inhibition of th1 immune response by glucocorticoids
TRANSCRIPT

of February 15, 2018.This information is current as
in T LymphocytesInhibits IL-12-Induced Stat4 PhosphorylationGlucocorticoids: Dexamethasone Selectively Inhibition of Th1 Immune Response by
Frucht, George P. Chrousos and John J. O'SheaRoberta Visconti, Y.-J. Zhou, Martin Aringer, David M. Denis Franchimont, Jérôme Galon, Massimo Gadina,
http://www.jimmunol.org/content/164/4/1768doi: 10.4049/jimmunol.164.4.1768
2000; 164:1768-1774; ;J Immunol
Referenceshttp://www.jimmunol.org/content/164/4/1768.full#ref-list-1
, 18 of which you can access for free at: cites 45 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2000 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on February 15, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on February 15, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

Inhibition of Th1 Immune Response by Glucocorticoids:Dexamethasone Selectively Inhibits IL-12-Induced Stat4Phosphorylation in T Lymphocytes1
Denis Franchimont,2*† Jérôme Galon,2* Massimo Gadina,* Roberta Visconti,* Y.-J. Zhou,*Martin Aringer,* David M. Frucht,* George P. Chrousos, † and John J. O’Shea3*
Glucocorticoids are widely used in the therapy of inflammatory, autoimmune, and allergic diseases. As the end-effectors of thehypothalamic-pituitary-adrenal axis, endogenous glucocorticoids also play an important role in suppressing innate and cellularimmune responses. Previous studies have indicated that glucocorticoids inhibit Th1 and enhance Th2 cytokine secretion. IL-12promotes Th1 cell-mediated immunity, while IL-4 stimulates Th2 humoral-mediated immunity. Here, we examined the regulatoryeffect of glucocorticoids on key elements of IL-12 and IL-4 signaling. We first investigated the effect of dexamethasone on IL-12-inducible genes and showed that dexamethasone inhibited IL-12-induced IFN-g secretion and IFN regulatory factor-1 expressionin both NK and T cells. This occurred even though the level of expression of IL-12 receptors and IL-12-induced Janus kinasephosphorylation remained unaltered. However, dexamethasone markedly inhibited IL-12-induced phosphorylation of Stat4 with-out altering its expression. This was specific, as IL-4-induced Stat6 phosphorylation was not affected, and mediated by theglucocorticoid receptor, as it was antagonized by the glucocorticoid receptor antagonist RU486. Moreover, transfection experi-ments showed that dexamethasone reduced responsiveness to IL-12 through the inhibition of Stat4-dependent IFN regulatoryfactor-1 promoter activity. We conclude that blocking IL-12-induced Stat4 phosphorylation, without altering IL-4-induced Stat6phosphorylation, appears to be a new suppressive action of glucocorticoids on the Th1 cellular immune response and may helpexplain the glucocorticoid-induced shift toward the Th2 humoral immune response. The Journal of Immunology,2000, 164:1768–1774.
Glucocorticoids are widely used in the therapy of inflam-matory, autoimmune, and allergic diseases (1). As end-effectors of the hypothalamic-pituitary-adrenal (HPA)4
axis, endogenous glucocorticoids also play an important role inrestraining the cellular immune response in several experimentalinflammatory diseases in rats and mice (2, 3). Although glucocor-ticoids generally suppress innate immunity, their action on the cel-lular and humoral immune responses is more complex (4). Thus,the cellular immune response (delayed-type hypersensitivity) isstrongly suppressed by glucocorticoids, whereas the humoral orallergic immune response is poorly inhibited or even enhanced(5–7). This is accompanied by a glucocorticoid-induced shift fromTh1 to Th2 cytokine secretion (8–11). Indeed, pre-exposure of
CD41 lymphocytes to glucocorticoids increased the secretion ofTh2-type cytokines, such as IL-4, IL-13, and IL-10, while it sup-pressed the secretion of Th1-type cytokines, such as IFN-g andTNF-a (9). Similarly, glucocorticoids caused a marked reductionof IL-12 secretion by human monocytes and, hence, a decreasedcapacity to produce IFN-g and an increased ability to induce IL-4secretion by T cells (10).
The differentiation of naive CD41 T cells into Th1 and Th2 celltypes is influenced by cytokines produced early in response to theAg triggering the immune response (12, 13). Specifically, IL-12promotes Th1 cell differentiation, which leads to cell-mediated im-munity, while IL-4 promotes Th2 cell differentiation, which drivesallergic and humoral-mediated immunity. The Th1 and Th2 im-mune responses in large measure depend upon the activation oftranscription factors Stat4 and Stat6, respectively (14). Stat4knockout mice have a deficient cellular immune response, whileStat6 knockout mice have an impaired humoral immune response(15–18).
Understanding the regulatory effects of glucocorticoids on cy-tokine signaling would help to further clarify how these hormonesinfluence Th1 cellular and Th2 humoral immune responses. Re-cently, it was reported that glucocorticoids suppressed the Th1immune response by inhibiting the responsiveness of activatedPBMC to IL-12, through down-regulation of the IL-12Rb1- andb2-chain expression (19). Here, we report that glucocorticoids de-crease IL-12 responsiveness through another mechanism, namelyinhibition of Stat4 phosphorylation. This novel mechanism con-tributes to the immunosuppressive action of glucocorticoids on theTh1 cellular immune response and the associated shift toward theTh2 humoral immune response.
*Lymphocyte Cell Biology Section, National Institute of Arthritis and Musculoskel-etal and Skin Diseases, and†Developmental Endocrinology Branch, National Instituteof Child Health and Human Development, National Institutes of Health, Bethesda,MD 20892
Received for publication August 30, 1999. Accepted for publication December3, 1999.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby markedadvertisementin accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by the Leon Fredericq Foundations (to D.F.). D.F. is aResearch Assistant of the Belgian National Foundation for Scientific Research.2 D.F. and J.G. contributed equally to this work.3 Address correspondence and reprint requests to Dr. J. J. O’Shea, National Institutesof Health, Building 10, Room 10N262, 10 Center Drive, Bethesda, MD 20892. E-mailaddress: [email protected] Abbreviations used in this paper: HPA, hypothalamic-pituitary-adrenal; IRF-1, IFNregulatory factor-1; Jak, Janus kinase; Tyk, tyrosine kinase; SOCS, suppressor ofcytokine signaling; CIS, cytokine inhibitor Src homology 2-containing protein; PIAS,protein inhibitor of activated Stat; SHP, Src homology protein.
Copyright © 2000 by The American Association of Immunologists 0022-1767/00/$02.00
by guest on February 15, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
Materials and MethodsCytokines, Abs, and cells
The following reagents were purchased: recombinant human IL-12 andIL-4 (R&D Systems, Minneapolis, MN), anti-phosphotyrosine Ab (4G10)(Upstate Biotechnology, Lake Placid, NY), polyclonal rabbit anti-Stat4 andanti-Stat6 Ab (Santa Cruz Biotechnology, Santa Cruz, CA), and polyclonalrabbit anti-Jak2, anti-Jak1, and anti-Tyk2 Ab (Upstate Biotechnology).Polyclonal rabbit anti-Jak3 and anti-Stat5a Abs were produced as previ-ously described by this laboratory (20). Human IL-2 was provided by Dr.C. Reynolds (National Cancer Institute, Frederick, MD). NK3.3 were pro-vided by Dr. J. Kornbluth (Arkansas Cancer Research Center, Little Rock,AR). NIH3T3 were provided by the American Type Culture Collection(Manassas, VA). PBMC from healthy donors were isolated by Ficoll-Paque gradient centrifugation, activated with PHA (2mg/ml) for 72 h andcultured for an additional day in the presence of IL-2 (40 IU/ml), as de-scribed previously (21, 22). Cell viability, as determined by trypan blueexclusion, thymidine incorporation, and FACS (annexin V/PI) analysis(Trevigen, Gaithersburg, MD), was not affected by dexamethasonetreatment.
IFN-g ELISA
NK3.3 cells and T lymphocytes (53 106/ml) were rested in 1% FCSmedium for 4 h, pretreated with 1027 M of dexamethasone for varioustimes, and then stimulated with IL-12 (20 ng/ml) for 10 h. The cell cultureswere centrifuged, and the supernatant was removed and stored at220 °C.IFN-g was measured using a specific immunoassay (R&D Systems) ac-cording to the manufacturer’s instructions.
RNase protection assay
NK3.3 cells and T lymphocytes were rested for 4 h, pretreated with dexa-methasone at 1027 M for various times, and then stimulated with IL-12 (20ng/ml) for 4 h. RNA extraction was performed (RNAgents; Promega, Mad-ison, WI), and mRNA expression was evaluated by RNase protection.RNase protection assay was performed as follows:32P-labeled RNA probeswere synthesized using SP6 RNA polymerase or T7 RNA polymerase forthe IFN regulatory factor (IRF)-1 probe and the multiprobe template set(Riboquant; PharMingen, San Diego, CA). DNA was digested with DNaseI (Boehringer Mannheim, Indianapolis, IN), and RNA probes were ex-tracted with phenol/chloroform and precipitated with ethanol. LabeledRNA probes were hybridized overnight with target RNA (5mg) at 56°Cand were digested with T1 RNase (Life Technologies, Gaithersburg, MD).The protected mRNA fragment was extracted with phenol and chloroform,precipitated with ethanol, resolved on a 6% denaturing polyacrylamide gel,and subjected to autoradiography. Gene transcripts were identified by thelength of the protected fragments. Equal loading of RNA was estimatedfrom the amounts of protected fragments of two housekeeping genes, L32and GAPDH.
Immunoprecipitation and immunoblotting
NK3.3 cells and T lymphocytes were pretreated with different concentra-tions of dexamethasone for various times, resuspended in 1 ml of serum-free medium (23 106 NK3.3 cells, 503 106 T cells), and stimulated witheither IL-12 (20 ng/ml) or IL-4 (20 ng/ml). Following stimulation, cellswere washed once in PBS and lysed on ice in a buffer containing 0.5%Triton X-100, 50 mM Tris-HCl, pH 7.5, 300 mM NaCl, 2 mM EDTA, 200mM Na3VO4, 10 mg/ml aprotinin, 10mg/ml leupeptin, and 2.5mM p-nitrophenylp-guanidinobenzoate on ice for 30 min. Immunoprecipitationwith anti-Stat or Jak Abs and subsequent SDS-PAGE were performed asdescribed previously (20–22) with detection by enhanced chemilumines-cence (LumiGLO, Kirkegaard & Perry Laboratories, Gaithersburg, MD).
Transfection experiments
NIH3T3 cells were seeded in six-well plates (1.83 105 per well). Eighteenhours later, the cells were transfected, using lipofectamine (Life Technol-ogies), according to the manufacturer’s protocol. The cells were transfected8 h with the following plasmids: p3xGAS-luciferase (kindly provided byDr. Richard Pine, Public Health Research Institute, New York, NY) (0.2mg), CMV-b-galactosidase (0.2mg), and, as indicated, with pEF-BOS-IL-12Rb1 (0.2mg), pEF-BOS-IL-12Rb2 (kindly provided by Dr. Uli Gubler,Roche Nutley, NJ) (0.6mg), pCDNA3-Stat 4 (kindly provided by Dr. JIhle, St. Jude Children’s Hospital, Memphis, TN) (0.2mg), and pCDNA3-Tyk2 (0.2 mg). The transfected cells were pretreated with 1027 M dexa-methasone (6 h), washed once, and then stimulated with IL-12 (10 ng/ml),where indicated, for another 6 h. Cells were lysed, and luciferase andb-galactosidase activities were determined using the Dual-Light Kit ac-
cording to manufacturer’s instruction (Tropix, Bedford, MA). The resultswere normalized for each sample by dividing luciferase activity byb-ga-lactosidase activity. The results are representative of three independentexperiments performed in triplicate.
ResultsDexamethasone inhibits IL-12-induced IFN-g and IRF-1expression
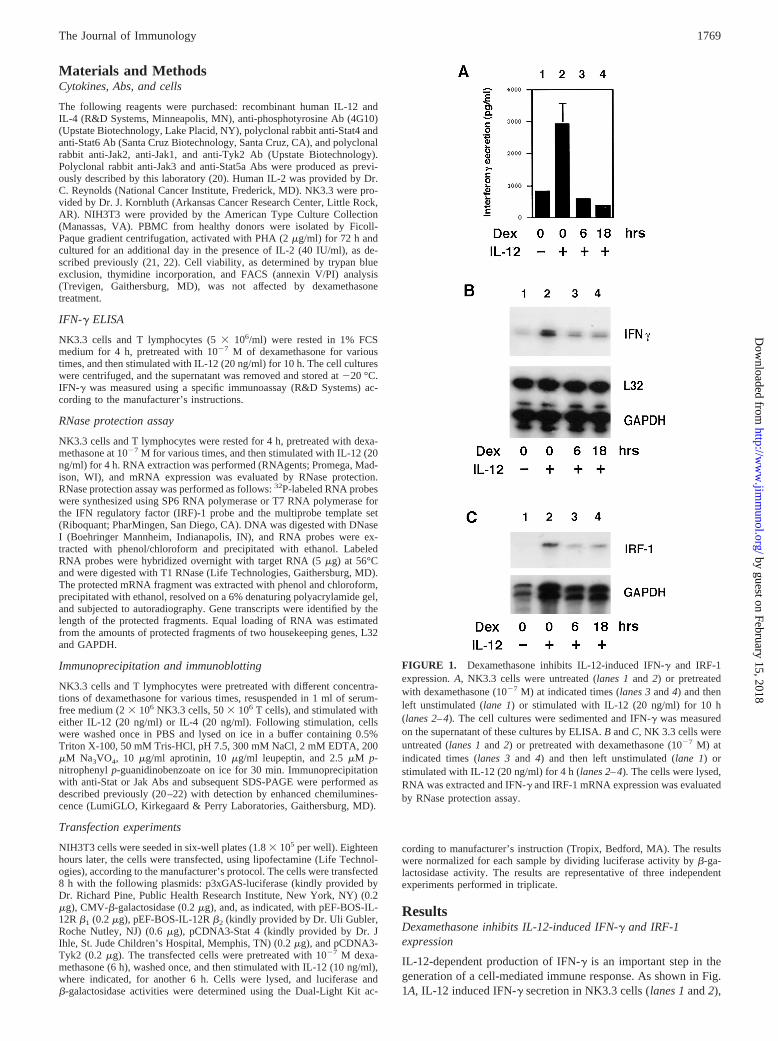
IL-12-dependent production of IFN-g is an important step in thegeneration of a cell-mediated immune response. As shown in Fig.1A, IL-12 induced IFN-g secretion in NK3.3 cells (lanes 1and2),
FIGURE 1. Dexamethasone inhibits IL-12-induced IFN-g and IRF-1expression.A, NK3.3 cells were untreated (lanes 1and 2) or pretreatedwith dexamethasone (1027 M) at indicated times (lanes 3and4) and thenleft unstimulated (lane 1) or stimulated with IL-12 (20 ng/ml) for 10 h(lanes 2–4).The cell cultures were sedimented and IFN-g was measuredon the supernatant of these cultures by ELISA.B andC, NK 3.3 cells wereuntreated (lanes 1and 2) or pretreated with dexamethasone (1027 M) atindicated times (lanes 3and 4) and then left unstimulated (lane 1) orstimulated with IL-12 (20 ng/ml) for 4 h (lanes 2–4).The cells were lysed,RNA was extracted and IFN-g and IRF-1 mRNA expression was evaluatedby RNase protection assay.
1769The Journal of Immunology
by guest on February 15, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

and dexamethasone pretreatment (1027 M) was found to block thisinduction (lanes 3and4). The inhibition of IL-12-induced IFN-gsecretion was observed after 6 h of dexamethasone pretreatmentand was maximal after 18 h. To better characterize the kinetics ofinhibition, we analyzed the levels of IFN-g mRNA expression. Asis evident in Fig. 1B, stimulation of NK3.3 with IL-12 for 4 hresulted in increased IFN-g expression (lanes 1and2). Notably,6 h and 18 h of dexamethasone pretreatment nearly completelyblocked the IL-12-induced IFN-g expression in NK3.3 cells (lanes3 and4). This result was also confirmed in T lymphocytes (data notshown).
To determine whether this was a general effect or limited to theIFN-g gene, we next analyzed the expression of another gene im-portant for Th1 differentiation, IRF-1 (Fig. 1C). We and othershave recently reported that IL-12 up-regulates IRF-1 expression(lanes 1and2) (23, 24). Interestingly, 6 and 18 h of pretreatmentwith dexamethasone substantially reduced IRF-1 expression (lanes3 and4). Equal amounts of GAPDH and L32 transcripts confirmedthat equal amounts of RNA were used (Fig. 1,B andC).
Dexamethasone does not alter the level of expression of IL-12receptors
To understand the possible mechanism of the decreased respon-siveness to IL-12 by dexamethasone, we first examined whetherdexamethasone influenced IL-12R (b1- andb2-chains) expressionin NK 3.3 cells. Dexamethasone was previously shown to decreaseIL-12 receptor expression in peripheral blood mononuclear cellsstimulated with immobilized anti-CD3 for 3 days (19). We eval-uated the IL-12R (b1- andb2-chains) expression after shorter in-cubations with dexamethasone (Fig. 2A). Treatment with dexa-methasone from 1 to 18 h did not alter the levels of these cytokinereceptors (lanes 1–6). We also analyzed IL-4R (a- and the com-mon g-chains) expression; it remained unchanged by dexametha-sone treatment (Fig. 2B,lanes 1–6). These results underscore thespecific decrease in expression of IFN-g and IRF-1. Equal amountsof GAPDH and L32 transcripts confirmed that equal amounts oftarget RNA was used. Similar results were also obtained in T lym-phocytes (data not shown).
Dexamethasone does not inhibit IL-12-induced Jak2 and Tyk2phosphorylation
The absence of modulation of the cytokine receptor mRNA ex-pression suggested that dexamethasone may be acting at anothersite, presumably downstream of IL-12 receptor engagement. Theearliest defined step of IL-12 signaling is the phosphorylation ofJak2 and Tyk2. Thus, we next looked at the effect of dexametha-sone treatment on the phosphorylation of these tyrosine kinases.As previously shown in Fig. 3A, IL-12 induced phosphorylation ofJak2 and Tyk2 within 20 min (lane 2) (22). However, preincuba-tion of the cells with dexamethasone (1027 M) for 6 or 18 h hadno effect on the phosphorylation of Jak2 and Tyk2 (Fig. 3A, lanes3 and 4). The levels of Jak2 and Tyk2 protein were also un-changed, as determined by reblotting with Abs to Jak2 and Tyk2.Thus, neither the phosphorylation nor the protein expression ofJak2 and Tyk2 were modulated by dexamethasone treatment. Wealso analyzed the effect of dexamethasone treatment on IL-4 sig-naling, namely activation of Jak1 and Jak3. As shown in Fig. 3B,IL-4 induced phosphorylation of these kinases within 20 min (lane2). Preincubation of the cells with dexamethasone (1027 M) for 6or 18 h had no effect on the phosphorylation of these kinases (Fig.3B, lanes 3and4). The levels of Jak1 and Jak3 protein were alsounchanged, as determined by reblotting with the relevant Abs.
Dexamethasone inhibits IL-12-induced phosphorylation of Stat4
Once Jaks are activated, they phosphorylate cytokine receptors al-lowing the recruitment and phosphorylation of Stat proteins, which
FIGURE 2. Dexamethasone does not affect IL-12 and IL-4 receptor ex-pression. NK3.3 cells were untreated (lane 1) or pretreated with dexameth-asone (1027 M) at indicated times (lanes 2–6).The cells were lysed, RNAwas extracted and receptor expression was evaluated by RNase protectionassay.A, IL-12R b1- andb2-chains.B, IL-4R a- and commong-chains.
FIGURE 3. Dexamethasone does not inhibit IL-12 or IL-4-induced Jakphosphorylation. NK3.3 cells were untreated (lanes 1and2) or treated withdexamethasone (1027 M) at indicated times (lanes 3and4). A, Cells werestimulated with IL-12 (20 ng/ml) for 20 min (lanes 2–4),lysed, and im-munoprecipitated with anti-Jak2 (upper panel) or anti-Tyk2 (lower panel)and then subjected to immunoblotting with anti-phosphotyrosine (top) andanti-Jak2 or anti-Tyk2 (bottom). B, Cells were stimulated with IL-4 (20ng/ml) for 20 min (lanes 2–4),lysed, and immunoprecipitated with anti-Jak3 (upper panel) or anti-Jak1 (lower panel) and then subjected to im-munoblotting with anti-phosphotyrosine (top) and anti-Jak3 or anti-Jak1(bottom).
1770 GLUCOCORTICOID-MEDIATED INHIBITION OF Stat4 PHOSPHORYLATION
by guest on February 15, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
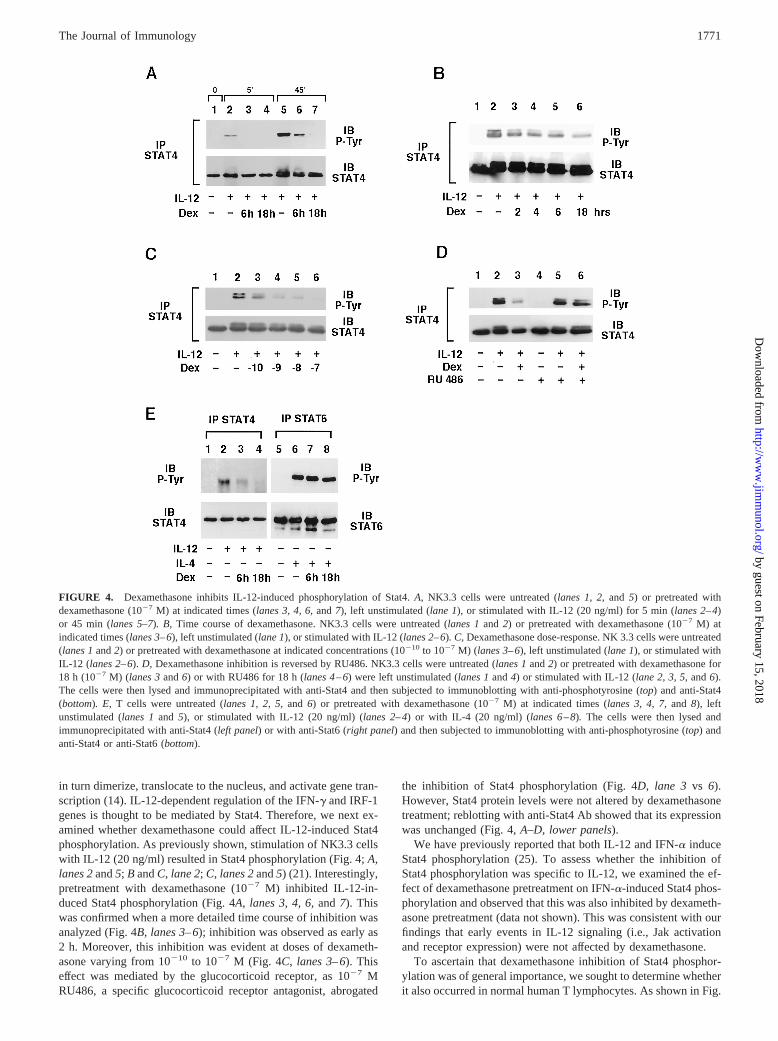
in turn dimerize, translocate to the nucleus, and activate gene tran-scription (14). IL-12-dependent regulation of the IFN-g and IRF-1genes is thought to be mediated by Stat4. Therefore, we next ex-amined whether dexamethasone could affect IL-12-induced Stat4phosphorylation. As previously shown, stimulation of NK3.3 cellswith IL-12 (20 ng/ml) resulted in Stat4 phosphorylation (Fig. 4;A,lanes 2and5; B andC, lane 2;C, lanes 2and5) (21). Interestingly,pretreatment with dexamethasone (1027 M) inhibited IL-12-in-duced Stat4 phosphorylation (Fig. 4A,lanes 3,4, 6, and7). Thiswas confirmed when a more detailed time course of inhibition wasanalyzed (Fig. 4B,lanes 3–6); inhibition was observed as early as2 h. Moreover, this inhibition was evident at doses of dexameth-asone varying from 10210 to 1027 M (Fig. 4C, lanes 3–6). Thiseffect was mediated by the glucocorticoid receptor, as 1027 MRU486, a specific glucocorticoid receptor antagonist, abrogated
the inhibition of Stat4 phosphorylation (Fig. 4D,lane 3 vs 6).However, Stat4 protein levels were not altered by dexamethasonetreatment; reblotting with anti-Stat4 Ab showed that its expressionwas unchanged (Fig. 4,A–D, lower panels).
We have previously reported that both IL-12 and IFN-a induceStat4 phosphorylation (25). To assess whether the inhibition ofStat4 phosphorylation was specific to IL-12, we examined the ef-fect of dexamethasone pretreatment on IFN-a-induced Stat4 phos-phorylation and observed that this was also inhibited by dexameth-asone pretreatment (data not shown). This was consistent with ourfindings that early events in IL-12 signaling (i.e., Jak activationand receptor expression) were not affected by dexamethasone.
To ascertain that dexamethasone inhibition of Stat4 phosphor-ylation was of general importance, we sought to determine whetherit also occurred in normal human T lymphocytes. As shown in Fig.
FIGURE 4. Dexamethasone inhibits IL-12-induced phosphorylation of Stat4.A, NK3.3 cells were untreated (lanes 1, 2,and 5) or pretreated withdexamethasone (1027 M) at indicated times (lanes 3, 4, 6,and7), left unstimulated (lane 1), or stimulated with IL-12 (20 ng/ml) for 5 min (lanes 2–4)or 45 min (lanes 5–7). B, Time course of dexamethasone. NK3.3 cells were untreated (lanes 1and 2) or pretreated with dexamethasone (1027 M) atindicated times (lanes 3–6), left unstimulated (lane 1), or stimulated with IL-12 (lanes 2–6). C, Dexamethasone dose-response. NK 3.3 cells were untreated(lanes 1and2) or pretreated with dexamethasone at indicated concentrations (10210 to 1027 M) (lanes 3–6), left unstimulated (lane 1), or stimulated withIL-12 (lanes 2–6).D, Dexamethasone inhibition is reversed by RU486. NK3.3 cells were untreated (lanes 1and2) or pretreated with dexamethasone for18 h (1027 M) (lanes 3and6) or with RU486 for 18 h (lanes 4–6) were left unstimulated (lanes 1and4) or stimulated with IL-12 (lane 2, 3, 5,and6).The cells were then lysed and immunoprecipitated with anti-Stat4 and then subjected to immunoblotting with anti-phosphotyrosine (top) and anti-Stat4(bottom). E, T cells were untreated (lanes 1, 2, 5,and 6) or pretreated with dexamethasone (1027 M) at indicated times (lanes 3, 4, 7,and 8), leftunstimulated (lanes 1and 5), or stimulated with IL-12 (20 ng/ml) (lanes 2–4) or with IL-4 (20 ng/ml) (lanes 6–8).The cells were then lysed andimmunoprecipitated with anti-Stat4 (left panel) or with anti-Stat6 (right panel) and then subjected to immunoblotting with anti-phosphotyrosine (top) andanti-Stat4 or anti-Stat6 (bottom).
1771The Journal of Immunology
by guest on February 15, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
4E, IL-12 induced Stat4 phosphorylation in activated T lympho-cytes within 20 min (lane 2). Consistent with the results in the NKcell line, dexamethasone pretreatment was also found to block theIL-12-induced Stat4 phosphorylation in T cells (lanes 3and4).
Th2 immune responses are poorly inhibited or even enhanced byglucocorticoids (5–11). In fact, glucocorticoids increase the syn-thesis of Ig E by IL-4-stimulated human lymphocytes (5–7). Itwould then be expected that IL-4-dependent phosphorylation ofStat6 should be resistant to dexamethasone. Therefore, we nexttested the specificity of the inhibition of IL-12-induced Stat4 phos-phorylation in T lymphocytes, examining the effect of dexameth-asone on IL-4-induced Stat6 phosphorylation. As expected, IL-4induced Stat6 phosphorylation (Fig. 4E, lane 6). In contrast toStat4, dexamethasone pretreatment did not affect IL-4-inducedStat6 phosphorylation (Fig. 4E,lane 7and8).
Dexamethasone pretreatment decreases IL-12-induced IRF-1promoter activity
To further characterize the specific inhibition of Stat4 activation bydexamethasone, we next examined its effect in a heterologous sys-tem using transfected NIH3T3 cells. In this model, we and othershave previously shown that the IRF-1 induction by IL-12 is de-pendent upon Stat4 (23, 24). This system allowed us to dissect theelements involved in the IL-12 signaling pathway and to study theeffect of dexamethasone.
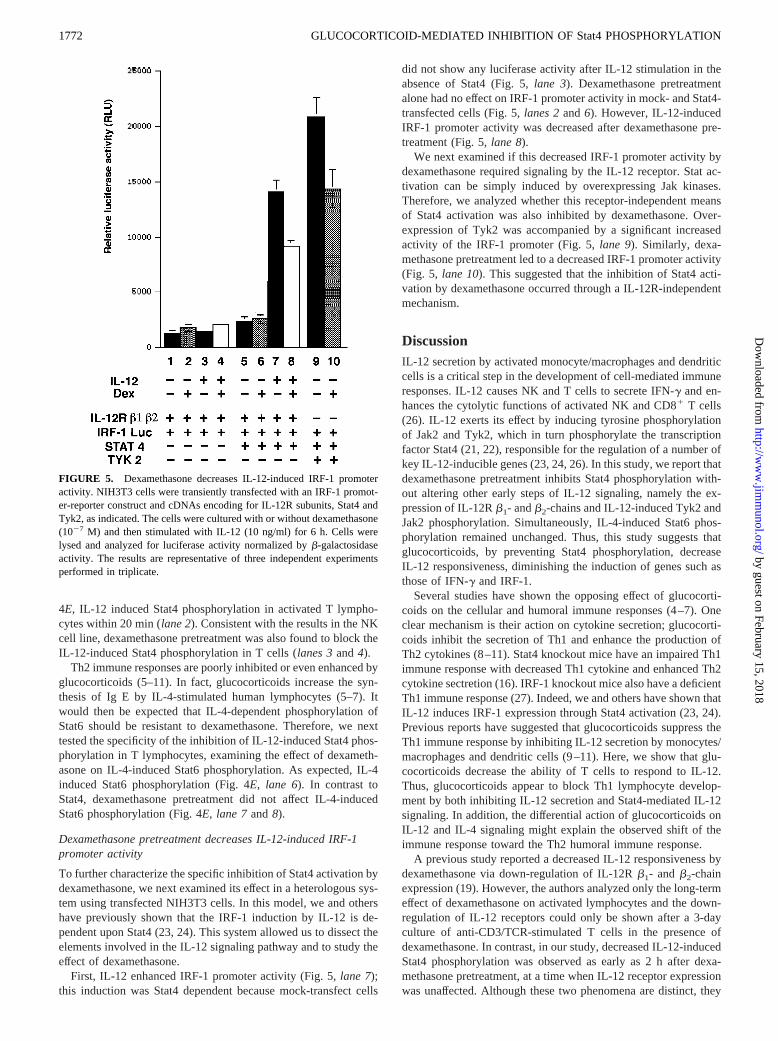
First, IL-12 enhanced IRF-1 promoter activity (Fig. 5,lane 7);this induction was Stat4 dependent because mock-transfect cells
did not show any luciferase activity after IL-12 stimulation in theabsence of Stat4 (Fig. 5,lane 3). Dexamethasone pretreatmentalone had no effect on IRF-1 promoter activity in mock- and Stat4-transfected cells (Fig. 5,lanes 2and6). However, IL-12-inducedIRF-1 promoter activity was decreased after dexamethasone pre-treatment (Fig. 5,lane 8).
We next examined if this decreased IRF-1 promoter activity bydexamethasone required signaling by the IL-12 receptor. Stat ac-tivation can be simply induced by overexpressing Jak kinases.Therefore, we analyzed whether this receptor-independent meansof Stat4 activation was also inhibited by dexamethasone. Over-expression of Tyk2 was accompanied by a significant increasedactivity of the IRF-1 promoter (Fig. 5,lane 9). Similarly, dexa-methasone pretreatment led to a decreased IRF-1 promoter activity(Fig. 5, lane 10). This suggested that the inhibition of Stat4 acti-vation by dexamethasone occurred through a IL-12R-independentmechanism.
DiscussionIL-12 secretion by activated monocyte/macrophages and dendriticcells is a critical step in the development of cell-mediated immuneresponses. IL-12 causes NK and T cells to secrete IFN-g and en-hances the cytolytic functions of activated NK and CD81 T cells(26). IL-12 exerts its effect by inducing tyrosine phosphorylationof Jak2 and Tyk2, which in turn phosphorylate the transcriptionfactor Stat4 (21, 22), responsible for the regulation of a number ofkey IL-12-inducible genes (23, 24, 26). In this study, we report thatdexamethasone pretreatment inhibits Stat4 phosphorylation with-out altering other early steps of IL-12 signaling, namely the ex-pression of IL-12Rb1- andb2-chains and IL-12-induced Tyk2 andJak2 phosphorylation. Simultaneously, IL-4-induced Stat6 phos-phorylation remained unchanged. Thus, this study suggests thatglucocorticoids, by preventing Stat4 phosphorylation, decreaseIL-12 responsiveness, diminishing the induction of genes such asthose of IFN-g and IRF-1.
Several studies have shown the opposing effect of glucocorti-coids on the cellular and humoral immune responses (4–7). Oneclear mechanism is their action on cytokine secretion; glucocorti-coids inhibit the secretion of Th1 and enhance the production ofTh2 cytokines (8–11). Stat4 knockout mice have an impaired Th1immune response with decreased Th1 cytokine and enhanced Th2cytokine sectretion (16). IRF-1 knockout mice also have a deficientTh1 immune response (27). Indeed, we and others have shown thatIL-12 induces IRF-1 expression through Stat4 activation (23, 24).Previous reports have suggested that glucocorticoids suppress theTh1 immune response by inhibiting IL-12 secretion by monocytes/macrophages and dendritic cells (9–11). Here, we show that glu-cocorticoids decrease the ability of T cells to respond to IL-12.Thus, glucocorticoids appear to block Th1 lymphocyte develop-ment by both inhibiting IL-12 secretion and Stat4-mediated IL-12signaling. In addition, the differential action of glucocorticoids onIL-12 and IL-4 signaling might explain the observed shift of theimmune response toward the Th2 humoral immune response.
A previous study reported a decreased IL-12 responsiveness bydexamethasone via down-regulation of IL-12Rb1- and b2-chainexpression (19). However, the authors analyzed only the long-termeffect of dexamethasone on activated lymphocytes and the down-regulation of IL-12 receptors could only be shown after a 3-dayculture of anti-CD3/TCR-stimulated T cells in the presence ofdexamethasone. In contrast, in our study, decreased IL-12-inducedStat4 phosphorylation was observed as early as 2 h after dexa-methasone pretreatment, at a time when IL-12 receptor expressionwas unaffected. Although these two phenomena are distinct, they
FIGURE 5. Dexamethasone decreases IL-12-induced IRF-1 promoteractivity. NIH3T3 cells were transiently transfected with an IRF-1 promot-er-reporter construct and cDNAs encoding for IL-12R subunits, Stat4 andTyk2, as indicated. The cells were cultured with or without dexamethasone(1027 M) and then stimulated with IL-12 (10 ng/ml) for 6 h. Cells werelysed and analyzed for luciferase activity normalized byb-galactosidaseactivity. The results are representative of three independent experimentsperformed in triplicate.
1772 GLUCOCORTICOID-MEDIATED INHIBITION OF Stat4 PHOSPHORYLATION
by guest on February 15, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

might coexist and together contribute to decreased IL-12 respon-siveness in T lymphocytes. Indeed, because the down-regulation ofSTAT4 activation results in decreased IFN-g secretion (Fig. 1) andIFN-g has been demonstrated to increase expression of IL-12Rb2
(28), the two observations may be directly linked.Glucocorticoids could inhibit Stat4 phosphorylation through a
number of potential mechanisms, several of which have alreadybeen identified. These include the modulation of expression oractivity of different factors that play a role in the attenuation ofcytokine signaling, such as phosphatases, suppressors of cytokinesignaling proteins (SOCS and cytokine inhibitor Src homology2-containing protein (CIS)), or protein inhibitors of activated Stats(PIAS). Finally, glucocorticoids might inhibit the expression ofsome factor that is required for the activation of Stat4. A potentialnegative regulation of a positive acting factor may also be envis-aged, although such a factor as not been described yet.
Src homology 2 domain-containing tyrosine phosphatases likeSrc homology protein (SHP)-1 and SHP-2 can attenuate cytokineand hormone signaling (29–34). Glucocorticoids might inhibit cy-tokine signaling by inducing or activating these or other tyrosinephosphatases (29). Presently, we cannot determine whether SHP-1or SHP-2 associate with IL-12Rb2, the subunit containing severaltyrosine residues, and whether glucocorticoids affect this associa-tion, as no reagents that immunoprecipitate IL-12Rb2 are cur-rently available. However, this mechanism of action seems un-likely, granted that the inhibition of Stat4 phosphorylation bydexamethasone appears not to be IL-12R dependent. Furthermore,the lack of inhibition of IL-4-induced Stat6 phosphorylation in Tcells suggests that this phosphatase may be specific for Stat4.While it is well recognized that Stat phosphorylation is specific,specific Stat phosphatases have yet to be identified.
Another alternative explanation would be the regulation of dif-ferent members of the family of SOCS (also known as JAK-bind-ing protein or STAT-induced STAT inhibitor) (35–37). For in-stance, IL-10 suppresses IFN-a- and IFN-g-dependent Stat1phosphorylation, and this is accompanied by the up-regulation ofSOCS-3 (38). However, the inhibition of IL-12-induced Stat4phosphorylation without altering Jak activation argues against aninvolvement of SOCS; SOCS-1 is thought to bind to Jaks, inhib-iting their activity (35–37). Nevertheless, the first identified inhib-itor of Stat signaling, CIS, was reported not to bind to Jaks butrather to interact with phosphotyrosine residues in the erythropoe-itin receptor and IL-3bc, thereby preventing Stat phosphorylation(37, 39). Thus, CIS might be a good candidate for a steroid in-ducible inhibitor of Stat4 phosphorylation.
Finally, the recently described PIAS1 and -3 interact with andblock Stat1 and -3 DNA-binding activities but do not alter Stat phos-phorylation (40, 41). No Stat4-specific PIAS family member has beencharacterized as yet; moreover, PIAS wihout altering Stat phosphor-ylation may not play a role in this condition. Further studies will beneeded to clarify the underlying mechanism responsible for the in-hibitory action of glucocorticoids on Stat4 phosphorylation.
Activation of the HPA axis by proinflammatory cytokines re-sults in the release of endogenous glucocorticoids, which in turnsuppress innate and specific immune responses (3). The inflam-matory response of streptococcus cell wall-induced arthritis in sus-ceptible Lewis and resistant Fischer rats is inversely related to themagnitude of their HPA axis response to inflammatory mediators.Moreover, the glucocorticoid receptor antagonist RU486 rendersFischer rats susceptible to arthritis (42, 43). Also, adrenalectomy inLewis rats with experimental allergic encephalomyelitis, a Th1cell-mediated autoimmune disease, leads to a chronically activedisease, while substitutive replacement of glucocorticoids inducesrecovery from the disease (44, 45). Thus, endogenous glucocorti-
coids might decrease susceptibility to Th1-type immune diseasesby lowering T cell responsiveness to IL-12.
Glucocorticoids have been used for 50 years as potent immu-nosuppressants in the therapy of Th1 inflammatory and autoim-mune diseases. Glucocorticoids render treated patients immuno-compromised, with an increased susceptibility to intracellularinfections. The ability of glucocorticoids to specifically suppressTh1 cellular immunity is well described but poorly understood.Our study suggests that glucocorticoid-mediated inhibition ofStat4 phosphorylation may represent a major underlying mecha-nism of the specific suppression of cellular immunity byglucocorticoids.
References1. Boumpas, D. T., G. P. Chrousos, R. L. Wilder, T. R. Cupps, and J. E. Balow.
1993. Glucocorticoid therapy for immune-mediated diseases: basic and clinicalcorrelates.Ann. Intern. Med. 119:1198.
2. Chrousos, G. P. 1995. The hypothalamic-pituitary-adrenal axis and immune-me-diated inflammation.N. Engl. J. Med. 20:1351.
3. Besedovsky, H. O., and A. Del Rey. 1996. Immune-neuro-endocrine interactions:facts and hypotheses.Endocr. Rev. 17:64.
4. Wilkens, T., and R. De Rijk. 1998. Glucocorticoids and immune function.Im-munol. Today 18:418.
5. Wu, C. Y., M. Sarfati, C. Heusser, S. Fournier, M. Rubio-Trujillo, R. Peleman,and G. Delespesse. 1991. Glucocorticoids increase the synthesis of immunoglob-ulin E by interleukin 4-stimulated human lynmphocytes.J. Clin. Invest. 87:870.
6. Akdis, C. A., T. Blesken, M. Akdis, S. S. Alkan, C. H. Heusser, and K. Blaser.1997. Glucocorticoids inhibit human antigen specific and enhance total IgE andIgG4 production due to differential effects on T and B cells in vitro.Eur. J. Im-munol. 9:2351.
7. Zieg, G., G. Lack, R. J. Harbeck, E. W. Gefland, and D. Y. Leung. 1994. In vivoeffects of glucocorticoids on IgE production.J. Allergy Clin. Immunol. 94:222.
8. Ramirez, F., D. J. Fowel, M. Puklavec, S. Simmonds, and D. Mason. 1996.Glucocorticoids promote a Th2 cytokine response by CD41 T cells in vitro.J. Immunol. 156:2406.
9. De Kruyff, R. H., Y. Fang, and D. T. Umetsu. 1998. Corticosteroids enhance thecapacity of macrophages to induce Th2 cytokines synthesis in CD41 lympho-cytes by inhibiting IL-12 production.J. Immunol. 160:2231.
10. Blotta, M. H., R. H. Dekruyff, and D. T. Umetsu. 1997. Corticosteroids inhibitIL-12 production in human monocytes and enhance their capacity to induce IL-4synthesis in CD41 lymphocytes.J. Immunol. 158:5589.
11. Elenkov, I. J., D. A. Papanicolaou, R. L. Wilder, and G. P. Chrousos. 1995.Modulatory effects of glucocorticoids and cathecolamines on human interleu-kin-12 and interleukin-10 production: clinical implications.Proc. Assoc. Am.Physicians 108:374.
12. Mosmann, T. R., and S. Sad. 1996. The expanding universe of T-cell subsets:Th1, Th2 and more.Immunol. Today 17:138.
13. Romagnani, S. 1997. The Th1/Th2 paradigm.Immunol. Today 18:263.14. Leonard, W. J., and J. J. O’Shea. 1998. Jaks and Stats: biological implications.
Annu. Rev. Immunol. 16:293.15. Thierfelder, W. E., J. M. van Deursen, K. Yamamoto, R. A. Tripp, S. R. Sarawar,
R. T. Carson, M. Y. Sangster, D. A. Vignali, P. C. Doherty, G. C. Grosveld, andJ. N. Ihle. 1996. Requirement for Stat4 in interleukin-12-mediated responses ofnatural killer and T cells.Nature 382:171.
16. Kaplan, M. H., S. Ya-Lin, T. Hoey, and M. J. Grusby. 1996. Impaired IL-12responses and enhanced development of Th 2 cells in Stat4-deficient mice.Na-ture 382:174.
17. Takeda, K., T. Tanaka, W. Shim, M. Matsumotom, M. Minami,S. I. Kashiwamura, K. Nakanishi, N. Yoshida, T. Kishimoto, and S. Akira. 1996.Essential role of Stat6 in IL-4 signalling.Nature 380:627.
18. Kaplan, M. H., U. Schindler, S. T. Smiley, and M. J. Grusby. 1996. Stat6 isrequired for mediating responses to IL-4 and for the development of Th2 cells.Immunity 4:313.
19. Wu, C. Y., K. Wangm, J. F. McDyerm, and R. A. Seder. 1998. Prostaglandin E2
and dexamethasone inhibit IL-12 receptor expression and IL-12 responsivness.J. Immunol. 161:2723.
20. Johnston, J. A., M. Kawamura, R. A. Kirken, Y. Q. Chen, T. B. Blake,K. Shibuya, J. R. Ortaldo, D. W. McVicar, and J. J. O’Shea. 1994. Phosphory-lation and activation of the Jak-3 Janus kinase in response to interleukin-2.Na-ture 370:151.
21. Bacon, C. M., E. F. Petricoin, J. R. Ortaldo, R. C. Rees, A. C. Larner,J. A. Johnston, and J. J. O’Shea. 1995. Interleukin 12 induces tyrosine phosphor-ylation and activation of Stat4 in human lymphocytes.Proc. Natl. Acad. Sci. USA92:7307.
22. Bacon, C. M., D. W. McVicar, J. R. Ortaldo, R. C. Rees, J. J. O’Shea, andJ. A. Johnston. 1995. Interleukin 12 (IL-12) induces tyrosine phosphorylation ofJAK2 and TYK2: differential use of Janus family tyrosine kinases by IL-2 andIL-12. J. Exp. Med. 181:399.
23. Coccia, E. M., M. Passini, A. Battistini, F. Sinigaglia, and L. Rogge. 1999. In-terleukin-12 induces expression of interferon regulatory factor-1 via signal trans-ducer and activator of transcription-4 in human T helper type 1 cells.J. Biol.Chem. 274:6698.
1773The Journal of Immunology
by guest on February 15, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

24. Galon, J., C. Sudarshan, I. Satochi, D. Finbloom, and J. J. O’Shea. 1999. IL-12induces interferon regulating factor-1 (IRF-1) gene expression in human NK andT cells.J. Immunol. 62:7256.
25. Cho, S. S., C. M. Bacon, C. Sudarshan, R.C. Rees, D. Finbloom, R. Pine, and J. J.O’Shea. 1996. Activation of Stat4 by IL-12 and IFNa: evidence for the involve-ment of ligand-induced tyrosine and serine phosphorylation.J. Immunol.157:4781.
26. Gately, M. K., L. M. Renzetti, J. Magram, A. S. Stern, L. Adorini, U. Gubler,D. Presky. 1998. The interleukin-12/interleukin-12-receptor system: role in nor-mal and pathologic immune responses.Annu. Rev. Immunol. 16:495.
27. Taki, S., T. Sato, K. Ogasawara, T. Fukuda, M. Sato, S. Hida, G. Suzuki,M. Mitsuyama, E. H. Shin, S. Kojima, T. Taniguchi, and Y. Asano. 1997. Mul-tistage regulation of Th 1-type immune responses by the transcription factorIRF-1. Immunity 6:673.
28. Szabo, S. J., A. S. Dighe, U. Gubler, and K. M. Murphy. 1997. Regulation of theinterleukin (IL)-12Rb2 subunit expression in developing T helper 1 (Th1) andTh2 cells.J. Exp. Med. 185:817.
29. Thomas, M. L. 1995. Positive and negative regulation of leukocyte activation byprotein tyrosine phosphatases.Semin. Immunol. 7:279.
30. Migone, T. S., N. A. Cacalano, N. Taylor, T. Yi, T. A. Waldmann, andJ. A. Johnston. 1998. Recruitment of SH2-containing protein tyrosine phospha-tase SHP-1 to the interleukin 2 receptor: loss of SHP-1 expression in humanT-lymphotropic virus type I-transformed T cells.Proc. Natl. Acad. Sci. USA95:3845.
31. Yi, T., A. L. Mui, G. Krystal, and J. N. Ihle. 1993. Hematopoietic cell phospha-tase associates with the interleukin-3 (IL-3) receptorb chain and down-regulatesIL-3-induced tyrosine phosphorylation and mitogenesis.Mol. Cell Biol. 13:7577.
32. Klingmuller, U., U. Lorenz, L.C. Cantley, B. G. Neel and H. F. Lodish. 1995.Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactiva-tion of JAK2 and termination of proliferative signals.Cell 80:729.
33. Petricoin, E. F., M. David, K. Igarashi, C. Benjamin, L. Ling, S. Goelz,D. S. Finbloom, and A. C. Larner. 1996. Inhibition ofa interferon but notginterferon signal transduction by phorbol esters is mediated by a tyrosine phos-phatase.Mol. Cell Biol. 16:1419.
34. You, M., D. H. Yu, and G. S. Feng. 1999. SHP-2 tyrosine phosphatase functionsas a negative regulator of the interferon-stimulated Jak/STAT pathway.Mol. CellBiol. 19:2416.
35. Naka, T., M. Narasazaki, M. Hirata, T. Matsumoto, S. Minamoto, A. Aono,N. Nishimoto, T. Kajita, T. Taga, K. Yoshizaki, S. Akira, and T. Kishimoto.
1997. Structure and function of a new STAT-induced STAT inhibitor.Nature387:924.
36. Starr, R., T. A. Wilson, E. M. Viney, L. J. L. Murray, J. R. Rayner, B. J. Jenkins,T. L. Gonda, W. S. Alexander, D. Metcalf, N. A. Nocola, and D. J. Hilton. 1997.A family of cytokine-inducible inhibitors of signaling.Nature 387:917.
37. Endo, T. A., M. Masuhara, M. Yocouchi, R. Suzuki, H. Sakamoto, K. Mitsul,A. Matsumoto, S. Tanimura, M. Ohtsubo, T. Miyazaki, et al. 1997. A new proteincontaining an SH2 domain that inhibits Jak kinases.Nature 387:921.
38. Ito, S., P. Ansari, M. Sakatsume, H. Dickensheets, N. Vazquez, R. P. Donnelly,A. C. Larner, and D. S. Finbloom. 1999. Interleukin 10 inhibits expression ofboth interferona- and interferong-induced gene by suppressing tyrosine phos-phorylation of Stat1.Blood 93:1456.
39. Yoshimura, A., T. Ohkubom, T. Kigushi, N. A. Jenkins, D. J. Gilbert,N. G. Copeland, T. Hara, and A. Miyajima. 1995. A novel cytokine-induciblegene encodes an SH2-containing protein that binds to tyrosine phosphorylatedinterleukin 3 and erythropoietin receptors.EMBO J. 14:2816.
40. Chung, C. D., J. Liao, B. Liu, X. Rao, P. Jay, P. Berta, and K. Shuai. 1997.Specific inhibition of Stat3 signal transduction by PIAS3.Science 278:1803.
41. Liu, B., X. Liao, S. A. Kushner, C. D. Chung, D. D. Chang, and K. Shuai. 1998.Inhibition of Stat1-mediated gene activation by PIAS1.Proc. Natl. Acad. Sci.USA 95:10226.
42. Sternberg, E. M., J. M. Hill, G. P. Chrousos, T. Kamilaris, S. J. Listwak,P. W. Gold, and R. L. Wilder. 1989. Inflammatory mediator-induced hypotha-lamic-pituitary-adrenal axis activation is defective in streptococcal cell wall ar-thritis-susceptible Lewis rats.Proc. Natl. Acad. Sci. USA 86:2374.
43. Sternberg, E. M., W. Young, R. Bernardini, A. E. Calogero, G. P. Chrousos,P. W. Gold, and R. L. Wilder. 1989. A central nervous system defect in biosyn-thesis of corticotropin-releasing hormone is associated with susceptibility tostreptococcal cell wall-induced arthritis in Lewis rats.Proc. Natl. Acad. Sci. USA86:4771.
44. MacPhee, I. A. M., F. A. Antoni, and D. W. Mason. 1989. Spontaneous recoveryof rats from experimental allergic encephalomyelitis is dependent on regulationof the immune system by endogenous adrenal corticosteroids.J. Exp. Med. 169:431.
45. Zamvil, S. S., and L. Steinman. 1990. The T lymphocyte in experimental allergicencephalomyelitis.Annu. Rev. Immunol. 8:579.
1774 GLUCOCORTICOID-MEDIATED INHIBITION OF Stat4 PHOSPHORYLATION
by guest on February 15, 2018http://w
ww
.jimm
unol.org/D
ownloaded from