kinetics of oxidation of l-histidine by tetrachloroaurate(iii) ion in perchloric acid solution
TRANSCRIPT

www.elsevier.com/locate/poly
Polyhedron 24 (2005) 1167–1174
Kinetics of oxidation of L-histidine by tetrachloroaurate(III) ionin perchloric acid solution
Vimal Soni, R.S. Sindal, Raj N. Mehrotra *
Department of Chemistry, JNV University, Pali Road, Jodhpur 342 005, India
Received 14 January 2005; accepted 9 March 2005
Available online 25 May 2005
Abstract
The oxidation of L-histidine by tetrachloroaurate(III) ion in HClO4 is first-order in both AuCl4� and histidine. The stoichiometry
ratio, D½AuCl4��=D½Histidine�, is 1.07 ± 0.10 and the oxidation product is b-imidazolylpuruvic acid. The kobs decreased with increase
in [H+] and [Cl�] at the constant concentration of the other. The rate dependence on [Cl�] is due to the equilibrium between the
reactive AuCl4� and AuCl3OH� species of Au(III); the reactivity being in the order: AuCl3OH� � AuCl4
� ion. The rate depen-
dence on [H+] is attributed to the reactive histidine species, RCH2-CHðNH3þÞCOO� which is in equilibrium with the non-reactive
R-CHðNH3þÞCOOH where R is the imidazole ring. An inner-sphere mechanism based on the NMR study on the complex formed
between [AuCl4]� with glycine is suggested. The outer-sphere mechanism is ruled out because the DS� values are very different for
the similarly charged species AuCl4� and AuCl3OH�.
� 2005 Elsevier Ltd. All rights reserved.
Keywords: Mechanism; Kinetics
1. Introduction
The reduction of Au(III)-complexes has been a sub-
ject of interest [1,2] in recent years. Substrates such as
dimethylsulfide [1c], thiocyanate [1d,2a], thiosulfate
[1e] and iodide [2d] (Y) ions attack the coordinated li-
gand X of the Au(III)-complex in the rate determining
step [3,4] which is followed by a fast bridged-two-elec-
tron transfer to the metal centre with the elimination
of XY. The [Pt(CN)4]2� and [Pt(NH3)4]
2+ ions are sim-ilarly oxidised [5]. There is a direct interaction between
the reducing nucleophile and the metal centre in the oxi-
dations of the sulfite [4] and thiosulfate ions [6]. In the
oxidation of thiocyanate [3b], dimethyl sulfide [7] and
thiosulfate [6] the substitution is faster than the reduc-
tion of [AuCl4]�, whereas for the iodide ion the redox
reaction is faster than the ligand substitution [8]. How-
0277-5387/$ - see front matter � 2005 Elsevier Ltd. All rights reserved.
doi:10.1016/j.poly.2005.03.057
* Corresponding author. Tel.: +912912721478.
E-mail address: [email protected] (R.N. Mehrotra).
ever, in the oxidation of thiocyanate by AuðNH3Þ43þthe rate controlling step is the substitution of an amineligand by thiocyanate, whereas for the oxidation by
AuðNH3Þ2X2þ, thiocyanate replaces halide (X) in two
rapid consecutive and reversible substitution steps prior
to the slower reduction [9].
The disulfide bond in the oxidation of cystine cleaved
to sulfonic acid [10–12], and the sulfur in methionine
was oxidised stereospecifically to the sulfoxide [13–15].
The study of the crystal structure of the complexes ob-tained by the slow reaction of AuCl4
� with dipeptide
glycyl-L-histidine [16], glycylglycyl-L-histidine [17] and
1,2-diaminoethane-N,N,N 0,N 0-tetra-(N-methylacetamide)
[18] suggested that these complexes are square planar.
There seems to be no kinetic study available on the
oxidation of amino acids, though NMR investigation
with [15N]glycine [1a], suggested the formation of the
[AuCl3N15H2CH2COOH] complex via the amino group
and subsequently a chelate in which N and O are
bonded with Au(III) is formed. The chelate, probably,

1168 V. Soni et al. / Polyhedron 24 (2005) 1167–1174
is inert to the oxidation of glycine. Two electrons within
the complex are transferred to Au(III) from glycine to
form a Au(I)-imine intermediate, which being unstable
undergoes hydrolysis to NH4þ and glyoxylic acid. An
excess of [AuCl4]� oxidizes glyoxylic acid to CO2 and
Au(o) through the intermediate formation of formicacid. Since the rate law was not given (dependence of
the rate on [glycine], [H+] and [Cl�]), it was of interest
to study the kinetics of the oxidation of histidine, an-
other amino acid.
2. Experimental
2.1. Chemicals and solutions
The solutions of AuCl4� (Johnson and Matthey) were
freshly prepared in perchloric acid (E. Merck, GR),
though such solutions are stable over 24 h. The concen-
trations were determined from the optical density
measured at 310 nm on a HP 8452A diode array spec-
trophotometer using carefully determined e = 4.82 ·103 dm3 mol�1 cm�1, which compared well with the lit.
value [19–21] 4.86 · 103 dm3 mol�1 cm�1. The solutions
of L-histidine (BioChemika Micro Select, Fluka) and
D-histidine (Puriss, Fluka) were freshly prepared by
weighing the samples. LiClO4 solution for adjusting
the ionic strength (l) was prepared and standardised
as described earlier [22]. All other chemicals were used
as received. The solutions were prepared in water, dis-tilled twice, and purged by nitrogen.
2.2. Reaction products
At the end of the reaction, the metal ion from the
reaction mixture was removed using an ion exchange
method. A portion of the filtrate was treated with a sat-
urated solution of 2,4-dinitrophenylhydrazine and thesame was left overnight in a refrigerator. The precipi-
tated 2,4-dinitrophenylhydrazone was separated by fil-
tration and recrystallised from a mixture of ethyl
acetate and light petroleum. The melting point of the
precipitated hydrazone, 190 ± 0.5 �C, was characteristicof b-imidazolyl pyruvic acid, indicating that the seat of
the reaction is not the imidazole ring since it remained
intact. Another portion of the eluent on treatment withNessler�s reagent confirmed the presence of the NH4
þ
ion.
2.3. Stoichiometry
Several reaction mixtures having excess of AuCl4�
over known histidine concentrations were prepared.
The optical density of the reaction mixtures was mea-sured at 310 nm at room temperature until a constant
value was recorded. The mean of several such estimates
yielded D½AuCl4��=D½Histidine� ¼ 1.07� 0.10. Hence, in
view of the characterised products and the stoichiometry
ratio, the equation of the reaction is expressible by
Eq. (1).
H2Oþ C3H3N2CH2CHðNH2ÞCOOH þAuCl4�
! C3H3N2CH2COCOOHþAuCl43� þNH4
þ þHþ
ð1Þ
2.4. Kinetics
The reaction mixtures were prepared so as to con-
form to the pseudo-first-order conditions (excess of
histidine over Au(III) ion) for studying the rate ofthe reaction that was followed at 360 nm (Beer�s law
was obeyed) with a Spectrochem MK II colorimeter
fitted with a thermostated reaction cell. The pseudo
first order rate constants kobs were calculated from
the slopes of the plots between log (At�A1) against
time ‘‘t’’, which were linear for more than two half-
lives (80–90%) where At and A1 are the optical
densities of the reaction mixture at any time ‘‘t’’and infinite time, respectively. These linear plots indi-
cated the monophasic character of the reaction and
that the products did not interfere with the rate pro-
file of the reaction. There was no trace of metallic
gold in the reaction mixture at the time of measuring
A1 after several half-lives. The EXCEL program was
used for plotting the log (At � A1)-time data and
evaluating the least square values of the slopes andintercepts of the linear plots. The pseudo first order
rate constants, kobs were generally reproducible
within 5%.
The repetitive spectra of the reaction mixtures for
several concentrations of histidine, recorded on a
HP8451A spectrophotometer, had not shown any
change from the spectral characteristics of the control
solution of AuCl4�. The absorbance of the spectra de-
creased with time and no isosbestic points appeared,
indicating that the spectra of the products of the reac-
tion do not overlap with that of the AuCl4 solution.
The absence of any transient absorbance peaks in the
spectra over a period of time, therefore, suggests that
the intermediates, if formed, are weak or only weak
bridged complexes are formed.
2.5. Test for free radical
The addition of acrylonitrile (6% v/v) to the reaction
mixture, degassed with nitrogen, produced cloudiness
due to the formation of polyacrylonitrile, indicating
the formation of free radicals. A similar solution of
acrylonitrile when added to similarly degassed blank
solutions of AuCl4 and histidine did not produce anycloudiness.

V. Soni et al. / Polyhedron 24 (2005) 1167–1174 1169
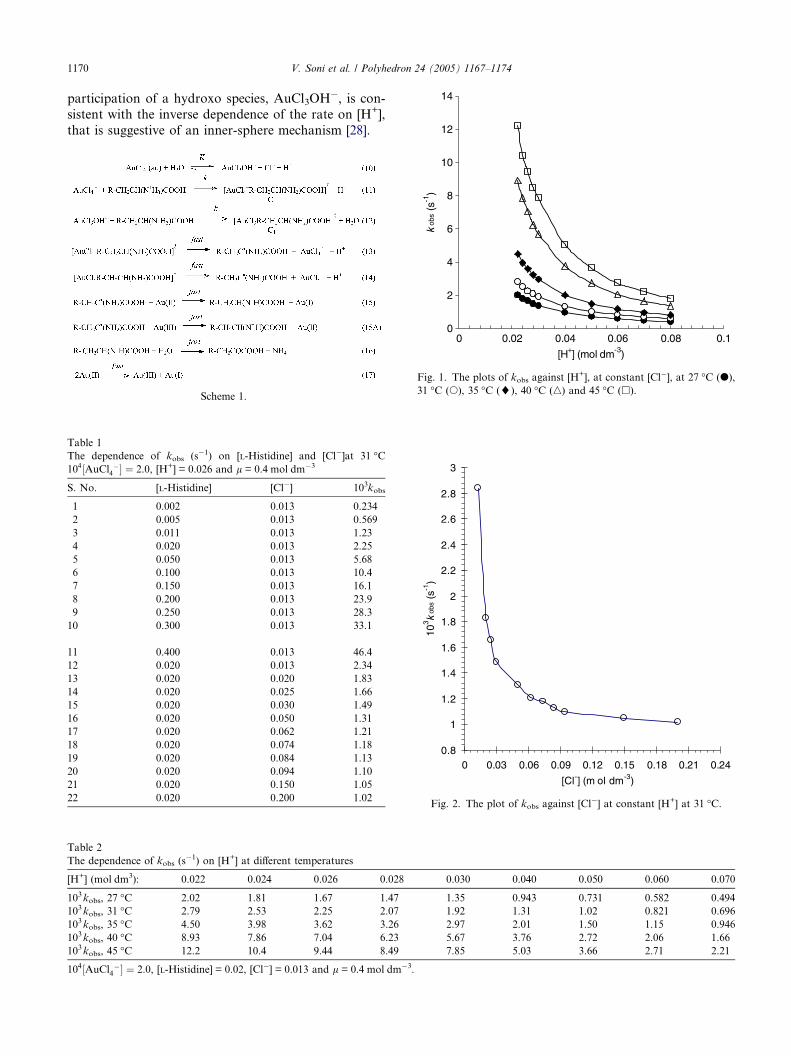
3. Results
kobs increased proportionately with [Histidine] at con-
stant [H+], [Cl�] and ionic strength (Table 1). The plot
of kobs against [Histidine]0 was linear and passed
through the origin, suggesting a first order dependencein histidine. The decreasing kobs with increasing [Cl�]
and [H+], at constant concentrations of the other, is re-
ported in Tables 1 and 2, respectively. The correlations
between kobs and [H+] or [Cl�] at constant concentration
of the other, are shown in Figs. 1 and 2, respectively.
These plots indicate the absence of a linear correlation
between kobs and the respective concentrations of H+
ð4Þ
ð5Þ
ð6Þ
and Cl� ions. A later analysis of the respective data indi-
cated that a linear correlation between kobs and [H+] or
[Cl�] is consistent with the empirical rate law, Eq. (2),
where k2 = kobs/[Histidine] and X is a constant. These re-
sults suggested the presence of an equilibria between
species that differ by a single proton and a [Cl�] ion
k2 � X ¼ aþ b½Hþ��1½Cl���1. ð2Þ
kobs decreased with an increase in the ionic strength,
Table 3, and the plot, in accordance with Eq. (3)
[23,24], is linear with a negative slope, Fig. 5, suggesting
the reactive species have opposite charges
log kobs ¼ log k0 þ 1.02ZaZb
ffiffiffil
p
1þ ffiffiffil
p � 0.05l
� �. ð3Þ
4. Mechanism and discussion
The following equilibria between various histidinespecies, A2+, B+, C and D� (the charge shown is the
net total charge on that species), and the respective
equilibrium constants [25] are known. The retarding ef-
fect of the H+ ion could be due to any one or all the
equilibria shown in Eqs. (4)–(6). However, these equi-
libria have no bearing on the retarding effect of the
Cl� ion. For an initial [Histidine] = 0.002 and[H+] = 0.02 mol dm�3, the calculated [B+] is
8.5 · 10�4 mol dm�3 using the given Ka1 value The con-
centrations of C and D�, using the respective Ka2 and
Ka3 values, can be shown to be negligibly small com-
pared to [B+]. The equilibria (7)–(9), involving Au(III)
species [19,26,27], can also explain the retarding effect
of both H+ and Cl� ions.
AuCl4� þH2O �
Khy
AuCl3H2Oþ Cl� ð7Þ
AuCl3H2O �Ka
AuCl3OH� þHþ ð8Þ
AuCl3ðOHÞ� þH2O �
Kaq
AuCl2ðH2OÞðOHÞ þ Cl� ð9Þ
The equilibrium (9) is excluded from consideration be-
cause the calculated [AuCl2(H2O)(OH)] is negligibly
small. The equilibria (7) and (8) can be combined to a
single equilibrium (10). A consequence of equilibrium
(10) is that the likely reactive Au(III) species are
AuCl4� and AuCl3OH� ions. The equilibrium constant
K (=KhyKa) can be calculated from the known Khy andKa values. The rate constants tend to limiting values at
the higher end of the [H+] (Fig. 1) and [Cl�] (Fig. 2) be-
cause the equilibrium constant K has a very small value.
The reactivity of AuCl4� and AuCl3OH� ions is consis-
tent with the analysis of the rate data that indicated two
parallel reactions. The reactivity of AuCl3OH� is �103
times the reactivity of AuCl4�ðaqÞ (Table 4). The
12
14
1170 V. Soni et al. / Polyhedron 24 (2005) 1167–1174
participation of a hydroxo species, AuCl3OH�, is con-
sistent with the inverse dependence of the rate on [H+],
that is suggestive of an inner-sphere mechanism [28].
Scheme 1.
Table 1
The dependence of kobs (s�1) on [L-Histidine] and [Cl�]at 31 �C104½AuCl4
�� ¼ 2.0, [H+] = 0.026 and l = 0.4 mol dm�3
S. No. [L-Histidine] [Cl�] 103kobs
1 0.002 0.013 0.234
2 0.005 0.013 0.569
3 0.011 0.013 1.23
4 0.020 0.013 2.25
5 0.050 0.013 5.68
6 0.100 0.013 10.4
7 0.150 0.013 16.1
8 0.200 0.013 23.9
9 0.250 0.013 28.3
10 0.300 0.013 33.1
11 0.400 0.013 46.4
12 0.020 0.013 2.34
13 0.020 0.020 1.83
14 0.020 0.025 1.66
15 0.020 0.030 1.49
16 0.020 0.050 1.31
17 0.020 0.062 1.21
18 0.020 0.074 1.18
19 0.020 0.084 1.13
20 0.020 0.094 1.10
21 0.020 0.150 1.05
22 0.020 0.200 1.02
0.8
1
1.2
1.4
1.6
1.8
2
2.2
2.4
2.6
2.8
3
0 0.03 0.06 0.09 0.12 0.15 0.18 0.21 0.24
[Cl-] (m ol dm-3)
103 k
obs
(s-1
)
Fig. 2. The plot of kobs against [Cl�] at constant [H+] at 31 �C.
0
2
4
6
8
10
0 0.02 0.04 0.06 0.08 0.1
[H+] (mol dm-3)
kob
s (s
-1)
Fig. 1. The plots of kobs against [H+], at constant [Cl�], at 27 �C (d),
31 �C (s), 35 �C (¤), 40 �C (n) and 45 �C (h).
Table 2
The dependence of kobs (s�1) on [H+] at different temperatures
[H+] (mol dm3): 0.022 0.024 0.026 0.028 0.030 0.040 0.050 0.060 0.070
103kobs, 27 �C 2.02 1.81 1.67 1.47 1.35 0.943 0.731 0.582 0.494
103kobs, 31 �C 2.79 2.53 2.25 2.07 1.92 1.31 1.02 0.821 0.696
103kobs, 35 �C 4.50 3.98 3.62 3.26 2.97 2.01 1.50 1.15 0.946
103kobs, 40 �C 8.93 7.86 7.04 6.23 5.67 3.76 2.72 2.06 1.66
103kobs, 45 �C 12.2 10.4 9.44 8.49 7.85 5.03 3.66 2.71 2.21
104½AuCl4�� ¼ 2.0, [L-Histidine] = 0.02, [Cl�] = 0.013 and l = 0.4 mol dm�3.

0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
0 10 20 30 40 50 60
[H+]
-1 (dm
3 mol
-1)
k2(
Ka1
+ [
H+ ])
/Ka1
Fig. 3. The linear plot of k2ðKa þ ½Hþ�ÞK�1a against [H+]�1 at constant
[Cl�] = 0.13 mol dm�3, consistent with the Eq. (21), at temperatures
27 �C (h), 31 �C (M), 40 �C (s) and 45 �C (d).
Table 4
The values of k and k1 at different temperatures and the related
thermodynamic parameters
Temperature (�C): 27 31 35 40 45
kc (dm3 mol�1 s�1) 0.0813 0.114 0.125 0.151 0.189
10�2kc1 (mol dm�3 s�1) 1.10 1.38 2.33 4.59 5.62
107 K (mol2 dm�6) 4.12 4.51 4.91 5.46 6.04
DH zk ¼ 32� 2 kJ mol�1; DH z
k1¼ 76� 5 kJ mol�1; DSzk ¼ �159�
8 J K�1 mol�1; DSzk1 ¼ 48� 17 J K�1 mol�1.
Table 3
The effect of the ionic strength (LiClO4) on the rate of the reaction at
35 �C
l (mol dm�3) 0.1 0.20 0.40 0.60 1.00 1.20
103kobs (s�1) 3.49 3.29 3.20 3.10 3.01 2.96
104½AuCl4�� ¼ 1.0, [Histidine] = 0.005, [H+] = 0.01 mol dm�3.
V. Soni et al. / Polyhedron 24 (2005) 1167–1174 1171
Though the formation of intermediates between
Au(III) and histidine is not indicated by the spectral
study of the reaction mixture in the UV region, their for-
mation in the proposed mechanism would be reasonable
in view of the NMR studies on glycine [1a], glycylglycyl-
L-histidine [17]. The observation that the imidazole ring
is not the seat of reaction because it is present in the final
oxidation product of the reaction is consistent with theview that the binding of a proton or a metal ion is very
unfavourable at the pyrrole nitrogen of histidine [17].
The results of the spectral studies would not be in con-
tradiction to the assumption of the formation of tran-
sient intermediates in the rate-determining step. The
mechanism in Scheme 1 is based on this assumption.
The probable free radical is R-CH2C�(NH2)COOH
where R is the imidazole ring. The presence of a freeradical is indicative of the one electron reduction of
Au(III) in the rate determining step. Transient Au(II)
is reported in several redox reactions [29–35]. The unsta-
ble Au(II) can oxidise the free radical instantaneously to
the imine (15). Imines with a proton on N are seldom
stable and undergo fast hydrolysis to the NH4þ ion
and the final product (17).
Alternately, Au(II) may disproportionate to Au(III)andAu(I) instantaneously, reaction (17), because the rate
of disproportionation [36], 1.67 · 108 dm3 mol�1 s�1, is
very near to the rate of diffusion. Au(II) is, therefore, very
likely removed through disproportionation that makes
the reactions (15) most unlikely. The rate law, deduced
from reactions (10)–(17), is expressible by Eq. (18) which
changes to Eq. (21) on substituting the proper values of
½AuCl4�� and [B+] (R–CH2CH(N+H3)COOH), given by
Eqs. (19) and (20), respectively, and applying the inequal-
ity K � [H+][Cl�] in view of [H+]and [Cl�] used where
k2 = kobs/[Histidine]0
�d½AuðIIIÞ�dt
¼ k þ k1K½Hþ�½Cl��
� �AuCl4
�� �½Bþ�; ð18Þ
AuCl4�� �
¼ ½AuðIIIÞ�½Hþ�½Cl��K þ ½Hþ�½Cl�� ; ð19Þ
½Bþ� ¼ Ka1½Histidine�0Ka1 þ ½Hþ� ; ð20Þ
k2ðKa1 þ ½Hþ�ÞKa1
¼ k þ k1K½Hþ�½Cl��
� �. ð21Þ
The reactivity of the species B+ in outer-sphere
oxidation with [Co(III)W]5� (E0 = 1.01 V [37],) and
[Mncdta]� (E0 = 0.81 V [38],) ions [39] is of the same
order as are the respective E0 values of these ions, and
so is the reactivity of the species D� with [Mncdta]�
and FeðCNÞ63� (E0 = 0.36 V) ions [40]. Though E0
(=0.95 V [41],) for the AuCl4�=AuCl2 couple is between
the E0 values of [Co(III)W]5� and [Mncdta(OH2)]� ions,
the reactivity of the species B+ with the Au(III) ion is
greater than that with the [Co(III)W]5� ion. This is
probably because the present reaction is inner-sphere.
The slope value, �0.3, of the plot in Fig. 5 is less than
the expected slope of �1 considering that the reactive
histidine species B+ has a net single positive charge
and the reactive Au(III) species have a single negative
charge. The deviation from the expected value couldbe due to the spreading of the net single positive charge
over the entire histidine molecule.
The Eq. (21) is consistent with the linear plots of the
left hand side against [H+]�1 at constant [Cl�] (Fig. 3),
and [Cl�]�1 at constant [H+] (Fig. 4) with intercepts on
the rate axis. The values of k (0.120 dm3 mol�1 s�1) and
k1 (139 dm3 mol�1 s�1) obtained from the plot in Fig. 4
are in close agreement with those (k =0.114 dm3 mol�1 s�1; k1 = 15.6 dm3 mol�1 s�1) obtained
from the plot in Fig. 3 at 31 �C. This close agreement

0.12
0.14
0.16
0.18
0.2
0.22
0.24
0.26
0.28
0.3
0.32
0 10 20 30 40 50 60 70 80[Cl-]-1 (dm3 mol-1)
k2 (
Ka1
+ [H
+ ])/K
a1 (
dm3 m
ol-1
s-1)
Fig. 4. The linear plot of k2ðKa þ ½Hþ�ÞK�1a against [Cl�]�1 at constant
[H+], consistent with the Eq. (21), at 31 �C.
0.4
0.42
0.44
0.46
0.48
0.5
0.52
0.54
0.56
0.58
0.6
0 0.1 0.
(√µ(1+√µ) − 0.05µ)2 0.3 0.4 0.5
3+lo
gk
obs
Fig. 5. The linear plot of log kobs against fl=ð1þplÞ � 0.05lg has an
intercept of �0.3. The ionic strength is varied using LiClO4.
KAuCl4
−(aq) + H2O AuCl3OH− + Cl− + H+ (10)
KcAuC l4
− + R-CH2CH(N+H3)COOH [AuCl4− R-CH2CH(NH2)COOH] + H+ (22)
C
Kc1AuCl3OH− + R-CH2CH(N+H3)COOH [AuCl3R-CH2CH(NH2)COOH] + H2O (23)
C1
kcC
AuCl42- +
NH2
COOH + H+CH2
.
C
N
NH
.
NH2CH(COOH)CH2-RCl
Cl ClAu
Cl
(24)
CH2 COOH
HN H
C
H
AuClCl
Cl
AuCl3- +
NH2
COOH + H+CH2
.
C
N
NH
N
NH
.
C1
kc1
(25)
fastR-CH2C
−(NH2)COOH + Au(II) R-CH2CH(N+H)COOH + Au(I) (15)
fast
R-CH2CH(N+H)COOH + H2O R-CH2COCOOH + NH4+ (16)
fast 2Au(II) Au(III) + Au(I) (17)
Scheme 2.
1172 V. Soni et al. / Polyhedron 24 (2005) 1167–1174
between the values of k and k1 obtained from two differ-
ent plots, not only support the validity of the rate law butthat of the proposed mechanism too. The values of k and
k1 at different temperatures are in Table 4. The values of
Ka1 [25], and K [20] (Table 4) at different temperatures,
were estimated from the published data. The large differ-
ence in the values of k and k1 (k1 � 103k) reflect the ease
of formation of the respective transient species.
The two intermediates are formed by equatorial coor-
dination of histidine with the respective Au3+ species.The rate of formation of the intermediate formed by
elimination of a water molecule is obviously faster than
that formed through a chloride bridge. This mechanism
is unlikely to show any difference in the rates of oxida-
tion of H1-histidine and H2-histidine (deuteriated mole-
cule). Since an authentic sample of H2-histidine was
unprocurable from a reliable source, the rates of H2-his-
tidine were not measurable. However, the most disturb-
ing aspect of the mechanism is the value of k1(110 dm3 mol�1 s�1 at 27 �C) which is inadmissible fora process in which a water molecule is eliminated be-
cause of the reaction between H+ and OH� ions. The
rate of such a process is diffusion controlled. This indi-
cated the presence of some other factor with k1 and thus
a modification in the mechanism is required.
The alternate mechanism presumes the formation of
the intermediates in equilibrium steps rather than in
the rate-limiting steps. The intermediates so formedcould be of a transitory nature defying spectrometric
detection if the rates of the formation and decomposi-
tion of the intermediates to the products approach the
rate of the backward reaction of the equilibrium, so that
there is no accumulation of the intermediates over a per-
iod of time. Yet another reason for the intermediates
defying spectrometric detection could be that the molar
absorptivity coefficient of the transition complexesmight not be very different from that of tetrachloroau-
rate(III) solution. The mechanism so modified is consid-
ered in Scheme 2 in which the reaction (15A) is
eliminated for the reasons given earlier.
The rate law, based on Scheme 2, is in Eq. (26). The
transformed form of (26) is Eq. (27) which is identical
with Eq. (21) where k = kcKc and k1 = kc1Kc1
�d½AuðIIIÞ�dt
¼ kcKc þkc1Kc1K½Hþ�½Cl��
� �AuCl4
��� �
½Bþ�;
ð26Þ
k2ðKa1þ½Hþ�ÞKa1
¼ kcKcþkc1Kc1K½Hþ�½Cl��
� �¼ kþ k1K
½Hþ�½Cl��
� �.
ð27Þ

V. Soni et al. / Polyhedron 24 (2005) 1167–1174 1173
The square planar AuCl4� is isoelectronic with the
square planar [Ag(OH)4]� ion. The latter is reported
to form intermediates with the H2PO2� ion [42] and
several others [43–50] in which the ligand is axially
coordinated. The high rates of S(IV) oxidation with
the AuCl4� ion is attributed to the axially coordinated
SO22� and HSO4
� ions in the intermediates [1e]. There-
fore histidine can coordinate with Au(III) either axially
(C1) or equatorially (C). In the equatorial coordina-
tion, the amide nitrogen can coordinate with AuCl4�
through a chloride bridge [1e] though glycine
substitutes for one of the chlorines. It may be men-
tioned that it is not clear if glycine had two simulta-
neous paths of oxidation as observed presently. Sincethe nature of coordination in the two intermediates is
different, their rate of decomposition are obviously ex-
pected to be different and there is no direct removal of
OH� by a proton as in Scheme 1.
In conclusion, histidine forms transitory intermedi-
ates with AuCl4� and AuCl3OH�, preferring an equato-
rial coordination with the AuCl4� ion, and the
substitution is likely to be through a chloride bridgewhereas an axial coordination is preferred with the
AuCl3OH� ion despite the sterically hindered large his-
tidine molecule. Since this coordinated species may be
under strain it obviously has an overall high rate of
decomposition compared to strain free equatorially
coordinated species. The intermediates defy spectromet-
ric detection either because the total rates of formation
and decomposition to the products have approachedthe rate of the backwards reaction of the equilibrium
step so that there is no accumulation of the intermedi-
ates over a period of time or the molar absorptivity coef-
ficient of the transition complexes is not very different
from that of the tetrachloroaurate(III) solution.
Acknowledgement
Sponsorship of this work by the University Grants
Commission (F.12-59/97 and F.12-147/01) is gratefully
acknowledged.
References
[1] (a) J. Zou, Z. Guo, J.A. Parkinson, Y. Chen, P.J. Sadler, Chem.
Commun. (1999) 1359;
(b) A. Ericson, J.C. Arthur, R.S. Coleman, L.I. Elding, S.K.C.
Elmroth, J. Chem. Soc., Dalton Trans. (1998) 1687;
(c) A. Ericson, L.I. Elding, S.K.C. Elmroth, J. Chem. Soc.,
Dalton Trans. (1997) 1159;
(d) S.K.C. Elmroth, L.I. Elding, Inorg. Chem. 35 (1996) 2337;
(e) J. Berglund, L.I. Elding, Inorg. Chem. 34 (1995) 513.
[2] (a) S. Elmroth, L.H. Skibsted, L.I. Elding, Inorg. Chem. 28
(1989) 2703;
(b) L. Drougge, L.I. Elding, Inorg. Chem. 26 (1987) 1073;
(c) L.I. Elding, L.H. Skibsted, Inorg. Chem. 25 (1986) 4084;
(d) L.I. Elding, L.F. Olsson, Inorg. Chem. 21 (1982) 779.
[3] (a) L.H. Skibsted, Adv. Inorg. Bioinorg. Mech. 4 (1986) 137;
(b) L.I. Elding, A.-B. Groning, O. Groning, J. Chem. Soc.,
Dalton Trans. (1981) 1093;
(c) R. Ettore, J. Chem. Soc., Dalton Trans. (1983) 2329.
[4] Refs. [1e–9] cited therein.
[5] Refs. [2b,12].
[6] G. Nord, L.H. Skibsted, A.S. Halonin, Acta Chem. Scand. A29
(1975) 505.
[7] A. Ericson, L.I. Elding, S.K.C. Elmroth, J. Chem. Soc., Dalton
Trans. (1997) 1159.
[8] L.I. Elding, L.F. Olsson, Inorg. Chem. 21 (1982) 779.
[9] S. Elmroth, L.H. Skibsted, L.I. Elding, Inorg. Chem. 28 (1989)
2703.
[10] C.F. Shaw, M.P. Cancro, P.L. Witkiewicz, J.E. Eldridge, Inorg.
Chem. 19 (1980) 3198.
[11] P.L. Wikiewicz, C.F. Shaw, J. Chem. Soc., Chem. Commun.
(1981) 1111.
[12] W.E. Smith, J. Reglinski, Perspect. Bioinorg. Chem. 1 (1991) 183.
[13] E. Bordignon, L. Catallini, G. Natile, A. Scatturi, J. Chem. Soc.,
Chem. Commun. (1973) 878.
[14] G. Natile, E. Bordignon, L. Catallini, Inorg. Chem. 15 (1976)
246.
[15] A.A. Isab, P.J. Sadler, Biochim. Biophys. Acta 492 (1979) 322.
[16] M. Wienken, B. Lippert, E. Zangrando, L. Randaccio, Inorg.
Chem. 31 (1992) 1983.
[17] S.L. Best, T.K. Chattopadhyay, M.I. Djuran, R.A. Palmer, P.J.
Sadler, J. Chem. Soc., Dalton Trans. (1997) 2587.
[18] A.A. Cornejo, A. Catineiras, A.I. Yanovsky, K.B. Nolan, Inorg.
Chim. Acta 349 (2003) 91.
[19] K.K. Sengupta, A. Sanyal, P.K. Sen, Transition Met. Chem. 19
(1994) 534.
[20] F.H. Fry, G.A. Hamilton, J. Turkevich, Inorg. Chem. 5 (1966)
1943.
[21] P. van Z. Bekker, W. Robb, Inorg. Chim. Acta 8 (1972) 849.
[22] A. Prakash Rekha, Raj N. Mehrotra, Can. J. Chem. 71 (1991)
2164.
[23] C.W. Davies, Ion Association, Butterworths, London, 1962, p. 39.
[24] C.W. Davies, Ion association, J. Chem. Soc. (1938) 2093.
[25] A.E. Martell, R.M. Smith, Critical Stability Constants, vol. 5,
Plenum, New York, 1982, p. 31 (First supplement).
[26] W. Robb, Inorg. Chem. 6 (1967) 382.
[27] B.I. Peshchevitskii, V.I. Belevantsev, N.V. Kubatova, Russ. J.
Inorg. Chem. (Engl. Transl.) 16 (1971) 1007.
[28] R.G. Wilkins, Kinetics and Mechanism of Reactions of Transition
Metal Complexes, 2nd ed., VCH, Weinheim, 1991, p. 262.
[29] K.K. Sengupta, B. Basu, Transition Met. Chem. 8 (1983) 6.
[30] K.K. Sengupta, B. Basu, S. Sengupta, S. Nandi, Polyhedron 2
(1983) 983.
[31] K.K. Sengupta, B. Basu, Polyhedron 3 (1984) 805.
[32] K.K. Sengupta, S. Das, S. Sengupta, Transition Met. Chem. 13
(1988) 261.
[33] A. Brown, W.C.E. Higginson, J. Chem. Soc., Dalton Trans.
(1972) 166.
[34] H.G. Foresberg, H. Widdel, L.G. Erwall, J. Chem. Educ. 37
(1960) 44.
[35] F. Basalo, R.G. Pearson, Mechanism of Inorganic Reactions, 2nd
ed., Wiley Eastern, New Delhi, 1977, p. 414.
[36] R.L. Rich, H. Taube, J. Phys. Chem. 58 (1954) 6.
[37] L. Eberson, J. Am. Chem. Soc. 105 (1983) 3192.
[38] R.E. Hamm, M.A. Suwyn, Inorg. Chem. 6 (1967) 139.
[39] S. Gangopadhyaya, M. Ali, P. Banerji, J. Chem. Soc., Perkin
Trans. 2 (1992) 781.
[40] D. Laloo, M.K. Mahanti, J. Chem. Soc., Dalton Trans. (1990)
311.
[41] R.L. Rich, H. Taube, J. Phys. Chem. 58 (1954) 1.

1174 V. Soni et al. / Polyhedron 24 (2005) 1167–1174
[42] RajN.Mehrotra, L.J.Kirschenbaum, Inorg. Chem. 28 (1989) 4327.
[43] L.J. Kirschenbaum, J.D. Rush, Inorg. Chem. 24 (1985) 744.
[44] L.J. Kirschenbaum, J. Inorg. Nucl. Chem. 38 (1976) 881.
[45] Kouadio, L.J. Kirschenbaum, Raj N. Mehrotra, J. Chem. Soc.,
Dalton Trans. (1990) 1929.
[46] R.K. Panda, L.J. Kirschenbaum, J. Chem. Soc., Dalton Trans.
(1989) 217.
[47] L.J. Kirschenbaum, L. Mrozowski, Inorg. Chem. 17 (1978)
3718.
[48] Kouadio, L.J. Kirschenbaum, Raj N. Mehrotra, J. Chem. Soc.,
Perkin Trans. 2 (1990) 2123.
[49] Y. Sun, L.J. Kirschenbaum, Issifou Kouadio, J. Chem. Soc.,
Dalton Trans. (1991) 2311.
[50] L.J. Kirschenbaum, Y. Sun, Inorg. Chem. 30 (1991) 2360.