l’ematologia pediatrica - ticino.labmed.ch · l’ematologia pediatrica. dal quadro normale alle...
TRANSCRIPT

L’ematologia pediatrica
Dal quadro normale alle patologie più frequenti
Formazione continua labmedSSMT Locarno - Sysmex
Dr. P. Brazzola

Sommario
• Organizzazione strutture pediatriche• Fisiologia e caratteristiche
pediatriche• Valori pediatrici «normali»• Particolarità tecniche• Patologie ematologiche più frequenti

Organizzazione
• Centralizzazione delle specialità a Bellinzona⇒ esami più «complessi»⇒ collaborazione con centri
universitari⇒ partecipazione a studi clinici⇒ più pressione sui laboratori

Caratteristiche pediatriche
• Il bambino non é un piccolo adulto

Caratteristiche pediatriche
Informazione ai genitori, raffronto
Rapporti interpersonali
Prelievi venosi / capillariPossibili artefatti in determinate analisi (p.es. coagulazione)
Prelievi «pensati», preparazione della preanaliticaSolo esami necessari
Prelievi «difficili»
Provette più piccole, sistemi differenti, apparecchiature con volume richiesto minore
Analisi di laboratorio con richiesta minima di sangue
Volumi dei campioni più piccoli
Hb, Ec, MCV, PTTValori normali relativi all’etàMalattie tipiche per l’età
Organismo in crescita
EsempioProcedereCaratteristica

Valori normali

Neonati• Volume sanguigno
– Prematuri 100-115 ml/kg, – Neonati 90 ml/kg– Adulti 65 ml/kg
• PM/NN fino a 1 sett.: Hctcapil > Hctvenoso – PM 27-30. settimana gravidanza (SG): >20% ↑– PM 31-35. SG: >10-15%– NN: >5-10% – Ca. dal 5. giorno di vita: Hct capillare = Hct venoso

Valori Hb

Valori LcLc 5.3–11.5 G/l
Neutrofili 34.3–72.9%Eosinofili 2.4–4.8%Basofili 1%Linfociti 13.5–52.8%Monociti 3.5–13.4%Linfociti atipici 2.6-5.6%
Valori fra 2 – 12 anniSoldin, S. J., Brugnara, C., & Wong, E. C. (2003). Pediatric reference ranges (4th ed.). Washington, DC: AACC Press.

Valori LcEtà Lc
(G/L)Seg PMN %
Band PMN%
ANC(G/L)
Eos%
Basos %
Lymphs%
ALYC(G/L)
Monos %
NRBCs (#/100 BCs)
0-3 giorni 9.0–35.0 32–62 0-18 3-28 0–2 0-1 19–29 2-10 5–7 0–24-9 giorni 5.0–21.0 19–49 0–15 1-13 0–2 0–1 26–36 2-8 5–7 010-14 giorni 5.0–20.0 14–34 0–14 1-10 0–2 0–1 36–45 2-9 6–10 015-30 giorni 5.0-19.5 15–35 0–12 1-9 0–2 0–1 43–53 2-10 7–11 01-6 mesi 6.0–17.5 13–33 0–11 1- 8 0–3 0–1 41–71 2-12 4–7 07-24 mesi 6.0–17.0 15-35 0–11 1-8 0–3 0–1 45–76 3-13 3–6 025-60 mesi 5.5–15.5 23-45 0–11 1.5–8.5 0–3 0–1 35–65 2-10 3–6 0
5-8 anni 5.0–14.5 32– 54 0–11 1.5–9.0 0–3 0–1 28–48 1.4–7.0 3–6 0
9-12 anni 4.5–13.5 33–61 0–11 1.5-9.5 0–3 0–1 28–48 1.3–6.5 3–6 0
13-18 anni 4.5–13.0 34–64 0–11 1.5–9.5 0–3 0–1 25–45 1.1–6.0 3–6 0
adulti 4.5–11.0 35–66 0–11 1.5–8.5 0–3 0–1 24–44 1.1–5.0 3–6 0
Nathan DG, and Oski, FA (1981) Hematology of Infancy and Childhood, ed 2, WB Saunders, pp 1552-74Dallman PR (1977) Pediatrics, 16th edition, Appleton-Century-Crofts, p 1111Pediatric Normal Ranges (1995) Children’s Hospital of Buffalo, Coulter VIEWPOINT, No. 17, p 8Pediatric Normal Range Study (1995) Children’s Hospitals, Minneapolis and St. Paul

Valori HbF

Valori Ferritina
Età Ferritina (µg/l)
prime 2-3 settimane > 90
fino ad 1 mese >144
fino a 2 mesi > 80
2-4 mesi > 35
4-6 mesi > 19
fino a 6-10 anni > 10

Emostasi
• Neonati• Vitamina K non ancora prodotta a
sufficienza dalla flora intestinale (INR ↑)• Minore produzione di fattori dela
coagulazione (PTT ↑)• Maggior quantità d‘acqua / kg di peso (INR ↑, PTT↑)

Emostasi
Età INR aPTT (sec.)
0 – 7 giorni 1.4 < 55
8 – 30 giorni 1.3 < 55
1 – 3 mesi 1.3 < 50
> 3 mesi 1.2 < 40
> 16 anni 1.2 < 40

Emostasi
0
0.5
1
1.5
Neona
ti
6 mes
i
1 - 5
anni
6 - 10
anni
11 - 1
6 anni
adult
i
Conc
entr
azio
ne U
/ml
FII
FVII
FIX
FX
Prot C
Prot S

Tecnica



Tecnica
• Accessi venosi difficili• Volumi piccoli (soprattutto neonati!)• Spesso scarsa collaborazione• Spesso un solo tentativo possibile senza poter
ripungere se manca qualcosa o prelievo non sufficiente o non conforme

Tecnica
• Strumentazione adeguata per prelievi capillari

Patologie

Caso
• Neonato di 3 giorni di vita in buone condizioni di salute
• Comparsa di ittero• Prelievo per valutare se fototerapia

Caso
• Hb 20 g/dl, MCV 100 fl, Lc 17 G/l, Tc 657 G/l

Caso
• ?
Neonato sano

Caso
• Lattante di 9 mesi in buone condizioni di salute
• Emocromo di routine presso il pediatra

Caso
• Hb 10 g/dl, MCV 74 fl, Lc 8,5 G/l, Tc 358 G/l

Caso
• ?
Lattante “sano”

Caso
• Bambino di 5 anni che ha febbre da 3 giorni con raffreddore e tosse
• Emocromo routine presso il pediatra

Caso
• Hb 10,6 g/dl, MCV 75 fl, Lc 13,6 G/l, Tc 420 G/l
• PCR 8 mg/l

Caso
• ?
Infezione virale con linfocitosi

Caso
• Bambino di 3 anni in controllo dal pediatra• Anamnesi famigliare blanda• Cresce molto bene, beve tanto latte

Caso
• Hb 8,5 g/dl, MCV 71 fl, MCH 22 pg, Lc 9,5 G/l, Tc 675 G/l
• PCR <5 mg/l

Caso
• ?
Anemia ferripriva


Regolazione ferro sistemico
Hepcidina
Fe
MacrofagiEnterociti
Fe
Fe
Nemeth E, et al. Science. 2004;306:2090-3.

• Hepcidina elevata in caso d‘infezione o infiammazione
• Produzione d‘hepcidina influenzata da tasso di transferrina nel sangue


• Indice sTfR/Ferritina• CHr: contenuto di emoglobina nei
reticolociti• CHr/Ferritina• Zinco-Protoporfirina eritrocitaria• Hepcidina-25 solubile

• A rischio– Prematuri
• Riserve scarse– Allattamento esclusivo al seno a lungo (?)– Fasi di crescita accellerata
• 0.5-1.5 anni e 11-14 anni– Alla comparsa del menarca
(50mg Fe/mestruazione)– Gravidanza

Caso
• Lattante di 4 mesi in controllo dal pediatra• Appare pallido• Alla nascita fototerapia per iperbilirubinemia• Anamnesi famigliare non chiara

Caso
• Hb 7,3 g/dl, MCV 75 fl, MCH 29 pg, MCHC 380 g/l, Lc 11,2 G/l, Tc 651 G/l
• PCR <5 mg/l

Caso
• ?
Sferocitosi

• Più comune forma di anemia emolitica da difetto di membrana dei globuli rossi
• Incidenza di 1:5000 • Colpisce > popolazione di origine Nord
Europea• Spettro di presentazione clinica
eterogeneo secondo gravità di malattia

Eziologia• 75% Autosomica Dominante
• 25% Mutazioni De novoAutosomica Recessiva
Mutazioni geni codificanti per proteine del citoscheletro
1. ANCHIRINA2. SPECTRINA 3. PROTEINA 3

Proteina Gravità Gene Pz con SE (%) ereditarietà
Anchirina-1 Da lieve a moderata
ANK1 50-67 Dominante e recessiva
Banda 3 Da lieve a moderata
AE1 (SLCA1) 15-20 > Dominante
β-spectrina Da lieve a moderata
SPTB 15-20 Dominante
α-spectrina Elevata SPTA1 <5 Recessiva
Proteina 4.2 Da lieve a moderata
EPB42 <5 Recessiva


Perdita di superficie di membrana senza perdita proporzionale di volume:- GR sferici- < deformabilità

La presentazione clinica varia in base alla gravità della malattia e all’età di presentazione:
• In utero: idrope fetale e morte intrauterina• Neonato:
- malattia emolitica con eventuale fototerapia ed exsanguinotrasfusione. (> emolisi per HbF che lega meno 2,3-DPG che aumenta e destabilizza ulteriormente le proteine del citoscheletro)
• Infanzia ed età adulta:- possibili crisi aplastiche in seguito a infezioni virali e in particolare da Parvovirus B19

A Sferocitosiereditaria
B Elissocitosiereditaria
C Piropoichilocitosiereditaria
D Stomatocitosiereditaria
E Acantocitosi F Emolisi da
frammentazione

Diagnosi
• Clinica • Anamnesi• Striscio
• Test diagnostici specifici per valutare la fragilità osmotica
• Immunofenotipizzazione

Caso
• Bambina di 7 anni in controllo dal pediatra• Buone condizioni di salute• Anamnesi famigliare con possibile talassemia

Caso
• Hb 10,4 g/dl, MCV 60 fl, MCH 19 pg, MCHC 330 g/l, Lc 8,2 G/l, Tc 384 G/l, Ec 5,38 T/l

Caso
• ?
Beta-Talassemia minor

Eziologia
• Thalassos = Mare• Thalassemia = Anemia che viene dal
mareMare = Mar Mediterraneo
• Cooley’s Anemia– Primo che ha descritto la malattia

Epidemiologia
• Mar mediterraneo, Africa, Cina, Indonesia, Medio Oriente
• Frequenza del gene 3-10%• Resistenza alla malaria degli
eterozigoti

Epidemiologia

Fisiologia (1)
• HbA = α2β2 > 95%• HbA2 = α2δ2 2-3.5%• HbF = α2γ2 < 2%• HBH = α -/- - 0%• HbBart = γ4 0%

Fisiologia (2)

Fisiologia (3)

Fisiologia (4)

Forme di talassemia (1)
• α-Talassemia (αα/α-, αα/--, α-/--, --/--)• β-Talassemia
– Talassemia minor• eterozigota
– Talassemia major• Produzione ridotta di globina β = β +
• Assenza di globina β = β °
• Omozigote β (β+/ β°, β+/ β+)

Forme di talassemia (2)
– Talassemia intermedia• β+/ β+, β°/ β+
• in generale nessun bisogno di trasfusioni

Patofisiologia (1)
• Eccesso di globina αcausa una distruzionecellulare e unaeritropoiesi inefficace
• Stabilità Ec ridotta• Espansione midollo causa
eritropoiesi inefficace• Deformità dello
scheletro causaespansione midollo
• Assorbimento Fe++
aumentato


Diagnostica
• Gold standard– Elettroforesi Hb
– Cromatografia Hb
– Analisi genetica

Talassemia minor• Anemia microcitica,
ipocromica• Cellule bersaglio• Nessuna risposta a
terapia con Fe++
• Emoglobina A2 aumentata
• Nessuna conseguenzaclinica!

Talassemia major• Presentazione < 2 anni• Anemia microcitica
severa (< 60 g/l)• Cellule bersaglio• Splenomegalia• Eritropoiesi
extramidollare (calotta, costole, fegato, milza)
• Trasfusioni!

Talassemia intermedia
• Anemia ipocromica, microcitica• Valori Hb > 70-80 g/l
• Nessuna trasfusione necessaria, salvo se infezioni intercorrenti
• Modificazioni strutture ossee!

Terapia (1)
• Regime trasfusionale– Valori medi Hb ca. 120 g/l– Valori pretrasfusionali Hb 95-100 g/l
(! Differenti regimi!)– Concentrati eritrocitari 4ml/kg =>
Aumento di 10 g/l

Terapia (2)
• Regime trasfusionale– Calo di Hb ca. 10g/settimana– ! Piccoli volumi se Hb < 50 g/l o problemi
cardiaci
Obiettivo: blocco dell‘ eritropoiesiinefficace

Terapia (3)
• Chelazione– Desferoxamine (Desferal®)
• t1/2 molto corto (15 min) => s.c. su 10-12 ore
– Deferiprone (Ferriprox®, L1)• 75 mg/kg/die PO
– Deferasirox (Exjade®, ICL670)• Una dose giornaliera sciolta in acqua o succo

Terapia (4)
• BMT– HLA identici, apparentati– Outcome migliore se carico in ferro non
troppo elevato e funzione cardiaca ed epatica non compromesse
– Risultati bambini > adulti (meno GvHD)

Complicazioni (1)
• Problemi di crescita (ossa, ipofisi)• Problemi di pubertà• Ipotireosi• Diabete mellito• Funzioni exocrine del pancreas
ridotte

Complicazioni (2)
• Ipertrofia cardiaca e dilatazione, fibrosi, aritmie, pericardite, insufficenza cardiaca
• Fibrosi epatica, cirrosi• Osteoporosi

Complicazioni (3)
• Infezioni– Yersiniosi (siderofagi)– Batteri incapsulati

Controlli (1)
• Ferritina < 2500 ng/ml, altrimenti cardiopatia 100% dopo 10 anni
• Valutazione funzione cardiaca e carica in ferro con RMN (T2*)
• Nessuna correlazione fra ferritina e carico Fe2+ cardiaco o epatico
• Nessuna correlazione fra carico cardiaco e epatico
• Crescita, DM, tiroide, FSH, LH

Caso
• Lattante di 1 anno in controllo dal pediatra• Buone condizioni di salute. Appare un po‘
gracilina• Anamnesi famigliare con madre con talassemia
major e padre eterozigote per anemia falciforme

Caso
• Hb 6,6 g/dl, Ec 2,86 T/l, MCV 74 fl, MCH 23 pg, MCHC 300 g/l, Lc 23,2 G/l, Tc 231 G/l

Caso
• ?
Doppio eterozigote HbS/Beta-Thal

Definizione
• Malattia sanguigna ereditaria autosomale recessiva
• Provocata da una mutazione puntiforme del gene per la β globina sul cromosoma 11: glutammato (aa.idrofilo) diventa valina (aa.idrofobo)
• Formazione di Hb S anormale• Hb S è meno solubile, soprattutto in
ambiente ipossico
• Precipitazione dell’Hb e formazione di globuli rossi a forma di “falce”

Epidemiologia• Trattasi
dell’emoglobinopatia nonché della malattia genetica più diffusa al mondo : 50 mio di persone affette
• Ogni anno in africa nascono 300.000 bambini malati di cui la metà non raggiungerà i 5 anni.
In Svizzera:

Epidemiologia

Patogenesi
Deossigenazione dell’HbS
Aggregazione in forma di polimero
Aumento dell’aderenza all’endotelio:
VASOOCCLUSIONE
Perdita dell’elasticità:VASOOCCLUSIONE
Diminuzione del trasporto e del rilascio di ossigeno in periferia
Distruzione precoce dei GR anormali:
ANEMIA EMOLITICA

Manifestazioni cliniche• Anemia
– Cronica, ben tollerata, normocromica, normocitaria, rigenerativa
• Crisi emolitica– Diminuzione acuta dell’Hb– Sequestro splenico (Attenzione! Ipovolemia,
Choc!) • Crisi aplastica
– Causa:• Infezioni, in particolare Parvovirus B19

Manifestazioni cliniche• Crisi vaso-occlusiva
– Ossea• Dactilite
– Cerebrale• Eventi ischemici• Eventi emorragici (formazione di neo vasi a causa
dell’ipossia)
– Polmonare• “Acute Chest Syndrome”

Manifestazioni cliniche
– Addominale• Micro-infarti mesenterici• Calcoli di bilirubina
– Renale• Ematuria (necrosi papillare)• IR
– Priapismo: URGENZA!

Complicazioni • Infezioni
– Infarti progressivi della milza provocano un’asplenismo funzionale: incapacità della milza a filtrare i microorganismi
– Suscettibilità ai batteri incapsulati• Streptococcus Pneumoniae +++• Haemophilus Influenzae• N. Menengitidis• Salmonella
– Osteomielite!• S. aureus

• Acute chest– Causa di mortalità– Eziologie
• Infettive– Chlamydia pneumoniae– Mycoplasma pneumoniae– Staph/pneumo/parvovirus
• Non infettive– Embolia grassosa su necrosi del midollo osseo
Complicazioni

• Ritardo di crescita• Deficit neurologico• Deformazioni scheletriche • Fibrosi polmonare• Insufficienza cardiaca• Impotenza
Complicazioni

Caso
• Neonata nata a termine• P 3340g, L 49cm, APGAR 9/9/10• pH 7,13/7,38• Gasometria con Hb 11,5 g/dl

Caso
• AF blanda• Esame clinico s.p.

Caso
• Al controllo del 1. mese esame clinico senza particolarità
• Nessun esame labor eseguito

Caso
• A sei settimane di vita visita in PS perché appare apatica, clonie arti inferiori e non più reattiva
• Labor: Hb 1,6g/dl
• ?

Caso
Anemia Blackfan-Diamond

Caso
• Gravidanza tramite FIVET• Parto indotto a termine• P 3150g, L 51cm, APGAR 9/9/10• pH 7,28/7,36
• Esame clinico s.p.• Difficoltà alimentari nei primi giorni

Caso
• A 20 giorni di vita consultazione perché mangia un po’ meno e ai genitori sembra pallido
• Esame clinico s.p.• Labor: Hb 8,0 g/dl

Caso
• A 27 giorni di vita mentre sono in vacanza in montagna notano che il piccolo geme. Contatto con il MC che suggerisce controllo in PS

Caso
• Colorito pallido, labbra pallide. FC 170/min, FR 50/min, gemiti e rientramenti diaframmatici. Vigile ma tono muscolare un po’ ridotto. Restante esame clinico s.p.

Caso
• Labor: Hb 4.8g/dL, HK 12%, MCV 87fL, MCH 36pg, reticolociti 25G/L, Ec 1.33T/L, Tc 240G/L, Lc 18.7G/L con diff citometrica normale.
• Gasometria: pH 7.129, pCO2 3.68kPa, Bic 8.8mmol/L, Lac 17mmol/L

Caso
Deficit enzimatico intracellulare del metabolismo della vitamina B12

Caso
• Bambina di 18 mesi• AF con casi di talassemia beta minor da
parte paterna, casi di celiachia da parte materna
• Fino a quel momento mai malata

Caso
• Controllo di routine dal MC con esame labor
• Labor: Hb 8,3 g/dl• Terapia sostitutiva con Aktiferrin® e
controllo dopo 2 settimane

Caso
• Al controllo eseguiti anche test per celiachia e ferritina
• Labor: Hb 5,9 g/dl, resto s.p.

Caso
• Esame clinico s.p., tranne che per pallore e soffio sistolico
• Hb 6.6 g/dl, Ec 2.39 T/l, Ht 0.19%, MCV 79 fl, MCH 28 pg, MCHC 350 g/l, Reticolociti in % 0.1%, Reticolociti assoluti 2 G/l, Tc 403 G/l, Lc 6.7 G/l (Neutrofili 0.86 G/l, Linfociti 4.98 G/l).
• Acido urico 151 µmol/l, Bilirubina totale 4.3 µmol/l, LDH 253 µmol/l.

Caso
• Nel decorso inizialmente osservazione e sostituzione con acido folico
• Controlli regolari Hb per 2 settimane con valori che si mantengono costanti attorno ai 6,0 g/dl
• Attività quotidiane non limitate

Caso
• Dopo due settimane si decide di procedere con esame midollo osseo– Il giorno dell’esame reticolociti in rialzo!– Annullato esame– Trasfusa perché comincia ad apparire
stanca

Caso
• Controlli regolari dal MC con valori dell’emoglobina e dei reticolociti sempre nella norma
• HbF e Adenosina deaminasi nella norma• Sierologie per Parvovirus B19 negative

Caso
Eritroblastopenia transitoria del lattante(TEC)

MCV, MCH, MCHC ↓ MCV, MCH, MCHC no MCV, MCH ↑
• Sideropenia
• Infezioni croniche
• Talassemie
• Problemi diassimilazione del ferro
• Intossicazioneda Pb
• Anemia aplastica
• Ec-penia transit.
• Toss./med.
• Renale
• Ipersplenismo
• Ipoproliferaz.
• Leucemia
Reti ↓
Emorragia acuta
Reti no
• Carenza B12• Carenza ac. folico• Neonati• Epatopatie• Ipotireosi• Anemia aplastica• SMD
Emorragia cronica
Reti ↑
Emolisi
Extracorpuscolare
• Immune
• Autoimmune
Coombs +
• Tossico
• CDI, SEU
• Meccanica
• Microangiopatia
Coombs -
Corpuscolare
• Sferocitosi
• Drepanocitosi
• Carenza-G6PD

Caso
• Ragazzo di 10 anni portato dal pediatra perché ai genitori appare un po‘ stanco
• Buone condizioni di salute. Afebbrile. Milza al costato, resto s.p.
• Anamnesi blanda

Caso
• Hb 8,4 g/dl, Ec 3,05 T/l, MCV 83 fl, MCH 27 pg, MCHC 329 g/l, Lc 7,6 G/l, Neutro 1,57 G/l, Linfo 5,53 G/l, LUC 3,9%, Tc 114 G/l

Caso
• ?
Leucemia linfoblastica acuta

Leucemia acuta linfoblastica

Leucemia acuta linfoblastica
• Epidemiologia– 25 % di tutti i tumori infantili– 75 % di tutte le leucemie– Picco all’età di 4 anni– M > F


Leucemia acuta linfoblastica
• Patogenesi– Fattori che favoriscono
• Raggi ionizzanti, anche in utero• Agenti alchilanti• Esposizione materna ad alcol, sigarette,
solventi• HTLV I e II, immunodeficienza congenita• Genetica (mutazioni gene p53, Bcl-2)



Leucemia acuta linfoblastica
• Patobiologia– Morfologia
• L1-L3• L3 prognosi peggiore
– Immunobiologia• Blasti presentano caratteristiche degli stadi
dell’evoluzione dei linfociti normali• Cellule B mature: morfologia L3• 90-95% CD10+ = common ALL• Cellule T

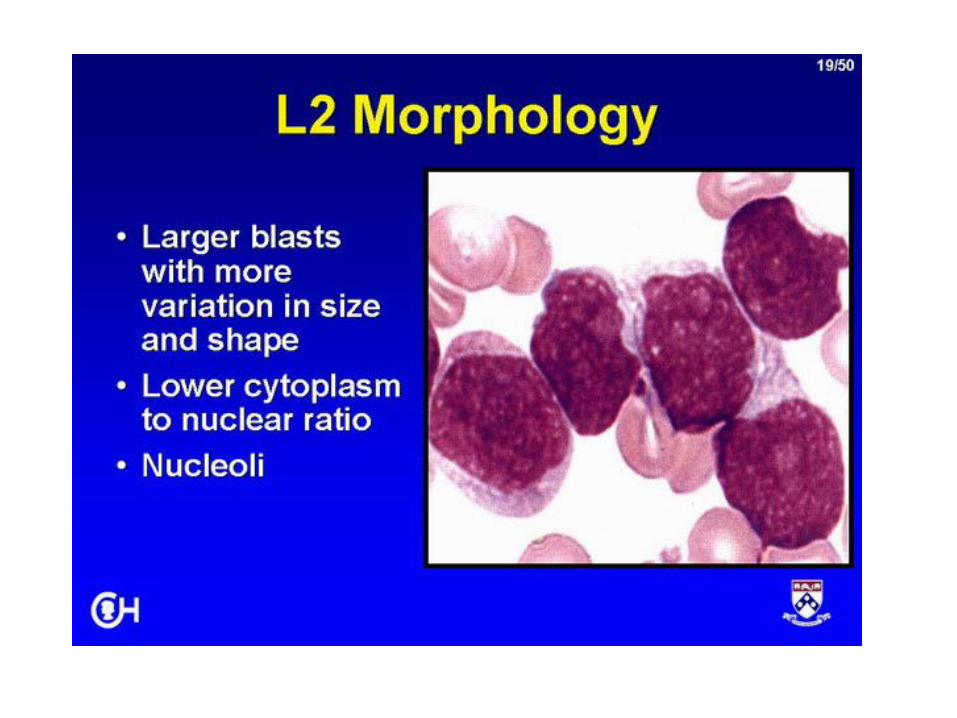



Leucemia acuta linfoblastica
• Genetica– Incidenza elevata in trisomia 21, Bloom’s
syndrome, anemia di Fanconi, Klinefelter, Shwachmann’s syndrome
– Traslocazioni nel 75% dei casi– Iperploidia = buona prognosi– Traslocazioni = in generale pessima
prognosi, tranne che per TEL/AML1



Leucemia acuta linfoblastica
• Clinica– Leucocitosi– Sintomi relativi alla soppressione
midollare– Epatosplenomegalia– Dolori ossei– Fatica , linfadenopatia (spesso in T-ALL
con aumento del timo)– Iperpotassemia, ipercalcemia,
iperfosfatemia con ipocalcemia

Leucemia acuta linfoblastica
• Clinica– SNC
• Segni di pressione intracranica aumentata• Deficit nervi cranici (III, IV, VII, VI),
iperacusia– Testicoli
• Spesso luogo di recidivi (10%)

Leucemia acuta linfoblastica
• Labor– Anemia, Tc-penia, leucocitosi – Reticolociti diminuiti– Aumento acido urico e LDH– Raramente coagulopatie


Leucemia acuta linfoblastica
• Prognosi– Cellule B > Cellule T– Cellule B
• Migliore se -> 1-9 annipochi Lc
• Peggiore se -> < 1 anno, adolescentiIperleucocitosi inizialet(9;22), t(4;11)

Leucemia acuta linfoblastica
• Prognosi– Peggiore
• Maschio• Assenza di remissione dopo 4-6 settimane• Prednisone good responder -> rischio recidivo
20%• Prednisone poor responder -> rischio recidivo
50%


Leucemia acuta linfoblastica
• Terapia– Multi-chemoterapia– Induzione
• Prednisone, Vincristina, L-Asparaginase, Daunorubicina– Consolidamento / CNS
• MTX i.th. • MTX-HD (sembra essere equivalente a RT)
– Re-induzione– Mantenimento
• 6-MP, MTX in dosi ottimali• in tutti i protocolli almeno 2 anni

Leucemia acuta linfoblastica
• Terapia– Remissione ematologica
• Blasti nel midollo < 5%• Assenza di blasti in periferia
– Remissione completa• Ematologica + assenza di manifestazioni
extramidollari (SNC, testicoli)– Remissione completa ad 1 mese nel 95%

Leucemia acuta linfoblastica
• Terapia– Trapianto cellule staminali
• Se t(9;22) o fattori per rischio elevato• Valutazione in base a MRD

MR
AIEOP LLA 2009
„ solo MRD-HR “„ solo No CR g+33 “
IB MIADXM
IB+
IBIAPDN
SCT
DNX-FLA + SCT
RHR
II
IIR2
PEG-ASP 20 SettimaneProt. IA con 2 dosi DNR (giorni 8 e 15)
Prot. IA con 4 dosi DNR (giorni 8, 15, 22 e 29)
E.coli ASP Medac 8 x 5000 U
HR
T/non-HR
pB#/non-HR
IISR
IB M
IA’PDN
IAPDNR1
IAPDN
Immunfenotipo non noto, opB-LLA + TEL/AML1 neg + e FCM-MRD g+15 >0.1%
# o immunofenotipo non noto* Tutti i pazienti con inziale malattia SNC (SNC 3) ricevono tRTC con 12 Gy o 18 Gy (Dose età-dipendente)
TEL/AML1 pos, e/oFCM-MRD g+15 <0.1%
IA‘D
IAD
53 104 S121
HR1‘
II
22 31 43
HR2‘
HR3‘
pRTC 12 Gy * se età≥ 2 aa/ In pazienti selezionati no pRTC ma 6x MTX IT
2010
II II
pRTC 12 Gy * se età≥ 2 aa/ In pazienti selezionati no pRTC ma 6x MTX IT
IB+

Caso
• Bambino di 6 anni portato dal pediatra perché da alcuni giorni é pieno di „blu“
• Buone condizioni di salute. Afebbrile. Esame clinico nella norma tranne che per suffusioni e petecchie
• Anamnesi blanda

Caso
• Hb 13,2 g/dl, Lc 7,4 G/l, Tc 6 G/l• LDH 235 U/l, acido urico 180 umol/l• CRP 6 mg/l

Caso
• ?
Trombocitopenia autoimmune

Purpura trombocitopenica
autoimmune

Storia (1)
• Nell‘antica grecia associazione con la peste
• Paul Gottlieb Werlhof (1735):– Descrizione clinica (Malattia di Werlhof)

Storia (2)• Kaznelson (1916):
– Studente in medicina– Postula che la milza é il sito d‘eliminazione dei
Tc e suggerisce la splenectomia• 1951:
– Scoperta del carattere immune della malattia– Primo tentativo di terapia con corticosteroidi
• 1981:– Utilizzo d‘immunoglobuline e.v. (Imbach,
D‘Apuzzo)

Sintomi (1)
• Classificazione– Asintomatica
• Nessun sintomo, scoperta per caso• Tc > 20x109/l
– Lieve• Petecchie ed ematomi• Lievi epistassi• Nessuna interferenza con la vita quotidiana• Tc > 10-20x109/l

Sintomi (2)
– Moderata• Manifestazioni cutanee più importanti• Lesioni delle mucose• Epistassi importanti, menorragie• Tc > 10x109/l
– Severa• Sanguinamenti che richiedono
l‘ospedalizzazione• Grave interferenza con la vita quotidiana• Tc < 10x109/l e/o calo Hb > 2 g/dl

Diagnostica (1)
• Striscio periferico con differenziazione– Pseudotrombopenia (aggregati Tc in EDTA)– Satellitismo
• Anamnesi familiare e remota– Emogrammi precedenti !
• Esame clinico– Adenopatie– Splenomegalia (ev. presente nel 10% dei casi!)– Esantemi– Articolazioni

Patofisiologia (1)
• Risposta immunitaria inadeguata a stimolo esterno– Grande maggioranza dei casi post-
infezione
• Ac anti-GPIIb/IIIa (in maggioranza)
• Tc con Ac alla superficie distrutti da macrofaghi (Recettore per frazione Fc -> FcRγIII)

Patofisiologia (2)
• Stimolazione linfociti B tramite linfociti T
• Presenza anomala di citochine– p.es. TGF-β


Terapia
• Immunoglobuline (IVIG)• Corticosteroidi• Anti-D

Complicazioni (1)
• Sanguinamenti– Intracerebrali 0,1%– 1/3 dopo inizio della terapia– Più a rischio se Tc < 10x109/l
=> Prestare sempre attenzione a sintomi potenzialmente indicativi per sanguinamento intracranico

Decorso (1)
• Normalizzazione Tc entro 12 mesi– Acuta
• Persistenza trombopenia > 12 mesi– Cronica

Decorso (2)
• Nei bambini 70% sono forme acute• Del restante 30% una parte si
normalizza spontaneamente• Adulti invece con prevalenza forme
croniche

Decorso (3)
• Nessun parametro clinico o di laboratorio che al momento può predire il decorso !
• La terapia iniziale non ha nessun influsso sul decorso !– Rende più breve la fase di trombopenia
severa