laura j. baughman ca state bar no. 263944 martin
TRANSCRIPT

Complaint – Page 1 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
Laura J. Baughman CA State Bar No. 263944 MARTIN | BAUGHMAN, PLLC 3141 Hood Street, Suite 600 Dallas, Texas 75219 Tel. 214-761-6614 Fax. 214-744-7590 Email: [email protected]
- and -
Ellen A. Presby Texas Bar No. 16249600 (Pro Hac Vice To Be Filed) Van Wey Law, PLLC 12720 Hillcrest Road, Suite 600 Dallas, Texas 75230 Tel. 214- 329-1350 Fax: 800-582-1042 Email: [email protected]
Attorneys for Plaintiff Nancy Kilmer
UNITED STATES DISTRICT COURT FOR THE EASTERN DISTRICT OF CALIFORNIA
FRESNO DIVISION
NANCY KILMER,
Plaintiff,
v.
MEDTRONIC, INC.; MEDTRONIC USA, INC.; MEDTRONIC LOGISTICS, LLC; and MEDTRONIC PUERTO RICO OPERATIONS CO.,
Defendants.
CIVIL ACTION NO:
COMPLAINT JURY TRIAL DEMANDED
COMES NOW Plaintiff Nancy Kilmer, by and through her undersigned attorneys, and files
this Complaint against Medtronic, Inc.; Medtronic USA, Inc.; Medtronic Logistics, LLC; and
Medtronic Puerto Rico Operations Co., and alleges as follows:
I. Jurisdiction.
1. This Court has personal jurisdiction over all Defendants pursuant to Cal. Civ. Proc.
Code § 410.10, under which a court in California may exercise jurisdiction on any basis not
1:20-at-00675
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 1 of 42

Complaint – Page 2 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
inconsistent with the Constitution of California or of the United States. Exercising jurisdiction
over Defendants is not inconsistent with the Constitution of California or of the United States,
because Defendants are, and all relevant times were, involved in the design, assembly,
manufacture, testing, packaging, labeling, marketing, distribution, sale, and/or promotion of,
and/or were otherwise involved in the placing in the stream of commerce, medical devices
including the SynchroMed II Programmable Implantable Infusion Pump System (hereinafter the
“SynchroMed II Device” or “Device”), and thus transacted business within California; committed
torts within California as pled herein; and/or committed torts outside of California as pled herein
while regularly doing and/or soliciting business in California and/or deriving substantial revenue
from interstate commerce within California, through their substantial and purposeful transactions
of business there, including but not limited to their sales of the SynchroMed II Device, for which
Defendants should reasonably expect their acts to have consequences in California.
2. This Court has diversity subject-matter jurisdiction over this action pursuant to 28
U.S.C. § 1332(a) because this is a civil action in which the matter in controversy exceeds the sum
or value of $75,000, exclusive of interests and costs, and is between citizens of different states as
well as between a citizen of a state and a citizen of a foreign state.
3. The Court also has supplemental jurisdiction pursuant to 28 U.S.C. § 1367(a).
4. Venue is proper in this Court pursuant to 28 U.S.C. § 1391(b)(2), because the
injuries giving rise to this action were suffered in this judicial district, which encompasses Fresno
County, of which Plaintiff is a resident.
II. Introduction.
5. This is a products liability action seeking damages for personal injuries sustained
by Nancy Kilmer arising from her use of a defective product designed, manufactured, labeled,
distributed, and/or otherwise placed into the stream of commerce by Defendants and/or each of
them. As set forth herein, Ms. Kilmer suffered severe injuries and hospitalization as a direct and
proximate result of defects in her Medtronic SynchroMed II Programmable Implantable Infusion
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 2 of 42

Complaint – Page 3 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
Pump System, which was implanted in her body for intrathecal drug delivery. Ms. Kilmer brings
this action to recover for the damages caused by Defendants’ conduct.
III. Parties.
6. Plaintiff Nancy Kilmer is, and at all relevant times was, a citizen of California and
resident of Fresno, Fresno County, California.
7. Defendant Medtronic, Inc. is, and at all relevant times was, a corporation or other
business entity and citizen of Minnesota, with its principal place of business at 710 Medtronic
Parkway, Minneapolis, Anoka County, Minnesota 55432.
8. Defendant Medtronic USA, Inc. is, and at all relevant times was, a corporation or
other business entity and citizen of Minnesota, with its principal place of business at 710 Medtronic
Parkway, Minneapolis, Anoka County, Minnesota 55432.
9. Defendant Medtronic Logistics, LLC is, and at all relevant times was, a limited
liability company organized under the laws of Minnesota with its principal place of business at
710 Medtronic Parkway, Minneapolis, Anoka County, Minnesota 55432. The sole member of
Medtronic Logistics, LLC is, and at all relevant times was, Medtronic USA, Inc., a corporation or
other business entity and citizen of Minnesota, with its principal place of business at 710 Medtronic
Parkway, Minneapolis, Anoka County, Minnesota 55432.
10. Defendant Medtronic Puerto Rico Operations Co. is, and at all relevant times was,
a corporation or other business entity and a wholly owned subsidiary of Defendant Medtronic,
Inc., and citizen of the Cayman Islands, with its principal place of business at Ceiba Norte
Industrial Park Road 31, Km. 24, HM 4 Call Box 4070, Juncos 00777-4070, Puerto Rico.
IV. Factual Allegations.
A. Background of the SynchroMed II Device.
11. The SynchroMed II Device is a programmable drug infusion system implanted in
the body for drug delivery. The SynchroMed II Device includes an infusion pump connected to a
thin, flexible catheter attached to the intrathecal space (spinal canal) of the patient, into which the
pump delivers medication.
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 3 of 42

Complaint – Page 4 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
12. The entire SynchroMed II Device is implanted and remains under the skin. A
clinician measures a precise amount of medication and injects the medication into the pump’s
reservoir fill port. The medication passes through a reservoir valve and into the pump reservoir.
At normal body temperatures, pressurized gas, used as a propellant, is stored below the reservoir
and it expands and exerts constant pressure on the reservoir. This pressure pushes the medication
into the pump tubing. The battery-powered electronics and motor gears deliver a programmed
dose of medication through the tubing out through a catheter port and into a catheter. Medication
delivery then continues through the catheter tubing and into the intrathecal space of a patient.
13. The intrathecal catheters and sutureless revision kits of the SynchroMed II Device
are designed to connect the pump with the patient’s intrathecal space. Each catheter has a pre-
attached strain relief sleeve, a connector pin, and a sutureless pump connector (also known as a
revision kit) that connects to the SynchroMed II pump.
14. The SynchroMed II Device is a Class III medical device, approved by the U.S. Food
and Drug Administration (FDA) through the Premarket Approval (PMA) process on September
12, 2003, PMA Supplement No. P860004 S056.
15. Since the initial approval, Medtronic has sought FDA approval of at least 303
supplements or changes to the originally approved Device.
16. The pump of the SynchroMed II Device is supplied in 20- and 40-ml reservoir sizes,
model nos. 8637-20 and 8637-40, respectively.
17. According to Medtronic’s SynchroMed II “System Components Sheet,” as well as
information identified through the FDA’s recall database, the catheter of the SynchroMed II
Device is supplied as one of the following brands and models, which are connected to the pump
using the following connector or revision kit models:
Brand Catheter Model No. Connector / Revision Kit Model No.
Indura 8709 8575, 8578
Indura 8709SC 8578
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 4 of 42

Complaint – Page 5 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
Brand Catheter Model No. Connector / Revision Kit Model No.
Indura 8711 Not specified
Not Specified 8731 8596, 8596SC, 8598, 8598A
Not Specified 8731SC 8596SC, 8598A
Ascenda 8780 8784
Ascenda 8781 8784
18. According to Medtronic’s SynchroMed II “Indications, Drug Stability, and
Emergency Procedures Reference Manual,” the SynchroMed II Device is FDA-approved solely
for the following uses:
a. The chronic intrathecal infusion of Infumorph (preservative-free morphine
sulfate sterile solution) in the treatment of chronic intractable pain, with a maximum
approved concentration of 25 mg/ml.
b. The chronic intrathecal infusion of Prialt (preservative-free ziconotide
sterile solution) for the management of severe chronic pain, with a maximum approved
concentration of 100 μg/ml.
c. The chronic intrathecal infusion of Lioresal Intrathecal (baclofen injection)
in the management of severe spasticity, with a maximum approved concentration of 2
mg/ml.
B. Nancy Kilmer’s Experience with the SynchroMed II Device.
19. Nancy Kilmer is a sixty-eight-year-old woman who injured her left knee, left
elbow, and low back on September 24, 1998 when she slipped and fell.
20. Ms. Kilmer suffers from lumbar disc displacement without myelopathy, post
lumbar spine surgery syndrome, and chronic intractable pain.
21. In or about April 2006, to treat her pain and reduce or eliminate the need for oral
medication, Ms. Kilmer was persuaded to have a SynchroMed II Device implanted in her body, to
administer a programmed amount of medication into the intrathecal space of her spine.
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 5 of 42

Complaint – Page 6 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
22. On April 19, 2006, Ms. Kilmer had a SynchroMed II Device, comprised of a model
no. 8637-20 pump with serial no. NGP021740N (hereinafter the “first pump”) and a model no.
8709 Indura-brand catheter with lot no. N005414230 (hereinafter the “first catheter”), implanted
into her body by Dr. Leonard Soloniuk of the Soloniuk Clinic, 2656 Edits Avenue, Suite B,
Redding CA 96001, at Mercy Medical Center, 914 Pine Street, Mt Shasta CA 96067.
23. The first pump was initially used to administer morphine; on March 29, 2007, the
Ms. Kilmer’s physicians discontinued morphine and began to use the pump to administer
hydromorphone and clonidine.
24. On August 19, 2008, the first pump malfunctioned, causing Ms. Kilmer to suffer
an onset of pain, a clammy feeling in her legs, vomiting, and symptoms of withdrawal.
25. In or about August 2012, to continue to treat her pain and reduce or eliminate the
need for oral medication, Ms. Kilmer was persuaded to have her SynchroMed II pump replaced.
26. On August 8, 2012, Ms. Kilmer had her first pump explanted and had a new
SynchroMed II pump, comprised of a model no. 8637-20 pump with serial no. NGP375709H
(hereinafter the “second pump”) implanted her body by Dr. Robert Salazar of Robert G. Salazar,
M.D., Inc, 7152 North Sharon, Suite 102, Fresno, CA 93720. The second pump was connected to
the first catheter.
27. The second pump was used to administer hydromorphone (Dilaudid), clonidine,
bupivacaine, and fentanyl.
28. On July 22, 2014, Ms. Kilmer underwent a pump refill procedure at Dr. Salazar’s
office, after which Ms. Kilmer started feeling light-headed, had a funny taste in her mouth, and
became tired, dizzy and short of breath. Dr. Salazar transferred Ms. Kilmer to St. Agnes Hospital,
1303 East Herndon Avenue, Fresno CA 93720, where she was admitted and diagnosed with an
overdose of Dilaudid.
29. That same day, after approximately four hours of hospitalization and observation,
Ms. Kilmer stabilized and was discharged from St. Agnes Hospital.
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 6 of 42

Complaint – Page 7 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
30. On September 7, 2018, Ms. Kilmer underwent a pump refill procedure at Dr.
Salazar’s office, after which Ms. Kilmer stated that she felt like there were “clouds in her head.”
Dr. Salazar administered the anti-overdose drug Narcan to Ms. Kilmer and transferred her to St.
Agnes Hospital, where she was admitted and diagnosed with an opiate overdose.
31. That same day, after approximately 90 minutes of hospitalization and observation,
Ms. Kilmer stabilized and was discharged from St. Agnes Hospital.
32. In or about December 2018, to continue to treat her pain and reduce or eliminate
the need for oral medication, Ms. Kilmer was persuaded to have her SynchroMed II pump replaced.
33. On December 20, 2018, Ms. Kilmer had her second pump explanted and had a new
SynchroMed II pump, comprised of a model no. 8637-20 pump with serial no. NGP001398H
(hereinafter the “third pump”) implanted her body by Dr. Robert Salazar of the Comprehensive
Pain Management Center, 7152 North Sharon, Suite 104, Fresno, CA 93720. The third pump was
connected to the first catheter using a model 8578 connector with lot no. HG2AXU507.
34. The third pump was used to administer hydromorphone (Dilaudid).
35. On March 19, 2019, Ms. Kilmer underwent a pump refill procedure at Dr. Salazar’s
office, after which Ms. Kilmer stated that she felt like she was “high.” Dr. Salazar administered
the anti-overdose drug Narcan to Ms. Kilmer, and monitored her for two hours, after which
overdose symptoms resolved.
36. On or about March 22, 2019, a Medtronic representative spoke with Ms. Kilmer
via telephone, and advised her that Medtronic was aware of pump overdoses occurring at refill
procedures and that Medtronic did not know why they were happening.
37. In or about July 2019, displeased with the performance of the SynchroMed II after
having suffered repeated overdoses, but still wanting to treat her pain and reduce or eliminate the
need for oral medication, Ms. Kilmer was persuaded to have her SynchroMed II pump replaced
with a Flowonix-brand pump.
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 7 of 42

Complaint – Page 8 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
38. On July 11, 2019, Ms. Kilmer had her third pump explanted and had a Flowonx
pump implanted her body by Dr. Robert Salazar of the Comprehensive Pain Management Center.
The indication for this replacement procedure was the malfunction of Ms. Kilmer’s third pump.
39. As a direct and proximate result of Medtronic’s conduct described herein, Ms.
Kilmer’s second pump, third pump, and first catheter failed to deliver the prescribed medication
as programmed, resulting in overinfusion and causing Ms. Kilmer to suffer damages including
pain and suffering; mental anxiety and anguish; pump removal and replacement; and medical bills
in amounts to be proven at trial.
C. Legal Requirements Following Premarket Approval of the SynchroMed II
Device.
40. Premarket approval (PMA) is the FDA process of scientific and regulatory review
to evaluate the safety and effectiveness of Class III medical devices. Class III medical devices are
those that support or sustain human life, are of substantial importance in preventing impairment of
human health, or which present a potential unreasonable risk of illness or injury. Due to the level
of risk associated with Class III devices, these devices require a premarket approval (PMA)
application under Section 515 of the Federal Food, Drug, and Cosmetic Act (FD&C Act) before
they can be sold in the United States. The SynchroMed II Device is a Class III medical device.
41. In a PMA application, the applicant is required to supply information to the FDA.
The information required includes device description, clinical safety trials, methods of its product
testing, design of the device and specific manufacturing controls, outcome evaluation, and
proposed labeling. The FDA does not conduct independent testing on a medical device in a PMA
application. The FDA reviews the documentation provided to them by the PMA applicant and
relies on the veracity of the company. The PMA applicant is solely responsible for submitting all
truthful and necessary documentation to the FDA.
42. Once an application for PMA is approved, the holder (here, Medtronic) must
comply with any and all post-approval requirements established by statute, the FDA, and federal
regulations.
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 8 of 42

Complaint – Page 9 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
43. In particular, federal regulations require a PMA holder such as Medtronic to comply
with the following requirements:
a. Adverse Events. Review, evaluate, and report to the FDA adverse events
associated with the medical device.
i. Report individual adverse events within 30 days after becoming
aware of an adverse event or aware of a reportable death, serious injury or
malfunction, 21 C.F.R. § 803.l0(c)(1); and
ii. Report individual adverse events no later than five workdays after
becoming aware of a “reportable event that requires remedial action to prevent an
unreasonable risk of substantial harm to the public health,” 21 C.F.R.
§ 803.10(c)(2)(i).
b. Quality System. Establish and maintain a quality system that is appropriate
for the specific medical devices designed or manufactured and that meets the requirement
of this part. 21 C.F.R. § 820.5.
c. Management Responsibility. Management with executive responsibility
shall establish its policy and objectives for, and commitment to quality. 21 C.F.R.
§ 820.20.
d. Qualified Personnel. Have sufficient personnel with the necessary
educational background, training, and experience to assure that all activities required by
this part are correctly performed. 21 C.F.R. § 820.25.
e. Corrective and Preventative Action (CAPA). Establish and maintain
procedures for implementing corrective and preventive actions and document all CAPA
activities. 21 C.F.R. § 820.100.
f. Complaint Files. Maintain complaint files, processed in a uniform and
timely manner, oral complaints must be documents and must be evaluated to determine
whether the complaint represents a reportable event under Medical Device Reporting. 21
C.F.R. § 820.198.
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 9 of 42

Complaint – Page 10 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
g. Statistical Techniques. Establish and maintain procedures for identifying
valid statistical techniques required for establishing, controlling and verifying the
acceptability of process capability and product characteristics. 21 C.F.R.§ 820.250.
h. Misbranded Drugs and Devices Prohibited. A device shall be deemed to be
“misbranded” if, among other things, there has been a failure or refusal to give required
notification or to furnish required material or information to the FDA. 21 U.S.C. § 352(t).
i. Adulterated Products Prohibited. If the manufacturer fails to ensure that the
methods used in, or the facilities or controls used for, their manufacture, packing, storage,
or installation are not in conformity with applicable requirements, including but not limited
to the Current Good Manufacturing Practice (CGMP) requirement of the Quality System
regulations found at Title 21 Code of Federal Regulations Section 820, then such products
are considered “adulterated.” 21 U.S.C. § 351(h).
j. Off-Label Promotion Prohibited. A product may not be manufactured
packaged, stored, labeled, distributed, advertised, or promoted in a manner that is
inconsistent with any conditions to approval specified in the PMA approval order for the
device. 21 C.F.R. § 814.80.
D. Violations of Federal Law Resulting in Plaintiff’s Defective and
Malfunctioning SynchroMed II Device.
1. Overview of FDA Inspections and Defendants’ Violations.
44. To ensure compliance with these statutes and regulations, the FDA conducts
inspections of medical device manufacturing and quality-control facilities. Following such
inspections, FDA inspectors issue FDA Form 483 documents, also known as Inspectional
Observations, which list conditions or practices that indicate potential violations of statutes or
regulations. The FDA may also issue a formal Warning Letter if, upon further review of the
Inspectional Observations, the FDA determines that serious statutory or regulatory violations exist
at a medical device manufacturing or quality-control facility.
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 10 of 42

Complaint – Page 11 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
45. Medtronic, in their manufacture of the SynchroMed II Device (including not only
the pump but also catheters), violated federal law governing manufacture and quality control of
PMA medical devices, which was discovered during a series of inspections by the FDA at
Medtronic’s manufacturing and quality control plants in Minneapolis, Minnesota and Juncos,
Puerto Rico.
46. The inspections were followed by a series of Warning Letters to Medtronic that
identify federal manufacturing and quality control violations at the plants that ultimately led to an
April 27, 2015 Complaint Requesting a Permanent Injunction filed against Medtronic by the U.S.
Department of Justice and U.S. Department of Health and Human Services, and a Court- Ordered
Consent Decree imposing a moratorium on the manufacture, sale, and distribution of the
SynchroMed II Device in violation of federal law.
47. In addition, since receiving PMA approval, the SynchroMed II Device and its
components associated with PMA No. P860004 have been subject to no fewer than 72 recalls.
48. These Warning Letters, recalls, and injunction, which include specific references
to the SynchroMed II pump as well as its affiliated intrathecal catheters, speak to the seriousness
of Defendants’ violations of federal law and negligence in the manufacture of the SynchroMed II
Device.
2. FDA Inspections and Warning Letters.
49. In 2006, 2007, 2008, 2009, 2012, and 2013, during the time Plaintiff’s SynchroMed
II Device was being manufactured by Medtronic, the FDA conducted numerous inspections of
Medtronic’s manufacturing and quality-control facilities in Minneapolis, Minnesota and Juncos,
Puerto Rico, discovering a multitude of significant violations of federal law governing the
manufacture and quality control of PMA medical devices including the SynchroMed II pump and
associated intrathecal catheters, as recorded in FDA Form 483s and Warning Letters issued to
Medtronic.
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 11 of 42

Complaint – Page 12 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
50. 2006 Inspection and 2006 Warning Letter.1
a. From May 18 to June 22, 2006, the FDA conducted an inspection of
Medtronic’s manufacturing plant located at 800 53rd Avenue NE, Minneapolis, Minnesota
55421, where Medtronic “manufacturers manufactures implantable drug
infusion . . . products to treat pain [and] movement disorders.”
b. On August 29, 2006, the FDA issued Medtronic a Warning Letter
concerning this inspection.
c. This inspection revealed that the SynchroMed II Device was “adulterated
under Section 501(h) of the Act [21 U.S.C. 351(h)], in that the methods used in, or the
facilities or controls used for, their manufacture, packing, storage, or installation are not in
conformance with the Current Good Manufacturing Practice (CGMP) requirements for
medical devices which are set forth in the Quality System regulation, found at Title 21,
Code of Federal Regulations (CFR), Part 820.”
d. The 2006 Warning Letter enumerated the following “significant deviations”
from the CGMP regulations with respect to catheters and pumps:
i. Violation of 21 C.F.R. § 820.30(c): Failure to implement procedures
to ensure that a device’s design input requirements are appropriate and address its
intended use, including user/patient needs, in that design input work for intrathecal
catheters had not resulted in development of a complete design specification for the
catheter tip bond.;
ii. Violation of 21 C.F.R. § 820.30(g): Failure to conduct design
validation using production units or their equivalents, in that design validation
testing of intrathecal catheters was conducted with catheters manufactured with a
tip marker bonding process that was different than that used in production;
1 See Ex. 1, FDA Warning Letter (Aug. 29, 2006). All quotations in the subparagraphs of this paragraph are sourced from this 2006 Warning Letter.
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 12 of 42

Complaint – Page 13 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
iii. Violation of 21 C.F.R. § 820.75(a): Failure to validate a process
whose results cannot be fully verified by subsequent inspections and tests, in that
the bonding process for the catheter has not been validated;
iv. Violation of 21 C.F.R. § 820.70(a): Failure to control production
processes to ensure that a device conforms to its specification, in that the bonding
manufacturing procedures contained nonconforming instructions.
v. Violation of 21 C.F.R. § 820.100(a)(2): Failure to implement CAPA
procedures addressing the investigation of the cause of nonconformities, including
closing CAPAs without proper root cause analyses, with incorrect conclusions, or
without evidence to support conclusions.
vi. Violation of 21 C.F.R. § 820.100(a)(5): Failure to implement
changes in methods and procedures needed to correct and prevent identified quality
problems, in that although a CAPA called for a catheter tip redesign, product
specification was not changed, the revised manufacturing process was not
validated, and no process monitoring was conducted.
vii. Violation of 21 C.F.R. § 820.100(a)(3): Failure to identify all of the
actions needed to correct and prevent the recurrence of nonconforming product and
other quality problems; and
viii. Violation of 21 C.F.R. § 820.184: Failure to implement procedures
to ensure that device history records for each batch, or unit are maintained to
demonstrate that the device is manufactured in accordance with regulations.
e. The Warning Letter concluded that these violations “may be symptomatic
of serious underlying problems in your firm’s manufacturing quality assurance systems”
and called for a follow-up inspection.
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 13 of 42

Complaint – Page 14 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
51. 2006–07 Inspection and 2007 Warning Letter.2
a. From November 21, 2006 to January 24, 2007, the FDA conducted a follow-
up inspection of Medtronic’s manufacturing plant located at 800 53rd Avenue NE,
Minneapolis, Minnesota 55421, where Medtronic “manufacturers implantable drug
infusion . . . products.”
b. On July 3, 2007, the FDA issued Medtronic a Warning Letter concerning
this inspection.
c. This inspection revealed that the SynchroMed II Device was “adulterated
within the meaning of section 501(h) of the Act [21 U.S.C. § 351(h)], in that the methods
used in, or the facilities or controls used for, their manufacture, packing, storage, or
installation are not in conformity with the Current Good Manufacturing Practice (CGMP)
requirements of the Quality System (QS) regulation found at Title 21, Code of Federal
Regulations, (21 CFR) Part 820.”
d. Specifically with respect to adulteration, the FDA found that Medtronic
violated 21 C.F.R. § 820.198(a)(3) through its “[f]ailure to implement complaint handling
procedures to ensure that all complaints are evaluated to determine whether the complaint
represents an event that must be filed as a Medical Device Report under 21 CFR Part 803.”
e. This inspection also revealed that the SynchroMed II Device was
“misbranded under section 502(t)(2) of the Act [21 U.S.C. § 352(t)(2)], in that [Medtronic]
failed or refused to furnish material or information respecting the device that is required by
or under section 519 of the Act (21 U.S.C. § 360i), and 21 CFR Part 803—Medical Device
Reporting (MDR) regulation.”
f. Specifically with respect to this misbranding, the FDA found that Medtronic
violated 21 C.F.R. § 803.50(a)(1) through its “[f]ailure to submit MDR reports within 30
2 See Ex. 2, FDA Warning Letter (July 3, 2007). All quotations in the subparagraphs of this paragraph are sourced from this 2007 Warning Letter. See also Ex. 3, FDA Form 483 (Jan. 24, 2007).
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 14 of 42

Complaint – Page 15 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
days of receiving or otherwise becoming aware of information that reasonably suggests
that a marketed device may have caused or contributed to a death or serious injury.”
Medtronic:
i. failed to report SynchroMed II Device’s intrathecal catheters
associated with granuloma or inflammatory masses at or near the distal tip, which
the FDA considers “serious injuries”;
ii. failed to report SynchroMed II Device’s intrathecal catheter
fractures;
iii. failed to report a malfunction MDR, required when a marketed
device malfunction would likely cause or contribute to a reportable death or serious
injury;
iv. failed to submit MDR reports within 30 days of learning of a
problem (pump malfunctions, catheter fracture or separation, inflammatory masses
and granulomas) with the SynchroMed II device in the medical literature; and
v. failed to report consumer self-reported adverse events.
g. The inspection further revealed that the SynchroMed II Device was also
“misbranded under section 502(t)(2) of the Act [21 U.S.C. § 352(t)(2)], in that [Medtronic]
failed or refused to furnish material or information respecting the device that is required by
or under section 519 of the Act, 21 U.S.C. § 360i, and 21 CFR Part 806—Reports of
Corrections and Removals.”
h. Specifically with respect to this additional misbranding, the FDA found that
Medtronic violated 21 C.F.R. § 806.10(a)(1) because a “correction or removal conducted
to reduce a risk to health posed by a device was not reported in writing to FDA” concerning
the risk of an inflammatory mass occluding intrathecal catheters.
i. The 2007 Warning Letter further warned Medtronic: “[Y]our firm has
several procedures for Medical Device Reporting and Adverse Drug Experience Reporting.
These procedures, in turn reference several other procedures. Your firm’s current problems
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 15 of 42

Complaint – Page 16 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
regarding MDR reporting, as discussed above in this Warning Letter, may be exacerbated
by the complexity of your procedures and might have contributed to your firm’s deviations
from the regulations regarding MDR reporting.”
j. The 2007 Warning Letter concluded by also revealing several ongoing
violations at Medtronic’s Minneapolis Plant’s Quality System that were noted in a Form
483, stating “[t]he specific violations noted in this letter and Form FDA 483 may be
symptomatic of serious underlying problems in your firm’s manufacturing and Quality
Assurance systems.” Specifically, the FDA warned that Medtronic failed to achieve
consistent compliance in areas such as design controls in violation of 21 C.F.R. § 820.30
and failed to achieve consistent CAPA compliance in violation of 21 C.F.R. § 820.100.
52. 2008 Inspection and 2009 Warning Letter.3
a. From November 12 to December 15, 2008, the FDA conducted an
inspection of Medtronic’s manufacturing plant located at Road 31, Km 24, Ceiba Norte
Industrial Park, Juncos, Puerto Rico, where Medtronic “manufacturers SynchroMed II
Pumps.”
b. On June 1, 2009, the FDA issued Medtronic a Warning Letter concerning
this inspection.
c. This inspection “revealed that the SynchroMed II Pumps are adulterated
within the meaning of section 501(h) of the Act (21 U.S.C. §351(h)), in that the methods
used in, or the facilities or controls used for, their manufacture, packing, storage, or
installation are not in conformity with the Current Good Manufacturing Practice (CGMP)
requirements of the Quality System (QS) regulation found at Title 21, Code of Federal
Regulations (C.F.R.), Part 820.”
d. The FDA enumerated the following violations in the 2009 Warning Letter:
3 See Ex. 4, FDA Warning Letter (June 1, 2009). All quotations in the subparagraphs of this paragraph are sourced from this 2009 Warning Letter. See also Ex. 5, FDA Form 483 (Dec. 15, 2008).
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 16 of 42

Complaint – Page 17 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
i. Violation of 21 C.F.R. § 820.70(a): “Failure to establish and
maintain process control procedures that describe any process controls necessary
to ensure conformance to specifications, which shall include monitoring and
control of process parameters and component and device characteristics during
production,” in that pumps were manufactured without propellant; “did not show
evidence of a perforated septum,” which is “performed to detect
obstruction . . . early in the manufacturing process”; and lacked “a safety
mechanism that serves to ensure that the pump is never overfilled.”
ii. Violation of 21 C.F.R. § 820.100(a): “Failure to establish and
maintain procedures for implementing corrective and preventive action that include
identifying the action(s) needed to correct and prevent recurrence of
nonconforming product and other quality problems,” in that a critical step was left
out of the pump manufacturing process concerning “critical internal functions such
as calculating drug reservoir levels and drug dispensing rates.” Despite numerous
complaints that Medtronic received regarding accuracy rates, Medtronic failed to
conduct any type of investigation into this problem.
iii. Violation of 21 C.F.R. § 820.184: “Failure to establish and maintain
procedures to ensure that Device History Records (DHR’s) for each batch, lot, or
unit are maintained to demonstrate that the device is manufactured in accordance
with the Device Master Record (DMR),” in that pump sterilization processes were
not performed in the order specified by Medtronic procedures; and
iv. Violation of 21 C.F.R. § 820.198(c): “Failure to review, evaluate,
and investigate complaints involving the possible failure of a device, labeling, or
packaging to meet any of its specifications,” in that, for several complaints of
infections from nonsterile pumps, “a copy of [Medtronic’s] investigation was not
included as part of the complaint record, there was no reference to a specific
investigation report number, . . . there was no documentation whether the
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 17 of 42

Complaint – Page 18 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
investigation was successfully closed, . . . [and] there was no record in the
complaint file that Medical Device Reports were filed by [Medtronic] with FDA.”
e. The Warning Letter concluded that these violations “may be symptomatic
of serious problems in your firm’s manufacturing quality assurance systems.”
53. 2012 Investigation and 2012 Warning Letter.4
a. From March 14 to May 9, 2012, the FDA conducted an inspection of
Medtronic’s manufacturing plant located at 7000 Central Avenue NE, Minneapolis,
Minnesota 55432, where Medtronic “manufactures implantable drug infusion systems.”
b. On July 17, 2012, the FDA issued Medtronic a Warning Letter concerning
this inspection.
c. This inspection revealed that Medtronic’s SynchroMed II Devices were
“adulterated within the meaning of section 501(h) of the Act, 21 U.S.C. § 351(h), in that
the methods used in, or the facilities or controls used for, their manufacture, packing,
storage, or installation are not in conformity with the Current Good Manufacturing Practice
(CGMP) requirements of the Quality System (QS) regulation found at Title 21, Code of
Federal Regulations (21 CFR), Part 820.”
d. The FDA enumerated the following violations in the 2012 Warning Letter:
i. Violation of 21 C.F.R. § 820.100(a): “Failure to establish adequate
procedures for corrective and preventive action,” in that Medtronic failed to
identify “the actions to correct and prevent recurrence of nonconforming product”
relating to pump motor stalls and relied on incomplete data when conducting CAPA
activities;
ii. Violation of 21 C.F.R. § 820.198(a): “Failure to establish adequate
procedures for receiving, reviewing, and evaluating complaints by a formally
4 See Ex. 6, FDA Warning Letter (July 17, 2012). All quotations in the subparagraphs of this paragraph are sourced from this 2012 Warning Letter.
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 18 of 42

Complaint – Page 19 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
designated unit,” in that “[c]omplaint information received during a call was not
documented”; and
iii. Violation of 21 C.F.R. § 820.198(c): “Failure to review, evaluate
and investigate, where necessary, complaints involving the possible failure of a
device to meet any of its specifications,” in that “Product Performance Specialists
did not adequately evaluate complaints,” “[c]oding of similar complaints is
inconsistent,” and “[t]rending of complaint data / coding for evaluation was not
completed per procedures.”
e. The FDA expressed its significant “concern[] that incomplete complaint
data and incorrect coding decisions . . . may have compromised Medtronic’s ability to
detect and investigate [safety] signals,” i.e., signs of safety problems.
f. The Warning Letter concluded that these violations “may be symptomatic
of serious problems in your firm's manufacturing and quality assurance systems.”
54. 2013 Inspection.5
a. From February 14 to April 3, 2013, the FDA conducted another inspection
of Medtronic’s manufacturing plant located at 7000 Central Avenue NE, Minneapolis,
Minnesota 55432.
b. On April 3, 2013, the FDA issued a Form 483 informing Medtronic that that
the plant failed to manufacture devices that adequately conform to specifications and
instead manufactured devices that are not adequately controlled. Specifically, Medtronic:
i. distributed nonconforming intrathecal catheters that were prone to
occlusion and
ii. failed to establish adequate CAPA procedures, in that “[a]ctions
needed to correct and prevent recurrence of a quality problem were identified but
not implemented” concerning electrical shorting leading to pump motor stalls and
5 See Ex. 7, FDA Form 483 (Apr. 3, 2013).
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 19 of 42

Complaint – Page 20 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
implementation of recommendations from the Risk Evaluation Board, “Health
Hazard Assessments for high priority CAPAs with the highest patient severity of
death were not completed in a timely fashion,” and “Health Hazard Assessments
have not been updated after CAPA effectiveness monitoring signaled an increase
in the rate of occurrence” of hazards involving intrathecal catheter occlusion.
55. Throughout the history of the manufacture of the SynchroMed II Device, the FDA
has repeatedly notified Medtronic that their manufacture of the SynchroMed II Device failed to
conform to manufacturing requirements enumerated in federal regulations and statutes. These
federal violations caused the aforementioned defects and malfunctions in Plaintiff’s SynchroMed
II pump and catheter, which caused her injuries and damages alleged herein.
56. As evidenced by the 2009 Warning Letter, Medtronic skipped a step in the
manufacturing process concerning “critical internal functions such as calculating drug reservoir
levels and drug dispensing rates,” which are crucial to ensuring the correct amount of medicine is
dispensed by the pump. As a result, second and third pumps were manufactured without necessary
steps designed to prevent overinfusion and to ensure accurate delivery of pain medication, which
resulted in Plaintiff’s pump miscalculating and overinfusing pain medication.
3. Recalls of the SynchroMed II Pump.
57. A recall is an action taken to address a problem with a medical device that violates
federal law.
58. Recalls are classified as either Class I, Class II, or Class III. A Class I recall is
issued for a situation in which there is a reasonable probability that the use of or exposure to a
violative product will cause serious adverse health consequences or death. A Class II recall is
issued for a situation in which use of or exposure to a violative product may cause temporary or
medically reversible adverse health consequences or where the probability of serious adverse
health consequences is remote. Finally, a Class III recall is issued for a situation in which use of
or exposure to a violative product is less likely to cause adverse health consequences.
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 20 of 42

Complaint – Page 21 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
59. The FDA has issued at least 19 Class I and II recalls specifically for SynchroMed
II pump models during the time the SynchroMed II Device has been on the market, as summarized
in the following table:
Recall No. Class Pump Model No. Recall Reason
Z-1040-04 2 8637-20 & 8637-40 Mislabeling of pump reservoir size, resulting in overfilling and overinfusion
Z-2181-2008 2 8637-20 Pumps manufactured without propellant, resulting in cessation of therapy, underinfusion, and withdrawal
Z-2182-2008 2 8637-40 Pumps manufactured without propellant, resulting in cessation of therapy, underinfusion, and withdrawal
Z-0591-2009 2 8637-20 MRI-related motor stall, resulting in cessation of therapy, underinfusion, and withdrawal
Z-0592-2009 2 8637-40 MRI-related motor stall, resulting in cessation of therapy, underinfusion, and withdrawal
Z-2276-2009 2 8637-20 & 8637-40 Battery failure, resulting in cessation of therapy, underinfusion, and withdrawal
Z-1060-2011 1 8637-20 & 8637-40 Inadequate instruction for filling/refilling of pumps, resulting in injection of some or all of the prescribed drug into the patient’s subcutaneous issue (an inadvertent "pocket fill") and corresponding overinfusion
Z-1061-2011 1 8637-20 & 8637-40 Inadequate instruction for filling/refilling of pumps, resulting in injection of some or all of the prescribed drug into the patient’s subcutaneous issue (an inadvertent "pocket fill") and corresponding overinfusion
Z-3043-2011 1 8637-20 & 8637-40 Battery failure, resulting in cessation of therapy, underinfusion, and withdrawal
Z-1338-2012 2 8637-20 & 8637-40 Software failure resulting in incorrect display of the scheduled pump replacement date
Z-0497-2013 1 8637-20 & 8637-40 Use of unapproved drugs in the pumps and corresponding motor stall, resulting in cessation of therapy, underinfusion, and withdrawal
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 21 of 42

Complaint – Page 22 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
Recall No. Class Pump Model No. Recall Reason
Z-1570-2013 1 8637-20 & 8637-40 Unintended delivery of drugs during the priming bolus procedure, resulting in life-threatening overdose and subsequent withdrawal
Z-1579-2013 1 8637-20 & 8637-40 Internal electrical shorting, resulting in a motor stall or battery failure, cessation of therapy, underinfusion, and withdrawal
Z-1570-2014 2 8637-20 & 8637-40 Overinfusion, resulting in life-threatening overdose and corresponding drug withdrawal
Z-1681-2015 2 8637-20 & 8637-40 Alarm failure, resulting in cessation of therapy, underinfusion, and withdrawal due to lack of audible warning of low or empty drug reservoir, pump end-of-service, pump motor stall, pump stoppage, or critical memory error
Z-0788-2017 1 8637-20 & 8637-40 Unintended delivery of drugs during the priming bolus procedure, resulting in life-threatening overdose and subsequent withdrawal
Z-1694-2017 2 8637-40 Software error preventing pump interrogation, resulting in cessation of therapy, underinfusion, and withdrawal due to inability to update or refill the pump
Z-0896-2018 2 8637-20 & 8637-40 Permanent motor stall due to corrosive wear, resulting in cessation of therapy, underinfusion, and withdrawal
Z-0508-2020 1 8637-20 & 8637-40 Permanent motor stall due to presence of a foreign particle inside the pump motor assembly, resulting in cessation of therapy, underinfusion, and withdrawal
60. The FDA has also issued at least 27 recalls specifically concerning SynchroMed
II catheters and catheter-pump connectors during the time the SynchroMed II Device has been on
the market, as summarized in the following table:
Recall No. Class Product Model No. (Brand) Recall Reason
Z-1414-06 1 Catheter 8731 Tip dislodgement during implantation
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 22 of 42

Complaint – Page 23 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
Recall No. Class Product Model No. (Brand) Recall Reason
Z-1415-06 1 Connector 8598 Tip dislodgement during implantation
Z-1308-2008 2 Connector 8596SC Packaging of the incorrect pin to connect the catheter to the pump
Z-1150-2008 1 Catheter All catheters used with SynchroMed II Pump model no. 8637-20
Formation of inflammatory masses near the tip of intrathecal catheters
Z-1151-2008 1 Catheter All catheters used with SynchroMed II Pump model no. 8637-40
Formation of inflammatory masses near the tip of intrathecal catheters
Z-2171-2008 2 Connector 8578 Inability to completely connect catheter to pump, resulting in leakage or disconnection of the catheter
Z-2172-2008 2 Connector 8596SC Inability to completely connect catheter to pump, resulting in leakage or disconnection of the catheter
Z-2173-2008 2 Catheter 8709SC (Indura) Inability to completely connect catheter to pump, resulting in leakage or disconnection of the catheter
Z-2174-2008 2 Catheter 8731SC Inability to completely connect catheter to pump, resulting in leakage or disconnection of the catheter
Z-2380-2008 1 Catheter 8709SC (Indura) Disconnection of catheters from the pump, or occlusion between the sutureless pump connector and the catheter port on the pump.
Z-2381-2008 1 Catheter 8731SC Disconnection of catheters from the pump, or occlusion between the sutureless pump connector and the catheter port on the pump.
Z-2382-2008 1 Connector 8578 Disconnection of catheters from the pump, or occlusion between the sutureless pump connector and the catheter port on the pump.
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 23 of 42

Complaint – Page 24 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
Recall No. Class Product Model No. (Brand) Recall Reason
Z-2383-2008 1 Connector 8596SC Disconnection of catheters from the pump, or occlusion between the sutureless pump connector and the catheter port on the pump.
Z-2073-2009 1 Catheter 8709SC (Indura) Labeling error incorrectly stating catheter-pump compatibility
Z-2074-2009 1 Catheter 8731SC Labeling error incorrectly stating catheter-pump compatibility
Z-2075-2009 1 Connector 8596SC Labeling error incorrectly stating catheter-pump compatibility
Z-2076-2009 1 Connector 8578 Labeling error incorrectly stating catheter-pump compatibility
Z-0334-2011 2 Catheter 8731SC Presence of endotoxin in excess of United States Pharmacopeial Convention (USP) limits
Z-0335-2011 2 Connector 8598A Presence of endotoxin in excess of United States Pharmacopeial Convention (USP) limits
Z-1573-2013 1 Connector 8578 Potential for catheter misalignment and occlusion at the catheter-to-pump interface.
Z-1574-2013 1 Connector 8596SC Potential for catheter misalignment and occlusion at the catheter-to-pump interface.
Z-1575-2013 1 Catheter 8709SC (Indura) Potential for catheter misalignment and occlusion at the catheter-to-pump interface.
Z-1576-2013 1 Catheter 8731SC Potential for catheter misalignment and occlusion at the catheter-to-pump interface.
Z-1723-2014 2 Catheter 8780 (Ascenda) Presence of endotoxin in excess of USP limits
Z-2172-2014 2 Catheter / Connector
8780 & 8781 (Ascenda) / 8784
Catheter retainer ring failed specification criteria, resulting in possible disconnection of the catheter from the pump
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 24 of 42

Complaint – Page 25 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
Recall No. Class Product Model No. (Brand) Recall Reason
Z-1271-2016 2 Catheter 8781 (Ascenda) Incorrect package labeling and lack of all components necessary to complete the implant procedure
Z-0537-2018 3 Catheter / Connector
8780 & 8781 (Ascenda) / 8784
Increased potential for kinking where the catheter connects to the pump
61. At least one of these pump and catheter recalls further explain and demonstrate the
manufacturing defects that caused Plaintiff’s malfunctioning SynchroMed II pump: Recall No. Z-
1570-2014.
a. On February 26, 2014, Medtronic initiated a Class II recall of both models
(8637-20 and 8637-40) of the SynchroMed II pump. This recall was posted by the FDA
on May 8, 2018 and terminated on September 28, 2018.6
b. In March 2014, as part of the recall, Medtronic issued an Urgent Medical
Device Correction letter to healthcare professionals, explaining that “Medtronic detected
an upward shift in reports of occurrence of overinfusion” (i.e., overdose), which “may lead
to emptying of the pump prior to a planned refill and therefore may present clinically as an
interruption of therapy including lack of therapeutic effect and withdrawal syndrome” (i.e.,
underdose or underinfusion).7
c. In September 2016, as part of the continuing recall, Medtronic issued an
Urgent Medical Device Correction Update letter to healthcare professionals, further
explaining that “[e]xamples of clinical use conditions that have been shown to increase the
likelihood of overinfusion are the use of nonindicated drug formulations,” among other
conditions.8
6 Ex. 8, Recall No. Z-1570-2014. 7 Ex. 9, Letter from Mike Crader, Vice President Quality, Medtronic Neuromodulation, to Healthcare Professional (Mar. 2014). 8 Ex. 10, Letter from Michael Ronningen, Vice President of Quality, Medtronic, to Healthcare Professional (Sept. 2016).
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 25 of 42

Complaint – Page 26 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
d. Plaintiff, who used nonindicated drug formulations, suffered two overdoses
with her second pump during the time when this recall was in effect, and suffered a another
overdose with her third pump shortly after this recall had terminated, all of which led to
life-threatening consequences and interruptions of therapy as identified in this recall.
4. Violations of the Permanent Injunction Resulting in the Manufacture,
Distribution, and Sale of Plaintiff’s Defective and Malfunctioning
SynchroMed II Device.
62. Throughout the history of the manufacture of the SynchroMed II Device, Medtronic
has shown an indifference to federal manufacturing requirements. Further, Medtronic, with full
knowledge that it was manufacturing the SynchroMed II Device in violation of law, nonetheless
demonstrated a pattern of delayed responses or complete failures to respond to reported and known
safety issues with the SynchroMed II Device.
63. Because of Medtronic’s years-long pattern of indifference to regulatory authority,
noncompliance with federal manufacturing requirements, and violations of federal law, the U.S.
Department of Justice and the U.S. Department of Health and Human Services on April 27, 2015
filed a Complaint against Medtronic requesting a Consent Decree for Permanent Injunction against
the manufacture, distribution, and sale of the SynchroMed II Device.9
64. The Complaint alleges that Medtronic is “well aware that their practices violate the
[FD&C] Act. FDA has repeatedly warned Defendants, both orally and in writing, about their
violative conduct, and has emphasized the importance of Defendants’ compliance with the Act.”10
65. In addition to the cited Warning Letters, the Complaint alleges that representatives
of Medtronic attended a meeting with FDA’s Center for Devices and Radiological Health and
Minneapolis District Office on January 31, 2013. At this meeting, “Defendants stated that they
were aware of the violations at their facilities and were taking steps to correct them.”11
9 Ex. 11, Complaint for Permanent Injunction, United States v. Medtronic, Inc., No. 15-cv-2168 (D. Minn. Apr. 27, 2015), ECF No. 1. 10 Id. ¶¶ 15–17. 11 Id. ¶ 18.
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 26 of 42

Complaint – Page 27 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
66. The Complaint further alleges Medtronic made promises to correct their violations
in written responses to each inspection; however, the Complaint alleged that none of the responses
contained adequate evidence that Medtronic corrected their deviations.12
67. The United States Attorney stated in the Complaint that, “[b]ased upon Defendants’
conduct, Plaintiff believes that, unless restrained by order of this Court, Defendants will continue
to violate 21 USC §§ 331(a) and (k)”—introducing into interstate commerce any article of device
that is adulterated or misbranded, or causing any article of device to become adulterated or
misbranded while such devices are held for sale after shipment in interstate commerce.13
68. The United States’ Complaint requested a permanent injunction to restrain
Medtronic in their manufacture, distribution, and sale of the SynchroMed II Device from their
continued violation of federal regulations, and specifically:
That the Court order Defendants and each of their directors, officers, agents, representatives, employees, attorneys, successors, and assigns, and any and all persons in active concert or participation with any of them, to cease directly and indirectly manufacturing, packing, labeling, and distributing (domestically and internationally) SynchroMed II implantable infusion pumps at or from its Medtronic’s Neuromodulation faculties, unless and until Defendants’ methods, facilities, and controls used to manufacture, process, pack, label, hold and distribute the SynchroMed II implantable infusion pumps are established, operated, and administered in compliance with 21 USC 360j(f)(1) and the Quality System regulation prescribed in 21 C.F.R. Part 820, and in a manner that has been found acceptable to FDA.14 69. On April 27, 2015, United States District Court Judge Joan N. Erickson signed a
Consent Decree of Permanent Injunction against Medtronic preventing the manufacture,
distribution, and sale of Medtronic SynchroMed Implantable Infusion Pump systems in violation
of the terms of the Consent Decree.15
70. Under the Consent Decree, Medtronic is “permanently restrained and enjoined,
pursuant to 21 U.S.C. § 332(a), from directly or indirectly designing, manufacturing, processing,
12 Id. ¶¶ 19–20. 13 Id. ¶ 21. 14 Id. at 8. 15 Ex. 12, Consent Decree of Permanent Injunction, United States v. Medtronic, Inc., No. 15-cv-2168 (D. Minn. Apr. 27, 2015), ECF No. 3.
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 27 of 42

Complaint – Page 28 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
packing, labeling, holding, storing, and distributing, importing into or exporting from the United
States of America, at or from any Medtronic Neuromodulation facilities, any model of, or
components or accessories for, its SynchroMed devices.”16
71. Under the Consent Decree, the permanent injunction would be lifted only in the
event that Medtronic complies with a series of enumerated requirements to ensure that it would
cease violating federal law in the production of its SynchroMed II Device. 17
72. Although there is an exception to the permanent injunction in cases of medical
necessity,18 Plaintiff’s SynchroMed II Device was not medically necessary and/or did not satisfy
the procedural requirements set forth in the Consent Decree for the medical-necessity exception to
apply.
73. Medtronic continues to produce, distribute, and sell their SynchroMed II Device in
violation of the Consent Decree, including Plaintiff’s Device, which was implanted nearly one
year after entry of the Consent Decree.
V. Causes of Action.
Count I: Strict Liability Manufacturing Defect
74. Plaintiff incorporates by reference, as if fully set forth herein, each and every
allegation contained in the preceding paragraphs of this Complaint.
75. The SynchroMed II Devices implanted in Plaintiff were manufactured in violation
of the Federal Food, Drug, and Cosmetic Act and federal regulations promulgated pursuant thereto,
and was manufactured in violation of California law that parallels federal requirements, in one or
more of the following ways:
76. Plaintiff’s SynchroMed II Devices were adulterated in violation of federal law.
a. The Quality-Control Requirements of the Current Good Manufacturing
Practices found in 21 C.F.R. Part 820 are designed to ensure Medtronic’s products conform
16 Id. ¶ 6. 17 Id. ¶ 6.A.–J. 18 Id. ¶ 9.A.
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 28 of 42

Complaint – Page 29 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
to manufacturing specifications, that non-conforming products do not reach the market,
and that problems with products in the field are properly monitored, tracked, and reported.
b. The SynchroMed II Devices implanted in Plaintiff were adulterated in
violation of federal law, because they were manufactured in deviation from the
manufacturing specifications approved by the FDA in Medtronic’s PMA application, in
violation of the CGMPs.
c. Specifically, as a result of numerous FDA inspections from 2006 through
2013 of Medtronic’s manufacturing plants in Minneapolis, Minnesota and Juncos, Puerto
Rico, as alleged herein, the FDA determined that Medtronic violated specific CGMPs as
previously pled (including 21 C.F.R.§§ 820.30(c), 820.30(g), 820.70(a), 820.75(a),
820.100(a), 820.100(a)(2), 820.100(a)(3), 820.100(a)(5), 820.184, 820.198(a),
820.198(a)(3), and 820.198(c)), rendering the SynchroMed II Devices implanted Plaintiff
adulterated.
d. A device that has been manufactured, monitored, packed, stored, inspected,
or installed in violation of the CGMPs is deemed to be adulterated under 21 U.S.C.
§ 351(h), and a manufacturer is prohibited from introducing, delivering, or selling an
adulterated device into interstate commerce under 21 U.S.C. § 331(a)–(c), (k).
e. The SynchroMed II Device was introduced or delivered for introduction
into interstate commerce and was adulterated in violation of 21 U.S.C. §§ 331(a), 351(h)
and 21 C.F.R. Part 820.
f. The SynchroMed II Device was adulterated in interstate commerce in
violation of 21 U.S.C. §§ 331(b), 351(h) and 21 C.F.R. Part 820.
g. The SynchroMed II Device was received in interstate commerce, was
adulterated, and was delivered for pay or otherwise in violation of 21 U.S.C. §§ 331(c),
351(h) and 21 C.F.R. Part 820.
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 29 of 42

Complaint – Page 30 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
h. The SynchroMed II Device was adulterated while held for sale after
shipment in interstate commerce in violation of 21 U.S.C. §§ 331(k), 351(h) and 21 C.F.R.
Part 820.
i. This adulteration contributed to the imposition of the Consent Decree
imposing a moratorium on the manufacture, sale, and distribution of the SynchroMed II
Device.
j. Specifically with respect to Plaintiff’s second and third pumps, as evidenced
by the 2009 Warning Letter, Medtronic skipped a step in the manufacturing process
concerning “critical internal functions such as calculating drug reservoir levels and drug
dispensing rates,” which are crucial to ensuring the correct amount of medicine is
dispensed by the pump. As a result, second and third pumps were manufactured without
necessary steps designed to prevent overinfusion and to ensure accurate delivery of pain
medication, which resulted in Plaintiff’s pump miscalculating and overinfusing pain
medication.
77. Plaintiff’s SynchroMed II Devices were misbranded in violation of federal law.
a. 21 U.S.C. § 360i and 21 C.F.R. Part 803 require Medtronic to evaluate
signals of unexpected or serious events of injury in the field and report to the FDA when a
device causes, or is suspected to cause, injury in the field.
b. A device for which there was a failure or refusal to furnish information to
the FDA as required by 21 U.S.C. § 360i is deemed misbranded under 21 U.S.C.
§ 352(t)(2).
c. The SynchroMed II Devices implanted in Plaintiff were misbranded in
violation of federal law because Medtronic failed to report required adverse-event
information to the FDA
d. As a result of numerous FDA inspections from 2006 through 2013 of
Medtronic’s manufacturing plants in Minneapolis, Minnesota and Juncos, Puerto Rico, as
alleged herein, the FDA determined that Medtronic violated 21 C.F.R. §§ 803.50(a)(1) and
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 30 of 42

Complaint – Page 31 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
806.10(a)(1), rendering the SynchroMed II Devices implanted in Plaintiff misbranded.
Specifically, the FDA found the following:
i. As evidenced by the 2007 Warning Letter, Medtronic failed to
implement complaint handling procedures to ensure that all complaints are
evaluated to determine whether the complaint represents an event that must be
reported to the FDA; failed to submit timely reports to the FDA of adverse events
relating to, among other things, inflammatory masses in intrathecal catheters and
fractures of intrathecal catheters.
ii. Further, as evidenced by the 2009 Warning Letter, Medtronic
continued to not properly document and report adverse events to the FDA.
iii. Further still, as evidenced by the 2012 Warning Letter, Medtronic
failed to properly record and code complaint information and failed to properly
evaluate complaint trending, compromising Medtronic’s ability to detect and
investigate safety signals presented by complaint data.
e. A manufacturer is prohibited from introducing, delivering, or selling a
misbranded device into interstate commerce under 21 U.S.C. § 331(a)–(c), (k).
f. The SynchroMed II Device was introduced or delivered for introduction
into interstate commerce and was misbranded in violation of 21 U.S.C. §§ 331(a), and
352(t)(2) and 21 C.F.R. Part 803.
g. The SynchroMed II Device was misbranded in interstate commerce in
violation of 21 U.S.C. §§ 331(b) and 352(t)(2) and 21 C.F.R. Part 803.
h. The SynchroMed II Device was received in interstate commerce, was
misbranded, and was delivered for pay or otherwise in violation of 21 U.S.C. §§ 331(c)
and 352(t)(2) and 21 C.F.R. Parts 803.
i. The SynchroMed II Device was misbranded while held for sale after
shipment in interstate commerce in violation of 21 U.S.C. §§ 331(k) and 352(t)(2) and 21
C.F.R. Part 803.
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 31 of 42

Complaint – Page 32 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
j. This misbranding contributed to the imposition of the Consent Decree
imposing a moratorium on the manufacture, sale, and distribution of the SynchroMed II
Device.
78. Plaintiff’s SynchroMed II Devices were adulterated in violation of California law.
a. Cal. Health & Safety Code § 111260 provides: “Any drug or device is
adulterated if the methods, facilities, or controls used for its manufacture, processing,
packing, or holding do not conform to, or are not operated or administered in conformity
with current good manufacturing practice to assure that the drug or device meets the
requirements of this part as to safety and has the identity and strength, and meets the quality
and purity characteristics that it purports or is represented to possess.”
b. The SynchroMed II Devices implanted in Plaintiff were manufactured in
deviation from the manufacturing specifications approved by the FDA in Medtronic’s
PMA application, in violation of Current Good Manufacturing Practices found in 21 C.F.R.
Part 820, and thus in violation of Cal. Health & Safety Code § 111260.
c. Cal. Health & Safety Code § 111295 provides: “It is unlawful for any
person to manufacture, sell, deliver, hold, or offer for sale any drug or device that is
adulterated.”
d. The adulterated SynchroMed II Devices implanted in Plaintiff were
manufactured, sold, held, and offered for sale by Defendants in violation of Cal. Health &
Safety Code § 111295.
79. Under California law, Defendants had a duty to individuals, including Plaintiff, to
use reasonable care in manufacturing the SynchroMed II Device, which includes complying with
state and federal laws and regulations designed to ensure the safe manufacture, assembly,
inspection, packaging, testing, and adverse-event reporting of medical devices.
80. As a result of their adulteration and misbranding under federal and state law, the
SynchroMed II Devices implanted in Plaintiff were not reasonably safe for their intended use as a
matter of law with respect to their manufacture.
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 32 of 42

Complaint – Page 33 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
81. As a direct and proximate result of the SynchroMed II Device’s aforementioned
defects, the SynchroMed II Devices implanted in Plaintiff failed and required removal and
replacement surgeries, causing Plaintiff to suffer injury and damages, including pain and suffering,
mental anxiety and anguish, and medical bills.
Count II: Negligent Manufacturing Defect
82. Plaintiff incorporates by reference, as if fully set forth herein, each and every
allegation contained in the preceding paragraphs of this Complaint.
83. The SynchroMed II Devices implanted in Plaintiff were manufactured in violation
of the Federal Food, Drug, and Cosmetic Act and federal regulations promulgated pursuant thereto,
and was manufactured in violation of California law that parallels federal requirements, in one or
more of the following ways.
84. Plaintiff’s SynchroMed II Devices were adulterated in violation of federal law, as
pled in paragraph 76, supra.
85. Plaintiff’s SynchroMed II Devices were misbranded in violation of federal law, as
pled in paragraph 77, supra.
86. Plaintiff’s SynchroMed II Devices were adulterated in violation of California law,
as pled in paragraph 78, supra.
87. Under California law, Defendants had a duty to individuals, including Plaintiff, to
use reasonable care in manufacturing the SynchroMed II Device, which includes complying with
federal state laws and regulations designed to ensure the safe manufacture, assembly, inspection,
packaging, testing, and adverse-event reporting of medical devices.
88. Defendants were negligent in failing to use reasonable care in manufacturing the
SynchroMed II Device, in that they failed to use reasonable care to ensure that Plaintiff’s
SynchroMed II Device complied with federal and state statutes and regulations, manufactured
Plaintiff’s SynchroMed II Device in a way that did not comply with federal and state statutes and
regulations, failed to test and inspect Plaintiff’s SynchroMed II Device before placing it into the
stream of commerce and making it available for sale to Plaintiff, and failed to report adverse events
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 33 of 42

Complaint – Page 34 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
to the FDA. In so doing, Defendants failed to comply with manufacturing requirements imposed
by the Device’s PMA requirements and post-approval regulations.
89. As a direct and proximate result of Defendants’ negligence, the SynchroMed II
Devices implanted in Plaintiff failed and required removal and replacement surgeries, causing
Plaintiff to suffer injury and damages, including pain and suffering, mental anxiety and anguish,
and medical bills.
Count III: Strict Liability Failure to Warn the FDA
90. Plaintiff incorporates by reference, as if fully set forth herein, each and every
allegation contained in the preceding paragraphs of this Complaint.
91. At all times relevant hereto, Medtronic, as a merchant of medical devices including
the SynchroMed II Device, was required by federal law to report to the FDA certain post-sale
adverse events. Specifically, a manufacturer must report whenever a medical device may have
caused or contributed to death or serious injury or malfunctioned in a manner that would likely
cause or contribute to death or serious injury if it recurred.
92. This requirement can be satisfied by conveying warnings to a third party (the FDA)
when the manufacturer has no effective way to convey a product warning to the ultimate consumer
(the patient receiving the SynchroMed II pump). Because implanted medical devices, such as the
SynchroMed II pump, are sold to healthcare providers as opposed to consumers directly,
Medtronic was required to report risks to the FDA, as Medtronic would have no effective way of
warning consumers like Plaintiff directly.
93. Under California law, a device manufacturer can be found strictly liable if it fails
to adequately warn of a particular risk that was known or knowable in light of the generally
recognized and prevailing best scientific and medical knowledge available at the time of
distribution.
94. Defendants failed to warn the FDA of adverse events in violation of 21 C.F.R.
§§ 803.50(a)(1) and 806.10(a)(1), as pled in paragraph 77.d., supra.
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 34 of 42

Complaint – Page 35 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
95. This failure to warn contributed to the imposition of the Consent Decree imposing
a moratorium on the manufacture, sale, and distribution of the SynchroMed II Device.
96. Had Defendants properly reported adverse events to the FDA, Plaintiff’s
physicians, and thus Plaintiff, would have learned of the risks associated with the SynchroMed II
Device, and Plaintiff would not have received a defective device and/or would have chosen an
alternative device.
97. As a direct and proximate result of Defendants’ failure to warn, Plaintiff elected to
have defectively manufactured SynchroMed II Devices implanted, which failed and required
removal and replacement surgeries, causing Plaintiff to suffer injury and damages, including pain
and suffering, mental anxiety and anguish, and medical bills.
Count IV: Negligent Failure to Warn the FDA
98. Plaintiff incorporates by reference, as if fully set forth herein, each and every
allegation contained in the preceding paragraphs of this Complaint.
99. At all times relevant hereto, Medtronic, as a merchant of medical devices including
the SynchroMed II Device, was required by federal law to report to the FDA certain post-sale
adverse events. Specifically, a manufacturer must report whenever a medical device may have
caused or contributed to death or serious injury or malfunctioned in a manner that would likely
cause or contribute to death or serious injury if it recurred.
100. Under California law, medical-device manufacturers have duty to convey warnings
to a third party (the FDA) when the manufacturer has no effective way to convey a product warning
to the ultimate consumer (the patient receiving the SynchroMed II pump). Because implanted
medical devices, such as the SynchroMed II pump, are sold to healthcare providers as opposed to
consumers directly, Medtronic was required to report risks to the FDA, as Medtronic would have
no effective way of warning consumers like Plaintiff directly.
101. Under California law, a device manufacturer can be found liable if it negligently
fails to adequately warn of a particular risk that was known or knowable in light of the generally
recognized and prevailing best scientific and medical knowledge available at the time of
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 35 of 42

Complaint – Page 36 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
distribution.
102. Defendants breached their duty by failing to warn the FDA of adverse events in
violation of 21 C.F.R. §§ 803.50(a)(1) and 806.10(a)(1), as pled in paragraph 77.d., supra.
103. This failure to warn contributed to the imposition of the Consent Decree imposing
a moratorium on the manufacture, sale, and distribution of the SynchroMed II Device.
104. Had Defendants properly reported adverse events to the FDA, Plaintiff’s
physicians, and thus Plaintiff, would have learned of the risks associated with the SynchroMed II
Device, and Plaintiff would not have received a defective device and/or would have chosen an
alternative device.
105. As a direct and proximate result of Defendants’ failure to warn, Plaintiff elected to
have defectively manufactured SynchroMed II Devices implanted, which failed and required
removal and replacement surgeries, causing Plaintiff to suffer injury and damages, including pain
and suffering, mental anxiety and anguish, and medical bills.
Count V: Negligence Per Se
106. Plaintiff incorporates by reference, as if fully set forth herein, each and every
allegation contained in the preceding paragraphs of this Complaint.
107. Under Cal. Evid. Code § 669(a), “[t]he failure of a person to exercise due care is
presumed if: (1) He violated a statute, ordinance, or regulation of a public entity; (2) The violation
proximately caused death or injury to person or property; (3) The death or injury resulted from an
occurrence of the nature which the statute, ordinance, or regulation was designed to prevent; and
(4) The person suffering the death or the injury to his person or property was one of the class of
persons for whose protection the statute, ordinance, or regulation was adopted.”
108. The SynchroMed II Devices implanted in Plaintiff violate the Federal Food, Drug,
and Cosmetic Act and federal regulations promulgated pursuant thereto, and violate California law
that parallels federal requirements, in one or more of the following ways.
109. Plaintiff’s SynchroMed II Devices were adulterated in violation of federal law, as
pled in paragraph 76, supra.
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 36 of 42

Complaint – Page 37 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
110. Plaintiff’s SynchroMed II Devices were misbranded in violation of federal law, as
pled in paragraph 77, supra.
111. Plaintiff’s SynchroMed II Devices were adulterated in violation of California law,
as pled in paragraph 78, supra.
112. As a direct and proximate result of Defendants’ violations of these federal and state
statutes and regulations, the SynchroMed II Devices implanted in Plaintiff failed and required
removal and replacement surgeries, causing Plaintiff to suffer injury and damages, including pain
and suffering, mental anxiety and anguish, and medical bills.
113. Defendants were negligent in failing to use reasonable care in manufacturing the
SynchroMed II Device, in that they failed to use reasonable care to ensure that Plaintiff’s
SynchroMed II Device complied with federal and state statutes and regulations, manufactured
Plaintiff’s SynchroMed II Device in a way that did not comply with federal and state statutes and
regulations, failed to test and inspect Plaintiff’s SynchroMed II Device before placing it into the
stream of commerce and making it available for sale to Plaintiff, and failed to report adverse events
to the FDA. In so doing, Defendants failed to comply with manufacturing requirements imposed
by the Device’s PMA requirements and post-approval regulations, as well as state statutes; the
harm complained of is therefore the same these statutes and regulations are intended to guard
against.
114. Defendants had a duty to individuals, including Plaintiff, to use reasonable care in
manufacturing the SynchroMed II Device, which includes complying with federal regulations and
state law designed to ensure the safe manufacture, assembly, inspection, packaging, testing and
adverse-event reporting of medical devices; Plaintiff therefore falls within the class of persons
these statutes and regulations were designed to protect, namely, consumers of medical devices.
Count VI: Breach of Express Warranty
115. Plaintiff incorporates by reference, as if fully set forth herein, each and every
allegation contained in the preceding paragraphs of this Complaint.
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 37 of 42

Complaint – Page 38 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
116. At all times relevant hereto, Medtronic expressly warranted and promised to
Plaintiff, by way of a written warranty provided to Plaintiff along with her SynchroMed II Device,
that if Plaintiff’s SynchroMed II pump “fail[s] to function within normal tolerances due to a defect
in materials or workmanship within . . . two (2) years commencing with the date of implantation,”
then “Medtronic will at its option: (a) issue a credit to the purchaser of the replacement Component
equal to the Purchase Price, . . . or (b) provide a functionally comparable replacement Component
at no charge.”19
117. This express warranty plainly relates to the SynchroMed II Device and became the
basis of the bargain because Plaintiff received and relied upon this warranty when deciding to have
the SynchroMed II Device implanted.
118. Defendants breached this express warranty because:
a. Plaintiff’s third pump failed fewer than two years after it was implanted,
due to manufacturing defects as pled herein;
b. Plaintiff met the qualifying conditions set forth in Section B of the warranty;
and
c. Medtronic has neither refunded nor replaced free of charge the defective
third pump.
119. As a direct and proximate result of this breach, Plaintiff has suffered damages
including medical bills consisting of the value of her defective pump.
Count VII: Breach of Implied Warranty of Merchantability
120. Plaintiff incorporates by reference, as if fully set forth herein, each and every
allegation contained in the preceding paragraphs of this Complaint.
121. The SynchroMed II Devices implanted in Plaintiff violate the Federal Food, Drug,
and Cosmetic Act and federal regulations promulgated pursuant thereto, and violates California
law that parallels federal requirements, in one or more of the following ways:
19 Ex. 13, Medtronic Limited Warranty Special Notice for Medtronic Pump System ¶ A(1).
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 38 of 42

Complaint – Page 39 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
122. Plaintiff’s SynchroMed II Devices were adulterated in violation of federal law, as
pled in paragraph 76, supra.
123. Plaintiff’s SynchroMed II Devices were misbranded in violation of federal law, as
pled in paragraph 77, supra.
124. Plaintiff’s SynchroMed II Devices were adulterated in violation of California law,
as pled in paragraph 78, supra
125. At all times relevant hereto, Medtronic, as a merchant of medical devices including
the SynchroMed II Device, impliedly warranted to Plaintiff that her SynchroMed II Devices were
fit for the ordinary purposes for which it would be used—the intrathecal administration of
medication.
126. Defendants breached their implied warranty of merchantability in violation of Cal.
Civ. Code § 1792 because the Defendants’ numerous violations of federal and state laws and
regulations resulted in the manufacture, sale, and distribution of defective SynchroMed II Devices
that were therefore unfit for their ordinary purpose.
127. As a direct and proximate result of Defendants’ breach of its implied warranty, the
SynchroMed II Devices implanted in Plaintiff failed and required removal and replacement
surgeries, causing Plaintiff to suffer injury and damages, including pain and suffering, mental
anxiety and anguish, and medical bills.
Count VIII: Breach of Implied Warranty of Fitness for a Particular Purpose
128. Plaintiff incorporates by reference, as if fully set forth herein, each and every
allegation contained in the preceding paragraphs of this Complaint.
129. The SynchroMed II Devices implanted in Plaintiff violate the Federal Food, Drug,
and Cosmetic Act and federal regulations promulgated pursuant thereto, and violates California
law that parallels federal requirements, in one or more of the following ways:
130. Plaintiff’s SynchroMed II Devices were adulterated in violation of federal law, as
pled in paragraph 76, supra.
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 39 of 42

Complaint – Page 40 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
131. Plaintiff’s SynchroMed II Devices were misbranded in violation of federal law, as
pled in paragraph 77, supra.
132. Plaintiff’s SynchroMed II Devices were adulterated in violation of California law,
as pled in paragraph 78, supra
133. At all times relevant hereto, Medtronic, as a merchant of medical devices including
the SynchroMed II Device, had reason to know that its SynchroMed Devices would be used for
the particular purpose of intrathecal administration of non-indicated medication.
134. At all times relevant hereto, Plaintiff and her healthcare providers relied on
Medtronic’s skill and judgment in selecting and furnishing SynchroMed II Devices for that
purpose, and Medtronic had reason to know of that reliance.
135. Medtronic therefore impliedly warranted to Plaintiff that her SynchroMed II
Devices were fit for the particular purpose for which they would be used—the intrathecal
administration of non-indicated medication.
136. Defendants breached their implied warranty of fitness for a particular purpose in
violation of Cal. Civ. Code § 1792.1, because Defendants’ numerous violations of federal and state
laws and regulations resulted in the manufacture, sale, and distribution of defective SynchroMed
II Devices that were therefore unfit for their particular purpose.
137. As a direct and proximate result of Defendants’ breach of its implied warranty, the
SynchroMed II Devices implanted in Plaintiff failed and required removal and replacement
surgeries, causing Plaintiff to suffer injury and damages, including pain and suffering, mental
anxiety and anguish, and medical bills.
Count IX: Punitive Damages
138. Plaintiff incorporates by reference as if fully set forth herein, each and every
allegation contained in the preceding paragraphs of this Complaint.
139. Defendants knew or should have known that the SynchroMed II Device was
defective and presented an unreasonable risk of harm to Plaintiff.
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 40 of 42

Complaint – Page 41 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
140. Defendants’ conduct as described in this Complaint, for which Plaintiff is entitled
to recover compensatory damages, manifested the entire want of care such that it demonstrated a
malicious, despicable, willful, and conscious disregard of the safety of those persons who might
foreseeably have been harmed by the SynchroMed II Device, including Plaintiff, justifying the
imposition of punitive damages pursuant to Cal. Civ. Code § 3294(a).
WHEREFORE, Plaintiff prays for the following:
(a) That Plaintiff recover from Defendants, jointly and severally, general and special
damages, all in an amount to be determined by a jury of Plaintiff’s peers;
(b) That Plaintiff recover against Defendants for their wrongful conduct such punitive
damages that will punish and deter similar conduct, all in an amount to be determined by a jury of
Plaintiff’s peers;
(c) That Plaintiff recover reasonable attorneys’ fees and expenses of litigation; and
(d) That Plaintiff has such other and further relief as this Honorable Court deems just
and proper under the circumstances.
Dated: September 8, 2020 Respectfully Submitted:
/s/ Laura J. Baughman Laura J. Baughman CA State Bar No. 263944 MARTIN | BAUGHMAN, PLLC 3141 Hood Street, Suite 600 Dallas, Texas 75219 Tel. 214-761-6614 Fax. 214-744-7590 Email: [email protected] - and -
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 41 of 42

Complaint – Page 42 of 42
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
Ellen A. Presby Texas Bar No. 16249600 (Pro Hac Vice To Be Filed) Van Wey Law, PLLC 12720 Hillcrest Road, Suite 600 Dallas, Texas 75230 Tel. 214- 329-1350 Fax: 800-582-1042 Email: [email protected] Attorneys for Plaintiff Nancy Kilmer
Case 1:20-cv-01277-AWI-JDP Document 1 Filed 09/08/20 Page 42 of 42

Exhibit 1
Case 1:20-cv-01277-AWI-JDP Document 1-1 Filed 09/08/20 Page 1 of 5

1/23/2020 2006 > Medtronic, Inc. 29-Aug-06
wayback.archive-it.org/7993/20170112201820/http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2006/ucm076047.htm# 1/4
Archived ContentThe content on this page is provided for reference purposes only. Thiscontent has not been altered or updated since it was archived.Search Archive
Medtronic, Inc. 29-Aug-06
Department of Health and Human Services Public Health Service Food and DrugAdministration
Minneapolis District OfficeCentral Region212 Third Avenue SouthMinneapolis, MN 55401Telephone: (612) 758-7133FAX: (612) 334-4142
August 29, 2006WARNING LETTER
CERTIFIED MAILRETURN RECEIPT REQUESTED
Refer to MIN 06- 35Arthur D . Collins, Jr.Chairman of the Board and Chief Executive OfficerMedtronic, Inc .710 Medtronic ParkwayMinneapolis, MN 55432Dear Mr. Collins:During a May 18 - June 22, 2006, inspection of your establishment, MedtronicNeurological, located at 800 - 53rd Avenue NE, Minneapolis, MN 55421, ourinvestigators determined that your firm manufactures implantable drug infusion andneurostimulation products to treat pain, movement disorders, and other medicalconditions. These products are devices as defined by Section 201(h) of the FederalFood, Drug, and Cosmetic Act (the Act) [21 U.S.C. 321(h)] because they areintended for use in the diagnosis of disease or other conditions or in the cure,mitigation, treatment, or prevention of disease, or are intended to affect thestructure or function of the body.This inspection revealed that these devices are adulterated under Section 501(h) ofthe Act [21 U.S.C. 351(h)], in that the methods used in, or the facilities or controlsused for, their manufacture, packing, storage, or installation are not in conformancewith the Current Good Manufacturing Practice (CGMP) requirements for medicaldevices which are set forth in the Quality System regulation, found at Title 21, Codeof Federal Regulations (CFR), Part 820. Significant deviations include, but are notlimited to, the following:
Inspections, Compliance, Enforcement, and Criminal InvestigationsHome Inspections, Compliance, Enforcement, and Criminal Investigations ComplianceActions and Activities Warning Letters 2006
Case 1:20-cv-01277-AWI-JDP Document 1-1 Filed 09/08/20 Page 2 of 5

1/23/2020 2006 > Medtronic, Inc. 29-Aug-06
wayback.archive-it.org/7993/20170112201820/http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2006/ucm076047.htm# 2/4
1. Failure to implement procedures to ensure that a device's design inputrequirements are appropriate and address its intended use, including user/patientneeds, as required by 21 CFR 820.30(c). Design input work for the 8731 IntrathecalCatheter has not resulted in development of a complete design specification for thePlatinum/ Iridium (Pt/Ir) catheter tip bond. (For more detail on this deviation, seeFDA-483 observation # 1 from the May 18 - June 22, 2006, inspection. Copy of FDA483 attached.)2 . Failure to conduct design validation using production units or their equivalents,as required by 21 CFR 820.30(g). Design validation testing of the Model 8731Catheter was conducted with catheters manufactured with a Pt/Ir tip markerbonding process that was different than the process eventually used in production.(See FDA-483 observation #2.)3. Failure to validate a process whose results cannot be fully verified by subsequentinspection and test as required by 21 CFR 820.75(a). For the 8731 Catheter, thePt/Ir tip bonding process has not been validated. (See FDA-483 observation #3.)4. Failure to control production processes to ensure that a device conforms to itsspecifications, as required by 21 CFR 820.70(a). For the 8731 Catheter, the tipbonding manufacturing procedures contained:
an [redacted] of the tip, andinstructions to [redacted] equipment that was no longer in service. (See FDA-483 observation #4.)
5. Failure to implement corrective and preventive action procedures addressing theinvestigation of the cause of nonconforrnities relating to product, processes, and thequality system as required by 21 CFR 820.100(a)(2). Examples include:a. Corrective / Preventive Action System (C/PAS) 747 (re: 8731 tip detachments)was closed with a root cause analysis that conflicts with information received incomplaints. No additional C/PAS was opened to address the complaints and failuresthat do not fit the root cause analysis in C/PAS 747. (See FDA-483 observation#5a.)b. Product Comment Report (PCR) 170998 reported an 8731 catheter tipdetachment and stated that " . . .post-operative the patient showed pain in the leftleg, which can be related with the remaining tip ." In conflict with this reportedevent, a Health Hazard Analysis and "TECH NOTE" concluded that none of the tipdetachments were associated with adverse clinical or neurological consequences.(See FDA-483 observation #5b.)c. System Correction Request (SCR) 877, which addresses pump motor stalls due[redacted] to failures in Synchromed EL implantable infusion pumps, was closedwithout evidence to support conclusions that were made. (See FDA-483 observation#5c.)6. Failure to-implement changes in methods and procedures needed to correct andprevent identified quality problems, as required by 21 CFR 820.100(a)(5). C/PAS747 called for a redesign of the catheter tip and a new product specification defininga requirement for [redacted]. However, the product specification was not changed,and as a result, the revised manufacturing process was not validated, and noprocess monitoring was conducted. As of the inspection, [redacted] complaints hadbeen received involving tip dislodgements in catheters produced after the redesignof the tip. (See FDA-483 observation #6.)7. Failure to identify all of the actions needed to correct and prevent the recurrenceof nonconforming product and other quality problems, as required by 21 CFR820.100(a)(3). In particular:a. C/PAS 747, which covered detachment of Pt/Ir tips in Model 8731 Catheters, didnot include an action to address 8731 Catheters that were in finished goods oralready distributed. (See FDA-483 observation #7a.) (NOTE: These Model 8731Intrathecal Catheters were eventually recalled by your firm on July 21, 2006.)b. A field corrective action was not conducted until June 6, 2006, to addressrecurring Catheter Access Port (CAP) detachment failures in Synchromed EL
Case 1:20-cv-01277-AWI-JDP Document 1-1 Filed 09/08/20 Page 3 of 5

1/23/2020 2006 > Medtronic, Inc. 29-Aug-06
wayback.archive-it.org/7993/20170112201820/http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2006/ucm076047.htm# 3/4
implantable infusion pumps. (See FDA-483 observation #7b.)8 . Failure to implement procedures to ensure that device history records for eachbatch, lot, or unit are maintained to demonstrate that the device is manufactured inaccordance with the device master record and the Quality Systems regulation asrequired by 21 CFR 820.184. Specifically:a. Traceability Cards for some Synchromed EL implantable infusion pumps did notinclude complete records of operations that were conducted under ManufacturingProcess Variances or Product Review Requests (PRR's). (See FDA-483 observation#8a.)b. A copy of process variance 1955, which covered [redacted] of Synchromed ELpumps, was not maintained in the documentation control system. (See FDA-483observation #8b.)This letter is not intended to be an all-inclusive list of deficiencies at your facility . Itis your responsibility to ensure compliance with the Act and regulations. The specificviolations noted in this letter and in the Form FDA-483 issued at the close of theinspection may be symptomatic of serious underlying problems in your firm'smanufacturing and quality assurance systems. You are responsible for investigatingand determining the causes of the violations identified by the FDA. You also mustpromptly initiate permanent corrective and preventive action to bring your productsinto compliance.Federal agencies are advised of the issuance of all Warning Letters about devices sothat they may take this information into account when considering the award ofcontracts. Additionally, no premarket approval applications for Class III devices towhich the Quality System regulation deficiencies are reasonably related will beapproved until the violations have been corrected. Also, no requests for Certificatesto Foreign Governments will be granted until the violations related to the subjectdevices have been corrected.You should take prompt action to correct the deviations described in this letter.Failure to promptly correct these deviations may result in regulatory action beinginitiated by the Food and Drug Administration without further notice. These actionsinclude, but are not limited to, seizure, injunction, and/or civil money penalties.On July 24, 2006, we received an undated letter from George Aram, Vice Presidentof Quality, Neurological Sector, which describes corrective actions taken and plannedby your firm to address the FDA-483 Inspectional Observations. Only two of thecorrective actions (for FDA-483 observations # 8 and 9) have been completed. Mr.Aram provided target completion dates for corrective actions to address theremaining FDA-483 Inspectional Observations, and he stated that monthly progressreports would be provided to our office beginning on August 28, 2006 . At this time,based on the limited information that has been provided, we are unable todetermine whether your corrective actions are appropriate. In order to fully assessthe implementation and effectiveness of the corrections, we will need to conduct afollow-up inspection.[Redacted]Please notify this office in writing within 15 working days to acknowledge receipt ofthis letter and to provide an update on the status of your corrective actions. Yourresponse should be sent to Timothy G. Philips, Compliance Officer, at the address onthis letterhead.Sincerely,/S/W. Charles BecoatDirectorMinneapolis District Page Last Updated: 07/08/2009 Note: If you need help accessing information in different file formats, see Instructions for Downloading
Case 1:20-cv-01277-AWI-JDP Document 1-1 Filed 09/08/20 Page 4 of 5

1/23/2020 2006 > Medtronic, Inc. 29-Aug-06
wayback.archive-it.org/7993/20170112201820/http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2006/ucm076047.htm# 4/4
Viewers and Players.Language Assistance Available: Español | 繁體中文 | Tiếng Việt | 한국어 | Tagalog | Русский | العربیة | KreyòlAyisyen | Français | Polski | Português | Italiano | Deutsch | 日本語 | فارسی | English
Accessibility Contact FDA Careers FDA Basics FOIA No FEAR Act Site Map Nondiscrimination WebsitePolicies
U.S. Food and Drug Administration10903 New Hampshire Avenue Silver Spring, MD 20993 Ph. 1-888-INFO-FDA (1-888-463-6332)Contact FDA
For Government For PressCombination Products Advisory Committees Science & Research Regulatory Information SafetyEmergency Preparedness International Programs News & Events Training and Continuing EducationInspections/Compliance State & Local Officials Consumers Industry Health Professionals FDA Archive
Links on this page:
Case 1:20-cv-01277-AWI-JDP Document 1-1 Filed 09/08/20 Page 5 of 5

Exhibit 2
Case 1:20-cv-01277-AWI-JDP Document 1-2 Filed 09/08/20 Page 1 of 6

1/23/2020 2007 > Medtronic, Inc. 03-Jul-07
wayback.archive-it.org/7993/20170112201127/http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2007/ucm076435.htm# 1/5
Archived ContentThe content on this page is provided for reference purposes only. Thiscontent has not been altered or updated since it was archived.Search Archive
Medtronic, Inc. 03-Jul-07
Department of Health and Human Services Public Health Service Food and DrugAdministration
Minneapolis District Office Central Region 212 Third Avenue South Minneapolis, MN 55401Telephone: (612) 758-7133 FAX: (612) 334-4142
July 3, 2007WARNING LETTER
CERTIFIED MAIL RETURN RECEIPT REQUESTED
Refer to MIN 07 - 18Arthur D. Collins, Jr. Chairman of the Board and Chief Executive OfficerMedtronic, Inc.710 Medtronic ParkwayMinneapolis, Minnesota 55432Dear Mr. Collins:During a limited inspection of your establishment, Medtronic Neuromodulation1, locatedat 800 53rd Avenue Northeast, Minneapolis, Minnesota, 55421, on November 21, 2006,through January 24, 2007, investigators from the Food and Drug Administration (FDA)determined that your establishment manufactures implantable drug infusion andneurostimulation products. Under section 201(h) of the Federal Food, Drug, andCosmetic Act (the Act), 21 U.S.C. § 321(h), these products are devices because they areintended for use in the diagnosis of disease or other conditions, or in the cure,mitigation, treatment, or prevention of disease, or are intended to affect the structure oany function of the body.Our inspection revealed that your devices are adulterated within the meaning of section501(h) of the Act [21 U.S.C. § 351(h)], in that the methods used in, or the facilities orcontrols used for, their manufacture, packing, storage, or installation are not inconformity with the Current Good Manufacturing Practice (CGMP) requirements of theQuality System (QS) regulation found at Title 21, Code of Federal Regulations, (21CFR) Part 820. We received responses from Mr. George Aram, Vice President of Qualityand Compliance, dated February 23, 2007, March 30, 2007, April 30, 2007, and June 4,2007, concerning our investigators' observations noted on the Form FDA 483, List ofInspectional Observations, that was issued to officials at your establishment. We addressthese responses below, in relation to each of the noted violations. These violationsinclude, but are not limited to:Failure to implement complaint handling procedures to ensure that allcomplaints are evaluated to determine whether the complaint represents an
Inspections, Compliance, Enforcement, and Criminal InvestigationsHome Inspections, Compliance, Enforcement, and Criminal Investigations Compliance Actionsand Activities Warning Letters 2007
Case 1:20-cv-01277-AWI-JDP Document 1-2 Filed 09/08/20 Page 2 of 6

1/23/2020 2007 > Medtronic, Inc. 03-Jul-07
wayback.archive-it.org/7993/20170112201127/http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2007/ucm076435.htm# 2/5
p p pevent that must be filed as a Medical Device Report under 21 CFR Part 803, asrequired by 21 CFR 820.198(a)(3).It is our understanding that your establishment documents product complaints in yourProduct Comment Reporting (PCR) system. During the inspection, our investigatorsfound on site several medical and/or scientific literature articles concerning adverseevents relating to your devices that had not been entered into your PCR system andevaluated for reportability under 21 CFR Part 803 (Medical Device Reporting). SeeObservation #4 in the Form FDA 483 issued on January 24, 2007. A manufacturer hasan obligation to submit an MDR report under Part 803 once it becomes aware ofinformation, from any source, that reasonably suggests that a device it markets mayhave caused or contributed to an MDR reportable event (21 CFR 803.50). Therefore,your firm should have considered whether the events described in these medical and/orscientific articles would represent reportable events under 21 CFR Part 803.In response to this observation, your firm drafted a new literature review SOP thatincludes proactive search methods for selecting relevant articles and reviewing them todetermine their reportability. As part of your response, you also provided a new workinstruction entitled "Medical Device Reporting" to facilitate the implementation of thenew literature review SOP. This portion of your response appears to be adequate and wilbe further evaluated at a future inspection of your facility.Your responses also state that Medtronic Neurological met with CDRH, Office ofSurveillance and Biometrics (OSB), on February 2, 2007, to discuss retrospectivereporting of MDR reports based on scientific literature. Your firm states that you[redacted]Our inspection revealed that your devices are misbranded under section 502(t)(2) of theAct [21 U.S.C. § 352(t)(2)], in that your firm failed or refused to furnish material orinformation respecting the device that is required by or under section 519 of the Act (21U.S.C. § 360i), and 21 CFR Part 803--Medical Device Reporting (MDR) regulation.Significant deviations include, but are not limited to:Failure to submit MDR reports within 30 days of receiving or otherwisebecoming aware of information that reasonably suggests that a marketeddevice may have caused or contributed to a death or serious injury, as requiredby 21 CFR 803.50(a)(1).Medtronic failed to submit MDR reports for serious injury adverse events that werereported by or confirmed by a health care professional, or that were reported by apatient or a patient's family member. Examples of this violation include, but are notlimited to, the following PCRs:58709, 235359, 258561, 234149, 183288, 202853, 267989, 55251, 94553, 119033,180984, 246172, 255091, 277026, 191620, 95901, 171432, 196649, 248557, 189519,167978, 61760, 95681, 170773, 186498, 187587, 190010, 196714, 202096, 206578,222730, 250677, 267713, 248978, 221032, 250099, and 269319.Many of these PCRs involve a granuloma or inflammatory mass at or near the distal tipof the intrathecal catheter used with the SynchroMed pump, which are reportable asserious injuries. Some of these were surgically removed and some of the patientsreported increased pain, tingling sensation in the legs, partial paralysis, total lower limbparalysis and other gait problems resulting from the granuloma or inflammatory mass.Some of the PCRs included a fracture of the intrathecal catheter. It is important to notethat the MDR regulation also provides for the submission of a malfunction MDR forevents in which the information reasonably suggests that a device you market hasmalfunctioned and would be likely to cause or contribute to a reportable death or seriousinjury if the malfunction were to recur. Your firm should have considered whether thefailures reported in the PCRs referenced above would have constituted reportable eventsunder 21 CFR Part 803.Your firm also failed to submit MDR reports within 30 days of becoming aware ofliterature articles that referenced problems to which your devices may have caused orcontributed. These include, but are not limited to, articles by Deer, McMillan et al., Hu etal., Kofler et al., and Loughrey et al. These articles included, among other things,information on pump malfunctions, catheter separation or fracture, and inflammatorymasses and granulomas.In addition, during the inspection of your facility, our investigators collected abstracts ofseveral literature articles. The articles associated with these abstracts must be reportedas MDRs if they discuss deaths, serious injuries, or malfunctions of your devices that
Case 1:20-cv-01277-AWI-JDP Document 1-2 Filed 09/08/20 Page 3 of 6

1/23/2020 2007 > Medtronic, Inc. 03-Jul-07
wayback.archive-it.org/7993/20170112201127/http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2007/ucm076435.htm# 3/5
would be likely to cause or contribute to a death or serious injury, if the malfunctionwere to recur.Your firm's responses indicate that you interpreted the MDR regulation to mean that anyconsumer self-reported events were not MDR reportable unless separately confirmed bya Health Care Professional (HCP). This interpretation of the MDR regulation is incorrect.Consumer self-reported events do not have to be confirmed by a HCP in order todetermine reportability. Under 21 CFR 803.50, a firm has 30 calendar days after the dayit receives or otherwise becomes aware of information, from any source, that reasonablysuggests that a device it markets may have caused or contributed to an MDR reportableevent. If, in the process of conducting an investigation, your firm contacts an HCP foradditional information, then the additional information can be used by the firm to helpmake a determination about the MDR reportability of the consumer complaint.Your responses also state that the MDR Work Instruction was revised to include arequirement to assess consumer self-reported events (whether or not confirmed by aHCP) and catheter events for MDR reportability. A copy of this revised procedure wasprovided as part of your responses. Your revised work instruction appears to adequatelyaddress our concern regarding the reporting of consumer self-reported events. However,this corrective action will be further assessed at a future inspection of your facility.Our inspection further revealed that your devices are misbranded under section 502(t)(2) of the Act [21 U.S.C. § 352(t)(2)], in that your firm failed or refused to furnishmaterial or information respecting the device that is required by or under section 519 ofthe Act, 21 U.S.C. § 360i, and 21 CFR Part 806 - Reports of Corrections and Removalsregulation. Significant deviations include, but are not limited to:A correction or removal conducted to reduce a risk to health posed by a devicewas not reported in writing to FDA, as required by 21 CFR 806.10(a)(1).In July 2003 your establishment sent a letter with an enclosed "EDUCATIONAL BRIEF,"entitled "Information about Inflammatory Mass," to SynchroMed customers (physicians)Also enclosed were reprints of two articles published in the December 2002 issue of PainMedicine and revised labeling for the SynchroMed Technical Manual. FDA defines a"correction" in 21 CFR 806.2(d) as " . . .the repair, modification, adjustment, relabeling,destruction, or inspection (including patient monitoring) of a device without its physicalremoval from its point of use to some other location." FDA believes that the July 2003Educational Brief, which was sent to all customers using SynchroMed pumps, meets thedefinition of "correction" in that the letter provided updated labeling to customers fordevices that were already in distribution.
The FDA also believes that the July 2003 Educational Brief is a reportable correctionunder 21 CFR 806.10(a) (1) in that the letter contained specific information intended toreduce the risk to health posed by the device. For example, the July 2003 EducationalBrief specifically states that "[i]f an inflammatory mass is detected in its clinical course,prompt discontinuation of opioid delivery into the mass may cause it to shrink ordisappear without the need for surgical removal." The letter also specificallyrecommends catheter replacement, repositioning, and other interventional procedures,depending on the patient's clinical condition. These recommendations were neitherincluded in the pump's original labeling, nor conveyed to customers in a January 2001communication regarding inflammatory masses.Additionally, the July 2003 Educational Brief contained new "Post implant" warnings thatsuggest that clinicians should routinely monitor patients for prodromal clinical signs orsymptoms of inflammatory mass such as change in character, quality or intensity ofpain; reports of new radicular pain, especially at or near the dermatomal level of thecatheter tip; frequent or large escalations of daily drug dose to maintain the analgesiceffect; and dose escalations that may only temporarily alleviate the patient's increasingpain. These new warnings were not included in the January 2001 letter or the pump'soriginal technical manual.Furthermore, the journal articles included with the July 2003 Educational Brief statedwith regard to adverse event reporting that 41 adverse events regarding inflammatorymass were identified as of November 2000 (conveyed to customers in the January 2001letter). The articles also state that an additional 51 events were identified after the 2001letter had been distributed to customers. The articles suggest that the number of newadverse events has more than doubled in one year of reporting. It is noteworthy thatduring the most recent inspection of your facility, your firm calculated the current rate ofinflammatory masses to be approximately [redacted] events per [redacted] implants.
Case 1:20-cv-01277-AWI-JDP Document 1-2 Filed 09/08/20 Page 4 of 6

1/23/2020 2007 > Medtronic, Inc. 03-Jul-07
wayback.archive-it.org/7993/20170112201127/http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2007/ucm076435.htm# 4/5
This figure, which has not yet been communicated to your customers, suggests that therisk of inflammatory masses occurring at or near the tip of intrathecal catheters usedwith SynchroMed pumps is [redacted] greater than the [redacted] rate indicated inthe January 2001 letter.Your firm's responses to this observation stated that the July 2003 Inflammatory Mass"Educational Brief" was based upon your judgment that the information presented in theBrief was an update to a January 19, 2001, "Dear Colleague" letter that had beenreviewed by FDA prior to its issuance. You further stated that the Agency did notconsider the 2001 "Dear Colleague" letter to be a correction or removal at that time. Inaddition, you stated that the revised labeling contained in the July 2003 EducationalBrief had been previously reviewed by FDA as part of PMA Supplement P860004/S053,which was approved by FDA on October 9, 2002. Your firm indicated that the July 2003Educational Brief did not constitute additional information beyond the approved labelingin the PMA Supplement.FDA disagrees with your conclusion that the July 2003 Educational Brief was not acorrection or removal. Although the Educational Brief contained language consistent withthe approved labeling in PMA Supplement P860004/S053, this new labeling had notbeen previously communicated to physicians whose patients already had a SynchroMedpump implanted within them. Note that the 21 CFR Part 806 definitions andrequirements do not depend upon whether the revised labeling in the July 2003Education Brief had gone through the PMA supplement process or that FDA had priorknowledge of the information through a PMA supplement. Your firm is required to revieweach corrective action and/or removal and determine whether the requirements of theregulation have been met and thus require a report. Providing the information to FDAvia another requirement does not abrogate your responsibility to comply with therequirements of 21 CFR Part 806. If your firm determines that the event in question isnot reportable, you must provide an explanation of your decision not to submit aCorrections and Removals report and keep a record of this justification, as required by21 CFR 806.20.Our inspection also revealed that your firm has several procedures for Medical DeviceReporting and Adverse Drug Experience Reporting. These procedures, in turn, referenceseveral other procedures. Your firm's current problems regarding MDR reporting, asdiscussed above in this Warning Letter, may be exacerbated by the complexity of yourprocedures and might have contributed to your firm's deviations from the regulationsregarding MDR reporting.In addition, the inspection revealed several ongoing violations in your quality systemthat were also noted in the 483. In particular, you have failed to achieve consistentcompliance in areas such as design controls (21 CFR 820.30) and corrective andpreventive action (21 CFR 820.100). These areas had previously been found not to be incompliance during the inspection performed from May 18 through June 22, 2006. Thesequality system violations were also cited in an August 29, 2006, Warning Letter that wassent to you. By letter dated June 4, 2007, George Aram, Vice President of Quality,Neurological Sector, provided an update on the status of the corrective actions taken andplanned by your firm to address these violations. In that letter, Mr. Aram stated that thelongest remediation activities extend into November 2007. We encourage you toexpedite your efforts to achieve full compliance and to keep us informed of yourprogress.In your firm's June 4, 2007 response, you also indicated that your Risk Evaluation Board(REB) met on May 10, 2007, to [redacted]This letter is not intended to be an all-inclusive list of deficiencies at your facility. It isyour responsibility to ensure compliance with the Act and regulations. The specificviolations noted in this letter and in the Form FDA 483 issued at the close of theinspection may be symptomatic of serious underlying problems in your firm'smanufacturing and quality assurance systems. You are responsible for investigating anddetermining the causes of the violations identified by he FDA. You also must promptlyinitiate permanent corrective and preventive action to bring your products intocompliance.Federal agencies are advised of the issuance of all Warning Letters about devices so thatthey may take this information into account when considering the award of contracts.Additionally, no premarket approval applications for Class III devices to which theQuality System regulation deficiencies are reasonably related will be approved until theviolations have been corrected. Also, no requests for Certificates to Foreign
Case 1:20-cv-01277-AWI-JDP Document 1-2 Filed 09/08/20 Page 5 of 6

1/23/2020 2007 > Medtronic, Inc. 03-Jul-07
wayback.archive-it.org/7993/20170112201127/http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2007/ucm076435.htm# 5/5
Governments will be granted until the violations related to the subject devices havebeen corrected.You should take prompt action to correct the deviations described in this letter. Failure topromptly correct these deviations may result in regulatory action being initiated by theFood and Drug Administration without further notice. These actions include, but are notlimited to, seizure, injunction, and/or civil money penalties.Please notify this office in writing within 15 working days to acknowledge receipt of thisletter and to provide an update on the status of your corrective actions. Your responseshould be sent to Timothy G. Philips, Compliance Officer, at the address on thisletterhead.Sincerely,/S/W. Charles Becoat Director Minneapolis District TGP/ccl______________________________________________________________________1At the time of the FDA's inspection, the establishment was known as MedtronicNeurological. Page Last Updated: 07/07/2009 Note: If you need help accessing information in different file formats, see Instructions for Downloading Viewersand Players.Language Assistance Available: Español | 繁體中文 | Tiếng Việt | 한국어 | Tagalog | Русский | العربیة | KreyòlAyisyen | Français | Polski | Português | Italiano | Deutsch | 日本語 | فارسی | English
Accessibility Contact FDA Careers FDA Basics FOIA No FEAR Act Site Map Nondiscrimination WebsitePolicies
U.S. Food and Drug Administration10903 New Hampshire Avenue Silver Spring, MD 20993 Ph. 1-888-INFO-FDA (1-888-463-6332)Contact FDA
For Government For PressCombination Products Advisory Committees Science & Research Regulatory Information Safety EmergencyPreparedness International Programs News & Events Training and Continuing EducationInspections/Compliance State & Local Officials Consumers Industry Health Professionals FDA Archive
Links on this page:
Case 1:20-cv-01277-AWI-JDP Document 1-2 Filed 09/08/20 Page 6 of 6

Exhibit 3
Case 1:20-cv-01277-AWI-JDP Document 1-3 Filed 09/08/20 Page 1 of 9

Case 1:20-cv-01277-AWI-JDP Document 1-3 Filed 09/08/20 Page 2 of 9

Case 1:20-cv-01277-AWI-JDP Document 1-3 Filed 09/08/20 Page 3 of 9

Case 1:20-cv-01277-AWI-JDP Document 1-3 Filed 09/08/20 Page 4 of 9

Case 1:20-cv-01277-AWI-JDP Document 1-3 Filed 09/08/20 Page 5 of 9

Case 1:20-cv-01277-AWI-JDP Document 1-3 Filed 09/08/20 Page 6 of 9

Case 1:20-cv-01277-AWI-JDP Document 1-3 Filed 09/08/20 Page 7 of 9

Case 1:20-cv-01277-AWI-JDP Document 1-3 Filed 09/08/20 Page 8 of 9

Case 1:20-cv-01277-AWI-JDP Document 1-3 Filed 09/08/20 Page 9 of 9

Exhibit 4
Case 1:20-cv-01277-AWI-JDP Document 1-4 Filed 09/08/20 Page 1 of 9

1/23/2020 2009 > Medtronic Puerto Rico Operations Company
wayback.archive-it.org/7993/20170112195621/http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2009/ucm168451.htm# 1/8
Archived ContentThe content on this page is provided for reference purposes only. Thiscontent has not been altered or updated since it was archived.Search Archive
Medtronic Puerto Rico Operations Company
Department of Health and Human Services
Public Health ServiceFood and DrugAdministration
San Juan DistrictCompliance Branch466 Fernandez JuncosAvenueSan Juan Puerto Rico00901-3223Telephone: 787-474-9500FAX: 787-729-6658
June 1, 2009
WARNING LETTERSJN-2009-08
Certified MailReturn Receipt Requested
Mr. William A. HawkinsCEO and PresidentMedtronic Inc.710 Medtronic ParkwayMinneapolis, MN 55432-5604
Dear Mr. Hawkins:
Food and Drug Administration
During an inspection of your firm located at Road 31 Km 24 Ceiba Norte IndustrialPark Juncos, Puerto Rico, on November 12, 2008, through December 15, 2008,investigators from the United States Food and Drug Administration (FDA)determined that your firm manufactures Synchromed® II Pumps and MiniMedParadigm® Insulin Pumps. Under section 201(h) of the Federal Food, Drug andCosmetic Act (the Act), 21 U.S.C. § 321(h), these products are devices because theyare intended for use in the diagnosis of disease or other conditions or in the cure,mitigation, treatment, or prevention of disease, or are intended to affect thestructure or function of the body.
Inspections, Compliance, Enforcement, and Criminal InvestigationsHome Inspections, Compliance, Enforcement, and Criminal Investigations ComplianceActions and Activities Warning Letters 2009
Case 1:20-cv-01277-AWI-JDP Document 1-4 Filed 09/08/20 Page 2 of 9

1/23/2020 2009 > Medtronic Puerto Rico Operations Company
wayback.archive-it.org/7993/20170112195621/http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2009/ucm168451.htm# 2/8
This inspection revealed that the Synchromed® II Pumps are adulterated within themeaning of section 501 (h) of the Act (21 U.S.C. §351 (h)), in that the methodsused in, or the facilities or controls used for, their manufacture, packing, storage, orinstallation are not in conformity with the Current Good Manufacturing Practice(CGMP) requirements of the Quality System (QS) regulation found at Title 21, Codeof Federal Regulations (C.F.R.), Part 820. We received written responses from Mr.Manuel Santiago, Vice President of Medtronic Puerto Rico Operations Company(MPROC), dated January 20, 2009, and March 31,2009, concerning ourinvestigators' observations noted on the form FDA 483, List of InspectionalObservations that was issued to your firm. We address these responses below, inrelation to each of the noted violations. These violations include, but are not limitedto, the following:
1) Failure to establish and maintain process control procedures that describe anyprocess controls necessary to ensure conformance to specifications, which shallinclude monitoring and control of process parameters and component and devicecharacteristics during production, as required by 21 CFR 820.70(a).
For example:
a) Multiple Synchromed® II Pumps were released for distribution and implanted inpatients even though they were not filled with propellant as required by yourProcess Operation Description (POD) (b) (4) Your firm's investigation,Nonconformance Report (NCR) (b) (4) which started in (b) (4) found that severalimplantable pumps, including serial numbers NGV300069H, NGV301133H,NGP302823H, NGV300225H, NGV401554H, NGV4022253H, NGP307091H,NGP301055H, and NGP304851H, were released to the market without being filledwith propellant and this was not discovered in the propellant weight check duringmanufacturing. Your firm's manufacturing step requires a (b) (4) after thepropellant is added to the pump. The 100% mass check was ineffective to identifythat devices lacked the propellant. You became aware of this situation afterconfirming two complaints receive on (b) (4) (Product Comment Report (PCR) (b)(4) and (b) (4) (PCR (b) (4) PCR (b) (4) states that the product had to beexplanted because of issues related to the lack of propellant. PCR (b) (4) created in(b) (4) also documented that two pumps had to be explanted on (b) (4) and (b)(4) due to lack of propellant.
b) On June 23, 2008, at the (b) (4) one Synchromed® II Pump was found that didnot show evidence of a perforated septum. The (b) (4) is performed at this station.The (b) (4) is performed to detect obstruction in the (b) (4) early in themanufacturing process. (b) (4)As part of your firm's assessment (NonconformanceEvaluation Request (NCER) (b) (4) that were at this manufacturing stage werevisually inspected. This inspection revealed that (b) (4) of the (b) (4)Synchromed® II Pumps did not contain the (b) (4) indicating that the (b) (4) wasnot conducted on these (b) (4) Synchromed® II Pumps.
c) On June 25, 2008, at the (b) (4) one Synchromed® II Pump was found withouta (b) (4) at the (b) (4) The (b) (4) needs to be perforated to test the (b) (4) The(b) (4) is a safety mechanism that serves to assure that the pump is neveroverfilled. As part of your firm's assessment (NCER (b) (4) ,the Synchromed® IIPumps in the firm's existing inventory at MPROC were visually inspected. (b) (4)were found without the (b) (4) However, the electronic device history record forthese devices showed entries indicating that the (b) (4) was conducted. Your firmexpanded the scope of the investigation (NCR (b) (4) and found (b) (4) additionalSynchromed® II Pumps where the (b) (4) pressure was not conducted and (b) (4)devices with testing discrepancies. Your firm's investigation further determined thata total of (b) (4) Synchromed® II Pumps had records that indicated that the (b)(4) was performed, when the test was not actually conducted. Of these affecteddevices, (b) (4) pumps were distributed to customers.
Case 1:20-cv-01277-AWI-JDP Document 1-4 Filed 09/08/20 Page 3 of 9

1/23/2020 2009 > Medtronic Puerto Rico Operations Company
wayback.archive-it.org/7993/20170112195621/http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2009/ucm168451.htm# 3/8
We have reviewed your responses dated January 20, 2009, and March 31, 2009, andour conclusions follow:
a) Regarding the corrective actions that your firm has taken to address theSynchromed® II Pumps with the missing propellant, you initially identified thisproblem in May 2006. You initiated a corrective and preventive action (CAPA)investigation in January 2007, determined the root cause to be related to the (b)(4) failing to properly fill propellant into the Synchromed® II Pump reservoir, andfailure of (b) (4) to verify the fill weight of devices after being processed throughthe filling equipment. Your firm conducted a Health Hazard Assessment in March2008. In May 2008, your firm conducted a voluntary recall of the Synchromed® IIPumps that did not contain any propellant, and notified the FDA. Your firm'sresponse indicates that MPROC has confirmed that the corrective actions regardingthe Synchromed® II Pumps with the missing propellant were completed andeffective. FDA is concerned with your failure to initiate a recall for devices affectedby the propellant problem in a timely manner. Based on the chronology identified inyour response, it took almost 2 years from when the missing propellant was initiallyidentified to conduct a recall. The adequacy of your response cannot be determinedat this time. FDA will assess the effectiveness of your firm's recall procedures andCAPA's during the next inspection.
b) Regarding the actions that your firm has taken to prevent recurrence ofSynchromed® II Pumps from being distributed without propellant, you conductedprocess validation for the manufacturing process changes between April and May2007. Subsequently, you updated your procedures and re-trained your personnel onthese procedures. The adequacy of your response cannot be determined at thistime. FDA will assess the effectiveness of your CAPA's during the next inspection.
c) Regarding the failure to conduct the and the (b) (4) and (b) (4) the adequacy othe response cannot be determined at this time. Based on your response, the rootcause was determined to be related to (b) (4) manufacturing instructions for theSynchromed® II Pumps. MPROC has performed detailed Health Hazard Analyses forthese two problems. Your firm has established additional checkpoints in themanufacturing process to verify the (b) (4) and (b) (4) are being completed;reviewed the manufacturing process to ensure that the steps were correct andspecific; retrained employees in performing the manufacturing steps; andestablished additional oversight by increasing the internal process audits of theSynchromed® II Pump manufacturing operation. Your firm identified otherimprovement actions that will be implemented within the next year, as identified bythe timetable in your responses. The adequacy of your corrective and preventiveactions will be determined during the next inspection.
2) Failure to establish and maintain procedures for implementing corrective andpreventive action that include identifying the action(s) needed to correct andprevent recurrence of nonconforming product and other quality problems, asrequired by 21 CFR 820.100(a).
For example:
On October 5, 2008, your firm performed a (b) (4) of data from the (b) (4)records (which stores the results of in-process testing) and the (b) (4)manufacturing records (which controls the manufacturing process for theSynchromed® II Pump). The intent of the (b) (4) was to provide another level ofoversight to ensure that in-process tests were actually being performed on devices,as they progressed through manufacturing. This report, however, revealed thatanother step, (b) (4) for each Synchromed® II Pump, was not performed duringmanufacturing. (b) (4) are unique to each device and have values that vary from(b) (4) This constant is used by the device in critical internal functions such as
Case 1:20-cv-01277-AWI-JDP Document 1-4 Filed 09/08/20 Page 4 of 9

1/23/2020 2009 > Medtronic Puerto Rico Operations Company
wayback.archive-it.org/7993/20170112195621/http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2009/ucm168451.htm# 4/8
calculating drug reservoir levels and drug dispensing rates. Our investigators foundover (b) (4) complaints in your firm's complaint handling system related toaccuracy rates. The (b) (4), report did not reference any NCR or other type ofinvestigation into this problem.
We have reviewed your responses dated January 20, 2009, and March 31, 2009, andour conclusions follow:
Your responses state that a comprehensive review of the CAPA procedures at MPROCwill be conducted by July 31, 2009. The adequacy of your response cannot bedetermined at this time. The adequacy of your firm's corrective actions will bedetermined during the next inspection.
3) Failure to establish and maintain procedures to ensure that Device HistoryRecords (DHR's) for each batch, lot, or unit are maintained to demonstrate that thedevice is manufactured in accordance with the Device Master Record (DMR), asrequired by 21 CFR 820.184.
Specifically, a review of thirteen (13) DHR's for the Synchromed® II Pumps revealedthat your firm's procedure entitled (b) (4) (Procedure POD (b) (4) Revision (b)(4) is not always followed. For example:
a) A comparison between DHR's for the Synchromed® II Pump serial numbersNGP319205H and NGV416698H, and the respective (b) (4) revealed that these twodevices were dispatched into the sterilizer after the (b) (4) Your procedures requirethat the devices be placed into the (b) (4)
b) DHR's for Synchromed® II Pump serial numbers NGV416743H, NGV404480H,NGV417063H, NGP306174H, NGV416451H, NGV416578H, NGV418943H, andNGP305847H show that the verification of the (b) (4) and (b) (4) and (b) (4)were recorded after the steam sterilization cycle had completed, and not prior toinitiating the cycle, as required by Procedure POD (b) (4)
We have reviewed your responses dated January 20,2009, and March 31, 2009, andour conclusions follow:
Your responses states that the devices described above went through the completesterilization process, and were determined to be sterile at the conclusion of thecycle. However, your firm acknowledges that the sterilization process was notperformed in the order specified by your procedures. The adequacy of your responsecannot be determined at this time. The adequacy of your firm's corrective andpreventive actions will be determined during the next inspection.
4) Failure to review, evaluate, and investigate complaints involving the possiblefailure of a device, labeling, or packaging to meet any of its specifications, asrequired by 21 CFR 820.198(c).
For example: (b) (4) received on (b) (4) and (b) (4) received on (b) (4) both describe eventswhere patients who were implanted with the Synchromed® II Pump developedinfections. A review of the DHR's for the devices identified in the PCR'sSynchromed® II Pump serial numbers NGP319205H and NGV416698H,respectively) show that the devices were dispatched into the sterilizer after the (b)(4) had already started. The complaint records stated that an investigation hadbeen opened to assess these complaints. However, a copy of this investigation wasnot included as part of the complaint record, there was no reference to a specific
Case 1:20-cv-01277-AWI-JDP Document 1-4 Filed 09/08/20 Page 5 of 9

1/23/2020 2009 > Medtronic Puerto Rico Operations Company
wayback.archive-it.org/7993/20170112195621/http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2009/ucm168451.htm# 5/8
investigation report number, and there was no documentation whether theinvestigation was successfully closed. Also, there was no record in the complaint filethat Medical Device Reports were filed by your firm with FDA for this complaint.
Your responses dated January 20, 2009 and March 31, 2009, did not address thischarge because it was not included in the FDA 483 issued to you on December 15,2008. The adequacy of your corrective and preventive actions will be determinedduring the next inspection.
Our inspection also revealed that your MiniMed Paradigm® Insulin Pumps aremisbranded under section 502(t)(2) of the Act [21 U.S.C. 352(t)(2)], in that yourfirm failed or refused to furnish material or information respecting the device that isrequired by or under section 519 of the Act, 21 U.S.C. 360i, and 21 C.F.R. Part 803 -Medical Device Reporting (MDR) regulation. Significant deviations include, but arenot limited to, the following:
5) Failure to report to FDA no later than 30 calendar days after the day that youreceive or otherwise become aware of information, from any source, that reasonablysuggests that a device that you market: (1) may have caused or contributed to adeath or serious injury; or (2) has malfunctioned and this device or a similar devicethat you market would be likely to cause or contribute to a death or serious injury, ifthe malfunction were to recur, as required by 21 CFR 803.50(a).
For example:
a) Complaint No. (b) (4) states that the reported complaint was not reportable asan MDR to the FDA based on testing of the returned MiniMed Paradigm® InsulinPump. Information in the complaint indicated that the patient was hospitalized fordiabetic ketoacidosis allegedly following battery problems with the pump. Thecomplaint file states that analysis of the pump did not find a battery problem. Yourfirm concluded that although "information does suggest that a device malfunctionoccurred," the malfunction was unlikely to result in death or injury if it were to recur
However, a review of the MDRs submitted by your firm to the FDA throughMedWatch shows that your firm has submitted serious injury MDRs with a diagnosisof diabetic ketoacidosis resulting from the use of the MiniMed Paradigm® InsulinPump. Since your firm has previously reported these MDRs where a patient hadbeen hospitalized for diabetic ketoacidosis from the use of the MiniMed Paradigm®Insulin Pump and your firm received a complaint of a similar nature, this devicemalfunction, if it were to recur, would be likely to cause or contribute to the sameserious injury. Furthermore, under 21 CFR 803.3, "Caused or contributed means thaa death or serious injury was or may have been attributed to a medical device, orthat a medical device was or may have been a factor in a death or serious injury...."
Based on the information in the complaint file, device failure or malfunction mayhave contributed to or caused the user's hospitalization and the device's malfunctionwould be likely to cause or contribute to a death or serious injury, if the malfunctionwere to recur. As a result, this serious injury is a reportable MDR event under 21CFR 803.50(a). Your firm did submit MDR (b) (4) for this complaint. The "Date ofEvent" and the "Date of Report" are listed as May 30, 2007. Your firm reported thisas a serious injury on the Mandatory Reporting Form, FDA-3500A, on November 14,2008, which is 18 months after the day that your firm received information of anMDR reportable event.
b) Complaint (b) (4) states that the reported complaint was not reportable as anMDR to the FDA based on testing of the returned MiniMed Paradigm® Insulin Pump.The information in the complaint indicated that the user contacted your firm becausethe user had a blood glucose level of 456, and that the user's MiniMed Paradigm®Insulin Pump had failed to alarm when it stopped delivering insulin. The user was
Case 1:20-cv-01277-AWI-JDP Document 1-4 Filed 09/08/20 Page 6 of 9

1/23/2020 2009 > Medtronic Puerto Rico Operations Company
wayback.archive-it.org/7993/20170112195621/http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2009/ucm168451.htm# 6/8
subsequently hospitalized and diagnosed with diabetic ketoacidosis. Follow-uprevealed that the user had trouble keeping the user's blood glucose level down, andwhen the user replaced infusion sets, the cannulas were bent. The complaint recordstates that, (b) (4) Under 21CFR 803.3, "Caused or contributed means that a death or serious injury was or mayhave been attributed to a medical device, or that a medical device was or may havebeen a factor in a death or serious injury...." In this instance, the patient hadcomplained of a potential device failure, and the patient was subsequentlyhospitalized for diabetic ketoacidosis. Based on the information in the complaint file,because your firm was aware of information that reasonably suggested that theuser's MiniMed Paradigm® Insulin Pump may have caused or contributed to aserious injury, you were required to report this event to FDA as an MDR within 30calendar days of receiving or otherwisebecoming aware of this information, under 21 CFR 803.50(a).
We have reviewed your responses dated January 20,2009, and March 31, 2009, andour conclusions follow:
Your responses state that MDR reports were submitted for the complaints identifiedabove. Your firm has also updated your procedure (b) (4) Medical Device Report (Effective Date: December 17, 2008), to reflect newcriteria for MDR reporting, and re-trained your employees on the new procedure onDecember 16, 2008. The adequacy of your corrective and preventive actions will bedetermined during the next inspection.
6) Failure to have a person who is qualified to make a medical judgment reasonablyconclude that a device did not cause or contribute to a death or serious injury, orthat a malfunction would not be likely to cause or contribute to a death or seriousinjury if it were to recur, as required by 21 CFR 803.20(c)(2). Persons qualified tomake a medical judgment include physicians, nurses, risk managers, and biomedicaengineers, under 21 CFR 803.20(c)(2).
For example:
Our investigators determined that a product reporting specialist was makingdecisions about MDR reportability for the MiniMed Paradigm® Insulin Pumps. Thetraining record for this particular employee showed that this person only had a highschool diploma with some additional in-house training.
Your responses dated January 20,2009 and March 31, 2009, did not address thischarge because it was not included in the FDA 483 issued to you on December 15,2008. The adequacy of your corrective and preventive actions will be determinedduring the next inspection.
You should take prompt action to correct the violations addressed in this letter.Failure to promptly correct these violations may result in regulatory action beinginitiated by the Food and Drug Administration without further notice. These actionsinclude, but are not limited to, seizure, injunction, and/or civil money penalties.Also, federal agencies are advised of the issuance of all Warning Letters aboutdevices so that they may take this information into account when considering theaward of contracts. Additionally, premarket approval applications for Class IIIdevices to which the Quality System regulation deviations are reasonably related wilnot be approved until the violations have been corrected. Requests for Certificates toForeign Governments will not be granted until the violations related to the subjectdevices have been corrected.
Please notify this office in writing within fifteen (15) working days from the date youreceive this letter of the specific steps you have taken to correct the notedviolations, including an explanation of how you plan to prevent these violations, or
Case 1:20-cv-01277-AWI-JDP Document 1-4 Filed 09/08/20 Page 7 of 9

1/23/2020 2009 > Medtronic Puerto Rico Operations Company
wayback.archive-it.org/7993/20170112195621/http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2009/ucm168451.htm# 7/8
, g p y p p ,similar violations, from occurring again. Include documentation of the correctiveaction you have taken. If your planned corrections will occur over time, pleaseinclude a timetable for implementation of those corrections. If corrective actioncannot be completed within 15 working days, state the reason for the delay and thetime within which the corrections will be completed.
Your response should be sent to:
U.S. Food and Drug AdministrationAttn: Mrs. Maridalia TorresDistrict Director466 Fernandez Juncos AvenueSan Juan, PR 00901-3223
If you have any questions about the content of this letter please contact Ms.Margarita Santiago, Compliance Officer, at (787) 474-4789.
Finally, you should know that this letter is not intended to be an all-inclusive list ofthe violations at your facility. It is your responsibility to ensure compliance withapplicable laws and regulations administered by FDA. The specific violations noted inthis letter and in the Inspectional Observations, Form FDA 483 (FDA 483), issued atthe closeout of the inspection may be symptomatic of serious problems in yourfirm's manufacturing and quality assurance systems. You should investigate anddetermine the causes of the violations, and take prompt actions to correct theviolations and to bring your products into compliance.
Regarding your firm's CAPA's for the Synchromed® II Pumps that did not have the(b) (4) test performed on them, your firm has not indicated how it will addressproduct that is currently distributed to customers. FDA's review of your firm'sinvestigation report(NCR (b) (4) did not reveal any evidence to demonstrate that(b) (4) was tested in subsequent manufacturing steps to verify that the safetymechanism performed as intended. As stated in the charges above, (b) (4)Synchromed® II Pumps on which the (b) (4) was not performed were distributedto customers. Should your firm undertake a voluntary correction or removal for theSynchromed® II Pumps where (b) (4) the was not performed, it must submit awritten report to FDA within 10 working days of initiating such an action, as specifiedby 21 CFR 806.10(a) & (b). See 21 CFR part 806 for additional information aboutcorrectives and removals.
In addition to the above charges, our inspection revealed that your firm uses onemanufacturing process system for both the Synchromed® II Pumps and the MiniMedParadigm® Insulin Pumps. To the extent that any of the above CGMP violations forthe Synchromed® II Pumps also implicate the MiniMed Paradigm® Insulin Pumps,your corrective actions should address and extend to the manufacturing proceduresof the MiniMed Paradigm® Insulin Pumps.
Sincerely,/S/Maridalia Torres IrizarryDistrict DirectorSan Juan District
Enclosure: Form FDA 483
cc: Mr. Manuel SantiagoVice PresidentMedtronic Puerto Rico Operations Company
Case 1:20-cv-01277-AWI-JDP Document 1-4 Filed 09/08/20 Page 8 of 9

1/23/2020 2009 > Medtronic Puerto Rico Operations Company
wayback.archive-it.org/7993/20170112195621/http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2009/ucm168451.htm# 8/8
Call Box 4070Juncos, PR 00777
cc: HFC-210 (electronic via CMS)HFZ-333 Nikhil Thakur, CDRHHFI-35 (redacted via CMS)HFR-SE1DD (MTI)DIB (VM)CSO (Marilyn Santiago)EF (3004369318)CBRFCB WL File
MS/meb: 06-01-2009
H:\Compliance Branch\Compliance Officers\Santiago\medtronic\Medtronic WL 2009-08 dated 06-01-2009.doc Page Last Updated: 03/16/2015 Note: If you need help accessing information in different file formats, see Instructions for DownloadingViewers and Players.Language Assistance Available: Español | 繁體中文 | Tiếng Việt | 한국어 | Tagalog | Русский | العربیة | KreyòlAyisyen | Français | Polski | Português | Italiano | Deutsch | 日本語 | فارسی | English
Accessibility Contact FDA Careers FDA Basics FOIA No FEAR Act Site Map Nondiscrimination WebsitePolicies
U.S. Food and Drug Administration10903 New Hampshire Avenue Silver Spring, MD 20993 Ph. 1-888-INFO-FDA (1-888-463-6332)Contact FDA
For Government For PressCombination Products Advisory Committees Science & Research Regulatory Information SafetyEmergency Preparedness International Programs News & Events Training and Continuing EducationInspections/Compliance State & Local Officials Consumers Industry Health Professionals FDA Archive
Links on this page:
Case 1:20-cv-01277-AWI-JDP Document 1-4 Filed 09/08/20 Page 9 of 9

Exhibit 5
Case 1:20-cv-01277-AWI-JDP Document 1-5 Filed 09/08/20 Page 1 of 7

(b) (4)
(b) (4)
(b) (4) (b) (4)
(b) (4) (b) (4)
(b) (4)(b) (4)
(b) (4)
Case 1:20-cv-01277-AWI-JDP Document 1-5 Filed 09/08/20 Page 2 of 7

(b) (4)
(b) (4)(b) (4)
(b) (4)(b) (4)
(b) (4) (b) (4)
(b) (4)(b) (4) (b)
(4)(b) (4) (b) (4)
(b) (4) (b) (4)
(b) (4) (b) (4)(b) (4)
(b) (4)(b) (4)
(b) (4)(b) (4)
(b) (4)
Case 1:20-cv-01277-AWI-JDP Document 1-5 Filed 09/08/20 Page 3 of 7

(b) (4)(b) (4)
(b) (4) (b) (4)
(b) (4) (b) (4)
(b) (4)
(b) (4) (b) (4)
(b) (4)(b) (4)
(b) (4)
Case 1:20-cv-01277-AWI-JDP Document 1-5 Filed 09/08/20 Page 4 of 7
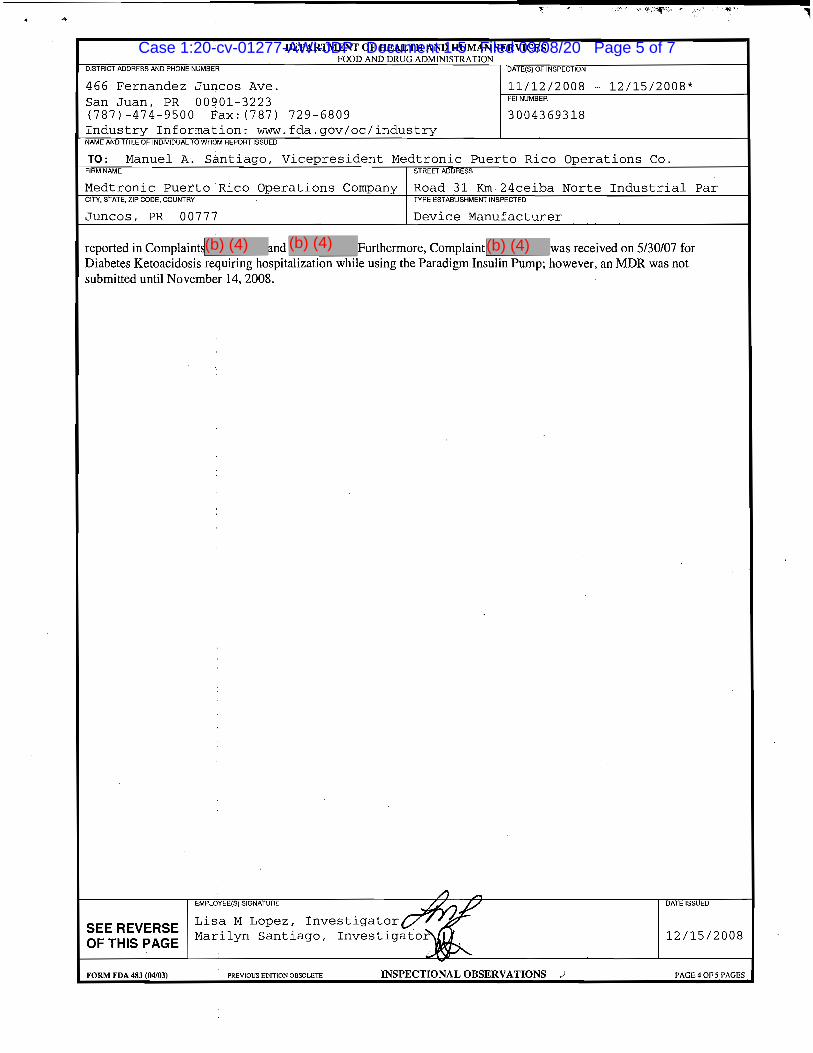
(b) (4) (b) (4) (b) (4)
Case 1:20-cv-01277-AWI-JDP Document 1-5 Filed 09/08/20 Page 5 of 7

Case 1:20-cv-01277-AWI-JDP Document 1-5 Filed 09/08/20 Page 6 of 7

Case 1:20-cv-01277-AWI-JDP Document 1-5 Filed 09/08/20 Page 7 of 7

Exhibit 6
Case 1:20-cv-01277-AWI-JDP Document 1-6 Filed 09/08/20 Page 1 of 10

1/23/2020 2012 > Medtronic, Inc. 7/17/12
wayback.archive-it.org/7993/20170406134449/https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2012/ucm314736.htm# 1/9
Medtronic, Inc. 7/17/12
Department of Health and Human Services
Public Health ServiceFood and DrugAdministration
Minneapolis District OfficeCentral Region250 Marquette Avenue,Suite 600Minneapolis, MN 55401Telephone: (612) 334-4100FAX: (612) 334-4142
July 17,2012WARNING LETTER
Refer to MIN 12- 39
CERTIFIED MAILRETURN RECEIPT REQUESTEDOmar S. IshrakChief Executive OfficerMedtronic, Inc.710 Medtronic ParkwayMinneapolis, Minnesota 55432Dear Mr. Ishrak:During an inspection of your firm, Medtronic Neuromodulation, located at 7000Central Avenue NE, in Minneapolis, Minnesota, from March 14 through May 9, 2012,investigators from the United States Food and Drug Administration (FDA)determined that your firm manufactures implantable drug infusion systems, deepbrain stimulation systems, spinal cord neurostimulation systems, nerve monitoringproducts, and other neurological medical/surgical products. Under section 201(h) ofthe Federal Food, Drug, and Cosmetic Act (the Act), 21 U.S.C. § 321(h), theseproducts are devices because they are intended for use in the diagnosis of diseaseor other conditions or in the cure, mitigation, treatment, or prevention of disease, orare intended to affect the structure or function of the body.This inspection revealed that these devices are adulterated within the meaning ofsection 501(h) of the Act, 21 U.S.C. § 351(h), in that the methods used in, or thefacilities or controls used for, their manufacture, packing, storage, or installation arenot in conformity with the Current Good Manufacturing Practice (CGMP)requirements of the Quality System (QS) regulation found at Title 21, Code ofFederal Regulations (21 CFR), Part 820. We received a response from Thomas M.Tefft, Senior Vice President and President, and Jill Smith, Vice President, Quality,dated May 30,2012 (and updated on June 29, 2012) concerning our investigators'observations noted on the Form FDA 483, List of lnspectional Observations, issued to
Inspections, Compliance, Enforcement, and Criminal InvestigationsHome Inspections, Compliance, Enforcement, and Criminal Investigations ComplianceActions and Activities Warning Letters 2012
Case 1:20-cv-01277-AWI-JDP Document 1-6 Filed 09/08/20 Page 2 of 10

1/23/2020 2012 > Medtronic, Inc. 7/17/12
wayback.archive-it.org/7993/20170406134449/https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2012/ucm314736.htm# 2/9
Mr. Tefft on May 9, 2012. We address the response below, in relation to each of thenoted violations. These violations include, but are not limited to, the following:1. Failure to establish adequate procedures for corrective and preventive action asrequired by 21 CFR 820.100(a). Specifically:
A) You have not identified the actions to correct and prevent recurrence of non-conforming product. GCAPA 1485, opened October 26, 2007, relates to motorcorrosion resulting in device field failure (motor stall). Within the InvestigationReport for SynchroMed II Pump Corrosion (NDHF1119-88863), it states"corrosion[ ... ] can result in partial or complete removal of gear teeth." Thiscan "seize" the motor altogether or "gear wheel [ ... ]will continue to rotate, buthere may be no drug delivery in the region of missing teeth." Identifiedcorrosion issues include wheel 3 corroded teeth, gear binding, gear shaftbinding, and bearing binding. This GCAPA includes 567 complaints and has notbeen closed.
FDA 483 Response: Your response describes actions taken to mitigate the risk ofdevice failure through communication to healthcare professionals and decreasedsusceptibility of the device to corrosion. However, we have concluded that yourresponse is not adequate. Health Hazard Analysis for SynchroMed II Pump MotorCorrosion (CAPA #1485), NDHF1119-101573, Version 4.0, predicts an additional (b)(4) patient injuries resulting from device failure due to motor corrosion. Thisanalysis was based only on confirmed failures (via returned product analysis) due tocorrosion; and thus, the number of additional patient injuries will likely be higherthan predicted.Your response also discusses the activities of your Corrosion Task Force (CTF) andyour planned in-depth review of SynchroMed II complaints alleging a motor stallwithout a product. CAPA 1485 and the Health Hazard will be updated. (b)(4)FDA requests a prompt meeting with you to discuss the pump motor corrosionfailure mode and the scope and timing of corrective actions to address this ongoingproblem. We propose Friday, September 7, 2012, at 10:00 a.m. EST for this meetingto be held at the Center for Devices and Radiological Health, 10903 New HampshireAvenue, Building 66, Silver Spring, Maryland. Please contact John Diehl, RegulatoryOperations Officer,· (301) 796-0993, to confirm your participation.
B) The "Corrective and Preventive Action (CAPA) Procedure," (QMS1861) states"assess quality issues, trends, and potential or actual product or processnonconformities." This was not completed in that data used for evaluation wasincomplete per citations 2 and 3 below.
FDA 483 Response: Your response states that you updated Product Event (PE)inclusion criteria for CAPA 1485 to include appropriate PEs associated with non-returned product. The CAPA 1485 Health Hazard Analysis will be updatedaccordingly, and the field corrective action decision will be re-evaluated.You also updated the form for PE inclusion criteria to require a documented rationalewhen PEs with non-returned product will not be assigned to the applicable CAPA.Further, you stated that upon completion of remediation activities to address FDA-483 observations 2 and 3, you will re-evaluate the impact to all open product-relatedCAPAs, monitors, and trends.We consider your proposed corrective actions to be appropriate; however, a follow-up inspection will be necessary to evaluate the implementation and effectiveness ofthe actions.2. Failure to establish adequate procedures for receiving, reviewing, and evaluatingcomplaints by a formally designated unit, which is required by 21 CFR 820.198(a).Specifically, Patient and Technical Services (PATS) did not document complaintinformation for incoming calls per the procedure "Customer Response Team Systems[CRTS]" (PTS6026). A complaint is defined as "Any written, electronic or oralcommunication that alleges deficiencies related to the identity, quality, durability,reliability, safety, effectiveness, or performance of a device ... " and the Patient andTechnical Consultant "Identifies and documents any report of a Complaint."Complaint information received during a call was not documented in the written callrecord for the following:
Case 1:20-cv-01277-AWI-JDP Document 1-6 Filed 09/08/20 Page 3 of 10

1/23/2020 2012 > Medtronic, Inc. 7/17/12
wayback.archive-it.org/7993/20170406134449/https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2012/ucm314736.htm# 3/9
Call Number Information Received in Phone CallNot Documented on ResultantWritten Call Record
2685890
A doctor requested information onwhether catheter removal is an optionwith a granuloma. This call was nothandled as a complaint for agranuloma/inflammatory mass.
2757084
Health care provider called to report amotor stall and that the patientexperienced withdrawal symptoms.Withdrawal symptoms were notdocumented on the written call record orresulting complaint.
2721299
Caller stated that Fentanyl was in pump. The drug was not documented on thewritten call record and the resultingcomplaint states drug description is“Unknown.”
2739594
Caller reported a motor stall with norecovery. Caller stated Baclofen as themedication in the pump. The drug wasnot documented on the written callrecord and the resulting complaint statesdrug description is “Unknown.”
2702294
Caller reported a vibration sensation andstated that “pump is not working.” Thepump not working was not documentedon the written call record or resultingcomplaint.
2724877
Caller reported a vibration sensation andthat pump is “not working for pain, like ithas all these years.” Pump not workingfor pain was not documented on thewritten call record or resulting complaint.
2694377
Caller reported that pain became worsesince device implantation which was notdocumented on the written call record orresulting complaint.
2579227
Caller reported Baclofen is in the pump. The drug was not recorded on the writtencall record and the resulting complaintstates drug description is “Unknown.”
2718965
Caller reported a granuloma and statedwithin the call that “the medicine workedin the beginning, but over time, it mademe worse. And I didn’t know it until itstopped working.” The information aboutthe medication was not captured on thewritten call record or resulting complaint.
FDA 483 Response: Your response states that you reviewed the audio call recordsand revised the written records accordingly. The events were reviewed again todetermine whether Medical Device Reports (MDRs) or Adverse Drug Experience
Case 1:20-cv-01277-AWI-JDP Document 1-6 Filed 09/08/20 Page 4 of 10

1/23/2020 2012 > Medtronic, Inc. 7/17/12
wayback.archive-it.org/7993/20170406134449/https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2012/ucm314736.htm# 4/9
Reports (ADRs) should be filed or supplemented. Reports were submitted whenrequired. Lastly, assigned codes were re-evaluated and revised if necessary.Broader corrective and preventive actions completed or promised include training,management review of calls and CRTS records, procedural changes, and audits ofPatient and Technical Services procedures and processes.Your corrective actions appear to be appropriate; however, a follow-up inspectionwill be necessary to evaluate implementation and effectiveness.3. Failure to review, evaluate and investigate, where necessary, complaints involvingthe possible failure of a device to meet any of its specifications. This is required by21 CFR 820.198(c). Specifically:
A) Product Performance Specialists did not adequately evaluate complaints.(1) Per the procedure "Product Performance Specialist Work Instruction,"(RPMWI1666) non-returned product with suspected non-conformance is tobe formally investigated. Eleven of 11 closed complaints involving motorstalls with unknown cause and no returned product were not formallyinvestigated nor was there an adequate explanation for why noinvestigation occurred. These complaints include:500073583: Motor stall, pain reported, volume discrepancy500099975: Motor stall, nausea, vomiting500047736: Motor stall, volume discrepancy, withdrawal, pump explanted500079921: Motor stall, volume discrepancy, pain500050534: Motor stall, underdose, pump explanted500031251: Motor stall, return of symptoms500054080: Motor stall, increased pain, underdose symptoms, pumpexplanted500024556: Motor stall, pain reported, pump explanted500022409: Motor stall, underdose, pump explanted700099823: Motor stall, no therapeutic effect700062012: Motor stall, withdrawal symptoms
FDA 483 Response: Your response states that the Neuromodulation ComplaintEvaluation Team (NCET) initiated an investigation and recommended that PEsalleging motor stall be assessed and dispositioned to open CAPAs, CAPA monitors,Data Monitors, and/ or PITCH Events. Additional broader corrective actions includedevelopment of improved criteria for complaint investigations and revisions to theRisk Evaluation Board (REB) and Product Performance Trend Reporting procedures.Your corrective actions appear to be appropriate; however, a follow-up inspectionwill be necessary to evaluate implementation and effectiveness.
(2) An investigation into reports of vibrating pumps entitled "WATCHLIST-Patient Reports of Pump Vibrations" was opened on March 30, 2007, and closedFebruary 7, 2008. This investigation included 19 separate complaints. It wasdetermined that "the likely cause for these vibrations is a physiologicalsensation due to surgery and the healing process."The following complaints involving "vibration" sensations were not investigatednor was there an adequate explanation for why no investigation occurred:
Complaint Number
Implant Date Notified Date Description
700074933 6/1/2006 12/2/2011 Inflammatorymass, vibratingsensation
500083053 3/9/2010 4/29/2011 Vibrating sensation,caller reportedpump "hasn't been
Case 1:20-cv-01277-AWI-JDP Document 1-6 Filed 09/08/20 Page 5 of 10

1/23/2020 2012 > Medtronic, Inc. 7/17/12
wayback.archive-it.org/7993/20170406134449/https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2012/ucm314736.htm# 5/9
working"
500078876 4/28/2007 7/11/2011
Vibration, callerreported pump "notworking like it usedto"
500047418 8/28/2007 10/6/2011
Abdominalvibration,withdrawal,catheter punctures
500205241 1/7/2010 10/3/2011
Vibration sensation
500167917 3/7/2011 8/10/2011
Painful vibration inabdomen
700074795 11/7/2007 12/1/2011
Vibration felt instomach
700078229 11/30/2005 12/14/2011
Vibration sensation,patient reportspump not working
700085549 2/28/2011 1/13/2012
Vibration sensation
500038321 1/17/2007 1/3/2011
Vibration sensation,increasedweakness
500037974 4/12/2004 12/16/2010
Vibration sensation,catheter kink
500073385 12/21/2007 4/23/2010
Vibration sensation
500091223 6/30/2009 1/18/2011
Vibration sensation
500046267 5/26/2010 10/6/2011
Feeling vibration,pain, blisters, andfluid in front ofpump
500184025 3/24/2011 6/29/2011
Vibration sensationin abdomen down tolower groin
500099975 5/22/2007 3/15/2010
Vibration sensation,3 months laterpatient experiencedmotorstall
FDA 483 Response: Your response states that Neuromodulation initiated a PITCH(Preliminary Investigation and Trending for Complaint Handling) event to investigatepotential causes and similarities I differences related to allegations of vibration withthe SynchroMed II pump.Your corrective actions appear to be appropriate; however, a follow-up inspectionwill be necessary to evaluate implementation and effectiveness.
(3) The procedure "Complaint Evaluation and Investigation Process" (RPM1234)states "assign appropriate functional area(s) to further investigate the issue."
Case 1:20-cv-01277-AWI-JDP Document 1-6 Filed 09/08/20 Page 6 of 10

1/23/2020 2012 > Medtronic, Inc. 7/17/12
wayback.archive-it.org/7993/20170406134449/https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2012/ucm314736.htm# 6/9
Complaint 500082715 was not assigned to the functional area of MedicalSafety. The complaint description states "HCP reports a death of a patient thathad a gastric stimulator implanted. He died on Monday, according to what wasreported to us he could not swallow, he had severe acid in his body."
FDA 483 Response: Neuromodulation re-reviewed the complaint and clearlydocumented the investigation activities. The complaint was reviewed by a MedicalSafety physician, and an MDR was filed for the event. In addition, you promised toimplement a more detailed process for medical review of complaints and develop aremediation plan for review of prior complaint flies.Your actions are appropriate; however, a follow-up inspection will be necessary toevaluate implementation and effectiveness.
(4) The procedure "Product Performance Specialist Work Instruction" (RPM1666) states "check for relationship of issue to existing investigations (e.g. [ ...] CAPA or Data monitor)."
a. Complaint 500037816 was a returned product due to volumediscrepancies at multiple refills. The analysis stated "corrosion and residuewere seen on both sides of gear wheel." This complaint was not added toGCAPA 1485 for motor corrosion.b. Complaint 500091325 stated the following on the Medical DeviceReport: "further information received from the healthcare providerindicated she believed the lead had migrated." This complaint was notadded to the Data monitor for "migration" for urinary InterStim.
FDA 483 Response: Your firm re-reviewed complaints 500037816 and 500091325and documented the investigations and conclusions. For complaint 500091325,coding was corrected and the monitor was updated.Your corrective actions appear to be appropriate; however, a follow-up inspectionwill be necessary to evaluate implementation and effectiveness.
B) Coding of similar complaints is inconsistent.Procedure "Complaint and Adverse Event Coding and Master Data ManagementProcess" (RPMWI1833) describes "what codes will be assigned in the PEs"(complaints) that could subsequently be used for trend analysis. Each complainis to receive a (b)(4) code defined as:(b)(4)Of the following 14 complaints relating to similar motor stall issues(700062012,500082653,500024556,500099975,500073583,500047736,500079921,500052853,500054080,500050534,500075490,500031526,700095413,500031251:
• 4 received a (b)(4)• 10 received (b)(4)• 2 received a (b)(4)• 9 received a (b)(4)• 3 received a (b)(4)Of the following 10 complaints relating to similar inflammatory mass issues(500166572,500054756,500050731,500071678,500093511,500075527,500093970, 500043194, 500074339, 700069121):• 5 received a (b)(4)• 1 received a (b)(4)• 2 received a (b)(4)• 2 received a (b)(4)• 6 received a (b)(4)• 3 received a (b)(4)• 1 received a (b)(4)
FDA 483 Response: Your response states that you implemented a secondaryreview of coding decisions to ensure accuracy and consistency (b)(4). Neuromodulation committed to a comprehensive assessment processes and to
Case 1:20-cv-01277-AWI-JDP Document 1-6 Filed 09/08/20 Page 7 of 10

1/23/2020 2012 > Medtronic, Inc. 7/17/12
wayback.archive-it.org/7993/20170406134449/https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2012/ucm314736.htm# 7/9
develop a revised coding strategy. Remediation of infusion system files will also beconducted. The specific complaints cited above involving motor stall andinflammatory mass were re-reviewed, and codes were revised if necessary.Your corrective actions appear to be appropriate; however, a follow-up inspectionwill be necessary to evaluate implementation and effectiveness.
C) Trending of complaint data/ coding for evaluation was not completed perprocedures:
(1) Devices that are not returned are trended per the procedure"Complaint and Adverse Event Trend Reporting" (RPMWI1832). This wasnot completed for 2011 and 2012 for the following products: infusionsystems, neurostimulation for movement disorder (DBS), neurostimulationfor pain, InterStim therapy, Enterra therapy, and Prostiva.
FDA 483 Response: Neuromodulation trended complaint PEs without an associatedproduct return. Your firm also developed a new analysis approach to replace thetrend "Device not returned, further investigation not possible without device,"previously required by RPMWI1832. An (b)(4) to perform statistical analysis ofpost-market surveillance data sources is being implemented.Your corrective actions appear to be appropriate; however, a follow-up inspectionwill be necessary to evaluate implementation and effectiveness.
(2) "Known Expected Events" are trended per the procedure Adverse EventTrend Reporting" (RPMWI1832), using a (b)(4) code. Due to a transition to anew complaint handling computer system, the following complaints weremissing an (b)(4) code and were not included in trending:
a. 99 complaints for inflammatory mass including, 500037107,500093511,500082334,500075104,500050731,500095044,500071809,500071678,500054756,500051396,500075527,500039586,500043194,500165916,700069121,500093970,500074339,500166572,500076576,and500081542.b. 88 complaints for Dysarthria. When this data was added to the system,three separate signals exceeded threshold.c. 11 complaints for Loculation.d. 104 complaints for Incision Pain.
FDA 483 Response: Your firm re-reviewed all complaints that were affected by thetransition/conversion issue, and missing (b)(4) codes were added to the files. New trending was conducted and resulting signals were investigated. On a broader scale,data conversion procedures were revised and implemented to address the rootcause of the problem.Your corrective actions appear to be appropriate; however, a follow-up inspectionwill be necessary to evaluate implementation and effectiveness.
(3) The threshold limit assigned to trends is not described in the procedure"Complaint and Adverse Event Trend Reporting" (RPMWI1832).
FDA483 Response: Your response states that you updated RPMWI1832 to includeinstructions for (b)(4)A follow-up inspection will be necessary to evaluate implementation andeffectiveness of this corrective action.
D) Data is not evaluated per procedure to determine if signals exist that wouldrequire further investigation.The procedure "Complaint and Adverse Event Trend Reporting" (RPMWI 1832)states "Evaluate the data and determine if any results meet the signalinvestigation requirement(s)." This was not completed due to incomplete datanoted above.
FDA 483 Response: Your response appears to be limited to the incomplete datacited above in 3. C) (2). The scope of this citation, however, is broader. We areconcerned that incomplete complaint data and incorrect coding decisions
Case 1:20-cv-01277-AWI-JDP Document 1-6 Filed 09/08/20 Page 8 of 10

1/23/2020 2012 > Medtronic, Inc. 7/17/12
wayback.archive-it.org/7993/20170406134449/https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2012/ucm314736.htm# 8/9
described elsewhere in this letter (e.g., citations 2 and 3) may have compromisedyour firm's ability to detect and investigate signals.In response to this letter, please describe the actions that your firm is taking toensure that you will appropriately detect and investigate all signals.Re: FDA 483 Response to Observations 4-6: The corrective actions reported andplanned appear to be adequate. Implementation and effectiveness will be evaluatedduring a follow-up inspection.You should take prompt action to correct the violations addressed in this letter.Failure to promptly correct these violations may result in regulatory action beinginitiated by the Food and Drug Administration without further notice. These actionsinclude, but are not limited to, seizure, injunction, and/ or civil money penalties.Also, federal agencies are advised of the issuance of all Warning Letters aboutdevices so that they may take this information into account when considering theaward of contracts. Additionally, premarket approval applications for Class IIIdevices to which the Quality System regulation deviations are reasonably related wilnot be approved until the violations have been corrected. Requests for Certificates toForeign Governments will not be granted until the violations related to the subjectdevices have been corrected.We are requesting that you submit to this office on the schedule below, certificationby an outside expert consultant that he/she has conducted an audit of yourestablishment's manufacturing and quality assurance systems relative to therequirements of the device Quality System regulation (21 CFR Part 820). You shouldalso submit a copy of the consultant's report and your certification that you havereviewed the consultant's report and that your establishment has initiated orcompleted all corrections called for in the report. The initial certifications of auditand corrections and subsequent certifications of updated audits and corrections (ifrequired) should be submitted to this office by the following dates:
• Initial certifications by consultant and establishment - by January 17,2013• Subsequent certifications of updated audits and corrections- by January17, 2014, and 2015
Please notify this office in writing within fifteen (15) working days from the date youreceive this letter with an update on the specific steps you have taken to correct thenoted violations, including an explanation of how you plan to prevent theseviolations, or similar violations, from occurring again. Include documentation of thecorrective actions you have taken. If your planned corrections will occur over time,please include a timetable for implementation. If corrective actions cannot becompleted within 15 working days, state the reason for the delay and the timewithin which the corrections will be completed.Your response should be sent to Timothy G. Philips, Compliance Officer, at theaddress on this letterhead. If you have any questions about the content of this letterplease contact Mr. Philips at (612) 758-7133.Finally, you should know that this letter is not intended to be an all-inclusive list ofthe violations at your facility. It is your responsibility to ensure compliance withapplicable laws and regulations administered by FDA. The specific violations noted inthis letter and in the Inspectional Observations, Form FDA 483, issued at thecloseout of the inspection may be symptomatic of serious problems in your firm'smanufacturing and quality assurance systems. You should investigate and determinethe causes of the violations and take prompt actions to correct the violations and tobring your products into compliance.Sincerely,/s/Michael Dutcher, DVMDirectorMinneapolis District
Case 1:20-cv-01277-AWI-JDP Document 1-6 Filed 09/08/20 Page 9 of 10

1/23/2020 2012 > Medtronic, Inc. 7/17/12
wayback.archive-it.org/7993/20170406134449/https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2012/ucm314736.htm# 9/9
Page Last Updated: 08/20/2012 Note: If you need help accessing information in different file formats, see Instructions for DownloadingViewers and Players.Language Assistance Available: Español | 繁體中文 | Tiếng Việt | 한국어 | Tagalog | Русский | العربیة | KreyòlAyisyen | Français | Polski | Português | Italiano | Deutsch | 日本語 | فارسی | English
Accessibility Contact FDA Careers FDA Basics FOIA No FEAR Act Site Map Nondiscrimination WebsitePolicies
U.S. Food and Drug Administration10903 New Hampshire Avenue Silver Spring, MD 20993 Ph. 1-888-INFO-FDA (1-888-463-6332)Contact FDA
For Government For PressCombination Products Advisory Committees Science & Research Regulatory Information SafetyEmergency Preparedness International Programs News & Events Training and Continuing EducationInspections/Compliance State & Local Officials Consumers Industry Health Professionals FDA Archive
Links on this page:
Case 1:20-cv-01277-AWI-JDP Document 1-6 Filed 09/08/20 Page 10 of 10

Exhibit 7
Case 1:20-cv-01277-AWI-JDP Document 1-7 Filed 09/08/20 Page 1 of 6

~----=-~=~------D-E_P_~_T_ME_NT __ O_F_HE_AL_m_AND_. __ HUMAN_. ___ S_ER_ ... VI-C_E_s"'"·----------------FOOD AND DRUG ADMINIS1RATION
DISlRICT ADDRESS AND PHONE NUMBER DATE(S) OF INSPECTION
250 Marquette Avenue, Suite 600 Minneapolis, MN 55401
02/14/2013 - 04/03/2013* FEINUMBER
(612) 334-4100 Fax: (612) 334-4134 2182207 Industry Information: www.fda.gov/oc/industry ~E ANO TTTI.E OF INDIVIDUAL TO WHOM REPORT ISSUED
TO: Omar S. Ishrak, Chairman and Chief Executive Officer FIRM NAME STREET ADDRESS
Medtronic Neurornodulation 7000 Central Ave NE CITY, STATE, ZIP CODE, COUNTR'f TYPE ESTABUSHMENT INSPECTED
Minneapolis, MN 55432-3568 Medical Device Manufacturer
This document lists observations made by the FDA representative(s) during the inspection of your facility. They are inspectional observations, and do not_represent a final Agency determination regarding your compliance. If you have an objection regarding an
-ob~rvation, or have implemented, or plan to implement, corrective action in response to an observation, you may discuss the objection or action with the FDA representative(s) during the inspection or submit this information to FDA at the address above. If you have any questions, please contact FDA at the phone number and address above.
The observations noted in this Form FDA-483 are not an exhaustive listing of objectionable conditions. Under the law, your firm is responsible for conducting internal self-audits to identify and correct arry and all violations of the quality system requirements.
DURING AN INSPECTION OF YOUR FIRM WE OBSERVED:
OBSERVATION 1
Products that do not conform to specifications are not adequately controlled.
Specifically,
A) Your firm distributed nonconforming SC catheters, and failures due to the nonconforming products have resulted in
serious adverse events. From September 10, 2012 to March 25, 2013, approximately- SC catheters that do not confirm
to the current product specifications have been distributed. Regulatory approval was received for Supplement -136 to PMA
P860004 on December 15, 2011 to change the design of SC Catheter models 8709SC, 8731 SC, 8596SC, and 8578 to mitigate
a known field issue associated with CAPA 1507- SC Catheter Occlusion. This design change was implemented via ECO 12-
00985, dated March 6, 2012, and the new revisions of Catheter models were released to the field in September 2012.
However, the previous SC catheter models which do not conform to the current design have continued to be distributed and
have attributed to 60 complaints of catheter occlusion since September 2012.
B) Your firm distributed approximately- lead kits containing nonconforming lead caps to the field from 19 NOV 2012
to 29 JAN 2013. On 31 OCT 2012 and 19 NOV 2012, your firm performed testing on the DBS lead cap that showed the
(b) (4) , The product specification contains mm, requirement ofllll -Per your procedure "QMS1340 TLP Escalating Quality Is$ues and Handling Nonconformances" ver. 9 .0 dated 1/11/12, vv:hen
SEE REVERSE OF THIS PAGE
FORM FDA 483 (09/08)
EMPLOYEE(S)SIGNAlURE ,_ d ~ . . ) Jessica L. Johnson, Investigatot~ · '2:7" t ',.-vf"'-/ Susan M. Matthias, Investigator f 1~ 11[: - /J
.. ~ NlfflvU6a -if. PREVIOUS EDmON OBSOIEIE INSPECTIONAL OBSERVATIONS
DATE ISSUED
~03
PAGE I OF5PAGES
Case 1:20-cv-01277-AWI-JDP Document 1-7 Filed 09/08/20 Page 2 of 6

OIS1RJCT ADDRESS AND PHONE NUMBER
DEPAR1 MENT OF ru.,ALTii ~l} HUMAN SERVICES FOOD AND DRUG ADMJNISTRATION
DATE($) OF fNSPECTlON
250 Marquette Avenue, Suite 600 Minneapolis, MN 55401
02/14/2013 - 04/03/2013* FEINUMBER
(612) 334-4100 Fax: (612) 334-4134 2182207 Industry Information: www.fda.gov/oc/industry NAME ANO TITLE OF INDIVIDUAL TO \MiOM REPORT ISSUED -
TO: Omar S. Ishrak, Chairman and Chief Executive Officer FIRM NAME STREET ADDRESS
Medtronic Neuromodulation 7000 Central Ave NE CITY. STATE, ZIP CODE, COUNTilY TYPE ESTABllSHMEITT INSPECTED
Minneapolis, MN 55432-3568 Medical Device Manufacturer
a product nonconformance is confirmed, the product is to be segregated and place on hold. If the product has been
distnbuted, the risk assessment decision must be documented within 30 days. The Risk Assessment for DBS Lead CAP
(b) (4) Issue (GCAPA 145631) was not completed until 28 JAN 2013.
In addition, your procedure also requrres an approved product deviation to distribute nonconforming product. A product
deviation for the nonconforming DBS lead kits was not authorized until 07 FEB 2013.
OBSERVATION 2
Procedures for corrective and preventive action have not been adequately established.
Specifically,
(A) Actions needed to correct and prevent recurrence of a quality problem were identified but not implemented. For
example,
(i) Feedthrough CAP A number 10594 identified actions on 02 APR 2008 via NDHFl 148-98756- "Feed
(ii)
Through Shorting, Effectiveness Report" to correct and prevent recurrence of
feedthrough shorting resulting in motor stalls in the SynchroMed II infusion pump. The recommended
action of has not been implemented. Since April 2008,
at least 298 serious adverse events have resulted from feedthrough shorting.
CAPA 110407 (b) (4) identified an action within the 21 JUN 2012 Risk
Evaluation Board meeting minutes. The recommended action was (b) (4)
. The NLT did not approve the
recommendation and delayed any action until the I-IHA was completed tii"9R ~ i:e~ during this ,,1,,__ I . · i "'/VI!, ~ 'fl 7fl'J, •
inspection. Since June 2012, at least 37 serious adverse events have been "possibly" related to thetG>iQI -CAPA.
(B) The Health Hazard Assessments for high priority CAP As with the highest patient severity of death were not completed
EMPLOYEE(S)SIGNATIJRE . t 1-/. !3/13 SEE REVERSE Jessica L. Johnson, Investigator OJ OF THIS PAGE Susan M. Matthias, Investigat<?r (} V- 4 ("'!:; ( / 3, ..
DATE ISSUED
04/03/2013 ·
FORM FDA 483 (09/0$) PREVIOUS EDITlON OBSOLE'l'E INSPECTIONAL OBSERVATIONS PAGE ZOF 5 PAGES
Case 1:20-cv-01277-AWI-JDP Document 1-7 Filed 09/08/20 Page 3 of 6

DEPARrMENl OF HEALTH AND HU.MAN SERVICES FOOD AND DRUG ADMINISTRATION
DISTRICT ADDRE$S AND PHONE NUMBER DATE(S) OF IN6PECTION
250 Marquette Avenµe, Suite 600 Minneapolis, MN 55401
02/14/2013 - 04/03/2013* FBNUMBER
(612) 334-4100 Fax: (612) 334-4134 2182207 Industry Information: www.fda.gov/oc/industry NAME AND TITLE OF INDIVIDUAL TO WHOM REPORT ISSUED
TO: Omar s. Ishrak, Chairman and Chief Executive Officer FIRM NAME STREET ADDRESS
Medtronic Neuromodulation 7000 Central Ave NE CITY, STAlE, ZIP CODE, COUNTRY TYPE ESTABLISHMENT INSPECTED
Minneapolis, MN 55432-3568 Medical Device Manufacturer
in a timely fashion. Your procedure, QMS1002 TLP Corrective and Preventive Actions requires an HHA for any high
priority CAP A with a patient risk. For example: .
(i)
(ii)
"CAPA 110407 (b) (4) "was opened on 01 NOV 2011. The HHA for this
CAP A was not completed until 11 MAR 13 ( during this inspection.)
(b) (4) "CAP A 132952
completed on 01 FEB 13.
was opened 26 June 2012. The BRA was
( C) Health Hazard Assessments have not been updated after CAP A effectiveness monitoring signaled an increase in the rate
ofoccurrence as evidenced by CAP As 3064, 7685, and 1507. QMSW114505 "CAPA Monitoring" states, "Update Health
Hazard Analysis document MEDN-0255, if required by identification of a new hazard / harm and or an in~ease in severity or
occurrence defined by a change in color on the Risk Index table."
(i) In February 2011, your firm detected a signal in the CAPA 1507 monitor showing .(b)(4)
(ii)
(iii)
SEE REVERSE OF THIS PAGE
FORM FDA 483 (09Xl8)
The 13 FEB 2012 High Priority CAPA Board recommended that the HHA for CAPA 1507 "SC Catheter
Occlusion" be updated. The HHA has not been updated since September 2008. At least 300 complaints for
this CAP A have been received since the HHA was last updated.
In February 2012, a signal was detected in the CAPA3064 monitor showing a (b) (4) . The
signal investigation was not completed until February 2013, _and the BRA has not been updated since
March 2009. At least 140 complaints forth.is CAPA have been received since the HHA was last updated.
In February 2011, your firm opened a CAP A monitor for CAP A 7685 (b) (4) . In December
2011, a decision was made to update the HHA for CAP A 7685; however, the HHA has not been updated
since September 2010. At least 40 complaints for this CAP A have been received since the HHA was last
updated.
EMPLOYEE(S) SlGNAl'URE
Jessica L. Johnson, Investigator Susan M. Matthias, Investigator
ffiEVIOUS EDIDON OBSOLE'IB INSPECTIONAL OBSERVATIONS
DATE ISSUED
04/03/2013
PAGE30FSPAGES
Case 1:20-cv-01277-AWI-JDP Document 1-7 Filed 09/08/20 Page 4 of 6

DEPARTMENT OF l:lEALTH AND HUMAN SERVICES FOOD AND DRUG ADMINISTRATION
DISTRICT ADORE'SS AND PHONE NUMBER
250 Marquette Avenue, Suite 600 Minneapolis, MN 55401 (612) 334-4100 Fax: (612) 334-4134
OATE(S) OF INSPECTION
02/14/2013 - 04/03/2013* FEINUMBER
2182207 Industry Information: www.fda.gov/oc/industry NAME AND TITlE OF INDIVIDUAL TO WHOM REPORT ISSUED
TO: Omar S. Ishrak, Chairman and Chief Executive Officer FIRM NAME STREET ADD!!ESS
Medtronic Neuromodulation ! 7000 Central Ave NE CITY, STATE, ZlP CODE. COUNlRY TYPE ESTABLISHMENT Il',ISPECTED
Minneapolis, MN 55432-3568 Medical Device Manufacturer
(D) Your firm did not perform a complaint search for CAPA 110407- (b) (4) from December
2011 until our request during this inspection. Your procedure, QMS 1861, Corrective and Preventive Action ( CAP A)
Procedure, versions 11.0 and 12.0 states, "NOTE: The first PE search must take place within 90 days after the CAP A Start
Date ... an additional PE search must be perfonned at least every 90 days during the investigation phase and documented in
the CAP A record."
OBSERVATION 3
Design verification does not confirm that design output meets design input requirements.
Specifically, design verification testing was never performed on the DBS lead cap to verify that the [mJQ requirement was met. A total of 103 complaints including l l serious adverse events have been reported since the lead cap
was released in May 2006.
OBSERvA·noN 4
Procedures for design change have not been adequately established.
Specifically, testing was not performed to verify that a design change did not adversely affect the product. Your firm . . -"·•
changed on the DBS lead extensions and lead caps from a[O)IQ to a[G)mJI in January 201 l. Seventy-five of the 103 complaints regarding connector block twisting and subsequent DBS
lead damage have been reported since the release of the in February 2011 .
SEE REVERSE OF THIS PAGE
FORM FDA 4113 (@/0&)
EMP!.OYEE(S) SIGNATURE
Jessica L. Johnson, Investigator Susan M. Matthias, Investigator
PREVIOUS EDITTON OBSOIEIE INSPECTIONAL OBSERVATIONS
ISSUED
13
PAGE4.0FSPAGES
Case 1:20-cv-01277-AWI-JDP Document 1-7 Filed 09/08/20 Page 5 of 6

I -
·' DEP Aih MEN 1 Or iiEAL 1 ti 'ANV HUMAN SERvlC-r;S FOOD AND DRUG ADMINISTRATION
DATE{S) OF INSPECTION DISTRICT ADDRESS AND PHON!a NUMBER
250-Marquette Avenue, Suite 600 Minneapolis, .MN 55401
02/14/2013 - 04/03/2013* FEINUMBER
(612) 334-4100 Fax:'(612) 334-4134 2182207 Industry Information: www.fda.gov/oc/industry NAME AND TITLE OF INDIVIDUAL TO \MiOM REPORT ISSUED
TO: Omar S. Ishrak, Chairman and Chief Executive Officer FIRM NAME STREET ADDRESS
Medtronic Neuromodulation 7000 Central Ave NE TYPE ESTABI.ISHMENT INSPECTED CITY, STATE, ZIP COOE, CCUNTRY
Minneapolis, MN 55432-3568 Medical Device Manufacturer
-'J, 9#f >l/~/f}
Observation/: Promised to correct
Observation Annotations 11.\ ~ 'I) "I/ a/13 "3 ~~ f#/'17
ObservationJ: Promised to correct Observation}; Promised to correct.
/./ ~11 ;.t/3/,J .QAr-'~, h 1J
* DATES OF INSPECTION: 02/14/2013(Thu), 02/15/2013(Fri), 02/19/2013(Tue), 02/20/2013(Wed), 02/22/2013(Fri), 02/25/2013(Mon), 02/26/2013(Tue), 02/28/2013(Thu), 03/01/2013(Fri), 03/04/2013(Mon), 03/07/2013(Thu), 03/ll/2013(Mon), 03/13/2013(Wed), 03/14/2013(Thu), 03/2I/20l3(Thu), 03/26/2013(Tue), 03/28/2013(Thu), 04/03/2013(Wed)
SEE REVERSE OF THIS PAGE
FORM FDA 483 (09"""
EMPLOYEE($) SIGNATURE . ro/11 . '»0f',V Jessica L. Joh?son, Inves~igator Susan M. Matthias, !nvestigator ·
PREVIOUS EDmON O!ISOUITE INSPECTIONAL OBSERVATIONS
'i/o/1 ) DATEISSUEO
04/03/2013
PAGE 5 OF 5 PAGES
Case 1:20-cv-01277-AWI-JDP Document 1-7 Filed 09/08/20 Page 6 of 6

Exhibit 8
Case 1:20-cv-01277-AWI-JDP Document 1-8 Filed 09/08/20 Page 1 of 6

2/6/2020 Class 2 Device Recall SynchroMed II Implantable Drug Infusion Pump
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfres/res.cfm?id=126144 1/5
6510(k)7 |DeNovo8|Registration &
Listing9|Adverse
Events10|Recalls11 |PMA12|HDE13|Classification14|Standards15
CFR Title
2116|Radiation-Emitting
Products17|X-Ray
Assembler18|Medsun
Reports19|CLIA20|TPLC21
New Search Back to Search Results
Class 2 Device RecallSynchroMed II Implantable DrugInfusion Pump
22
Date Initiated by Firm February 26, 2014
Date Posted May 08, 2014
Recall Status1 Terminated 3 on September 28, 2018
Recall Number Z-1570-2014
Recall Event ID 6772023
PMA Number P860004S05624
Product Classification Pump, infusion, implanted, programmable25 - Product Code LKK26
Product Medtronic SynchroMed¿ II Implantable Drug Infusion Pump, Model 8637-20,8637-40.
The implantable Medtronic SynchroMed II programmable pumps are part of aninfusion system that stores and delivers a prescribed drug to a specific site. Theimplanted infusion system consists of a SynchroMed II pump and a catheter.
Code Information This Medical Device Correction notification affects all SynchroMed II pumps.
Recalling Firm/Manufacturer
Medtronic Neuromodulation7000 Central Ave NE
Class 2 Device Recall SynchroMed II Implantable Drug Infusion PumpFDA Home3 Medical Devices4 Databases5
Case 1:20-cv-01277-AWI-JDP Document 1-8 Filed 09/08/20 Page 2 of 6

2/6/2020 Class 2 Device Recall SynchroMed II Implantable Drug Infusion Pump
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfres/res.cfm?id=126144 2/5
Minneapolis MN 55432-3568
For AdditionalInformation Contact
Donna Marquard763-526-6248
Manufacturer Reasonfor Recall
This recall provides important new information regarding overinfusion associatedwith the Medtronic SynchroMed II Implantable Pump. Overinfusion can result in alife-threatening overdose and can also result in drug withdrawal due to premature emptying of the pump. Due tothe low reported rate of occurrence of this issue and the inability to predict whichpumps may be at risk, Medtronic is not re
FDA DeterminedCause 2
Under Investigation by firm
Action Medtronic sent a "Urgent Medical Device Correction" letter dated March 2014.The letter was sent to all affected customers. The letter identified the product theproblem and the action needed to be taken by the customer. The letter provided the Explanation of the Issue, Scope and Severity,Recommendations, and Important Guidelines. Customer visits were started by Medtronic field Representatives on February26th, 2014. Medtronic is communicating this information to the appropriate regulatoryagencies globally, including the U.S. Food and Drug Administration. We arecommitted to continuing to improve our product performance and services toenable you to manage your patients in a safe and effective manner. If you havequestions, please contact Medtronic Neuromodulation Technical Services at 1-800-707-0933 weekdays 7am- 6pm CST.
Quantity in Commerce 195,198 pumps (146,435 US, 48,763 OUS)
Distribution Worldwide Distribution - All states in USA. OUS: List not provided at this time.
Total Product Life CycleTPLC Device Report27
1 A record in this database is created when a firm initiates a correction or removal action. The record isupdated if the FDA identifies a violation and classifies the action as a recall, and it is updated for a final timewhen the recall is terminated. Learn more about medical device recalls28.
2 Per FDA policy, recall cause determinations are subject to modification up to the point of termination of therecall.
Case 1:20-cv-01277-AWI-JDP Document 1-8 Filed 09/08/20 Page 3 of 6

2/6/2020 Class 2 Device Recall SynchroMed II Implantable Drug Infusion Pump
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfres/res.cfm?id=126144 3/5
3 For details about termination of a recall see Code of Federal Regulations (CFR) Title 21 §7.5529.
PMA Database PMAs with Product Code = LKK and Original Applicant = MEDTRONIC Inc.30
Links on this page:1. http://www.addthis.com/bookmark.php?
u508=true&v=152&username=fdamain
2. http://www.addthis.com/bookmark.php
3. https://www.fda.gov/
4. http://www.fda.gov/MedicalDevices/default.htm
5. http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/Databases/default.htm
6. /scripts/cdrh/devicesatfda/index.cfm
7. /scripts/cdrh/cfdocs/cfPMN/pmn.cfm
8. /scripts/cdrh/cfdocs/cfpmn/denovo.cfm
9. /scripts/cdrh/cfdocs/cfRL/rl.cfm
10. /scripts/cdrh/cfdocs/cfMAUDE/TextSearch.cfm
11. /scripts/cdrh/cfdocs/cfRES/res.cfm
12. /scripts/cdrh/cfdocs/cfPMA/pma.cfm
13. /scripts/cdrh/cfdocs/cfHDE/hde.cfm
14. /scripts/cdrh/cfdocs/cfPCD/classification.cfm
15. /scripts/cdrh/cfdocs/cfStandards/search.cfm
16. /scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm
17. /scripts/cdrh/cfdocs/cfPCD_RH/classification.cfm
18. /scripts/cdrh/cfdocs/cfAssem/assembler.cfm
19. /scripts/cdrh/cfdocs/Medsun/searchReportText.cfm
20. /scripts/cdrh/cfdocs/cfClia/Search.cfm
21. /scripts/cdrh/cfdocs/cfTPLC/tplc.cfm
22. http://www.fda.gov/safety/recalls/enforcementreports/default.htm
23. /scripts/cdrh/cfdocs/cfRES/res.cfm?start_search=1&event_id=67720
24. /scripts/cdrh/cfdocs/cfpma/pma.cfm?ID=P860004S056
25. /scripts/cdrh/cfdocs/cfPCD/classification.cfm?ID=LKK
26. /scripts/cdrh/cfdocs/cfPCD/classification.cfm?ID=LKK
27. /scripts/cdrh/cfdocs/cfTPLC/tplc.cfm?id=LKK
Case 1:20-cv-01277-AWI-JDP Document 1-8 Filed 09/08/20 Page 4 of 6

2/6/2020 Class 2 Device Recall SynchroMed II Implantable Drug Infusion Pump
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfres/res.cfm?id=126144 4/5
28. http://www.fda.gov/MedicalDevices/Safety/ListofRecalls/ucm329946.htm
29. /scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm?fr=7.55
30. /scripts/cdrh/cfdocs/cfPMA/pma.cfm?start_search=1&productcode=LKK&applicant=MEDTRONIC%20Inc%2E
Page Last Updated: 02/05/2020Note: If you need help accessing information in different file formats, see Instructions forDownloading Viewers and Players. Language Assistance Available: Español | 繁體中文 | Tiếng Việt | 한국어 | Tagalog | Русский | العربیة |Kreyòl Ayisyen | Français | Polski | Português | Italiano | Deutsch | 日本語 | فارسی | English
Accessibility Contact FDA Careers FDA Basics FOIA No FEAR Act Nondiscrimination WebsitePolicies
U.S. Food and Drug Administration10903 New Hampshire Avenue Silver Spring, MD 20993 Ph. 1-888-INFO-FDA (1-888-463-6332)Contact FDA
For Government For PressCombination Products Advisory Committees Science & Research Regulatory Information SafetyEmergency Preparedness International Programs News & Events Training and ContinuingEducation Inspections/Compliance State & Local Officials Consumers Industry HealthProfessionals FDA Archive
Links on this page:1. http://www.addthis.com/bookmark.php?
u508=true&v=152&username=fdamain
2. http://www.addthis.com/bookmark.php
3. https://www.fda.gov/
4. http://www.fda.gov/MedicalDevices/default.htm
5. http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/Databases/default.htm
6. /scripts/cdrh/devicesatfda/index.cfm
7. /scripts/cdrh/cfdocs/cfPMN/pmn.cfm
8. /scripts/cdrh/cfdocs/cfpmn/denovo.cfm
9. /scripts/cdrh/cfdocs/cfRL/rl.cfm
10. /scripts/cdrh/cfdocs/cfMAUDE/TextSearch.cfm
11. /scripts/cdrh/cfdocs/cfRES/res.cfm
12. /scripts/cdrh/cfdocs/cfPMA/pma.cfm
13. /scripts/cdrh/cfdocs/cfHDE/hde.cfm
14. /scripts/cdrh/cfdocs/cfPCD/classification.cfm
15. /scripts/cdrh/cfdocs/cfStandards/search.cfm
Case 1:20-cv-01277-AWI-JDP Document 1-8 Filed 09/08/20 Page 5 of 6

2/6/2020 Class 2 Device Recall SynchroMed II Implantable Drug Infusion Pump
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfres/res.cfm?id=126144 5/5
16. /scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm
17. /scripts/cdrh/cfdocs/cfPCD_RH/classification.cfm
18. /scripts/cdrh/cfdocs/cfAssem/assembler.cfm
19. /scripts/cdrh/cfdocs/Medsun/searchReportText.cfm
20. /scripts/cdrh/cfdocs/cfClia/Search.cfm
21. /scripts/cdrh/cfdocs/cfTPLC/tplc.cfm
22. http://www.fda.gov/safety/recalls/enforcementreports/default.htm
23. /scripts/cdrh/cfdocs/cfRES/res.cfm?start_search=1&event_id=67720
24. /scripts/cdrh/cfdocs/cfpma/pma.cfm?ID=P860004S056
25. /scripts/cdrh/cfdocs/cfPCD/classification.cfm?ID=LKK
26. /scripts/cdrh/cfdocs/cfPCD/classification.cfm?ID=LKK
27. /scripts/cdrh/cfdocs/cfTPLC/tplc.cfm?id=LKK
28. http://www.fda.gov/MedicalDevices/Safety/ListofRecalls/ucm329946.htm
29. /scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm?fr=7.55
30. /scripts/cdrh/cfdocs/cfPMA/pma.cfm?start_search=1&productcode=LKK&applicant=MEDTRONIC%20Inc%2E
Case 1:20-cv-01277-AWI-JDP Document 1-8 Filed 09/08/20 Page 6 of 6

Exhibit 9
Case 1:20-cv-01277-AWI-JDP Document 1-9 Filed 09/08/20 Page 1 of 4

@, Mnll:ranic
Urgent: Medical Device Correction SynchroMed® II Implantable Drug Infusion Pump
Overinfusion
Dear Healthcare Professional,
March 2014
This letter provides important new information regarding overinfusion associated with the SynchroMed® II Implantable Pump. Overinfusion can result in a life-threatening overdose and can also result in drug withdrawal due to premature emptying of the pump. Due to the low reported rate of occurrence of this issue and the inability to predict which pumps may be at risk, Medtronic is not recommending prophylactic replacement of pumps.
This communication is based on information available to date and was developed in collaboration with clinical experts. Medtronic continues to investigate this issue and we are committed to providing updates as more information becomes available.
Explanation of the Issue:
Medtronic detected an upward shift in reports of occurrence for overinfusion. Overinfusion is defined as an infusion rate exceeding the programmed infusion rate by more than 14.5% as described in the labeling (see enclosed flow rate accuracy section from the SynchroMed II Implant Manual). When overinfusion occurs, it will result in a volume discrepancy at pump refill. where the volume withdrawn from the pump is less than the volume expected. The cause(s) for pump malfunction leading to overinfusion remains under investigation and has not been linked to any specific pump lot, drug used, or geographical area. Based on reports, the onset of overinfusion has occurred as early as five months after implant and throughout the service life of the pump. Reports received indicate that once a pump has started to overinfuse, infusion rates can continue to increase, in some cases abruptly.
Scope and Severity:
Based on current data from Medtronic's prospective, long-term multi-center registry study (ISPR), the occurrence rate for overinfusion is less than 0.16o//
As of November 18, 2013, 76 pumps have been confirmed for overinfusion through returned product analysis since the introduction of the device in 2003:
• 44 were explanted for reasons consistent with overinfusion. o 14 reports of life-threatening overdose o 27 reports of non-life threatening overdose and/or withdrawal o 3 reports of volume discrepancy without overinfusion symptoms
• 32 were explanted for reasons other than overinfusion. However, routine testing of returned pumps found these pumps to be overinfusing.
Adverse events associated with overinfusion will vary depending on the drug being infused, but may include confusion or altered mental state, sleepiness, nausea, respiratory depression and coma, with the risk of death. Overinfusion may lead to emptying of the pump prior to a planned refill and therefore may present clinically as an interruption of therapy including lack of
Page 1 of 3
Case 1:20-cv-01277-AWI-JDP Document 1-9 Filed 09/08/20 Page 2 of 4

~ Medtronic March 2014
therapeutic effect and withdrawal syndrome. There has not been a report of a patient death associated with this issue.
The low reservoir alarm in the SynchroMed II is designed to activate based on programmed flow rates and starting volumes. The device does not measure actual reservoir volume and in the context of overinfusion the reservoir may empty entirely without activating an alarm. It is not possible to detect the issue other than by following the recommendations below.
Recommendations (Developed in collaboration with clinical experts):
• Medtronic does not recommend prophylactic removal of SynchroMed II pumps. • Educate patients, caregivers and family members to recognize the signs and symptoms
associated with intrathecal drug therapy overdose, underdose or withdrawal. • At every refill visit, question and examine the patient for signs and symptoms of overdose,
underdose or withdrawal. • Follow the labeled refill instructions, so that any volume discrepancy can be detected based
on the amount of medication withdrawn prior to refill (see guidelines below). • At every refill visit record the actual and expected reservoir volume. • Review prior refill data to identify any changes in volume discrepancy over time. If there are
increases in volume discrepancy over time (volume withdrawn from the pump is less than expected) or if there is a volume discrepancy of more than 2ml:
o Evaluate for other causes, such as unrecognized partial pocket fill, self-aspiration of reservoir medication, and less than full reservoir at prior refill .
o If overinfusion is strongly suspected clinically monitor the patient and consider pump replacement. The decision to replace the pump should take the following factors into consideration: history of pump volumes, magnitude of the volume discrepancy, presence/severity of overdose symptoms, and the individual patient situation.
o To stop delivery of drug from a pump suspected of overinfusion, program a "therapy stop", which sets the pump to minimum rate, and remove any remaining drug from the reservoir to avoid continued drug delivery.
o Reducing the dose and/or concentration will not correct overinfusion because infusion rates may increase over time.
Important Guidelines: Always follow pump refill instructions per the device labeling. The following steps should be conducted during each pump refill procedure to allow detection of an overinfusing pump:
• Aspirate all fluid from the reservoir until air bubbles no longer appear in the syringe, and record as the amount withdrawn.
• Compare the amount withdrawn from the pump reservoir with the expected volume displayed by the pump programmer. The amount withdrawn should approximately equal the expected volume.
• Determine fill volume (fill with no more than the labeled reservoir volume, 20 or 40 ml). • Accurately measure the volume to be instilled. • If you are unsure whether drug was injected correctly into the pump, completely aspirate
the pump to verify that the entire injected volume of drug has been removed. • Ensure that refill dates are chosen sufficiently in advance of the low reservoir alarm date
so the pump does not run dry.
Page 2 of 3
Case 1:20-cv-01277-AWI-JDP Document 1-9 Filed 09/08/20 Page 3 of 4

~ Mnll:ronic March 2014
Inform Medtronic Neuromodulation Technical Services if overinfusion is strongly suspected. Please return any explanted products to Medtronic for mechanical and functional analysis. Your local Medtronic representative can assist you.
Medtronic is communicating this information to the appropriate regulatory agencies globally, including the U.S. Food and Drug Administration. We are committed to continuing to improve our product performance and services to enable you to manage your patients in a safe and effective manner. If you have questions, please contact Medtronic Neuromodulation Technical Services at 1-800-707-0933 weekdays 7am - 6pm CST.
Please report any malfunction or adverse event related to a device to Medtronic Neuromodulation Technical Services and to FDA's MedWatch Program (www.fda.gov/medwatch).
Sincerely,
Mike Crader Vice President Quality Medtronic Neuromodulation
Enclosed: Flow rate accuracy as described in the SynchroMed II Implant Manual.
; There have been four reports of overinfusion in 5,765 SynchroMed II pumps included in Medtronic's prospective, long-term multi-center registry study (ISPR), providing a 95% confidence that the occurrence rate is less than 0.0016 (0.16%).
Page 3 of 3
Case 1:20-cv-01277-AWI-JDP Document 1-9 Filed 09/08/20 Page 4 of 4

Exhibit 10
Case 1:20-cv-01277-AWI-JDP Document 1-10 Filed 09/08/20 Page 1 of 3

September 2016
Page 1 of 2
Urgent Medical Device Correction Update SynchroMed® II Implantable Drug Infusion Pump
Update to the March 2014 Communication on Overinfusion
Dear Healthcare Professional:
This communication is an update to Medtronic’s March 2014 notification regarding the potential for SynchroMed II pump overinfusion. This notification updates information related to contributing causes, occurrence rate and patient management recommendations. Consistent with the previous communication, Medtronic is not retrieving SynchroMed II pumps from the field or recommending prophylactic replacement of the pumps. Please share the enclosed recommendations and this update with personnel responsible for the management of patients implanted with a SynchroMed II pump.
Explanation of the Issue: “Overinfusion” is defined as the delivery of more drug volume than the programmed rate, exceeding the pump’s flow rate accuracy specification. Pump reservoir contents aspirated during a refill procedure that are less than expected may indicate that the pump has overinfused. Overinfusion may or may not be associated with clinically relevant symptoms. When the pump delivers more drug volume than the programmed rate, patients may experience overdose symptoms, and the pump reservoir will deplete more quickly than expected. Patients may experience underdose or withdrawal symptoms if the drug is depleted prior to the scheduled refill date from an overinfusing pump.
The low reservoir alarm of an overinfusing pump will not sound if the pump reservoir is prematurely depleted. The low reservoir alarm is calculated from the pump’s programmed delivery rate and is not a direct measurement of the actual drug volume in the pump reservoir. Therefore, it is important to follow the enclosed recommendations.
Investigation Results: Medtronic’s investigation has not identified any single factor that results in overinfusion; rather the interaction of several variables increases the likelihood that a given pump will overinfuse. Some risk factors are associated with normal variability with pump components and manufacturing processes, while other factors are associated with clinical use conditions. Examples of clinical use conditions that have been shown to increase the likelihood of overinfusion are the use of nonindicated drug formulations, overfilling of the pump reservoir, operation of the pump with no fluid in the reservoir, catheter occlusion, and pump stops or motor stalls lasting more than 48 hours.
Occurrence Rate Based on Registry Data: Five occurrences of overinfusion have been identified in Medtronic’s prospective, long-term multi-center registry study (Product Surveillance Registry) as of January 2016, resulting in a rate estimate of less than 0.14%1 (approximately 1-in-700). All 5 occurrences of overinfusion noted in
1 Through 31 January 2016, there have been five reports of overinfusion in 7,505 SynchroMed II pumps included in Medtronic’s prospective, long-term multi-center registry study (PSR, formerly ISPR), providing an upper 95% confidence bound on the occurrence rate of 0.0014 (0.14%). Based on investigation results, this rate is not significantly changed from the 0.16% upper 95% confidence bound reported in the March 2014 communication.
Case 1:20-cv-01277-AWI-JDP Document 1-10 Filed 09/08/20 Page 2 of 3

September 2016
Page 2 of 2
the Registry were associated with pumps used to infuse drug formulations that were not indicated for use with the SynchroMed II pump.
Reports of Adverse Events: Since commercial release of the SynchroMed II pump, over 238,000 pumps have been implanted. During Medtronic’s investigation of overinfusion, complaint data and returned product analysis were assessed, resulting in 103 pumps with related adverse events through 05 July 2016. Medtronic has been unable to establish a definitive causal relationship between the adverse events and overinfusion due to potential contributing factors. However, it is reasonable to conclude that overinfusion was a contributing factor in these cases. Other factors that may have contributed to an adverse event are: infused drug dosage, the patient’s medical history, and the concomitant use of other drugs, such as oral opioids and other central nervous system (CNS) depressants.
Reported patient outcomes associated with these adverse events ranged from temporary discomfort to life threatening overdose and/or withdrawal as well as two reports of death. While the full drug history of these pumps is unknown, 99 of the 103 pumps were associated with nonindicated drug formulations in use at the time of the pump’s last refill. The estimated implant duration for the 103 pumps is 3.7 years (with a range of 0.4 – 6.4 years).
Recommendations: See the enclosed Recommendations and Guidelines.
Additional Information: Medtronic is communicating this information to the appropriate regulatory agencies globally, including the U.S. Food and Drug Administration.
We are committed to continuing to improve our product performance and services to enable you to manage your patients in a safe and effective manner. If you have questions, please contact Medtronic Neuromodulation Technical Services at 1-800-707-0933 weekdays 7am - 6pm CT. Please report any malfunction or adverse event related to a device to Medtronic Neuromodulation Technical Services and to FDA’s MedWatch Program (www.fda.gov/medwatch).
Sincerely, Michael Ronningen Vice President of Quality Medtronic Enclosures:
• Recommendations and Guidelines • Physician Reply Form
Case 1:20-cv-01277-AWI-JDP Document 1-10 Filed 09/08/20 Page 3 of 3

Exhibit 11
Case 1:20-cv-01277-AWI-JDP Document 1-11 Filed 09/08/20 Page 1 of 11

UNITED STATES DISTRICT COURT FOR THE DISTRICT OF MINNESOTA
Civil No. 15 - 2168
UNITED STATES OF AMERICA, ) )
Plaintiff ) )
v. ) ) COMPLAINT FOR
MEDTRONIC INC., a corporation, and ) PERMANENT INJUNCTION S. OMAR ISHRAK and ) THOMAS M. TEFFT, individuals, )
) )
Defendants. ) _________________________________)
INTRODUCTION
Plaintiff, the United States of America, by its undersigned attorneys, respectfully
represents to this Court as follows:
1. This statutory injunction proceeding is brought under the Federal Food,
Drug, and Cosmetic Act (the "Act"), 21 U.S.C. § 332(a), to enjoin Medtronic Inc.
(“Medtronic”), a corporation, and S. Omar Ishrak, and Thomas M. Tefft, individuals
(hereinafter, collectively, “Defendants”) from violating:
A. 21 U.S.C. § 331(a), by introducing or delivering for introduction into
interstate commerce, or causing the introduction or delivery for introduction into
interstate commerce, articles of devices, as defined by 21 U.S.C. § 321(h), that are
adulterated within the meaning of the Act, 21 U.S.C. § 351(h), in that the methods used
in, or the facilities or controls used for, their manufacture, packing, storage, and
CASE 0:15-cv-02168 Document 1 Filed 04/27/15 Page 1 of 10Case 1:20-cv-01277-AWI-JDP Document 1-11 Filed 09/08/20 Page 2 of 11

2
installation are not in conformity with current good manufacturing practice requirements
prescribed at 21 C.F.R. Part 820;
B. 21 U.S.C. § 331(k), by causing devices to become adulterated within
the meaning of 21 U.S.C. § 351(h), as described in paragraph A above, while such
devices are held for sale after shipment in interstate commerce.
JURISDICTION AND VENUE
2. This Court has jurisdiction under 21 U.S.C. § 332(a) and 28 U.S.C. §§ 1331
and 1345.
3. Venue in this District is proper pursuant to 28 U.S.C. § 1391(b) and (c).
DEFENDANTS
4. Medtronic is incorporated under the laws of Minnesota. Medtronic
Neuromodulation (“Medtronic Neuro”), a business unit of Medtronic, manufactures
medical devices, including but not limited to, SynchroMed II implantable infusion
pumps. The headquarters of Medtronic Neuro is located at 7000 Central Ave. NE,
Minneapolis, MN 55432, and its manufacturing facility is located at 53rd Avenue, NE,
Columbia Heights, MN 55421.
5. S. Omar Ishrak is Medtronic’s Chairman and CEO. He is the most
responsible person at the firm, and oversees the firm's product development, product
management, and international relations and sales. He performs his duties at 710
Medtronic Parkway, Minneapolis, MN 55432.
6. Thomas M. Tefft is the Senior Vice President of Medtronic, and the
President of Medtronic Neuro. He is the most responsible person at Medtronic Neuro,
CASE 0:15-cv-02168 Document 1 Filed 04/27/15 Page 2 of 10Case 1:20-cv-01277-AWI-JDP Document 1-11 Filed 09/08/20 Page 3 of 11

3
and oversees the business unit’s product development, research, regulatory compliance
and marketing. He performs his duties at 7000 Central Ave. NE, Minneapolis, MN
55432.
7. Defendants have been, and are now, manufacturing and distributing in
interstate commerce various articles of devices, as defined by 21 U.S.C. § 321(h),
including, but not limited to, SynchroMed II implantable infusion pumps, the subject of
this injunction.
8. Defendants’ products are devices, within the meaning of 21 U.S.C. §
321(h), in that they are intended to affect the structure or any function of the body of
man.
LEGAL STANDARDS
9. A device must be manufactured, packed, stored, and installed in conformity
with good manufacturing practice to ensure its safety and effectiveness. 21 U.S.C.
§ 360j(f). The statutory good manufacturing practice requirement is set out in the quality
system ("QS") regulation for devices, 21 C.F.R. Part 820. A device that has been
manufactured, packed, stored, or installed in violation of this requirement is deemed to be
adulterated. 21 U.S.C. § 351(h).
10. The introduction or delivery for introduction into interstate commerce of an
adulterated article of device is a violation of the Act, 21 U.S.C. § 331(a).
11. The adulteration of a device while it is held for sale after shipment in
interstate commerce constitutes a violation of the Act, 21 U.S.C. § 331(k).
CASE 0:15-cv-02168 Document 1 Filed 04/27/15 Page 3 of 10Case 1:20-cv-01277-AWI-JDP Document 1-11 Filed 09/08/20 Page 4 of 11

4
APRIL 2013 INSPECTION
12. FDA inspected Medtronic Neuro’s manufacturing facility on February 14 –
April 3, 2013 (“April 2013 inspection”). During the April 2013 inspection, the FDA
investigators documented numerous violations of the QS regulation at Medtronic Neuro.
Many of these violations related directly to the manufacture of the SynchroMed II
implantable infusion pump. FDA investigators observed the following violations of the
QS regulation set forth in 21 C.F.R. Part 820:
A. Defendants fail to establish and maintain adequate design validation
procedures to ensure that devices conform to defined user needs and intended uses, to
complete proper risk analysis, and to document the results of the validation, in violation
of 21 C.F.R. § 820.30(g);
B. Defendants fail to establish and maintain adequate procedures to
include requirements for identifying the action(s) needed to correct and prevent
recurrence of nonconforming product and other quality problems, in violation of 21
C.F.R. § 820.100(a)(3);
C. Defendants fail to establish and maintain adequate procedures to
include requirements for verifying or validating the corrective and preventive action to
ensure that such action is effective and does not adversely affect the finished device, in
violation of 21 C.F.R. § 820.100(a)(4);
D. Defendants fail to establish and maintain procedures for
implementing corrective and preventive action, in violation of 21 C.F.R. § 820.100(a);
CASE 0:15-cv-02168 Document 1 Filed 04/27/15 Page 4 of 10Case 1:20-cv-01277-AWI-JDP Document 1-11 Filed 09/08/20 Page 5 of 11

5
E. Defendants fail to establish and maintain procedures for verifying
the device design, in violation of 21 C.F.R. § 820.30(f);
F. Defendants fail to establish and maintain procedures for the
identification, documentation, validation or where appropriate verification, review, and
approval of design changes before their implementation, in violation of 21 C.F.R.
§ 820.30(i); and
G. Defendants fail to establish and maintain procedures to control
product that does not conform to specified requirements, in violation of 21 C.F.R.
§ 820.90(a).
PRIOR INSPECTIONS
13. FDA inspected Medtronic Neuro’s facilities previously in May 2012,
January 2011, January 2007, and June 2006. At these inspections, FDA repeatedly
observed and documented violations of the QS regulations similar to those cited above
during the April 2013 inspection, including, but not limited to, violations involving:
design controls (21 C.F.R. § 820.30) and corrective and preventive action (21 C.F.R.
§ 820.100).
14. At the conclusion of each of the prior inspections, the FDA investigators
issued a Form FDA 483 detailing Defendants' numerous violations of the Act to
Defendants, and discussed the documented observations with them. Defendants
promised corrections at the conclusion of each inspection.
CASE 0:15-cv-02168 Document 1 Filed 04/27/15 Page 5 of 10Case 1:20-cv-01277-AWI-JDP Document 1-11 Filed 09/08/20 Page 6 of 11

6
PRIOR NOTICE OF VIOLATIONS
15. Defendants are well aware that their practices violate the Act. FDA has
repeatedly warned Defendants, both orally and in writing, about their violative conduct,
and has emphasized the importance of Defendants' compliance with the Act.
16. FDA issued a Warning Letter dated July 17, 2012 to Defendants, following
the May 2012 inspection of the Medtronic Neuro facility. The letter discussed the QS
violations involving corrective and preventive actions and complaint handling (21 C.F.R.
§ 820.198) observed at the inspection. The letter also warned Defendants that further
enforcement actions, including injunction, could occur if they did not correct the
violations.
17. Defendants also received Warning Letters, dated July 3, 2007 and August
29, 2006, following the January 2007 and June 2006 inspections. These letters also
addressed the numerous QS violations, including but not limited to design controls and
corrective and preventive action, observed during the inspections and warned of further
enforcement actions if corrections were not made.
18. Representatives of Medtronic also attended a meeting with FDA’s Center
for Devices and Radiological Health and Minneapolis District Office on January 31,
2013. At this meeting, Defendants stated that they were aware of the violations at their
facilities and were taking steps to correct them.
19. At the conclusion of each of FDA's inspections of the firm, the FDA
investigators issued a Form FDA 483 detailing Defendants' various violations of the Act
CASE 0:15-cv-02168 Document 1 Filed 04/27/15 Page 6 of 10Case 1:20-cv-01277-AWI-JDP Document 1-11 Filed 09/08/20 Page 7 of 11

7
to a responsible individual at the firm and discussed the documented observations with
the recipient.
20. Defendants made promises to correct their violations in written responses to
the April 2013 inspection, dated April 24, and several follow-up responses, detailing
how and when the corrections promised in the April 24 letter had been made. None of
these responses contained adequate evidence that Defendants have corrected their
deviations.
21. Based on Defendants' conduct, Plaintiff believes that, unless restrained by
order of this Court, Defendants will continue to violate 21 U.S.C. §§ 331(a) and (k).
WHEREFORE, Plaintiff prays:
I. That Defendants and each of their directors, officers, agents,
representatives, employees, attorneys, successors, and assigns, and any and all persons in
active concert or participation with any of them, be permanently restrained and enjoined
pursuant to 21 U.S.C. § 332(a) from directly or indirectly:
A. violating 21 U.S.C. § 331(a), by introducing or delivering for
introduction into interstate commerce, or causing the introduction or delivery for
introduction into interstate commerce, any article of device that is adulterated within the
meaning of 21 U.S.C. § 351(h); or
B. violating 21 U.S.C. § 331(k), by causing any article of device to
become adulterated within the meaning of 21 U.S.C. § 351(h) while such devices are held
for sale after shipment in interstate commerce.
CASE 0:15-cv-02168 Document 1 Filed 04/27/15 Page 7 of 10Case 1:20-cv-01277-AWI-JDP Document 1-11 Filed 09/08/20 Page 8 of 11

8
II. That the Court order Defendants and each of their directors, officers,
agents, representatives, employees, attorneys, successors, and assigns, and any and all
persons in active concert or participation with any of them, to cease directly and
indirectly manufacturing, packing, labeling, and distributing (domestically and
internationally) SynchroMed II implantable infusion pumps at or from its Medtronic
Neuro facilities, unless and until Defendants' methods, facilities, and controls used to
manufacture, process, pack, label, hold, and distribute the SynchroMed II implantable
infusion pumps are established, operated, and administered in compliance with 21 U.S.C.
§ 360j(f)(1) and the Quality System regulation prescribed in 21 C.F.R. Part 820, and in a
manner that has been found acceptable to FDA; and
III. That the Court authorize FDA, pursuant to this injunction, to inspect
Defendants' Medtronic Neuro facility to ensure continuing compliance with the terms of
this injunction, with the costs of such inspections to be borne by Defendants at the rates
prevailing at the time the inspections are performed.
CASE 0:15-cv-02168 Document 1 Filed 04/27/15 Page 8 of 10Case 1:20-cv-01277-AWI-JDP Document 1-11 Filed 09/08/20 Page 9 of 11

9
IV. That Plaintiff be granted judgment for its costs herein, and that this Court
grant such other and further relief as it deems just and proper.
ANDREW M. LUGER United States Attorney s/ Chad A. Blumenfield CHAD BLUMENFIELD Assistant U.S. Attorney Attorney ID 387296 600 Courthouse 300 South Fourth St. Minneapolis, MN 55415 Ross S. Goldstein Trial Attorney Consumer Protection Branch U.S. Department of Justice Civil Division P.O. Box 386 Washington, DC 20044 OF COUNSEL: WILLIAM B. SCHULTZ Acting General Counsel ELIZABETH DICKINSON Associate General Counsel Food and Drug Division ANNAMARIE KEMPIC Deputy Chief Counsel, Litigation
CASE 0:15-cv-02168 Document 1 Filed 04/27/15 Page 9 of 10Case 1:20-cv-01277-AWI-JDP Document 1-11 Filed 09/08/20 Page 10 of 11

10
TARA BOLAND Associate Chief Counsel United States Department of Health and Human Services Office of the General Counsel 10903 New Hampshire Ave. Silver Spring, MD 20993-0002 (301) 796-8549
CASE 0:15-cv-02168 Document 1 Filed 04/27/15 Page 10 of 10Case 1:20-cv-01277-AWI-JDP Document 1-11 Filed 09/08/20 Page 11 of 11

Exhibit 12
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 1 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 1 of 27
UNITED STAT ES DISTRICT COURT FOR THE DISTRICT OF MINNESOTA
U ITED STATES O F AI\IERICA,
Plaintiff.
0 .
IEDTRONIC, I C .. a corporation. and S. 0 !AR ISi IRAK and T HO~ IAS I. TE F FT, indi viduals,
Defe ndants.
Case No. ________ _
CO NSENT D EC REE O F P E RM ANENT IN JUNCTIO N
Plaintiff, the Uni ted States of America, by its undersigned attorneys, having fi led a
complaint for permanent injunction against t-.kd tronic, Inc. (''t-.kdtronic"), a corporation,
and S. Omar lshrak and T homas ~I. Te fft, individuals (collect ively, "Defendants"). and
Defendants, havi ng appeared and havi ng conscnced tu e ntry of chis Decree without concc;sc,
"·ithout admitting or denyi ng the allcgacions in the Complaint, and disclaiming any liability
in connection therewith and before any testimony has been cakcn, and the.: United Scates
having consented to th is Decree.:,
IT IS I IE REBY ORDERED, ADJUDG ED, A D DECREED AS FOLLOWS:
I. This Court has jurisdiction over the subject matter of this action and has
personal jurisdiction over all parties to chis action.
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 2 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 2 of 27
2. The Complaint for Pe rmane nt Injunc tion states a cause of action aga inst
Defendants under the Federa l Food, Drug, and Cosmetic Act (the "Act"), 21 U.S.C. § 301 el .
seq.
3. The Compla int a lleges that Defendants violate the Act, 21 U.S.C. § 331(a), by
introduc ing or d elivering for introduction into interstate commerce, o r ca us ing the
introduction or de livery fo r introd uction into inte rstate commerce, articles of device, as
defined by 21 U.S.C. § 32l(h), name ly Synchrol'v1ecl Implantable Infusion Pump Systems,
that are ad ulterated with in the meaning of 21 lT.S.C. § 351(h), in that the methods used in, or
the faci lities or controls used fo r, the ir manufacture, packing, and storage are not in
conformity with current good manufactu ring practice requirements prescribed at 21 C.F.R.
Part 820.
4. The Compla int a lso a lleges that Defendan ts vio late the Act, 21 U.S.C. §
331(k ), by caus ing the SynchroMcd Implantable Infusion Pump systems to become
adulterated within the meaning of 21 lJ.S.C. § 351(h) while such devices arc held for sale
after shipment in interstate commerce.
DEFINITIONS
S. For the pu rposes of this D ecree, the fol lowing definitions ap ply:
J\. "Synchrol'vJed device" s hall mean all implan table infusion pumps and
thei r accessories that are designed, manufactured, processed, packed, labele d, held, scored,
installed , and distributed at o r from any Medtronic Neuromodulation fac ility.
B. "l'vJedcronic Ncuromodulation" s hall mean the l\,fedtronic
Ncuromodulation Busi ness Unit of Medtronic, Inc., which is responsible fo r des ign ing,
2
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 3 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 3 of 27
manufacturing, process ing. packing, label ing, holding, scoring, and distributing, among other
devices, the SynchroMed devices.
C. "i\,kdtronic Neuromodulation facilit ies" sha ll mean Medtronic
Neuromodulation's headquarte rs, located at 7000 Centra l Ave. NE, l''vlinneapolis. lVIN. and
the manufacturing facil ity located at 53'0 Avenue NE, Columbia Heights, !VIN.
D . A Synchrol'vl ed device is " medically necessary" if (i) it is used to treat
one or more of the following condi tions for which the benefits of us ing rhe Synchrol\lecl
device outwe igh the risks: (a) severe spasricity; (b) chronic intractable pain; (c) severe
chronic pain; and/or (d) primary or me tastatic cancer; and (ii ) rhe physician, after reviewing
the notitication lecter actached he reto as Exhibit A, sig ns a fo rm approved by FDA, attached
hereto as Exhibit B, certifying that s/he is aware of FDJ\'s findings and deems the
Synchro!'vled d evice necessary to treat his/he r patient under the cond itions referred co in
th is paragraph (he reafte r, "Certificate of Med ica l Necessity'').
E. Days shall refer to calendar days un less othe rwise stated.
I N J UNCTIVE PROV ISTO S
6. Upon e ntry of th is D ecree, except as described in paragraph 9, Defe ndants,
and each and all of their d irectors, officers, agents, re presentatives, employees, a ttorneys.
successors, and assigns, and any and all pe rsons in active concert or participation with any of
them (including franch isees, affiliates, and "doing business as" entities) who have received
actual notice of the contents of this D ecree by personal service or otherwise are
pe rmanen tly restrained and enjoine d, pursuant co 21 U.S.C. § 332(a), from directly or
ind irectly designing, manufacturing, processing, packing, labe ling, holding, scoring, and
distributing, importing into or exporting from the United States of America, at or from any
3
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 4 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 4 of 27
Me dtronic Neuromodulation facilities, any model of, or compone nts or accessories for, its
Synchroi'vle d devices, unless and until:
A. De fe ndants' methods, facilities, and controls used to d es ign,
manufacture, p rocess, pack, la bel, hold, store, and distribute SynchroJlvled devices are
esta blished, ope rated, and admini ste re d in compliance with 21 U.S.C . § 360j (f)(I) and the
Quality Syste m ("QS") regulation sec fo rth in 21 C.ER. Pare 820.
B. Defendants select and re tain at l\lcdcronic 's expe nse, within thirty (30)
days of the e ntry of this Decree, an inde pende nt pe rson or pe rsons (the " Expe rt"), co
conduce inspections of D efe ndants' opera tions and co review De fe ndants' procedures and
me thods for designing, manufacturing, processing, packing, la be ling, holding, scoring, and
distributing Synchrol'vlcd devices, to de te rmine whe the r their m ethods, facilities, and
controls a re operated and admini ste red in conformity with the Act, its imple me nting
regulations, and this Decree. The Expe rt sha ll be qualifie d by education, training, and
expe rie nce to conduc t such inspecti ons, and shall be without pe rsona l or financia l ties (other
than a consulting agreement be tween the Ex pert and Medtronic or l\1ledtronic
Neuromodulation) to Defe nda nts' office rs o r e mployees o r t he ir immediate families.
De fendants shall no tify FDA in writing of the identity of the E xpe rt within ten (10) days o f
re taining such Expe rt.
C. The Expe rt s hall pe rform comprehe nsive inspections of Medtronic
Neuromodulati on facilities chat design, manu faccure, process, pack, labe l, hold , store, o r
distribute the SynchroMed devices or any component the reof and certify in writing
simultaneously co De fe ndants and FDA: (i) that he o r she has inspected De fendants'
fac iliti es, processes, and controls; (ii) whe ther De fe ndants ha,·e corrected a ll findings and
4
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 5 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 5 of 27
violations sec forth in FDA's lnspccti ona l Observations ("Forms FDA 483") and Warning
Letters issued to i\,fedtronic Neuromodulacion fac ilities from a ll FDA inspections since
January 2011; and (iii ) based upon these comprehensive inspections, whethe r Defendants'
operations arc operated in conformity with chc Ace, its imple me nting regul ations, and chis
Decree. The Expert's certification re port shall e ncompass, but not be limited to, an
eva luation of the following as they relate to Synchroi'vled devices:
(i) D efendants' compliance with 21 U.S.C. § 35l(h) and 21 C .FR.
Part 820;
(ii) D efendants' procedures for their Corrective and Preventive
Action ("CAPA") sys tem. incl uc..ling, but not limited to, analyzing quality data to identify,
correct, and prevent existing ancJ potential causes of nonconfo rming product and other
quality problems;
(i ii) Defendants' procedures for the ir design control system,
including, but no t limited to, establ ish ing and implementing adequate design and
development plans, inputs, outputs, design reviews, verification, validation, risk ana lyses,
design change controls, and a design history file for each rype of device;
(iv) D efendants' procedures for their noncon forming produce,
inc luding, but not limited to. the identification, documentation, evaluation, segrega tion, and
disposition, including rework, of noncon forming product; and
(v) D efendants' design verification and design va lidation
documents for the SynchroMed device to e nsure that the approved product specifications
a re being mer. In circumstances where the Defendants have identified a design defect that
ca uses tht: SynchroMed device to no t perform according to the approved product
5
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 6 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 6 of 27
sr ecifications, the Expe rt shall review the design defect analys is docume ntation. The
design defect analysis documentation hould include a descrirtion of the design defect. the
pote ntial risk to patie nts associated with the defect, a timc line of actions take n during the
defect in\"estigation, proposed corrective actions, design changes being considered,
developed. and /or tested, and actions that have been take n or will be taken to potenti ally
correct the design defect. The Expert shall also re,·ie \\· design changes made to the
Synchro~ led device in the previous fi,·c (5) years to verify that rhe changes previously
implemented arc effective and do not adve rsely affect the device .
D . Wi thin forcy-fi,·c (-tS) days of receiving the Expert's inspection report
under pa ragraph 6.C, Defendants sha ll submit a wri tten report ("work plan") co FDA
derailing the specific action Defendants ha,·e taken and/or will cake co address the Expert 's
observations and co bring th e methods, fac il ities, processes, and controls used to design,
man ufac ture, process, pack. label, hold, store, and distribute the Synchro led device into
compliance with the requirements of this Decree, che Act, and the QS regu lation. The
specific actions in the " ·ork pl:111 shall be set forth in numbered steps and, where
appropriate, the numbered steps may include s ubordinate lettered steps. The work plan
shall incluue a timetable \\"ith a specific dace for completing each numbered seep and may
include, whe re appropriate, inte rim daces for comple ting su bord ina te le tte red steps. The
\\'Ork pl::in , including its proposed specific actions and timetable, shall be subject to FD/\
approval, and D efendants shall e nsure the implementation of the numbered steps in the
work plan in accordance with the timetable approved by F DA. F DA shall approve or
disapprove in writing the proposed work plan wi thin sixty (60) days.
6
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 7 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 7 of 27
E. Defendants may bcgin implementing the.: work plan as soon as they
recei ,·e written F DA approval. Under no c ircum ranees may F DA's s ilence be construed as
approva l. As the actions deta iled in the work plan are comple ted, Defendants shall notify
the Expert in \\'riting. \\'ho shall promptly inspect and ,·erify whether chose actions have
hcen completed in a manncr char complies with th e requirements of chis Decree. the Acc.
and rhe QS regulation co the Expert's satisfaction and in accordance with the work p lan
timetable.
F. If the.: Expert dcrcrmincs that an action has nor been completed to his
or he r satisfaction. the Expert s hall promptly notify Defendants in writing. Beginning thirty
(30) days after imple me ntation of the work plan, and quarterly thereafter, the Expert shall
s ubmit to FDA a cable that summarizes the Expcrc·s findings regarding " ·hethcr the actions
have been comple ted to the Expert's satisfaction and in accordance with the numbered
steps in the work plan timetable. F DA may, at its discretion and " ·irhout prior notice,
periodically inspect l\ ledtronic Neuromodularion facili ties ,111d undertake such add itiona l
examinations, revicws, and analyses as FDA deems appropriate to verify whethe r the
actions reported tu the Expcrr as completed have in face bccn adequate ly completed on
rime . In che e,·ent that F DA determines char an action that has been reported co be
comple ted is inadequate, F DA shall notify Defe ndants in writing, and Defendants shall
cake appropriate action in accorda nce \\'ith a timetable approved by FDA.
G. When rhe Expert determines that a ll of the actions identified in thc
work p lan have bee n completed co his or her sa ci sfaccion, the Expert shall provide
Defendants and FDA with a written certification chat all of the actions have be1.:n
completed and that, based on the inspections cond ucted unde r paragra ph 6.C and on the
7
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 8 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 8 of 27
sati factory completion of the actions in the work plan identified under paragraph 6. D,
De fendants' methods, facilities , processes. and controls used to design, manufacture,
process. pack, label. hold, score, and distribute the Synchrot\ lcd devices, a re aml, if properly
maintained and implemented by Defe ndants. will continuously remain in conformity with
the requirements of this Decree, the Act, and the QS n.:gulation. The Expert's certification
shall incluck a full and complete derailed report of the results of his or her inspection.
11. Within thirty (30) bu ·iness days of F OA's receiving the Expert's
ccrcifieation under paragraph 6.(;, Jul y authorized FDA representatives may inspect, as
FDA deems necessary and without prior notice, the 1\lcdcronic curomodulation facilities.
inc luding buildings. equipment, personnel, finished and unfinished materials, conta iners.
and labeling, and al l records relating to the methods used in, and the facilities and controls
used for, the manufacwre, design, processing. packing, labeling. holding. storage. and
distribution of Synchrot\lcd de\·ices. to determine whether the requirements of paragraphs
6.A-G of this Decree ha\·e been met, c1nd whether Defe ndants are o therwise operating in
conformitv with this Decree. the Act, and the QS rcgulc1tion.
I. If FD/\ determines that Defendants arc not operating in conformi ty
with the requirements of this Decree, the Act, and the QS regulation with regard to the
SynchroMed de,·ices, F DA will notify Defendants of the deficiencies it obserYetl and will
rake any other action FDA deems appropriate (e.g., issuing an order pursuant to paragraph
11 ). Within thirty (30) days of receiving this notifi cation from F DA Defendants shall subrnic
co f.'DA a plan describing the actions Defendants propose co rake and a timetable for
com;cting the deficiencie!i. The timetable and p lan shall be !iubject to FDA approval.
D efendants shall promptly correct all deficiencies noted by FDt\ in accordance with the
8
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 9 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 9 of 27
FD/\ a pproved timetable and plan , and sha ll cause the Expe rc to rei nspect the conditions
relevant co the de ficie nc ies no te d by FD/\ and ei ther:
( i) c<..:rtify that the defic ie ncies have been corrected co c nsure chat
De fendants' methods, faci lities, processes, and controls used fo r manufacturing, processing,
packing, labeling, holding, storing, and distributing the Synchrol\ led de,·iccs arc in
con formity with che require ments of chis Decree, rhe Ace, and the QS regulation : or
(ii) notify Defendants and FDA in wri ting cha r one o r m ore
defic iencies rem ain uncorrected. If one o r more deficie nc ies have not heen corrected.
Dcfcndancs sha ll correcc the deficie nc ies co the E xpert's satisfaction, at which point the
Expe rt sha ll issue the certification simultaneous ly co De fendants and FDA. Withi n forty
five (-l-5) business days after FDA rece ives rhe ccrrificacion. 1: oA may reinspect as it dccms
necessary, without prior notice.
J. FDA notifies Defendants in writi ng that Defendants ap pear to be in
compl iance with che requireme nts sec fo rth in paragraphs 6.A-L Such notice sha ll not be
depe ndent upon De fe ndants' complt.: cion o f the Synchro led Pump Re mediation Plan
describe d in paragraph 7.
7. No late r than twenty (20) days after e ntry of this Decree, Defendants shall
submi t co F DA in writing a Pump Re mediation Plan to e nsure that the Synchrol\lcd
dc,·iccs currently produced in the United S tares arc in compl iance with the Act, its
imple me nting regulations, and this Decree ("Synchro~lcd P RP").
/\. The Synchro~ lcd PRP shall include, among orhe r things:
9
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 10 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 10 of 27
(i) the identificat ion of the root causes or, if not precise ly known,
the prohable root causes. of failures in the Synchro led dc\·ices D efendants arc proposing
to correct;
(ii) a description of and the supporting documentation for
upgrades, modi fications. and/or actions necessary co correc t the ide ntified failures;
(i ii ) the testing conducted or to be conducted to verify and validate
such upgrades anJ/or modifications:
(iv) the projected dates on which Defe ndants will implement and
complete th e Sym:hrol\led PRP;
(v) the mannt.:r in which the upgrades and/or mod ifications wil l be
made ro th e Synchrol\lcd dc\'ices: and
(vi) a clear state ment whe ther Defendants believe that premarket
apprm·al by FDA is required fo r the proposed upgrades and/or modifications to the
Synchrol\ led devices proposed in t he SynchroM cd P RP, and the reason for that bel ie f.
13. Defendants shall not initiate the Synchrol\Ied PRP until FDA has firsr
provided Defendants with wriccen acknowledgement co proceed with a ll or a portion of the
Synchrol\lcd PRP. FDA shall respond in writing \\'ithin thirty (30) days of FDA's receipt of
Defendants' Synchrol\led P RP and notify Defendants in writing whether the proposed plan
is acceptable. If FDA finds some or all of the Synchrol\ Jed PRP unacceptable, it shall state
in writing the basis for finding specific portions of the proposed Synchrol\ led PRP
unacceptable, and Defendants shall submit a revised SynchroMed PRP in writing within
twenty (20) days of receipt of FDA's re ponsc. FDA shall re pond in writing \\·i chin twenty
(20) days of F OA's receipt of Defendants' revised Synchrol\lcd P RP and notify Defendants
JO
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 11 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 11 of 27
in writ ing whether the re\'isecl plan is acceptable; and, if specific portions of the revised plan
are un acceptable, FDA sha ll s tate the basis in its wrim.:n respo nse.
C. Defendants shall commence those portions of the initial a nd/or revised
Synchrol\ lcc.l PRP that were found acceptable by FDA \\'i th in th irty (30) days of recei,·ing
FDA's writte n auchorization of the initia l and/or revised Synchro l\,fcd PRP. Defendants
sha ll , beginn ing one month after the dace on wh ich imple mentatio n of the Synchrol\l e d
PRP. in who le or in part. has begun. and continuing until its completion. subm it to FDA
quarterly written progress reports tha t descri be the status o f the:.: Synch rol\kd PRP. If
Defendants have not obtained FDA' a uthorization for the Synchrol\led PRP within six (6)
months after the da te chis Decree is e nte red. FDA may rake a ny ac.:cio n(s) it deems
approp riate to the extent permitted unde r parag raph 11 of this Decree.
D. PRP documentation. described above in paragraph 7.A, sha ll be
ava ilable fo r Expert and FDA revic\\' in accordance w ith paragra ph 6.
8. Upo n e ntry of chi s Decree, except as permitted in parag raph 9, Defendants
and each and all of their directors, officers, agc:.:ncs, reprcsencati\'(.:s. employees, attorneys,
successors. and assigns. and any a nd all persons in acti ve concert o r pa rticipa tio n with a ny of
chem (inc luding franch isees. affiliates. a nd "doing business as" e ntities). w ho have receive d
acw al no tice of this Decree hy persona l sen ·icc or othCr\\'ise, arc permanently e njoi ned
under the provisions o f 21 U.S.C. § 332(a) from directly o r inuirccrly do ing o r causing to be
done any act that:
A. Vio lates 21 U.S.C. § 331(a), hy introd uci ng or deliH:ring fo r
introduc tio n into interstate commerce, or causing the introduction o r delive ry for
introduction into interstate commerct.: of, Synchrol\ led devices, o r any oche r l\Jcdcronic
11
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 12 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 12 of 27
devices of a s imilar design or fo r a similar use, as definccJ by 21 l 1.S.C. § 32J(h ), chat arc
adulterated within the meaning of 21 ll.S.C. § 351(h).
B. Violates 21 U.S.C. § .Bl(k), by causing the Synchrot\kd devices, or any
ocher l\ lcdcronic devices of a similar design or for a similar use, co become ad ulwrated
" ·ithin the meaning of 21 U.S.C. § 351(h}, while such devices nre held for sale after shipment
in intc rscatc commerce.
EXCLLJSIO 15
9. Paragraphs 6 and 8 of chis Decree shall noc apply co the fo llowing:
/\. l\Janufacturing. processing, packing. labeling, holding. scoring. and
distributing Synchrol\·lc d devices that arc inte nded for use in medica lly necessary cases, as
defined in paragra ph 5.D. i\kdtronic may provide ::i medically necessa ry SynchroMed
device only if the fol lowing requirements have been and continue co be, or will be, met: (i)
the patient's phy ician has completed the Certificate of led ical cccssity (Cl\11 ),
referenced in paragraph 5.0 and attached he reto as Exhibit 8 ; (i i) lcdtronic prornpcl y
provides FDA with copies of all Cl\ lNs for the first three (.3) months following e ntry of this
Decree; (iii) l\,fcdtroni<.: maintains and promptly provides co FDA upon request copies of
any additional CMNs executed after the tirst three (3) months; and (iv) .tvlcdtronic provides
reports of granted Gl\ lNs to FDA every three (J) months for a period of one (1) year and not
less than every six (6) months fo r a pe riod of four (-1-) years the reafte r. In circumstances
where the Synchrol\led pump is required fo r use in an eme rgency case and it is impractical
or thcre is insufficient time co obtain a C I in adva nce of the procedure, lcdtronie may
p rovide the Synchrol\lcd device for such usc so long as the patient's physician (i) complctes
the C l\ l fo llowing the proced ure, and (ii ) submits the completccl Ct\ lN co l\lccltroni<.: as
12
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 13 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 13 of 27
soon as possible following the proceuure. The parries agn.:e chat such si tuations will be
infrequent. In those cases in which prior approval is nor feasible, lcdtronic will s upply
FDA with a copy of cornpieted Ct\ l within three (3) busim:ss days o f receiving the CI\ I
from the ph ysic ian.
13. t\ lanu faccuring, process ing, packi ng, labeling, holding. stori ng, and
distributing Synchro leu ueviccs intcnucd for patients seeki ng a replacement Synchrol\ led
dc,·icc. l\ lcdcronic sha ll pro,·ide a replacement Synchro kd device to a patient only if the
fo llowi ng requirements have been and continue to be . or will be, met: (i) the patie nt's
physic ian has completed the Replacement Pump Certificate ("RPC''), attached he reto as
Exhibit C; (ii) lcdcronic prompcly provides f.DA with copies of all RPCs fo r the first three
months fo llowing entry of th is Decree; (iii ) lcdtronic maintains and p rompcly provide to
FDA upon request copies of any RPCs executed after the fi rst three (3) months; and (iv)
t\ fe dtronic provides re ports of granted RPCs co FDA every three (3) months for a pe riod of
one (1) year and not less than every six (6) months fo r a pcriod of four (4 ) years the reafter. In
ci rcumstances where a replacement Synch rol\ led pump is needed for use in an e mergency
case and it is im pracric~ll o r the re is insuffic ie nt time to obtain an RPC in advance of the
procedure, the Defe ndants may di stribute the replacement Synchrol\,lcu device for s uch
use. p rovided t hat the patient's physician (i) completes the RPC fo llowing the procedure,
and (ii) submits the completed RPC co lcdcronic as soon as possible following the
procedure. The parties agree chat such situations will be infrequent. In each case in wh ich
prio r approval is nor feas ible, t\ lcdtronic will s upply FDA with a copy o f the completed
RPC withi n three (3) business days of receiving the RPC from the physician.
13
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 14 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 14 of 27
C. t-.·lanufaccuring, process ing, packing, label ing, holding, storing, and
distributing any component, part, raw material, accessory, refill kit, o r sub-assembly, solely
for the purpose of providing service or repair to a Synchrol\ lcd device implanted prior to the
date of the entry of this Decree, or that was providcd purs uant to paragraph 9.A, 9. B, or 9.l
of this Decree. i\lcdtronic may pro,·ide replacement compone nts, pares, raw material s,
accessories. refill ki ts, and sub-assemblies to patients, their physicians. healthca re pro,·iders,
and facilities for sen·ice or repai r of Synchrol\ led de,·ices and components on ly if the
fo llowi ng requirements have been met: (i) Medtronic .~ends a copy of the notification kttcr
attached hereto as Exhibit A to the ph ys ic ians, hea lthcare providers, or fac ilities co whom
ledtronic pro\'idcs such ite ms; and (ii ) i\lc<lrronic maintains records, and allows FDA
access co such records upon r<.:quest, of a ll serv ice and repair components, pares, raw
mate ria ls, accessories, refi ll kits and s ub-assemblies prov i<kd un<lcr this paragraph,
including copies of chc notifica tion letters sent co physicians, healthcare provide rs, and
facilitic .
D. l\ lan ufacrnring. processing. packing, label ing. holding. scoring, an<l
distributi ng limited quanti ti es of Synchrol\ lcd de\'iccs that are not intended for human use
and arc inte nded for use in development. testing. verification, val idncion. or qualification
aeti,·ities nece sary co comple te (i) design changes in support of the Synchrol\Jcd PRP. (ii )
changes to prod uction and process controls. (iii) changes ro manufac turing procedures, (i, ·)
corrective and preventive actions, and/or (v) changes co componen ts, parts, or suppliers.
£. Testing, verifying, or va lidating design changes of Synchro led
devices, including any component or accessory, and subsequentl y manufac turing and
14
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 15 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 15 of 27
distributing the Synchrol\ led dcvicc.:s, components, or accessories. for the sole purpose o f
implementing a correction o r removal as defi ned in 21 C.F.R § 806.
F. Desig n work related to remediation of ex isting safety iss ues with chc
Synchrol\kd devices, or relate d to safety issues with chc Synchrol\lcd devices di scovered
during the impkmentation of chis Decree.
G. lanufaccuring, processing. packing, labeling, ho lding, scoring, and
distributing Synchrol\ lcd d e\'ices for dc,·elopment acti,·itics and di cribucing such devices
for demonstration and research purposes on ly, suc h as use in produce de mo nscracio ns and
resc.:arch in laboratories. including preclinical animal research , provided chat the devices are
labeled "NOT FOR l llJi•vIA USE."
11. l\fanufaccuring, process ing, packing. labeling, ho lding, s toring, and
distributing Synchrol\ lcd devices solely for the purpose of pe rmitting c linical tria ls to be
cond ucted in accordance with 21 C. ER. Part 312 or 812, or for international clinical trials
conducted in accordance wich Good Cl inica l Practices. pro,·idcd that Defendants comply
with all applicable laws and rc.:gu lacio ns relating co the manu facture and distribucion of
investiga ci onal devices.
I. l\,lanufac curing, processing, packing. labe ling. ho lding. storing, a nd
dis tributing Synchrol\kd devices that were ordered or provided for cases char were
scheduled prior to entry of this Decree.
J. Importing compone nts and accessories necessa ry to manufacture and
distribute Sync hro led devices, pares, components, and accessories as permitted by
paragraphs 9.A-J of this Decree.
15
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 16 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 16 of 27
ADD ITI O A L RE QU IREJ\IENTS
10. Afcer Defendants ha,·e complied wi th paragraphs 6.A-l and FDA has notified
Defendants in writing pursuant to paragraph 6.J, Defendants shall rerain an independent
person or pe rsons (the "Auditor") at l\lcdtronie's expe nse to conduce aud it inspections of
Defendants' operations not less than once every six (6) months fo r a period of one (1 ) yea r
and not less than once every twelve (12) months for a period of two (2) years thereafter. T he
Audicor sha ll be qualified by education, training, and experience co conduct such
inspections. and shall be without personal or fi nancial t ies (other than a consult ing
agreement e nw red in to by the Aud itor and l\kdtron ic or l\ ledtronic Neurornoclu lation) to
Defcnda ncs· officers or employees or their immediate fam il ies. T he Audi ror may be the
amc person or persons described as the Expert in paragraph 6.
t\ . Ac the conclusion of each audit inspection, the Auditor shall prepare a
writte n audit report (the "Audie Re port") analyzing whethe r l'vleclcronic Ncuromodulation is
operated and administered in compliance with the Act, its implementing regulations, and
chis Decree, and ide ntifying in derail any clc,·iat ions from the foregoi ng (''Aud ie Report
Findings"). As part of 1..;very Audit Re port, except the fi rst, the Auditor shall assess the
adequacy of correcti ve actions ta ke n by Defendants co correct all previous Audit Report
Find ings. The Audit Reports shall be delivered conte mporaneously co Defenda nts and
FDA by courier service or o,·e rnighc deli, ·cry sen·ice, no lacer than twenty (20) days after
the dace each audit inspection is completed. If any Audi t Rcporc(s) identify any deviations
from the Act, its imple me nting regulations, and/or this Decree, FDA may, in its discre tion,
requi re that the two (2) year auditing cycle be extc nded or begin anew. In add ition,
Defendants shall maintain complete Audit Reports and all of their underlying data in
16
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 17 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 17 of 27
separate files at th ei r fac ilities :ind shall promptly ma ke the Audit Reports a nd unde rlyi ng
data a\·ailable to FDA upo n request.
13. If an Audi t Re po rt co ntai ns a ny ad verse Audit Report Findings,
Defenda nts sha ll, withi n fo rty-five (45) da ys o f receipt of the Aud it Report. correct t hose
Findings, unless FDA notifies D efendants t hat a sho rter time period is necessary. If, afte r
recei\·ing the udit Report, D e fenda nts bel ieve tha t correction of a ny ad\·erse Aud it Report
Finding \\'ill rake longer th:.1n fo r~·-li\·e (45) clays, D e fendants sh:1 11, w ithin fiftee n (15) days
o f receipt of the Audit Report, propose a schedule for completing correctio ns ("Correction
Schedule") and provide justificatio n for ch t: addi tio nal time. D ef<.: ndants sha ll comp lete a ll
correctio ns according to the Correction Sche d ule. Within forty-ti\·c (4S) days of Defendants'
receipt of a n Aud it Re port, or within the time period prm·ided in a Correctio n Schedule, the
Auditor shall re view t he actions taken by D e fe ndants to correct th e adverse Audie Report
Finding(s). With in te n bus iness days of rhe completion of that review, the Auditor s ha ll
report in writing to FDA w hether each of the ad,·crse Audi t Report Findi ngs has been
correcte d a nd , if no t. which ad\·crse Audie Re port Findings re main uncorrected.
11. I f, at any rime a fter this Decree has been ente red, FDA determi n<;s, bas<;d o n
rhe resu lts of a n inspection; the ana lysis of samples; a re po rt or darn prepared or submitte d
by D e fe ndants. the Expert, o r the Audi tor pursuant to ch is Decree; or any ocher information,
chat Ddc nda ncs ha\·e fa iled to comply with any provision o f this Decree. o r haYc violated
the Act o r its imple menting regu lations, o r that additio na l correc tive actio ns are necessa ry to
achieve complia nce with chis Decree, the Ac t, or its implementing regulations, FDA may, as
and whe n it deem s necessary. o rder Defendants in writing co take appropriate actio ns with
17
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 18 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 18 of 27
respect to Synchrol\kd devices. Such actions may inc lude, but arc not limited to, the
fo llowing:
1. Cease designi ng, manufacturing. processing, packing. labeling,
holding, storing, distributing, importing and/or exporting Synchrol\ lcd devices produced at
rhe l\kdtronic Neuromodula rion facilities;
11. Revise. modify, or expand any rcport( s) prepared pursuant to
the D ecree;
111. Submit au<l itiona l notifications, re ports. or any other matcri,1 ls
or information co FDA with respect ro Synchrol'vled devices;
I\' . Recall and/or prm·ide refunds for, at l\ ledtronic's sole ex pen c,
adulterated or m isbranded dc\'ices or components manufactured . distributed, and/or sold by
Defendants or char arc under the custody and control of D e fendants' agents, distri butors.
custome rs, or consume rs;
v. Issue a safety a le rt. public hea lth advisory and/or press release
with respect co the Synchrol\ lcd devices; and/or
v1. ' Eike any other corrective action(s) with respcct co the
SynchroMed devices as FDA. in its discre tion, dee ms necessary to protect the public hea lth
or to bring D efendants into compliance with the Act, its imple me nting regulations, and this
l)ceree.
12. The fo llowing process and procedures sha ll apply in the event that FDA
issues an orde r unde r paragraph 11:
Unless a different timeframe i specified by f-DA in its order, with in
ten (10) business days afte r receiving s uch order, Defendants s ha ll notify FDA in writing
18
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 19 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 19 of 27
either cha t: (i) Defendants arc undertaking or h,l\'C undertaken corrective action, in which
cvcnc Defendants s hall also ucscribe the specific action taken or proposed co be taken and
thc proposcd schedule for completing the action; or ( ii) Defendants do not agree with FDA's
order. If Defendants notify FD/\ chat they do not agree with FD/\'s order, Defendants s ha ll
explain in writing the basis for their disagreement; in so doing. Oeft:ndants may also
propose s pecific altcrn:ni,·e actions and time frames for achie,·ing FDA's objectives.
B. If Defendants notify F DA that they do not agree with FOJ\'s ordcr.
FD1\ will review De fendants ' notification, and thereafter, in writing, :iffirm, modify, or
withdraw its order, as FDA deems appropriate. If FDA affirms or modifies its order, it sha ll
explain the basis for its decision in writing. The writtcn noticc of affirmation or modification
shall constitutc fina l agency nction.
C. If FDA affirms or modifies its ordcr, Defendants shall, upon receipt of
FDA's order. immediately implement the order (as modified, if app licablc), and may, if rhcy
so choose, bring the matter bcfore chis Court on an expedited basis. While seek ing Court
re ,·ie \\'. Dcfe ndants shall continue to diligently implement FO/\'s order. unless the Court
stays. secs aside, or modifies FD/\'s order. Judicial review of FD/\'s ordcr shall be made
pursuant to paragraph 24.
D. The process and proccdures sec forth in paragraphs 12.A-C shall not
apply to any order issued pursuant to paragraph 11 if s uch order scares chat, in FO/\'s
judgment, the order raises a significant public health concern. In such case, Defendants
sha ll. upon receipt of s uch orde r. immediately and fully comply wich chc ccrms of the orde r.
S hould Defendants seek co cha llenge any such order, they ma y petition chis Court for re lief
19
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 20 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 20 of 27
whi le they impleme nt FDA's order. Jud icial review of FDA's decision under chi s paragrap h
shall he made pursuant co paragraph 2-t
1.1. Any cessation of operations or other action as desc rihed in paragraph 11 sha ll
continue until Defendants: (a) recei,·e wri tte n notification from F DA chat t\ ledtron ic
Neuromodulacion appears co he in compl iance with this D ecree, the Acc. and its
implementing regu lations or (b) receive written authorization from che Court. After a
cessation of operation . and \\'h ile determining whether Defendants are in com p liance wich
t hi s Decree , che Ace, and its impleme nting rt.:gulacions, F DA may req uire Defendants co re
institute o r re-implement any of the requi rements of chis Decree. Defenda nt l\ lcdcron ic
shall pay the costs of F D A supen·ision, inspections, investigations, analyses, examinations,
re,·iews, sampl ing, testi ng. cra,·el time. and ubsistence expenses to imple me nt the remedies
set forth in pa ragraph 11 , at the.; rates specific.;d in paragraph 15.
14. Re presentatives of F DA sha ll be pe rmitted. without prior notice and as and
when FDA deems necessary, co make inspections of Defendants' operations at the
Icdcronic Ne11romod 11 lacion facil ities and, without prior notice, cake any other mc.;asures
necessary ro monito r and to c nsure continuing compliance with the te rms of th is Decree.
During s uch inspections, FDA re presentati ves shall be pe rmitted: access to bu ild ings,
equipment, in-proces and finisheu materials, conta iners, anu labe ling the re in; to cake
photographs and make \'ideo recordings; to cake sam ples of Defendant:;' materia ls and
produces. conta iners, and labe ling; and to examine and copy all records relati ng co che
receipt, manufac ture . processi ng, packing. la be ling. holuing. and uiscrib ution of the
Synch ro led devices and the design of the Synch rot\ led devices. F D A will p rovide
Defendants wi th a receipt for any samples taken pursuant to 21 l 1.S.C. § .174 anu with copies
20
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 21 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 21 of 27
of any photographs or video recordings, upon the receipt of a written request by
Defe ndants, and ac i\ lcdtronic's expense. The inspections sha ll be pe rmitte d upon
presenting a copy of chis Decree and appropriate c redentia ls. The inspection authority
granted by th is Decree is separate from, and in addition to. the authority ro make
inspections under tht: Ace, 21 U.S.C. § 374.
15. Dcfencl:int fedcronic shall reimburse FDA fo r the coses of all FDA
inspec tions, im·cscigacions, supe n ·is ion, re,·icws. examinations, and analyses that FDA
deems necessary to evaluate Defendants' compliance with this Decree. T he costs of such
inspec tions shall be borne by l\ledtronic a t the preva il ing rates in e ffect ac the time the coses
arc incurred. As of the dace th at chis Decree is signed by the pa rries. these races a re: $88.45
per hour and fract ion thereof per representati,·c for inspection work; $106.03 per hou r or
fraction thereo f per re presentative for analytical or re ,·ie,,· work: $0.56 per mile fo r travel
expe nses by automobile; government rate or the equivalent for travel by air or othe r m eans;
and the published government pe r diem rate or the cqui,·alent for the areas in which the
ins pec tions arc performed per-day, per-representative fo r subsisct:nce expenses. FD/\ sha ll
submit a bi ll of costs to Defendant l\ ledcronic. In the event that the standard rates
applicable to FDA supe rvision of cou rt-ordered t:ompliance are modified. these races sha ll
be increased or decreased in accordance with the n"lOdified races withou t further order of the
Court.
16. Within tive (5) hus iness days o f the e ntry of chis Decree. De fendants s hall
post a copy of chis Decree in the employee common areas at rhc fVlcdtron ic
eu romodu lacion facilities and on l\ lcdrronic's intranet website in such a manner as to
ensure that it will be viewed by employees at the lcdcronic euromodulacion faci lities.
21
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 22 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 22 of 27
D efendants s hall ensure that the D ecree rema ins poste<l in its employee common a reas and
on its inrranet website for as long as the Decree remains in effect.
17. Wit hin te n (10) days after the enrry of chis Decree. Defendants shall provide a
copy o f chis D ecree, by personal service, electronic mai l, o r certified mai l ( restricted
delivery. re turn receipt requested), to each a n<l all of its directors. officers. agents,
representati ,·es. employees, attorneys. successors. and assigns, and any and all persons in
active concert or participation with any of chem (includ ing; franchisees. affi liates, and "doing
business as" entities). with respons ibility for the design, manufacture and/or distri bution of
the SynchroJ\ Jcd devices at or from the rvkdtron ic e uromodulation faci lities (he reinafter,
collecti,·ely referred to as "Associated Persons''). f or interna tional /\ssoci:ned Persons.
f\lcdtronic Neuromodulation sha ll prm·ide a copy of the Decree by personal service,
d cccronic ma il. o r certifie d ma il ( restricted de livery, re turn receipt requested ) withi n
twenty-five (25) days afcer thc e ntry of chis Decree. Within thirty (30) days after the entry of
this D ecree, l\ lcdcronic shall provide to FDA an aftida,·ic seating the face and manne r of
com pliance with this paragraph, identifying che names, addresses. and positions of all
persons o r e n cities who hav<.: been provided a copy of chis Decree purs uant co chis parag raph
and a ttaching doeumcnracion of t he manne r in which copies of the Decree were provided.
18. In the event chat t\ ledrronic Neuromodulation becomes as ociated, at any
rime after th<.: e ncry of ch is Decree. with any new Associated Person, led cronic sha ll " ·ichin
fifteen business days of the commencement of such association: (a) provide a copy of this
Decree to each such Associated Person by persona l service, c lec rronic mail, o r ccrcified ma il
(restricted delivery, rerurn receipt req uested); and (b) on a quarterly basis, notify FDA in
writi ng. in accordance with paragra ph 20, when, how, and to whom the Decree was provided.
22
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 23 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 23 of 27
Defendants shall pro,·ide co FDA an affidavit stating the fac t and manner of compl iance
with this paragraph, ide ntifyi ng the names, add resses, and positions of a ll persons or entit ies
that have been provided a copy of t his Decree purs uant co ch is paragraph, and
docume ntation of the manne r in wh ich copies of th e Decree were provided.
19. Defendant Medtronic sha ll notify the District D irector, FDA l\,f in neapol is
D isrricr Office, in writing at lease fifteen (15) days before: (i) any change in ownershi p,
characte r, or name of the l'vledtronic Neuromodu lation business, such as d issolution.
ass ignment, or sale resulting in t he em ergence of a successor corporation that, in each case,
may affect complia nce with this Decree; (ii ) the creation or d issolution of subs id ia ri es,
franchisees, affiliates, or ''doing bus iness as" e ntities, o r any ocher change in the corporate
structure of j\,f edtronic Neuromodulacion or in the sale or ass ignme nt of any b usiness assets,
such as build ings, equip ment, or inventory, that, in each case. may affect com pliance wi th
th is Decree. Medtronic sha ll provide a copy of this Decree co any pote ntial successor or
assignee ac lease fiftee n (15) clays before any sale or assignme nt. Medtronic sha ll fu rn ish
FDA with an affidavit of compl iance with chi s paragraph no lace r than ten (10) days prior to
such assignme nt or change in owners hip.
20. All notifications, correspondence, anJ comm unications required co be sent to
FDA by the terms of ch is D ecree shall be addressed co the District Director, M inneapolis
District Office, 250 Marq ue tte Ave., Suite 600, Mi nneapolis, J'vlN 55401. All notifications,
corresponde nce, and communications required to be sent to Defendants by the te rms of this
Decree s hall be addressed to Director of Consen t Decree Compliance Task F orce,
l\kdrronic Ncuromodul ation, 7000 Centra l Avenue NE, Mi nneapolis, MN 55432.
23
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 24 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 24 of 27
FINAN C IAL PROVISIONS
21. I n the event that Defendants fa il. as dete rm ined by F DA. to comply with any
time frame or provision of this Decree, then FDA sha ll have the sole and unrev ie wablc
d iscre tion to ord e r i\ ledtronic to pay the Uni ted States Treasury as liquidate d damages the
sum of fi fteen thousand dollars ($15,000.00) per violation of chis D ecree and an additiona l
sum of fifteen thousand dollars ($15,000.00) fo r each day such \·io lation continues.
22. In the event De fe ndants fa il, as de te rmined by FDA, to satisfactorily
comple te one or more of rhe numbere d ste ps, including the comple tion date for all
numbe re d steps, in the \\"ork p lan refere nced in paragraph 6.D, FDA may order le dtronic
co pay the United Scates Treasury as liq uidated damages the sum of fi fteen thousand dolla rs
($15.000.00) for each incomplete num bered ste p. per busi ness day (e.f!; .. if two ste ps are not
time ly complied with for two business days, the n liquidated damages may be assessed up co
$60,000.00), until t he numbered see p is full y imple me nted and comple ted co FDA's
satisfaction. The amount of liq uidated damages imposed unde r paragraphs 21 and/or 22 sha ll
not exceed ten (1 0) million dolla rs ($10,000,000.00) in any one cal endar year.
23 . The..: re medy under paragraphs 21-22 sha ll be in addition co any othe r
re medies a\·a ilable to the United Scate s unde r this D ecree or the law. De fe ndants
unde rstand and agree that t he imposition of lici uidaced damages untler paragraphs 21- 22
docs nor in any way limit the ability of che United Scares co seek, or the power of the Court
to impo c, additiona l crimina l or ci\·il penalties or remed ies based on conduct chat may a l o
be the basis for payment of liquidated damagc .s pursuant w paragraphs 21-22.
2-1-
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 25 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 25 of 27
GENERAL PROVISIONS
2-1-. Defendants shall abid<.: by the decisions of FDA, and FDA's dec isions shall be
final. Al l tkcis ions con ferr<.:d upon FDA in this D<.:crec shall be vested in FOA's discretion
and, if contested. shall be re,·iewcd by this Court under the arbitrary and capric ious standard
set forth in 5 U.S.C. § 706(2)(A). Rc ,·ic w by the Court of a ny FDA decis ion rendered unde r
thi s Decree sha ll be based exclusively on the written record before FDA at the rime the
decision was made. No d isco ,·ery shal l be taken by a ny parry.
Should the Unite d States bring, and preYail in, a contempt action co enforce
the terms of this Decree. l\kdtronic sha ll, in addition co o ther remedi<.:s. reimburse the
U nited Scates for its arcorn<.:ys' fees , in,·estigational expenses, expert witness fc<.:s, travel
expenses incurred by atto rneys a nd witnesses, and administrati ve court costs rela ting co
such conte mpt proceedings.
26. The pa rties may at any time petition each other in writing co modify any
deadline provided he re in ; :tnd if the pa rties mutuall y agree in writing to modify a deadline,
such modification may be ~ranted and may become effecci,·e wi thout lca,·e of the Court.
27. If, and for so long as, an individua l defendant ceases co be e mployed by and
to act on behal f of l\lcdcronic o r any of its suh-;idia ries, franchisees. affiliates and/or ''tlo ing
bus iness as" entities, then that individua l shall not be subjec t to this Decree, except as to
such individual's act(s) or foi lure(s) to act under this Dec ree prior ro the time such
individua l ceased to be employed by and to act on beha lf of l\ledtronic or a ny of its
subsidi:irics, franchisees, affiliates, a nd/or "doing business as" entities.
25
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 26 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 26 of 27
28. This Court retains jurisd ict ion o,·cr chis action and the p:trrics chereco for the
purpose of enforcing and modifying chis Decree and for che purpose of wanc ing suc h
additional relief as may be ncccssary or appropriate. SO O RDERED:
This _ _ _ _ <.byof __ . 20 15.
T he undersigned hereby consent co the entry of the foregoing Deere<.;:
UNIT ED ST /\TES D ISTRICT JLJDCE
26
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 27 of 28

CASE 0:15-cv-02168 Document 3 Filed 04/27/15 Page 27 of 27
For the Defendant~:
S. Oi\ It\ R ISi I RAK lndivicl11ally and on behalf of \h.:dtrnnic. Inc .. as its Chairman and CEO
Individually and on behalf of \kdtronic. Inc .• as its Senior \ 'ice Presic.knt. i\ lcdtronic \!e11romod11l;.1tion B11sine-;s l ' nit
D11111sdfor . If ,,di m11 if·. I 11r. King & Spalding I ,LP 1700 Pcnn-;yh·ania ,.\,·enue. 1\\"
Washington. D C 20006
~i~ r:ou11Jd.for .1/,: /4,ml r111fl. ll,: 1i:[(I \\'ill iams & Connolly I ,LP 725 Twelfth Street. N\\' \\'a:.hingwn. DC 20005
!.7
F or th e Plaintiff:
ANDRE\\' ;\I. Ll !GER l 'nited States ,\ttorney
;\-;sist.mt l fnite c.l Scares :\tmrney
~~ ROSS S. (iOLDSTEli\ Trial :\ttorlH.:Y Consumer Protection Br.rneh t · nitcd States Departmcnr of J usri<:c PO. Box 386 \\'ashingwn. DC: 20044-0386
\YILl.l :\ ~I B. SCI ll l l.Z C.ener.tl C:ounsel
ELIZABET! I I I. DICKINSON Chief Counsel Food and Drug Di,·ision
1\NN,\~IARIE KE\IPIC: Deputy Chief Counsel for L itigat ion
T :\R·\ BOI .:\ ')) .\ ssol·iarc Chief C:111111-;cl l IniteJ States Dcparcn1cnr of I lcalth and
l lunrnn Ser.ices Ofticc of the Ccncral Counscl l()lJ(I.\ '.\:cw I lampshin.: .\,·enuc Silver Spring. ~ID 209<JJ-0002
Case 1:20-cv-01277-AWI-JDP Document 1-12 Filed 09/08/20 Page 28 of 28

Exhibit 13
Case 1:20-cv-01277-AWI-JDP Document 1-13 Filed 09/08/20 Page 1 of 5

A04611001 Rev X
A04611_001.fm 2/24/04 11:13 amUCxxxxxx EN 4.5 x 6 inches (114 mm x 152 mm)
Medtronic ConfidentialNeuroIFP_R00
LIMITED WARRANTYSPECIAL NOTICEfor Medtronic® Pump System
Case 1:20-cv-01277-AWI-JDP Document 1-13 Filed 09/08/20 Page 2 of 5

A04611001 Rev X
A04611_001.fm 2/24/04 11:13 amUCxxxxxx EN 4.5 x 6 inches (114 mm x 152 mm)
Medtronic ConfidentialNeuroIFP_R00
Medtronic Neurological Implantable Pump SystemLimited Warranty1
(U.S. Customers Only)A. This Limited Warranty provides the following assurance to the patient who receives
a Medtronic Neurological Implantable Pump System. The Pump System includes pumps, catheters, refill kits, and accessories, hereafter referred to as Components, unless specifically noted.(1) Should the Components fail to function within normal tolerances due to a defect
in materials or workmanship within these periods:■ In the case of any pump model except IsoMed, two (2) years commencing with
the date of implantation;■ In the case of the IsoMed pump, during the life of the patient into whom it is
implanted;■ In the case of the catheters and accessories, one (1) year commencing with
the date of implantation;■ In the case of the refill kits, prior to its “Use By” date.
Medtronic will at its option: (a) issue a credit to the purchaser of the replacement Component equal to the Purchase Price, as defined in Subsection A(3), against the purchase of any same Component requested as its replacement, or (b) provide a functionally comparable replacement Component at no charge.
(2) Pump battery cell depletion for any model except IsoMed (which does not use batteries) will occur with time and is not considered to be a defect in materials or workmanship. The batteries have a specified capacity that may deplete at different rates depending on settings and individual requirements for pump functions. Therefore, no representation is made that the pump batteries will last the entire term of this Limited Warranty.
(3) As used herein, Purchase Price shall mean the lesser of the net invoiced price of the original or current functionally comparable or replacement Component.
B. To qualify for this Limited Warranty, these conditions must be met:(1) The Components must be implanted prior to their “Use By” date.(2) The Components must be used in conjunction with components compatible with
the Medtronic Neurological Pump System.(3) All device registration materials must be completed and returned to Medtronic
within thirty (30) days of implantation of the Components.
1 This Limited Warranty is provided by Medtronic Inc, 710 Medtronic Parkway, Minneapolis, MN 55432-5604. It applies only in the United States. Areas outside the United States should contact their local Medtronic representative for exact terms of the Limited Warranty.
Case 1:20-cv-01277-AWI-JDP Document 1-13 Filed 09/08/20 Page 3 of 5

A04611001 Rev X
A04611_001.fm 2/24/04 11:13 amUCxxxxxx EN 4.5 x 6 inches (114 mm x 152 mm)
Medtronic ConfidentialNeuroIFP_R00
(4) Replaced pumps must be returned to Medtronic within thirty (30) days of explantation and shall be the property of Medtronic. The catheter, refill kits, or accessory, or portion thereof, must be returned to Medtronic within thirty (30) days after discovery of the defect and shall be the property of Medtronic, and if not explanted, the serial number or lot number must be provided to Medtronic instead.
(5) The use of medication with the Components must be used in accordance with the labeling and instructions for use provided with the Components.
C. This Limited Warranty is limited to its express terms. In particular:(1) Except as expressly provided by this Limited Warranty, MEDTRONIC IS NOT
RESPONSIBLE FOR ANY DIRECT, INCIDENTAL OR CONSEQUENTIAL DAMAGES BASED ON ANY DEFECT, FAILURE OR MALFUNCTION OF THE COMPONENTS TO FUNCTION WITHIN NORMAL TOLERANCES WHETHER THE CLAIM IS BASED ON WARRANTY, CONTRACT, NEGLIGENCE, STRICT LIABILITY, OR OTHER TORT OR OTHERWISE.
(2) This Limited Warranty is made only to the patient in whom the Components are implanted. AS TO ALL OTHERS, MEDTRONIC MAKES NO WARRANTY, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO, ANY IMPLIED WARRANTY OF MERCHANTABILITY OR FITNESS FOR A PARTICULAR PURPOSE, WHETHER ARISING FROM STATUTE, COMMON LAW, CUSTOM OR OTHERWISE. NO SUCH EXPRESS OR IMPLIED WARRANTY TO THE PATIENT SHALL EXTEND BEYOND THE PERIOD SPECIFIED IN A(1) ABOVE. THIS LIMITED WARRANTY SHALL BE THE EXCLUSIVE REMEDY AVAILABLE TO ANY PERSON.
(3) The exclusions and limitations set out above are not intended to, and should not be construed so as to contravene mandatory provisions of applicable law. If any part or term of this Limited Warranty is held to be illegal, unenforceable or in conflict with applicable law by a court of competent jurisdiction, the validity of the remaining portions of the Limited Warranty shall not be affected, and all rights and obligations shall be construed and enforced as if this Limited Warranty did not contain the particular part or term held to be invalid. This Limited Warranty gives the patient specific legal rights. The patient may also have other rights that vary from state to state.
(4) No person has any authority to bind Medtronic to any representation, condition, or warranty, except this Limited Warranty.
Case 1:20-cv-01277-AWI-JDP Document 1-13 Filed 09/08/20 Page 4 of 5

A04611001 Rev X
A04611_001.fm 2/24/04 11:13 amUCxxxxxx EN 4.5 x 6 inches (114 mm x 152 mm)
Medtronic ConfidentialNeuroIFP_R00
© Medtronic, Inc. 2004All Rights Reserved
A04611001
*A04611001*
Medtronic, Inc.710 Medtronic ParkwayMinneapolis, MN 55432-5604USAInternet: www.medtronic.comTel. 1-763-505-5000Fax 1-763-505-1000
Special Notice for Neurological Implantable Pumps, Catheters, and Refill KitsMedtronic Neurological Pump Systems consist of implantable pumps, catheters, refill kits, and accessories designed to contain and administer medications. Pump Systems are implanted in the extremely hostile environment of the human body. This environment places severe demands on their design and function. Reasons for failure of the Pump System include, but are not limited to: body rejection phenomena; change in performance characteristics due to component changes or failures; unusual physiological variations in patients; medical complications; complete or partial catheter occlusion; catheter dislodgment; catheter leakage; catheter breakage; migration; or erosion of the area around the pump. In addition, despite the exercise of all due care in design, component selection, manufacture, and testing prior to sale, the Pump System may be damaged before, during, or after implantation by improper handling or filling; by drugs or uses not described in the user manual; or by other intervening acts. The pump includes a nonseparable power source which will ultimately cease to function due to exhaustion or premature failure, thereby necessitating removal of the pump. Consequently, no representation or warranty is made that failure or cessation of function of the Pump System will not occur, or that the body will not react adversely to their implantation. (This paragraph does not apply to the IsoMed pump because it does not have a power source.)No representation is made that any one Pump System (except the IsoMed pump as stated in the limited warranty) will last the entire lifetime of any user or for any specific length of time. Inherent uncertainties regarding the longevity of the components make any such assurance impossible. For further information regarding safety information or possible complications resulting from the use of a Pump System, consult your patient manual. For additional copies of the patient manual, contact Patient Services at 1-800-510-6735.
Case 1:20-cv-01277-AWI-JDP Document 1-13 Filed 09/08/20 Page 5 of 5