ir-spektroskopie organischer moleküle · spektrenkurs ir-spektroskopie organischer moleküle 6 c-c...
TRANSCRIPT

Spektrenkurs IR-Spektroskopie organischer Moleküle
1
IR-Spektroskopie organischer Moleküle
Die Infrarot-Spektroskopie dient in der Organischen Chemie der Ermittlung von
Strukturelementen, funktionellen Gruppen und ggf. von Isomeren und Konformeren. Ein
Schwingungsspektrum eines organischen Moleküls lässt sich in substanzspezifische
Schwingungen (a) und charakteristische Schwingungen (b) aufteilen.
a) Im Bereich 1500 cm-1 liegen Absorptionsbanden, die durch starke
Schwingungskopplung hervorgerufen werden. Gekoppelte Schwingungen sind stark
vom Molekülrest abhängig, weswegen der Wellenlängenbereich <1500 cm-1
Fingerprint-Bereich genannt wird, da er für die Substanz spezifisch ist.
b) Charakteristische Schwingungen sind nahezu unabhängig von der Molekülstruktur,
die Schwingungen treten also quasi „lagenkonstant“ auf. D.h. die Absorptionsbanden
können einer bestimmten Atomgruppe zugeordnet werden.
1. Schwingungsarten Unter einer Schwingung versteht man die Bewegung der Massenträger relativ zum
Schwerpunkt. Der Schwerpunkt selbst ändert die Lage nicht. Man unterscheidet hauptsächlich
die Valenzschwingung und die Deformationsschwingung.
Valenzschwingung Deformationsschwingung
Abbildung 1: Schwingungsarten

Spektrenkurs IR-Spektroskopie organischer Moleküle
2
2. Lage und Intensität charakteristischer Schwingun gen Die Intensität der Bande ist proportional zu der Größe der Änderung des Dipolmomentes. Die
Lage der Schwingungen im IR-Spektrum hängt von der Kraftkonstanten, f, und der
reduzierten Masse, µ, der beteiligten Atome ab.
µπf
vosz ⋅=2
1
21
21
mm
mm
+⋅=µ
Die Kraftkonstante steigt mit
a) der Bindungsordnung ( fC-C < fC=C)
b) zunehmenden s-Charakter im Hybrid-Orbital (f=C-H (sp2) > f-C-H (sp3)
c) der effektiven Kernladungszahl
Bei großen Massendifferenzen ist die reduzierte Masse klein, somit hat die Schwingung
zwischen Atomgruppen mit großer Massendifferenz eine hohe Frequenz.
Abbildung 2: Übersicht über die Lage der Absorptionsbanden der funktionellen Gruppen.

Spektrenkurs IR-Spektroskopie organischer Moleküle
3
In den folgenden Tabellen sind die charakteristischen Wellenzahlen für verschiedene
funktionelle Gruppen zusammengestellt. Die Auflistung hat keinen Anspruch auf
Vollständigkeit (siehe auch ausführliche Tabellenwerke z.B. Hesse, Meier, Zeeh
Spektroskopische Methoden in der Organischen Chemie).
Übersicht über charakteristische Schwingungen
Tabelle 1: Charakteristische Gruppenschwingungen
Schwingung νννν [cm -1] Bemerkung
OH-, NH- und CH
OH – Valenzschwingung (Tabelle 2)
NH – Valenzschwingung (Tabelle 3)
CH - Valenzschwingung (sp2) (Tabelle 5)
CH - Valenzschwingung (sp3) (Tabelle 4)
3650 – 2800
3650 - 3000
3500 - 3300
3100 - 3010
2960 – 2850
Meist breite Bande
Meist breite Bande
Dreifachbindung u. kummulierte Doppelbindung
C≡C Valenzschwingung
C≡N Valenzschwingung
-N=C=O Cyanate, Valenzschwingung
-N=C=S Thiocyanate, Valenzschwingung
C=C=C Allene, Valenzschwingung
2300 - 1900
2300 – 2100
2250
2270
2150
1950
Scharfe Bande
Doppelbindungen Valenzschwingung
C=O – Valenzschwingung (Tabelle 7)
C=C – Valenzschwingung (Tabelle 6)
C=N – Valenzschwingung
1850 – 1500
1850 – 1650
1600 – 1500
1690 – 1630
Intensive Bande
Fingerprintbereich
Schwingungskopplungen führen zu einem Absorp-tionsmuster, das für eine bestimmte Substanz charakteristisch ist.
CH- Deformationsschwingungen in Alkanen (Tabelle 4)
CH – Out of plane Schwingungen bei Alkenen (Tabelle 5)
CH - Deformationsschwingung am Aromaten (Tabelle 6)
< 1500
1465 – 1355
990 – 660
900 - 690
Intensive Banden

Spektrenkurs IR-Spektroskopie organischer Moleküle
4
Schwingungen der OH-Gruppe
Tabelle 2: OH - Valenz- und Deformationsschwingungen
Schwingung νννν [cm -1] Bemerkung
OH – Valenzschwingungen
Freie OH-Valenz (Gasphase, hoch verd. Lösungen)
Spezifische Einzelbrücken
Höhere Assoziate
Carbonsäure-Dimere
3650 – 3000
3650 - 3520
3550 - 3450
3400 – 3200
um 3000
Scharfe Bande
Breite Bande
Extrem breite Bande
OH - Deformationsschwingungen
In plane bending
Out of plane bending (Alkohole und Phenole)
Out of plane bending (Carbonsäure-Dimere)
1420 – 650
1420 - 1330
770 - 650
920
Schwingungen der NH-Gruppe
Tabelle 3: NH - Valenz- und Deformationsschwingungen
Schwingung νννν [cm -1] Bemerkung
NH - Valenzschwingungen
Primäre Amine
Sekundäre Amine
Höhere Arylamine und heterocycl. N-H
Primäre Amide
Sekundäre Amide
3500 – 3300
3500
3400
3350 - 3310
3490 - 3450
3520
3400
3460 - 3400
Asymmetrische Schw.
Symmetrische Schw.
Asymmetrische Schw.
Symmetrische Schw
NH - Deformationsschwingungen
Primäre Amine
Primäre Amide
Sekundäre Amide
1650 - 1500
1650 - 1580
1620 - 1590
1570 - 1510
Verd. Lösungen

Spektrenkurs IR-Spektroskopie organischer Moleküle
5
CH-Schwingungen
Tabelle 4: CH - Valenz- und Deformationsschwingungen - Alkane
Schwingung νννν [cm -1] Bemerkung
CH-Valenzschwingungen
CH3 (Alkane)
CH2 (Alkane)
CH (Alkane)
2960 – 2850
2962 u. 2872
2926 u. 2852
2890
je ± 10; νa > νs
je ± 10; νa > νs
meist nur schwach
CH – Deformationsschwingungen
CH3 (Alkane)
CH2 (Alkane)
1465 – 1355
1450 u. 1375
1465
νa > νs
Tabelle 5: CH - Valenz- und Deformationsschwingungen – Alkene, Alkine
Schwingung νννν [cm -1] Bemerkung
CH-Valenzschwingungen
-CH=CH2
R2C=CH2
R2C=CHR
-CΞCH
3300 – 3010
3040–3010 u.
3095 - 3075
3095 - 3075
3040 – 3010
3300
Alle Banden sind zumeist in Multipletts aufgespalten
Scharf, wenig intensiv
CH - Deformationsschwingungen
RCH=CH2
R2C=CH2
R-CH=CH-R´ Alkene (trans)
R-CH=CH-R´ Alkene (cis)
R2C=CHR
990 – 660
990 u. 915
890
980 – 960
730 – 665
840 - 790
Tabelle 4: CH - Valenz- und Deformationsschwingungen – Aromaten
Schwingung νννν [cm -1] Bemerkung
CH-Valenzschwingungen ~ 3030 Scharf, wenig intensiv
CH - Deformationsschwingungen
5 benachbarte H (Aromat)
4 benachbarte H (Aromat)
3 benachbarte H (Aromat)
2 benachbarte H (Aromat)
1 isoliertes H (Aromat)
900 – 690
770 – 690
770 – 735
810 – 750
860 – 800
900 - 860
zwei Banden (mono)
zwei Banden (meta)
Spektrenkurs IR-Spektroskopie organischer Moleküle
6
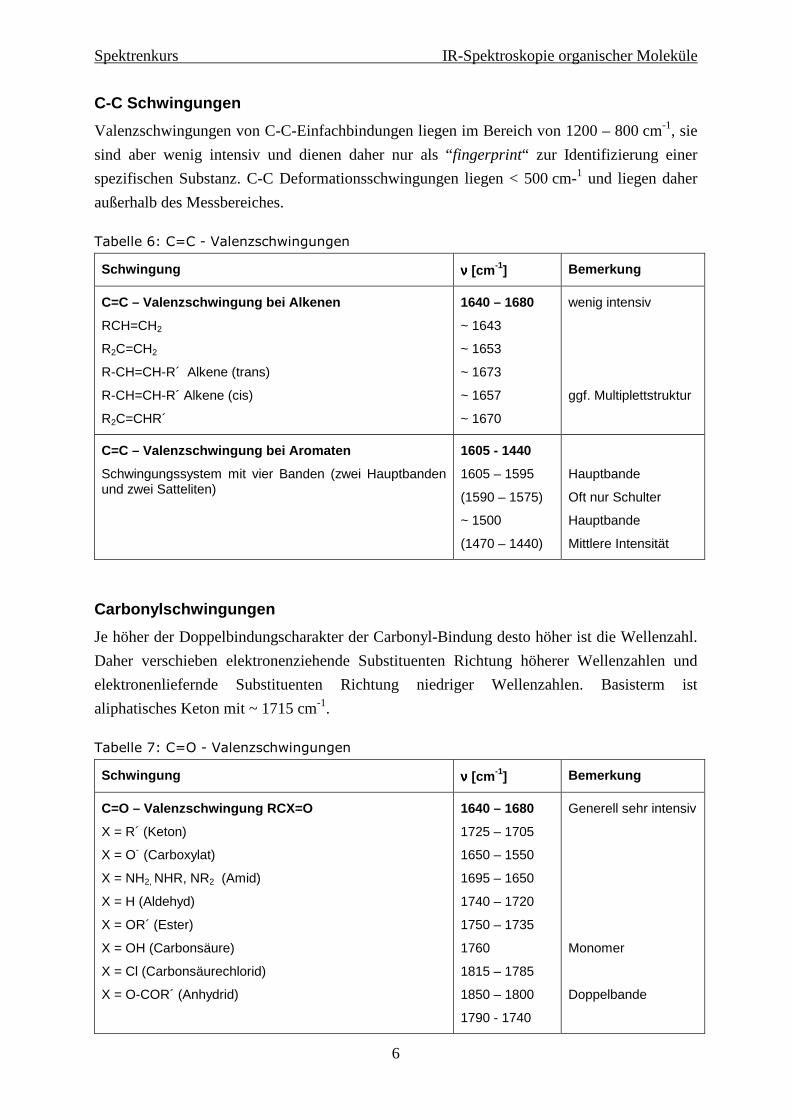
C-C Schwingungen
Valenzschwingungen von C-C-Einfachbindungen liegen im Bereich von 1200 – 800 cm-1, sie
sind aber wenig intensiv und dienen daher nur als “fingerprint“ zur Identifizierung einer
spezifischen Substanz. C-C Deformationsschwingungen liegen < 500 cm-1 und liegen daher
außerhalb des Messbereiches.
Tabelle 6: C=C - Valenzschwingungen
Schwingung νννν [cm -1] Bemerkung
C=C – Valenzschwingung bei Alkenen
RCH=CH2
R2C=CH2
R-CH=CH-R´ Alkene (trans)
R-CH=CH-R´ Alkene (cis)
R2C=CHR´
1640 – 1680
~ 1643
~ 1653
~ 1673
~ 1657
~ 1670
wenig intensiv
ggf. Multiplettstruktur
C=C – Valenzschwingung bei Aromaten
Schwingungssystem mit vier Banden (zwei Hauptbanden und zwei Satteliten)
1605 - 1440
1605 – 1595
(1590 – 1575)
~ 1500
(1470 – 1440)
Hauptbande
Oft nur Schulter
Hauptbande
Mittlere Intensität
Carbonylschwingungen
Je höher der Doppelbindungscharakter der Carbonyl-Bindung desto höher ist die Wellenzahl.
Daher verschieben elektronenziehende Substituenten Richtung höherer Wellenzahlen und
elektronenliefernde Substituenten Richtung niedriger Wellenzahlen. Basisterm ist
aliphatisches Keton mit ~ 1715 cm-1.
Tabelle 7: C=O - Valenzschwingungen
Schwingung νννν [cm -1] Bemerkung
C=O – Valenzschwingung RCX=O
X = R´ (Keton)
X = O- (Carboxylat)
X = NH2, NHR, NR2 (Amid)
X = H (Aldehyd)
X = OR´ (Ester)
X = OH (Carbonsäure)
X = Cl (Carbonsäurechlorid)
X = O-COR´ (Anhydrid)
1640 – 1680
1725 – 1705
1650 – 1550
1695 – 1650
1740 – 1720
1750 – 1735
1760
1815 – 1785
1850 – 1800
1790 - 1740
Generell sehr intensiv
Monomer
Doppelbande

Spektrenkurs IR-Spektroskopie organischer Moleküle
7
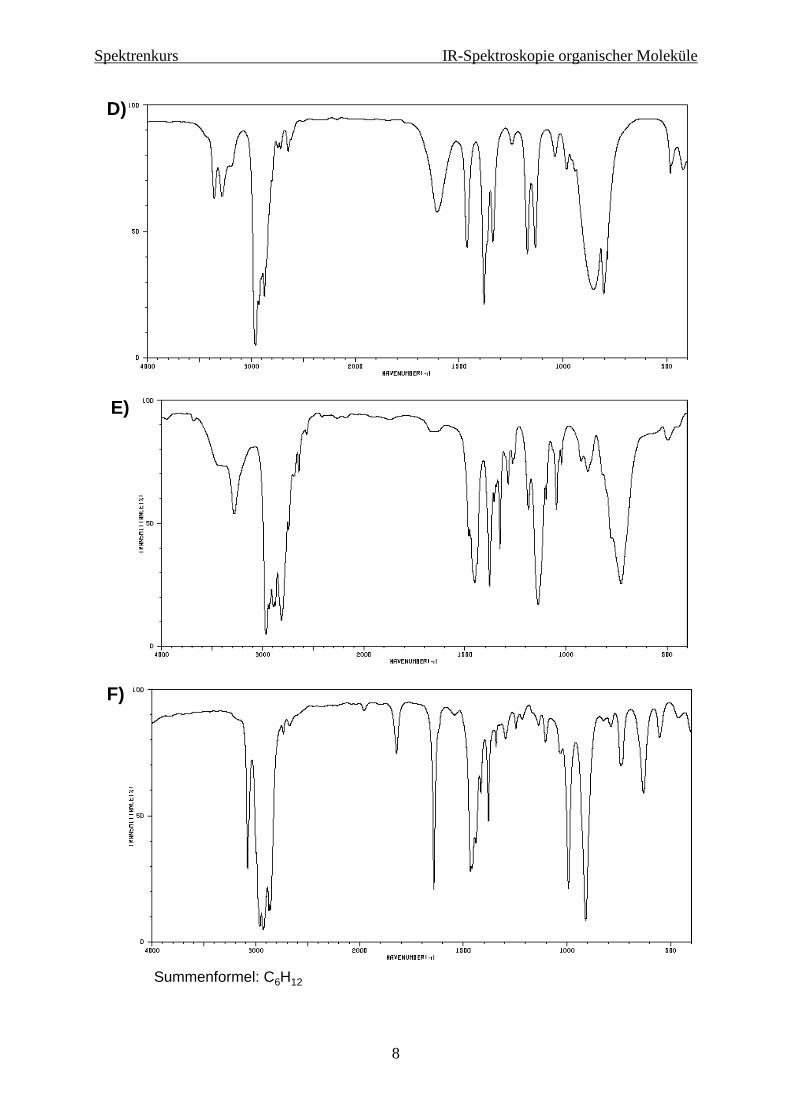
Beispielspektren
A)A)
B)B)
C)C)
Spektrenkurs IR-Spektroskopie organischer Moleküle
8
D)
E)
F)
Summenformel: C6H12

Spektrenkurs IR-Spektroskopie organischer Moleküle
9
G)G)
H)H)
I)I)

Spektrenkurs IR-Spektroskopie organischer Moleküle
10
J)J)
K)K)
L)L)

Spektrenkurs IR-Spektroskopie organischer Moleküle
11
3. Knobelaufgaben a) Aus der Elementaranalyse ergibt sich eine Summenformel von C8H10. Um welche
Verbindung handelt es sich?
b) Ordnen Sie die beiden Spektren cis-2-Penten bzw. trans-2-Penten zu

Spektrenkurs IR-Spektroskopie organischer Moleküle
12
c) Um welche Verbindung mit der Summenformel C7H8O handelt es sich?
d) Sie haben Essigsäurechlorid mit Ethanol umgesetzt, hat sich das gewünschte Produkt
gebildet?