mcb1_mutations_jan12(2)
DESCRIPTION
mutationsTRANSCRIPT

Mutation & DiseaseHow changes in DNA result in disease
Read & Donnai, Chapter 6
MCB @ AUC
Details on many of the disorders described can be found in the MCB handout and by searching at the following siteshttp://www.ncbi.nlm.nih.gov/sites/GeneTests/review?db=GeneTestshttp://www.ncbi.nlm.nih.gov/omim

Mutation
Heritable change in DNAi.e. not necessarily pathologic
Base substitution - replacement of (usually) single base
Deletion - one or more nucleotides removed
Insertion - one or more nucleotides introduced
Remember - mutations can be somatic or germ line
Think about whether a change in DNA sequence will affect the synthesis or function of protein – transcription, splicing, folding etc.

Polymorphisms, Variants & Mutations
Polymorphism - rare allele exists at a frequency of ≥ 0.01 in a human population
Variant - a change in DNA sequence that may or may not be polymorphic, and may or may not be pathogenic
Pathologic mutation - change in DNA sequence that has an effect on the encoded protein or on gene expression that results in disease

Mutation rates
Average human lifetime ~1017 cell divisionsi.e. 6 x 1026 insertions onto a growing DNA strand
Uncorrected replication errors ~ 10-9 - 10-11
Taking the average coding regions as 1.65kb, coding region mutations occur at a frequency of 1.65 x 10-6 - 1.65 x 10-8 per gene per cell division
~10-9 per haploid genome per generation5-6 new mutations per person

Mutagenesis
Spontaneous mutations - occur all the time- result in de novo cases of many disorders
The rate at which mutations occur is increased by mutagens and limited by DNA repair
Mutagens radiation - X-rays & -rays chemicals - mustard gas, formaldehyde, benzene
DNA repair (Dr. van Oost’s lectures)
reduces the mutation rate by ~100 fold

Null mutations - no protein product from mutant allele

11_03.jpg
DNA Sequence Conservation
Comparison of > 3,000 human and mouse genesChanges in conserved sequence more likely to be deleterious
Is sequence more prone to change in some parts of gene than others?

11_11.jpg
Splice mutations

Splice mutations
Whether an exon is skipped or an intron retained depends on the order of splicing - which exons are spliced together firste.g. on previous slide, if there is an SA mutation in intron 1, but exons 2 & 3 have already spliced together, then intron 1 will be retained. Conversely, if the splicing of exon 1 to exon 3 is energetically more favourable than splicing of exon 2 to exon 3, exon 2 will be skipped.

11_11_2.jpg

11_12.jpg
Note: frameshift

11_13.jpg

Insertion/deletion mutations (indels)

11_05_2.jpg
- obviously, an indel that is not a multiple of 3bp will result in a frameshift mutation if in the coding region
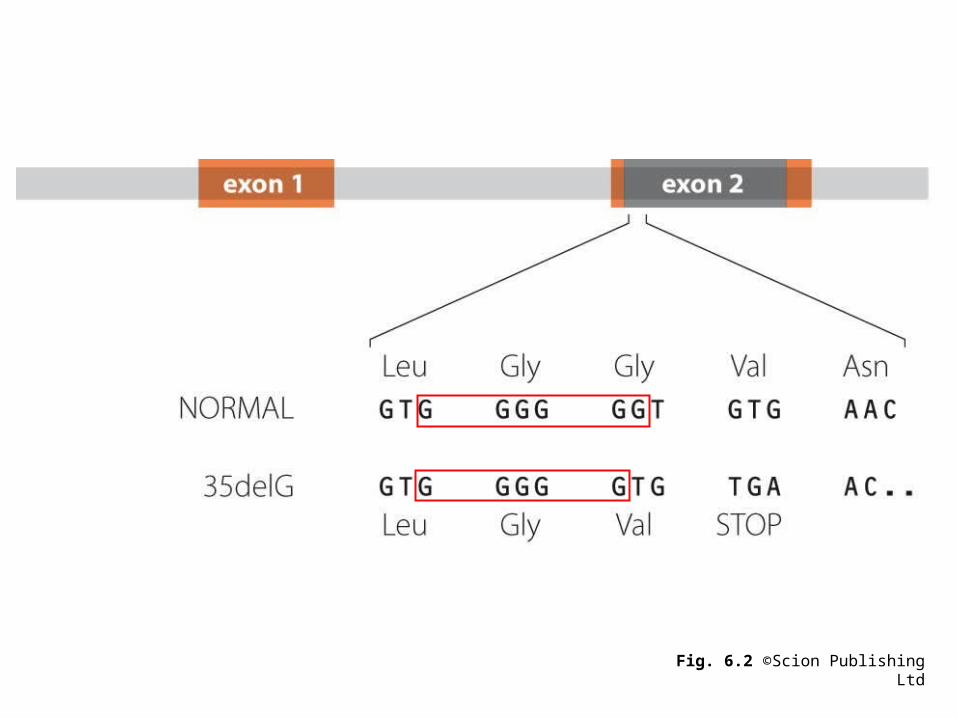
Fig. 6.2 ©Scion Publishing Ltd

Remember: single base substitutions are the most common changes in DNA and may be silent, missense or nonsense. Insertions/deletions (indels) are more common w/i regions of repetitive sequence.

Change Example
Delete:(i) the entire gene Most -thalassemia mutations(ii) part of the gene 60% of Duchenne muscular dystrophy
Insert a sequence into the gene Insertion of LINE-1 repetitive sequence into F8C gene in hemophilia A
Disrupt the gene structure:(i) by a translocation X-autosome translocations in women with DMD(ii) by an inversion Inversion in F8C gene
Prevent the promoter working:(i) by mutation -Globin -29A G mutation(ii) by methylation Fragile-X full mutation (FMR1)
Destabilize the mRNA:(i) by a polyadenylation site mutation -globin AATAAA AATAGA mutation(ii) by nonsense-mediated RNA decay Fibrillin mutations (FBN1)
Examples of Mutations

Change Example
Prevent correct splicing:(i) by inactivating donor splice site PAX3 451 + 1G T mutation(ii) by inactivating acceptor splice site PAX3 452-2A G mutation(iii) by activating a cryptic splice site -Globin intron 1 -110G A mutation
Introduce a frameshift in translation PAX3 874_875insG mutation
Convert a codon into a stop codon PAX3 Q254X mutation
Replace an essential aminoacid PAX3 R271C mutation
Prevent post-transcriptional Cleavage-resistant collagen N-terminal propeptide processing in Ehlers Danlos VII syndrome
Prevent correct cellular localization p.F508del (F508) mutation in cystic fibrosis

Single gene disorders
Mendelian inheritance - AD, AR, X-linked
Becoming relatively more common
Mendelian disorders account for 20% of infant mortality, 10% paediatric hospitalisations
2-3% of neonates have a congenital abnormality
~1% due to a single gene disorder
50% of childhood blindness
50% of childhood deafness
50% of severe learning difficulties
1% of malignancy
Genetic

Definitions
Incidence - the rate at which new cases occur
Prevalence - proportion of population affected (usually less than incidence)
Frequency - not strictly defined, usually synonymous with incidence
Congenital - present at birth
See other definitions in glossary (handout)

Relationship between mode of inheritance and encoded protein
Recessive Enzymes
Transporters
Dominant Structural proteins
Transcription factors
A trend - not an absolute correlation

Transcription factors
Receptors
Modifiers of protein function
Enzymes
Jimenez-Sanchez, Childs & Valle.Nature 409:853, 2001

Examples
Cystic fibrosis Nail Patella SyndromeHaemophilia AchondroplasiaMuscular dystrophy CollagenopathiesThalassaemias Marfan SyndromeInborn errors Huntington Disease
Remember – think about the context in which we discuss a disease as an example of a principle. Also know the mode of inheritance and the gene.

Duchenne Muscular Dystrophy (DMD)
Progressive symmetrical muscular weakness, proximal greater than distal, calf hypertrophy
Symptoms present < 5yoa
Wheelchair dependent by 13yoa
~ 1 in 5,000 males

Becker Muscular Dystrophy (BMD)
Progressive symmetrical muscular weakness, proximal greater than distal, calf hypertrophy
Activity-induced cramping
Wheelchair dependent after 16yoa
Preservation of neck flexor muscle strength
~ 1 in 18,000 males

Histology photos courtesy of Dr Richard Charlton.
Normal
DMD
XDMDBMD
Both Duchenne & Becker MD result from mutations in dystrophin - a large protein connecting the cytoskeletal elements of muscle cells with the extracellular matrix
Sarcoglycans
DMD

DMD BMD
Near absence of dystrophin
Deletions
Frameshift
Nonsense
Partially functional dystrophin
In-frame deletions
DMD/BMD Mutations
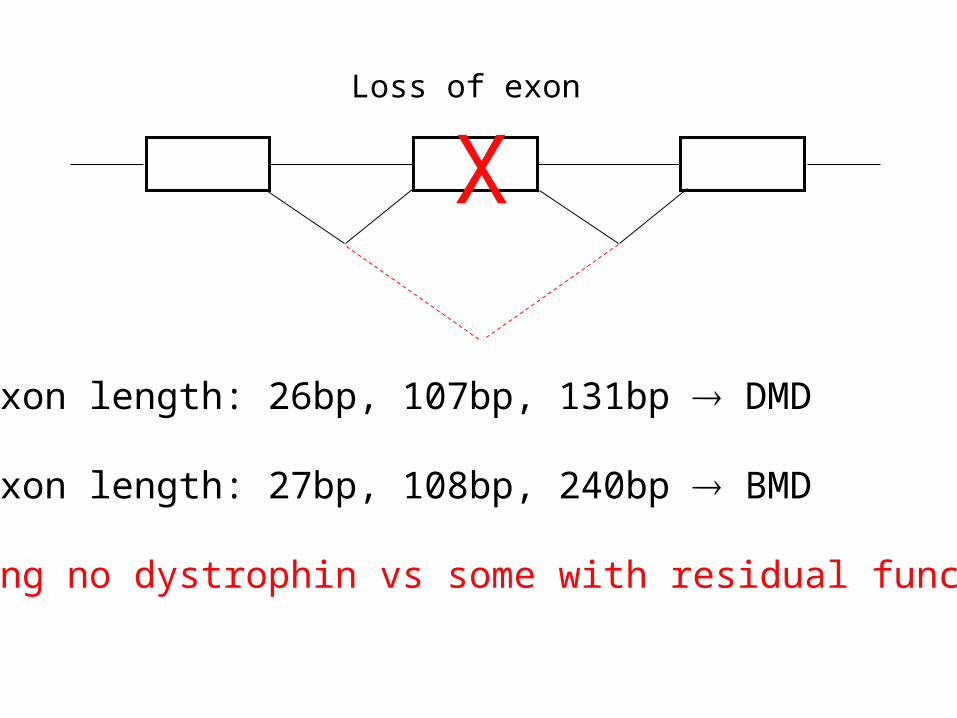
If exon length: 26bp, 107bp, 131bp DMD
If exon length: 27bp, 108bp, 240bp BMD
Making no dystrophin vs some with residual function
XLoss of exon

Haemoglobinopathies
Emery, Chapter 10
See Handout
http://www.ommbid.com

The importance of haemoglobinopathies
~ 650-700 new cases born every day
Lot of experimental material!
Illustrate multiple pathogenic mechanisms
With a few, RARE, exceptions, haemoglobinopathies exhibit autosomal recessive inheritance

Carrier Frequencies of Hb alleles
Allele Population Carrier frequency
Hb S African AmericanWest Africa
1/12up to 1/4
--/αα SE Asia & Chinese 1/25
β0 Italian/Greek 1/30
β+ African American 1/70
β+ SE Asia 1/30 – 1/50

Haemoglobin

Downloaded from: StudentConsult (on 23 January 2006 07:35 PM)
© 2005 Elsevier
Different haemoglobins formed from different combinations of globin gene products
The - and -globin regions on chromosomes 16 and 11 showing the structural genes and pseudogenes () and the various haemoglobins produced. (Adapted from Carrell R W, Lehman H 1985 The haemoglobinopathies. In: Dawson A M, Besser G, Compston N (eds) Recent advances in medicine 19. Churchill Livingstone, Edinburgh, pp. 223-225.)
You should KNOW the composition of embryonic (ζ2ε2), foetal (α2γ2) and adult (A & A2; α2β2 & α2δ2 ) haemoglobin - don’t worry about the others

Downloaded from: StudentConsult (on 23 January 2006 07:35 PM)
© 2005 Elsevier
Haemoglobin switching
ζ2ε2
α2γ2 α2β2
α2δ2

LCR controls expression across the cluster
LCR = locus control region (an enhancer)

Thalassaemias
Imbalance of globin chain production results in the accumulation of free globin chains in the RBC precursors, which, being insoluble, precipitate, resulting in haemolysis of RBCs (haemolytic anaemia) with consequent compensatory hyperplasia of the bone marrow.

Downloaded from: StudentConsult (on 23 January 2006 07:35 PM)
© 2005 Elsevier
Hydrops foetalis(Hb Barts, 4)
No -globin synthesis severe anaemia heart failure oedema

Hb HMilder than Hb Barts- some -globin synthesis- majority of Hb is -tetramer (4, unstable)
Hb Constant Spring
Phenotypically similar to Hb H- mutation of stop codon in -globin geneleads to longer, unstable protein
4, 4 and CS have O2 affinities comparable to myoglobin => no release to tissues

Downloaded from: StudentConsult (on 23 January 2006 07:35 PM)
© 2005 Elsevier
-thalassaemias
Structure of the normal and deleted -globin structural genes in the various forms of -thalassemia. (Adapted from Emery A E H 1984 An introduction to recombinant DNA. John Wiley, Chichester.)
+-thal

16_03.jpgDeletion of -globin genes in -thalassaemia


Downloaded from: StudentConsult (on 23 January 2006 07:35 PM)
© 2005 Elsevier
Haemoglobin electrophoresis
Changes in amino acid sequence affect the mobility of proteins

-thalassaemia(Cooley’s anaemia)
Usually presents as severe anaemia before 12moa
β-Thalassaemia Intermedia in a 13-Year-Old Girl. A. Typical thalassaemic facies with enlargement of cheek bones and maxilla. B. Distinct abdominal swelling from hepatosplenomegaly.
β-Thalassaemia Major. “Hair-on-end” appearance of the skull.

-31 -30 -29 -28 -31 -30 -29 -28 A T A A G T A A

Point mutations in -globin(few deletions of entire gene)
Remember - Missense mutations of -globin generally result in specific phenotypes (e.g. Sickle) and not -thalassaemia Note - promoter mutations (more later)
Location and some of the types of mutation in the -globin gene and flanking region that result in -thalassaemia. (Adapted from Orkin S H, Kazazian H H 1984 The mutation and polymorphism of the human -globin gene and its surrounding DNA. Annu Rev Genet 18: 131-171.)

Downloaded from: StudentConsult (on 23 January 2006 07:35 PM)
© 2005 Elsevier
Hb-Lepore
Mechanism of unequal crossing over which generates Hb Lepore and anti-Lepore. (Adapted from Weatherall D J, Clegg J B 1981 The thalassaemia syndromes. Blackwell, Oxford.)
Note that in Hb-Lepore, transcription is from the -globin promoter so the hybrid gene is expressed at the same low level as . Hb-Lepore is a -thalassaemia, anti-Lepore is essentially normal.
Downloaded from: StudentConsult (on 23 January 2006 07:35 PM)
© 2005 Elsevier
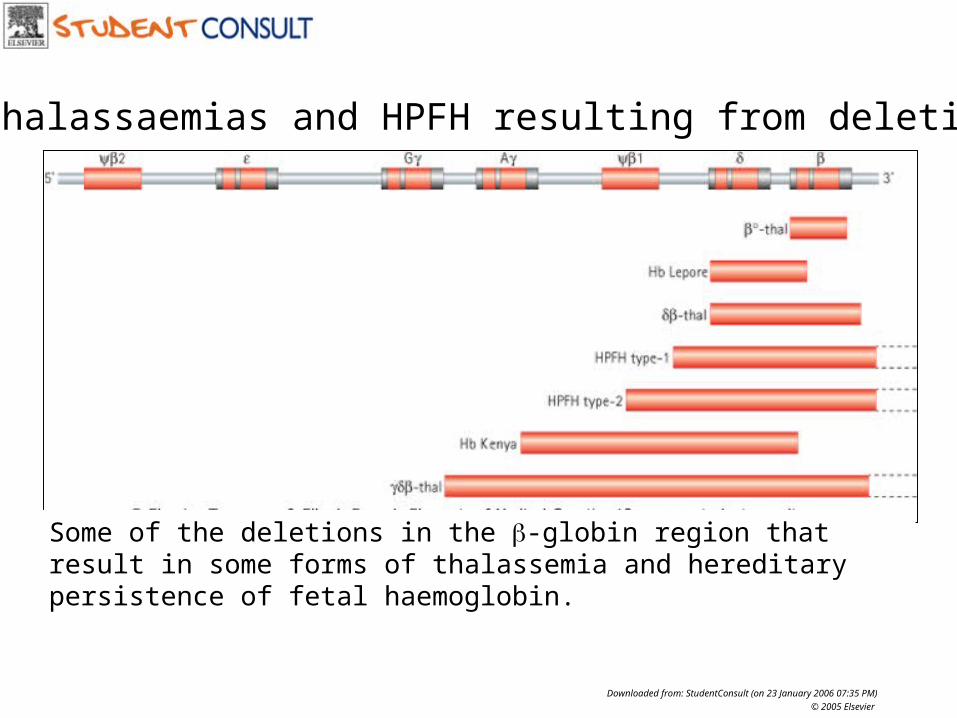
-thalassaemias and HPFH resulting from deletions
Some of the deletions in the -globin region that result in some forms of thalassemia and hereditary persistence of fetal haemoglobin.

Hereditary Persistence of Foetal Haemoglobin (HPFH)
Deletion of - & - genes removes promoter elements that LCR would interact with- no competition for -gene promoter so chain production continues post-natally
Normal phenotype unless exposed to low O2
What would happen if a woman with HPFH becomes pregnant? Any thoughts about therapeutic approaches for β-thalassaemia?
HPFH can also result from mutations in silencer elements around the genes.

Ye Olde Thalassaemia Nomenclature - thalassaemia+-thal - 2 of 4 normal -globin genesHb-H disease - 1 normal -globin geneHydrops foetalis - 0 normal -globin genes
-thalassaemia +-thal - residual -globin synthesis (leaky mutations) 0-thal - no -globin synthesis (major mutations)
Interesting point: +/- / +-thal compound milder due to lower -chain excess
- remember, a lack of -globin results in formation of 4-tetramers which precipitate causing cell lysis; if there is less -globin, there will be fewer 4-tetramers and less lysis so milder disease

Mutations in Dominantly Inherited Diseases
1) Haploinsufficiency Loss of function
Missense
Nonsense
Splice
Frameshift
Deletion
Translocation
Generally, all mutations in the same gene result in the same phenotype
How to explain a phenotype in the presence of a normal allele?

Nail Dysplasia Absent/hypoplastic patellae
Exostoses of the ilia (iliac horns)
Classic Tetrad of Congenital Abnormalities
Elbow dysplasia
Nail Patella Syndrome - another excellent example of pleiotropy
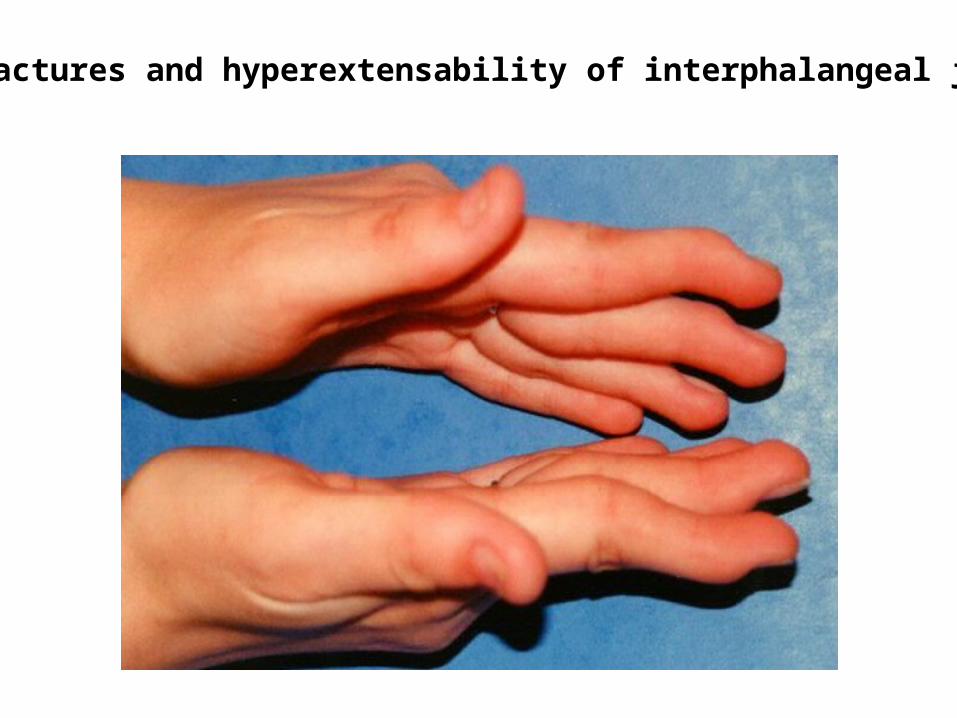
Contractures and hyperextensability of interphalangeal joints

Patellar aplasiaPatellar dysplasia
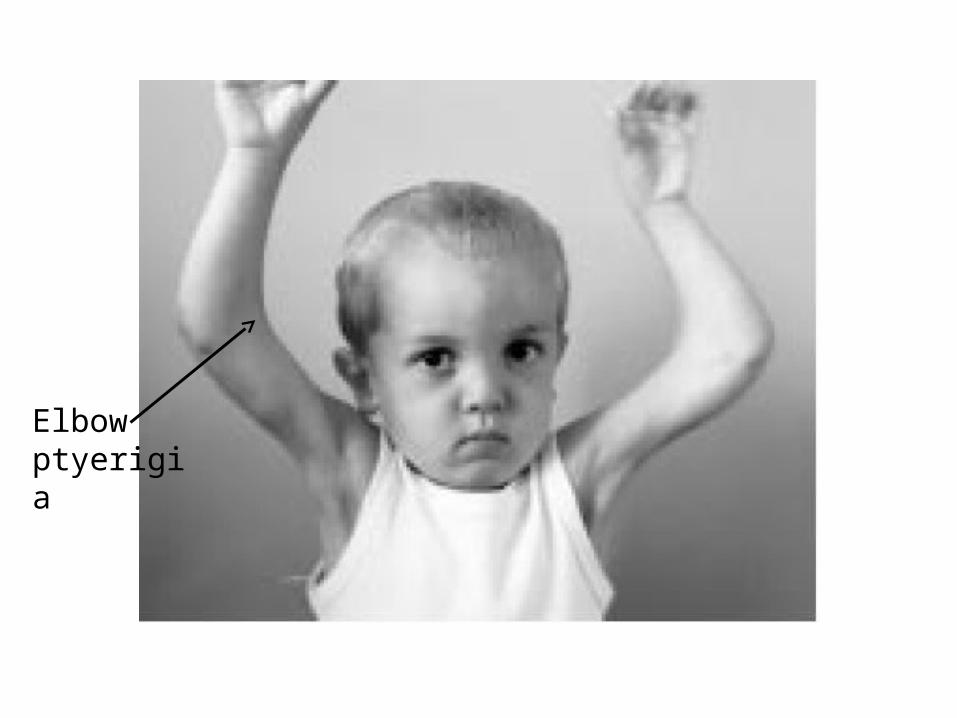
Elbow ptyerigia

The Kidney Phenotype in lmx1b-/- Micelmx1b+/+ lmx1b-/-
Foot processes
Aberrantcell-celljunctions
Miner et al., 2002

Anterior chamber defects in lmx1b KO mice
WT KO
Pressman et al., 2000

HDLIM-A LIM-B
Distribution of 134 NPS mutations within LMX1B
March 2004
Missense mutations clustered in LIM & HD domains
XX X X X XXXX XX XX XX X X
X X X X
XX X
X X X
X
Nonsense, frameshift and splice mutations}
Deletion of part or all of the gene result in the same phenotype

Mutations in transcription factors often cause a range of clinical features because the same factor is used in different cell types at different stages in development. Note that most, not all, mutations act via haplo-insufficiency

Haploinsufficiency
Literally - “half is not enough”
50% reduction in the amount of (functional) protein results in phenotype
Usually see a number of different mutations (types) resulting in the same phenotype

Mutations in Dominantly Inherited Diseases
2) Gain of Function Up-regulation
Novel function
Missense (usually)
Different mutations in the same gene may result in different phenotypese.g. FGFRs

FGFR3 Spectrum
Severity
Thanatophoric Dysplasia Achondroplasia Hypochondroplasia

AchondroplasiaAutosomal dominant, 80% cases are sporadic, ~1:20,000 live births, rhizomelic (proximal) shortening of long bones, depressed nasal bridge, pronounced lumbar lordosis, p.G380R mutation
Thanatophoric Dysplasia
Thanatophoric = “death loving”, always sporadic, ~1:20,000 births (usually stillborn), micromelic limb shortening, severe frontal bossing, v. small chest
TD-I - curved femurs, +/- kleeblattschaedel (cloverleaf skull)TD-II - straight femurs w/ kleeblattschaedel

Major complication in achondroplasia is foramen magnun compression

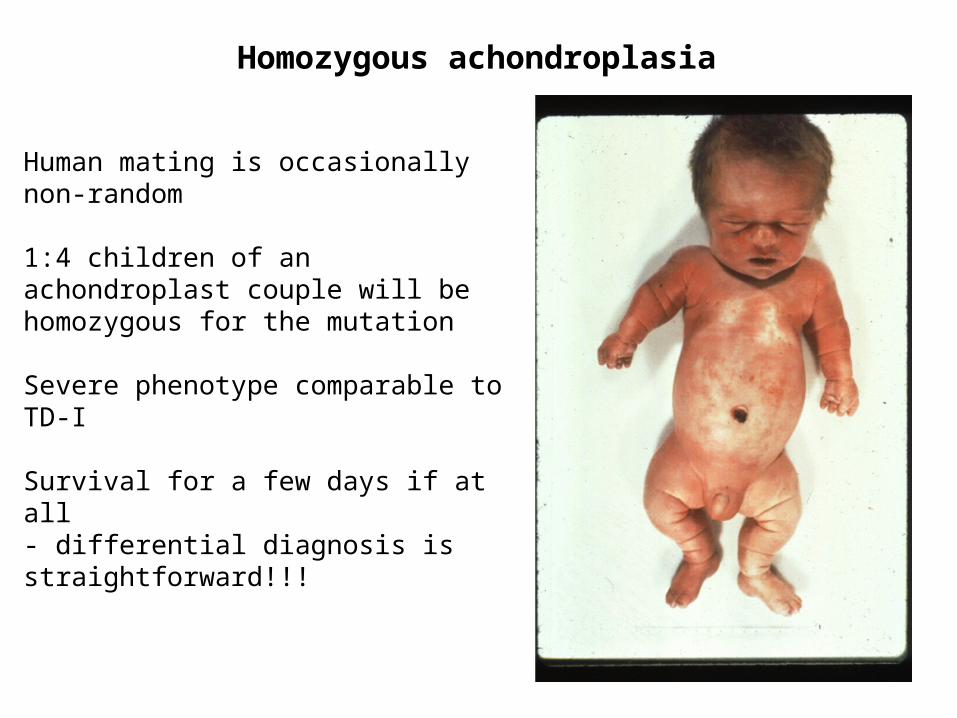
Homozygous achondroplasia
Human mating is occasionally non-random
1:4 children of an achondroplast couple will be homozygous for the mutation
Severe phenotype comparable to TD-I
Survival for a few days if at all- differential diagnosis is straightforward!!!

FGFR3
TD2
TD1
ACHIg I Ig II Ig III
-s-s-
TM TK1 TK2
-s-s- -s-s-
HCHHCH
HCH
ACH: G380R, G375C, S279C, Y278CHCH: N540K, K650N, K650Q, N540S, N540T, I538V, N328I, G268C, R200C, N262H, S84L, V381E … TD1: R248C. S249C. G370C, S371C, Y373C, J807G, R, C, LTD2: K650E
© Gary Bellus
John Wasmuth

Fig. Box 6.3 ©Scion Publishing Ltd
Different mutations of the same gene can result in different phenotypesSimilar mutations in similar genes can result in different phenotypes if the genes are expressed in different cell types or at different times in development
FGFR2 is expressed predominantly in the developing skull so mutations result primarily in skull defects with minor limb defectsFGFR3 plays a greater role in limb development
Apert syndrome

How do mutations in FGFR3 result in these phenotypes?
Experiments:
1. Knock out the gene – if we see the same phenotype(s) in ko mice then must be haploinsufficiency
2. “Knock in” mice – if the phenotype is mutation-specific then we are looking at gain of function

Fgfr3 mutant mice
Fgfr3-/-
Normal Normal Fgfr3-/-
TibiaH&E
col2a1
Normal Fgfr3-/-
Wang et al., 1999

Fgfr3G374R mice
Normal FgfrG374R/+Wang et al., 1999

Normal FgfrG374R/+
Tibial growth plates
col2a1
Alk phos
Wang et al., 1999

Col 2,9,11Comp
Col10
Karsenty
Osteogenesis- & action of FGFR3

Fibrodysplasia ossificans progressiva (FOP)
Congenital malformation of big toesProgressive heterotopic ossification of skeletal muscles, tendons, ligaments and fasciaCharacteristic progression- dorsal ventral - axial appendicular- cranial caudal- proximal distal
Rare, autosomal dominant, most cases sporadic

Shore et al., 2006
FOP candidate region contains the ACVR1 gene
ACVR1 is a BMP receptor

Every FOP patient has the same mutation (R206H)
Shore et al., 2006

Modeling predicts effect of R206H
Shore et al., 2006

BMP/TGF- pathway

Gain of Function Mutations
Specific missense mutations result in specific phenotypes
A mutant protein is synthesised
Remember – FOP is a disease distinct from achondroplasia etc. but the mutational mechanism is the same – gain of function mutation – receptor activation

Mutations in Dominantly Inherited Diseases
3) Dominant-negative Mutant protein interferes with normal
MissenseSplice
Generally applies to structural proteins e.g. collagens
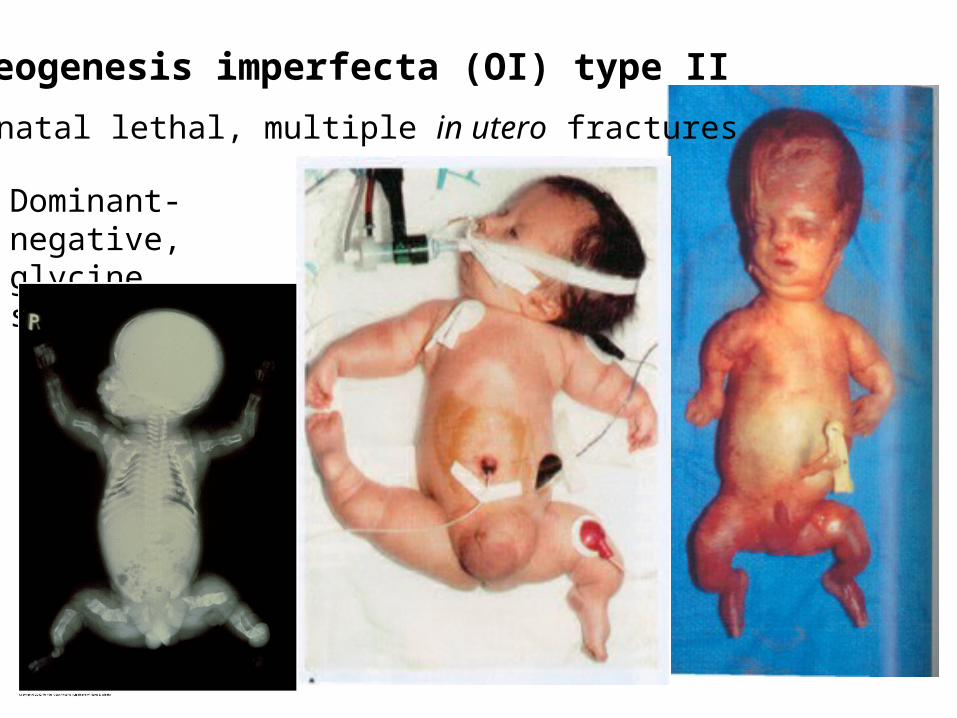
Osteogenesis imperfecta (OI) type II
Neonatal lethal, multiple in utero fractures
Dominant-negative, glycine substitutions

Osteogenesis imperfecta (OI) type III
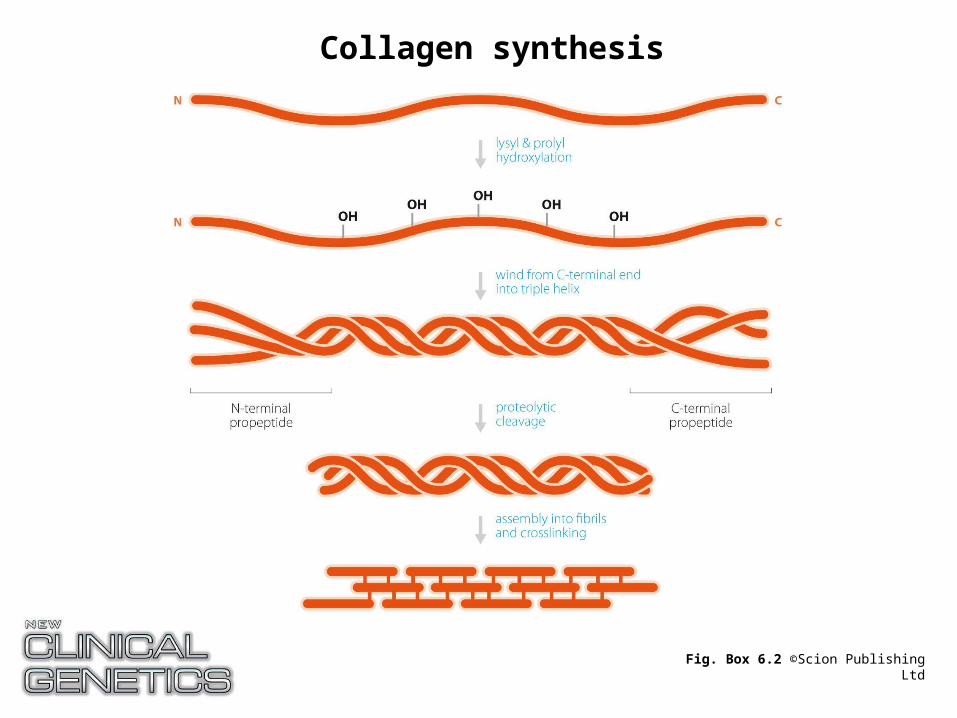
Fig. Box 6.2 ©Scion Publishing Ltd
Collagen synthesis


16_04.jpg
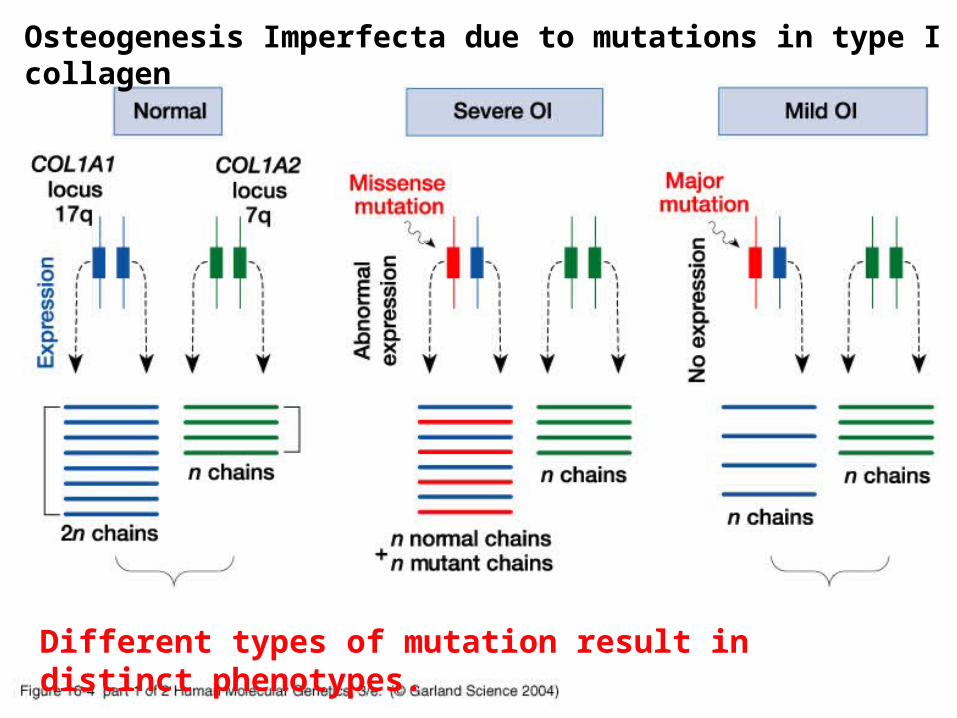
Different types of mutation result in distinct phenotypes.
Osteogenesis Imperfecta due to mutations in type I collagen

16_04_2.jpg
Glycine substitutions disrupt triple helix formation and expose all 3 chains to hydroxylation and glycosylation
Normal Severe OI Mild OI

Fig. 6.8 ©Scion Publishing Ltd
Dominant-negative mutations act via the mutant protein interfering with the normal gene product

Osteogenesis imperfecta (OI) type I

Osteogenesis imperfecta (OI) type I
Blue sclerae
16_04.jpg
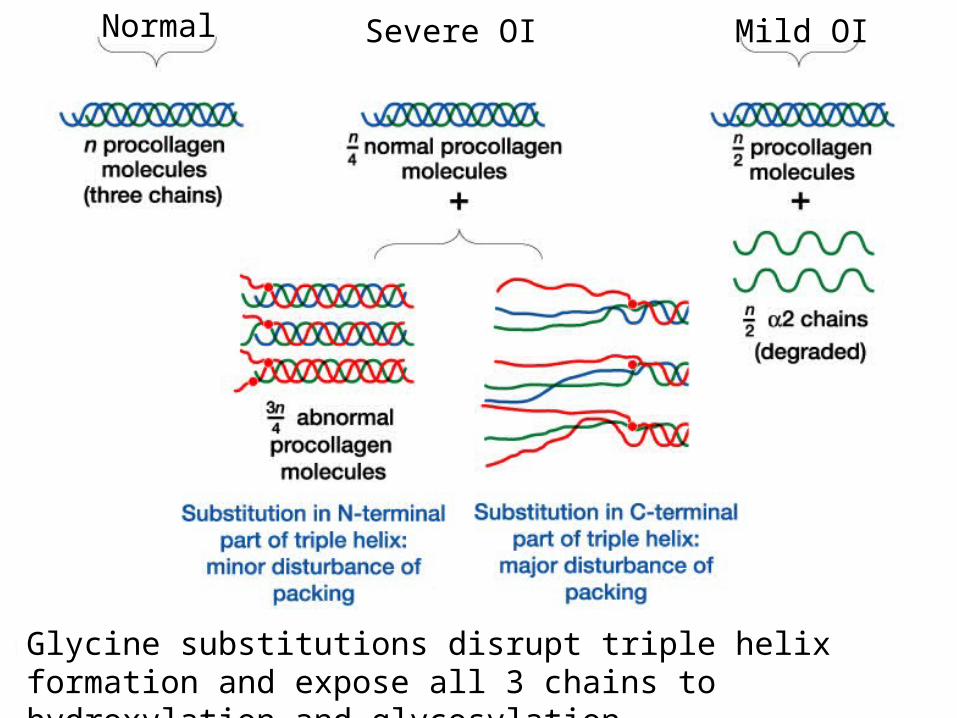
Different types of mutation result in distinct phenotypes.
Osteogenesis Imperfecta due to mutations in type I collagen
16_04_2.jpg
Glycine substitutions disrupt triple helix formation and expose all 3 chains to hydroxylation and glycosylation
Normal Severe OI Mild OI

Osteogenesis imperfecta (OI)
Type I - null mutations, haploinsufficiencyTypes II-IV - missense, dominant negative
- severity approximately correlated with size of mutant amino acid and position of substitution
Bisphosphonates - as used for osteoporosis - potential treatment for OI-type I

Autosomal Recessive OI
Some cases of OI show abnormal modification of collagen but no COL1A1 or COL1A2 mutationEvidence for recessive inheritanceThese patients are deificient for enzymes essential for the post-translational modification of collagen – depending on the enzyme the disease severity can vary from neonatal lethal to progressive to mild
The important point is that very similar phenotypes arise by different means- Failure to synthesise a protein- Failure to secrete a protein- Failure to modify a protein to functional form
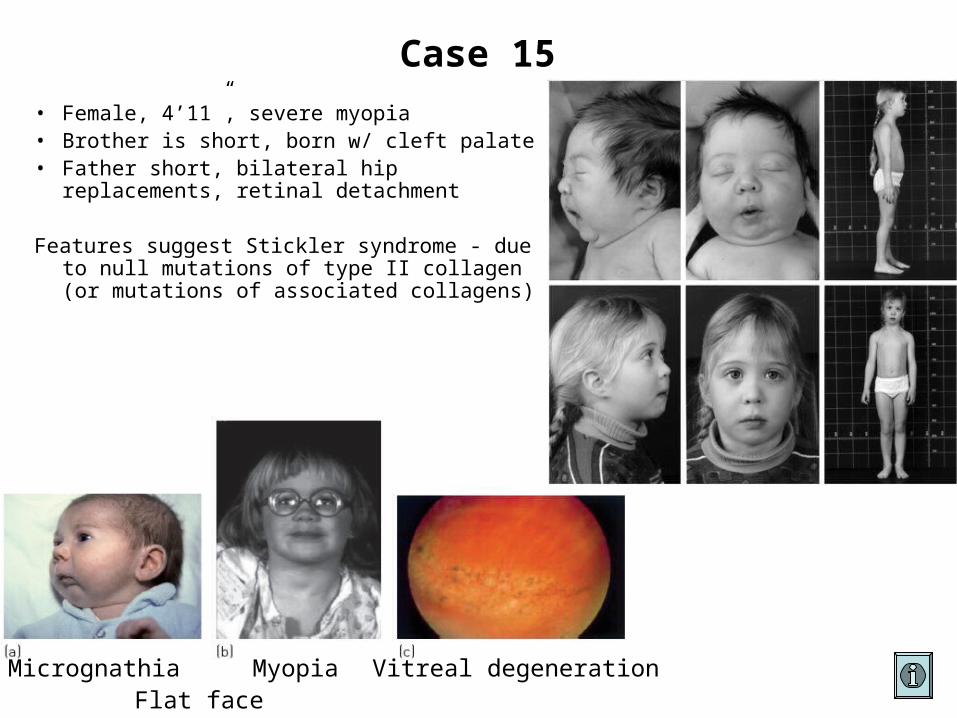
Case 15
• Female, 4’11”, severe myopia• Brother is short, born w/ cleft palate• Father short, bilateral hip replacements,
retinal detachment
Features suggest Stickler syndrome - due to null mutations of type II collagen (or mutations of associated collagens)
Micrognathia Myopia Vitreal degenerationFlat face

Fig. 6.3 ©Scion Publishing Ltd
Frameshift mutation in COL2A1 gene resulting in premature termination codon (PTC)
Ribosome
Ribosomes stall at termination codonsNonsense-mediated mRNA decay (NMD) machinery investigatesIf there are EJCs downstream then it is a PTC - because real stop codons are almost always in the last exon - and the mRNA will be degradedIf no downstream EJCs then it is the real stop codon and the ribosome falls off to reinitiate on this or another transcript
(EJCs)

Therefore, mutations that result in PTCs are NULL mutations and do not result in the significant production of truncated polypeptide
- OI type I vs OI type II (mutations of type I collagen)- Stickler syndrome vs more severe chondrodysplasias
(mutations of type II collagen)
Older texts (and some exam questions) will state (infer) that PTCs do result in synthesis of truncated polypeptides - we now know this to be the exception rather than the rule in almost all eukaryotes

Dominant-negative Mutations
The mutant gene product interferes with the function of the normal gene product
Again, a mutant protein has to be synthesised

Summary
Recessive disorders (either autosomal or X-linked)- mutations reduce or abolish protein function- missense, nonsense, splice, frameshift, chromosomal
Dominant disorders- disease in presence of one normal copy- haploinsufficiency (“half is NOT enough”)
- same type of mutations as for recessive- gain of function
- almost always missense- dominant-negative
- mostly missense
Mutant protein exerts effect
PTCs DO NOT result in truncated polypeptides!!!

Mode of inheritance?
Recessive
Loss of function mutations
Dominant
Null alleles observed?
Yes
Haploinsufficiency
No
Do different missense mutations yield distinct phenotypes?
Dominant-negative
No* Yes
Gain of function* If null mutations are observed in the same gene and result in a milder phenotype, missense mutations probably dominant-negative e.g. collagen

100%
50%
25%
Dominant-negative mutationsIf a MISSENSE mutation acts in a dominant-negative manner, a NULL mutation in the same gene will result in a milder version of the same phenotype.Note the sliding scale from normal through haploinsufficiency to the severe dominant-negative.

Gain of Function MutationsIf a MISSENSE mutation acts to cause a gain of function, a NULL mutation in the same gene will result in the opposite phenotype.
A specific missense mutation of MSX2 results in excessive DNA binding by the transcription factor and premature closure of sutures
A heterozygous loss of function mutation in MSX2 reduces DNA binding and inhibits bone growth in the skull
Craniosynostosis Parietal foramina

Mutations outwith the coding region
Recall promoter structure of eukaryotic genes
If there is no functional promoter will protein be synthesised?
Mutations in proximal promoter elements prevent transcriptione.g. -globin
Distal elements & enhancers are more interesting- Polydactyly- Campomelic dysplasia- Lactose tolerance

Remember how enhancers work
This distance can be 500bp - 1Mb

Polydactyly
Common birth defect, part of many different syndromes, multiple animal models, marked locus heterogeneity

Lettice et al., 2002; 2003
Linkage studies in a large family segregating polydactyly as an AD characteristic suggested a mutation in or near the SHH gene.
Mutations of SHH are one cause of holoprosencephaly in humans - these polydactyly patients do not have SHH mutationsA mouse polydactyly mutant mapped to the same region - found to be an insertion w/i an intron of the gene upstream from SHH Insertion disrupts a region that is highly conserved during mammalian evolution

X
~1,000kb
This conserved region functions as an enhancer of SHH in the distal limb
Enhancer mutation far upstream of SHH results in polydactyly and not holoprosencephaly
SHH
Proximal promoter
Enhancer
A similar situation is the cause of blue/brown eye colour variation- single base change w/i intron of gene upstream from OCA2
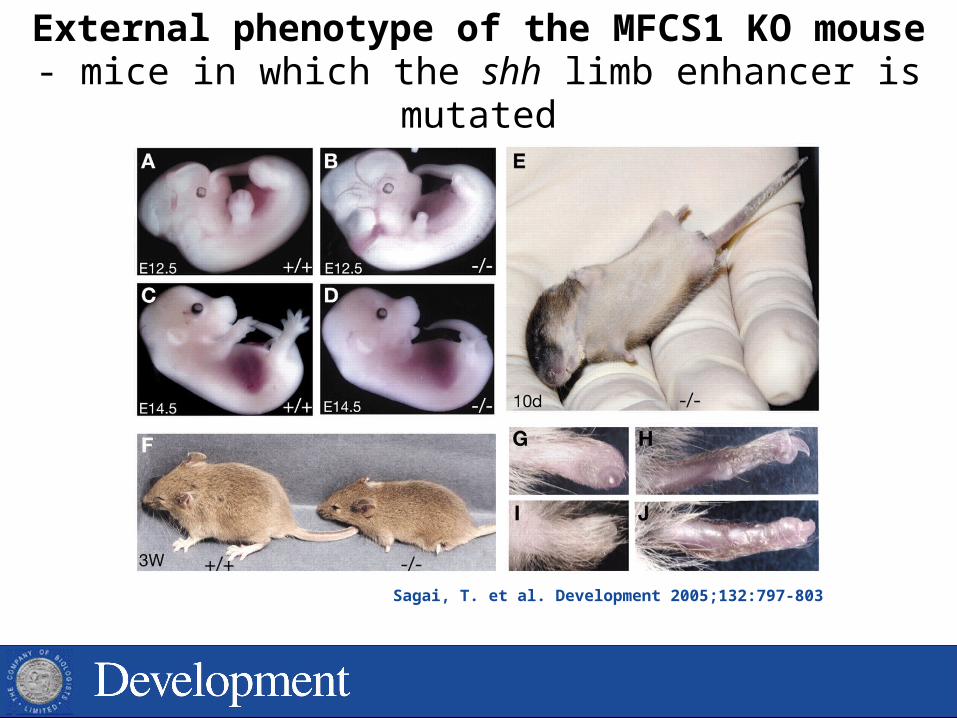
Sagai, T. et al. Development 2005;132:797-803
External phenotype of the MFCS1 KO mouse- mice in which the shh limb enhancer is mutated

DIFFERENT enhancers can direct expression of the SAME gene in DIFFERENT tissues
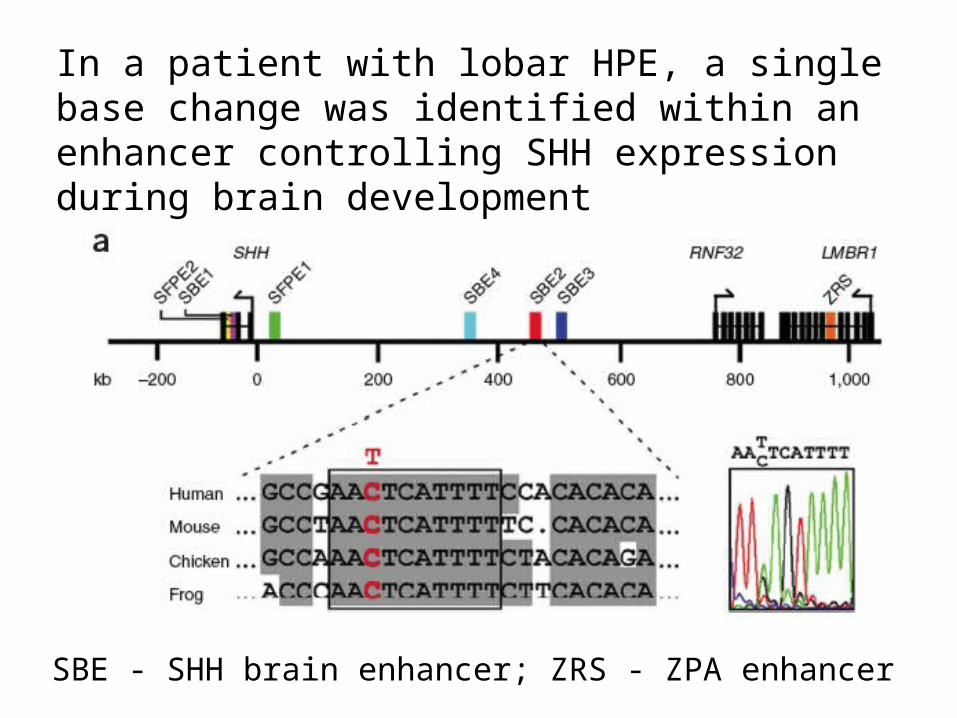
In a patient with lobar HPE, a single base change was identified within an enhancer controlling SHH expression during brain development
SBE - SHH brain enhancer; ZRS - ZPA enhancer

Normal Mutation Deletion
SBE2 controls brain expression (b,e,h,k)
The mutation identified restricts expression (c,f,i,l)
Deletion almost eliminates expression (d,g,j,m)

Distant chromosomal rearrangements candisrupt gene expression
Campomelic dysplasia

http://biology.plosjournals.org/archive/1545-7885/3/1/figure/10.1371_journal.pbio.0030007.g002-L.jpg
Conserved non-coding sequences are clustered close to genes controlling differentiation and development.
Remember:5% of our genome sequence has been conserved by selection - but only 1.5% codes for protein, 3.5% is directing the expression of 1.5%

Lactose intolerance
Inability to metabolise lactose - lactase not expressed- normal mammalian phenotype
Lactase persistence (LP) is a mutant phenotype
LCT
14kb
CT @ -13910 binding of OCT-1 and maintains intestinal expression of lactase in Europeans
Other variants in this region associated with LP in other populations exhibiting LP - suggests selection for different variants as dairy production developed at different times in human historyDo you think LP is AD or AR??

Variation in sequences controlling gene expression are important causes of human variation and disease
- as more and more variants are discovered that play a role in common disorders such as heart disease, hypertension, diabetes etc., many are found to be in sequences controlling gene expression rather than changing the protein sequence

Genomic Disorders
Disorders due to gross changes in DNA
- duplication/deletion
Usually due to recombination between non-homologous repeat sequences
e.g. 22q11 deletion syndrome
-thalassaemia
Williams syndrome
Smith-Magenis syndrome

Imbalance in protein production causes problems
Yellow = deletion, blue = duplication

Williams Syndrome

Williams-Beuren Syndrome (WBS)
AbnormalitiesGrowth Mild pre- and postnatal growth deficiency
Facies Medial eyebrow flare, short palpebral fissure, epicanthal folds, stellate iris, antiverted nares, everted lower lip.
Cardiac Supravalvular stenosis, PS, septal defects
GU Nephrocalcinosis, small or ectopic kidney, urethral stenosis, reflux
Mental IQ range 40 to 80. Mean IQ 56. Loquacious personality. Language > cognitive ability, mild neurologic dysfunction

Deletion vs DuplicationWilliams Syndrome
IQ < 80; Supravalvular aortic stenosis; VSD and/or ASD; Long philtrum, full lips
Bicycle Elephant Details vs Big picture
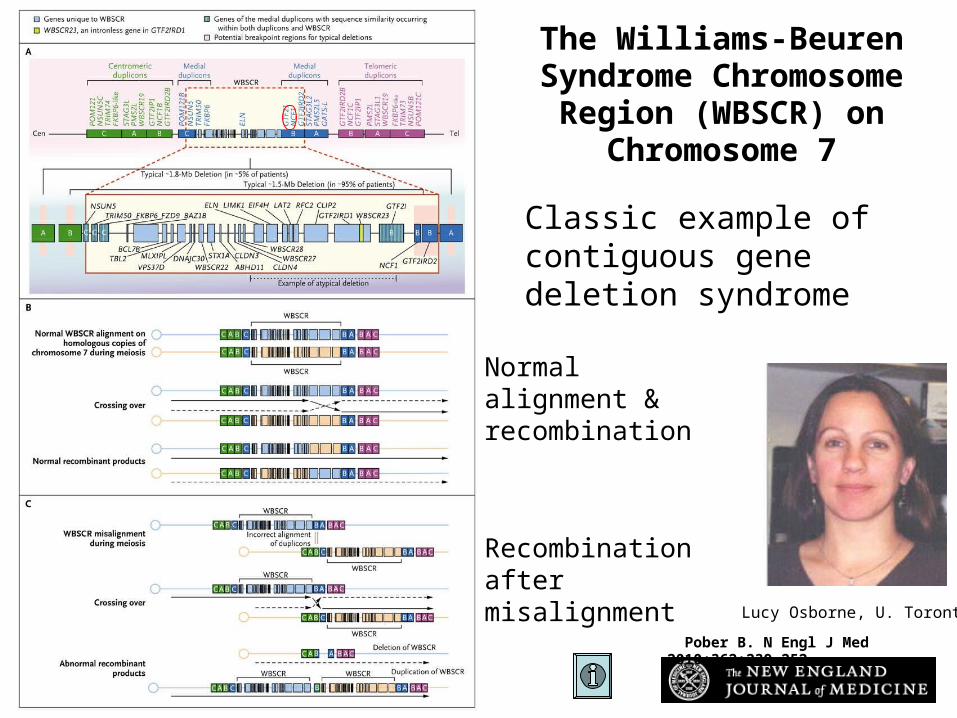
Pober B. N Engl J Med 2010;362:239-252
The Williams-Beuren Syndrome Chromosome
Region (WBSCR) on Chromosome 7
Lucy Osborne, U. Toronto
Normal alignment & recombination
Recombination after misalignment
Classic example of contiguous gene deletion syndrome

Recombination between flanking repeats results in one gamete deleted for intervening sequence and one duplicated for same.Deletions seem to result in a phenotype more often (c.f. monosomy vs trisomy)Deletion and duplication phenotypes may be approx opposite of each other
For every deletion there should be a corresponding duplication
Genes
Genes
Genes Genes
Rpts

Somerville et al., 2005
Duplication of WBS region associated with speech delay
ADHD Language delay
Duplication of WBSCR observed in boy with language delayTriplication results in a more severe phenotype

Somerville et al., 2005
Similar phenotypes in deletion and duplication syndromesPhenotype in deletion, duplication normalOpposite phenotypes
Phenotypes may be opposite or similar comparing deletion with duplication

Deletion
Deletion and Duplication of 7q11.23 Result Deletion and Duplication of 7q11.23 Result in Contrasting Phenotypesin Contrasting Phenotypes
Severe expressive language delay
Relative strength in expressive language
Relative strength in spatial skills
Very weak spatial skills
Short philtrumThin lips
Larger teethArched palate
Narrow foreheadNormal periorbital
area
Long philtrumFull lips
Small teethNormal palate
Broad foreheadPeriorbital
fullness
Duplication Deletion
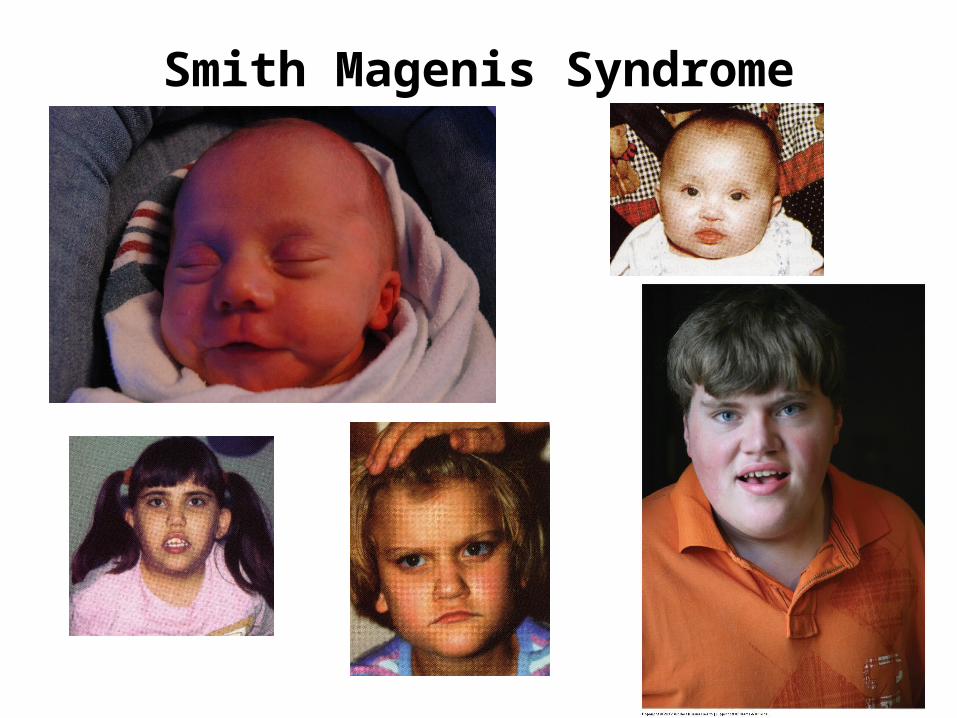
Smith Magenis Syndrome

Smith Magenis SyndromeClinical Features
Distinctive facial appearance, Broad square-shaped face, Brachycephaly, Prominent forehead, Synophrys, Upslanting palpebral fissures Deep set eyes, Broad nasal bridge, Midface hypoplasia, Short full tipped nose Micrognathia progressing to prognathia with age, Distinctive mouth with tented appearance to upper lip.
Hypotonia, Short stature, Brachydactyly, Eye and Ear abnormalities, Speech delay, Scoliosis, Moderate mental retardation, occasional cardiac and renal abnormalities
Etiology - Deletion 17p11.2 3700 kb
duplication causes Potocki-Lupski syndrome (PLS)

Charcot-Marie-Tooth Disease
Genetically heterogenous (> 20 loci)Neuropathies characterised by chronic motor & sensory polyneuropathy
CMT1Aautosomal dominantslowly progressive weakness and atrophy of distal
leg muscleshyporeflexiamuscular denervation 2º to axonal degenerationuniform slowing of nerve conduction velocity (NCV)

Hereditary Neuropathy with liability to Pressure Palsies (HNPP)
Autosomal dominantRepeated focal pressure neuropathies
(e.g. carpal tunnel syndrome)Prolongation of distal nerve conduction latenciesEvidence for demyelination on biopsy
Both HNPP and CMT1A map to chr. 17p11.2 (but distinct from SMS deletion region)
PMP22 - major component of myelin - expressed in myelinated fibers in PNS - predominantly produced by Schwann cells- role during Schwann cell growth & differentiation

PMP22 in a myelinated axon

CMT1A HNPP
17p11.2
Missense mutations in PMP22 generally more severe than CMT1ANull mutations in PMP22 generally cause HNPP

Summary
Chromosomal rearrangements are very often the result of recombination between very similar sequences
Which technique might be used to detect the deletion?
For every deletion there should be a corresponding duplication
Depending on the genes involved, the phenotypes resulting from duplication and deletion may be distinct or quite similar
e.g. Williams syndrome vs speech delayCMT1A vs HNPP-thalassaemia