mice with mutations impairing cerebellar LTD (Kishimoto,Hirono, et al., 2001; Kishimoto, Kawahara, et al., 2001), raise thepossibility that the cerebellar cortex is bypassed in trace eyeblinkconditioning. Input from hippocampus and/or prefrontal cortexmay replace or supersede cerebellar cortical input to interpositusnucleus.
Speculation about hippocampal input to the cerebellar deepnuclei highlights the limitations of our knowledge about pathwaysbetween hippocampus and cerebellum in genetically normal mam-mals. Investigation of relevant cerebellar-hippocampal circuitry islimited (e.g., Berger, Weikart, Bassett, & Orr, 1986). Connectionsbetween the two structures are indirect and involve at least foursynapses. Could the direct Purkinje neuron input to deep cerebellarnuclei that optimizes acquisition in delay eyeblink conditioningreally be superseded by an indirect pathway from the hippocampusin trace conditioning? Another possibility is that there are prefron-tal cortical pathways to cerebellum that are more direct than thosefrom hippocampus. An alternative hypothesis is that mutant andgenetically altered mice with abnormalities in cerebellar cortexdevelop compensatory connections between deep cerebellar nucleiand higher cortical structures that are not found in geneticallyintact mammals. The result that reduced Purkinje neuron firingimpairs delay but not trace eyeblink classical conditioning identi-fies a gap in knowledge about cerebellar–hippocampal interactionsand involvement in trace eyeblink conditioning that requires ad-ditional investigation.
References
Aman, T. K., Grieco, T. M., Khaliq, Z. M., Levin, S. I., Meisler, M. H., &Raman, I. M. (2004). Electrophysiological correlates of motor disordersproduced by the deletion of the Na channel Scn8a exclusively in cere-bellar Purkinje cells (Program No. 397.2). 2004 Abstract Viewer/Itinerary Planner, Washington, DC: Society for Neuroscience, Online.
Berger, T. W., Weikart, C. L., Bassett, J. L., & Orr, W. B. (1986). Lesionsof the retrosplenial cortex produce deficits in reversal learning of therabbit nictitating membrane response: Implications for potential interac-tions between hippocampal and cerebellar brain systems. BehavioralNeuroscience, 100, 802–809.
Bracha, V. (2004). Role of the cerebellum in eyeblink conditioning.Progress in Brain Research, 143, 331–339.
Burgess, D. L., Kohrman, D. C., Galt, J., Plummer, N. W., Jones, J. M.,Spear, B., & Meisler, M. H. (1995, August). Mutation of a new sodiumchannel gene, Scn8a, in the mouse mutant ‘motor endplate disease’.Nature Genetics, 10, 461–465.
Burwell, R. D., Saddoris, M. P., Bucci, D. J., & Wiig, K. A. (2004).Corticohippocampal contributions to spatial and contextual learning.Journal of Neuroscience, 24, 3826–3836.
Caldwell, J. H., Schaller, K. L., Lasher, R. S., Peles, E., & Levinson, S. R.(2000). Sodium channel Na(v)1.6 is localized at nodes of Ranvier,dendrites, and synapses. Proceedings of the National Academy of Sci-ences, USA, 97, 5616–5620.
Chen, C., Kano, M., Abeliovich, A., Chen, L., Bao, S., Kim, J. J., et al.(1995, December). Impaired motor coordination correlates with persis-tent multiple climbing fiber innervation in PKC gamma mutant mice.Cell, 83, 1233–1242.
Chen, G., & Steinmetz, J. E. (1998). A general-purpose computer system
238 WOODRUFF-PAK, GREEN, LEVIN, AND MEISLER

for behavioral conditioning and neural recording experiments. Behav-ioral Research Methods, Instruments, & Computers, 30, 384–391.
Chen, L., Bao, S., Lockard, J. M., Kim, J. J., & Thompson, R. F. (1996).Impaired classical eyeblink conditioning in cerebellar lesioned and Pur-kinje cell degeneration (pcd) mutant mice. Journal of Neuroscience, 16,2829–2838.
Christian, K. M., & Thompson, R. F. (2003). Neural substrates of eyeblinkconditioning: Acquisition and retention. Learning & Memory, 11, 427–455.
Clark, R. E., & Squire, L. R. (1998, April). Classical conditioning and brainsystems: The role of awareness. Science, 280, 77–81.
Delis, F., Mitsacos, A., & Giompres, P. (2004). Dopamine receptor andtransporter levels are altered in the brain of Purkinje cell degenerationmutant mice. Neuroscience, 125, 255–268.
D’Hooge, R., & De Deyn, P. P. (2001). Application of the Morris watermaze in the study of learning and memory. Brain Research: BrainResearch Review, 36, 60–90.
Dick, D. J., Boakes, R. J., Candy, J. M., Harris, J. B., & Cullen, M. J.(1986). Cerebellar structure and function in the murine mutant “jolting”.Journal of Neuroscience, 76, 255–267.
Freeman, J. H., Jr., & Nicholson, D. A. (2001). Ontogenetic changes in theneural mechanisms of eyeblink conditioning. Integrative Physiologicaland Behavioral Science, 36, 15–35.
Gasbarri, A., Pompili, A., Pacitti, C., & Cicirata, F. (2003). Comparativeeffects of lesions to the ponto-cerebellar and olivo-cerebellar pathwayson motor and spatial learning in the rat. Neuroscience, 116, 1131–1140.
Ghetti, B., Norton, J., & Triarhou, L. C. (1987). Nerve cell atrophy and lossin the inferior olivary complex of “Purkinje cell degeneration” mutantmice. Journal of Comparative Neurology, 260, 409–422.
Goodlett, C. R., Hamre, K. M., & West, J. R. (1992). Dissociation ofspatial navigation and visual guidance performance in Purkinje celldegeneration (pcd) mutant mice. Behavioural Brain Research, 47, 129–141.
Green, J. T., & Steinmetz, J. E. (2005). Purkinje cell activity in thecerebellar anterior lobe after rabbit eyeblink conditioning. Learning &Memory, 12, 260–269.
Green, J. T., & Woodruff-Pak, D. S. (2000). Eyeblink classical condition-ing: Hippocampus is for multiple associations as cerebellum is forassociation–response. Psychological Bulletin, 126, 138–158.
Harvey, J. A., Quinn, J. L., Liu, R., Aloyo, V. J., & Romano, A. G. (2003).Selective remodeling of rabbit frontal cortex: Relationship between5-HT2A receptor density and associative learning. Psychopharmacology(Berlin), 172, 435–442.
Kearney, J. A., Buchner, D. A., De Haan, G., Adamska, M., Levin, S. I.,Furay, A. R., et al. (2002). Molecular and pathological effects of amodifier gene on deficiency of the sodium channel Scn8a (Nav1.6).Human Molecular Genetics, 11, 2765–2775.
Kim, J. J., Clark, R. E., & Thompson, R. F. (1995). Hippocampectomyimpairs the memory of recently, but not remotely, acquired trace eye-blink conditioned responses. Behavioral Neuroscience, 109, 195–203.
Kishimoto, Y., Hirono, M., Sugiyama, T., Kawahara, S., Nakao, K.,Kishio, M., et al. (2001). Impaired delay but normal trace eyeblinkconditioning in PLC�4 mutant mice. NeuroReport, 12, 2919–2922.
Kishimoto, Y., Kawahara, S., Suzuki, M., Mori, H., Mishina, M., & Kirino,Y. (2001). Classical eyeblink conditioning in glutamate receptor subunit�2 mutant mice is impaired in the delay paradigm but not in the traceparadigm. European Journal of Neuroscience, 13, 1249–1253.
Kleim, J. A., Freeman, J. H., Bruneau, R., Nolan, B. C., Cooper, N. R.,Zook, A., & Walters, D. (2002). Synapse formation is associated withmemory storage in the cerebellum. Proceedings of the National Acad-emy of Sciences, USA, 99, 13228–13231.
Kronforst-Collins, M. A., & Disterhoft, J. A. (1998). Lesions of the caudalarea of rabbit medial prefrontal cortex impair trace eyeblink condition-ing. Neurobiology of Learning and Memory, 69, 147–162.
Krupa, D. J., Thompson, J. K., & Thompson, R. F. (1993, May). Local-ization of a memory trace in the mammalian brain. Science, 260,989–991.
Lalonde, R., & Strazielle, C. (1999). Motor performance of spontaneousmurine mutations with cerebellar atrophy. In W. E. Crusio & R. T.Gerlai (Eds.), Handbook of molecular–genetic techniques for brain andbehavior research (pp. 627–637). New York: Elsevier Science.
Levin, S. I. (2004). Pathophysiology of movement disorders resulting fromconditional inactivation of the voltage-gated sodium channel Scn8a.Unpublished doctoral dissertation, University of Michigan, Ann Arbor.
Levin, S. I., Aman, T. K., Khaliq, Z. M., Grieco, T. M., Raman, I. M., &Meisler, M. H. (2005). Impaired motor function in mice with cell-specific knockout of Na channel Scn8a (Nav1.6) in cerebellar Purkinjeneurons and granule cells. Manuscript submitted for publication.
Levin, S. I., & Meisler, M. H. (2004a). Conditional inactivation of thevoltage-gated sodium channel Scn8a (Nav1.6) in cerebellar Purkinje andgranule cells (No. 397.1). 2004 Abstract Viewer/Itinerary Planner,Washington, DC: Society for Neuroscience, Online.
Levin, S. I., & Meisler, M. H. (2004b). Floxed allele for conditionalinactivation of the voltage-gated sodium channel Scn8a (Nav1.6). Gen-esis, 40, 234–239.
Linden, D. J., & Connor, J. A. (1995). Long-term synaptic depression.Annual Review of Neuroscience, 18, 319–357.
Mamounas, L. A., Thompson, R. F., & Madden, J., IV. (1987). CerebellarGABAergic processes: Evidence for critical involvement in a form ofsimple associative learning in the rabbit. Proceedings of the NationalAcademy of Sciences, USA), 84, 2102–2105.
McCormick, D. A., & Thompson, R. F. (1984). Neuronal responses of therabbit cerebellum during acquisition and performance of a classicallyconditioned nictitating membrane-eyelid response. Journal of Neuro-science, 4, 2811–2822.
Medina, J. F., Nores, W. L., Ohymama, T., & Mauk, M. D. (2000).Mechanisms of cerebellar learning suggested by eyelid conditioning.Current Opinion in Neurobiology, 10, 717–724.
Meisler, M. H., Kearney, J., Escayg, A., MacDonald, B. T., & Sprunger,L. K. (2001). Sodium channels and neurological disease: Insights fromScn8a mutations in the mouse. Neuroscientist, 7, 136–146.
Miyata, M., Kim, H.-T., Hashimoto, K., Lee, T.-K., Cho, S.-Y., Jiang, H.,et al. (2001). Deficient long-term synaptic depression in the rostralcerebellum correlated with impaired motor learning in phospholipaseC�4 mutant mice. European Journal of Neuroscience, 13, 1945–1954.
Morris, R. G., Garrud, P., Rawlins, J. N., & O’Keefe, J. (1982, June). Placenavigation impaired in rats with hippocampal lesions. Nature, 297,681–683.
Moyer, J. R., Deyo, R. A., & Disterhoft, J. F. (1990). Hippocampectomydisrupts trace eyeblink conditioning in rabbits. Behavioral Neuro-science, 104, 243–252.
Nicholson, D. A., & Freeman, J. H., Jr. (2004). Developmental changes ineyeblink conditioning and simple spike activity in the cerebellar cortex.Developmental Psychobiology, 44, 45–57.
Powell, D. A., Skaggs, H., Churchwell, J., & McLaughlin, J. (2001).Posttraining lesions of the medial prefrontal cortex impaired perfor-mance of Pavlovian eyeblink conditioning but have no effect on con-comitant hear rate changes in rabbits (Oryctolagus cuniculus). Behav-ioral Neuroscience, 115, 1029–1038.
Raman, I. M., Sprunger, L. K., Meisler, M. H., & Bean, B. P. (1997).Altered subthreshold sodium currents and disrupted firing patterns inPurkinje neurons of Scn8a mutant mice. Neuron, 19, 881–891.
Sakamoto, T., Takatsuki, K., Kawahara, S., Kirino, Y., Niki, H., &Mishina, M. (2005). Role of hippocampal NMDA receptors in traceeyeblink conditioning. Brain Research, 1039, 130–136.
Schaller, K. L., Krzemien, D. M., Yarowsky, P. J., Krueger, B. K., &Caldwell, J. H. (1995). A novel, abundant sodium channel expressed inneurons and glia. Journal of Neuroscience, 15, 3231–3242.
239LEARNING IN PURKINJE SODIUM CHANNEL KNOCKOUT MICE

Schmaltz, L. W., & Theios, J. (1972). Acquisition and extinction of aclassically conditioned response in hippocampectomized rabbits (Oryc-tolagus cuniculus). Journal of Comparative and Physiological Psychol-ogy, 79, 328–333.
Shibuki, K., Gomi, H., Chen, L., Bao, S., Kim, J. J., Wakatuski, H., et al.(1996). Deficient cerebellar long-term depression, impaired eyeblinkconditioning, and normal motor coordination in GFAP mutant mice.Neuron, 16, 587–599.
Solomon, P. R., Vander Schaaf, E. R., Thompson, R. F., & Weisz, D. J.(1986). Hippocampus and trace conditioning of the rabbit’s classicallyconditioned nictitating membrane response. Behavioral Neuroscience,100, 729–744.
Sprunger, L. K., Escayg, A., Tallaksen-Greene, S., Albin, R. L., & Meisler,M. H. (1999). Dystonia associated with mutation of the neuronal sodiumchannel Scn8a and identification of the modifier locus Scnm1 on mousechromosome 3. Human Molecular Genetics, 8, 471–479.
Stanton, M., E. (2000). Multiple memory systems, development, andconditioning. Behavioural Brain Research, 110, 25–37.
Steinmetz, J. E. (2000). Brain substrates of classical eyeblink conditioning:A highly localized but also distributed system. Behavioural Brain Re-search, 110, 13–24.
Takehara, K., Kawahara, S., & Kirino, Y. (2003). Time-dependent reor-ganization of the brain components underlying memory retention intrace eyeblink conditioning. Journal of Neuroscience, 23, 9897–9905.
Thach, W. T., Jr. (1967). Somatosensory receptive fields of single units incat cerebellar cortex. Journal of Neurophysiology, 30, 675–696.
Thompson, R. F. (1986, August). The neurobiology of learning and mem-ory. Science, 233, 941–947.
Thompson, R. F. (1990). Neural mechanisms of classical conditioning inmammals. Philosophical Transactions of the Royal Society of London,B, 329, 161–170.
Thompson, R. F. (2005). In search of memory traces. Annual Review ofPsychology, 56, 1–23.
Tseng, W., Guan, J. F., Disterhoft, J. F., & Weiss, C. (2004). Traceeyeblink conditioning is hippocampally dependent in mice. Hippocam-pus, 14, 58–65.
Weible, A. P., McEchron, M. D., & Disterhoft, J. F. (2000). Corticalinvolvement in acquisition and extinction of trace eyeblink conditioning.Behavioral Neuroscience, 114, 1058–1067.
Weiss, C., Bouwmeester, H., Power, J. M., & Disterhoft, J. E. (1999).Hippocampal lesions prevent trace eyeblink conditioning in the freelymoving rat. Behavioural Brain Research, 99, 123–132.
Woodruff-Pak, D. S. (in press). Stereological estimation of Purkinje neuronnumber in CS7BL/6 mice and its relation to associative learning. Neu-roscience.
Woodruff-Pak, D. S., Lavond, D. G., & Thompson, R. F. (1985). Traceconditioning: Abolished by cerebellar nuclear lesions but not lateralcerebellar cortex aspirations. Brain Research, 348, 249–260.
Woodruff-Pak, D. S., & Trojanowski, J. Q. (1996). The older rabbit as ananimal model: Implications for Alzheimer’s disease. Neurobiology ofAging, 17, 283–290.
Received October 26, 2005Revision received December 9, 2005
Accepted December 27, 2005 �
240 WOODRUFF-PAK, GREEN, LEVIN, AND MEISLER