mechanisms of action of novel influenza a/m2 viroporin …molpharm.aspetjournals.org › content ›...
TRANSCRIPT

1521-0111/90/2/80–95$25.00 http://dx.doi.org/10.1124/mol.115.102731MOLECULAR PHARMACOLOGY Mol Pharmacol 90:80–95, August 2016Copyright ª 2016 by The American Society for Pharmacology and Experimental Therapeutics
Mechanisms of Action of Novel Influenza A/M2 ViroporinInhibitors Derived from Hexamethylene Amiloride s
Pouria H. Jalily, Jodene Eldstrom, Scott C. Miller, Daniel C. Kwan, Sheldon S. -H. Tai,Doug Chou, Masahiro Niikura, Ian Tietjen, and David FedidaDepartment of Anesthesiology, Pharmacology, and Therapeutics, Faculty of Medicine, University of British Columbia, Vancouver(P.H.J., J.E., S.C.M., D.C.K., D.C., I.T., D.F.), and Faculty of Health Sciences, Simon Fraser University, Burnaby (S.S.-H.T., M.N.,I.T.), British Columbia, Canada
Received December 7, 2015; accepted May 12, 2016
ABSTRACTThe increasing prevalence of influenza viruses with resistance toapproved antivirals highlights the need for new anti-influenzatherapeutics. Here we describe the functional properties of hexam-ethylene amiloride (HMA)–derived compounds that inhibit the wild-type and adamantane-resistant forms of the influenza A M2 ionchannel. For example, 6-(azepan-1-yl)-N-carbamimidoylnicotina-mide (9) inhibits amantadine-sensitive M2 currents with 3- to6-fold greater potency than amantadine or HMA (IC50 5 0.2 vs.0.6 and 1.3 mM, respectively). Compound 9 competes withamantadine for M2 inhibition, andmolecular docking simulationssuggest that 9binds at site(s) that overlapwith amantadine binding.In addition, tert-butyl 49-(carbamimidoylcarbamoyl)-29,3-dinitro-
[1,19-biphenyl]-4-carboxylate (27) acts both on adamantane-sensitive anda resistantM2variant encodinga serine to asparagine31 mutation (S31N) with improved efficacy over amantadine andHMA (IC505 0.6mMand4.4mM, respectively).Whereas 9 inhibitedin vitro replication of influenza virus encodingwild-typeM2 (EC5052.3 mM), both 27 and tert-butyl 49-(carbamimidoylcarbamoyl)-29,3-dinitro-[1,19-biphenyl]-4-carboxylate (26) preferentially inhibitedviruses encoding M2(S31N) (respective EC50 5 18.0 and 1.5 mM).This finding indicates thatHMAderivativescanbedesigned to inhibitviruses with resistance to amantadine. Our study highlights thepotential of HMA derivatives as inhibitors of drug-resistant influenzaM2 ion channels.
IntroductionViroporins are virally encoded transmembrane proteins that
facilitate conduction of ions or small molecules and arerequired for efficient viral replication (Nieva et al., 2012).Despite their small size (frequently ,100 amino acids),viroporins in many cases have evolved to function as highlyregulated ion channels, which makes them attractive mini-malist models of ion conductance and ion channel evolution(Pinto et al., 1992; Stouffer et al., 2008; Thiel et al., 2011;OuYang et al., 2013; OuYang and Chou, 2014). The M2viroporin of influenza A is a 97-amino-acid, type I transmem-brane domain protein that forms a tetrameric ion channel that
is proton-gated and proton-selective (Nieva et al., 2012). Afterviral entry into host cells, M2 conducts protons from acidichost-cell endosomes to the virion interior to allow for uncoatingand release of viral RNA. M2 on host-cell endosomal mem-branes is also observed in some cases to conduct protonsto elevate secretory vesicle pH, thereby delaying egress ofnascent virion particles and preventing viral hemagglutininfrom adopting a nonfunctional, low pH conformation (Sugrueet al., 1990; Alvarado-Facundo et al., 2015). The M2 ionchannel of influenza B (B/M2) is a functional homolog ofA/M2. It is 109 residues long and forms a homotetramer inthe membrane like A/M2. Furthermore, B/M2 exhibits higherchannel activity but shows a similar pH dependence in terms ofits proton conductance; however, major differences exist be-tween the two channels. Other than the HXXXW sequencemotif crucial for channel activity, the two proteins share nearlyno sequence homology, and unlike A/M2, the B/M2 protonconductance activity is entirely insensitive to amantadine andrimantadine (Mould et al., 2003; Wang et al., 2009).The compounds amantadine and rimantadine (Fig. 1A) are
potent inhibitors of A/M2 proton conductance and licensed
This work was funded by a Canadian Institutes of Health ResearchIndustry-Partnered Collaborative Research Operating Grant [IPR-124291]which was cofunded by Cardiome Pharma Corp., Vancouver, Canada. We alsowish to acknowledge the support from Grand Challenges Canada [0487-01-10].Grand Challenges Canada is funded by the Government of Canada and isdedicated to supporting Bold Ideas with Big Impact in global health.
dx.doi.org/10.1124/mol.115.102731.s This article has supplemental material available at molpharm.
aspetjournals.org.
ABBREVIATIONS: B/M2, M2 ion channel of influenza B; Boc, tert-butyloxycarbonyl; BIT-225, N-(5-(1-methyl-1H-pyrazol-4-yl)naphthalene-2-carbonyl)guanidine; EIPA, 5-(N-ethyl-N-isopropyl)-amiloride; GFP, green fluorescent protein; HMA, hexamethylene amiloride; I-V, current-voltagerelationship; IC50, half-maximal inhibitory concentration; LM, ltk- murine fibroblast; MDCK, Madin-Darby canine kidney cell line; MEM, modifiedEagle’s medium; MES, 4-morpholineethanesulfonic acid; M2WJ352, N-((5-(thiophen-2-yl)isoxazol-3-yl)methyl)adamantan-1-amine; M2WJ332, N-((5-(thiophen-2-yl)isoxazol-3-yl)methyl)adamantan-1-amine; NMDG, N-methyl-D-glucamine; NMR, nuclear magnetic resonance; N31, asparagine atposition 31; PCR, polymerase chain reaction; pHi, internal solution pH; pHo, external solution pH; RMSD, root mean square deviation; SAR,structure-activity relationship; SARS-COV, severe acute respiratory syndrome coronavirus; S31, serine at position 31; TEVC, two-electrode voltageclamp; T27, threonine at position 27; V27, valine at position 27; WT, wild type.
80
http://molpharm.aspetjournals.org/content/suppl/2016/05/18/mol.115.102731.DC1Supplemental material to this article can be found at:
at ASPE
T Journals on July 23, 2020
molpharm
.aspetjournals.orgD
ownloaded from

influenza antivirals (Hay et al., 1985; Pinto et al., 1992;Chizhmakov et al., 1996); however, M2 sequence changes thatrender resistance to adamantanes are now so prevalent thatthese compounds are no longer recommended for use (Fioreet al., 2011). For example, more than 90% of transmissibleadamantane-resistant influenza strains encode an M2 serineto asparagine mutation at position 31 (S31N). This mutationdisrupts adamantane interactions within theM2 pore withoutadversely affecting ion channel activity (Hay et al., 1986;Belshe et al., 1988; Pinto et al., 1992; Bright et al., 2005;Stouffer et al., 2008; Balannik et al., 2010). Thus, new smallmolecules that inhibit adamantane-resistant M2 are neededfor both improved understanding of the chemical space andmechanisms by which M2 activity can be modified in additionto development of new influenza antivirals.To date, few compounds are reported to act on the S31N
form of M2 from influenza A. Furthermore, several proposedM2 inhibitors that are derived from established M2 inhibitorsand act on subsets of drug-resistant influenza viruses in vitroinstead confer antiviral activity through alternative mecha-nisms (Wang et al., 2013a; Kolocouris et al., 2014), indicatingthat direct screening of M2 ion channel activity is necessary toidentify bona fideM2(S31N) inhibitors. One of themost potentadamantane derivative for which detailed M2 current anal-ysis is available, N-((5-(thiophen-2-yl)isoxazol-3-yl)methyl)-adamantan-1-amine (M2WJ352; Fig. 1A), inhibits M2(S31N)but not wild-type (WT) M2 proton currents as measured bytwo electrode voltage clamp (TEVC) electrophysiology, withan IC50 of 14 mM (Wang et al., 2013b).A separate class of compounds that remains poorly explored
for anti-M2 activity is the acylguanidines (Kleyman andCragoe, 1988; Gazina and Petrou, 2012). Initially identified
as potassium-sparing diuretics, acylguanidine-containingamilorides, and hexamethylene amiloride (HMA) in particular(Fig. 1B), are inhibitors of multiple viroporins including thoseof hepatitis virus, HIV-1, and severe acute respiratory syn-drome coronavirus (SARS-CoV) (Kleyman and Cragoe, 1988;Ewart et al., 2002; Premkumar et al., 2004; Pervushin et al.,2009; Gazina and Petrou, 2012). HMA has also been report-ed to inhibit M2(WT) (IC50 5 1.1 mM by TEVC) but notM2(S31N), whereas a related compound N-(5-(1-methyl-1H-pyrazol-4-yl)naphthalene-2-carbonyl)guanidine (BIT-225; Fig.1B) inhibits currents from the viroporins of hepatitis C andHIV-1 and shows promising activity in early clinical trials(Khoury et al., 2010; Luscombe et al., 2010; Gazina andPetrou, 2012). Although additional acylguanidines, includingethyl-isopropyl amiloride (EIPA) and (6-(1-methylpryazol-4-yl)-2-napthoyl)guanidinium (BIT-314; Fig. 1B), are alsoreported to have antiviral activity against multiple viroporinsand viruses, their effects on M2 currents are not yet reported(Ewart et al., 2009; Gazina and Petrou, 2012). Herein wedescribe an electrophysiology-driven approach to characterizethe mechanism of and pharmacologically evaluate a series ofacylguanidines and HMA-like derivatives on inhibition of WTand adamantane-resistant influenza M2 viroporins.
Materials and MethodsChemistry. Detailed information for the synthesis and character-
ization of compounds 1–33 can be found in the SupplementalMaterial.M2WJ352 was synthesized as described previously (Wang et al.,2013b). Amantadine hydrochloride, 5-(N,N-hexamethylene)amiloride,5-(N-ethyl-N-isopropyl)amiloride, and 1-benzoylguanidine were pur-chased from Sigma-Aldrich (St. Louis, MO).
Electrophysiology. The tsA-201 cells, a derivative of the HEK293T cell line, or ltk- murine fibroblast (LM) cells were cultured inmodified Eagle ’s medium plus 10% fetal calf serum, 100 U/mlpenicillin, and 100 mg/ml streptomycin (MEM 1 medium). cDNAsequences encoding full-length M2 were derived from the A/HongKong/1073/99(H9N2) or B/Lee/1940 references sequence and con-tained an N-terminal FLAG-tag plus 3 (Gly) repeat linker. This tagwas used to confirm M2(WT) expression on the cell surface oftransfected cells by immunocytochemistry (Supplemental Fig. 1).M2 sequences were cloned into the pcDNA3 plasmid and tran-siently cotransfected with a pcDNA3 plasmid encoding greenfluorescent protein (GFP) into tsA-201 cells using standard trans-fection protocols (Lipofectamine 2000; Life Technologies, Waltham,MA); 24–48 hours after transfection, single GFP-positive cells wereperfused continuously at 3–5 ml/min with external (bath) solutioncontaining (in mM): 150 NMDG, 10 HEPES, 10 D-glucose, 2 CaCl2,1 MgCl2 buffered at pH 7.4. For low external pH (pHo 5.9 or 5.5)solution, HEPES was replaced by MES. Patch electrodes were pulledfrom thin-walled borosilicate glass (World Precision Instruments,Sarasota, FL) and fire-polished before filling with standard pipettesolution containing (in mM): 140 NMDG, 10 EGTA, 10 HEPES, and1MgCl2 buffered at pH 7.2 or pH 6.0 (10 mMMES). M2 currents weredetected in .90% of GFP-positive cells assayed during the course ofthe study.
Voltage-clamp experiments were performedwith anAxopatch 200Bamplifier (Molecular Devices, Sunnyvale, CA) connected to a Digida-ta1322A 16-bit digitizer. Pipettes typically had a resistance of 3-5MV.Data were acquired with pCLAMP9.2 software (Molecular Devices)sampled at 10-kHz and low-pass filtered at 5 kHz. The standardvoltage protocol consisted of holding a cell at 240 mV, followed by a100-millisecond pulse to280 mV, a 300-millisecond ramp to140 mV,and a 200-millisecond step to 0 mV before stepping back to 240 mVrepeated every 4 seconds at 20–22°C.
Fig. 1. Examples of reported M2 inhibitors. (A) Adamantane inhibitors:amantadine, rimantadine, and N-((5-(thiophen-2-yl)isoxazol-3-yl)methyl)-adamantan-1-amine (M2WJ352) (Wang et al., 2013b). (B) Acylguanidines:HMA (Gazina and Petrou, 2012), BIT-225 (Khoury et al., 2010; Luscombeet al., 2010), EIPA, and BIT-314.
Mechanisms of Action of Novel Influenza A/M2 Viroporin Inhibitors 81
at ASPE
T Journals on July 23, 2020
molpharm
.aspetjournals.orgD
ownloaded from

Compound Screening. Compounds were prepared in dimethyl-sulfoxide at 100 mM and diluted with external pH 5.5 solution todesired concentrations. To measure inhibition of M2 currents bycompounds, cells were exposed repeatedly to pHo 7.4 and pHo 5.5solutions until stable, pH-dependent inward currents were reproduc-ibly observed. Cells were then treated with compounds at definedconcentrations in pHo 5.5 solution for$2 minutes. Data are presentedas percent current inhibition at a given concentration of compound orconcentration required for IC50. All compounds were initially tested at100 mM. Compounds that showed greater than 50% inhibition at100 mM were further tested at lower concentrations to estimate theIC50. This was calculated from nonlinear regression fitting of percent-age inhibition at minimum of three concentrations, and experimentswere performed at least three times at each concentration.
Current-Voltage Relations. M2-transfected ltk-murine fibro-blast (LM) cells were used 18–48 hours post-transfection. Cells werevoltage clamped using the whole-cell patch-clamp configuration pre-viously described herein. To maximize internal pH (pHi) control, highconcentrations of pH buffer were used as impermeant ions: the patchpipette contained 90 mM N-methyl-D-glucamine (NMDG), 10 mMEGTA, and 180mMN-2-hydroxyethylpiperazine-N9-2-ethanesulphonicacid (HEPES). The bath contained a similar solution with 2mMCaCl2replacing EGTA. The pH of all solutions was lowered to desired valuesby addition of 5 M aqueous HCl. The osmolality of both bath solutions(pHo 7.4 and 5.6) as well as the patch pipette solution were preciselyadjusted to 300mOsmol/kg by the addition of glucose andmeasured byan osmometer (OsmetteII, Precision Systems Inc., MA). Membranecurrent was recorded at 20–22°C, digitized at 10 kHz. Current wasmeasured using a modified voltage ramp protocol that extended thevoltage range from280 to1120mV. The pHi was held constant at 7.2,whereas pHo was changed by fast perfusion close to the cell, andwashout was by slow perfusion. M2 current voltage relations weredetermined by subtracting current at pHo 7.4, obtained during theramp phase of the voltage protocol, from the activated M2 currents atpHo 5.6 or from current after exposure to compound 9 or amantadineat pHo 5.6. Reversal potentials (Erev) were estimated by linearinterpolation of data points of the I–V relation on each side of zerocurrent.
Molecular Docking. Blind docking was carried out using Auto-Dock4.2 software (Morris et al., 1998) using the default parameters,the Lamarckian genetic algorithm with local search, and 25 millionenergy evaluations per run; 150 runs were performed for 9, 120 runsfor 26, and 100 for 27. The solution nuclearmagnetic resonance (NMR)structure ofM2 (A/HongKong/156/97; PDB 2LY0) (Wang et al., 2013b)with N-((5-(thiophen-2-yl)isoxazol-3-yl)methyl)adamantan-1-amine(M2WJ332) removed, was used as the receptor. Autodock4.2 allowsfor 32 flexible bonds to bemodeled; twowere used for compound 9, sixfor compound 26, and seven for compound 27. The remaining bondswere used to allow flexibility of the side chains of Val27, Ser31, andHis37. The remaining residues of the channel were held rigid duringthe docking process. The grid box size in initial runs were 20.25 � 30�20.25 Å in the x, y, and z dimensions, which covered Ser23 to Arg45 ofthe crystal structure, and was expanded to 17.62 � 45 � 18.75 Å insubsequent runs to include Asp21 to Lys49, both centered around theinner pore. Figures were generated using PyMOL (Schrödinger, Inc.,NY) with the PyMOL Autodock Plugin (Seeliger and De Groot,2010).
Cytotoxicity. The tsA-201 cells were assessed for cell viabilityusing the MTT reagent kit (Life Technologies). Cells were plated in96-well plates (2.5 × 104 cells / well) and incubated for 24 hours,followed by treatment with drug for 24 hours. Cells were thenincubated with MTT reagent for 4 hours and treated with an equalvolume of 10% SDS 1 0.01 M HCl. Cell cultures were read byspectrophotometry at an absorbance wavelength of 570 nm (A570).A570 background values from wells without cells were subtracted fromtheA570 cell culture values and normalized to the averageA570 value ofcell cultures in the absence of compounds. Data were obtained from atleast three experiments and at least five concentrations.
Generation of Amantadine-Sensitive and -Resistant Influ-enza Viruses. Recombinant influenza A viruses were generatedusing the reverse genetic system based on the A/Puerto Rico/8/34(PR8) strain (Neumann et al., 1999) provided by Dr. Y. Kawaoka(University of Wisconsin, Madison). The endogenous M2 sequence ofPR8 encodes both a threonine at position 27 (T27) and asparagine atposition 31 (N31) that render amantadine resistance. Amantadine-sensitive PR8 viruses were therefore generated by mutating T27 andN31 to valine (V27) and serine (S31), respectively. Amantadine-resistant M2 encoding V27 and N31 was also generated. Nucleotidesubstitutions were introduced by modifying the sequence of pPol-PR8-MG-M (one of the 12 plasmids that make up the reverse geneticssystem) using two-step overlap extension polymerase chain reaction(PCR) and cloning of subsequent DNA fragments into the pHH21vector. A DNA fragment containing the S31 point mutation wasgenerated using the primers TSH289, TSH293, TSH294, and TSH292(Supplemental Table 1) and pPol-PR8-HG-M (M2 T27 1 N31) as thetemplate. The PCR fragment and pHH21 vector were then treatedwith restriction endonuclease BsmBI and ligated to generate pPol-PR8-HG-M-V27-N31 (M2 V271N31). A DNA fragment encoding V27and S31 was generated using the same primers and pPol-PR8-HG-M-A27-S31 (M2A271S31) as the template, which in turnwas generatedusing primers TSH289, TSH290, TSH291, and TSH292 (Supplemen-tal Table 1) and pPol-PR8-HG-M (M2 T27 1 N31) as the template,resulting in generation of pPol-PR8-HG-M-V27-S31 (M2 V27 1 S31).PCRs were performed using the Expand High Fidelity PCR System(Roche), and all constructs were confirmed by sequencing usingprimers TSH284 and TSH285 (Supplemental Table 1). Viruses weregenerated by transfecting one of the recombinant pPol-PR8-HG-Mplasmids with the other 11 plasmids of the reverse genetics system to293FT cells (Life Technologies). Cells were plated in a six-well plateand transfectedwith the plasmidmixture (250 ng each) using TransITLT-1 (Mirus). Culture supernatants were collected 48 hours aftertransfection and passed through a 0.45-mm filter. After propagatingvirus in Madin-Darby canine kidney (MDCK) cells for three to fourpassages, virus stocks were stored at 280°C.
Viral Cytopathic Assay. MDCK cells were cultured in Dulbecco’sMEM (Life Technologies) plus 10%heat-inactivated fetal bovine serum(Hyclone), 100 U/ml penicillin, and 100 mg/ml streptomycin (LifeTechnologies) except during generation of virus stocks and plaqueassays, when serum was removed from the media. Plaque reductionassays were carried out in triplicate as described (Song et al., 2005)with minor modifications. Briefly, approximately 100 plaque-formingunits of either PR8 recombinantwithM2V271S31 (PR8M2(WT)) or onewith M2 V271N31 (PR8M2(S31N)) mixed with different concentrationsof the compounds, ranging from 100 mM to 1 mM, and inoculated onconfluent MDCK monolayers in six-well plates. After a 1-houradsorption at 37°C, the inocula were removed, and cells were washedtwice with phosphate-buffered saline (PBS). The cells were thenoverlaid with Dulbecco’s MEM containing 1% Noble agar (Affymetrix,Santa Clara, CA), 10 mM HEPES buffer (Lonza, Ververs, Belgium),0.00075%Difco Trypsin 250 (BD Biosciences, San Jose, CA), 100 U/mlpenicillin, 100 mg/ml streptomycin, and each compound at test con-centration. After incubation in 5%CO2 at 37°C for 3 days, plaqueswerevisualized and counted by staining the cells with 0.01% Neutral Red(Sigma-Aldrich). The concentration that reduces plaque number by50% compared with the dimethylsulfoxide control (EC50) was calcu-lated by regression analysis of the dose–response curves.
ResultsDetection of M2 Currents by Whole-Cell Patch-
Clamp Electrophysiology. We initially investigated base-line M2 ion channel activity by cotransfecting tsA-201cells with plasmids encoding GFP and M2(WT) (A/HongKong/1073/99(H9N2)) and recording pH-dependent ion cur-rents in GFP-positive cells by whole-cell patch-clamp
82 Jalily et al.
at ASPE
T Journals on July 23, 2020
molpharm
.aspetjournals.orgD
ownloaded from

electrophysiology (Fig. 2; Chizhmakov et al., 1996; Kolocouriset al., 2014). Cells were held at a constant membranepotential of 240 mV, and currents were recorded every4 seconds by applying 100-millisecond pulses to 280 mV.When M2-expressing (i.e., GFP-positive) cells were incubatedin solution at pHo 7.4 (designated by the horizontal black linesabove each panel in Fig. 2), minimal negative or inwardcurrent was observed (,10 pA), which was normalized to0.0 pA after all leak and initial current changes had settled.In contrast, exposure to pHo 5.9–5.5 (designated by thehorizontal white rectangles above each panel) resulted inan initially large inward current that decayed ∼50% duringcontinued exposure to low pHo. Return to pHo 7.4 caused thesteady inward current to deactivate. Subsequent repeatedexposures to extracellular acid solutions reversibly activatedthe relatively constant level of inward current (Fig. 2A),consistent with previous observations (Pinto et al., 1992;Kolocouris et al., 2014). Currents were also detected in cellstransfected with M2(WT) lacking the N-terminal FLAGepitope (data not shown) but not in cells transfected with onlyGFP control vector (Supplemental Fig. 2).Also consistent with previous reports (Pinto et al., 1992;
Chizhmakov et al., 1996), extracellular administration ofamantadine inhibited M2(WT) currents at acidic pHo in adose-dependent manner (Fig. 2B), indicating that this pro-tocol, which differs in cell type, M2 strain, and pulse protocolfrom previous electrophysiology studies (Pinto et al., 1992;Chizhmakov et al., 1996), could be used to screen test agentsfor M2 current inhibition. No obvious differences were ob-served at pHo 5.5–5.9 in efficacy or rate of M2 inhibition byamantadine and subsequent compounds (data not shown). Forall compounds tested in this study, we obtained dose-responserelationships if $50% inhibition of M2 current was observedat 100 mM. For amantadine, we determined an IC50 of 0.6 60.2 mM (Table 1).Using this protocol, we then screened four acylguanidine-
containing molecules (Fig. 1B; Table 1). The most potentcompound was HMA, which blocked M2(WT) currents with anIC50 of 1.36 0.3mM(Fig. 2C), followed byEIPA, which blocked
M2(WT) currents with an IC50 of 526 8 mM. In contrast, bothBIT-225 and BIT-314 showed only modest activity againstM2(WT) (30% 6 5% and 14% 6 2% inhibition of M2(WT) at100 mM, respectively) and were not explored further.Synthesis of a Novel HMA Derivative with Improved
Activity against M2(WT). As HMA exhibited the mostactivity of the acylguanidines tested against M2(WT), we nextaskedwhich functional groups of HMAwere responsible for itsinhibitory activity. To test initially the role of central ringsubstitutions, we began by replacing the central pyrazine witha phenyl ring to generate compound 1. Despite removal of thering nitrogens, the exocyclic amino groups and the chloroatom1 exhibited improved activity against M2(WT) (IC50 5 0.4 60.1 mM). In contrast, an analog of 1 with complete removalof the seven-membered azepane ring (1-benzoylguanidine)had almost no activity (,10% block; Table 1), indicating that adistal moiety is required. We did find that substitution of theazepane ring with cyclohexane was somewhat tolerated foranti-M2(WT) activity (e.g., 4; IC50 5 5.0 6 1.0 mM); however,this activity was sharply reduced when the cyclohexane wasreplaced with a more distally polar morpholine ring (6; 33%63% inhibition at 100 mM). Moreover, substitution with apyrrolidine, benzene, or methoxybenzene was also less toler-ated (IC50 5 506 10 mM for 5; 33% inhibition at 100 mM for 2and 3). Furthermore, the residual activity of 2 was abolishedwhen the distal ring was shifted to the ortho or meta positionof the central ring relative to acylguanidine (i.e.,,10%block ofM2(WT) by 100 mM of compounds 7 or 8), emphasizing theimportance of ligand linearity and consistent with our obser-vations of limited anti-M2(WT) efficacy of nonlinearmoleculesBIT-225 and BIT-314 (Table 1).Wenext substituted a single phenyl carbon of 1withnitrogen
and synthesized compound 9. Like 1, compound 9 was alsohighly potent and inhibited M2(WT) current at pHo 5.5 (Fig.3A), with an IC50 of 0.2 6 0.1 mM (Table 1). In contrast, noinhibition of pH-dependent currents from theM2 ion channel ofinfluenza B (B/Lee/1940) (Mould et al., 2003) was observedwith up to 100 mM of 9 (Fig. 3B), demonstrating that theinhibitory activity of 9 is specific toM2 encoded by influenza A.
Fig. 2. Representative diary plots ofpH-dependent currents detected at 280 mVfrom single tsA-201 cells transiently coex-pressing GFP and M2(WT) [A/HongKong/1073/99 (H9N2)]. (A) RepresentativeM2 current. Dots denote currents recordedat280 mV every 4 seconds. In the presence ofpHo 7.4 solution (denoted by thin black linesabove the traces), minimal inward or negativecurrent is observed. When pHo 5.9 solution isapplied (denoted by white bars above thetraces), an initially large inward current isobserved, which eventually normalizes and isreversible with repeated pulses of pHo 7.4 orpHo 5.9 solution. H9N2 M2(WT) currents aredose dependently inhibited by amantadine(B), and HMA (C). Data from each panel arerepresentative of at least three independentexperiments.
Mechanisms of Action of Novel Influenza A/M2 Viroporin Inhibitors 83
at ASPE
T Journals on July 23, 2020
molpharm
.aspetjournals.orgD
ownloaded from
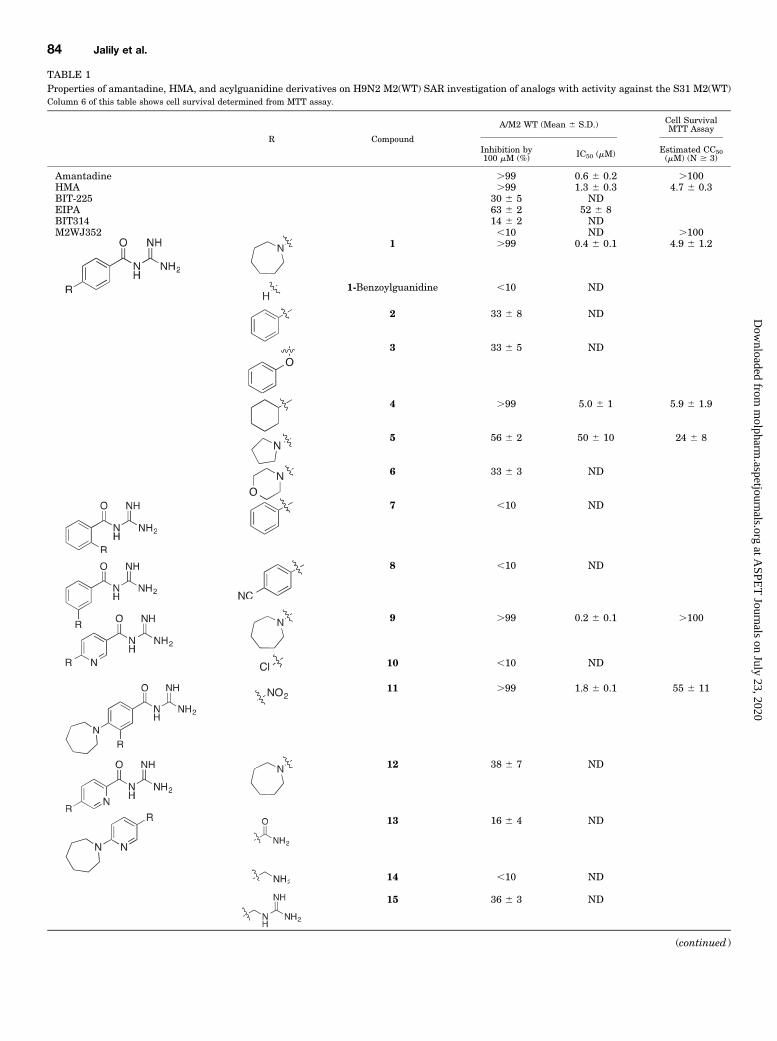
TABLE 1Properties of amantadine, HMA, and acylguanidine derivatives on H9N2 M2(WT) SAR investigation of analogs with activity against the S31 M2(WT)Column 6 of this table shows cell survival determined from MTT assay.
R Compound
A/M2 WT (Mean 6 S.D.) Cell SurvivalMTT Assay
Inhibition by100 mM (%) IC50 (mM) Estimated CC50
(mM) (N $ 3)
Amantadine .99 0.6 6 0.2 .100HMA .99 1.3 6 0.3 4.7 6 0.3BIT-225 30 6 5 NDEIPA 63 6 2 52 6 8BIT314 14 6 2 NDM2WJ352 ,10 ND .100
1 .99 0.4 6 0.1 4.9 6 1.2
1-Benzoylguanidine ,10 ND
2 33 6 8 ND
3 33 6 5 ND
4 .99 5.0 6 1 5.9 6 1.9
5 56 6 2 50 6 10 24 6 8
6 33 6 3 ND
7 ,10 ND
8 ,10 ND
9 .99 0.2 6 0.1 .100
10 ,10 ND
11 .99 1.8 6 0.1 55 6 11
12 38 6 7 ND
13 16 6 4 ND
14 ,10 ND
15 36 6 3 ND
(continued )
84 Jalily et al.
at ASPE
T Journals on July 23, 2020
molpharm
.aspetjournals.orgD
ownloaded from
Further structure activity studies were performed using 9.With respect to the central ring, replacing the pyridyl nitrogenwith an exocyclic nitrogen in the form of a nitro group (11) alsolargely retained activity against M2(WT) (IC50 5 1.8 6 0.1mM), but shifting the central pyridyl nitrogen to the orthoposition relative to the acylguanidine moiety (12) substan-tially reduced activity (38% 6 7% inhibition at 100 mM).Similar to the compound 1 to 1-benzoylguanidine transition,removal of the distal seven-membered azapane ring of 9 alsoeliminated activity (10). Moreover, removal of the guanidi-nium and/or carbonyl groups of the acylguanidine moiety (13,14, 15) completely abolished or strongly reduced M2(WT)current inhibition at 100 mM (e.g., maximum 36% 6 3%inhibition for 15; Table 1).Compound 9 exhibited the most activity against M2(WT)
currents, with almost an order of magnitude improved efficacyover the parent compound HMA. Moreover, and consistentwith the docking model described as follows, molecule linear-ity combined with the presence of acylguanidine and distalring moieties were necessary for M2(WT) activity, althoughsome modifications to the central and distal rings weretolerated.Effects of Compound 9 on M2(WT) Current-Voltage
Relationships. We were interested in understanding thevoltage dependence of 9 effects on pure M2 conductance inmammalian cells and so carried out voltage clamp experi-ments in LM cells expressing M2(WT) protein. H1 whole-cellcurrents were measured in the absence of other monovalentcations (Na1 and K1) using pipette and external mediathat contained only impermeant organic ions N-methyl D-glucamine(NMDG1) and HEPES- or MES-. Consistent withresults obtained from tsA-201 cells, in LM cells transfectedwith M2(WT), a change in pHo from 7.4 to 5.6 induced aninward current at 280 mV that was sensitive to both 9 (Fig.4A) and amantadine (Supplemental Fig. 3). Current-voltagerelationships obtained during the ramp phase of the protocol
between280 and1120mV in pHo 7.4, 5.6, and at pHo 5.6 plus100 mM 9 are shown in Fig. 4B. It can be seen that all threerelationships cross close to 180 mV. Subtraction of therelationship obtained at pHo 7.4, which we assume containslittle M2 current, from those at pHo 5.6 places the reversalpotential of the pHo 5.6 relations on the zero current axis andgives an Erev of 180 mV (Fig. 4C), which is close to thepredicted reversal potential of 192 mV for a pure, M2-dependent H1 current. Similarly, subtraction of the tworelationships obtained at pHo 5.6 gave the 9-sensitive current(Fig. 4D), which showed an Erev of185 mV, again close to thatpredicted for a pure H1 current. The S.D. of the data pointsaround the best-fit line through the current-voltage relationsat their reversal potentials was 68 mV, indicating the overallerror in estimating Erev. These results suggest that loweredpHo activates a relatively pure M2H1 current and that 9 (Fig.4) and amantadine (Supplemental Fig. 3) both directly inhibitM2 H1 currents in these cells.Molecular Modeling of M2(WT) Block. To gain insight
into how 9, 26, and 27 (described below)might bind and inhibitthe M2 channel, we performed molecular docking analyses(Morris et al., 1998; Huey et al., 2007; Seeliger and De Groot,2010) with the M2 transmembrane domain tetramer (PDBentry 2LY0, the NMR structure of residues 19–49 of M2 ofA/Chiba/5/71(H3N2) in dodecylphosphocholine micelles, com-putationally modified to include S31 when necessary; Wanget al., 2013b). In addition, we ran a compound that did notblock in electrophysiologic studies to observe the predictionsmade by the program. Because of the constraints of theAutoDock 4 software that limit the number of flexible bondsin the ligand plus the receptor to 32 for accuracy (Trott andOlson, 2010), the M2 structure was largely held rigid duringdocking simulations, with the exception of three flexibleresidues per tetramer subunit (Val27, Ser31 or Asn31, andHis37). These residues were selected based on 9’s interactionsduring a trial molecular docking simulation where M2(WT)
TABLE 1—Continued
R Compound
A/M2 WT (Mean 6 S.D.) Cell SurvivalMTT Assay
Inhibition by100 mM (%) IC50 (mM) Estimated CC50
(mM) (N $ 3)
26a 20 6 5 ND 25 6 527a .99 0.6 6 0.1 55 6 1728a ,10 ND .100
CC50, 50% cytotoxic concentration; HMA, hexamethylene amiloride; ND, not determined; R, substitution.aStructures are shown in Table 2.
Fig. 3. Effects of increasing concentra-tions of compound 9 on currents from tsA-201 cells expressing (A) M2 (H9N2)(WT).(B) B/M2. As for Fig.2, these are diary plotsof currents measured at 280 mV every4 seconds.
Mechanisms of Action of Novel Influenza A/M2 Viroporin Inhibitors 85
at ASPE
T Journals on July 23, 2020
molpharm
.aspetjournals.orgD
ownloaded from

A B
C D
Fig. 4. Current-voltage relations of H+-activated current and block by 9. (A) Diary plot of M2 (WT) current against protocol pulse number. Cells werepulsed every 6 seconds from a holding potential of 240 mV, first to 80 mV for 200 milliseconds and then ramped for 600 milliseconds to +120 mV, a400-millisecond repolarization to 0 mV, and then back to240 mV. A current reading was taken at280 mV and plotted against pulse number. Cells wereexposed to pHo 7.4 solution twice as denoted by the thin black bar above the graph, to pHo 5.6 during the periods denoted by the thick bars, and also to pHo5.6 + 100 mM compound 9 (gray thick bar). Current voltage relations obtained under the three conditions at the times indicated in (A) by the symbols areplotted in (B). All three relationships showmild inward rectification but cross at ∼+80 mV. (C) Current activated at pHo 5.6, before and in the presence of100mMcompound 9, obtained by subtracting current at pHo 7.4. (D) Compound 9-sensitive current only, obtained by subtracting relationships in pHo 5.6,before and after compound 9 exposure, Erev = +85 mV.
86 Jalily et al.
at ASPE
T Journals on July 23, 2020
molpharm
.aspetjournals.orgD
ownloaded from

Fig. 5. Mechanism of inhibition by 9. (A–C) Models of 9 interacting with the transmembrane pore of M2(WT) (PDB entry 2LY0; residues 19–49 of M2 ofA/Chiba/5/71(H3N2)) (Wang et al., 2013b). Illustrated as a stepwise process, (A) the compound first encounters the turret but does not usually block at thisstage; (B) the seven-membered azepane ring passes through the narrow ring created by valines at position 27, (C andD) and then the rest of the compoundfollows to bind within the inner vestibule. Predicted binding energies were 215.06 kcal/mol (A), 212.54 kcal/mol (B), 214.61 kcal/mol (C and D). (D)Interacting residues of M2 are shown in stick format; the rest of M2 is in line format. The –A or –D after each numbered residue in this and later figuresdenotes which of the four tetramer subunit fromwhich the amino acid originates. Only interacting residues are labeled. Hydrogens are hidden for clarity.Figure was created using AutoDock and PyMOL with the PyMOL Autodock Plugin (Seeliger and De Groot, 2010). (E–H) Plots of normalized H9N2M2(WT) currents in cells exposed to extracellular pH 5.5 solution with the addition of either amantadine (E), 9 (F), amantadine added in presence of 9 (G),or amantadine followed by and in the presence of 9 (H). (E–H) Data are representative of four independent experiments.
Mechanisms of Action of Novel Influenza A/M2 Viroporin Inhibitors 87
at ASPE
T Journals on July 23, 2020
molpharm
.aspetjournals.orgD
ownloaded from

TABLE 2Properties of acylguanidine derivatives on H9N2 M2(S31N)SAR investigation of analogs with activity against the N31 mutant. In column 5, where an IC50 has been measured it is explicitly stated. Last column of table shows cellsurvival determined from MTT assay.
R X Compound A/M2 S31N (Mean 6 S.D.)Inhibition by 100 mM (%)
Cell SurvivalMTT Assay
Estimated CC50(mM) (n $ 3)
Amantadine 24 6 1 .100HMA 10 6 3 4.7 6 0.3M2WJ352 75 6 5 .100
IC50 = 44 6 49a ,10 .10011a 10 6 8 55 6 11
16 16 6 4
17 10 6 5
5 ,10
6 ,10
— 18 ,10
— 19 ,10
20 ,10
21 ,10
22 17 6 8
23 ,10
24 ,10
25 33 6 15 58 6 11
26 60 6 7IC50 = 42 6 5
25 6 5
27 .99IC50 = 4.4 6 0.5
55 6 17
—28 32 6 5 .100
(continued )
88 Jalily et al.
at ASPE
T Journals on July 23, 2020
molpharm
.aspetjournals.orgD
ownloaded from

was held entirely rigid, in addition to a visual scan for residueswith side chains that were most obviously pointing into theinner vestibule and could provide steric hindrance if left rigid.Using these parameters, we identified several binding sites
for 9 (Fig. 5, A–D). Many of the lowest energy bindingconformations were in the turret (Fig. 5A), and many did notactually block the channel, suggesting this might not be theblock site. This result is consistent with the fact that 9 does notblock N31 (Table 2 and described as follows), and the twoshare identical sequence in the turret. Autodock also predictedin some conformations that the hydrophobic seven-memberedazepane ring of 9 could pass through the constriction createdby the valines at position 27, whereas the acyl guanidineportion maintained polar interactions with Ser22 and Asp24,creating an effective plug of the pore (Fig. 5B). Much of 9 blockis readily reversible upon washout (Fig. 5F), and this bindingconfiguration might explain this reversibility since most of 9remains outside the inner vestibule. The 20% block that is notreversible might then be a result of full entry of the drug intothe inner vestibule (Fig. 5, C and D). Approximately 50% ofpredicted binding conformations were in the inner vestibule,but all these fell 1 or 2 kcal/mol higher in energy than mostturret binding conformations (213.61 to 211.85 kcal/mol).These could be with either the acyl guanidine pointing towardthe valines (Fig. 5, C and D) or toward the histidines atposition 37 (Supplemental Fig. 6) with similar bindingenergies. If the initial trajectory of the passage involves ahydrophobic interaction between the seven-membered aze-pane ring and the valines, this would favor an inner vestibuleorientation as shown in Fig. 5C (shown in more detail in Fig.5D). In this conformation, the seven-membered azepane ringis reaching down to interact with histidine 37 from twosubunits as well as the backbone of leucine 38. In addition,the azepane ring and the central pyridyl ring make hydropho-bic interactions with Gly34 and Ala30, respectively, and theacyl guanidine interacts with valines that define the roof of theinner vestibule (Fig. 5D).Interestingly, mutations at multiple M2 residues predicted
to interact with 9, including Val27, Ala30, and Gly34, lead to
adamantane resistance in vitro or in transmissible viruses(Hay et al., 1985, 1986; Belshe et al., 1988). Facets of thedocking approach, such as use of an empirical hydration forcefield for the drug binding in the water-filled lumen and use of amicellar protein structure and homology model thereof, re-quire a conservative interpretation confined to assessment ofthe steric fit of the drug in the pore. Nevertheless, takentogether, these observations further support that 9 inhibitsM2(WT), but potentially not M2(S31N), by a pore-blockingmechanism. That 9 does not block M2–N31 leads us tospeculate that the extra bulk and hydrophilicity of theasparagine side chain hinders, or it makes energeticallyunfavorable, the entry of the bulky hydrophobic 7-memberedazepane ring of 9 into the inner vestibule of the mutantchannel.As 9 and amantadine are predicted to interact with many of
the same M2 pore-lining residues, we next tested whether 9and amantadine compete for M2(WT) inhibition (Fig. 5, E–H).Treatment of M2(WT)-expressing cells with 10 mM amanta-dine at pHo 5.5 reduced pH-dependent currents by 63.5% (Fig.5E). After removal of amantadine from extracellular solution,however, little recovery of baseline M2(WT) current wasobserved after either at least 3.3 minutes at pHo 5.5 (Fig.5E) or iterative treatment with pHo 7.4 and pHo 5.5 solutions(data not shown), consistent with previous observations thatamantadine does not readily dissociate from blocked M2(WT)channels (Balannik et al., 2009, 2010). In contrast, although10 mM of 9 reduced M2(WT)-dependent currents by 80.5% inFig. 5F, these currents recovered 57.1% within 3.3 minutes ofremoval of 9, indicating reversible inhibition and rapid re-covery of M2(WT) currents. The different M2(WT) currentrecovery rates observed after removal of amantadine and 9from pHo 5.5 solution let us test which compound preferen-tially blockedM2 (WT) when applied in competition. As shownin Fig. 5G, when 10 mM of 9 was administered at pHo 5.5 toinhibit M2(WT) (90.5% inhibition in this example), no obviousfurther inhibition by 10 mM amantadine was observed. Afterremoval of both compounds, M2(WT) current recovered by58.0% after 5minutes, consistent with amodel whereM2(WT)
TABLE 2—Continued
R X Compound A/M2 S31N (Mean 6 S.D.)Inhibition by 100 mM (%)
Cell SurvivalMTT Assay
Estimated CC50(mM) (n $ 3)
— 29 11 6 0
— 30 ,10
— 31 ,10
— 32 ,10
33 ,10
CC50, 50% cytotoxic concentration; HMA, hexamethylene amiloride; R, substitution; SAR, structure activity relationship.aStructures are shown in Table 1.
Mechanisms of Action of Novel Influenza A/M2 Viroporin Inhibitors 89
at ASPE
T Journals on July 23, 2020
molpharm
.aspetjournals.orgD
ownloaded from
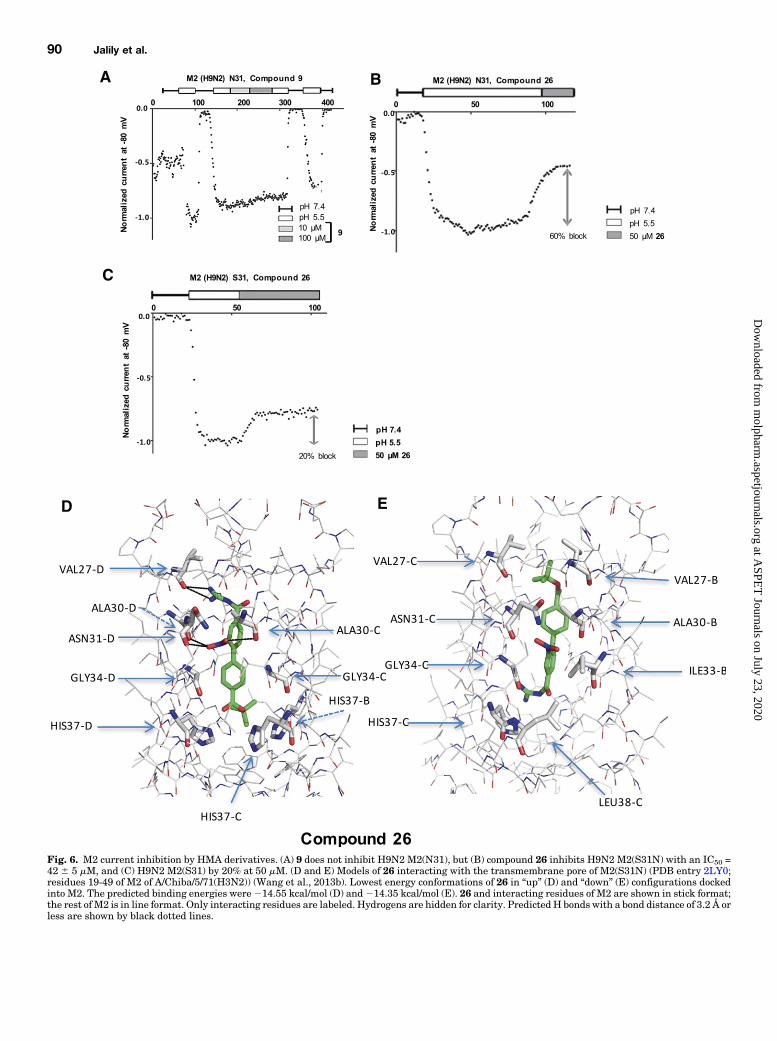
Fig. 6. M2 current inhibition by HMA derivatives. (A) 9 does not inhibit H9N2 M2(N31), but (B) compound 26 inhibits H9N2 M2(S31N) with an IC50 =42 6 5 mM, and (C) H9N2 M2(S31) by 20% at 50 mM. (D and E) Models of 26 interacting with the transmembrane pore of M2(S31N) (PDB entry 2LY0;residues 19-49 of M2 of A/Chiba/5/71(H3N2)) (Wang et al., 2013b). Lowest energy conformations of 26 in “up” (D) and “down” (E) configurations dockedinto M2. The predicted binding energies were214.55 kcal/mol (D) and214.35 kcal/mol (E). 26 and interacting residues of M2 are shown in stick format;the rest of M2 is in line format. Only interacting residues are labeled. Hydrogens are hidden for clarity. PredictedH bonds with a bond distance of 3.2 Å orless are shown by black dotted lines.
90 Jalily et al.
at ASPE
T Journals on July 23, 2020
molpharm
.aspetjournals.orgD
ownloaded from
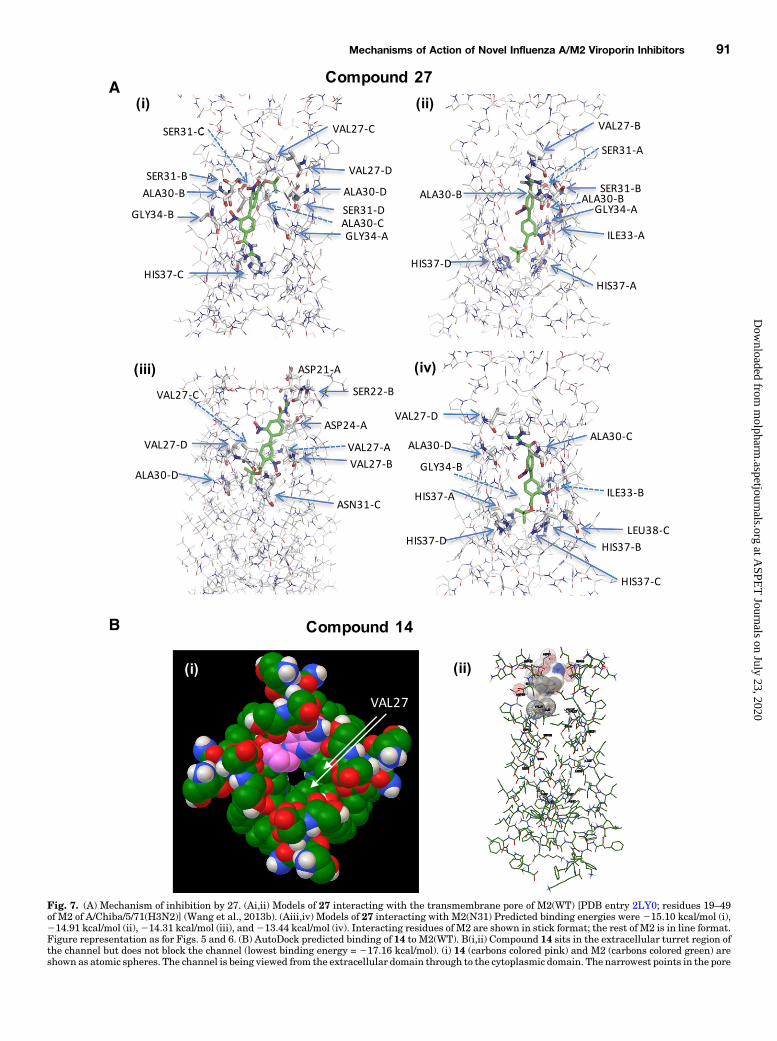
Fig. 7. (A) Mechanism of inhibition by 27. (Ai,ii) Models of 27 interacting with the transmembrane pore of M2(WT) [PDB entry 2LY0; residues 19–49of M2 of A/Chiba/5/71(H3N2)] (Wang et al., 2013b). (Aiii,iv) Models of 27 interacting with M2(N31) Predicted binding energies were 215.10 kcal/mol (i),214.91 kcal/mol (ii),214.31 kcal/mol (iii), and213.44 kcal/mol (iv). Interacting residues of M2 are shown in stick format; the rest of M2 is in line format.Figure representation as for Figs. 5 and 6. (B) AutoDock predicted binding of 14 to M2(WT). B(i,ii) Compound 14 sits in the extracellular turret region ofthe channel but does not block the channel (lowest binding energy = 217.16 kcal/mol). (i) 14 (carbons colored pink) and M2 (carbons colored green) areshown as atomic spheres. The channel is being viewed from the extracellular domain through to the cytoplasmic domain. The narrowest points in the pore
Mechanisms of Action of Novel Influenza A/M2 Viroporin Inhibitors 91
at ASPE
T Journals on July 23, 2020
molpharm
.aspetjournals.orgD
ownloaded from

was inhibited primarily by 9, and amantadine was largelyunable to dislodge it from the pore. These recovered M2(WT)currents were subsequently inhibited by 10 mM amantadine(data not shown), showing that removal of 9 from the poreallowed amantadine access. When 5 mM of 9 was used (Fig.5H), which resulted in 61% block, subsequent exposure to10mMamantadine caused a further reduction in current. Thissuggests that amantadine can access M2(WT) channels leftunblocked by the lower concentration of 9. Taken together,these experiments suggest that 9 and amantadine likelycompete for the same or highly overlapping binding sites toinhibit M2(WT).Activity of Compound 9 against Adamantane-
Resistant M2 Sequence. To assess the inhibitory potentialof 9 against mutant M2 viroporins, we examined inhibition ofpHo-dependent currents in tsA-201 cells expressing anA/Hong Kong/1073/99(H9N2) M2 sequence encoding S31N.pH-dependent currents from M2(S31N)-expressing cells wereidentical to those of cells expressing M2(WT) (Fig. 6). Consis-tent with this mutation conferring adamantane resistance(Balannik et al., 2010; Williams et al., 2013), we observed thatM2(S31N) was inhibited only 24% 6 1% by 100 mM amanta-dine and 10% 6 3% by 100 mM HMA in patch-clamp experi-ments (Table 2). Similarly, sequential exposure to 10 and100 mM of 9 at pHo 5.5 had almost no activity onM2(S31N), asshown in Fig. 6A and Table 2 (,10% inhibition).Discovery of HMA Derivatives with Activity against
M2(S31N). M2(S31N) is the most prevalent adamantane-resistant mutation observed in transmissible influenza(Bright et al., 2005), so we next asked if derivatives of 9could inhibit M2(S31N). As S31N affects both the polarityand the size of the hydrophobic adamantane binding site(Wang et al., 2011; Williams et al., 2013), we hypothesizedthat modifications to the hydrophobic terminus of 9 incorpo-rating more polar groups might improve M2(S31N) inhibi-tion, perhaps by stabilizing proximal or distal interactionswithin the M2(S31N) pore. We first measured whetherM2(S31N) could be inhibited by compounds 6, 16, 18, and20, all of which contain polar substituents in the distal ring;however, these compounds had little or no activity at 100 mM(e.g., maximum 16% 6 4% inhibition for 16; Table 2).Furthermore, 100 mM 1 and 11 also had limited or no activityagainst M2(S31N) (e.g., maximum 10% 6 1% inhibition for11; Table 2), indicating that these central ring modifications,on their own, did not substantially improve activity againstM2(S31N).We then assessed the combined effects of a more polar, tert-
butyloxycarbonyl (Boc)-conjugated distal piperazine (i.e., 16)with a polar nitro group on the central ring (i.e., 11). In-terestingly, this compound (28) inhibited M2(S31N) withslightly more than additive effects relative to 16 and 11(32%6 5% at 100 mM; Table 2), indicating that both the nitroand terminal Boc groups of the piperazine are needed forM2(S31N) inhibition; however, 28 did not inhibit M2(WT).This observation, combined with activity against M2(S31N),which is only slightly improved over amantadine, prompted us
to continue exploring 28 derivatives with improved M2(S31N)and M2(WT) activities.After our identification of 28 as an early lead for M2(S31N)
inhibition, we next found that substitution of the central ringnitro group with either a chlorine (23) or nitrile (22) reducedactivity against M2(S31N) (,10% and 17% 6 8% at 100 mM,respectively). Shifting of the nitro group to ortho-position withregard to the acylguanidine moiety (17) also reduced activity(10% 6 5% at 100 mM). Activity against M2(S31N) was alsoeliminated by increasing the size of the second ring frompiperazine (28) to homopiperazine (21), with inhibition lessthan 10% at 100 mM. Finally, compounds with different sizecarboxylates (29 and 32) and sulfonamide (31) did not exhibitpotent activity against M2(S31N) (maximum 11% 6 8% at100 mM); however, switching the Boc-conjugated piperazine(28) with a piperidine ring with a slightly more polar terminalgroup (25) resulted in a compound having the same level ofefficacy. Replacing the heterocyclic ring with an aromatic ring(26) substantially improved activity againstM2(S31N) (60%67% at 100 mM; IC50 5 42 6 5 mM; Fig. 6B). Unlike 28,compound 26 also had observable, albeit weak activity againstM2(WT) (20% 6 5% inhibition at 100 mM; Fig. 6C). Furthersubstitution of a polar nitro group into the distal ring resultedin compound 27, which had even more activity against bothM2(WT) and M2(S31N), with IC50s of 0.6 mM and 4.4 mM,respectively.In this way, we identified multiple derivatives of 9 that
inhibit M2(S31N) currents, with lead compounds 26 and 27inhibitingM2(S31N)with greatly improved activity overHMAand amantadine while retaining submicromolar M2(WT)activity in the case of 27. As whole-cell patch clamp electro-physiology and TEVC are not directly comparable, we synthe-sized M2WJ352 (Wang et al., 2013b), a potent adamantanederivative, and found its IC50 to be 44 mM (Table 2), some10-fold weaker than 27.Molecular Docking Studies of M2(S31N) Block by
26 and 27. When molecular docking studies were performedwith 26 within the M2(S31N) pore, two confirmations withinverse orientations were again observed to interact withVal27, Ala30, Gly34, His37, and Leu38, alongwith Asn31 (Fig.6, D and E); however, similar to 9, the orientation of theacylguanidine differed by interacting with either one or moreof the valines at position 27 (Fig. 6D) or by interacting with thehistidines of the proton sensor (Fig. 6E). Both the guanidineamines and the oxygens of the nitro-substituted phenyl ringcould form hydrogen bonds, as shown in Fig. 6D. Also, in someconformations (still within 2.0 RMSD), the oxygens of the Bocterminal group contributed to hydrogen bondingwithHis37 orAsn31 (data not shown). Taken together, in comparison with9, the longer molecule of 26, with added bulk and polarity ofboth the nitro and Boc group, may help stabilize interactionsacross more of the pore, thereby improving block againstM2(S31N).Whereas 26 does not bind in the turret, the variety of
conformations observed for this compound was no less im-pressive than for 9 (Supplemental Fig. 4). The compound could
are the valines at position 27 (indicated by arrows for two subunits) and the histidines at position 37 that create the narrowing in the distance. (Bii) Sameconformation as in (i). This time, residues of the channel interacting with 14 are represented bymesh spheres, and the rest of the channel is in stick form.Labeled residues are those that are flexible (Val27, Asn31, and His37) for orientation purposes, as well as those interacting with 14.
92 Jalily et al.
at ASPE
T Journals on July 23, 2020
molpharm
.aspetjournals.orgD
ownloaded from

be found in an orientation with the acyl guanidine, pointingtoward the extracellular domain (Fig. 6D; Supplemental Fig.4B) or cytoplasmically (Fig. 6E; Supplemental Fig. 4A) and atvarying depths within theM2 pore. It is conceivable that sincethe lowest energy conformations are deeper in the pore (Fig. 6,D and E; Supplemental Fig. 4A, B, far left), the otherconformations are intermediate states of the compound as itprogresses to a preferred binding site, although all of theconformations lead to block. That 26 does not wash out uponsolution change (data not shown) leads one to speculate thatonce at least the bulk of the drug is past the valine ring, it isdifficult to reverse the entry process.The binding patterns for 27 were fairly similar whether
there was a serine at position 31 or an asparagine. Averagedover many runs for each channel construct, Val27 wasoccupied ∼67% of the time, Ala30 ∼47%, Ser/Asn31 ∼27%,and Gly34 ∼16%. Again, compound 27 was found to bind thechannel in “Boc up” or “Boc down” conformations. WhenSerine was present at position 31, the lower-energy clusterwas Boc up (Fig. 7Ai); Supplemental Fig. 5A), although 63% ofthe runs fell into one of the Boc down conformations. Whenasparagine was present, the lower energy clusters were Bocdown, and 54% of runs fell into these two clusters (Fig. 7Aiii,iv); Supplemental Fig. 5B). The lowest-energy conformationfound in M2(N31) was almost a kcal/mol more than the lowestenergy conformation found when serine was present, which isconsistent with the electrophysiologic data showing 27 to bealmost 10 times more effective in blocking the WT channel(Tables 1 and 2).The nitro-substituted phenyl rings tended to interact with
the hydrophobic wall of the inner vestibule, Val27, Ala30,Ile33, and Gly34. The nitro groups on these phenyl ringsinteracted with serine or asparagine and even histidine if thedrug bound low enough in the pore, and occasionally hydrogenbonds were predicted (Fig. 7Aiv). When 27 bound residues inthe turret, hydrogen bonds were much more common betweenthe acyl guanidine hydrogens and the oxygens of Asp21,Ser22, Ser23, and Asp24. The Boc group made hydrophobicinteractions, depending on the depth and orientation, with thecarbons in the histidine side chains (Fig. 7Aii,iv), or withAla30 and Val27 (Fig. 7Ai,iii). Given that the Boc grouppresents a hydrophobic terminus, it is tempting to speculatethat it is this end that would pass through the valine ringmorereadily and that the Boc-down orientation might be favored.Adding the bulkier, more hydrophilic asparagine just belowthe valine might make entry slightly less favorable.Molecular Docking of Compound 14 onto M2. Com-
pound 14 is one that failed to blockM2(WT) at 100 mM (,10%,Table 1), and modeling frequently predicted that 14 wouldbind in the extracellular turret (Fig. 7B) without blocking thechannel with very low binding energy (217.16 kcal/mol).Interactions were made with Asp21, Ser22, Ser23, Asp24,and sometimes with one of the valines, often with the aminehydrogens forming hydrogen bonds with the aspartic acidoxygens. It is likely possible that more than onemolecule of 14could in fact bind in the turret given the 4-fold symmetry of thechannel. Even in this situation, given that the compound is offthe central axis of the pore, there would be no block. Where abinding site was found in the internal vestibule, the lower-energy conformations were almost 2 kcal/mol higher in energythan the turret site (215.41 kcal/mol; not shown). Migrationinto the inner vestibule would require the high-affinity turret
site to remain vacant to allow an additional molecule of 14 topass; 14 is a smaller molecule than 9, 26, and 27. It lacks theacyl guanidine of 9, 26, and 27 that provides bulk and lengthand the nitro groups bulking up the rings of 26 and 27. It islikely for this reason thatwhen 14 binds the channel, it can failto block the channel (Fig. 7B).In Vitro Cytotoxicity. Compounds that inhibited .50%
of M2(WT) or .30% of M2(S31N) currents at 100 mM wereassessed for cytotoxicity. Using a standard MTT-based cellmetabolic assay, no obvious effects were observed in tsA-201cells incubated for 24 hours with up to 100 mM amantadine,M2WJ352, or compound 9 (Table 1). In contrast, HMAinhibited growth of tsA-201 cells with a 50% cytotoxicconcentration of 4.7 6 0.3 mM. Thus, although HMA inhibitsM2(S31) currents with efficacy comparable to that of theestablished inhibitor amantadine and 9, it also has undesir-able cellular toxicity consistent with previous observations(Balgi et al., 2013). Intermediate levels of cytotoxicity betweenHMA and 9were observed for both inhibitors ofM2(S31N) andother 9 derivatives (e.g., 50% cytotoxic concentration 5 25 65 mM for 26, 55, 6 17 mM for 27; Table 2).In Vitro Activities against Influenza Viruses in a
Cytopathic Assay. As functional experiments were primar-ily performed up to this point on heterologously expressed M2ion channels, we next determined the effects of select com-pounds on replication of influenza A virus in vitro using a viralcytopathic assay with MDCK cells (Table 3). Here, we used apreviously described reverse genetic system based on theA/Puerto Rico/8/34 (PR8) strain (Neumann et al.., 1999) togenerate PR8 strains encoding M2 with exclusively S31(PR8M2(WT), containing both V27 and S31) or N31 (PR8M2(S31N)
containing V27 and N31). As expected, amantadine exhibitedantiviral activity against PR8M2(WT) but not PR8M2(S31N) inthese assays EC50 ,1 mM for PR8M2(WT) vs. .100 mM forPR8M2(S31N); Table 3). Compound 9 also inhibited PR8M2(WT)
(EC50 5 2.3 6 0.1 mM), consistent with results from patch-clamp studies using M2(WT). Notably, 9 also exhibited ∼10-fold weaker activity against PR8M2(S31N) (EC50 5 23 6 5 mM).As 9 did not inhibit M2(S31N) currents at up to 100 mM, theseobservations suggest that compound 9may have an additionalantiviral mechanism besides 2(WT) blockade. Althoughoff-target antiviral effects are frequently observed for multipleM2 inhibitors (Kolocouris et al., 2014), the disproportionateability of 9 to inhibit PR8M2(WT) relative to PR8M2(S31N)
replication is nevertheless consistent with its ability toexclusively inhibit M2(WT) in patch-clamp studies and further
TABLE 3Antiviral activities from viral cytopathic assay of selected derivatives ofacylguanidine against S31 and N31 forms of the A/PR/8/1934(H1N1) V27influenza virus
Compound
Viral Cytopathic Assay
Estimated EC50 (mM)
A/PR/8/1934(H1N1) V27
S31 N31
Amantadine ,1 .100M2WJ352 34 6 6 1.8 6 0.79 2.3 6 0.1 23 6 526 6.9 6 1.6 1.5 6 0.127 40 6 1 18 6 1
Mechanisms of Action of Novel Influenza A/M2 Viroporin Inhibitors 93
at ASPE
T Journals on July 23, 2020
molpharm
.aspetjournals.orgD
ownloaded from

suggests that the additional efficacy of compound 9 againstPR8M2(WT) relative to PR8M2(S31N) is due to M2 blockade.The previously described M2(S31N) inhibitor M2WJ352
(Wang et al., 2013b) inhibited PR8M2(S31N) with an EC50 51.8 6 0.7 mM, and with a ∼19-fold reduced activity againstPR8M2(WT) (EC50 5 34 6 6 mM). As M2WJ352 did not inhibitM2(WT) currents at up to 100 mM, these observations suggestthat M2WJ352, like 9, may have antiviral mechanisms inaddition to M2(S31N) blockade. Similarly, PR8M2(S31N) repli-cation was blocked by both 26 and 27 (EC50 vs. PR8M2(S31N) 51.56 0.1 mMand 186 1 mM, respectively) but weaker activityagainst PR8M2(WT) was also observed (EC50 vs. PR8M2(WT) 56.96 1.6 mM and 406 1 mM, respectively; Table 3). Althoughthe antiviral activity of 26 is consistent with electrophysiologystudies, antiviral and electrophysiology profiles of 27 arediscordant; thus, the antiviral activity of 27 observed here isunlikely to be due primarily to M2 blockade. Finally, noobvious cellular toxicities in MDCK cells were observed forany tested compound with the exception of 26, where clear celldeath was observed only at higher concentrations (100 mM).
DiscussionHere we showed that of four acylguanidine compounds
previously reported to act against multiple viroporins orviruses (Gazina and Petrou, 2012), HMA had the most potentefficacy against influenza AM2(WT) currents, as measured bywhole-cell patch-clamp electrophysiology. We then showedthat the HMA derivative 9 inhibits M2(WT) and has improvedefficacy over amantadine and HMA, whereas inhibition ofadamantane-resistant M2(S31N) can be achieved with otherderivatives of this chemical class, such as 26 and 27. Using acombination of electrophysiologic, molecular docking andstructure-activity relationship studies, we identified the trans-membrane pore of the M2 tetramer as the likely interactionsite of 9, 26, and 27, which leads toM2 current block in a regionthat overlaps the reported adamantane interaction site.Mechanisms of Block of M2(WT). The M2 protein can
transfer protons selectively across membranes with a H1
electrochemical gradient, a property consistent with its rolein modifying virion and trans-Golgi pH during virus infection(Sugrue et al., 1990; Alvarado-Facundo et al., 2015). In LMcells expressing the M2 protein, with pHi 7.2 and pHo 5.6, theI-V plots were similar in conductance and shape across alltransfected cells. The inhibitory effect of amantadine (Sup-plemental Fig. 3) and 9 (Fig. 4) brought about an identicalcompression of the current across all voltages, whereas thepH- and drug-dependent currents throughM2 demonstrated areversal potential close to the equilibrium potential for thetransmembrane pH gradient at pHo 5.6, suggesting that 9 andamantadine inhibited proton currents similarly in our exper-iments. Our initial molecular docking studies predicted that,in the lowest-energy state, 9 interacts within theM2(WT) porein two different orientations (Fig. 5; Supplemental Fig. 6) in amanner reminiscent of the previously described orientationsof rimantadine and amantadine. Whereas alternative modelsof 9-M2(WT) interactions are clearly possible, our model issupported by the competition of 9 with amantadine forM2(WT) inhibition (Fig. 5), similar effects on current-voltagerelations, and the inability of 9 to inhibit currents fromadamantane-resistant M2(S31N) (Fig. 6A). Our structure-activity relationship study also identifies distinct moieties of
9 that are required to maintain anti-M2(WT) activity. Forexample, derivatives of 9 lacking portions of the acylguanidinegroup (e.g., 13, 14, and 15) were less effective in inhibitingM2(WT), whereas removal of the azepane ring (compounds 10and 1-benzoylguanidine) or substitution with smaller rings(compounds 2, 3, 4, 5, and 6) did not improve on the activity of9 (Table 1). A linear molecule was also required for activity, asshifting of the distal ring to either ortho or meta positionrelative to the acylguanidine also eliminated activity (7, 8).Some, but not all, compounds with substitutions in the centralpyridyl ring maintained activity against M2(WT) (e.g., 1 and11), but 9 retained themost potent inhibition ofM2(WT) of thenovel compounds tested here; however, 9 was ineffective inblocking the adamantane-resistant M2(S31N) variant. Wethen envisioned that the polarity changes brought about bythe S31Nmutation would require amolecule with both amoredistal polar end, as well as a polar substitution on the centralring, and 28 bearing a Boc distal end and a nitro-substitutedcentral ring appeared to bind and block the S31N mutant.Mechanisms of Block of M2(S31N). Compared with
amantadine, 28, and 25 somewhat improved blockade ofcurrents from adamantane-resistant M2(S31N) (32%–33%inhibition at 100 mM). We did, however, identify two otherHMA derivatives, 26 and 27, of which 26 showed improvedpotency against M2(S31N) with an IC50 of 42 6 5 mM andweak activity against M2(WT), whereas 27 exhibited a potentdual inhibitory effect against both M2(WT), as well asM2(S31N), with an IC50 of 4.4 6 0.5 mM and 0.6 6 0.1,respectively, as measured by whole-cell patch clamp (Tables 1and 2). Autodock predicts binding of 26 in S31 that would leadto block, and so the failure to see significant block in theelectrophysiologic experiments suggests that forces drivingthe drug into the pore are different between S31 and N31. Theaddition of the second set of oxygens on the second ringstructure in 27 appears to overcome this. The overall orien-tation of 26 and 27withM2(S31N) is, to a first approximation,similar to that of 9withM2(WT); 26, 27, and 9 are predicted toenter their binding site in two orientations to interact variablywith both the selectivity filter (Val27) and the proton sensor(His37) (Fig. 5, A–D and Fig. 6, D and E). The binding site for26 and 27 is predicted to span fromVal27 to His37 (Figs. 6 and7; Supplemental Figs. 4 and 5), which also overlaps to a degreewith the binding site previously reported by NMR for the lesspotent inhibitor M2WJ332 (Wang et al., 2013b). Notably, theacylguanidine carbonyl of 9, 26, and 27 may contribute to anH-bond with Ser31 in M2(WT) (for 9) or Val27 in M2(S31N)(for 26 and 27), and removal of this acylguanidine carbonyl, asseen in compounds 14 and 15, significantly reduced activityagainst M2(WT). Thus the ability of 26 and 27 to act onM2(S31N) appears to be driven primarily by the presence ofthe acylguanidine moiety, the second aromatic ring, anadditional distal Boc group, and the central ring nitro, whichincrease bulk and perhaps stabilize deeper interactions withinthe expanded M2(S31N) pore. The second aromatic ring of 26(compared with 28 with a piperazine ring) appears to play acritical role in further stabilizing hydrophobic interactionswith the walls of the amphipathic pore.Correlation of Viral and Electrophysiology Assays.
Interestingly, in viral assays (Table 3), both amantadine and9 displayed significant activity against PR8M2(WT), but notPR8M2(S31N) virus, supporting that 9 targets this virus, at leastin part, by inhibiting proton flux throughM2. In contrast, both
94 Jalily et al.
at ASPE
T Journals on July 23, 2020
molpharm
.aspetjournals.orgD
ownloaded from

M2WJ352 and 26 were more potent against PR8M2(S31N),further supporting their action against M2(S31N). We notethat the limited ability of 9 to inhibit PR8M2(S31N) and 26 toinhibit PR8M2(WT) electrophysiologically, but significant anti-viral activity against these isoforms, suggests that effects ofthe M2 block may be amplified in vitro or indeed that theseacylguanidine-based compounds may exhibit additional anti-viral mechanisms, as previously suggested for adamantanes(Kolocouris et al., 2014). Notably, compounds 9, 26, and 27also exhibited markedly reduced cytotoxicity compared withthe parent compound HMA in MTT assays with tsA-201 cells,further suggesting that these compounds may be improvedstarting points for future antivirals compared with HMA.In summary, we describe novel acylguanidine-containing
inhibitors of M2 viroporins with binding mechanisms similarto adamantanes, with potent activity against WT andadamantane-resistantM2 ion channels and viruses. The novelHMA derivatives, including compounds 26 and 27 as dualinhibitors of both M2(WT) and M2(S31N), provide scaffoldsthat may aid in development of further non-adamantanecompounds with improved inhibitory activity against drug-resistant forms of M2. These promising leads for additionalmedicinal chemistry optimization will also require furtherin vivo studies to assess their antiviral efficacy, stability, andpharmacokinetic parameters in animal disease models.
Acknowledgments
The authors thank Luping Yan, Hannah E. Boycott, Shunping Lin,Zhuren Wang, and Daniel Mendonca for superb technical assistance;Raymond Andersen and Luping Yan for assistance with massspectrometry; and Zabrina Brumme and Mark Brockman for use offacilities and critical review of the manuscript.
Authorship Contributions
Participated in research design: Jalily, Eldstrom, Miller, Tai,Niikura, Tietjen, Fedida.
Conducted experiments: Jalily, Eldstrom, Miller, Kwan, Tai, Chou,Niikura, Tietjen.
Preformed data analysis: Jalily, Eldstrom, Tietjen, Fedida.Wrote or contributed to the writing of the manuscript: Jalily,
Eldstrom, Tai, Tietjen, Fedida.
References
Alvarado-Facundo E, Gao Y, Ribas-Aparicio RM, Jiménez-Alberto A, Weiss CD,and Wang W (2015) Influenza virus M2 protein ion channel activity helps tomaintain pandemic 2009 H1N1 virus hemagglutinin fusion competence duringtransport to the cell surface. J Virol 89:1975–1985.
Balannik V, Carnevale V, Fiorin G, Levine BG, Lamb RA, Klein ML, Degrado WF,and Pinto LH (2010) Functional studies and modeling of pore-lining residue mu-tants of the influenza a virus M2 ion channel. Biochemistry 49:696–708.
Balannik V, Wang J, Ohigashi Y, Jing X, Magavern E, Lamb RA, Degrado WF,and Pinto LH (2009) Design and pharmacological characterization of inhibitors ofamantadine-resistant mutants of the M2 ion channel of influenza A virus. Bio-chemistry 48:11872–11882.
Balgi AD, Wang J, Cheng DY, Ma C, Pfeifer TA, Shimizu Y, Anderson HJ, Pinto LH,Lamb RA, and DeGrado WF et al. (2013) Inhibitors of the influenza A virus M2proton channel discovered using a high-throughput yeast growth restoration assay.PLoS One 8:e55271.
Belshe RB, Smith MH, Hall CB, Betts R, and Hay AJ (1988) Genetic basis of re-sistance to rimantadine emerging during treatment of influenza virus infection. JVirol 62:1508–1512.
Bright RA, Medina MJ, Xu X, Perez-Oronoz G, Wallis TR, Davis XM, Povinelli L, CoxNJ, and Klimov AI (2005) Incidence of adamantane resistance among influenza A(H3N2) viruses isolated worldwide from 1994 to 2005: a cause for concern. Lancet366:1175–1181.
Chizhmakov IV, Geraghty FM, Ogden DC, Hayhurst A, Antoniou M, and Hay AJ(1996) Selective proton permeability and pH regulation of the influenza virusM2 channel expressed in mouse erythroleukaemia cells. J Physiol 494:329–336.
Ewart GD, Luscombe CA, and Miller M (2009) inventors, Biotron Limited, assignee.Hepatitis c antiviral compositions and methods. U.S. patent WO2009018609A1.Aug 4, 2008.
Ewart GD, Mills K, Cox GB, and Gage PW (2002) Amiloride derivatives block ionchannel activity and enhancement of virus-like particle budding caused by HIV-1protein Vpu. Eur Biophys J 31:26–35.
Fiore AE, Fry A, Shay D, Gubareva L, Bresee JS, and Uyeki TM; Centers for DiseaseControl and Prevention (CDC) (2011) Antiviral agents for the treatment and che-moprophylaxis of influenza: recommendations of the Advisory Committee on Im-munization Practices (ACIP). MMWR Recomm Rep 60:1–24.
Gazina EV and Petrou S (2012) Viral targets of acylguanidines. Drug Discov Today17:1039–1043.
Hay AJ, Wolstenholme AJ, Skehel JJ, and Smith MH (1985) The molecular basis ofthe specific anti-influenza action of amantadine. EMBO J 4:3021–3024.
Hay AJ, Zambon MC, Wolstenholme AJ, Skehel JJ, and Smith MH (1986) Molecularbasis of resistance of influenza A viruses to amantadine. J Antimicrob Chemother18(Suppl B):19–29.
Huey R, Morris GM, Olson AJ, and Goodsell DS (2007) A semiempirical free energyforce field with charge-based desolvation. J Comput Chem 28:1145–1152.
Khoury G, Ewart G, Luscombe C, Miller M, and Wilkinson J (2010) Antiviral efficacyof the novel compound BIT225 against HIV-1 release from human macrophages.Antimicrob Agents Chemother 54:835–845.
Kleyman TR and Cragoe EJ, Jr (1988) Amiloride and its analogs as tools in the studyof ion transport. J Membr Biol 105:1–21.
Kolocouris A, Tzitzoglaki C, Johnson FB, Zell R, Wright AK, Cross TA, Tietjen I,Fedida D, and Busath DD (2014) Aminoadamantanes with persistent in vitro ef-ficacy against H1N1 (2009) influenza A. J Med Chem 57:4629–4639.
Luscombe CA, Huang Z, Murray MG, Miller M, Wilkinson J, and Ewart GD (2010) Anovel Hepatitis C virus p7 ion channel inhibitor, BIT225, inhibits bovine viraldiarrhea virus in vitro and shows synergism with recombinant interferon-a-2b andnucleoside analogues. Antiviral Res 86:144–153.
Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, and Olson AJ(1998) Automated docking using a Lamarckian genetic algorithm and an empiricalbinding free energy function. J Comput Chem 19:1639–1662.
Mould JA, Paterson RG, Takeda M, Ohigashi Y, Venkataraman P, Lamb RA,and Pinto LH (2003) Influenza B virus BM2 protein has ion channel activity thatconducts protons across membranes. Dev Cell 5:175–184.
Neumann G, Watanabe T, Ito H, Watanabe S, Goto H, Gao P, Hughes M, Perez DR,Donis R, and Hoffmann E et al. (1999) Generation of influenza A viruses entirelyfrom cloned cDNAs. Proc Natl Acad Sci USA 96:9345–9350.
Nieva JL, Madan V, and Carrasco L (2012) Viroporins: structure and biologicalfunctions. Nat Rev Microbiol 10:563–574.
OuYang B and Chou JJ (2014) The minimalist architectures of viroporins and theirtherapeutic implications. Biochim Biophys Acta 1838:1058–1067.
OuYang B, Xie S, Berardi MJ, Zhao X, Dev J, Yu W, Sun B, and Chou JJ (2013)Unusual architecture of the p7 channel from hepatitis C virus.Nature 498:521–525.
Pervushin K, Tan E, Parthasarathy K, Lin X, Jiang FL, Yu D, Vararattanavech A,Soong TW, Liu DX, and Torres J (2009) Structure and inhibition of the SARScoronavirus envelope protein ion channel. PLoS Pathog 5:e1000511.
Pinto LH, Holsinger LJ, and Lamb RA (1992) Influenza virus M2 protein has ionchannel activity. Cell 69:517–528.
Premkumar A, Wilson L, Ewart GD, and Gage PW (2004) Cation-selective ionchannels formed by p7 of hepatitis C virus are blocked by hexamethylene ami-loride. FEBS Lett 557:99–103.
Seeliger D and de Groot BL (2010) Ligand docking and binding site analysis withPyMOL and Autodock/Vina. J Comput Aided Mol Des 24:417–422.
Song JM, Lee KH, and Seong BL (2005) Antiviral effect of catechins in green tea oninfluenza virus. Antiviral Res 68:66–74.
Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V,Nanda V, Stayrook S, and DeGradoWF (2008) Structural basis for the function andinhibition of an influenza virus proton channel. Nature 451:596–599.
Sugrue RJ, Bahadur G, Zambon MC, Hall-Smith M, Douglas AR, and Hay AJ (1990)Specific structural alteration of the influenza haemagglutinin by amantadine.EMBO J 9:3469–3476.
Thiel G, Baumeister D, Schroeder I, Kast SM, Van Etten JL, and Moroni A (2011)Minimal art: or why small viral K(1) channels are good tools for understandingbasic structure and function relations. Biochim Biophys Acta 1808:580–588.
Trott O and Olson AJ (2010) AutoDock Vina: improving the speed and accuracy ofdocking with a new scoring function, efficient optimization, and multithreading. JComput Chem 31:455–461.
Wang J, Ma C, Fiorin G, Carnevale V, Wang T, Hu F, Lamb RA, Pinto LH, Hong M,and Klein ML et al. (2011) Molecular dynamics simulation directed rational designof inhibitors targeting drug-resistant mutants of influenza A virus M2. J Am ChemSoc 133:12834–12841.
Wang J, Ma C, Wang J, Jo H, Canturk B, Fiorin G, Pinto LH, Lamb RA, Klein ML,and DeGrado WF (2013a) Discovery of novel dual inhibitors of the wild-type andthe most prevalent drug-resistant mutant, S31N, of the M2 proton channel frominfluenza A virus. J Med Chem 56:2804–2812.
Wang J, Pielak RM, McClintock MA, and Chou JJ (2009) Solution structure andfunctional analysis of the influenza B proton channel. Nat Struct Mol Biol 16:1267–1271.
Wang J, Wu Y, Ma C, Fiorin G, Wang J, Pinto LH, Lamb RA, Klein ML, and DegradoWF (2013b) Structure and inhibition of the drug-resistant S31N mutant of the M2ion channel of influenza A virus. Proc Natl Acad Sci USA 110:1315–1320.
Williams JK, Tietze D, Wang J, Wu Y, DeGrado WF, and Hong M (2013) Drug-induced conformational and dynamical changes of the S31N mutant of the in-fluenza M2 proton channel investigated by solid-state NMR. J Am Chem Soc 135:9885–9897.
Address correspondence to: Dr. David Fedida, 2.301 Life Sciences Institute,2350 Health Sciences Mall, Vancouver, BC Canada V6T 1Z3. E-mail: [email protected]
Mechanisms of Action of Novel Influenza A/M2 Viroporin Inhibitors 95
at ASPE
T Journals on July 23, 2020
molpharm
.aspetjournals.orgD
ownloaded from