large-scale proteomics analysis of the human kinome
TRANSCRIPT

Large-scale Proteomics Analysis of theHuman Kinome□S
Felix S. Oppermann‡, Florian Gnad§, Jesper V. Olsen§, Renate Hornberger‡,Zoltan Greff¶, Gyorgy Keri¶�, Matthias Mann§, and Henrik Daub‡**
Members of the human protein kinase superfamily are themajor regulatory enzymes involved in the activity controlof eukaryotic signal transduction pathways. As proteinkinases reside at the nodes of phosphorylation-based sig-nal transmission, comprehensive analysis of their cellularexpression and site-specific phosphorylation can provideimportant insights into the architecture and functionalityof signaling networks. However, in global proteome stud-ies, low cellular abundance of protein kinases often re-sults in rather minor peptide species that are occluded bya vast excess of peptides from other cellular proteins.These analytical limitations create a rationale for kinome-wide enrichment of protein kinases prior to mass spec-trometry analysis. Here, we employed stable isotope la-beling by amino acids in cell culture (SILAC) to comparethe binding characteristics of three kinase-selective affin-ity resins by quantitative mass spectrometry. The evalu-ated pre-fractionation tools possessed pyrido[2,3-d]py-rimidine-based kinase inhibitors as immobilized captureligands and retained considerable subsets of the humankinome. Based on these results, an affinity resin display-ing the broadly selective kinase ligand VI16832 was em-ployed to quantify the relative expression of more than170 protein kinases across three different, SILAC-en-coded cancer cell lines. These experiments demonstratedthe feasibility of comparative kinome profiling in a com-pact experimental format. Interestingly, we found highlevels of cytoplasmic and low levels of receptor tyrosinekinases in MV4–11 leukemia cells compared with the ad-herent cancer lines HCT116 and MDA-MB-435S. TheVI16832 resin was further exploited to pre-fractionate ki-nases for targeted phosphoproteomics analysis, whichrevealed about 1200 distinct phosphorylation sites onmore than 200 protein kinases. This hitherto largest sur-vey of site-specific phosphorylation across the kinomesignificantly expands the basis for functional follow-upstudies on protein kinase regulation. In conclusion, thestraightforward experimental procedures described here
enable different implementations of kinase-selective pro-teomics with considerable potential for future signaltransduction and kinase drug target analysis. Molecular& Cellular Proteomics 8:1751–1764, 2009.
Reversible protein phosphorylation represents the mostcommon type of post-translational modification (PTM)1 in eu-karyotic organisms. A plethora of studies on a large variety ofproteins have established that site-specific phosphorylationevents fulfill key functions in the activity control of signalingcascades and networks (1). Cellular protein phosphorylationis controlled by more than 500 members of the protein kinasesuperfamily, which comprises one of the largest enzyme fam-ilies encoded by the human genome (2). Protein kinases rep-resent the key elements in phosphorylation-based signaltransmission. Aberrant protein kinase expression and/or ac-tivity, often because of gene amplification or mutationalchanges, is involved in pathological processes leading tomalignant transformation and tumor development (3). There-fore, protein kinases have emerged as a major class of drugtargets for therapeutic intervention (4–6). Given the diversityof molecular mechanisms related to de-regulated kinase func-tion in human cancers, proteomic approaches could signifi-cantly enhance our understanding of disease-relevant kinasefunction and also help to optimize and adjust therapeuticstrategies. In addition to assessing protein expression, theanalysis of site-specific phosphorylations on protein kinasesis of particular relevance, as these PTMs can be indicative oftheir cellular catalytic activities (7, 8). Protein kinases can notonly modulate each other’s functions and activities throughsite-specific phosphorylation events, but often also undergosite-specific autophosphorylation once they get activated(9). Thus, the comprehensive assessment of kinase-derivedphosphopeptides can provide important insights into theregulation of these key players in phosphorylation-con-trolled signaling.
Regulatory enzymes such as protein kinases are often ex-pressed at low cellular levels. This can impede their detection
From the ‡Cell Signaling Group, Department of Molecular Biology,§Department of Proteomics and Signal Transduction, Max PlanckInstitute of Biochemistry, Am Klopferspitz 18, 82152 Martinsried,Germany, ¶Vichem Chemie Ltd., Herman Otto u. 15., Budapest,1022, Hungary, and �Pathobiochemistry Research Group of the Hun-garian Academy of Science, Semmelweis University, Puskin u. 9.,Budapest, 1088, Hungary
Received, December 18, 2008, and in revised form, April 9, 2009Published, MCP Papers in Press, April 15, 2009, DOI 10.1074/
mcp.M800588-MCP200
1 The abbreviations used are: AML, acute myelogenous leukemia;FLT3, FMS-like tyrosine kinase 3; GO, gene ontology; IPI, InternationalProtein Index; LC-MS, liquid chromatography-mass spectrometry;NEK, NIMA-related expressed kinase; PTK, protein tyrosine kinase;PTM, post-translational modification; SILAC, stable isotope labeling byamino acids in cell culture; ACN, acetonitrile; FDR, false-discovery rate.
Research
© 2009 by The American Society for Biochemistry and Molecular Biology, Inc. Molecular & Cellular Proteomics 8.7 1751This paper is available on line at http://www.mcponline.org
by guest on April 10, 2019
http://ww
w.m
cponline.org/D
ownloaded from

by LC-MS in highly complex peptide mixtures derived fromtotal cell or tissue extracts. These analytical challenges arefurther aggravated in phosphoproteomic experiments due tothe fact that many phosphopeptide species result from sub-stoichiometric phosphorylation events (10). Consequently,phosphopeptide isolation methods have proven to be es-sential. Among others, techniques such as immobilizedmetal affinity chromatography or enrichment by means oftitanium dioxide (TiO2)-coated beads have found wide-spread use in MS-based phosphoproteomics (11–13). Inaddition, to reduce initial sample complexity, either proteinfractionation by gel electrophoresis or peptide separationby strong cation exchange chromatography is typically in-cluded in contemporary phosphoproteomics workflows(14–16). These separation techniques in combination withLC-MS on state-of-the-art mass spectrometers enabled theidentification of thousands of phosphorylation sites fromtotal cellular extracts (15, 17, 18). Despite these impressiveadvances, such large-scale efforts require considerable in-strument time, and the current methodology is still notcomprehensive across the full dynamic range of the entirephosphoproteome. This creates the rationale for sub-pro-teome analyses to achieve high coverage and analyticalsensitivity, which is particularly relevant for members of theprotein kinase enzyme family.
To date, the only pre-fractionation techniques permittingthe enrichment of more than a few protein kinases areaffinity capture methods relying on immobilized and kinase-selective small molecule inhibitors (19–21). We and othershave demonstrated that combinations of such kinase inhib-itor resins efficiently pre-fractionate kinases for subsequentphosphorylation analysis (7, 22, 23). Ideally, capture mole-cules for kinase proteomics have two properties. First, theyshould exhibit high non-selectivity within the kinase super-family. Second, they should efficiently discriminate betweenprotein kinases and other classes of cellular proteins un-der the biochemical conditions of the pre-fractionationprocedure.
In our efforts to characterize affinity reagents fulfillingthese criteria, we quantitatively compared a selection ofimmobilized pyrido[2,3-d]pyrimidine-based inhibitors withrespect to their proteome-wide kinase binding properties.Based on this assessment, an affinity matrix displaying thesmall molecule VI16832 was used as an enrichment tool forthe comparative expression analysis of protein kinases indifferent cancer cell lines. The highly efficient VI16832 af-finity resin further enabled a large-scale phosphoproteom-ics survey resulting in the identification and confident as-signment of about 1200 phosphorylation sites on more than200 distinct protein kinases.
EXPERIMENTAL PROCEDURES
Cell Culture—For quantitative kinase inhibitor studies MV4–11(ATCC, CRL-9591) were grown in RPMI 1640 medium (Invitrogen)
containing 20% fetal bovine serum (Invitrogen), MDA-MB-435S(435S, ATCC, HTB-129) (24), and HCT116 (ATCC, CCL-247) weregrown in Dulbecco’s modified Eagle’s medium (Invitrogen) containing10% fetal bovine serum. All media were supplemented with penicillinand streptomycin (Invitrogen). For quantitative MS-based studies,cells were labeled with either L-arginine and L-lysine (Arg0, Lys0),L-[U-13C6,14N4]arginine, and L-[2H4]lysine (Arg6, Lys4) or L-[U-13C6,15N4] and L-[U-13C6,15N2]lysine (Arg10, Lys8) (Cambridge IsotopeLaboratories or Sigma) for six cell doublings to achieve completelabeling of cellular proteins. The adherent HCT116 and 435S cellswere lysed directly on cell culture plates, MV4–11 cells were har-vested by centrifugation before cell lysis.
Generation of Kinase Inhibitor Resins—The kinase inhibitorVI16832 was prepared as described (7). VI16741 and VI16743 weresynthesized accordingly, except that 8-ethyl-2-methanesulfonyl-8H-pyrido[2,3-d]pyrimidine-7-one and 8-cyclopentyl-2-methanesulfonyl-8H-pyrido[2,3-d]pyrimidine-7-one were used as starting materialinstead of 8-bicyclo-[2.2.1]hept-2-yl-2-methanesulfonyl-8H-pyrido-[2,3-d]pyrimidine-7-one in case of VI16832 (25). For preparation of theaffinity resins used in the SILAC experiments, 2 volumes of a 3 mM
inhibitor solution prepared in 50% dimethylformamide, 50% EtOHwere mixed with 1 volume of drained ECH-Sepharose beads (GEHealthcare) and then subjected to carbodiimide-catalyzed immobili-zation according to described procedures (22). Coupling efficiencieswere similar for all three inhibitors, resulting in a concentration of 1.5mM immobilized ligand on Sepharose beads as determined by UV-Vismeasurements (data not shown). Kinase enrichment in the phospho-proteomics experiments was performed with VI16832 covalently im-mobilized on epoxy-activated Sepharose (GE Healthcare) accordingto a reported coupling protocol (26). Here, 2 volumes of 1.5 mM
VI16832 solution prepared in 50% DMSO, 50% 50 mM Na2CO3, pH11 were subjected to 1 volume of drained epoxy-activated Sepharoseto initiate the coupling reaction.
Cell Lysis and Affinity Enrichment—Cells were lysed in 50 mM
Hepes-NaOH, pH 7.5, 150 mM NaCl, 0.5% Triton X-100, 1 mM EDTA,1 mM EGTA, 1 mM phenylmethylsulfonyl fluoride, 10 mM NaF, 2.5 mM
Na3VO4, 50 ng/ml calyculin A (Alexis Biochemicals, San Diego, CA),10 �g/ml aprotinin, 10 �g/ml leupeptin, and 1% phosphatase inhibitormixture 1 and 2 (Sigma) for 1 h at 4 °C. The cell debris was removedby centrifugation (20 min at 13,000 rpm) and by filtering through0.22-�m mixed esters of cellulose membranes (Millipore). Proteinconcentration was measured using the BCA assay (Pierce). For com-parative SILAC analysis of different inhibitor resins, 1.5 mg from eachdifferentially labeled MV4–11 lysate was subjected to in vitro associ-ation with the respective kinase inhibitor resins. 30 �l of drainedbeads coupled with the respective kinase inhibitor were washed threetimes with lysis buffer and further three times with lysis buffer con-taining 1 M NaCl. Washed beads were incubated for 2.5 h at 4 °C inthe dark with the lysates that had been adjusted to 1 M NaCl in a finalvolume of 650 �l. In each experiment, aliquots of the three differen-tially labeled lysates were pooled to determine the initial SILAC ratiosand resulting correction factors for the quantification after affinityenrichment. Beads were washed twice with lysis buffer containing 1 M
NaCl and twice with lysis buffer containing 150 mM NaCl. For elution,resin-bound proteins were incubated for 10 min with 50 �l 0.5% LDSbuffer (Invitrogen) containing 50 mM dithiothreitol at 70 °C. Elutionfractions were pooled and concentrated by a factor of three in avacuum concentrator (Eppendorf). Moreover, aliquots of the differentelution fractions were compared by immunoblotting with kinase-spe-cific antibodies.
For SILAC-based comparison of protein kinases in MV4–11,HCT116, and 435S cells, total cell lysates were prepared as describedabove and all adjusted to 1.5-mg protein in a volume of 500 �l. Thisamount of protein was obtained upon lysis of 17 � 106 MV4–11,
Kinase-selective Proteomics
1752 Molecular & Cellular Proteomics 8.7
by guest on April 10, 2019
http://ww
w.m
cponline.org/D
ownloaded from

7.3 � 106 HCT116, and 5.3 � 106 435S cells, respectively. The threelysates were pooled prior to incubation with 90 �l of drained VI16832beads according to the same protocol as used for the inhibitor resincomparisons.
For immunoblotting of either different affinity-purified fractionsfrom MV4–11 cells or of total cell lysates from MV4–11, HCT116, and435S cells, the following antibodies were used: rabbit anti-CDC2,mouse anti-Met and rabbit anti-PAK4 (Cell Signaling Technology,Inc.), mouse anti-PLK1 (7), rabbit anti-Fer (27), rabbit anti-PYK2 (Mil-lipore), goat anti-Axl, goat anti-CK1�, rabbit anti-DDR1 (C-20), rabbitanti-FAK (C-20), goat anti-Fes (C-19), rabbit anti-HCK (N-30), rabbitanti-JAK1 (HR-785), and rabbit anti-Syk (N-19) (all from Santa CruzBiotechnology, Inc.).
Protein kinase enrichment for phosphorylation site mapping wasperformed using an AKTA explorer system and Tricorn 5/20 chro-matography columns (GE Healthcare) packed with 500 �l ofVI16832 resin. Cells were lysed in a volume of 35–40 ml perexperiment. The protein amounts of the starting extracts used in thefirst and second experiments were: 435S, 85 and 120 mg; HCT116,240 and 175 mg; MV4–11, 180 and 120 mg. Lysates were adjustedto 1 M NaCl prior to loading onto the VI16832 column at a flow rateof 0.07 ml/min. Subsequent washing and elution steps were per-formed as described previously (22). Protein-containing elutionfractions were lyophilized, re-suspended in one tenth of the initialvolume, and then desalted by protein precipitation prior to gelelectrophoresis (28).
Sample Preparation and MS Analysis—For gel electrophoresis,ready-made 10% NuPAGE� Bis-Tris gels (Invitrogen) were used ac-cording to the manufacturer’s instructions. Resolved proteins werestained using the Collodial Blue staining kit (Invitrogen). In all SILACexperiments, gels were cut into three slices followed by in-gel diges-tion with trypsin and peptide purification with StageTips as described(29, 30).
For phosphopeptide identifications, gels were cut in either three(experiment 1) or 6 (experiment 2) molecular weight regions prior toin-gel proteolysis with trypsin (29). Phosphopeptides were specifi-cally enriched using titanium dioxide (TiO2) microspheres (31, 32).The TiO2 beads (GL Science, Tokyo, Japan) were first equilibratedby consecutive incubations with 20 mM NH4OH in 20% acetonitrile(ACN), pH 10.5, washing buffer (50% ACN, 0.1% trifluoroaceticacid) and loading buffer (5 g/liter 2,5-dihydrobenzoic acid in 55%ACN). Fractions of extracted peptides were adjusted to loadingconditions and incubated for 30 min with 5 mg TiO2 beads at roomtemperature on a rotating wheel. Afterward, beads were washedonce with 100 �l of loading buffer, three times with 1.5 ml ofwashing buffer, and phosphopeptides were eluted by incubatingtwice with 30 �l of 20 mM NH4OH in 20% ACN, pH 10.5. Eluateswere combined and passed through C8 StageTips followed by a30-�l rinse with 80% ACN, 0.5% acetic acid. After adjusting to a pHof 6, samples were concentrated to �3 �l and mixed with an equalvolume of 4% ACN, 0.2% trifluoroacetic acid. MS analyses weredone as described previously (7, 15). Briefly, peptide separationswere done on 15-cm analytical columns (75-�m inner diameter)in-house packed with 3-�m C18 beads (Reprosil-AQ Pur, Dr.Maisch) using a nanoflow high pressure liquid chromatographysystem (Agilent Technologies 1100), which was coupled online to aLTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific) via ananoelectrospray ion source (Proxeon Biosystems). The LTQ-Orbi-trap was operated in the data-dependent mode to automaticallyswitch between full scan MS in the orbitrap analyzer (with resolutionr � 60,000 at m/z 400) and the fragmentation of the five mostintense peptide ions by either MS/MS or multi-stage activation in theLTQ part of the instrument, the latter being triggered on neutral lossspecies at 97.97, 48.99, or 32.66 m/z below the precursor ion for 30 ms
(33). For all measurements with the orbitrap detector, a lock-massstrategy was used for internal calibration as described (34).
Peptide Identification, Quantitation, and Data Analysis—Raw MS filesacquired from individual experiments were merged using the Raw2msmsoftware (34), and the resulting msm files were searched against con-catenated forward and reversed versions of the human IPI proteindatabase version 3.13 (SILAC-based inhibitor comparison), version3.19 (phosphorylation site mapping), or version 3.24 (SILAC-basedkinome profiling) containing 57,032, 60,397, and 66,921 proteinentries, using the MASCOT search engine (Matrix Science). Alldatabases contained frequently occurring contaminants includinghuman keratins, porcine trypsin, and endopeptidase Lys-C. Searchparameters were set to up to three missed cleavages, mass toler-ances of 25 ppm for MS, and 0.5 Da for MS/MS scans. Carbam-idomethylation of cysteine was set as fixed modification; variablemodifications included oxidized methionine, phosphorylation ofserine, threonine and tyrosine, N-acetyl protein, N-pyroglutamineand in the SILAC experiments, the isotopic variants Lys4, Lys8,Arg6, and Arg10.
The html output files generated by MASCOT together with the rawdata files were then further processed using the MSQuant software,version 1.4.0 used for SILAC-based inhibitor comparison and version1.4.3 for SILAC-based kinome profiling and phosphorylation siteidentification). Prior to peptide quantification or computation of PTMscores, peptide datasets were filtered for a false-discovery rate (FDR)of less than 1% (p � 0.01) according to a target/decoy databasesearching strategy. To achieve a FDR of less than 1%, filtering criteriasuch as a peptide length � 6 and a mass error � 5ppm were appliedtogether with a minimal MASCOT score that ranged from 21 to 29depending on the experiment.
MSQuant determines the average ratio over the peptide elutionprofile, and all precursor ion assignments used for quantitation weremanually validated (15). Upon normalization for the initial SILAC pool-ing error, protein ratios were calculated as the mean of all ratios fromuniquely assigned peptides.
To identify highly significant differences in relative protein abun-dance, the relative ratios of the protein quantifications from the twobiological replicate experiments were analyzed for their normal dis-tribution to account for the combined biological and technical varia-tion in the quantitative MS analyses. Protein abundance was consid-ered as significantly different (p � 0.01) in case ratios differed from themean by 2.58 � as determined from the “ratio of ratios” distributionsof the biological replicate analyses.
The assignment of phosphorylation sites in identified phosphopep-tides was done with the PTM scoring algorithm implemented in MS-Quant as described previously by Olsen et al. (15). In our presentstudy, phosphorylation sites were rated as class I in case of a local-ization probability of at least 0.95. The localization p values for allidentified phosphopeptides as well as the corresponding annotatedMS/MS spectra can be accessed online (35).For enrichment analysisof gene ontology (GO) categories, Cytoscape (36) together with theBinGO plugin (36, 37) was used to identify statistically over-repre-sented GO molecular function terms compared with a reference data-set consisting of all IPI entries and their respective GO identifiersessentially as described (38).
RESULTS
Comparative Target Profiling for Kinase-selective Pre-frac-tionation Reagents—To enable broad kinase enrichment, im-mobilized kinase inhibitors should ideally exhibit considerablenon-selectivity in conjunction with high affinity for many mem-bers of the protein kinase superfamily. The previously de-scribed pyrido[2,3-d]pyrimidine-based capture molecule
Kinase-selective Proteomics
Molecular & Cellular Proteomics 8.7 1753
by guest on April 10, 2019
http://ww
w.m
cponline.org/D
ownloaded from

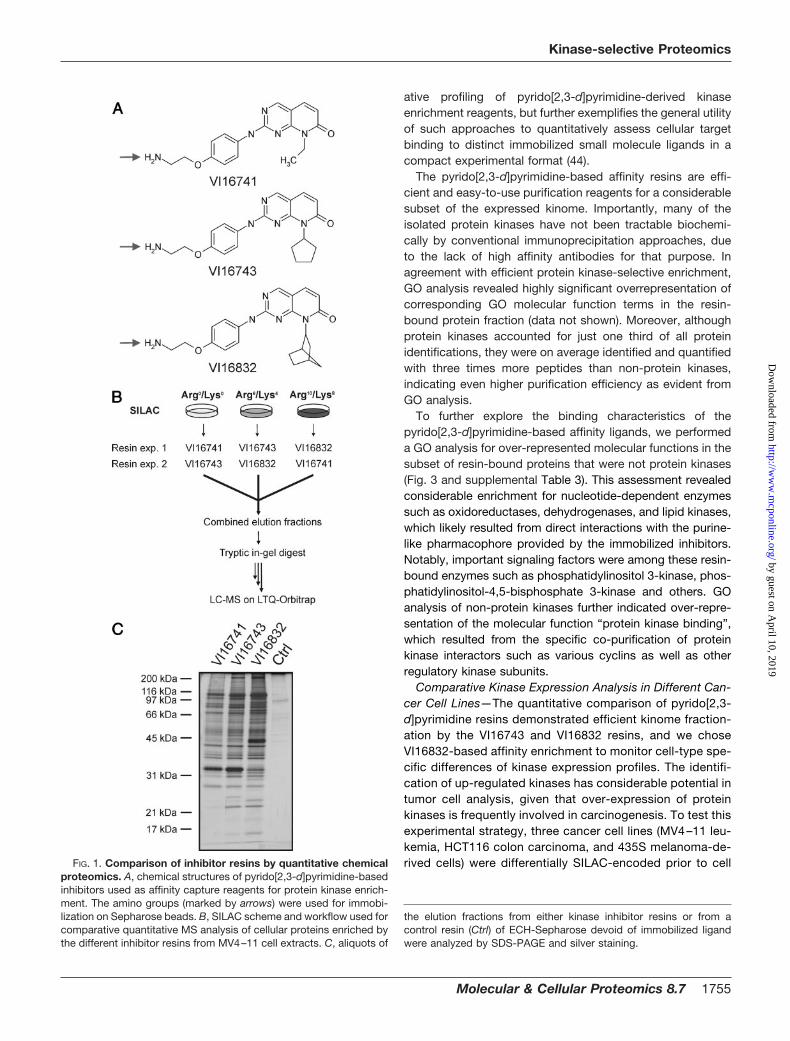
PP58 exhibited high potency and non-selectivity for a subsetof protein kinases comprising about 25% of the human ki-nome (39). These kinases possess a small amino acid (often athreonine residue) at a critical “gate-keeper” position, whichdoes not interfere with the positioning of the inhibitor’s dichlo-rophenyl moiety in a hydrophobic pocket located at their ATPbinding sites (40). In contrast, this cavity is rather inaccessiblein the majority of protein kinases with more spacious “gate-keeper” residues (40). We initially reasoned that this part ofthe kinome might be targeted by a PP58-related compoundthat lacks the dichlorophenyl group but is otherwise identicalin structure. However, such a capture molecule exhibitedfairly weak kinase affinity (data not shown). Therefore, basedon previously described structure-activity relationship data forCdk4 inhibition by pyrido[2,3-d]pyrimidine kinase antagonists(25), we have recently introduced compounds with largercyclopentyl and norbornyl moieties (designated VI16743 andVI16832) at the N8 position to compensate for the observeddrop in potency (7)2. As expected, these capture moleculesretained considerable numbers of kinases. However, the ac-tual effect of the N8 substituent on cellular target bindingprofiles has not been systematically analyzed. Such informa-tion would help to adjust pre-fractionation toward the cellularkinases that are of interest in individual projects. Therefore,we prepared pyrido[2,3-d]pyrimidine ligands with the cyclo-pentyl and norbornyl moieties (VI16743 and VI16832) as wellas a derivative with a smaller ethyl moiety in the N8 position(VI16741) and immobilized all three compounds through theirprimary amino groups (Fig. 1A) (7).
To compare the VI16741, VI16743, and VI16832 resins, weperformed stable isotope labeling by amino acids in cell culture(SILAC) with the acute myelogenous leukemia (AML) cell lineMV4–11 to enable quantitative MS analysis (41). These cellsharbor an internal tandem duplication in the juxtamembranedomain of the FMS-like tyrosine kinase 3 (FLT3). This mutationis also present in a subset of AML patients and results inconstitutive up-regulation of growth-promoting FLT3 tyrosinekinase activity (42). Upon quantitative incorporation of eithernormal arginine and lysine (Arg0/Lys0) or their isotopic variants(Arg6/Lys4 or Arg10/Lys8), lysates from three differentiallySILAC-encoded MV4–11 cell populations were incubated withthe affinity resins carrying covalently immobilized VI16741,VI16743, or VI16832 as capture ligands (Fig. 1, A and B). After invitro association and elution of retained proteins, we first ana-lyzed small aliquots of the elution fractions by gel electrophore-sis and silver staining. Most proteins appeared to be present inall three fractions, albeit typically at somewhat higher levels inthe VI16743 and VI16832 resin eluates (Fig. 1C). In addition,some protein bands showed specific resin binding, such as aprominent VI16741 and VI16743 resin-interacting 35 kDa pro-tein identified as mitochondrial delta(3,5),delta(2,4)-dienoyl-CoA
isomerase and a VI16832 resin-bound 50 kDa protein identifiedas the multifunctional protein ADE2 (data not shown), indicatingthat different hydrophobic moieties in the N8 position can con-fer selectivity as evident for these abundant purine-binding non-protein kinases.
The large remainders of the elution fractions were com-bined prior to gel electrophoresis and tryptic digestion ofproteins from three different molecular weight regions. Theresulting mixtures of proteolytically derived peptides wereanalyzed by LC-MS on a LTQ-Orbitrap hybrid mass spec-trometer. After database searching and filtering for an identi-fication certainty greater than 99%, relative peptide abun-dances between VI16741-, VI16743-, or VI16832-enrichedfractions were determined by SILAC-based quantification.Quantitative peptide data was then used to calculate therelative protein levels in the different resin eluates (supple-mental Tables 1 and 2). In total, more than 130 distinct proteinkinases could be identified and quantified in the inhibitor resineluates. This demonstrates the capacity of the pyrido[2,3-d]pyrimidine-based affinity ligands to pre-fractionate and de-tect almost half of the expressed kinome, which is estimatedto consist of up to 300 distinct protein kinases in a givenmammalian cell (43). A considerable subset of protein kinasesinteracted more strongly with the VI16743 and V16832 resinsthan with the VI16741-containing beads, indicating that thespace-filling cyclopentyl and norbonyl moieties in the N8 po-sition resulted in an overall increase in potency with respect tokinase binding (Fig. 2A). In a few cases, bulky N8-substituentswere not well tolerated, notably by various NIMA-related ex-pressed kinase (NEK) family members (NEK3, 6, and 7) thatwere found in higher abundance in the VI16741 resin eluates.The differences between the VI16743 and V16832 resins wereless pronounced, with relatively small subsets of protein ki-nases preferentially bound by either affinity matrix. Similarcomparisons were also made for the more than 250 non-protein kinase proteins quantified from the affinity resin elu-ates (supplemental Fig. 1A). To verify the quantitative MSapproach with a second assay, immunoblotting was donewith a selection of kinase-specific antibodies. The outcome ofthis analysis was found in excellent agreement with the MSresults (Fig. 2, B and C). To evaluate the reproducibility of theSILAC-based quantification, the protein kinase ratios ob-tained in biological replicate analysis were visualized in scat-ter blots (Fig. 2D). Notably, independent experimental ratiosfor VI16743 versus VI16741 as well as for VI16832 versusVI16741 resin binding were similar, demonstrating the accu-racy and reliability of the quantitative MS approach. Compa-rable results were obtained for the identified non-protein ki-nases (supplemental Fig. 1B). Moreover, as determined fromthe distribution of the ratios of the replicate, log2-trans-formed protein quantifications, values of more than 2.65 orless than 0.38 indicated differential binding with high con-fidence (p � 0.01) (supplemental Fig. 2). Hence, the SILAC-enabled strategy applied here not only enabled the compar-
2 T. Reinl, M. Nimtz, G. Keri, J. Wehland, H. Daub, and L. Jansch,submitted for publication.
Kinase-selective Proteomics
1754 Molecular & Cellular Proteomics 8.7
by guest on April 10, 2019
http://ww
w.m
cponline.org/D
ownloaded from
ative profiling of pyrido[2,3-d]pyrimidine-derived kinaseenrichment reagents, but further exemplifies the general utilityof such approaches to quantitatively assess cellular targetbinding to distinct immobilized small molecule ligands in acompact experimental format (44).
The pyrido[2,3-d]pyrimidine-based affinity resins are effi-cient and easy-to-use purification reagents for a considerablesubset of the expressed kinome. Importantly, many of theisolated protein kinases have not been tractable biochemi-cally by conventional immunoprecipitation approaches, dueto the lack of high affinity antibodies for that purpose. Inagreement with efficient protein kinase-selective enrichment,GO analysis revealed highly significant overrepresentation ofcorresponding GO molecular function terms in the resin-bound protein fraction (data not shown). Moreover, althoughprotein kinases accounted for just one third of all proteinidentifications, they were on average identified and quantifiedwith three times more peptides than non-protein kinases,indicating even higher purification efficiency as evident fromGO analysis.
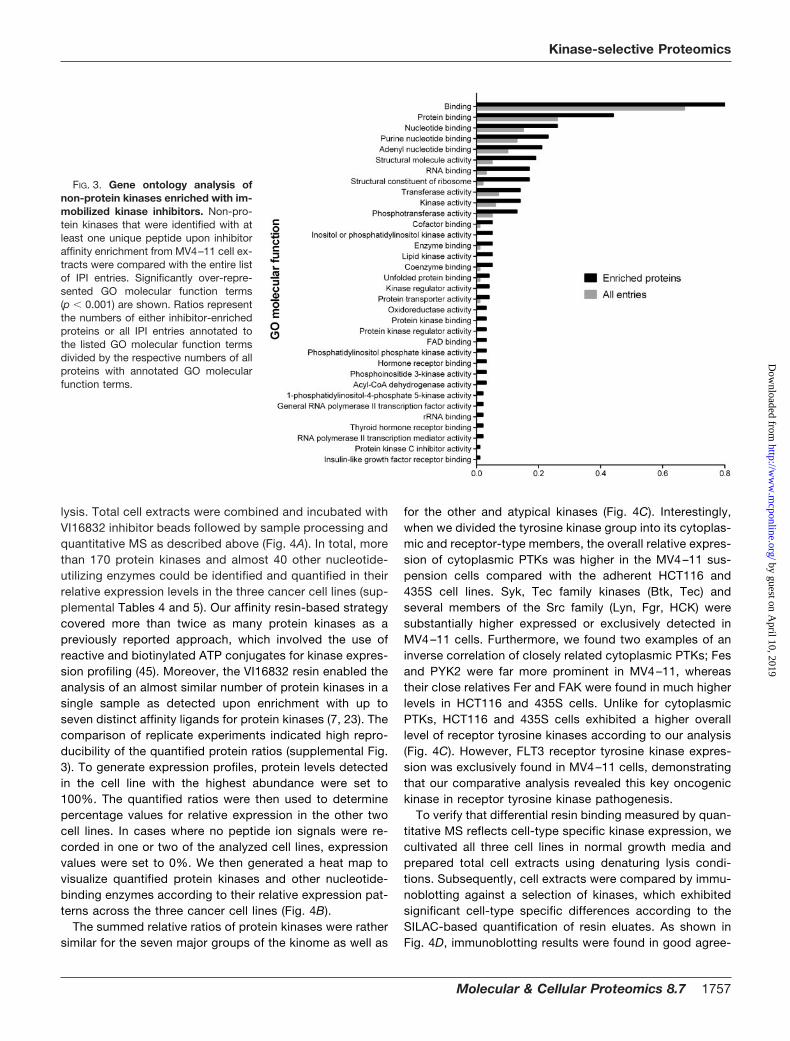
To further explore the binding characteristics of thepyrido[2,3-d]pyrimidine-based affinity ligands, we performeda GO analysis for over-represented molecular functions in thesubset of resin-bound proteins that were not protein kinases(Fig. 3 and supplemental Table 3). This assessment revealedconsiderable enrichment for nucleotide-dependent enzymessuch as oxidoreductases, dehydrogenases, and lipid kinases,which likely resulted from direct interactions with the purine-like pharmacophore provided by the immobilized inhibitors.Notably, important signaling factors were among these resin-bound enzymes such as phosphatidylinositol 3-kinase, phos-phatidylinositol-4,5-bisphosphate 3-kinase and others. GOanalysis of non-protein kinases further indicated over-repre-sentation of the molecular function “protein kinase binding”,which resulted from the specific co-purification of proteinkinase interactors such as various cyclins as well as otherregulatory kinase subunits.
Comparative Kinase Expression Analysis in Different Can-cer Cell Lines—The quantitative comparison of pyrido[2,3-d]pyrimidine resins demonstrated efficient kinome fraction-ation by the VI16743 and VI16832 resins, and we choseVI16832-based affinity enrichment to monitor cell-type spe-cific differences of kinase expression profiles. The identifi-cation of up-regulated kinases has considerable potential intumor cell analysis, given that over-expression of proteinkinases is frequently involved in carcinogenesis. To test thisexperimental strategy, three cancer cell lines (MV4–11 leu-kemia, HCT116 colon carcinoma, and 435S melanoma-de-rived cells) were differentially SILAC-encoded prior to cellFIG. 1. Comparison of inhibitor resins by quantitative chemical
proteomics. A, chemical structures of pyrido[2,3-d]pyrimidine-basedinhibitors used as affinity capture reagents for protein kinase enrich-ment. The amino groups (marked by arrows) were used for immobi-lization on Sepharose beads. B, SILAC scheme and workflow used forcomparative quantitative MS analysis of cellular proteins enriched bythe different inhibitor resins from MV4–11 cell extracts. C, aliquots of
the elution fractions from either kinase inhibitor resins or from acontrol resin (Ctrl) of ECH-Sepharose devoid of immobilized ligandwere analyzed by SDS-PAGE and silver staining.
Kinase-selective Proteomics
Molecular & Cellular Proteomics 8.7 1755
by guest on April 10, 2019
http://ww
w.m
cponline.org/D
ownloaded from
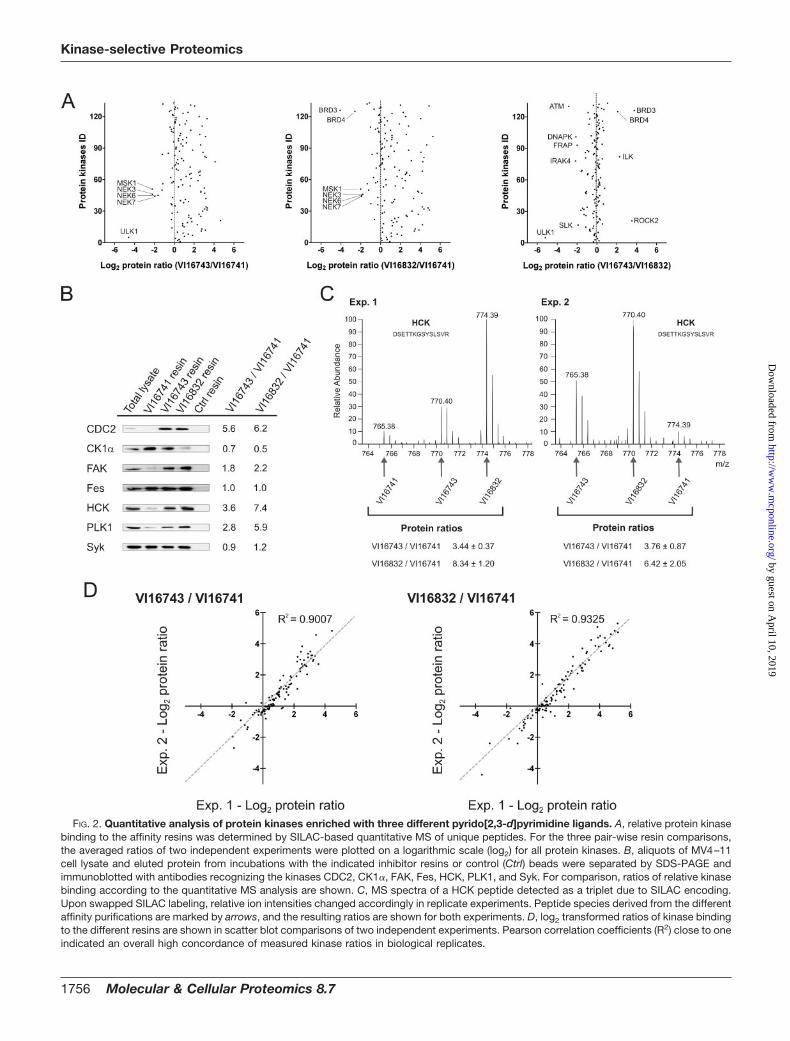
FIG. 2. Quantitative analysis of protein kinases enriched with three different pyrido[2,3-d]pyrimidine ligands. A, relative protein kinasebinding to the affinity resins was determined by SILAC-based quantitative MS of unique peptides. For the three pair-wise resin comparisons,the averaged ratios of two independent experiments were plotted on a logarithmic scale (log2) for all protein kinases. B, aliquots of MV4–11cell lysate and eluted protein from incubations with the indicated inhibitor resins or control (Ctrl) beads were separated by SDS-PAGE andimmunoblotted with antibodies recognizing the kinases CDC2, CK1�, FAK, Fes, HCK, PLK1, and Syk. For comparison, ratios of relative kinasebinding according to the quantitative MS analysis are shown. C, MS spectra of a HCK peptide detected as a triplet due to SILAC encoding.Upon swapped SILAC labeling, relative ion intensities changed accordingly in replicate experiments. Peptide species derived from the differentaffinity purifications are marked by arrows, and the resulting ratios are shown for both experiments. D, log2 transformed ratios of kinase bindingto the different resins are shown in scatter blot comparisons of two independent experiments. Pearson correlation coefficients (R2) close to oneindicated an overall high concordance of measured kinase ratios in biological replicates.
Kinase-selective Proteomics
1756 Molecular & Cellular Proteomics 8.7
by guest on April 10, 2019
http://ww
w.m
cponline.org/D
ownloaded from
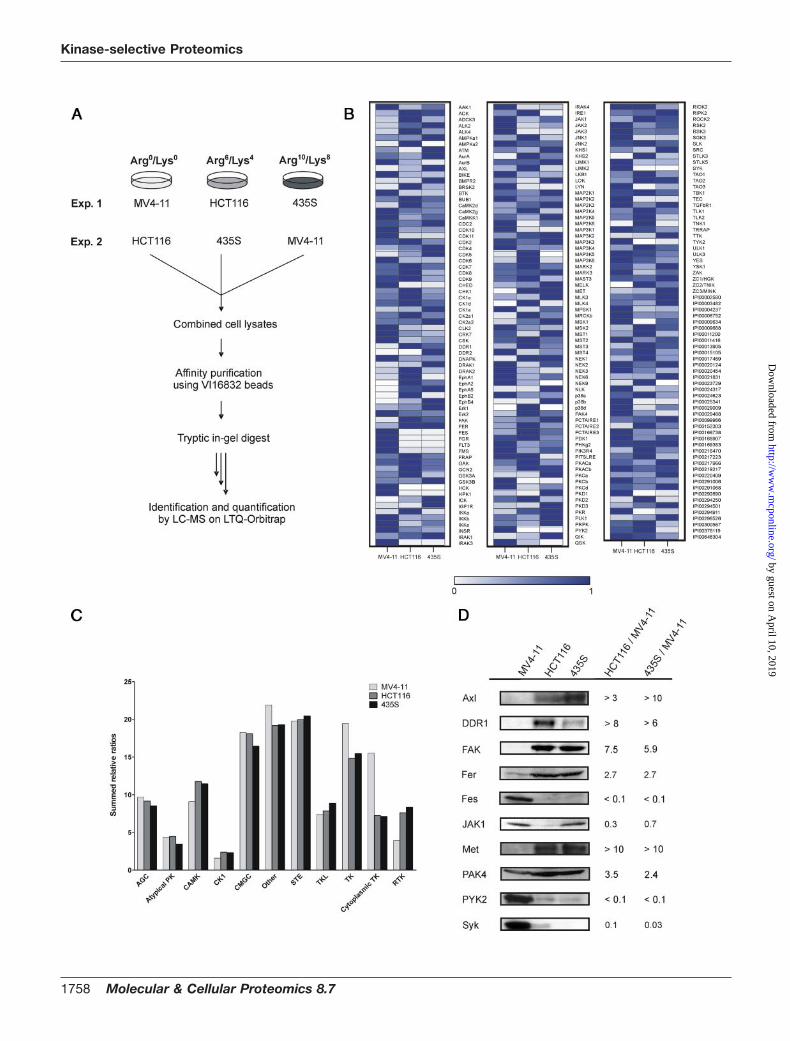
lysis. Total cell extracts were combined and incubated withVI16832 inhibitor beads followed by sample processing andquantitative MS as described above (Fig. 4A). In total, morethan 170 protein kinases and almost 40 other nucleotide-utilizing enzymes could be identified and quantified in theirrelative expression levels in the three cancer cell lines (sup-plemental Tables 4 and 5). Our affinity resin-based strategycovered more than twice as many protein kinases as apreviously reported approach, which involved the use ofreactive and biotinylated ATP conjugates for kinase expres-sion profiling (45). Moreover, the VI16832 resin enabled theanalysis of an almost similar number of protein kinases in asingle sample as detected upon enrichment with up toseven distinct affinity ligands for protein kinases (7, 23). Thecomparison of replicate experiments indicated high repro-ducibility of the quantified protein ratios (supplemental Fig.3). To generate expression profiles, protein levels detectedin the cell line with the highest abundance were set to100%. The quantified ratios were then used to determinepercentage values for relative expression in the other twocell lines. In cases where no peptide ion signals were re-corded in one or two of the analyzed cell lines, expressionvalues were set to 0%. We then generated a heat map tovisualize quantified protein kinases and other nucleotide-binding enzymes according to their relative expression pat-terns across the three cancer cell lines (Fig. 4B).
The summed relative ratios of protein kinases were rathersimilar for the seven major groups of the kinome as well as
for the other and atypical kinases (Fig. 4C). Interestingly,when we divided the tyrosine kinase group into its cytoplas-mic and receptor-type members, the overall relative expres-sion of cytoplasmic PTKs was higher in the MV4–11 sus-pension cells compared with the adherent HCT116 and435S cell lines. Syk, Tec family kinases (Btk, Tec) andseveral members of the Src family (Lyn, Fgr, HCK) weresubstantially higher expressed or exclusively detected inMV4–11 cells. Furthermore, we found two examples of aninverse correlation of closely related cytoplasmic PTKs; Fesand PYK2 were far more prominent in MV4–11, whereastheir close relatives Fer and FAK were found in much higherlevels in HCT116 and 435S cells. Unlike for cytoplasmicPTKs, HCT116 and 435S cells exhibited a higher overalllevel of receptor tyrosine kinases according to our analysis(Fig. 4C). However, FLT3 receptor tyrosine kinase expres-sion was exclusively found in MV4–11 cells, demonstratingthat our comparative analysis revealed this key oncogenickinase in receptor tyrosine kinase pathogenesis.
To verify that differential resin binding measured by quan-titative MS reflects cell-type specific kinase expression, wecultivated all three cell lines in normal growth media andprepared total cell extracts using denaturing lysis condi-tions. Subsequently, cell extracts were compared by immu-noblotting against a selection of kinases, which exhibitedsignificant cell-type specific differences according to theSILAC-based quantification of resin eluates. As shown inFig. 4D, immunoblotting results were found in good agree-
FIG. 3. Gene ontology analysis ofnon-protein kinases enriched with im-mobilized kinase inhibitors. Non-pro-tein kinases that were identified with atleast one unique peptide upon inhibitoraffinity enrichment from MV4–11 cell ex-tracts were compared with the entire listof IPI entries. Significantly over-repre-sented GO molecular function terms(p � 0.001) are shown. Ratios representthe numbers of either inhibitor-enrichedproteins or all IPI entries annotated tothe listed GO molecular function termsdivided by the respective numbers of allproteins with annotated GO molecularfunction terms.
Kinase-selective Proteomics
Molecular & Cellular Proteomics 8.7 1757
by guest on April 10, 2019
http://ww
w.m
cponline.org/D
ownloaded from
Kinase-selective Proteomics
1758 Molecular & Cellular Proteomics 8.7
by guest on April 10, 2019
http://ww
w.m
cponline.org/D
ownloaded from

ment with the measured SILAC ratios. This indicated thatother potential sources of variation that could affect theaffinity purification approach, such as cell-type specific ex-pression changes upon SILAC or differential solubilizationof kinases due to non-denaturing cell lysis, apparently hadno major influence on the quantitative cell line comparisons.
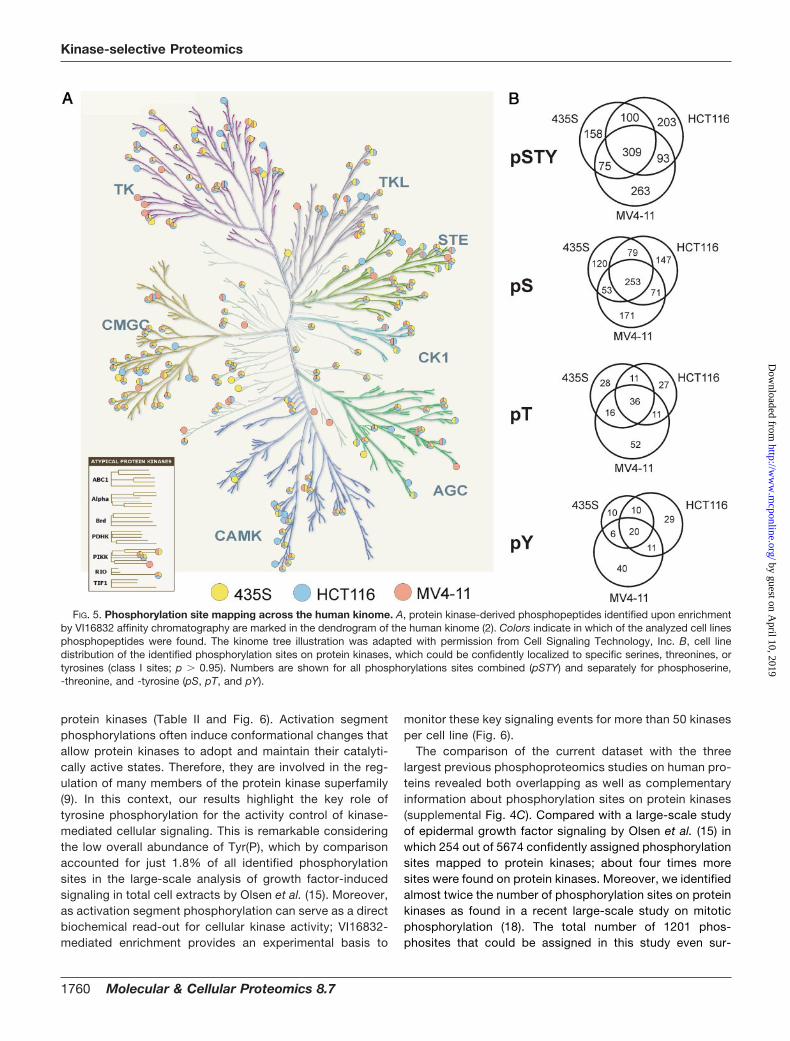
Large-scale Phosphoproteomics Analysis of Cancer CellLines upon Kinase Affinity Enrichment—The efficient pre-frac-tionation of protein kinases from total cell extracts provides anexperimental basis for the analysis of post-translational mod-ifications with high analytical sensitivity. To exploit this poten-tial for phosphoproteomics analysis, we employed theVI16832 resin for protein kinase enrichments from MV4–11,HCT116, and 435S cell lysates. In contrast to the batch puri-fication protocol described above, a column chromatographyset-up was employed to enable processing of larger amountsof starting material and thereby enhance the sensitivity ofphosphopeptide detection. Affinity-purified proteins from thedifferent cancer cell lysates were separated by gel electro-phoresis prior to in-gel digestion with trypsin and subsequentphosphopeptide enrichment with TiO2 microspheres (31, 32).Raw data from the LC-MS analyses were filtered for phos-phopeptide identifications within a false-discovery rate of lessthan 1% for each individual experiment. Phosphoproteomicsanalysis of VI16832-enriched fractions from MV4–11,HCT116, or 435S cells resulted in more than 8500 phos-phopeptide identifications. These translated into almost 1700distinct phosphopeptide species derived from 212 differentmembers of the protein kinase superfamily. We further iden-tified more than 1300 distinct phosphopeptides on 563 non-protein kinases (Table I). Notably, about 30% of the proteinkinases and 50% of the other proteins were not detected inour previous analysis of kinase-enriched fractions from HeLa
S3 cells. Using computational PTM scoring, more than 1200phosphorylation sites on kinases and 900 on other proteinscould be localized with high confidence (Table I and supple-mental Table 6). All identified phosphopeptides can be ac-cessed through the Phosida database, which also provideslinks to annotated fragmentation spectra harboring the iden-tified phosphopeptides (35). The more than 200 identifiedprotein kinases were rather evenly distributed in the dendro-gram of the human kinome (Fig. 5A). This indicated thatVI16832 did not select for sequence-related determinantsrestricted to certain subsets of the kinome and further high-lighted the utility of this reagent as broadly kinase-selectiveenrichment tool for sensitive PTM analysis. The Venn dia-grams show the cell line distribution of the identified phos-phorylation sites on protein kinases. Importantly, the anal-ysis of VI16832-retained proteins from the three cancer celllines considerably increased the overall number of identifiedphosphorylation sites on protein kinases (Fig. 5B). However,as kinase-enriched fractions from the different cell extractswere subjected to individual, qualitative phosphopeptidemapping experiments, selective identifications did not nec-essarily indicate cell-type specific differences but could alsobe due to run-to-run variability inherent to LC-MS in thedata-dependent acquisition mode. Phosphorylation siteswere also identified on various nucleotide-binding enzymesas well as other proteins including regulatory subunits ofprotein kinases (Table I and supplemental Fig. 4A and sup-plemental Table 6). Although the retained non-kinase phos-phoproteins accounted for 60–70% of all identifications,they were typically found with fewer phosphopeptides perprotein. Moreover, on average, peptides from these proteinsexhibited considerably lower signal intensities than proteinkinase-derived peptides. Considering the sum of all phos-phopeptide intensities as a measure for VI16832-enrichedprotein amount, more than 80% was derived from proteinkinases. This value demonstrates the remarkable kinaseselectivity of the phosphoproteomics workflow presentedhere (supplemental Fig. 4B).
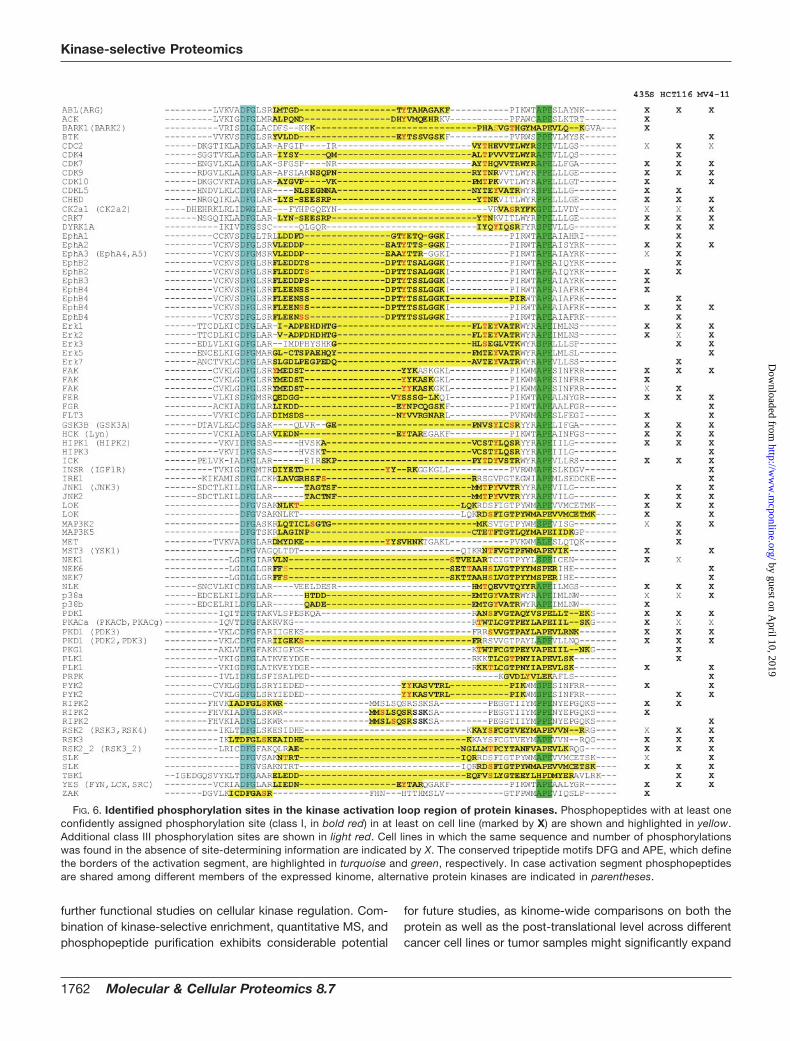
When we analyzed the distribution of phosphoserine,-threonine, and -tyrosine sites, we found tyrosine phospho-rylation to be more frequent on protein kinases than onother identified proteins (Table II). Even more intriguingly,tyrosine phosphorylation accounted for more than one thirdof the identified sites in the activation segment regions of
FIG. 4. Comparative kinase expression analysis across different cancer cell lines. A, scheme illustrating the SILAC-based chemicalproteomics workflow for quantitative comparison of VI16832-interacting sub-proteomes in three cancer cell lines. B, quantified ratios forprotein kinases and other nucleotide-binding proteins were transformed into relative expression levels with the highest expression in anyof the three cell lines set to 1. Based on these values, a heat map was generated to visualize relative expression levels across the analyzedcancer cell lines. C, summed relative expression values for the seven major kinase groups as well as the atypical and other kinasesaccording to Manning et al. (2) were compared for MV4–11, HCT116, and 435S cells. In addition, members of the tyrosine kinase (TK)group were subdivided into cytoplasmic and receptor tyrosine kinase in the cell line comparison. D, total cell lysates of MV4–11, HCT116,and 435S cells were immunoblotted with antibodies recognizing the kinases Axl, DDR1, FAK, Fer, Fes, JAK1, Met, PAK4, PYK2, and Syk.For comparison, binding ratios according to the SILAC-based quantification of VI16832 resin-bound protein kinases are shown.
TABLE IOverview of results from phosphokinome analysis
Numbers of identified phosphoproteins, phosphopeptide se-quences, phosphopeptides, and confidently assigned phosphoryla-tion sites (p � 0.95) are shown either for protein kinases or for all otherproteins analyzed upon VI16832 affinity chromatography.
Protein kinases Other proteins
Phosphoproteins 212 563Phosphopeptide sequences 1431 1224Phosphopeptides 1695 1329Phosphosites (class I) 1201 904
Kinase-selective Proteomics
Molecular & Cellular Proteomics 8.7 1759
by guest on April 10, 2019
http://ww
w.m
cponline.org/D
ownloaded from
protein kinases (Table II and Fig. 6). Activation segmentphosphorylations often induce conformational changes thatallow protein kinases to adopt and maintain their catalyti-cally active states. Therefore, they are involved in the reg-ulation of many members of the protein kinase superfamily(9). In this context, our results highlight the key role oftyrosine phosphorylation for the activity control of kinase-mediated cellular signaling. This is remarkable consideringthe low overall abundance of Tyr(P), which by comparisonaccounted for just 1.8% of all identified phosphorylationsites in the large-scale analysis of growth factor-inducedsignaling in total cell extracts by Olsen et al. (15). Moreover,as activation segment phosphorylation can serve as a directbiochemical read-out for cellular kinase activity; VI16832-mediated enrichment provides an experimental basis to
monitor these key signaling events for more than 50 kinasesper cell line (Fig. 6).
The comparison of the current dataset with the threelargest previous phosphoproteomics studies on human pro-teins revealed both overlapping as well as complementaryinformation about phosphorylation sites on protein kinases(supplemental Fig. 4C). Compared with a large-scale studyof epidermal growth factor signaling by Olsen et al. (15) inwhich 254 out of 5674 confidently assigned phosphorylationsites mapped to protein kinases; about four times moresites were found on protein kinases. Moreover, we identifiedalmost twice the number of phosphorylation sites on proteinkinases as found in a recent large-scale study on mitoticphosphorylation (18). The total number of 1201 phos-phosites that could be assigned in this study even sur-
FIG. 5. Phosphorylation site mapping across the human kinome. A, protein kinase-derived phosphopeptides identified upon enrichmentby VI16832 affinity chromatography are marked in the dendrogram of the human kinome (2). Colors indicate in which of the analyzed cell linesphosphopeptides were found. The kinome tree illustration was adapted with permission from Cell Signaling Technology, Inc. B, cell linedistribution of the identified phosphorylation sites on protein kinases, which could be confidently localized to specific serines, threonines, ortyrosines (class I sites; p � 0.95). Numbers are shown for all phosphorylations sites combined (pSTY) and separately for phosphoserine,-threonine, and -tyrosine (pS, pT, and pY).
Kinase-selective Proteomics
1760 Molecular & Cellular Proteomics 8.7
by guest on April 10, 2019
http://ww
w.m
cponline.org/D
ownloaded from

passed the 1007 confidently localized phosphorylationevents reported in our previous analysis of cell cycle-regu-lated changes in kinase-enriched fractions (7). Moreover,despite an overlap of 555 phosphosites between these twostudies, we find almost 650 additional site-specific phos-phorylations on protein kinases. In contrast to these earlierquantitative studies using more complex, SILAC-encodedsamples, the analyses in our current study were done in aqualitative manner with the goal to promote comprehensivephosphorylation site mapping, which, to the best of ourknowledge, resulted in the most extensive phosphopro-teomics analysis of protein kinases reported to date.
DISCUSSION
In our present study, we immobilized kinase inhibitorsfrom the pyrido[2,3-d]pyrimidine class of compounds togenerate affinity resins for the pre-fractionation of proteinkinase-enriched sub-proteomes (25). Cellular target capturewas compared for three immobilized pyrido[2,3-d]pyrimi-dine derivatives using SILAC-based quantitative MS (41).The VI16743 and VI16832 resins were particularly efficientas purification tools, as these affinity resins were capable ofretaining more than 130 distinct protein kinases from asingle cell extract. Thus, they represent straightforward andeasy-to-use purification reagents for a considerable subsetof the expressed human kinome. Due to a lack of affineantibodies, many protein kinases are difficult to study byconventional immunoprecipitation approaches. In suchcases, small molecule-based isolation can provide astraightforward alternative for targeted analysis; for exam-ple when sample processing and MS analysis is restricted tothe molecular weight region comprising the kinase-of-interest.Thus, the datasets reported here specify a large number ofprotein kinases amenable for focused signal transduction
analysis, which might involve quantitative MS to monitor PTMregulation upon different types of cell treatment.
The quantitative MS strategy for the comparison ofpyrido[2,3-d]pyrimidine derivatives represents a generic ap-proach. It can easily be extended to characterize other ki-nase-selective capture molecules, which retain subsets of theexpressed kinome not efficiently purified by the VI16743 andVI16832 resins. Such comparative analyses should be usefulto further improve on previously described multi-resin ap-proaches, which combine immobilized kinase inhibitors withdistinct target profiles for maximal coverage of the expressedkinome (7, 22, 23).
Kinase-selective proteomics focuses on a subset of theproteome, which is of high relevance for targeted therapeuticintervention in diseases such as human cancer. Our compar-ative analysis of three different cancer cell lines demonstratesquantitative profiling of kinase expression in a compact ex-perimental format. Although only three cell lines can be com-pared by SILAC in a single experiment, further multiplexing ispossible by merging data from parallel triple-labeling experi-ments through a shared reference sample (15). SILAC-basedkinase profiling across larger collections of cancer cell linescould reveal subgroup-specific expression patterns, whichmight help to adjust targeted therapeutic interventions to thekinases involved in disease progression. Conceptually similarstrategies could also be applied to the analysis of proteinkinase-enriched fractions from primary tumor specimens; forexample by employing chemical tagging with iTRAQ reagentsas an alternative to SILAC-based quantification. In a recentlypublished large-scale study by Rikova et al. (8), non-small celllung cancer cell lines and tumors were comparatively ana-lyzed for their phosphotyrosine-containing proteomes. Re-markably, this survey allowed clustering of the analyzed sam-ples into different groups with distinct tyrosine kinasepatterns. Compared with phosphotyrosine-directed ap-proaches, proteomics of kinase-enriched sub-proteomes canbe expected to provide both overlapping information (withrespect to tyrosine kinases) as well as complementary dataregarding serine/threonine kinases (and also other nucleotide-binding proteins). Protein expression data from the serine/threonine kinase branches of the human kinome likely revealfurther insights into cancer cell biology, as exemplified byfindings that overexpression of mitotic serine/threonine ki-nases, such as Aurora A and B, polo-like kinase1 and NEK2,can result in chromosomal instability and has been implicatedin malignant transformation (46, 47). The pyrido[2,3-d]pyrimi-dine inhibitor resins described in this study are particularlyuseful for such sub-proteome surveys due to their ability tocapture these key mitotic enzymes as well as many otheradditional serine/threonine kinases. Here, we have furtherexploited the enrichment abilities of the pyrido[2,3-d]pyrimi-dine-based capture ligand VI16832 to extensively map phos-phorylation sites on protein kinases. Results from these large-scale analyses provide a multitude of new starting points for
TABLE IIDistribution of Ser(P), Thr(P), and Tyr(P)
Distributions of phosphorylation sites by amino acid are shownfor affinity purified fractions from 435S, HCT116, and MV4–11 cells.Percentages are shown for protein kinases, for the activation loopregions of protein kinases, and for all other phosphoproteins.
Ser(P) Thr(P) Tyr(P)
% % %
Protein kinases435S 78.5 14.1 7.4HCT116 77.7 12.1 10.2MV4–11 73.9 15.5 10.6
PK activation loop435S 32.2 28.8 39.0HCT116 28.6 27.0 44.4MV4–11 34.4 28.1 37.5
Other phosphoproteins435S 88.8 9.9 1.3HCT116 90.1 9.5 1.4MV4–11 86.3 9.2 4.5
Kinase-selective Proteomics
Molecular & Cellular Proteomics 8.7 1761
by guest on April 10, 2019
http://ww
w.m
cponline.org/D
ownloaded from
further functional studies on cellular kinase regulation. Com-bination of kinase-selective enrichment, quantitative MS, andphosphopeptide purification exhibits considerable potential
for future studies, as kinome-wide comparisons on both theprotein as well as the post-translational level across differentcancer cell lines or tumor samples might significantly expand
FIG. 6. Identified phosphorylation sites in the kinase activation loop region of protein kinases. Phosphopeptides with at least oneconfidently assigned phosphorylation site (class I, in bold red) in at least on cell line (marked by X) are shown and highlighted in yellow.Additional class III phosphorylation sites are shown in light red. Cell lines in which the same sequence and number of phosphorylationswas found in the absence of site-determining information are indicated by X. The conserved tripeptide motifs DFG and APE, which definethe borders of the activation segment, are highlighted in turquoise and green, respectively. In case activation segment phosphopeptidesare shared among different members of the expressed kinome, alternative protein kinases are indicated in parentheses.
Kinase-selective Proteomics
1762 Molecular & Cellular Proteomics 8.7
by guest on April 10, 2019
http://ww
w.m
cponline.org/D
ownloaded from

our knowledge about kinase drug targets and their oncogenicactivities on a system-wide level.
Acknowledgments—We thank Axel Ullrich for his generous supportof the present study with funding from the Department of MolecularBiology, Max Planck Institute of Biochemistry. The Fer-specific anti-body was kindly provided by Peter A. Greer. The modified kinomedendrogram shown in Fig. 5A was reproduced with permission of CellSignaling Technology, Inc.).
□S The on-line version of this article (available at http://www.mcp.org) contains supplemental data.
** To whom correspondence should be addressed: E-mail:[email protected]. Tel.: 49-89-8578-3773; Fax: 49-89-8578-2454.
REFERENCES
1. Ubersax, J. A., and Ferrell, J. E., Jr. (2007) Mechanisms of specificity inprotein phosphorylation. Nat. Rev. Mol. Cell Biol. 8, 530–541
2. Manning, G., Whyte, D. B., Martinez, R., Hunter, T., and Sudarsanam, S.(2002) The protein kinase complement of the human genome. Science298, 1912–1934
3. Blume-Jensen, P., and Hunter, T. (2001) Oncogenic kinase signaling. Na-ture 411, 355–365
4. Strebhardt, K., and Ullrich, A. (2008) Paul Ehrlich’s magic bullet concept:100 years of progress. Nat. Rev. Cancer 8, 473–480
5. Krause, D. S., and Van Etten, R. A. (2005) Tyrosine kinases as targets forcancer therapy. N. Engl. J. Med. 353, 172–187
6. Faivre, S., Demetri, G., Sargent, W., and Raymond, E. (2007) Molecularbasis for sunitinib efficacy and future clinical development. Nat. Rev.Drug Discov. 6, 734–745
7. Daub, H., Olsen, J. V., Bairlein, M., Gnad, F., Oppermann, F. S., Korner, R.,Greff, Z., Keri, G., Stemmann, O., and Mann, M. (2008) Kinase-selectiveenrichment enables quantitative phosphoproteomics of the kinomeacross the cell cycle. Mol. Cell 31, 438–448
8. Rikova, K., Guo, A., Zeng, Q., Possemato, A., Yu, J., Haack, H., Nardone,J., Lee, K., Reeves, C., Li, Y., Hu, Y., Tan, Z., Stokes, M., Sullivan, L.,Mitchell, J., Wetzel, R., Macneill, J., Ren, J. M., Yuan, J., Bakalarski,C. E., Villen, J., Kornhauser, J. M., Smith, B., Li, D., Zhou, X., Gygi, S. P.,Gu, T. L., Polakiewicz, R. D., Rush, J., and Comb, M. J. (2007) Globalsurvey of phosphotyrosine signaling identifies oncogenic kinases in lungcancer. Cell 131, 1190–1203
9. Nolen, B., Taylor, S., and Ghosh, G. (2004) Regulation of protein kinases;controlling activity through activation segment conformation. Mol. Cell15, 661–675
10. Steen, H., Jebanathirajah, J. A., Rush, J., Morrice, N., and Kirschner, M. W.(2006) Phosphorylation analysis by mass spectrometry: myths, facts,and the consequences for qualitative and quantitative measurements.Mol. Cell. Proteomics 5, 172–181
11. Collins, M. O., Yu, L., and Choudhary, J. S. (2007) Analysis of proteinphosphorylation on a proteome-scale. Proteomics 7, 2751–2768
12. Schreiber, T. B., Mausbacher, N., Breitkopf, S. B., Grundner-Culemann, K.,and Daub, H. (2008) Quantitative phosphoproteomics - an emerging keytechnology in signal-transduction research. Proteomics 8, 4416–4432
13. Macek, B., Mann, M., and Olsen, J. V. (2009) Global and site-specificquantitative phosphoproteomics: principles and applications. Annu. Rev.Pharmacol. Toxicol. 49, 199–221
14. Li, X., Gerber, S. A., Rudner, A. D., Beausoleil, S. A., Haas, W., Villen, J.,Elias, J. E., and Gygi, S. P. (2007) Large-scale phosphorylation analysisof alpha-factor-arrested Saccharomyces cerevisiae. J. Proteome Res. 6,1190–1197
15. Olsen, J. V., Blagoev, B., Gnad, F., Macek, B., Kumar, C., Mortensen, P.,and Mann, M. (2006) Global, in vivo, and site-specific phosphorylationdynamics in signaling networks. Cell 127, 635–648
16. Beausoleil, S. A., Jedrychowski, M., Schwartz, D., Elias, J. E., Villen, J., Li,J., Cohn, M. A., Cantley, L. C., and Gygi, S. P. (2004) Large-scalecharacterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad.Sci. U.S.A. 101, 12130–12135
17. Villen, J., Beausoleil, S. A., Gerber, S. A., and Gygi, S. P. (2007) Large-scalephosphorylation analysis of mouse liver. Proc. Natl. Acad. Sci. U.S.A.
104, 1488–149318. Dephoure, N., Zhou, C., Villen, J., Beausoleil, S. A., Bakalarski, C. E.,
Elledge, S. J., and Gygi, S. P. (2008) A quantitative atlas of mitoticphosphorylation. Proc. Natl. Acad. Sci. U.S.A. 105, 10762–10767
19. Daub, H. (2005) Characterization of kinase-selective inhibitors by chemicalproteomics. Biochim. Biophys. Acta 1754, 183–190
20. Godl, K., Wissing, J., Kurtenbach, A., Habenberger, P., Blencke, S., Gut-brod, H., Salassidis, K., Stein-Gerlach, M., Missio, A., Cotten, M., andDaub, H. (2003) An efficient proteomics method to identify the cellulartargets of protein kinase inhibitors. Proc. Natl. Acad. Sci. U.S.A. 100,15434–15439
21. Rix, U., Hantschel, O., Durnberger, G., Remsing Rix, L. L., Planyavsky, M.,Fernbach, N. V., Kaupe, I., Bennett, K. L., Valent, P., Colinge, J., Kocher,T., and Superti-Furga, G. (2007) Chemical proteomic profiles of theBCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinaseand nonkinase targets. Blood 110, 4055–4063
22. Wissing, J., Jansch, L., Nimtz, M., Dieterich, G., Hornberger, R., Keri, G.,Wehland, J., and Daub, H. (2007) Proteomics analysis of protein kinasesby target class-selective prefractionation and tandem mass spectrome-try. Mol. Cell. Proteomics 6, 537–547
23. Bantscheff, M., Eberhard, D., Abraham, Y., Bastuck, S., Boesche, M.,Hobson, S., Mathieson, T., Perrin, J., Raida, M., Rau, C., Reader, V.,Sweetman, G., Bauer, A., Bouwmeester, T., Hopf, C., Kruse, U.,Neubauer, G., Ramsden, N., Rick, J., Kuster, B., and Drewes, G. (2007)Quantitative chemical proteomics reveals mechanisms of action of clin-ical ABL kinase inhibitors. Nat. Biotechnol. 25, 1035–1044
24. Rae, J. M., Creighton, C. J., Meck, J. M., Haddad, B. R., and Johnson, M. D.(2007) MDA-MB-435 cells are derived from M14 melanoma cells–a lossfor breast cancer, but a boon for melanoma research. Breast Cancer Res.Treat. 104, 13–19
25. Barvian, M., Boschelli, D. H., Cossrow, J., Dobrusin, E., Fattaey, A., Fritsch,A., Fry, D., Harvey, P., Keller, P., Garrett, M., La, F., Leopold, W.,McNamara, D., Quin, M., Trumpp-Kallmeyer, S., Toogood, P., Wu, Z.,and Zhang, E. (2000) Pyrido[2,3-d]pyrimidin-7-one inhibitors of cyclin-dependent kinases. J. Med. Chem. 43, 4606–4616
26. Brehmer, D., Greff, Z., Godl, K., Blencke, S., Kurtenbach, A., Weber, M.,Muller, S., Klebl, B., Cotten, M., Keri, G., Wissing, J., and Daub, H. (2005)Cellular targets of gefitinib. Cancer Res. 65, 379–382
27. Senis, Y. A., Craig, A. W., and Greer, P. A. (2003) Fps/Fes and Fer protein-tyrosinekinases play redundant roles in regulating hematopoiesis. Exp.Hematol. 31, 673–681
28. Wessel, D., and Flugge, U. I. (1984) A method for the quantitative recoveryof protein in dilute solution in the presence of detergents and lipids. Anal.Biochem. 138, 141–143
29. Shevchenko, A., Tomas, H., Havlis, J., Olsen, J. V., and Mann, M. (2006)In-gel digestion for mass spectrometric characterization of proteins andproteomes. Nat. Protoc. 1, 2856–2860
30. Rappsilber, J., Mann, M., and Ishihama, Y. (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides forproteomics using StageTips. Nat. Protoc. 2, 1896–1906
31. Larsen, M. R., Thingholm, T. E., Jensen, O. N., Roepstorff, P., and Jør-gensen, T. J. (2005) Highly selective enrichment of phosphorylated pep-tides from peptide mixtures using titanium dioxide microcolumns. Mol.Cell. Proteomics 4, 873–886
32. Pinkse, M. W., Uitto, P. M., Hilhorst, M. J., Ooms, B., and Heck, A. J. (2004)Selective isolation at the femtomole level of phosphopeptides from pro-teolytic digests using 2D-NanoLC-ESI-MS/MS and titanium oxide pre-columns. Anal. Chem. 76, 3935–3943
33. Schroeder, M. J., Shabanowitz, J., Schwartz, J. C., Hunt, D. F., and Coon,J. J. (2004) A neutral loss activation method for improved phosphopep-tide sequence analysis by quadrupole ion trap mass spectrometry. Anal.Chem. 76, 3590–3598
34. Olsen, J. V., de Godoy, L. M., Li, G., Macek, B., Mortensen, P., Pesch, R.,Makarov, A., Lange, O., Horning, S., and Mann, M. (2005) Parts permillion mass accuracy on an Orbitrap mass spectrometer via lock massinjection into a C-trap. Mol. Cell. Proteomics 4, 2010–2021
35. Gnad, F., Ren, S., Cox, J., Olsen, J. V., Macek, B., Oroshi, M., and Mann,M. (2007) PHOSIDA (phosphorylation site database): management,structural and evolutionary investigation, and prediction of phosphosites.Genome Biol. 8, R250
36. Maere, S., Heymans, K., and Kuiper, M. (2005) BiNGO: a Cytoscape plugin
Kinase-selective Proteomics
Molecular & Cellular Proteomics 8.7 1763
by guest on April 10, 2019
http://ww
w.m
cponline.org/D
ownloaded from

to assess overrepresentation of gene ontology categories in biologicalnetworks. Bioinformatics 21, 3448–3449
37. Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D.,Amin, N., Schwikowski, B., and Ideker, T. (2003) Cytoscape: a softwareenvironment for integrated models of biomolecular interaction networks.Genome Res. 13, 2498–2504
38. Adachi, J., Kumar, C., Zhang, Y., Olsen, J. V., and Mann, M. (2006) Thehuman urinary proteome contains more than 1500 proteins, including alarge proportion of membrane proteins. Genome Biol. 7, R80
39. Wissing, J., Godl, K., Brehmer, D., Blencke, S., Weber, M., Habenberger,P., Stein-Gerlach, M., Missio, A., Cotten, M., Muller, S., and Daub, H.(2004) Chemical proteomic analysis reveals alternative modes of actionfor pyrido[2,3-d]pyrimidine kinase inhibitors. Mol. Cell. Proteomics 3,1181–1193
40. Blencke, S., Zech, B., Engkvist, O., Greff, Z., Orfi, L., Horvath, Z., Keri, G.,Ullrich, A., and Daub, H. (2004) Characterization of a conserved struc-tural determinant controlling protein kinase sensitivity to selective inhib-itors. Chem. Biol. 11, 691–701
41. Mann, M. (2006) Functional and quantitative proteomics using SILAC. Nat.Rev. Mol. Cell Biol. 7, 952–958
42. Gilliland, D. G., and Griffin, J. D. (2002) The roles of FLT3 in hematopoiesisand leukemia. Blood 100, 1532–1542
43. Su, A. I., Cooke, M. P., Ching, K. A., Hakak, Y., Walker, J. R., Wiltshire, T.,Orth, A. P., Vega, R. G., Sapinoso, L. M., Moqrich, A., Patapoutian, A.,Hampton, G. M., Schultz, P. G., and Hogenesch, J. B. (2002) Large-scaleanalysis of the human and mouse transcriptomes. Proc. Natl. Acad. Sci.U.S.A. 99, 4465–4470
44. Zhang, Y. X., Knyazev, P. G., Cheburkin, Y. V., Sharma, K., Knyazev, Y. P.,Orfi, L., Szabadkai, I., Daub, H., Keri, G., and Ullrich, A. (2008) AXL is apotential target for therapeutic intervention in breast cancer progression.Cancer Res. 68, 1905–1915
45. Patricelli, M. P., Szardenings, A. K., Liyanage, M., Nomanbhoy, T. K., Wu,M., Weissig, H., Aban, A., Chun, D., Tanner, S., and Kozarich, J. W.(2007) Functional interrogation of the kinome using nucleotide acyl phos-phates. Biochemistry 46, 350–358
46. Li, J. J., and Li, S. A. (2006) Mitotic kinases: the key to duplication,segregation, and cytokinesis errors, chromosomal instability, and onco-genesis. Pharmacol. Ther. 111, 974–984
47. Strebhardt, K., and Ullrich, A. (2006) Targeting polo-like kinase 1 for cancertherapy. Nat. Rev. Cancer 6, 321–330
Kinase-selective Proteomics
1764 Molecular & Cellular Proteomics 8.7
by guest on April 10, 2019
http://ww
w.m
cponline.org/D
ownloaded from