mapping the enzyme specificities of intestinal maltase
TRANSCRIPT

MAPPING THE ENZYME SPECIFICITIES OF
INTESTINAL MALTASE-GLUCOAMYLASE AND SUCRASE-ISOMALTASE
by
Razieh Eskandari
B.Sc. (Chemistry), Yasouj University, 2003 M.Sc. (Chemistry), Shiraz University, 2006
THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY
in the Department of Chemistry
Faculty of Science
© Razieh Eskandari 2012 SIMON FRASER UNIVERSITY
Spring 2012
All rights reserved. However, in accordance with the Copyright Act of Canada, this work may be reproduced, without authorization, under the conditions for “Fair Dealing.” Therefore, limited reproduction of this work for the purposes of private
study, research, criticism, review and news reporting is likely to be in accordance with the law, particularly if cited appropriately.

ii
APPROVAL
Name: Razieh Eskandari
Degree: Doctor of Philosophy
Title of Thesis: Mapping the Enzyme Specificities of Intestinal Maltase-Glucoamylase and Sucrase-Isomaltase Examining Committee: Chair: Dr. Tim Storr Assistant Professor Dr. B. Mario Pinto Professor Senior Supervisor Dr. Steven Holdcroft Professor Supervisor Dr. Peter D. Wilson Associate Professor Supervisor Dr. Andrew J. Bennet Professor Internal Examiner, Department of Chemistry Dr. Jeffrey W. Keillor Professor External Examiner , Department of Chemistry University of Ottawa
Date Defended/Approved: January 20, 2012

Last revision: Spring 09
Declaration of Partial Copyright Licence The author, whose copyright is declared on the title page of this work, has granted to Simon Fraser University the right to lend this thesis, project or extended essay to users of the Simon Fraser University Library, and to make partial or single copies only for such users or in response to a request from the library of any other university, or other educational institution, on its own behalf or for one of its users.
The author has further granted permission to Simon Fraser University to keep or make a digital copy for use in its circulating collection (currently available to the public at the “Institutional Repository” link of the SFU Library website <www.lib.sfu.ca> at: <http://ir.lib.sfu.ca/handle/1892/112>) and, without changing the content, to translate the thesis/project or extended essays, if technically possible, to any medium or format for the purpose of preservation of the digital work.
The author has further agreed that permission for multiple copying of this work for scholarly purposes may be granted by either the author or the Dean of Graduate Studies.
It is understood that copying or publication of this work for financial gain shall not be allowed without the author’s written permission.
Permission for public performance, or limited permission for private scholarly use, of any multimedia materials forming part of this work, may have been granted by the author. This information may be found on the separately catalogued multimedia material and in the signed Partial Copyright Licence.
While licensing SFU to permit the above uses, the author retains copyright in the thesis, project or extended essays, including the right to change the work for subsequent purposes, including editing and publishing the work in whole or in part, and licensing other parties, as the author may desire.
The original Partial Copyright Licence attesting to these terms, and signed by this author, may be found in the original bound copy of this work, retained in the Simon Fraser University Archive.
Simon Fraser University Library Burnaby, BC, Canada

iii
ABSTRACT
In humans, maltase-glucoamylase (MGAM) and sucrase-isomaltase (SI)
are the small intestinal glucosidases responsible for catalyzing the last glucose-
releasing step in starch digestion. MGAM and SI are each composed of
duplicated catalytic domains, N-terminal membrane domains (ntMGAM and ntSI)
and C-terminal luminal domains (ctMGAM and ctSI). They display
complementary substrate specificities for the mixture of short, linear and
branched oligosaccharide substrates that typically make up terminal starch-
digestion products. As they are involved in the breakdown of dietary starch and
sugars into glucose, regulating their activities with α-glucosidase inhibitors is an
attractive approach to control blood glucose levels for the prevention and
treatment of type-2 diabetes.
This thesis work deals with mapping (determination of selectivity and
specificity) of MGAM and SI with synthetic inhibitors. The syntheses and
enzymatic evaluation of sulfonium-ion glucosidase inhibitors, with potent
inhibitory activities against intestinal glucosidases are the main topics of this
thesis. First, an alternative route for the synthesis of kotalanol, a naturally-
occurring sulfonium-ion glucosidase inhibitor isolated from Salacia reticulata, and
its 6'-epimer are described, and the inhibitory activities of these compounds
against ntMGAM are reported. Second, the total syntheses of de-O-sulfonated

iv
ponkoranol, another naturally-occurring sulfonium-ion glucosidase inhibitor
isolated from the same species, its 5'-epimer, and their selenium analogues are
described. The synthetic route is also extended to obtain 3'-O-methylponkoranol.
The inhibitory activities of these latter compounds against the four human
intestinal glucosidase enzymes, ntMGAM, ctMGAM, ntSI, and ctSI are examined.
Finally, from the structural studies of ntMGAM, it was postulated that ctMGAM
might have an extended binding site compared to ntMGAM, which favours
binding of longer inhibitors such as acarbose (an antidiabetic drug that is
currently in use for the treatment of type-2 diabetes). Based on this difference,
the syntheses of candidate inhibitors containing maltose extensions at 3'- and 5'-
of de-O-sulfonated ponkoranol are described.
The inhibition of maltose hydrolysis suggests that selective inhibition of
one enzyme unite over the others is possible despite relatively small structural
changes in the inhibitor. This panel of inhibitors can now be used to turn off
certain enzymes while probing the action of others with respect to starch
digestion.
Keywords: Glucosidase inhibitors; Salacia reticulata; kotalanol; ponkoranol; maltase-glucoamylase; sucrase-isomaltase; type-2 diabetes; sulfonium-ions; selenonium ions.

v
DEDICATION
This work is dedicated to my respected parents and my teachers who
helped me every step
of the way so that I can get to where I am today.
It is also dedicated to my lovely husband, Mehdi
who was extremely patient with me all these years.

vi
ACKNOWLEDGMENTS
All praise is to God Almighty Who continues to bless my life.
I would like to thank my senior supervisor, Dr. B. Mario Pinto, for his
support and guidance and giving me the opportunity to work in his laboratory.
I would like to thank my supervisory committee: Dr. Peter Wilson and Dr.
Steven Holdcroft and my examining committee: Dr. Andrew J. Bennet and Dr.
Jeffery W. Keiller for their valuable advice and feedback. I would also like to
thank our collaborators, Dr. David R. Rose, Ms. Kyra Jones, and Dr. Douglas A.
Kuntz for the enzyme inhibition data. I am grateful to Dr. Andrew Lewis and Mr.
Colin Zhang for the NMR help and Mr. Hongwen Chen for acquiring the high
resolution mass Spectra.
I thank Dr. H. Sharghi, my M.Sc. supervisor, who taught me how to work
in a chemistry laboratory. I would like to thank Dr. Silvia Borrelli for proof reading
my introduction chapter and Dr. Niloufar Choubdar and Dr. Rehana Hossany for
helping me in the lab during the first year of my PhD program and being
wonderful friends. I would like to thank Dr. Jayakanthan Kumarasamy, Dr.
Ravinder Reddy, with whom I collaborated and Dr. Sankar Mohan for his help.
Last but not least, my deepest gratitude to my parents, my sister for their
support and love. My deepest thanks go to my husband, Mehdi, for his support,
patience and encouragement.

vii
TABLE OF CONTENTS
Approval .............................................................................................................................ii Abstract ............................................................................................................................. iii Dedication ......................................................................................................................... v Acknowledgments .............................................................................................................vi Table of Contents ............................................................................................................. vii List of Figures.................................................................................................................... x List of Schemes ............................................................................................................... xiii List of Tables ................................................................................................................... xiv Abbreviations ...................................................................................................................xv
CHAPTER 1: General introduction ................................................................................ 1 1.1 Carbohydrates .......................................................................................................... 1 1.2 Carbohydrates in health and disease ....................................................................... 4 1.3 Starch digesting enzymes ......................................................................................... 5
1.3.1 Glycoside hydrolases .................................................................................... 8 1.3.2 Alpha-amylases ............................................................................................. 8 1.3.3 Brush-border hydrolases ............................................................................... 8 1.3.1 Disaccharidase deficiency in heath and disease ........................................ 12
1.4 Glycosidase mechanism of action .......................................................................... 13 1.4.1 Mechanism of retaining glycosidases ......................................................... 13 1.4.2 Mechanism of inverting glycosidases .......................................................... 15
1.5 Transition-state mimics ........................................................................................... 16 1.6 Glycosidase inhibitors ............................................................................................. 19
1.6.1 Iminosugars................................................................................................. 20 1.6.2 Carbasugars................................................................................................ 24 1.6.3 Marine organosulfates ................................................................................. 25 1.6.4 Sulfonium-sulfate thiosugars ....................................................................... 27
1.7 Thesis overview ...................................................................................................... 34 1.8 References ............................................................................................................. 37
CHAPTER 2: Synthesis of a biologically active isomer of kotalanol, a naturally-occurring glucosidase inhibitor .................................................................. 44 2.1 Keywords ................................................................................................................ 46 2.2 Abstract ................................................................................................................... 46 2.3 Introduction ............................................................................................................. 46 2.4 Results and discussion ........................................................................................... 50 2.5 Experimental ........................................................................................................... 55
2.5.1 General ....................................................................................................... 55 2.5.2 Enzyme inhibition assays ............................................................................ 55

viii
2.5.3 Compound characterization data ................................................................ 56 2.6 Acknowledgments ................................................................................................... 66 2.7 References ............................................................................................................. 67 2.8 Supporting Information ........................................................................................... 69
CHAPTER 3: Potent glucosidase inhibitors: de-O-sulfonated ponkoranol and its stereoisomer ..................................................................................................... 82 3.1 Keywords ................................................................................................................ 84 3.2 Abstract ................................................................................................................... 84 3.3 Introduction ............................................................................................................. 84 3.4 Results and discussion ........................................................................................... 88 3.5 Experimental ........................................................................................................... 93
3.5.1 General methods......................................................................................... 93 3.5.2 Compound characterization data ................................................................ 94
3.6 Acknowledgments ................................................................................................... 98 3.7 References ............................................................................................................. 98 3.8 Supporting Information ......................................................................................... 101
CHAPTER 4: The effect of heteroatom substitution of sulfur for selenium in glucosidase inhibitors on intestinal α-glucosidase activities ................................ 108 4.1 Keywords .............................................................................................................. 110 4.2 Abstract ................................................................................................................. 110 4.3 Introduction ........................................................................................................... 110 4.4 Results and discussion ......................................................................................... 113 4.5 Experimental ......................................................................................................... 118
4.5.1 General methods....................................................................................... 118 4.5.2 Enzyme kinetics ........................................................................................ 118 4.5.3 Compound characterization data .............................................................. 119
4.6 Acknowledgments ................................................................................................. 122 4.7 References ........................................................................................................... 123 4.8 Supporting Information ......................................................................................... 126
CHAPTER 5: Probing the active-site requirements of human intestinal N-terminal maltase-glucoamylase: The effect of replacing the sulfate moiety by a methyl ether in ponkoranol, a naturally-occurring α-glucosidase inhibitor ........................................................................................................................ 132 5.1 Keywords .............................................................................................................. 134 5.2 Abstract ................................................................................................................. 134 5.3 Introduction ........................................................................................................... 135 5.4 Results and discussion ......................................................................................... 139 5.5 Experimental ......................................................................................................... 144
5.5.1 General methods....................................................................................... 144 5.5.2 Enzyme kinetics ........................................................................................ 145 5.5.3 Compound characterization data .............................................................. 145
5.6 Acknowledgments ................................................................................................. 152 5.7 References ........................................................................................................... 152

ix
5.8 Supporting Information ......................................................................................... 155
CHAPTER 6: Selectivity of 3'-O-methylponkoranol for inhibition of N- and C-terminal maltase-glucoamylase and sucrase-isomaltase, potential therapeutics for digestive disorders or their sequelae ........................................... 165 6.1 Keywords .............................................................................................................. 167 6.2 Abstract ................................................................................................................. 167 6.3 Introduction ........................................................................................................... 167 6.4 Results and discussion ......................................................................................... 173 6.5 Acknowledgments ................................................................................................. 176 6.6 References ........................................................................................................... 177
CHAPTER 7: Probing the intestinal α-glucosidase enzyme specificities of starch-digesting maltase-glucoamylase and sucrase-isomaltase: Synthesis and inhibitory properties of 3′- and 5′- maltose-extended de-O-sulfonated ponkoranol ................................................................................................................... 179 7.1 Keywords .............................................................................................................. 181 7.2 Abstract ................................................................................................................. 181 7.3 Introduction ........................................................................................................... 182 7.4 Results and discussion ......................................................................................... 187 7.5 Experimental ......................................................................................................... 199
7.5.1 Compound characterization data .............................................................. 199 7.6 Acknowledgments ................................................................................................. 211 7.7 References ........................................................................................................... 212 7.8 Supporting Information ......................................................................................... 216
CHAPTER 8: Conclusions and future work .............................................................. 231 8.1 Conclusions .......................................................................................................... 231 8.2 Future work ........................................................................................................... 235 8.3 References ........................................................................................................... 239

x
LIST OF FIGURES
Figure 1-1: Glucose (1), lactose (2), sucrose (3). ........................................................ 2 Figure 1-2: Components of starch: amylose and amylopectin. .................................... 3 Figure 1-3: Starch digestion by the action of salivary and pancreatic α–
amylases and small intestinal α–glucosidases. ......................................... 7 Figure 1-4: Pictorial representation of brush-border membrane-bound MGAM
and SI. ....................................................................................................... 9 Figure 1-5: Surface representation of the ntSI and ntMGAM active sites
(Reproduced from J. Biol. Chem. 2010, 285, 17763–17770. © the American Society for Biochemistry and Molecular Biology). ................... 11
Figure 1-6: Transition state proposed for glycosidase catalyzed reactions. .............. 17 Figure 1-7: Proposed half chair and boat conformations of pyranosyl cations
in glycosidase-mediated hydrolysis reactions. ........................................ 18 Figure 1-8: Proposed envelope conformations of furanosyl cations in
glycosidase mediated-hydrolysis reactions. ............................................ 18 Figure 1-9: Positive charge build-up at the exocyclic oxygen, endocyclic
oxygen, and the anomeric carbon during glycosidase-mediated hydrolysis reactions. ................................................................................ 19
Figure 1-10: Skeletal frameworks most commonly found in naturally-occurring iminosugar glycosidase inhibitors and their synthetic derivatives. .......... 20
Figure 1-11: Nojirimycin (12), 1-deoxynojirimycin (13), and isofagomine (14). ............ 21 Figure 1-12: Naturally-occurring polyhydroxylated pyrrolidine-based
glycosidase inhibitors. ............................................................................. 22 Figure 1-13: Structures of selected, naturally-occurring pyrrolizidine alkaloids,
which display α-glucosidase inhibitory activities. ..................................... 23 Figure 1-14: Castanospermine (23), swainsonine (24). ............................................... 24 Figure 1-15: Examples of nortropanes (25, 26). .......................................................... 24 Figure 1-16: Examples of naturally-occurring carbasugar α-glucosidase
inhibitors and the antibiotic validamycin. ................................................. 25 Figure 1-17: Structures of organosulfate α-glucosidase inhibitors isolated from
marine invertebrates. ............................................................................... 26 Figure 1-18: Structures of organosulfate α-glucosidase inhibitors isolated from
marine invertebrates. ............................................................................... 27

xi
Figure 1-19: Structure of an N-oxide analogue of castanospermine and a sulfonium-ion analogue. .......................................................................... 28
Figure 1-20: Structure of compounds 40-43. ............................................................... 29 Figure 1-21: Naturally-occurring zwitterionic sulfonium-sulfate glucosidase
inhibitors. ................................................................................................. 29 Figure 1-22: Initially proposed structure of a naturally-occurring sulfoxide α-
glucosidase inhibitor. ............................................................................... 31 Figure 1-23: Initially proposed structure of the naturally-occurring α-
glucosidase inhibitor neosalacinol. .......................................................... 32 Figure 2-1: Components isolated from Salacia species. ............................................ 48 Figure 2-2: Kotalanol stereoisomer. ........................................................................... 48 Figure 3-1: Components isolated from Salacia species. ............................................ 85 Figure 3-2: Proposed structure of neosalacinol. ........................................................ 86 Figure 3-3: De-O-sulfonated ponkoranol and its 5’-stereoisomer. ............................. 88 Figure 3-4: 1D-NOESY correlations of selected protons in compounds 18 and
21. ............................................................................................................ 92 Figure 4-1: Components isolated from Salacia species. .......................................... 111 Figure 4-2: Structure of the 5′-stereoisomer of de-O-sulfonated ponkoranol. .......... 112 Figure 4-3: Superimposition of the ring carbon atoms of the proposed
intermediate in glucosidase-catalyzed reactions (in green) and the selenonium ion (in blue). ....................................................................... 113
Figure 4-4: Selenium analogues of de-O-sulfonated ponkoranol and its 5’-stereoisomer. ......................................................................................... 113
Figure 4-5: 2D-NOESY correlations of selected protons in compound 13. .............. 116 Figure 5-1: Components isolated from Salacia species. .......................................... 136 Figure 5-2: De-O-sulfonated ponkoranol and its 5’-stereoisomer. ........................... 137 Figure 5-3: Effect of removing the sulfate group. Superposition of kotalanol (4)
(orange) and de-O-sulfonated kotalanol (5) (purple) structures. Double-headed arrows show the proximities of the sulfate group to the surrounding hydrophobic residues Y299, W406 and F575. (Reproduced with permission from Biochemistry, 2010, 49, 443–451. Copyright American Chemical Society). ........................................ 138
Figure 5-4: 3’-O-Methylponkoranol. ......................................................................... 138 Figure 5-5: 1D-NOESY correlations of selected protons in compound 9. ................ 143 Figure 6-1: Components of starch: amylose and amylopectin. ................................ 168 Figure 6-2: Schematic diagram of MGAM and SI indicating their hydrolytic
activities. ................................................................................................ 169 Figure 6-3: Sulfonium-ion α-glucosidase inhibitors 1-7. ........................................... 171

xii
Figure 6-4: Representative Lineweaver-Burk plot of ctMGAM-N2 inhibited by 3 at concentrations of 0 nM, 75 nM, 125 nM, and 200 nM. ................... 174
Figure 7-1: Schematic diagram of MGAM and SI indicating hydrolytic activity. ....... 183 Figure 7-2: Components 1-8 isolated from Salacia species. ................................... 184 Figure 7-3: Structure of acarbose 9, an α-glucosidase inhibitor currently used
in the treatment of type-2 diabetes. ....................................................... 185 Figure 7-4: 3′-O-β-maltosyl-de-O-sulfonated ponkoranol 10 and 5′-O-β-
maltosyl-de-O-sulfonated ponkoranol 11. .............................................. 187 Figure 7-5: Structure of 3'-O-methylponkoranol 26. ................................................. 192 Figure 7-6: NOESY correlations in compound 24. ................................................... 194 Figure 8-1: Structures of compounds 1-8. ............................................................... 232 Figure 8-2: Structures of compounds 9-11. ............................................................. 235 Figure 8-3: Proposed selective inhibitors of ntSI. .................................................... 236

xiii
LIST OF SCHEMES
Scheme 1-1: Proposed mechanism for retaining glycosidases. ............................... 15 Scheme 1-2: Proposed mechanism for inverting glycosidases. ................................ 16 Scheme 2-1: First attempted synthesis of kotalanol 4. ............................................. 49 Scheme 2-2: Synthesis of kotalanol 4. ...................................................................... 50 Scheme 2-3: Retrosynthetic analysis. ....................................................................... 51 Scheme 2-4: Synthesis of the diols 18 and 19. ......................................................... 52 Scheme 2-5: Synthesis of the cyclic sulfates 12 and 22. .......................................... 53 Scheme 2-6: Coupling reactions. .............................................................................. 54 Scheme 3-1: Retrosynthetic analysis. ....................................................................... 89 Scheme 3-2: First attempted synthesis of 8. ............................................................. 89 Scheme 3-3: Second attempted synthesis of 8. ........................................................ 90 Scheme 3-4: Synthesis of compound 8. ................................................................... 91 Scheme 3-5: Synthesis of compound 9. ................................................................... 92 Scheme 4-1: Retrosynthetic analysis. ..................................................................... 114 Scheme 4-2: Synthesis of compounds 7 and 8. ..................................................... 115 Scheme 5-1: Retrosynthetic analysis. ..................................................................... 139 Scheme 5-2: First attempted synthesis of 9. ........................................................... 140 Scheme 5-3: Second attempted synthesis of 9. ..................................................... 141 Scheme 5-4: Synthesis of compound 9. ................................................................. 143 Scheme 7-1: Retrosynthetic analysis. ..................................................................... 188 Scheme 7-2: Synthesis of benzyl 6-O-trifluoromethanesulfonyl-D-
glucopyranoside derivatives 17 and 22, with benzylated maltose units at C-2 or C-4. .............................................................. 190
Scheme 7-3: Coupling reactions to give sulfonium-ions. ........................................ 191 Scheme 7-4: Synthesis of compounds 10 and 11. ................................................. 193 Scheme 8-1: Proposed synthetic route for target compound 12. ............................ 237

xiv
LIST OF TABLES
Table 1-1: Comparison of inhibitory activities (IC50 in µM) of naturally-occurring sulfonium-ion glucosidase inhibitors 44-46, 48 and 49 against rat intestinal α-glucosidases. ............................................................................... 33
Table 3-1: Experimentally determined Ki values (nM) of compounds 2-5, 8 and 9.. ........ 87 Table 4-1: Experimentally determined Ki values (µM) of compounds 5-8 ...................... 117 Table 6-1: IC50 (µM) against disaccharidases. ............................................................... 172 Table 6-2: Experimentally determined Ki values (µM) of 3 and 7. ................................. 175 Table 7-1: Comparison of inhibition profiles against MGAM and SI subunits, Ki
(µM) .............................................................................................................. 198

xv
ABBREVIATIONS
Å Ångström, 10-10 m
AcOH acetic acid
aq aqueous
Ala alanine
Ar aromatic
B boat
Bn benzyl
br broad
C chair
c concentration
Calcd calculated
CAZy carbohydrate-active enzyme database
COSY correlation spectroscopy
ct Carboxyl terminal
ctMGAM C-terminal domain of MGAM
ctMGAM-N2 C-terminal domain of MGAM, isoform N2
ctMGAM-N20 C-terminal domain of MGAM, isoform N20
ctSI C-terminal domain of SI
d doublet

xvi
dd doublet of doublets
ddd doublet of doublets of doublets
dt doublet of triplets
DMF N, N-dimethylformamide
DAB 1,4-dideoxy-1,4-imino-D-arabinitol
DGDP 2,5-dideoxy-2,5-imino-D-glucitol
DMDP 2,5-dideoxy-2,5-imino-D-mannitol
E envelope
F575 phenylalanine575
EtOAc ethyl acetate
GH glycoside hydrolase
Glu glutamic acid
h hour
H half chair
HFIP 1,1,1,3,3,3-hexafluoro-2-propanol
HRMS high resolution mass spectrometry
HSQC heteronuclear single quantum coherence
IC50 concentration required to inhibit 50% of the enzyme activity
J coupling constant in Hertz
KIE kinetic isotope effect
Ki inhibition constant
Km Michaelis constant

xvii
Kmobs Km observed in presence of inhibitor
Leu leucine
m multiplet
Me methyl
MeOH methanol
MGAM maltase-glucoamylase
NMR nuclear magnetic resonance
NOESY nuclear Overhauser effect spectroscopy
nt amino terminal
ntMGAM N-terminal domain of MGAM
ntSI N-terminal domain of SI
Ph phenyl
PMB para-methoxybenzyl
pr propyl
psi lb/inch2
s singlet
SI sucrase-isomaltase
t triplet
TFA trifluoroacetic acid
THF tetrahydrofuran
Thr threonine
TLC thin layer chromatography

xviii
Trp tryptophan
TS transition state
Tyr tyrosine
Tris tris(hydroxymethyl)-aminomethane
V max enzyme saturated velocity
Val valine
W406 tryptophan406
Y 299 tyrosine299

1
CHAPTER 1: GENERAL INTRODUCTION
1.1 Carbohydrates
Carbohydrates are the major source of metabolic energy in the modern
human diet. During the nineteenth century carbohydrates were defined as
molecules made of carbon, oxygen and hydrogen atoms, with the general
formula Cn(H2O)n. However, this definition has been modified to include
derivatives of carbohydrates as well as nitrogen containing carbohydrates that do
not fit into this formula. Today in general, polyhydroxy aldehydes or ketones,
alcohols, acids, their simple analogues and their heterocyclic analogues are also
considered to be carbohydrates.
In the modern human diet, carbohydrates are found in many food sources
such as cereals, fruits and vegetables. Plants, the main source of carbohydrates,
use water and carbon dioxide in the photosynthesis process for the production of
carbohydrates. The most known of the carbohydrate family is glucose (1, Figure
1-1). A single carbohydrate molecules such as glucose (1), is called a
monosaccharide.
When a monosaccharide is covalently linked to another carbohydrate
molecule, it is called a disaccharide. The most common disaccharides are
lactose (2, Figure 1-1), and sucrose (3, Figure 1-1). Lactose (milk sugar) (2), is
the primary source of carbohydrates for nursing infants, and sucrose (table

2
sugar) (3) is found mainly in fruits, vegetables or as an added sweetener for food
and beverages.1
Figure 1-1: Glucose (1), lactose (2), sucrose (3).
Polysaccharides are composed of repeating units of either mono- or di-
saccharides, and are also called glycans.
Carbohydrates can be classified as digestible or non-digestible,
nutritionally.2,3 Digestible carbohydrates include starch and simple mono and
disaccharide sugars. Disaccharides and larger molecules from this class can
break down into their monosaccharide components that can be absorbed in the
upper intestinal tract. Non-digestible carbohydrates such as fibers, inulin and
polyols are resistant to digestion by intestinal enzymes but are instead processed
by bacterial enzymes in the colon.
Starch is the most abundant storage polysaccharide and a major source of
metabolic energy in plants. Starch has been used by humans, even before they
discovered how to write. The Egyptians used it to bake their bread 5000 years
ago and to glue papyrus despite a lack of knowledge of the fermentation
Glucose (1) Lactose (milk sugar, 2)
O
OHOHHO
OHHO
O
OHOHO
OHHO
O
OHHO
OH
HO
O
OHOH
OH
O
OH
O
HO
HO
OH
OH
Sucrose (table sugar, 3)

3
process. Persians and Indians used it to make dishes like wheat halva. Rice
starch as surface treatment of paper has been used in paper production in China,
from 700 AD onwards. Starch is made up of two main structural components:
amylose, a long linear chain joined by α-1,4-linked glucose units and
amylopectin, a larger branched molecule with linear α-1,4-linked glucose chains
with approximately 5% having additional α-1,6-linked branch points (Figure 1-2).4
In contrast to the digestion of simpler carbohydrates such as sucrose and lactose
that require the activities of sucrase and lactase, respectively, starch digestion
requires the action of six enzymes. This is discussed further in section 1.3. The
multiplicity of animal starch-digesting enzymes mirrors the multiplicity of the
starch synthetic enzymes of plants.5
Figure 1-2: Components of starch: amylose and amylopectin.
Amylose
Amylopectin
α-1,4α-1,6

4
Sugar alcohols are another group of carbohydrates which have been used
in recent years as part of “sugar-free” and low-calorie diets. Sugar alcohols such
as sorbitol, xylitol and maltitol are reduced forms of sugars, naturally found in
some fruits and vegetables, and can be used in place of sucrose to sweeten
foods. They are partially hydrolyzed by intestinal enzymes and thus are
incompletely absorbed into the bloodstream.
1.2 Carbohydrates in health and disease
In order to meet the needs of humans for energy produced from glucose
oxidation, carbohydrate rich diets are important.6 The rapid change in diet and
lifestyle of humans, has not occurred without side effects.
In the diet of early humans, when an excess of carbohydrates was
available their storage by conversion to fats was important to provide energy
requirements during times of low food availability.7 However, the present rapid
change in patterns of carbohydrate consumption by humans involves ingestion of
large quantities of carbohydrates in short periods of time.7
In many cases, the energy intake exceeds the requirement, leading to the
storage of carbohydrates in the form of body fat and causing development of
obesity. 8 In addition, high intake of carbohydrates can result the development of
type-2 diabetes. Blood glucose levels are tightly regulated through the secretion
of insulin from β-cells of the pancreas, which, in turn, stimulates uptake of
glucose into muscle cells. The digestion and absorption of large quantities of
carbohydrates, imposes pressure on β-cells for secretion of insulin. In individuals,

5
this chronic stress has been implicated as a damaging factor for pancreatic β-
cells and cellular response mechanisms that contribute to the development of
type-2 diabetes.9 An approach to control high blood sugar levels, is to delay
intestinal glucose absorption through the inhibition of starch-digesting enzymes
with α-glucosidase inhibitors. This is further discussed in Section 1.6. To
understand and manipulate the physiologic responses of the human organism to
carbohydrate feeding, studies to identify the multiple enzyme/starch (as the
highest consumption by the human population) interactions and the glucosidic
activities of the human gastrointestinal tract are necessary.
1.3 Starch digesting enzymes
The starch digesting enzymes belong to the class of enzymes known as
Glycoside Hydrolases (GH). Glycoside hydrolases, also known as glycosidases,
are a prevalent class of enzymes that cleave the glycosidic bond between two
carbohydrate molecules, one of the strongest bonds found in natural polymers.
These enzymes are capable of breaking glycosidic bonds 1017 times faster than
the uncatalyzed reaction.10 The mechanism of action of glycosidases will be
discussed in Section 1.4.
Depending on their point of action in a carbohydrate chain, glycosidases
are classified as endo- or exohydrolases: endohydrolases hydrolyze internal
glycosidic linkages of a carbohydrate chain to give smaller chains, whereas,
exohydrolases cleave the glycosidic linkage at the non-reducing end of a glycan
structure, releasing monomer units.11,12

6
In humans, four different enzymes mediate the digestion of ingested
carbohydrates. First two endo-acting glucosidases, salivary and pancreatic α-
amylases, break down the complex starch molecules into smaller linear and
branched oligomers (limit dextrins) which are then further hydrolyzed into glucose
by two small-intestinal, brush-border exohydrolases, maltase-glucoamylase
(MGAM) and sucrase-isomaltase (SI); glucose is then absorbed into the
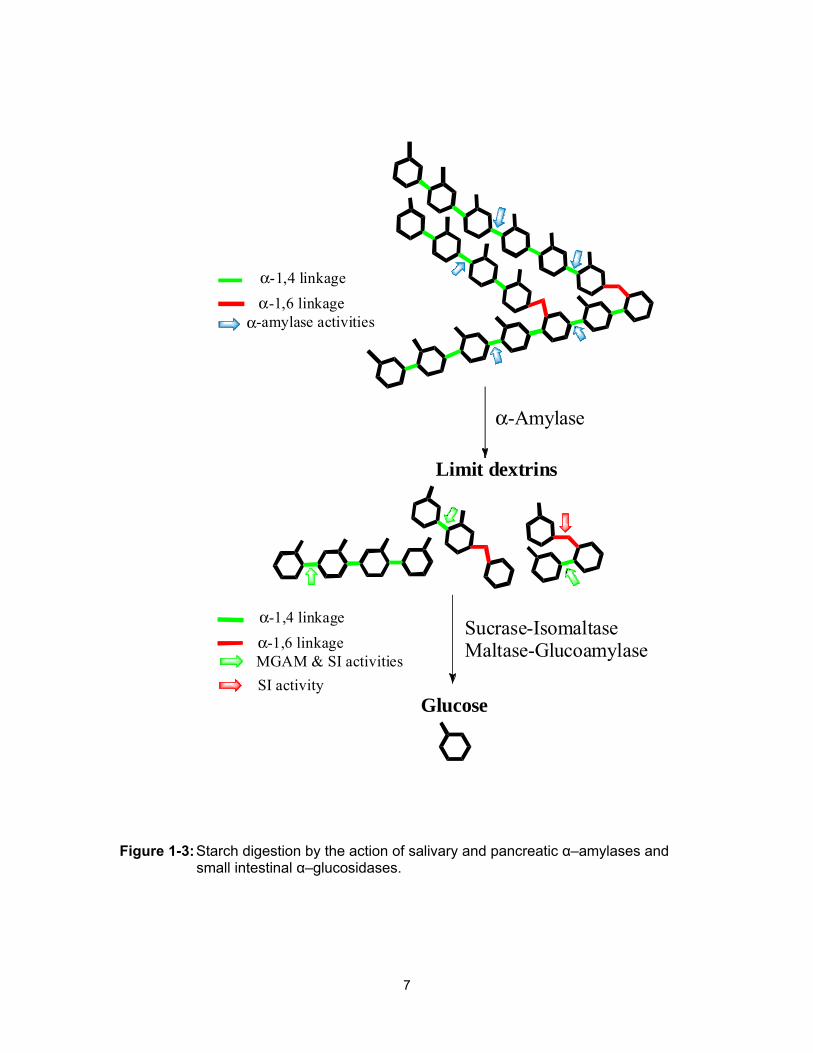
bloodstream (Figure 1-3).
7
Figure 1-3: Starch digestion by the action of salivary and pancreatic α–amylases and small intestinal α–glucosidases.
α-Amylase
Limit dextrins
Sucrase-IsomaltaseMaltase-Glucoamylase
Glucose
α-1,4 linkageα-1,6 linkage
α-amylase activities
α-1,4 linkageα-1,6 linkageMGAM & SI activitiesSI activity

8
1.3.1 Glycoside hydrolases
There are currently 125 different GH families, which are defined according
to amino acid sequence similarity, according to the “Carbohydrate Active Enzyme
database (CAZY).11 Salivary and pancreatic α–amylases belong to GH13
whereas, two small-intestinal, brush-border maltase-glucoamylase (MGAM) and
sucrase-isomaltase (SI) belong to GH31.12,13
1.3.2 Alpha-amylases
Amylase was the first enzyme identified by biochemists. The enzymes
require Ca2+ and Cl− ions to display their full activity.14,15 After birth, full production
of amylases occur as a weaning adaptation to carbohydrate feeding, and this
stimulus remains during life as the primary stimulus leading to the synthesis,
processing, and secretion of pancreatic α-amylase and other digestive enzymes.
GH13 salivary and pancreatic α-amylases are believed to catalyze the hydrolysis
of starch via the retaining mechanism (discussed in Section 1.4.1) and cleave the
internal α-1,4 bonds of amylose and amylopectin (Figure 1-3).
1.3.3 Brush-border hydrolases
For effective release of free glucose, glucose oligomers resulting from the
amylase activities have to be further hydrolyzed by four exoglucosidic activities of
the small intestine. These activities consist of the enzymes sucrase-isomaltase
(SI) and maltase-glucoamylase (MGAM), each composed of two subunits
containing catalytic sites that act on the non-reducing ends of linear glucose
oligomers, with substantial release of free glucose monomers (Figure 1-3).16

9
MGAM and SI share the common ancestry therefore, they share many similar
features. The individual MGAM and SI domains were referred to as either N-
terminal (ntSI and ntMGAM) for the domain closest to the membrane anchor or
C-terminal (ctSI and ctMGAM) for the luminal domain to avoid the confusion of
the associated domain activities (Figure 1-4).
Figure 1-4: Pictorial representation of brush-border membrane-bound MGAM and SI.
MGAM is less abundant than SI and comprises about 2% of brush-border
membrane proteins.13 MGAM has very little activity for starch itself but shows
greatly enhanced hydrolytic activities with α-amylase pre-treated starch.17

10
The four mucosal maltase enzymes are referred to as α-glucosidase
activities because they hydrolyze the nonreducing end of all α-1,4-glucose
oligomers to free glucose. In addition, the isomaltase subunit of SI also shows α-
1,6-glucosidic activity that cleaves the linkages present in the branching points of
intermediate branched oligomers and isomaltose (Figure 1-3). To understand the
structural basis for differential substrate specificity observed between ntMGAM
and ntSI, their active sites were compared.
1.3.3.1 Crystal structures of brush-border hydrolases
Thus far, of the four α-glucosidase activities only the crystal structures of
ntMGAM and ntSI have been solved.18,19
From kinetic studies, it was observed that maltose (Glu-α-(1,4)-Glu) could
be hydrolyzed by both ntMGAM and ntSI whereas isomaltose (Glu-α-(1,6)-Glu)
could only be hydrolyzed efficiently by ntSI. The different substrate specificities of
ntMGAM and ntSI seem consistent with their roles in terminal starch digestion.19
The active site of ntMGAM and ntSI were found to be composed of a
substrate-binding pocket comprising –1 and +1 subsites.18,20 Substrates bind to
the pocket via their non-reducing end, with the non-reducing sugar ring
interacting with the –1 subsite and the reducing ring interacting with the +1
subsite. Substrate cleavage occurs between –1 and +1 subsites, following a
catalytic mechanism that results in retention of configuration at the anomeric
center (discussed in Section 1.4.1).

11
Structural superposition of the ntMGAM and ntSI kotalanol-bound
structures (Section 1.6.4) revealed identical conservation of -1 subsite residues.
Therefore, substrate discrimination is most likely mediated by the +1 subsite and
there are accordingly structural differences observed between the ntSI and
ntMGAM in this region. A notable structural difference between ntMGAM and ntSI
subsite architecture includes substitution of smaller residues in ntMGAM (Thr205,
Ala576, Tyr299 and Thr204) by larger residues in ntSI (Leu233, Val605, Trp327
and Gln232) that results in a wide and open ntMGAM +1 subsite compared to the
narrow groove ntSI +1 subsite (Figure 1-5).19
Figure 1-5: Surface representation of the ntSI and ntMGAM active sites (Reproduced from J. Biol. Chem. 2010, 285, 17763–17770. © the American Society for Biochemistry and Molecular Biology).
It seems counterintuitive that the narrow +1 subsite of ntSI would
accommodate both its α-1,4 and α-1,6 substrates, whereas a wider +1 subsite in
ntMGAM would account for specificity for the α-1,4 substrate. However, perhaps
the substrate with α-1,6 branch points requires constraining prior to hydrolysis.

12
Based on sequence comparison of the ntMGAM and SI domains, in ntSI Trp327
may be important in conferring α-1,6 specificity since it is conserved as Tyr in
ntMGAM, ctMGAM and ctSI.20
1.3.1 Disaccharidase deficiency in heath and disease
In the three-month-old human embryo, all of the intestinal disaccharidases
are already active.21 The α-disaccharidase activities reach the normal adult level
in the sixth to seventh month of fetal life. The only exception is maltase, which is
still low in the newborn. But carbohydrate malabsorption is not rare in children.
The symptoms vary and are dependent on diet and age. The common symptoms
are abdominal pain and watery diarrhea especially for lactase deficiency and
congenital sucrase-isomaltase deficiency (CSID) due to the osmotic force of the
undigested sugar that attracts fluid into the small intestinal lumen. Intraluminal
fluid accumulation increases peristalsis and decreases small intestinal transit
time.22, 23 The symptoms of maltase-glucoamylase (MGAM) deficiency are poorly
defined. Glucoamylase deficiency may also cause chronic diarrhea in young
children.22 The osmotic force of starch is less than that of sucrose or lactose
because of its larger molecular weight.24
Congenital sucrase-isomaltase deficiency (CSID) is characterized by
absence or deficiency of the mucosal sucrase-isomaltase enzyme. Specific
diagnosis requires upper gastrointestinal biopsy. Recently, 13C-sucrose breath
tests have been used as a safe, simple and non-invasive method of CSID
diagnosis. In simple terms, 13C-sucrose breath tests involve the oral ingestion of
labelled 13C-glucose and 13C-sucrose as substrates to overnight-fasting patients

13
on two separate days, followed by collecting breath samples. These 13C-labeled
sugars are cleaved by the action of the specific glycosidases. Further oxidation
gives 13C-enriched 13CO2 which is excreted in a patient's breath. 13CO2 breath
enrichments are assayed using an infrared spectrometer. Sucrose digestion and
oxidation are calculated as a mean percent coefficient of glucose oxidation
averaged between 30 and 90 minutes.25
CSID, also known as disaccharide intolerance, in patients is relieved if
sucrose is removed from the diet or an oral supplement of sucrase enzyme is fed
with the sugar.
1.4 Glycosidase mechanism of action
Glycosidase mediated hydrolysis can occur via two main mechanisms:
retaining or inverting. In a retaining glycosidase-catalyzed reaction, the hydrolysis
product will have retention of configuration at the anomeric carbon, whereas the
product from an inverting glycosidase-catalyzed reaction will have inversion of
configuration at the anomeric center. The sugar substrate can be a five-
membered ring (furanose) or a six-membered ring (pyranose), thus leading to the
classification of furanoside or pyranoside hydrolases.26 In 1953, Koshland
proposed the mechanism for these two main groups of glycosidases which are
still widely supported.27
1.4.1 Mechanism of retaining glycosidases
This mechanism is illustrated using α-glycosidase as an example (Scheme
1-1). In this mechanism, glycosidic bond breakage occurs via the double bond-

14
displacement involving the formation and subsequent hydrolysis of a glycosyl-
enzyme intermediate. The two carboxyl groups involved in the mechanism are
5.5 Å apart. 28-30 In the first step, one of the two carboxylic acid residues acts as
a general acid catalyst and protonates the aglycon to make it a better leaving
group, whereas the second carboxylate residue acts as a nucleophile, forming
the glycosyl-enzyme intermediate. In the second step, the carboxylate side chain
acts as a general base to assist deprotonation of an incoming water molecule,
which attacks at the anomeric center of the glycosyl-enzyme intermediate,
liberating the hydrolyzed product with net retention of configuration (α). Both
steps, the formation of the glycosyl-enzyme intermediate and its conversion into
product, occurs via oxacarbenium-ion like transition states as shown below
(Scheme 1-1).

15
Scheme 1-1: Proposed mechanism for retaining glycosidases.
1.4.2 Mechanism of inverting glycosidases
This mechanism is illustrated using a β-glycosidase as an example
(Scheme 1-2). In this mechanism, the hydrolysis reaction takes place via a single
displacement. The two carboxyl groups involved in the mechanism are suitably
placed (10.5 Å apart on average),28-30 allowing for the substrate and water
molecules to bind between them (Scheme 1-2). One carboxyl group serves as a
OHOHO
OR
OH
OH
O O
δ+
δ−
H
OO OHOHO
OR
OH
O O
δ+
H
OO
HO
δ+
δ−
δ−
++
OHOHO
OH
OH
O O
ROH
O
O
glycosyl-enzyme intermediateOHO
HO
OH
OH
O O
δ−
H
OO
HO
δ+
δ−
δ−
++
OHOHO
OH
OH
OH
HO O
OO
oxacarbenium-ion like transition state
oxacarbenium-ion like transition state
nucleophile
general acid
general base
HOH

16
general acid and protonates the aglycon, the other residue acts as a general
base directing a water molecule to attack at the anomeric carbon, forming the
product with inversion of configuration (α) at the anomeric center. This catalytic
mechanism also proceeds through an oxacarbenium-ion like transition state
(Scheme 1-2).
Scheme 1-2: Proposed mechanism for inverting glycosidases.
1.5 Transition-state mimics
Both inverting and retaining glycosidase enzymes mechanisms involve a
transition state (TS) with substantial oxacarbenium ion-like character (Figure 1-
6).31,32 The positive charge on the endocyclic oxygen atom and bond between
this oxygen and the anomeric carbon atom has partial double bond character
(Figure 1-6).
OHOHO
OH
OH
O O
δ−
H
OOOHO
HO
O
OH
O O
δ−
H
OO
HO
δ+
δ−
δ−
++
oxacarbenium-ion like transition state
ORH
O H
δ+
H
Oδ+
R
HOHO
HO
OH
OH
HO O
OO
HO
ROH
general acid
general base

17
Figure 1-6: Transition state proposed for glycosidase catalyzed reactions.
Formation of the oxacarbenium ion has been supported by kinetic isotope
effect (KIE) measurements which indicated various degrees of sp2 character at
the anomeric carbon.33
In enzyme-catalyzed reactions, the transition state is stabilized by
electrostatic and hydrophobic interactions with the enzyme active site. These
interactions are optimized at the transition state so that the activation energy for
the enzyme-catalyzed reactions is less than that of the non-catalyzed reaction.34
Therefore, a highly effective inhibitor for these enzymes can be a stable molecule
which can mimic both charge and shape of the oxacarbenuim-ion transition state.
Design efforts focusing on mimicking the assumed geometry of the
transition state of the oxacarbenium-ion, in the case of a pyranosyl cation (six-
membered ring glycosyl cation) have focused on either half chair (4H3 or 3H4) or
boat conformations (2,5B or B2,5) which enable the C-1, C-2, O-5 and C-5 atoms
to be in a coplanar arrangement, as shown below (Figure 1-7).35
OHOHO
O
OH
δ−
H
HO
δ+
++
H
O
δ+
R
R
δ+
δ−

18
Figure 1-7: Proposed half chair and boat conformations of pyranosyl cations in glycosidase-mediated hydrolysis reactions.
In the case of the furanosyl cation (five-membered ring glycosyl cation),
the ring probably possesses an envelope conformation in which the C-1, C-2, O-
4 and C-4 atoms adopt a coplanar arrangement and hence, the furanose ring is
presumed to adopt an envelope conformation (3E or E3), as shown below for the
ribofuranosyl cation (Figure 1-8).35
Figure 1-8: Proposed envelope conformations of furanosyl cations in glycosidase mediated-hydrolysis reactions.
Designs focusing on charge have mimicked charge build up at three
different positions, the exocyclic oxygen, endocyclic oxygen, and anomeric
OHO
OH
OH
OH1
2
3
4
5
OHO25
OH
OH
HO
3
4
1
3H4
O OHOH
OH
HO
OOH
OHHO
HO
2,5B B2,5
12
34
5 1
2345
4H3
O
OH
HO
HO
O
OH
OHOH3E E3
12
3
4
123
4

19
center. Some inhibitors mimic charge by protonation at the exocyclic oxygen
which is typical of an early transition state and resembles closely the conjugate
acid of the glycoside 4 (Figure 1-9); others have charge build up at the endocyclic
oxygen atom along with its resonance form which is typical of a late transition
state and resemble more closely the resonance structures of an oxacarbenium-
ion intermediate 5 and 6, as shown below (Figure 1-9).36
Figure 1-9: Positive charge build-up at the exocyclic oxygen, endocyclic oxygen, and the anomeric carbon during glycosidase-mediated hydrolysis reactions.
1.6 Glycosidase inhibitors
Molecules that can inhibit the activity of an enzyme are very important in
controlling many biological activities. Glycosidase inhibitors have many potential
therapeutic applications in the treatment of diseases such as diabetes, cancer
and viral infections.37
The number of glycosidase inhibitors is continually growing and it is
outside the scope of this thesis to review them all. Instead, the general features
representative of naturally-occurring glucosidase inhibitors will be described.
OOH
HOHO
HO
4
OH
OOH
HOHO
HO
OOH
HOHO
HO
5 6R

20
1.6.1 Iminosugars
Since the discovery of nojirimycin (12) in 1966 (Figure 1-11),38 the most
popular way of designing glycosidase inhibitors has been a nitrogen atom in the
saccharide ring. Iminosugar-based glycosidase inhibitors have been the subject
of intense study,39 because of their profound effect on glycosidases.
Iminosugars (aza sugars) are moderately basic and are believed to be
protonated in the endocyclic nitrogen atom under physiological conditions;
therefore, they might mimic the positive charge on the ring oxygen at the
transition state, but not the flattened conformation.40,41,36 Alternatively they may
just bind electrostatically to carboxylate residues in the active site as ground-
state analogues.
Iminosugars can generally be categorized into five major structural
classes, namely piperidines (7), pyrrolidines (8), pyrrolizidines (9), indolizidines
(10), and nortropanes (11) (Figure 1-10).
Figure 1-10: Skeletal frameworks most commonly found in naturally-occurring iminosugar glycosidase inhibitors and their synthetic derivatives.
N
N
7
10
N N
N
8 9
11

21
1.6.1.1 Piperidines
Polyhydroxylated piperidines constitute the largest subgroup of α-
glycosidase inhibitors. Nojirimycin (12) was the first natural saccharide isolated
from several strains of Bacillus, Streptomyces, and mulberry tree leaves, and
was identified as an antibiotic. Although capable of inhibiting α- and β-
glucosidases, the compound is relatively unstable.42 Therefore, reduction of
nojirimycin results in a more stable compound, 1- deoxynojirimycin (13), which
has been shown to inhibit glucosidases in a reversible and competitive manner
(Figure 1-11).39
Isofagomine (14) is another example of the same family with about 440
times more inhibitory activity against β-glucosidases compared to 1-
deoxynojirimycin, but is only a moderate inhibitor of α-glucosidases (Figure 1-
11).43
Figure 1-11: Nojirimycin (12), 1-deoxynojirimycin (13), and isofagomine (14).
1.6.1.2 Pyrrolidines
Polyhydroxylated pyrrolidine alkaloids, including 2,5-dideoxy-2,5-imino-D-
mannitol (DMDP, 15), 2,5-dideoxy-2,5-imino-D-glucitol (DGDP, 16), 1,4-dideoxy-
N
OHOHHO
OH
HN
OHOHHO
OH
HHO
Nojirimycin (12) 1-Deoxynojirimycin (13)
NH
OH
HOH2C
HO
Isofagomine (14)
22
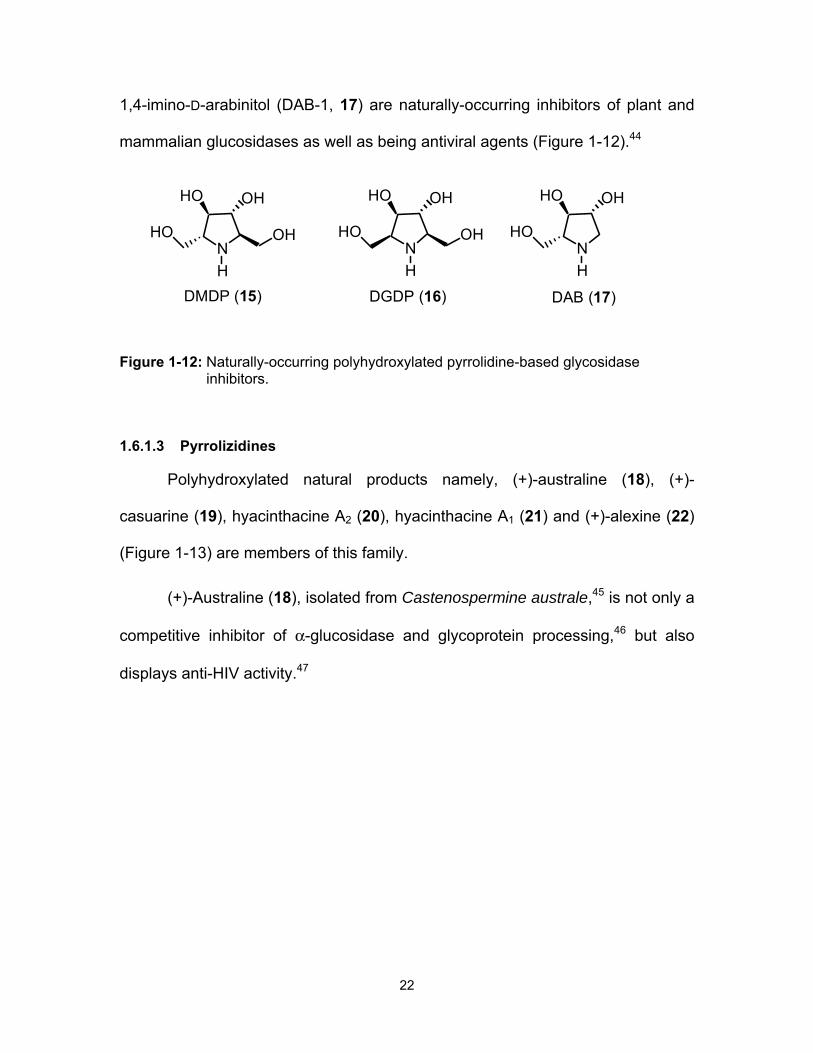
1,4-imino-D-arabinitol (DAB-1, 17) are naturally-occurring inhibitors of plant and
mammalian glucosidases as well as being antiviral agents (Figure 1-12).44
Figure 1-12: Naturally-occurring polyhydroxylated pyrrolidine-based glycosidase inhibitors.
1.6.1.3 Pyrrolizidines
Polyhydroxylated natural products namely, (+)-australine (18), (+)-
casuarine (19), hyacinthacine A2 (20), hyacinthacine A1 (21) and (+)-alexine (22)
(Figure 1-13) are members of this family.
(+)-Australine (18), isolated from Castenospermine australe,45 is not only a
competitive inhibitor of α-glucosidase and glycoprotein processing,46 but also
displays anti-HIV activity.47
N
OHHO
H
OHHON
OHHO
H
OHHON
OHHO
H
HO
DMDP (15) DGDP (16) DAB (17)

23
Figure 1-13: Structures of selected, naturally-occurring pyrrolizidine alkaloids, which display α-glucosidase inhibitory activities.
1.6.1.4 Indolizidines
The indolizidine alkaloid (+)-castanospermine (23, Figure 1-14) isolated from the
seeds of the Moreton Bay chestnut tree (Castanospermum australe),48 is one
example of this family that has shown potent inhibition of endoplasmic reticulum
α-glucosidase, and intestinal maltase and sucrase.49 Importantly,
castanospermine (23) exhibits activity against a range of human viral pathogens,
including parainfluenza,50 dengue virus,51 HSV-252 and HIV-1.53
Swainsonine (24, Figure 1-14), another example of this group of compounds,
was isolated from Swansona canescens, Astragalus lentiginosus, and Ipomoea
carnea, and is the first alkaloid with strong inhibition (nM) toward α-mannosidase
II.54
N
H OH
OH
HO
OH(+)-Australine (18)
N
H OH
OH
HO
OH(+)-Casuarine (19)
HON
H OH
OH
OH(+)-Hyacinthacine A2 (20)
N
H OH
OH
HO
OH(+)-Alexine (22)
N
H OH
OH
OH(+)-Hyacinthacine A1 (21)

24
Figure 1-14: Castanospermine (23), swainsonine (24).
1.6.1.5 Nortropanes
This class of compounds is called calystegins and is isolated from plants
such as Calystegia sepium, Ipomoea carnea, and Physalis alkekengi var.
francheti. Calystegin A3 (25) and calystegin B1 (26) are examples of this class
that have shown α,β-glucosidase inhibitory activity (Figure 1-15).55
Figure 1-15: Examples of nortropanes (25, 26).
1.6.2 Carbasugars
Carbasugars (or pseudosugars) are carbocyclic analogues of monosaccharides
in which the ring-oxygen atom has been replaced by a methylene group.56 5a-
Carba-α-D-galactopyranose (27) (Figure 1-16)57 was discovered as the first
carbasugar natural product. Carbasugars and aminocyclitols occur more
N
OHHO
OH
HO
H
Castanospermine (23)
N
OHOHH
Swainsonine (24)
OH
NH
OH
HO
OH
OH
Calystegin B1 (26)
NH
OHOH
OH
Calystegin A3 (25)
HO

25
commonly as components of more complex natural products. Examples include
the aminocyclitols valienamine (28), valiolamine (29), validamine (30) and their
derivatives voglibose (31) and acarbose (32), some with potent α-glucosidase
inhibitory activity. Acarbose (32) and voglibose (31), are approved for the clinical
treatment of type-2 diabetes. Voglibose (31) was shown to possess 20 to 30
times more potent α-glucosidase inhibitory activity than acarbose (32),58 with
fewer side effects (Figure 1-16).59
Figure 1-16: Examples of naturally-occurring carbasugar α-glucosidase inhibitors and the antibiotic validamycin.
1.6.3 Marine organosulfates
During the last decade there has been increasing interest in the discovery
of inhibitors from marine organisms.60 In this regard, a number of novel α-
glucosidase inhibitors have been isolated which resemble iminosugars.61
OH
OHOH
HO
HO
27
NH2
OHOH
HO
HO
Valienamine (28)
NH2
OHOH
HO
HO
Valiolamine (29)
OH
NH
OHOH
HO
HO
Voglibose (31)
OH
OH
OH
NH2
OHOH
HO
HO
Validamine (30)
OHN
HO
OOH
H3C
O
OOH
OH
O
HO OH
HO
HOHO
OH
HO
HO
OHAcarbose (32)

26
Penarolides (33 and 34)62 (Figure 1-17) and the schulzeine family (35–37)
(Figure 1-18)63 are members of this group that structurally comprise an alkaloid
or amino acid residue coupled with an O-sulfated long-chain fatty acid through an
amide group.
1.6.3.1 Penoralide sulfates
Penarolide sulfate A1 (33) and A2 (34) are structurally unique 30- and 31-
membered macrolides which include a trisulfated, lipophilic chain encircled by a
proline residue (Figure 1-17).62 Isolated from a marine sponge Penares sp.,
these compounds inhibit α-glucosidase with IC50 values of 1.2 and 1.5 mg/mL,
making them approximately 30–40 times more active than 1-deoxynojirimycin
(13).
Figure 1-17: Structures of organosulfate α-glucosidase inhibitors isolated from marine invertebrates.
OON
O
NaO3SO
OSO3Na
OSO3Na
Penarolide A1 (33)
OON
O
Penarolide A2 (34)
OSO3Na
OSO3NaNaO3SO

27
1.6.3.2 Schulzeines
The schulzeines (35–37), represent a new class of marine alkaloids,
isolated by Fusetani and co-workers from extracts of the marine sponge Penares
schulzei, they show potent α-glucosidase-inhibitory activity (IC50 = 48–170 nM)
(Figure 1-18).61
Figure 1-18: Structures of organosulfate α-glucosidase inhibitors isolated from marine invertebrates.
1.6.4 Sulfonium-sulfate thiosugars
Castanospermine (23) is thought to derive its glycosidase inhibition properties
from an ability to mimic the charge and the shape of the oxacarbenium ion-like
transition state. A requirment for this transition state mimicry is protonation of the
ring nitrogen atom at physiological pH within the active site of the enzyme.
()5
()9NH
NaO3SO
OSO3Na
ON O
HO
OH
OSO3Na
R1 R2
35 (+) schulzeines A R1= H, R2 = Me36 (+) schulzeines C R1=H, R2 = H
()5
()9NH
NaO3SO
OSO3Na
ON O
HO
OH
OSO3Na
37 (-) schulzeines C

28
Additional efforts to design glycosidase inhibitors involved the synthesis of
candidates which carry a permanent positive charge in order to provide the
required electrostatic interactions between inhibitor and the active site. In this
regard, the N-oxide analogue of castanospermine (38, Figure 1-19) has been
synthesized and was shown to be a weak inhibitor of β-glucosidase in
comparison with the parent compound, castanospermine (23).63
For the purpose of establishing such a permanent positive charge,
introduction of a sulfur atom in the castanospermine scaffold was considered
next. Our group reported the synthesis of a bridgehead sulfonium salt analogue
of the indolizidine alkaloid castanospermine (39) as well as its conformational
study, as a model to test the theory that formation of a sulfonium salt carrying a
permanent positive charge might be advantageous in providing the necessary
electrostatic interactions between the inhibitor and the active site carboxylate
residues (Figure 1-19).64,65
Figure 1-19: Structure of an N-oxide analogue of castanospermine and a sulfonium-ion analogue.
The syntheses of bicyclic sulfonium ion 40 and 41, analogues of a
swainsonine (24) and sulfonium compounds 42 and 43 with structures related to
australine (18) has also been reported by our group.66
S ClO4
OHHO
HON
OHHO
HOO
HO
38 39

29
Figure 1-20: Structure of compounds 40-43.
This approach was strongly validated by Yoshikawa’s discovery, beginning
in 1997, of a series of naturally-occurring glucosidase inhibitors that encompass
a zwitterionic sulfonium-sulfate structure 44-49 (Figure 1-21).67
Figure 1-21: Naturally-occurring zwitterionic sulfonium-sulfate glucosidase inhibitors.
S
OHHO
OSO3 OH
OH OH
HO
OHOH
S
OHHO
OSO3 OH
OH OH
HO
OH
S
OHHO
OSO3
OH OH
HO
S
OHHO
OSO3
OH
HO
Kotalanol (45)Salacinol (44)
Ponkoranol (49)Salaprinol (48)
S
OHHO
OH OH
OH OH
HO
OHOH
CH3OSO3
De-O-sulfonated Kotalanol (46)
S
OHHO
OH
OH OH
HOHCO2
De-O-sulfonated Salacinol (47)
S S
S S
OH OH
OH
OH OH
OHOHOH OH
OHH OHHR R
Cl Cl
OTf OTf
40 41
42 43R = H or OH

30
The compounds were Isolated from the aqueous extracts of the roots and
stems of Salacia reticulata and related plant species and were structurally related
in having a 1,4-anhydro-4-thio-D-arabinitol unit and a polyhydroxylated acyclic
chain of varying length.68-70
1.6.4.1 Salacinol
The first of the sulfonium-sulfate inhibitors to be discovered was salacnol,
isolated by Yoshikawa and co-workers in 1997 from the extracts of the dried
roots and stems of Salacia reticulata, known as kotalahimbutu’ in Singhalese, a
large climbing plant found throughout the forests of Southern India and Sri Lanka
(Figure 1-21).68 Aqueous extracts of this plant, have been traditionally used in
Indian medicine for treating type-2 diabetes. Prepared extracts by soaking the
bark and roots in water overnight, have been employed for the treatment of type-
2 diabetes.71
Salacinol (44) shows strong blood glucose level control in rats72 and
inhibits intestinal α-glucosidases such as maltase, sucrase, and isomaltase
(Table 1-1).
1.6.4.2 Kotalanol
In addition to the isolation of salacinol (44), the aqueous extracts of
Salacia reticulata have yielded a number of other bioactive components,
including kotalanol (45),69 which displays more potent α-glucosidase inhibitory
activity than 44 (Table 1-1). From a structural perspective, kotalanol (45) differs
from salacinol (44) in the length of the polyhydroxylated side chain.

31
Although degradation studies69 of kotalanol (45) led to establishment of the
absolute stereochemistry of the 1-deoxy-4-thiopentofuranosyl moiety, the
absolute stereochemistry of the heptitol side chain remained unknown until our
group reported the structural elucidation and total synthesis of this medicinally
important natural product.73
1.6.4.3 De-O-sulfonated kotalanol
In 2008, Ozaki and co-workers reported the isolation of another α-
glucosidase inhibitor from S. reticulata using the bioassay-guided isolation
technique similar to the one used by Yoshikawa et al. and assigned its structure
as the unusual 13-membered cyclic sulfoxide 50 (Figure 1-22).74 Subsequent
reevaluation of Ozaki’s data by Muraoka et al. has led to the conclusion that this
compound is in fact de-O-sulfonated kotalanol (46),70 which Yoshikawa et al. had
previously prepared via the desulfonation of kotalanol.74 This compound was the
most potent inhibitor of rat intestinal α-glucosidase isolated from S. reticulata.
Figure 1-22: Initially proposed structure of a naturally-occurring sulfoxide α-glucosidase inhibitor.
S
HO
HO
HO
OH
OH
OH
OHOH
O
50

32
1.6.4.4 Neosalacinol
Neosalacinol (51) isolated from the medicinal plant Salacia oblanga by
Asano et al.76 On the basis of NMR spectroscopic analysis, mass spectrometry,
and a degradation study, the identity of this compound was established as a de-
O-sulfonated derivative of salacinol, and its structure was formulated as the
sulfonium–alkoxide zwitterion 51 (Figure 1-23).76 Subsequent synthetic studies by
Muraoka et al. have revealed that this is indeed de-O-sulfonated salacinol (47),
and it is not an inner salt.77 The inhibitory activity was found to be the same as
salacinol (44).
Figure 1-23: Initially proposed structure of the naturally-occurring α-glucosidase inhibitor neosalacinol.
1.6.4.5 Salaprinol
Salaprinol (48) is the least structurally complex of the zwitterionic
thiosugar α-glucosidase inhibitors discovered to date,76 isolated from the
methanolic extract of the roots and stems of the Sri Lankan plant Salacia
prinoides.
S
OHHO
O
OH OH
HO
51

33
1.6.4.6 Ponkoranol
Isolated from Salacia reticulata by Kitamura et al.,78 as a structurally
similar zwitterionic sulfonium sulfate. Ponkoranol is a six-carbon polyhydroxylated
side chain homologue of salacinol (44). The compound was initially named
reticulanol, and was then found to occur in Salacia prinoides by Yoshikawa et al.
In common with other members of the thiosugar sulfonium sulfate family,
ponkoranol (49) inhibits maltase, sucrase and isomaltase with IC50 values in the
low micromolar range (Table 1-1).70 Interestingly this compound was synthesized
in our laboratory prior to its isolation.79
Table 1-1: Comparison of inhibitory activities (IC50 in µM) of naturally-occurring sulfonium-ion glucosidase inhibitors 44-46, 48 and 49 against rat intestinal α-glucosidases.a
aValues in parentheses indicate Ki values (µM).
Inhibitor Maltase Sucrase Isomaltase Ref.
Salacinol (44) 5.2 (0.97) 1.6 (0.2) 1.3 (1.1) 75, 80
Kotalanol (45) 7.2 (0.52) 0.75 (0.42) 5.7 (4.2) 75, 80
De-O-sulfonated kotalanol (46) 0.227 (0.11) 0.186 (0.52) 0.099 (0.42) 77
Salaprinol (48) >100 >100 - 75
Ponkoranol (49) 3.2 0.29 2.6 75

34
In this thesis work an alternative route for the synthesis of kotalanol as
well as the design and synthesis of the 6'-epimer of kotalanol, de-O-sulfonated
ponkoranol, its 5'-epimer, and their selenium analogues will be described. The
thesis will also describe the design and synthesis of 3’-O-methylponkoranol and
C-3′- and C-5′-β-O-maltose-extended de-O-sulfonated ponkoranol. These
candidates will be used to probe the active-site requirements of the intestinal α-
glucosidase enzyme specificities of starch-digesting maltase-glucoamylase and
sucrase-isomaltase. Such understanding is intended in the long term to either
use these inhibitors for controlled release of glucose from starch digestion or use
the enzymes themselves as therapeutic agents in individuals lacking these
enzyme activities.
1.7 Thesis overview
This thesis work is presented primarily in journal article style, with Chapter
1 serving as a general introduction, followed by Chapters 2-7 as journal articles,
and Chapter 8 describing conclusions and future work.
Chapter 1, presents an introduction to carbohydrates, related diseases,
glycosidase enzymes, starch-digesting enzymes, the crystal structure of brush
border enzymes, their deficiency in health and disease, and their mechanism of
action. This is followed by a discussion on the transition states of glycosidase-
mediated hydrolysis reactions. Some examples of glucosidase inhibitors which
belong to one of the categories of iminosugars, carbasugars, marine
organosulfates, thiosugars, and sulfonium salts are then presented.

35
Chapter 2 presents the manuscript (Eskandari, R.; Jayakanthan, K.;
Kuntz, D. A.; Rose, D. R.; Pinto B. M. Bioorg. Med. Chem. 2010, 18, 2829-2835)
that describes the design and synthesis of the naturally-occurring glucosidase
inhibitor, kotalanol and its stereoisomer and their inhibitory activities against
recombinant human N-terminal maltase-glucoamylase (ntMGAM).
Chapter 3 presents the manuscript (Eskandari, R.; Kuntz, D. A.; Rose, D.
R.; Pinto, B. M. Org. Lett. 2010, 12, 1632-1635) that describes the synthesis of
de-O-sulfonated ponkoranol and its 5'-epimer and their evaluation as glucosidase
inhibitors against ntMGAM.
Chapter 4 presents the manuscript (Eskandari, R.; Jones, K.; Rose, D. R.;
Pinto, B. M. J. Chem. Soc., Chem. Commun. 2011, 47, 9134-9136) that
describes the synthesis of the selenium analogues of de-O-sulfonated
ponkoranol and their evaluation as glucosidase inhibitors against four human
intestinal glucosidase enzymes maltase-glucoamylase MGAM (ntMGAM,
ctMGAM) and sucrase-isomaltase (ntSI, ctSI).
Chapter 5 presents the manuscript (Eskandari, R.; Jones, K.; Rose, D.
R.; Pinto, B. M. Bioorg. Med. Chem. Lett. 2010, 20, 5686-5689) that describes
the synthesis of 3'-O-methylponkoranol and its evaluation as an inhibitor of
ntMGAM.
Chapter 6 presents the manuscript (Eskandari, R.; Jones, K.; Rose, D. R.;
Pinto, B. M. Bioorg. Med. Chem. Lett. 2011, 21, 6491-6494) that describes
biological evaluation of 3'-O-methylponkoranol against human intestinal

36
glucosidase enzymes, namely ntMGAM, ctMGAM and sucrase-isomaltase (ntSI,
ctSI).
Chapter 7 presents the manuscript (Eskandari, R.; Jones, K.; Reddy K.
R.; Jayakanthan, K.; Chaudet, M.; Rose, D. R.; Pinto, B. M. Chem. Eur. J. 2011,
17, 14817-14825) that describes the synthesis of two C-3′- and C-5′-β-O-
maltose-extended analogues of the naturally-occurring sulfonium ion inhibitor,
de-O-sulfonated ponkoranol. Evaluation of inhibitory activities against ntMGAM,
ctMGAM, ntSI, ctSI is also reported.
In Chapter 8, the general conclusions and future work are presented.

37
1.8 References
1. Robayo-Torres, C. C.; Quezada-Calvillo, R.; Nichols, B. L. Clin.
Gastroenterol. Hepatol. 2006, 4, 276–287.
2. Asp, N. G. Am. J. Clin. Nutr. 1995, 61, 930S-937S.
3. Englyst, H. N.; Kingman, S. M.; Cummings, J. H. Eur. J. Clin. Nutr. 1992, 46, S33-50.
4. Annison, G.; Topping, D. L. Annu. Rev. Nutr. 1994, 14, 297-320.
5. Quezada-Calvillo, R.; Robayo-Torres, C. C.; Ao, Z.; Hamaker, B. R.;
Quaroni, A.; Brayer, G. D.; Sterchi, E. E.; Baker, S. S.; Nichols, B. L. J.
Pediatr. Gastroenterol. Nutr. 2007, 45, 32–43. b) Quezada-Calvillo, R.;
Robayo-Torres, C. C.; Opekun, A. R.; Sen, P.; Ao, Z.; Hamaker, B. R.;
Quaroni, A.; Brayer, G. D.; Wattler, S.; Nehls, M. C.; Sterchi, E. E.;
Nichols, B. L. J. Nutr. 2007, 137, 1725–1733.
6. Chugani, H. T. Prev. Med., 1998, 27, 184-188.
7. Pond, W. G.; Nichols, B. L.; Brown, D. L. Adeqaute Food for All, Culture,
Science, and Technology of Food in the 21st Century, CRC Press, NY,
2009.
8. Cordain, L.; Eaton, S. B.; Sebastian, A.; Mann, N.; Lindeberg, S.; Watkins,
B. A..; O'Keefe, J. H.; Brand-Miller, J. Am. J. Clin. Nutr. 2005, 81(2), 341-
354.
9. Livesey, G. Proc. Nutr. Soc. 2005, 64, 105–113.
10. Yip, V. L.; Withers, S. G. Org. Biomol. Chem. 2004, 2, 2707-2713.
11. Henrissat, B. Biochemistry J. 1991, 280, 309-316.
12. Marks, V.; Flatt, P.; Dobbing, J.; Ed. Springer-Verlag: Berlin Heidelberg,
1989; pp 135-146.

38
13. Van Beers, E. H.; Buller, H. A.; Grand, R. J.; Einerhand, A. W.; Dekker, J.
Crit. Rev. Biochem. Mol. Biol. 1995, 30, 197-262.
14. Brayer, G. D.; Sidhu, G.; Maurus, R.; Rydberg, E. H.; Braun, C.; Wang,
Y.; Nguyen, N. T.; Overall, C. M.; Withers S. G. Biochemistry 2000, 39,
4778–4791.
15. Maurus, R.; Begum, A.; Kuo, H. H.; Racaza, A.; Numao, S.; Andersen, C.;
Tams, J. W.; Vind, J.; Overall, C. M.; Withers, S. G.; Brayer G. D. Protein
Sci. 2005, 14, 743–755.
16. Gray, G. M. J. Nutr. 1992, 122, 172-177. b) Gray, G. M. N. Engl. J. Med.
1975, 292, 1225-1230. c) Fujita, S.; Fuwa, H. J. Nutr. Sci. Vitaminol.
(Tokyo) 1984, 30,135-142. d) Jones, B. J.; Brown, B. E.; Loran, J. S.;
Edgerton, D.; Kennedy J. F.; Stead, J. A.; Silk D. B. A. Gut 1983, 24,
1152-1160.e) Corring, T. Reprod. Nutr. Dev. 1980, 20, 1217-1235.
17. Ao, Z.; Quezada-Calvillo, R.; Sim, L.; Nichols, B. L.; Rose, D. R.; Sterchi,
E. E.; Hamaker, B. R. FEBS Lett. 2007a, 581, 2381-2388.
18. Sim, L.; Jayakanthan, J.; Mohan, S.; Nasi, R.; Johnston, B.D.; Pinto, B. M.;
Rose, D. R. Biochemistry 2010, 49, 443-451.
19. Sim, L.; Willemsma, C.; Mohan, S.; Naim, H. Y.; Pinto, B. M.; Rose, D. R.
J. Biol. Chem. 2010, 285, 17763-17770.
20. Sim, L.; Quezada-Calvillo, R.; Sterchi, E. E.; Nichols B. L.; Rose, D. R. J.
Mol. Biol. 2008, 375, 782-792.
21. Auricchio, S.; Rubino, A.; Murset, G. Pediatrics 1965, 35, 944-954.
22. Enattah, N. S.; Sahi, T.; Savilahti, E.; Terwilliger, J. D.; Peltonen, L.;
Jarvela, I. Nature Genet. 2002, 30, 233–237.
23. Savilahti, E.; Launiala, K.; Kuitunen, P. Arch. Dis. Childhood 1983, 58,
246–252.

39
24. Lebenthal, E.; Khin-Muang, U.; Zheng B. J. Pediatr. 1994, 124, 541–546.
25. Robayo-Torres, C. C.; Opekun, A. R.; Quezada-Calvillo, R.; Villa, X.;
Smith, E. O.; Navarrete, M.; Baker, S. S.; Nichols, B. L. J. Pediatr.
Gastroenterol. Nutr. 2009, 48, 412–418.
26. Sinnott, M. L. Chem. Rev. 1990, 90, 1171-1202.
27. Koshland, D. E. Biol. Rev. 1953, 28, 416-436.
28. McCarter, J. D.; Withers, S. G. Curr. Opin. Struct. Biol. 1994, 4, 885-892.
29. Davies, G.; Henrissat, B. Structure 1995, 3, 853-859.
30. Wang, Q. P.; Graham, R. W.; Trimbur, D.; Warren, R. A. J.; Withers, S. G.
J. Am. Chem. Soc. 1994, 116, 11594-11595.
31. Namchuk, M. N.; Withers, S. G. Biochemistry 1995, 34, 16194-16202.
32. McCarter, J. D.; Adam, M. J.; Withers, S. G. Biochem. J. 1992, 286, 721-
727.
33. Bennet, A. J.; Sinnott, M. L. J. Am. Chem. Soc. 1986, 108, 7287-7294.
34. Meister, A. Adv. Enzymol. 1975, 43, 219-410.
35. Sinnott, M. L. Chem. Rev. 1990, 90, 1171-1202.
36. Lillelund, V. H.; Jensen, H. H.; Liang, X.; Bols, M. Chem. Rev. 2002, 102,
515-553.
37. Holman, R. R.; Cull, C. A.; Turner, R. C. Diabetes Care 1999, 22, 960-964.
(b) Jacob, G. S. Curr. Opin. Struct. Biol. 1995, 5, 605-611. (d) Dwek, R. A.
Chem. Rev. 1996, 96, 683-720.
38. Ishida, N.; Kumagai, K.; Niida, T.; Tsuroka, T.; Yumoto, H. J. Antibiot.
1967, 20, 66-71.
39. Stutz, A. E. Wiley-VCH, New York, 1999, pp. 292-295 (b) Asano, N. Curr.
Top. Med. Chem., 2003, 3, 471-484.

40
40. Sears, P.; Wong, C. H. Angew. Chem. Int. Ed. 1999, 38, 2300-2324.
41. Heightman, T. D.; Vasella, A. T. Angew. Chem. Int. Ed. 1999, 38, 750-
770.
42. Inouye, S. E.; Tsuruoka, Y.; Ito, T.; Niida, T. Tetrahedron 1968, 24, 2125-
2144.
43. Pandey, G.; Kapur, M.; Khan, M. I.; Gaikwad, S. M. Org. Biomol. Chem.
2003, 1, 3321-3326.
44. Yu, C.; Asano, N.; Ikeda, K.; Wang, M.; Butters, T. D.; Wormald, M. R.;
Raymond, A. L. W.; Dwek, A.; Nash R. J.; Fleet, G. W. T. Chem.
Commun. 2004, 1936-1937.
45. Molyneux, R. J.; Benson, M.; Wong, R. Y.; Tropea, J. E.; Elbein, A. D. J.
Nat. Prod. 1988, 51, 1198-1206.
46. Tropea, J. E.; Molyneux, R. J.; Kaushal, G. P.; Pan, Y. T.; Mitchell, M.;
Elbein, A. D. Biochemistry 1989, 28, 2027-2034.
47. Vlietinck, A. J.; De Bruyne, T.; Apers, S.; Pieters, L. A. Planta Med.
1998, 64, 97-109.
48. Hohenschutz, L. D.; Bell, E. A.; Jewess, P. J.; Leworthy, D. P.; Pryce, R.
J.; Arnold, E.; Clardy, J. Phytochemistry 1981, 20, 811-814.(b) Nash, R. J.;
Fellows, L. E.; Dring, J. V.; Stirton, C. H.; Carter, D.; Hegarty, M. P.; Bell,
E. A. Phytochemistry, 1988, 27, 1403-1406.
49. Michael, J. P. Nat. Prod. Rep. 2007, 24, 191-222.
50. Tanaka, Y.; Kato, J.; Kohara, M.; Galinski, M. S. Antiviral Res., 2006, 72,
1-9.
51. Whitby, K.; Pierson, T. C.; Geiss, B.; Lane, K.; Engle, M.; Zhou, Y.; Doms,
R. W.; Diamond, M. S. J. Virol., 2005, 79, 8698-8706.

41
52. Ahmed, S. P.; Nash, R. J.; Bridges, C. G.; Taylor, D. L.; Kang, M. S.;
Porter, E. A.; Tyms, A. S. Biochem. Biophys. Res. Commun. 1995, 208,
267-273.
53. Bridges, C. G.; Brennan, T. M.; Taylor, D. L.; McPherson, M.; Tyms, A. S.
Antiviral Res. 1994, 25, 169-175.
54. Colegate, S. M.; Dorling, P. R.; Huxtable, C. R. Aust. J. Chem. 1979, 32,
2257-2264.
55. Molyneux, R. J.; Pan, Y. T.; Goldmann, A.; Tepfer, D. A.; Elbein, A. D.
Arch. Biochem. Biophys. 1993, 304, 81-88.
56. McCasland, G. E.; Futura, S.; Durham, L. J. J. Org. Chem., 1966, 31,
1516-1521. (b) McCasland, G. E.; Futura, S.; Durham, L. J. J. Org. Chem. 1968, 33, 2835-2841.
57. Miller, T. W.; Arison, B. H.; Albers-Schonberg, G. Biotechnol. Bioeng.
1973, 15, 1075-1080.
58. Yasuda, K.; Shimowada, K.; Uno, M.; Odaka, H.; Adachi, T.; Shihara, N.;
Suzuki, N.; Tamon, A.; Nagashima, K.; Hosokawa, M.; Tsuda, K.; Seino,
Y. Diabetes Res. Clin. Pract. 2003, 59, 113-122.
59. Vichayanrat, A.; Ploybutr, S.; Tunlakit, M.; Watanakejorn, P. Diabetes Res.
Clin. Pract. 2002, 55, 99-103.
60. For selected examples of marine α-glucosidase inhibitors, see: (a) Heo, S.
J.; Hwang, J. Y.; Choi, J. I.; Han, J. S.; Kim, H. J.; Jeon, Y. J. Eur. J.
Pharmacol. 2009, 615, 252-256. (b) Ingavat, N.; Dobereiner, J.;
Wiyakrutta, S.; Mahidol, C.; Ruchirawat, S.; Kittakoop, P. J. Nat. Prod.
2009, 72, 2049-2059. (c) Saludes, J. P.; Lievens, S. C.; Molinski, T. F. J.
Nat. Prod. 2007, 70, 436-638. (d) Nakao, Y.; Uehara, T.; Matsunaga, S.;
Fusetani N.; Van Soest, R. W. J. Nat. Prod. 2002, 65, 922-924.

42
61. Takada, K.; Uehara, T.; Nakao, Y.; Matsunaga, S.; Van Soest R. W. M.;
Fusetani, N. J. Am. Chem. Soc. 2004, 126, 187-193.
62. Nakao, Y.; Maki, T.; Matsunaga, S.; Van Soest R. W. M.; Fusetani, N.
Tetrahedron 2000, 56, 8977-8987.
63. Kajimoto, T.; Liu, K. K. C.; Pederson, R. L.; Zhong, Z. Y.; Ichikawa, Y.;
Porco, J. A.; Wong, C. H. J. Am. Chem. Soc. 1991, 113, 6187–6196.
64. Svansson, L.; Johnston, B. D.; Gu, J. H.; Patrick, B.; Pinto, B. M. J. Am.
Chem. Soc. 2000, 122, 10769-10775.
65. Johnson, M. A.; Jensen, M. T.; Svensson, B.; Pinto, B. M. J. Am. Chem.
Soc. 2003, 125, 5663-5670.
66. (a) Kumar, N. S.; Pinto. B. M. J. Org. Chem. 2006, 71, 1262-1264. (b)
Kumar, N. S.; Pinto. B. M. J. Org. Chem. 2006, 71, 2935-2943. (c) Kumar,
N. S.; Pinto. B. M. Carbohydr. Res. 2006, 341, 1685-1691.
67. Matsuda, H.; Morikawa, T.; Yoshikawa, M. Pure Appl. Chem. 2002, 74,
1301-1308.
68. Yoshikawa, M.; Murakami, T.; Shimada, H.; Matsuda, H.; Yamahara, J.;
Tanabe, G.; Muraoka, O. Tetrahedron Lett. 1997, 38, 8367-8370.
69. Matsuda, H.; Li, Y. H.; Murakami, T.; Matsumura, N.; Yamahara, J.;
Yoshikawa, M. Chem. Pharm. Bull. 1998, 46, 1399-1403.
70. Muraoka, O.; Xie, W. J.; Tanabe, G.; Amer, M. F. A.; Minematsu, T.;
Yoshikawa, M. Tetrahedron Lett. 2008, 49, 7315-7317.
71. Li, Y.; Huang T. H. W.; Yamahara, J. Life Sci. 2008, 82, 1045-1049.
72. Shimoda, H.; Fujimura, T.; Makino, K.; Yoshijima, K.; Naitoh, K.; Ihota, H.;
Miwa, Y. J. Food Hyg. Soc. Jpn. 1999, 40, 198-205.
73. Jayakanthan, K.; Mohan, S.; Pinto, B. M. J. Am. Chem. Soc. 2009, 131,
5621-5626.

43
74. Ozaki, S.; Oe, H.; Kitamura, S. J. Nat. Prod. 2008, 71, 981-984.
75. Yoshikawa, M.; Xu, F.; Nakamura, S.; Wang, T.; Matsuda, H.; Tanabe, G.;
Muraoka, O. Heterocycles 2008, 75, 1397-1405.
76. Minami, Y.; Kuriyama, C.; Ikeda, K.; Kato, A.; Takebayashi, K.; Adachi, I.;
Fleet, G. W. J.; Kettawan, A.; Okamoto, T.; Asano, N. Bioorg. Med.
Chem. 2008, 16, 2734-2740.
77. Tanabe, G.; Xie, W.; Ogawa, A.; Cao, C.; Minematsu, T.; Yoshikawa, M.;
Muraoka, O. Bioorg. Med. Chem. Lett. 2009, 19, 2195-2198.
78. Asada, M.; Kawahara, Y.; Kitamura, S. US Patent 0037870, 2007.
79. Johnston, B. D.; Jensen, H. H.; Pinto, B. M. J. Org. Chem. 2006, 71, 1111-
1118.
80. Matsuda, H.; Yoshikawa, M.; Morikawa, T.; Tanabe, G.; Muraoka, O. J.
Trad. Med. 2005, 22, 14-153.

44
CHAPTER 2: SYNTHESIS OF A BIOLOGICALLY ACTIVE ISOMER OF KOTALANOL, A NATURALLY-OCCURRING
GLUCOSIDASE INHIBITOR
This Chapter comprises the manuscript “Synthesis of a biologically
active isomer of kotalanol, a naturally-occurring glucosidase inhibitor”
which was published in Bioorganic and Medicinal Chemistry (2010, 18, 2829-
2835).
Razieh Eskandari,a Kumarasamy Jayakanthan,a Douglas A. Kuntz,b David R.
Rose,b B. Mario Pintoa
aDepartment of Chemistry, Simon Fraser University, Burnaby, British Columbia,
Canada V5A 1S6
b Department of Biology, University of Waterloo, Waterloo, Ontario,
Canada N2L 3G1

45
Kotalanol is one of the active principles in the aqueous extracts of Salacia
reticulata, a climbing tree shrub native to Sri Lanka and Southern India, used in
the treatment of diabetes. In this Chapter, kotalanol and its 6'-epimer were
synthesized and their glucosidase inhibitory activities against N-terminal maltase-
glucoamylase (ntMGAM) were studied. The thesis author performed all the
experimental synthetic work and the characterization of the compounds and
wrote and edited the manuscript with Dr. B. Mario Pinto. Dr. Jayakanthan
Kumarasamy assisted in the synthetic design. Drs. Douglas A. Kuntz and David
R. Rose performed the enzyme inhibition studies.
Graphical abstract:
O O
OSO3OMOM
OMOM
OMOMS
OPMBPMBO
PMBOS
OHHO
OSO3 OH
OH OH
HO
OHOH
O O
OS O
OOOMOM
OMOM
OMOMS
OPMBPMBO
PMBO +

46
2.1 Keywords
Kotalanol, isomer of kotalanol, D-mannitol, cyclic sulfate, glucosidase
inhibitors, human maltase-glucoamylase, type-2 diabetes.
2.2 Abstract
The syntheses of an isomer of kotalanol, a naturally-occurring glucosidase
inhibitor, and of kotalanol itself are described. The target compounds were
synthesized by nucleophilic attack of PMB-protected 1,4-anhydro-4-thio-D-
arabinitol at the least hindered carbon atom of two 1,3-cyclic sulfates, which were
synthesized from D-mannose. Methoxymethyl ether and isopropylidene were
chosen as protecting groups. The latter group was critical to ensure the facile
deprotection of the coupled products in a one-step sequence to yield kotalanol
and its isomer. The stereoisomer of kotalanol, with the opposite stereochemistry
at the C-6’ stereogenic centre, inhibited the N-terminal catalytic domain of
intestinal human maltase-glucoamylase (ntMGAM) with a Ki value of 0.20 ± 0.02
µM; this compares to a Ki value for kotalanol of 0.19 ± 0.03 µM. The results
indicate that the configuration at C-6’ is inconsequential for inhibitory activity
against this enzyme.
2.3 Introduction
Glycosidases are enzymes that are involved in the catabolism of
glycoproteins and glycoconjugates and the biosynthesis of oligosaccharides.
Disruption in regulation of glycosidases can lead to diseases.1,2 Over the years,
glycosidase inhibitors have received considerable attention in the field of

47
chemical and medicinal research3 because of their effects on quality control,
maturation, transport, secretion of glycoproteins, and cell-cell or cell-virus
recognition processes. This principle has potential for many therapeutic
applications, such as in the treatment of diabetes, cancer and viral infections.1
Bioactive components isolated from medicinal plants that are used in
traditional medicine or folk medicine often provide the lead structures for modern
drug-discovery programs. For example, the large woody climbing plant Salacia
reticulata, known as Kothalahimbutu in Singhalese, is used in traditional medicine
in Sri Lanka and Southern India for treatment of type-2 diabetes.4,5 A person
suffering from diabetes was advised to drink water stored overnight in a mug
carved from Kothalahimbutu wood.6 Several potent glucosidase inhibitors have
been isolated from the water soluble fraction of this plant extract and also other
plants that belong to the Salacia genus such as Salacia chinensis, Salacia
prinoides, and Salacia oblonga which explain, at least in part, the antidiabetic
property of the aqueous extract of this plant.7-9 All these compounds share a
common structural motif that comprises a 1,4-anhydro-4-thio-D-arabinitol and a
polyhydroxylated side chain. So far, five components have been isolated, namely
salaprinol (1),9 salacinol (2),7 ponkoranol (3),9 kotalanol (4),8 and de-O-sulfonated
kotalanol (5)10 (Figure 2-1). The absolute stereostructure for these compounds,
except salacinol, was not determined at the time of isolation, but synthetic work
has led to their stereochemical structure elucidation.11,12

48
S
OHHO
OSO3 OH
OH OH
HO
OHOH
S
OHHO
OSO3 OH
OH OH
HO
OH
S
OHHO
OSO3
OH OH
HOS
OHHO
OSO3
OH
HO
Kotalanol (4)
Salacinol (2) Ponkoranol (3)Salaprinol (1)
S
OHHO
OH OH
OH OH
HO
OHOH
CH3OSO3
De-O-sulfonated kotalanol (5)
Figure 2-1: Components isolated from Salacia species.
The synthesis of kotalanol 4 and its stereoisomer 6 (Figure 2-2) are of
interest here.
S
OHHO
OSO3 OH
OH OH
HO
OHOH
6
Figure 2-2: Kotalanol stereoisomer.

49
Our first attempt employed the reaction of the cyclic sulfates 8 in the
coupling reaction (Scheme 2-1).12 However, attempts to remove the methylene
acetal in the coupled products required forcing conditions and resulted in de-O-
sulfonation (Scheme 2-1).12 We have also reported a successful synthesis of
kotalanol using a cyclic sulfate derived from a naturally-occurring heptitol, D-
perseitol (Scheme 2-2).12
S
OPMBPMBO
PMBO
7
HFIP, K2CO3
S
OPMBPMBO
PMBO
OO
OSO3OBn
OBn
OBn
9
O O
OS O
OO
OBnOBn
OBn
8
+
then MeOH
S
OHHO
OH OH
OH OH
HO
OHOH
CH3OSO3
5
1.0 M BCl3
Scheme 2-1: First attempted synthesis of kotalanol 4.

50
O O O
PMBO OPMB OPMB
O
PhS
O O10
7HFIP, K2CO3
OSO3 O
PMBO OPMB OPMB
O
Ph
S
OPMBPMBO
PMBO
80% TFA 4
11
D-perseitol
Scheme 2-2: Synthesis of kotalanol 4.
However, it was of interest to develop a synthesis of the isomer of
kotalanol 6 in view of the fact that the C-6’ stereoisomer of de-O-sulfonated
kotalanol was just as active an inhibitor as de-O-sulfonated kotalanol 5 itself
against a key intestinal enzyme, human maltase-glucoamylase.13 We report here
a general synthetic route to this isomer 6 and also an alternative synthesis of
kotalanol 4.
2.4 Results and discussion
We chose to replace the problematic methylene acetal group of compounds 8
with an isopropylidene acetal (compound 12) to ensure not only its facile removal
after the coupling reaction but also to maintain some rigidity in the cyclic sulfate.
We chose also to replace the benzyl ethers with methoxymethyl (MOM) ethers,
because the latter can survive the hydrogenolysis conditions required for removal

51
of the benzylidene acetal. The cyclic sulfate 12 could be synthesized from D-
mannitol as shown in the retrosynthetic analysis (Scheme 2-3).
12
O O
OS O OMOM
OMOM
OMOM
OO
O O
OO OMOM
Ph
D-Mannitol
13
14
O O
OO O
Ph
O
Ph
Scheme 2-3: Retrosynthetic analysis.
Thus, the D-mannitol-derived diol 15,14 was protected as the acetonide to
give the C2-symmetric compound 14 in 73% yield. Mild hydrolysis of this
compound using catalytic PTSA in methanol effected the removal of one
benzylidene group to give the corresponding diol in 70% yield based on
recovered starting material. Selective protection of the primary hydroxyl group as
its TBDMS ether followed by sequential protection of the secondary hydroxyl
group as its MOM ether and removal of the TBDMS group with
tetrabutylammonium fluoride (TBAF) gave 17 in 73% yield over three steps.
Treatment of this alcohol with Dess-Martin periodinane provided the aldehyde
which was reacted with methyltriphenylphosphonium bromide to yield the olefinic
product 13 in 61% yield over two steps (Scheme 2-4).
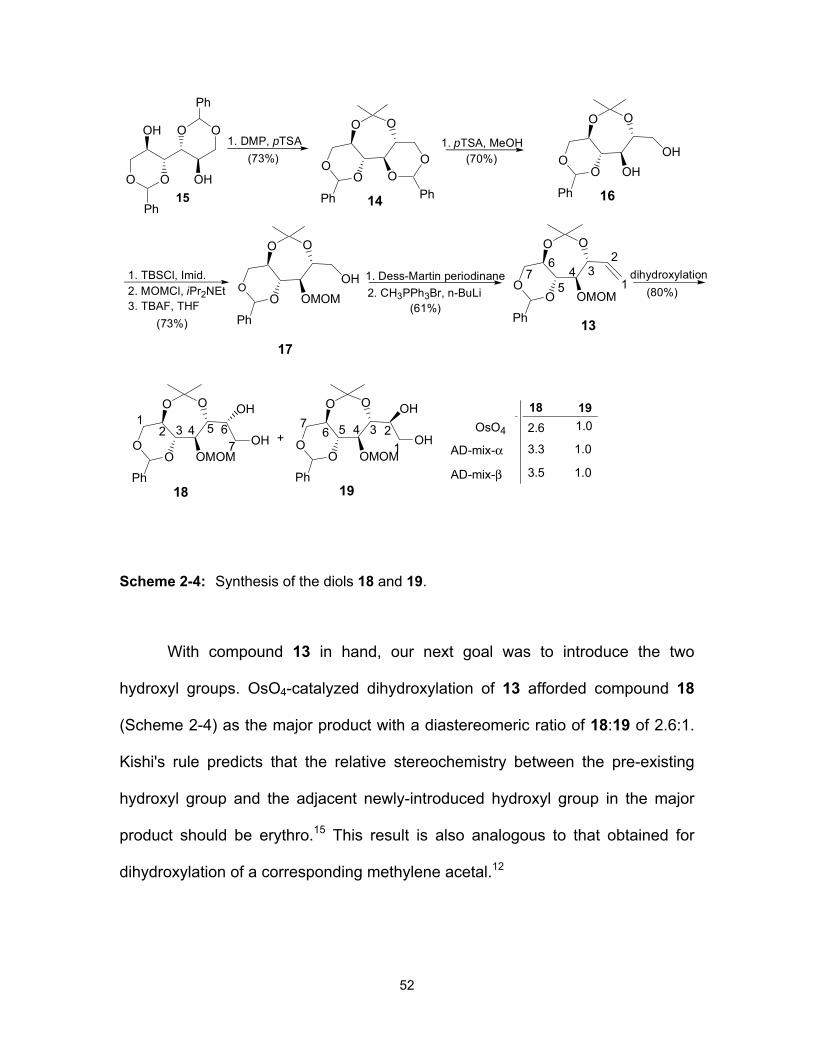
52
O O
OH O
OH
O
Ph
Ph
O O
OO O
Ph
O1. DMP, pTSA 1. pTSA, MeOH
1. TBSCl, Imid.2. MOMCl, iPr2NEt
O O
OO OMOM
Ph
OH
3. TBAF, THF2. CH3PPh3Br, n-BuLi1. Dess-Martin periodinane
15 14
17
(70%)
(73%)(61%)
Ph
(73%)
O O
OO OMOM
Ph 13
O O
OO OMOM
Ph
OH
OH
18
12 3 4 5 6
7
O O
OO OMOM
Ph
OH
OH
19
123456
7+
dihydroxylation(80%)
OsO4
AD-mix-α
AD-mix-β
18 192.6 1.0
3.3
3.5
1.0
1.0
O O
OO OH
Ph
OH
16
1
234
5
67
Scheme 2-4: Synthesis of the diols 18 and 19.
With compound 13 in hand, our next goal was to introduce the two
hydroxyl groups. OsO4-catalyzed dihydroxylation of 13 afforded compound 18
(Scheme 2-4) as the major product with a diastereomeric ratio of 18:19 of 2.6:1.
Kishi's rule predicts that the relative stereochemistry between the pre-existing
hydroxyl group and the adjacent newly-introduced hydroxyl group in the major
product should be erythro.15 This result is also analogous to that obtained for
dihydroxylation of a corresponding methylene acetal.12

53
Interestingly, AD-mix-α and AD-mix-β also afforded compound 18 as a
major product, with a diastereomeric ratio of 3.3:1 and 3.5:1 (determined by 600
MHz 1H NMR), respectively. The unsatisfactory selectivity can be explained by
the steric hindrance imposed by the bicyclic structure, observed previously with a
similar structure.16 The two isomers were separated by column chromatography
and each was converted into its cyclic sulfate 12 or 22 as follows. The hydroxyls
in 18 were protected with MOM groups and the product was subjected to
hydrogenolysis to effect removal of the benzylidene group and to yield the
corresponding diol 20 in 72% yield over 2 steps. The cyclic sulfate 12 was then
obtained by treatment of 20 with thionyl chloride in the presence of triethylamine
to give the mixture of diastereomeric sulfites, followed by their oxidation with
sodium periodate and ruthenium (III) chloride as a catalyst. A similar sequence of
reactions with the diol 19 yielded the cyclic sulfate 22 (Scheme 2-5).
O O
OS O
OO
OMOMOMOM
OMOM
2. Pd(OH)2, MeOH1. SOCl2, Et3N, CH2Cl22. NaIO4, RuCl3 CCl4:CH3CN
O O
HOHO OMOM
OMOM
OMOM
21 22
(75%)(76%)
1. MOMCl, i-Pr2NEt19
O O
OS O
OO
OMOMOMOM
OMOM
2. Pd(OH)2, MeOH
1. SOCl2, Et3N, CH2Cl22. NaIO4, RuCl3 CCl4:CH3CN
O O
HOHO OMOM
OMOM
OMOM18
20 12
(72%)(70%)
1. MOMCl, i-Pr2NEt
Scheme 2-5: Synthesis of the cyclic sulfates 12 and 22.

54
The target compounds were prepared by opening of the cyclic sulfates 12
and 22 by nucleophilic attack of the sulfur atom in 2,3,5-tri-O-p-methoxybenzyl-
1,4-anhydro-4-thio-D-arabinitol 7.11 Reactions were carried out at 72 °C in
1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) containing K2CO317 for 6 days to give
the sulfonium salts 23 and 24 in 65 and 57% yield, respectively. Finally,
deprotection of the coupled products 24 and 23 using aqueous 30%
trifluoroacetic acid (TFA) at 50 °C gave the desired compounds 4 and 6 in 91 and
93% yields, respectively (Scheme 2-6).
12, HFIP,K2CO3 65%
7
22, HFIP,K2CO3 57%
O O
OSO3 OMOM
OMOM
OMOMS
OPMBPMBO
PMBO
O O
OSO3OMOM
OMOM
OMOMS
OPMBPMBO
PMBO
24
23
30% TFA
30% TFAS
OHHO
OSO3 OH
OH OH
HO
OHOH
S
OHHO
OSO3 OH
OH OH
HO
OHOH
4
6
93%
91%
1' 2'
1' 2'
3'
3'
4'
4'5'
5'
6'
6'
Scheme 2-6: Coupling reactions.
Comparison of the 1H and 13C NMR spectra of kotalanol 4 with those
reported13 revealed identical data and served, therefore, to confirm the
stereochemistry at C-6', and, by inference, the stereochemistry at C-2 in each of
18 and 19.

55
Finally, the inhibitory activities of compounds 4 and 6 were examined
against the N-terminal catalytic domain of recombinant human maltase-
glucoamylase (ntMGAM), a critical intestinal glucosidase for processing starch-
derived oligosaccharides into glucose. The stereoisomer of kotalanol 4 inhibited
ntMGAM with a Ki value of 0.20 ± 0.02 µM; this compares to a Ki value for
kotalanol of 0.19 ± 0.03 µM,18 and Ki values of 0.10 ± 0.02 µM and 0.13 ± 0.02
µM for other stereoisomers of 4 with opposite configurations at C-5’ or both C-5’
and C-6’, respectively.16 It is clear, therefore, that the configurations at C-5’ and
C-6’ are not critical for dictating enzyme inhibitory activity against ntMGAM.
2.5 Experimental
2.5.1 General
Optical rotations were measured at 23 °C. 1H and 13C NMR spectra were
recorded at 600 and 150 MHz, respectively. All assignments were confirmed with
the aid of two-dimensional 1H, 1H (COSYDFTP) or 1H, 13C (INVBTP) experiments
using standard pulse programs. Column chromatography was performed with
Silica 60 (230-400 mesh). High resolution mass spectra were obtained by the
electrospray ionization method, using an Agilent 6210 TOF LC/MS high
resolution magnetic sector mass spectrometer.
2.5.2 Enzyme inhibition assays
Compounds 4 and 6 were tested for inhibition of ntMGAM, as previously
described.16

56
2.5.3 Compound characterization data
2.5.3.1 1,3,4,6-Di-O-benzylidene-2,5-O-isopropylidene-D-mannitol (14)
Compound 15 (9.30 g, 26.00 mmol) was dissolved in 2,2-di-
methoxypropane (150 mL), PTSA (1.50 g, 0.3 eq) was added, and the mixture
was rotated on a rotary evaporator at room temperature under reduced pressure
for 1 h. The reaction mixture was quenched by addition of Et3N to pH > 9. The
reaction mixture was concentrated under vacuum to give a white solid which was
dissolved in CHCl3 (200 mL) and washed with water (3×50 mL). The separated
organic layer was dried over Na2SO4, concentrated, and the residue was purified
by column chromatography with EtOAc/Hexanes (1:4) as eluent to afford 14 as a
white solid (7.55 g, 73%). Mp 160-162 °C; [α] 23D = -83°, (c = 1.1, CH2Cl2). 1H
NMR (CDCl3) δ 7.54-7.37 (10H, m, Ar), 5.54 (2H, s, 2CH-Ph), 4.24 (2H, dd, J1a,1b
= 10.8, J2,1 = 5.3 Hz, H-1), 3.95-3.91 (2H, m, H-6a, H-5), 3.84-3.80 (2H, m, H-3,
H-4), 3.74 (2H, t, J1,2 = J2,3 = J5,6b = J6a,6b = 10.5, H-2, H-6b), 1.42 (6H, s, 2Me).
13C NMR (CDCl3) δ 137.5 (CMe2), 129.9-126.2 (m, Ar), 100.7 (CH-Ph), 82.2 (C-3,
C-4), 69.4 (C-1, C-6), 61.7 (C-2, C-5), 24.4 (2Me). HRMS Calcd for C23H27O6
(M+H): 399.1802. Found: 399.1809.
2.5.3.2 1,3-O-Benzylidene-2,5-O-isopropylidene-D-mannitol (16)
To a solution of compound 14 (7.50 g, 18.84 mmol) in MeOH (300 mL),
was added PTSA (300 mg), and the reaction was stirred at room temperature for
30 min. The reaction mixture was then quenched by addition of Et3N to pH > 9,
and the solvent were removed under vacuum to give a solid. The solid was
dissolved in CH2Cl2 (100 mL) and washed with water (50 mL). The organic

57
solution was dried (Na2SO4), concentrated, and the crude product was purified
through a silica column with EtOAc/Hexanes (1:1) as eluent to yield 16 as a foam
(4.1g, 70%). [α] 23D = -15°, (c = 1 , CH2Cl2). 1H NMR (CDCl3) δ 7.42-7.30 (5H, m,
Ar), 5.40 (1H, s, CH-Ph), 4.12 (1H, dd, J1a,1b = 10.8, J1a,2 = 5.5 Hz, H-1a), 3.81
(1H, dd, J6a,6b = 10.9, J6a,5 = 4.3 Hz, H-6a), 3.76-3.72 (2H, m, H-3, H-5), 3.66 (1H,
m, H-6b), 3.60-3.53 (2H, m, H-4, H-1b), 3.43 (1H, t, J1,2 = 8.9 Hz, H-2), 2.23 (2H,
br, 2OH), 1.30 (6H, s, 2Me). 13C NMR (CDCl3) δ 137.3 (CMe2), 129.3- 101.7 (m,
Ar), 101.1 (CH-Ph), 85.2 (C-2), 73.9 (C-4), 70.3 (C-5), 69.3 (C-1), 63.6 (C-6),
61.2 (C-3), 24.8, 24.6 (2Me). HRMS Calcd for C16H23O6 (M+H): 311.1489. Found:
311.1487.
2.5.3.3 1,3-O-Benzylidene-2,5-O-isopropylidene-4-O-methoxymethyl-D-mannitol (17)
To a solution of 16 (6.80 g, 21.93 mmol) in DMF (125 mL) was added
imidazole (4.47g, 65.81 mmol). The reaction was cooled in an ice bath,
TBDMSCl (3.79 g, 24.13 mmol) was added portionwise, and the mixture was
stirred at 0 °C under N2 for 2 h. The reaction was quenched by the addition of ice-
water, and the reaction mixture was extracted with Et2O (3×75 mL). The
combined organic solvents were dried (Na2SO4) and concentrated to give the
crude product which was used directly in the next step without further purification.
The crude product was dissolved in DMF (60 mL), and i-Pr2NEt (26 mL, 150.75
mmol) and MOMCl (5.7 mL, 75.38 mmol) were added. The reaction mixture was
heated at 60 °C overnight, then quenched with ice, and extracted with ether
(3×50 mL). The organic solution was dried (Na2SO4) and concentrated to give a

58
crude product. The crude residue was dissolved in THF (100 mL), TBAF (1.0 M
solution in THF, 13.8 mL, 24.12 mmol) was added, and the reaction mixture was
stirred at room temperature. After 4 h it was concentrated and the residue was
purified by flash chromatography (EtOAc/Hexanes (1:3)) to yield 17 as a white
solid (5.67 g, 73%). Mp 65-67 °C; [α] 23D = +22° (c = 1, MeOH). 1H NMR (CDCl3)
δ 7.49-6.37 (5H, m, Ar), 5.50 (1H, s, CH-Ph), 4.93, 4.73 (2H, 2d, JA,B = 6.4 Hz,
CH2OMe), 4.20 (1H, dd, J1a,1b = 10.9, J1a,2 = 5.5 Hz, H-1a), 3.89-3.79 (4H, m, H-
2, H-5, H-6a,b), 3.74 (1H, t, J3,4 = J5,4 = 8.1 Hz, H-4), 3.69-3.65 (2H, m, H-1b, H-
3), 3.40 (3H, s, OMe), 2.69 (1H, t, J6,OH = 8.5 Hz, OH), 1.41, 1.38 (6H, 2s, 2Me).
13C NMR (CDCl3) δ 137.5 (CMe2), 128.9-101.5 (m, Ar), 100.9 (CH-Ph), 98.6
(CH2-OMe) 85.3 (C-3), 78.2 (C-4), 70.4 (C-5), 69.5 (C-1), 63.1 (C-6), 61.3 (C-2),
56.4 (OMe), 24.7, 24.4 (2Me). HRMS Calcd for C18H27O7 (M+H): 355.1751.
Found: 355.1741.
2.5.3.4 5,7-O-Benzylidene-1,2-dideoxy-3,6-O-isopropylidene-4-O-methoxymethyl-D-manno-hep-1-enitol (13)
Compound 17 (2.60 g, 7.34 mmol) was dissolved in CH2Cl2 (50 mL) and
NaHCO3 (2.77 g, 33.03 mmol) and Dess Martin periodinane (3.73 g, 8.81 mmol)
were added. The reaction mixture was stirred for 2h at room temprature, diluted
with ether (100 mL), and poured into saturated aqueous NaHCO3 (100 mL)
containing a seven fold excess of Na2S2O3. The mixture was stirred to dissolve
the solid, and the ether layer was separated and dried over Na2SO4. The ether
was removed to give the aldehyde that was further dried under high vacuum for 1
h. Methyltriphenylphosphonium bromide (2.99 g, 8.80 mmol) in dry THF (15 mL),

59
was cooled to -78 °C and n-BuLi (n-hexane solution, 14.67 mmol) was added
dropwise under N2. The reaction mixture was stirred at the same temperature for
1h, and a solution of the previously made aldehyde in THF (10 mL) was added.
The resulting mixture was allowed to warm to rt and was stirred overnight. The
reaction was quenched by the addition of acetone (1.5 mL), and the mixture was
extracted with ether (3×100 mL). The combined organic layers were washed with
brine, dried (Na2SO4), and concentrated in vacuo. Chromatographic purification
of the crude product (EtOAc/Hexanes (1:10)) gave 13 as a foam (1.56 g, 61%).
[α] 23D = +4° (c = 0.5, CH2Cl2). 1H NMR (CDCl3) δ 7.50-7.36 (5H, m, Ar), 6.05 (1H,
ddd, J5,6 = 6.1, J6,7b = 10.5, J6,7a = 16.6 Hz, H-6), 5.51 (1H, s, CH-Ph), 5.39 (1H,
ddd, J7b,7a = 17.1, J6,7a = 3.3, J5,7a = 1.5 Hz, H-7a), 5.36 (1H, ddd, J7a,7b = 10.7,
J6,7b = 3.1, J5,7b = 1.5 Hz, H-7b), 5.27, 5.26 (2H, 2d, JA,B = 6.25 Hz, CH2OMe),
4.25 (1H, m, H-5), 4.20 (1H, dd, J1a,1b = 10.8, J1a,2 = 5.4 Hz, H-1a), 3.90 (1H, dt,
J2,3 = 5.4, J2,1 = 9.9 Hz, H-2), 3.68 (2H, m, H-3, H-1b), 3.56 (1H, dd, J3,4 = 8.1 ,
J4,5 = 9.7 Hz, H-4), 3.33 (3H, s, OMe), 1.40, 1.37 (6H, 2s, 2Me). 13C NMR
(CDCl3) δ 137.6 (CMe2), 136.2 (C-6), 128.9-101.3 (m, Ar), 116.8 (C-7), 100.7
(CH-Ph), 97.9 (CH2OMe), 85.5 (C-3), 80.2 (C-4), 71.1 (C-5), 69.6 (C-1), 61.4 (C-
2), 56.4 (OMe), 24.8,24.1 (2Me). HRMS Calcd for C19H26NaO6 (M+Na):
373.1622. Found: 373.1606.
2.5.3.5 1,3-O-Benzylidene-2,5-O-isopropylidene-4-O-methoxymethyl-D-glycero-D-manno-heptitol (18)
To a solution of 13 (2.00 g, 5.71 mmol) in acetone:water (9:1, 6 mL) at rt
were added NMO (N-methylmorpholine-N-oxide) (735 mg, 6.28 mmol) and OsO4

60
(40 mg, 2.5 wt % solution in 2-methyl-2-propanol). The reaction mixture was
stirred at rt for 48 h before it was quenched with a saturated solution of NaHSO3.
After being stirred for an additional 15 min the reaction mixture was extracted
with ethyl acetate and the organic layer was washed with water and brine, dried
(Na2SO4), and concentrated in vacuo. The crude material was purified by column
chromatography on silica gel (MeOH/CH2Cl2 (1:100)) to give 18 (1.27 g, 58%)
and 19 (0.48 g, 22%) as foams. [α] 23D = +5.8° (c = 4.6, MeOH). 1H NMR (MeOD)
δ 7.49-7.36 (5H, m, Ar), 5.54 (1H, s, CH-Ph), 4.82 (1H, s, CH2OMe ), 4.13 (1H,
dd, br, H-1a), 4.00 (1H, br, q, H-6), 3.87-3.77 (3H, m, H-4, H-5, H-2), 3.68-3.55
(4H, H-1b, H-3, H-7a, H-7b), 3.32 (3H, s, OMe), 1.39, 1.34 (6H, 2s, 2Me). 13C
NMR (MeOD) δ 138.0 (CMe2), 128.4-101.1 (m, Ar), 100.8 (CH-Ph), 97.7
(CH2OMe), 85.3 (C-4), 77.1 (C-2), 69.2 (C-6), 69.1 (C-5), 69.0 (C-1), 62.3 (C-7),
61.1 (C-3), 55.3 (OMe), 23.5, 23.4 (2Me). HRMS Calcd for C19H29O8 (M+H):
385.1857. Found: 385.1875.
2.5.3.6 5,7-O-Benzylidene-3,6-O-isopropylidene-4-O-methoxymethyl-D-glycero-D-galacto-heptitol (19)
[α] 23D = -20° (c = 0.1, MeOH). 1H NMR (MeOD) δ 7.48-7.34 (5H, m, Ar),
5.51(1H, s, CH-Ph), 4.49, 4.47 (2H, 2d, JA,B = 6.2 Hz, CH2OMe), 4.13 (1H, dd,
J7a,7b = 10.7, J6,7b = 5.4 Hz, H-7a), 4.08 (1H, m, H-2), 3.95 (1H, dd, J3,4 = 9.7, J5,4
= 2.8 Hz, H-4), 3.85 (1H, dd, J1a,1b = 11.4, J2,1a = 3.6 Hz, H-1a), 3.78 (1H, dt, J6,7
= 9.9, J5,6 = 5.4 Hz, H-6), 3.67-3.60 (4H, m, H-5, H-7b, H-1b, H-3), 3.35 (3H, s,
OMe), 1.37, 1.36 (6H, 2s, 2Me). 13C NMR (MeOD) δ 137.9 (CMe2), 128.5 -101.3
(m, Ar), 100.6 (CH-Ph), 97.7 (CH2OMe), 86.0 (C-5), 78.2 (C-3), 72.1 (C-4), 71.3

61
(C-2), 69.1 (C-7), 61.3 (C-1), 61.0 (C-6), 55.6 (OMe), 23.6, 23.4 (2Me). HRMS
Calcd for C19H29O8 (M+H): 385.1857. Found: 385.1865.
2.5.3.7 2,5-O-Isopropylidene-4,6,7-tri-O–methoxymethyl-D-glycero-D-manno-heptitol (20)
Compound 18 (580 mg, 1.51 mmol), was dissolved in DMF (20 mL) and i-
Pr2NEt (4.21 mL, 24.16 mmol) and MOMCl (0.9 mL, 12.08 mmol) were added.
The reaction mixture was heated at 60 °C for 2 h, then quenched with ice, and
extracted with ether (3×30 mL). The organic solution was dried (Na2SO4) and
concentrated to give a crude product that was further dried under high vacuum
for 1 h. The crude product was dissolved in MeOH (50 mL) and the solution was
stirred with Pd(OH)2 20 wt% on carbon (520 mg) under 100 Psi of H2 for 1h. The
catalyst was removed by filtration through a bed of Celite, then washed with
methanol. The solvents were removed under reduced pressure and the residue
was purified by flash column chromatography (EtOAc/Hexanes (1.5:1)) to give 20
as a colorless syrup (420 mg, 72%). [α] 23D = +48.0° (c = 0.1, MeOH). 1H NMR
(MeOD) δ 4.90-4.63 (6H, m, 3CH2OMe), 4.20 (1H, dd, br, H-6), 3.95 (1H, d, br,
J4,5 = 8.6 Hz, H-5), 3.86-380 (2H, m, H-1a, H-7a), 3.68-3.58 (3H, m, H-2, H-7b,
H-1b), 3.45, 3.42, 3.36 (9H, 3s, 3OMe), 3.34 (2H, m, H-4, H-3), 1.35 (6H, s,
2Me). 13C NMR (CDCl3) δ 100.7 (CMe2), 98.4, 96.3, 95.5 (3CH2OMe), 83.9 (C-4),
75.0 (C-6), 74.9 (C-3), 71.2 (C-2), 70.5 (C-5), 66.2 (C-7), 62.5 (C-1), 55.3, 54.5,
54.1 (3OMe), 22.6, 22.4 (2Me). HRMS Calcd for C16H33O10 (M+H): 385.2068.
Found: 385.2083.

62
2.5.3.8 3,6-O-Isopropylidene-1,2,4-tri-O–methoxymethyl-D-glycero-D-galacto-heptitol (21)
Compound 21 was obtained as a colorless syrup (285 mg, 75%) from 19
(380 mg, 1 mmol) using the same procedure that was used to obtain 20. [α] 23D = -
30° (c = 0.4, MeOH). 1H NMR (MeOD) δ 4.84-4.61 (6H, m, 3CH2OMe), 4.08 (1H,
ddd, J3,2 = 1.3 , J2,1a = 5.6, J2,1b = 7.2 Hz, H-2), 3.86-3.84 (2H, m, H-7a, H-3),
3.74 (1H, dd, J1a,1b = 9.5, J1a,2 = 5.6 Hz, H-1a), 3.69 (1H, ddd, J6,5 = 2.9, J6,7b
=6.8, J6,7a = 9.8 Hz, H-6), 3.60-3.55 (2H, m, H-1b, H-7b), 3.45(1H, m, H-5), 3.44,
3.38, 3.35 (9H, 3s, 3OMe), 3.34 (1H, m, H-4), 1.36, 1.32 (6H, 2s, 2Me). 13C NMR
(CDCl3) δ 100.1 (CMe2), 97.8, 96.8, 95.9 (3CH2OMe), 83.3 (C-5), 75.1 (C-2),
73.8 (C-4), 70.3 (C-6), 67.8 (C-3), 65.9 (C-1), 61.8 (C-7), 54.3, 54.1, 53.7
(3OMe), 22.9, 22.8 (2Me). HRMS Calcd for C16H33O10 (M+H): 385.2068. Found:
385.2067.
2.5.3.9 2,5-O-Isopropylidene-4,6,7-tri-O–methoxymethyl-D-glycero-D-manno-heptitol-1,3-cyclic sulfate (12)
A mixture of 20 (400 mg, 1.04 mmol) and Et3N (0.57 mL, 4.16 mmol) in
CH2Cl2 (10 mL) was stirred in an ice bath. Thionyl chloride (0.12 mL, 1.56 mmol)
in CH2Cl2 (2 mL) was then added dropwise over 15 min, and the mixture was
stirred for an additional 30 min. The mixture was poured into ice-cold water and
extracted with CH2Cl2 (3×30 mL). The combined organic layers were washed
with brine and dried over Na2SO4. The solvent was removed under reduced
pressure and the residue was dried under high vacuum for 1 h. The
diasteromeric mixture of cyclic sulfites was dissolved in a mixture of CH3CN:CCl4
(1:1, 25 mL) and sodium periodate (333 mg, 1.56 mmol) and RuCl3 (10 mg) were

63
added, followed by water (2 mL). The reaction mixture was stirred for 2 h at rt,
then filtered through a bed of Celite, and washed with ethyl acetate. The volatile
solvents were removed, and the aqueous solution was extracted with EtOAc
(2×30 mL). The combined organic layers were washed with brine, dried over
Na2SO4, concentrated under reduced pressure, and the residue purified by flash
column chromatograghy (EtOAc/Hexanes (1:2)) to give 12 as a colorless syrup
(325 mg, 70%). [α] 23D = +1.2° (c = 0.85, CH2Cl2). 1H NMR (CDCl3) δ 4.77-4.65
(6H, m, 3CH2OMe), 4.62 (1H, t, J2,3 = J4,3 = 9.1 Hz, H-3), 4.54 (1H, t, J1a,1b = J2,1a
= 11.1 Hz, H-1a), 4.37 (1H, dd, J2,1a = 5.4, J1a,1b = 11.1 Hz, H-1b), 4.16 (2H, m,
H-2, H-6), 3.98 (1H, d, J4,5 = 9.8 Hz, H-5), 3.80 (1H, dd, J6,7a = 4.8, J7a,7b = 10.8
Hz, H-7a), 3.75 (1H, t, J3,4 = J4,5 = 8.6 Hz, H-4), 3.64 (1H, t, J7a,7b = J6,7b = 8.9 Hz,
H-7b), 3.44, 3.41, 3.39 (9H, 3s, 3OMe), 1.38, 1.36 (6H, 2s, 2Me). 13C NMR
(CDCl3) δ 102.3 (CMe2), 97.9, 96.7, 96.1 (3CH2OMe), 89.2 (C-3), 76.9 (C-4),
74.6 (C-6), 72.2 (C-1), 71.0 (C-5), 66.8 (C-5), 66.8 (C-7), 56.5 (C-2), 56.6, 55.7,
55.3 (3OMe), 24.4, 23.9 (2Me). HRMS Calcd for C16H31O12S (M+H): 447.1531.
Found: 447.1516.
2.5.3.10 3,6-O-Isopropylidene-1,2,4-tri-O–methoxymethyl-D-glycero-D-galacto-heptitol-5,7-cyclic sulfate (22)
Compound 22 was obtained as a colorless syrup (220 mg, 76%) from 21
(250 mg, 0.65 mmol) using the same procedure that was used to obtain 12. [α] 23D
= -32° (c = 0.46, CH2Cl2). 1H NMR (CDCl3) δ 4.83-4.63 (6H, m, 3CH2OMe), 4.70
(1H, m, H-5), 4.55 (1H, t, J7a,7b = J6,7a = 11.1 Hz, H-7a), 4.39 (1H, dd, J6,7a = 4.9,
J7a,7b = 10.7 Hz, H-7b), 4.24 (1H, td, J5,6 = 5.7, J6,7 = 10.5 Hz, H-6), 4.09 (1H,

64
ddd, J1a,2 = 6.9, J1b,2 = 5.3, J3,2 = 1.4 Hz, H-2), 3.97 (1H, dd, J4,3 = 10.0, J3,2 = 1.5
Hz, H-3), 3.89 (1H, dd, J3,4 = 10.0, J5,4 = 7.7 Hz, H-4), 3.80 (1H, dd, J2,1a = 5.4,
J1a,1b = 9.8 Hz, H-1a), 3.58 (1H, t, J1a,1b = J2,1b = 9.2 Hz, H-1b), 3.45, 3.41, 3.39
(9H, 3s, 3OMe), 1.43, 1.37 (6H, 2s, 2Me). 13C NMR (CDCl3) δ 102.32 (CMe2),
98.1, 98.0, 97.9 (3CH2OMe), 89.6 (C-5), 76.6 (C-4), 75.1(C-2), 72.0 (C-7), 68.9
(C-3), 66.2 (C-1), 59.5 (C-6), 56.4, 56.0, 55.7 (3OMe), 24.8, 23.8 (2Me). HRMS
Calcd for C16H30NaO12S (M+Na): 470.1383. Found: 470.1399.
2.5.3.11 2,3,5-Tri-O–p–methoxybenzyl-1,4-dideoxy-1,4-[[2S,3S,4R,5R,6R]-2,5-isopropylidene-4,6,7-tri-O–methoxymethyl-3-(sulfooxy)heptyl]-(R)-epi-sulfoniumylidine-D-arabinitol inner salt (23)
The cyclic sulfate 12 (260 mg, 0.58 mmol) and the thiosugar 7 (360 mg,
0.70 mmol) were dissolved in HFIP (1.5 mL), containing anhydrous K2CO3 (10
mg). The mixture was stirred in a sealed reaction vessel in an oil bath at 72 °C
for 6 days. The progress of the reaction was followed by TLC analysis
(developing solvent EtOAc:MeOH, 10:1). The mixture was cooled, then diluted
with EtOAc and evaporated to give a syrupy residue. Purification by column
chromatograghy (EtOAc/MeOH 99:1) gave the sulfonium salt 23 as a syrup (360
mg, 65%). [α] 23D = +62° (c = 0.85, CH2Cl2). 1H NMR (acetone-d6) δ 7.32-
6.91(12H, m, Ar), 5.12-4.52 (12H, m, 3CH2OMe, 3CH2-Ph), 4.69 (1H, m, H-2),
4.55 (1H, m, H-3), 4.39-4.30 (4H, m, H-1'a, H-2', H-3', H-6'), 4.08 (1-H, t, J3,4 =
J5,4 = 7.4 Hz, H-4), 4.02-3.90 (4H, m, H-1a, H-1'b, H-5'), 3.85-3.78 (3H, m, H-5a,
H-7'a, H-1b), 3.82 (9H, s, 3Ph-OMe), 3.60 (1H, t, J7'a,7'b = J6',7'b = 9.1 Hz, H-7'b),
3.42 (1H, m, H-4'), 3.39, 3.36, 3.33 (9H, 3s, 3CH2OMe) 1.37, 1.32 (6H, 2s, 2Me).
13C NMR (acetone-d6) δ 159.8-129 (m, Ar), 101.6 (CMe2), 98.7, 96.5, 95.2

65
(3CH2OMe), 83.5 (C-3), 81.2 (C-2), 79.7 (C-2'), 78.6 (C-4'), 74.0 (C-6'), 72.7,
71.6, 71.4 (3CH2Ph), 71.3 (C-5'), 66.9 (C-7'), 66.6 (C-3'), 66.5 (C-5), 65.1 (C-4),
55.9-54.2 (6OMe), 51.5 (C-1'), 47.4 (C-1), 24.4, 23.5 (2Me). HRMS Calcd for
C45H65O18S2 (M+H): 957.3607. Found: 957.3604.
2.5.3.12 1,4-Dideoxy-1,4-[[2S,3S,4R,5R,6R-2,4,5,6,7-pentahydroxy-3-(sulfooxy)heptyl]-(R)-epi-sulfoniumylidine]-D-arabinitol inner salt (6)
The protected sulfonium salt 23 (150 mg, 0.16 mmol) was dissolved in
30% aqueous solution of TFA (25 mL) and the mixture was stirred at 50 °C for
5h. The solvent was removed under reduced pressure and the residue was
dissolved in water (5 mL) and washed with CH2Cl2 (3×5 mL). The water layer
was evaporated to give the crude product that was purified on silica gel with
EtOAc/MeOH/H2O 6:3:1 (v/v) as eluent to give compound 6 as a colorless solid
(61 mg, 93%). Mp 82-84 °C [α] 23D = +5.5° (c = 0.55, CH2Cl2). 1H NMR (D2O) δ
4.67 (1H, dd, J1a,2 = 3.7, J1b,2 = 7.4 Hz, H-2), 4.56 (1H, d, J2',3' = 8.2 Hz, H-3'),
4.39 (1H, t, J2,3 = J3,4 = 3.1 Hz, H-3), 4.35 (1H, dt, J2’,3’ = 3.3, J2’,1’ = 7.8 Hz, H-2'),
4.02 (3H, m, H-5a, H-1'a, H-4), 3.91-3.83 ( 5H, m, H-6', H-5', H-5b, H-4', H-1’b),
3.81 (2H, d, J1,2 = 3.9 Hz, H-1a,b), 3.71 (1H, dd, J7'a,7'b = 3.2, J7'b,6' = 11.9 Hz, H-
7'b), 3.62 (1H, dd, J7'b,7'a = 7.8, J7'a,6' = 11.6 Hz, H-7'a). 13C NMR (D2O) δ 78.3 (C-
3'), 77.7 (C-3), 76.7 (C-2), 72.9 (C-6'), 70.7 (C-5'), 70.0 (C-4), 69.1 (C-4'), 66.0
(C-2'), 61.6 (C-7'), 59.2 (C-5), 50.7 (C-1'), 47.7 (C-1). HRMS Calcd for
C12H25O12S2 (M+H): 425.0782. Found: 425.0778.

66
2.5.3.13 1,4-Dideoxy-1,4-[[2S,3S,4R,5R,6S-2,4,5,6,7–pentahydroxy-3-(sulfooxy)heptyl]-(R-)epi-sulfoniumylidine]-D-arabinitol inner salt (4)
A mixture of the thiosugar 7 (100 mg, 0.224 mmol) and the cyclic sulfate
22 (137 mg, 0.269 mmol) in HFIP (1 mL) containing K2CO3 (5 mg) was placed in
a sealed reaction vessel and heated at 72 °C with stirring for 6 days. The
progress of the reaction was followed by TLC analysis (developing solvent
EtOAc:MeOH, 10:1). The mixture was cooled, then diluted with EtOAc and
evaporated to give a syrupy residue. Purification by column chromatography
(EtOAc/MeOH 95:5) gave the protected sulfonium salt as a foam (120 mg, 57%).
The protected sulfonium salt 24 (100 mg, 0.11 mmol) was dissolved in 30%
aqueous TFA (10 mL) and stirred at 50 °C for 5h. The solvents were removed
under reduced pressure and the residue was dissolved in water (5 mL) and
washed with CH2Cl2 (3×5 mL). The water layer was evaporated to give the crude
product that was purified on silica gel column with EtOAc/MeOH/H2O 6:3:1 (v/v)
as eluent to give compound 4 as a colorless solid (40 mg, 91%).12
2.6 Acknowledgments
We are grateful to the Canadian Institutes for Health Research (FRN79400) and
the Heart and Stroke Foundation of Ontario (NA-6305) for financial support.

67
2.7 References
1. Dwek, R. A. Chem. Rev. 1996, 96, 683-720.
2. Varki, A.; Cummings, R.; Esko, J.; Freeze, H.; Hart, G.; Marth, J.
Essentials of Glycobiology; Cold Spring Harbor Laboratory Press: Cold
Spring Harbor, NY, 1999.
3. For a recent review on glycosidase inhibitors: de Melo, E. B.; Gomes, A.
D.; Carvalho, I. Tetrahedron 2006, 62, 10277-10302.
4. Chandrasena, J. P. C. The Chemistry and Pharmacology of Ceylon and
Indian medicinal plants, H&C Press, Colombo, Sri Lanka, 1935.
5. Jayaweera, D. M. A. Medicinal Plants Used in Ceylon-Part 1, National
Science Council of Sri Lanka: Colombo, 1981, p. 77.
6. Vaidyartanam, P. S. In Indian Medicinal Plants: a compendium of 500
species, Warrier, P. K.; Nambiar, V. P. K.; Ramankutty, C.; Eds. Orient
londman: India 1993, pp. 47-48.
7. Yoshikawa, M.; Murakami, T.; Shimada, H.; Matsuda, H.; Yomahara, J.;
Tanabe, G.; Muraoka, O. Tetrahedron Lett. 1997, 38, 8367-8370.
8. Yoshikawa, M.; Murakami, T.; Yashiro, K.; Matsuda, H.; Chem. Pharm.
Bull. 1998, 46, 1339-1340.
9. Yoshikawa, M.; Xu, F.; Nakamura, S.; Wang, T.; Matsuda, H.; Tanabe, G.;
Muraoka, O. Heterocycles 2008, 75, 1397-1405.
10. Muraoka, O.; Xie, W.; Tanabe, G.; Amer, M. F. A.; Minematsu, T.;
Yoshikawa, M. Tetrahedron Lett. 2008, 49, 7315-7317.
11. For reviews see: a) Mohan, S.; Pinto, B. M. Carbohydr. Res. 2007, 342,
1551-1580. b) Mohan, S.; Pinto, B. M. Collect. Czech. Chem. Commun.
2009, 74, 1117-1136.

68
12. Jayakanthan, K.; Mohan, S.; Pinto, B. M. J. Am. Chem. Soc. 2009, 131,
5621-5626.
13. Mohan, S.; Jayakanthan, K.; Nasi, R.; Kuntz D. A.; Rose, D. R.; Pinto, B.
M. Organic Lett. 2010, 12, 1088-1091.
14. Baggett, N.; Stribblehill, P. J. Chem. Soc., Perkin Trans. 1, 1977, 1123-
1126.
15. Harris, J. M.; Keranen, M. D.; O'Doherty, G. A. J. Org. Chem. 1999, 64,
2982-2983.
16. Nasi, R.; Patrick, B. O.; Sim, L.; Rose, D. R.; Pinto, B. M. J. Org. Chem.
2008, 73, 6172-6181.
17. Ghavami, A.; Sadalapure, K. S.; Johnston, B. D.; Lobera, M.; Snider, B.
B.; Pinto, B. M. Synlett 2003, 9, 1259-1262.
18. Sim, L.; Jayakanthan, J.; Mohan, S.; Nasi, R.; Johnston, B.D.; Pinto, B. M.;
Rose, D. R. Biochemistry 2010, 49, 443-451.

69
2.8 Supporting Information
Synthesis of a biologically active isomer of kotalanol, a naturally-
occurring glucosidase inhibitor
Razieh Eskandari, Kumarasamy Jayakanthan, Douglas A. Kuntz, David R.
Rose, B. Mario Pinto
70
ppm (t1)50100
ppm (t1)2.03.04.05.06.07.0
O O
OO O
Ph
O
14Ph
O O
OO O
Ph
O
14Ph
71
ppm (t1)2.03.04.05.06.07.0
O O
OO OH
Ph
OH
16
ppm (t1)50100
O O
OO OH
Ph
OH
16
72
ppm (t1)50100
ppm (t1)2.03.04.05.06.07.0
O O
OO OMOM
Ph
OH
17
O O
OO OMOM
Ph
OH
17

73
ppm (t1)50100
ppm (t1)2.03.04.05.06.07.0
O O
OO OMOM
Ph 13
O O
OO OMOM
Ph 13

74
ppm (t1)50100
ppm (t1)2.03.04.05.06.07.0
O O
OO OMOM
Ph
OH
OH
18
12 3 4 5 6
7
O O
OO OMOM
Ph
OH
OH
18
12 3 4 5 6
7

75
ppm (t1)50100
ppm (t1)2.03.04.05.06.07.0
O O
OO OMOM
Ph
OH
OH
19
123456
7
O O
OO OMOM
Ph
OH
OH
19
123456
7

76
ppm (t1)30405060708090100
ppm (t1)1.502.002.503.003.504.004.50
O O
HOHO OMOM
OMOM
OMOM
20
O O
HOHO OMOM
OMOM
OMOM
20

77
ppm (t1)30405060708090100
ppm (t1)1.502.002.503.003.504.004.50
O O
OS O
OOOMOM
OMOM
OMOM
12
O O
OS O
OOOMOM
OMOM
OMOM
12

78
ppm (t1)30405060708090100
ppm (t1)1.502.002.503.003.504.004.50
O O
HOHO OMOM
OMOM
OMOM
21
O O
HOHO OMOM
OMOM
OMOM
21

79
ppm (t1)30405060708090100
ppm (t1)1.502.002.503.003.504.004.50
O O
OS O
OOOMOM
OMOM
OMOM
22
O O
OS O
OOOMOM
OMOM
OMOM
22

80
ppm (t1)2.03.04.05.06.07.0
ppm (t1)50100150
O O
OSO3OMOM
OMOM
OMOMS
OPMBPMBO
PMBO
23
O O
OSO3OMOM
OMOM
OMOMS
OPMBPMBO
PMBO
23

81
ppm (t1)50.055.060.065.070.075.0
ppm (t1)4.004.50
S
OHHO
OSO3 OH
OH OH
HO
OHOH
6
S
OHHO
OSO3 OH
OH OH
HO
OHOH
6

82
CHAPTER 3: POTENT GLUCOSIDASE INHIBITORS: DE-O-SULFONATED PONKORANOL AND ITS
STEREOISOMER
This Chapter comprises the manuscript “Potent glucosidase inhibitors:
de-O-sulfonated ponkoranol and its stereoisomer” which was published in
Organic Letters (2010, 12, 1632-1635).
Razieh Eskandari,a Douglas A. Kuntz,b David R. Rose,b B. Mario Pintoa
aDepartment of Chemistry, Simon Fraser University, Burnaby, British Columbia,
Canada V5A 1S6
b Department of Biology, University of Waterloo, Waterloo, Ontario,
Canada N2L 3G1

83
Ponkoranol is a naturally-occurring glucosidase inhibitor isolated from the
plant Salacia reticulata. The compound comprises a sulfonium ion with an
internal sulfate counter ion. Since de-O-sulfonated compounds of naturally-
occurring sulfonium-ion glucosidase inhibitors isolated from Salacia species show
better inhibitory activities against α-glucosidases, we report in this Chapter, an
efficient synthetic route to de-O-sulfonated ponkoranol and its stereoisomer, and
show that these compounds are potent glucosidase inhibitors that inhibit a key
human intestinal glucosidase, the N-terminal domain of maltase glucoamylase.
The thesis author performed all the experimental synthetic work and the
characterization of the compounds, and wrote and edited the manuscript with Dr.
B. Mario Pinto. Drs. Douglas A. Kuntz and David R. Rose performed the enzyme
inhibition studies.
Graphical abstract:
O
HOOH
OBnTsO
S
OHHO
HO
OH
OH
OH
OH
ClS
OBnBnO
BnO+
R1 = OH, R2 = HR1 = H, R2 = OH
R2R1
8. R1 = OH, R2 = H9. R1 = H, R2 = OH
R2 R1

84
3.1 Keywords
Ponkoranol, sulfonium-ion, de-O-sulfonated ponkoranol, glucosidase
inhibitors.
3.2 Abstract
Ponkoranol, a glucosidase inhibitor isolated from the plant Salacia
reticulata, comprises a sulfonium-ion with an internal sulfate counterion. An
efficient synthetic route to de-O-sulfonated ponkoranol and its 5’-stereoisomer is
reported, and it is shown that these compounds are potent glucosidase inhibitors
that inhibit a key intestinal human glucosidase, the N-terminal catalytic domain of
maltase-glucoamylase, with Ki values of 43 ± 3 and 15 ± 1 nM, respectively.
3.3 Introduction
Compounds isolated from medicinal plants can provide the lead structures
for drug development programs.1,2 For example, the aqueous extracts of the
roots and stems of the large woody climbing plant Salacia reticulata, known as
Kothalahimbutu in Singhalese, have been used in the Ayurvedic system of Indian
medicine in Sri Lanka and Southern India for the treatment of type-2 diabetes.3,4
Several glucosidase inhibitors have been isolated from the water-soluble fraction
of this plant extract and also other plants that belong to the Salacia genus such
as Salacia chinensis, Salacia prinoides, and Salacia oblonga which explain, at
least in part, the antidiabetic property of the aqueous extracts of these plants.5-7
Thus far, six components have been isolated from the plant S. reticulate, namely
salaprinol (1), 7 salacinol (2), 6 ponkoranol (3), 7 kotalanol (4),5 de-O-sulfonated

85
kotalanol (5),8 and de-O-sulfonated salacinol (6)9 (Figure 3-1), all of which
possess a common structural motif that comprises a 1,4-anhydro-4-thio-D-
arabinitol and a polyhydroxylated side chain.
Figure 3-1: Components isolated from Salacia species.
We have carried out extensive research on the synthesis of higher
homologues of salacinol (2) which has led to the stereochemical structure
elucidation of compounds 3-5.10-12 Interestingly, ponkoranol (3), the recently
isolated7 six-carbon-chain homologue of salacinol, was synthesized by us several
years earlier with the expectation that it would be an effective glucosidase
inhibitor.13
S
OHHO
OSO3 OH
OH OH
HO
OHOH
S
OHHO
OSO3 OH
OH OH
HO
OH
S
OHHO
OSO3
OH OH
HOS
OHHO
OSO3
OH
HO
Kotalanol (4)
Salacinol (2) Ponkoranol (3)Salaprinol (1)
S
OHHO
OH OH
OH OH
HO
OHOH
CH3OSO3
De-O-sulfonated kotalanol (5)
S
OHHO
OH
OH OH
HOHCO2
De-O-sulfonated salacinol (6)

86
Recently, Minami et al . 9 reported the isolation of a thiosugar sulfonium-
alkoxide inner salt (7), neosalacinol (Figure 3-2), from S. reticulata; however,
Tanabe et al.14 have shown that this compound is de-O-sulfonated salacinol (6).
Figure 3-2: Proposed structure of neosalacinol.
Comparison of the inhibitory activities of de-O-sulfonated salacinol (6) vs
salacinol (2) and de-O-sulfonated kotalanol (5) vs kotalanol (4) against rat
intestinal α-glucosidases (maltase, sucrase, and isomaltase) revealed that the
desulfonated analogues were either equivalent or better inhibitors than the parent
compounds.7,15,16 Furthermore, we have shown recently that de-O-sulfonated
kotalanol (5) (Ki = 0.03 ± 0.01 µM) is more potent an inhibitor of the N-terminal
catalytic domain of human intestinal maltase-glucoamylase (ntMGAM) than
kotalanol (4) itself (Ki = 0.19 ± 0.03 µM) (Table 3-1).17
S
OHHO
O
OH OH
HO
7

87
Table 3-1: Experimentally determined Ki values (nM)a of compounds 2-5, 8 and 9.
Inhibitor Ki (nM)
2
190 ± 2019
3
170 ± 3019
4
190 ± 3017
5
30 ± 1017
8
43 ± 1
9
15 ± 1
a Analysis of MGAM inhibition was performed using maltose as the substrate.
S
OHHO
OSO3 OH
OH OH
HO
OHOH
S
OHHO
OH OH
OH OH
HO
OHOH
CH3OSO3
S
OHHO
HO
OH
OH
OH
OH
OHCl
S
OHHO
HO
OH
OH
OH
OH
OHCl
S
OHHO
HO
OH
OH
OSO3
OH
OH
S
OHHO
HO
OH
OSO3
OH

88
In view of these findings, it was of interest to question whether de-O-
sulfonated ponkoranol 8 and its 5’-stereoisomer 9 (Figure 3-3) would be more
potent inhibitors than ponkoranol itself.
Figure 3-3: De-O-sulfonated ponkoranol and its 5’-stereoisomer.
Our previous work with kotalanol analogues had suggested that the
configuration at C-6’ was not critical for inhibitory activity.17,18 We report here an
efficient synthetic route to de-O-sulfonated ponkoranol 8 and its 5’-stereoisomer
9, and show that they are very potent inhibitors of the amino terminal catalytic
domain of human maltase-glucoamylase (ntMGAM).19
3.4 Results and discussion
The sulfonium-ions A could be synthesized by alkylation of an
appropriately protected 1,4-anhydro-4-thio-D-arabinitol B at the ring sulfur atom
with agent C. The desired stereochemistry at C-5' could be readily obtained by
choice of either D-glucose or D-mannose as starting material (Scheme 3-1).
S
OHHO
OH OH
OH OH
HO
OH
S
OHHO
OH OH
OH OH
HO
OH
Cl Cl
8 9
1'2'
3'4'
5'6'
1'2'
3'4'
5'6'

89
Scheme 3-1: Retrosynthetic analysis.
Initially, the S-alkylation of thioarabinitol 1220 with methyl 6-iodo-β-D-
glucopyranoside 1121 in CH3CN using AgBF4 at 65 °C was examined, based on
the procedure that has been reported for S-alkylation with simple alkyl halides
(Scheme 3-2).22 No product formation and decomposition of the starting material
were observed by TLC; the reaction in 1,1,1,3,3,3-hexafluoroisopropyl alcohol
(HFIP)23 as a solvent was also unsuccessful.
Scheme 3-2: First attempted synthesis of 8.
O
HOOH
OPL
S
OPPO
POS
OHHO
HO
OH
OH
OH
OH
L
L = leaving groupP = protecting group
+
A B C
R2 R1
R1R2
R1 = OH, R2 = H R1 = H, R2 = OH
8. R1 = OH, R2 = H, L = Cl9. R1 = H, R2 = OH, L = Cl
O
HOOH
OH
OMeI
AgBF4, CH3CN, 65oC
11
No reaction
PPh3, I2, imidazole,THF, reflux, 2 h
S
BnO OBn
BnO
12
O
HOOH
OH
OMeHO
10

90
In contrast, the coupling reaction with the p-toluenesulfonyl ester 1324 in
HFIP at 70 °C proceeded smoothly and yielded the sulfonium ion 14 (Scheme 3-
3). The benzyl groups of compound 14 were removed by treatment with boron
trichloride at -78 °C in CH2Cl2. However, attempts to hydrolyze the methyl
glycoside 15 with 2 M HCl were not successful, and decomposition of the product
was observed.
Scheme 3-3: Second attempted synthesis of 8.
Therefore, a benzyl glycoside was chosen as a protecting group at the
anomeric position to ensure its facile removal after the coupling reaction. Thus,
benzyl 6-O-p-toluenesulfonyl-α-D-gluco 17 or manno- pyranoside 20 were readily
prepared from D-glucose and D-mannose, respectively, according to literature
procedures.25-27 The thioether 12 was reacted with 17 in HFIP containing K2CO323
to give the protected sulfonium-ion 18 in 52% yield (Scheme 3-4). The benzyl
O
HOOH
OH
OMeTsO
HFIP, 70 oC,4 d, 45%
O
HOOH
OH
OMeS
BnO OBn
BnO
OTs
1314
15
S
BnO OBn
BnO
1. BCl3, CH2Cl2,, -78 oC
2. 2M HCl, -70oC, 20 h
12
O
HOOH
OH
OMeS
HO OH
HO
OTs

91
groups were then removed by treatment with boron trichloride at -78 °C in
CH2Cl2. During the course of deprotection, the p-toluenesulfonate counterion was
partially exchanged with chloride ion. Similar results were observed in previous
work from our laboratory.22 Hence, after removal of the benzyl groups, the
product was subsequently treated with Amberlyst A-26 resin (chloride form) to
completely exchange the p-toluenesulfonate counterion with chloride ion. Finally,
the crude product was reduced with NaBH4 to provide the desired de-O-
sulfonated ponkoranol 8 in 48% yield over three steps (Scheme 3-4).
Scheme 3-4: Synthesis of compound 8.
The other diastereomer was obtained similarly. Thus, compound 20 was
reacted with the thioether 12 to give the protected sulfonium-ion 21 in 47% yield
which was converted, as before, to the desired compound 9 in 41% yield over
three steps (Scheme 3-5).
1.BnOH, p-TSOH,70 oC, 4 h, 36%2.TsCl, Pyridine,-10 oC, 4 h, 60%
O
HOOH
OH
OBnTsO
HFIP, 70 oC, 4 d, 52%
O
HOOH
OH
OBnS
BnO OBn
BnO
1. BCl3, CH2Cl2,
-78 oC, 6 h
2. Amberlyst A-26,
H2O, 3 h
3. NaBH4, H2O, 3 h
S
OHHO
HO
OH
OH
OH
OH
OHOTs Cl
17
18 8
1' 1' 2'2'
3'
3'4'
4'
5'5'6'
6'
48%
S
BnO OBn
BnOO
HOOH
OH
OHHO
16
12

92
Scheme 3-5: Synthesis of compound 9.
The absolute stereochemistry at the stereogenic sulfur center in 18 and 21
was established by means of 1D-NOESY experiments (Figure 3-4) which showed
H-4 to H-6' correlations, implying that these atoms are syn-facial with respect to
the sulfonium salt ring.
Figure 3-4: 1D-NOESY correlations of selected protons in compounds 18 and 21.
1.BnOH, HCl,rt, 1 h, 35%2.TsCl, Pyridine,-10 oC, 15 h, 97%
O
HOOH
OH
OBnTsO
HFIP, 70 oC,4 d, 47%
O
HOOH
OH
OBnS
BnO OBn
BnOS
OHHO
HO
OH
OH
OH
OH
OHOTs Cl
20
21 9
1'
2'3'
3'
4'4'
5'5'6'
6'1' 2'
41%
S
BnO OBn
BnOO
HOOH
OH
OHHO
1. BCl3, CH2Cl2,
-78 oC, 6 h
2. Amberlyst A-26,
H2O, 3 h
3. NaBH4, H2O, 3 h
19
12
O
OH
OH
OHBnO
S
BnO OBn
18
H
H2C
4
BnO
O
OH
OH
OHBnO
S
BnO OBn
21
H
H2C
4
BnO

93
Finally, we comment on the inhibitory activities of compounds 8 and 9
against the N-terminus of recombinant human maltase-glucoamylase
(ntMGAM),19 a critical intestinal glucosidase for postamylase processing of
starch-derived oligosaccharides into glucose. The de-O-sulfonated ponkoranol 8
and its 5’-stereoisomer 9 inhibited ntMGAM with Ki values of 43 ± 3 and 15 ± 1
nM, respectively, both significantly lower than that (170 ± 30)19 for ponkoranol (3)
itself (Table 3-1). Thus, it would appear that de-O-sulfonation is beneficial. We
have attributed this fact previously to alleviation of steric compression of the
sulfate anion in a hydrophobic pocket within the active site of ntMGAM.17 The Ki
values for 8 and 9 compare to a Ki value for de-O-sulfonated kotalanol of 30 ± 1
nM.17 It would appear, therefore, that the configuration at C-5’ is not critical for
dictating enzyme inhibitory activity against ntMGAM and, furthermore, that
extension of the acyclic carbon chain beyond six carbons is not essential. We
note that 9 is the most potent compound to date in this class of molecules.
3.5 Experimental
3.5.1 General methods
Optical rotations were measured at 23 °C. 1H and 13C NMR spectra were
recorded at 600 and 150 MHz, respectively. All assignments were confirmed with
the aid of two-dimensional 1H, 1H (COSYDFTP) or 1H, 13C (INVBTP) experiments
using standard pulse programs. Column chromatography was performed with
Silica 60 (230-400 mesh). Reverse column chromatography was performed with
Silica C-18 cartridges. High resolution mass spectra were obtained by the

94
electrospray ionization method, using an Agilent 6210 TOF LC/MS high
resolution magnetic sector mass spectrometer.
3.5.2 Compound characterization data
3.5.2.1 Methyl 6-deoxy-6-[2,3,5-tri-O-benzyl-1,4-dideoxy-episulfoniummylidene-D-arabinitol]-α-D-glycopyranoside-p-toluenesulfonate (14)
The mixture of the thioether 1220 (270 mg, 0.64 mmol) and methyl 6-O-p-
toluenesulfonyl-β-D-glucopyranoside 1324 (200 mg, 0.57 mmol) in HFIP (1 mL)
were placed in a sealed tube in an oil bath at 70 oC for 4 days. The mixture was
cooled, then diluted with EtOAc, and evaporated to give a syrupy residue.
Purification by column chromatography (CHCl3/MeOH 10:1) gave the sulfonium
salt 14 as a syrup (150 mg, 45%). [α] 23D = + 19 (c = 1.4, MeOH). 1H NMR (MeOD)
δ 7.72-7.20 (19H, m, Ar), 4.69 (2H, m, H-1', H-3), 4.67-4.50 (6H, m, 3CH2-Ph),
4.47 (1H, br, H-2), 4.40 (1H, dd, J3,4 = 5.9, J4,5 = 10 Hz, H-4 ), 4.13 (1H, d, H-1a),
3.96 (2H, m, H-6'a , H-5'), 3.84 (2H, m, H-5a, H1b), 3.74 (2H, m, H-6'b, H-5b),
3.62 (1H, t, J2',3' = J3',4' = 8.9 Hz, H-3'), 3.39 (1H, dd, J1',2' = 3.7, J2',3' = 9.5 Hz, H-
2'), 3.36 (3H, s, OMe), 3.27 (1H, t, J4',5' = J3',4' = 8.9 Hz, H-4'), 2.35 (3H, s, Me).
13C NMR (MeOD) δ 142.3-125.6 (m, Ar), 100.5 (C-1'), 83.3 (C-3), 83.2 (C-2),
73.1, 72.0, 71.9 (3CH2-Ph), 73.0 (C-3'), 72.9 (C-4'), 71.7 (C-2'), 68.3 (C-5'), 66.9
(C-4), 66.4 (C-5), 55.1 (OMe), 48.6.0 (C-1), 48.2 (C-6'), 19.9 (Me). HRMS Calcd
for C33H41O8S (M+.): 599.2542. Found: 599.2564.

95
3.5.2.2 Attempted hydrolysis of the methyl glycoside (15)
To a solution of the sulfonium-ion 15 (150 mg, 0.274 mmol) in CH2Cl2 (5
mL) at -78 oC under N2 atmosphere was added BCl3 (1M, in CH2Cl2, 2.20 mmol).
The reaction mixture was stirred at the same temperature for 30 min, and then
allowed to warm to 5 oC for 6 h. The reaction was quenched by addition of MeOH
(5 mL), the solvents were removed, and the residue was co-evaporated with
MeOH (2 × 5 mL) to give a solid residue. The crude product was dissolved in 2 M
HCl (5 mL) and the solution was stirred at 70 °C for 20 h. The progress of the
reaction was monitored by 1H NMR spectroscopy which showed decomposition
of the starting material without hydrolysis of the methyl group.
3.5.2.3 Benzyl 6-deoxy-6-[2,3,5-tri-O-benzyl-1,4-dideoxy-episulfoniummylidene-D-arabinitol]-α-D-glycopyranoside-p-toluenesulfonate (18)
Benzyl 6-O-p-toluene-sulfonyl-β-D-glucopyranoside 17 25,27 (470 mg, 1.11
mmol) and the thioether 12 (558 mg, 1.33 mmol) were dissolved in HFIP (1.5
mL), containing anhydrous K2CO3 (10 mg). The mixture was stirred in a sealed
reaction vessel in an oil bath at 70 oC for 4 days. The mixture was cooled, then
diluted with EtOAc, and evaporated to give a syrupy residue. Purification by
column chromatograghy (EtOAc/MeOH 92:8) gave the sulfonium salt 18 as a
syrup (388 mg, 52%). [α] 23D = + 16 (c = 0.8, MeOH). 1H NMR (MeOD) δ 7.67-7.17
(24H, m, Ar), 4.87 (1H, d, J1',2' = 3.6 Hz, H-1'), 4.63 (1H, m, H-3), 4.64-4.46 (8H,
m, 4CH2-Ph), 4.41 (1H, br, H-2), 4.28 (1H, dd, J3,4 = 5.7 J4,5 = 9.4 Hz, H-4 ), 3.93
(2H, m, H-1a, H-5'), 3.81 (1H, dd, J6'a,5' = 3.1, J6'a,6'b = 13.2 Hz, H-6'a), 3.77 (1H,
dd, J5a,4 = 6.0, J5a,5b = 10.5 Hz, H-5a), 3.72 (1H, dd, J1,2 = 3.6, J1a,1b = 13.3 Hz,

96
H-1b), 3.67-3.62 (3H, m, H-5b, H-6b, H-3'), 3.35 (1H, dd, J1',2' = 3.6, J2',3' = 9.8
Hz, H-2') 3.21 (1H, t, J4',5' = J3',4' = 8.9 Hz, H-4'), 2.32 (3H, s, Me). 13C NMR
(MeOD) δ 142.2-125.6 (m, Ar), 99.3 (C-1'), 83.2 (C-3), 83.0 (C-2), 73.1, 72.0,
71.9, 70.7 (4CH2-Ph), 73.0 (C-4'), 72.9 (C-3'), 71.7 (C-2'), 68.8 (C-5'), 66.9 (C-4),
66.5 (C-5), 49.0 (C-1), 48.2 (C-6'), 19.9 (Me). HRMS Calcd for C39H45O8S (M+.):
673.2830. Found: 673.2831.
3.5.2.4 1,4-Dideoxy-1,4-[[2S, 3S, 4R, 5S]-2,3,4,5,6-pentahydroxy-hexyl]-(R)-epi-sulfoniumylidine]-D-arabinitol chloride (8)
Compound 18 (300 mg, 0.36 mmol) was dissolved in CH2Cl2 (25 mL), the
mixture was cooled to -78 oC, and BCl3 (1M solution in CH2Cl2, 3.56 mmol) was
added under N2. The reaction mixture was stirred at the same temperature for 30
min, and then allowed to warm to 5 oC for 6 h. The reaction was quenched by
addition of MeOH (5 mL), the solvents were removed, and the residue was co-
evaporated with MeOH (2 × 5 mL). The crude residue was dissolved in H2O (10
mL), Amberlyst A-26 resin (200 mg) was added, and the reaction mixture was
stirred at room temprature for 3 h. Filtration through cotton, followed by solvent
removal gave the crude hemiacetal. The crude product was dissolved in water (8
mL), and the solution was stirred at room temperature while NaBH4 (67 mg, 1.78
mmol) was added in small portions over 30 min. Stirring was continued for
another 3 h and the mixture was acidified to pH < 4 by dropwise addition of 2 M
HCl. The mixture was evaporated to dryness and the residue was co-evaporated
with anhydrous MeOH (3 × 30 mL). Treatment of the solid residue with 50%
EtOAc:MeOH (5-10 mL) resulted in precipitation of most of the borate salt.

97
Filtration through cotton, followed by solvent removal gave the crude compound.
The residue was purified by reverse phase column chromatography (MeOH/H2O
(2:100)) to give 8 as a colorless solid (60 mg, 48%). [α] 23D = + 4o, (c = 0.5 , H2O).
1H NMR (D2O) δ 4.64 (1H, m, H-2), 4.35 (1H, t, br, H-3), 4.14 (1H, td, J1',2' = 9.1,
J2',3' = 3.0 Hz, H-2'), 4.02 (2H, m, H-5a, H-4), 3.87-3.77 (4H, m, H-5b, H-1'a, H-
1a, H-1b), 3.72-3.66 (3H, m, H-4', H-5', H-1'b), 3.62 (2H, m, H-6'a, H-3'), 3.50
(1H, dd, J6'a,6,b = 11.7, J5',6'b = 5.6 Hz, H-6'b). 13C NMR (D2O) δ 77.5 (C-3), 76.9
(C-2), 73.1 (C-3'), 72.7 (C-5'), 70.0 (C-4), 69.3 (C-4'), 67.4 (C-2'), 62.2 (C-6'),
59.2 (C-5), 50.0 (C-1'), 48.2 (C-1). HRMS Calcd for C11H23O8S (M+.): 315.1108.
Found: 315.1117.
3.5.2.5 Benzyl 6-deoxy-6-[2,3,5-tri-O-benzyl-1,4-dideoxy-episulfoniummylidene-D-arabinitol]-α-D-mannopyranoside-p-toluenesulfonate (21)
Reaction of the thioether 12 (590 mg, 1.41 mmol) with benzyl 6-O-p-
toluenesulfonyl-β-D-mannopyranoside 2026 (500 mg, 1.18 mmol) in HFIP (1.5
mL), containing anhydrous K2CO3 (10 mg) at 70 oC for 4 days gave the sulfonium
salt 21 as a foam (370 mg, 47%) after purification by column chromatography
(EtOAc/MeOH (92:8)). [α] 23D = +8o, (c = 0.5, MeOH). 1H NMR (MeOD) δ 7.73-7.23
(24H, m, Ar), 4.87 (1H, m, H-1'), 4.70 (1H, m, H-2), 4.69-4.52 (8H, m, 4CH2-Ph),
4.49 (1H, m, H-3), 4.31 (1H, t, J3,4 = J4,5 = 9.6 Hz, H-4 ), 4.04 (1H, d, br, J1,2 =
13.1 Hz, H-1a), 3.94 (2H, m, H-6'a, H-4'), 3.90-3.85 (3H, m, H-2', H-1b, H-5a),
3.79-3.73 (3H, m, H-6'b, H-5b, H-3'), 3.61 (1H, t, J4',5' = J5',6' = 9.3 Hz, H-5'), 2.38
(3H, s, Me). 13C NMR (MeOD) δ 142.2-125.6 (m, Ar), 100.5 (C-1'), 83.3 (C-2),
82.9 (C-3), 73.1, 72.0, 71.8, 70.1(4CH2-Ph), 70.6 (C-2'), 70.4 (C-3'), 66.7 (C-4),

98
66.5 (C-5), 48.8 (C-1), 48.2 (C-6'), 19.9 (Me). HRMS Calcd for C39H45O8S (M+.):
673.2830. Found: 673.2828.
3.5.2.6 1,4-Dideoxy-1,4-[[2S, 3S, 4R, 5R]-2,3,4,5,6-pentahydroxy-hexyl]-(R)-epi-sulfoniumylidine]-D-arabinitol chloride (9)
Compound 9 was obtained as a colorless solid (51 mg, 41%) from 21 (300
mg, 0.36 mmol) using the same procedure that was used to obtain 8. [α] 23D = +
11o, (c = 0.3 , H2O). 1H NMR (D2O) δ 4.65 (1H, d, br, H-2), 4.35 (1H, t, br, H-3),
4.12 (1H, td, J1',2' = 9.1, J2',3' = 3.0 Hz, H-2'), 4.02 (2H, m, H-5a, H-4), 3.89-3.60
(9H, m, H-1'a, H-5b, H-1a, H-1b, H-3', H-6'a, H-1'b, H-4', H-5'), 3.55 (1H, dd,
J6'a,6,b = 11.7, J5',6'b = 5.8 Hz, H-6'b). 13C NMR (D2O) δ 77.5 (C-3), 76.9 (C-2),
71.5 (C-3'), 70.5 (C-5'), 70.0 (C-4), 68.8 (C-4'), 67.3 (C-2'), 63.0 (C-6'), 59.2 (C-
5), 50.4 (C-1'), 48.1 (C-1). HRMS Calcd for C11H23O8S (M+.): 315.1108. Found:
315.1122.
3.6 Acknowledgments
We are grateful to the Canadian Institutes for Health Research
(FRN79400) and the Heart and Stroke Foundation of Ontario (NA-6305) for
financial support.
3.7 References
1. Harvey, A. L. Natural products in drug discovery. Drug Discov. Today
2008, 13, 894-901.
2. Butler, M. S. Natural products to drugs: natural product-derived
compounds in clinical trials. Nat. Prod. Rep. 2008, 25, 475-516.

99
3. Chandrasena, J. P. C. The Chemistry and Pharmacology of Ceylon and
Indian Medicinal Plants; H&C Press: Colombo, Sri Lanka, 1935.
4. Jayaweera, D. M. A. Medicinal Plants Used in Ceylon-Part 1: National
Science Council of Sri Lanka: Colombo, 1981.
5. Matsuda, H.; Li, Y. H.; Murakami, T.; Matsumura, N.; Yamahara, J.;
Yoshikawa, M. Chem. Pharm. Bull. 1998, 46, 1399-1403.
6. Yoshikawa, M.; Murakami, T.; Shimada, H.; Matsuda, H.; Yamahara, J.;
Tanabe, G.; Muraoka, O. Tetrahedron Lett. 1997, 38, 8367-8370.
7. Yoshikawa, M.; Xu, F. M.; Nakamura, S.; Wang, T.; Matsuda, H.; Tanabe,
G.; Muraoka, O. Heterocycles 2008, 75, 1397-1405.
8. Muraoka, O.; Xie, W. J.; Tanabe, G.; Amer, M. F. A.; Minematsu, T.;
Yoshikawa, M. Tetrahedron Lett. 2008, 49, 7315-7317.
9. Minami, Y.; Kurlyarna, C.; Ikeda, K.; Kato, A.; Takebayashi, K.; Adachi, I.;
Fleet, G. W. J.; Kettawan, A.; Karnoto, T.; Asano, N. Bioorg. Med. Chem. 2008, 16, 2734-2740.
10. Jayakanthan, K.; Mohan, S.; Pinto, B. M. J. Am. Chem. Soc. 2009, 131,
5621-5626.
11. Mohan, S.; Pinto, B. M. Carbohydr. Res. 2007, 342, 1551-1580.
12. Mohan, S.; Pinto, B. M. Collect. Czech. Chem. Commun. 2009, 74, 1117-
1136.
13. Johnston, B. D.; Jensen, H. H.; Pinto, B. M. J. Org. Chem. 2006, 71, 1111-
1118.
14. Tanabe, G.; Xie, W. J.; Ogawa, A.; Cao, C. N.; Minematsu, T.; Yoshikawa,
M.; Muraoka, O. Bioorg. Med. Chem. Lett. 2009, 19, 2195-2198.
15. Matsuda, H.; Yoshikawa, M.; Murakami, T.; Tanabe, G.; Muraoka, O. J.
Trad. Med. 2005, 22, 145-153.

100
16. Ozaki, S.; Oae, H.; Kitamura, S. J. Nat. Prod. 2008, 71, 981-984.
17. Sim, L.; Jayakanthan, K.; Mohan, S.; Nasi, R.; Johnston, B. D.; Pinto, B.
M.; Rose, D. R. Biochemistry 2010, 49, 443-451.
18. Nasi, R.; Patrick, B. O.; Sim, L.; Rose, D. R.; Pinto, B. M. J. Org. Chem.
2008, 73, 6172-6181.
19. Rossi, E. J.; Sim, L.; Kuntz, D. A.; Hahn, D.; Johnston, B. D.; Ghavami, A.;
Szczepina, M. G.; Kumar, N. S.; Sterchi, E. E.; Nichols, B. L.; Pinto, B. M.;
Rose, D. R. FEBS J. 2006, 273, 2673-2683.
20. Satoh, H.; Yoshimura, Y.; Sakata, S.; Miura, S.; Machida, H.; Matsuda, A.
Bioorg. Med. Chem. Lett. 1998, 8, 989-992.
21. Skaanderup, P. R.; Poulsen, C. S.; Hyldtoft, L.; Jorgensen, M. R.;
Madsen, R. Synthesis 2002, 1721-1727.
22. Sankar, M.; Sim, L.; David, R. R.; Pinto, B. M. Carbohydr. Res. 2007, 342,
901-912.
23. Ghavami, A.; Sadalapure, K. S.; Johnston, B. D.; Lobera, M.; Snider, B.
B.; Pinto, B. M. Synlett 2003, 1259-1262
24. Pellowska-Januszek, L.; Dmochowska, B.; Skorupa, E.; Chojnacki, J.;
Wojnowski, W.; Wiśniewski, A. P. Carbohydr. Res. 2004, 339, 1537-1544.
25. Andreana, P. R.; Sanders, T.; Janczuk, A . ; Warrick, J. I.; Wang, P. G.
Tetrahedron Lett. 2002, 43, 6525-6528.
26. Branchaud, B. P.; Meier, M. S. J. Org. Chem. 1989, 54, 1320-1326.
27. Koto, S.; Inada, S.; Yoshida, T.; Toyama, M.; Zen, S. Can. J. Chem. 1981,
59, 255-259.

101
3.8 Supporting Information
Potent glucosidase inhibitors: de-O-sulfonated ponkoranol and its stereoisomer
Razieh Eskandari, Douglas A. Kuntz, David R. Rose, B. Mario Pinto

102
ppm (t1)3.04.05.06.07.0
3.33
1.00
0.99
2.35
2.02
2.08
1.31
1.03
1.22
2.83
4.27
2.20
0.97
4.49
13.7
7
3.93
O
HOOH
OH
OMeS
BnO OBn
BnO
OTs
14600 MHz, MeOD
ppm (t1)50100
O
HOOH
OH
OMeS
BnO OBn
BnO
OTs
14150 MHz, MeOD

103
ppm (t1)3.04.05.06.07.01.
071.
04
3.03
1.00
1.95
23.1
3
5.15
1.20
1.68
2.20
1.02
1.01
2.05
1.20
3.16
0.98
1.17
O
HOOH
OH
OBnS
BnO OBn
BnO
OTs
18
1'
2'3'4'5'6'
600 MHz, MeOD
ppm (t1)4.004.50
H-6’a H-4
H-6’b
O
HOOH
OH
OBnS
BnO OBn
BnO
OTs
18
1'
2'3'4'5'6'
150 MHz, MeOD

104
ppm (t1)3.504.004.50
2.22
3.66
2.46
1.57
1.00
1.09
4.54
ppm (t1)50.055.060.065.070.075.0
S
OHHO
HO
OH
OH
OH
OH
OHCl
8
1' 2'3'
4'5'
6'
150 MHz, D2O
S
OHHO
HO
OH
OH
OH
OH
OHCl
8
1' 2'3'
4'5'
6'
600 MHz, D2O
105
ppm (t1)3.04.05.06.07.0
3.00
2.21
1.20
3.89
4.19
2.60
1.24
1.20
5.40
3.37
4.98
25.0
6
O
HOOH
OH
OBnS
BnO OBn
BnO
OTs
21
1'2'3'4'
5'6'
600 MHz, MeOD
ppm (t1)50100
O
HOOH
OH
OBnS
BnO OBn
BnO
OTs
21
1'2'3'4'
5'6'
150 MHz, MeOD

106
ppm (t1)3.504.004.50
0.83
0.88
1.00
1.80
1.23
9.03
S
OHHO
HO
OH
OH
OH
OH
OHCl
9
3'4'
5'6'1' 2'
600 MHz, D2O
ppm (t1)4.004.50
H-4H-6’a
H-6’b
O
HOOH
OH
OBnS
BnO OBn
BnO
OTs
21
1'2'3'4'
5'6'
500 MHz, MeOD

107
ppm (t1)45.050.055.060.065.070.075.080.0
S
OHHO
HO
OH
OH
OH
OH
OHCl
9
3'4'
5'6'1' 2'
150 MHz, D2O

108
CHAPTER 4: THE EFFECT OF HETEROATOM SUBSTITUTION OF SULFUR FOR SELENIUM IN GLUCOSIDASE INHIBITORS ON INTESTINAL α-
GLUCOSIDASE ACTIVITIES
This Chapter comprises the manuscript “The effect of heteroatom
substitution of sulfur for selenium in glucosidase inhibitors on intestinal α-
glucosidase activities” which was published in Journal of Chemical Society
Chemical Communications (2011, 47, 9134-9136).
Razieh Eskandari,a Kyra Jones, b David R. Rose,b B. Mario Pintoa
aDepartment of Chemistry, Simon Fraser University, Burnaby, British Columbia,
Canada V5A 1S6
b Department of Biology, University of Waterloo, Waterloo, Ontario,
Canada N2L 3G1

109
After we had successfully synthesized de-O-sulfonated ponkoranol and its
5'-epimer, we turned our attention to the structure-activity relationship (SAR)
studies of ponkoranol. In this Chapter, we describe the effect of selenium
substitution on the inhibitory activities of de-O-sulfonated ponkoranol and its 5'-
epimer. Thus, the selenium analogues of de-O-sulfonated ponkoranol were
synthesized and their glucosidase inhibitory activities against four human
intestinal glucosidase enzymes: maltase-glucoamylase MGAM (ntMGAM,
ctMGAM) and sucrase-isomaltase (ntSI, ctSI) were studied and compared with
the activities of the previously synthesized (Chapter 3) de-O-sulfonated
ponkoranol and its 5'-epimer. The thesis author performed all the experimental
synthetic work and the characterization of the compounds, and wrote and edited
the manuscript with Dr. B. Mario. Pinto. Mrs. Kyra Jones and Dr. David R. Rose
performed the enzyme inhibition studies.
Graphical abstract:
O
HOOH
OBnTsO Se
OHHO
HO
OH
OH
OH
OH
ClSe
OBnBnO
BnO+
R1 = OH, R2 = HR1 = H, R2 = OH
R2R1
7. R1 = OH, R2 = H8. R1 = H, R2 = OH
R2 R1

110
4.1 Keywords
Selenium analogue, maltase-glucoamylase, sucrase-isomaltase, de-O-
sulfonated ponkoranol.
4.2 Abstract
The synthesis of selenium analogues of de-O-sulfonated ponkoranol, a
naturally occuring sulfonium-ion glucosidase inhibitor isolated from Salacia
reticulata, and their evaluation as glucosidase inhibitors against two recombinant
intestinal enzymes maltase-glucoamylase (MGAM) and sucrase-isomaltase (SI)
are described.
4.3 Introduction
The roots and stems extract of Salacia reticulata, a large woody climbing
plant found in Sri Lanka and southern India, have traditionally been used for
treatment of type-2 diabetes.1 Human clinical trials on patients with type-2
diabetes, conducted with extract of Salacia reticulata, have shown effective
treatment of type-2 diabetes with minimal side effects.2 Our efforts in recent years
have focused on this novel class of α-glucosidase inhibitors comprising
sulfonium-ions as putative mimics of the oxacarbenium ion intermediates in
glucosidase-mediated hydrolysis reactions.3 Thus, we have described the
synthesis and stereochemical structure elucidation of the naturally-occurring
glucosidase inhibitors salacinol (1),4 ponkoranol (2),5 kotalanol (3),6 de-O-
sulfonated kotalanol (4)6 (Figure 4-1) from the plant extract of Salacia reticulata.

111
Interestingly, one of our synthetic compounds, de-O-sulfonated ponkoranol (5),7
(Figure 4-1), was isolated recently from the same plant.8
Figure 4-1: Components isolated from Salacia species.
The antidiabetic property of these herbal extract can be attributed, at least
in part, to the inhibition of intestinal α-glucosidases by these sulfonium-ion
components.9-11 Crystallographic studies of the N-terminal domain of human
intestinal maltase-glucoamylase (ntMGAM), a family 31 glycoside hydrolase
(GH31),12 with several candidate inhibitors suggested that the permanent positive
charge of these entities may mimic the oxacarbenium-ion like transition state of
the glucosidase-mediated hydrolysis reactions.13 We also suggested that the ring
conformation adopted by the thiocyclitol in kotalanol (3), a 3T2 conformation,
S
OHHO
OSO3 OH
OH OH
HO
OHOH
S
OHHO
OSO3 OH
OH OH
HO
OH
S
OHHO
OSO3
OH OH
HO
Kotalanol (3)Salacinol (1) Ponkoranol (2)
S
OHHO
OH OH
OH OH
HO
OHOH
CH3OSO3
De-O-sulfonated kotalanol (4) De-O-sulfonated ponkoranoll (5)
S
OHHO
OH OH
OH OH
HO
OH
Cl

112
closely resembles the proposed 4H3 conformation of the oxacarbenium-ion like
transition state.13
We have also reported the synthesis of the 5′-stereoisomer of de-O-
sulfonated ponkoranol 6 (Figure 4-2), and have shown that it is a potent (Ki = 15
nM) inhibitor of ntMGAM.7
Figure 4-2: Structure of the 5′-stereoisomer of de-O-sulfonated ponkoranol.
As part of our continuing interest in evaluating the effect of heteroatom
substitution in the sugar ring on glucosidase inhibitory activity, we have also
synthesized selenium congeners of several candidates as potential glucosidase
inhibitors. 14-16
Comparison of the proposed intermediate in the glucosidase-catalyzed
reaction and the selenonium ion, inferred from the X-ray crystal structures of
complexes of kotalanol, miglitol, and acarbose with ntMGAM,13 shows that the
selenonium ion in a 3T2 conformation, superimposes well on the 4H3
conformation of the oxacarbenium ion (Figure 4-3).
S
OHHO
OH OH
OH OH
HO
OH
Cl
6

113
Figure 4-3: Superimposition of the ring carbon atoms of the proposed intermediate in glucosidase-catalyzed reactions (in green) and the selenonium ion (in blue).
We now report the synthesis and enzyme inhibitory activity of analogues
of 5 and 6, in which the sulfur atom has been replaced by the heavier cognate
atom selenium to give 7 and 8, respectively (Figure 4-4).
Figure 4-4: Selenium analogues of de-O-sulfonated ponkoranol and its 5’-stereoisomer.
4.4 Results and discussion
Retrosynthetic analysis indicated that 7 or its analogue 8 could be
obtained by alkylation of an appropriately protected 1,4-anhydro-4-seleno-D-
arabinitol B at the ring heteroatom with agent C (Scheme 4-1).7
Se
OHHO
OH OH
OH OH
HO
OH
Se
OHHO
OH OH
OH OH
HO
OH
Cl Cl
7 8
1'2'
3'4'
5'6'
1'2'
3'4'
5' 6'
OHO
OH
OH
HO
4H3
1 2
3
4
5
O
HO
OH
OH
HO 123
4
5
SeHO OH
HO
3T2
1
2
3
4
Half chair
Twist
RSe
OHHO
HO
R
5
66
1
23
45 C3
C1 C2
C3
C4
C1
C4
C6
C5
C5 C2

114
Scheme 4-1: Retrosynthetic analysis.
The required benzyl (Bn)-protected D-selenoarabinitol (10)14 and benzyl 6-
O-p-toluenesulfonyl-α-D-gluco 9 or manno pyranoside 127 were obtained by
literature methods. The selenonium salts 11 and 13 were synthesized by
alkylation of 10 with 9 or 12 (1.2 equiv) in hexafluoroisopropanol (HFIP),
containing K2CO3, to give 11 and 13 in 55% and 45%, yield, respectively
(Scheme 4-2). In the case of compound 11, just one isomer was obtained at the
stereogenic selenium atom, whereas in the case of compound 13, a mixture of
isomers was obtained; 1H NMR spectroscopy showed the presence of two
isomers in a ratio of 10:1. The benzyl protecting groups were removed with boron
trichloride at -78 °C to give the desired sulfonium salts. During the course of
deprotection, some of the tosylate counterion was exchanged with the chloride
ion. Hence, the deprotected sulfonium salts were treated with Amberlyst A-26
(chloride form) to completely exchange the tosylate counterion with chloride ion.7
Finally, the products were reduced with NaBH4 to provide the desired selenium
analogues of de-O-sulfonated ponkoranol 7 and its 5’ epimer 8 in 52% and 45%
yield, respectively, over three steps (Scheme 4-2).
O
HOOH
OPL
Se
OPPO
POSe
OHHO
HO
OH
OH
OH
OH
L
L = leaving groupP = protecting group
+
A B C
R2 R1
R1R2
R1 = OH, R2 = H R1 = H, R2 = OH
7. R1 = OH, R2 = H, L = Cl8. R1 = H, R2 = OH, L = Cl

115
Scheme 4-2: Synthesis of compounds 7 and 8.
The stereochemistry at the selenium centre for the major isomer of 13 was
confirmed with the aid of a 2D-NOESY experiment, which showed a correlation
between H-4 and H-6′a, thus indicating that these atoms are syn-facial with
respect to the sulfonium salt ring (Figure 4-5). In the case of 11, the absolute
O
HOOH
OH
OBnTsO
HFIP, 70 oC, 4 d, 55%
O
HOOH
OH
OBnSe
BnO OBn
BnO
1. BCl3, CH2Cl2,
-78 oC, 6 h
2. Amberlyst A-26,
H2O, 3 h
3. NaBH4, H2O, 3 h
Se
OHHO
HO
OH
OH
OH
OH
OH
OTs
Cl
9 11
7
1'
1' 2'
2'
3'
3'
4'
4'
5'
5'
6'
6'
52%
Se
BnO OBn
BnO
10
O
HOOH
OH
OBnTsO
HFIP, 70 oC,4 d, 45%
O
HOOH
OH
OBnSe
BnO OBn
BnO
Se
OHHO
HO
OH
OH
OH
OH
OH
OTs
Cl
12 13
8
1'
2'3'
3'
4'
4'
5'
5'
6'
6'1' 2'
45%
Se
BnO OBn
BnO
1. BCl3, CH2Cl2,
-78 oC, 6 h
2. Amberlyst A-26,
H2O, 3 h
3. NaBH4, H2O, 3 h
10
d.r, ~ 10:1
d.r, ~ 10:1

116
configuration at the selenium centre was assigned by analogy since a NOESY
experiment was not possible owing to overlapping signals for H-4 and H-6′. This
assignment is consistent with our previous work.3
Figure 4-5: 2D-NOESY correlations of selected protons in compound 13.
Finally, we comment on the inhibitory activities of the compounds
synthesized in this study and previous study7 against the two recombinant
intestinal enzymes, maltase-glucoamylase (MGAM) and sucrase-isomaltase (SI),
critical for postamylase processing of starch-derived oligosaccharides into
glucose. Each of these enzymes exhibits two separate catalytic units (by gene
duplication) at their C- and N-terminal domains, resulting in ntMGAM, ctMGAM,
ntSI, and ctSI enzyme activities.17-22 There are also various alternative splicing
patterns of ctMGAM in mammals; two spliceforms from mice are discussed in this
paper: ctMGAM-N2 and ctMGAM-N20.23,24 The experimentally determined
inhibition constants (Kis) for the competitive inhibitors de-O-sulfonated
ponkoranol (5) and its synthetic analogues (6-8) are listed in Table 4-1.
O
OH
OH
OHBnO
Se
BnO OBn
13
H
H2C
4
BnO

117
Table 4-1: Experimentally determined Ki values (µM)a of compounds 5-8.
a Analysis of inhibition was performed using maltose as the substrate.
Comparison of the data in Table 4-1 indicates that substitution of the ring
sulfur atom in de-O-sulfonated ponkoranol (5) and its 5’ epimer (6) by selenium
results in a 13-fold and 23-fold improvement, respectively, in inhibition of ntSI.
NtSI demonstrates the broadest substrate specificity of all four subunits and
hydrolyses both α (1-4) and α (1-6) linkages.25 Similarly, substitution of sulfur by
selenium leads to an increase in inhibitory activity against ctSI (cf. 5 and 6 vs. 7
and 8). In contrast, little discrimination is observed between the congeners for
inhibition of ntMGAM or ctMGAM-N20. Interestingly, compounds 5-8 differentiate
between the spliceforms of ctMGAM, and none shows inhibition of ctMGAM-N2;
the reason for this selectivty must await structural analysis.
ctMGAM-N2 ctMGAM-N20 ntMGAM ctSI ntSI
5 No Inhibition 0.096 ± 0.015 0.043 ± 0.0017 0.103 ± 0.037 0.302 ± 0.123
6 No Inhibition 0.138 ± 0.068 0.015 ± 0.0017 0.132 ± 0.047 0.138 ± 0.012
7 No Inhibition 0.047 ± 0.014 0.038 ± 0.008 0.018 ± 0.004 0.013 ± 0.008
8 No Inhibition 0.041 ±0.027 0.025 ± 0.014 0.019 ±0.010 0.010 ± 0.002

118
4.5 Experimental
4.5.1 General methods
Optical rotations were measured at 23 °C. 1H and 13C NMR spectra were
recorded at 600 and 150 MHz, respectively. All assignments were confirmed with
the aid of two-dimensional 1H, 1H (COSYDFTP) or 1H, 13C (INVBTP) experiments
using standard pulse programs. Column chromatography was performed with
Silica 60 (230-400 mesh). High resolution mass spectra were obtained by the
electrospray ionization method, using an Agilent 6210 TOF LC/MS high
resolution magnetic sector mass spectrometer.
4.5.2 Enzyme kinetics
Kinetic parameters were determined by measuring the amount of glucose
produced upon the addition of enzyme at increasing maltose concentrations
(from 0.5 to 30 mM) in the presence of increasing inhibitor concentration (0-200
nM) by a two-step glucose oxidase assay in a 96-well plate. The enzyme was
allowed to act on the maltose substrate in the presence of inhibitor for 45 minutes
at 37°C. The reactions were then quenched with Tris-HCl to a final concentration
of 1M. Glucose oxidase reagent (Sigma-Aldrich) was then added to each well
(125µl) and the reactions were allowed to develop for 30 minutes at 37°C.
Reactions were performed in quadruplicate and absorbance measured at 405 nM
by a SpectraMax 190 Plate Reader (Molecular Devices). Absorbance readings
were averaged to give the final value, which was compared to a glucose standard
curve to determine the amount of glucose released by the enzyme from the
substrate. The program KaleidaGraph 4.1 was used to fit the data to the

119
Michaelis-Menten equation and estimate Km, Kmobs (Km in the presence of the
inhibitor) and Vmax of the catalytic subunits. Ki values for each inhibitor were
determined using the equation Ki = [I]/(Kmobs/Km)-1). The Ki values reported for
each inhibitor were determined by averaging the Ki values from three different
inhibitor concentrations. The weight of compounds 8 was adjusted for the
presence of major isomer. The data was also plotted on Lineweaver-Burk plots to
verify that the inhibitors were acting as competitive inhibitors. The methods used
for kinetic assays were reported previously.25
4.5.3 Compound characterization data
4.5.3.1 Benzyl 6-deoxy-6-[2,3,5-tri-O-benzyl-1,4-dideoxy-(R)-epi-seleniumylidene-D-arabinitol]-β-D-glycopyranoside-p-toluenesulfonate (11).
Benzyl 6-O-p-toluenesulfonyl-β-D-glucopyranoside 97 (310 mg, 0.73 mmol)
and 1,4-dideoxy-2,3,5-tri-O-benzyl-1,4-anhydro-4-seleno-D-arabinitol 1014 (409
mg, 0.88 mmol) were dissolved in HFIP (1.5 mL), containing anhydrous K2CO3
(10 mg). The mixture was stirred in a sealed reaction vessel in an oil bath at 65-
70 oC for 4 days. The mixture was cooled, then diluted with EtOAc, and
evaporated to give a syrupy residue. Purification by column chromatography
(CHCl3/MeOH 95:5) gave the sulfonium salt 11 as a white amorphous solid (358
mg, 55%). [α] 23D = + 22o. 1H NMR (MeOD) δ 7.74-7.24 (24H, m, Ar), 4.78 (1H,
dd, J1,2 = 7.8, J2,3 = 4.2 Hz, H-2), 4.68-4.49 (8H, m, 4CH2-Ph), 4.55 (1H, m, H-3),
4.48 (1H, m, H-4), 4.41 (1H, d, J1′,2′ = 7.8 Hz, H-1′), 3.98 (1H, m, H-6′a), 3.91 (1H,
m, H-1a), 3.84 (1H, dd, J5a,4 = 6.3, J5a,5b = 10.3 Hz, H-5a), 3.75 (3H, m, H-6′b, H-
3′, H-5b), 3.70 (1H, dd, J1,2 = 2.8, J1a,1b = 12.5 Hz, H-1b), 3.40 (1H, t, J4′,5′ =

120
J6′,5′ = 9.1 Hz, H-5′), 3.28 (2H, m, H-2′, H-4′), 2.38 (3H, s, Me). 13C NMR (MeOD)
δ 142.2-125.5 (m, Ar), 103.0 (C-1′), 83.8 (C-2), 83.5 (C-3), 75.8 (C-5′), 73.4 (C-
4′), 73.3 (C-2′), 73.1, 72.0, 71.7, 71.3 (4CH2-Ph), 71.8 (C-3′), 66.5 (C-5), 66.2 (C-
4), 45.5 (C-1), 45.2 (C-6′), 19.8 (Me). HRMS Calcd for C39H45O8Se (M+.):
721.2278. Found: 721.2279.
4.5.3.2 1,4-Dideoxy-1,4-[[2S, 3S, 4R, 5S]-2,3,4,5,6-pentahydroxyhexyl]-(R/S)-epi-seleniumylidine]-D-arabinitol chloride (7).
Compound 11 (200 mg, 0.22 mmol) was dissolved in CH2Cl2 (20 mL), the
mixture was cooled to -78 oC, and BCl3 (1M solution in CH2Cl2, 3.6 mmol) was
added under N2. The reaction mixture was stirred at the same temperature for 30
min, and then allowed to warm to -5 oC for 6 h. The reaction was cooled to -78 oC
and quenched by addition of MeOH (5 mL), the solvents were removed, and the
residue was co-evaporated with MeOH (2 × 5 mL). The crude residue was
dissolved in H2O (10 mL), Amberlyst A-26 resin (200 mg) was added, and the
reaction mixture was stirred at room temperature for 3 h. Filtration through
cotton, followed by solvent removal gave the crude hemiacetal. The crude
product was dissolved in water (8 mL), and the solution was stirred at room
temperature while NaBH4 (34 mg, 0.9 mmol) was added in small portions over 30
min. Stirring was continued for another 3 h and the mixture was acidified to pH <
4 by dropwise addition of 2M HCl. The mixture was evaporated to dryness and
the residue was co-evaporated with anhydrous MeOH (3 × 30 mL). Treatment of
the solid residue with 50% EtOAc:MeOH (5-10 mL) resulted in precipitation of
most of the borate salt. Filtration through cotton, followed by solvent removal

121
gave the crude compound. The residue was purified by crystallization with
minimum amount of MeOH to give 7 as a colorless solid (46 mg, 52%).
[α] 23D = + 12.8 o, (c = 0.5 , H2O). 1H NMR (D2O) δ 4.76 (1H, dd, J1,2 = J2,3
= 3.7 Hz, H-2), 4.46 (1H, t, br, H-3), 4.20 (1H, m, H-2′), 4.15 (1H, m, H-4), 4.05
(1H, dd, J4,5a = 12.2, J5a,5b = 4.8 Hz, 5a) 3.92-3.87 (2H, m, H-5b, H-1′a), 3.78-
3.74 (5H, m, H-5′, H-4′, H-1′b, H-1a, H-1b), 3.69-3.65 (2H, m, H-3′, H-6′a ), 3.55
(1H, dd, J6′a,6,b = 10.7, J5′,6′b = 5.9 Hz, H-6′b). 13C NMR (D2O) δ 77.8 (C-3), 77.2
(C-2), 73.2 (C-3′), 72.2 (C-5′), 69.1 (C-4), 69.0 (C-4′), 67.0 (C-2′), 61.8(C-6′),
558.9 (C-5), 47.0 (C-1′), 44.7 (C-1). HRMS Calcd for C11H23O8Se (M+.):
363.0551. Found: 363.0559.
4.5.3.3 Benzyl 6-deoxy-6-[2,3,5-tri-O-benzyl-1,4-dideoxy-(R)-epi-seleniumylidene-D-arabinitol]-α-D-mannopyranoside-p-toluenesulfonate (13).
Reaction of 1,4-dideoxy-2,3,5-tri-O-benzyl-1,4-anhydro-4-seleno-D-
arabinitol 10 (660 mg, 1.4 mmol) with benzyl 6-O-p-toluenesulfonyl-β-D-
mannopyranoside 13 (500 mg, 1.2 mmol) in HFIP (1.5 mL), containing
anhydrous K2CO3 (10 mg) at 65-70 oC for 4 days gave the selenonium salt 13
as a foam (473 mg, 45%) after purification by column chromatography
(CHCl3/MeOH (95:5)). Analysis by NMR showed that the product was a mixture
of two isomers (~10:1) at the stereogenic selenium centre. [α] 23D = + 13.6 o Data
for the major diastereomer: 1H NMR (MeOD) δ 7.74-7.24 (24H, m, Ar), 4.86 (1H,
m, H-1′), 4.81 (1H, m, H-2), 4.71-4.50 (8H, m, 4CH2-Ph), 4.58 (1H, m, H-3), 4.42
(1H, dd, J3,4 = 6.8, J4,5 = 9.4 Hz, H-4 ), 4.03 (1H, d, J1,2 = 12.8 Hz, H-1a), 3.94
(2H, m, H-6′a, H-4′), 3.88 (1H, dd, J1′,2′ =2.0 J2′,3′ = 2.7 Hz, H-2′) 3.83 (1H, dd, J4,5

122
= 6.7, J5a,5b = 10.3 Hz, H-5a), 3.78-3.73 (4H, m, H-1b, H-5b, H-3′, H-6b), 3.59
(1H, t, J4′,5′ = J5′,6′ = 9.3 Hz, H-5′), 2.39 (3H, s, Me). 13C NMR (MeOD) δ 141.7-
125.1 (m, Ar), 100.0 (C-1′), 83.7 (C-2), 83.2 (C-3), 72.6, 71.5, 71.2, 69.5 (4CH2-
Ph), 70.2 (C-2′), 70.0(C-5′), 69.9 (C-3′), 68.9 (C-4′), 66.1 (C-5),65.7 (C-4), 45.9
(C-1), 45.6 (C-6′), 19.4 (Me). HRMS Calcd for C39H45O8Se (M+.): 721.2278.
Found: 721.2278.
4.5.3.4 1,4-Dideoxy-1,4-[[2S, 3S, 4R, 5R]-2,3,4,5,6-pentahydroxy-hexyl]-(R/S)-epi-seleniumylidene]-D-arabinitol chloride (8).
Compound 8 was obtained as a colorless solid (40 mg, 45%) from 13 (200
mg, 0.22 mmol) using the same procedure that was used to obtain 7. [α] 23D = + 4o.
1H NMR (D2O) δ 4.85 (1H, dd, J1,2 =7.6, J2,3 = 3.85, H-2), 4.54 (1H, t, J3,4 = J2,3 =
3.5 Hz,, H-3), 4.30-4.23 (2H, m, H-2′, H-4), 4.13 (1H, dd, J4,5a = 12.4, J5a,5b = 5.2
Hz, 5a) 4.02-3.97 (2H, m, H-1′a,H-5b), 3.94-3.82 (5H, m, H-6′a, H-3′, H-1′b, H-
1a, H-1b), 3.78-3.73 (2H, m, H-4′, H-5′b) 3.68 (1H, dd, J6′a,6,b = 11.7, J5′,6′b = 5.6
Hz, H-6′b). 13C NMR (D2O) δ 78.3 (C-3), 77.9 (C-2), 72.1 (C-3′), 70.7(C-5′), 69.6
(C-4), 69.2 (C-4′), 67.5 (C-2′), 63.1 (C-6′), 59.4 (C-5), 48.1 (C-1′), 45.3 (C-1).
HRMS Calcd for C11H23O8Se (M+.): 363.0553. Found: 363.0544.
4.6 Acknowledgments
We thank Buford L. Nichols, Roberto Quezada-Calvillo and Hassan Naim
for reagents for recombinant expression, and Lyann Sim for MGAM and SI
enzyme purification. Work by K.J. was funded by CIHR and the Canadian
Digestive Health Foundation (CDHF). We also thank the Canadian Institutes of

123
Health Research and the Heart and Stroke Foundation of Ontario (NA-6305) for
financial support.
4.7 References
1. (a) J. P. C. Chandrasena, The Chemistry and Pharmacology of Ceylon
and Indian Medicinal Plants; H&C press: Colombo, Sri Lanka, 1935. (b) D.
M. A. Jayaweera, Medicinal Plants Used in Ceylon-Part 1;
NationalScience Council of Sri Lanka: Colombo, 1981; p 77. (c) P. S.
Vaidyartanam, In Indian Medicinal Plants: a compendium of 500 species,
P. K. Warrier V. P. K. Nambiar and C. Ramankutty, Eds.; Orient Longman:
Madras, India, 1993, pp. 47-48.
2. M. H. S. Jayawardena, N. M. W. de Alwis, V. Hettigoda and D. J. S.
Fernando, J. Ethnopharmacol. 2005, 97, 215–218.
3. For reviews see: a) S. Mohan and B. M. Pinto, Carbohydr. Res. 2007, 342,
1551-1580. b) S. Mohan and B. M. Pinto, Collect. Czech. Chem. Commun.
2009, 74, 1117-1136. c) S. Mohan and B. M. Pinto, Nat. Prod. Rep. 2010,
27, 481-488.
4. A. Ghavami, B. D. Johnston and B. M. Pinto, J. Org. Chem. 2001, 66,
2312–2317.
5. B. D. Johnston, H. H. Jensen and B. M. Pinto, J. Org. Chem. 2006, 71,
1111–1118.
6. K. Jayakanthan, S. Mohan and B. M. Pinto, J. Am. Chem. Soc. 2009, 131,
5621-5626.
7. R. Eskandari, D. A. Kuntz, D. R. Rose and B. M. Pinto, Org. Lett. 2010, 12, 1632-1635.

124
8. W. Xie, G. Tanabe, J. Akaki, T. Morikawa, K. Ninomiya, T. Minematsu, M.
Yoshikawa, X. Wu and O. Muraoka, Bioorg & Med. Chem. 2011, 19, 2015-
2022.
9. M. Yoshikawa, T. Murakami, H. Shimada, H. Matsuda, J. Yamahara, G.
Tanabe and O. Muraoka, Tetrahedron Lett. 1997, 38, 8367–8370.
10. M. Yoshikawa, T. Murakami, K. Yashiro and H. Matsuda, Chem. Pharm.
Bull. 1998, 46, 1339–1340.
11. S. Ozaki, H. Oe and S. Kitamura, J. Nat. Prod. 2008, 71, 981–984. (b) O.
Muraoka, W. Xie, G. Tanabe, M. F. A. Amer, T. Minematsu and M.
Yoshikawa, Tetrahedron Lett. 2008, 49, 7315–7317.
12. L. Sim, R. Quezada-Calvillo, E. E. Sterchi, B. L. Nichols and D. R. Rose, J.
Mol. Biol. 2008, 375, 782-792.
13. L. Sim, K. Jayakanthan, S. Mohan, R. Nasi, B. D. Johnston, B. M. Pinto,
and D. R. Rose, Biochemistry 2010, 49, 443–451.
14. B. D. Johnston, A. Ghavami, M. T. Jensen, B. Svensson and B. M. Pinto,
J. Am. Chem. Soc. 2002, 124, 8245–8250.
15. H. Liu, R. Nasi, K. Jayakanthan, L. Sim, H. Heipel, D. R. Rose and B.M.
Pinto, J. Org. Chem. 2007, 72, 6562–6572.
16. S. Mohan, K. Jayakanthan, R. Nasi, D. A. Kuntz, D. R. Rose and B. M.
Pinto, Org. Lett. 2010, 12, 1088.
17. B. L. Nichols, J. Eldering, S. E. Avery, D. Hahn, A. Quaroni and E. E.
Sterchi, J. Biol. Chem. 1998, 273, 3076-3081.
18. R. Quezada-Calvillo, L. Sim, Z. Ao, B. R. Hamaker, A. Quaroni, G. D.
Brayer, E. E. Sterchi, C. C. Robayo-Torres, D. R. Rose and B. L. Nichols,
J. Nutr. 2008, 138, 685-692.

125
19. B. L. Nichols, S. E. Avery, P. Sen, D. M. Swallow, D. Hahn and E. E.
Sterchi, PNAS 2003, 100, 1432-1437.
20. H. Heymann, D. Breitmeier and S. Günther, Biol. Chem. Hoppe-Seyler
1995, 376, 249-253.
21. H. Heymann and S. Günther, Biol. Chem. Hoppe-Seyler 1994, 375, 451-
455.
22. C. Robayo-Torres, R. Quezada-Calvillo and B. L. Nichols, Clin.
Gastroenterol. Hepatol. 2006, 4, 276-287.
23. Naumoff, D. G. Mol. Biol. (Mosk). 2007, 41, 1056-1068.
24. K. Jones, L. Sim, S. Mohan, K. Jayakanthan, H. Liu, S. Avery, H. H.
Naim, R. Quezada-Calvillo, B. L. Nichols, B. M. Pinto and D. R. Rose,
Bioorg. Med. Chem. 2011, 19, 3929-3934.
25. L. Sim, C. Willemsma, S. Mohan, H. Y. Naim, B.M. Pinto and D. R. Rose,
J. Biol. Chem. 2010, 285, 17763-17770.

126
4.8 Supporting Information
The effect of heteroatom substitution of sulfur for selenium
in glucosidase inhibitors on intestinal α-glucosidase activities
Razieh Eskandari, Kyra Jones, David R. Rose, B. Mario Pinto

127
ppm (t1)2.03.04.05.06.07.08.0
ppm (t1)50100
O
HOOH
OH
OBnSe
BnO OBn
BnO
OTs
11
1'
2'3'4'
5'6'
O
HOOH
OH
OBnSe
BnO OBn
BnO
OTs
11
1'
2'3'4'
5'6'

128
ppm (t1)3.504.004.50
Se
OHHO
HO
OH
OH
OH
OH
OHCl
7
1' 2'3'
4'5'
6'
ppm (t1)45.050.055.060.065.070.075.080.0
Se
OHHO
HO
OH
OH
OH
OH
OHCl
7
1' 2'3'
4'5'
6'

129
ppm (t1)2.03.04.05.06.07.08.0
O
HOOH
OH
OBnSe
BnO OBn
BnO
OTs
13
1'
2'3'4'
5'6'
ppm (t1)50100150
O
HOOH
OH
OBnSe
BnO OBn
BnO
OTs
13
1'
2'3'4'
5'6'

130
H-4 H-6’a H-6’b
O
OH
OH
OHBnO
Se
BnO OBn
13
H
H2C
4
BnO
ppm (t1)4.004.505.00
Se
OHHO
HO
OH
OH
OH
OH
OHCl
8
3'4'
5'6'1' 2'

131
Representative Lineweaver-Burk plot of ntSI inhibited by 5 at concentrations of 0 nM, 75 nM, 125 nM, and 200 nM.
ppm (t1)4050607080
Se
OHHO
HO
OH
OH
OH
OH
OHCl
8
3'4'
5'6'1' 2'

132
CHAPTER 5: PROBING THE ACTIVE-SITE REQUIREMENTS OF HUMAN INTESTINAL N-TERMINAL
MALTASE-GLUCOAMYLASE: THE EFFECT OF REPLACING THE SULFATE MOIETY BY A METHYL
ETHER IN PONKORANOL, A NATURALLY-OCCURRING α-GLUCOSIDASE INHIBITOR
This Chapter comprises the manuscript “Probing the active-site requirements of human intestinal N-terminal maltase-glucoamylase: The effect of
replacing the sulfate moiety by a methyl ether in ponkoranol, a naturally-
occurring α-glucosidase inhibitor” which was published in Bioorganic and
Medicinal Chemistry Letters (2010, 20, 5686-5689).
Razieh Eskandari,a Kyra Jones,b David R. Rose,b B. Mario Pintoa
aDepartment of Chemistry, Simon Fraser University, Burnaby, British Columbia,
Canada V5A 1S6
b Department of Biology, University of Waterloo, Waterloo, Ontario,
Canada N2L 3G1

133
The crystal structures of N-terminal maltase-glucoamylase (ntMGAM) in
complex with kotalanol and de-O-sulfonated kotalanol have shown that removing
the sulfate group enhances the inhibitory activity presumably due to alleviation of
interaction between the polar sulfate group and hydrophobic groups in the active
site. In this Chapter, we set out to probe the active-site requirements of ntMGAM
using 3’-O-methylponkoranol. We describe here the synthesis of this compound
and the results obtained from the evaluation of its inhibitory activity against N-
terminal maltase-glucoamylase (ntMGAM). The thesis author performed all the
experimental synthetic work and the characterization of the compound, and wrote
and edited the manuscript with Dr. B. Mario Pinto. Mrs. Kyra Jones and Dr. David
R. Rose performed the enzyme inhibition studies.
Graphical abstract:
O
MeOO
OBnTsO S
OHHO
HO
OH
OH
OMe
OH
ClS
OBnBnO
BnO+ O
OH
MeO
OMe

134
5.1 Keywords
α-Glucosidase inhibitors, 3’-O-methylponkoranol, maltase-glucoamylase,
thiosugar, sulfonium-ion.
5.2 Abstract
Ponkoranol is a naturally-occurring glucosidase inhibitor isolated from the
plant Salacia reticulata. The compound comprises a sulfonium-ion with an
internal sulfate counter ion. We report here an efficient synthetic route to 3'-O-
methylponkoranol to test the hypothesis that occupation of a hydrophobic pocket
by a methyl group instead of the polar sulfate ion within the active site of human
N-terminal maltase-glucoamylase would be beneficial. The synthetic strategy
relies on the nucleophilic attack of 2,3,5-tri-O-benzyl-1,4-anhydro-4-thio-D-
arabinitol at the C-6 position of benzyl 6-O-p-toluenesulfonyl β-D-
glucopyranoside, followed by deprotection using boron trichloride and reduction
with sodium borohydride. The target compound inhibited the N-terminal catalytic
domain of intestinal human maltase-glucoamylase (ntMGAM) with a Ki value of
0.50 ± 0.04 µM, higher than those of de-O-sulfonated ponkoranol (Ki = 43 ± 3
nM), or its 5’-stereoisomer (Ki = 15 ± 1 nM). We conclude that the interaction of
the methyl group with hydrophobic residues in the active site is not as beneficial
to inhibition of ntMGAM as the other interactions of the polyhydroxylated chain
with active-site residues.

135
5.3 Introduction
Glycosidase inhibitors have many potential therapeutic applications
because glycosidase enzyme-catalyzed hydrolysis of complex carbohydrates is
biologically widespread, and has been implicated in several disease states.1,2 For
example, inhibition of starch-hydrolyzing enzymes such as pancreatic α-amylase
and intestinal α-glucosidases that leads to a delay in digestion of ingested
carbohydrates is one of the therapeutic approaches for the treatment of type-2
diabetes.3,4 Bioactive components isolated from medicinal plants often provide
the lead structures for drug development programs.5,6 For example, a relatively
new and interesting class of inhibitors is the sulfonium-ion containing inhibitors,
which were first isolated from Salacia reticulata, a plant that is used in traditional
Ayurvedic medicine in Sri Lanka and South India for treating type-2 diabetes.7-9
The active components in Salacia reticulata were found to include salaprinol
(1),10 salacinol (2),11 ponkoranol (3), 10 kotalanol (4),12 de-O-sulfonated kotalanol
(5),13 and de-O-sulfonated salacinol (6)14 (Figure 5-1), whose structures comprise
a 1,4-anhydro-4-thio-D-arabinitol core and polyhydroxylated acyclic chain (Figure
5-1).

136
Figure 5-1: Components isolated from Salacia species.
Comparison of the inhibitory activities against the human N-terminal
catalytic domain of maltase-glucoamylase (ntMGAM) of de-O-sulfonated
kotalanol (5) and some of its stereoisomers vs kotalanol (4) and the
corresponding sulfated stereoisomers, respectively, revealed that the de-O-
sulfonated analogues were more potent inhibitors than the parent
compounds.15,16 Furthermore, we have shown recently that de-O-sulfonated
ponkoranol (7) (Ki = 0.043 ± 0.01 µM) and its 5’-stereoisomer (8) (Ki = 0.015 ±
0.01 µM) are more potent inhibitors of ntMGAM than ponkoranol (3) itself (Ki =
0.17 ± 0.03 µM) (Figure 5-2).17
S
OHHO
OSO3 OH
OH OH
HO
OHOH
S
OHHO
OSO3 OH
OH OH
HO
OH
S
OHHO
OSO3
OH OH
HOS
OHHO
OSO3
OH
HO
Kotalanol (4)
Salacinol (2) Ponkoranol (3)Salaprinol (1)
S
OHHO
OH OH
OH OH
HO
OHOH
CH3OSO3
De-O-sulfonated kotalanol (5)
S
OHHO
OH
OH OH
HOHCO2
De-O-sulfonated salacinol (6)

137
Figure 5-2: De-O-sulfonated ponkoranol and its 5’-stereoisomer.
Our previous X-ray crystallographic studies of ntMGAM in complex with
kotalanol (4) and de-O-sulfonated kotalanol (5) had indicated that removal of the
sulfate group affects the conformation of the rest of the polyhydroxylated chain.18
We concluded that although the stereoconfiguration at C3’ does not affect
inhibitory activity, the proximity of the sulfate group to the large hydrophobic
groups (Y299, W406 and F575) likely restricts its conformational freedom.
Therefore, by removing the sulfate group, the positional constraint imposed by
the bulky hydrophobic residues surrounding the C3’ group is relieved, allowing
the rest of the polyhydroxylated chain to make optimal contacts with the ntMGAM
active site (Figure 5-3).18
S
OHHO
OH OH
OH OH
HO
OH
S
OHHO
OH OH
OH OH
HO
OH
Cl Cl
7 8
1'2'
3'4'
5'6'
1'2'
3'4'
5' 6'

138
Figure 5-3: Effect of removing the sulfate group. Superposition of kotalanol (4) (orange) and de-O-sulfonated kotalanol (5) (purple) structures. Double-headed arrows show the proximities of the sulfate group to the surrounding hydrophobic residues Y299, W406 and F575. (Reproduced with permission from Biochemistry, 2010, 49, 443–451. Copyright American Chemical Society).
In view of these findings, it was of interest to question whether replacing
the sulfate group with a hydrophobic methyl ether in ponkoranol (9) (Figure 5-3)
would increase its inhibitory properties through hydrophobic interactions in the
site compared to de-O-sulfonated ponkoranol 7.
Figure 5-4: 3’-O-Methylponkoranol.
S
OHHO
OMe OH
OH OH
HO
OH
Cl
9
1'2'
3'4'
5'6'

139
5.4 Results and discussion
We report here an efficient synthetic route to 3’-O-methylponkoranol (9).
Our synthetic strategy involved the alkylation of an appropriate protected
anhydrothioarabinitol B at the ring sulfur atom with agent C. Agent C could be
obtained by methylation of protected D-glucose at C-4’(Scheme 5-1).
Scheme 5-1: Retrosynthetic analysis.
Thus, benzyl 2,3-di-O-benzyl-4-O-methyl-6-O-tosyl-β-D-glucopyranoside
(11) was obtained from 1019 by treatment with a mixture of methyl iodide in
aqueous sodium hydroxide and dimethyl sulfoxide (50% w/w) 20 to afford 11. Our
initial attempt at the coupling reaction employed 11 with the anhydrothioarabinitol
(12)21 in 1,1,1,3,3,3-hexafluoroisopropyl alcohol (HFIP) at 70 °C, as in our
previous work (Scheme 2).22 No product formation and decomposition of the
starting material were observed by TLC; the coupling reaction of benzyl 4-O-
methyl-6-O-tosyl-β-D-glucopyranoside (13) with the anhydrothioarabinitol (12)
was also unsuccessful, the same result being observed (Scheme 5-2).
O
MeOOP
OPL
S
OPPO
POS
OHHO
HO
OH
OH
OMe
OH
L
L = leaving groupP = protecting group
+
A B C
OHOP
O
HOOP
OPL
OP

140
Scheme 5-2: First attempted synthesis of 9.
Next, a trifluoromethanesulfonyl (OTf) group was chosen as a leaving
group to ensure milder conditions for the coupling reaction. Thus, the primary
hydroxyl group in the diol 1419 was protected as its TBDMS ether followed by
sequential methylation of the secondary hydroxyl group to give 15 in 53% over
two steps (Scheme 5-3). Removal of the TBDMS group using
tetrabutylammonium fluoride (TBAF) gave the alcohol which was treated with
trifluoromethanesulfonyl anhydride to yield the glycoside 16 in 76% yield over two
steps. The coupling reaction of the OBn-protected thioether (12) with benzyl 2,3-
di-O-benzyl-4-O-methyl-6-O-trifluoromethanesulfonyl-α-D-glucopyranoside 16
was carried out in dry CH2Cl2 at room temperature to give the corresponding
O
MeOOBn
OBn
OBnTsO
HFIP, 70oC
11
Starting material decompositon
MeI, NaOH, DMSO, rt. 85%
S
BnO OBn
BnO
12
O
HOOBn
OBn
OBnTsO
10
O
MeOOH
OH
OBnTsO
HFIP, 70oC
13
S
BnO OBn
BnO
12 Starting material decompositon

141
protected sulfonium-ion 17 as a 4:1 mixture of diastereomers at the stereogenic
sulfur center. However, attempts to separate the mixture of diastereomers, even
after deprotection and reduction, were unsuccessful.
Scheme 5-3: Second attempted synthesis of 9.
In another attempt, the tosylate 18, containing a butane diacetal (BDA)
protecting group, was chosen as the coupling partner. As shown in Scheme 5-4,
the tosylate 18 was synthesized in two steps from benzyl 2,3-O-[(2R,3R)-2,3-
dimethoxybutane-2,3-diyl]-β-D-glucopyranoside 19 which was, in turn, prepared
from D-glucose according to literature procedures.23 Thus, compound 19 was
treated with p-toluenesulfonyl chloride to afford the p-toluenesulfonyl ester 20 in
1.TBDMSCl,Imid,2.MeI, NaOHDMSO, rt, 53%
O
MeOOBn
OBn
OBnTBSO
CH2Cl2, rt, 90%
O
MeOOBn
OBn
OBnS
BnO OBn
BnO
S
OHHO
HO
OH
OH
OMe
OH
OHOTfCl
15
179
1'
2'3'
3'
4'
4'5'5'6'
6'1' 2'
57%
S
BnO OBn
BnO
O
HOOBn
OBn
OBnHO
1. BCl3, CH2Cl2,
-78 oC, 6 h
2. Amberlyst A-26,H2O, 3 h3. NaBH4, H2O, 3 h
14
1. TBAF, THF2. TfO2, Pyridine, CH2Cl2, 76%
O
MeOOBn
OBn
OBnTfO
16
12

142
75%, which was methylated with methyl iodide to give 18 in 91% yield. The
coupling reactions of the OBn-protected 1,4-anhydro-4-thio-D-arabinitol (12) with
the p-toluenesulfonyl ester 18 were carried out in HFIP to give the protected
sulfonium-ion 21 as a single diastereomer in 70% yield (Scheme 5-4).
Deprotection of the coupled product 21 was carried out in a two-step
procedure. The benzyl group was first removed by treatment with boron
trichloride at -78 °C in CH2Cl2 ,followed by sequential BDA deprotection with 80%
TFA. During the course of benzyl group deprotection with BCl3, the p-
toluenesulfonate counterion was partially exchanged with chloride ion. Similar
results were observed in our previous work.18 Hence, after deprotection, the
product was treated with Amberlyst A-26 resin (chloride form) to completely
exchange the p-toluenesulfonate counterion with chloride ion. Finally, the crude
product was reduced with NaBH4 to provide the desired 3'-O-methylponkoranol 9
in 51% yield over three steps (Scheme 5-4).
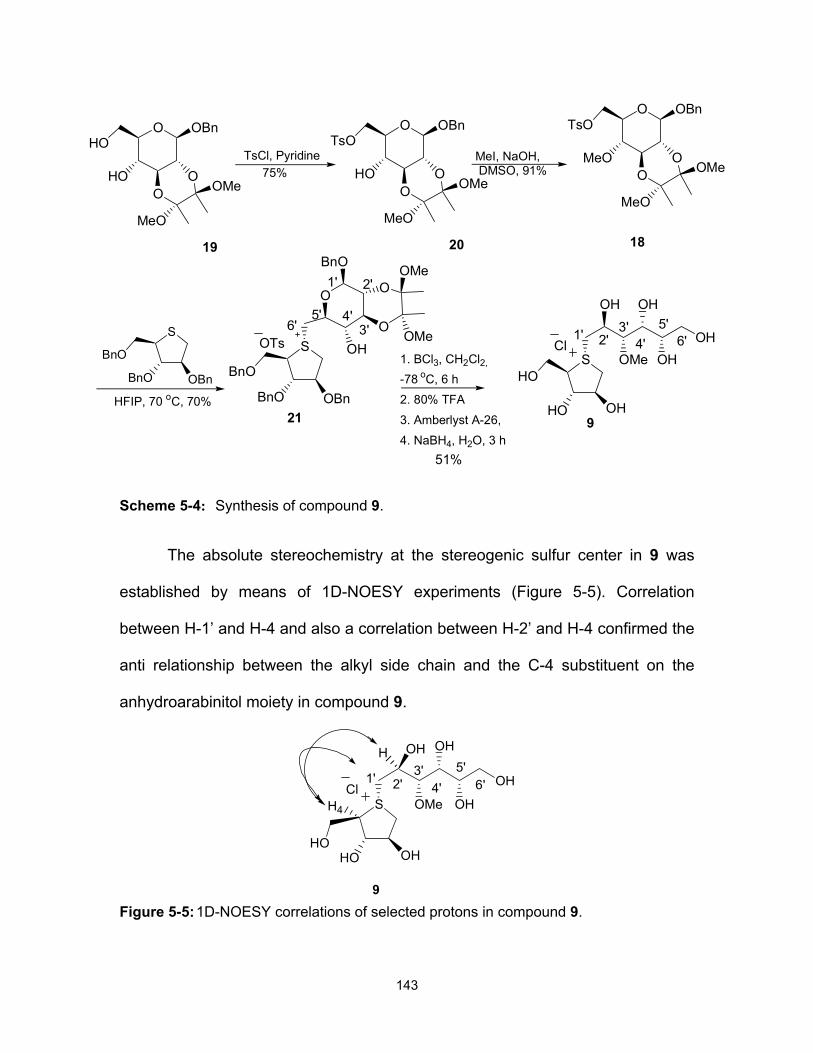
143
Scheme 5-4: Synthesis of compound 9.
The absolute stereochemistry at the stereogenic sulfur center in 9 was
established by means of 1D-NOESY experiments (Figure 5-5). Correlation
between H-1’ and H-4 and also a correlation between H-2’ and H-4 confirmed the
anti relationship between the alkyl side chain and the C-4 substituent on the
anhydroarabinitol moiety in compound 9.
Figure 5-5: 1D-NOESY correlations of selected protons in compound 9.
HFIP, 70 oC, 70%
S
BnO OBn
BnOS
OHHO
HO
OH
OH
OMe
OH
OHOTs Cl
21 9
3'4'
5'6'1' 2'
51%
S
BnO OBn
BnO
O
HOO
O
OBnHO
1. BCl3, CH2Cl2,
-78 oC, 6 h
2. 80% TFA
3. Amberlyst A-26,4. NaBH4, H2O, 3 h
19
OMe
MeO
O
MeOO
O
OBnTsO
OMe
MeO
TsCl, Pyridine
1' 2'
3'4'5'
6'
O
OHO
O
BnOOMe
OMe
18
MeI, NaOH, DMSO, 91%
O
HOO
O
OBnTsO
20
OMe
MeO
75%
S
OHHOHO
OH
OH
OMe
OH
OHCl
9
3'4'
5'6'1' 2'
H4
H

144
Finally, the inhibitory activity of compound 9 was examined against the N-
terminal catalytic domain of recombinant human maltase-glucoamylase
(ntMGAM), a critical intestinal glucosidase for processing starch-derived
oligosaccharides into glucose. The 3’-O-methylponkoranol 9 inhibited ntMGAM
with a Ki value of 0.50 ± 0.04 µM; by comparison, de-O-sulfonated ponkoranol 7
and its 5’-stereoisomer 8 inhibited ntMGAM with Ki values of 43 ± 3 and 15 ± 1
nM, respectively.17. We conclude, therefore, that the hydrophobic interactions
between the methyl group and the hydrophobic residues Y299, W406 and F575
in the active site are not as optimal as the the interactions of the latter groups
with the rest of polyhydroxylated chain in the absence of the methyl ether; a
similar situation is also observed in the binding of the sulfated compound,
ponkoranol (3) (Ki = 0.17 ± 0.03 µM).
5.5 Experimental
5.5.1 General methods
Optical rotations were measured at 23 °C. 1H and 13C NMR spectra were
recorded at 600 and 150 MHz, respectively. All assignments were confirmed with
the aid of two-dimensional 1H, 1H (COSYDFTP) or 1H, 13C (INVBTP) experiments
using standard pulse programs. Column chromatography was performed with
Silica 60 (230-400 mesh). Reverse column chromatography was performed with
Silica C-18 cartridges. High resolution mass spectra were obtained by the
electrospray ionization method, using an Agilent 6210 TOF LC/MS high
resolution magnetic sector mass spectrometer.

145
5.5.2 Enzyme kinetics
Activity of recombinant N-terminal domain of maltase-glucoamylase
(ntMGAM) was determined using the glucose oxidase assay17 to follow the
production of glucose from maltose upon addition of the enzyme (0.8 nM). A no-
inhibitor control and five different inhibitor concentrations were used in
combination with 7 different maltose concentrations (ranging from 1.5 to 24 mM).
A reaction time of 60 min at 37 ºC was employed. Reactions were linear within
this time frame. Values of Ki and standard deviations were determined by the
program GraFit 4.0.14 (Erithacus Software) which employs nonlinear fitting of the
data for each inhibitor concentration to the Michaelis-Menten equation.
5.5.3 Compound characterization data
5.5.3.1 General Procedure for Methylation
To a vigorously stirred solution of the glucopyranoside in dimethyl
sulfoxide (DMSO) was added 50% aqueous sodium hydroxide (1.7 equiv.) to
form a gel-like suspension. Methyl iodide (1.5 equiv), was added dropwise in one
portion, and the mixture was further stirred for 2-15 min, then quenched with 2:1
ether:water. The organic layer was separated and the aqueous layer was
extracted with ether. The organic solution was dried (Na2SO4) and concentrated
to give the crude product.
5.5.3.2 Benzyl 2,3-di-O-benzyl-4-O-methyl-6-O-p-toluenesulfonyl-β-D-glucopyranoside 11
Reaction of 1019 (150 mg, 0.25 mmol) with methyl iodide (0.026 mL) in
presence of 50% aqueous sodium hydroxide (6 drops) in DMSO (3.0 mL) for 5

146
min at room temperature gave compound 11 as a white, amorphous foam (130
mg, 85%). [α] 23D = + 3 (c = 1, CH2Cl2). 1H NMR (CDCl3) δ 7.76-7.18 (19H, m, Ar),
4.82, 4.78, 4.75, 4.65, 4.59, 4.48 (6H, 6d, JA,B = 12.1 Hz, 3CH2Ph), 4.33 (1H, d,
J1,2 = 7.7 Hz, H-1), 4.23 (1H, dd, J6a,6b = 2.3, J5,6a = 10.7 Hz, H-6a), 4.16 (1H, dd,
J6a,6b = 5.3, J5,6b = 10.5 Hz, H-6b), 3.44 (1H, t, J2,3 = J3,4 = 9.6 Hz, H-3 ), 3.40
(3H, s, OMe), 3.31 (2H, m, H-2, H-5), 3.08 (1H, dd, J3,4 = 8.9, J4,5 = 10.1 Hz, H-
4), 2.34 (3H, s, Me). 13C NMR (CDCl3) δ 144.9-127.7 (m, Ar), 102.1 (C-1), 84.3
(C-3), 81.8 (C-2), 79.1 (C-4), 75.5, 74.9, 71.0 (3 CH2Ph), 72.8 (C-5), 68.7 (C-6),
60.8 (OMe). HRMS Calcd for C35H39O8S (M+H): 619.236. Found: 619.2363.
5.5.3.3 Benzyl 4-O-methyl-6-O-p-toluenesulfonyl-β-D-glucopyranoside 13
Compound 20 (report in this paper) (200 mg, 0.36 mmol) was dissolved in
80% TFA (5 mL) and stirred at room temperature for 1 h. The solvents were
removed under reduced pressure, and the residue was purified by flash
chromatography (EtOAc/Hexanes (1:2)) to yield 13 as a white foam (150 mg,
95%). [α] 23D = - 22.8 (c =0.4, MeOH). 1H NMR (CDCl3) δ 7.84-7.28 (9H, m, Ar),
4.75, 4.74 (2H, 2d, JA,B = 11.5 Hz, CH2Ph), 4.26 (3H, m, H-1, H-6a, H6b), 3.49
(3H, s, OMe), 3.40 (2H, m, H-3, H-5), 3.19 (1H, t, J2,3 = J1,2 = 8.5 Hz, H-2 ), 3.04
(1H, t, J4,5 = J3,4 = 9.0 Hz, H-4 ), 2.42 (3H, s, Me). 13C NMR (CDCl3) δ 145.2-
127.3 (m, Ar), 101.7 (C-1), 79.0 (C-4), 76.6 (C-3), 73.6 (C-2), 72.5 (C-5), 70.3
(CH2Ph), 68.9 (C-6), 59.5 (OMe), 120.2 (Me). HRMS Calcd for C12H26NaO8S
(M+Na): 463.1248. Found: 463.1242.

147
5.5.3.4 Benzyl 2,3-di-O-benzyl-6-O- tert-butyldimethylsilyl-4-O-methyl-6-O-p-toluenesulfonyl-α- D-glucopyranoside 15
To a solution of 1419 (500 mg, 1.1 mmol) in DMF (15 mL) was added
imidazole (235 mg, 3.4 mmol). The reaction was cooled in an ice bath, TBDMSCl
(190 mg, 1.2 mmol) was added portionwise, and the mixture was stirred at 0 °C
under N2 for 2 h. The reaction was quenched by the addition of ice-water, and the
reaction mixture was extracted with Et2O (3×30 mL). The combined organic
solvents were dried (Na2SO4), and concentrated to give the crude product which
was used directly in the next step without further purification. The residue was
dissolved in DMSO (10 mL) and methyl iodide (0.12 mL) and 50% aqueous
sodium hydroxide (12 drops) were added. The reaction mixture was stirred for 5
min at room temperature to yield 15 as a syrup (340 mg, 53%). [α] 23D = + 98 (c =
1, CH2Cl2). 1H NMR (CDCl3) δ 7.35-7.17 (15H, m, Ar), 4.89, 4.76, 4.64, 4.60,
4.49, 4.47 (6H, 6d, JA,B = 12.1 Hz, 3CH2Ph), 4.75 (1H, d, J1,2 = 2.8 Hz, H-1), 3.89
(1H, t, J2,3 = J3,4 = 9.3 Hz, H-3), 3.72 (1H, dd, J6a,6b = 4.3, J5,6a = 11.1 Hz, H-6a),
3.67 (1H, dd, J6a,6b = 1.8, J5,6b = 11.1 Hz, H-6b), 3.54 (1H, ddd, J4,5 = 1.8, J6a,5 =
4.3, J6b,5 = 5.8 Hz, H-5), 3.50 (3H, s, OMe), 3.40 (1H, dd, J2,3 = 3.6, J1,2 = 10.1
Hz, H-2 ), 3.21 (1H, t, J3,4 = J4,5 = 9.4 Hz, H-4), 0.84 (9H, s, 3Me), 0.01, 0.00 (6H,
2s, 2Me). 13C NMR (CDCl3) δ 139.0-127.6 (m, Ar), 95.2 (C-1), 82.2 (C-3), 80.1
(C-2), 79.5 (C-4), 75.8, 73.0, 68.7 (3 CH2Ph), 71.9 (C-5), 62.2 (C-6), 60.8 (OMe),
26.0, -5.1, -5.3 (5Me). HRMS Calcd for C34H46NaO6Si (M+Na): 601.2956. Found:
601.2954.

148
5.5.3.5 Benzyl 2,3-di-O-benzyl-4-O-methyl-6-O-tert-butyldimethylsilyl-α-D-glucopyranoside 16
To a solution of 15 (600 mg, 1.04 mmol) in THF (50 mL), TBAF (1.0 M
solution in THF, 1.2 mL, 1.2 mmol) was added, and the reaction mixture was
stirred at room temperature. After 2 h it was concentrated and further dried under
high vacuum for 1 h. The crude product was dissolved in CH2Cl2 (25 mL), and
pyridine (0.1 mL, 1 eq) was added. The mixture was cooled to -10 °C, and Tf2O
(0.2 mL, 1.1 eq) was added under N2. After 5-10 min it was concentrated and the
residue was purified by flash chromatography (EtOAc/Hexanes (1:5)) to yield 16
as a white foam (620 mg, 76%). [α] 23D = +75 (c = 0.1, CH2Cl2). 1H NMR (CDCl3) δ
7.40-7.25 (15H, m, Ar), 4.98, 4.77 (2H, 2d, JA,B = 11.1 Hz, CH2Ph), 4.84 (1H, d,
J1,2 = 3.6 Hz, H-1), 4.68-4.49 (6H, m, 2CH2Ph, H-6a, H-6b), 3.95 (1H, t, J2,3 = J3,4
= 9.2 Hz, H-3 ), 3.80 (1H, ddd, J4,5 = 1.8, J6a,5 = 4.6, J6b,5 = 6.6 Hz, H-5), 3.55
(3H, s, OMe), 3.48 (1H, dd, J1,2 = 3.5, J2,3 = 9.4 Hz, H-2), 3.17 (1H, dd, J3,4 = 8.8,
J4,5 = 10.1 Hz, H-4). 13C NMR (CDCl3) δ 138.5 (CF3), 137.9-127.7 (m, Ar), 95.6
(C-1), 81.7 (C-3), 79.6 (C-2), 78.7 (C-4), 75.6, 73.1, 69.7 (3 CH2Ph), 74.8 (C-6),
68.5 (C-5), 60.9 (OMe). HRMS Calcd for C29H31F3NaO8S (M+Na): 619.1584.
Found: 619.1585.
5.5.3.6 Benzyl 2,3-O-[(2R,3R)-2,3-dimethoxybutane-2,3-diyl]-6-O-p-toluenesulfonyl -β-D-glucopyranoside 20
To a solution of 1923 (634 mg, 1.65 mmol) in pyridine (10 mL) was added
p-toluenesulfonyl chloride (TsCl) (380 mg, 3.27 mmol), and the mixture was
stirred at room temperature overnight. Another portion of TsCl (35 mg, 0.16
mmol) was added, the mixture was stirred for 4h, and the pyridine was removed

149
by distillation. The residue was purified by flash chromatography
(EtOAc/Hexanes (1:2)) to yield 20 as a white foam (660 mg, 75%). [α] 23D = - 208
(c = 1, CH2Cl2). 1H NMR (CDCl3) δ 7.85-7.32 (9H, m, Ar), 4.86, 4.63 (2H, 2d, JA,B
= 12.1 Hz, CH2Ph), 4.58 (1H, d, J1,2 = 8.0 Hz, H-1), 4.38 (1H, dd, J6a,6b = 1.7, J5,6a
= 10.9 Hz, H-6a), 4.28 (1H, dd, J6a,6b = 5.5, J5,6b = 10.7 Hz, H-6b), 3.66 (2H, m,
H-3, H-4), 3.55 (2H, m, H-2, H-5), 3.29 (6H, s, OMe), 2.43 (3H, s, Me), 1.97 (1H,
br, OH), 1.33, 1.34 (6H, 2s, 2Me). 13C NMR (CDCl3) δ 144.9-127.4 (m, Ar), 99.8
(C-1), 99.6, 99.5 (2MeCOMe), 74.1 (C-5), 72.4 (C-3), 70.7 (CH2Ph), 69.0 (C-2),
68.7 (C-6), 67.3 (C-4), 48,0 47.9 (2OMe), 21.6 (OMe), 17.6 (2Me). HRMS Calcd
for C26H34NaO10S (M+Na): 561.1765. Found: 561.1772.
5.5.3.7 Benzyl 2,3-O-[(2R,3R)-2,3-dimethoxybutane-2,3-diyl]-4-O-methyl-6-O-p-toluenesulfonyl-β-D-glucopyranoside 18
Reaction of 20 (710 mg, 1.3 mmol) with methyl iodide (0.13 mL) in
presence of 50% aqueous sodium hydroxide (25 drops) in DMSO (5.0 mL) for 3
min at room temperature gave compound 18 as a white, amorphous foam (660
mg, 91%). [α] 23D = -111 (c =1, CH2Cl2). 1H NMR (CDCl3) δ 7.84-7.29 (9H, m, Ar),
4.82, 4.61 (2H, 2d, JA,B = 12.7 Hz, CH2Ph), 4.51 (1H, d, J1,2 = 7.8 Hz, H-1), 4.31
(1H, dd, J6a,5 = 2.0, J6b,6b = 10.6 Hz, H-6a), 4.25 (1H, dd, J6b,5 = 5.4, J6b,6b = 10.6
Hz, H-6b), 3.78 (1H, t, J2,3 = J3,4 = 9.9 Hz, H-3 ), 3.56 (1H, dd, J1,2 = 8.0, J2,3 =
10.1 Hz, H-2), 3.55 (3H, s, OMe), 3.45 (1H, ddd, J4,5 = 2.0, J6a,5 = 5.4, J6b,5 = 7.6
Hz, H-5), 3.31, 3.30 (6H, 2s, 2OMe), 3.27 (1H, dd, J3,4 = 9.2, J4,5 = 18.6 Hz, H-4),
2.42 (3H, s, OMe), 1.34, 1.33 (6H, 2s, 2Me). 13C NMR (CDCl3) δ 144.8-127.4 (m,
Ar), 99.6 (C-1), 99.4 (MeCOMe), 75.6 (C-4), 73.7 (C-3), 73.4 (C-5), 70.6

150
(CH2Ph), 69.1 (C-2), 68.6 (C-6), 60.6 (OMe), 48,0 47.9 (2OMe), 21.6 (OMe),
17.8, 17.7 (2Me). HRMS Calcd for C27H36NaO10S (M+Na): 575.1921. Found:
575.1931.
5.5.3.8 Benzyl 2,3-O-[(2R,3R)-2,3-dimethoxybutane-2,3-diyl]-4-O-methyl-6-deoxy-6-[2,3,5-tri-O-benzyl-1,4-dideoxy-episulfoniumylidene-D-arabinitol]-β-D-glucopyranoside-p-toluenesulfonate 21
The mixture of the thioether 1221 (540 mg, 1.3 mmol) and benzyl 2,3-O-
[(2R,3R)-2,3-dimethoxybutane-2,3-diyl]-4−Ο−methyl-6−Ο−p-toluenesulfonyl−β-D-
glucopyranoside 20 (235 mg, 0.43 mmol) in HFIP (1 mL) were placed in a sealed
tube in an oil bath at 70 °C for 6 days. The mixture was cooled, then diluted with
EtOAc, and evaporated to give a syrupy residue. Purification by column
chromatography (EtOAc/MeOH 20:1) gave the sulfonium salt 21 as a syrup (310
mg, 70% based on 20). [α] 23D = -22.5 (c = 0.1, CH2Cl2). 1H NMR (CDCl3) δ 7.78-
7.01 (24H, m, Ar), 4.69, 4.56 (2H, 2d, JA,B = 12.1 Hz, CH2Ph), 4.49-4.35 (6H, m,
3 CH2Ph), 4.45 (1H, m, H-1’), 4.44 (1H, m, H-3), 4.41 (1H, m, H-4), 4.28 (1H, br,
H-2), 4.13 (1H, t, J6a’,5 = J6b’,6b’ = 11.2 Hz, H-6a’), 4.01 (1H, dd, J6b’,5 = 7.7, J6’b,6’b
= 13.3 Hz, H-6b’), 3.83 (1H, dd, J4,5a =7.6 J5a,5b = 10.0 Hz, H-5a ), 3.79-3.69 (5H,
m, H-1a, H-1b, H-5b, H-5’, H-3’), 3.49 (3H, s, OMe), 3.47 (1H, dd, J1’,2’ = 2.1,
J2’,3’ = 5.1 Hz, H-2’), 3.23, 3.19 (6H, 2s, 2OMe), 3.18 (1H, m, H-4’), 2.23 (3H, s,
OMe), 1.25, 1.24 (6H, 2s, 2Me). 13C NMR (CDCl3) δ 144.1-126.2 (m, Ar), 100.7
(C-1’), 99.5, 99.4 (2MeCOMe), 83.1 (C-2), 82.7 (C-3), 78.0 (C-4’), 73.5, 72.2,
71.9, 71.6 (4CH2Ph), 73.1 (C-3’), 71.8 (C-5’), 68.9 (C-2’), 66.9 (C-5), 65.3 (C-4),
60.7 (OMe), 48.1 (C-1) 48,0 47.9 (2OMe), 47.6 (C-6’), 21.2 (Me), 17.7, 17.5
(2Me). HRMS Calcd for C46H57O10S (M+.): 801.3667. Found: 801.3676.

151
5.5.3.9 1,4-Dideoxy-1,4-[[2S, 3S, 4R, 5S]-2,4,5,6-tetrahydroxy-3-(methyl) hexyl]-(R)-epi-sulfoniumylidine]-D-arabinitol chloride 9
Compound 21 (150 mg, 0.15 mmol) was dissolved in CH2Cl2 (15 mL), the
mixture was cooled to -78 °C, and BCl3 (1M solution in CH2Cl2, 2.3 mmol) was
added under N2. The reaction mixture was stirred at the same temperature for 30
min, and then allowed to warm to 5 °C for 6 h. The reaction was quenched by
addition of MeOH (5 mL), the solvents were removed, and the residue was co-
evaporated with MeOH (2 × 5 mL). The crude residue was dissolved in 80% TFA
(5 mL) and stirred at room temperature for 1h. The solvents were removed under
reduced pressure, the residue was dissolved in water (5 mL), and washed with
CH2Cl2 (3×5 mL). The water layer was evaporated to give the crude product. The
residue was dissolved in H2O (10 mL), Amberlyst A-26 resin (200 mg) was
added, and the reaction mixture was stirred at room temperature for 2 h.
Filtration through cotton, followed by solvent removal gave the crude hemiacetal.
The crude product was dissolved in water (8 mL), and the solution was stirred at
room temperature while NaBH4 (23 mg, 0.6 mmol) was added in small portions
over 30 min. Stirring was continued for another 3 h and the mixture was acidified
to pH < 4 by dropwise addition of 2M HCl. The mixture was evaporated to
dryness and the residue was co-evaporated with anhydrous MeOH (3 × 20 mL).
Treatment of the solid residue with 50% EtOAc:MeOH (5-10 mL) resulted in
precipitation of most of the borate salt. Filtration through cotton, followed by
solvent removal gave the crude compound. The residue was purified by reverse
phase column chromatography (H2O) to give 9 as a colorless solid (29 mg, 51%).
[α] 23D = + 3.3, (c = 0.6, H2O). 1H NMR (D2O) δ 4.66 (1H, m, H-2), 4.36 (1H, t, br,

152
H-3), 4.25 (1H, td, J1',2' = 3.5, J2',3' = 8.9 Hz, H-2'), 4.05 (1H, dd, J4,5a = 4.9, J5a,5b
= 12.0 Hz, H-5a), 3.97 (1H, m, H-4), 3.87-3.73 (5H, m, H-5b, H-1a, H-1b, H-1'a,
H1'b), 3.70-3.63 (3H, m, H-4', H-5', H-6'a), 3.54 (1H, dd, J5',6'b = 6.6, J6'a,6'b = 11.7
Hz, H-6'b), 3.46 (3H, s, OMe), 3.43 (1H, m, H-3'). 13C NMR (D2O) δ 83.4 (C-3'),
77.5 (C-3), 76.9 (C-2), 71.9 (C-5'), 70.2 (C-4'), 69.9 (C-4), 67.9 (C-2'), 62.5 (C-6'),
60.6 (OMe), 59.2 (C-5), 49.3 (C-1'), 48.2 (C-1). HRMS Calcd for C12H25O8S
(M+.): 329.1265. Found: 329.126.
5.6 Acknowledgments
We are grateful to the Canadian Institutes for Health Research and the
Heart and Stroke Foundation of Ontario grant # NA-6305 for financial support of
this work.
5.7 References
1. Lillelund, V. H.; Jensen, H. H.; Liang, X.; Bols, M. Chem. Rev. 2002, 102,
515.
2. Gloster, T. M. Davies, G. J. Org. Biomol. Chem., 2010, 8, 305.
3. Holman, R. R.; Cull, C. A.; Turner, R. C. Diabetes Care 1999, 22, 960.
4. Jacob, G. S. Curr. Opin. Struct. Biol. 1995, 5, 605.
5. Harvey, A. L. Drug Discov. Today 2008, 13, 894.
6. Butler, M. S. Nat. Prod. Rep. 2008, 25, 475.
7. Chandrasena, J. P. C. The Chemistry and Pharmacology of Ceylon and
Indian Medicinal Plants; H&C Press: Colombo, Sri Lanka, 1935.

153
8. Jayaweera, D. M. A. Medicinal Plants Used in Ceylon-Part 1, National
Science Council of Sri Lanka: Colombo, 1981.
9. Matsuda, H.; Yoshikawa, M.; Murakami, T.; Tanabe, G.; Muraoka, O. J.
Trad. Med. 2005, 22, 145.
10. Yoshikawa, M.; Xu, F. M.; Nakamura, S.; Wang, T.; Matsuda, H.; Tanabe,
G.; Muraoka, O. Heterocycles 2008, 75, 1397.
11. Yoshikawa, M.; Murakami, T.; Shimada, H.; Matsuda, H.; Yamahara, J.;
Tanabe, G.; Muraoka, O. Tetrahedron Lett. 1997, 38, 8367-8370.
12. Matsuda, H.; Li, Y. H.; Murakami, T.; Matsumura, N.; Yamahara, J.;
Yoshikawa, M. Chem. & Pharm. Bull. 1998, 46, 1399.
13. Muraoka, O.; Xie, W. J.; Tanabe, G.; Amer, M. F. A.; Minematsu, T.;
Yoshikawa, M. Tetrahedron Lett. 2008, 49, 7315.
14. Minami, Y.; Kurlyarna, C.; Ikeda, K.; Kato, A.; Takebayashi, K.; Adachi, I.;
Fleet, G. W. J.; Kettawan, A.; Karnoto, T.; Asano, N. Bioog. & Med. Chem.
2008, 16, 2734.
15. Jayakanthan, K.; Mohan, S.; Pinto, B. M. J. Am. Chem. Soc. 2009, 131,
5621.
16. Mohan, S.; Jayakanthan, K.; Nasi, R.; Kuntz, D. A.; Rose, D. R.; Pinto, B.
M. Org. Lett., 2010, 12, 1088.
17. Eskandari, R.; Kuntz, D. A.; Rose, D. R.; Pinto, B. M. Org. Lett. 2010, 12,
1632.
18. Sim, L.; Jayakanthan, K.; Mohan, S.; Nasi, R.; Johnston, B. D.; Pinto, B.
M.; Rose, D. R. Biochemistry 2010, 49, 443.
19. Shinkiti, K.; Kusunoki, A.; Hirooka, M. Bull. Chem. Soc. Jpn. 2000, 73,
967.

154
20. Wang, H.; Sun, L.; Glazebnik, S.; Zhao, K. Tetrahedron Lett. 1995, 36,
2953.
21. Satoh, H.; Yoshimura, Y.; Sakata, S.; Miura, S.; Machida, H.; Matsuda, A.
Bioorg & Med. Chem. Lett. 1998, 8, 989.
22. Ghavami, A.; Sadalapure, K. S.; Johnston, B. D.; Lobera, M.; Snider, B.
B.; Pinto, B. M. Synlett 2003, 1259.
23. Liu, H.; Nasi, R.; Jayakanthan, K.; Sim, L.; Heipel, H.; Rose, D. R.; Pinto,
B. M. J. Org. Chem. 2007, 72, 6562.

155
5.8 Supporting Information
Probing the active-site requirements of human intestinal N-terminal
maltase-glucoamylase: The effect of replacing the sulfate moiety by a
methyl ether in ponkoranol, a naturally-occurring α-glucosidase inhibitor
Razieh Eskandari, Kyra Jones, David R. Rose, B. Mario Pinto

156
ppm (t1)3.04.05.06.07.0
ppm (t1)50100
O
MeOOBn
OBn
OBnTsO
11
O
MeOOBn
OBn
OBnTsO
11

157
ppm (t1)3.04.05.06.07.0
ppm (t1)50100150
O
MeOOH
OH
OBnTsO
13
O
MeOOH
OH
OBnTsO
13

158
ppm (t1)0.01.02.03.04.05.06.07.0
ppm (t1)050100
O
MeOOBn
OBn
OBnTBSO
15
O
MeOOBn
OBn
OBnTBSO
15

159
ppm (t1)4.05.06.07.0
ppm (t1)60708090100110120130140
O
MeOOBn
OBn
OBnTfO
16
O
MeOOBn
OBn
OBnTfO
16

160
ppm (t1)1.02.03.04.05.06.07.0
ppm (t1)50100
O
HOO
O
OBnTsO
OMe
MeO20
O
HOO
O
OBnTsO
OMe
MeO20
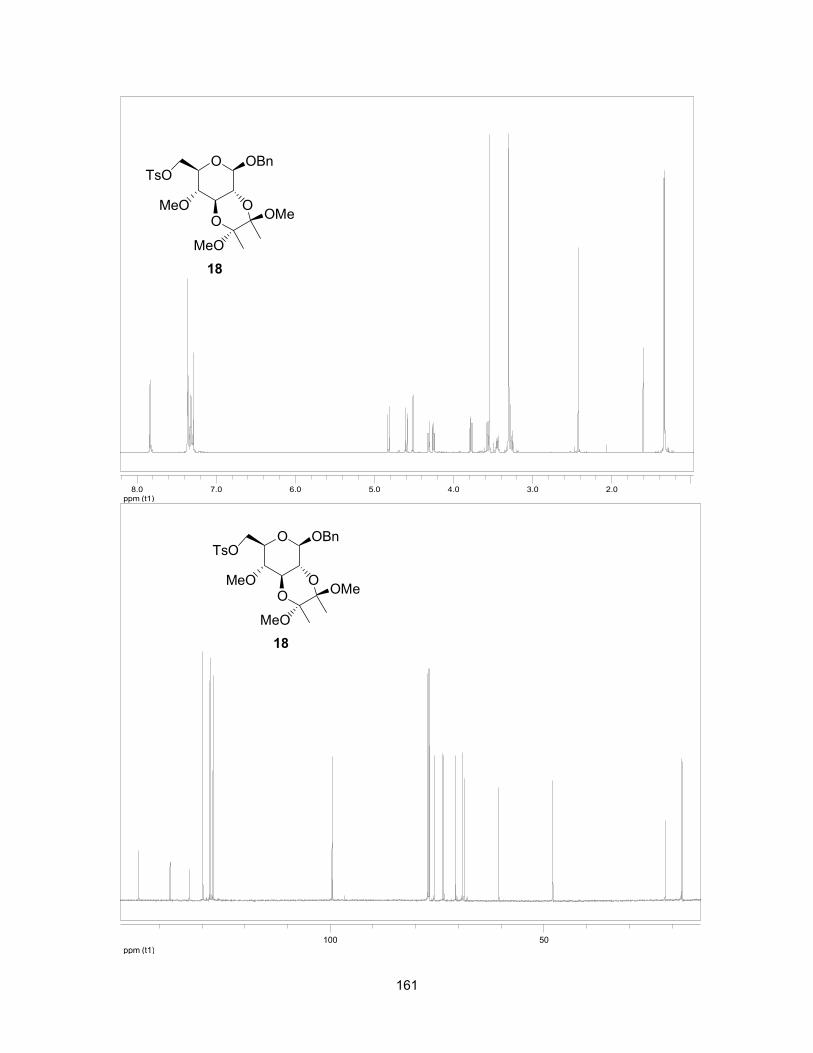
161
ppm (t1)2.03.04.05.06.07.08.0
ppm (t1)50100
O
MeOO
O
OBnTsO
OMe
MeO
18
O
MeOO
O
OBnTsO
OMe
MeO
18

162
ppm (t1)2.03.04.05.06.07.0
ppm (t1)50100
S
BnO OBn
BnO
OTs
1' 2'
3'4'5'
6'
O
OHO
O
BnO OMe
OMe
21
S
BnO OBn
BnO
OTs
1' 2'
3'4'5'
6'
O
OHO
O
BnO OMe
OMe
21
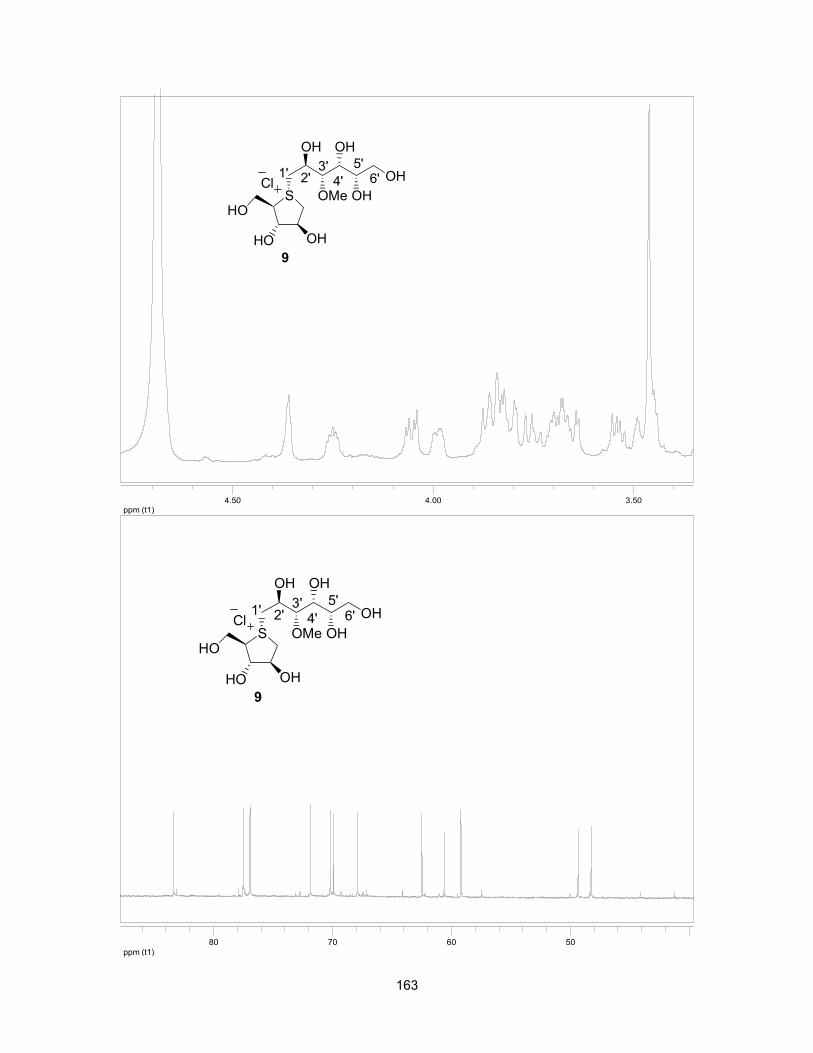
163
ppm (t1)3.504.004.50
ppm (t1)50607080
S
OHHO
HO
OH
OH
OMe
OH
OHCl
3'4'
5'6'1' 2'
9
S
OHHO
HO
OH
OH
OMe
OH
OHCl
3'4'
5'6'1' 2'
9
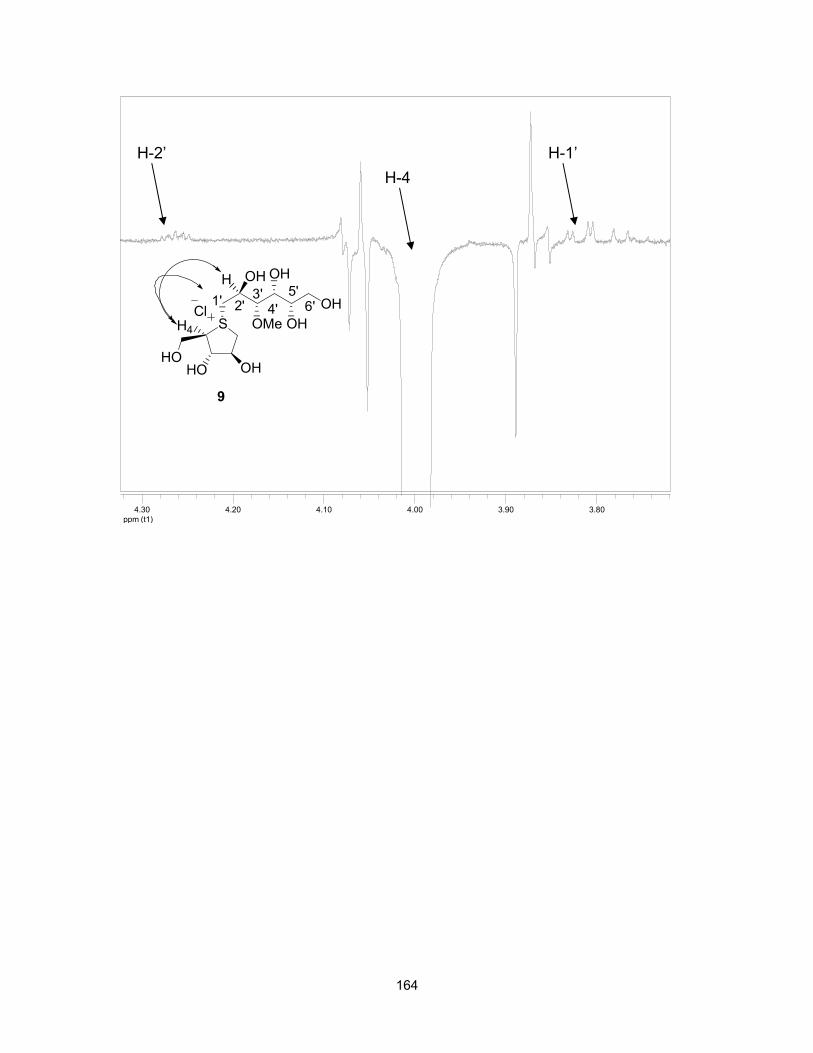
164
ppm (t1)3.803.904.004.104.204.30
H-4H-2’ H-1’
S
OHHOHO
OH
OH
OMe
OH
OHCl
9
3'4'
5'6'1' 2'
H4
H

165
CHAPTER 6: SELECTIVITY OF 3'-O-METHYLPONKORANOL FOR INHIBITION OF N- AND C-
TERMINAL MALTASE-GLUCOAMYLASE AND SUCRASE-ISOMALTASE, POTENTIAL THERAPEUTICS FOR DIGESTIVE DISORDERS OR THEIR SEQUELAE
This Chapter comprises the manuscript “Selectivity of 3'-O-methylponkoranol for inhibition of N- and C-terminal maltase-glucoamylase and sucrase-
isomaltase, potential theraputics for digestive disorders or their sequelae”
which was published in Bioorganic and Medicinal Chemistry Letters (2011, 21,
6491-6494).
Razieh Eskandari,a Kyra Jones,b David R. Rose,b B. Mario Pintoa
aDepartment of Chemistry, Simon Fraser University, Burnaby, British Columbia,
Canada V5A 1S6
b Department of Biology, University of Waterloo, Waterloo, Ontario,
Canada N2L 3G1

166
In this Chapter, the selectivity of 3'-O-methylponkoranol for the different
intestinal enzymes N- and C- terminal maltase-glucoamylase and sucrase-
isomaltase is reported. The thesis author wrote and edited the manuscript with
Dr. B. Mario Pinto. Mrs. Kyra Jones and Dr. David R. Rose performed the
enzyme inhibition studies.
Graphical abstract:
Starch

167
6.1 Keywords
α-Glucosidase inhibitors, 3'-O-methylponkoranol, maltase-glucoamylase,
sucrase-isomaltase, selective inhibition, therapeutics of digestive disorders.
6.2 Abstract
Human maltase-glucoamylase (MGAM) and sucrase-isomaltase (SI) are
two human intestinal glucosidases responsible for the final step of starch
hydrolysis.
MGAM and SI are anchored to the small intestinal brush border epithelial
cells and contain two catalytic N-terminal and C-terminal subunits. In this study,
we report the inhibition profile of 3'-O-methylponkoranol for the individual
recombinant N and C terminal enzymes and compare the inhibitory activities of
this compound with de-O-sulfonated ponkoranol. We show that 3'-O-
methylponkoranol inhibits the different subunits to different extents, with
extraordinary selectivity for C-terminal SI (Ki = 7 ± 2 nM). The enzymes
themselves could serve as therapeutic targets for the treatment of digestive
disorders or their sequelae.
6.3 Introduction
Starches constitute the main source of energy for adult humans and have
particular nutritional importance in children. Ingestion of starch in the human diet
has received particular attention in recent years due to their nutritional value. The
imbalance between energy consumption and dietary intake of energy-rich foods
has a direct relationship to the development of energy metabolism diseases,

168
such as obesity or diabetes.1-3 Such adverse consequences have led to the study
of enzyme/substrate or enzyme/inhibitor interactions in the starch digestion
process.
Starch is present in two main molecular structures: mostly linear α-1,4-D-
glucopyranose linked polymers known as amyloses, and those with a mixture of
α-1,4- and α-1,6-branched α-D-glucopyranose linkages, known as amylopectins
(Figure 6-1).
Figure 6-1: Components of starch: amylose and amylopectin.
The manner in which mammals derive glucose molecules from starch
involves multiple enzymes.4-8 Two luminal α-1,4 endoglucosidases, namely
salivary and pancreatic amylases, hydrolyze linear, unbranched glucose
sequences present in amylose and amylopectin molecules into soluble glucose
Amylose
AmylopectinO
O
OH
HOHO
OH O
O
HOOH
OH
1
4
6
α-1,4 linkage
O
OH
HOHO
OH
1 α-1,6 linkage
O
O
OH
HOHO
OH O
OH
HOOH
OH
1
12
34 5
6
α-1,4 linkage

169
oligomers with both linear and branched structures,9-12 and the membrane bound
enzymes maltase-glucoamylase (MGAM) and sucrase-isomaltase (SI) are then
responsible for the hydrolysis of these oligomers into glucose.13,14 The enzymes
each contain two catalytic subunits: an N-terminal subunit (ntMGAM and ntSI)
near the membrane-bound end of the enzyme and a C-terminal luminal subunit
(ctMGAM and ctSI) (Figure 6-2).15
Figure 6-2: Schematic diagram of MGAM and SI indicating their hydrolytic activities.
Since MGAM and SI genes arose from the duplication of an ancestral
gene,16 the N-terminal catalytic subunits of MGAM and SI are more similar to one
another in sequence, as are the C-terminal domains (60% sequence identity),
than the N- and C-terminal subunits within the same protein.17 There are also
multiple spliceforms of C-terminal MGAM in mammals, two of which are studied
in this paper (ctMGAM-N2 and ctMGAM-N20).17,18
From a therapeutic point of view, it is important to understand the
thermodynamics and kinetics of action of the enzymes involved in the starch
digestion pathway, which will in turn inform the treatment of digestive disorders
and their sequelae. The individual roles of the four catalytic subunits comprising

170
the human intestinal MGAM and SI complexes in the digestion of food starches,
have not been investigated in depth, until recently. Compounds are being
developed to regulate the activities of the catalytic subunits independently. Small
modifications in a compound can have profound effects on its inhibitory
properties and ability to inhibit one catalytic subunit selectively in comparison to
the other enzymes. The characterization of these enzymes, using glucosidase
inhibitors, for example, is an essential prelude to their use in the treatment of
digestive disorders and their sequelae. Certain clinical indications might also
benefit from specific inhibition of one particular enzyme, but not others, to reduce
unwanted side-effects.
The crystal structure of human ntMGAM in complex with inhibitors such as
kotalanol (1) and de-O-sulfonated kotalanol (2)19 (Figure 6-3) had indicated that
removal of the sulfate group did not significantly affect the conformation of the
rest of the polyhydroxylated chain,20 although the C3'-sulfate anion seemed to be
constrained by the hydrophobic residues of the enzyme, and made no hydrogen
bonding interactions with other residues. Removal of the sulfate group allowed
the rest of the polyhydroxylated chain to make optimal contacts with the ntMGAM
active site,20 and resulted in an approximately 7-fold decrease in Ki values.19 In
order to further probe the accommodation by this hydrophobic pocket of other
groups, we synthesized 3'-O-methylponkoranol (3). Disappointingly, the Ki value
(0.50 ± 0.04 µM) was higher than that of 2, indicating that the hydrophobic
interactions between the methyl group and the hydrophobic residues in the active

171
site of ntMGAM are not as optimal as the interactions of the latter groups with the
rest of polyhydroxylated chain in the absence of the methyl ether.21
Very recently Tanabe et al. have synthesized four 3'-O-alkylated salacinol
derivatives (4a–4d), in which the 3'-sulfate moiety was replaced by OCH3,
OC2H5, OC13H27, or OCH2Ph groups, and their inhibitory activities were evaluated
against several disaccharidases (Figure 6-3).22
Figure 6-3: Sulfonium-ion α-glucosidase inhibitors 1-7.
Compared to salacinol (5) and de-O-sulfonated salacinol (6) all the
molecules synthesized (4a-4d) showed equal or higher inhibitory activities (Table
6-1). Compound 4b had the highest inhibitory activity against sucrase and
compound 4d was the most potent inhibitor of maltase and isomaltase among
this type of molecules. The enhanced inhibitory activity of 4d was attributed to
S
OHHO
OSO3 OH
OH OH
HO
OHOH
S
OHHO
OSO3
OH OH
HO
Kotalanol (1)
Salacinol (5)
S
OHHO
OH OH
OH OH
HO
OHOH
CH3OSO3
De-O-sulfonated kotalanol (2)
S
OHHO
OR
OH OH
HOX
3'-O-Alkylated salacinol R-4a: Me, 4b: Et, 4c: C13H27, 4d: Bn
S
OHHO
OMe OH
OH OH
HO
OH
3'-O-Methyl ponkoranol (3)
Cl
S
OHHO
OH
OH OH
HO
De-O-sulfonated salacinol (6)
HCO2 S
OHHO
OH OH
OH OH
HO
OH
Cl
De-O-sulfonated ponkoranol (7)

172
π/π or CH/π interactions of the phenyl ring at 3' with surrounding aromatic
residues of the active site in the enzyme. It is clear from this work and our
previous work that subtle structural modifications of the ligands can differentiate
between the active sites of the various glucosidase enzymes.
Table 6-1: IC50 (µM) against disaccharidases.
In the present work, we report the glucosidase inhibitory activity of 3'-O-
methylponkoranol (3) against recombinant human maltase-glucoamylase
(ntMGAM and ctMGAM) and sucrase-isomaltase (ntSI and ctSI), in comparison
with the parent compound, de-O-sulfonated ponkoranol (7) (Figure 6-3).24
Compound Sucrase Maltase Isomaltase
4a 0.46 5.3 0.39
4b 0.12 1.7 0.27
4c 1.3 1.0 0.95
4d 0.32 0.44 0.14
5 1.619 5.223 1.323
6 1.319 8.023 0.323

173
6.4 Results and discussion
Enzyme kinetics. Kinetic parameters of the four domains were determined
by measuring the amount of glucose produced upon the addition of enzyme at
increasing maltose concentrations (from 0.5 to 30 mM) in the presence of
increasing inhibitor concentration (0-200 nM) by a two-step glucose oxidase
assay. The methods used for kinetic assays were reported previously.25,26
Reactions were performed in quadruplicate and absorbance measured at 405 nM
with a SpectraMax 190 Plate Reader (Molecular Devices). Absorbance readings
were averaged to give the final value, which was compared to a glucose standard
curve to determine the amount of glucose released by the enzyme from the
substrate. The program KaleidaGraph4.1 was used to fit the data to the
Michaelis-Menten equation and estimate Km, Kmobs (Km in the presence of the
inhibitor) and Vmax of the catalytic subunits. Ki values for each inhibitor were
determined using the equation Ki = [I]/(Kmobs/Km)-1). The Ki values reported for
each inhibitor were determined by averaging the Ki values from three different
inhibitor concentrations. The data were also plotted on Lineweaver-Burk plots to
verify that the inhibitors were acting as competitive inhibitors (Figure 6-4).

174
Figure 6-4: Representative Lineweaver-Burk plot of ctMGAM-N2 inhibited by 3 at concentrations of 0 nM, 75 nM, 125 nM, and 200 nM.
Kinetic Analysis. Inhibition constants (Ki), calculated for each catalytic
subunit with compound 3, as described above, are shown in Table 6-2.
Compound 3 was a potent inhibitor of all the catalytic subunits. With regard to
MGAM, it was found to inhibit both ctMGAM spliceforms similarly, demonstrating
inhibitory activity in the nanomolar range for both ctMGAM-N2 (Ki = 0.060 ± 0.015
µM) and ctMGAM-N20 (Ki = 0.055 ± 0.014 µM). Unlike the other catalytic
subunits, compound 3 is a weak inhibitor of ntMGAM (Ki = 0.5 ± 0.04 µM);21 it is,
therefore, ten times more potent an inhibitor of ctMGAM-N2 and ctMGAM-N20

175
than ntMGAM. With respect to ctSI, Compound 3 exhibited remarkable inhibitory
activity in the nanomolar range (Ki = 0.007 ± 0.002 µM). This compound
demonstrates the most potency against ctSI, approximately seventy times more
active than with ntMGAM. Compound 3 inhibited ntSI in the nanomolar range as
well, with a Ki value of 0.035 ± 0.013 µM. Furthermore, there is some
differentiation between ctSI and ntSI as compound 3 is five times more potent
against ctSI compared to ntSI. These results demonstrate the variation in
biochemical and structural properties of the enzymes despite their similarity in
amino acid sequence.
Finally, we compare the inhibitory activities of compounds 3 and de-O-
sulfonated pokoranol (7) against ntMGAM, ctMGAM, ntSI, and ctSI. The results
(Table 6-2) indicate that addition of the methyl group at the 3' position of de-O-
sulfonated ponkoranol (7) results significant inhibition of ctMGAM-N2 (Ki = 0.060
± 0.015 µM), in contrast to 7 which does not inhibit ctMGAM-N2.24
Table 6-2: Experimentally determined Ki values (µM) of 3 and 7.a
a analysis of inhibition was performed with maltose as the substrate.
ctMGAM-N2 ctMGAM-N20 ntMGAM ctSI ntSI
3 0.060 ± 0.015 0.055 ± 0.014 0.50 ± 0.0421 0.007 ± 0.002 0.035 ± 0.013
724 No Inhibition 0.096 ± 0.015 0.043 ± 0.001 0.103 ± 0.037 0.302 ± 0.123

176
Both compounds 3 and 7 inhibited ctMGAM-N20 with similar Ki values.
However, an order of magnitude greater inhibition of ntSI by 3 over 7 was
observed. Strikingly, compound 3 inhibited ctSI with Ki values of 7 ± 2 nM,
significantly lower than that (103 ± 30)24 for de-O-sulfonated ponkoranol (7) itself
(Table 6-2). We note that 3 is the most potent compound against ctSI to date in
this class of molecules. We speculate that ctSI will have a hydrophobic pocket in
the catalytic site that better accommodates the methyl group and provides
favorable hydrophobic interactions, relative to the other enzymes in which steric
interactions between the methyl group and active-site residues might lead to
unfavorable contacts. Such unfavorable contacts are also seen in the binding of
the sulfated derivative, kotalanol (1) relative to its de-O-sulfonated analogue
(2).20
6.5 Acknowledgments
We thank Buford L. Nichols, Roberto Quezada-Calvillo and Hassan Naim
for reagents for recombinant expression, and Lyann Sim for MGAM and SI
enzyme purification. K. J. was supported by a scholarship from the Canadian
Institutes of Health Research (CIHR) and the Canadian Digestive Health
Foundation. We also thank CIHR (MOP111237), NSERC, and the Heart and
Stroke Foundation of Ontario (NA-6305) for financial support.

177
6.6 References
1. Peters, A.; Schweiger, U.; Pellerin, L.; Hubold, C.; Oltmanns, K. M.;
Conrad, M.; Schultes, B.; Born J.; Fehm, H. L. Neurosci. Biobehav. Rev.
2004, 28, 143.
2. Livesey, G. Proc. Nutr. Soc. 2005, 64, 105.
3. McCall, A. L. Eur. J. Pharmacol. 2004, 490,147.
4. Corring, T. Reprod. Nutr. Dev. 1980, 20, 1217.
5. Jones, B. J.; Brown, B. E.; Loran, J. S.; Edgerton, D.; Kennedy J. F.;
Stead, J. A.; Silk D. B. A. Gut 1983, 24, 1152.
6. Lee, P.C. J. Pediatr. Gastroenterol. Nutr. 1983, 2, S227.
7. Fujita, S.; Fuwa, H. J. Nutr. Sci. Vitaminol. (Tokyo) 1984, 30,135.
8. Gray, G. M. J. Nutr. 1992, 122, 172.
9. Gray, G. M. N. Engl. J. Med. 1975, 292, 1225.
10. Brayer, G. D.; Sidhu, G.; Maurus, R.; Rydberg, E. H.; Braun, C.; Wang, Y.;
Nguyen, N. T.; Overall, C. M.; Withers, S. G. Biochemistry 2000, 39, 4778.
11. Horvathova, V.; Janecek, S.; Sturdik, E. Gen. Physiol. Biophys. 2001, 20,
7.
12. Robyt, J.; French, D. J. Biol. Chem. 1970, 15, 3917.
13. Van Beers, E. H.; Buller, H. A.; Grand, R. J.; Einerhand, A. W.; Dekker, J.
Crit. Rev. Biochem. Mol. Boil. 1995, 30, 197.
14. Semenza, G. Ann. Rev. Cell Biol. 1986, 2, 255.
15. Ernst, H. A.; Leggio, L. L.; Willemoës, M.; Leonard, G.; Blum, P.; Larsen,
S. J. Mol. Biol. 2006, 358, 1106.

178
16. Quezada-Calvillo, R.; Sim, L.; Ao, Z.; Hamaker, B. R.; Quaroni, A.;
Brayer, G. D.; Sterchi, E. E.; Robayo-Torres, C. C.; Rose, D. R.; Nichols,
B. L. J. Nutr. 2008, 138, 685.
17. Jones, K.; Sim, L.; Mohan, S.; Jayakanthan, K.; Liu, H.; Avery, S.; Naim,
H. H.; Quezada-Calvillo, R.; Nichols, B. L.; Pinto B.M.; Rose, D. R. Bioorg.
Med. Chem. 2011, 19, 3929.
18. Naumoff, D. G. Mol. Biol. (Mosk). 2007, 41, 1056.
19. Jayakanthan, K.; Mohan, S.; Pinto, B. M. J. Am. Chem. Soc. 2009, 5621.
20. Sim, L.; Jayakanthan, K.; Mohan, S.; Nasi, R.; Johnston, B. D.; Pinto, B.
M.; Rose, D. R. Biochemistry 2010, 49, 443.
21. Eskandari, R.; Jones, K.; Rose, D. R.; Pinto, B. M. Bioorg. Med. Chem.
Lett. 2010, 20, 5686.
22. Tanabe, G.; Otani, T.; Cong, W.; Minematsu, T.; Ninomiya, K.; Yoshikawa,
M.; Muraoka, O. Bioorg. Med. Chem. Lett. 2011, 21, 3159.
23. Yoshikawa, M.; Xu, F.; Nakamura, S.; Wang, T.; Matsuda, H.; Tanabe, G.;
Muraoka, O. Heterocycles 2008, 75, 1397.
24. a) Eskandari, R.; Kuntz, D. A.; Rose, D. R.; Pinto, B. M. Org. Lett. 2010,
12, 1632; b) Eskandari, R.; Jones, K.; Rose, D. R.; Pinto, B. M. Chem.
Commun. 2011, 47, 9134.
25. Sim, L.; Willemsma, C.; Mohan, S.; Naim, H. Y.; Pinto, B.M.; Rose, D. R.
J. Biol. Chem. 2010, 285, 17763.
26. Nasi, R.; Sim, L.; Rose, D. R.; Pinto, B. M. J. Org. Chem. 2007, 72, 180.

179
CHAPTER 7: PROBING THE INTESTINAL α-GLUCOSIDASE ENZYME SPECIFICITIES OF STARCH-
DIGESTING MALTASE-GLUCOAMYLASE AND SUCRASE-ISOMALTASE: SYNTHESIS AND INHIBITORY PROPERTIES OF 3′- AND 5′- MALTOSE-EXTENDED DE-
O-SULFONATED PONKORANOL
This Chapter comprises the manuscript “Probing the intestinal α-glucosidase enzyme specificities of starch-digesting maltase-glucoamylase and
sucrase-isomaltase: Synthesis and inhibitory properties of 3′- and 5′- maltose-extended de-O-sulfonated ponkoranol” which was published in
Chemistry A European Journal (2011, 17, 14817-14825).
Razieh Eskandari,a Kyra Jones,b Kongara Ravinder Reddy, a Kumarasamy
Jayakanthan, a Marcia Chaudet, b David R. Rose,b B. Mario Pintoa
aDepartment of Chemistry, Simon Fraser University, Burnaby, British Columbia,
Canada V5A 1S6
b Department of Biology, University of Waterloo, Waterloo, Ontario,
Canada N2L 3G1

180
An approach to controlling blood glucose level in individuals with type-2
diabetes is to inhibit intestinal glucosidases using α-glucosidase inhibitors.
Maltase-glucoamylase and sucrase-isomaltase are two intestinal glucosidases
that can be targeted for this purpose. The design and synthesis of specific
domain inhibitors that exploit differences between MGAM and SI subsites will be
helpful to reduce unwanted side effects. The design of domain-specific inhibitors
will also aid in the characterization of these domains and help to better define the
complementary roles of MGAM and SI in the process of terminal starch digestion.
In this Chapter, C-3′- and C-5′-β-O-maltose-extended de-O-sulfonated
ponkoranol were synthesized to probe the intestinal α-glucosidase enzyme
specificities of MGAM and SI. We have evaluated the inhibitory activities of
these compounds against recombinant human maltase-glucoamylase (ntMGAM
and ctMGAM) and sucrase-isomaltase (ntSI and ctSI).
The thesis author performed all the experimental synthetic work and
characterization of the C-3′-β-maltose-extended de-O-sulfonated ponkoranol. Dr.
Kongara Ravinder Reddy performed part of the experimental synthetic work and
characterization of the C-5′-β-maltose-extended de-O-sulfonated ponkoranol
compound. Dr. Jayakanthan Kumarasamy assisted in the synthetic design and
part of the experimental synthetic work and characterization of the C-5′-β-O-
maltose-extended de-O-sulfonated ponkoranol compound. The thesis author
wrote and edited the manuscript with Dr. B. Mario Pinto. Mrs. Kyra Jones, Ms.
Marcia Chaudet and Dr. David R. Rose performed the enzyme inhibition studies.

181
Graphical abstract:
7.1 Keywords
Intestinal glucosidase inhibition, maltose-extended-de-O-sulfonated
ponkoranol, sulfonium-ions, glucosidase inhibitors.
7.2 Abstract
The synthesis and glucosidase inhibitory activities of two C-3′- and C-5′-β-
maltose-extended de-O-sulfonated analogues of the naturally-occurring
sulfonium-ion inhibitor, de-O-sulfonated ponkoranol, are described. The
compounds are designed to test the specificity towards four intestinal glycoside
hydrolase family 31 enzyme activities, responsible for the hydrolysis of terminal
starch products and sugars into glucose, in humans. The target sulfonium-ion
compounds were synthesized by means of nucleophilic attack of benzyl
protected 1,4-anhydro-4-thio-D-arabinitol at the C-6 position of 6-O-
trifluoromethanesulfonyl trisaccharides as alkylating agents. The alkylating
S
OHHO
OH O
OH OH
HO
OH
S
OHHO
OH
OH OH
HO
OH
O
O OH
OH
O OH
O
HOOH
HO
O OH
OH
O OH
O
HOOH
HO
Cl
HO HO
Cl

182
agents were synthesized from D-glucose via glycosylation at C-4 or C-2 with
maltosyl trichloroacetimidate. Deprotection of the coupled products using a two-
step sequence, followed by reduction, afforded the final compounds. Evaluation
of the target compounds for inhibition of the four glucosidase activities indicated
that selective inhibition of one enzyme over the others is possible.
7.3 Introduction
Type-2 (noninsulin-dependent) diabetes, is a metabolic disorder
characterized by elevated blood glucose levels with defects in insulin secretion,
insulin action or both. In the treatment of type-2 diabetes, controlling blood
glucose levels is critical. One strategy is to slow down the breakdown of ingested
carbohydrates and starches and thus delay glucose absorption by inhibiting the
enzymes that are involved in the breakdown of dietary starches and sugars into
glucose. In humans, six enzyme activities are involved in the complete digestion
of dietary starches and sugars into glucose. Two endohydrolases, salivary and
pancreatic α-amylases are responsible for digestion of starch into shorter linear
and branched dextrin chains and four exohydrolase α-glucosidase activities,
maltase-glucoamylase (MGAM) and sucrose-isomaltase (SI), are responsible for
the hydrolysis of terminal starch products and sugars into glucose.1,2 MGAM and
SI are anchored to the brush-border epithelial cells of the small intestine each
containing two catalytic subunits classified under glycosyl hydrolase family 31
(GH31): an N-terminal subunit (ntMGAM and ntSI) near the membrane bound
end of the enzyme and a C-terminal luminal subunit (ctMGAM and ctSI) (Figure
7-1).3

183
Figure 7-1: Schematic diagram of MGAM and SI indicating hydrolytic activity.
SI exhibits hydrolytic activity on branched α-1,6 linkages, complemented
by the hydrolytic activity of both SI and MGAM on α-1,4 linkages.4 These
complementary activities of the human enzymes permit digestion of starches of
plant origin comprising two-thirds of most diets; however, the main substrate of SI
is that with α-1,4 linkages.5
SI is more abundant than MGAM; however, to counteract this deficit in
abundance, MGAM displays higher hydrolytic activities.6-8 With respect to
similarity in sequence, SI and MGAM show 59% amino acid sequence similarity.
The catalytic subunits of MGAM and SI are 40-60% identical in amino acid
sequence.3 N-terminal catalytic subunits and the C-terminal catalytic subunits of
MGAM and SI are more closely related in sequence to one another than the N-
and C-terminal subunits within the same protein.9 There are also multiple
spliceforms of C-terminal MGAM in mammals, two of which are studied in this
paper (ctMGAM-N2 and ctMGAM-N20).9,10 In recent years, the aqueous extracts
of the plant Salacia reticulata found in Sri Lanka and Southern India, have been
used by patients as a remedy for the treatment of type-2 diabetes.11 The active
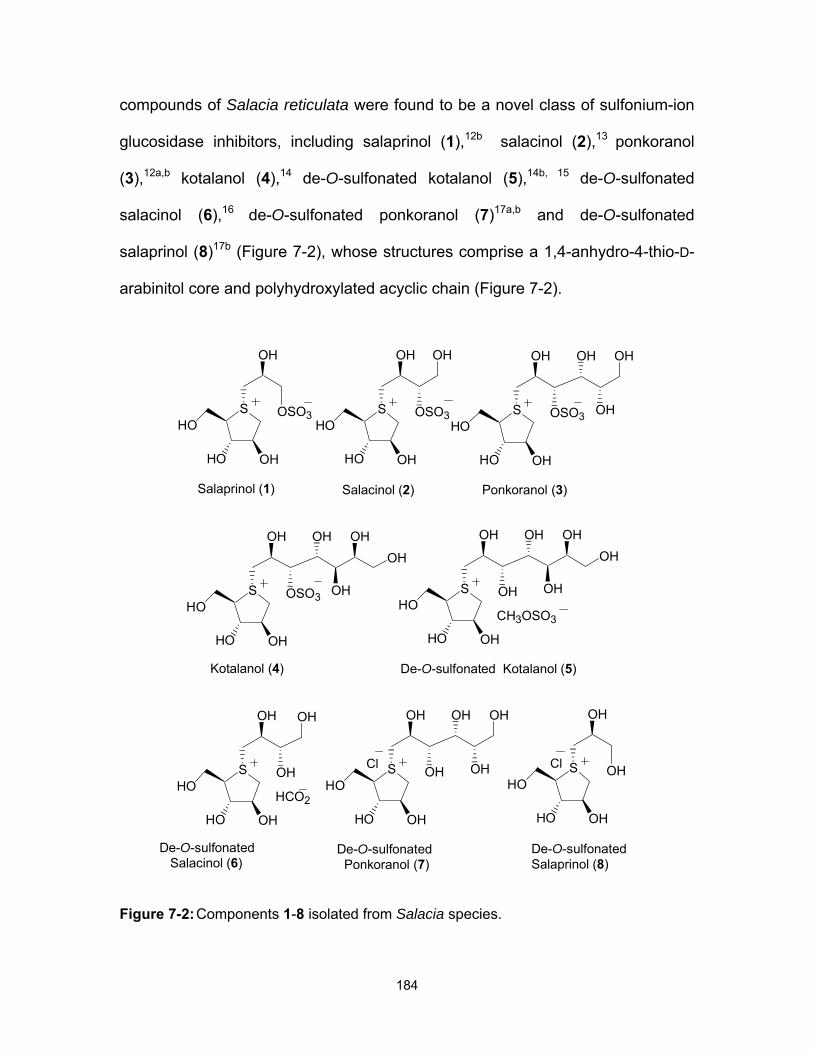
184
compounds of Salacia reticulata were found to be a novel class of sulfonium-ion
glucosidase inhibitors, including salaprinol (1),12b salacinol (2),13 ponkoranol
(3),12a,b kotalanol (4),14 de-O-sulfonated kotalanol (5),14b, 15 de-O-sulfonated
salacinol (6),16 de-O-sulfonated ponkoranol (7)17a,b and de-O-sulfonated
salaprinol (8)17b (Figure 7-2), whose structures comprise a 1,4-anhydro-4-thio-D-
arabinitol core and polyhydroxylated acyclic chain (Figure 7-2).
Figure 7-2: Components 1-8 isolated from Salacia species.
S
OHHO
OSO3 OH
OH OH
HO
OHOH
S
OHHO
OSO3 OH
OH OH
HO
OH
S
OHHO
OSO3
OH OH
HOS
OHHO
OSO3
OH
HO
Kotalanol (4)
Salacinol (2) Ponkoranol (3)Salaprinol (1)
S
OHHO
OH OH
OH OH
HO
OHOH
CH3OSO3
De-O-sulfonated Kotalanol (5)
S
OHHO
OH
OH OH
HOHCO2
De-O-sulfonated Salacinol (6)
S
OHHO
OH OH
OH OH
HO
OH
De-O-sulfonated Ponkoranol (7)
S
OHHO
OH
OH
HO
De-O-sulfonated Salaprinol (8)
ClCl

185
They have been shown to be stronger inhibitors of ntMGAM, with Ki values
in the sub micromolar range (i.e., 0.03-0.19 µM) compared to acarbose (Ki =
62±13 µM), the naturally-occurring α-glucosidase inhibitor currently in use for the
treatment of type-2 diabetes.18-23 Interestingly, de-O-sulfonated ponkoranol (7)
was one of our synthetic compounds,17a isolated recently from the same plant.17b
Acarbose (9) (Figure 7-3) has been shown to be an efficient inhibitor of α-
amylase with a reported Ki of 15 nM24 and of the C-terminal domain of MGAM-N2
(Ki = 0.009±0.002 µM),4, 9 but a weaker inhibitor of the N-terminal domain (Ki =
62±13 µM) (Table 7-1).4, 19
Figure 7-3: Structure of acarbose 9, an α-glucosidase inhibitor currently used in the treatment of type-2 diabetes.
The study of ntMGAM in complex with acarbose had indicated that
acarbose was bound to the ntMGAM active site primarily through side chain
interactions with its acarvosine unit and almost no interactions were made with its
other sugar rings. Since it is suggested that additional subsite interactions with
the acarbose sugar rings would significantly increase its inhibitory properties for
OHN
HO
OOH
H3C
O
OOH
HO
O
HO OH
HO
HOHO
OH
HO
HO
OH9
Acarvosine
Maltose

186
ctMGAM4, 25 it is of interest to examine whether glucose residues appended to the
polyhydroxylated carbon chain of salacinol-based compounds would lead to
differential inhibition of the four enzyme activities. We have shown recently that
extension of the acyclic carbon chain beyond six carbons in salacinol-based
compounds is not essential for activity; and furthermore that the de-O-sulfonated
analogues were more potent inhibitors than the parent compounds.17a Therefore,
de-O-sulfonated ponkoranol has been chosen for modification at C-3′-OH and C-
5′-OH of its side chain in our preliminary studies. We report here the synthesis of
C-3′-β-maltose-extended de-O-sulfonated ponkoranol analogue 10 and C-5′-β-
maltose extended-de-O-sulfonated ponkoranol 11 (Figure 7-4) to investigate the
individual roles of the four catalytic subunits comprising human intestinal MGAM
and SI. These candidates were chosen to test whether occupying the subsite
with glucosyl units would suffice, irrespective of their orientation (reducing or non-
reducing positional substitution) or α- or β-stereochemistry at the anomeric
centre. The β-isomers were chosen as initial candidates because their syntheses
were more tractable.

187
Figure 7-4: 3′-O-β-maltosyl-de-O-sulfonated ponkoranol 10 and 5′-O-β-maltosyl-de-O-sulfonated ponkoranol 11.
7.4 Results and discussion
Synthesis. Retrosynthetic analysis (Scheme 7-1) revealed that the target
molecules could be synthesized by alkylation at the sulfur atom of suitably
protected alkylating agents. The alkylating agents (B) could be obtained by
glycosylation of protected D-glucose (C) at C-4 or C-2 with maltosyl
trichloroacetimidate (12). The required maltosyl trichloroacetimidate donor (12)
could be prepared from maltose according to the literature procedure.26
S
OHHO
OH O
OH OH
HO
OH
S
OHHO
OH
OH OH
HO
OH
O
O OH
OH
O OH
O
HOOH
HO
O OH
OH
O OH
O
HOOH
HO
X
HO HO
X
10 11

188
Scheme 7-1: Retrosynthetic analysis.
To synthesize the desired β-(1→4) and β-(1→2)- maltosyl-linked
glucopyranosides, we chose to use benzyl 2,3-O-[(2R,3R)-2,3-dimethoxybutane-
2,3-diyl]-6-O-tert-butyldimethylsilyl-β-D-glucopyranoside (14) and benzyl 3,4-O-
[(2R,3R)- 2,3 -dimethoxybutane- 2,3 -diyl]- 6 -O- tert -butyldimethylsilyl- β - D -
glucopyranoside (19) as glycosyl acceptors. The acceptors 14 and 19 were
synthesized by selective protection of their primary hydroxyl groups as tert-
butyldimethylsilyl (TBDMS) ethers from butane-2,3-diacetal (BDA) protected
glucopyranosides 13 and 18 which were readily obtained from benzyl β-D-
S
OPPO
POS
OHHO
OH OH
HO
OH
X5'
O
R2O
R1O
PO
L
OP
OOAcO
OC(NH)CCl3AcO
OAc
D-Glucose Maltose
3'
OR2OR1
10. R1 = Maltose, R2 = H11. R1 = H, R2 = Maltose
R1 = Maltose, R2 = P R1 = P, R2 = Maltose
L = leaving groupP = protecting group
O
R2O
R1O
PO
OTBDS
OP
A B
C 12
OAcOAcO
AcO
OAc

189
glucopyranoside by a known procedure.27 The syntheses of the desired
trisaccharides 15 and 20 were achieved by treatment of maltosyl
trichloroacetimidate 12 with the acceptors 14 and 19 in dichloromethane, using a
catalytic amount of boron trifluoride etherate as promoter at room temperature to
give the compounds maltosyl-(1 4)-β- linked 6-O-tert-butyldimethylsilyl-D-
glucopyranoside 15 and maltosyl-(1 2)-β-linked 6-O-tert-butyldimethylsilyl-D-
glucopyranoside 20 in 45% and 52% yield, respectively. The O-deacetylation of
15 and 20 was carried out using NaOMe, followed by protection of the hydroxyl
groups as benzyl ethers to afford benzyl-protected maltosyl-(1 4)-β- linked 6-O-
tert-butyldimethylsilyl-D-glucopyranoside 16 and benzyl protected maltosyl-(1
2)-β- linked 6-O-tert-butyldimethylsilyl-D-glucopyranoside 21. 6- O- Desilylation
of trisaccharides 16 and 21 with TBAF in THF and subsequent triflation of the
free hydroxyl group afforded maltosyl-(1 4)-β- linked 6-O-
trifluoromethanesulfonyl-D-glucopyranoside 17 and maltosyl-(1 2)-β- linked 6-O-
trifluoromethanesulfonyl-D-glucopyranoside 22 in 41% and 86% yield,
respectively (Scheme 7-2).

190
Scheme 7-2: Synthesis of benzyl 6-O-trifluoromethanesulfonyl-D-glucopyranoside derivatives 17 and 22, with benzylated maltose units at C-2 or C-4.
OAcO
AcO
OOAc O
O
OAc
AcO
AcOHO
O
OO
HO
OBn
OMe
OMe
TBDMSCl, Imid. DMF, 0 oC-RT
HOO
OO
TBDMSO
OBn
OMe
OMe
12, BF3.Et2O, DCM,
4oA, 30 min.
O
OO
OTBDMS
OBn
OMe
OMe
OBnOBnO
OOBn O O
OBn
BnO
BnO
O
OO
TBDMSO
OBn
OMe
OMe
1. NaOMe, MeOH, RT, 24h
2. BnBr, NaH, DMF, RT, 2h,
1. TBAF, THF, 12h, RT
2. Tf2O, DCM, -10oC, pyridine
OBnO
BnO
OOBn O
O
OBn
BnO
BnO
O
OO
OTf
OBn
OMe
OMe
13 14 15
16 17
OAc
OBnOBn
O
OAcOAc
O
OAc
O OAc
AcOOAc
O
OH
OH
OBn
TBDMSCl, Imid. DMF, 0 oC-RT
12, BF3.Et2O, DCM,
4oA, 30 min.
1. NaOMe, MeOH,
RT, 24h
2. BnBr, NaH, DMF,
RT, 1h,
1. TBAF, THF,
4h, RT
2. Tf2O, DCM,
-10oC, pyridine
18
20 21 22
OO
OMe
OMe
O
OH
OTBDMS
OBn
19
OO
OMe
OMe
O
O
OTBDMS
OBnOO
OMe
OMe
O
OBnOBn
O
OBn
O OBn
BnOOBn
O
O
OTBDMS
OBnOO
OMe
OMe
O
OBnOBn
O
OBn
O OBn
BnOOBn
O
O
OTf
OBnOO
OMe
OMe
AcO BnO BnO

191
The coupling reaction of the benzyl-protected anhydrothioarabinitol 2328
with 6-O-trifluoromethanesulfonyl trisaccharides 17 and 22 as alkylating agents
was carried out in dichloromethane at room temperature to give the
corresponding sulfonium-ions 24 and 25, respectively, as a 5:1 and 2.8:1 mixture
of diastereomers at the stereogenic sulfur centre (Scheme 7-3).
Scheme 7-3: Coupling reactions to give sulfonium-ions.
SBnO
BnO BnO
OOBnO
OBn
OBnOBnO
BnOOBn
OBn
OOO
OOMe
OMe
SBnO
BnOBnO
OTf
17
S
OBnBnO
BnO
DCM, 24h., RT, 95%
O
O
OO
OMe
OMe
OBn
O
OOBn
OBn
BnO
O
OBnOBn
OBn
BnO
OTf
OBn
22
S
OBnBnO
BnO
DCM, 24h., RT, 90%
24 d.r, 5:1
25a:25bd.r, 2.8:1

192
The diastereoisomers of 24 and 25 were found to be inseparable, even by
HPLC, although repeated attempts to separate the mixture of diastereomers of
25 were partially successful. Prior to choosing 6-O-trifluoromethanesulfonyl
trisaccharides 17 and 22 as alkylating agents, the coupling reaction of 6-O-p-
toluenesulfonyl trisaccharides was carried out in 1,1,1,3,3,3-hexafluoro-2-
propanol (HFIP), based on the procedure that has been reported for synthesis of
3′-O-methylponkoranol (26) (Figure 7-5)29 but the reaction failed and no product
formation was observed. Temperature variation to -78 °C and -25 °C failed to
give a single isomer.
Figure 7-5: Structure of 3'-O-methylponkoranol 26.
The reaction was also carried out at 0 °C and reflux conditions in
dichloromethane but the same ratio was observed. Deprotection of the coupled
products 24 and 25 was carried out by a two-step procedure, first hydrogenolysis
of the benzylethers followed by BDA deprotection. The corresponding crude
materials were then treated with Amberlyst A-26 resin (chloride form) to
completely exchange their triflate counterions to chloride ions.29 Finally, the
corresponding hemiacetals were reduced with NaBH4 in water to provide the
desired C-3′-β-maltose-extended de-O-sulfonated ponkoranol 10 (dr, 5:1) and C-
S
OHHO
OMe OH
OH OH
HO
OH
Cl
26

193
5′-β- maltose-extended de-O-sulfonated ponkoranol 11 (dr, 2.8:1) in 70% and
64% yield, respectively, over four steps (Scheme 7-4).
Scheme 7-4: Synthesis of compounds 10 and 11.
The absolute stereochemistry at the stereogenic sulfur center for the major
isomer of 24 was established by means of a 2D-NOESY experiment (Figure 7-5).
A NOESY correlation was observed between the H-4 proton and H-6′a, implying
that these two hydrogens are syn-facial with respect to the sulfonium ring. In the
case of 25, the absolute configuration at the sulfonium centre for the major
isomer was assigned to be the same as in 24, since a NOESY experiment was
not possible owing to overlapping signals for H-4 and H-6′.
1. Pd(OH)2, 80% AcOH:H2O
2. 85% TFA3. Amberlyst A-26,H2O, 3 h4. NaBH4, 70%
24
1. Pd(OH)2, 80% AcOH:H2O
2. 85% TFA3. Amberlyst A-26,H2O, 3 h4. NaBH4, 64%
25S
OHHO
OH O
OH OH
HO
OH
S
OHHO
OH
OH OH
HO
OH
O
O OH
OH
O OH
O
HOOH
HO
O OH
OH
O OH
O
HOOH
HO
Cl
HO HO
Cl
10 11

194
Figure 7-6: NOESY correlations in compound 24.
Enzyme kinetics. Kinetic parameters were determined by measuring the
amount of glucose produced upon the addition of enzyme at increasing maltose
concentrations (from 0.5 to 30 mM) in the presence of increasing inhibitor
concentration (0-200 nM) by a two-step glucose oxidase assay in a 96-well plate.
The enzyme was allowed to act on the maltose substrate in the presence of
inhibitor for 45 minutes at 37°C. The reactions were then quenched with Tris-HCl
to a final concentration of 1M. Glucose oxidase reagent (Sigma-Aldrich) was then
added to each well (125µl) and the reactions were allowed to develop for 30
minutes at 37°C. Reactions were performed in quadruplicate and absorbance
measured at 405 nM by a SpectraMax 190 Plate Reader (Molecular Devices).
Absorbance readings were averaged to give the final value, which was compared
to a glucose standard curve to determine the amount of glucose released by the
enzyme from the substrate. The program KaleidaGraph4.1 was used to fit the
data to the Michaelis-Menton equation and estimate Km, Kmobs (Km in the
presence of the inhibitor) and Vmax of the catalytic subunits. Ki values for each
inhibitor were determined using the equation Ki = [I]/(Kmobs/Km)-1). The Ki values
OOBnO
OBn
OBnOBnO
BnOOBn
OBn
OOH2C
O
OOMe
OMe
SBnO
BnOOBn
OTf
OBn
24
H4
6'

195
reported for each inhibitor were determined by averaging the Ki values from three
different inhibitor concentrations. The weights of compounds 10 and 11 were
adjusted for the presence of major isomer. The data were also plotted on
Lineweaver-Burk plots to verify that the inhibitors were acting as competitive
inhibitors (see Supporting Information). The methods used for kinetic assays
were reported previously.30, 31
Kinetic Analysis. The inhibition constants (Ki) of acarbose,19, 25, 30 C-3′-β-
maltose-extended de-O-sulfonated ponkoranol 10 and C-5′-β-maltose-extended
de-O-sulfonated ponkoranol 11 against MGAM and SI were determined using the
glucose oxidase assay and maltose as a substrate. There are also multiple,
alternatively-spliced isoforms of ctMGAM; the two studied in this context, referred
to as ctMGAM-N2 and ct-MGAM-N20, occur in humans.9 The experimentally
determined inhibition constants for acarbose,19, 25, 30 10, and 11 are listed Table
7-1. The current anti-diabetic compound, acarbose, was found to be a
micromolar inhibitor of both ntMGAM (Ki = 62 ±13 µM)19 and ntSI (Ki = 14 ± 1
µM).30 In contrast, acarbose is a 1000-fold better inhibitor of ctMGAM-N2 (Ki =
0.009 ± 0.002 µM), ctMGAM-N20 (Ki = 0.028 ± 0.005 µM) and 100-fold better of
ctSI (Ki = 0.246 ± 0.005 µM). C-3′-β-maltose-extended de-O-sulfonated
ponkoranol 10, generally shows some selectivity for inhibiting N-terminal MGAM,
as compared to C-terminal enzymes: ntMGAM (Ki = 0.039 ± 0.025) over
ctMGAM-N2 (Ki = No Inhibition) and ctMGAM-N20 (Ki = 0.655 ± 0.063µM), ctSI
(Ki = 0.062 ± 0.005µM) and ntSI (Ki = 0.046 ± 0.018µM). Surprisingly, C-5′-β-
maltose-extended de-O-sulfonated ponkoranol 11, shows improved inhibition of

196
ntMGAM (Ki = 0.008 ± 0.002 µM), with little distinction between the other catalytic
enzyme subunits: ctMGAM-N20 (Ki = 0.067 ± 0.012 µM), ctSI (Ki = 0.045 ± 0.001
µM), ctMGAM-N2 (Ki = 0.077 ± 0.015), and ntSI (Ki 0.019 ± 0.008 µM).
In this study, we report the design, synthesis, and glucosidase inhibitory
activities of two ponkoranol-based compounds 10 and 11 against recombinant
human maltase-glucoamylase (ntMGAM and ctMGAM) and sucrase-isomaltase
(ntSI and ctSI). These compounds were intended to probe whether one could
differentiate between the different enzymes, i.e. to toggle between the different
enzymes even though they are not strictly analogous to acarbose. Kinetic
analysis confirmed the enzyme activity of the recombinant proteins, and inhibition
analysis confirmed classic competitive inhibition by the α-glucosidase inhibitors.
The results (Table 7-1) indicate that despite the overall similarity between the
subunits in terms of amino-acid sequence, they exhibit different biochemical and
structural properties. Compound 11 demonstrates the ability to inhibit all of the
catalytic subunits very well, especially ntMGAM. In fact, compound 11 is the most
potent inhibitor of ntMGAM to date. Compound 10 is also a good inhibitor of ctSI,
ntSI, and ntMGAM, with Ki values in the nanomolar range. Although elongation of
the scaffold does not result in a significant gain in binding energy, nonetheless it
does result in some interesting selectivities in inhibitory activities. A significant
finding is that 10 differentiates between the two C-terminal catalytic subunits.
Compound 10 is a very poor inhibitor of ctMGAM-N20 and shows no inhibition
against ctMGAM-N2. By confirming the higher potency of 10 against other
subunits as compared with ctMGAM, our results suggest that this inhibitor shows

197
specificity towards these different α-glucosidases. Since ctMGAM is expressed in
more than one spliceform,9 it provides further complexity to the system. It is
hypothesized that these units act in a complementary manner depending on the
organism’s nutritional sources and requirements, as well as its physical and
environmental conditions. Therefore, with compound 10, we are able to keep the
ctMGAM activity on and dampen the others, and we are in a position to test this
hypothesis. According to previous studies, acarbose is a very powerful inhibitor of
ctMGAM over ntMGAM and SI subunits.9 Therefore, we are now also in a
position to turn off the ctMGAM unit and study the effect of others subunits in
starch digestion. The results also demonstrate that the ability to selectively inhibit
one enzyme unit over the others can result from relatively small changes in the
structure of the compound and we are closer to being able to independently
toggle each subunit on and off. This observation is important clinically because
the design of α-glucosidase inhibitors for the treatment of type-2 diabetes might
require specificity for enzymes later in the starch digestion pathway in order to
reduce unwanted side-effects. The initial results of the inhibition assays are
promising; at this point, analysis of structure-activity relationships can only be
somewhat speculative. However, the very interesting and unprecedented enzyme
selectivity observed in this study set the stage for improvement of the specificity
and affinity of these compounds for their potential development as antidiabetic
agents, irrespective of whether or not the binding of these compounds occurs
through a mechanism analogous to that of acarbose. It is noteworthy that de-O-
sulfonated ponkoranol 7 (Figure 7-2) 32 and 3’-O-methylponkoranol (26) (Figure

198
7-5)33 also show an interesting selectivity profile for the different subunits (Table
7-1). Further confirmation of the importance of inhibitor structure and how it
affects binding in the ctMGAM active site will be possible with an analysis of the
atomic structure of the ctMGAM binding site in the presence of bound inhibitor.
Determination of this structural information will be a valuable tool in future design
and synthesis of α-glucosidase inhibitors effective against and specific to MGAM
and SI subunits. These inhibitors should be promising lead candidates as oral
agents for the treatment and prevention of type-2 diabetes.
Table 7-1: Comparison of inhibition profiles against MGAM and SI subunits, Ki (µM).
ctMGAM-N2 ctMGAM-N20 ntMGAM ctSI ntSI
732 no Inhibitiona 0.096 ± 0.015 0.043 ± 0.001 0.103 ± 0.037 0.302 ± 0.123
9 0.009±0.002 0.028±0.005 62±13 0.246±0.005 14±1
10 no Inhibitiona 0.655±0.063 0.039±0.025 0.062±0.005 0.046±0.018
11 0.077 ± 0.015 0.067±0.012 0.008±0.002 0.045±0.001 0.019±0.008
2633 0.060 ± 0.015 0.055 ± 0.014 0.50 ± 0.0429 0.007 ± 0.002 0.035 ± 0.013
a at 200 nM

199
7.5 Experimental
7.5.1 Compound characterization data
7.5.1.1 Benzyl 6-O-tert-butyldimethylsilyl-2,3-O-[(2R,3R)-2,3-dimethoxybutane-2,3-diyl]-β-D-glucopyranoside 14.
To a solution of 13 (500 mg, 1.3 mmol) in DMF (15 mL) was added
imidazole (270 mg, 3.9 mmol). The reaction was cooled in an ice bath, TBDMSCl
(226 mg, 1.4 mmol) was added portionwise, and the mixture was stirred at 0 °C
for 15 min and at room temperature 1 h. The reaction was quenched by the
addition of ice-water, and the reaction mixture was extracted with Et2O (3×30
mL). The combined organic solvents were washed with water (15 mL) and brine
(15 mL), dried (Na2SO4), and concentrated to give the crude. The crude was
purified by column chromatography (Hexanes/EtOAc 2:1) to afford 14 as foam
(564 mg, 87%). [α] 23D = - 124.5 (c = 2.3, CH2Cl2). 1H NMR (CDCl3) δ 7.28-7.15
(5H, m, Ar), 4.79, 4.55 (2H, 2d, JA,B = 12.3 Hz, CH2Ph), 4.51 (1H, d, J1,2 = 8.0 Hz,
H-1), 3.83 (1H, dd, J6a,6b = 5.5, J5,6a = 10.3 Hz, H-6a), 3.75 (1H, dd, J6a,6b = 5.8,
J5,6b = 10.7 Hz, H-6b), 3.63 (2H, m, H-3, H-4), 3.49 (1H, t, J1,2 = J2,3 = 8.5 Hz, H-
2 ), 3.30 (1H, ddd, J5,6a = 5.7, J5,6b = 8.6, J4,5 = 10.7 Hz, H-5), 3.21, 3.19 (6H, 2s,
2OMe), 3.06 (1H, br, OH), 1.25, 1.23 (6H, 2s, 2Me), 0.81 (9H, s, 3Me), 0.00 (6H,
s, 2Me). 13C NMR (CDCl3) δ 137.7-127.5 (m, Ar), 99.9 (C-1), 99.5, 99.4
(2MeOCMe), 74.9 (C-5), 72.5 (C-3), 70.8 (CH2Ph), 70.3 (C-4), 69.2 (C-2), 64.8
(C-6), 48.0, 47.9 (2OMe), 25.9 (3Me), 18.32 (CMe3), 17.7 (2Me), -5.4, -5.5 (2Me).
HRMS Calcd for C25H42NaO8Si (M+Na): 521.2541. Found: 521.2537.

200
7.5.1.2 Benzyl 6-O-tert-butyldimethylsilyl-3,4-O-[(2R,3R)-2,3-dimethoxybutane-2,3-diyl)]-β-D-glucopyranoside 19
The compound was obtained as white solid (654.4 mg, 82 %) from 18 (614
mg, 1.6 mmol) using the same procedure that was used to obtain 14. M.P. 145-
147 oC. [α] 23D = + 81.8 (c = 1.0, CH2Cl2). 1H NMR (CDCl3): δ 7.38−7.27 (5H, m,
Ar), 4.90 (1H, d, JAB = 11.4 Hz, CH2Ph), 4.60 (1H, d, JAB = 11.4 Hz, CH2Ph), 4.39
(1H, d, J1, 2 = 7.4 Hz, H-1), 3.90 (1H, dd, J6a, 6b = 11.6, J6a, 5 = 2.2 Hz, H-6a), 3.85
(1H, dd, J6b, 6a = 11.6, J6b, 5 = 3.8 Hz, H-6b), 3.77−3.68 (2H, m, H-3, H-4), 3.59
(1H, t, J1, 2 = J2, 3 = 9.6 Hz, H-2 ), 3.44 (1H, ddd, J5, 6a = 2.2, J5, 6b = 3.7, J5, 4 =
9.4 Hz, H-5), 3.29, 3.27 (6H, 2s, 2OMe), 2.53 (1H, br, OH), 1.33, 1.29 (6H, 2s,
2Me), 0.90 (9H, s, 3 Me), 0.10, 0.08 (6H, 2s, 2Me). 13C NMR (CDCl3) δ
137.0−127.9 (m, Ar), 101.9 (C-1), 99.5, 99.0 (2MeOCMe), 74.7 (C-5), 71.9 (C-3),
71.2 (C-2), 70.7 (CH2Ph), 65.1 (C-4), 61.3 (C-6), 48.0, 47.9 (2OMe), 25.8 (3 Me),
18.3 (CMe3), 17.6 (2 Me) -5.0, -5.4 (2Me). HRMS Calcd for C25H42NaO8Si
(M+Na): 521.2541. Found: 521.2546.
7.5.1.3 Benzyl 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl-(1 4)-2,3,6-tri-O-acetyl-β-D-glucopyranosyl-(1 4)-6-O-tert-butyldimethylsilyl-2,3-O-[(2R,3R)-2,3-dimethoxybutane-2,3-diyl]-β-D-glucopyranoside 15
To a solution of the acceptor glycoside 14 (0.7 g, 1.4 mmol) and maltosyl
trichloroacetimidate 12 (1.3 g, 1.68 mmol) in dry CH2Cl2 (10 mL) under N2,
BF3.Et2O (0.02 mL, 0.2 eq) was added at room temperature. After stirring for 30
min, the mixture was quenched with NEt3 (0.2 eq.) with vigorous stirring. The
mixture was concentrated and purified by column chromatography
(Hexanes/EtOAc 1:1) to give the coupled product 15 as white foam (0.7g, 45%).

201
[α] 23D = - 9 (c = 0.7, CH2Cl2). 1H NMR (CDCl3) δ 7.39-7.28 (5H, m, Ar), 5.42 (1H,
d, J1′′,2′′ = 4.1 Hz, H-1′′), 5.38 (1H, t, J2′′,1′′ = J2′′,3′′ = 10.4 Hz, H-2′′), 5.19 (1H, t, J3′,4′
= J2′,3′ = 7.7 Hz, H-3′), 5.08 (1H, t, J3′′,4′′ = J4′′,5′′ = 9.9 Hz, H-4′′), 4.90, 4.66 (2H, 2d,
JA,B = 12.0 Hz, CH2Ph), 4.87 (1H, t, J3′′,4′′ = J2′′,3′′ = 4.1 Hz, H-3′′), 4.83 (2H, m, H-
1′, H-2′), 4.55 (1H, d, J1,2 = 7.8 Hz, H-1), 4.42 (1H, dd, J6′a,6′b = 3.1, J5′,6′a = 12.5
Hz, H-6′a), 4.29 (1H, dd, J6′a,6′b = 3.6, J5′,6′b = 12.2 Hz, H-6′b), 4.24 (1H, dd, J6′′a,6′′b
= 3.6, J5′′,6′′a = 12.5 Hz, H-6′′a), 4.06 (2H, m, H-6′′b, H-4′), 3.94 (1H, ddd, J5′′,6a′′ =
3.1, J5′′,6b′′ = 2.6, J4′′,5′′ = 10.0 Hz, H-5′′), 3.88 (1H, dd, J6a,6b = 1.5, J5,6a = 11.5 Hz,
H-6a), 3.82 (1H, t, J3,4 = J2,3 = 9.6 Hz, H-3), 3.71 (2H, m, H-4, H-6b), 3.67 (1H,
ddd, J5′,6a′ = 3.2, J5′,6b′ = 3.0, J4′,5′ = 9.5 Hz, H-5′), 3.59 (1H, dd, J1,2 =7.8, J2,3 =
10.0 Hz, H-2 ), 3.34 (1H, ddd, J5,6a = 1.5, J5,6b = 5.1, J4,5 = 6.8 Hz, H-5), 3.32,
3.30 (6H, 2s, 2OMe), 2.14, 2.12, 2.06, 2.05, 2.04, 2.02, 2.01 (21H, 7s, 7OAc),
1.34, 1.32 (6H, 2s, 2Me), 0.94 (9H, s, 3Me), 0.11, 0.10 (6H, 2s, 2Me). 13C NMR
(CDCl3) δ 170.4-169.3 (m, CO), 137.4-127.4 (m, Ar), 100.0 (C-1′), 99.4, 99.3
(2MeOCMe), 99.2 (C-1), 95.4 (C-1′′), 76.1 (C-5), 75.5 (C-3′), 75.4 (C-4), 72.8 (C-
2′), 72.3 (C-4′), 71.9 (C-5′), 71.4 (C-3), 70.3 (CH2Ph), 69.8 (C-3′′), 69.4 (C-2),
69.2 (C-2”), 68.3 (C-5”), 67.8 (C-4”), 62.9 (C-6′), 61.9 (C-6), 61.2 (C-6”), 47.8,
47.7 (2OMe), 25.8 (3Me), 20.7-20.4 (m, OAc), 18.22 (CMe3), 17.5, 17.4 (2Me), -
5.1, -5.3 (2Me). HRMS Calcd for C51H76NaO25Si (M+Na): 1139.4337. Found:
1139.4330.

202
7.5.1.4 Benzyl 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl-(1 4)-2,3,6-tri-O-acetyl-β-D-glucopyranosyl-(1 2)-6-O-tert-butyldimethylsilyl-3,4-O-[(2R,3R)-2,3-dimethoxybutane-2,3-diyl]-β-D-glucopyranoside 20
The compound was obtained as white solid (1.7 g, 52 %) from 19 (1.5 g,
2.95 mmol) using the same procedure that was used to obtain 15. M.P. 95-98 oC.
[α] 23D = + 64.4 (c = 0.1, CH2Cl2). 1H NMR (CDCl3) δ 7.40−7.29 (5H, m, Ar), 5.37
(1H, d, J1′′, 2′′ = 4.0 Hz, H-1′′), 5.32 (1H, t, J3′′,2′′ = J3′′,4′′ = 9.5 Hz, H-3′′), 5.16 (1H,
t, J3′,2′ = J3′,4′ = 9.1 Hz, H-3′), 5.03 (1H, t, J4′′,3′′ = J4′′,5′′ = 10.1 Hz, H-4′′), 4.94−4.91
(2H, m, H-1′, CH2Ph), 4.85 (1H, dd, J2′′,1′′ = 7.5, J2′′,3′′ = 4.0 Hz, H-2′′), 4.81 (1H,
dd, J2′,1′ = 7.5, J2′,3′ = 2.1 Hz, H-2′), 4.62 (1H, d, JAB = 11.7 Hz, CH2Ph), 4.50 (1H,
d, J1, 2 = 6.9 Hz, H-1), 4.28 (1H, dd, J6′b,6′a = 12.2, J6′b,5′ = 2.6 Hz, H-6′b), 4.23 (1H,
dd, J6′′a, 6′′b = 12.6, J6′′a,5′′ = 3.8 Hz, H-6′′a), 4.08−3.96 (3H, m, H-6′a, H-6′′b, H-4′),
3.89 (1H, ddd, J5′′,4′ ′ = 10.1, J6′′a,5′′ = 3.3, J6′′b,5′′ = 2.3 Hz , H-5′′), 3.86−3.84 (2H, m,
H-6a, H-6b), 3.75−3.64 (3H, m, H-2, H-4, H-5), 3.49 (1H, ddd, J5′,4′ = 9.7, J5′, 6′a =
3.7, J5′, 6′b = 2.4, H-5′), 3.37 (1H, dd, J3,2 = 3.4, J3,4 = 1.8, H-3), 3.24, 3.20 (6H, 2s,
2OMe), 2.07−2.00 (21H, 7s, 7OAc), 1.26, 1.25 (6H, 2s, 2Me), 0.88 (9H, s, 3Me),
0.07, 0.05 (6H, 2s, 2Me). 13C NMR (CDCl3, 100MHz) δ 170.5−169.4 (m, CO),
137.6−127.4 (m, Ar), 101.8 (C-1), 99.9 (C-1′), 99.6, 99.4 (2MeOCMe), 95.6 (C-
1′′), 77.9 (C-2), 75.8 (C-3′), 74.4 (C-3), 73.3 (C-2′), 72.6 (C-4′), 72.2 (C-5′), 71.2
(C-5), 70.7 (CH2Ph), 69.9 (C-2′′), 69.4 (C-3′′), 68.4 (C-5′′), 68.0 (C-4′′), 65.2 (C-4),
63.0(C-6′), 61.4 (C-6′′), 61.3 (C-6), 48.0, 47.9 ( 2OMe), 25.8 (3Me), 21.0−20.6 (m,
OAc), 18.4 (CMe3), 17.6, 17.5 (2Me), -5.0, -5.4 (2Me). HRMS Calcd. for
C51H76NaO25Si (M+Na): 1139.4337.Found: 1139.4325.

203
7.5.1.5 Benzyl 2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl-(1 4)-2,3,6-tri-O-benzyl-β-D-glucopyranosyl-(1 4)-6-O-tert-butyldimethylsilyl-2,3-O-[(2R,3R)-2,3-dimethoxybutane-2,3-diyl]-β-D-glucopyranoside 16
To a solution of compound 15 (500 mg, 4.5 mmol) in MeOH (15 mL), was
added catalytic amount of 1M NaOMe and the reaction was stirred at room
temperature overnight. The solvent were removed under vacuum to give the
crude product which was used directly in the next step without further purification.
The crude product was dissolved in DMF (20 mL), and NaH (130 mg, 5.4 mmol)
and BnBr (0.5 mL, 4 mmol) were added at 0 °C. The reaction mixture was stirred
for 2 h at room temperature, then quenched with ice, and extracted with ether
(3×50 mL). The organic solution was washed with water (30 mL) and brine (30
ml), dried (Na2SO4) and concentrated and the residue was purified by flash
chromatography (EtOAc/Hexanes (1:5)) to yield 16 as white foam (475 mg, 73%).
[α] 23D = - 15 (c = 0.8, CH2Cl2). 1H NMR (CDCl3) δ 7.40-7.11 (40H, m, Ar), 5.73
(1H, d, J1′′,2′′ = 3.7 Hz, H-1′′), 4.96-4.52 (14H, m, 7CH2Ph), 4.63 (1H, m, H-1′),
4.60 (1H, m, H-1), 4.45, 4.26 (2H, 2d, JA,B = 12.1 Hz, CH2Ph), 4.18 (1H, t, J3′′,4′′ =
J4′′,5′′ = 8.9 Hz, H-4′′), 4.01 (1H, t, J3′,4′ = J4′,5′ = 9.5 Hz, H-4′), 3.95-3.77 (8H, m, H-
6a, H-3′′, H-4′, H-6′′a, H-3, H-6b, H-6b′′, H-3′), 3.66 (2H, m, H-5′, H-2), 3.54-3.49
(3H, m, H-5′′, H-6′a, H-2′′), 3.46 (1H, dd, J2′,1′ = 8.0 J2′,3′ = 9.0 Hz, H-2′), 3.38 (1H,
dd, J6′a,6′b = 1.8, J5′,6′b = 10.9 Hz, H-6′b), 3.33 (1H, ddd, J5,6a = 1.8, J5,6b = 3.5,
J4,5 = 5.3 Hz, H-5), 3.31, 3.28 (6H, 2s, 2OMe), 1.34, 1.32 (6H, 2s, 2Me), 1.32,
1.21 (6H, 2s, 2Me), 0.91 (9H, s, 3Me), 0.07, 0.06 (6H, 2s, 2Me). 13C NMR
(CDCl3) δ 138.3-126.1 (m, Ar), 101.3 (C-1′), 99.2 (C-1), 99.1, 99.0 (2MeOCMe),
96.2 (C-1′′), 84.6 (C-3′), 82.3 (C-2′), 81.6 (C-4′), 78.8 (C-5′′), 77.3 (C-5′), 76.3 (C-

204
5), 75.0, 74.5, 74.2, 73.6, 73.2, 73.0, 72.8, 69.8 (8CH2Ph), 74.4 (C-2”), 72.5 (C-
4), 71.6 (C-3′′), 70.4 (C-3), 70.3 (C-4”), 69.4 (C-2), 68.1 (C-6′′), 67.7 (C-6′), 61.2
(C-6), 47.5, 47.4 (2OMe), 25.5 (3Me), 17.93 (CMe3), 17.2, 17.1 (2Me), -5.4, -5.6
(2Me). HRMS Calcd for C86H104NaO18Si (M+Na): 1477.6941. Found: 1477.6941.
7.5.1.6 Benzyl 2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl-(1 4)-2,3,6-tri-O-benzyl-β-D-glucopyranosyl-(1 2)-6-O-tert-butyldimethylsilyl-3,4-O-[(2R,3R)-2,3-dimethoxybutane-2,3-diyl]-β-D-glucopyranoside 21
The compound was obtained as white solid (585 mg, 76 %) from 20 (592
mg, 0.53 mmol) using the same procedure that was used to obtain 16. M.P. 90-
92 oC. [α] 23D = + 76.8 (c = 1.0, CH2Cl2). 1H NMR (CDCl3) δ 7.43−7.17 (40H, m,
Ar), 5.77 (1H, d, J1′′,2′′ = 3.6 Hz, H-1′′), 5.05-4.84 (8H, m, H-1′, 7CH2Ph),
4.71−4.51 (9H, m, H-1, 8CH2Ph), 4.40 (1H, d, JA,B = 12.1 Hz, CH2Ph), 4.24 (1H,
t, J4′,3′ = J4′,5′ = 9.2 Hz, H-4′), 4.18 (1H, t, J4′′,3′′ = J4′′,5′′ = 8.9 Hz, H-4′′), 4.13−3.80
(8H, m, H-6a, H-6b, H-3, H-3′, H-3′′, H-4, H-6′a, H-6′′a), 3.76−3.66 (4H, m, H-6′′b,
H-6′b, H-2, H-5′′), 3.58−3.48 (4H, m, H-2′′, H-2′, H-5′, H-5), 3.35, 3.17 (6H, 2s,
2OMe), 1.33, 1.30 (6H, 2s, 2Me), 0.98 (9H, s, 3Me), 0.18, 0.16 (6H, 2s, 2Me).
13C NMR (CDCl3) δ 138.8−126.5 (m, Ar), 101.9 (C-1′), 101.2 (C-1), 99.5, 99.4
(2MeOCMe), 96.7 (C-1′′), 84.8 (C-5′′), 83.0 (C-2′), 81.9 (C-3′′), 79.2 (C-2′′), 77.7
(C - 4′′), 75.4, 74.9, 74.6 (3 CH2Ph), 74.4 (C-5, C-5′), 73.8, 73.4 (CH2Ph), 73.3
(C-3), 73.1 (CH2Ph), 72.3 (C-4′), 70.8 (C-3′), 70.9 (C-2), 69.7 (CH2Ph), 68.2 (C-
6′′), 68.1 (C-6′), 65.1 (C-4), 61.4 (C-6), 48.0 (2OMe), 25.8, 25.5 (3Me), 18.3
(CMe3), 17.8, 17.6 (2Me), -5.0, -5.4 (2Me). HRMS Calcd. for C86H104NaO18Si
(M+Na): 1477.6941. Found: 1476.6938.

205
7.5.1.7 Benzyl 2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl-(1 4)-2,3,6-tri-O-benzyl-β-D-glucopyranosyl-(1 4)-6-O-trifluoromethanesulfonyl-2,3-O-[(2R,3R)-2,3-dimethoxybutane-2,3-diyl]-β-D-glucopyranoside 17
To a solution of 16 (205 mg, 0.14 mmol) in THF (15 mL), TBAF (1.0 M
solution in THF, 0.28 mL, 0.28 mmol) was added, and the reaction mixture was
stirred at room temperature. After 12 h it was concentrated and further dried
under high vacuum for 1 h. The crude product was dissolved in CH2Cl2 (10 mL),
and pyridine (0.013 mL, 1.2 eq) was added. The mixture was cooled to -10 °C,
and Tf2O (0.036 mL, 1.5 eq) was added under N2. After 30 min the reaction was
quenched by the addition of cold saturated NaHCO3 (1.5 mL). The organic layers
were washed with 1N HCl (5 mL), water (5 mL) and brine (5 mL), dried (Na2SO4),
and concentrated in vacuo. Chromatographic purification of the crude product
(EtOAc/Hexanes (1:10)) gave 17 as a foam (85 mg, 41%). [α] 23D = - 5.5 (c = 0.2,
CH2Cl2). 1H NMR (CDCl3) δ 7.38-7.11 (40H, m, Ar), 5.62 (1H, d, J1′′,2′′ = 3.7 Hz,
H-1′′), 4.91- 4.32 (16H, m, 8CH2Ph), 4.85 (1H, m, H-6a), 4.60 (1H, m, H-6b), 4.59
(1H, m, H-1′), 4.53 (1H, m, H-1), 4.05 (2H, m, H-5′′, H5′), 3.91 (1H, t, J3,4 = J4,5 =
9.7 Hz, H-4), 3.85 (1H, t, J3′,4′ = J4′,5′ = 9.6 Hz, H-4′), 3.76 (4H, m, H-4′′, H-6′a,b,
H-3′), 3.66 (2H, m, H-2, H-3), 3.56-3.48 (4H, m, H-6′′a, H-5, H-2′, H-2′′), 3.42 (1H,
dd, J6′′a,6′′b = 1.8, J5′′,6′′b = 10.5 Hz, H-6′′b), 3.30, 3.29 (6H, 2s, 2OMe), 1.32, 1.23
(6H, 2s, 2Me). 13C NMR (CDCl3) δ 138.6-126.5 (m, Ar), 118.4 (m, CF3), 100.6 (C-
1′), 99.7, 99.5 (2MeOCMe), 99.4 (C-1), 96.9 (C-1′′), 84.9 (C-4′′), 82.2 (C-2′), 81.9
(C-4), 79.1 (C-5), 77.6 (C-3), 75.5, 75.0, 74.7, 74.3, 73.9, 73.4, 73.3, 73.2
(8CH2Ph), 74.6 (C-2′′), 74.5 (C-4′), 72.8 (C-3′), 72.6 (C-5′′), 71.0 (C-3′′), 70.7 (C-
6), 69.6 (C-2), 69.1 (C-6′), 68.4 (C-5′), 68.1 (C-6′′), 48.6, 47.9 (2OMe), 17.5
(2Me). HRMS Calcd for C81H89F3NaO20S (M+Na): 1493.5512. Found: 1493.5543.

206
7.5.1.8 Benzyl 2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl-(1 4)-2,3,6-tri-O-benzyl-β-D-glucopyranosyl- (1 2)-6-O-triflouromethanesulfonyl-3,4-O-[(2R,3R)-2,3-dimethoxybutane-2,3-diyl]-β-D-glucopyranoside 22
Compound was obtained as white solid (227 mg, 86 %) from 21 (262 mg,
0.18 mmol) using the same procedure that was used to obtain 17. M.P. 80-82 oC.
[α] 23D = + 51.4. (c = 0.9, CH2Cl2). 1H NMR (CDCl3): δ 7.34−7.10 (40H, m, Ar), 5.67
(1H, d, J1′′,2′′ = 3.7 Hz, H-1′′), 4.95−4.73 (9H, m, H-1′, H-6b, 7CH2Ph), 4.63−4.45
(10H, m, H-1, H-6a, 8CH2Ph), 4.33 (1H, d, JA,B = 12.1, CH2Ph), 4.14 (1H, t, J4′,3′ =
J4′,5′ = 9.2 Hz, H-4′), 4.05 (1H, t, J5,4 = J5, 6a = 9.3 Hz, H-5), 3.91−3.84 (2H, m, H-
3′′, H-4), 3.78−3.73 (4H, m, H-5′′, H-6′′a, H-3′, H-2), 3.70−3.64 (3H, m, H-3, H-4′′,
H-6′′b), 3.60 (1H, dd, J6′b,6′a =10.7 Hz, J6′b,5′ = 2.7 Hz, H-6′b), 3.50−3.42 (4H, m, H-
2′′, H-2′, H-5′, H-6′a), 3.24, 3.07 (6H, 2s, 2OMe), 1.26, 1.21 (6H, 2s, 2Me). 13C
NMR (CDCl3) δ 138.6-126.5 (m, Ar), 121.8, 119.6, 117.5, 115.4 (m, CF3), 102.0
(C-1′), 101.3(C-1), 99.8, 99.7 (2MeOCMe), 96.8 (C-1′′), 84.8 (C-5′′), 82.9 (C-2′),
82.0 (C-3′′), 79.2 (C-2′′), 77.7 (C-4′′), 75.5, 75.0 (CH2Ph), 74.7 (C-6, C-5), 74.5
(C-5′), 73.9, 73.8, 73.5, 73.3, 73.2 (6CH2Ph), 72.6 (C-4), 72.4 (C-4′), 71.0 (C-3′),
70.9 (C-2), 68.3 (C-6′′), 68.2 (C-6′), 65.3 (C-3), 48.1 (2OMe), 17.7,17.5 (2Me).
HRMS Calcd. for C81H89F3NaO20S (M+Na): 1493.5512. Found: 1493.5487.
7.5.1.9 Benzyl 2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl-(1 4)-2,3,6-tri-O-benzyl-β-D-glucopyranosyl-(1 4)-6-deoxy-6-[2,3,5-tri-O-benzyl-1,4-dideoxy-episulfoniumylidene-D-arabinitol]-2,3-O-[(2R,3R)-2,3-dimethoxybutane-2,3-diyl]-β-D-glucopyranoside trifluoromethanesulfonate 24
The mixture of the thioether 23 (64 mg, 0.15 mmol) and 17 (75 mg, 0.051
mmol) in CH2Cl2 (1 mL) were stirred overnight at room temperature for 24 h. The
mixture was concentrated and purified by column chromatography (CHCl3/MeOH
20:1) to give the sulfonium salt 24 as syrup (90 mg, 95%). Analysis by NMR

207
showed that the product was a mixture of two isomers (~5:1) at the stereogenic
sulfur center. The major component of the mixture was assigned to be the
diastereomer with a cis relationship between H-4 and H-6′ on the basis of
analysis of the NOESY spectrum.
Data for the major diastereomer (trans-24) follow. 1H NMR (CDCl3) δ 7.36-
7.11 (55H, m, Ar), 5.56 (1H, d, J1′′′,2′′′ = 3.6 Hz, H-1′") 4.90-4.36 (22H, m, CH2Ph),
4.67 (1H, m, H-1"), 4.56 (1H, m, H-1′), 4.46 (1H, m, H-2), 4.27 (1H, m, H-3), 4.11-
4.01 (4H, m, H-5′, H-4, H-4′, H-5′′′), 3.92 (1H, t, J3′′′,2′′′ = J3′′′,4′′′ = 9.5 Hz, H-3′′′),
3.83-3.70 (8H, m, H-1a, H-5′′, H-3′′, H-5a, H-5b, H-6′a, H-3, H-6′′′′a), 3.68-3.56
(6H, m, H-4′′′, H-6′b, H-4′′, H-6′′a, H-6′′′b, H-2′), 3.52-3.47 (3H, m, H-2′′′, H-2′′, H-
6′′b), 3.43 (1H, dd, J1a,1b = 4.0, J1b,2 = 13.4 Hz, H-1b), 3.31, 3.29 (6H, 2s, 2OMe),
1.34, 1.23 (6H, 2s, 2Me). 13C NMR (CDCl3) δ 138.6-126.6 (m, Ar), 120.8 (m,
CF3), 100.7 (C-1′), 100.3 (H-1′′), 99.7, 99.6 (2MeOCMe), 99.0 (C-1′′′), 84.6 (C-5′′),
82.4 (C-3), 82.3 (C-2), 81.8 (C-3′′′), 81.5 (C-2′′), 79.2 (C-2′′′), 77.6 (C-4′′′), 76.5 (H-
3′′), 75.5, 75.0, 74.0, 73.6, 73.5, 73.2, 73.0, 72.4, 72.1, 71.8 (m, CH2Ph), 74.5 (H-
2′), 73.3 (H-4′), 71.5 (C-5′), 71.1 (C-3′), 69.5 (C-4′′), 69.3 (C-5), 68.1 (C-6′′), 67.7
(C-5′′′), 67.1 (C-4), 66.4 (C-6′′′), 48,8 47.9 (2OMe), 47.2 (C-1), 46.9 (C-6′), 17.5,
17.4 (2Me). HRMS Calcd for C106H118O20S (M+H):1742.7932. Found: 1742.7932.
7.5.1.10 Benzyl 2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl-(1 4)-2,3,6-tri-O-benzyl-β-D-glucopyranosyl-(1 2)-6-deoxy-6-(2,3,5-tri-O-benzyl-1,4-dideoxy-episulfoniumylidene-D-arabinitol)-3,4-O-[(2R,3R)-2,3-dimethoxybutane-2,3-diyl]-β-D-glucopyranoside trifluoromethanesulfonate 25
The mixture of the thioether 23 (64 mg, 0.15 mmol) and 22 (80mg, 0.054
mmol) in CH2Cl2 (1 mL) were stirred overnight at room temperature for 24 h. The

208
mixture was concentrated and purified by column chromatography (CHCl3/MeOH
20:1) to give the sulfonium salts 25a and 25b as syrups (91.8 mg, 90%). Analysis
by crude NMR showed that the product was a mixture of two isomers (~2.8:1) at
the stereogenic sulfur center.
Data for the major diastereomer (25a): [α] 23D = + 62.5 (c = 0.2, CH2Cl2). 1H
NMR (CDCl3) δ 7.34−7.08 (55H, m, Ar), 5.64 (1H, d, J1′′′,2′′′ = 3.6 Hz, H-1′") ,
4.93−4.72 (8H, m, H-1′′, 7CH2Ph), 4.64 (1H, d, JA,B = 12.6, CH2Ph), 4.60−4.40
(15H, m, H-2, H-1′, 13CH2Ph), 4.39 (1H, brd, J = 7.5, H-4), 4.31−4.27 (2H, m, H-
3, CH2Ph), 4.09 (1H, t, J4′′′,3′′′ = J4′′′, 5′′′ = 9.2 Hz, H- 4′′), 3.99−3.92 (3H, m, H-2′, H-
6′b, H-1b), 3.90−3.85 (1H, m, H-5′, H-3′′′), 3.83−3.73 (2H, m, H-3′, H-1a),
3.77−3.71 (6H, H-5′′′, H-6′′′a, H-6′′′b, H-3′′, H-6′′a, H-6′a), 3.65−3.62 (2H, m, H-
6′′b, H-4′′′), 3.57 −3.54 (2H, m, H-4′, H-5a), 3.49−3.40 (4H, m, H-2′′′, H-2′′, H-5′′,
H-5b), 3.19, 3.06 (6H, 2s, 2OMe), 1.25, 1.20 (6H, 2s, 2Me). 13C NMR (CDCl3) δ
138.7− 126.5 (m, Ar), 121.9, 119.7 (CF3), 102.5 (C-1′), 102.1 (C-1′′), 100.1, 99.8
(2 MeOCMe), 96.8 (C-1′′′), 84.8 (C-5′′′, C-2′′), 82.7 (C-2, C-3), 82.0 (C-3′′′), 79.2
(C-2′′′), 77.7 (C-4′′′), 75.5, 75.0, 74.7(3CH2Ph), 74.5 (C-5′′), 74.4 (C-2′), 73.9,
73.7, 73.5, 73.4, 73.3 (5CH2Ph), 72.6 (C-4′′), 72.5, 72.2 (CH2Ph), 72.0 (C-3′),
71.3 (CH2Ph), 71.0 (C-3′′), 70.0 (C-5′), 68.8 (C-6′′), 68.2 (C-5, C-4′ ), 66.5 (C-4,
C-6′′′), 48.6 (OMe), 48.2 (C-1), 48.1 (OMe), 46.6 (C-6′), 17.7, 17.3 (2Me). HRMS
Calcd for C106H118O20S (M+H): 1742.7932. Found: 1742.7882.
Data for the minor diastereomer (25b): [α] 23D = - 125.0 (c = 0.04, CH2Cl2).
1H NMR (CDCl3) δ 7.38−7.12 (55H, m, Ar), 5.68 (1H, d, J1′′′,2′′′ = 3.6 Hz, H-1′"),
4.97−4.88 (4H, m, H-1′′, 3CH2Ph), 4.83−4.78 (5H, m, 5CH2Ph), 4.66 (1H, d, J1′,2′

209
= 7.5 Hz, H-1′), 4.63−4.46 (12H, m, H-3, 11CH2Ph), 4.48−4.46 (2H, m, 2CH2Ph),
4.44 (1H, m, H-2), 4.37−4.33 (2H, m, H-4, CH2Ph), 4.13 (1H, t, J4′′,3′′ = J4′′,5 ′′ = 9.2
Hz, H-4′′), 4.08 (1H, dd, J6′′a, 5′′ = 4.1, J6′′a, 6′′b = 11.7 Hz, H-6′′a), 3.98 (1H, dd, J2′, 1′
= 9.7 Hz, J2′, 3′ = 2.2 Hz, H-2′), 3.95−3.89 (4H, m, H-3′′′, H-5′, H-6′′b, H-6′a), 3.84
(1H, t, J3′,4′ = J3′,2′ = 9.6 Hz, H-3′), 3.82−3.76 (4H, m, H-5′′′,H-6′′′a, H-3′′, H-6′b),
3.75−3.70 (2H, m, H-6′′′b, H-1a), 3.67 (1H, t, J3′′′,4′′′ = J5′′′,4′′′ = 9.5 Hz, H-4′′′),
3.63−3.59 (2H, m, H-1b, H-5a), 3.55 (1H, t, J4′,3′ = J5′,4′ = 9.7 Hz, H-4′),
3.52−3.45 (4H, H-2′′′, H-2′′, H-5′′, H-5b), 3.07, 2.97 (6H, 2s, 2OMe ), 1.19, 1.06
(6H, 2s, 2Me). 13C NMR (CDCl3) δ 138.7-126.5 (m, Ar), 121.9, 119.8 (CF3), 102.8
(C-1′), 102.1 (C-1′′), 100.0, 99.7 (2 MeOCMe), 96.8 (C-1′′′), 84.8 (C-5′′′), 83.2 (C-
2), 83.5 (C-3), 82.7 (C-2′′), 82.0 (C-3′′′), 79.2 (C-2′′′), 77.7 (C-4′′′), 75.5, 75.0
(CH2Ph), 74.7 (C-2′), 74.6 (C-5′′), 73.8 - 72.9 (9CH2Ph), 72.6 (C-4′′), 71.9 (C-3′),
71.0 (C-3′′), 69.1(C-5′), 68.8 (C-6′′′), 68.2 (C-5), 67.1 (C-4′), 65.0 (C-6′′), 63.3 (C-
4), 48.0, 48.1 (2OMe), 46.5 (C-1), 38.6 (C-6′), 17.7, 17.3 (2Me). HRMS Calcd. for
C106H118O20S (M+H): 1742.7932. Found: 1742.7882.
7.5.1.11 1,4-Dideoxy-1,4-[[2S, 3S, 4R, 5S]-2,4,5,6-tetrahydroxy-3-(4-O-α-D-Glucopyranosyl-D-glucosyl) hexyl]-(R/S)-epi-sulfoniumylidine]-D-arabinitol chloride 10
To a solution of sulfonium salt 24 (85 mg, 0.045 mmol, 5:1 mixture of
isomers) in 85% AcOH:H2O (40 mL) was added Pd(OH)2, 20 % weight on carbon
(200 mg) and the mixture was stirred under 100 Psi of H2 for 24h. The catalyst
was removed by filtration through a bed of Celite, and then washed with water.
The solvents were removed under reduced pressure and the residue was
dissolved in 80% TFA (5 mL) and stirred at room temperature for 2h. The

210
solvents were removed under reduced pressure; the residue was dissolved in
water (5 mL), and washed with CH2Cl2 (2×5 mL). The water layer was
evaporated to give the crude product. The residue was dissolved in H2O (10 mL),
Amberlyst A-26 resin (100 mg) was added, and the reaction mixture was stirred
at room temperature for 2 h. Filtration through cotton, followed by solvent
removal gave the crude hemiacetal. The crude product was dissolved in water (5
mL), and the solution was stirred at room temperature while NaBH4 (7 mg, 0.18
mmol) was added. Stirring was continued for another 3 h and the mixture was
acidified to pH < 4 by dropwise addition of 2M HCl. The mixture was evaporated
to dryness and the residue was co-evaporated with anhydrous MeOH (3 × 10
mL). The residue was purified by crystallization with EtOAc:MeOH:H2O (10:3:1)
followed by reverse phase chromatography (H2O) to give 10 (d.r, 5:1) as a
colorless solid (21 mg, 70%).
Data for the major diastereomer 10 follow. 1H NMR (D2O) δ 5.34 (1H, d,
J1′′′,2′′′ = 3.7 Hz, H-1′′′), 4.68 (1H, m, H-2), 4.61 (1H, d, J1′′,2′′ = 7.6 Hz, H-1′′), 4.36
(2H, m, H-3, H-2′), 4.08 (1H, dd, J4,5a = 5.0, J5a,5b = 12.2 Hz, H-5a), 4.02 (2H, m,
H-1a, H-3′′′), 3.92 (1H, m, H-3′), 3.89-3.57 (16H, m, H-4′′, H-5b, H-1′a, H-1′b,
H1b, H-6′′′a, H-4, H-6′′′b, H-6′′a, H-6′′b, H-4′, H-3′′, H-6′a, H-5′′, H-5′, H-6′b), 3.51
(2H, m, H-4′′′, H-2′′′), 3.33 (2H, m, H-5′′′, H-2′′). 13C NMR (D2O) δ 102.9 (H-1′′),
99.5 (C-1′′′), 81.5 (C-3′), 77.3 (C-3), 76.9 (H-2), 66.2 (C-5′′), 76.0 (C-3′′), 74.4 (C-
4′′′), 73.1 (C-2′′), 72.7 (C-5′), 72.6 (C-4′), 71.9 (C-4), 71.5 (H-2′′′), 69.8 (H-3′′′),
69.3 (H-5′′′), 69.2 (H-2′′), 67.9 (C-2′), 62.3 (C-6′), 60.4 (C-6′′′), 60.3 (H-6′′), 59.2

211
(C-5), 49.2 (C-1), 48.2 (C-1′). HRMS Calcd for C23H43O18S (M+ ): 639.2165.
Found: 639.2152.
7.5.1.12 1,4-Dideoxy-1,4-[[2S, 3S, 4R, 5S]-2,3,4,6-tetrahydroxy-5-(4-O-α-D-Glucopyranosyl-D-glucosyl) hexyl]-(R/S)-epi-sulfoniumylidine]-D-arabinitol chloride 11
Compound was obtained as white solid (18 mg, 64 %, 2.8:1 mixture of
isomers) from mixture of 25a and 25b (79 mg, 0.042 mmol, 2.8:1 mixture of
isomers) using the same procedure that was used to obtain 10.
Data for the major isomer (11) : 1H NMR (D2O) : δ 5.33 (1H, d, J1′′′, 2′′′ = 2.6
Hz, H-1′′′), 4.70 (1H, m, H-2), 4.57 (1H, d, J1′′,2′′ = 7.9 Hz, H-1′′), 4.39 (1H, brs, H-
3), 4.19 (1H, brs, H-2′), 4.08−4.04 (2H, m, H-4, H-6′a), 3.98−3.50 (20H, m, H-6′′b,
H-1a, H-1b, H-5a, H-5b, H-1′a, H-3′,H-4′, H-6′b, H-6′′′a, H-6′′′b, H-3′′, H-6′′a, H-1′b,
H-5′, H-3′′′, H-5′′′, H-4′′, H-2′′′, H-5′′), 3.36−3.26 (2H, m, H-4′′′, H-2′′). 13C NMR
(D2O) δ 103.0 (C-1′′), 99.5 (C-1′′′), 83.1 (C-5′), 77.0 (C-3, C-2), 76.5 (C-3′, C-4′′),
74.5 (C-5′′), 73.2 (C-2′′), 72.8 (C-5′′′), 72.6 (C-3′′′), 72.5 (C-3′′), 71.6 (C-2′′′), 70.0
(C-4), 69.3 (C-4′′′), 68.5 (C-4′), 67.5 (C-2′), 61.1 (C-5), 60.6 (C-6′′), 60.4 (C-6′′′),
59.2 (C-6′), 50.1 (C-1′), 48.2(C-1) HRMS Calcd. for C23H43O18S (M+): 639.2165.
Found: 639.2160.
7.6 Acknowledgments
We thank Buford L. Nichols, Roberto Quezada-Calvillo and Hassan Naim
for reagents for recombinant expression, and Lyann Sim for MGAM and SI
enzyme purification. K. J. was supported by a scholarship from the Canadian
Institutes of Health Research (CIHR) and the Canadian Digestive Health

212
Foundation. We also thank the CIHR (111237) and the Heart and Stroke
Foundation of Ontario (NA-6305) for financial support.
7.7 References
1. E. H. Van Beers, H. A. Buller, R. J. Grand, A. W. Einerhand J. Dekker,
Crit. Rev. Biochem. Mol. Bio. 1995, 30, 197-262.
2. G. Semenza, Annu. Rev. Cell Biol. 1986, 2, 255-313.
3. H. A. Ernst, L. L. Leggio, M. Willemoës, G. Leonard, P. Blum, S. Larsen, J.
Mol. Biol. 2006, 358(4), 1106-1124.
4. R. Quezada-Calvillo, L. Sim, Z. Ao, B. R. Hamaker, A. Quaroni, G. D.
Brayer, E. E. Sterchi, C. C. Robayo-Torres, D. R. Rose, B. L. Nichols, J.
Nutr. 2008, 138, 685-692.
5. B. L. Nichols, S. Avery, P. Sen, D. M. Swallow, D. Hahn, E. Sterchi, Proc.
Natl. Acad. Sci. U S A 2003, 100(3), 1432-1437.
6. H. Heymann, D. Breitmeier, S. Günther, Biol. Chem. Hoppe-Seyler 1995,
376, 249-253.
7. H. Heymann, S. Günther, Biol. Chem. Hoppe-Seyler 1994, 375, 451-456.
8. C. Robayo-Torres, R. Quezada-Calvillo, B. L. Nichols, Clin. Gastroenterol.
Hepatol. 2006, 4, 276-287.
9. K. Jones, L. Sim, S. Mohan, K. Jayakanthan, H. Liu, S. Avery, H. H.
Naim, R. Quezada-Calvillo, B. L. Nichols, B. M. Pinto, D. R. Rose, Bioorg.
Med. Chem. 2011, 19, 3929-3934.
10. Naumoff, D. G. Mol. Biol. (Mosk). 2007, 41, 1056-1068.

213
11. a) J. P. C. Chandrasena, The Chemistry and Pharmacology of Ceylon
and Indian Medicinal Plants; H&C press: Colombo, Sri Lanka, 1935; b) D.
M. A. Jayaweera, Medicinal Plants Used in Ceylon-Part 1;
NationalScience Council of Sri Lanka: Colombo, 1981; p 77; c) P. S.
Vaidyartanam, In Indian Medicinal Plants: a compendium of 500 species,
P. K. Warrier V. P. K. Nambiar and C. Ramankutty, Eds.; Orient Longman:
Madras, India, 1993, pp. 47-48.
12. a) B. D. Johnston, H. H. Jensen, B. M. Pinto, J. Org. Chem. 2006, 71,
1111-1118; b) M. Yoshikawa, F. M. Xu, S. Nakamura, T. Wang, H.
Matsuda, G. Tanabe, O. Muraoka, Heterocycles 2008, 75, 1397-1405.
13. a) M. Yoshikawa, T. Murakami, H. Shimada, H. Matsuda, J. Yamahara, G.
Tanabe and O. Muraoka, Tetrahedron Lett. 1997, 38, 8367–8370; b) H.
Yuasa, J. Takada, H. Hashimoto, Tetrahedron Lett. 2000, 41, 6615-6618;
c) A. Ghavami, B. D. Johnston, B. M. Pinto, J. Org. Chem. 2001, 66,
2312-2317.
14. a) H. Matsuda, Y. H. Li, T. Murakami, N. Matsumura, J. Yamahara, M.
Yoshikawa, Chem. & Pharm. Bull. 1998, 46, 1399-1403; b) K.
Jayakanthan, S. Mohan, B. M. Pinto, J. Am. Chem. Soc. 2009, 131, 5621-
5626; c) R. Eskandari, K. Jayakanthan, D. A. Kuntz, D. R. Rose, B. M.
Pinto, Bioorg. Med. Chem. 2010, 18, 2829-2835.
15. a) S. Ozaki, H. Oe, S. Kitamura, J. Nat. Prod. 2008, 71, 981-984; b) H. Oe,
S. Ozaki, Biosci. Biotechnol. Biochem. 2008, 72, 1962-1964; c) O.
Muraoka, W. Xie, G. Tanabe, M. F. A. Amer, T. Minematsu, M.
Yoshikawa, Tetrahedron Lett. 2008, 49, 7315–7317.
16. a) Y. Minami, C. Kurlyarna, K. Ikeda, A. Kato, K. Takebayashi, I. Adachi,
G. W. J. Fleet, A. Kettawan, T. Karnoto, N. Asano, Bioorg. & Med. Chem.
2008, 16, 2734-2740; b) G. Tanabe, W. Xie, A. Ogawa, C. Cao, T.
Minematsu, M. Yoshikawa, O. Muraoka, Bioorg. Med. Chem. Lett. 2009,
19, 2195-2198.

214
17. a) R. Eskandari, D. A. Kuntz, D. R. Rose, B. M. Pinto, Org. Lett. 2010, 12,
1632-1635; b) W. Xie, G. Tanabe, J. Akaki, T. Morikawa, K. Ninomiya, T.
Minematsu, M. Yoshikawa, X. Wu, O. Muraoka, Bioorg. Med. Chem. 2011, 19, 2015-2022.
18. E. J. Rossi, L. Sim, D. A. Kuntz, D. Hahn, B. D. Johnston, A. Ghavami, M.
G. Szczepina, N. S. Kumar, E. E. Sterchi, B. L. Nichols, B. M. Pinto, D. R.
Rose, FEBS J. 2006, 273, 2673-2683.
19. L. Sim, K. Jayakanthan, S. Mohan, R. Nasi, B. D. Johnston, B. M. Pinto,
and D. R. Rose, Biochemistry 2010, 49, 443–451.
20. S. Mohan, B. M. Pinto, Carbohydr. Res. 2007, 342, 1551-1580.
21. S. Mohan, B. M. Pinto, Collect. Czech. Chem. Commun. 2009, 74, 1117-
1136.
22. S. Mohan, B. M. Pinto, Nat. Prod. Rep. 2010, 27, 481-488.
23. J. D. Wardrop, S. L. Waidyarachchi, Nat. Prod. Rep. 2010, 27, 1431-1468.
24. C. Li, A. Begum, S. Numao, K. H. Park, S. G. Withers, G. D. Brayer,
Biochem. 2005, 44(9), 3347-3357.
25. L. Sim, R. Quezada-Calvillo, E. E. Sterchi, B. L. Nichols, D. R. Rose, J.
Mol. Biol. 2008, 375, 782-792.
26. Y. Su, J. Xie, Y. Wang, X. Hu, X. Lin, Eur. J. Med. Chem. 2010, 45, 2713-2718.
27. H. Liu, R. Nasi, K. Jayakanthan, L. Sim, H. Heipel, D. R. Rose, B. M.
Pinto, J. Org. Chem. 2007, 72, 6562-6572.
28. H. Satoh, Y. Yoshimura, S. Sakata, S. Miura, H. Machida, A. Matsuda,
Bioorg. Med. Chem. Lett. 1998, 8, 989-992.
29. R. Eskandari, K. Jones, D. R. Rose, B. M. Pinto Bioorg. Med. Chem. Lett.
2010, 20, 5686-5689.

215
30. L. Sim, C. Willemsma, S. Mohan, H. Y. Naim, B.M. Pinto, D. R. Rose, J.
Biol. Chem. 2010, 285, 17763-17770.
31. R. Nasi, L. Sim, D. R. Rose, B. M. Pinto, J. Org. Chem. 2007, 72, 180-186
32. R. Eskandari, K. Jones, D. R. Rose, B. M. Pinto, Chem. Commun. 2011,
47, 9134-9136.
33. R. Eskandari, K. Jones, D. R. Rose, B. M. Pinto, Bioorg. Med. Chem. Lett.
2011, 21, 6491-6494.

216
7.8 Supporting Information
Probing the intestinal α-glucosidase enzyme specificities of starch-
digesting maltase-glucoamylase and sucrase-isomaltase: Synthesis and
inhibitory properties of 3′- and 5′- maltose-extended de-O-sulfonated
ponkoranol
Razieh Eskandari, Kyra Jones, Kongara Ravinder Reddy, Kumarasamy
Jayakanthan, Marcia Chaudet, David R. Rose, B. Mario Pinto

217
ppm (t1)050100
ppm (t1)0.01.02.03.04.05.06.07.0
O
OBn
O
O OMe
OMe
HO
600 MHz, CDCl314
OTBDMS
O
OBn
O
O OMe
OMe
HO
600 MHz, CDCl314
OTBDMS

218
ppm (t1)
050100
OO
O
OMe
OMe
OBnOH
19
OTBDMS
100 MHz, CDCl3
ppm (t1)0.01.02.03.04.05.06.07.0
400 MHz, CDCl3
OOO
TBDMSOOMe
OMeOBn
OH
17
OO
O
OMe
OMe
OBnOH
19
OTBDMS
400 MHz, CDCl3
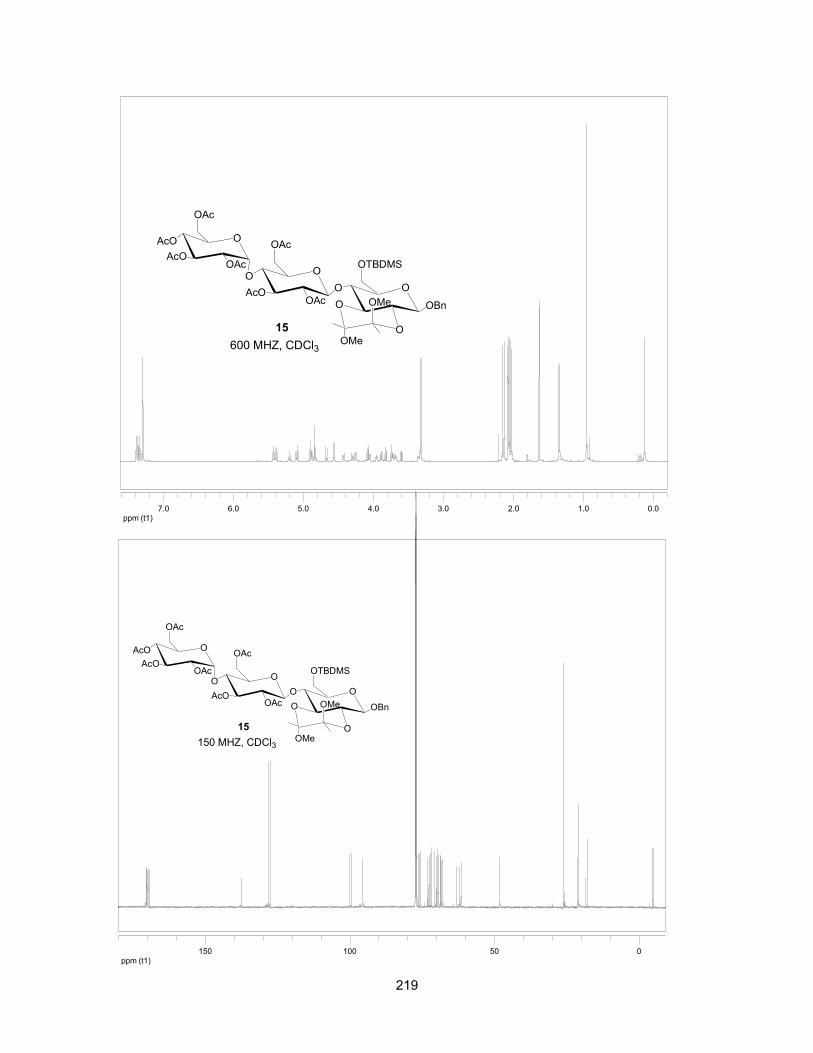
219
ppm (t1)0.01.02.03.04.05.06.07.0
O
OBn
O
O OMe
OMe
OO
OAcAcO
O
OAcO
AcOAcO OAc
OTBDMS
OAc
15600 MHZ, CDCl3
ppm (t1)050100150
O
OBn
O
O OMe
OMe
OO
OAcAcO
O
OAcO
AcOAcO OAc
OTBDMS
OAc
15150 MHZ, CDCl3

220
ppm (t1)0.01.02.03.04.05.06.07.0
ppm (t1)050100150
OO
O
MeO
OMe
OBn
OOAc
O
OAc
OOAc
O
OAcOAc
20
OTBDMS
AcO
AcO
400 MHz, CDCl3
OO
O
MeO
OMe
OBn
OOAc
O
OAc
OOAc
O
OAcOAc
20
OTBDMS
AcO
AcO
100 MHz, CDCl3

221
ppm (t1)0.01.02.03.04.05.06.07.0
O
OBn
O
O OMe
OMe
OO
OBnBnO
O
OBnO
BnOBnO OBn
OTBDMS
OBn
16600 MHZ, CDCl3
ppm (t1)050100
O
OBn
O
O OMe
OMe
OO
OBnBnO
O
OBnO
BnOBnO OBn
OTBDMS
OBn
16150 MHz, CDCl3

222
ppm (t1)0.01.02.03.04.05.06.07.0
OO
O
MeO
OMe
OBn
OOBn
O
OBn
OOBn
O
OBnOBn
OTBDMS
BnO
BnO
21400 MHz, CDCl3
ppm (t1)050100
OO
O
MeO
OMe
OBn
OOBn
O
OBn
OOBn
O
OBnOBn
OTBDMS
BnO
BnO
21100 MHz, CDCl3

223
ppm (t1)2.03.04.05.06.07.0
ppm (t1)050100
O
OBn
O
O OMe
OMe
OO
OBnBnO
O
OBnO
BnOBnO OBn
OTf
OBn
17150 MHz, CDCl3
O
OBn
O
O OMe
OMe
OO
OBnBnO
O
OBnO
BnOBnO OBn
OTf
OBn
17600 MHz, CDCl3
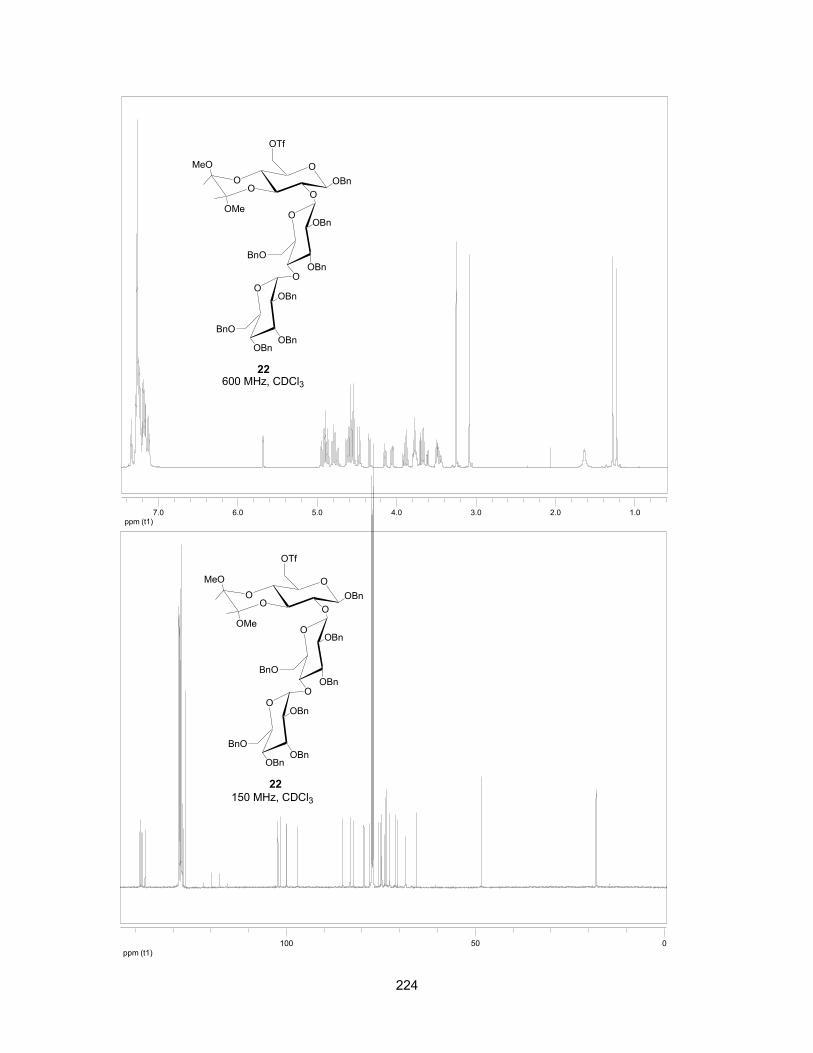
224
ppm (t1)1.02.03.04.05.06.07.0
OO
O
MeO
OMe
OBn
OOBn
O
OBn
OOBn
O
OBnOBn
OTf
BnO
BnO
22600 MHz, CDCl3
ppm (t1)050100
OO
O
MeO
OMe
OBn
OOBn
O
OBn
OOBn
O
OBnOBn
OTf
BnO
BnO
22150 MHz, CDCl3

225
ppm (t1)2.03.04.05.06.07.0
SBnO
BnO OBn
OTf
24 d.r, 5:1
OOBn
O
O OMe
OMe
OO
OBnBnO
O
OBnO
BnOBnO OBn
OBn
600 MHz, CDCl3
ppm (t1)50100
SBnO
BnO OBn
OTf
24 d.r, 5:1
OOBn
O
O OMe
OMe
OO
OBnBnO
O
OBnO
BnOBnO OBn
OBn
150 MHz, CDCl3

226
ppm (t2)3.503.603.703.803.904.00
3.50
4.00
ppm (t1
compound 24 NOESY mix t, 500 ms
ppm (t1)1.02.03.04.05.06.07.0
SBnO
BnO BnO
OTf
OO
O
MeO
OMe
OBn
OOBn
O
OBn
OOBn
O
OBnOBn
BnO
BnO
25a600 MHz, CDCl3
H-4 H-6’a

227
ppm (t1)50100
SBnO
BnO BnO
OTf
OO
O
MeO
OMe
OBn
OOBn
O
OBn
OOBn
O
OBnOBn
BnO
BnO
25a150 MHz, CDCl3
ppm (t1)1.02.03.04.05.06.07.0
SBnO
BnO BnO
OTf
OO
O
MeO
OMe
OBn
OOBn
O
OBn
OOBn
O
OBnOBn
BnO
BnO
25b600 MHz, CDCl3

228
ppm (t1)050100
SBnO
BnO BnO
OTf
OO
O
MeO
OMe
OBn
OOBn
O
OBn
OOBn
O
OBnOBn
BnO
BnO
25b150 MHz, CDCl3
ppm (t1)3.504.004.505.005.50
S
OHHO
OH
OH OH
HO
OH
O
10
O OH
HO
O OH
O
HOOH
HO
Cl
HO
600 MHz, D2O

229
ppm (t1)5060708090100
S
OHHO
OH
OH OH
HO
OH
O
10
O OH
HO
O OH
O
HOOH
HO
Cl
HO
150 MHz, D2O
ppm (t1)3.504.004.505.005.50
S
OHHO
OH O
OH OH
HO
OH
11
O OH
HO
O OH
O
HOOH
HO
HO
Cl
600 MHz, D2O

230
Representative Lineweaver-Burk plot of ntMGAM inhibited by 11 at concentrations of 0 nm, 30 nM, 125 nM, and 200 nM.
ppm (t1)405060708090100110
S
OHHO
OH O
OH OH
HO
OH
11
O OH
HO
O OH
O
HOOH
HO
HO
Cl
150 MHz, D2O

231
CHAPTER 8: CONCLUSIONS AND FUTURE WORK
8.1 Conclusions
The work described in this thesis focused on the design and syntheses of
α-glucosidase inhibitors. Since the isolation of kotalanol in late 1990s, the
absolute stereostructure of kotalanol 1 (Figure 8-1) had not been determined.
Our group successfully established the absolute stereostructure of kotalanol (1)
and also completed its total synthesis and that of its de-O-sulfonated derivative 2
(Figure 8-1).1 It was interesting to note that de-O-sulfonated kotalanol (2) showed
higher α-glucosidase inhibitory activities than the original sulfate (1). In Chapter 2
of this thesis, an alternative route for the synthesis of kotalanol (1) as well as the
synthesis of its 6'-epimer (3) were shown.2 As part of these extended studies, the
syntheses of de-O-sulfonated ponkoranol (4) and its 5'-epimer (5)3 (Figure 8-1),
and their selenium analogues,4 (6 and 7, Figure 8-1) were completed by the
thesis author as shown in Chapters 3 and 4. Biological evaluation of compounds
4-7 as glucosidase inhibitors against two recombinant intestinal enzymes
maltase-glucoamylase (MGAM) and sucrase-isomaltase (SI) were also described
(Chapter 4).4 The de-O-sulfonated ponkoranol (4) was also used as a scaffold for
designing additional inhibitors because extension of the polyhydroxylated side
chain beyond six carbon was shown not to be beneficial.
The crystal structures of ntMGAM in complex with kotalanol (1) and de-O-
sulfonated kotalanol (2),5 obtained by our collaborator Dr. David R. Rose, showed

232
that the 3'-O-sulfate anion in kotalanol was surrounded by hydrophobic residues
in the enzyme active site, and made no hydrogen bonding interactions with other
residues. Therefore, 3'-O-methylponkoranol (8) (Figure 8-1) was synthesized
(Chapter 5) to see whether the methyl group in this compound would make
hydrophobic interactions within the ntMGAM active site and lead to a tighter
inhibitor.6 In Chapter 6, the inhibition profile of 3'-O-methylponkoranol (8) for the
individual recombinant N- and C- terminal MGAM and SI were reported.7 The
results indicated that the interaction of the methyl group with hydrophobic
residues in the active site is not beneficial to inhibition of ntMGAM but resulted in
significant inhibition of ctSI.
Figure 8-1: Structures of compounds 1-8.
To better define the individual roles of the MGAM and SI domains in the
process of terminal starch digestion, we proposed further modification to the
S
OHHO
OSO3 OH
OH OH
HO
OHOH
1
S
OHHO
OH OH
OH OH
HO
OHOH
CH3OSO3
2
S
OHHO
OSO3 OH
OH OH
HO
OHOH
3
S
OHHO
OH OH
OH OH
HO
OH
X: S=4X: Se=6
ClS
OHHO
OH
OH OH
HO
OH
X: S=5X: Se=7
ClOH S
OHHO
OMe OH
OH OH
HO
OH
8
Cl

233
ponkoranol scaffold to toggle their activities on and off with domain specific
inhibitors.
From the structural and biological studies of ntMGAM and ctMGAM, it was
postulated that ctMGAM might have an extended binding site compared to
ntMGAM, which favors binding of longer inhibitors such as acarbose (9, Figure 8-
2).8 Based on this difference we synthesized extended structures of de-O-
sulfonated ponkoranol (4) by linking maltose units at the C3’ (10) or C5’ (11)
position of the hydroxylated side chain; these additional sugars were intended to
provide further contacts with the subsites within the active site. The inhibitory
activities of these compounds against individual subunits of MGAM and SI
indicated that C-3′-β-maltose-extended de-O-sulfonated ponkoranol 10 showed
some selectivity for inhibiting ntMGAM compared to ctMGAM, ntSI, and ctSI; C-
5′-β-maltose-extended de-O-sulfonated ponkoranol 11 showed the ability to
inhibit all the subunits very well especially, ntMGAM.9
Examination of the enzyme inhibitory activities showed for the first time
that de-O-sulfonated ponkoranol (4), its 5'-epimer (5), and their selenium
analogues (6, 7) could differentiate between ctMGAM spilceforms. We also
showed that substitution of the ring sulfur atom by selenium leads to 13-fold and
23-fold improvement in inhibition of ntSI; and 6-fold and 7-fold improvement in
inhibition of ctSI (4 and 5 vs. 6 and 7, respectively). Interestingly, 3'-O-
methylponkoranol (8) could not differentiate between ctMGAM spliceforms and
inhibited both spliceforms; however, it showed extraordinary selectivity for ctSI (7
± 2 nM). The inhibitory activities of C-3′-β-maltose-extended de-O-sulfonated

234
ponkoranol 10 and 5′-β-maltose-extended de-O-sulfonated ponkoranol 11
indicated that 10 showed higher potency against other subunits as compared
with ctMGAM, whereas 11 demonstrated the ability to inhibit all subunits,
especially ntMGAM. A significant finding was that 10 also could differentiate
between ctMGAM spliceforms and showed selectivity towards the different
enzyme activities. Based on a previous study acarbose (9) was shown to be a
very powerful inhibitor of ctMGAM over ntMGAM and SI subunits. Therefore, we
are now in a position to turn on and off the ctMGAM unit and study the effect of
other subunits in starch digestion.
Ultimately, small structural changes in a compound can result in
significant changes in inhibitory activity and selective inhibition of one enzyme
over the others, despite the overall similarity between the subunits in term of
amino acid sequence.

235
Figure 8-2: Structures of compounds 9-11.
8.2 Future work
It was observed from kinetic studies, that maltose (1,4-glycosidic linkage)
could be hydrolyzed by both ntMGAM and ntSI, whereas isomaltose (1,6-
glycosidic linkage) could only be hydrolyzed efficiently by ntSI.10 With respect to
MGAM and SI roles in terminal starch digestion, the different substrate
specificities of ntMGAM and ntSI seem consistent. The redundancy in α-1,4
OHN
HO
OOH
H3C
O
OOH
HO
O
HO OH
HO
HOHO
OH
HO
HO
OH9
S
OHHO
OH O
OH OH
HO
OH
S
OHHO
OH
OH OH
HO
OH
O
O OH
HO
O OH
O
HOOH
HO
O OH
HO
O OH
O
HOOH
HO
X
HO HO
X
10 11

236
activity seen in both enzymes (also in ctMGAM and ctSI) is beneficial due to the
higher distribution of α-1,4 linkages (95%) in starch molecules. In contrast, α-1,6
linkages make up a small fraction of the linkages (5%) in starch molecules.
Thus, only one subunit (ntSI) is likely necessary for processing these structures.
Close inspection of the active site complexes of kotalanol with ntMGAM and ntSI,
indicates that it might be possible to increase the inhibitory potency towards ntSI
through the addition of a side chain with an isomaltose extension, as shown in
compound 12 (Figure 8-3). Hence, we propose compound 12 as a potential
inhibitor of ntSI, with selectivity over ntMGAM. Selective inhibitors of ntSI could
then be used to further our understanding of the critical role of different subunits
of SI and MGAM in the starch digestion process and the structural basis of
substrate specificity in these intestinal α-glucosidases.
Figure 8-3: Proposed selective inhibitors of ntSI.
For the synthesis of target compounds 12, we propose a synthetic route
as shown in Scheme 8-1.
S
OHHO
OH
HO
12HO
O
HO OH
O
HOO
OH
O
HO
OMe
OHOH
OH
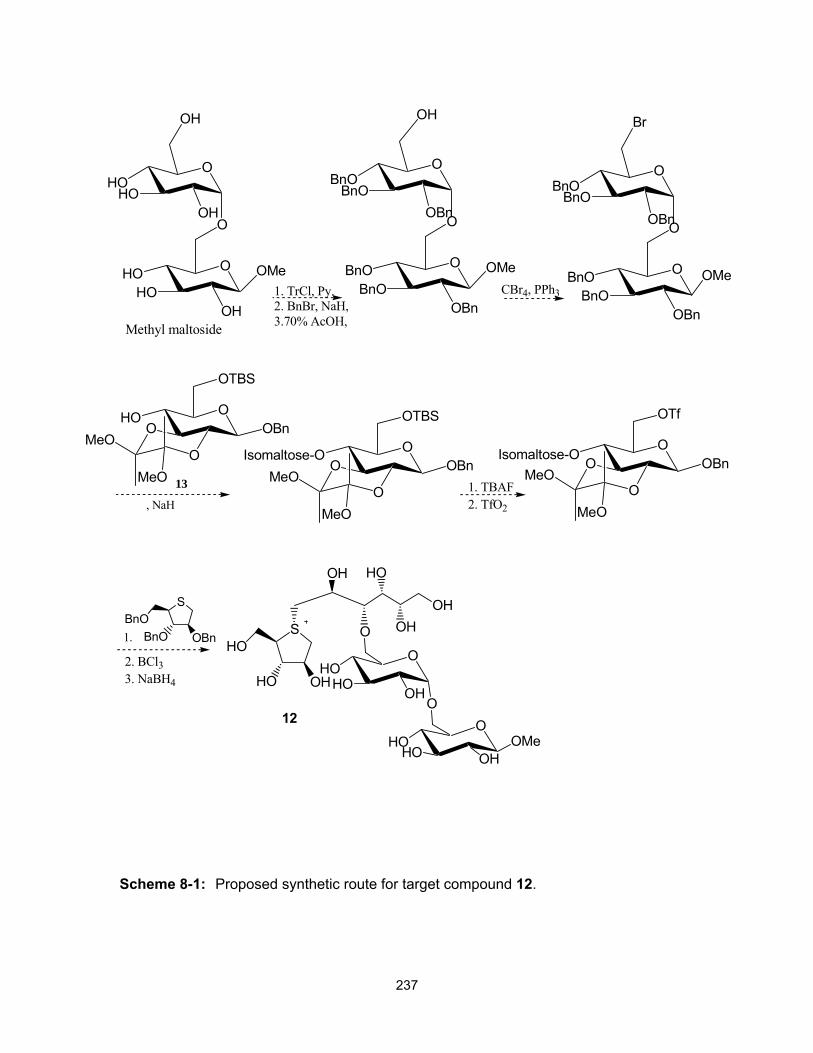
237
Scheme 8-1: Proposed synthetic route for target compound 12.
O
O
O
OH
HOHO
OH
HOHO
OH
OMe
O
O
O
OH
BnOBnO
OBn
BnOBnO
OBn
OMe1. TrCl, Py.2. BnBr, NaH,3.70% AcOH,
CBr4, PPh3
O
O
O
Br
BnOBnO
OBn
BnOBnO
OBn
OMe
OOBnO
HO
OTBS
OMeO
MeO
, NaH
OOBnO
Isomaltose-O
OTBS
OMeO
MeO
1. TBAF2. TfO2
OOBnOIsomaltose-O
OTf
OMeO
MeO
S
BnO OBnBnO
2. BCl33. NaBH4
1.
Methyl maltoside
13
S
OHHO
OH
HO
12HO
O
HO OH
O
HOO
OH
O
HO
OMe
OHOH
HO

238
For the synthesis of the target compound 12, we propose a synthetic route
that takes advantage of the same key intermediate 139 from the synthesis of our
3'-O-maltose-extended de-O-sulfonated ponkoranol (see Scheme 8-1).
Since α-amylase starch-digesting enzymes in the small-intestine break
down starch to α-limit dextrins, SI and MGAM are exposed to α-limit dextrins as a
substrate rather than maltose. The true in vivo substrate composition of SI and
MGAM is likely to change throughout the entire length of the small intestine, as
starch is broken down gradually as it passes through. Therefore, an important
avenue to be explored is the role of our different inhibitors on MGAM and SI
enzyme activities using dextrin structures as substrates to determine how our
inhibitors inhibit real starch (α-limit dextrins) and whether they have preferences
for inhibition of one subunit over others. Finally, experiments in which the
different inhibitors are mixed with α-limit dextrins and the mixtures are used as
substrates for the different intestinal mucosal enzymes, ntMGAM, ctMGAM, ntSI,
and ctSI should be explored. The thesis that one should be able to control starch
digestion to affect slow release of glucose should be explored further.

239
8.3 References
1. Jayakanthan, K.; Mohan, S.; Pinto, B. M. J. Am. Chem. Soc. 2009, 131,
5621-5626.
2. Eskandari, R.; Jayakanthan, K.; Kuntz, D. A.; Rose, D. R.; Pinto B. M.
Bioorg. Med. Chem. 2010, 18, 2829-2835.
3. Eskandari, R.; Kuntz, D. A.; Rose, D. R.; Pinto, B. M. Org. Lett. 2010, 12,
1632-1635.
4. Eskandari, R.; Jones, K.; Rose, D. R.; Pinto, B. M. J. Chem. Soc., Chem.
Commun. 2011, 47, 9134-9136.
5. Sim, L.; Jayakanthan, K.; Mohan, S.; Nasi, R.; Johnston, B. D.; Pinto, B.
M.; Rose, D. R. Biochemistry 2009, 49, 443-451.
6. Eskandari, R.; Jones, K.; Rose, D. R.; Pinto B. M. Bioorg. Med. Chem.
Lett. 2010, 20, 5686-5689.
7. Eskandari, R.; Jones, K.; Rose, D. R.; Pinto, B. M. Bioorg. Med. Chem.
Lett. 2011, 21, 6491-6494.
8. Sim, L.; Quezada-Calvillo, R.; Sterchi, E. E.; Nichols, B. L.; Rose, D. R. J.
Mol. Biol. 2008, 375, 782-792.
9. Eskandari, R.; Jones, K.; Reddy, K. R.; Jayakanthan, K. Chaudet, M.;
Rose, D. R.; Pinto, B. M. Chem. Eur. J. 2011, 17, 14817-14825.
10. Sim, L.; Willemsma, C.; Mohan, S.; Naim, H. Y.; Pinto, B. M.; Rose, D. R.
J. Biol. Chem. 2010, 285, 17763-17770.