master aba di i e ii livello: appliedbehavioranalysis ... e... · epilessia ed autismo incluse le...
TRANSCRIPT

Consorzio Universitario Humanitas© - tutti i diritti riservati, è vietata la riproduzione e/o l’utilizzo non autorizzato
MASTER ABA di I e II livello: Applied Behavior Analysis
Epilessia ed autismo incluse le manifestazioni non epilettiche
Prof. Federico Vigevano
Dipartimento di Neuroscienze, Ospedale Pediatrico Bambino Gesù

613 children enrolled with newly diagnosed epilepsy and followed a median of 10.5 years:
Global cognitive function: Normal = 451 (73,6%)Borderline. = 31 ( 5,1%)Mild MR = 21 ( 3,4%)Severe MR = 45 (7,3%)Devastated = 29 (4,7%)NFC = 36 (5.9%)
Variable associated to subnormal global cognitive function:Age at onset < 5 yearsSymptomatic etiologyEpileptic encephalopathyRemission statusCurrent AED treatment

Intellectual deterioration in childhood epilepsy
Apparent
¡ delay in cognitive development
¡ performance against age-related norms appears to drop
Real
¡ due to concomitant degenerative neurological disease of which epilepsy is also a symptom
¡ consequence of one or more of a number of other factors
Brown S., Epilepsia 2006

AEDs reduce neuronal irritability but also reduce neuronal excitability and , so, may impair cognition

The risk of side effects increases with :- higher drug load- higher doses and serum levels- polytherapy- speed of uptitration

¡ “Condition in which the epileptiformabnormalities are believed to contribute to progressive disturbance in cerebral function.” (Engel, 2001)
Epileptic encephalopathy - Definitions
¡ “Evidence suggests or supports the notion that there is an epilepsy-dependentneurodevelopmental or neurodegenerative process involved in the evolution of the syndrome (as opposed to an underlying metabolic, degenerative, or encephalitic process),”
(Engel, 2006)
¡ Epileptic encephalopathy embodies the notion that the epileptic activity itself may contribute to severe cognitive and behavioral impairments above and beyond what might be expected from the underlying pathology alone (e.g., cortical malformation), and that these can worsen over time.
¡ …..and…. may potentially occur in association with any form of epilepsy. (Berg et al. 2010)
¡ .. the term epileptic encephalopathyrefers to conditions characterized by epilepsy associated with psychomotor impairment, the latterbeing potentially reversible once epileptic activity is controlled

Epileptic Encephalopathies
THERAPEUTIC GOALS
1- Seizures control
2- Decrease or resolution of EEG abnormalities
3- Developmental outcome
ETIOLOGIES - Brain malformations- Chromosomal or genetic
abnormalities - Neurocutaneous diseases- Hypoxic ischemic injuries- Postnatal causes (vascular
or infectious insults)
¡ “Condition in which the epileptiform abnormalitiesare believed to contribute to progressive disturbance in cerebral function.” (Engel, 2001)

¡ Etiologies are variable.
¡ Peculiar evolution of epilepsy towards a syndrome specific electro-clinical picture.
¡ Quantifiable cognitive and motor regression, characterized by an evident worsening of the neuropsychological profile when compared to pre-onset neurodevelopmental phenotype.
¡ Variable evolution, ranging from complete remission to very severe conditions, such as drug resistant epilepsy and severe mental retardation.
West syndromeLennox-Gastaut syndromeEpilepsy with continuous spike-waves during slow-wave sleep (CSWS)

Severe epileptic encephalopathy characterized by:
¡ epileptic spasms
¡ hypsarrhythmic EEG pattern
¡ developmental delay
West Syndrome

West Syndrome,Infantile Spasms
- Cryptogenic
- Malformation
- Genetic
- Hypoxic-ischemic
- Metabolic
- Seizure-free
-Drug-resistance
-Cognitive decline
Syndrome
Etiology Prognosis
Fusco L.

Before ACTH
After 4 weeks of treatment

¡ 1-10% of childhood epilepsies
¡ 7% of children with ID ( 55% LGS IQ <50)
¡ Prevalence: 4% of childhood epilepsies – incidence in new onset epilepsy : 0,6% ( Trevathan et al, 1997; Camfield et al, 1996)
¡ Onset: 2-8yrs ( most commonly 3-5yrs); very rare late onset ( Down’s syndrome)
¡ Persist through adolescence and on into adulthood
¡ Males have 5.3 relative risk vs female
¡ De Novo : 10% ( ? )
¡ Prior West Syndrome: 30 - 65%
¡ Preceding history other than West syndrome: 70 - 80%
Lennox-Gastaut Syndrome: Epidemiology and Etiology

¡ Cryptogenic 33%
¡ Symptomatic 66%:
¡ - Brain malformation ( LIS1, DCX,GPR56)
¡ - Infection
¡ - Tumor
¡ - TSC (TSC1, TSC2)
¡ - HHE
¡ - Gene mutations
Lennox-Gastaut Syndrome: Epidemiology and Etiology
( CHD2 – FOXG1 )

LGS Characteristic EEG:
American Epilepsy Society 2015
Slow Spike and Wave
Paroxysmal Fast Activity

Epilepsy with continuous spikes and waves during slow sleep
• Focal seizures, negative myoclonus and drop attacks
• ESES
• Cognitive decline, attention deficit, behavioralproblems

ML, 7 yrs onset, awake
Epilepsy
• Onset of rolandic seizures
• After 6 months new focal motor seizures without impairment of counsciousness
• Drop attacks and abcences
• Cognitive and attention deficits
• CLB and LEV ineffective
• Video-EEG: multiple myoclonicseizures, CSWS
ML, 8 yrs, Myoclonic Seizure
ML, 8 yrs, Sleep
M.L. , 9 yrs, F
Epilepsy
•ACTH (20 IU)
• disappearance of CSWS, recovery
ML, 9 yrs outcome
Rolandic Epilepsy

EEG Abnormalities
+Seizures
Cognitive deterioration
EPILEPTIC ENCEPAHOPATHY
Normal/abnormaldevelopment
Epilepsy
SuppressImprovement of
RESOLUTION OF
• Epileptic encephalopathy is a dynamic condition not depending from the etiology that may persist over time causing increasingly severe functional effects
• It may improve and remit, either spontaneously or with treatment which suppresses the proposed causative epileptic activity

Delta power Associatedpattern
. Although spikes caused a cortical activation pattern similar to that in focal epilepsies, slow wave activity produced a hypsarrhythmia-specific activation in cortex and subcortical structures such as brainstem, thalamus, and putamen.

EEG-fMRI reveals activation of brainstem and thalamusin patients with Lennox-Gastaut syndrome
*Michael Siniatchkin, *Diana Coropceanu, *Friederike Moeller, yRainer Boor,and *yUlrich Stephani
*Department of Neuropediatrics, Christian-Albrechts-University, Kiel, Germany; and yNorthern German Epilepsy Center,
Raisdorf, Germany
SUMMARY
Purpose: Even if etiologies of Lennox-Gastaut syndrome(LGS) are diverse, the multiple causes converge into afinal common pathway that results in this specific epilepsyphenotype. There is little knowledge, however, aboutneuronal networks that may be a part of this pathway.Methods: To investigate these networks, 11 children withLGS and 9 control children with multifocal epileptic activ-ity were investigated using simultaneous recordings ofEEG and functional MRI (EEG-fMRI) in a 3 Tesla scanner.Key Findings: Individual and group analyses revealed sig-nificant activation of brainstem and thalamus (especially
centromedian and anterior thalamus) associated withepileptiform discharges in patients with LGS. None of thepatients with multifocal epileptic activity presented withthe same hemodynamic activation pattern.Significance: Because brainstem activation has been asso-ciated with infantile spasms, which often evolve into LGS,and thalamus activation has been observed in patientswith primary (idiopathic generalized syndromes) andsecondary (focal epilepsies) bilateral synchrony, thedescribed network in LGS may represent the commonpathogenetic pathway of these different conditions.KEY WORDS: Lennox-Gastaut syndrome, EEG-fMRI,Brainstem, Thalamus, Children.
Lennox-Gastaut syndrome (LGS) is an epileptic encepha-lopathy characterized by different types of seizures (tonic,tonic–clonic, and atonic seizures as well as atypicalabsences), typical electroencephalographic changes (slowspike-wave complexes ranging from 1–2.5/s, runs of rapidspikes and polyspikes), and accompanying mental retarda-tion (Arzimanoglou et al., 2009). LGS is a disabling disor-der often associated with pharmacoresistance and poorprognosis (Beaumanoir & Blume, 2005). In analogy toinfantile spasms (West syndrome), which frequently evolveinto LGS, it may be caused by different structural, func-tional, and genetic abnormalities. Although a common neu-ronal network responsible for clinical manifestations andelectroencephalographic features of LGS has been sug-gested, it has not yet been described (Blume, 2001).
Simultaneous recording of electroencephalography(EEG) and blood oxygenation level dependent (BOLD)functional magnetic resonance imaging (EEG-fMRI) is anew noninvasive technique that allows the evaluation of
hemodynamic changes in the brain associated with interictalepileptiform discharges (IEDs) (Gotman et al., 2006; Laufs& Duncan, 2007). This technique can characterize the com-plex neuronal network underlying a particular type of IED.Specific networks have been demonstrated for primary andsecondary generalized IEDs, hypsarrhythmia, and malfor-mations of cortical development (MCDs) (Aghakhani et al.,2004; Hamandi et al., 2006; Jacobs et al., 2008a; Kobayashiet al., 2006b; Siniatchkin et al., 2007a). In this study, weperformed EEG-fMRI recordings in children with LGS andcompared them to studies of patients with focal epilepsywho presented with multifocal epileptic activity in order tocharacterize the specificity of the neuronal network associ-ated with LGS.
MethodsSubjects
Between 2005 and 2009, 11 children with LGS (group 1,mean age 6.45 € 2.42 years) and 9 children with focal epi-lepsy (group 2, mean age 10.88 € 3.55 years) wererecruited from the Department of Neuropediatrics, Kiel,Germany. The inclusion criteria consisted of (1) multifocaland diffuse epileptic activity; (2) for group 1: diagnosis ofpharmacoresistent LGS based on clinical features of epi-lepsy, and presence of mental retardation (all patients with
Accepted November 24, 2010; Early View publication January 28,2011.
Address correspondence to Michael Siniatchkin, Department of Neuro-pediatrics, Pediatric Hospital, Christian-Albrechts-University, Schwanenweg20, D-24105 Kiel, Germany. E-mail: [email protected]
Wiley Periodicals, Inc.ª 2011 International League Against Epilepsy
Epilepsia, 52(4):766–774, 2011doi: 10.1111/j.1528-1167.2010.02948.x
FULL-LENGTH ORIGINAL RESEARCH
766
stronger and more significant if a common regressor for alltypes of IEDs was applied. It seems likely that all IEDs,independent of IED localization, were associated with acommon neuronal network. The same is true for positiveBOLD signal changes in the thalamus. Activation in thisstructure was found only in association with polyspikes inthree patients. However, this activation became stronger ifall IEDs were included in the analysis. For all IEDs, activa-tion in the thalamus was observed in eight children withLGS (73%). Another consistent finding in patients withLGS was the detection of positive BOLD signal changes inthe cerebellum. Again, significant activation in the cerebel-lum was associated with polyspikes in three patients; withpolyspikes and right- or left-sided IEDs in three children;with right- or left-sided IEDs in only two children; and withall IEDs together in eight children (73%). In addition,patients with LGS presented with significant, bilateral acti-vation in various cortical regions. There was no consistencyin these findings, however, and may be part of an individualnetwork associated with different types of IED that weremerged in the analysis. The most significant z-values for allrelevant structures involved in the activated network areshown in Table S1. Note that activation of brainstem, thala-mus and, cerebellum was equally significant in both symp-tomatic and cryptogenic cases of patients with LGS.
All but one patient with multifocal epileptic activityand partial epilepsy (group 2) showed significant, individ-ual patterns of positive BOLD signal changes (p < 0.05,FWE corrected, z-values ranged from 5.5 to 17.5). Mostof these changes were localized in different corticalregions and, in one patient, in the thalamus. Except forone patient (Patient 19), who showed significant activa-tion in the thalamus and cerebellum only after summationof all IEDs, no summation effect was seen in all otherpatients. In these patients, analysis of all IEDs togetheryielded activations in cortical areas that resembled theactivation pattern of the most frequent IEDs. In contrastto children with LGS, there were no consistent, positiveBOLD signal changes in subcortical structures in childrenwith multifocal partial epilepsy.
In both groups, negative BOLD responses were less sig-nificant (z-values ranged from )5.1 to )7.9) and did notshow any consistency in their localization.
Random effect group analysisIn patients with LGS, group analyses that were performed
for right- and left-sided IEDs, as well as for polyspikes sepa-rately, did not reveal significant results on the second level.Only the group analysis, which was performed for all IEDsmerged in one regressor, demonstrated significant, positive
Figure 1.EEG pattern and fMRI results of all IEDs together in a boy with LGS (Patient 1).Epilepsia ILAE
769
EEG-fMRI in Lennox-Gastaut Syndrome
Epilepsia, 52(4):766–774, 2011doi: 10.1111/j.1528-1167.2010.02948.x
Because LGS as a clinical entity can be associated withdifferent etiologies, one may suggest that multiple causescan activate a syndrome-specific neuronal network. Brain-stem nuclei and reticular formation have been discussed assignificant parts of this network (Blume, 2001; Hayashi,2001). It seems likely that the brainstem plays a substantialrole in mechanisms of tonic axial seizures, an importantfeature of the LGS. The following evidences support thishypothesis: Tonic-automatic seizures have been observedin a hydranencephalic patient (Velasco et al., 1997); tonicseizures fail to respond to callosotomy (Rougier et al.,1997); and tonic seizures are less responsive to resection ofapparently epileptogenic cortical areas (Bladin, 1985). Ithas been hypothesized that clinical seizures might resultfrom intermittent interference of descending brainstempathways controlling spinal reflex activity and tonic neuro-
nal bursting, whereas activity in the ascending pathwaysfrom the same brainstem areas that project broadly to thecerebral cortex might produce the characteristic EEGfeatures in LGS (Beaumanoir & Blume, 2005). Specificsignificance of the brainstem in LGS may be supported bypositron emission tomography (PET) and EEG-fMRI stud-ies performed in patients with West syndrome. West syn-drome is another epileptic encephalopathy presenting withhypsarrhythmia, infantile spasms, and developmentalabnormalities (Hrachovy & Frost, 2003). In a large propor-tion of patients, West syndrome evolves into LGS (as itwas the case in five patients in our study). In patients withcortical lesions/abnormalities such as lissencephaly andhypoxic ischemic encephalopathy with West syndrome andLGS, Hayashi (2001) found specific morphologic changesin the brainstem, whereas abnormalities in the limbic sys-tem in these patients were heterogeneous and seemed tohave no interrelationship with the described syndromes.Irrespective of the etiology, autopsy studies in patients withWest syndrome and LGS demonstrated similar distur-bances in specific brainstem nuclei containing monoaminesand neuropeptides (Hayashi, 2001). Taking into accountresults of the present study as well as PET and EEG-fMRIstudies performed in children with West syndrome, whichhave revealed areas of hypermetabolim and significantlyincreased activation in brainstem (Chugani et al., 1992;Siniatchkin et al., 2007a), we argue that brainstem activa-tion (especially in the region of the reticular formation) is a
Table 3. Results of the random effect group analysisfor LGS patients. Coordinates and t-values for
positive BOLD signal changes are shown (p < 0.001,noncorrected)
t-value x, y, z
Thalamus centromedian part (left) 12.3 )10, )28, 3Thalamus centromedian part (right) 9.97 9, )27, 3Anterior thalamus (left) 9.32 )9, )10, 3Anterior thalamus (right) 10.11 7, )9, 3Brainstem 8.25 )1, )16, )29
Figure 2.Results of the group analysis in patients with LGS (p < 0.001, noncorrected).Epilepsia ILAE
771
EEG-fMRI in Lennox-Gastaut Syndrome
Epilepsia, 52(4):766–774, 2011doi: 10.1111/j.1528-1167.2010.02948.x
Even if etiologies of Lennox-Gastaut syndrome (LGS) are diverse, the multiple causes converge into a final common pathway that results in this specific epilepsy phenotype.
Significant activation of brainstem and thalamus (especially centromedian and anterior thalamus) associated with epileptiform discharges in patients with LGS.

BRAINA JOURNAL OF NEUROLOGY
Neuronal networks in children with continuousspikes and waves during slow sleepMichael Siniatchkin,1 Kristina Groening,1 Jan Moehring,1 Friederike Moeller,1 Rainer Boor,2
Verena Brodbeck,3 Christoph M. Michel,3 Roman Rodionov,4 Louis Lemieux4 and Ulrich Stephani1
1 Neuropaediatric Department, Christian-Albrechts-University, Kiel, Germany2 Northern German Epilepsy Centre, Raisdorf, Germany3 Functional Brain Mapping Laboratory, University of Geneva, Switzerland4 Department of Clinical and Experimental Epilepsy, UCL Institute of Neurology, Queen Square, London, UK
Correspondence to: Michael Siniatchkin,Neuropaediatric Department,Paediatric Hospital,Christian-Albrechts-University,Schwanenweg 20,D-24105 Kiel,GermanyE-mail: [email protected]
Epileptic encephalopathy with continuous spikes and waves during slow sleep is an age-related disorder characterized by the
presence of interictal epileptiform discharges during at least 485% of sleep and cognitive deficits associated with this elec-
troencephalography pattern. The pathophysiological mechanisms of continuous spikes and waves during slow sleep and neuro-
psychological deficits associated with this condition are still poorly understood. Here, we investigated the haemodynamic
changes associated with epileptic activity using simultaneous acquisitions of electroencephalography and functional magnetic
resonance imaging in 12 children with symptomatic and cryptogenic continuous spikes and waves during slow sleep. We
compared the results of magnetic resonance to electric source analysis carried out using a distributed linear inverse solution
at two time points of the averaged epileptic spike. All patients demonstrated highly significant spike-related positive (activa-
tions) and negative (deactivations) blood oxygenation-level-dependent changes (P50.05, family-wise error corrected). The
activations involved bilateral perisylvian region and cingulate gyrus in all cases, bilateral frontal cortex in five, bilateral parietal
cortex in one and thalamus in five cases. Electrical source analysis demonstrated a similar involvement of the perisylvian brain
regions in all patients, independent of the area of spike generation. The spike-related deactivations were found in structures of
the default mode network (precuneus, parietal cortex and medial frontal cortex) in all patients and in caudate nucleus in four.
Group analyses emphasized the described individual differences. Despite aetiological heterogeneity, patients with continuous
spikes and waves during slow sleep were characterized by activation of the similar neuronal network: perisylvian region, insula
and cingulate gyrus. Comparison with the electrical source analysis results suggests that the activations correspond to both
initiation and propagation pathways. The deactivations in structures of the default mode network are consistent with the
concept of epileptiform activity impacting on normal brain function by inducing repetitive interruptions of neurophysiological
function.
Keywords: continuous spikes and waves during slow sleep (CSWS); EEG-fMRI; EEG source analysis; perisylvian region;remote inhibition; children
doi:10.1093/brain/awq183 Brain 2010: 133; 2798–2813 | 2798
Received December 8, 2009. Revised January 1, 2010. Accepted June 2, 2010. Advance Access publication August 5, 2010! The Author (2010). Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved.For Permissions, please email: [email protected]
by guest on Novem
ber 14, 2013http://brain.oxfordjournals.org/
Dow
nloaded from • The spike-related deactivations were found in structures of the default mode network (precuneus, parietal cortex and medial frontal cortex)in all patients and in caudate nucleus in four.
propagated epileptic activity (peak) was able to describe electrical
sources in the left and right perisylvian region, left and right tem-
poral cortices (mostly in the superior temporal gyrus), temporo-
parietal junction and the left prefrontal and parietal cortices.
The results of the source analysis on the group level show good
correspondence with results of the random effect group analysis of
fMRI data, especially with activation in the perisylvian region and
deactivation in the parietal cortex and temporoparietal junction.
This positive correspondence between fMRI results and EEG pat-
tern supports the contention that fMRI is able to reveal a physio-
logically meaningful pathological network.
DiscussionThis study revealed the following main findings: (i) independent of
aetiology, a common pattern of CSWS-related BOLD increases
bilaterally in the perisylvian region; (ii) this pattern was confirmed
using EEG source reconstruction showing a common bilateral pat-
tern of propagation in the perisylvian region associated with dif-
ferent individual localization of the initial epileptic activity; (iii)
there is frequent involvement of the prefrontal cortex and cingu-
late gyrus in both EEG-fMRI and source analysis; (iv) common
BOLD increases in the thalamus and BOLD decreases in the caud-
ate nuclei are associated with CSWS, especially revealed by the
group analysis; and (v) there is a common pattern of BOLD de-
creases in the default mode network.
Figure 6 Results of the random effect group analysis for both positive and negative BOLD signal changes (false discovery rate corrected,P50.05).
Table 3 Results of the group analysis (maximum ofactivation in the cluster)
Structures t-value Voxels MNIcoordinates
BOLD increases
Right perisylvian region and insula 7.27 293 36/0/!9
Left perisylvian region and insula 6.69 143 !51/!6/15
Right prefrontal cortex andcingulate gyrus
7.80 252 10/!9/51
Left prefrontal cortex andcingulate gyrus
6.19 154 !39/!15/54
Thalamus 4.11 54 12/!12/6
BOLD decreases
Precuneus 4.40 271 5/!56/30
Right parietal cortex 4.40 364 51/!60/27
Left parietal cortex 4.54 117 !51/!69/30
Caudate nucleus 4.22 45 !8/15/5
2806 | Brain 2010: 133; 2798–2813 M. Siniatchkin et al.
by guest on Novem
ber 14, 2013http://brain.oxfordjournals.org/
Dow
nloaded from
• Despite aetiological heterogeneity, patients with CSWS were characterized by activation of the similar neuronal network: perisylvian region, insula and cingulate gyrus.
• The deactivations in structures of the default mode network are consistent with the concept of epileptiform activity impacting on normal brain function by inducing repetitive interruptions of neurophysiological function.

CSWS
West syndrome
Lennox-Gastaut
Absence seizures
Deactivation in theDefault mode network
Multiple causes may activate a syndrome specific neuronal networkCourtesy of Siniatchkin
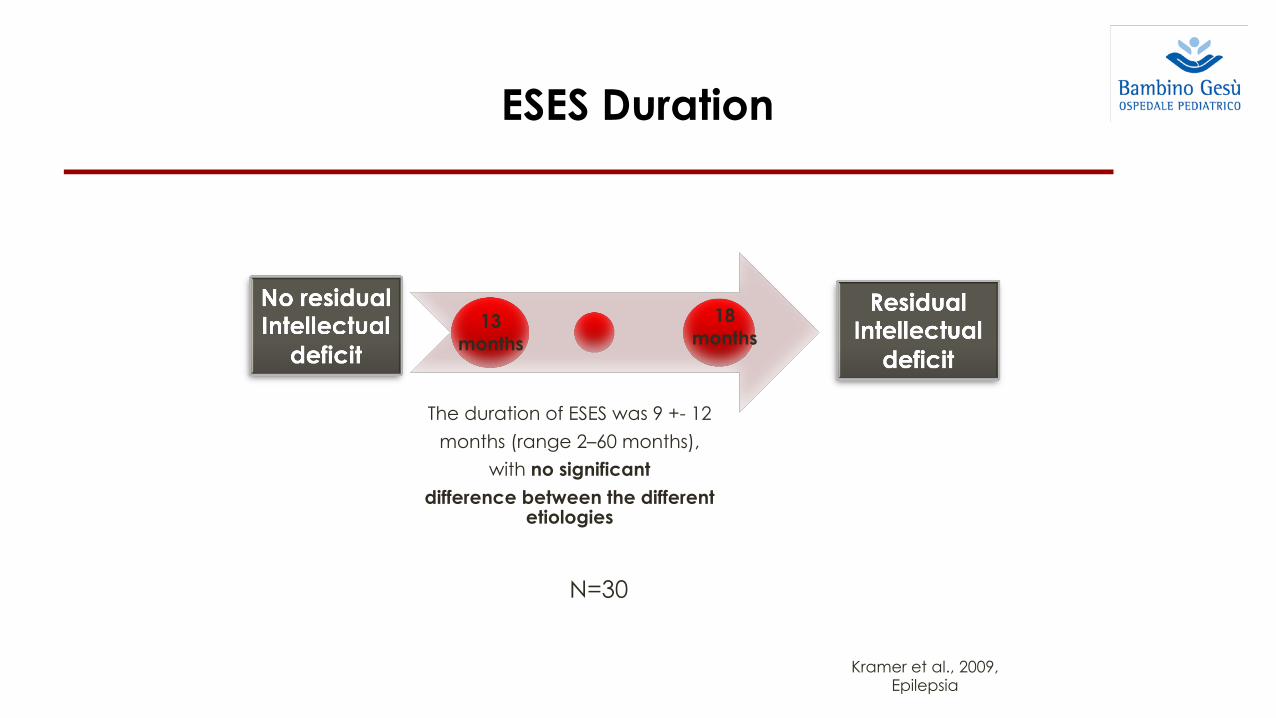
Kramer et al., 2009, Epilepsia
13 months
18months
ESES Duration
The duration of ESES was 9 +- 12 months (range 2–60 months),
with no significantdifference between the different
etiologies
N=30


Dravet syndrome
v Epileptic encephalopathy
v Onset: 1st year of lifeFebrile and afebrile seizures Febrile and afebrile status epilepticusMyoclonic seizures Absences Focal seizures Early photosensitivityCognitive Delay

25
Cognitive and adaptive evaluation of 21 consecutive patients withDravet syndrome
Nathalie Villeneuve a,b,c, Virginie Laguitton a, Marine Viellard b, Anne Lépine a,c, Brigitte Chabrol c,Charlotte Dravet d, Mathieu Milh c,e,⁎a CINAPSE, Hôpital Henri Gastaut Centre Saint Paul, 13009 Marseille, Franceb Centre Ressource Autisme, Hopital Sainte Marguerite, 13009 Marseille, Francec APHM, Service de neurologie pédiatrique, Hôpital de la Timone, 13005 Marseille, Franced Child Neurology and Psychiatry, Catholic University, Rome, Italye INSERM, UMR 910, Aix-Marseille Université, 13005 Marseille, France
a b s t r a c ta r t i c l e i n f o
Article history:Received 13 June 2013Revised 15 November 2013Accepted 21 November 2013Available online 8 January 2014
Keywords:Dravet syndromeCognitive evaluationEpilepsySCN1A
In order to assess the cognitive and adaptive profiles of school-aged patientswith Dravet syndrome (DS),we pro-posed to evaluate the intelligence and adaptive scores in twenty-one 6- to 10-year-old patients with DS followedin our institution between 1997 and 2013. Fourteen patients were tested using the Wechsler Intelligence Scalefor Children (WISC) and the Vineland Adaptive Behavioral Scales (VABS); 6 patients could not be tested withthe WISC and were tested with the VABS only, and one was tested with the WISC only. Data regarding theepilepsy were retrospectively collected. Statistical analysis (Spearman rank order and Pearson correlationcoefficient) was used to correlate early epilepsy characteristics with the cognitive and adaptive scores.Sodium channel, neuronal alpha-subunit type 1 (SCN1A) was mutated in 19 out of 21 patients. After the age of6 years, none of the DS patients had a normal intelligence quotient (IQ) using WISC (age at the testing period:mean = 100 ± 5; median = 105 months; mean total IQ = 47 ± 3; n = 15). Only five patients had a verbaland/or a non verbal IQ of more than 60 (points). Their cognitive profile was characterized by an attention deficit,an inability to inhibit impulsive responses, perseverative responses and deficit in planning function. Administer-ing the Vineland Adaptive Behavioral Scales in the same period, we showed that socialization skills were signif-icantly higher than communication and autonomy skills (age at the testing period: mean = 100 ± 4;median = 100 months; n = 20).We did not find any significant correlation between the IQ or developmental quotient assessed between 6 and10 years of age and the quantitative and qualitative parameters of epilepsy during the first two years of life inthis small group of patients.Despite an overall moderate cognitive deficit in this group of patients, the Vineland Adaptive Behavioral Scalesdescribed an adaptive/behavioral profile with low communication and autonomy capacities, whereas the social-ization skills were more preserved. This profile was different from the one usually found in young patients withautism and may require specific interventions.
© 2013 Elsevier Inc. All rights reserved.
1. Introduction
Dravet syndrome (DS), previously known as severe myoclonic epi-lepsy in infancy (SMEI), is a rare, chronic epileptic syndrome occurringwithin the first years of life in a previously normal infant. Patients en-dure recurrent and prolonged hemiclonic or generalized tonic–clonicseizures,mostly febrile ones, and usually develop other seizure types in-cluding atypical absence seizures, focal seizures, and myoclonic jerksthat usually emerge between 1 and 4 years of age [1]. SCN1A encoding
a voltage sensitive sodium channel has been found mutated or deletedin 70–80% of the patients with DS [2].
A singular time course of psychomotor development has been classi-cally described in DS [3]. Before the age of two years, childrenwho havebeen tested by the Brunet–Lezine scale [4] or the Griffiths scale [5]displayed a normal or subnormal developmental quotient (DQ). Inolder children, cognitive evaluation indicated a low DQ in most cases,characterized by poor visuomotor skills, heterogeneous language, andbehavioral disorders such as hyperactivity, poor relational capacitiesand gestural stereotypies [4,5]. Behavioral disturbance such as hyperac-tivity and autistic behavior was frequently observed, as well as speechdisturbance [4–6]. The cognitive outcome was usually poor, and sei-zures remained anti-epileptic drugs even in adulthood [7,8], although
Epilepsy & Behavior 31 (2014) 143–148
⁎ Corresponding author at: Service de neurologie pédiatrique, Hopital Timone-Enfants,264 Rue Saint Pierre, 13005 Marseille, France. Fax: +33 491 38 68 09.
1525-5050/$ – see front matter © 2013 Elsevier Inc. All rights reserved.http://dx.doi.org/10.1016/j.yebeh.2013.11.021
Contents lists available at ScienceDirect
Epilepsy & Behavior
j ourna l homepage: www.e lsev ie r .com/ locate /yebeh
uSCN1A was mutated in 19 out of 21 patients
uAfter the age of 6 years, none of the DS patients had a normal intelligence
quotient (IQ):mean total IQ = 47 ± 3
uThey did not find any significant correlation between the IQ or developmental
quotient assessed between 6 and 10 years of age and the quantitative and
qualitative parameters of epilepsy during the first two years of life in this small
group of patients.

Encephalopathy in children with Dravetsyndrome is not a pure consequence of epilepsy
Patients with severe delayand neurological featuresin the mutated group
- Lack of psychomotor deterioration- No correlation between the severity of the epilepsy and the severity of the cognitive delay- Main troubles in visual motor integration and visual perception- Additional direct role for SCN1A mutation ?
N Chemaly

u Epileptic encephalopathy due to a single etiological factor (SCN1A – interneuron abnormalities).
u It is characterized by a constant electro-clinical pattern, evolving only in terms of intensity and severity, without any possibility of complete resolution.
u It may be considered as a real disease and a specific nosographicentity.
Dravet syndrome

¡ Developmental encephalopathy where there is just developmental impair- mentwithout frequent epileptic activity associated with regression or further slowing of development;
¡ Epileptic encephalopathy where there is no preexisting developmental delay and the genetic mutation is not thought to cause slowing in its own right;
¡ Developmental and epileptic encephalopathy where both factors play a role
¡ Where a genetic mutation is identified, the well recognized developmental and epileptic encephalopathies can be called by their gene name together with the word “encephalopathy” : KCNK2 Encephalopathy.
“Developmental and epileptic encephalopathies”
Scheffer et al., 2017

¡ 30% of patients with Autism have epilepsy.
¡ 30% of patients with epilepsy have Autism.
¡ Highest risk for Autism is seen in those whose seizures that start in the 1st year of life.
¡ Risk for epilepsy in children with Autism is higher in those with greater intellectual disability, symptomatic vs. unknown cause, and history of regression
¡ 35-65% of patients with Autism have EEG abnormalities
¡ Epilepsy in autism confers increased mortality
How common is epilepsy in patients with autism?

¡Difficulty in distinguishing a behavioral episodes (staring, repetitive movement) from an epileptic seizure.
¡EEG may be abnormal in patients who do not have epileptic seizures.
Difficulty in Diagnosing Epilepsy in Children with Autism

¡ Atypical neuronal networks
- Phases of synaptogenesis in human brain
Do seizures cause autism?Does autism lead to seizures?
were found to alter the property of the NLGN to triggersynapse formation in cultured neuronal cells [17]. NLGNmutations probably concern a limited number of cases(<1% of the individuals), but following these initialresults, mutations in other synaptic proteins such asSHANK3, NRXN1, CNTNAP2, CNTN3/4, and PCDH9/10 were identified in patients with ASD [18–25]. Inter-estingly, NRXN1 codes for the presynaptic binding part-ner of NLGNs, CNTNAP2 (Caspr2) possess stronghomology to NRXN and SHANK3 is a scaffolding proteinof the postsynaptic density that binds to NLGN andregulates the size and shape of dendritic spines [26].
Only limited data are available for understanding the roleof these proteins in the human brain, but studies usingneuronal cell culture and animal models have providedcrucial information. Firstly, NLGNs and NRXNsenhance synapse formation in vitro [27!!], but are notrequired for the generation of synapses in vivo [28!!].Therefore, NLGNs may not establish synapses, but maycontribute to the activity-dependent formation of neuralcircuits [29!]. Secondly, NLGNs and NRXNs are emer-ging as central organizing molecules for excitatory gluta-matergic and inhibitory GABAergic synapses in themammalian brain [30,31]. The mutant mice carrying aR451C Nlgn3 mutation identified in two brothers withASD displays an increased number of GABAergicsynapses and inhibitory currents [32]. An imbalance ofinhibition and excitation was also observed in MeCP2
knockout [33] and in several mice proposed as model ofautism such as the Caps2 knockout [34] or mice subject toprenatal valproate treatment [35]. Interestingly, the linkbetween GABA function and spine pruning has beenidentified during a critical period of brain developmentwhen individual experience is essential for the normaldevelopment of the neuronal network [36]. Therefore,impaired inhibitory–excitatory balance can be manifest asa shifted critical period for brain development [37] or analteration of sensory processing, such as reduced gammaoscillations in FMRP knockout mice [38] as seen also inASD [39]. Taken together, these results strongly suggestthat synapse homeostasis and specificity play an import-ant role in the susceptibility to ASD.
Atypical neuronal networks in ASDIn the human cerebral cortex, the first synapses areevident at the 40th day after conception. Thereafter,the rate of synapse formation and pruning exhibit distinctphases, the most dramatic change takes place during theperinatal period (Figure 1). During the first three years oflife, synaptic contacts are formed, but only some will bestabilized. This selection process represents a key step inthe cognitive development of the child. The NLGN–NRXN–SHANK pathway is probably required duringthis stabilization phase of the synapse in response toneuronal activity. Strikingly, the role of the NLGN–NRXN–SHANK pathway in the development of socialinteraction seems to be conserved in other species.
232 Development
Figure 1
Schematic representation of the different phases of synaptogenesis in the human brain. During the first three years of life, an excess of cell/synapticgrowth rate and inhibitory currents could increase the risk of ASD. Mutations within the mTOR/PI3K pathway lead to an excess of synaptic/cell growth.Mutations within the NRXN–NLGN–SHANK pathway lead to abnormal synaptogenesis and excess of inhibitory currents. The arrows entering the redzone illustrate the excess of synaptic/cell growth and inhibitory currents during early brain development.
Current Opinion in Neurobiology 2009, 19:231–234 www.sciencedirect.com
ü During the first three years of life, an excess of cell/synaptic growth rate and inhibitory currents could increase the risk of ASD.
ü Mutations within the mTOR/PI3K pathway lead to an excess of synaptic/cell growth.
ü Mutations within the NRXN–NLGN–SHANK pathway lead to abnormal synaptogenesis and excess of inhibitory currents.
Bourgeon, 2009

Altered genes in ASD with epilepsy
Page 3 of 6
Licensee OAPL (UK) 2014. Creative Commons Attribution License (CC-BY)
FOR CITATION PURPOSES: Canitano R. Autism with epilepsy: A neurodevelopmental association. OA Autism 2014 Apr 08;2(1):7.
Review
Com
petin
g in
tere
sts:
Non
e de
clar
ed.
Conf
lict o
f int
eres
ts:
Non
e de
clar
ed.
All a
utho
rs c
ontr
ibut
ed to
conc
eptio
n an
d de
sign,
man
uscr
ipt p
repa
ratio
n, re
ad a
nd a
ppro
ved
the
final
man
uscr
ipt.
Al
l aut
hors
abi
de b
y th
e As
soci
atio
n fo
r Med
ical
Eth
ics (
AME)
eth
ical
rule
s of d
isclo
sure
.
linked neuroligin mutations have been associated with familial ASD22. All these studies highlighted abnormal synaptic GABAergic signaling, establishing the vanguard of current research on molecular abnormalities in ASD and epilepsy. Alterations of neocortical minicolumns (namely, morphological changes of GABA interneurons associated with reduced width of minicolumns) have been demonstrated in ASD. Minicolumns are anatomically characterized by vertical arrays of pyramidal neurons with dendrite and axon projections. Pyramidal cell arrays are accompanied by their GABAergic interneurons that establish synapses with pyramidal cell bodies, axons and dendrites. A narrowing of these cortical minicolumns has been demonstrated in ASD patients. It was hypothesized that this reduced intercolumnar distance was dependent on structural/anatomical defects in GABAergic interneurons surrounding principal pyramidal cortical neurons23,24. In addition, a significant decrease in GABAA receptor α4, α5, β1 and β3 subunits25 in ASD brains has been observed. GABAB receptors were also reduced in restricted regions of the cerebral cortex from ASD patients. However, current research in humans is still at the stage of preliminary investigations. Advanced neuroimaging techniques are developing this field. For example, 1H-magnetic resonance spectroscopy (1H-
MRS) is a non-invasive neuroimaging technique that allows for the estimation of specific in vivo neurochemical metabolites of GABA. In a recent study, creatine-normalized GABA+ ratios (GABA+/Cr) were measured in a group of 17 children with ASD, and in a control group of 17 typically developing children, for motor, auditory and visual regions of interest. In the ASD group, deficits in GABA+/Cr were demonstrated at approximately 11% in motor regions and at approximately 22% in auditory regions26. These findings support a model of regional brain differences in GABA+/Cr in ASD with an imbalance in the inhibitory component. Clinical characteristics of epilepsy in ASD On clinical grounds, the occurrence of seizures in children and adolescents with ASD is unpredictable, and seizures typically occur in otherwise healthy children/adolescents. The exception is the occurrence of early onset seizures in infants that have an early epileptic syndrome and who later develop an ASD. Seizures appear as an additional clinical burden to ASD in the first case and, as a rule, neither trigger factors nor concomitant relevant events are identified. Epilepsy is an additional problem in ASD that must be considered by clinicians and family members, since some interventions should be made readily available to the child1,27.
A set of assistance measures should be explained to the family, including emergency and prophylactic norms that are the same as those used in the general care of children with epilepsy. In the evolution of seizures, the causes and clinical course remain elusive and they are not helpful in the choice of treatment, nor in clinical consultation. Seizures are heterogeneous and they vary individually, as any kind of seizure may appear in young people with ASD with an unpredictable course. There is a wide range of possible evolutions, from rare to only one lifetime seizure, or to intractable epilepsy, as is commonly found in epilepsy without autism. Thus, caution is always warranted in management and outcome prediction in each individual case. Epilepsy is a further burden for ASD individuals with already varying degrees of adaptive difficulties, warranting a careful approach by clinicians. Epileptiform abnormalities without seizures are as frequent as 20-30% in individuals with ASD and epilepsy, however, their role in the development of the nuclear disturbances of autism is controversial7. Paroxysmal epileptiform abnormalities are thought to be involved in cognitive disturbances and in the social core difficulties of ASD, but the issue is still unclear. An initial overgrowth of white matter in the first 2 years of life is followed by arrested/abnormal growth of the dendritic tree.
Table 1: Altered genes in ASD with epilepsy. Modified by Amiet 2013 and Betancur 2011. Gene Locus Type of mutation Transmission Molecular abnormalities SCN1A 2q24 Point mutation De novo Na-channel
SCN2A 2q23–q24.3 Deletion De novo Na-channel KCNMA1 10q22 Point mutation Dominant inheritance K-channel NLGN4X Xp22.31 Point mutation Inherited Synapse formation NRXN1 2p16.3 Deletion
Point mutation Recessive inheritance De novo
Synapse formation
CNTNAP2 7q35 Deletion Point mutation
Recessive inheritance De novo
Synapse formation
SYNGAP1 6p21.3 Point mutation De Novo Synapse RasGAP ARX Xp22.13 Duplication Inherited
De novo Aristaless-related homeobox protein
Modified from Amiet 2013 and Betancur 2011

Disorders of synaptic plasticity
(TSC), neuroligin mutations and “interneuronopathies”resulting from anstaless-related homeobox, X-linked(ARX) and Neuropilin 2 (NRP2) gene mutations. Inaddition, the process of epilepsy development (i.e., epi-leptogenesis) and/or spontaneous seizures themselvesmay result in maladaptive synaptic plasticity producingimbalances of excitation and inhibition that contributeto learning and behavioral difficulties. Abnormalitiesin synaptic plasticity can arise from alterations in recep-tors, signaling molecules or neurotropins. Alterations inmultiple of these molecules are known to occur afterearly-life seizures and with genetic conditions knownto be associated with both ASD and epilepsy (Fig. 1).
Synaptic plasticity describes the process whereby syn-apses, the connections between 2 neurons, get strength-ened by experience or practice. When synapses areactivated, depolarization mediated by AMPA receptorsallows release of magnesium blockade and calcium entrythrough NMDA receptors. This triggers calcium depen-dent activation of kinases and other signaling pathwaysresulting in enhanced gene transcription and traffickingof receptors that result in faster and stronger synapticconnections. This is known as long-term potentiationand is thought be the cellular basis of learning. Synapticplasticity depends on a variety of proteins whose genesare disrupted in genetic conditions associated withautism and epilepsy. These include cyclin-dependent
kinase-like 5 (CDKL5) in West syndrome, MeCP2 inRett syndrome, FMRP in fragile X mental retardationsyndrome, mTOR in tuberous sclerosis, and reelin in lis-sencephaly. These are discussed in more detail below.
2. Fragile X syndrome
Fragile X syndrome (FXS) is the most frequent formof inherited mental retardation and often presents withautism spectrum disorder and epilepsy. The hallmarkof FXS pathology is the hyperabundance of dendriticspines with a long, thin, and otherwise immature mor-phology [2,3]. Fragile X results from an expanded tripletrepeat in the FMR1 gene. Fmr1 knock-out mice (an ani-mal model for FXS) exhibits a similar excess of long,thin spines [4] and display altered learning and behavior,greater susceptibility to seizures, and altered synapticplasticity [5]. The FMR1 gene is located at the cytoge-netic fragile X site on the X-chromosome [6]. ExpandedCGG repeats within the 50-untranslated region of FMR1result in FXS (normal length of !30 triplets, 55–200triplets in premutations (in FXS carriers), 200–800 trip-lets in affected FXS individuals). FMR1 gene codes forthe fragile X mental retardation protein (FMRP).FMRP is an mRNA-binding protein abundant in thebrain, that binds to and regulates 4% of brain mRNA,including many RNAs important for synaptic plasticity.FMRP regulates mRNA transport in dendrites and isassociated with polyribosomes in neuronal dendritesand spines where it regulates local protein synthesisimportant for spine development and synaptic plasticity.In the absence of FMRP, excess and dysregulatedmRNA translation leads to altered synaptic functionand loss of protein synthesis-dependent plasticity.FMRP is also involved in axonal development, synapseformation, and the development and wiring of neuronalcircuits [6–14].
FMRP regulates metabotropic glutamate receptor(mGluR)-induced long-term depression (LTD), and inthe absence of FMRP, there is excessive AMPA receptor(AMPAR) internalization and exaggerated LTD leadingto impaired synaptic excitatory function (“The mGluRtheory of fragile X”; [15]). Stimulation of group1mGluRs activates protein phosphatase 2-A (PP2A)and dephosphorylates FMRP, allowing rapid transla-tion of FMRP-associated mRNAs that lead to removalof AMPARs from the cell membrane (LTD). Soon after,the mammalian target of rapamycin (mTOR) is acti-vated and inhibits PP2A and leads to FMRP phosphor-ylation via S6 kinase. Inhibition of FMRPphosphorylation leads to excessive AMPA receptorinternalization, disrupted synaptic function and exag-gerated Ltd. This is known as the “mGluR theory offragile X”, and although it adds to our understandingof intellectual impairment in fragile X syndrome, it doesnot seem to readily explain the CNS hyperexcitability
Genetic conditions causing disrupted synaptic plasticity,
abnormal Inhibition/Excitation
ASD Early life seizures
Fig. 1. There is evidence that both epilepsy and ASD arise fromabnormal excitability and disrupted synaptic plasticity in the develop-ing brain. This abnormal plasticity can result from genetic conditions.In addition, epilepsy development (epileptogenesis) and/or seizuresduring early post-natal development may alter synaptic plasticity andcontribute to ASD. Abnormalities in synaptic plasticity can arise fromalterations in receptors, signaling molecules or neurotrophins. Multipleof these molecules are known to be altered by early-life seizures andgenetic conditions associated with both ASD and epilepsy.
732 A. Brooks-Kayal / Brain & Development 32 (2010) 731–738
Abnormal plasticityü genetic conditionsü seizures during early post-natal
development may alter synaptic plasticity and contribute to ASD
Abnormalities in synaptic plasticity ü alterations in receptorsü signaling moleculesü neurotrophins
Brooks-Kayal, 2010