interval mapping basics
TRANSCRIPT

ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 1
2 Key Statistical Issues for QTL• general notation and data structure• recombination model
– two linked markers– flanking markers to a QTL– map distance and map functions
• modelling the phenotype– phenotype model– model likelihood– Bayesian posterior
• missing data concepts and algorithms• model selection
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 2
interval mapping basics• observed measurements
– Y = phenotypic trait– X = markers & linkage map
• i = individual index 1,…,n• missing data
– missing marker data– Q = QT genotypes
• alleles QQ, Qq, or qq at locus• unknown genetic architecture
– λ = QT locus (or loci)– θ = genetic action– m = number of QTL
• pr(Q|X,λ,m) recombination model– grounded by linkage map, experimental cross– recombination yields multinomial for Q given X
• pr(Y|Q,θ,m) phenotype model– distribution shape (assumed normal here) – unknown parameters θ (could be non-parametric)
observed X Y
missing Q
unknown λ θafter
Sen Churchill (2001)

ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 3
2.1 general notation and data structure
• Y = phenotype values– as concept and realized (observed) values
• X = marker genotypes– type of experimental cross– linkage map construction
• marker orders, positions, linkage phases– observed marker genotypes (possibly with error)
• pr(Y,X) = joint probability– what we “know” about Y and X for this experiment– usually assume linkage map is “known”
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 4
conditional data likelihood
• condition on markers and linkage map
• pr(X) comprises information on linkage map– not influenced by phenotype– thus can “ignore” for QTL purposes
)(pr),(pr)|(pr
XXYXY =

ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 5
unknown QTL genotypes• usually have sparse linkage map of markers
– want to condition on actual QTL genotype Qpr(Y|Q)
– but actual QTL affecting phenotype not known• need to consider all possibilities
– average pr(Y|Q) over all possible genotypes Q– weight by recombination pr(Q|X)
∑=Q
XQQYXY )|(pr)|(pr)|(pr
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 6
enter the (Greek) parameters• θ = genetic effects, or gene action
– additive, dominance, epistasis– may include reference values
• grand mean (µ), environmental variance (σ2)
• λ = location(s) of QTL– measured along “linear” genome– related to recombination and map distance
∑==Q
XQQYXYXYL ),|(pr),|(pr),,|(pr),|,( λθλθλθ

ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 7
2.2 recombination model• locus λ is distance along linkage map
– identifies flanking marker region• flanking markers provide good approximation
– map assumed known from earlier study– inaccuracy slight using only flanking markers
• extend to next flanking markers if missing data– could consider more complicated relationship
• but little change in results
pr(Q|X,λ) = pr(geno | map, locus) ≈pr(geno | flanking markers, locus)
kX 1+kXQ?
λ
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 8
• backcross design– n individuals, 2 markers– follow one gamete
• recombinants– Ab, aB– nR = n12 + n21
• non-recombinants– ab, AB– nNR = n11 + n22
• recombination rate
2.2.1 two linked markersA B
22211211
2112ˆnnnn
nnnnr R
++++==
r
21122211
,,nnnnnn
aBAbABabRNR
RNR +=+=
n21

ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 9
no linkage?• test for no linkage: r = 1/2• assumption: all individuals have same rate
– implies binomial variation
• normal or chi-square test statisticn
rrrnnnn
nnnnr R )ˆ1(ˆ)ˆ( var,ˆ
22211211
2112 −≈+++
+==
21
22 ~
)/()2/(Zor )1,0(~
)ˆ(var2/1ˆ χ
nnnnnN
rrZ
NRR
R −=−=
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 10
binomial probabilitiesbinomial probn = 30,100r = 0.4,0.2
normal approx
knkR rr
kn
kn −−
== )1()(pr

ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 11
likelihood ratio and LOD test• likelihood for linked markers
• likelihood for unlinked markers
• likelihood ratio and LOD
nCL )()( 21
21 =
22
10
21
2
217.)10log(2
)(log
~)log(2,)ˆ1()ˆ(2
GGLRLOD
LRGrrLR NRR nnn
===
=−= χ
NRR nnR rCrrnnrL )1(),|(pr)( −==
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 12
test statistic: distribution
• Z2 and G2 are generally close to each other– Z2 based on properties of counts– G2 and LOD based on likelihood principle– both have approximate chi-square distribution
• (non)central chi-square distribution
22;1
22
21
22
)5.0(4,~,:5.0
~,:5.0
rnncpGZr
GZr
ncp −=<
=
χχ

ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 13
backcross examples
• n=100 individuals, nR= 40 recombinants– r = 0.4, se(r) = 0.049– Z = -2.04, Z2 = 4.17, p-value = 0.041– G2 = 4.03, LOD = 0.874, p-value = 0.045
• n=100 individuals, nR= 20 recombinants– r = 0.2, se(r) = 0.04– Z = -7.5, Z2 = 56.25, p-value < 0.0001– G2 = 38.55, LOD = 8.37, p-value < 0.0001
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 14
backcross examples
• n=30 individuals, nR= 12 recombinants– r = 0.4, se(r) = 0.089– Z = -1.12, Z2 = 1.25, p-value = 0.26– G2 = 1.21, LOD = 0.262, p-value = 0.27
• n=30 individuals, nR= 6 recombinants– r = 0.2, se(r) = 0.073– Z = -4.11, Z2 = 16.87, p-value < 0.0001– G2 = 11.56, LOD = 2.51, p-value < 0.0001

ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 15
simulations of LOD distribution
n=100,r=0.3,0.51000 sampleshistogram
chi-square curverescaled by 2log(10)
centralr = 0.5
non-centralr = 0.3
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 16
LOD and LR over possible rn = 30nR=12 or 6evaluate at
all possible rnot just “best”
LR like a densityLOD is basis for
hypothesis test estimate interval
0.0 0.1 0.2 0.3 0.4 0.5
0.0
e+00
5.0
e-19
1.0
e-18
1.5
e-18
recombination rate
likel
ihoo
d ra
tio
0.0 0.1 0.2 0.3 0.4 0.5
-15
-10
-50
recombination rate
LOD
0.0 0.1 0.2 0.3 0.4 0.5
0.0
e+00
1.0
e-16
2.0
e-16
recombination rate
likel
ihoo
d ra
tio
0.0 0.1 0.2 0.3 0.4 0.5
-4-2
02
recombination rate
LOD

ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 17
LR, LOD and p-values
p-value p-valueLR LOD 1 d.f. 2 d.f.10 1 0.0319 0.131.6 1.5 0.0086 0.0316100 2 0.0024 0.011000 3 0.0002 0.00110000 4 <0.0001 0.0001
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 18
LOD-based interval estimate for rpoint estimate interval estimate
from LOD peakdown 1.5 LOD(or 1 or 2 or …)
n = 30, nR = 12, 6
nnr R /ˆ =
0.20 0.25 0.30 0.35 0.40 0.45 0.50
-1.5
-1.0
-0.5
0.0
n=30,r=0.4
LOD
0.1 0.2 0.3 0.4
0.5
1.0
1.5
2.0
2.5
n=30,r=0.2
LOD

ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 19
LOD-based interval calculationsconfidence 96.8% 99.1% 99.76%n nR r 1 LOD 1.5 LOD 2 LOD
30 3 0.1 0.03-0.25 0.02-0.29 0.01-0.33100 10 0.1 0.05-0.17 0.04-0.19 0.04-0.2130 6 0.2 0.08-0.37 0.06-0.42 0.05-0.46100 20 0.2 0.13-0.29 0.11-0.31 0.10-0.3330 12 0.4 0.23-0.50 0.19-0.50 0.17-0.50100 40 0.4 0.30-0.50 0.28-0.50 0.26-0.50
Note skew in intervals for small recombination rates.Note upper boundary of 0.5.
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 20
LOD-based intervals
0.00 0.10 0.20 0.30
3.0
3.5
4.0
4.5
n=30,r=0.1
LOD
0.1 0.2 0.3 0.4
0.5
1.0
1.5
2.0
2.5
n=30,r=0.2
LOD
0.20 0.30 0.40 0.50
-1.5
-1.0
-0.5
0.0
n=30,r=0.4
LOD
0.05 0.10 0.15 0.20
14.0
14.5
15.0
15.5
16.0
n=100,r=0.1
LOD
0.10 0.15 0.20 0.25 0.30
6.5
7.0
7.5
8.0
n=100,r=0.2
LOD
0.30 0.35 0.40 0.45 0.50
-1.0
-0.5
0.0
0.5
n=100,r=0.4
LOD

ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 21
likelihood & Bayesian posterior• recall the likelihood and likelihood ratio:
• posterior turns likelihood into a density– assume r may be any value prior to seeing data– posterior = likelihood x prior / constant
NRR
NRR
nnn
nnR
rrrLR
rCrrnnrL
)1(2)(
)1(),|(pr)(
−=
−==
1),|(pr sumcurve or likelihoodunder area
/)(or /)(),|(pr
==
==
Rr
R
nnrLRA
ArLRArLnnr
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 22
LR and Bayes posteriorimagine LR as density
area under curve = 1pr(r | nR ) = LR(r) / A
what is probability that ris between 0.25 and 0.5?
where is interval with highest posterior mass? (HPD region)
example: n=30, nR =12,695% HPD regions
0.0 0.1 0.2 0.3 0.4 0.5
0.0
e+00
1.0
e-18
recombination rate
likel
ihoo
d ra
tio
0.0 0.1 0.2 0.3 0.4 0.5
0.00
00.
010
0.02
0
recombination rate
dens
ity
0.0 0.1 0.2 0.3 0.4 0.5
0.0
e+00
1.5
e-16
recombination rate
likel
ihoo
d ra
tio
0.0 0.1 0.2 0.3 0.4 0.5
0.00
00.
010
0.02
0
recombination rate
dens
ity
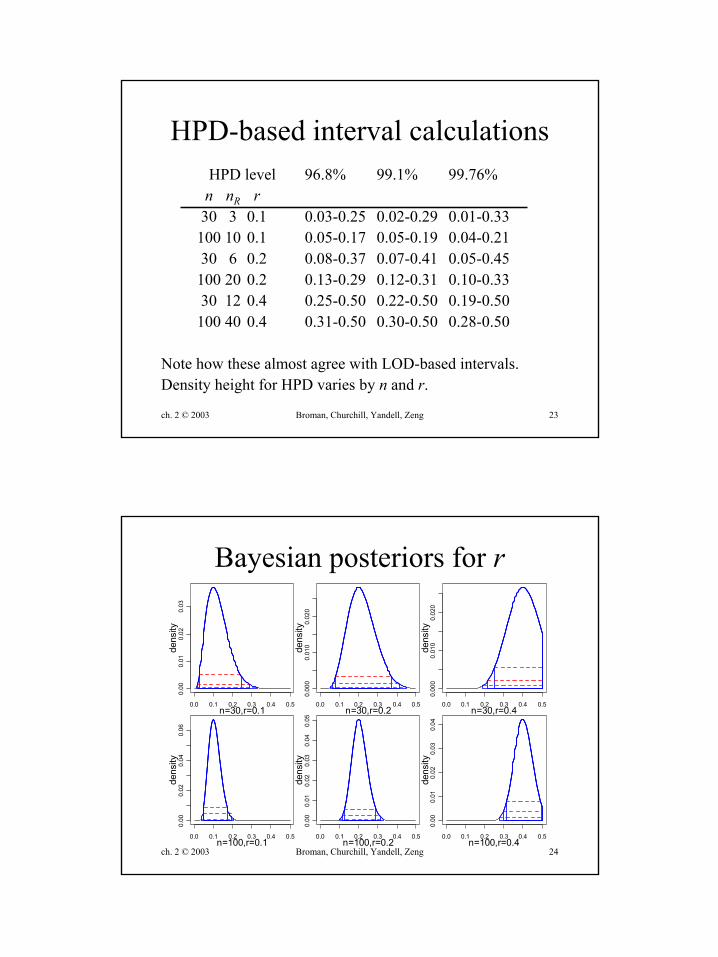
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 23
HPD-based interval calculationsHPD level 96.8% 99.1% 99.76%n nR r
30 3 0.1 0.03-0.25 0.02-0.29 0.01-0.33100 10 0.1 0.05-0.17 0.05-0.19 0.04-0.2130 6 0.2 0.08-0.37 0.07-0.41 0.05-0.45100 20 0.2 0.13-0.29 0.12-0.31 0.10-0.3330 12 0.4 0.25-0.50 0.22-0.50 0.19-0.50100 40 0.4 0.31-0.50 0.30-0.50 0.28-0.50
Note how these almost agree with LOD-based intervals.Density height for HPD varies by n and r.
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 24
Bayesian posteriors for r
0.0 0.1 0.2 0.3 0.4 0.5
0.00
0.01
0.02
0.03
n=30,r=0.1
dens
ity
0.0 0.1 0.2 0.3 0.4 0.5
0.00
00.
010
0.02
0
n=30,r=0.2
dens
ity
0.0 0.1 0.2 0.3 0.4 0.5
0.00
00.
010
0.02
0
n=30,r=0.4
dens
ity
0.0 0.1 0.2 0.3 0.4 0.5
0.00
0.02
0.04
0.06
n=100,r=0.1
dens
ity
0.0 0.1 0.2 0.3 0.4 0.5
0.00
0.01
0.02
0.03
0.04
0.05
n=100,r=0.2
dens
ity
0.0 0.1 0.2 0.3 0.4 0.5
0.00
0.01
0.02
0.03
0.04
n=100,r=0.4
dens
ity

ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 25
• Reverend Thomas Bayes (1702-1761)– part-time mathematician– buried in Bunhill Cemetary, Moongate, London– famous paper in 1763 Phil Trans Roy Soc London– was Bayes the first with this idea? (Laplace)
• billiard balls on rectangular table– two balls tossed at random (uniform) on table– where is first ball if the second is to its left (right)?
who was Bayes?
prior pr(θ) = 1likelihood pr(Y | θ)= θY(1- θ)1-Y
posterior pr(θ |Y)= ?
θ
Y=1 Y=0
firstsecond
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 26
where is the first ball?
=−=
=
=−=
=
∫ −
0)1(212
)|(pr
21)1()(pr
)(pr)(pr)|(pr)|pr(
1
0
1
YY
Y
dY
YYY
YY
θθ
θ
θθθ
θθθ
prior pr(θ) = 1likelihood pr(Y | θ)= θY(1- θ)1-Y
posterior pr(θ |Y)= ?
θ
Y=1 Y=0
first ball
second ball
0.0 0.2 0.4 0.6 0.8 1.0
0.0
0.5
1.0
1.5
2.0
Y = 1
Y = 0
θ
pr(θ
|Y)
(now throw second ball n times)

ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 27
what is Bayes theorem?• before and after observing data
– prior: pr(θ) = pr(parameters)– posterior: pr(θ|Y) = pr(parameters|data)
• posterior = likelihood * prior / constant– usual likelihood of parameters given data– normalizing constant pr(Y) depends only on data
• constant often drops out of calculation
)(pr)(pr)|(pr
)(pr),(pr)|(pr
YY
YYY θθθθ ×==
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 28
Bayes rule for recombination r
∫=
×=
≤≤≤−=≤≤
−==
2/1
02),|(pr)|(pr
:constant g normalizin)|(pr
)(pr),|(pr),|(pr
:rule Bayes2/10
)(2)pr(: ion recombinat onprior
)1(),|(),|(pr
:likelihood
drrnnnn
nnrrnnnnr
baabbra
rrCrnnrLrnn
RR
R
RR
nnRR
NRR
0.0 0.1 0.2 0.3 0.4 0.5
01
23
45
posterior & prior
rela
tive
frequ
ency n=30
nR=12
0.0 0.1 0.2 0.3 0.4 0.5
01
23
45
posterior & prior
rela
tive
frequ
ency n=30
nR=6

ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 29
two markers in F2 intercross• two meioses
– follow both gametes– 16 possibilities
• ambiguity with AaBb– 0 or 2 recombinations
• log likelihood ratio:lkjlj
( ))5.0(/)(logsumlog xxxx frfnLR =
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 30
two markers in F2 intercross• γ = probability of double recombinant
– for AaBb genotype, haplotype not known– need to “guess” the recombinant fraction of n11 offspring
• γ and r are inter-related– no “closed” solution, need to iterate– guess γ , use to estimate r, improve γ , etc.
[ ]) ˆ(2)(21ˆ
)1(AaBb
aBAbpr
11200221121001
22
2|nnnnnnn
nr
rrr
γ
γ
++++++=
+−=
=

ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 31
EM algorithm for F2 recombination• initial guess: r = 0.5, γ = 0.5• Expectation (E) step
– substitute expected values for nuisance γ– update γ given current value of r
• Maximization (M) step– maximize likelihood for parameter r– update r given current value of γ
• iterate E-step and M-step until “convergence”– stop when change in log-likelihood is small
– usually change in r is small at this point( ))5.0(/)(logsumlog xxxx frfnLR =
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 32
2.2.2 flanking markers to QTL
• most genotype information is local– linkage drops off with distance– approximate by using only flanking markers– exception: linkage disequilibrium
• different chromosome regions could be correlated• due to selection, etc.• not a problem for backcross or F2 intercross
• missing marker data: use next flanking marker
A QrAQBrQB
rAB

ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 33
backcross QTL & flanking markers1 meiosis8 possible genotypes3 recombination ratessmall distances & rates?
no double crossoversρ = rAQ /rAB
A QrAQBrQB
rAB
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 34
F2 QTL & flanking markers2 meioses27 possible genotypes3 recombination ratesEM steps on γ and rABsmall distances & rates?
no double crossovers
A QrAQBrQB
rAB
22
2
)1(,/
ABAB
ABABAQ rr
rrr+−
== γρ

ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 35
2.2.3 map distance & map functions• How to relate genetic linkage to physical distance?
– math assumptions = crude approximations– critical for map building, minor effect on QTL
• x = genetic map distance (1Morgan = 100cM)– expected number of crossovers per meiosis between
two loci on a single chromatid strand (Sturtevant 1913)
• typical map functions– Morgan: interference rAB = rAQ + rQB
– Kosambi: intermediate rAB = (rAQ + rQB)/(1+4rAQ rQB)– Haldane: no interference rAB = rAQ + rQB - 2rAQ rQB
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 36
2.3 modelling the phenotype• trait = mean + genetic + environment• pr( trait Y | genotype Q, effects θ )
EGYGQY
Q
Q
++=
=
µσθ ),(normal),|(pr 2
8 9 10 11 12 13 14 15 16 17 18 19 20
qq QQ

ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 37
caution: don’t trust raw histograms!
8 9 10 11 12 13 14 15 16
qqQq
no QTL?
8 9 10 11 12 13 14 15 16
qqQq
skewed?
8 9 10 11 12 13 14 15 16 17 18 19 20 21
qqQq
dominance?
0 1 2 3 4 5 6 7 8 9
QqQQ
zeros?
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 38
2.3.1 phenotype model• how is phenotype related to genotype?• typical assumptions
– normal environmental variation• residuals e (not Y!) have bell-shaped histogram
– genetic value GQ is composite of a few QTL• other polygenic effects same across all individuals
– genetic effect uncorrelated with environment
effects ),,(
),|(var,),|(
),0(~,
2
2
2
σµθ
σθµθ
σµ
Q
Q
Q
G
QYGQYE
NeeGY
=
=+=
++=

ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 39
F2 intercross phenotype model• here assume only one QTL• genotypes QQ, Qq, qq• genotypic values GQQ, GQq, Gqq
• decompose as additive, dominance effects
222:Cockerham-Fisher:Jinx-Mather
qqQqQQ: genotype
δδδ αµµαµαµδµαµ−−+−+=
−++==
Q
Q
GG
Q
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 40
2.3.2 model likelihood• why study the likelihood?
– uncover hidden aspects of QTL– loci λ, effects θ, given data (Y,X)
• what is evidence to support a QTL?• where are the QTL? • how precise can estimate the loci & effects?• what genetic architecture is supported?

ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 41
building the model likelihood• likelihood links phenotype & recombination
– through unknown QTL genotypes Q– mixture over all possible genotypes
• contribution from individual i
• product over all individuals
),,|(pr prod),|,(
),|(pr),|(prsum prod),|,(
),|(pr),|(pr sum),,|(pr
λθλθλθλθ
λθλθ
iii
iiQi
iiQii
XYXYL
XQQYXYL
XQQYXY
=
=
=
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 42
and if there are no QTL?
• Y = µ + e, or L(µ | Y )• no relationship with markers & map X• for normal data, maximum likelihood yields
nYYs
nYYYNYL
ii
ii
/)(sumˆ/sumˆ
),|()|(
222
2
−==
===
•
σµ
σµµ

ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 43
maximum likelihood & LOD• likelihood peaks at some (θ,λ)
– use “hat” (^) to signify value at maximum• LOD profiles likelihood peak
– find θ to maximize likelihood for each λ– profile (scan) loci λ over entire genome
=
==
)|(max),|,(maxlog),|(
),,|(pr prod),,|(pr),|,(
10 YLXYLXYLOD
XYXYXYL iii
µλθλ
λθλθλθ
µ
θ
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 44
2.3.3 Bayesian posterior
• treat unknowns as random– build “uncertainty” into model framework– genetic architecture: gene action θ, QTL locus λ
• interpret weighted likelihood as a density– weights based on prior “beliefs”
)|(pr)(pr)|,(pr)|(pr
)|,(pr),,|(pr),|,(pr
XXXY
XXYXY
λθλθ
λθλθλθ
=
=

ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 45
choice of Bayesian priors• elicited priors
– higher weight for more probable parameter values• based on prior empirical knowledge
– use previous study to inform current study• weather prediction, previous QTL studies on related organisms
• conjugate priors– convenient mathematical form– essential before computers, helpful now to simply computation– large variances on priors reduces their influence on posterior
• non-informative priors – may have “no” information on unknown parameters– prior with all parameter values equally likely
• may not sum to 1 (improper), which can complicate use• always check sensitivity of posterior to choice of prior
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 46
incorporate missing genotypes Q• augment data with missing genotypes Q
– use recombination model to state uncertainty– avoid likelihood mixture by augmentation
• marginal posterior on unknowns of interest– average over fully augmented posterior
),|,,(pr sum),|,(pr)|(pr
)|,(pr),|(pr),|(pr),|,,(pr
XYQXYXY
XXQQYXYQ
Q λθλθ
λθλθλθ
=
=

ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 47
Bayesian parameter estimates• estimates are posterior means or modes
– mean = weighted average of all possible values– mode = maximum
• can get joint or marginal estimates
( )),|,(pr sum argmaxˆ),|,(pr sumˆ
mode
,mean
XY
XY
λθθ
λθθθ
λθ
λθ
=
=
ch. 2 © 2003 Broman, Churchill, Yandell, Zeng 48
2.4 missing data concepts
• missing QTL genotype Q--see section 2.3• missing marker data X
– errors in genotyping– difficulty reading signal (on gel)– marker not fully informative
• distinguish full data X from observed Xobs– weighted average over all possible marker values
)|(pr),|(prsum),|(pr obsobs XXXQXQ X λλ =