ligand field theory, density ... - uni-heidelberg.de · dft the density functional theorem —...
TRANSCRIPT

Ligand Field Theory, Density Functional Theory and Molecular Mechanics:
Adventures with d-electronsDr Rob Deeth
Inorganic Computational Chemistry Group

Overview• The Density Functional Theory revolution• Practical applications• Beyond DFT: Classical modelling for TM systems
and ‘smart’ approaches• LFT v. CFT• LFT v. DFT• Acknowledgements

DFTThe Density Functional Theorem — (Hohenberg-Kohn, 1964) the ground state total energy, E0, is a unique functional of the electron density, ρ.
E0 = E[ρ]
The theorem includes ALL electron correlation (Quantum Mechanics).

Pre-1988• Slater exchange-only Xα model• Vx ∝ ρ1/3
• Fast, accurate electronic structure• No automatic geometry optimisation• [CuCl4]2-, [VOCl2(urea)2]
d-d transitions energiesmolecular orbitalscharge distributions

Energy Gradients and Functionals
• Versluis and Ziegler: J Chem Phys, 1988• Exact DFT solution for a Uniform Electron Gas• Slater exchange plus fitted correlation – LOCAL
DENSITY APPROXIMATION• LDA overbinds• GENERALISED GRADIENT APPROXIMATION (e.g.
BP86, B3LYP)• GGA includes ∇ρ corrections

Structural Chemistry: 1994
TcO
ONNH
NHN
OHO
[TcO2Pent(AO)2]
199458 atoms297 basis functions10 hrs per geometry step on DEC 3000/700 workstation
Overlay: Obs/Calc(RMS ~0.03 Å)

Structural Chemistry: 2002
Overlay: Obs/Calc(RMS ~0.04 Å)
Zr
OBzBz
ONN
iPr
iPr2002113 atoms586 basis functions2 hrs per geometry step on 4 CPUs of Pentium III cluster
• DFT gives ~ X-ray structure accuracy for TM complexes

Aziridination
Cu
N
N
N
Ts
*
*
R'NR
R = TsR' = H, CO2Me
A Cu+-nitrene diimine is the proposed catalyst.
No experimental structure— it’s a catalyst!! — the structure must be computed.

Real Substrates: CinnimatesOnly the ester group was modelled.Quadrant 2 system spontaneously gives a Cu-O=C interaction at 2.26 Å and a structure 20 kJ mol-1more stable than the comparable styrene complex.DFT predicts much better e.e.s (Obs. e.e. >95%) Obs. and calc. absolute configurations identical
2.26 Å

Mechanism
2.00
2.08
2.3382°
Pd
‡2.01
2.1995°
1.99
‡
DFT provides direct access to Transition States

Activation barriersExchange on d8 centres: H2O, MeCN, MeNC, C2H4,
CO, CN-
Excellent correlation with ∆H‡ but GGA energies vital!
Activation Energies
-80
-60
-40
-20
0
20
40
60
80
100
120
0 20 40 60 80 100 120
Energy (kJ/mole)
Ener
gy (k
J/m
ole)
LDA
GC
Exp
4
3
1
6
52
89
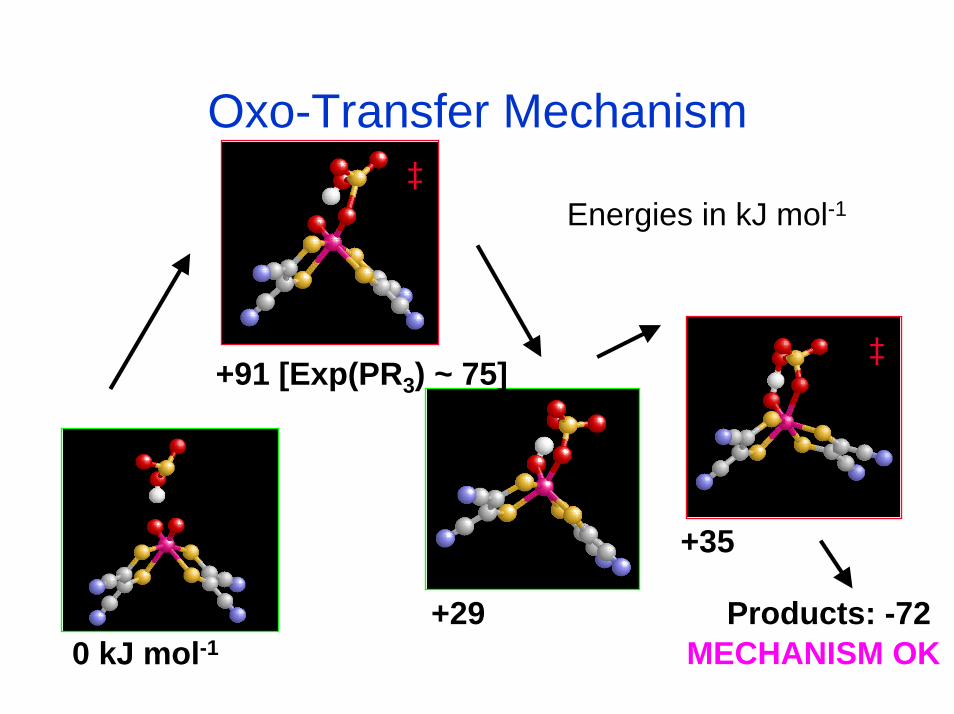
Oxo-Transfer Mechanism
Products: -72
+91 [Exp(PR3) ~ 75]
+29
+35
0 kJ mol-1
‡
‡Energies in kJ mol-1
MECHANISM OK

Conclusions: DFT• DFT is the best QM model for TM systems.• Excellent structures and direct access to Transition
States• “Static” DFT modelling a good starting point for catalyst
design• Functionals not specifically designed for TM systems
(too covalent) so require validation• DFT (or any QM method) is too slow for really big
molecules, VHTS or MD

Need for Speed
• DFT is accurate but ALL QM methods, including DFT, are relatively slow
• QM poorly suited to:Conformational searchingVirtual high-throughput screeningBiomoleculesMolecular Dynamics

Molecular Mechanics• Etot = ΣEstr + ΣEbend + ΣEtor + ΣEvdw + ΣEC
Fast (big systems, dynamics)Accurate (experimental information built in to Force Field parameters)Works well for organics and TM complexes with “regular” coordination environmentsConventional MM has fundamental problems with Transition Metal systems

Challenges• Coordination numbers > 4
• Multiple oxidation states
• Multiple spin states
• Organometallic versus classical Werner complexes
• Electronic effects (Jahn-Teller)

Electronic Effects: d-electronsThe d electrons are structurally and energetically non-innocent.The effect can be correlated with changes in the LIGAND FIELD STABILISATION ENERGY (LFSE)E.g.: d9 [CuL6]: ∆EJT electronic driving force
eg
t2g
∆EJT
∆EJT
L
CuL L
L
L
L+2δ
-δ
dx2-y2
dz2

Extending MM to the d-block• Many TM properties have a LFSE component
(‘double hump’ behaviour)• Add LFSE directly to MM
Ligand Field Molecular Mechanics (LFMM)
• Etot = ΣEstr + ΣEbend + ΣEtor + ΣEvdw + ΣEC+ LFSE
• LFMM captures d electronic effects directly
Dr Veronica Paget (nee Burton)

Getting LF Parameters• The LFSE is computed using the Angular Overlap Model
(AOM).• Each M-L bond described by LOCAL parameters — eσ, eπx, eπy
• Values from ‘d-d’ spectra: e.g. ∆oct = 3eσ- 4eπ
• Fit to general expression:eλ = a0 + a1 r + a2 r-2 + a3 r-3 + a4 r-4 + a5 r-5 + a6 r-6
L
eσ
dz2
deπx
dxz
d eπyd
L
M
L
MX
Y
Z
dyz
M

LFSE Gradients• Ε ’ = tr(NW’)• W’ = Q’VQT + QV’QT + QVQT
• = tr(NQ QT)
• LFSE includes σ and π parameters and d-s mixing effects
δV δx(λκ)
i
δΕ δx(λκ)
i
Dr Dave Foulis

Parameter FittingCannot simply take existing TM FF parametersBond length, r, is a balance of conventional MM terms (e.g. Morse function D0, α and r0) and LFSELow spin d6 has maximum LFSE: hence r < r0
-120
-80
-40
0
40
80
120
160
1.50 1.70 1.90 2.10 2.30 2.50
Bond Length
Ener
gy
MorseCLFSETotal
[Co(NH3)6]3+
r0r
r = 1.93 År0 = 2.11 Å

Spectroscopic AccuracyParameter a0 a1
eσ (Cl) 14400 -4000 eπ(Cl) 4300 -1500 eds(Cl) 9500 -3500
[CuCl4]2- Calca (no π or d-s) Calc Obs Cu-Cl /Å 2.26 2.26 2.26 dxy → dx2-y2 10050 12440 12500 dxz/yz → dx2-y2 10050 14260 14300 dz2 → dx2-y2 6700 17080 17000 [CuCl6]4- Cu-Cl(eq) /Å - 2.28 2.30 Cu-Cl(ax) /Å - 2.86 2.90 dz2 → dx2-y2 - 10720 10800 dxy → dx2-y2 - 12320 12220 dxz/yz → dx2-y2 - 14080 13300
Morse parameters: D0, r0, α: 80.0 kcal mol-1, 2.50 Å, 0.30

LFMM: d9 Cu(II)
MOE parametersAll Cu-N 1.93Å
Molecular OperatingEnvironmentDOMMIMOE
LFMM parameters(MMFF94-TM)Cu-Nax 2.29Å (2.32)Cu-Neq 2.05Å (2.06)Dr Natalie Fey
Ben Williams-Hubbard

Auto J-T Effect: d9
Cu
NH3
NH3
NH3
NH3
NH3
NH3
Cu
N
N
NN
NN
Cu
N
N
NN
N
N
CuN
NNN
N
N
Cu
NN
N
N
N
N
1 2 3 4 5
Cu-Neq (av) 2.042.15
1.992.04
2.052.06
2.062.07
2.012.07
Cu-Nax (av) 2.482.45
2.462.41
2.292.32
2.412.41
2.282.35
cis N-Cu-N (av) 9090
8081
8181
8081
8997
trans N-Cu-N (av) 180180
170169
177178
164166
180180
No other MM scheme gives J-T distortions automatically

High-spin/low-spin d8
eg
t2g
L
NiL L
L
L
L
dx2-y2
dz2
The structures of d8
Ni(II) complexes are determined by the LFSE
eg
t2g
2∆EJT
L
NiL L
L
L
L
dx2-y2
dz2

High and Low Spin States: d8
NH3H3N
H3N NH3
Ni
NH3
NH3
NN
NN
N
N
Ni Ni
N
NN
N
N NNi
N
NN
N
N N
Ni
N
NN
N
N N NN
NN
N
N
NiNN
N NNi
NN
N NNi
NN
N NNi
NN
N NNi
RMS ErrorsNi-N: 0.01ÅN-Ni-N: 0.6°
Ni-N(hs) ~2.1ÅNi-N(ls) ~1.9Å
JACS, 117, 8407, (1995)
One set of M-L Force Field parameters handles widely different M-L bond lengths

Relative Energies‘Conventional’ MM has different parameters for each
spin state.Cannot compare MM energies directly
MM cannot predict lowest energy spin stateLFSE term in LFMM can model spin-states
E.g.:[CoF6]3-: 5T2g [Co(CN)6]3-: 1A1g
Test case: d6 Co3+ octahedral in high and low spin
Ben Williams-Hubbard

Spin State Energies
5T2
1I
3H
3T15D
E/B
∆/B
3T1
3T2
1T1
5T2
1T2
3T1
3E
1T2,1E
5E
1A2
1A1
5T2
d6 Tanabe-Sugano
Both the LFSE and the interelectron repulsion needed.Theoretical d6 spin-crossover point:
∆E(ls-hs) = 2∆oct - (5F2 + 255F4)
F2 and F4 are ier parameters
The 5T2g-1A1g splitting from DFT is ~2400 which is consistent with d-d spectroscopy and full LFT.

Getting Other Parameters:DFT to the Rescue
Experimental data are not always available
DFT can access actual and hypothetical systems with and without constrained geometries and/or spin states
Use DFT to develop ‘smart’ LFMM parameters
DFT for high-symmetry ML6 complexes very fast

DFT Protocol for Bond Lengths• Optimised Bond lengths for [CoL6]3-
Co-F Co-CNLDA(hs) 1.97 2.12Exp 1.94 -
LDA(ls) 1.88 1.88Exp - 1.89
• Can rapidly tune LFMM parameters to reproduce CoL6 DFT data for BOTH spin states
Experimentalvalidation
Access tounobservables

The ResultsLFMM energies can be compared directlyParameters for homoleptic complexes applied unchanged to mixed-ligand systems - 10 for 2[Co(CN)nF6-n]3-, spin crossover at n = 1.
-1560
-1540
-1520
-1500
-1480
-1460
-1440
F6CN0
F5CN1
F4CN2c
isF4C
N2tran
sF3C
N3fac
F3CN3m
erF2C
N4cis
F2CN4m
erF1C
N5F0C
N6
Ener
gy (k
cal/m
ole)
DFT(high)DFT(low)LFMM(high)LFMM(low)
-100
-80
-60
-40
-20
0
20
Ener
gy (k
cal/m
ole)
DFTLFMM

Conclusions: LFMM• A fast scheme is needed for TM systems which
handles the important d-electron effectsLFMM
• designed for TM systems for all those cases where LFSE important
• captures the essential physics around the metal and facilitates calculations which would otherwise require full-blown QM approaches - ‘smart’ parameters
• automatic Jahn-Teller distortions• single parameter sets for multiple coordination
numbers and spin states• energies can be compared directly• efficient enough for large scale simulations

LFT v. CFT• Amines σ-bonding only• AOM: no π bonding = degenerate ‘t2g’ = two ‘d-d’ bands, • CFT: electrostatic model = four bands in rhombic D2h
• [Cu(dien)2]2+: 8800, 9900, 15400, 15900 cm-1
• AOM wrong?• DFT to the rescue!?!
eg
t2g
dx2-y2
dxy
dyz
dz2
tetragonalelongation
Oh D4h
tetragonalelongation
dx2-y2
'dπ' (dxy/dxz/dyz)
dz2
D4h
[M(NH3)6]n+: σ onlyAOM Electrostatic CFT
D2h D2h
rhombicdistortion
rhombicdistortion
dxz

[Cu(dien)2]2+
• DFT optimised structure agrees with X-ray• DFT d orbitals 1:3 pattern, EPR g-values too low• DFT too covalent (SOMO: Calc 43% d, Exp (EPR)
65-70% d)
• Tune DFT g-values by optimising Cu nuclear charge -best q = 28.2. SOMO now 69% d
• Good g-values - d orbitals still 1:3 pattern• DFT agrees with AOM, not with CFT
d lig d lig
Wrong covalent/ionicbalance
Correct covalent/ionicbalance
Mostly ligandMostly metal

New Interpretation• [Cu(dien)2]2+ has two possible elongation axes• ‘d-d’ bands arise from ~6:1 mixture of two
complexes aligned at ~90° • Exp. solid state structure obtained from DFT in
vacuo geometry• Even get rhombic equatorial geometry from
superimposing asymmetric axial elongation
Cu NNN
N
N
N
Cu NNN
NN
N
Cu NNN
N
N
N
85% 15%+ = Obs
Asymmetricaxial
elongation
2.13 2.07

LFT v. DFT• DFT seems to be replacing LFT• Lots of useful knowledge derived from
Ligand Field Theory• Would be a shame to lose it all• Does the Ligand Field description of
metal-ligand bonding map onto Density Functional Theory?

Tetragonal Distortion
• Classical (point charge) CFT model predicts t2gsplitting
• Qualitative MO model for [PtCl4]2-
concurs• What about DFT?
eg
t2g
b1g (dx2-y2)
b2g (dxy)
eg (dxz/dyz)
a1g (dz2)
tetragonal elongation
Oh D4h

Planar [MCl4]2-x
y
y
xz
b2g
eg
eg*
b2g*
eg
b2g
eg
b2g
b2g*
eg*
qualitative MO DFT
d
s
ClM ClM
• Experiment places dxy above dxz/dyz for M= Cu/Pd/Pt• DFT inverts energies of nominal dπ orbitals• Is DFT wrong?

Planar [M(NH3)4]2+
• NH3 σ-bonding only, simple MO theory and CLF model predict degenerate ‘t2g’ d orbitals
• EHMO actually gives degenerate dxy/dxz/dyz
• Ground state DFT calculations for M=Cu/Pd place dxy~4000 cm-1 lower than dxz/dyz
• DFT implies NH3 is a net π acceptor!• Point charge model always splits ‘t2g’ set but dxy is
always higher than dxz/dyz
• Is DFT wrong?

Can DFT really be wrong?• DFT gives excellent description of ground state properties
(geometries, frequencies, multipole moments etc.)• Relative energies of bonding MOs imply M-Cl π donor • DFT charge distributions reasonable• But ground state DFT d-orbital sequence still looks wrong• LFT treats both ground and excited d-d states so maybe it’s an
excited state issue• DFT excited state energies are qualitatively correct
(ADF multiplet states relative to AOCs)• DFT is OK so where is the problem?
[PdCl4]2- Exp. DFT1A1g → 1A2g 21700 164691A1g → 1Eg 23200 180211A1g → 1B1g 28910 21067
[CuCl4]2- Exp. DFT2B1g → 2B2g 12500 138412B1g → 2Eg 14300 161612B1g → 2A1g 17000 22625

DFT is OK, but...• Hoffmann and Baerends argue that Khon-Sham
orbitals provide a good basis for discussing bonding• LFT naturally focuses on the (anti-bonding) d orbitals
and assumes their energies are ‘mirrored’ by equal and opposite movements of the matching bonding functions - c.f. zero-overlap approximation -antibonding orbital increases by the same amount that the bonding MO decreases
• DFT is far more sophisticated and consequently we loose this simple relationship between antibonding d orbitals and their bonding counterparts
• Thus, ground state DFT d orbital energies cannot be interpreted in the ‘usual’ way

Implications for LFMM• Cannot use ground state DFT ‘d’ orbital energies directly for FF
development• DFT includes d-d interelectron repulsion explicitly and within the
full molecular symmetry• LFT separates d orbitals from interelectron repulsion and treats
the latter within a spherical central field approximation• If we define an approximate ‘spherical average’ configuration
where each d orbital has roughly the same population - E.g.: t2g
3.6eg2.4 for an octahedral d6 complex - then LFT and DFT
agree(N.B. Software issues. Jaguar versus ADF)

Acknowledgements• Inorganic Computational Chemistry Group
Dr Veronica Paget (LFMM) — now with AcelrysDr David Foulis (LFMM development)Dr Natalie Fey (LFMM/MOE, Diels-Alder)Ben Williams-Hubbard (LFMM/MOE,Co spin states and Cu proteins)Nicola Waite (acrylate polymers)Jack Smith (DFT and Heck reaction)Joanne Hanna (Binding energies)James Burnside (acrylate polymers)
• Peter Scott, WarwickLars Ivar Elding, LundDominic Ryan, Millenium
• ££££sEPSRCUniversity of WarwickChemical Computing Group