malattie genetiche - users.unimi.itusers.unimi.it/minucci/patologialezioni_2018-19/malattie...
TRANSCRIPT

Mutazioni Genomiche (n° Cromosomi # 46)
• Cromosomi Autosomici • Cromosomi Sessuali
NON EREDITARIE
Mutazioni Cromosomiche (Alterazione Struttura Cromosomi) • Traslocazioni • Delezioni
EREDITARIE E NON EREDITARIE
MALATTIE GENETICHE
Mutazioni Geniche • Delezioni, Inserzioni, Mutazioni Puntiformi, Espansione di Triplette ) Possono essere EREDITATE in modo : • MENDELIANO: singolo gene dominate/recessivo, cromosomi sessuali • NON MENDELIANO: Espansione triplette, mitocondriali

PATOLOGIA GENETICA
Studia i fenomeni patologici che riconoscono come causa un’alterazione del genoma
Differenza tra:
• Malattie genetiche
• Malattie ereditarie
• Malattie congenite

Malattie genetiche: comprendono tutte quelle condizioni patologiche a carico del patrimonio genetico (ereditarie e non)
Malattie ereditarie: Derivano dai genitori, sono trasmesse attraverso le cellule germinali nelle diverse generazioni e sono quindi familiari Non tutte le malattie ereditarie si manifestano al momento della nascita (ex. Corea di Huntington)
Malattie congenite: significa “Nato con”. I sintomi sono riscontrabili al momento della nascita. Non tutte le malattie congenite sono ereditarie (ex. Sindrome di Down, raramente lo e’) oppure determinate geneticamente (ex. Toxoplasmosi). Derivano da fattori patogeni di natura fisica/chimica/biologica che agendo durante la vita intrauterina inducono alterazioni organiche

• L’insorgenza di mutazioni é dovuta ad una interazione fra componenti ambientali (esempio: radiazioni ionizzanti, raggi UV, composti chimici) e una non perfetta efficienza dei sistemi di riparazione dei danni al DNA
• Le mutazioni peró possono insorgere anche spontaneamente: una classe di DNA polimerasi é detta “a bassa fedeltá” proprio per la sua tendenza ad inserire errori: vi é pressione selettiva per la riduzione, non per la scomparsa delle mutazioni
Mutazioni ed Evoluzione

NON TUTTE LE MUTAZIONI RISULTANO ESSERE CAUSA DI MALATTIE…
HIV-1 e ∆CCR5
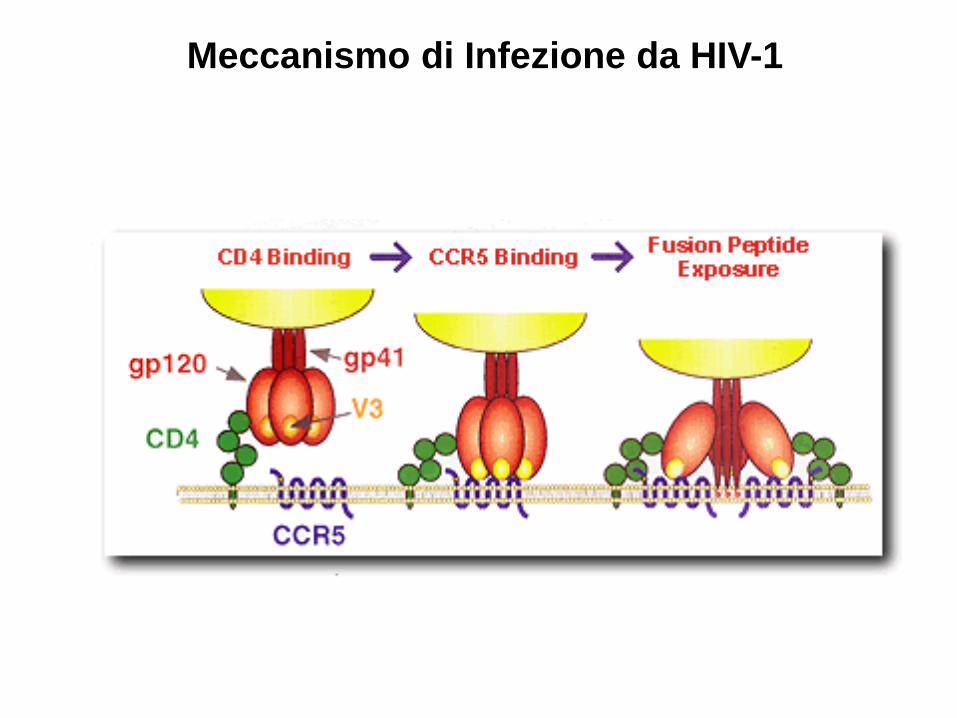
Meccanismo di Infezione da HIV-1

Il Recettore CCR5 (CD195)
• Localizzato sul cromosoma 3 • Recettore per le chemiochine RANTES, Mp1alfa e Mip1 beta • Espresso su linfociti T, Macrofagi, cellule dendritiche e microgliali • Esiste una mutazione ∆32 (32 nucleotidi) che produce una proteina non capace di raggiungere la superficie cellulare • L’allele ∆32 è comune nella popolazione Caucasica (frequeunza 10%) ma quasi assente in Africa e Asia
• I portatori Omozigoti di questa mutazione non hanno CCR5 sulla superficie cellulare • Sono resistenti all’infezione da HIV-1 (R5) • Hanno un fenotipo normale
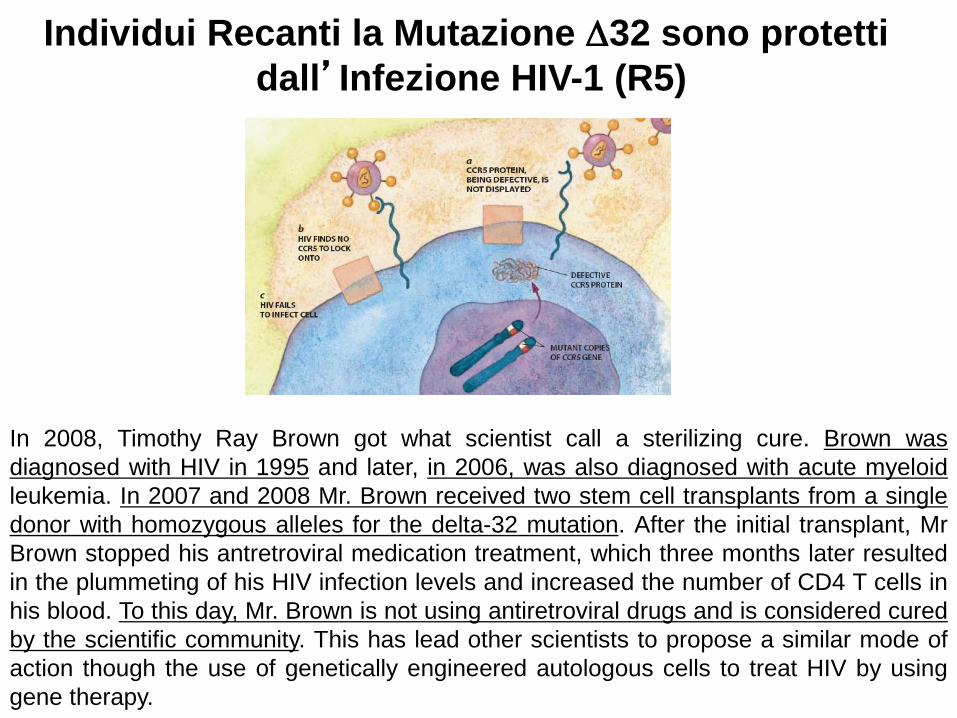
Individui Recanti la Mutazione ∆32 sono protetti dall’Infezione HIV-1 (R5)
In 2008, Timothy Ray Brown got what scientist call a sterilizing cure. Brown was diagnosed with HIV in 1995 and later, in 2006, was also diagnosed with acute myeloid leukemia. In 2007 and 2008 Mr. Brown received two stem cell transplants from a single donor with homozygous alleles for the delta-32 mutation. After the initial transplant, Mr Brown stopped his antretroviral medication treatment, which three months later resulted in the plummeting of his HIV infection levels and increased the number of CD4 T cells in his blood. To this day, Mr. Brown is not using antiretroviral drugs and is considered cured by the scientific community. This has lead other scientists to propose a similar mode of action though the use of genetically engineered autologous cells to treat HIV by using gene therapy.

In funzione dell’estensione della mutazione posso essere suddivise in: 1) Mutazioni genomiche (ANEUPLOIDIA): perdita o acquisto di interi
cromosomi (ex. Sindrome di Down)
2) Mutazioni cromosomiche (riarrangiamento di materiale genomico, ex. traslocazioni)
Definite anche come: Aberrazioni Cromosomiche 3) Mutazioni Geniche
Malattie Genetiche

Trisomia: 2n+1
Monosomia: 2n-1
• A carico dei cromosomi autosomici sono rare • Più frequenti a livello dei cromosomi sessuali
Mutazioni genomiche

Mutazioni genomiche (Cause)
• Alterazione della meiosi durante la gametogenesi • Alterazioni mitotiche durante le prime fasi dello sviluppo (MOSAICISMO) (46,XY/47,XY,+21) In entrambi i casi il fenomeno più frequente è di: • NON DISGIUNZIONE (meiotica/mitotica) Raramente SEGREGAZIONE (ritardo nella migrazione del cromosoma)

• NON-DISGIUNZIONE: incapacità dei cromatidi fratelli appaiati di separarsi durante la divisione meiotica. I due cromosomi o cromatidi congiunti migrano ad un polo e vengono inclusi in una sola cellula figlia, mentre l’altra avrà materiale genetico in meno
• RITARDO ANAFASICO: ritardata migrazione del
cromosoma durante l’anafase, conseguente perdita del cromosoma. Mancata incorporazione di un cromosoma nel nucleo di una delle cellule figlie.

Meiosi: è un processo di divisione mediante il quale una cellula eucariotica con corredo cromosomico diploide dà origine a quattro cellule con corredo cromosomico aploide.
Aneuploidia per non disgiunzione Meiotica

Sindrome di Down: trisomia 21 (incidenza 1/700) • causata prevalentemente da non disgiunzione meiotica, a carico dell’ oocita • aumenta con l’eta’ della madre (over 35 amniocentesi) 1/1550 sotto 20 anni, 1/25 sopra i 45 anni • Più raramente (3-4% casi) causata da non disgiunzione mitotica in una fase più o meno precoce dello zigote (Mosaicismo) con fenotipo più lieve del precedente
Mutazioni genomiche (Cromosomi Autosomici)
• Traslocazione del braccio lungo del cromosoma 21 sul 14 (rara)
(QUESTA E’ EREDITARIA)

Sindrome di Down

La SD si associa sovente a complicanze malformative che richiedono interventi chirurgici rilevanti nel corso dei primi anni di vita: • il 50% presenta malformazioni cardiache, • il 30% stenosi duodenale, • l’1% atresia esofagea, • il 2% malformazioni anorettali. • La chirurgia oftalmica è richiesta nel 12% dei casi per problemi di cataratta. Oltre alle malformazioni congenite descritte, il soggetto con SD ha la tendenza a sviluppare patologie secondarie per deficit nel sistema immunitario con particolare predisposizione ad infezioni batteriche; nell’1% poi dei casi compare leucemia acuta. Nel corso della vita il soggetto Down tende anche a sviluppare ipotiroidismo e diabete mellito.
Sindrome di Down

Edwards Syndrome: Trisomia 18 (1/6000). La sindrome ha un tasso molto basso di sopravvivenza, derivante da anomalie cardiache, malformazioni renali e altri problemi negli organi interni.
Mutazioni genomiche (Cromosomi Autosomici)
MANIFESTAZIONI •Ritardo mentale •Ipotonia/Ipertonia •Alterazioni flessioni dita •Vizi cardiaci •Anormalita’ piedi •Occipite prominente

Patau Syndrome: trisomia 13 (1/15000) common abnormalites include: Nervous system Musculoskeletal and cutaneous Urogenital
Mutazioni genomiche (Cromosomi Autosomici)
•Derivano quasi sempre da non disgiunzione meiotica nell’oocita. • La probabilità Incrementa con l’età della madre • A causa della severità del fenotipo, in entrambi i casi, la morte sopraggiunge nel primo anno di età
MANIFESTAZIONI •Ritardo mentale •Microcefalia microftalmo •Labbro leporino e palatoschisi •Vizi cardiaci •Anormalita’ piedi •Polidattilia

Mutazioni genomiche (Cromosomi Sessuali)
Più numerose perchè meglio tollerate:
• inattivazione di tutti i cromosomi X in eccesso tranne uno
• scarsa quantità di informazione genica presente sul cromosoma Y
Tutte le malattie: • Causano problemi di sviluppo sessuale e fertilità • Sono normalmente evidenziabili durante la pubertà • Possono essere associate a ritardo mentale

Mutazioni genomiche (Cromosomi Sessuali)
Lyonization: (Mary Frances Lyon 1961) solo uno dei due cromosomi X rimane attivo (in modo casuale paterno/materno), l’altro viene represso durante le prime fasi dell’embriogenesi e la repressione persiste nella progenie. Le femmine hanno mosaicismo derivante da cellule che hanno attivo il cromosoma X da parte del padre o da parte della madre . -Evidenziabile dal corpo di Barr nei nuclei in interfase (massa al lato della membrana nucleare)
- Oggi si sa che non tutti i geni del X inattivo sono veramente inattivi (ex. Sindrome di Turner femmine con un solo X) hanno severe anomalie somatiche e sessuali

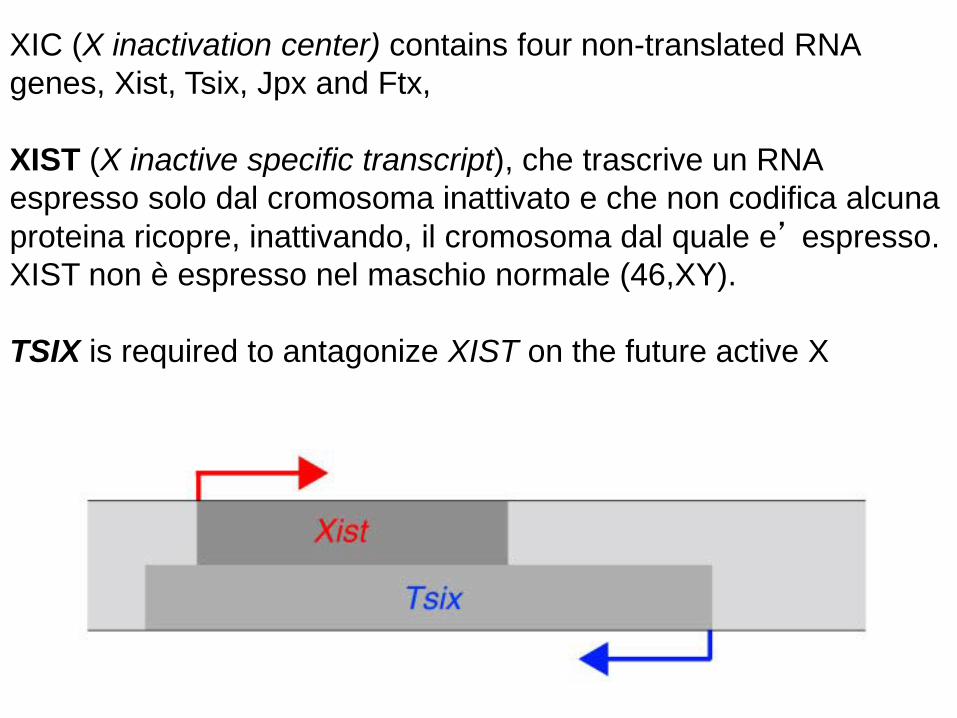
XIC (X inactivation center) contains four non-translated RNA genes, Xist, Tsix, Jpx and Ftx, XIST (X inactive specific transcript), che trascrive un RNA espresso solo dal cromosoma inattivato e che non codifica alcuna proteina ricopre, inattivando, il cromosoma dal quale e’ espresso. XIST non è espresso nel maschio normale (46,XY). TSIX is required to antagonize XIST on the future active X


The X-inactive specific transcript (Xist) gene encodes a large non-coding RNA that is responsible for mediating the specific silencing of the X chromosome from which it is transcribed. The inactive X chromosome is coated by Xist RNA, whereas the Xa is not . The Xist gene is the only gene which is expressed from the Xi but not from the Xa. X chromosomes which lack the Xist gene cannot be inactivated. Chromosomal translocations which place the XIC on an autosome lead to inactivation of the autosome, and X chromosomes lacking the XIC are not inactivated.
The existence of genes along the inactive X which are not silenced explains the defects in humans with abnormal numbers of the X chromosome, such as Turner syndrome (X0) or Klinefelter syndrome (XXY).
Compared to the Xa, the Xi has high levels of DNA methylation, low levels of histone acetylation, low levels of histone H3 lysine-4 methylation, and high levels of histone H3 lysine-9 methylation, all of which are associated with gene silencing.


X Inactivation

• Klinefelter syndrome: 47,XXY (1/1000)
•Turner syndrome: 45,X (1/5000)
• Multi-X Females: 47,XXX; 49,XXXXX
• XYY Sindrome: 47,XYY (1/1000)
Esempi di malattie causate da anomalie nel numero dei cromosomi sessuali
Un solo cromosoma Y e’ in grado di determinare il sesso, su di esso e’ presente il gene SRY (Sex-determining Region Y) che determina la soppressione dei caratteri primari femminili (utero, tube e ovaio) e promuove lo sviluppo dei caratteri sessuali maschili Codifica per un fattore trascrizionale (HMG-box family) che inizia la determinazione sessuale maschile
Klinefelter syndrome: 47,XXY (1/1000)

• mancato sviluppo dei caratteri sessuali secondari • microrchidia e aspermatogenesi • tendenza all'alta statura • L'analisi dei cromosomi sui linfociti è lo standard genetico di diagnosi • deficit di androgeni • solo il 10% presenta un ritardo mentale. • ridotto sviluppo del linguaggio, con problemi di espressività • sul piano comportamentale si possono riscontrare immaturità, poca sicurezza, timidezza
Klinefelter syndrome: 47,XXY (1/1000)

Sintomi della sindrome di Turner sono: • ipogonadismo con fenotipo femminile • bassa statura • torace a scudo (piatto) • orecchie a basso impianto; • il viso può avere un aspetto da persona anziana • sterilità dovuta a malformazioni dell'ovaio • amenorrea primaria, cioè, l'assenza della mestruazione avviene in età precoce
Turner syndrome: 45,X (1/5000)

Multi-X Females: 47,XXX; 49,XXXXX
Le principali caratteristiche standard sono: • altezza (maggiore che nella media) • circonferenza cranica inferiore alla norma (soprattutto alla nascita, poi c’è un ridotto recupero). • sviluppo motorio generalmente un po’ ritardato. Inizio della pubertà tendenzialmente ritardato. • sviluppo cognitivo. In media di poco inferiore alla norma. •sviluppo comunicativo e linguistico. Tendenzialmente allo stesso livello o inferiore rispetto alle prestazioni intellettive generali.

XYY Sindrome: 47,XYY (1/1000)
I maschi XYY non presentano particolari fenotipi patologici. Si tratta infatti di persone normali, con sviluppo intellettivo normale, vita normale e prole normale. Alcuni vecchi studi, riportano un aumento della statura media dei maschi XYY. In una percentuale variabile tra il 25 ed il 50% possono andare incontro a problemi nella sfera del linguaggio

(Anomalie strutturali dei cromosomi ex. Traslocazioni/Delezioni)
Mutazioni cromosomiche

• (A) acrocentrici : centromero in posizione terminale • (B) telocentrici: centromero in posizione subterminale • (C) submetacentrici: centromero in posizione submediana • (D) metacentrici: centromero in posizione mediana

Mutazioni cromosomiche
Rottura
Rottura e fusione delle estremita’ problemi in meiosi/mitosi
Perdita di un braccio e duplicazione di quello rimanente

Acute Promyelocytic Leukemia •APL is a subtype of acute myeloid leukemia (M3 variant according to the FAB classification) with distinctive biological and clinical features.
•Cells from 90% of the APL cases have a balanced translocation between chromosomes 15 and 17 which generates the fusion transcript joining the PML and RARα genes.
PML RARα
PML RARα
enhanced survival
Insinga A et al, EMBO J (2004)
Impairment of p53 pathway
repression of RA target genes
36
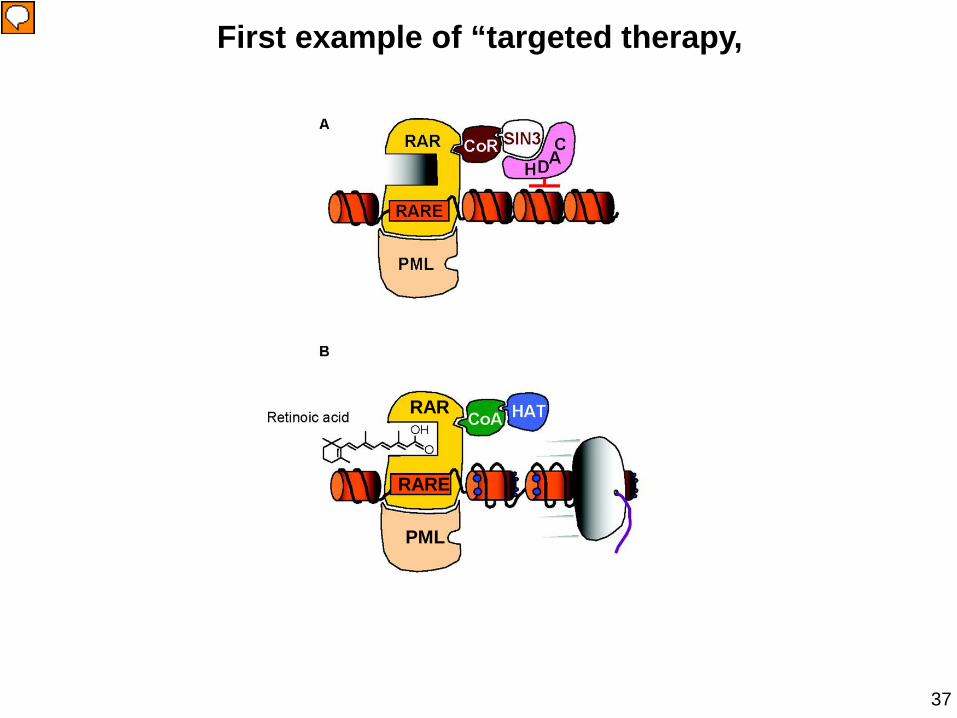
First example of “targeted therapy,
37
RAR
RARE
PML

Mutazioni cromosomiche

La persona portatrice di questa anomalia è perfettamente sana. È da considerare, però, che un’alta percentuale dei gameti prodotti dalle persone con traslocazioni bilanciate/ robertsoniane può essere “sbilanciata”. Di conseguenza, le persone che hanno una traslocazione, nella loro vita riproduttiva, hanno un rischio superiore, rispetto a quello di chi non è portatore di traslocazioni, di avere figli con patologia malformativa o di avere gravidanze interrotte da aborti
TRASLOCAZIONE ROBERTSONIANA
Not Viable

Malattie da Genomic Imprinting
(Prader-Willi, Angelman)

• Per imprinting genomico si intende una modificazione “epigenetica” di uno specifico allele nel gamete o nello zigote, responsabile per l’espressione differenziale dei due alleli del gene nelle cellule somatiche della progenie
• For the vast majority of autosomal genes, expression occurs from both alleles simultaneously. In mammals however, a small proportion (<1%) of genes are imprinted, meaning that gene expression occurs from only one allele.The expressed allele is dependent upon its parental origin. For example, the gene encoding Insulin-like growth factor 2 (Igf2) is only expressed from the allele inherited from the father.
Genomic imprinting

• Per modificazione epigenetica, si intende una modificazione ereditaria genomica, che non é associata ad un cambio della sequenza di DNA.
EPIGENETICA

Epigenetics: the bridge between Genotype and Phenotype
Epigenetic modifications modulate genetic information defining
the cellular Transcriptome and therefore Cell identity
Neu
ral c
ells
B
lood
Cel
ls
Transcriptome B Epigenome B
Transcriptome A Epigenome A
43

Epigenetic regulation is a dynamic process. Epigenetic writers such as histone acetyltransferases (HATs), histone methyltransferases (HMTs), protein arginine methyltransferases (PRMTs) and kinases lay down epigenetic marks on amino acid residues on histone tails. Epigenetic readers such as proteins containing bromodomains, chromodomains and Tudor domains bind to these epigenetic marks. Epigenetic erasers such as histone deacetylases (HDACs), lysine demethylases (KDMs) and phosphatases catalyse the removal of epigenetic marks. Addition and removal of these post-translational modifications of histone tails leads to the addition and/or removal of other marks in a highly complicated histone code. Together, histone modifications regulate various DNA-dependent processes, including transcription, DNA replication and DNA repair.
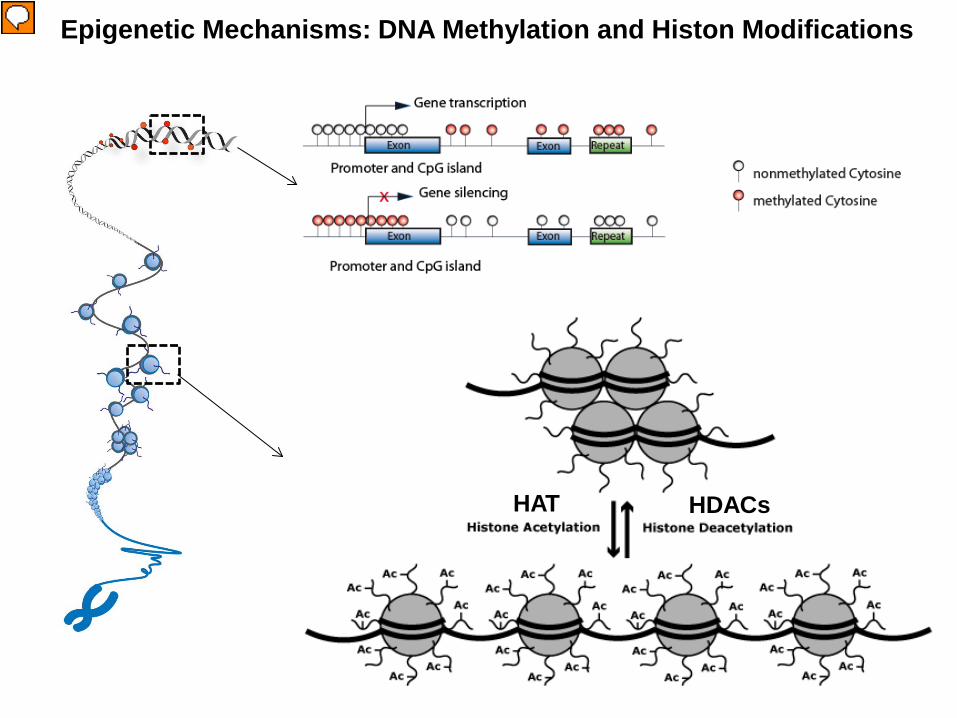
Epigenetic Mechanisms: DNA Methylation and Histon Modifications
• DNA Methylation of promoter and CpG Islands leads to
transcriptional silencing.
• The N-terminal of the core histone are subjected to several
types of post-translational modifications, including Acetylation,
Methylation, Phosporylation, Ubiquitylation, etc. 45
HAT HDACs



Imprinting control region
La delezione di ICR porta all’espressione materna di igf2
Demetilated maternal region
Differentially methylated region

Malattie da Imprinting
• Prader-Willi syndrome
• Angelman syndrome

• E’ una malattia genetica rara (colpisce 1 su 15.000-25.000 nati vivi) caratterizzata dall'alterazione del cromosoma 15 •Il 70% degli individui affetti da PWS hanno una delezione (∼4 Mb) sul cromosoma paterno 15q11-q13, circa il 25% hanno disomia uniparentale materna e il 3% mostra mutazioni nell’ Imprinting Center • Ipotonia nell' infanzia, obesità dovuta ad iperfagia, ipogonadismo, mani e piedi piccoli
• E’ la più comune tra le sindromi di microdelezione cromosomica • Avviene per due diverse cause accertate, entrambe di tipo genetico: Delezione sul cromosoma 15 paterno Disomia uniparentale materna del cromosoma 15
Prader-Willi Syndrome

I geni SNRPN, MKRN3 e NDN son stati individuati e risultano espressi solo dall'allele ereditato dal padre. • SNRP (small nuclear ribonucleoprotein complex) è coinvolto nello splicing del pre-mRNA (trascritto primario) • MKRN3 codifica per una proteina zinc finger (funzione ignota) • NDN espresso a livello dei neuroni (terminal differentiation of neurons) • MAGEL2 (ubiquitin ligase regulators) espresso nell’ipotalamo (funzione simile a NDN?) Tutti questi geni hanno un'isola CpG al 5', che non è metilata nell'allele paterno espresso, mentre viene metilata in quello sotto imprinting (materno).

• Nella PWS il gene materno è silenziato perché sotto imprinting,
mentre quello paterno è deleto
• La regione in questione è sul cromosoma 15 (15q11-q13).
• La PWS è strettamente correlata con la Sindrome di Angelman (AS),
che è causata da imprinting paterno e delezione del gene materno
caratterizzata da movimenti ripetitivi, simmetrici, atassici e da una
disposizione all' allegria, al riso frequente
Prader-Willi/Angelman Syndrome

Genomic Imprinting (Prader-Willi and Angelman Syndromes)
Sono attivi solo: • Un set di geni Prader-Willi (paterni) • Un set di geni Angelman (materni)
Sono attivi solo: • Un set di geni Prader-Willi (paterni)
Sono attivi solo: •Un set di geni Angelman (materni)
Uniparental Disomy
Stesso risultato delle delezione
Delezione banda q12 Cromosoma 15
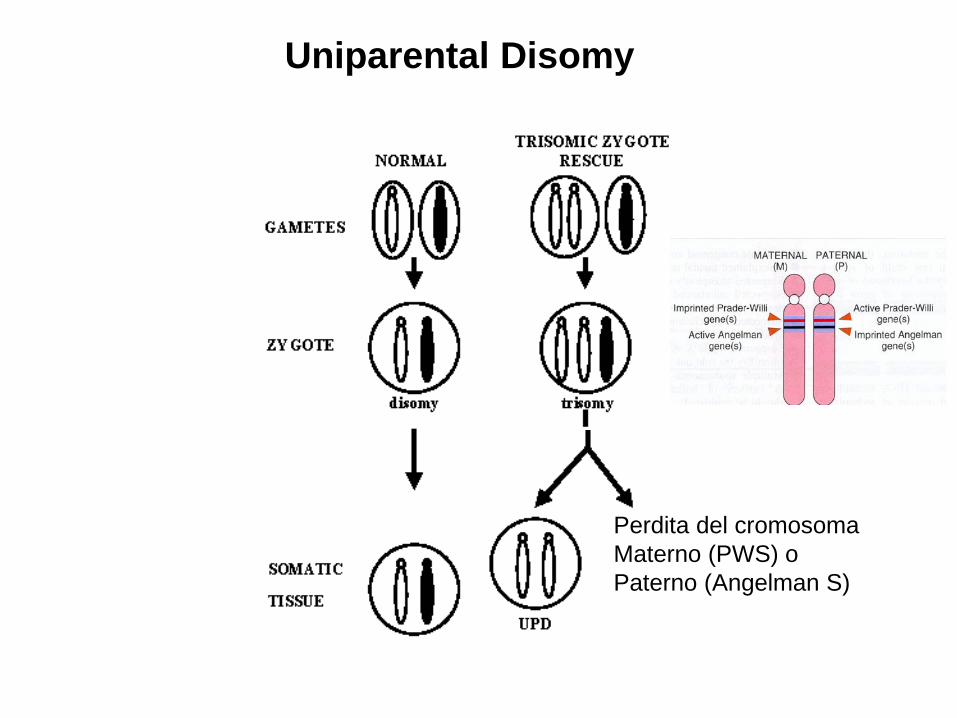
Uniparental Disomy
Perdita del cromosoma Materno (PWS) o Paterno (Angelman S)

MUTAZIONI GENICHE EREDITARIE
Tutte quelle malattie derivanti da alterazioni genetiche che: • possono essere trasmesse per via parentale perché presenti nelle cellule germinali Normalmente: • non alterano la capacità riproduttiva • sono compatibili con la vita

DEFINIZIONE DI MUTAZIONE
Permanente cambio nella sequenza del DNA Tutte le mutazioni che avvengo a livello delle cellule germinali possono essere trasmesse alla progenie e dare luogo a malattie ereditarie Le mutazioni a carico di cellule somatiche non vengono trasmesse per via ereditaria ma sono importanti nello sviluppo di cancro e malattie congenite

Possono verificarsi nelle regioni: codificanti, regolatorie (ex promotori) e nei siti di splicing • Mutazioni puntiformi: sostituzione di aa che causano cambio di sequenza o stop codon (talassemia) • Frame shift: delezione o inserzione di nucleotidi (cambio intera sequenza aa a valle della mutazione)
Tipi di Mutazioni Geniche (I)
• Inserzione/delezione di triplette: aggiunta o eliminazione di aa
• Espansione di triplette: caratterizzate dall’essere dinamiche perchè aumentano durante la gametogenesi (ex.X-Fragile e Corea Huntington)

Introduzione stop
Autosomica recessiva beta° talasemia
Mutazioni puntiformi
uracil-DNA glicosilasi

Inserzione di 4 nts e cambio della sequenza aa a valle della mutazione
Mutazioni “frame-shift”
Autosomico recessivo (esosaminidasi A) Malattia da accumulo lisosomiale nel cervello

Eliminazione di aa con alterazione della struttura/funzione della proteina) Fibrosi cistica autosomica recessiva
Delezioni di triplette
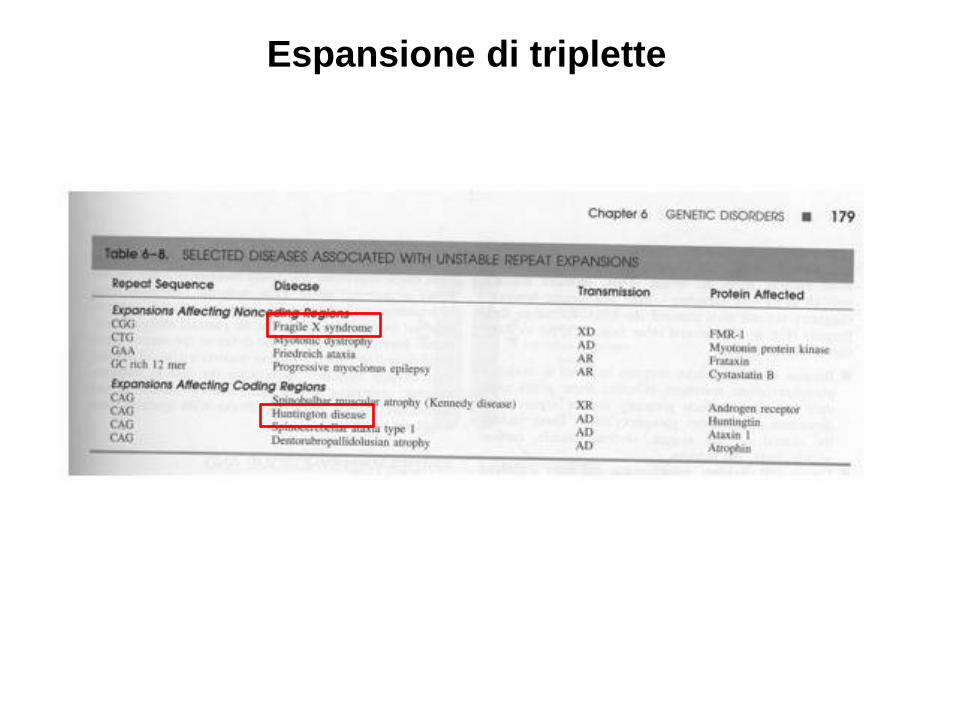
Espansione di triplette