intestinal barriers to oral drug absorption - diva portal
TRANSCRIPT


Dissertation for the Degree of Doctor of Philosophy (Faculty of Pharmacy) in Pharmaceutics presented at Uppsala University in 2003
ABSTRACT
Engman, H., 2003. Intestinal Barriers to Oral Drug Absorption: Cytochrome P450 3A and ABC-Transport Proteins. Acta Universitatis Upsaliensis. Comprehensive Summaries of Uppsala Dissertations from the Faculty of Pharmacy 296. 61 pp. Uppsala.ISBN 91-554-5752-5.
The subject of this thesis was to study two intestinal barriers to oral drug bioavailability, drug efflux proteins of the ABC-transporter family, and in particular ABCB1/P-glycoprotein (Pgp), and the drug metabolizing enzyme cytochrome P450 (CYP) 3A4. At the onset of this thesis, similarities between CYP3A4 and Pgp in terms of their tissue distribution and gene regulation, along with overlapping substrate specificities, had generated the hypothesis that CYP3A4 and Pgp may have a complementary function and thus form a coordinated intestinal barrier to drug absorption and gut wall metabolism. In the first part of this thesis, a cell culture model of the intestinal epithelium that expressed both functional Pgp and CYP3A4 was developed. This model was then used to investigate the steroselective drug efflux and metabolism of R/S-verapamil. In summary, the results indicated that the two barriers in the cell culture model were in agreement with those in the human intestine. Both ABC-transporters and CYPs are regulated by drugs that interact with nuclear receptors. However, while the regulation of CYPs is quite well understood, less is known about how repeated drug administration regulates the most abundantly expressed ABC-transporters. Therefore, in the second part of this thesis, the effects of repeated drug administration on the gene regulation of four ABC-transporters and CYP3A4 were studied in intestinal epithelial cell lines in vitro and in the perfused human jejunum in vivo. The in vitro studies revealed that the ABC-transporters are induced by drugs that interact with slightly different sets of nuclear receptors. The in vivo study showed that repeated oral administration of St John’s wort decreased the bioavailability of verapamil, predominantly by induction of intestinal CYP3A4. This part of the thesis provides new information about the regulation of ABC-transporters, shows that the intestinal metabolism is the most significant barrier to oral bioavailability of verapamil and provides evidence for a clinically significant interaction between verapamil and St John’s wort in vivo.
Helena Engman, Department of Pharmacy, Uppsala Biomedical Centre, Box 580,SE-751 23 Uppsala, Sweden
© Helena Engman 2003
ISSN 0282-7484 ISBN 91-554-5752-5 Printed in Sweden by University Printers, Uppsala 2003

CONTENTS
PAPERS DISCUSSED 5
ABBREVIATIONS 6
1. INTRODUCTION 71.1 FACTORS INFLUENCING ORAL DRUG ABSORPTION AND BIOAVAILABILITY 8 1.2 THE INTESTINAL EPITHELIUM 9 1.3 MECHANISMS OF INTESTINAL DRUG TRANSPORT 10
1.3.1 Passive transcellular transport 1.3.2 Passive paracellular transport 11 1.3.3 Carrier-mediated transport
1.4 THE ATP-BINDING CASSETTE (ABC) SUPERFAMILY OF TRANSPORTERS 13 1.4.1 Structure of ABC-transporters 141.4.2 ABCB1/MDR1 P-glycoprotein 15 1.4.3 ABCC2/MRP2 161.4.4 ABCG2/BCRP
1.5 INTESTINAL DRUG METABOLISM 17 1.5.1 Cytochrome P450 (CYP) 3A 1.5.2 Phase II metabolism 18
1.6 FUNCTIONAL INTERPLAY BETWEEN ABCB1 AND CYP3A4 19 1.7 REGULATION OF ABC-TRANSPORTERS AND CYP450 ENZYMES
VIA NUCLEAR RECEPTOR PATHWAYS 201.8 DRUG-DRUG INTERACTIONS INVOLVING INDUCTION OF ABCB1 AND
CYP3A4 AT THE INTESTINAL LEVEL 21 1.8.1 Induction of ABCB1 1.8.2 Induction of CYP3A4 22 1.8.3 Induction of ABCB1 and CYP3A4 23 1.8.4 Clinical relevance 24
1.9 METHODS FOR STUDYING INTESTINAL DRUG TRANSPORT AND METABOLISM 25 1.9.1 Intestinal drug transport 1.9.2 Intestinal drug metabolism 26 1.9.3 Cell culture models for the assessment of passive drug transport
and drug efflux 27 1.9.4 Cell culture models for simultaneous studies of drug transport
and CYP3A4 metabolism 1.9.5 In vivo methods for studying transport and metabolism 28
2. AIMS OF THE THESIS 29
3. MATERIALS AND METHODS 303.1 DRUGS AND RADIOLABELED MARKERS3.2 CELL CULTURE 3.3 REAL-TIME QUANTITATIVE PCR ANALYSIS 31

3.4 IMMUNOBLOT ANALYSIS3.5 IN VITRO DRUG TRANSPORT STUDIES3.6 IN VITRO DRUG METABOLISM STUDIES 32 3.7 IN VIVO SINGLE-PASS PERFUSION OF THE HUMAN JEJUNUM3.8 ENANTIOSELECTIVE HPLC ANALYSIS OF R/S-VERAPAMIL AND
R/S-NORVERAPAMIL 33 3.8.1 In vitro cell culture samples and jejunal perfusates 3.8.2 Plasma samples
3.9 DATA ANALYSIS 34 3.9.1 Apparent permeability coefficients 3.9.2 Absorption variables from jejunal perfusate data3.9.3 Pharmacokinetic analysis of plasma data 35 3.9.4 Statistical analysis
4. RESULTS AND DISCUSSION 364.1 CYP3A4, CYP3A5, AND MDR1 (ABCB1) IN HUMAN SMALL AND LARGE INTESTINAL CELL LINES4.2 ENANTIOSELECTIVE TRANSPORT AND CYP3A4-MEDIATED METABOLISM OF
R/S-VERAPAMIL IN CACO-2 CELL MONOLAYERS 38 4.3 DRUG-INDUCED GENE REGULATION AND FUNCTION OF ABC-TRANSPORTERS
IN HUMAN INTESTINAL EPITHELIAL CELL LINES 41 4.4 ST JOHN’S WORT DECREASES THE BIOAVAILABILITY OF R- AND S-VERAPAMIL
THROUGH INDUCTION OF THE FIRST-PASS METABOLISM 43
5. CONCLUDING REMARKS 46
6. ACKNOWLEDGEMENTS 47
7. REFERENCES 49

5
PAPERS DISCUSSED
This thesis is based on the following papers, which will be referred to by their roman numeral in the text:
I. Engman, H., Lennernäs, H., Taipalensuu, J., Otter, C., Leidvik, B., Artursson, P. CYP3A4, CYP3A5, and MDR1 in human small and large intestinal cell lines suitable for drug transport studies. J. Pharm. Sci. 2001, 90, 1736-1751.Reproduced with permission. 2001 John Wiley, Inc. and the American Pharmaceutical Association
II. Engman, H., Tannergren, C., Artursson, P., Lennernäs, H. Enantioselective transport and CYP3A4-mediated metabolism of R/S-verapamil in Caco-2 cell monolayers. Eur. J. Pharm. Sci. 2003, 19, 57-65Reproduced with permission. 2003 Elsevier Science
III. Engman, H., Svensson, A.-C., Artursson, P. Drug-induced gene regulation and function of ABC-transporters in human intestinal epithelial cell lines. In manuscript.
IV. Tannergren, C., Engman, H., Knutson, L., Hedeland, M., Bondesson, U., Lennernäs, H. St John's wort decreases the bioavailabilty of R- and S-verapamil through induction of the first-pass metabolism. Submitted.

6
ABBREVIATIONS
a-b apical-to-basolateral ABC adenosine triphosphate (ATP) Binding Cassette Ar appearance ratio b-a basolateral-to-apical BCRP breast cancer resistance protein CAR constitutive androstane receptor CYP cytochrome P450 D3 1 ,25-dihydroxy vitamin D3, calcitriol EG extraction ratio in the gut wall EH extraction ratio in the liver F systemic bioavailability fa fraction dose absorbed FCS fetal calf serum GR glucocorticoid receptor HBSS Hank’s balanced salt solution HNF4 hepatocyte nuclear factor 4HPLC high performance liquid chromatography i.v. intravenous MDR multidrug resistance MDR1 Pgp multidrug resistant gene 1 P-glycoprotein MRP multidrug resistance associated protein Papp apparent permeability coefficient Peff effective permeability coefficient PXR pregnane X receptor RT-PCR reverse transcription polymerase chain reaction SD standard deviation TER transepithelial electrical resistance VDR vitamin D receptor Å Ångström

Introduction
7
1. INTRODUCTION Oral administration of drugs is strongly preferred because of for its convenience, the relatively low production cost and the high level of patient safety. However, a considerable proportion of compounds do not display the characteristics required for oral administration. The clinical development of new drugs is frequently hampered by unfavourable pharmacokinetics, such as poor or variable bioavailability of the drug after oral administration.
A prerequisite for a drug to be efficient after oral administration is that it, largely, avoids a sequential series of barriers in the gastrointestinal tract and in the liver. Until recently, systemic bioavailability of orally administered drugs has, primarily been considered to be a function of intestinal drug absorption and subsequent phase I metabolism in the liver. However, the human small intestine has increasingly been recognized as an important site for first-pass extraction. Both cytochrome P450 (CYP) 3A isoenzymes (1, 2) and efflux proteins, belonging to the ATP-binding cassette (ABC) transporter family (e.g., ABCB1/P-glycoprotein, Pgp) (3-5), are abundantly expressed in the human small intestine and contribute to a reduced bioavailability of many drugs (6-8). Among the members of the CYP3A subfamily, CYP3A4 is the most important, both quantitatively and qualitatively (9). Recently, similarities between the tissue distribution and gene regulation of CYP3A4 and Pgp, along with their overlapping specificities for substrates, inhibitors, and inducers (10) generated the hypothesis that CYP3A4 and Pgp have a complementary function and thus form a coordinated intestinal barrier to drug absorption and gut wall metabolism (11). The lack of direct evidence supporting the concertede action between CYP3A4 and Pgp is partly due to the lack of inhibitors specific for the enzyme and transporter, respectively, but also to difficulties in separating the relative contribution of the two proteins in vivo. For this purpose, reproducible in vitro models allowing detailed mechanistic studies would be useful. The aim of the first part of this thesis was therefore to establish and characterize a cell culture model of the intestinal epithelium for application in studies of both CYP3A4-mediated drug metabolism and Pgp-mediated efflux activity.
Induction of CYP3A and, potentially also ABC-transporters in the human small intestine is an important source of drug-drug interactions and variations in oral bioavailability (8, 12-14). Many clinically relevant drugs are substrates for both ABC-transporters and CYP3A4. Several nuclear receptors, such as PXR, CAR, and VDR have recently been shown to be important participants in the transcriptional pathways that control the adaptive response of ABC-transporters and CYP450 to both exogenous and endogenous compounds (15-19). These compounds include many clinically relevant drugs. While the induction of CYP3A in the intestine has been thoroughly investigated, much less is known about how drugs regulate intestinal ABC-transporters. Thus, the aim of the second part of this thesis was therefore to investigate the effects of repeated drug administration on the gene regulation and function of ABC-transporters and CYP3A4 in the human intestine. These studies were performed in two human intestinal epithelial cell models in vitro, and in the human intestine in vivo.

8
Permeability
Disintegration
DissolutionLiver extraction
Degradation,complexation
Drug in solution
Drug in systemic
circulation
Gas
troin
test
inal
trans
it
CYP3A
Permeability
Disintegration
DissolutionLiver extraction
Degradation,complexation
Drug in solution
Drug in systemic
circulation
Gas
troin
test
inal
trans
it
CYP3ACYP3A
1.1 FACTORS INFLUENCING ORAL DRUG ABSORPTION AND BIOAVAILABILITY
The bioavailability (F) of a drug is defined as the fraction of the dose that appears intact in the systemic circulation (20). The various factors that influence the systemic bioavailability following oral administration can be described by the following equation:
F = fa (1-EG) (1-EH) (1)
where fa is the fraction of the dose absorbed over the mucosal membrane of the enterocyte, (1-EG) is the fraction of the drug that escapes metabolism in the gut wall, and (1-EH) is the fraction of the drug that escapes metabolism and biliary excretion in the liver (21).
As can be seen in Figure 1, the intestinal absorption and bioavailability after oral drug administration is governed by several factors (20, 22, 23).
Figure 1. Schematic illustration of factors influencing oral drug absorption and bioavailability.
Drugs designed to be systemically active must be absorbed from the site of administration in order to be efficient. Furthermore, to allow passage through biological membranes, the drug must be in solution. Since most drugs are administered as solid dosage forms, disintegration of the formulation must precede dissolution of the drug in the surrounding media. The disintegration rate is influenced by characteristics of the pharmaceutical formulation and by physiological factors such as the gastric emptying rate, the transit time and the pH in the gastrointestinal fluids. The dissolution rate is not only dependent on the physico-chemical properties of the particle and the drug molecule in itself (e.g., pKa, molecular size, lipohilicity and hydrogen bonding), but also on the luminal pH and the composition of the luminal contents. Once in solution, the drug is susceptible to both chemical and enzymatic degradation. The oral bioavailability may be

Introduction
9
Extrusion
Matureabsorptive
cells
Differentiating
Proliferating
Stem cells
Cac
o-2
Cry
ptV
illus
Extrusion
Matureabsorptive
cells
Differentiating
Proliferating
Stem cells
Cac
o-2
Cry
ptV
illus
further reduced by efflux mechanisms or first-pass metabolism in the intestinal epithelium and/or the liver.
The intestinal solubility of and permeability to a drug are considered to be the two most important determinants of oral absorption e.g., (24, 25). The latter is defined as the transport of the unchanged drug molecule from its dosage form across the apical membrane of the enterocyte (20). In this thesis, the focus is on drugs in solution, and hence the effects of drug solubility and dissolution are not considered. If, however, one wished to consider aspects of solubility and permeability, these parameters have been incorporated into the biopharmaceutical classification system, BCS (26), which allows e.g., predictions of the oral drug absorption to be made in humans. The interplay between permeability and solubility can also be roughly assessed by calculating the maximum absorbable dose, MAD (24). However, valid use of MAD requires the assumption that complicating factors such as luminal degradation are negligible.
1.2 THE INTESTINAL EPITHELIUM
Once in solution, a drug molecule encounters a system of sequential barriers during its transport from the intestinal lumen into the blood, figure 2. The rate and extent of intestinal absorption is dependent on the epithelial permeability to the drug (27, 28) and the epithelial surface area. The epithelial surface available for contact with the luminal contents is greatly amplified by the folds, villi, and microvilli structures (29).
Figure 2. The crypt-villus functional unit of the human small intestine. The mucosal epithelium is rapidly renewing, and the proliferative cells, which arise from the stem cells located at the bottom of the crypts, lose their ability to proliferate. They start to differentiate in the upper third of the crypt and then migrate toward the tip of the villus, before being exfoliated into the intestinal lumen. Caco-2 cells are commonly used as a model of the human intestinal epithelium. Their loss of proliferating and acquisition of differentiating characteristics appears gradually along the crypt-villus axis. Redrawn from (30).

10
The functional unit in the intestinal epithelium is the crypt-villus axis, figure 2. Within the axis, the epithelium is spatially separated into proliferating and differentiating cells (in the lower and upper crypt regions, respectively) cells, with functional, absorptive cells (called enterocytes) situated on the villus tip (30). The intestinal epithelium contains three types of cells with distinct functions: endocrine, exocrine and absorptive cells. The endocrine cells secrete digestive hormonal peptides, while the exocrine ones secrete mucus (goblet cells) or antimicrobial peptides (Paneth cells). The enterocytes are the most abundant cells accounting for 80-90% of the total number of epithelial cells. The three groups of differentiated cells originate from the same multipotential stem cell that proliferate near the bottom of the crypt (29).
The properties that are relevant for drug absorption differ between the cells in one region and another along the crypt-villus axis. For instance, in several species, the paracellular space between the cells seems to have a lower permeability at the villus tips than further down the crypt-villus axis (31, 32). High permeability drugs are believed to be absorbed mainly at the tips of the villi (33), while low-permeability drugs may diffuse down the length of the crypt-villus axis to be absorbed over a larger absorptive surface area (34).
1.3 MECHANISMS OF INTESTINAL DRUG TRANSPORT
Drug transport across absorptive epithelia is mediated by one or several of the following mechanisms: passive transcellular or paracellular processes, carrier-mediated absorptive or secretory flux and transcytosis, figure 3.
1.3.1 Passive transcellular transport A drug molecule must penetrate the membrane surrounding the epithelial cell in order to transverse the cell. Thus, the passive transport is largely determined by the biophysical properties of the membrane. Cell membrane consist of a double layer of phospholipids and cholesterol, with proteins embedded in the layer (35). The type and relative quantities of lipids and proteins differ between different cell types, thus creating a basis for unique permeability properties. Like other epithelial cells, the enterocytes are polarised with marked differences in membrane composition of the apical and basolateral membranes (36).
The first step of passive transcellular transport is the penetration of the apical membrane by the drug, which is generally assumed to be followed by diffusion through the cytoplasm of the cell interior, and subsequent permeation of the basolateral membrane. Very lipophilic drugs may become trapped in the apical membrane, or their transport may involve lateral diffusion in the cell interior. However, diffusion of small molecules in the cytoplasm is normally a rapid process, and therefore the apical membrane is usually considered to be the rate-limiting barrier to passive transcellular transport (37).
For the distruibution to the apical membrane and the subsequent transcellular diffusion to be effective, the drug must be sufficiently lipophilic and moderate in size. Nevertheless, several studies suggest that the majorityof completely absorbed drugs are transcellularly transported, some of these studies are reviewed in (38). Thus, the rather complex process of intestinal drug absorption can often be satisfactorily described by

Introduction
11
1 2 3 41 2 3 4
just considering the passive transport across the apical membrane only. Thereby explaining why experimental and theoretical models describing transport by this mechanism have received particular attention (39, 40). Different theoretical models have been developed for passive transcellular permeability. Irrespective of which model is used, the rate of passive permeation largely depends on simple molecular descriptors such as the hydrogen bond capacity, lipophilicity, and the size and charge of the molecule (41). For instance, it has been estimated that oral drug absorption via the passive transcellular route is unlikely if the polar surface area (a measure of the hydrogen bonding capacity) of the drug molecule is more than 120Å2 (42, 43).
1.3.2 Passive paracellular transport Transport via water-filled pores between the cells is a process known as paracellular transport. This transport route is generally considered to be passive, although it appears to be more permeable to cationic drugs than to anionic or neutral species (44, 45). Drugs that are relatively hydrophilic and of small to moderate size (e.g., atenolol and furosemide) can permeate the intestinal epithelium via this route in significant amounts, at least in the upper part of the small intestine, where the paracellular route is more leaky than in the lower parts of the small intestine and colon (46). However, this type of drug is usually incompletely absorbed since the paracellular pores represent only 0.01-0.1% of the total surface area of the intestine. Moreover, the apical and basolateral membrane domains are separated by tight junctions, providing a seal between adjacent epithelial cells that further restricts transport via this route (47, 48). The paracellular permeability is dynamically regulated, a fact that has been exploited to enhance drug delivery via this route (49, 50).
Figure 3. Schematic representation of routes and mechanisms for drug transport across the intestinal epithelium. 1. passive transcellular and 2. paracellular transport, 3. carrier-mediated efflux, and 4. carrier-mediated active transport. Membrane proteins involved in carrier-mediated transport or efflux may also be localized in the basolateral membrane of the enterocyte.
1.3.3 Carrier-mediated transport Although a vast number of drugs are transported by means of passive diffusion, recent studies suggest that carrier-mediated transport has an even larger impact than had been believed previously. An increasing number of active transporters such as the ABC-transporters that may have an impact on oral absorption are being identified, although their influence on the in vivo drug absorption is still largely unexplored. The mapping of the human genome identified nearly 1300 genes coding for ion channels and transporters, which might be directly or indirectly involved in the absorption process

12
(51), and a qualitative cDNA array analysis of the number of transporters in the human jejunum indicated the presence of mRNA for approximately 200 transporters (52). Langmann et al. (4) provided the mRNA expression profiles for 47 of the 48 known ABC transporters in 20 human tissues, and showed that human tissues vary greatly in these profiles. This variability may have an impact on regional kinetics displayed by different organs within the body, especially since additional variability in ABC-transporter expression may occur during disease or as a response to administered drugs.
Intestinal absorptive cells express a number of carrier systems which play a key role in determining the exposure of cells to a variety of solutes including nutrients (e.g., peptides, amino acids and sugars) and cellular by-products. Transport by means of carriers can be directly or indirectly energy dependent, referred to as active transport, or independent on energy, i.e., facilitated diffusion (53). The process of carrier-mediated transport is saturable, i.e., drugs that are substrates may show non-linear pharmacokinetics.
In order to prevent unwanted entry of compounds into the body, carriers are substrate specific. However, the substrate specificity is not absolute and carrier-mediated transport is available to a limited number of drugs with nutrient-like molecular structures. The most promiscuous drug transporter known to date that enhances oral drug absorption is the H+-coupled oligopeptide transporter PEPT1, which is abundantly expressed in the small intestine (54, 55), and participates in the absorption of -lactamantibiotics, renin inhibitors and angiotensin converting enzyme (ACE) inhibitors (56).
Organic anion and cation transporters have recently received considerable attention. For instance, the organic cation/carnitine transporter 2 (OCTN2), transports physiologically important carnitine in an Na+-dependent manner, as well as organic cations in an Na+-independent manner (57-59). Most importantly, Tsuji and collegues provided evidence that primary systemic carnitine deficiency is caused by loss of OCTN2 function (60). In addition, the uptake of L-carnitine was shown to be primarily mediated by this transporter in differentiated Caco-2 cells (61). OCTN2 also plays a pharmacologicalrole, since it mediates the transport of cations such as tetraethylammonium (TEA) and drugs such as pyrilamine, valproate, and verapamil (58).
Members of the organic anion transporting polypeptide (OATP) family are involved in the transport of various endogenous and xenobiotic compounds, such as conjugated metabolites of steroid hormones, thyroid hormones, bile acids, bilirubin, pravastatin,benzylpenicillin, and digoxin. So far, nine members of the OATP family have been reported in humans (62) of which OATP-B is the first OATP member shown to be expressed at the apical membrane of human enterocytes. OATP-B might therefore play arole in the pH-dependent transport of anionic drugs in the human intestine (63).
In the process known as receptor-mediated transcytosis, the solute binds to a receptor on the cell surface where it is internalised by endocytosis, and then a fraction of the internalised vesicle proceeds towards the opposite membrane. This complex route has a very low capacity and is only of relevance for highly potent macromolecular drugs that are active at low concentrations (64).

Introduction
13
Apical
Basolateral
ABCB1 ABCC1 ABCC2 ABCC3 ABCG2
Apical
Basolateral
ABCB1 ABCC1 ABCC2 ABCC3 ABCG2
1.4 THE ATP-BINDING CASSETTE (ABC) SUPERFAMILY OF TRANSPORTERS
Multidrug resistance is a phenomenon whereby tumor cells in vitro that have been exposed to one cytotoxic agent develop cross-resistance to a range of structurally and functionally unrelated compounds. Multidrug resistance is, in part, the result of an increased expression of efflux proteins such as ABCB1/MDR1 P-glycoprotein, which is a member of the ATP-binding cassette (ABC) superfamily of transporters. These efflux proteins limit the intracellular exposure to xenobiotics by pumping agents out of the cell (65). However, ABC-transporters are also expressed in normal human tissues including the gastrointestinal tract (3, 4, 66, 67).
The human genome contains 48 ABC genes, of which 16 have a known function and 14 are associated with a defined human disease, as reviewed by (65, 68). Physiological functions of the ABC transporters include the transport of lipids, bile acids, peptides and toxic compounds (68). The role of ABC transporters in disease is exemplified by mutations in the ABCC2/MRP2 gene, which causes the Dubin-Johnson syndrome, in which biliary excretion of conjugated bilirubin by ABCC2 is impaired (69). Most importantly, multidrug resistance is a significant obstacle to the success of chemotherapy in many cancers (70). In addition, polymorphisms in the ABCB1 gene result in kinetic alterations on the transporter level that may have an impact on the therapeutic efficacy of many drugs (71, 72). The polymorphism in the ABCB1 gene is further discussed below.
Figure 4. Schematic representation of the localization of ABCB1, ABCC1-3 and ABCG2 in polarized cells in the intestinal epithelium. See text for further details. Redrawn from (73).
Efflux proteins have the potential to affect oral bioavailability by pumping agents out of the enterocyte. Export of these compounds occurs in an active ATP-dependent manner, and can take place against considerable concentration gradients. ATP-hydrolysis provides the energy for this process (74, 75). Membrane proteins shown to transport clinically relevant drugs belong to the ABC-transporter family, and more specifically, include ABCB1/MDR1 Pgp, multidrug resistance proteins ABCC/MRP1-5, and breast cancer resistance protein ABCG2/BCRP (76). As shown in figure 4, these transporters are localised either to the apical membrane of the intestinal enterocytes (such as ABCB1, ABCC2 and ABCG2), where they function as a total organism defence mechanism by pumping drugs out of the cell back to the intestinal lumen, or to the basolateral membrane of the enterocyte (such as ABCC1 and ABCC3) where they constitute a form of cellular defence (74, 76, 77).

14
1.4.1 Structure of ABC-transporters The variation in the subunit structure of ABC-transporters is demonstrated in Figure 5. The basic structure, exemplified by that found in P-glycoprotein (ABCB1), is thought to consist of 12 transmembrane segments and two ATP-binding sites in a protein consisting of about 130 amino acids. ABCG2/BCRP functions as a homodimer i.e., the basic structure is assembled from two equal halves (78, 79) .
Figure 5. Predicted topology of three major classes of mammalian ABC-transporters. The intracellular nucleotide-binding domains NBDs, and the N and C termini of the protein are indicated in this simplified scheme. Redrawn from (65).
How ABC-transporters work is not yet known in detail. For instance, there is still doubt about the position and number of substrate-binding sites in ABC-transporters, such as Pgp (65). Nevertheless, several models have been presented for the transport mechanisms (80). However, a recently published crystallographic structure of the bacterial multidrug efflux pump AcrB reveals a large drug binding cavity with separate drug-binding sites for each of the four substrates studied. If the Pgp binding cavity resembles that of AcrB, this could explain the difficulties encountered when attempting to describe the substrate binding process in Pgp (81).
ABCB1/MDR1 Pgp, ABCC2/MRP2 and, more recently, ABCG2/BCRP have come to be considered as potentially important barriers to the intestinal absorption of a large variety of drugs (76, 82, 83). These transporters were, therefore, selected for a more detailed discussion.
Table 1. Nomenclature for some ABC transporters that are expressed in the human intestine and studied in Paper III.
Symbola Conventional name Abbreviation
ABCB1 Multidrug resistant gene 1 P-glycoprotein MDR1 Pgp
ABCC2 Multidrug resistance associated protein 2 MRP2
ABCC3 Multidrug resistance associated protein 3 MRP3
ABCG2 Breast cancer resistance protein BCRPa Standardized names adopted by the Human Gene Nomenclature committee http://www.gene.ucl.ac.uk/nomenclature/genefamily/abc.html
N NBDNBD
C
NBD
N C C
NBD NBDN NBDNBD
C
N NBDNBD
C
NBD
N CN C C
NBD NBD
C
NBD NBDNBD
ABCB1 ABCG2 ABCC1
N NBDNBD
C
NBD
N C C
NBD NBDN NBDNBD
C
N NBDNBD
C
NBD
N CN C C
NBD NBD
C
NBD NBDNBD
ABCB1 ABCG2 ABCC1

Introduction
15
1.4.2 ABCB1/MDR1 P-glycoprotein P-glycoprotein (Pgp), encoded by the ABCB1 gene, was discovered by Juliano and Ling (84) and is the most studied ABC drug efflux transporter to date. Proof-of-concept was provided by Sparreboom et al. (85) who tested the fate of orally and i.v.-administered paclitaxel in wild-type (wt) and mdr1a( / ) mice. This study showed that Pgp in the intestinal epithelium limits the oral bioavailability of paclitaxel, and that it is likely that a similar protection is afforded to many other orally administered compunds that are Pgp-substrates. It was also concluded that intestinal Pgp contribute to the elimination of parenterally administered substrate drugs by a direct secretion of drug into the intestinal lumen.
The most striking property of ABCB1 Pgp include its broad substrate specificity, and the fact that it transports a large number of structurally diverse drugs used in a range of clinical applications (76). Hydrophobicity, planar aromatic rings and the presence of tertiary amino groups favour substrate interaction with Pgp, but no highly conserved elements of recognition have been found (86-88). Recently, pharmacophore models of Pgp substrate affinity have been proposed, generally containing ring aromatic or hydrophobic functionalities as well as hydrogen bond acceptor functional groups (89, 90). Identification of compounds that are ABCB1/Pgp substrates can aid drug candidate selection and optimization. In this respect, ultimately, the generation of 3D-structures of ABC-transporters would help us to understand how the ABC-transporters work, and ideally, to predict when and whether, and if so, how a drug candidate will interact with the transporter in question.
Overexpression of Pgp in cancer cells confers high levels of resistance to Vinca alkaloids, anthracyclines, taxanes, etoposide and a vast number of other compounds (91, 92). Several studies suggest that the physiological role of Pgp is to protect the organism from toxic compounds. This suggestion is partly based on the fact that Pgp is expressed at barriers involved in drug excretion, including the epithelium lining the gastrointestinal tract (66, 93). In addition, evidence is derived from in vivo knock-out mouse models in which the murine orthologue for Pgp has been deleted or disrupted. These mice are considered to be healthy and reproduce normally in a protected environment, but show altered sensitivity to and excretion of compounds that are ABCB1 Pgp substrates (94, 95).
Variations in the expression levels and the activity of ABCB1-encoded Pgp may have a major impact on the therapeutic efficacy of many drugs. As the functionality of Pgp is defined by the amino acid sequence that is encoded by the ABCB1 allele, the ability to detect relevant MDR1 alleles is of potential importance for the treatment of patients with drugs that are substrates of Pgp (96). The first two naturally occurring ABCB1 polymorphisms were described and correlated with possible clinical effects by Mickley et al. (97). More extensive studies followed, with ninteen polymorphisms being identified in Germany (71, 98) and a further nine in Japan (99, 100).
The most interesting mutation, C3435T, is associated with a lower level of ABCB1 expression in duodenum and higher digoxin plasma levels (71), which were suggested to result from a weaker restriction of absorption. However, this finding was opposed by

16
recent investigations, which also suggested the importance of the C3435T, but the relationship with the phenotype did not always agree with the report by Hoffmeyer et al. (71). Plasma or serum concentrations of digoxin (101, 102), fexofenadine (103) and efavirenz and nelfinavir (104) were lower in subjects with the mutant T/T allele. However, another recent clinical study (Kurata et al., 2002) supported the observations by Hoffmeyer et al. (71). The discrepancy was proposed to be related to e.g., pharmacokinetic profiles of the probe drug used, ethnic differences and mode of administration (102).
The influence on the oral bioavailability of the pharmaceutical formulation and by physiological factors such as gastric emptying rate, and pH in the gastrointestinal fluids were illustrated in a clinical study where the effects of ABCB1 genotype on the duodenal absorption of digoxin were investigated (102). When digoxin was administered as a conventional tablet, delayed absorption caused by disintegration of the tablet, dissolution of digoxin, and by gastric emptying resulted, and no significant influence of the ABCB1 genotype was observed. In contrast, when digoxin was applied directly in the duodenum as a solution, a marked effect of the genotype-dependent serum digoxin levels was observed. Thus, in subjects expressing the T/T allele, the bioavailability of digoxin was markedly reduced (102).
1.4.3 ABCC2/MRP2 ABCC2 transports a large range of organic anions and endogenous compounds, including glutathione- , glucuronide-, and sulfate conjugates. ABCC2 affects the pharmacokinetics of clinically important drugs including cancer chemotherapeutics (irinotecan, methotrexate, vinblastine), antibiotics (ampicillin, ceftriaxone, and rifampin), and angiotensin-converting enzyme inhibitors, as well as many toxins and their conjugates (105). Many of these substrates are common with ABCB1 and ABCG2 (76). ABCC2 is able to confer resistance against e.g., Vinca alkaloids, anthracyclines, mitoxantrone and cisplatin in vitro, but its role in anticancer drug resistance in patients remains to be verified (106, 107).
ABCC2 is not only acting as a protective barrier in the intestine, but also in the liver, kidney, brain and placenta (106). Most importantly, ABCC2 has an important function in the biliary excretion of endogenous compounds such as conjugates of bilirubin, steroids and leukotrienes (65) thus, patients lacking functional ABCC2 have hyperbilirubinemia (105).
1.4.4 ABCG2/BCRP ABCG2 was first discovered by Doyle et al. because of its overexpression in a highly doxorubicin-resistent breast cancer cell line (108). The range of drugs to which ABCG2 can confer resistance is somewhat more limited than that for ABCB1, but includes mitoxantrone, topotecan derivatives and anthracyclines, but not Vinca alkaloids and taxanes (108, 109). ABCG2 is a half-size transporter that is presumed to require dimerisation to form a functional unit. ABCG2 is highly expressed in the human small intestine, colon, placenta and in the bile ducts of the liver (3, 110).

Introduction
17
ABCG2 was recently shown to transport sulfated conjugates of both steroids and xenobiotics; and estrone-3-sulfate (E1S) and dehydroepiandrosterone sulfate were suggested to be the potential physiological substrates for this transporter (111). In addition. ABCG2 has been demonstrated to be expressed in a variety of stem cells and to be a molecular determinant of the side-population phenotype (112).
1.5 INTESTINAL DRUG METABOLISM
Compounds entering the body can be modified by oxidation, through phase I metabolism e.g., by the CYP450 system, and/or made more water soluble by conjugation with glutathione (GSH), sulfate or glucuronic acid through phase II metabolism.
In a survey on the elimination pathways of over 400 drugs marketed in Europe and the United States, it was shown that P450-mediated metabolism represented 55% of the total elimination of these drugs (113). CYP3A appears to be involved in the metabolism of nearly 50% of all the drugs currently prescribed (114, 115). Hence, alteration in the activity or expression of this enzyme is a key predictor of drug responsiveness and toxicity.
Of the 35 P450 enzymes hitherto described in man, only P450’s in families 1, 2 and 3 appear to be responsible for the metabolism of drugs and are, therefore, potential sites for drug-drug interactions. P450 enzymes from other families are generally involved in endogenous processes, particularly hormone biosynthesis (113).
1.5.1 Cytochrome P450 3AAmong adults, CYP3A4 is the dominant CYP3A isoform in human small intestine (2, 116) and liver (117). CYP3A5 is also found in the liver and intestinal mucosa (118, 119) and other extrahepatic tissues (120, 121), but its expression is clearly polymorphic, with some individuals exhibiting relatively high or low levels of the protein (118, 122). It has been suggested that, for most individuals, CYP3A5 only plays a minor role in the first-pass extraction of CYP3A substrates in vivo (113) and CYP3A7 is primarily a fetal enzyme (123). Recently, human CYP3A43 has been identified and cloned (124),although its contribution to hepatic or extrahepatic CYP3A-dependent drug clearance is believed to be negligible (125).
Many CYP3A-substrates undergo substantial metabolism in the mucosal lining of the small intestine , examples being midazolam (6), felodipine (126, 127), verapamil (128), nifedipine (129), tacrolimus (130), saquinavir (131) and cyclosporine (21). CYP3A constitutes only 30% of total human CYP content (132), but accounts for approximately 70% of the CYP content in the human intestine (1, 133, 134). The situation of CYP3A in the villus tip enterocytes (2) renders a very large surface area over which the enzyme can encounter its substrates, thus providing conditions for an extensive metabolism by CYP3A4 in the intestine.

18
Interindividual differences in the oral bioavailability and systemic clearance of CYP3A substrates can be attributed, in large part, to variable expression of CYP3A in the mucosal epithelium of the small intestine (6, 119) and in the liver (135, 136). This variation is manifested by differences greater than 10-fold in the in vivo metabolism of drugs that are substrates for CYP3A enzymes e.g., (136). This variation can be further enhanced by inhibition or induction of CYP3A4 and may result in clinically important differences in toxicity and pharmacological response.
Genetic factors appear to be of great importance for the interindividual variability in constitutive expression and activity of CYP3A, but the underlying mechanisms remain largely unknown (137). The functional polymorphism of the CYP3A4 gene has been studied, and different CYP3A4 alleles have been found (138). However, these mutations are rare and only a few of them influence function. Thus, variable expression of other members in the CYP3A family or of the human pregnane X receptor (PXR) might influence the overall variability in CYP3A4. It has been suggested that polymorphic expression of CYP3A5 is a significant contributor to CYP3A-dependent drug metabolism in individuals with a high hepatic expression (139), and a single nucleotide polymorphism has been identified, explaining the lack of CYP3A5 in the majority of Caucasian livers. In contrast, however, a more recent study showed that the contribution of CYP3A5 to hepatic CYP3A activity in Caucasians is insignificant. Furthermore, it was concluded that regulatory factors common to the CYP3A4, CYP3A5, CYP3A43 and PXR genes seem to be important for their expression and for the interindividual variability in CYP3A4 (140).
The vitamin D receptor (VDR) was recently identified as a potentially important regulator of intestinal CYP3A4 (18, 141). Interindividual differences in expression of intestinal CYP3A4 could arise from mutations in the VDR (142) and from differences in intestinal VDR content and circulating 1 , 25-dihydroxy vitamin D3 (D3) (18). VDR, D3, and CYP3A4 have been shown to participate in the enteric defence against colon cancer (143).
1.5.2 Phase II metabolism Several phase II enzymes in the intestine are expressed at levels that are comparable to or exceed those in the liver. Specific forms of phase II enzymes have been identified in the intestine, including glutathione S-transferase A1-1 which may contribute to variable oral clearance of busulfan (144), and dopamine sulfotransferase SULT1A3 (145). The UDP-glucuronosyltransferases (UGT) catalyse the addition of a –glucuronic acid moiety to a variety of sites (146) e.g., UGT1A1, 1A8 and 1A10 (147-149).
The conjugates formed through phase II metabolism are often too hydrophilic to diffuse out of the cell and therefore require transporters such as members of the ABCC subfamily, to assist their exit. Thus, conjugative enzymes and efflux transporters such as ABCC2 may act synergistically to reduce intestinal absorption of organic anions and conjugates (105). Transport of typical substrates for ABCC2, such as 2,4-dinitrophenyl-S-glutathione and 17 estradiol-17 -D-glucuronide, has been demonstrated in Caco-2 cells (150, 151).

Introduction
19
Drug
CYP3A4
Metabolite
Drug
Intestinallumen
Blood
AB
CB
1
Drug
CYP3A4
Metabolite
Drug
Intestinallumen
Blood
AB
CB
1
1.6 FUNCTIONAL INTERPLAY BETWEEN ABCB1 AND CYP3A4It is believed that intestinal CYP3A4 and ABCB1 act in a concerted manner to control the absorption of their substrates (131, 152-155). This suggestion is based on a considerable overlap in the substrate specificity of the two proteins and the proximity of their expression in the intestine. Furthermore, it was demonstrated that modulators and substrates of ABCB1 and CYP3A up-regulate these proteins in human colon carcinoma cells in a coregulatory manner (67). At the onset of this thesis, it was hypothesized that ABCB1 effectively recycles its substrates over the apical membrane, thereby maximizing the exposure to metabolising CYP3A4 in the intestine, and decreasing the importance of enzyme quantity, Figure 6 (154). Although ABCB1 and CYP3A4 may be functionally coordinated in order to minimize the exposure of the organism to xenobiotics, they appear to be regulated separately (156).
Figure 6. A schematic representation of the events occurring after oral administration of drugs, which are substrates for both ABCB1 and CYP3A4. A drug may be directly effluxed by ABCB1 back to the intestinal lumen, and, if the drug is subjected to CYP3A4-mediated metabolism, the formed metabolite(s) may be effluxed by ABCB1 and/or they may proceed through the basolateral membrane.
Several studies have been performed to test the ‘apical recyling’ hypothesis, using cell culture or rat models. Hochman et al. investigated the metabolism of indinavir in D3-induced Caco-2 cell monolayers, and showed that the amount of metabolite formed per molecule of transported indinavir was higher when ABCB1 was active than when it was inhibited (157). When the metabolism of verapamil across excised rat intestine was evaluated at increasing substrate concentrations, it was found that the cellular residence time and intestinal metabolism were higher at low concentrations of verapamil when ABCB1 was active, than at higher concentrations where saturation had occurred (158). Further, the bioavailability for the cystein protease inhibitor K02 (a dual substrate for CYP3A and ABCB1) increased from 3 to 30% in rats dosed with oral ketoconazole, with no corresponding change in intravenous clearance (159). However, in none of the studies was the relative importance of CYP3A and Pgp on intestinal metabolism clarified, since the experiments were performed at saturation levels and the substrates/inhibitors were not specific for CYP3A and/or ABCB1.
Recently, a rat single-pass intestinal perfusion model was used) to demonstrate that the extraction ratio of K77, a dual CYP3A/Pgp-substrate, decreased when ABCB1 was inhibited, while it stayed the same for midazolam, an exclusive CYP3A substrate (160).

20
The rat data were in agreement with in vitro data from CYP3A4-transfected Caco-2 cells (155). In summary, although a number of studies have been performed to investigate the suggested apical recycling by Pgp further studies are needed. Moreover, support for an in vivo link between enterocytic efflux transport and gut wall metabolism in humans still remains to be provided.
1.7 REGULATION OF ABC-TRANSPORTERS AND CYP450 ENZYMES VIA NUCLEAR RECEPTOR PATHWAYS
Many drugs in clinical use have been shown to, directly or indirectly, regulate ABC-transporters as well as CYP450 enzymes through nuclear receptor pathways. Importantly, the orphan nuclear pregnane X receptor (PXR) has been shown to coregulate genes for CYP450 enzymes (e.g., CYP3A4) and ABC-transporters (e.g, ABCB1) in the intestine (19), Figure 7.
Figure 7. Cross-talk between the PXR and CAR signaling pathways. Both CAR and PXR are modulated by both xenobiotics and endogenous compounds and bind as heterodimers with RXR to response elements in the regulatory regions of genes encoding proteins involved in metabolism and transport. Other regulatory pathways involving nuclear receptors exist in parallel with those described for PXR and CAR.
For instance, PXR is activated by the anti-cancer drug paclitaxel and induce the expression of both CYP3A4 and ABCB1/Pgp. Since paclitaxel is both metabolised by CYP3A4 and transported by Pgp, induction of these proteins leads to its enhanced clearence (19) .
Hyperforin, the active component in the anti-depressant Saint John’s wort (SJW), is a highly potent PXR-activator (161), and has lately gained much attention for its involvement in several severe interactions with orally coadministered drugs. Hyperforin induction of ABCB1/Pgp and CYP3A4 in the intestine have been reported to reduce the oral bioavailability of drugs such as e.g., cyclosporine and digoxin (162, 163).
Xenobiotics
Natural steroids
Xenobiotic and steroid metabolism
PXR RXR CAR RXR
Phase I enzymesPhase II enzymesTransporters
Xenobiotics
Natural steroids
Xenobiotic and steroid metabolism
PXR RXR CAR RXRPXR RXR CAR RXR
Phase I enzymesPhase II enzymesTransporters

Introduction
21
Rifampicin, another PXR-ligand, substantially reduced the plasma concentrations of orally administered digoxin by induction of intestinal ABCB1 (8). Further, the constitutive androstane receptor (CAR), the glucocorticoid receptor (GR) and the vitamin D receptor (VDR) have been reported to be important transcriptional regulators and coregulators of the ABC-transporter and CYP450 gene expression (164-169). Interestingly, the orphan nuclear receptor hepatocyte nuclear factor4 (HNF4 ) seems to be directly involved in PXR and CAR-mediated transactivation of CYP3A4, and may also participate in cell type dependent upregulation of CYP3A4 (17).
However, although a great deal is known about the regulation of basal and induced CYP450 and ABCB1 gene expression, comparatively little is known about how various drugs regulate the expression of other ABC-transporters. For instance, the adaptive response by ABCG2/BCRP to drug exposure has yet to be understood. Further, although it is clear that PXR plays a key role in the induction of Pgp, the inductive effects of a single drug may be mediated by more than one mechanism. Therefore, a detailed knowledge of the inductive patterns for each drug is required before one can understand its implications and consequences. The aim of the second part of this thesis was therefore to study potential effects of repeated drug administration on the gene regulation and activity of different ABC-transporters and CYP3A4 in the human intestine.
1.8 DRUG-DRUG INTERACTIONS INVOLVING INDUCTION OF ABCB1 ANDCYP3A4 AT THE INTESTINAL LEVEL
Inhibition and induction of CYP enzymes, particular CYP3A4, are the most common causes documented for drug-drug interactions (170). However, there is an increasing awareness that the pharmacokinetics of drugs that are not subject to metabolism, but to carrier-mediated transport mechanisms such as ABCB1, may have a substantial potential for drug-drug interactions (171). The pharmacokinetic consequences of ABCB1 induction are similar to those observed for induction of CYP3A4, that is, induction of ABCB1 results in a decrease in systemic exposure. Thus, data on how drugs regulate transporter expression are helpful in predicting pharmacokinetics and drug-drug interactions at the transporter level (172, 173).
In the section below, the discussion will focus on drug-drug interactions involving induction of ABCB1. This is followed by a presentation of drug-drug interactions based on induction of CYP3A4 and, finally, interactions involving induction of both ABCB1 and CYP3A4 are considered.
1.8.1 Drug-drug interactions caused by induction of ABCB1 Greiner et al. provided compelling evidence that ABCB1 induction can be the cause of drug-drug interactions in a clinical study comparing the pharmacokinetics of digoxin before and after ten days pre-treatment with rifampin (600 mg daily) in eight healthy volunteers (8). The plasma AUC value of oral digoxin was significantly lower during rifampin treatment while the effect was less pronounced after intravenous administration. The renal clearance and half-life of digoxin were unaltered by rifampin. In addition, the ABCB1 content in duodenal biopsies was increased by a factor of 3.5 by

22
rifampin, an increase which correlated with the AUC value after oral but not iv administration of digoxin (8). Digoxin is mainly eliminated by renal excretion in humans, and administered orally at a very low dose (0.5-1 mg). It was therefore concluded that the decreased plasma concentration of digoxin was caused by a reduced absorption and bioavailability of digoxin, arising from induction of ABCB1 at the intestinal level.
The effects of rifampin of nine days pretreatment (600 mg daily) on the pharmacokinetics of the P-glycoprotein substrate talinolol, a 1-blocker without appreciable metabolism, but extensive intestinal secretion, was studied in healthy volunteers (174). The AUC values for intravenously and orally administered talinolol were significantly lower (than their control levels during rifampin treatment. A 4-fold increase in the expression of duodenal P-glycoprotein was observed, which was significantly correlated with the total clearence of talinolol. On the basis of these results, Westphal et al. concluded that the talinolol-rifampin interaction is attributable, mainly, to thecombination of a decrease in absorption and an increase in elimination from induced Pgp expression in the intestine (174).
The bioavailability of fexofenadine was also decreased by rifampin pre-treatment (600 mg daily for six days) in twenty volunteers (13). The authors assumed that the metabolism of fexofendine is negligible in humans, and suggested that the decrease in the plasma concentration of fexofenadine relied on an induction of intestinal Pgp. However, the basis for this interaction have been claimed to be more complex, involving factors such as metabolism and hepatic uptake by rifampin-induced CYP and OATP, respectively (170, 175, 176).
St John’s wort (SJW) is one of the most popular herbal drugs for treating mild depression without a prescription. Since 1999, there has been a growing concern regarding the clinical significance of drug interactions involving SJW. Hence, the Food and Drug Administration (177) and the Medical Products Agency (178), recently issued a warning about the use of SJW in combination with other drugs. For instance, Durr et al. showed that administration of SJW for 14 days in healthy volunteers resulted in a 18% decrease in digoxin plasma AUC after a single dose (12). The decrease in AUC was accompanied by an increase in the duodenal expression of Pgp. Further, the coadministration of SJW and the HIV protease inhibitor indinavir reduced the systemic exposure of indinavir by nearly 60% (14), the induction of intestinal Pgp by SJW was assumed to play a role in this.
Interestingly, SJW also displayed inhibitory properties. The effect of SJW on Pgp-activity in vivo was recently examined using fexofenadine as a selective Pgp probe. A single dose of SJW resulted in a significant inhibition of intestinal Pgp, while this effect was not observed following repeated administration of SJW (179).
1.8.2 Drug-drug interactions caused by induction of CYP3A4 Midazolam is an established in vivo probe drug for CYP3A4 activity, and there are several examples of interactions involving this compound (180). For instance, two weeks of treatment with SJW (300 mg three times a day) in healthy volunteers

Introduction
23
Drug
CYP3A enzyme,ABCB1 Pgp
PXR RXR
CYP3A, ABCB1
Metabolism Efflux
Drug
CYP3A enzyme,ABCB1 Pgp
PXR RXR
CYP3A, ABCB1
PXR RXR
CYP3A, ABCB1CYP3A, ABCB1
Metabolism Efflux
increased the oral clearance of midazolam by more than 50%, while a 20% increase was observed after intravenous administration. Since midazolam is not a substrate for ABCB1, it was concluded that the increase in clearance was caused by induced CYP3A4-levels (181).
Figure 8. The molecular basis for a drug-drug interaction involvinginduction of CYP3A and/or ABCB1. Redrawn from (182).
The effect of enzyme induction on prehepatic and hepatic metabolism of R/S-verapamil after simultaneous oral and intravenous administration of rifampin for 11 days (600 mg daily) was examined by Fromm et al. (128). Rifampin increased the apparent oral clearance of S-verapamil 32-fold and decreased its bioavailability 25-fold. It was concluded that prehepatic metabolism, presumably in the intestinal epithelium, was preferentially induced compared with hepatic metabolism. Other orally administered CYP3A4 substrates that show a similar reduction in AUC during rifampin treatment include nifedipine (129), buspirone (183), and tamoxifen (184).
Certain drugs act as both inhibitors and inducers, an example being ritonavir, for which interactions with CYP3A substrates will be time-dependent. Initial exposure to ritonavir inhibited CYP3A, but as the duration of exposure proceeded, CYP3A was induced. Thus, the clinical outcome after administration of ritonavir is very complex and varies amongst individuals (185).
1.8.3 Drug-drug interactions caused by induction of ABCB1 and CYP3A4 Owing to the overlap substrate specificities of ABCB1 and CYP3A4, and because many inducers affect both proteins, many drug-drug interactions may involve both enzyme and transporter systems. The interaction between rifampin and cyclosporine resulted in an increased clearance and decreased bioavailability of cyclosporine A (7). Since cyclosporin A is a substrate for both ABCB1 and CYP3A4, and rifampin is an inducer of both proteins, the basis for the interaction is most likely a combined effect of induced levels of both ABCB1 and CYP3A4. Similarly, coadministration of SJW with cyclosporine has been found to reduce the level of cyclosporine in the blood by 30% to 60% after oral administration; and a number of cases of organ transplant rejection have been associated with this interaction (186, 187).
The effects of administration of SJW for 12 days (300 mg three times a day) on the disposition of three in vivo probe drugs were explored in 21 volunteers (Dresser et al., 2003). Midazolam was used after oral and intravenous administration to assess CYP3A activity in both the intestine and the liver.The disposition of fexofenadine after an oral

24
dose provided a measure of ABCB1 function; and the oral plasma concentration-time profile of cyclosporine was considered to reflect both CYP3A and ABCB1 activitiy. SJW markedly affected the disposition of all three drugs. The effect of induction measured as oral clearance was more pronounced on CYP3A activity (midazolam) than on ABCB1 activity (fexofenadine). Despite the fact that the disposition of cyclosporine involves both CYP3A and ABCB1, the change in oral clearance of cyclosporine appeared to be more closely related to the increase in ABCB1-function. The lower than expected effect of SJW on the oral clearance of cyclosporine (as compared with the CYP3A-substrate midazolam) suggested that ABCB1 was the rate-limiting step for the overall intestinal uptake an metabolism (188). In conclusion, one can say that the quantitative aspects of induction of CYP3A4 and ABCB1 are complex and vary from one drug to another.
1.8.4 Clinical relevance According to the Food and Drug Administration industry guidelines (189), a drug interaction is associated with clinically significant induction when there is a greater than 30% decrease in plasma drug concentrations and this decrease has the potential to alter the drug response . However, although many drug-drug interactions are of clinical relevance, it is important to keep in mind that the usual outcome is that the pharmacokinetic disposition and clinical activity of each drug proceed independently of each other. In the process of deciding what interactions are of real concern in the course of drug therapy, some general guidelines have been applied. That is, drug interactions are more likely to be important when: 1) drug A produces a very large change in the kinetics and plasma levels of drug B, that is, drug A is a potent inhibitor or inducer (e.g., rifampin or ritonavir coadministered with CYP3A-substrates); 2) the therapeutic index of drug B is narrow (e.g., digoxin) (185). Furthermore, the intrinsic pharmacokinetic properties of the drugs involved influence the potential consequences of an interaction.
As discussed in previous sections, the overlap in substrate specificities between ABCB1 and CYP3A4, along with the fact that many inducers affect both proteins, means that many drug-drug interactions may involve both enzyme and transporter systems. However, there is still no straightforward method by which these two systems can be quantified owing to the complexity involved when both intestinal and hepatic CYP3A4 and Pgp and potentially also other ABC-transporters, are being considered. Care should be taken when evaluating the underlying mechanism of drug-drug interactions until the relative contribution of Pgp-transport and CYP3A4-metabolism to the overall interaction can be quantitatively estimated.
Recently, it was argued that the role of transporters as an intestinal barrier to oral bioavailability may have been overemphasized, especially when evaluated in in vitro systems such as the Caco-2 cell model e.g., (170). This argument was motivated by the fact that high secretory efflux over cell monolayers in vitro do not necessarily correspond to an unsatisfactory oral bioavailability in vivo. For instance, although digoxin and cyclosporin show high efflux ratios in vitro, they show sufficiently high oral bioavailability in vivo.

Introduction
25
Apical
Basolateral
Porous filter membraneExtracellular matrix
Apical
Basolateral
Porous filter membraneExtracellular matrix
In summary, CYP3A mediated metabolism as an intestinal barrier to oral drug bioavailability and as a cause of important drug-drug interactions is well established. However, although the importance of transporters as an intestinal barrier to oral bioavailability may have been overemphasized, the significance of efflux proteins such as Pgp as a cause for drug-drug interactions is being appreciated and should be considered during drug development. Similarly, the interplay between ABCB1 and CYP3A4 remains a complex issue to study; new methods are required to assist with this. Moreover, ABC-transporters localised to other sites in the body than the intestine,including the liver, kidney, and the blood-brain-barrier and the maternal-fetal barrier, may also have a major impact on the disposition of drugs (65, 170). Studies on the role of ABC-transporters as a cause for drug-drug interactions and reduced drug bioavaiability are therefore warranted not only in the intestine, but also in other major organs.
1.9 METHODS FOR STUDYING INTESTINAL DRUG TRANSPORT AND METABOLISM
Studies of intestinal drug absorption and metabolism can be performed using models of increasing complexity in the order in vitro < in situ < in vivo. Detailed mechanistic studies are usually easier to interpret when using the less complex in vitro models and require a much smaller amount of drug, than evaluations done in vivo. However, once a mechanism has been revealed in vitro, its relevance in an in vivo situation must still be shown (190).
1.9.1 Methods for studying intestinal drug transport In the characterization of intestinal drug transport, a number of experimental approaches can be applied. Simple uptake/efflux studies can be performed in whole cells either adherent or in suspension or in membrane vesicles for a rapid screening of substrates and/or inhibitors involved in drug efflux interactions (191). In such assays, fluorescent probes, that are substrates for well-studied ABC-transporters such as ABCB1 are often used. Examples of such probes are rhodamine-123 (192) or calcein-AM (193). Monitoring ATPase activity in cell membrane preparations or purified membrane proteins represents another method of identifying compounds that interact with ATP-consuming ABC-transporters . This method is readily adapted to high throughput format, and ABCB1 membrane preparations are commercially available for this purpose (193).
Figure 9. Schematic representation of an epithelial cellmonolayer grown on a permeable support. Cell monolayers used in Paper I-III were cultured on permeable supports coated with a laminin-rich extracellular matrix. The dotted line represents the surfaces of the experimental medium in the apical and basolateral chambers (194).

26
Drug permeability studies in polarized cell monolayers have been extensively used to determine the effect of drug efflux on the permeability of a drug. A major advantage with this approach is the ability to study the directionality of the efflux. Popular epithelial expression systems for studies of drug transport by a selected ABC-transporter include MDCK and LLC-PK1 cells, which are canine and porcine kidney epithelial cell lines, respectively. These cells require relatively short time in culture and grow as monolayers, thus enabling drug transport studies in both absorptive and secretory directions. MDCK and LLC-PK1 cells transfected with efflux transporters have been used for over a decade, e.g.,(195, 196). Double-transfectants with one uptake- and one efflux transporter were recently demonstrated (197, 198). These systems allow for the study of the interplay between uptake and efflux transporters. Wild-type cells are used as controls for the transfected variant. A commonly used alternative to the epithelial expression systems is the Caco-2 cell line, which originates from human colon carcinoma (199) and spontaneously differentiate into tight monolayers. Caco-2 cells express ABC-transporters in amounts comparable to the human jejunum (3), at least at the mRNA level.
In situ intestinal perfusion techniques allow simultaneous measurements of drug transport, secretion and metabolism to be made. Perfusions are generally performed in anaesthetized experimental animals such as rats. An intestinal segment is isolated by surgery, tubes are connected and the segment is perfused with a buffer containing the drug. Since the blood supply is intact in this model, the viability of the tissue is high. In situ perfusions of rat jejunum has been used for simultaneous determinations of passive permeability, carrier-mediated transport, for instance by ABCB1, and metabolism (200-202).
Current transgenic animal models for assessing drug efflux transporter activity include single, double and triple knock-out mouse models (94, 203, 204). These animals are obtained by the removal or silencing of a gene through homologous recombination. In a seminal study by Sparrebom et al. (85), wild-type (wt) and mdr1a( / ) mice were used to show that Pgp in the intestinal epithelium limits the oral bioavailability of paclitaxel. Although knock-out models are invaluable tools in transporter science (205), removal or silencing of one or more genes will certainly have the potential to affect the organism in different ways. For instance, Schuetz et al. (203) demonstrated compensatory effects in hepatic P-450s and mdr1b following deletion of mdr1a. This must be taken into account when interpreting data obtained from knock-out models.
1.9.2 Methods for studying intestinal drug metabolism In the screening of phase I biotransformation of a new drug, the preferable sequence is to start with microsomes, then progress to CYP supersomes, followed by the use of stable or transfected cell lines and excised tissue (206). Methods with a higher degree of complexity include in situ perfusion techniques performed in anaesthetized animals (200), and transgenic animal models (207, 208).
Subcellular fractions, supersomes (209) and other sources of artificially expressed human CYP are commercially available and have become an integrated part of the drug discovery process. By using different subcellular fractions it is possible to differentiate

Introduction
27
between phase I and II reactions (206). Since microsomes are derived from the endoplasmatic reticulum they are very rich in drug metabolising enzymes such as oxidases like CYPs, and are therefore used for the evaluation of phase I metabolism. Intestinal microsomes can be prepared from different segments of the intestine, thereby enabling regional characterization (119). The cytosolic fraction may used for evaluating certain phase II reactions catalysed by soluble enzymes (206).
A disadvantage with subcellular fractions is that they contain a mixture of enzymes, so it may be difficult to determine which enzyme that catalyses a particular reaction. Thus, the contribution of individual CYPs can be investigated in the presence of specific inhibitors (210), or by using expression systems such as supersomes (209). Supersomes consist of the microsomal fraction of insect cells transfected with the drug metabolizing enzymes of choice. Insect cells lack endogenous CYP expression, and nontransfected insect cells can therefore be used as controls.
Below, the two methods used for the drug transport and metabolism studies in this thesis are presented.
1.9.3 Cell culture models for the assessment of passive drug transport and drug efflux Caco-2 cells grown as monolayers on permeable supports are extensively used in studies of both passive and active drug transport. Studies in this cell culture model are easily performed under controlled conditions, which makes it possible to extract detailed mechanistic information on the various transport processes (50, 194). Since Caco-2 cells are unusually well differentiated, they are functionally similar to the human small intestinal enterocyte in many respects, despite the fact that they originate from a human colorectal carcinoma (30, 199, 211). A variety of transport systems and enzymes are expressed in Caco-2 cells (3, 52, 212). Thus, the Caco-2 cell model may be considered as a multifunctional transport model that allows the study of the interplay between different e.g., ABC-transporters, and also of the relative active and passive contributions to the overall transport.
However, the simultaneous expression of several parallel transport and metabolism processes is also a drawback since: 1) the expression levels in the cell lines do not always mimic the in vivo counterparts, and 2) the study of a single transport or metabolic pathway in a multifunctional system require that specific substrates/inhibitors are available, which is not always the case. Therefore, when various transporters are to be studied in isolation, they are usually expressed in cell lines or other expression systems with a lower background than, for instance, Caco-2 cells.
1.9.4 Cell culture models for simultaneous studies of drug transport and CYP3A4 metabolism In principle, three approaches have been used to develop intestinal cell models for studies of CYP3A4-mediated drug transport and metabolism: 1) use of the CYP3A5 expressing Caco-2 clone TC7 as a substitute for CYP3A4 expression (213) 2) development of CYP3A4-expressing Caco-2 cells using heterologous expression techniques (214-216)

28
Proximal drainage
Stomach drainage
Jejunal segment
Proximal drainage
Stomach drainage
Jejunal segment
and 3) 1 ,25-dihydroxy vitamin D3-mediated induction of CYP3A4 in the Caco-2 cell line (157, 217). The third approach is the most physiological system, in which endogenous CYP3A4 is hormonally upregulated and therefore that system was selected for the studies in this thesis.
In its original version, the D3-induced Caco-2 cell model is based on a specific Caco-2 clone, which requires 14 days growth in a complex differentiation medium, in which D3 is the supplement responsible for the upregulated CYP3A4 expression and activity (217). It was reasoned that a more accessible model, based on a widely available cell line such as the parental Caco-2 cells, would find wider application.
1.9.5 In vivo methods for studying transport and metabolism Direct measurements of intestinal absorption, metabolism and secretion of drugs in humans in vivo may be performed using regional perfusion techniques. Perfusions of the proximal part of the human jejunum have now been successfully performed using a large number of drugs by a few laboratories e.g., (218-220). By collecting shed enterocytes during the single-pass perfusion, analysis of the expression and function of human transporters and enzymes may be combined with the determination of intestinal transport and presystemic metabolism (219, 221).
Figure 10. The Loc-I-Gut instrument consists of a multilumen tube that is introduced from the mouth. Two ballons occlude a 10 cm long intestinal segment, which is perfused with a buffered solution of the drug. Redrawn from Lennernas et al. (218).
In this thesis, the Loc-I-Gut technique (218), shown in Figure 10, was used to investigate the effect of repeated oral administration of St John’s wort (SJW) on jejunal transport and presystemic metabolism of R/S-verapamil. By combining measurements of jejunal permeability and of plasma concentrations of drug and metabolite before and after SJW administration, conclusions could be drawn on the potential interaction between St John’s wort and R/S-verapamil.

Aims
29
2. AIMS OF THE THESIS
The overall aim of this thesis was to study the role of intestinal barriers to oral drug bioavailability. Specific aims were to:
establish and characterize a cell culture model of the human intestinal epithelium for use in studies of ABCB1-mediated drug efflux and CYP3A4-mediated drug metabolism in vitro (Paper I)
use the new cell culture model to study enantioselective efflux and CYP3A4-mediated metabolism of R/S-verapamil (Paper II)
study the effects of repeated drug administration on the gene regulation of ABC-transporters and CYP3A4 in human intestinal cell lines in vitro (Paper III)
explore the effects of repeated oral administration of the herbal drug St John’s wort on the intestinal absorption and presystemic metabolism of R/S-verapamil in humans(Paper IV)

30
3. MATERIALS AND METHODS 3.1 DRUGS AND RADIOLABELED MARKERS
Testosterone, 6 -hydroxytestosterone, clotrimazole, 9-cis-retinoic acid, dexamethasone, rifampicin, verapamil, R-methoxyverapamil, tetrahexylammonium, 17 -estradiol, hyperforin, digoxin and mitoxantrone were purchased from Sigma Chemical Co. (St. Louis, MO, USA). 1 ,25-dihydroxy vitamin D3 (D3) was purchased from Solvay Duphar (Weesp, the Netherlands). Phenobarbital was obtained through Apoteket AB, Sweden. The hydrochloride salts of racemic verapamil and racemic norverapamil, respectively were kindly provided by Knoll AG (Darnstadt, Germany). GF120918 was a generous gift from Glaxo Wellcome (Hertfordshire, UK). Androstenedione and 2 -hydroxytestosterone were kindly provided by Dr. Tommy B Andersson (AstraZeneca, Mölndal, Sweden), and the recombinant human growth hormone was a gift from Dr. Jonas Fransson (Pharmacia Corp., Stockholm, Sweden). [14C]celiprolol (37.3 Ci mg-1)and [14C]mannitol (52 mCi mmol-1) were purchased from Rhone-Poulenc Rorer Pharmaceutical Inc. (Collegeville, PA, USA), and New England Nuclear (Boston, MA, USA), respectively. [3H ]digoxin (17.0 Ci/mmol) and [3H]mitoxantrone (3.0 Ci/mmol), were obtained from Perkin Elmer Life Sciences (Sweden) and Biochemicals Larodan (Sweden), respectively.
In the in vivo study (Paper IV), racemic verapamil hydrochloride (120 mg/L, 244 M,Knoll AG, Darnstadt, Germany) and St John’s wort tablets (300 mg, Movina ,Boehringer Ingelheim, Germany) were used. A low perfusate concentration (10 mg/L, 53 M) of antipyrine (Astra Läkemedel AB, Södertälje, Sweden) was used as a marker for passive transcellular diffusion in all of the perfusion experiments. 14C labelled polyethylene glycol 4000 (14C-PEG 4000) (2.5 Ci/L, Amersham Pharmacia Biotech, Little Chalfont, England) was used as a non-absorbable volume marker.
3.2 CELL CULTURE (PAPER I-III)Three cell lines (BN, LG and WT) originating from biopsies taken from healthy human duodenum were generous gifts from Dr Gerald Pang, cultured according to (222) and used at passage numbers 17-30. The parental population of Caco-2 cells, Caco-2p (199), and the morphologically well-differentiated Caco-2 clone, BBe-1 (223), were obtained from American Type Culture Collection (ATCC; Rockville, MD, USA), while the CYP3A-expressing Caco-2 clone TC7 was a generous gift from Dr. Alain Zweibaum (224). The Caco-2 cells were cultured as described in detail previously (211, 225). Caco-2 cells were used at passage intervals 20-30 and 92-105, while TC7 and BBe-1 were used at passage intervals 22-30 and 48-58, respectively. In Paper I and II, the cell lines were seeded at a density of 400 000 cells/cm2 onto Transwell cell culture inserts (filter diameter 24 mm, mean pore size 0.45 m, Costar, The Netherlands) coated with a mixture of extracellular matrix proteins (Matrigel, 15 g/cm2). At confluency, the culture medium was supplemented with 0.5 M of D3 (217).
The LS180 cell line (226) was also obtained from ATCC, cultured at 37 C, 10% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FCS, 1% sodium pyruvate, 1% L-glutamine, and 1% non-essential amino acids and used at

Materials and methods
31
passage 39 to 43. In Paper III, Caco-2p and LS180 cells were seeded at a density of 150 000 cells/cm2 in plastic culture dishes (12-well Transwell plates) coated with Matrigel 15 g/cm2. The cell culture medium was replaced with fresh medium inducer every second day and always 24 h before an experiment. The exposure to the drugs took place at concentrations recommended previously in published cell culture studies (67, 167, 227).
3.3 REAL-TIME QUANTITATIVE PCR ANALYSIS (PAPER I AND III)Total-RNA was isolated using the RNeasy mini kit (QIAGEN) according to the instructions provided by the manufacturer, with an additional on-column DNase treatment step. RNA was quantified using the RiboGreen reagent from Molecular Probes. Quantitative real-time PCR assay of transcripts was carried out using gene-specific double fluorescence labelled probes and the TaqMan® Universal PCR Master Mix in a 7700 Sequence Detector (PE Applied Biosystems, Norwalk, CT). 6-Carboxy fluorescein (FAM) was used as the 5´fluorescent reporter while quenching tetramethylrhodamine (TAMRA) or non-quenching major grove binding (MGB) probes were added to the 3´end. Gene-specific primers for ABCB1, ABCC2, ABCC3, ABCG2, CYP3A4, CYP3A5 and CYP2B6 (FAM) were obtained from PE Applied Biosystems (Foster City, CA). For primers and probe sequences, see Paper I and III. To account for variations in the amount of RNA added to each reaction and for variations in the efficiency of the reverse transcription, data were normalized against the number of human acidic ribosomal phosphoprotein (huPO) or 18S rRNA transcripts in each sample. The relative fold of induction was calculated using the comparative CT method ( CT, Paper I) or the standard curve method (Paper III), using huPO or 18S rRNA, respectively, as the endogenous control reference in Paper I and III, respectively. The untreated samples were designated the calibrator and assigned the value of 1x for each of the CYP or ABC-transporter targets. The quantity of each mRNA in each treated sample was given relative to the calibrator sample.
3.4 IMMUNOBLOT ANALYSIS (PAPER I)In Paper I, cell lysates from Caco-2 and TC7 cell monolayers were resuspended in a sodium dodecyl sulfate reducing buffer and heated for 5 min at 95 ºC. The samples underwent electrophoresis on a 12% SDS-polyacrylamide gel and were transferred to nitrocellulose membranes, which were sequentially incubated with a monoclonal antibody to human CYP3A4 (Xenotech LLC, Cambridge, KS, USA) and an alkaline phosphatase conjugated anti-rabbit IgG. The immunoblots were then visualised using the CDP-Star Western Blot Chemiluminescence Reagent (NEN Life Science Products, Inc.).
3.5 IN VITRO DRUG TRANSPORT AND EFFLUX STUDIES (PAPER I AND II)The in vitro drug transport experiments in Papers I and II were performed as described previously (211, 225). Briefly, the cell monolayers were kept at 37 C and agitated on a plate shaker throughout the experiment. Samples were taken from the receiver chamber

32
at given times and the extracted volume was compensated with inhibitor solution. At the end of the experiment a sample was drawn from the donor chamber. Drugs were dissolved in Hanks’ Balanced Salt Solution (HBSS, pH 7.4). The integrity of the monolayers was routinely checked with the hydrophilic integrity marker [14C]-mannitol and by measuring the transepithelial electrical resistance. In order to detect any significant carrier-mediated transport of the compound, and to evaluate any possible effects of D3 on transport protein functionality, the R/S-verapamil and [14C]celiprolol transport rates were determined in both apical-to-basolateral (a-b) and basolateral-to-apical (b-a) directions in the untreated (control) and D3-treated monolayers. The efflux activity was expressed as the ratio b-a/a-b. Inhibition of carrier-mediated transport across both control and D3-treated Caco-2 cell monolayers was investigated after the addition of inhibitors to the apical and basolateral side.
The uptake and efflux of 3H-digoxin and 3H-mitoxantrone in Paper III were studied in Caco-2 and LS180 cells grown in Matrigel-coated wells and exposed to hyperforin, GF120918 or vehicle for 5 days. After an initial 30 min washing step at 37 C, the cells were incubated for 30 min at 37 C with the respective drug solution. The cells were then placed on ice and rinsed three times in ice-cold PBS. Efflux medium containing unlabelled drug was added, and at regular time intervals, samples were removed. Finally, the cells were lysed and samples were taken again for scintillation counting and for protein analysis.
3.6 IN VITRO DRUG METABOLISM STUDIES (PAPER I AND II)In Paper I, the formation of 6 -hydroxy testosterone from testosterone was used as a measure of CYP3A activity in filter-grown Caco-2 and TC7 cell monolayers after exposure to D3.26 The experiments were perfomed at 37ºC in HBSS, pH 7.4. Uninduced cell monolayers were used as the controls. The metabolite 6 -OH-testosterone was analysed by a reversed phase HPLC method modified from Kostrubsky et al. (228).
In Paper II, the level of CYP3A4-mediated metabolism of R/S-verapamil was determined in CYP3A4-expressing (D3-treated) Caco-2 cell monolayers, using untreated cells as controls. The cell monolayers were washed and equilibrated 15 min in pre-warmed HBSS, pH 7.4. Thereafter, racemic verapamil dissolved in HBSS was added to the apical side of the monolayer, while HBSS without the drug was added to the receiver side. The monolayers were incubated at 37ºC for up to 240 min depending on whether time- or concentration-dependent metabolic studies were performed. The concentrations of verapamil and the metabolite norverapamil in apical, basolateral and intracellular samples were quantified by an enantioselective HPLC-analysis described below.
3.7 IN VIVO SINGLE-PASS PERFUSION OF THE HUMAN JEJUNUM (PAPER IV)The study described in Paper IV was performed at the Clinical Research Department, University Hospital, Uppsala, Sweden and was approved by both the Ethics Committee of the Medical Faculty, Uppsala University and the Swedish Medical Product Agency,

Materials and methods
33
Uppsala, Sweden. The study consisted of two sequential parts, a control and a treatment phase. In the control phase, a regional single-pass perfusion of the proximal jejunum with R/S-verapamil was performed in eight healthy male volunteers using the Loc-I-Gut® perfusion instrument, figure 10. The subjects had fasted overnight (10 hours) when they arrived at the clinic. After local anesthesia with lidocaine (Xylocain ,AstraZeneca, Södertälje, Sweden) in the upper throat, the tube was introduced orally and positioned in the proximal part of the jejunum by fluoroscopic control. The two balloons were inflated with approximately 26-30 ml of air, creating a 10 cm long jejunal segment. To prevent nausea, gastric juice was drained by a separate tube positioned in the stomach. The jejunal segment was continuously perfused with the test solution containing 120 mg/L racemic verapamil at a flow rate of 2.0 mL/min. The total dose of verapamil given in each 100 minute perfusion experiment was 24 mg. Perfusate leaving the jejunal segment was collected and the drug absorption process was terminated by rinsing the segment with isotonic saline immediately after the perfusion. Venous blood samples were collected immediately before and at intervals until 12 hrs after the start of the perfusion.
The treatment phase was initiated following a washout period of four weeks. Each subject was treated orally with St John’s wort for 14 days (300 mg three times a day) before undergoing a procedure identical to that described for the control phase. In order to prevent any acute inhibitory effects of SJW during the absorption phase, the last dose of St John’s wort was scheduled for the day before the second jejunal perfusion experiment.
3.8 ENANTIOSELECTIVE HPLC ANALYSIS OF R/S-VERAPAMIL AND R/S-NORVERAPAMIL
3.8.1 In vitro cell culture samples and jejunal perfusates (Paper II and IV)R/S-verapamil and R/S-norverapamil in samples from in vitro transport and metabolism experiments in HBSS, pH 7.4 (Paper II) and from jejunal perfusion experiments in perfusion buffer, pH 6.5 (Paper IV) were quantified by enantioselective reversed phase HPLC with fluorescence detection using a modified method from Sandström et al. (229). The analytical column was a Chiral-AGP® (150 x 4.0 mm I.D.). R- and S-verapamil and R- and S-norverapamil were eluted with phosphate buffer (pH .6; ionic strength 0.01)/acetonitrile/methanol (74/18/8 v/v) at a flow rate of 1.0 mL/min. To determine intracellular amounts of verapamil and norverapamil in Caco-2 cell samples, intact cell monolayers were washed in ice-cold PBS, cut and frozen. R/S-verapamil and norverapamil were extracted in ethanol and R-methoxyverapamil was added as an internal standard. After reconstitution in the mobile phase, samples were analyzed as above.
3.8.2 Plasma samples (Paper IV) In Paper IV, the plasma sample preparation was performed using liquid-liquid extraction as described by Hedeland et al. (230). Analysis was performed with LC-MS/MS in the Selected Reaction Monitoring (SRM) mode, using a Chiral-AGP® (150 x

34
4.0 mm I.D) analytical column, and a mobile phase consiting of (pH 7.4 aqueous ammonium acetate buffer/acetonitrile (85/15 v/v). The LC-MS/MS system consisted of an Agilent 1100 liquid chromatograph and a Quattro LC triple quadrupole mass spectrometer (Micromass, Manchester, UK).
3.9 DATA ANALYSIS
3.9.1 Apparent permeability coefficients (Paper I and II)
The apparent permeability coefficient (Papp, cm/s) of mannitol and celiprolol was determined according to the following equation:
AVK=P r
app (2)
where K is the time-dependent steady state rate of change in concentration in the receiver chamber (Ct/C0), Ct is the concentration in the receiver compartment at the end of each time interval, C0 is the initial concentration in the apical chamber for each time interval (mole/ml), Vr is the volume of the receiver chamber (ml) and A is the surface area of the filter membrane (cm2). For rapidly transported compounds such as verapamil and norverapamil, for which sink conditions could not be maintained during the experiments in Paper II, the apparent permeability coefficient (Papp) was calculated using the non-sink condition analysis described previously (231).
3.9.2 Absorption variables from jejunal perfusate data (Paper IV) Calculations were performed from steady-state concentrations in the outlet jejunal perfusate (Paper IV). The concentration of each compound in the perfusate leaving the segment was corrected for water flux in the subsequent calculations of the fraction of drug absorbed (fabs), and the effective permeability (Peff). The fraction of the drug absorbed in the segment during the perfusion (fabs) was calculated according to (232):
outin
inoutabs PEGC
PEGC-1f (3)
where Cin and Cout are the concentrations entering and leaving the jejunal segment, respectively, while PEGin and PEGout are the concentrations of 14C-PEG 4000 (dpm/mL) entering and leaving the segment, respectively. The effective jejunal permeability (Peff)of each drug was calculated according to a well-mixed tank model
rL2Q
CCC=P in
out
outineff (4)

Materials and methods
35
where the cylindrical area representing the jejunal segment (2 rL) was calculated using the intestinal radius (r = 1.75 cm). The calculation of Peff is based on the disappearance ofdrug from the jejunal segment, and is therefore not dependent on the mechanisms involved in the drug transport across the intestinal epithelium (26). The appearance of each enantiomer of the metabolite in the perfusate was expressed as the appearance ratio (Ar) for each enantiomer.
absin
out
f)(verapamilCmil)(norverapaCAr (5)
where Cout (norverapamil) is the concentration of R- or S-norverapamil in the perfusate leaving the segment, and the product Cin (verapamil) . fabs represents the amount of verapamil enantiomer absorbed in the segment (233).
3.9.3 Pharmacokinetic analysis of plasma data (paper IV) In Paper IV, the area under the plasma concentration time curve from time zero to infinity (AUC0- ) corresponding to the enantiomers of verapamil and norverapamil was calculated for each subject using the linear and the logarithmic trapezoidal rules for ascending and descending plasma concentrations, respectively, up to the last time point. The plasma AUC beyond the last point in time to infinity was estimated by dividing the predicted plasma concentration at the last point by the calculated terminal elimination rate constant, k. The linear trapezoidal method was used to calculate the plasma AUC up to 100 minutes (AUC0-100). The (apparent) terminal half-life (t1/2) was obtained from k. The maximum plasma concentration (Cmax) of verapamil and norverapamil, and the time at which it occurred (tmax), were obtained by visual inspection.
3.9.4 Statistical analysis
The differences between two mean values were analyzed with either a two-tailed paired or unpaired Student’s t-test, as appropriate. For multiple comparisons, a one-way analysis of variance was conducted, followed by Tukey’s, Dunnett’s or Newmann-Keuls’ post test, as appropriate. After a variance stabilizing logarithmic transformation, the plasma pharmacokinetic data in Paper IV was analyzed by analysis of variance using the SAS statistical package (SAS Institute Inc., PROC GLM). Differences between the control and treatment phase and the enantiomers of norverapamil and verapamil, were estimated and then transformed back to the original scale. Values are expressed as means one standard deviation (SD) if not stated otherwise. P-values of < 0.05 were considered to be significant.

36
4. RESULTS AND DISCUSSION
4.1 CYP3A4, CYP3A5, AND MDR1/ABCB1 IN HUMAN SMALL AND LARGE INTESTINAL CELL LINES
The aim of Paper I was to develop a cell culture model that would allow investigation of the relative contribution of and interplay between CYP3A, preferably CYP3A4, and MDR1 (ABCB1) in studies of intestinal drug uptake and gut wall metabolism.
At the onset of the work conducted for this thesis, the 1 ,25-dihydroxy vitamin D3(D3)-induced Caco-2 cell model presented by Schmiedlin-Ren et al. (217) had not been fully characterized and could, therefore, be improved upon. For instance, Caco-2 cells originate from the colon, where the expression of CYP is lower than in the small intestine and may be differently regulated (157) In an attempt to obtain a cell model with higher CYP3A4 expression initial studies on three cell lines isolated from the human duodenum were performed (222) These cell lines were the first monolayer-forming intestinal epithelial cell lines to be obtained from healthy human intestine. Analysis of the CYP3A4, CYP3A5 and MDR1 mRNA expression in the duodenal cell lines and in three cell lines of human colonic origin revealed that the two duodenal cell lines BN and LG expressed CYP3A4 and two colonic Caco-2 cell lines (parental Caco-2, Caco-2p, and Caco-2 TC7) expressed CYP3A5. These four cell lines were investigated to determine their monolayer formation on ECM-coated permeable supports.
The two duodenal cell lines BN and LG formed cell layers with extremely high and/or variable TEER values. Transmission electron microscopy (TEM) photographs showed that the BN cells formed tight monolayers of cuboid, partially differentiated epithelial cells with a few small patches in which bilayers were formed. In contrast, LG cells formed continuous bi- to multilayers, making this cell line unsuitable for transport studies.
Cell lines forming monolayers with constitutive expression of CYP3A and MDR1 were induced with D3. CYP3A in the duodenal cell line BN was not induced by D3, and was therefore excluded from further studies. One reason for the low expression of CYP3A4 may be the crypt origin of these cells since, the expression of CYP3A4 follows a gradient along the crypt-villus axis from low expression in the mid-crypts to high expression on the villus tips (234). However, exposure of both Caco-2p and TC7 cells to D3 in monolayers resulted in strongly induced CYP3A4 expression, both at the mRNA and protein levels. In addition, MDR1/ABCB1 mRNA levels were increased by D3.
CYP3A5 has been shown to display a comparable catalytic activity towards several CYP3A4 substrates (235), and the two isoenzymes may also produce different product ratios from the same substrates (1). Thus, expression of CYP3A5 is not ideal in a cell model designed for studies of CYP3A4 metabolism. Preliminary studies of a series of Caco-2 clones revealed that the different clones (TC7, BBe-1, clone 12, and Caco-2p of low and high passage numbers) displayed highly variable expression of CYP3A4 and CYP3A5 mRNA both before and after D3 treatment.

Results and discussion
37
For instance, the strongest D3-induction of CYP3A4 was observed in Caco-2p at high passage numbers (used throughout Paper I-III). Unexpectedly, CYP3A5 mRNA was markedly induced in both Caco-2p and TC7 cells. Since the increase of CYP3A5 was less pronounced than that of CYP3A4 in the high passage Caco-2p cells, we identified this cell line as having the most advantageous CYP3A4/CYP3A5 ratio.
Figure 11. Concentration-dependent CYP3A metabolic activity in a) Caco-2 and b) TC7 control ( ) and D3-treated ( ) cell monolayers measured as 6 -hydroxylation of testosterone. Cell monolayers were incubated for 120 min in HBSS containing 0-200
M testosterone. Samples from the apical and basolateral sides were pooled. Each value represents the mean SD of three cultures.
Human CYP3A5 has not generally been considered to be an inducible enzyme (118, 236), and there is now evidence that CYP3A5 lacks the distal PXR response element responsible for enhanced transcription of CYP3A4 (237). However, Schuetz and collegues were able to induce CYP3A5 in the human intestinal cell line, LS180, using clotrimazole and reserpine as inducing agents (67). The discrepancy regarding CYP3A5 induction might be due to tissue-specific regulation (17, 238).
Certain Caco-2 cell clones have been reported to partially loose their integrity after D3 treatment (217, 239), which may bias the interpretation of transport studies in D3 induced cells. Such a large reduction in integrity will seriously affect the permeability of the Caco-2 cell monolayers to low permeability drugs (240). In Paper II, it was found that the D3-treated Caco-2p cell line retained monolayer integrity, suggesting that this polyclonal cell line is superior for transport studies in CYP3A4-expressing cell monolayers. Clone-specific effects (e.g., a loss of integrity after D3 induction) would be evened out in a polyclonal cell population, while they would be readily detectable in isolated subclones.
6bet
a-O
H-te
stos
tero
ne
(pm
ol/m
in/m
g ce
llula
r pro
tein
)
0
50
100
150
200
250
0 50 100 150 2000
50
100
150
200
0 50 100 150 200 Testosterone ( M) Testosterone ( M)
a. b.

38
Figure 12. Effect of D3 treatment on Pgp activity in the Caco-2 cell line. The cumulative fraction of celiprolol transported across a) control and b) D3 treated Caco-2 monolayers was determined in the apical-to-basolateral ( ) and the basolateral-to-apical ( ) directions, respectively. D3 treatment resulted in a small increase in Pgp activity. Each value represents the mean SD of three cultures.
Caco-2p and TC7 both expressed functional CYP3A4 after D3 induction (Fig 12). Thus, after D3 induction, the rate of 6 -hydroxylation of testosterone in Caco-2p cell monolayers increased from below the level of detection to 149 12 pmol/min/mg protein, Figure 11a and in TC7 cells from 3 to 86 4 pmol/min/mg protein, Figure 11b. The 6 -hydroxytestosterone formation rate was comparable to results reported previously for D3 induced Caco-2 cells or stable CYP3A4 expression systems (157, 214). Functional MDR1 Pgp expression in Caco-2p cells, displaying the most advantageous CYP3A4/CYP3A5 ratio, was indicated by celiprolol permeability being five-fold higher in the secretory direction than in the absorptive direction. This efflux ratio was only slightly affected by the D3-treatment, Figure 12.
In conclusion, the absence of CYP3A4 function in uninduced Caco-2p cells as compared to TC7 cells makes the Caco-2 cell line more suitable for studies of intestinal CYP3A4 metabolism, since the CYP3A4 expression can be turned on and off. Caco-2p cells also showed less variability in CYP3A4 activity than the TC7 cells. D3-induced Caco-2p cells were therefore selected for the studies of concomitant drug transport and metabolism.
4.2. ENANTIOSELECTIVE TRANSPORT AND CYP3A4-MEDIATED METABOLISM OF R/S-VERAPAMIL IN CACO-2 CELL MONOLAYERS
In Paper II, the cell culture model established in Paper I was used to simultaneously investigate the enantioselective aspects of both transport and metabolism in Caco-2 cells, applying clinically relevant concentrations of a commonly used and well-studied drug, R/S-verapamil. . Verapamil was chosen as the model drug, since at the onset of the study, the following information was available: After oral administration, the
a. b.
Cel
ipro
lol t
rans
port
(cum
ulat
ive
frac
tion
abso
rbed
)
0.00
0.02
0.04
0.06
0.08
0.10
0.12
30 60 90 1200.00
0.02
0.04
0.06
0.08
0.10
0.12
30 60 90 120Time (minutes) Time (minutes)
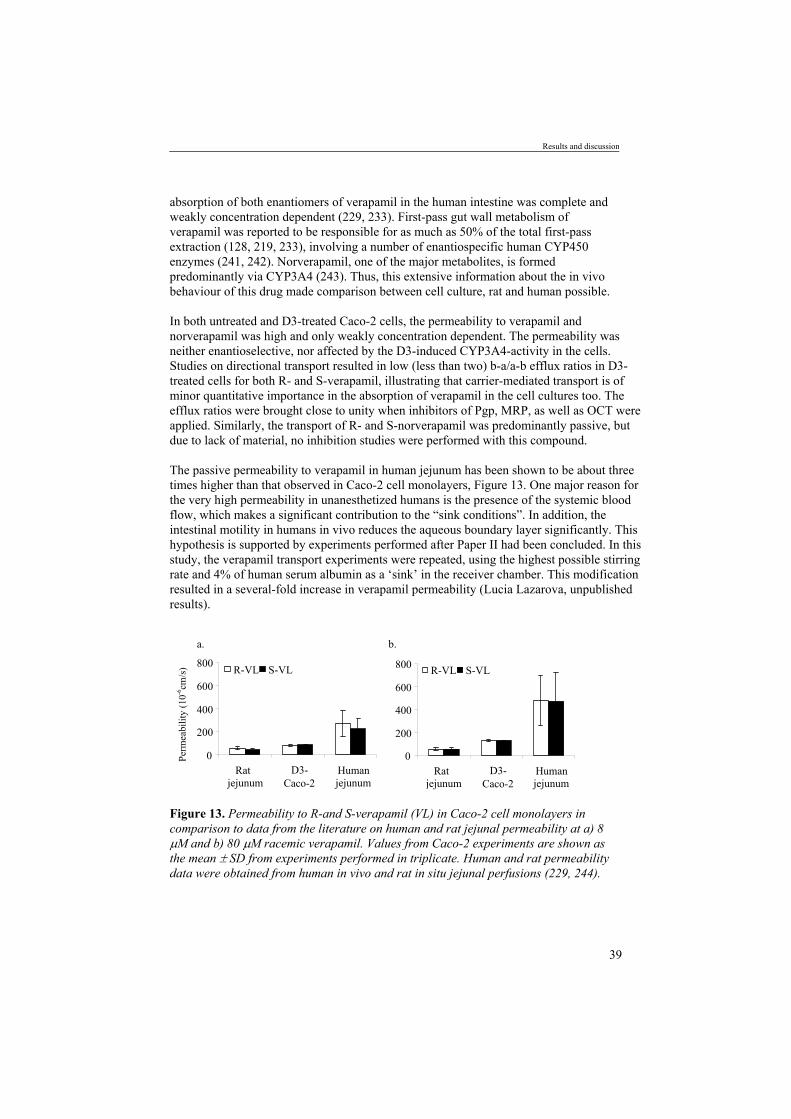
Results and discussion
39
absorption of both enantiomers of verapamil in the human intestine was complete and weakly concentration dependent (229, 233). First-pass gut wall metabolism of verapamil was reported to be responsible for as much as 50% of the total first-pass extraction (128, 219, 233), involving a number of enantiospecific human CYP450 enzymes (241, 242). Norverapamil, one of the major metabolites, is formed predominantly via CYP3A4 (243). Thus, this extensive information about the in vivo behaviour of this drug made comparison between cell culture, rat and human possible.
In both untreated and D3-treated Caco-2 cells, the permeability to verapamil and norverapamil was high and only weakly concentration dependent. The permeability was neither enantioselective, nor affected by the D3-induced CYP3A4-activity in the cells. Studies on directional transport resulted in low (less than two) b-a/a-b efflux ratios in D3-treated cells for both R- and S-verapamil, illustrating that carrier-mediated transport is of minor quantitative importance in the absorption of verapamil in the cell cultures too. The efflux ratios were brought close to unity when inhibitors of Pgp, MRP, as well as OCT were applied. Similarly, the transport of R- and S-norverapamil was predominantly passive, but due to lack of material, no inhibition studies were performed with this compound.
The passive permeability to verapamil in human jejunum has been shown to be about three times higher than that observed in Caco-2 cell monolayers, Figure 13. One major reason for the very high permeability in unanesthetized humans is the presence of the systemic blood flow, which makes a significant contribution to the “sink conditions”. In addition, the intestinal motility in humans in vivo reduces the aqueous boundary layer significantly. This hypothesis is supported by experiments performed after Paper II had been concluded. In this study, the verapamil transport experiments were repeated, using the highest possible stirring rate and 4% of human serum albumin as a ‘sink’ in the receiver chamber. This modification resulted in a several-fold increase in verapamil permeability (Lucia Lazarova, unpublished results).
Figure 13. Permeability to R-and S-verapamil (VL) in Caco-2 cell monolayers in comparison to data from the literature on human and rat jejunal permeability at a) 8
M and b) 80 M racemic verapamil. Values from Caco-2 experiments are shown as the mean SD from experiments performed in triplicate. Human and rat permeability data were obtained from human in vivo and rat in situ jejunal perfusions (229, 244).
Perm
eabi
lity
(10-6
cm/s
)
0
200
400
600
800
Rat jejunum
D3-Caco-2
Human jejunum
R-VL S-VL
0
200
400
600
800
Rat jejunum
D3-Caco-2
Human jejunum
R-VL S-VL
a. b.

40
In contrast, the permeability to verapamil in Caco-2 cell monolayers and in rat jejunum were of similar magnitude, although, at some concentrations, the Caco-2 cell permeability was more than twice as high (Figure 13). The relationship between Caco-2 and rat permeability has been demonstrated to be valid for several other rapidly transported compounds (245).
CYP3A4-mediated demethylation of R/S-verapamil to R- and S-norverapamil in D3-treated Caco-2 cell monolayers was enantioselective, i.e., S-norverapamil formation was higher than that of R-norverapamil, Figure 15, and the levels of R- and S-norverapamil formed constituted 2.8% and 3.4% of the apically applied dose, respectively. The distribution of verapamil and the metabolite formed was comparable for the apical and basolateral sides, which is in accordance with previously reported data (246). The conversion of verapamil to norverapamil was regarded as a CYP3A4-specific reaction, since norverapamil was not formed in untreated Caco-2 cells lacking CYP3A4-activity (247). The CYP3A4-mediated metabolism of verapamil in the human jejunum (229) was significantly higher than that in Caco-2 cells, illustrating a higher level of CYP3A4 expression (3) and activity in vivo (128, 219).
In conclusion, this study shows that the enantioselectivity in the CYP3A4-catalyzed reactions clearly demonstrated a qualitative correlation between the Caco-2 cell model and the human jejunum. The evaluation of this cell culture model against human and rat jejunal data makes it particularly useful in studies of drug transport and metabolism in the preclinical evaluation of new drugs. Since passive transport mechanisms dominate for both verapamil and norverapamil at the clinical concentrations applied in this investigation, it was not possible to study the interplay between drug efflux and metabolism in greater depth.
Figure 14. Concentration-dependent demethylation of R/S-verapamil to a) R-and b) S-norverapamil (NV) in D3-treated Caco-2 cells. Samples were drawn separately from the apical (apical) and the basolateral (basolat) compartments. Data from duplicate determinations are shown for each substrate concentration.
R-norverapamil ( M) S-norverapamil ( M)
0
200
400
600
0 30 60 90 120 150
S-NV apical S-NV basolat
0
200
400
600
0 30 60 90 120 150
R-NV apical R-NV basolateral
Nor
vera
pam
il (p
mol
/inse
rt)
a. b.

Results and discussion
41
Control levels in Caco-2
0.0E+00
1.0E+07
2.0E+07
3.0E+07
4.0E+07
ABCB1 ABCC2 ABCC3 ABCG2
Cop
ies/
ugto
tal R
NA 3 days
4 days5 days7 days16 days
Control levels in LS180
0.0E+00
1.0E+07
2.0E+07
3.0E+07
4.0E+07
ABCB1 ABCC2 ABCC3 ABCG2
Cop
ies/
ugto
tal R
NA 3 days
4 days5 days7 days16 days
Control levels in Caco-2
0.0E+00
1.0E+07
2.0E+07
3.0E+07
4.0E+07
ABCB1 ABCC2 ABCC3 ABCG2
Cop
ies/
ugto
tal R
NA 3 days
4 days5 days7 days16 days
Control levels in LS180
0.0E+00
1.0E+07
2.0E+07
3.0E+07
4.0E+07
ABCB1 ABCC2 ABCC3 ABCG2
Cop
ies/
ugto
tal R
NA 3 days
4 days5 days7 days16 days
4.3 DRUG-INDUCED GENE REGULATION AND FUNCTION OF ABC-TRANSPORTERS IN HUMAN INTESTINAL EPITHELIAL CELL LINES
In Paper III, the gene regulation and function of the four ABC-transporters expressed in the human jejunum (ABCB1, ABCC2, ABCC3, and ABCG2) were studied. The effects of repeated exposure to a panel of drugs that are known to interact with a variety of nuclear receptors such as PXR, VDR and CAR, were evaluated in the two human intestinal epithelial cell lines, Caco-2 and LS180. These two cell lines differ in that Caco-2 cells express VDR and CAR (164, 248), but not PXR (18), whilst LS180 cells express VDR and PXR (18), but not CAR (164). Studies of the regulation of CYP3A4 and CYP2B6, known to be coregulated through cross-talk between PXR and CAR signaling pathways (167, 169), were used as a control of the transcriptional regulation in the two cell lines.
ABC-transporter and CYP expression in untreated intestinal epithelial cell linesABCB1, ABCC2, ABCC3 and ABCG2 were expressed in both Caco-2 and LS180 cell lines. For Caco-2 cells, an increase in transcript levels was observed with time in cell culture for ABCC2 and ABCC3, while the expression of ABCB1 and ABCG2 remained relatively constant over time, Figure 15. The expression of the four transporters was much lower in LS180 cells and no clear time dependence was observed. None of the two investigated CYPs were found in the untreated cell lines.
CYP450 expression in intestinal epithelial cells after repeated drug exposure D3-treatment, previously shown to have a pronounced effect on the expression CYP3A4 in intestinal epithelial cells, served as a positive control for the induction capacity in the two cell lines (217, 249), Paper I. CYP3A4 was strongly induced by D3 (a ligand for VDR) in both cell lines, and in LS180 cells also by the PXR activators rifampicin and hyperforin. These results were in agreement with the presence of VDR in both cell lines, and the presence and absence of PXR in LS180 and Caco-2 cells, respectively (18, 248, 250). CYP2B6 was upregulated by the CAR-activator phenobarbital (251) in Caco-2, but not in LS180 cells, which is in agreement with the presence and absence, respectively, of CAR.
Figure 15. Changes in ABCB1, ABCC2, ABCC3 and ABCG2 transcript levels with time after seeding in untreated Caco-2p (left), and right in LS180 cells. Data are shown as mean values S.D.

42
Effect of drug exposure time on ABC-transporter expressionIn general, ABCB1 and ABCG2 but not ABCC2 and ABCC3 were upregulated following repeated exposure to drugs that activated the various transcriptional pathways. Phenobarbital, which has been shown to activate both CAR and PXR pathways (167, 169),increased the expression of both ABCB1 and ABCG2 significantly in Caco-2 cells. Since CAR has been shown to regulate the basal expression of ABCB1, the upregulation of ABCB1 by phenobarbital could be mediated by CAR. This is supported by that phenobarbital upregulated CYP2B6 transcript levels in Caco-2 cells. LS180 cells exposed to phenobarbital displayed a specific 10- to 30-fold increase in ABCB1 transcript levels, an effect almost certainly mediated by PXR. The absence of CAR in LS180cells was supported by the lack of effect on CYP2B6 by phenobarbital (not shown). In Caco-2 cells, the ABCB1 transcript levels were also significantly increased by the clinically used drugs verapamil (substrate/inhibitor for ABCB1/Pgp and ABCC/MRP), and GF120918 (inhibitor of ABCB1/Pgp and ABCG2/BCRP). In general, the effects of the PXR-activators hyperforin and rifampicin were modest in Caco-2 cells (except for the 3-fold induction of ABCG2), while both compounds were potent inducers of ABCB1 in LS180. These results are compatible with PXR being expressed in LS180 but not in Caco-2 cells (18, 250). Nevertheless, hyperforin mediated an up to 3-fold increase in the ABCG2 expression in Caco-2 cells, presumably via some pathway other than PXR.
Interestingly, long-term repeated exposure to dexamethasone (a ligand for PXR and GR) (252) selectively induced ABCG2 8-fold in Caco-2 cells. Dexamethasone is a well know mediator of cellular differentiation (253) and, although, it is not yet known if dexamethasone is a substrate for ABCG2, it can be speculated that continuous and long-term exposure of Caco-2 cells to dexamethasone might trigger quiescent ABCG2 activity in less differentiated Caco-2 cells as a protective barrier against the differentiating effects of dexamethasone, in analogy with that found in stem cells (112).
Effects of drug exposure on ABC-transporter functionSince it was mainly ABCB1 and ABCG2 transcript levels that were upregulated, selected functional studies were performed using digoxin and mitoxantrone as substrates for ABCB1/Pgp (254) and ABCG2/BCRP (255), respectively. ABCB1/Pgp and ABCG2/BCRP function was upregulated in Caco-2 cells exposed to GF120918 and hyperforin, as observed by an increased efflux of 3H-digoxin and 3H-mitoxantrone, respectively, Figure 16. In contrast, no effects on drug efflux were observed in the LS180 cells (not shown). Digoxin is specifically transported by ABCB1, and a good correlation exists between ABCB1 transcript, protein and transporter function (254). Thus, the increased efflux of digoxin in Caco-2 cells was presumably caused by an increased expression of functional ABCB1.

Results and discussion
43
Figure 16. Effects of drug exposure on the ABCB1 mRNA levels and transporter function. The ABCB1 transcript levels in Caco-2 cells were analysed after exposure to hyperforin or GF120918 for up to 14 days (left). Functional effects of ABCB1 on the efflux of digoxin was evluated in Caco-2 cells exposed to GF120918, hyperforin or vehicle for five days (right). Data are shown as mean values S.D.
Figure 17. Effects of drug exposure on the ABCG2 mRNA levels and transporter function. The ABCB1 transcript levels in Caco-2 cells were analysed after exposure to hyperforin or GF120918 for up to 14 days (left). Functional effects of ABCG2 on the efflux of mitoxantrone was evluated in Caco-2 cells exposed to GF120918, hyperforin or vehicle for five days (right). Data are shown as mean values S.D.
In conclusion, it is not only ABCB1 but also ABCG2 that is upregulated by repeated exposure to drugs that interact with common transcriptional pathways. These findings formed a platform for further investigations of drug-induced changes in the expression and function of ABC-transporters in vitro and in vivo. Thus, it was considered worthwile to investigate the effects of hyperforin on the oral bioavailability of verapamil in humans. This study has been conducted and the results are presented in Paper IV.
4.4 ST JOHN’S WORT DECREASES THE BIOAVAILABILITY OF R- AND S-VERAPAMIL THROUGH INDUCTION OF THE FIRST-PASS METABOLISM
In Paper IV, a jejunal perfusion technique was used to investigate the effect of repeated administration of a widely used herbal drug, St John’s wort (SJW), on the systemic exposure of the enantiomers of verapamil and one its major metabolites, norverapamil. Single-pass perfusions with 120 mg/L R/S-verapamil were performed in eight healthy
0
1
2
3
4
5
6
0 5 10 15 20Time (min)
pmol
/ m
g pr
otei
n
CONTROL CELLS HYP- CELLS GF-CELLS
0
1
2
3
4
5
6
1 2 3 5 14Time (days)
Fold
-indu
ctio
n
CONTROL CELLS HYP CELLS GF CELLS
0
6
12
18
24
0 5 10 15 20Time (min)
pmol
/ m
g pr
otei
n
CONTROL CELLS HYP- CELLS GF-CELLS
0
1
2
3
4
5
6
1 2 3 5 14Time (days)
Fold
-indu
ctio
n
CONTROL CELLS HYP CELLS GF CELLS

44
male volunteers before and after 14 days of oral treatment with St John’s wort (300 mg three times a day).
Following repeated administration of SJW, the area under the plasma concentration-time curve (AUC) for R- and S-verapamil decreased by 78 and 80%, respectively, Figure 18. The corresponding decrease in the maximum concentration (Cmax) was 76 and 78%, Figure 19, respectively, while the terminal half-life was unchanged. S-verapamil was more extensively metabolized than R-verapamil and the AUC for R-verapamil was six times higher than that of S-verapamil in the control phase. This is in excellent agreement with data reported previously (128). Interestingly, SJW did not appear to preferentially induce the metabolism of any of the enantiomers, which makes sense if one consider that the AUC is expected to decrease after induction, irrespective of whether the drug has a high or low high extraction ratio (256).
Figure 18. Plasma concentration-time profiles (mean ± standard error) for (left) R- (squares) and S-verapamil (triangles) and (right) R- (squares) and S-norverapamil (triangles) after 100 minutes jejunal perfusion with racemic verapamil before (Control) (open symbols) and after 14 days of treatment with St John’s wort (+ SJW) (closed symbols) (300 mg three times a day).
Verapamil undergoes extensive presystemic and systemic metabolism, mainly mediated by CYP3A4. Hyperforin, the active component in SJW, is a potent activator of PXR, which has been shown to mediate the induction of intestinal and hepatic CYP3A4 and ABCB1 (161, 250). Norverapamil is mainly formed via CYP3A4 (241, 257) and it can be speculated that the strongest inducing effect was evoked in intestinal CYP3A4 since the enterocytes are exposed to higher concentrations of SJW than are the hepatocytes. Based on these facts, it was concluded that SJW mediates its effects on the verapamil bioavailability through induction of intestinal CYP3A4. Similarly, the PXR-ligand and CYP3A4-inducer rifampicin decreased the bioavailability of R- and S-verapamil mainly by increasing the gut wall extraction ratio (128).
The results from this study also show that induction of ABCB1 or other intestinal transporters is a less likely explanation for the reduction in oral bioavailbility of verapamil, since the jejunal permeability and the terminal half-life were unchanged for both enantiomers. This is in agreement with previous studies, which concluded that the
0
2
4
6
8
10
12
0 100 200 300 400 500 600 700 800
Time (minutes)
Ver
apam
il co
ncen
tratio
n (n
g/m
L)
mean R-Vera controlmean R-Vera SJW
mean S-Vera control
mean S-Vera SJW
Nor
vera
pam
ilco
ncen
tratio
n (n
g/m
L)
0
2
4
6
8
10
12
0 100 200 300 400 500 600 700 800
Time (minutes)
mean R-NV controlmean R-NV SJW
mean S-NV control
mean S-NV SJW
0
2
4
6
8
10
12
0 100 200 300 400 500 600 700 800
Time (minutes)
Ver
apam
il co
ncen
tratio
n (n
g/m
L)
mean R-Vera controlmean R-Vera SJW
mean S-Vera control
mean S-Vera SJW
mean R-Vera controlmean R-Vera SJW
mean S-Vera control
mean S-Vera SJW
Nor
vera
pam
ilco
ncen
tratio
n (n
g/m
L)
0
2
4
6
8
10
12
0 100 200 300 400 500 600 700 800
Time (minutes)
mean R-NV controlmean R-NV SJW
mean S-NV control
mean S-NV SJW
mean R-NV controlmean R-NV SJW
mean S-NV control
mean S-NV SJW

Results and discussion
45
human intestinal permeability of both R- and S-verapamil was unaffected by concomitant administration of ketoconazole, an inhibitor of CYP3A4 and ABCB1 (233). In addition, although a small but significant increase in the secretion of S-norverapamil was detected after treatment with SJW, the effect was modest and may just mirror an increased metabolic activity in the enterocytes.
St John’s wort decreased the plasma levels of the metabolite R/S-norverapamil. Since the pharmacokinetics of norverapamil are rate limited by elimination, it is plausible that this route too is upregulated by SJW. The observed effect of SJW on norverapamil pharmacokinetics was less pronounced than that on verapamil, which could be explained by an increase in the elimination rate partly counteracted by an increase in the formation rate. The metabolism of R/S-norverapamil in vivo has not been well characterized, but in vitro induction studies indicate that both CYPs and phase II enzymes may be involved (258, 259).
Figure 19. Individual AUC0- values for R-verapamil (left) and (right) S-verapamil after a 100 minutes jejunal perfusion with racemic verapamil before (Control) and after 14 days of treatment with St John’s wort (+ SJW), 300 mg three times a day.
In summary, repeated administration of St John’s wort significantly decreased the bioavailability of verapamil. It was concluded that induction of intestinal CYP3A4 played a major role in causing this effect. In contrast, the absorption of the drug was not affected by SJW-treatment, suggesting that intestinal transporters such as ABCB1, play a minor role in limiting the oral bioavailability of verapamil. St John’s wort also affected the pharmacokinetics of norverapamil, which is in line with an upregulation of the CYP3A4 metabolism.
0
1000
2000
3000
4000
5000
6000
Control + SJW0
500
1000
1500
AU
C (n
g/m
l * m
in)
Control + SJW
AU
C (n
g/m
l * m
in)
0
1000
2000
3000
4000
5000
6000
Control + SJW0
500
1000
1500
AU
C (n
g/m
l * m
in)
Control + SJW
AU
C (n
g/m
l * m
in)

46
5. CONCLUDING REMARKS
Two intestinal barriers to oral drug bioavailability have been studied in this thesis: drug efflux proteins of the ABC-transporter family, in particular ABCB1, and the drug-metabolizing enzyme CYP3A4. The main conclusions from this investigation are:
A cell culture model of the human intestinal epithelium that expressed both functional Pgp and CYP3A4 was established. The cell culture model was based on the generally available parental Caco-2 cell line, which, in contrast to normal duodenal cells formed tight cell monolayers that were suitable for drug transport studies.
The new cell culture model was successfully applied in an investigation of the steroselective transport and metabolism of verapamil. The results in the cell culture model, which indicated that active efflux and metabolism play a minor role as barriers to the intestinal epithelial permeability to verapamil, were in agreement with those in the rat and human jejunum. This makes this model potentially useful in studies of drug transport and metabolism during the preclinical evaluation of new drugs.
The effects of repeated drug exposure on the regulation of the ABCB1, ABCC2, ABCC3 and ABCG2 transporters follow complex patterns in intestinal epithelial cell lines, suggesting that both parallel and diverse nuclear receptor pathways participate in the regulation. Not only ABCB1 but also other ABC-transporters, in particular ABCG2 were upregulated, and the effects were observed on both the transcriptional and the functional level. Results from these experiments also indicate that GR and/or CAR might regulate ABCG2 expression.
Repeated oral administration of the herbal drug St John’s wort to healthy volunteers decreased the bioavailability of verapamil, providing evidence for a clinically significant herb-drug interaction. The predominant cause of the decreased oral bioavailability was induction of intestinal CYP3A4, since the oral absorption of verapamil was not affected by treatment with St John’s wort. It was concluded, therefore, that whilst metabolism plays a major role in limiting the oral bioavailability of verapamil, the role of active efflux was far less significant.

Acknowledgements
47
6. ACKNOWLEDGEMENTS
The studies in this thesis were carried out at the Divison of Pharmaceutics, Department of Pharmacy, Faculty of Pharmacy, Uppsala University.
I wish to express my sincere gratitude to:
Mina handledare, professor Per Artursson och professor Hans Lennernäs, för utmärkt vetenskaplig ledning.
Prof. Göran Alderborn och Prof. Christer Nyström för tillhandahållande av ändamålsenliga lokaler och gemytlig atmosfär.
Dr. Anna-Lena Ungell, Dr. Ulf Bredberg, Dr. Tommy B Andersson, och Dr. Carl G Regårdh för givande råd och vetenskapliga diskussioner.
Christer Tannergren, medförfattare och god vän.
Övriga medförfattare, Ann-Cathrin Svensson, Jan Taipalensuu, Charlotta Otter, Brith Leidvik, Lars Knutson, Mikael Hedeland och Ulf Bondesson för era värdefulla bidrag till mitt arbete.
Johan Gråsjö, Göran Ocklind, Ulla Wästberg-Galik för er förmåga att lösa problem av allehanda slag.
Eva Nises-Ahlgren för att du är en sådan klippa!
”Gamla” och nya medlemmar i ”Cellgruppen” för roliga stunder på och utanför lab.
Gamla och nya medlemmar i ”ABS-gruppen” för fantastiskt skojiga fester och trevligt resesällskap.
Dr. Lucia Lazarova, du är en pärla! Tack för all fantastisk hjälp under de här åren!
Dr Staffan Tavelin för mer eller mindre vetenskapliga diskussioner, allmänna retsamheter och glada tillrop.

48
“Mina” studenter Gunilla Bäckström, Pär Mattson, David Rungstad och Sara Ahlgren för era bidrag till mitt avhandlingsarbete!
Dr. Rikard Sandström och Britt Jansson för nyttiga tips om kirala kolonner och enantioselektiva analyser i allmänhet
Alla mina övriga kollegor för att ni gör institutionen till en så trivsam arbetsplats.
Mina rumskamrater och fantastiska vänner, Sofia Berggren och Dr. Eva Krondahl.
Kicki, Evur, Jonas, Soffan, Niclas P, Ulrikur, Björn, Christer, Niclas R, Pär, Helene, och Denny för alla glada och galna upptåg (som bara kan bli fler!)
Mina föräldrar, Gunnar & Maud, för er orubbliga tro på mig genom åren.
Min älskade Tobias, för ditt fantastiska stöd och för att du gör mig så glad!
Financial support from AstraZeneca; The Swedish Academy of Pharmaceutical Sciences, The Swedish Medical Research Council, The Swedish National Board for Laboratory Animals and The Swedish Foundation for Strategic Research is gratefully acknowledged.

References
49
7. REFERENCES
1. Kolars, J.C., Lown, K.S., Schmiedlin-Ren, P., Ghosh, M., Fang, C., Wrighton, S.A., Merion, R.M., and Watkins, P.B. 1994. CYP3A gene expression in human gut epithelium. Pharmacogenetics 4:247-259.
2. Kolars, J.C., Schmiedlin-Ren, P., Schuetz, J.D., Fang, C., and Watkins, P.B. 1992. Identification of rifampin-inducible P450IIIA4 (CYP3A4) in human small bowel enterocytes. J Clin Invest90:1871-1878.
3. Taipalensuu, J., Tornblom, H., Lindberg, G., Einarsson, C., Sjoqvist, F., Melhus, H., Garberg, P., Sjostrom, B., Lundgren, B., and Artursson, P. 2001. Correlation of gene expression of ten drug efflux proteins of the ATP-binding cassette transporter family in normal human jejunum and in human intestinal epithelial Caco-2 cell monolayers. J Pharmacol Exp Ther 299:164-170.
4. Langmann, T., Mauerer, R., Zahn, A., Moehle, C., Probst, M., Stremmel, W., and Schmitz, G. 2003. Real-Time Reverse Transcription-PCR Expression Profiling of the Complete Human ATP-Binding Cassette Transporter Superfamily in Various Tissues. Clin Chem 49:230-238.
5. Fojo, A., Ueda K, Slamon DJ, Poplack DG, Gottesman MM, Pastan I. 1987. Expression of a multidrug-resistance gene in human tumors and tissues. Proc Natl Acad Sci U S A 84:265-269.
6. Paine, M.F., Shen, D.D., Kunze, K.L., Perkins, J.D., Marsh, C.L., McVicar, J.P., Barr, D.M., Gillies, B.S., and Thummel, K.E. 1996. First-pass metabolism of midazolam by the human intestine. Clin Pharmacol Ther 60:14-24.
7. Hebert MF, R.J., Prueksaritanont T, Benet LZ. 1992. Bioavailability of cyclosporine with concomitant rifampin administration is markedly less than predicted by hepatic enzyme induction. Clin Pharmacol Ther 52: 453-457.
8. Greiner, B., Eichelbaum, M., Fritz, P., Kreichgauer, H.P., von Richter, O., Zundler, J., and Kroemer, H.K. 1999. The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J Clin Invest 104:147-153.
9. Jones, C.a. 2002. Human cytochromes P450 and their role in metabolism-based drug-drug interactions. In Drug-Drug Interactions. A. Rodrigues, editor. New York: Marcel Dekker, Inc.
10. Benet, L.a.C., CL. 2001. The drug efflux-metabolism alliance: biochemical aspects. Adv Drug Deliv Rev 50, Suppl 1:S3-11.
11. Wacher, V.J., Wu, C.Y., and Benet, L.Z. 1995. Overlapping substrate specificities and tissue distribution of cytochrome P450 3A and P-glycoprotein: implications for drug delivery and activity in cancer chemotherapy. Mol Carcinog 13:129-134.
12. Durr, D., Stieger, B., Kullak-Ublick, G.A., Rentsch, K.M., Steinert, H.C., Meier, P.J., and Fattinger, K. 2000. St John's Wort induces intestinal P-glycoprotein/MDR1 and intestinal and hepatic CYP3A4. Clin Pharmacol Ther 68:598-604.
13. Hamman, M.A., Bruce, M.A., Haehner-Daniels, B.D., and Hall, S.D. 2001. The effect of rifampin administration on the disposition of fexofenadine. Clin Pharmacol Ther 69:114-121.
14. Piscitelli, S.C., Burstein, A.H., Chaitt, D., Alfaro, R.M., and Falloon, J. 2000. Indinavir concentrations and St John's wort. Lancet 355:547-548.
15. Kliewer, S.A., Moore, J.T., Wade, L., Staudinger, J.L., Watson, M.A., Jones, S.A., McKee, D.D., Oliver, B.B., Willson, T.M., Zetterstrom, R.H., et al. 1998. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell 92:73-82.
16. Maglich, J.M., Stoltz, C.M., Goodwin, B., Hawkins-Brown, D., Moore, J.T., and Kliewer, S.A. 2002. Nuclear Pregnane X Receptor and Constitutive Androstane Receptor Regulate Overlapping but Distinct Sets of Genes Involved in Xenobiotic Detoxification. Mol Pharmacol62:638-646.
17. Tirona, R.G., Lee, W., Leake, B.F., Lan, L.B., Cline, C.B., Lamba, V., Parviz, F., Duncan, S.A., Inoue, Y., Gonzalez, F.J., et al. 2003. The orphan nuclear receptor HNF4alpha determines PXR- and CAR-mediated xenobiotic induction of CYP3A4. Nat Med 9:220-224.
18. Thummel, K.E., Brimer, C., Yasuda, K., Thottassery, J., Senn, T., Lin, Y., Ishizuka, H., Kharasch, E., Schuetz, J., and Schuetz, E. 2001. Transcriptional control of intestinal cytochrome P-4503A by 1alpha,25-dihydroxy vitamin D3. Mol Pharmacol 60:1399-1406.
19. Synold, T.W., Dussault, I., and Forman, B.M. 2001. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat Med 7:584-590.
20. Rowland, M.T., T.N. 1995. Clinical pharmacokinetics: Concepts and applications.

50
21. Wu, C.Y., Benet, L.Z., Hebert, M.F., Gupta, S.K., Rowland, M., Gomez, D.Y., and Wacher, V.J. 1995. Differentiation of absorption and first-pass gut and hepatic metabolism in humans: studies with cyclosporine. Clin Pharmacol Ther 58:492-497.
22. Dressman JB, B.P., Ritschel WA, Friend DR, Rubinstein A, Ziv E. 1993. Gastrointestinal parameters that influence oral medications. J Pharm Sci 82:857-872.
23. Kararli, T. 1989. Gastrointestinal absorption of drugs. Crit Rev Ther Drug Carrier Syst. 6:39-86. 24. Curatolo, W. 1998. Physical chemical properties of oral drug candidates in the discovery and
exploratory dvelopment settings. PSTT 1:387-393. 25. Lipinski, C.A. 2000. Drug-like properties and the causes of poor solubility and poor
permeability. J Pharmacol Toxicol Methods 44:235-249. 26. Amidon, G.L., Lennernas, H., Shah, V.P., and Crison, J.R. 1995. A theoretical basis for a
biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res 12:413-420.
27. Artursson, P., and Karlsson, J. 1991. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem Biophys Res Commun 175:880-885.
28. Fagerholm, U., and Lennernas, H. 1995. Experimental estimation of the effective unstirred water layer thickness in the human jejunum and its importance in oral drug absorption. Eur J Pharm Sci 3:247-253.
29. Madara JL, T.J. 1994. The functional morphology of the small intestine. In Physiology of the gastrointestinal tract. New York: Raven Press. pp.1577-1622.
30. Pageot, L.P., Perreault, N., Basora, N., Francoeur, C., Magny, P., and Beaulieu, J.F. 2000. Human cell models to study small intestinal functions: recapitulation of the crypt-villus axis. Microsc Res Tech 49:394-406.
31. Fihn, B.M., Sjoqvist, A., and Jodal, M. 2000. Permeability of the rat small intestinal epithelium along the villus-crypt axis: effects of glucose transport. Gastroenterology 119:1029-1036.
32. Marcial, M.A., Carlson, S.L., and Madara, J.L. 1984. Partitioning of paracellular conductance along the ileal crypt-villus axis: a hypothesis based on structural analysis with detailed consideration of tight junction structure-function relationships. J Membr Biol 80:59-70.
33. Strocchi, A., Levitt MD. 1993. Role of villous surface area in absorption. Science versus religion. Dig Dis Sci 38:385-387.
34. Artursson, P., Palm, K., and Luthman, K. 2001. Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv Drug Deliv Rev 46:27-43.
35. Gorter, E., Grendel, F. 1925. On bimolecular layers of lipoids on the chromocytes of the blood. JExp Med 41:439-443.
36. Alberts, B., Bray, D, Lewis, J et al. 1989. The plasma membrane. In Molecular biology of the cell. New York: Garland Publishing , Inc. 275-340.
37. Muranishi, S. 1990. Absorption enhancers. Crit Rev Ther Drug Carrier Syst 7:1-33. 38. Stenberg, P., Luthman, K., and Artursson, P. 2000. Virtual screening of intestinal drug
permeability. J Control Release 65:231-243. 39. Clark, D.E., and Pickett, S.D. 2000. Computational methods for the prediction of 'drug-likeness'.
Drug Discov Today 5:49-58. 40. Pickett, S.D., McLay, I.M., and Clark, D.E. 2000. Enhancing the hit-to-lead properties of lead
optimization libraries. J Chem Inf Comput Sci 40:263-272. 41. Stenberg, P., Bergstrom, C.A., Luthman, K., and Artursson, P. 2002. Theoretical predictions of
drug absorption in drug discovery and development. Clin Pharmacokinet 41:877-899. 42. Palm, K., Stenberg, P., Luthman, K., and Artursson, P. 1997. Polar molecular surface properties
predict the intestinal absorption of drugs in humans. Pharm Res 14:568-571. 43. Kelder, J., Grootenhuis, P.D., Bayada, D.M., Delbressine, L.P., and Ploemen, J.P. 1999. Polar
molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm Res 16:1514-1519.
44. Adson, A., Raub, T.J., Burton, P.S., Barsuhn, C.L., Hilgers, A.R., Audus, K.L., and Ho, N.F. 1994. Quantitative approaches to delineate paracellular diffusion in cultured epithelial cell monolayers. J Pharm Sci 83:1529-1536.
45. Karlsson, J., Ungell, A., Grasjo, J., and Artursson, P. 1999. Paracellular drug transport across intestinal epithelia: influence of charge and induced water flux. Eur J Pharm Sci 9:47-56.

References
51
46. Artursson, P., Ungell, A.L., and Lofroth, J.E. 1993. Selective paracellular permeability in two models of intestinal absorption: cultured monolayers of human intestinal epithelial cells and rat intestinal segments. Pharm Res 10:1123-1129.
47. Diamond, J. 1977. Twenty-first Bowditch lecture. The epithelial junction: Bridge, gate and fence. Physiologist 20:10-18.
48. Madara, J. 1989. Loosening tight junctions: lessons from the intestine. J Clin Invest 83:1089-1094.
49. Fasano, A. 1998. Innovative strategies for the oral delivery of drugs and peptides. Trends Biotechnol 16:152-157.
50. Tavelin, S., Hashimoto, K., Malkinson, J., Lazarova, L., Toth I., and Artursson, P. 2003. A new principle for tight junction modulation based on occludin peptides. Mol Pharmacol submitted.
51. Venter, J.C., Adams, M.D., Myers, E.W., Li, P.W., Mural, R.J., Sutton, G.G., Smith H.O., Yandell, M., Evans, C.A., Holt, R.A., et al. 2001. The sequence of the human genome. Science291.
52. Sun, D., Lennernas, H., Welage, L.S., Barnett, J.L., Landowski, C.P., Foster, D., Fleisher, D., Lee, K.D., and Amidon, G.L. 2002. Comparison of human duodenum and Caco-2 gene expression profiles for 12,000 gene sequences tags and correlation with permeability of 26 drugs. Pharm Res 19:1400-1416.
53. Marks, D.L., Gores, G.J., and LaRusso, N.F. 1995. Heaptic processing of peptides. In Peptide based drug design: controlling transport and metabsolism. Washington DC: American Chemical Society. pp. 222-248.
54. Ganapathy, M.E., Huang, W., Wang, H., Ganapathy, V., and Leibach, F.H. 1998. Valacyclovir: a substrate for the intestinal and renal peptide transporters PEPT1 and PEPT2. Biochem Biophys Res Commun 246:470-475.
55. Ganapathy, V., and Leibach, F.H. 1996. Peptide transporters. Curr Opin Nephrol Hypertens5:395-400.
56. Tsuji, A., and Tamai, I. 1996. Carrier-mediated intestinal transport of drugs. Pharm Res 13:963-977.
57. Tamai, I., Ohashi, R., Nezu, J., Yabuuchi, H., Oku, A., Shimane, M., Sai, Y., and Tsuji, A. 1998. Molecular and functional identification of sodium ion-dependent, high affinity human carnitine transporter OCTN2. J Biol Chem 273:20378-20382.
58. Wu, X., Huang, W., Prasad, P.D., Seth, P., Rajan, D.P., Leibach, F.H., Chen, J., Conway, S.J., and Ganapathy, V. 1999. Functional characteristics and tissue distribution pattern of organic cation transporter 2 (OCTN2), an organic cation/carnitine transporter. J Pharmacol Exp Ther290:1482-1492.
59. Ohashi, R., Tamai, I., Yabuuchi, H., Nezu, J.I., Oku, A., Sai, Y., Shimane, M., and Tsuji, A. 1999. Na(+)-dependent carnitine transport by organic cation transporter (OCTN2): its pharmacological and toxicological relevance. J Pharmacol Exp Ther 291:778-784.
60. Nezu, J., Tamai, I., Oku, A., Ohashi, R., Yabuuchi, H., Hashimoto, N., Nikaido, H., Sai, Y., Koizumi, A., Shoji, Y., et al. 1999. Primary systemic carnitine deficiency is caused by mutations in a gene encoding sodium ion-dependent carnitine transporter. Nat Genet 21:91-94.
61. Elimrani, I., Lahjouji, K., Seidman, E., Roy, M.J., Mitchell, G.A., and Qureshi, I. 2003. Expression and localization of organic cation/carnitine transporter OCTN2 in Caco-2 cells. Am J Physiol Gastrointest Liver Physiol 284:G863-871.
62. Hagenbuch, B., and Meier, P.J. 2003. The superfamily of organic anion transporting polypeptides. Biochim Biophys Acta 1609:1-18.
63. Kobayashi, D., Nozawa, T., Imai, K., Nezu, J., Tsuji, A., and Tamai, I. 2003. Involvement of human organic anion transporting polypeptide OATP-B (SLC21A9) in pH-dependent transport across intestinal apical membrane. J Pharmacol Exp Ther 306:703-708.
64. Swaan, P.W. 1998. Recent advances in intestinal macromolecular drug delivery via receptor-mediated transport pathways. Pharm Res 15:826-834.
65. Borst, P., and Elferink, R.O. 2002. MAMMALIAN ABC TRANSPORTERS IN HEALTH AND DISEASE. Annu. Rev. Biochem. 71:537-592.
66. Cordon-Cardo, C., O'Brien, J.P., Boccia, J., Casals, D., Bertino, J.R., and Melamed, M.R. 1990. Expression of the multidrug resistance gene product (P-glycoprotein) in human normal and tumor tissues. J Histochem Cytochem 38:1277-1287.

52
67. Schuetz, E.G., Beck, W.T., and Schuetz, J.D. 1996. Modulators and substrates of P-glycoprotein and cytochrome P4503A coordinately up-regulate these proteins in human colon carcinoma cells. Mol Pharmacol 49:311-318.
68. http://www.nutrigene.4t.com/humanabc.htm. 69. Gotoh, Y., Suzuki, H., Kinoshita, S., Hirohashi, T., Kato, Y., and Sugiyama, Y. 2000.
Involvement of an organic anion transporter (canalicular multispecific organic anion transporter/multidrug resistance-associated protein 2) in gastrointestinal secretion of glutathione conjugates in rats. J Pharmacol Exp Ther 292:433-439.
70. Bates, S.E. 2003. Solving the problem of multidrug resistance: ABC transporters in clinical oncology. In ABC protein from bacteria to man. London: Academic Press Elsevier Science Ltd.
71. Hoffmeyer, S., Burk, O., von Richter, O., Arnold, H.P., Brockmoller, J., Johne, A., Cascorbi, I., Gerloff, T., Roots, I., Eichelbaum, M., et al. 2000. Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc Natl Acad Sci U S A 97:3473-3478.
72. Kerb, R., Hoffmeyer, S., and Brinkmann, U. 2001. ABC drug transporters: hereditary polymorphisms and pharmacological impact in MDR1, MRP1 and MRP2. Pharmacogenomics2:51-64.
73. Kruijtzer, C.M.F., Beijnen, J.H., and Schellens, J.H.M. 2002. Improvement of Oral Drug Treatment by Temporary Inhibition of Drug Transporters and/or Cytochrome P450 in the Gastrointestinal Tract and Liver: An Overview. Oncologist 7:516-530.
74. van Helvoort, A., Smith, A.J., Sprong, H., Fritzsche, I., Schinkel, A.H., Borst, P., and van Meer, G. 1996. MDR1 P-glycoprotein is a lipid translocase of broad specificity, while MDR3 P-glycoprotein specifically translocates phosphatidylcholine. Cell 87:507-517.
75. Shapiro, A., Ling, V. 1998. The mechanism of ATP-dependent multidrug transport by P-glycoprotein. Acta Physiol Scand Suppl. 643.
76. Schinkel, A.H., and Jonker, J.W. 2003. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv Drug Deliv Rev 55:3-29.
77. Borst, P., Schinkel, A.H., Smit, J.J., Wagenaar, E., Van Deemter, L., Smith, A.J., Eijdems, E.W., Baas, F., and Zaman, G.J. 1993. Classical and novel forms of multidrug resistance and the physiological functions of P-glycoproteins in mammals. Pharmacol Ther 60:289-299.
78. Zhang, M., Wang, G., Shapiro, A., and Zhang, J.-T. 1996. Topological Folding and Proteolysis Profile of P-glycoprotein in Membranes of Multidrug-Resistant Cells: Implications for the Drug-Transport Mechanism. Biochemistry 35:pp 9728 - 9736.
79. Linton, R., Kerr, and Higgins. 2003. Structure of ABC Transporters. In ABC protein from bacteria to man. London: Academic Press Elsevier Science Ltd.
80. Litman, T., Druley, T.E., Stein, W.D., and Bates, S.E. 2001. From MDR to MXR: new understanding of multidrug resistance systems, their properties and clinical significance. CellMol Life Sci 58:931-959.
81. Yu, E.W., McDermott, G., Zgurskaya, H.I., Nikaido, H., and Koshland, D.E., Jr. 2003. Structural Basis of Multiple Drug-Binding Capacity of the AcrB Multidrug Efflux Pump. Science 300:976-980.
82. Jonker, J.W., Smit, J.W., Brinkhuis, R.F., Maliepaard, M., Beijnen, J.H., Schellens, J.H., and Schinkel, A.H. 2000. Role of breast cancer resistance protein in the bioavailability and fetal penetration of topotecan. J Natl Cancer Inst 92:1651-1656.
83. Hirohashi, T., Suzuki, H., Chu, X.-Y., Tamai, I., Tsuji, A., and Sugiyama, Y. 2000. Function and Expression of Multidrug Resistance-Associated Protein Family in Human Colon Adenocarcinoma Cells (Caco-2). J Pharmacol Exp Ther 292:265-270.
84. Juliano, R.L., and Ling, V. 1976. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta 455:152-162.
85. Sparreboom, A., van Asperen, J., Mayer, U., Schinkel, A.H., Smit, J.W., Meijer, D.K., Borst, P., Nooijen, W.J., Beijnen, J.H., and van Tellingen, O. 1997. Limited oral bioavailability and active epithelial excretion of paclitaxel (Taxol) caused by P-glycoprotein in the intestine. Proc Natl Acad Sci U S A 94:2031-2035.
86. Seelig, A., and Landwojtowicz, E. 2000. Structure-activity relationship of P-glycoprotein substrates and modifiers. Eur J Pharm Sci 12:31-40.
87. Seelig, A. 1998. A general pattern for substrate recognition by P-glycoprotein. Eur J Biochem251:252-261.

References
53
88. Ambudkar, S.V., Dey, S., Hrycyna, C.A., Ramachandra, M., Pastan, I., and Gottesman, M.M. 1999. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol 39:361-398.
89. Ekins, S., Kim, R.B., Leake, B.F., Dantzig, A.H., Schuetz, E.G., Lan, L.B., Yasuda, K., Shepard, R.L., Winter, M.A., Schuetz, J.D., et al. 2002. Application of three-dimensional quantitative structure-activity relationships of P-glycoprotein inhibitors and substrates. Mol Pharmacol61:974-981.
90. Pajeva, I.K., and Wiese, M. 2002. Pharmacophore model of drugs involved in P-glycoprotein multidrug resistance: explanation of structural variety (hypothesis). J Med Chem 45:5671-5686.
91. Gottesman, M.M., and Pastan, I. 1993. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu Rev Biochem 62:385-427.
92. Scala, S., Akhmed, N., Rao, U.S., Paull, K., Lan, L.B., Dickstein, B., Lee, J.S., Elgemeie, G.H., Stein, W.D., and Bates, S.E. 1997. P-glycoprotein substrates and antagonists cluster into two distinct groups. Mol Pharmacol 51:1024-1033.
93. Thiebaut F, T.T., Hamada H, Gottesman MM, Pastan I, Willingham MC. 1987. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci U S A. 84:7735-7738.
94. Schinkel, A.H., Mayer, U., Wagenaar, E., Mol, C.A., van Deemter, L., Smit, J.J., van der Valk, M.A., Voordouw, A.C., Spits, H., van Tellingen, O., et al. 1997. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc Natl Acad Sci U S A 94:4028-4033.
95. Borst, P., and Schinkel, A.H. 1996. What have we learnt thus far from mice with disrupted P-glycoprotein genes? Eur J Cancer 32A:985-990.
96. Brinkmann, U., Roots, I., and Eichelbaum, M. 2001. Pharmacogenetics of the human drug-transporter gene MDR1: impact of polymorphisms on pharmacotherapy. Drug Discovery Today6:835-839.
97. Mickley, L.A., Lee, J.S., Weng, Z., Zhan, Z., Alvarez, M., Wilson, W., Bates, S.E., and Fojo, T. 1998. Genetic polymorphism in MDR-1: a tool for examining allelic expression in normal cells, unselected and drug-selected cell lines, and human tumors. Blood 91:1749-1756.
98. Cascorbi, I., Gerloff, T., Johne, A., Meisel, C., Hoffmeyer, S., Schwab, M., Schaeffeler, E., Eichelbaum, M., Brinkmann, U., and Roots, I. 2001. Frequency of single nucleotide polymorphisms in the P-glycoprotein drug transporter MDR1 gene in white subjects. Clinical Pharmacology & Therapeutics 69:169-174.
99. Tanabe, M., Ieiri, I., Nagata, N., Inoue, K., Ito, S., Kanamori, Y., Takahashi, M., Kurata, Y., Kigawa, J., Higuchi, S., et al. 2001. Expression of P-glycoprotein in Human Placenta: Relation to Genetic Polymorphism of the Multidrug Resistance (MDR)-1 Gene. J Pharmacol Exp Ther297:1137-1143.
100. Ito, S., Ieiri, I., Tanabe, M., Suzuki, A., Higuchi, S., Otsubo, K. 2001. Polymorphism of the ABC transporter genes, MDR1, MRP1 and MRP2/cMOAT, in healthy Japanese subjects. Pharmacogenetics 11:175-184.
101. Sakaeda, T., Nakamura, T., Horinouchi, M., Kakumoto. M., Ohmoto, N., Sakai. T., Morita, Y., Tamura, T., Aoyama, N., Hirai, M., Kasuga, M., Okumura, K. 2001. MDR1 genotype-related pharmacokinetics of digoxin after single oral administration in healthy Japanese subjects. Pharm Res.
102. Morita, Y., Sakaeda, T., Horinouchi, M., Nakamura, T., Kuroda., K., Miki, I., Yoshimura, K., Sakai, T., Shirasaka, D., Tamura, T., Aoyama, N., Kasuga, M., Okumura, K. 2003. MDR1 Genotype-Related Duodenal Absorption Rate of Digoxin in Healthy Japanese Subjects. Pharmaceutical Research 206:552-555.
103. Kim, R.B., Leake, B.F., Choo, E.F., Dresser, G.K., Kubba, S.V., Schwarz, U.I., Taylor, A., Xie, H.G., McKinsey, J., Zhou, S., et al. 2001. Identification of functionally variant MDR1 alleles among European Americans and African Americans. Clin Pharmacol Ther 70:189-199.
104. Fellay, J., Marzolini, C., Meaden, E.R., Back, D.J., Buclin, T., Chave, J.P., Decosterd, L.A., Furrer, H., Opravil, M., Pantaleo, G., et al. 2002. Response to antiretroviral treatment in HIV-1-infected individuals with allelic variants of the multidrug resistance transporter 1: a pharmacogenetics study. Lancet 359:30-36.
105. Gerk, P.M., and Vore, M. 2002. Regulation of Expression of the Multidrug Resistance-Associated Protein 2 (MRP2) and Its Role in Drug Disposition. J Pharmacol Exp Ther 302:407-415.

54
106. Borst, P., Evers, R., Kool, M., and Wijnholds, J. 1999. The multidrug resistance protein family. Biochim Biophys Acta 1461:347-357.
107. Borst, P., Kool, M., and Evers, R. 1997. Do cMOAT (MRP2), other MRP homologues, and LRP play a role in MDR? Semin Cancer Biol 8:205-213.
108. Doyle, L.A., Yang, W., Abruzzo, L.V., Krogmann, T., Gao, Y., Rishi, A.K., and Ross, D.D. 1998. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci U S A 95:15665-15670.
109. Litman, T., Brangi, M., Hudson, E., Fetsch, P., Abati, A., Ross, D.D., Miyake, K., Resau, J.H., and Bates, S.E. 2000. The multidrug-resistant phenotype associated with overexpression of the new ABC half-transporter, MXR (ABCG2). J Cell Sci 113 ( Pt 11):2011-2021.
110. Maliepaard, M., Scheffer, G.L., Faneyte, I.F., van Gastelen, M.A., Pijnenborg, A.C., Schinkel, A.H., van De Vijver, M.J., Scheper, R.J., and Schellens, J.H. 2001. Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res 61:3458-3464.
111. Suzuki, M., Suzuki, H., Sugimoto, Y., and Sugiyama, Y. 2003. ABCG2 transports sulfated conjugates of steroids and xenobiotics. J Biol Chem 278:22644-22649.
112. Zhou, S., Schuetz, J.D., Bunting, K.D., Colapietro, A.M., Sampath, J., Morris, J.J., Lagutina, I., Grosveld, G.C., Osawa, M., Nakauchi, H., et al. 2001. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med 7:1028-1034.
113. Clarke, S.J., BC. 2002. Human cytochromes P450 and their role in metabolism-based drug-drug interactions. In Drug-drug interactions. New York: Marcel Dekker, Inc. 55-88.
114. Guengerich, F.P. 1997. Role of cytochrome P450 enzymes in drug-drug interactions. Adv Pharmacol 43:7-35.
115. Guengerich, F.P. 1999. Cytochrome P-450 3A4: regulation and role in drug metabolism. AnnuRev Pharmacol Toxicol 39:1-17.
116. Krishna, D.R., and Klotz, U. 1994. Extrahepatic metabolism of drugs in humans. Clin Pharmacokinet 26:144-160.
117. Shimada, T., and Guengerich, F.P. 1989. Evidence for cytochrome P-450NF, the nifedipine oxidase, being the principal enzyme involved in the bioactivation of aflatoxins in human liver. Proc Natl Acad Sci U S A 86:462-465.
118. Wrighton, S.A., Brian, W.R., Sari, M.A., Iwasaki, M., Guengerich, F.P., Raucy, J.L., Molowa, D.T., and Vandenbranden, M. 1990. Studies on the expression and metabolic capabilities of human liver cytochrome P450IIIA5 (HLp3). Mol Pharmacol 38:207-213.
119. Paine, M.F., Khalighi, M., Fisher, J.M., Shen, D.D., Kunze, K.L., Marsh, C.L., Perkins, J.D., and Thummel, K.E. 1997. Characterization of interintestinal and intraintestinal variations in human CYP3A-dependent metabolism. J Pharmacol Exp Ther 283:1552-1562.
120. Kaminsky, L.a.D.X. 2003. HUMAN EXTRAHEPATIC CYTOCHROMES P450: Function in Xenobiotic Metabolism and Tissue-Selective Chemical Toxicity in the Respiratory and Gastrointestinal Tracts*. Annual Review of Pharmacology and Toxicology 43:pp. 149-173.
121. Kivisto, K.T., Bookjans, G., Fromm, M.F., Griese, E.U., Munzel, P., Kroemer, H.K. 1996. Expression of CYP3A4, CYP3A5 and CYP3A7 in human duodenal tissue. Br J Clin Pharmacol42:387-389.
122. Tateishi, T., Watanabe, M., Moriya, H., Yamaguchi, S., Sato, T., and Kobayashi, S. 1999. No ethnic difference between Caucasian and Japanese hepatic samples in the expression frequency of CYP3A5 and CYP3A7 proteins. Biochemical Pharmacology 57:935-939.
123. Kitada M, K.T. 1994. Cytochrome P450 in human fetal liver: significance and fetal-specific expression. Drug Metab Rev. 26:3035-3323.
124. Domanski, T.L., Finta, C., Halpert, J.R., and Zaphiropoulos, P.G. 2001. cDNA Cloning and Initial Characterization of CYP3A43, a Novel Human Cytochrome P450. Mol Pharmacol59:386-392.
125. Westlind, A., Malmebo, S., Johansson, I., Otter, C., Andersson, T.B., Ingelman-Sundberg, M., and Oscarson, M. 2001. Cloning and tissue distribution of a novel human cytochrome p450 of the CYP3A subfamily, CYP3A43. Biochem Biophys Res Commun 281:1349-1355.
126. Regardh CG, E.B., Olsson R, Kendall M, Collste P and Shansky C. 1989. Pharmacokinetics of felodipine in patients with liver disease. Eur J Clin Pharmacol 36:437-439.
127. Wang, S., Sutfin, T., Baarnhielm, C., and Regardh, C. 1989. Contribution of the intestine to the first-pass metabolism of felodipine in the rat. J Pharmacol Exp Ther 250:632-636.

References
55
128. Fromm, M.F., Busse, D., Kroemer, H.K., and Eichelbaum, M. 1996. Differential induction of prehepatic and hepatic metabolism of verapamil by rifampin. Hepatology 24:796-801.
129. Holtbecker, N., Fromm, M.F., Kroemer, H.K., Ohnhaus, E.E., and Heidemann, H. 1996. The nifedipine-rifampin interaction. Evidence for induction of gut wall metabolism. Drug Metab Dispos 24:1121-1123.
130. Lampen, A., Christians, U., Guengerich, F.P., Watkins, P.B., Kolars, J.C., Bader, A., Gonschior, A.K., Dralle, H., Hackbarth, I., and Sewing, K.F. 1995. Metabolism of the immunosuppressant tacrolimus in the small intestine: cytochrome P450, drug interactions, and interindividual variability. Drug Metab Dispos 23:1315-1324.
131. Wacher, V.J., Silverman, J.A., Zhang, Y., and Benet, L.Z. 1998. Role of P-glycoprotein and cytochrome P450 3A in limiting oral absorption of peptides and peptidomimetics. J Pharm Sci87:1322-1330.
132. Shimada, T., Yamazaki, H., Mimura, M., Inui, Y., and Guengerich, F.P. 1994. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther 270:414-423.
133. Kolars, J.C., Schmiedlin-Ren, P., Dobbins, W.O., 3rd, Schuetz, J., Wrighton, S.A., and Watkins, P.B. 1992. Heterogeneity of cytochrome P450IIIA expression in rat gut epithelia. Gastroenterology 102:1186-1198.
134. Watkins PB, W.S., Schuetz EG, Molowa DT, Guzelian PS. 1987. Identification of glucocorticoid-inducible cytochromes P-450 in the intestinal mucosa of rats and man. J Clin Invest 80:1029-1036.
135. Lown, K.S., Kolars, J.C., Thummel, K.E., Barnett, J.L., Kunze, K.L., Wrighton, S.A., and Watkins, P.B. 1994. Interpatient heterogeneity in expression of CYP3A4 and CYP3A5 in small bowel. Lack of prediction by the erythromycin breath test. Drug Metab Dispos 22:947-955.
136. Thummel KE, S.D., Podoll TD, Kunze KL, Trager WF, Bacchi CE, Marsh CL, McVicar JP, Barr DM, Perkins JD, et al. 1994. Use of midazolam as a human cytochrome P450 3A probe: II. Characterization of inter- and intraindividual hepatic CYP3A variability after liver transplantation. J Pharmacol Exp Ther. 271:557-566.
137. Ozdemir, V., Kalowa, W., Tang, B.K., Paterson, A.D., Walker, S.E., Endrenyi, L., and Kashuba, A.D. 2000. Evaluation of the genetic component of variability in CYP3A4 activity: a repeated drug administration method. Pharmacogenetics 10:373-388.
138. www.imm.ki.se/CYPalleles. 139. Kuehl, P., Zhang, J., Lin, Y., Lamba, J., Assem, M., Schuetz, J., Watkins, P.B., Daly, A.,
Wrighton, S.A., Hall, S.D., et al. 2001. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat Genet 27:383-391.
140. Westlind-Johnsson, A., Malmebo, S., Johansson, A., Otter, C., Andersson, T.B., Johansson, I., Edwards, R.J., Boobis, A.R., and Ingelman-Sundberg, M. 2003. Comparative analysis of CYP3A expression in human liver suggests only a major role for CYP3A5 in drug metabolism. Drug Metab Dispos 31:755-761.
141. Drocourt, L., Ourlin, J.C., Pascussi, J.M., Maurel, P., and Vilarem, M.J. 2002. Expression of CYP3A4, CYP2B6, and CYP2C9 is regulated by the vitamin D receptor pathway in primary human hepatocytes. J Biol Chem 277:25125-25132.
142. Hughes MR, M.P., Kieback DG, Kesterson RA, Pike JW, Feldman D, O'Malley BW. 1988. Point mutations in the human vitamin D receptor gene associated with hypocalcemic rickets. Science 242:1702-1705.
143. Makishima, M., Lu, T.T., Xie, W., Whitfield, G.K., Domoto, H., Evans, R.M., Haussler, M.R., and Mangelsdorf, D.J. 2002. Vitamin D Receptor As an Intestinal Bile Acid Sensor. Science296:1313-1316.
144. Gibbs, J.P., Yang, J.-S., and Slattery, J.T. 1998. Comparison of Human Liver and Small Intestinal Glutathione S-Transferase-Catalyzed Busulfan Conjugation in Vitro. Drug Metab Dispos 26:52-55.
145. Eisenhofer, G., Coughtrie, M.W., and Goldstein, D.S. 1999. Dopamine sulphate: an enigma resolved. Clinical And Experimental Pharmacology & Physiology. Supplement 26:S41-S53.
146. Meech, R., and Mackenzie, P.I. 1997. Structure and function of uridine diphosphate glucuronosyltransferases. Clin Exp Pharmacol Physiol 24:907-915.

56
147. Paine, M.F., and Fisher, M.B. 2000. Immunochemical Identification of UGT Isoforms in Human Small Bowel and in Caco-2 Cell Monolayers. Biochemical and Biophysical Research Communications 273:1053-1057.
148. Cheng, Z., Radominska-Pandya, A., and Tephly, T.R. 1999. Studies on the substrate specificity of human intestinal UDP- lucuronosyltransferases 1A8 and 1A10. Drug Metabolism And Disposition: The Biological Fate Of Chemicals 27:1165-1170.
149. Fisher MB, V.M., Findlay K, Burchell B, Thummel KE, Hall SD, Wrighton SA. 2000. Tissue distribution and interindividual variation in human UDP-glucuronosyltransferase activity: relationship between UGT1A1 promoter genotype and variability in a liver bank. Pharmacogenetics:727-739.
150. Suzuki, H., and Sugiyama, Y. 1998. Excretion of GSSG and glutathione conjugates mediated by MRP1 and cMOAT/MRP2. Semin Liver Dis 18:359-376.
151. Konig, J., Nies, A.T., Cui, Y., Leier, I., and Keppler, D. 1999. Conjugate export pumps of the multidrug resistance protein (MRP) family: localization, substrate specificity, and MRP2-mediated drug resistance. Biochimica et Biophysica Acta (BBA) - Biomembranes 1461:377-394.
152. Gan, L.S., Moseley, M.A., Khosla, B., Augustijns, P.F., Bradshaw, T.P., Hendren, R.W., and Thakker, D.R. 1996. CYP3A-like cytochrome P450-mediated metabolism and polarized efflux of cyclosporin A in Caco-2 cells. Drug Metab Dispos 24:344-349.
153. Suzuki, H., and Sugiyama, Y. 2000. Role of metabolic enzymes and efflux transporters in the absorption of drugs from the small intestine. Eur J Pharm Sci 12:3-12.
154. Hebert, M.F. 1997. Contributions of hepatic and intestinal metabolism and P-glycoprotein to cyclosporine and tacrolimus oral drug delivery. Adv Drug Deliv Rev 27:201-214.
155. Cummins, C.L., Jacobsen, W., and Benet, L.Z. 2002. Unmasking the dynamic interplay between intestinal P-glycoprotein and CYP3A4. J Pharmacol Exp Ther 300:1036-1045.
156. Lown, K.S., Mayo, R.R., Leichtman, A.B., Hsiao, H.L., Turgeon, D.K., Schmiedlin-Ren, P., Brown, M.B., Guo, W., Rossi, S.J., Benet, L.Z., et al. 1997. Role of intestinal P-glycoprotein (mdr1) in interpatient variation in the oral bioavailability of cyclosporine. Clin Pharmacol Ther62:248-260.
157. Hochman, J.H., Chiba, M., Nishime, J., Yamazaki, M., and Lin, J.H. 2000. Influence of P-glycoprotein on the transport and metabolism of indinavir in Caco-2 cells expressing cytochrome P-450 3A4. J Pharmacol Exp Ther 292:310-318.
158. Johnson BM, C.W., Porter CJ. 2001. The impact of P-glycoprotein efflux on enterocyte residence time and enterocyte-based metabolism of verapamil. J Pharm Pharmacol 53:1611-1619.
159. Zhang Y, B.L. 1998. Characterization of P-glycoprotein mediated transport of K02, a novel vinylsulfone peptidomimetic cysteine protease inhibitor, across MDR1-MDCK and Caco-2 cell monolayers. Pharm Res 15:1520-1524.
160. Cummins, C.L., Salphati, L., Reid, M.J., and Benet, L.Z. 2003. In vivo modulation of intestinal CYP3A metabolism by P-glycoprotein: studies using the rat single-pass intestinal perfusion model. J Pharmacol Exp Ther 305:306-314.
161. Moore, L.B., Goodwin, B., Jones, S.A., Wisely, G.B., Serabjit-Singh, C.J., Willson, T.M., Collins, J.L., and Kliewer, S.A. 2000. St. John's wort induces hepatic drug metabolism through activation of the pregnane X receptor. PNAS 97:7500-7502.
162. Johne, A., Brockmoller, J., Bauer, S., Maurer, A., Langheinrich, M., and Roots, I. 1999. Pharmacokinetic interaction of digoxin with an herbal extract from St John's wort (Hypericum perforatum). Clin Pharmacol Ther 66:338-345.
163. Bauer, S., Stormer, E., Johne, A., Kruger, H., Budde, K., Neumayer, H.H., Roots, I., and Mai, I. 2003. Alterations in cyclosporin A pharmacokinetics and metabolism during treatment with St John's wort in renal transplant patients. Br J Clin Pharmacol 55:203-211.
164. Burk, O., Geick, A., Arnold, K., Tegude, H., and Eichelbaum, M. 2001. Regulation of intestinal MDR1 expression by the nuclear receptor CAR. Drug Metab Rev 33:Abstract 127.
165. Cherrington, N.J., Hartley, D.P., Li, N., Johnson, D.R., and Klaassen, C.D. 2002. Organ distribution of multidrug resistance proteins 1, 2, and 3 (Mrp1, 2, and 3) mRNA and hepatic induction of Mrp3 by constitutive androstane receptor activators in rats. J Pharmacol Exp Ther300:97-104.
166. Pascussi, J.M., Gerbal-Chaloin, S., Fabre, J.M., Maurel, P., and Vilarem, M.J. 2000. Dexamethasone enhances constitutive androstane receptor expression in human hepatocytes: consequences on cytochrome P450 gene regulation. Mol Pharmacol 58:1441-1450.

References
57
167. Moore, L.B., Parks, D.J., Jones, S.A., Bledsoe, R.K., Consler, T.G., Stimmel, J.B., Goodwin, B., Liddle, C., Blanchard, S.G., Willson, T.M., et al. 2000. Orphan nuclear receptors constitutive androstane receptor and pregnane X receptor share xenobiotic and steroid ligands. J Biol Chem275:15122-15127.
168. Kast, H.R., Goodwin, B., Tarr, P.T., Jones, S.A., Anisfeld, A.M., Stoltz, C.M., Tontonoz, P., Kliewer, S., Willson, T.M., and Edwards, P.A. 2002. Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor, and constitutive androstane receptor. J Biol Chem 277:2908-2915.
169. Goodwin, B., Moore, L.B., Stoltz, C.M., McKee, D.D., and Kliewer, S.A. 2001. Regulation of the human CYP2B6 gene by the nuclear pregnane X receptor. Mol Pharmacol 60:427-431.
170. Lin JH, Y.M. 2003. Role of P-glycoprotein in pharmacokinetics: clinical implications. Clin Pharmacokinet 42:59-98.
171. Mizuno, N., Niwa, T., Yotsumoto, Y., and Sugiyama, Y. 2003. Impact of Drug Transporter Studies on Drug Discovery and Development. Pharmacol Rev 55:425-461.
172. Moore JT, K.S. 2000. Use of the nuclear receptor PXR to predict drug interactions. Toxicology16:1-10.
173. Raucy, J.L., Mueller, L., Duan, K., Allen, S.W., Strom, S., and Lasker, J.M. 2002. Expression and Induction of CYP2C P450 Enzymes in Primary Cultures of Human Hepatocytes. JPharmacol Exp Ther 302:475-482.
174. Westphal, K., Weinbrenner, A., Zschiesche, M., Franke, G., Knoke, M., Oertel, R., Fritz, P., von Richter, O., Warzok, R., Hachenberg, T., et al. 2000. Induction of P-glycoprotein by rifampin increases intestinal secretion of talinolol in human beings: a new type of drug/drug interaction. Clin Pharmacol Ther 68:345-355.
175. Cvetkovic, M., Leake, B., Fromm, M.F., Wilkinson, G.R., and Kim, R.B. 1999. OATP and P-Glycoprotein Transporters Mediate the Cellular Uptake and Excretion of Fexofenadine. Drug Metab Dispos 27:866-871.
176. Tirona, R.G., Leake, B.F., Wolkoff, A.W., and Kim, R.B. 2003. Human Organic Anion Transporting Polypeptide-C (SLC21A6) Is a Major Determinant of Rifampin-Mediated Pregnane X Receptor Activation. J Pharmacol Exp Ther 304:223-228.
177. http://www.fda.gov/cder/drug/advisory/stjwort.htm. 178. Henderson, L., Yue, Q.Y., Bergquist, C., Gerden, B., and Arlett, P. 2002. St John's wort
(Hypericum perforatum): drug interactions and clinical outcomes. Br J Clin Pharmacol 54:349-356.
179. Wang, Z., Hamman, M.A., Huang, S.M., Lesko, L.J., and Hall, S.D. 2002. Effect of St John's wort on the pharmacokinetics of fexofenadine. Clin Pharmacol Ther 71:414-420.
180. Thummel, K.E., and Wilkinson, G.R. 1998. In vitro and in vivo drug interactions involving human CYP3A. Annu Rev Pharmacol Toxicol 38:389-430.
181. Wang, Z., Gorski, J.C., Hamman, M.A., Huang, S.-M., Lesko, L.J., and Hall, S.D. 2001. The effects of St John's wort (Hypericum perforatum) on human cytochrome P450 activity. Clinical Pharmacology & Therapeutics 70:317-326.
182. Willson, T.M., and Kliewer, S.A. 2002. PXR, CAR and drug metabolism. Nat Rev Drug Discov1:259-266.
183. Lamberg, K., Neuvonen. 1998. Concentrations and effects of buspirone are considerably reduced by rifampicin. British Journal of Clinical Pharmacology 45:381.
184. Kivisto, K.T., Villikka, K., Nyman, L., Anttila, M., Neuvonen, P.J. 1998. Tamoxifen and toremifene concentrations in plasma are greatly decreased by rifampin. Clin Pharmacol Ther64:648-654.
185. Greenblatt, D.J.a.v.M., L.L. 2002. Drug-drug interactions: Clinical perspective. In Drug-drug interactions. New York: Marcel Dekker, Inc.
186. Barone, G.W., Gurley, B.J., Ketel, B.L., and Abul-Ezz, S.R. 2001. Herbal supplements: a potential for drug interactions in transplant recipients. Transplantation 71:239-241.
187. Moschella C, J.B. 2001. Interaction between cyclosporine and Hypericum perforatum (St. John's wort) after organ transplantation. Am J Kidney Dis. 38:1105-1107.
188. Greenblatt DJ, v.M.L., Daily JP, Harmatz JS, Shader RI. 1999. Extensive impairment of triazolam and alprazolam clearance by short-term low-dose ritonavir: the clinical dilemma of concurrent inhibition and induction. J Clin Psychopharmacol 19:293-296.
189. http://www.fda.gov/cder/guidance/index.htm. 1999.

58
190. Kremers, P. 2002. In vitro tests for predicting drug-drug interactions: the need for validated procedures. Pharmacol Toxicol 91:209-217.
191. Zhang, Y., Bachmeier, C., and Miller, D.W. 2003. In vitro and in vivo models for assessing drug efflux transporter activity. Adv Drug Deliv Rev 55:31-51.
192. Lee JS, P.K., Alvarez M, Hose C, Monks A, Grever M, Fojo AT, Bates SE. 1994. Rhodamine efflux patterns predict P-glycoprotein substrates in the National Cancer Institute drug screen. Mol Pharmacol:627-638.
193. Polli, J.W., Wring, S.A., Humphreys, J.E., Huang, L., Morgan, J.B., Webster, L.O., and Serabjit-Singh, C.S. 2001. Rational Use of in Vitro P-glycoprotein Assays in Drug Discovery. JPharmacol Exp Ther 299:620-628.
194. Artursson, P., and Magnusson, C. 1990. Epithelial transport of drugs in cell culture. II: Effect of extracellular calcium concentration on the paracellular transport of drugs of different lipophilicities across monolayers of intestinal epithelial (Caco-2) cells. J Pharm Sci 79:595-600.
195. Horio, M., Chin, K., Currier, S., Goldenberg, S., Williams, C., Pastan, I., Gottesman, M., and Handler, J. 1989. Transepithelial transport of drugs by the multidrug transporter in cultured Madin-Darby canine kidney cell epithelia. J. Biol. Chem. 264:14880-14884.
196. Tanigawara Y, O.N., Hirai M, Yasuhara M, Ueda K, Kioka N, Komano T, Hori R. 1992. Transport of digoxin by human P-glycoprotein expressed in a porcine kidney epithelial cell line (LLC-PK1). J Pharmacol Exp Ther. 263:840-845.
197. Cui, Y., Konig, J., and Keppler, D. 2001. Vectorial Transport by Double-Transfected Cells Expressing the Human Uptake Transporter SLC21A8 and the Apical Export Pump ABCC2. Mol Pharmacol 60:934-943.
198. Sasaki, M., Suzuki, H., Ito, K., Abe, T., and Sugiyama, Y. 2002. Transcellular Transport of Organic Anions Across a Double-transfected Madin-Darby Canine Kidney II Cell Monolayer Expressing Both Human Organic Anion-transporting Polypeptide (OATP2/SLC21A6) and Multidrug Resistance-associated Protein 2 (MRP2/ABCC2). J. Biol. Chem. 277:6497-6503.
199. Fogh, J., Fogh, J.M., and Orfeo, T. 1977. One hundred and twenty-seven cultured human tumor cell lines producing tumors in nude mice. J Natl Cancer Inst 59:221-226.
200. Sandstrom, R., and Lennernas, H. 1999. Repeated oral rifampicin decreases the jejunal permeability of R/S-verapamil in rats. Drug Metab Dispos 27:951-955.
201. Krondahl, E., Orzechowski, A., Ekstrom, G., and Lennernas, H. 1997. Rat jejunal permeability and metabolism of mu-selective tetrapeptides in gastrointestinal fluids from humans and rats. Pharm Res 14:1780-1785.
202. Svensson, U.S., Sandstrom, R., Carlborg, O., Lennernas, H., and Ashton, M. 1999. High in situ rat intestinal permeability of artemisinin unaffected by multiple dosing and with no evidence of P-glycoprotein involvement. Drug Metab Dispos 27:227-232.
203. Schuetz, E.G., Umbenhauer, D.R., Yasuda, K., Brimer, C., Nguyen, L., Relling, M.V., Schuetz, J.D., and Schinkel, A.H. 2000. Altered Expression of Hepatic Cytochromes P-450 in Mice Deficient in One or More mdr1 Genes. Mol Pharmacol 57:188-197.
204. Wijnholds, J., de Lange, E.C.M., Scheffer, G.L., van den Berg, D.-J., Mol, C.A.A.M., van der Valk, M., Schinkel, A.H., Scheper, R.J., Breimer, D.D., and Borst, P. 2000. Multidrug resistance protein 1 protects the choroid plexus epithelium and contributes to the blood-cerebrospinal fluid barrier. J. Clin. Invest. 105:279-285.
205. Schinkel, A.H. 1998. Pharmacological insights from P-glycoprotein knockout mice. Int J Clin Pharmacol Ther 36:9-13.
206. Brandon, E.F.A., Raap, C.D., Meijerman, I., Beijnen, J.H., and Schellens, J.H.M. 2003. An update on in vitro test methods in human hepatic drug biotransformation research: pros and cons. Toxicology and Applied Pharmacology 189:233-246.
207. Robertson, G.R., Field, J., Goodwin, B., Bierach, S., Tran, M., Lehnert, A., and Liddle, C. 2003. Transgenic Mouse Models of Human CYP3A4 Gene Regulation. Mol Pharmacol 64:42-50.
208. Granvil, C.P., Yu, A.-M., Elizondo, G., Akiyama, T.E., Cheung, C., Feigenbaum, L., Krausz, K.W., and Gonzalez, F.J. 2003. Expression of the Human CYP3A4 Gene in the Small Intestine of Transgenic Mice: In Vitro Metabolism and Pharmacokinetics of Midazolam. Drug Metab Dispos 31:548-558.
209. www.gentest.com. 210. Birkett, D.J., Mackenzie, P.I., Veronese, M.E., Miners, J.O. 1993. In vitro approaches can
predict human drug metabolism. Trends Pharmacol Sci 14:292-294.

References
59
211. Artursson, P. 1990. Epithelial transport of drugs in cell culture. I: A model for studying the passive diffusion of drugs over intestinal absorptive (Caco-2) cells. J Pharm Sci 79:476-482.
212. Carriere, V., Barbat, A., Rousset, M., Brot-Laroche, E., Dussaulx, E., Cambier, D., De Waziers, I.D., Beaune, P., and Zweibaum, A. 1996. Regulation of sucrase-isomaltase and hexose transporters in Caco-2 cells: a role for cytochrome P-4501A1? Am J Physiol 270:G976-986.
213. Raeissi, S.D., Guo, Z., Dobson, G.L., Artursson, P., and Hidalgo, I.J. 1997. Comparison of CYP3A activities in a subclone of Caco-2 cells (TC7) and human intestine. Pharm Res 14:1019-1025.
214. Crespi, C.L., Penman, B.W., and Hu, M. 1996. Development of Caco-2 cells expressing high levels of cDNA-derived cytochrome P4503A4. Pharm Res 13:1635-1641.
215. Hu, M., Li, Y., Davitt, C.M., Huang, S.M., Thummel, K., Penman, B.W., and Crespi, C.L. 1999. Transport and metabolic characterization of Caco-2 cells expressing CYP3A4 and CYP3A4 plus oxidoreductase. Pharm Res 16:1352-1359.
216. Brimer, C., Dalton, J.T., Zhu, Z., Schuetz, J., Yasuda, K., Vanin, E., Relling, M.V., Lu, Y., and Schuetz, E.G. 2000. Creation of polarized cells coexpressing CYP3A4, NADPH cytochrome P450 reductase and MDR1/P-glycoprotein. Pharm Res 17:803-810.
217. Schmiedlin-Ren, P., Thummel, K.E., Fisher, J.M., Paine, M.F., Lown, K.S., and Watkins, P.B. 1997. Expression of enzymatically active CYP3A4 by Caco-2 cells grown on extracellular matrix-coated permeable supports in the presence of 1alpha,25-dihydroxyvitamin D3. Mol Pharmacol 51:741-754.
218. Lennernas, H., Ahrenstedt, O., Hallgren, R., Knutson, L., Ryde, M., and Paalzow, L.K. 1992. Regional jejunal perfusion, a new in vivo approach to study oral drug absorption in man. Pharm Res 9:1243-1251.
219. von Richter, O., Greiner, B., Fromm, M.F., Fraser, R., Omari, T., Barclay, M.L., Dent, J., Somogyi, A.A., and Eichelbaum, M. 2001. Determination of in vivo absorption, metabolism, and transport of drugs by the human intestinal wall and liver with a novel perfusion technique. Clin Pharmacol Ther 70:217-227.
220. Petri, N., Lennernas, H. 2003. In vivo permeability studies in the GI tract. In Drug bioavailability. Estimation of solubility, permeability, absorption, and bioavailability. Berlin: Wiley-VCH.
221. Glaeser, H., Drescher, S., van der Kuip, H., Behrens, C., Geick, A., Burk, O., Dent, J., Somogyi, A., Von Richter, O., Griese, E.U., et al. 2002. Shed human enterocytes as a tool for the study of expression and function of intestinal drug-metabolizing enzymes and transporters. Clin Pharmacol Ther 71:131-140.
222. Pang, G., Buret, A., O´Loughlin, E., Smith, A., Batey, R., Clancy, R. 1996. Immunologic, functional and morphologfical characterization of of three new human intestinal epitheliial cell lines. Gastroenterology 111:8-18.
223. Peterson, M.D., and Mooseker, M.S. 1992. Characterization of the enterocyte-like brush border cytoskeleton of the C2BBe clones of the human intestinal cell line, Caco-2. J Cell Sci 102 ( Pt 3):581-600.
224. Carriere, V., Lesuffleur, T., Barbat, A., Rousset, M., Dussaulx, E., Costet, P., de Waziers, I., Beaune, P., and Zweibaum, A. 1994. Expression of cytochrome P-450 3A in HT29-MTX cells and Caco-2 clone TC7. FEBS Lett 355:247-250.
225. Artursson, P., Karlsson, J., Ocklind, G., and Schipper, N. 1996. Cell culture models of epithelial tissues - A practical approach. New York: Oxford University press.
226. Tom, B.H., Rutzky, L.P., Jakstys, M.M., Oyasu, R., Kaye, C.I., and Kahan, B.D. 1976. Human colonic adenocarcinoma cells. I. Establishment and description of a new line. In Vitro 12:180-191.
227. Moore, L.B., Goodwin, B., Jones, S.A., Wisely, G.B., Serabjit-Singh, C.J., Willson, T.M., Collins, J.L., and Kliewer, S.A. 2000. St. John's wort induces hepatic drug metabolism through activation of the pregnane X receptor. Proc Natl Acad Sci U S A 97:7500-7502.
228. Kostrubsky, V.E., Ramachandran, V., Venkataramanan, R., Dorko, K., Esplen, J.E., Zhang, S., Sinclair, J.F., Wrighton, S.A., and Strom, S.C. 1999. The use of human hepatocyte cultures to study the induction of cytochrome P-450. Drug Metab Dispos 27:887-894.
229. Sandstrom, R., Karlsson, A., Knutson, L., and Lennernas, H. 1998. Jejunal absorption and metabolism of R/S-verapamil in humans. Pharm Res 15:856-862.
230. Hedeland, M., Fredriksson, E., Lennernas, H., Bondesson, U. 2003. Simultaneous determination of the nenatiomers of verapamil and its N-demethylated metabolite in human plasma using liquid

60
chromatography tandem mass spectrometry - with appliacation to an absorption study. submitted.
231. Palm, K., Luthman, K., Ros, J., Grasjo, J., and Artursson, P. 1999. Effect of molecular charge on intestinal epithelial drug transport: pH-dependent transport of cationic drugs. J Pharmacol Exp Ther 291:435-443.
232. Lennernas, H., Lee, I.D., Fagerholm, U., and Amidon, G.L. 1997. A residence-time distribution analysis of the hydrodynamics within the intestine in man during a regional single-pass perfusion with Loc-I-Gut: in-vivo permeability estimation. J Pharm Pharmacol 49:682-686.
233. Sandstrom, R., Knutson, T.W., Knutson, L., Jansson, B., and Lennernas, H. 1999. The effect of ketoconazole on the jejunal permeability and CYP3A metabolism of (R/S)-verapamil in humans. Br J Clin Pharmacol 48:180-189.
234. de Waziers, I., Cugnenc, P.H., Yang, C.S., Leroux, J.P., Beaune, P.H. 1990. Cytochrome P 450 isoenzymes, epoxide hydrolase and glutathione transferases in rat and human hepatic and extrahepatic tissues. J Pharmacol Exp Ther. 253:387-394.
235. Gibbs, M.A., Thummel, K.E., Shen, D.D., and Kunze, K.L. 1999. Inhibition of cytochrome P-450 3A (CYP3A) in human intestinal and liver microsomes: comparison of Ki values and impact of CYP3A5 expression. Drug Metab Dispos 27:180-187.
236. Wrighton, S., Ring, B., Watkins, P., and VandenBranden, M. 1989. Identification of a polymorphically expressed member of the human cytochrome P-450III family. Mol Pharmacol36:97-105.
237. Lin, Y.S., Dowling, A.L., Quigley, S.D., Farin, F.M., Zhang, J., Lamba, J., Schuetz, E.G., and Thummel, K.E. 2002. Co-regulation of CYP3A4 and CYP3A5 and contribution to hepatic and intestinal midazolam metabolism. Mol Pharmacol 62:162-172.
238. Lehmann, J.M., McKee, D.D., Watson, M.A., Willson, T.M., Moore, J.T., and Kliewer, S.A. 1998. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest 102:1016-1023.
239. Fisher, J.M., Wrighton, S.A., Watkins, P.B., Schmiedlin-Ren, P., Calamia, J.C., Shen, D.D., Kunze, K.L., and Thummel, K.E. 1999. First-pass midazolam metabolism catalyzed by 1alpha,25-dihydroxy vitamin D3-modified Caco-2 cell monolayers. J Pharmacol Exp Ther289:1134-1142.
240. Lindmark, T., Kimura, Y., and Artursson, P. 1998. Absorption enhancement through intracellular regulation of tight junction permeability by medium chain fatty acids in Caco-2 cells. J Pharmacol Exp Ther 284:362-369.
241. Kroemer, H.K., Gautier, J.C., Beaune, P., Henderson, C., Wolf, C.R., and Eichelbaum, M. 1993. Identification of P450 enzymes involved in metabolism of verapamil in humans. Naunyn Schmiedebergs Arch Pharmacol 348:332-337.
242. Busse, D., Cosme, J., Beaune, P., Kroemer, H.K., and Eichelbaum, M. 1995. Cytochromes of the P450 2C subfamily are the major enzymes involved in the O-demethylation of verapamil in humans. Naunyn Schmiedebergs Arch Pharmacol 353:116-121.
243. Eichelbaum, M., Ende, M., Remberg, G., Schomerus, M., and Dengler, H.J. 1979. The metabolism of DL-[14C]verapamil in man. Drug Metab Dispos 7:145-148.
244. Sandstrom, R., Karlsson, A., and Lennernas, H. 1998. The absence of stereoselective P-glycoprotein-mediated transport of R/S-verapamil across the rat jejunum. J Pharm Pharmacol50:729-735.
245. Palm, K., Luthman, K., Ungell, A.L., Strandlund, G., and Artursson, P. 1996. Correlation of drug absorption with molecular surface properties. J Pharm Sci 85:32-39.
246. Pauli-Magnus, C., von Richter, O., Burk, O., Ziegler, A., Mettang, T., Eichelbaum, M., and Fromm, M.F. 2000. Characterization of the major metabolites of verapamil as substrates and inhibitors of P-glycoprotein. J Pharmacol Exp Ther 293:376-382.
247. Engman, H.A., Lennernas, H., Taipalensuu, J., Otter, C., Leidvik, B., and Artursson, P. 2001. CYP3A4, CYP3A5, and MDR1 in human small and large intestinal cell lines suitable for drug transport studies. J Pharm Sci 90:1736-1751.
248. Giuliano, A.R., Franceschi, R.T., and Wood, R.J. 1991. Characterization of the vitamin D receptor from the Caco-2 human colon carcinoma cell line: effect of cellular differentiation. Arch Biochem Biophys 285:261-269.
249. Schmiedlin-Ren, P., Thummel, K.E., Fisher, J.M., Paine, M.F., and Watkins, P.B. 2001. Induction of CYP3A4 by 1 alpha,25-dihydroxyvitamin D3 is human cell line-specific and is unlikely to involve pregnane X receptor. Drug Metab Dispos 29:1446-1453.

References
61
250. Geick, A., Eichelbaum, M., and Burk, O. 2001. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J Biol Chem 276:14581-14587.
251. Sueyoshi, T., Kawamoto, T., Zelko, I., Honkakoski, P., and Negishi, M. 1999. The repressed nuclear receptor CAR responds to phenobarbital in activating the human CYP2B6 gene. J Biol Chem 274:6043-6046.
252. Pascussi, J.M., Drocourt, L., Gerbal-Chaloin, S., Fabre, J.M., Maurel, P., and Vilarem, M.J. 2001. Dual effect of dexamethasone on CYP3A4 gene expression in human hepatocytes. Sequential role of glucocorticoid receptor and pregnane X receptor. Eur J Biochem 268:6346-6358.
253. Quaroni, A., Tian, J.Q., Goke, M., and Podolsky, D.K. 1999. Glucocorticoids have pleiotropic effects on small intestinal crypt cells. Am J Physiol Gastrointest Liver Physiol 277:G1027-1040.
254. Taipalensuu, J., Tavelin, S., Lazorova, L., Svensson, A.C., and Artursson, P. 2003. Exploring the quantitative relationship between the level of MDR1 transcript, protein and function using digoxin as a marker of MDR1-dependent drug efflux activity. European Journal of Pharmaceutical Sciences In Press.
255. Ross, D.D., Yang, W., Abruzzo, L.V., Dalton, W.S., Schneider, E., Lage, H., Dietel, M., Greenberger, L., Cole, S.P., and Doyle, L.A. 1999. Atypical multidrug resistance: breast cancer resistance protein messenger RNA expression in mitoxantrone-selected cell lines. J Natl Cancer Inst 91:429-433.
256. Rowland, M. 2002. Introducing pharmacokinetics and pharmacokinetic concepts. In Drug-drug interactions. New York: Marcel Dekker, Inc. 1-31.
257. Kroemer, H.K., Echizen, H., Heidemann, H., and Eichelbaum, M. 1992. Predictability of the in vivo metabolism of verapamil from in vitro data: contribution of individual metabolic pathways and stereoselective aspects. J Pharmacol Exp Ther 260:1052-1057.
258. Tracy, T.S., Korzekwa, K.R., Gonzalez, F.J., and Wainer, I.W. 1999. Cytochrome P450 isoforms involved in metabolism of the enantiomers of verapamil and norverapamil. Br J Clin Pharmacol47:545-552.
259. Xie, W., Yeuh, M.F., Radominska-Pandya, A., Saini, S.P., Negishi, Y., Bottroff, B.S., Cabrera, G.Y., Tukey, R.H., and Evans, R.M. 2003. Control of steroid, heme, and carcinogen metabolism by nuclear pregnane X receptor and constitutive androstane receptor. Proc Natl Acad Sci U S A100:4150-4155.
