l-cysteine reversibly inhibits glucose-induced biphasic ... · l-cysteine reversibly inhibits...
TRANSCRIPT

L-cysteine reversibly inhibits glucose-induced biphasicinsulin secretion and ATP production byinactivating PKM2Daiki Nakatsua,1, Yuta Horiuchia,1, Fumi Kanoa,b, Yoshiyuki Noguchia, Taichi Sugawaraa, Iseki Takamotoc,Naoto Kubotac,d,e, Takashi Kadowakic, and Masayuki Murataa,2
aDepartment of Life Sciences, Graduate School of Arts and Sciences, University of Tokyo, 3-8-1 Komaba, Meguro-ku, Tokyo 153-8902, Japan; bPrecursoryResearch for Embryonic Science and Technology (PRESTO), Japan Science and Technology Agency, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan;cDepartment of Diabetes and Metabolic Diseases, Graduate School of Medicine, University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8655, Japan;dDepartment of Clinical Nutrition Therapy, University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8655, Japan; and eLaboratory for MetabolicHomeostasis, RIKEN Center for Integrative Medical Sciences, 1-7-22 Suehiro-cho, Turumi-ku, Yokohama, Kanagawa 230-0045, Japan
Edited by David W. Russell, University of Texas Southwestern Medical Center, Dallas, TX, and approved January 29, 2015 (received for review September21, 2014)
Increase in the concentration of plasma L-cysteine is closely asso-ciated with defective insulin secretion from pancreatic β-cells,which results in type 2 diabetes (T2D). In this study, we investi-gated the effects of prolonged L-cysteine treatment on glucose-stimulated insulin secretion (GSIS) from mouse insulinoma 6 (MIN6)cells and from mouse pancreatic islets, and found that the treat-ment reversibly inhibited glucose-induced ATP production andresulting GSIS without affecting proinsulin and insulin synthe-sis. Comprehensive metabolic analyses using capillary electropho-resis time-of-flight mass spectrometry showed that prolongedL-cysteine treatment decreased the levels of pyruvate and itsdownstream metabolites. In addition, methyl pyruvate, a mem-brane-permeable form of pyruvate, rescued L-cysteine–induced in-hibition of GSIS. Based on these results, we found that both in vitroand in MIN6 cells, L-cysteine specifically inhibited the activity ofpyruvate kinase muscle isoform 2 (PKM2), an isoform of pyruvatekinases that catalyze the conversion of phosphoenolpyruvate topyruvate. L-cysteine also induced PKM2 subunit dissociation (tet-ramers to dimers/monomers) in cells, which resulted in impairedglucose-induced ATP production for GSIS. DASA-10 (NCGC00181061,a substituted N,N′-diarylsulfonamide), a specific activator for PKM2,restored the tetramer formation and the activity of PKM2, glucose-induced ATP production, and biphasic insulin secretion in L-cysteine–treated cells. Collectively, our results demonstrate that impairedinsulin secretion due to exposure to L-cysteine resulted from itsdirect binding and inactivation of PKM2 and suggest that PKM2is a potential therapeutic target for T2D.
type 2 diabetes | insulin secretion | metabolomics | L-cysteine | PKM2
Ametabolite, L-cysteine, is found in blood plasma, and itsconcentration is closely associated with an increase in fat
mass and the body-mass index. These values are used as an indexof obesity (1, 2), which is a major risk factor for type 2 diabetes(T2D) (3). The relationship between L-cysteine and diabetes hasattracted attention because there is increasing evidence for apositive correlation between increases in plasma L-cysteine con-centrations and the development and progression of diabetes.For example, increased plasma L-cysteine concentrations wereassociated with T2D in African American women (4), renal in-sufficiency [reduced glomerular filtration rate (GFR)] in T2Dpatients (5), obstructive sleep apnea [a risk factor for diabetes(6, 7)], and insulin resistance among Europeans (8).Reduced insulin secretion from pancreatic β-cells is the major
cause of T2D (9, 10). Many investigators have studied the mo-lecular mechanisms of glucose-stimulated insulin secretion (GSIS),which have been elucidated in detail. Elevated extracellular glucoseconcentration results in the enhancement of ATP production, anincreased ATP/ADP ratio, the closure of ATP-sensitive K channels
(KATP channels), and depolarization (11). The resulting activa-tion of voltage-dependent Ca2+ channels (VDCCs) induces aninflux of calcium ions and elevated intracellular Ca2+ concen-trations, which triggers insulin secretion (11). Perifusion experi-ments have shown that insulin secretion could be categorizedinto two phases. The first phase involves a sharp increase in in-sulin secretion within ∼5 min, followed by a second phase, duringwhich moderate insulin secretion lasts for hours (9, 12). A loss ofthe GSIS first phase is closely associated with the future de-velopment of T2D (9, 13, 14).Many recent studies have reported that L-cysteine is involved
in GSIS. In mouse pancreatic islets and mouse insulinoma 6(MIN6) cells, L-cysteine treatment decreased both intracellularATP levels and insulin secretion (15). Ammon et al. alsoreported that the total amount and GSIS second phase werespecifically inhibited by L-cysteine for rat pancreatic islets (16).Several groups showed that an increase in the H2S moiety, whichis generated from L-cysteine in cells (17), was one possible causefor L-cysteine–induced impairment of GSIS by inhibiting KATPchannels and VDCCs (18–20). However, opposite results were
Significance
There is increasing evidence for a positive correlation betweenincreases in plasma L-cysteine concentrations and the devel-opment and progression of type 2 diabetes (T2D) caused byobesity. Here, we show that prolonged treatment of mouseinsulinoma 6 (MIN6) cells and mouse pancreatic islets withL-cysteine inhibited glucose-stimulated insulin secretion (GSIS).L-cysteine reversibly inactivated pyruvate kinase muscle isoform 2(PKM2), an isoform of the rate-limiting enzyme pyruvate kinasein glycolysis, by inducing PKM2 subunit dissociation (tetramersto dimers/monomers), which resulted in impairment of glucose-induced ATP production and subsequent GSIS. Our findings inthis work will raise caution about using a variety of L-cysteinecontaining supplements to diabetic patients and shed a light on apreviously unidentified physiological role of L-cysteine and PKM2.
Author contributions: D.N., Y.H., F.K., I.T., N.K., T.K., and M.M. designed research; D.N.,Y.H., F.K., Y.N., T.S., and I.T. performed research; D.N., Y.H., F.K., and M.M. contributednew reagents/analytic tools; D.N., Y.H., F.K., Y.N., and M.M. analyzed data; D.N., Y.H.,F.K., and M.M. wrote the paper; and D.N., Y.H., F.K., I.T., N.K., T.K., and M.M. revisedthe manuscript.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Freely available online through the PNAS open access option.1D.N. and Y.H. contributed equally to this work.2To whom correspondence should be addressed. Email: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1417197112/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1417197112 PNAS | Published online February 23, 2015 | E1067–E1076
CELL
BIOLO
GY
PNASPL
US
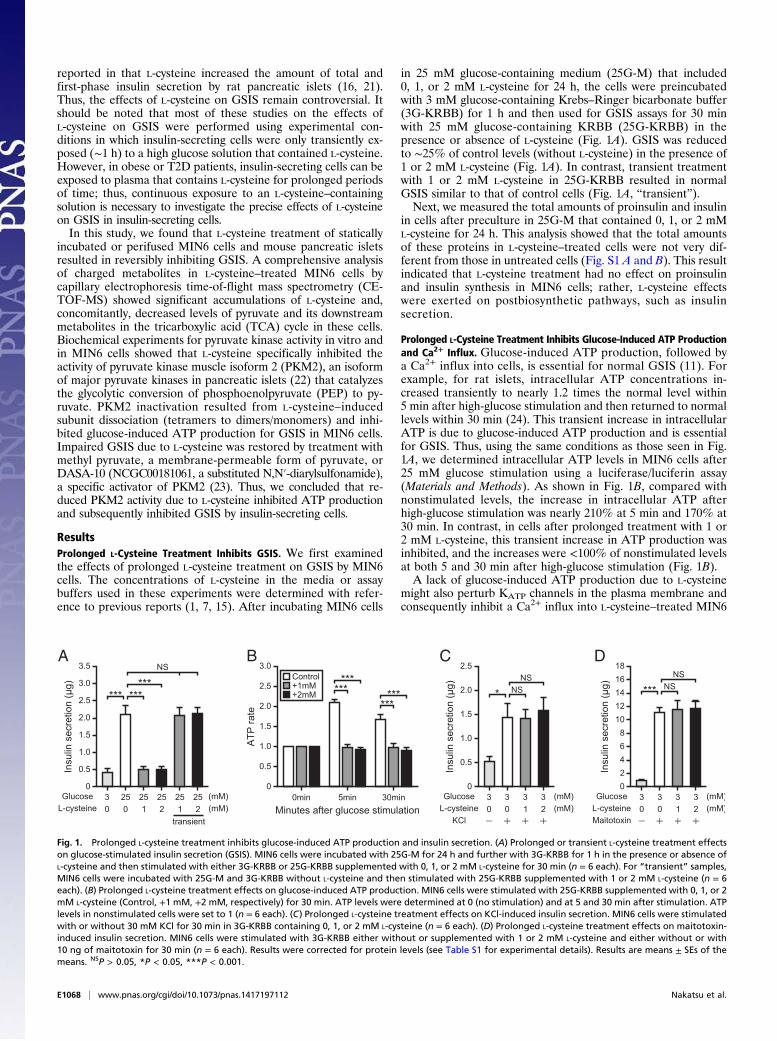
reported in that L-cysteine increased the amount of total andfirst-phase insulin secretion by rat pancreatic islets (16, 21).Thus, the effects of L-cysteine on GSIS remain controversial. Itshould be noted that most of these studies on the effects ofL-cysteine on GSIS were performed using experimental con-ditions in which insulin-secreting cells were only transiently ex-posed (∼1 h) to a high glucose solution that contained L-cysteine.However, in obese or T2D patients, insulin-secreting cells can beexposed to plasma that contains L-cysteine for prolonged periodsof time; thus, continuous exposure to an L-cysteine–containingsolution is necessary to investigate the precise effects of L-cysteineon GSIS in insulin-secreting cells.In this study, we found that L-cysteine treatment of statically
incubated or perifused MIN6 cells and mouse pancreatic isletsresulted in reversibly inhibiting GSIS. A comprehensive analysisof charged metabolites in L-cysteine–treated MIN6 cells bycapillary electrophoresis time-of-flight mass spectrometry (CE-TOF-MS) showed significant accumulations of L-cysteine and,concomitantly, decreased levels of pyruvate and its downstreammetabolites in the tricarboxylic acid (TCA) cycle in these cells.Biochemical experiments for pyruvate kinase activity in vitro andin MIN6 cells showed that L-cysteine specifically inhibited theactivity of pyruvate kinase muscle isoform 2 (PKM2), an isoformof major pyruvate kinases in pancreatic islets (22) that catalyzesthe glycolytic conversion of phosphoenolpyruvate (PEP) to py-ruvate. PKM2 inactivation resulted from L-cysteine–inducedsubunit dissociation (tetramers to dimers/monomers) and inhi-bited glucose-induced ATP production for GSIS in MIN6 cells.Impaired GSIS due to L-cysteine was restored by treatment withmethyl pyruvate, a membrane-permeable form of pyruvate, orDASA-10 (NCGC00181061, a substituted N,N′-diarylsulfonamide),a specific activator of PKM2 (23). Thus, we concluded that re-duced PKM2 activity due to L-cysteine inhibited ATP productionand subsequently inhibited GSIS by insulin-secreting cells.
ResultsProlonged L-Cysteine Treatment Inhibits GSIS. We first examinedthe effects of prolonged L-cysteine treatment on GSIS by MIN6cells. The concentrations of L-cysteine in the media or assaybuffers used in these experiments were determined with refer-ence to previous reports (1, 7, 15). After incubating MIN6 cells
in 25 mM glucose-containing medium (25G-M) that included0, 1, or 2 mM L-cysteine for 24 h, the cells were preincubatedwith 3 mM glucose-containing Krebs–Ringer bicarbonate buffer(3G-KRBB) for 1 h and then used for GSIS assays for 30 minwith 25 mM glucose-containing KRBB (25G-KRBB) in thepresence or absence of L-cysteine (Fig. 1A). GSIS was reducedto ∼25% of control levels (without L-cysteine) in the presence of1 or 2 mM L-cysteine (Fig. 1A). In contrast, transient treatmentwith 1 or 2 mM L-cysteine in 25G-KRBB resulted in normalGSIS similar to that of control cells (Fig. 1A, “transient”).Next, we measured the total amounts of proinsulin and insulin
in cells after preculture in 25G-M that contained 0, 1, or 2 mML-cysteine for 24 h. This analysis showed that the total amountsof these proteins in L-cysteine–treated cells were not very dif-ferent from those in untreated cells (Fig. S1 A and B). This resultindicated that L-cysteine treatment had no effect on proinsulinand insulin synthesis in MIN6 cells; rather, L-cysteine effectswere exerted on postbiosynthetic pathways, such as insulinsecretion.
Prolonged L-Cysteine Treatment Inhibits Glucose-Induced ATP Productionand Ca2+ Influx. Glucose-induced ATP production, followed bya Ca2+ influx into cells, is essential for normal GSIS (11). Forexample, for rat islets, intracellular ATP concentrations in-creased transiently to nearly 1.2 times the normal level within5 min after high-glucose stimulation and then returned to normallevels within 30 min (24). This transient increase in intracellularATP is due to glucose-induced ATP production and is essentialfor GSIS. Thus, using the same conditions as those seen in Fig.1A, we determined intracellular ATP levels in MIN6 cells after25 mM glucose stimulation using a luciferase/luciferin assay(Materials and Methods). As shown in Fig. 1B, compared withnonstimulated levels, the increase in intracellular ATP afterhigh-glucose stimulation was nearly 210% at 5 min and 170% at30 min. In contrast, in cells after prolonged treatment with 1 or2 mM L-cysteine, this transient increase in ATP production wasinhibited, and the increases were <100% of nonstimulated levelsat both 5 and 30 min after high-glucose stimulation (Fig. 1B).A lack of glucose-induced ATP production due to L-cysteine
might also perturb KATP channels in the plasma membrane andconsequently inhibit a Ca2+ influx into L-cysteine–treated MIN6
2.0
1.5
1.0
0.5
0
Insu
lin s
ecre
tion
(μg)
2.5
3.5
3.0
transient
0
0.5
1.0
1.5
2.0
3.0
2.5
ATP
rate
0min 5min 30minMinutes after glucose stimulation
2.5
2.0
1.5
1.0
0.5
0
Insu
lin s
ecre
tion
(μg)
16141210
468
20
Insu
lin s
ecre
tion
(μg)
18DCBA
Fig. 1. Prolonged L-cysteine treatment inhibits glucose-induced ATP production and insulin secretion. (A) Prolonged or transient L-cysteine treatment effectson glucose-stimulated insulin secretion (GSIS). MIN6 cells were incubated with 25G-M for 24 h and further with 3G-KRBB for 1 h in the presence or absence ofL-cysteine and then stimulated with either 3G-KRBB or 25G-KRBB supplemented with 0, 1, or 2 mM L-cysteine for 30 min (n = 6 each). For “transient” samples,MIN6 cells were incubated with 25G-M and 3G-KRBB without L-cysteine and then stimulated with 25G-KRBB supplemented with 1 or 2 mM L-cysteine (n = 6each). (B) Prolonged L-cysteine treatment effects on glucose-induced ATP production. MIN6 cells were stimulated with 25G-KRBB supplemented with 0, 1, or 2mM L-cysteine (Control, +1 mM, +2 mM, respectively) for 30 min. ATP levels were determined at 0 (no stimulation) and at 5 and 30 min after stimulation. ATPlevels in nonstimulated cells were set to 1 (n = 6 each). (C) Prolonged L-cysteine treatment effects on KCl-induced insulin secretion. MIN6 cells were stimulatedwith or without 30 mM KCl for 30 min in 3G-KRBB containing 0, 1, or 2 mM L-cysteine (n = 6 each). (D) Prolonged L-cysteine treatment effects on maitotoxin-induced insulin secretion. MIN6 cells were stimulated with 3G-KRBB either without or supplemented with 1 or 2 mM L-cysteine and either without or with10 ng of maitotoxin for 30 min (n = 6 each). Results were corrected for protein levels (see Table S1 for experimental details). Results are means ± SEs of themeans. NSP > 0.05, *P < 0.05, ***P < 0.001.
E1068 | www.pnas.org/cgi/doi/10.1073/pnas.1417197112 Nakatsu et al.

cells. To test this hypothesis, we assessed the effects of L-cysteineon glucose-induced Ca2+ influx in MIN6 cells using fluorescencemicroscopy. In Fluo4-loaded cells without L-cysteine treatment,there was a sharp increase in fluorescence intensity after glucoseexposure (Fig. S2A). In contrast, Fluo4 fluorescence was notincreased in cells treated with L-cysteine, which suggested nearlycomplete inhibition of glucose-induced Ca2+ influx due toL-cysteine treatment (Fig. S2A).Closure of KATP channels and opening of VDCCs play a cru-
cial role in Ca2+ influx that triggers GSIS upon high-glucosestimulation (11). KCl, a membrane depolarizer, and maitotoxin,a calcium channel opener, induce Ca2+ influx and subsequent in-sulin secretion (11, 25–28). Thus, we examined the effects ofL-cysteine treatment on KCl- and maitotoxin-induced insulin se-cretion by MIN6 cells; this treatment had little effect on insulinsecretion (Fig. 1 C and D). Furthermore, whereas H2S genera-tion by L-cysteine treatment was reported to inhibit KATPchannels and VDCCs and the subsequent insulin secretion, wefound that prolonged L-cysteine treatment had no significanteffect on H2S production in MIN6 cells (Fig. S2B). These resultsshowed that prolonged L-cysteine treatment was restricted tosome components of the insulin secretion machinery upstreamof Ca2+-channel activation (such as glucose uptake and ATPproduction).
Prolonged L-Cysteine Treatment Results in Metabolic Alterationsin Pyruvate and Its Downstream Metabolites. Aerobic glycolysisand the TCA cycle are essential for glucose-induced ATP pro-duction, opening VDCCs and Ca2+ influx (11). We investigatedfor any metabolic alterations in MIN6 cells after prolongedL-cysteine treatment. After incubating MIN6 cells in 25G-M thatcontained 0, 1, or 2 mM L-cysteine for 24 h, the steady-statemetabolite levels in these cells were analyzed by CE-TOF-MS.As expected, compared with the levels in untreated cells, L-cysteineconcentrations were significantly increased by 4.8 and 7.6 times incells treated with 1 and 2 mM L-cysteine, respectively (Fig. 2).Compared with untreated cells, pyruvate concentrations were
dramatically decreased by 0.46 and 0.24 times in cells treatedwith 1 and 2 mM L-cysteine, respectively (Fig. 2). In addition,L-cysteine treatment resulted in reduced concentrations of themetabolites in the TCA cycle that were downstream metabolitesof pyruvate, including citrate, cis-aconitate, isocitrate, α-keto-
glutarate, succinate, fumarate, and malate (11) (Fig. 2). In con-trast, there were almost no alterations in the levels of upstreampyruvate metabolites, such as glucose 6-phosphate, fructose6-phosphate, fructose 1,6-bisphosphate, glyceraldehyde 3-phos-phate, 3-phosphoglycerate, 2-phosphoglycerate, and PEP (9)(Fig. 2). These results indicated that L-cysteine treatment atten-uated glucose-mediated production of pyruvate and its downstreammetabolites.Indeed, L-cysteine treatment resulted in reduced NADH and
NAD+ levels, which are downstream pyruvate metabolites andpositive regulators of GSIS first phase (11, 29), to ∼0.6–0.9 timesof those in untreated cells, and that of malonyl-CoA, which isa downstream pyruvate metabolite and a positive regulator ofGSIS second phase (11, 30), to ∼40% of that in untreated cells(Fig. 2). Unlike the L-cysteine–induced inhibition of glucose-induced ATP production shown in Fig. 1B, ATP concentrationsin L-cysteine–treated cells were indistinguishable from those inuntreated cells, which indicated that L-cysteine treatment hadspecifically inhibited glucose-stimulated ATP production but hadlittle effect on steady-state ATP levels. In fact, we also examinedthe effect of L-cysteine on the oxygen consumption rate, which iscorrelated with the activation of the TCA cycle, and foundthat glucose-stimulated oxygen consumption was inhibited byL-cysteine (Fig. S2C).
Inhibitory Effect of L-Cysteine on GSIS Is Rescued by Methyl Pyruvate.Our comprehensive metabolic analysis of L-cysteine–treated cellsshowed that L-cysteine treatment resulted in reduced amounts ofpyruvate and its downstream metabolites without significantlyaltering the levels of its upstream metabolites (Fig. 2). It waspreviously reported that pyruvate and its downstream metabo-lites were closely involved in GSIS (11). Thus, we first investi-gated the effects of methyl pyruvate, a membrane-permeableform of pyruvate that is used to generate these downstreammetabolites, on insulin secretion. After incubating MIN6 cells in25G-M that contained 2 mM L-cysteine for 24 h, the cells werepreincubated with 3G-KRBB that contained 2 mM L-cysteine for1 h, and were then used for GSIS assays using 25G-KRBB thatcontained either 2 mM L-cysteine or a 2-mM L-cysteine/2-mMmethyl pyruvate mixture for 30 min. As shown in Fig. S3A, 2 mMmethyl pyruvate significantly restored the GSIS response ofMIN6 cells under static incubation, even in the presence of
Glucose 6-phosphate (G6P)Fructose 6-phosphate (F6P)Fructose 1,6-bisphosphate (F1,6P)Glyceraldehyde 3-phosphate (G3P)3-phosphoglycerate (3-PG)2-phosphoglyserate (2-PG)Phosphoenolpyruvate (PEP)PyruvateAcetyl-CoA (AcCoA)Malonyl-CoA (MalCoA)Oxaloacetate (OA)Citratecis-Aconitate (cis-Aco)Isocitrateα-ketoglutarate (α-KG)Succinyl-CoA (SucCoA)SuccinateFumarateMalateNADHNAD+
ADPATPL-Cysteine
1.001.001.00
1.001.001.001.001.001.00
1.001.001.001.00
1.001.001.001.001.001.001.001.00
1.071.191.04
0.750.700.770.461.330.35
0.820.640.630.69
0.730.740.690.670.770.890.964.76
1.191.311.11
0.830.951.030.241.010.45
0.880.690.710.49
0.750.730.670.730.901.081.107.63
1.40E-032.96E-045.26E-04
4.97E-048.18E-051.98E-041.38E-036.36E-051.01E-04
2.64E-026.23E-043.66E-041.43E-03
4.32E-033.32E-033.11E-024.38E-046.80E-039.89E-038.70E-025.08E-04
1.50E-033.53E-045.49E-04
3.75E-045.74E-051.53E-046.37E-048.45E-053.56E-05
2.15E-023.97E-042.31E-049.87E-04
3.14E-032.46E-032.15E-022.94E-045.23E-038.83E-038.34E-022.42E-03
1.68E-033.89E-045.83E-04
4.13E-047.75E-052.04E-043.38E-046.46E-054.55E-05
2.32E-024.28E-042.61E-046.97E-04
3.25E-032.43E-032.09E-023.20E-046.15E-031.07E-029.54E-023.87E-03
relative amount(ratio)
intracellular concentration(pmol/10^6 cells)
Glucose Glycolysis
Fig. 2. Prolonged L-cysteine treatment reduces cell pyruvate and its downstream metabolites’ levels. MIN6 cells were incubated with 25G-M supplementedwith 0 (Control), 1 (+1 mM L-cys), or 2 mM L-cysteine (+2 mM L-cys) for 24 h, after which metabolite levels per 106 cells were determined (intracellularconcentration). Metabolite levels in control cells were set to 1 [relative amount (ratio)]. Results are mean values (n = 3 each).
Nakatsu et al. PNAS | Published online February 23, 2015 | E1069
CELL
BIOLO
GY
PNASPL
US
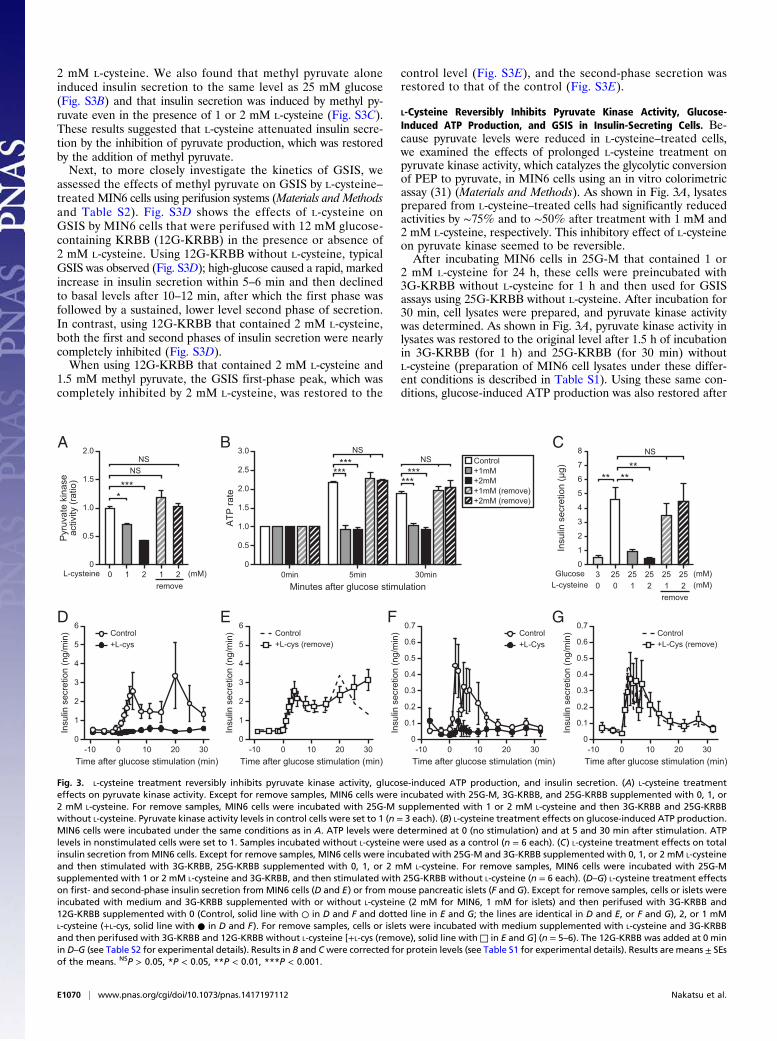
2 mM L-cysteine. We also found that methyl pyruvate aloneinduced insulin secretion to the same level as 25 mM glucose(Fig. S3B) and that insulin secretion was induced by methyl py-ruvate even in the presence of 1 or 2 mM L-cysteine (Fig. S3C).These results suggested that L-cysteine attenuated insulin secre-tion by the inhibition of pyruvate production, which was restoredby the addition of methyl pyruvate.Next, to more closely investigate the kinetics of GSIS, we
assessed the effects of methyl pyruvate on GSIS by L-cysteine–treated MIN6 cells using perifusion systems (Materials and Methodsand Table S2). Fig. S3D shows the effects of L-cysteine onGSIS by MIN6 cells that were perifused with 12 mM glucose-containing KRBB (12G-KRBB) in the presence or absence of2 mM L-cysteine. Using 12G-KRBB without L-cysteine, typicalGSIS was observed (Fig. S3D); high-glucose caused a rapid, markedincrease in insulin secretion within 5–6 min and then declinedto basal levels after 10–12 min, after which the first phase wasfollowed by a sustained, lower level second phase of secretion.In contrast, using 12G-KRBB that contained 2 mM L-cysteine,both the first and second phases of insulin secretion were nearlycompletely inhibited (Fig. S3D).When using 12G-KRBB that contained 2 mM L-cysteine and
1.5 mM methyl pyruvate, the GSIS first-phase peak, which wascompletely inhibited by 2 mM L-cysteine, was restored to the
control level (Fig. S3E), and the second-phase secretion wasrestored to that of the control (Fig. S3E).
L-Cysteine Reversibly Inhibits Pyruvate Kinase Activity, Glucose-Induced ATP Production, and GSIS in Insulin-Secreting Cells. Be-cause pyruvate levels were reduced in L-cysteine–treated cells,we examined the effects of prolonged L-cysteine treatment onpyruvate kinase activity, which catalyzes the glycolytic conversionof PEP to pyruvate, in MIN6 cells using an in vitro colorimetricassay (31) (Materials and Methods). As shown in Fig. 3A, lysatesprepared from L-cysteine–treated cells had significantly reducedactivities by ∼75% and to ∼50% after treatment with 1 mM and2 mM L-cysteine, respectively. This inhibitory effect of L-cysteineon pyruvate kinase seemed to be reversible.After incubating MIN6 cells in 25G-M that contained 1 or
2 mM L-cysteine for 24 h, these cells were preincubated with3G-KRBB without L-cysteine for 1 h and then used for GSISassays using 25G-KRBB without L-cysteine. After incubation for30 min, cell lysates were prepared, and pyruvate kinase activitywas determined. As shown in Fig. 3A, pyruvate kinase activity inlysates was restored to the original level after 1.5 h of incubationin 3G-KRBB (for 1 h) and 25G-KRBB (for 30 min) withoutL-cysteine (preparation of MIN6 cell lysates under these differ-ent conditions is described in Table S1). Using these same con-ditions, glucose-induced ATP production was also restored after
1.5
1.0
0.5
0
Pyr
uvat
e ki
nase
activ
ity (r
atio
)
2.0
remove
0
0.5
1.0
1.5
2.0
3.0
2.5
ATP
rate
0min 5min 30minMinutes after glucose stimulation
5
4
3
2
1
0In
sulin
sec
retio
n (μ
g)
6
8
7
remove
Insu
lin s
ecre
tion
(ng/
min
)
Time after glucose stimulation (min)
+L-cysControl
0
2
1
3
4
5
6
0-10 10 20 30
2
1
3
4
5
6
Insu
lin s
ecre
tion
(ng/
min
)
Time after glucose stimulation (min)
00-10 10 20 30
+L-cys (remove)Control
Insu
lin s
ecre
tion
(ng/
min
)
Time after glucose stimulation (min)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0-10 10 20 30
+L-CysControl
0.1
0.2
0.3
0.4
0.5
+L-Cys (remove)Control
Insu
lin s
ecre
tion
(ng/
min
)
Time after glucose stimulation (min)
0
0.7
0.6
0-10 10 20 30
A B C
GFED
Fig. 3. L-cysteine treatment reversibly inhibits pyruvate kinase activity, glucose-induced ATP production, and insulin secretion. (A) L-cysteine treatmenteffects on pyruvate kinase activity. Except for remove samples, MIN6 cells were incubated with 25G-M, 3G-KRBB, and 25G-KRBB supplemented with 0, 1, or2 mM L-cysteine. For remove samples, MIN6 cells were incubated with 25G-M supplemented with 1 or 2 mM L-cysteine and then 3G-KRBB and 25G-KRBBwithout L-cysteine. Pyruvate kinase activity levels in control cells were set to 1 (n = 3 each). (B) L-cysteine treatment effects on glucose-induced ATP production.MIN6 cells were incubated under the same conditions as in A. ATP levels were determined at 0 (no stimulation) and at 5 and 30 min after stimulation. ATPlevels in nonstimulated cells were set to 1. Samples incubated without L-cysteine were used as a control (n = 6 each). (C) L-cysteine treatment effects on totalinsulin secretion fromMIN6 cells. Except for remove samples, MIN6 cells were incubated with 25G-M and 3G-KRBB supplemented with 0, 1, or 2 mM L-cysteineand then stimulated with 3G-KRBB, 25G-KRBB supplemented with 0, 1, or 2 mM L-cysteine. For remove samples, MIN6 cells were incubated with 25G-Msupplemented with 1 or 2 mM L-cysteine and 3G-KRBB, and then stimulated with 25G-KRBB without L-cysteine (n = 6 each). (D–G) L-cysteine treatment effectson first- and second-phase insulin secretion from MIN6 cells (D and E) or from mouse pancreatic islets (F and G). Except for remove samples, cells or islets wereincubated with medium and 3G-KRBB supplemented with or without L-cysteine (2 mM for MIN6, 1 mM for islets) and then perifused with 3G-KRBB and12G-KRBB supplemented with 0 (Control, solid line with ○ in D and F and dotted line in E and G; the lines are identical in D and E, or F and G), 2, or 1 mML-cysteine (+L-cys, solid line with ● in D and F). For remove samples, cells or islets were incubated with medium supplemented with L-cysteine and 3G-KRBBand then perifused with 3G-KRBB and 12G-KRBB without L-cysteine [+L-cys (remove), solid line with□ in E and G] (n = 5–6). The 12G-KRBB was added at 0 minin D–G (see Table S2 for experimental details). Results in B and Cwere corrected for protein levels (see Table S1 for experimental details). Results are means ± SEsof the means. NSP > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001.
E1070 | www.pnas.org/cgi/doi/10.1073/pnas.1417197112 Nakatsu et al.
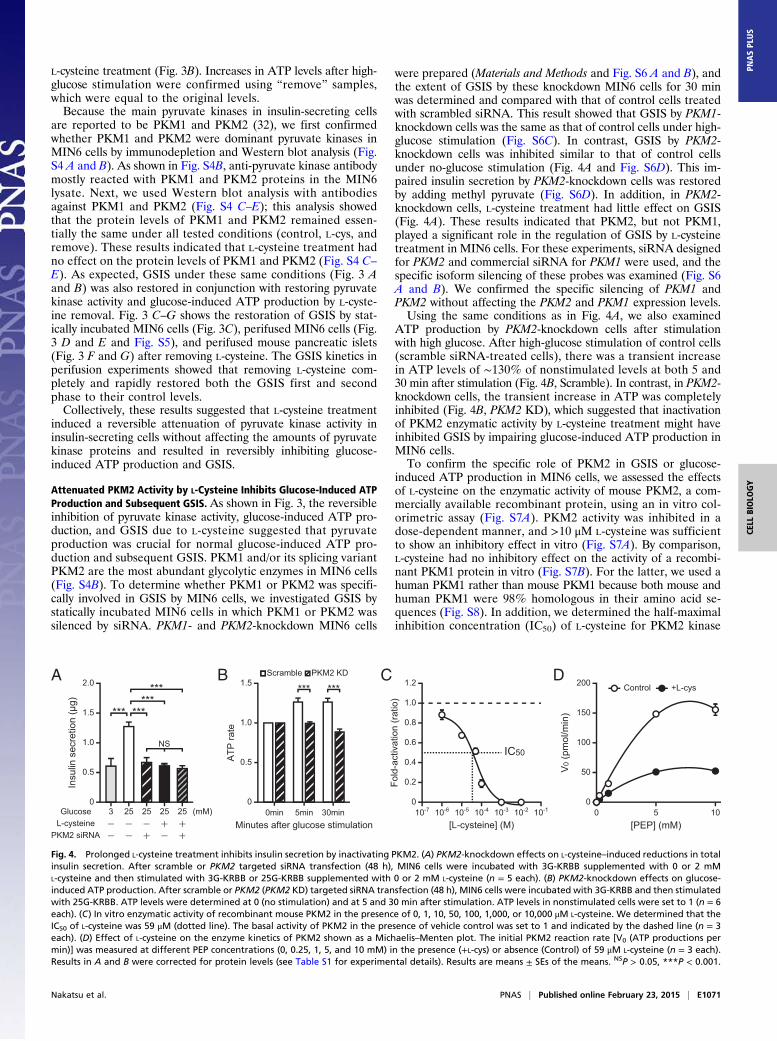
L-cysteine treatment (Fig. 3B). Increases in ATP levels after high-glucose stimulation were confirmed using “remove” samples,which were equal to the original levels.Because the main pyruvate kinases in insulin-secreting cells
are reported to be PKM1 and PKM2 (32), we first confirmedwhether PKM1 and PKM2 were dominant pyruvate kinases inMIN6 cells by immunodepletion and Western blot analysis (Fig.S4 A and B). As shown in Fig. S4B, anti-pyruvate kinase antibodymostly reacted with PKM1 and PKM2 proteins in the MIN6lysate. Next, we used Western blot analysis with antibodiesagainst PKM1 and PKM2 (Fig. S4 C–E); this analysis showedthat the protein levels of PKM1 and PKM2 remained essen-tially the same under all tested conditions (control, L-cys, andremove). These results indicated that L-cysteine treatment hadno effect on the protein levels of PKM1 and PKM2 (Fig. S4 C–E). As expected, GSIS under these same conditions (Fig. 3 Aand B) was also restored in conjunction with restoring pyruvatekinase activity and glucose-induced ATP production by L-cyste-ine removal. Fig. 3 C–G shows the restoration of GSIS by stat-ically incubated MIN6 cells (Fig. 3C), perifused MIN6 cells (Fig.3 D and E and Fig. S5), and perifused mouse pancreatic islets(Fig. 3 F and G) after removing L-cysteine. The GSIS kinetics inperifusion experiments showed that removing L-cysteine com-pletely and rapidly restored both the GSIS first and secondphase to their control levels.Collectively, these results suggested that L-cysteine treatment
induced a reversible attenuation of pyruvate kinase activity ininsulin-secreting cells without affecting the amounts of pyruvatekinase proteins and resulted in reversibly inhibiting glucose-induced ATP production and GSIS.
Attenuated PKM2 Activity by L-Cysteine Inhibits Glucose-Induced ATPProduction and Subsequent GSIS. As shown in Fig. 3, the reversibleinhibition of pyruvate kinase activity, glucose-induced ATP pro-duction, and GSIS due to L-cysteine suggested that pyruvateproduction was crucial for normal glucose-induced ATP pro-duction and subsequent GSIS. PKM1 and/or its splicing variantPKM2 are the most abundant glycolytic enzymes in MIN6 cells(Fig. S4B). To determine whether PKM1 or PKM2 was specifi-cally involved in GSIS by MIN6 cells, we investigated GSIS bystatically incubated MIN6 cells in which PKM1 or PKM2 wassilenced by siRNA. PKM1- and PKM2-knockdown MIN6 cells
were prepared (Materials and Methods and Fig. S6 A and B), andthe extent of GSIS by these knockdown MIN6 cells for 30 minwas determined and compared with that of control cells treatedwith scrambled siRNA. This result showed that GSIS by PKM1-knockdown cells was the same as that of control cells under high-glucose stimulation (Fig. S6C). In contrast, GSIS by PKM2-knockdown cells was inhibited similar to that of control cellsunder no-glucose stimulation (Fig. 4A and Fig. S6D). This im-paired insulin secretion by PKM2-knockdown cells was restoredby adding methyl pyruvate (Fig. S6D). In addition, in PKM2-knockdown cells, L-cysteine treatment had little effect on GSIS(Fig. 4A). These results indicated that PKM2, but not PKM1,played a significant role in the regulation of GSIS by L-cysteinetreatment in MIN6 cells. For these experiments, siRNA designedfor PKM2 and commercial siRNA for PKM1 were used, and thespecific isoform silencing of these probes was examined (Fig. S6A and B). We confirmed the specific silencing of PKM1 andPKM2 without affecting the PKM2 and PKM1 expression levels.Using the same conditions as in Fig. 4A, we also examined
ATP production by PKM2-knockdown cells after stimulationwith high glucose. After high-glucose stimulation of control cells(scramble siRNA-treated cells), there was a transient increasein ATP levels of ∼130% of nonstimulated levels at both 5 and30 min after stimulation (Fig. 4B, Scramble). In contrast, in PKM2-knockdown cells, the transient increase in ATP was completelyinhibited (Fig. 4B, PKM2 KD), which suggested that inactivationof PKM2 enzymatic activity by L-cysteine treatment might haveinhibited GSIS by impairing glucose-induced ATP production inMIN6 cells.To confirm the specific role of PKM2 in GSIS or glucose-
induced ATP production in MIN6 cells, we assessed the effectsof L-cysteine on the enzymatic activity of mouse PKM2, a com-mercially available recombinant protein, using an in vitro col-orimetric assay (Fig. S7A). PKM2 activity was inhibited in adose-dependent manner, and >10 μM L-cysteine was sufficientto show an inhibitory effect in vitro (Fig. S7A). By comparison,L-cysteine had no inhibitory effect on the activity of a recombi-nant PKM1 protein in vitro (Fig. S7B). For the latter, we used ahuman PKM1 rather than mouse PKM1 because both mouse andhuman PKM1 were 98% homologous in their amino acid se-quences (Fig. S8). In addition, we determined the half-maximalinhibition concentration (IC50) of L-cysteine for PKM2 kinase
0
2.0
1.5
1.0
0.5
Insu
lin s
ecre
tion
(μg)
0
0.5
1.0
1.5
ATP
rate
0min 5min 30minMinutes after glucose stimulation
Scramble PKM2 KD
[L-cysteine] (M)
Fold
-act
ivat
ion
(rat
io)
1.2
0.8
0.6
0.4
0.2
1.0
010-7 10-6 10-110-210-310-410-5
IC50
200
50
0010 5
100
150
V0
(pm
ol/m
in)
[PEP] (mM)
+L-cysControlA B C D
Fig. 4. Prolonged L-cysteine treatment inhibits insulin secretion by inactivating PKM2. (A) PKM2-knockdown effects on L-cysteine–induced reductions in totalinsulin secretion. After scramble or PKM2 targeted siRNA transfection (48 h), MIN6 cells were incubated with 3G-KRBB supplemented with 0 or 2 mML-cysteine and then stimulated with 3G-KRBB or 25G-KRBB supplemented with 0 or 2 mM L-cysteine (n = 5 each). (B) PKM2-knockdown effects on glucose-induced ATP production. After scramble or PKM2 (PKM2 KD) targeted siRNA transfection (48 h), MIN6 cells were incubated with 3G-KRBB and then stimulatedwith 25G-KRBB. ATP levels were determined at 0 (no stimulation) and at 5 and 30 min after stimulation. ATP levels in nonstimulated cells were set to 1 (n = 6each). (C) In vitro enzymatic activity of recombinant mouse PKM2 in the presence of 0, 1, 10, 50, 100, 1,000, or 10,000 μM L-cysteine. We determined that theIC50 of L-cysteine was 59 μM (dotted line). The basal activity of PKM2 in the presence of vehicle control was set to 1 and indicated by the dashed line (n = 3each). (D) Effect of L-cysteine on the enzyme kinetics of PKM2 shown as a Michaelis–Menten plot. The initial PKM2 reaction rate [V0 (ATP productions permin)] was measured at different PEP concentrations (0, 0.25, 1, 5, and 10 mM) in the presence (+L-cys) or absence (Control) of 59 μM L-cysteine (n = 3 each).Results in A and B were corrected for protein levels (see Table S1 for experimental details). Results are means ± SEs of the means. NSP > 0.05, ***P < 0.001.
Nakatsu et al. PNAS | Published online February 23, 2015 | E1071
CELL
BIOLO
GY
PNASPL
US

activity. Recombinant PKM2 protein was incubated for 20 minwith pyruvate kinase reaction mix in the presence of 0, 1, 10, 50,100, 1,000, or 10,000 μM L-cysteine, and then its kinase activitywas measured with a pyruvate kinase activity assay kit. As shownin Fig. 4C, we determined that the IC50 of L-cysteine was 59 μM.Furthermore, we constructed a Michaelis–Menten plot and aLineweaver–Burk plot of PKM2 activity (Fig. 4D and Fig. S7C,respectively) and determined that the maximum velocity (Vmax) orthe Michaelis constant (Km) without L-cysteine was 180 pmol/minor 3.9 mM, respectively. In addition, L-cysteine lowered Vmax(84 pmol/min) with little effect on Km (4.5 mM). These resultsindicate that L-cysteine suppresses PKM2 enzymatic activity bynoncompetitive inhibition and strongly support our conclusionthat L-cysteine inhibits PKM2 activity through the dissociation ofPKM2 tetramers.
The PKM2 Activator DASA-10 Restores L-Cysteine–Induced Inhibition ofGlucose-Induced ATP Production and Subsequent GSIS. To strengthenthe evidence for our hypothesis that PKM2 was a pivotal enzymefor insulin secretion in MIN6 cells, we examined whether PKM2activation restored the L-cysteine–induced impairments in glucose-induced ATP production and GSIS. To test this hypothesis, weassessed the effects of DASA-10, a specific activator of PKM2,for restoring glucose-induced ATP production and GSIS inL-cysteine–treated cells. After the cells were precultured in 25G-Mthat contained 0 or 2 mM L-cysteine for 24 h, these cells werepreincubated with 3G-KRBB that contained either DMSO only,2 mM L-cysteine/DMSO, or 2 mM L-cysteine/20 μM DASA-10for 1 h, and were then used for GSIS and ATP assays using25G-KRBB that contained the same concentrations of re-agents as for preincubation to initiate GSIS. As shown in Fig. 5A,glucose-induced ATP production was completely inhibited with25G-KRBB that contained 2 mM L-cysteine/DMSO whereasATP production was normal when using 25G-KRBB that con-
tained DMSO or 2 mM L-cysteine/20 μM DASA-10; upon glucosestimulation, ATP levels increased to almost 210% of nonstimulatedlevels at 5 min after stimulation, and to 200% or 180% at 30 min,respectively (Fig. 5A). These results suggested that PKM2 enzymaticactivity, which was activated by adding DASA-10 to L-cysteine–treated cells, could ameliorate the L-cysteine–induced impair-ment in glucose-induced ATP production. As expected, underthe same conditions, L-cysteine–induced impairment of GSIS byMIN6 cells under static incubation was also restored by DASA-10treatment (Fig. 5B).Next, we investigated the effects of DASA-10 on GSIS by
L-cysteine–treated MIN6 cells and mouse pancreatic islets usingperifusion systems. After MIN6 cells were precultured in 25G-Mthat contained 2 mM L-cysteine for 24 h, these cells were pre-incubated and perifused with 3G-KRBB that contained 2 mML-cysteine/DMSO or 2 mM L-cysteine/5 μM DASA-10 for 30 min,and then GSIS by perifused MIN6 cells was assessed using12G-KRBB in the presence of either DMSO only, 2 mM L-cysteine/DMSO, or 2 mM L-cysteine/5 μM DASA-10. When 12G-KRBBthat contained 2 mM L-cysteine/DMSO was used, typical GSISwas completely inhibited (Fig. 5C, “+L-cys”). In contrast, as shownin Fig. 5D, DASA-10 treatment restored GSIS first and secondphases to their control levels, as was also shown when using12G-KRBB without L-cysteine. L-cysteine–mediated inhibitionof GSIS and its restoration by DASA-10 were also observed inmouse pancreatic islets (Fig. 5 E and F).Collectively, these results indicated that prolonged L-cysteine
treatment might have caused inactivation of PKM2 enzymatic ac-tivity both in MIN6 cells and in mouse pancreatic islets and resultedin impaired glucose-induced ATP production and subsequent GSIS.
DASA-10 Restores L-Cysteine–Induced PKM2 Inactivation Resultingfrom Its Subunit Dissociation in MIN6 Cells. Next, we investigatedhow L-cysteine induced the impaired PKM2 enzymatic activity
0
0.5
1.0
1.5
2.0
3.0
2.5
ATP
rate
0min 5min 30minMinutes after glucose stimulation
0
2.0
1.5
1.0
0.5
Insu
lin s
ecre
tion
(μg)
Insu
lin s
ecre
tion
(ng/
min
)
Time after glucose stimulation (min)
+L-cysControl
0
2
1
3
4
0-10 10 20 30
2
1
3
4
Insu
lin s
ecre
tion
(ng/
min
)
Time after glucose stimulation (min)
00-10 10 20 30
+L-cys+DASA-10Control
Insu
lin s
ecre
tion
(ng/
min
)
Time after glucose stimulation (min)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0-10 10 20 30
+L-CysControl
0.1
0.2
0.3
0.4
0.5
+L-Cys+DASA-10Control
Insu
lin s
ecre
tion
(ng/
min
)
Time after glucose stimulation (min)
0
0.7
0.6
0-10 10 20 30
MIN6 in vitro
PKM2monomer
PKM2dimer
PKM2 tetramer(active form)
A B C D
E F G
Fig. 5. DASA-10 restores L-cysteine–induced reductions in PKM2 activity, glucose-induced ATP production, and glucose-stimulated insulin secretion. (A) MIN6cells were stimulated with 25G-KRBB supplemented with DMSO (Control), DMSO/2 mM L-cysteine (L-cys), or 20 μM DASA-10/2 mM L-cysteine (L-cys + DASA-10).ATP levels were determined at 0 (no stimulation) and at 5 and 30 min after stimulation. ATP levels in nonstimulated cells were set to 1 (n = 6 each). (B) MIN6cells were stimulated with 3G-KRBB or 25G-KRBB supplemented with DMSO, DMSO/2 mM L-cysteine, or 20 μM DASA-10/2 mM L-cysteine (n = 6 each).(C–F) DASA-10 treatment effects on L-cysteine–treated MIN6 cells (C and D) or mouse pancreatic islets (E and F). Cells or islets were incubated and perifusedwith 3G-KRBB supplemented with DMSO, DMSO/L-cysteine, or 5 μM DASA-10/L-cysteine and further perifused with 12G-KRBB supplemented with DMSO(Control, solid line with ○ in C and E and dotted line in D and F; the lines are identical in C and D or in E and F), DMSO/L-cysteine (+L-cys, solid line with ● inC and E), or 5 μM DASA-10/L-cysteine (+L-cys+DASA-10, solid line with □ in D and F) (n = 4–6). The 12G-KRBB was added at 0 min in C–F (see Table S2 forexperimental details). (G) L-cysteine or DASA-10 treatment effects on PKM2 tetramer formation in vitro and in MIN6 cells. MIN6 cell lysates or recombinantmouse PKM2 proteins were added to DMSO only, DMSO/1 mM L-cysteine, or 10 μM DASA-10/1 mM L-cysteine and then incubated. After incubation, PKM2tetramer, dimer, and monomer forms were separated by native SDS/PAGE and immunoblotted with an anti-PKM2 antibody. Results in A and Bwere correctedfor protein levels. For assays that included DASA-10, we used the same amount of DMSO as a negative control (see Tables S1 and S2 for experimental details).Results are means ± SEs of the means. NSP > 0.05, **P < 0.01, ***P < 0.001.
E1072 | www.pnas.org/cgi/doi/10.1073/pnas.1417197112 Nakatsu et al.

and found that DASA-10 was sufficient to rescue the L-cysteine(1 mM)-induced impairment of mouse recombinant PKM2protein enzymatic activity in a dose-dependent manner usingan in vitro colorimetric assay (Fig. S7D). PKM2 subunit dis-sociation (tetramers to dimers/monomers) results in down-regulating its activity (33–35). Thus, we investigated the effect ofL-cysteine and DASA-10 treatment on the oligomerization statesof PKM2 protein in MIN6 cells using native SDS polyacrylamidegel electrophoresis (SDS/PAGE) based on the method ofNowakowski, et al. (36). As shown in Fig. 5G, three PKM2 states,monomer, dimer, and tetramer, were readily recognizable onnative SDS/PAGE.The tetramer form, which is known to be an active form of
PKM2, eventually disappeared in L-cysteine–treated cells com-pared with untreated cells (Fig. 5G). In contrast, when cells wereincubated with L-cysteine in the presence of 10 μM DASA-10,tetramer formation was observed even in the presence of 1 mML-cysteine (Fig. 5G). We also investigated the PKM1 state inL-cysteine–treated MIN6 cells and found that L-cysteine orDASA-10 showed no effect on the tetramer–dimer–monomerratio of PKM1 (Fig. S7E).In addition, we performed an in vitro assay using recombinant
PKM2 and PKM1 to determine whether L-cysteine directly in-hibited the tetramer formation. Recombinant PKM2 or PKM1protein was incubated with DMSO, DMSO/1 mM L-cysteine, or10 μM DASA-10/1 mM L-cysteine for 1 h and then was subjectedto native SDS/PAGE and Western blotting using antibodiesagainst PKM2 or PKM1. We found that L-cysteine induced thedissociation of PKM2 tetramer in vitro and that it was restoredby DASA-10 (Fig. 5G). We detected the time delay of 20 minbefore the reactivation of PKM2 enzymatic activity by DASA-10(Fig. S7D), which might be required for the interaction betweenPKM2 and DASA-10 and the subsequent tetramer formation tofully activate PKM2 activity. In contrast, no significant changewas observed in the tetramer–dimer–monomer equilibrium ofPKM1 with L-cysteine or DASA-10 (Fig. S7E). These resultsstrongly suggest that L-cysteine directly inhibited the tetramerformation of PKM2, leading to the specific inhibition of enzymaticactivity of PKM2, but not of PKM1.Taken together, these data suggested that L-cysteine directly
impaired PKM2 activity (Fig. 5G and Fig. S7A) and that PKM2activity in L-cysteine–treated MIN6 cells might have been down-regulated (by subunit dissociation) in a reversible manner toinhibit the glycolytic pathway toward pyruvate production.
DiscussionPyruvate kinase (EC 2.7.1.40) has four identified isoforms; L, R,M1, and M2 (31). PKM2 is expressed in embryonic, fetal, adultdividing (e.g., adipocytes, lung, pancreatic islet, and spleen), andtumor cells (22, 31, 37, 38) and plays a crucial role in catalyzingthe transphosphorylation from PEP to pyruvate as the rate-lim-iting step of glycolysis for ATP production. PKM2 also has manynonglycolytic functions. For example, nuclear PKM2 has proteinkinase activity and phosphorylates tyrosine residues using PEP asa phosphate donor, which plays a role in promoting cell pro-liferation (34). PKM2 is also a phosphotyrosine-binding protein,and the phosphotyrosine-binding form of PKM2 is a criticalfactor for the rapid growth of cancer cells (39). In addition,EGFR-induced protein interactions between ERK2 and PKM2result in PKM2 nuclear translocation, which functions as acoactivator for gene transcription and promotes the Warburgeffect, during which aerobic glycolysis rather than mitochondrialoxidative phosphorylation is enhanced to produce energy in cancercells (40). Pyruvate kinase activity of PKM2 is optimal when itforms tetramers and is down-regulated by subunit dissociationinto dimers or monomers (33–35) whereas the dimeric form is aprotein kinase (34). These reports indicated that the multi-functional enzymatic activity of PKM2 depended not only on its
expression level but also on the allosteric regulation betweentetramers, dimers, and monomers.In this study, we found that L-cysteine inhibited pyruvate ki-
nase activity of PKM2 due to L-cysteine–induced dissociation ofPKM2 tetramers to dimers/monomers in vitro and in insulin-secreting cells (Fig. 5G), inhibited the transient but crucial in-crease in intracellular ATP levels upon high-glucose stimulation,and resulted in serious defects in GSIS by insulin-secreting cells(Fig. 5). The proposed mechanisms are summarized in Fig. 6.It has been shown that the amount of intracellular pyruvate,
which is produced from PEP by pyruvate kinase, plays a crucialrole in GSIS (11). However, the precise role of pyruvate in theL-cysteine–induced inhibition of GSIS has remained unclearbecause the amounts of pyruvate in cells cannot be determinedunambiguously. L-cysteine is one of the inhibitors of pyruvatekinase and is also a substrate for cystathionine γ-lyase (CSE) forgenerating pyruvate (41–43). Our comprehensive analysis of themetabolites in L-cysteine–treated cells showed that the amountof pyruvate in these cells eventually decreased compared withcontrol cells, probably because demand may have overtakensupply (Fig. 2). In addition, defective GSIS induced by L-cysteinewas restored by exogenously added methyl pyruvate (Fig. S3 Aand E) or by PKM2 activation with DASA-10 (Fig. 5 B, D, andF). This result suggested that L-cysteine had induced seriouslydefective GSIS, in which both the first and second phases ofinsulin secretion had been abolished in insulin-secreting cells dueto the decreased levels of pyruvate and production of its down-stream metabolites.The impaired PKM2 activity by L-cysteine was likely attribut-
able to the direct interaction between L-cysteine and PKM2. Asshown in Fig. S7A, the enzymatic activity of a recombinantPKM2 protein in vitro was inhibited in the presence of L-cysteinein a concentration-dependent manner. Furthermore, in vitro orin MIN6 cells, L-cysteine induced the dissociation of PKM2tetramers (active form) to dimers/monomers (inactive forms),which resulted in decreased PKM2 enzymatic activity for pyru-vate production (Fig. 5G). We also found that L-cysteine had noeffect on the equilibrium between tetramer and dimer/monomerof the PKM1 protein either in vitro or in MIN6 cells (Fig. S7E).These results strongly suggested that L-cysteine specifically in-hibits PKM2 enzymatic activity in MIN6 cells by directly regu-lating the equilibrium between the tetramer and the dimer/monomer through noncompetitive inhibition. Although the precisemechanism of tetramer dissociation by L-cysteine remains unclear,one possible mechanism is that L-cysteine might affect the major
Glucose
Increase of L-cysteineconcentrations in
extracellular environment
Decrease of L-cysteineconcentrations in
extracellular environment
Active PKM2(tetramer forms)
Inactive PKM2(dissociation of tetramers
to dimers/monomers)
PEP
Pyruvate ATPproduction
Insulinsecretion
PKM2(tetramer)
PKM2(dimer/monomer)
L-cysteineInsulin granulePKM2 monomer
ATPproduction
Glucose
PEP
Pyruvate
Insulinsecretion
Fig. 6. Model for glucose-stimulated insulin secretion (GSIS) inhibition due toincreased L-cysteine concentrations in the extracellular environment. IncreasedL-cysteine concentrations in the extracellular environment are associated withincreased intracellular levels, which induce PKM2 inactivation by disruptingPKM2 tetramer formation (pyruvate kinase active form) due to increaseddirect binding of L-cysteine to PKM2. PKM2 inactivation results in inhibitingglucose-induced ATP production and insulin secretion. Thus, reduced L-cysteineconcentrations in the extracellular environment readily restore glucose-inducedATP production and insulin secretion to normal levels.
Nakatsu et al. PNAS | Published online February 23, 2015 | E1073
CELL
BIOLO
GY
PNASPL
US

intersubunit contact domain, which was anticipated from the modelresulting from X-ray crystallographic analyses (44). Based on this3D structural analysis, an alternate possibility is that L-cysteine issubstituted for L-serine, which binds to PKM2 to activate it (44),and results in impaired PKM2 activity through either attenuationof its tetramer dissociation or direct structural PKM2 modifications.Further studies are needed to clarify the role of L-cysteine inimpaired PKM2 activity.Perifusion analysis of GSIS by MIN6 cells and mouse pan-
creatic islets revealed that both the first and second phases wereinhibited by L-cysteine treatment (Figs. 3 D and F and 5 C and Eand Figs. S3D and S5A). To our knowledge, this report is the firstto show a negative L-cysteine effect on the first phase of insulinrelease that is commonly recognized as the early phenotypefound in impaired glucose tolerance, which is closely associatedwith further progression to T2D (45–47). To inhibit first-phasesecretion, it is quite possible that L-cysteine primarily inhibitsglucose-induced ATP production. As shown in Fig. 2, the de-crease in pyruvate levels in L-cysteine–treated cells depressed theTCA cycle and resulted in a reduced NADH supply from theTCA cycle, which subsequently inhibited transient ATP pro-duction upon high-glucose treatment. Although H2S productionfrom L-cysteine was shown to limit GSIS by impairing KATPchannels and VDCCs (18–20), this effect could be ruled outbecause adding methyl pyruvate and DASA-10 restored GSIS byL-cysteine–treated cells (Fig. 5 and Fig. S3). In fact, we detectedno increase in H2S concentration in MIN6 cells by prolongedtreatment with L-cysteine (Fig. S2B). The reason why the pro-longed L-cysteine treatment in our study had no effect on H2Sproduction in MIN6 cells, in contrast to the transient L-cysteinetreatment in (15), remains unclear, and further studies will beneeded for clarifying the reason. We speculate that the in-consistency is due to the difference between the prolonged andtransient treatments with L-cysteine. The degradation of H2Smay be enhanced by the activities of catalytic enzymes during theprolonged (∼24 h) treatment of MIN6 cells.The necessity of the prolonged treatment of L-cysteine for
inhibiting insulin secretion may be due simply to the slowness ofL-cysteine uptake (or accumulation) in MIN6 cells. As shown inFig. 3, the accumulated L-cysteine in MIN6 cells seems tobe metabolized or to flow readily out of cells, at least within90 min of incubation with 3G-KRBB and 25G-KRBB withoutL-cysteine. So it will take a long time to accumulate a sufficientamount of L-cysteine in MIN6 cells for the inhibition ofPKM2 activity.The precise reason why second-phase insulin secretion was
inhibited in L-cysteine–treated cells remains unclear. One pos-sible mechanism is that decreased amounts of malonyl-CoA dueto a shortage of pyruvate in L-cysteine–treated cells resulted inreduced concentrations of long-chain acyl-CoA, which was pre-dicted to be necessary for activating the GSIS second phase dueto the inhibition of carnitine palmitoyl transferase by malonyl-CoA (30). However, opposite results have also been reported:i.e., malonyl-CoA and long-chain acyl-CoA had no effects onGSIS (48, 49). Furthermore, we could not rule out that othermetabolites in L-cysteine–treated cells might affect GSIS. Forexample, oxaloacetate, which is a TCA-cycle metabolite, wassuggested to inhibit serine/threonine protein phosphatase re-sulting in activation of GSIS (50). In addition, previous reportsdemonstrated that decreased oxaloacetate production from py-ruvate by inactivation of pyruvate carboxylase attenuated GSISfirst and second phases (11, 24). Although the amounts of oxa-loacetate were not determined in this analysis (Fig. 2), otherTCA-cycle metabolites were found to be decreased and mighthave had an effect on GSIS through a metabolic amplifyingpathway, another insulin secretion pathway (Fig. 2). Thus, theL-cysteine effects on inhibiting second-phase secretion will re-quire further experimental validation.
One of our interesting findings was that L-cysteine–inducedinhibition of GSIS in insulin-secreting cells was reversible, atleast under our experimental conditions (Fig. 3 C–G andFig. S5). Although there have been many reports that showedthat L-cysteine had an inhibitory effect on GSIS, to our knowledge,our results are the first to show a reversible L-cysteine effect onGSIS. This reversibility aspect will be important for elucidatingthe physiological role of L-cysteine in blood. As shown in Fig. 3C–G and Fig. S5, even after prolonged exposure to 1–2 mML-cysteine by MIN6 cells and mouse pancreatic islets, pre-incubation for 1 h without L-cysteine followed by stimulationwith high glucose for 30 min was sufficient for these cells tohave restored normal GSIS, which showed that this L-cysteine–induced inhibition was reversible. This result indicates that anincreased L-cysteine concentration in the blood not only can beone possible cause of impaired insulin secretion from islet cellsbut also is possibly involved in homeostatic regulation to preventexcessive insulin secretion.Increased L-cysteine in the blood is a reported biomarker of
obstructive sleep apnea, which is an independent risk factor fordiabetes (6, 7). In addition, an increase in the total free cysteineconcentration in blood of more than twofold was associated withan increase in body mass (1), which is another risk factor fordiabetes (3). Furthermore, improved drugs for diabetes control,such as bis(ethylmal-tolato)oxovanadium(IV) (BEOV) and bis(maltolato)oxovanadium(IV) (BMOV), decreased blood con-centrations of L-cysteine in Zucker rats, a well-known obesitymodel animal (51). These reports support that exposure to highconcentrations of L-cysteine by islet cells is one of the crucialregulatory factors for GSIS and that ameliorating these highblood concentrations of L-cysteine might be a therapeutic modalityfor diabetes.Our results suggest that exposure to elevated L-cysteine levels
by pancreatic β-cells can cause a significant perturbation of bi-phasic insulin secretion and ATP production upon high-glucosestimulation due to the impaired glycolytic enzyme activity ofPKM2 accompanying the subunit dissociation of tetramer form,probably by a direct interaction between L-cysteine and PKM2.Moreover, removing L-cysteine or treating it with a PKM2 activatorrestores PKM2 activity, ATP production, and insulin secretion, thusproposing that these inhibitions are reversible. Because PKM2 hasreceived much attention recently as a biomarker for various cancers(52, 53), the PKM2 regulatory mechanism of L-cysteine suggestsa potential therapeutic target for both T2D and cancer.
Materials and MethodsCell Culture. MIN6 cells were a kind gift from Jun-ichi Miyazaki (Osaka Uni-versity, Osaka, Japan). MIN6 cells were cultured in DMEM that contained25 mM glucose (Wako) supplemented with 10% (vol/vol) FCS (GIBCO),penicillin/streptomycin (GIBCO), and 72 μM β-mercaptoethanol (Wako)(hereafter, 25G-M) in humidified 5% CO2 at 37 °C.
MIN6 Cells Precultured in Medium With and Without L-Cysteine. For conditionsof transient or prolonged L-cysteine treatment, MIN6 cells were preculturedin 25G-M supplemented with 0, 1, or 2 mM L-cysteine for 24 h at 37 °C,before being used in experiments. See Table S1 and Table S2 for detailedexperimental conditions and procedures.
Isolation and Culture of Mouse Pancreatic Islets. Pancreatic islets were iso-lated from male C57BL/6J mice (CLEA) aged 8 wk by the collagenasedigestion method as described previously (54). The animal care and ex-perimental procedures were approved by the Animal Care Committee ofthe University of Tokyo. Groups of 20 size-matched islets were hand-picked and precultured in RPMI 1640 that contained 11 mM glucose (Nissui)supplemented with 10% (vol/vol) FCS and penicillin/streptomycin with0 or 1 mM L-cysteine for 24 h in humidified 5% CO2 at 37 °C, before beingused for the insulin secretion assay. See Table S2 for the detailed exper-imental conditions and procedures.
E1074 | www.pnas.org/cgi/doi/10.1073/pnas.1417197112 Nakatsu et al.

Antibodies and Reagents. We used the following primary antibodies: rabbitanti-actin antibody (A5060; Sigma-Aldrich), rabbit anti–PKM1-specific an-tibody (15821-1-AP; Proteintech), rabbit anti–PKM2-specific antibody(15822-1-AP; Proteintech), rabbit anti-pyruvate kinase antibody (AV41699;Sigma-Aldrich), mouse anti–β-tubulin antibody (T8328; Sigma-Aldrich),and mouse anti-proinsulin/insulin antibody (L6B10; Cell Signaling). Thefollowing secondary antibodies were used: HRP-conjugated anti-mouseIgG antibody (W4021; Promega) and HRP-conjugated anti-rabbit IgGantibody (7074; Cell Signaling). The following reagents were also used:L-cysteine (Wako), DASA-10 (Millipore), methyl pyruvate (Sigma Aldrich),and maitotoxin (Wako).
RNA Interference. Small interfering RNAs (siRNAs) directed against mousePKM1 or PKM2 were from Sigma-Aldrich. These siRNAs were designed tospecifically knockdown a PKM1 or a PKM2 splicing variant of the mousePKM gene using the Rosetta siRNA Design Algorithm (Sigma-Aldrich). ThesiRNA sequences were as follows: PKM2 siRNA, 5′-GUGCGAGCCUCCAGU-CACUdTdT-3′; and PKM1 siRNA, 5′-GGCAGAGGCUGCCAUCUACdTdT-3′.Scrambled siRNA was from Ambion. A total of 1.8 × 105 MIN6 cells weretransfected with 100 pmol of siRNA using Lipofectamine RNAi Max (Invi-trogen) according to the manufacturer’s instructions. After incubation for48 h, the cells were used for insulin secretion, ATP production, and immuno-blotting assays. When RNA interference was combined with L-cysteine treat-ment, L-cysteine was added to culture medium after 24 h of siRNA transfection.
In Vitro Pyruvate Kinase Activity Assay. Human PKM1 or mouse PKM2 proteinwas purchased from Sigma or from Abcam, respectively. PKM1 and PKM2enzymatic activity and half-maximal inhibition concentration (IC50) of L-cysteinewere determined using pyruvate kinase activity assay kits (Biovision)according to the manufacturer’s instruction. Briefly, 10 ng of PKM2 or 1 ngof PKM1 protein was incubated with pyruvate kinase reaction mix (Bio-vision) containing different concentrations of L-cysteine (0–10,000 μM) for20 min (IC50) or 0–90 min (enzymatic activity). After the incubation, pyruvatekinase activity of PKM1 or PKM2 was calculated by measuring pyruvate contentsin the mixture.
We determined the maximum velocity (Vmax) and the Michaelis constant(Km) of PKM2 enzymatic activity and the effect of L-cysteine on Vmax and Km
according to the method of Chaneton et al. (44) with slight modifications.Because pyruvate kinase catalyzes the reaction of phosphoenolpyruvate(PEP) + ADP → pyruvate + ATP, its activity could be estimated by the ATPcontent in the reaction mixture. Recombinant PKM2 protein was incubatedwith 50 mM Tris, 100 mM KCl, 10 mM MgCl2, 200 mM ADP, 3% (vol/vol)DMSO, and 0–10 mM PEP in the presence or absence of 59 μM L-cysteine for20 min. Then, the ATP content was measured with an ATP determinationkit (Molecular Probes) and was normalized to the control without PKM2proteins. We constructed a Michaelis–Menten plot and a Lineweaver–Burkplot and determined Vmax and Km.
In Vivo Pyruvate Kinase Activity Assay. After MIN6 cells were washed twicewith KRBB (140 mM NaCl, 3.6 mM KCl, 0.5 mM NaH2PO4, 0.5 mM MgSO4,1.5 mM CaCl2, 10 mM Hepes, and 2 mM NaHCO3, pH 7.4), these cells werepreincubated with 3 mM glucose-containing KRBB (3G-KRBB) supplementedwith 0, 1, or 2 mM L-cysteine at 37 °C for 1 h and then washed with KRBB.Subsequently, the cells were incubated with 25 mM glucose-containingKRBB (25G-KRBB) supplemented with 0, 1, or 2 mM L-cysteine at 37 °C for30 min. After incubation, the cells were washed twice with ice-cold PBS andlysed with 4 volumes of pyruvate kinase assay buffer (Biovision). Cell lysateswere centrifuged at 20,400 × g at 4 °C for 5 s, after which supernatants werecollected. Pyruvate kinase activity in a supernatant was determined using apyruvate kinase activity assay kit according to the manufacturer’s instructions.Total protein in a supernatant was adjusted to 3 μg.
SDS/PAGE and Western Blotting. Native SDS/PAGE was performed using a5–20% wt/vol acrylamide gradient gel (Wako) and gel running buffer (0.25 MTris-base and 1.92 M glycine) that contained 0.05% SDS under constantcurrent conditions (20 mA/gel) at 4 °C for 180 min, after which proteins weretransferred to a polyvinylidene fluoride (PVDF) membrane using the wet-tank method. Other experiments were performed according to methodsdescribed by Nowakowski et al. (36) with slight modifications. SDS/PAGEused a 5–20% wt/vol acrylamide gradient gel and gel running buffer thatcontained 0.1% SDS under constant current conditions (20 mA/gel) at roomtemperature for 80 min, after which proteins were transferred to a PVDFmembrane using a Trans-Blot SD cell (Bio-Rad). A membrane was blockedwith 5% (wt/vol) BSA or 5% (wt/vol) nonfat dry milk in TBST (TBS containing0.1% Tween 20) at room temperature for 60 min, incubated with appro-
priate primary antibodies at 4 °C overnight and then with HRP-conjugatedsecondary antibodies at room temperature for 60 min. Antigen–antibodycomplexes were detected using enhanced chemiluminescence reagents(Western Lightning Plus-ECL; Perkin-Elmer). Band intensities were quantifiedusing an LAS-4000 mini imaging system (GE Healthcare).
Insulin Secretion from Statically Incubated Cells. Under conditions of pro-longed L-cysteine treatment, after MIN6 cells were washed twice with KRBB,they were preincubated in 3G-KRBB supplemented with 0, 1, or 2 mML-cysteine at 37 °C for 1 h and then washed with KRBB. Subsequently, thecells were incubated in 3G-KRBB or 25G-KRBB supplemented with 0, 1, or2 mM L-cysteine at 37 °C for 30 min. Under conditions of transient L-cysteinetreatment, cells precultured without L-cysteine were washed twice with KRBB,preincubated in 3G-KRBB at 37 °C for 1 h, and then washed with KRBB. Sub-sequently, the cells were incubated in 25G-KRBB supplemented with 0, 1, or2 mM L-cysteine at 37 °C for 30 min. When methyl pyruvate treatment wascombined with L-cysteine treatment, 2 mM methyl pyruvate was addedto 25G-KRBB for incubation. When DASA-10 treatment was combinedwith L-cysteine treatment, 20 μM DASA-10 or DMSO was added to3G-KRBB and 25G-KRBB for preincubation and incubation. After in-cubation, the buffers used during incubation were collected and centri-fuged at 20,400 × g at 4 °C for 1 min. Subsequently, insulin in thesesupernatants was determined using an AlphaLISA insulin kit (Perkin-Elmer). See Table S1 for detailed experimental conditions and procedures.
Insulin Secretion from Perifused MIN6 Cells and Mouse Pancreatic Islets. MIN6cells or groups of 20 size-matched mouse pancreatic islets were washed twicewith KRBB and preincubated in 3G-KRBB supplemented with 0, 1, or 2 mML-cysteine at 37 °C for 30 min. These cells were then perifused at a rate of0.5 mL/min (for MIN6 cells) or 50 μL/min (for mouse pancreatic islets) withthe same buffer at 37 °C for 30 min for equilibration, and the glucoseconcentration was increased to 12 mM (12 mM glucose-containing KRBB,12G-KRBB) with or without L-cysteine and then perifused for an additional30 min. When methyl pyruvate treatment was combined with L-cysteinetreatment, 1.5 mM methyl pyruvate was added to 12G-KRBB for the dura-tion of high-glucose stimulation. When DASA-10 treatment was combinedwith L-cysteine treatment, 5 μM DASA-10 or DMSO was added to 3G-KRBBand 12G-KRBB. During perifusion experiments, effluent buffer was collectedat 1-min intervals for insulin measurements. The buffers were centrifuged at20,400 × g at 4 °C for 1 min, after which insulin in supernatants was deter-mined using an AlphaLISA insulin kit. See Table S2 for detailed experimentalconditions and procedures.
Quantification of Intracellular ATP Levels. The cells were washed twice withKRBB and lysed with passive lysis buffer (Promega). After two freeze/thawcycles and centrifugation (20,400 × g, 4 °C, 1 min), ATP levels in supernatantswere measured using an ATP determination kit (Molecular Probes) accordingto the manufacturer’s instructions.
Analysis of Ionic Metabolites by CE-TOF-MS. Analysis of ionic metabolites wascarried out by HumanMetabolome Technologies, Inc. as described previously(55, 56). Detailed experimental conditions are described in SI Materialsand Methods.
Statistical Analysis. For the results shown in Figs. 1 and 3 A–C and Figs. S1 Aand B, S2B, S3C, S4 D and E, and S6C, statistical comparisons were madeusing Dunnett’s post hoc test. For results in Fig. 4A and Fig. S3 A and B,statistical comparisons were made using Bartlett’s test and Tukey–Kramer orSteel–Dwass test. For results in Fig. 5A and B and Fig. S6D, statistical com-parisons were made using F-tests and Student’s t tests with Bonferronicorrections. For results in Fig. 4B and Figs. S2C and S6 A and B, statisticalcomparisons were made using F-tests and Student’s t tests. Results are givenas means ± SEs of the means (SEMs). Significant differences were consideredat *P < 0.05, **P < 0.01, and ***P < 0.001.
ACKNOWLEDGMENTS. We thank Eishin Hirata, Ayumi Nagano, andDr. Yoshitaka Sakurai (Department of Diabetes and Metabolic Diseases,Graduate School of Medicine, University of Tokyo) for assistance withpreparation of mouse pancreatic islet; Kaichiro Endo and Prof. Hajime Wada(Department of Life Sciences, Graduate School of Arts and Sciences, Uni-versity of Tokyo) for assistance with measurement of oxygen consumption;and Kishiko Osaka and Naomi Okamoto for experimental assistance. Thiswork was in part supported by grants from Precursory Research forEmbryonic Science and Technology (PRESTO), Japan Science and TechnologyAgency (JST) (to F.K.), by a grant from the Technology DevelopmentProgram for Advanced Measurement and Analysis, JST (to M.M.), and bygrants from The Canon Foundation (to F.K. and M.M.).
Nakatsu et al. PNAS | Published online February 23, 2015 | E1075
CELL
BIOLO
GY
PNASPL
US

1. Elshorbagy AK, Smith AD, Kozich V, Refsum H (2012) Cysteine and obesity. Obesity(Silver Spring) 20(3):473–481.
2. Elshorbagy AK, Kozich V, Smith AD, Refsum H (2012) Cysteine and obesity: Consis-tency of the evidence across epidemiologic, animal and cellular studies. Curr Opin ClinNutr Metab Care 15(1):49–57.
3. Vazquez G, Duval S, Jacobs DR, Jr, Silventoinen K (2007) Comparison of body massindex, waist circumference, and waist/hip ratio in predicting incident diabetes: Ameta-analysis. Epidemiol Rev 29:115–128.
4. Fiehn O, et al. (2010) Plasma metabolomic profiles reflective of glucose homeostasis innon-diabetic and type 2 diabetic obese African-American women. PLoS ONE 5(12):e15234.
5. Herrmann W, et al. (2005) Disturbed homocysteine and methionine cycle inter-mediates S-adenosylhomocysteine and S-adenosylmethionine are related to degreeof renal insufficiency in type 2 diabetes. Clin Chem 51(5):891–897.
6. Botros N, et al. (2009) Obstructive sleep apnea as a risk factor for type 2 diabetes. Am JMed 122(12):1122–1127.
7. Cintra F, et al. (2011) Cysteine: A potential biomarker for obstructive sleep apnea.Chest 139(2):246–252.
8. Gall WE, et al.; RISC Study Group (2010) Alpha-hydroxybutyrate is an early biomarkerof insulin resistance and glucose intolerance in a nondiabetic population. PLoS ONE5(5):e10883.
9. Cheng K, Andrikopoulos S, Gunton JE (2013) First phase insulin secretion and type 2diabetes. Curr Mol Med 13(1):126–139.
10. Potter KJ, Westwell-Roper CY, Klimek-Abercrombie AM, Warnock GL, Verchere CB(2014) Death and dysfunction of transplanted β-cells: lessons learned from type 2diabetes? Diabetes 63(1):12–19.
11. Prentki M, Matschinsky FM, Madiraju SR (2013) Metabolic signaling in fuel-inducedinsulin secretion. Cell Metab 18(2):162–185.
12. Fehse F, et al. (2005) Exenatide augments first- and second-phase insulin secretion inresponse to intravenous glucose in subjects with type 2 diabetes. J Clin EndocrinolMetab 90(11):5991–5997.
13. Del Prato S, Tiengo A (2001) The importance of first-phase insulin secretion: Im-plications for the therapy of type 2 diabetes mellitus. Diabetes Metab Res Rev 17(3):164–174.
14. Bacha F, Lee S, Gungor N, Arslanian SA (2010) From pre-diabetes to type 2 diabetes inobese youth: Pathophysiological characteristics along the spectrum of glucose dys-regulation. Diabetes Care 33(10):2225–2231.
15. Kaneko Y, Kimura Y, Kimura H, Niki I (2006) L-cysteine inhibits insulin release fromthe pancreatic beta-cell: Possible involvement of metabolic production of hydrogensulfide, a novel gasotransmitter. Diabetes 55(5):1391–1397.
16. Ammon HP, Abdel-hamid M (1981) Potentiation of the insulin-releasing capacity oftolbutamide by thiols: Studies on the isolated perfused pancreas. Naunyn Schmie-debergs Arch Pharmacol 317(3):262–267.
17. Yu XH, et al. (2014) Hydrogen sulfide as a potent cardiovascular protective agent. ClinChim Acta 437:78–87.
18. Tang G, Zhang L, Yang G, Wu L, Wang R (2013) Hydrogen sulfide-induced inhibitionof L-type Ca2+ channels and insulin secretion in mouse pancreatic beta cells. Dia-betologia 56(3):533–541.
19. Yang W, Yang G, Jia X, Wu L, Wang R (2005) Activation of KATP channels by H2S inrat insulin-secreting cells and the underlying mechanisms. J Physiol 569(Pt 2):519–531.
20. Ali MY, Whiteman M, Low CM, Moore PK (2007) Hydrogen sulphide reduces insulinsecretion from HIT-T15 cells by a KATP channel-dependent pathway. J Endocrinol195(1):105–112.
21. Ammon HP, Hehl KH, Enz G, Setiadi-Ranti A, Verspohl EJ (1986) Cysteine analoguespotentiate glucose-induced insulin release in vitro. Diabetes 35(12):1390–1396.
22. MacDonald MJ, Chang CM (1985) Pancreatic islets contain the M2 isoenzyme of py-ruvate kinase: Its phosphorylation has no effect on enzyme activity. Mol Cell Biochem68(2):115–120.
23. Boxer MB, et al. (2010) Evaluation of substituted N,N’-diarylsulfonamides as acti-vators of the tumor cell specific M2 isoform of pyruvate kinase. J Med Chem 53(3):1048–1055.
24. Fransson U, Rosengren AH, Schuit FC, Renström E, Mulder H (2006) Anaplerosis viapyruvate carboxylase is required for the fuel-induced rise in the ATP:ADP ratio in ratpancreatic islets. Diabetologia 49(7):1578–1586.
25. Soergel DG, Gusovsky F, Yasumoto T, Daly JW (1990) Stimulatory effects of maito-toxin on insulin release in insulinoma HIT cells: Role of calcium uptake and phos-phoinositide breakdown. J Pharmacol Exp Ther 255(3):1360–1365.
26. Soergel DG, Yasumoto T, Daly JW, Gusovsky F (1992) Maitotoxin effects are blockedby SK&F 96365, an inhibitor of receptor-mediated calcium entry. Mol Pharmacol41(3):487–493.
27. Meucci O, et al. (1992) Maitotoxin-induced intracellular calcium rise in PC12 cells:Involvement of dihydropyridine-sensitive and omega-conotoxin-sensitive calciumchannels and phosphoinositide breakdown. J Neurochem 59(2):679–688.
28. Musgrave IF, Seifert R, Schultz G (1994) Maitotoxin activates cation channels distinctfrom the receptor-activated non-selective cation channels of HL-60 cells. Biochem J301(Pt 2):437–441.
29. Jitrapakdee S, Wutthisathapornchai A, Wallace JC, MacDonald MJ (2010) Regulationof insulin secretion: Role of mitochondrial signalling. Diabetologia 53(6):1019–1032.
30. Straub SG, Sharp GW (2002) Glucose-stimulated signaling pathways in biphasic insulinsecretion. Diabetes Metab Res Rev 18(6):451–463.
31. Mazurek S (2011) Pyruvate kinase type M2: A key regulator of the metabolic budgetsystem in tumor cells. Int J Biochem Cell Biol 43(7):969–980.
32. Martens GA, et al. (2010) Protein markers for insulin-producing beta cells with higherglucose sensitivity. PLoS ONE 5(12):e14214.
33. Wang HJ, et al. (2014) JMJD5 regulates PKM2 nuclear translocation and reprogramsHIF-1α-mediated glucose metabolism. Proc Natl Acad Sci USA 111(1):279–284.
34. Gao X, Wang H, Yang JJ, Liu X, Liu ZR (2012) Pyruvate kinase M2 regulates genetranscription by acting as a protein kinase. Mol Cell 45(5):598–609.
35. Anastasiou D, et al. (2012) Pyruvate kinase M2 activators promote tetramer formationand suppress tumorigenesis. Nat Chem Biol 8(10):839–847.
36. Nowakowski AB, Wobig WJ, Petering DH (2014) Native SDS-PAGE: High resolutionelectrophoretic separation of proteins with retention of native properties includingbound metal ions. Metallomics 6(5):1068–1078.
37. Clower CV, et al. (2010) The alternative splicing repressors hnRNP A1/A2 and PTBinfluence pyruvate kinase isoform expression and cell metabolism. Proc Natl Acad SciUSA 107(5):1894–1899.
38. Imamura K, Tanaka T (1972) Multimolecular forms of pyruvate kinase from rat andother mammalian tissues. I. Electrophoretic studies. J Biochem 71(6):1043–1051.
39. Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC (2008) Pyruvate kinaseM2 is a phosphotyrosine-binding protein. Nature 452(7184):181–186.
40. Yang W, et al. (2012) ERK1/2-dependent phosphorylation and nuclear translocationof PKM2 promotes the Warburg effect. Nat Cell Biol 14(12):1295–1304.
41. Julian D, Statile JL, Wohlgemuth SE, Arp AJ (2002) Enzymatic hydrogen sulfide pro-duction in marine invertebrate tissues. Comp Biochem Physiol A Mol Integr Physiol133(1):105–115.
42. Yilmaz S, Ozan S, Ozercan IH (2003) Comparison of pyruvate kinase variants frombreast tumor and normal breast. Arch Med Res 34(4):315–324.
43. Carbonell J, Felíu JE, Marco R, Sols A (1973) Pyruvate kinase: Classes of regulatoryisoenzymes in mammalian tissues. Eur J Biochem 37(1):148–156.
44. Chaneton B, et al. (2012) Serine is a natural ligand and allosteric activator of pyruvatekinase M2. Nature 491(7424):458–462.
45. Snehalatha C, et al. (2009) Changes in insulin secretion and insulin sensitivity in re-lation to the glycemic outcomes in subjects with impaired glucose tolerance in theIndian Diabetes Prevention Programme-1 (IDPP-1). Diabetes Care 32(10):1796–1801.
46. Tuomilehto J, et al.; Finnish Diabetes Prevention Study Group (2001) Prevention oftype 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucosetolerance. N Engl J Med 344(18):1343–1350.
47. Gerstein HC, et al.; DREAM (Diabetes REduction Assessment with ramipril and rosi-glitazone Medication) Trial Investigators (2006) Effect of rosiglitazone on the fre-quency of diabetes in patients with impaired glucose tolerance or impaired fastingglucose: A randomised controlled trial. Lancet 368(9541):1096–1105.
48. Antinozzi PA, Segall L, Prentki M, McGarry JD, Newgard CB (1998) Molecular orpharmacologic perturbation of the link between glucose and lipid metabolism iswithout effect on glucose-stimulated insulin secretion: A re-evaluation of the long-chain acyl-CoA hypothesis. J Biol Chem 273(26):16146–16154.
49. Mulder H, et al. (2001) Overexpression of a modified human malonyl-CoA de-carboxylase blocks the glucose-induced increase in malonyl-CoA level but has noimpact on insulin secretion in INS-1-derived (832/13) beta-cells. J Biol Chem 276(9):6479–6484.
50. Sjöholm A, et al. (2002) Glucose metabolites inhibit protein phosphatases and directlypromote insulin exocytosis in pancreatic beta-cells. Endocrinology 143(12):4592–4598.
51. Wasan KM, Risovic V, Yuen VG, McNeill JH (2006) Differences in plasma homo-cysteine levels between Zucker fatty and Zucker diabetic fatty rats following 3weeks oral administration of organic vanadium compounds. J Trace Elem Med Biol19(4):251–258.
52. Christofk HR, et al. (2008) The M2 splice isoform of pyruvate kinase is important forcancer metabolism and tumour growth. Nature 452(7184):230–233.
53. Israelsen WJ, et al. (2013) PKM2 isoform-specific deletion reveals a differential re-quirement for pyruvate kinase in tumor cells. Cell 155(2):397–409.
54. Takamoto I, et al. (2014) TCF7L2 in mouse pancreatic beta cells plays a crucial role inglucose homeostasis by regulating beta cell mass. Diabetologia 57(3):542–553.
55. Ohashi Y, et al. (2008) Depiction of metabolome changes in histidine-starved Escherichiacoli by CE-TOFMS. Mol Biosyst 4(2):135–147.
56. Mitsuishi Y, et al. (2012) Nrf2 redirects glucose and glutamine into anabolic pathwaysin metabolic reprogramming. Cancer Cell 22(1):66–79.
E1076 | www.pnas.org/cgi/doi/10.1073/pnas.1417197112 Nakatsu et al.