mechanisms underlying the regulatory function of tumor
TRANSCRIPT

Mechanisms underlying the regulatory function of tumor necrosis factor-α in skin inflammation
Dissertation
Zur Erlangung des akademischen Grades
Doktor rerum naturalium
(Dr. rer. nat)
im Fach Biologie
eingereicht an der
Lebenswissenschaftlichen Fakultät
der Humboldt-Universität zu Berlin
von
M.Sc. Vandana Kumari
Präsident der Humboldt-Universität zu Berlin Prof. Dr. Jan-Hendrik Olbertz
Dekan der Lebenswissenschaftlichen Fakultät Prof. Dr. Richard Lucius
Gutacher/innen: 1. Prof. Dr. A. Radbruch 2. Prof. Dr. M. Worm 3. Prof. Dr. P. Franken Tag der mündlichen Prüfung: 21.04.2015

ALL THAT WE ARE IS THE RESULT OF ALL THAT
WE HAVE THOUGHT.
- BUDDHA

Table of contents
TABLE OF CONTENTS LIST OF ABBREVIATIONS .......................................................................................... 6
ABSTRACT ................................................................................................................. 10
ZUSAMMENFASSUNG .............................................................................................. 11
1. INTRODUCTION ..................................................................................................... 12
1.1. ANATOMICAL SKIN STRUCTURE .............................................................................. 12
1.2. SKIN BARRIER AND IT’S DISRUPTION IN SKIN PATHOLOGY .................................. 14
1.2.1 Physical and chemical irritants ......................................................................... 15
1.2.2 Contact dermatitis (CD) and Atopic dermatitis (AD) ......................................... 16
1.3. KERATINOCYTES ........................................................................................................ 21
1.3.1 Role of keratinocytes in skin irritation ............................................................... 22
1.3.2 Role of keratinocytes in AD .............................................................................. 23
1.4 TUMOR NECROSIS FACTOR-α (TNF-α) ...................................................................... 24
1.4.1 TNF-α – a proinflammatory cytokine................................................................. 24
1.4.2 Role of TNF-α in skin irritation .......................................................................... 25
1.4.3 Role of TNF-α in AD ......................................................................................... 26
1.5 THYMIC STROMAL LYMPHOPOIETIN (TSLP) ............................................................. 27
1.5.1 Role of TSLP in skin irritation ........................................................................... 28
1.5.2 Role of TSLP in AD .......................................................................................... 30
1.6 OBJECTIVES ................................................................................................................. 31
2. MATERIAL AND METHODS .................................................................................. 32
2.1 MATERIALS ................................................................................................................... 32
2.2 METHODS ..................................................................................................................... 36
2.2.1 Animal experiments .......................................................................................... 36
2.2.2 Cell culture methods ......................................................................................... 41
2.2.3 TSLP enzyme linked immunosorbent assay (ELISA) ....................................... 43
2.2.4 RNA isolation .................................................................................................... 44
2.2.5 Reverse transcription ........................................................................................ 44
2.2.6 Real-time polymerase chain reaction ............................................................... 45
3

Table of contents
2.2.7 Isolation and culture of bone marrow cells and generation of bone marrow-
derived mast cells (BMcMCs) .................................................................................... 47
2.2.8 Flow cytometry ................................................................................................. 48
2.2.9 Stimulation of BMcMCs .................................................................................... 49
2.2.10 Histology and immunohistochemistry ............................................................. 49
2.3 STATISTICAL ANALYSIS .............................................................................................. 52
3. RESULTS ................................................................................................................ 53
3.1 SKIN IRRITATION LEADS TO TSLP PRODUCTION ..................................................... 53
3.1.1 Physical or chemical irritation of the skin leads to production of TSLP in vivo . 53
3.1.2 Pro-inflammatory cytokines elevate TSLP production in murine KCs ............... 55
3.1.3 Skin biopsies from mouse and human produce TSLP ex vivo .......................... 57
3.1.4 IL-1 contributes to SDS-mediated TSLP induction ........................................... 58
3.2 AGGRAVATED AD IN TNF-/- MICE ................................................................................ 59
3.3 ROLE OF TSLP IN AD AGGRAVATION UPON TNF DEFICIENCY ............................... 60
3.3.1 Increased TSLP levels in lesional skin of TNF-/- mice and correlation with AD
severity ...................................................................................................................... 60
3.3.2 Anti-TSLP protect TNF-/- regarding AD onset ................................................... 62
3.4. ENDOGENOUS TNF-α DOES NOT CONTRIBUTE TO TSLP PRODUCTION ............. 63
3.5 MAST CELLS CONTRIBUTE TO TSLP PRODUCTION ................................................ 65
3.5.1 MCs are increased in lesional skin of TNF-/- mice and correlate with AD and
TSLP ......................................................................................................................... 65
3.5.2 Anti c-Kit is protective for AD development in TNF-/- mice ................................ 66
3.5.3 MCs do not produce a relevant amount of TSLP .............................................. 67
3.5.4 MCs as instructors of TSLP production by KCs ................................................ 68
4. DISCUSSION .......................................................................................................... 71
4.1 SKIN IRRITATION LEADS TO RAPID INDUCTION OF TSLP, INDEPENDENT FROM
TNF-α, BUT PARTIALLY DEPENDS ON IL-1 ...................................................................... 71
4.2 TNF-/- MICE DEVELOP AGGRAVATED AD AND DISPLAY INCREASED TSLP
EXPRESSION AND MCs NUMBERS CORRELATING WITH DISEASE SEVERITY ........... 76
4.3 ENHANCED TSLP LEADS TO AD MANIFESTATION ................................................... 79
4.4 MCs SEEM TO PLAY A ROLE BETWEEN TNF-DEFICIENCY AND TSLP .................... 81
4

Table of contents
4.5 CONCLUSION AND OUTLOOK .................................................................................... 84
REFERENCES ............................................................................................................ 87
APPENDIX .................................................................................................................. 99
ACKNOWLEDGEMENTS ......................................................................................... 101
SELBSTÄNDIGKEITSERKLÄRUNG / DECLARATION .......................................... 103
5

List of abbreviations
LIST OF ABBREVIATIONS
-/-
αh
αm
ANOVA
AD
e.c
β-Me
BMcMCs
bp
BSA
C57BL/6
CASY
CCL
CD
DNA
cDNA
dsDNA
CLA
CT
CXCL8
DC
dDCs
EDTA
ELISA
ERK
FACS
FBS
Fc
FcεRI
Fig
FITC
Knockout
Anti-human
Anti-mouse
Analysis of variance
Atopic dermatitis
Epicutaneous
β-mercaptoethanol
Bone marrow cultured mast cells
Base pair
Bovine serum albumin
C57 black 6
CASY® Cell Counter
Chemokine ligand
Cluster of differentiation
Desoxyribonucleic acid
Copy desoxyribonucleic acid
Double-Stranded DNA
Cutaneous lymphocyte-associated antigen
Threshold cycle value
CXC ligand 8
Dendritic cell
Dermal dendritic cells
Ethylenediaminetetraacetic acid
Enzyme linked immunosorbent assay
Extracellular signal-regulated kinase
Fluorescence activated cell sorter
Fetal Bovine Serum
Fragment crystallizable of Ig
Fc epsilon receptor I
Figure
Fluorescein IsoThioCyanate
6

List of abbreviations
g
GM-CSF
H1R
H2O2
H4R
HCl
HMGB1
HPRT
hrs
HRP
IFNγ
Ig
ICAM-1
IL-
IL-7Rα
IL-1Ra
IMDM
i.p
i.d
LSAB2
JAK
JNK
KCs
kDa
LTα
LTC4
MΦ
MACS
MAP
MCs
MDM2
MgCl2
Acceleration of gravity
Granulocyte-macrophage colony-stimulating factor
Histamine 1 receptor
Hydrogen peroxide
Histamine 4 receptor
Hydrochloric acid
High mobility group box chromosomal Protein 1
Hypoxanthine-guanine phosphoribosyltransferase
Hours
Horseradish peroxidase
Interferon gamma
Immunoglobulin
Intercellular adhesion molecule-1
Interleukin-
Interleukin-7 receptor alpha
Interleukin-1 receptor antagonist
Iscove's Modified Dulbecco's Medium
Intraperitoneal
Intradermal
Labelled Streptavidin-Biotin2 System-
Janus Activated Kinase
c-Jun N-terminal kinases
Keratinocytes
Kilodalton
Lymphotoxin α
Leukotriene C4
Macrophage
Magnetic Cell Sorting
Mitogen-activated protein
Mast cells
Murine double minute 2
Magnesium Chloride
7

List of abbreviations
mMCP6
mRNA
NF-κB
NHBE
NK
O.C.T
OVA
p38
PBS
PBST
PCR
PE
Pen/Strep
PGD2
Plcb 3
PMA
Poly I:C
RANTES
rh
rm
RNA
rpm
RT
SB
SEM
SC
SCF
SCORAD
SDS
SG
SLS
SS
Mouse Mast Cell Protease 6
Messenger ribonucleic acid
Nuclear factor kappa-light-chain-enhancer of activated B cells
Normal Human Bronchial Epithelial
Natural killer
Optimal Cutting Temperature
Ovalbumin
Phospho 38
Phosphate buffered saline
Phosphate buffered saline + Tween-20
Polymerase chain reaction
phycoerythrin
Penicillin and streptomycin
Prostaglandin D2
Phospholipase C-Beta 3
Phorbol Myristate Acetate
Polyinosinic:polycytidylic acid
Regulated on Activation Normal T Cell Expressed and Secreted
Recombinant human
Recombinant mouse
Ribonucleic acid
Revolutions per minute
Reverse transcriptase
Stratum basale
Standard error of the mean
Stratum corneum
Stem cell factor
Severity Scoring of Atopic Dermatitis
Sodium dodecyl sulphate
Stratum granulosum
Sodium lauryl sulphate
Stratum spinosum
8

List of abbreviations
STAT6
TAE
TBS
TEWL
TGF-β
Th
TLR
TNF-α
TNFR
TPA
Treg
TSLP
TSLPR
qPCR
UTR
UV
wt
Signal Transducers and Activators of Transcription 6
TRIS-Acetat-EDTA
Tris-buffered saline
Transepidermal water loss
Transforming growth factor beta
T-helper
Toll like receptor
Tumor necrosis factor-α
Tumor necrosis factor receptor
12-o-Tetradecanoylphorbol-13- acetate
Regulatory T cell
Thymic stromal lymphopoietin
Thymic stromal lymphopoietin receptor
quantitative PCR
Untranslated region
Ultraviolet
Wildtype (C57BL/6)
9

Abstract
ABSTRACT
The skin is the largest organ of an individuum and builds the barrier for a host
against the environment. Skin barrier disruption by exogenous or endogenous
stimuli can lead to skin inflammation. As a consequence, irritant or atopic eczema,
frequent skin diseases, may evolve. Tumor necrosis factor-α (TNF-α) is a pleiotropic
cytokine which plays a central role in inflammatory processes.
The main aim of this thesis was to clarify whether and how endogenous TNF-α is
contributing to skin inflammation driven by exogenous and endogenous triggers.
The role of endogenous TNF-α was studied using TNF knockout (-/-) mice. In an
irritation model, chemical and physical stimuli were applied on to mouse skin.
Thymic stromal lymphopoietin (TSLP) was significantly induced by the used
irritants. This TSLP induction was independent from endogenous TNF-α proven by
using TNF-/- mice.
Next the role of TNF-α in atopic dermatitis (AD) promoting an allergic skin
inflammation was investigated. TNF-/- mice developed more severe AD compared to
the wildtype mice and TSLP was significantly increased and correlated with the
severity of the eczema. To prove the pathophysiological role of TSLP for AD
progression, TNF-/- mice were pretreated with an TSLP antibody. Indeed, these
mice developed less AD symptoms compared to the control mice.
Mast cells (MCs) were also significantly increased in lesional skin in the AD model
and moreover, correlated with AD severity, but also with TSLP expression.
10

Zusammenfassung
ZUSAMMENFASSUNG
Die Haut ist das größte Organ des Menschen und bildet die Barriere gegenüber
Einwirkungen aus der Umwelt. Die Störung der Hautbarriere durch exogene und
endogene Reize führt zu einer Entzündungsreaktion in der Haut. In der Folge
können Hauterkrankungen wie die irritative oder Atopische Dermatitis entstehen.
Der Tumor Nekrose Faktor-α (TNF-α) ist ein pleiotrop wirksames Zytokin, das eine
zentrale Rolle bei entzündlichen Prozessen spielt.
Ziel der vorgelegten Promotionsarbeit war zu untersuchen, ob und wie TNF-α zu
Entzündungsgeschehen, ausgelöst durch exogene und endogene Faktoren,
beiträgt.
Die Bedeutung von TNF-α wurde in TNF-ko Mäusen in verschiedenen
Hautmodellen untersucht. Für das Irritationsmodell wurden chemische und
physikalische Reize verwendet. TSLP (Thymic stromal lymphopoietin) wurde durch
die verschiedenen Stimuli signifikant induziert. Diese Induktion war unabhängig von
der endogenen TNF-α Produktion, gezeigt durch den Einsatz von TNF- ko Mäusen .
Da endogenes TNF-α für die Hautirritation keine notwendige Bedingung darstellte,
wurde die Bedeutung von TNF-α bei der atopischen Dermatitis (AD) untersucht.
TNF-α defiziente Mäuse zeigen verstärkt Ekzeme im Vergleich zu Wildtyp Mäusen.
Die Behandlung von TNF-ko Mäusen mit einem TSLP Antikörper führte zu einer
Verminderung des Ekzems.
Mastzellen wurden vermehrt in läsionaler Haut gefunden und korrelierten mit dem
Schweregrad des atopischen Ekzems sowie der TSLP-Expression. Schlagwörter: Tumor Nekrose Faktor-α, Thymic stromal lymphopoietin, Hauterkrankungen,
Atopischen Dermatitis, Mastzellen
Keywords: Tumor necrosis factor-α, Thymic stromal lymphopoietin, skin inflammation, Atopic
dermatitis, Mast cell
11

Introduction
1. INTRODUCTION
1.1. ANATOMICAL SKIN STRUCTURE
The skin provides a protective barrier between the inner and outer environment to
protect an individuum from various potential dangerous microbes1. The skin is
composed of three layers, the epidermis, dermis and subcutis2. The epidermis is of
highest importance for the skin barrier integrity and provides an individuum with
physical, chemical or biochemical barriers. The epidermis is formed by several layers
of keratinocytes which undergo a differentiation process. These are the stratum
basale, the stratum spinosum, the stratum granulosum and the stratum corneum
(Fig. 1)1,3,4. The stratum basale is the layer which contains basal stem cells that are
capable to proliferate into keratinocytes and can amplify the cell numbers5. The
stratum spinosum is characterized by visible desmosomes, which contribute to the
appearance of spindle shaped cells. These cells express the early differentiation
marker cytokeratin 10. The differentiation of cells can be seen from bottom to top, by
the presence of intermediate differentiation marker involucrin in the upper spinous
cell layers but not in the lower ones5. The skin core is mainly composed of a
continuous sheet of flat anucleated corneocytes which represent differentiated
keratinocytes of the outer layer of stratum granulosum containing keratin
filaments1,3,4. The stratum granulosum consist of 3–5 cell layers and is characterized
by lamellar bodies and keratohyalin granules. These layered of cells express and
process the two late differentiation markers filaggrin and loricrin6. The primary skin
barrier is mainly provided by stratum corneum layer as a robust barrier against the
percutaneous penetration of chemicals and microbes, but also mechanical injuries1,7.
Cells in the stratum corneum layers are connected together by lipid bilayers, which
forms a brick-like structure which form an insoluble, rigid structure referred to as
cornified envelope. The stratum corneum is also responsible in different active
processes such as regulation of water loss from the skin to the outer atmosphere,
known as transepidermal water loss (TEWL)1,7.
12

Introduction
Figure 1: Anatomical skin structure including the epidermal layers. The skin structure is complex and enables to build a barrier against environment. The epidermis contains
stratum corneum followed by stratum lucidum, stratum granulosum, stratum spinosum and stratum basale. The
dermis is mainly composed of collagen, elastic tissue and reticular fibres. It contains many different cell types
such as dendritic cells (DCs), T cells subsets, fibroblast, macrophages and mast cells (MC) (not shown). The
subcutis is composed of the adipose tissue.
Adopted from Skin barrier function and its importance at the start of the atopic march, Mary Beth Hogan, Kathy
Peele, and Nevin W. Wilson, Journal of Allergy (2012).
The dermis forms the thickest structure of the skin containing sebaceous glands,
sweat glands and hair follicles8,9. The dermis is formed by connective tissue and a
network of capillaries and blood vessels. Dilatation or constriction of these blood
vessels and capillaries provides thermoregulation to the body10. The dermis also
provides elasticity to the skin as it contains elastin fibers and collagen11. By
contrast, the epidermis contains tight junctions, adherens junctions, desmosomes,
gap junctions and keratins filaments to form the skin barrier12. Tight junctions are
the cell to cell junctions which regulate paracellular activities of molecules and are
responsible for the separation of the apical from the basolateral part of the cell
membrane, reducing the diffusion of proteins and lipids between the cells. Tight
junctions and desmosomes play a vital role in the stabilization of the cell to cell
adhesion, to maintain the cell shape and the tissue integrity. Gap junctions are
13

Introduction
important for cell to cell interaction. The major components of gap junctions are
connexins, which homo- or heteromerize to connexons to form channels, which
allow the passage of ions and small molecules between cells1. Keratins are the
most abundant structural proteins synthesized by keratinocytes that assemble
throughout the cytoplasm and terminate at desmosomes1,9.
1.2. SKIN BARRIER AND IT’S DISRUPTION IN SKIN PATHOLOGY
The skin is a metabolically active organ. Different physiological processes support
to maintain the skin barrier10. The primary function of the skin is to protect inner
body from physical, chemical, thermal or mechanical hazards as well as the
invasion of microorganisms (Fig. 2)1. It also reduces the harmful effects of UV
radiation and acts as a sensory organ (Fig. 2)10. To maintain the function of the skin
barrier, a large number of factors are required. These include an cell to cell
interaction within epidermis, the prevention of excessive water loss, the
communication with the immune system and the renewal of the skin cells. When the
epidermal skin barrier is disrupted, the initial response to cellular damage of the
epidermal cells is a stimulation signal to replace the damaged cells13 and to
maintain the skin homeostasis. The skin-resident immune cells such as epidermal
langerhans cells or dendritic cells are key players in restoring the homeostasis14.
Upon skin injury, KCs start producing pro-inflamamatory cytokines such as
Interleukin-1β (IL-1β), IL-6, IL-18 and TNF-α, which further activate dermal dendritic
cells (DCs) in the presence or absence of antigen. Upon stress signalling, KCs gets
activated and contribute to dermal DC activation by releasing interferon-α (IFN-α)
(Fig. 2). Activated dermal DCs promote the proliferation of skin-resident T cells i.e.
CD4+ or CD8+ T cells (Fig. 2). Stimulated T cell further produce pro-inflammatory
cytokines and chemokines which stimulate epithelial and mesenchymal cells e.g.
keratinocytes and fibroblasts thus amplifying the inflammatory reaction in the skin
(Fig. 2)14.
14

Introduction
Figure 2: Disrupted skin barrier leads to inflammatory response skin. Exposure to irritants, UV light or infections agent’s leads to barrier disruption is triggering the immune response
to retain the skin homeostasis. Upon stimulation keratinocytes produce proinflammatory cytokines such as TNF-
α, IL-1β, TSLP, which further promote the transition of dermal dendritic cells (dDCs) and activate MCs and T-
cells.
Adapted from Skin immune sentinels in health and disease. Frank O. Nestle, Paola Di Meglio, Jian-Zhong Qin
and Brian J. Nickoloff, Nat Rev Immunol. Oct 2009; 9(10): 679–691.
1.2.1 Physical and chemical irritants
Exposure of the skin to different irritants can lead to an impairment of the barrier
function and a consecutive damage of the epidermal cells15. Many studies have
been done to understand the mechanism of acute and chronic irritation16. As it is
difficult for ethical reasons to study the pathogenesis of irritation at a cellular level in
humans, mouse models have been used to study the physico-chemical events
during these reactions. Many studies have been performed using different irritants
such as sodium dodecyl sulphate (SDS), acetone, croton oil or tape stripping17.
Measurements to assess a disturbed skin barrier include TEWL, electrical
15

Introduction
capacitance (stratum corneum hydration), percutaneous drug transport, and skin
color reflectance (erythema)17,18. Willis CM et al. observed that irritation with 5%
SDS for 48 hrs resulted a strong inflammatory response with the onset of increased
numbers of infiltrating cells consisting polymorphonuclear leukocytes and
mononuclear cells19. Another group has shown that higher concentrations of SDS
resulted in a down regulation of HLA-DR expression on Langerhans cells20. Another
common irritation method which is widely used for the induction of barrier disruption
with less cytopathic effects on keratinocytes is tape stripping. With the aid of
adhesive tape strips, the layers of the stratum corneum were removed after 30times
tape stripping21. Disruption of stratum corneum leads to an increase of the TEWL
and induces the production of different inflammatory mediators17,22. Such induction
of a proinflammatory immune response in human keratinocytes has been shown by
different irritants such as croton oil, phenol and SLS as published by Wilmer et al.
(1994)23. In particular croton oil and phenol directly induce the expression of IL-18
without the intermediate production of IL-1α and TNF-α23.
1.2.2 Contact dermatitis (CD) and Atopic dermatitis (AD)
Contact dermatitis Contact dermatitis is an inflammatory response of the skin characterized by
erythematous and pruritic skin lesions that occur after direct contact with exogenous
substances24. Contact dermatitis is frequent and a main cause of occupational
dermatitis25. Based on the pathophysiology contact dermatitis is classified in two
subtypes: irritant contact dermatitis (ICD) and allergic contact dermatitis (ACD)24.
Even though it is possible to differentiate between ICD from ACD at clinical levels,
both manifestations can have similar clinical and histological presentations26.
Irritant contact dermatitis (ICD) Irritant contact dermatitis is considered as the most common type of contact
dermatitis26. It is the consequence of an activated innate immune response of skin
to various physical and chemical stimuli. It occurs in response of skin injury by
foreign particle without prior immunological sensitization of the skin. The
16

Introduction
development of ICD depends on a complex interplay between endo- and
exogenous factors27. Intrinsic factors which influence development of ICD include
genetic predisposition eg. age, sex and body area, whereas extrinsic factor include
the type of the irritant, the irritant concentration and the time of exposure27. An
impairment of the skin horny layer and epidermal cell damage are considered to be
the main factors in the pathogenesis of ICD. The underlying mechanism of ICD
includes an activation of the innate immune response with the release of IL-1α, IL-
1β, TNF-α, GM-CSF and IL-8 (Fig. 3A)28. Consecutively, these cytokines activate
Langerhans cells (LC), dDCs and endothelial cells, which further support the cellular
recruitment at the site of damage e.g. lymphocytes, macrophages, neutrophilis (Fig.
3A). These cellular infiltrates further promote the inflammatory pathway (Fig. 3A)28.
Allergic contact dermatitis (ACD) Allergic contact dermatitis is a delayed hypersensitivity reaction mediated by
antigen-specific T cells29. It occurs only in sensitized patients i.e. individuals who
have build an immunological memory response upon a prior contact. The
concentration of an allergen is important to initiate an ACD26. ACD is characterized
by pruritic papules and vesicles on an erythematous base, in the chronic condition
lichenified pruritic plaques can be present. Individuals with a history of ACD develop
the symptoms a few days after exposure in the area that was in direct contact with
the allergen30. Similar to the scenario in ICD the allergen exposure result in an
activation of the innate immune system through a release of proinflammatory
cytokines by KC including IL-1α, IL-1β, TNF-α, GM-CSF, IL-8 and IL-18 with in
consequence the onset of vasodilation and cellular recruitment (Fig. 3B)28. Upon
contact with allergens, LCs and dDCs migrate to the draining lymph nodes, where
they activate allergen-specific T cells e.g. Th1, Th2, Th17 and regulatory T (Treg)
cells (Fig. 3B)28. Activated T cells further proliferate and enter into the circulation
and reach to the site of initial exposure, along with other immune cell such as mast
cells and eosinophils (Fig. 3B). Once an individual is re-exposed to an allergen, the
allergen-specific T cells, along with other inflammatory cells, enter the site of
exposure and release proinflammatory cytokines which consequently stimulate the
KCs to induce an inflammatory cascade (Fig. 3B)28.
17

Introduction
Figure 3. Pathogenesis of irritant contact dermatitis (ICD) and allergic contact dermatitis (ACD). A) In ICD, encounter with an irritant stimulate KCs by activating innate immunity with the release of
pronflammatory cytokines such as IL-1α, IL-1β, TNF-α etc. from epidermal KCs. These cytokines further
activate inflammatory cells e.g. LCs, dDCs, and endothelial cells, all of which contribute to cellular recruitment to
the site of KC damage and further initiate the inflammatory cascade.
B) During sensitization phase of ACD, allergens activate innate immunity through KC activation and
proinflammatory cytokines release as well as with vasodilation, cellular recruitment, and infiltration. Upon
exposure to allergen, LCs and dDCs migrate to the lymph nodes, where they activate allergen-specific T cells
e.g. Th1, Th2, Th17, and regulatory T (Treg) cells. Activated T cells proliferate and reach to the site of infection
along with other cell types such as mast cells and eosinophils. Upon re-encountering with allergen, the hapten-
specific T cells get activated and along with other inflammatory cells, enter the site of exposure and release
proinflammatory cytokines and subsequently stimulate KCs to induce an inflammatory cascade. Reprinted from Dhingra et al. 2013: Mechanisms of contact sensitization offer insights into the role of barrier
defects vs. intrinsic immune abnormalities as drivers of atopic dermatitis, J Invest Dermatol.2311-4. (Oct 1,
2013.). Copyright (2014), with permission from Nature publishing group.
18

Introduction
Atopic dermatitis AD is a chronic-relapsing, eczematous skin disease clinically characterized by
erythema, edema, excoriation, xerosis, intense pruritus and a typical localization
pattern31. Commonly, AD initiates early in childhood (i.e. early-onset AD)31,32.
Epidemiological studies point towards an increase in AD prevalence in the last
decades affecting around 10-20% of children and 1-3% of the adult population
worldwide32-34.
Pathophysiology of atopic dermatitis
AD is a highly complex inflammatory skin disease which depends on the interplay
between genetic and environmental factors35. The understanding of AD
development is still not completely clear especially at the molecular level36. It is still
not certain whether AD is a consequence of an immune dysfunctioning or due to
genetic defects or both31,32,36,37. A defect of the skin barrier function plays a crucial
role in the pathogenesis of the disease. It leads to an increase of the epidermal
water loss and a promotion of an invasion of allergens, microbes or any other
irritants (Fig. 4)38. Different studies have shown that a defect of skin barrier
promotes skin inflammation in AD patients34,39. Filaggrin an important skin barrier
protein was identified to play a significant role in AD progression. Around 20% of AD
patients display a null mutation in the gene encoding for filaggrin34,35,40. The
presence of the filaggrin gene mutation has shown to increase skin dryness in AD
patients41. Different cytokines such as IL-4, IL-13 and TNF-α have been shown to
reduce the expression level of filaggrin in AD patients as well42. Among filaggrin
several other proteins are involved in forming the skin barrier and may be relevant
in AD as well. Moreover patients even though carrying filaggrin mutations can
outgrow the disease suggesting that breakdown in the skin barrier is not sufficient
for the development of AD43,44.
Various studies have shown that different immune cells are involved in the AD
progression apart from the skin barrier. T cell plays a major role in the AD
development especially at the early stage of the disease where an increased Th2
response is responsible for the major immune dysbalance45. Data from both human
19

Introduction
and mouse studies show that CD4+ T cells are involved in the development of
AD31,37,46. Specific DCs in the skin including epidermal Langerhans cells and
inflammatory dendritic cells activate T cells38. In acute and chronic AD lesions, the
expression levels of T cell induced cytokines i.e. IL-4, IL-5 and IL-13 were
significantly increased (Fig. 4). Several studies indicate that also the other T-cell
types such as T-reg, Th17, Th 9 and Th 22 are involved in the pathogenesis of AD
but their exact role in the AD progression is still not clear (Fig. 4)47,48. Keratinocytes
in the skin are regarded to be the key contributors or initiators of the disease. An
increased production of TSLP by keratinocytes from atopic skin has been reported
to further activate dendritic cells to drive Th2 polarization (Fig. 4)31.
Even though T cells which were previously described to be crucial for AD
pathogenesis are dispensable under certain conditions and can be “replaced” by
innate immune cells which include MCs, eosinophil’s and macrophages (Fig. 4)49-51.
Likewise, AD can develop in the absence of IL-4, signal transducers and activators
of transcription 6 (STAT6) and IgE, although the overexpression of IL-4 can trigger
AD development in the skin52,53. Thus, AD seems to have highly superfluous
mechanisms which converge furthermore with barrier impairment, xerosis and itch.
Findings showing that AD may be present in of two different immunological forms,
the extrinsic AD (atopic eczema) and the intrinsic AD (non-atopic eczema)34,40 are
underlining this complexity of AD. Generally, 20-30% of the patients are affected by
intrinsic AD. These patients have no increased levels of allergen specific or total IgE
nor eosinophil numbers; yet, the two subtypes are indistinguishable in their clinical
presentation. Thus, based on the heterogeneity of AD, it is likely that immune
deviations and aberrations in skin cells both can contribute to AD independently and
set off its development54.
20

Introduction
Figure. 4: Pathogenesis of atopic dermatitis. In AD, barrier disruption leads to entry of antigens, which encounter langerhans cells, dendritic cells and
activating Th2 cells. T cells produces IL-4 and IL-13 which stimulate keratinocytes to produce TSLP. Activated
TSLP express OX40 ligand to induce Th2 cells. Cytokines and chemokines, such as IL-4, IL-5 and IL-13
produced by Th2 cells and DCs stimulate skin infiltration by inducing DCs, mast cells, and eosinophils. Reprinted from Dhingra et al. 2013: Mechanisms of contact sensitization offer insights into the role of barrier
defects vs. intrinsic immune abnormalities as drivers of atopic dermatitis, J Invest Dermatol.2311-4. (Oct 1,
2013.). Copyright (2014), with permission from Nature publishing group.
1.3. KERATINOCYTES
Keratinocytes are the highly specialized epithelial cells which maintain the physical
and biochemical barrier integrity of the skin55,56. To form the skin barrier and to
maintain the skin integrity, keratinocytes continuously undergo a complex
differentiation process. The most relevant morphological and cytostructural changes
of keratinocytes occur during differentiation in the spinous and granular layers.
21

Introduction
During this process many different differentiation-dependent proteins are produced
such as involucrin, filaggrin, transglutaminase, claudin etc.55. A dysregulation of
these genes can lead to the skin disease and diminishment of skin barrier47,57-59.
Studies have shown that cytokines produced by keratinocytes play a critical role in
maintaining the immune response, cellular communication and in the pathogenesis
of disease28,44,60. For the barrier function of the skin, cytokine signaling can result in
multiple consequences e.g. proliferation and differentiation of keratinocytes which
are influenced by cytokines production and are partly modulated by gene
expression in these cells60. An increased expression of certain cytokines can result
in an activation of complex network of signaling molecules which can disrupt the
physiology of keratinocytes and the quality of the skin barrier3. Upon skin disruption,
keratinocytes are stimulated and the production of different proinflammatory
cytokines such as TSLP, TNF-α, IL-1α is initiated (Fig. 5)14.
1.3.1 Role of keratinocytes in skin irritation
As indicated above, keratinocytes are the most important cell type for maintaining
the homeostasis of the skin. They provide a rigid structure by undergoing a
differentiation process. During differentiation, numerous genes (e.g. loricrin,
involucrin, pro-filaggrin etc.) are expressed and finally the cells enters into a cell
cycle arrest61.
Keratinocytes are the main producers of many different inflammatory mediators
during skin irritation. IL-1α is considered as one of the primary alarm signals
followed upon skin disruption in the inflammatory cascade (Fig. 5)62. Several, in
vitro studies have shown that different irritants are capable to induce IL-1α in
keratinocytes61,63-65. The production of IL-1α further activates the release of other
pro-inflammatory cytokines or chemokines such as IL-1β, TNF-α, IL-6, IL-8 by other
epidermal and dermal cells66. IL-1β is produced in an inactive form by keratinocytes
and cleaved into the active form by proteases which are not generally present in the
resting keratinocytes. Proteases are activated upon irritation of keratinocytes with
phorbol myristate acetate (PMA) or sodium lauryl sulphate (SLS)67. IL-1α along with
22

Introduction
IL-1β has pleiotropic effects and is involved in the activation of dendritic cells and T
cells67.
Figure 5: Role of keratinocytes in skin inflammation. Skin barrier disruption allows microbes or irritant to enter in the skin which stimulates the keratinocytes and
initiates the immune responses. Stimulated keratinocytes produces different proinflammatory cytokines such as
TNF-α, TSLP, IL-1α etc. which leads to skin inflammation and further eczema development.
Adapted from Skin immune sentinels in health and disease. Frank O. Nestle, Paola Di Meglio, Jian-Zhong Qin
and Brian J. Nickoloff, Nat Rev Immunol. Oct 2009; 9(10): 679–691.
1.3.2 Role of keratinocytes in AD
AD is characterized by itch and the onset of chronic or relapsing eczematous skin
lesions68. A range of different factors and cell types are known to contribute to the
pathogenesis of AD69. Keratinocytes are considered to be the primary source of
barrier deficiency in AD development70. Since a decade, there has been better
understanding in the role of keratinocytes in AD. Under AD environment,
keratinocytes produces a unique pattern of cytokines and chemokine’s such as
increased levels of chemokine ligand (CCL)5 (RANTES) after stimulation with TNF-
α and IFN-γ71. It has been also shown that keratinocytes driven from AD patients
produce more granulocytes- macrophage colony- stimulation factor and TNF-α 72.
23

Introduction
Other studies with stimulated keratinocytes of nonlesional skin from AD patients
have shown a lower expression of beta-defensin-2, an antimicrobial peptide which
chemoattracts Th17 cells compared to healthy or psoriasis controls73. More recent
studies, showing the contribution of keratinocyte-derived cytokines such as TSLP
on the inflammatory response provide a greater appreciation for the active role of
keratinocytes not only as barriers to the environment74, but also as perpetuating
cells with activating DCs to prime T cells to further produce IL-4 and IL-1371. TSLP
activated DCs also produce chemokines such as CCL17 and macrophage derived
CCL22, which further leads to the infiltration of Th2 cells in AD lesions38. Studies
have also shown that activated keratinocytes produce IL-25 and IL-33 which than
act on mast cells and antigen presenting cells (DCs and LCs)38,44.
1.4 TUMOR NECROSIS FACTOR-α (TNF-α)
1.4.1 TNF-α – a proinflammatory cytokine
Figure 6: Different forms of TNF-α. Two forms of TNF-α present i.e. a) Soluble TNF-α (or secreted form) and b) Membrane TNF-α (or cell
associated). Binding of TNF-α to its receptors TNFR1 and TNFR1 triggers intracellular signaling cascade. Upon
activation, TNF receptor forms trimer which binds to the monomer of TNF-α which leads to the conformational
change in to the structure of receptor.
Reprinted from Palladino et al. 2003: Anti-TNF-α therapies: the next generation: Nature Reviews Drug
Discovery 2, 736-746 (September 2003). Copyright (2014), with permission from Nature publishing group.
24

Introduction
TNF-α was first identified as an endotoxin-induced glycoprotein which causes
haemorrhagic necrosis of sarcomas in a mouse model. In 1984, the cDNA of TNF-α
was first cloned and shown to have the structural and functional homology to
lymphotoxin (LT) β and was described as (LT) α75,76. TNF proteins are ubiquitously
expressed by different cell types of the innate and acquired immunity such as B cells,
T cells, NK cells, DCs, and monocytes3. TNF-α is expressed in two different forms,
one is the cell-associated or membrane TNF-α (26-kDa) and the other one is the
secreted or soluble TNF-α (17-kDa) form 77(Fig. 6). Both forms of TNF-α are
biologically active. The cell-membrane bound form of TNF-α is thought to be
responsible for juxtacrine signalling whereas secreted form for the direct cell-to-cell
contact, though the exact functions of these two forms are still controversial 77,78.
Based on numerous studies, TNF-α is considered as one of the best known
proinflammatory cytokine having a crucial role in host defense and inflammatory
diseases79,80. It has been associated with the development of many autoimmune
disorders such as rheumatoid arthritis, psoriatic arthritis and inflammatory bowel
disease77. TNF-α is also known to enhance disease severity by its capability to
induce different proinflammatory cytokines, such as IL-1 and different chemokines81.
The administration of TNF-α antibodies and its interference with the TNF pathway are
widely used for controlling pathogenesis of many diseases such as rheumatoid
arthritis, psoriasis, inflammatory bowel disease 77,81. Since the last 10 years,
monoclonal antibodies against TNF-α or its receptor are widely used in the clinic for
the blockage of TNF pathway81 for the treatment of autoimmune diseases like
rheumatoid arthritis, but also psoriasis.
1.4.2 Role of TNF-α in skin irritation
The exposure of the skin to various irritants or chemicals results in skin irritation. Skin
irritation is a complex process which involves a series of responses such as skin
damage, cell death and activation of keratinocytes and other cells82. Keratinocytes
are well known to produce large amounts of proinflammatory cytokines such as TNF-
α, IL-1β, IL-6 (Fig. 5)14. The upregulation of TNF-α in the skin during irritation has
been shown by different irritants e.g. dimethyl sulfoxide, PMA, formaldehyde,
25

Introduction
tributyltin, and SLS67. TNF-α has pleiotropic effects on keratinocytes and endothelial
cells, where it increases the expression of major histocompatibility complex class II
molecules and upregulates cell adhesion molecules e.g ICAM-1. TNF-α is also
capable of inducing inflammatory factors such as IL-1, IL-6, IFN-γ, granulocyte-
macrophage colony-stimulating factor (GM-CSF) and CXC ligand 8 (CXCL8)56.
During irritation, TNF-α has common functions with IL-1α as a primary alarm signal to
other cell types, to further initiate the release of CCL20 and CXCL8 chemokines
production from macrophages. An increased expression level of CCL20 and CXCL8
leads to the migration of cells to the site of injury. T-cells, but also immature DCs are
activated83. The important role of IL-1α and TNF-α in the pathogenesis of skin
irritation has been proven at genetic levels. It has been shown, that certain genetic
polymorphisms of both TNF-α and IL-α are linked with an altered risk of skin irritation.
Individuals with TNFA-308 polymorphisms have a lower risk to develop ICD whereas
TNFA-238 alleles have an increased risk to ICD. Likewise, IL1A-889 C/T alleles are
protective for the development of ICD, clearly indicating that these genetic
polymorphisms are associated with an increased or decreased risk of ICD
development67. Hanel et al 2013 have shown the involvement of TNF-α in barrier
repair. TNF-α inhibited the expression of skin barrier genes such as filaggrin and
loricrin, TNF-α thereby weakening the skin barrier3. The central role of TNF-α in skin
irritation was further confirmed by the direct administration of TNF neutralizing
antibodies in vivo. These studies show, that the skin inflammation was reduced upon
antibody administration84,85.
1.4.3 Role of TNF-α in AD
The direct role of TNF-α for the development of AD is not completely understood. A
detailed analysis of the literature revealed a negative association between TNF and
AD development86-89. The most remarkable evidence for a functionally relevant
inverse association between TNF and AD comes from different clinical studies, which
have reported the onset of possible AD as a side effect upon anti-TNF therapy in
single patients with rheumatoid arthritis, Crohn's disease and psoriasis 90,91. On the
other hand few reports show a beneficial effect of TNF-α directed therapy in single
26

Introduction
AD patients92. These patients suffered from AD subsets (long-lasting and/or
combined with contact dermatitis). Another evidence of defective TNF production in
AD patients came from an analysis of peripheral blood leukocytes, in which
decreased TNF-α production was consistently reported in AD patients87-89. Recent
studies indicated that cytokines like IL-1β, IL-4, IL-5, IL-12, and IFN-γ are enhanced,
whereas TNF-α levels are reduced in AD skin compared to healthy controls88.
Although TNF-α is undoubtedly one of the best-characterized proinflammatory
cytokines, it can also exert anti-inflammatory effects and contribute to the resolution
of inflammatory diseases by various mechanisms, e.g. by promoting cluster of
differentiation (CD) 4+CD25+ T regulatory cells93, by mediating apoptosis of auto-
reactive effector T cells94 and by inducing local glucocorticoid production95.
1.5 THYMIC STROMAL LYMPHOPOIETIN (TSLP)
TSLP is an IL-7 like cytokine and has been first discovered in the culture
supernatants of mouse thymic stromal cells which gave rise for this nomenclature.
TSLP supports the growth and differentiation of B cells but also the proliferation of T
cells96,97. Different groups throughout the world demonstrated that high affinity TSLP
binding requires the combined binding to the IL-7 receptor α-chain and TSLP
receptor (TSLPR)97-99. TSLP is mainly expressed by epithelial cells from the thymus,
the skin, the lung, the intestine and tonsils as well as by stromal cells and mast
cells100-103. In the thymus, TSLP is responsible for the differentiation of Treg cells by
instructing thymic DCs104. Interestingly, human TSLP does not exert the same
functions as its murine counterparts; however it does activate immature CD11c+
myeloid DCs101,103. Thus, DCs can activate naïve CD4+ T cell proliferation and
initiate the production of IL-4, IL-5, IL-13 and TNF-α (Fig. 7). In contrast, the
production of the anti-inflammatory cytokines IL-10 and IFN-γ is inhibited by TSLP-
induced DCs103. TSLP is known to activate the upstream component of JAK1 and
JAK2, which bind to IL-7Rα and TSLPR chain8. Subsequently JAK1/2 are
phosphorylated and activate STAT5105. TSLP binding may also lead to an activation
of the subsequent STAT family members 1, 3, 4 and 6106,107. Recent
27

Introduction
phosphoproteomic data show that TSLP is also involved in a number of additional
signalling pathways. It was shown that often signal transduction like Erk1/2,
JNK1/2and p38 were phosphorylated after TSLP dependent activation108. TSLP
exerts its effects on a broad range of cells. Therefore it has been implicated to play
an important role in many diseases like infections, cancer and inflammatory bowel
diseases109-111. However, an even more important role of TSLP has been anticipated
in allergic diseases like AD and asthma112. TSLP has been shown to be upregulated
in an OVA-driven mouse model of airway inflammation113. These observations were
confirmed in an OVA-induced murine model of allergic asthma and AD with TSLPR-/-
mice which show a defective airway inflammation and allergic skin
inflammation114,115.
1.5.1 Role of TSLP in skin irritation
An acute insult against the stratum corneum results in perturbation of the barrier
integrity and induces a process of positive and negative alarm signals which initiate
both homeostatic and proinflammatory responses in the skin22,116. The compromised
barrier integrity further triggers the production of critical cytokines to initiate skin
inflammation117-119. TSLP is one of the cytokines which is expressed by keratinocytes
in response to physical injury and inflammatory cytokine stimulation (Fig. 7)74. The
crucial role of TSLP in allergic inflammation is well established but the underlying
mechanisms behind the trigger of TSLP production by different factors are still
unknown50,120,121. Primary human keratinocytes and skin explants were shown to
produce TSLP upon bacterial, viral or inflammatory stimuli or physical trauma 122,123.
Angelova-Fischer et al. (2010) investigated the role of tape stripping and SLS on skin
irritation and show that the stratum corneum of the epidermis is damaged, which is
associated with an increased TSLP expression117. They also observed that
keratinocytes express TSLP in the suprabasal cell layers of the epidermis. Among
these layers it is mainly localised in the granular and spinous
28

Introduction
layer and is not expressed by keratinocytes in the basal layer. These data are in
alignment with previous observations which have shown that TSLP expression is a
characteristic sign of keratinocytes which are undergoing a differentiation
process103,124. As previously described, human TSLP can induce synergistic effects
between proinflammatory and Th2 cytokines123. On the other hand keratinocytes
from Notch-deficient mice show an increased level of TSLP expression and an
eczema-like phenotype in skin upon barrier disruption 123,125,126 indicating that there
is a link between barrier integrity and TSLP production.
Figure 7: TSLP induction in keratinocytes. Skin barrier disruption, allergen or Th2 derived cytokines triggers the epithelium cells for TSLP production.
TSLP activates DCs for the further recruitment of T cells for further production of proinflammatory cytokines or
chemokine’s such as IL-4, IL-5, and TNF-α. TSLP also activates mast cells to produce other cytokines e.g. IL-
13, IL-5 and TSLP itself (not shown).
Reprinted from Hamida Hammad et al. 2008: DCs and epithelial cells: linking innate and adaptive immunity in
asthma: Nature Reviews Immunology 8, 193-204 (March 2008), Copyright © 2008, with permission from Nature
Publishing Group (2014).
29

Introduction
1.5.2 Role of TSLP in AD
Many factors can elicit AD when overexpressed, though not being absolutely
essential. The role of TSLP in AD development was not clear until studies showed
that an overexpression of TSLP in the skin of mice leads to the development of a
“spontaneous” dermatitis, the most characteristics feature of human AD49,103. Since
TSLP is primarily produced by epithelial cells, this provided further evidence to the
theory of KCs as the “initiators” of AD (Fig. 7)127. Later on various groups confirmed
TSLP as a major initiator of AD50,51,128. Another study has shown that a direct
administration of TSLP into the skin leads to AD-like lesions74. Although this thesis
is focusing on the skin, similar results were obtained for atopic asthma models60,129.
TSLP is involved in the proliferation and differentiation of Th2 cells and the
subsequent production of IL-4, IL-5, IL-13 and TNF-α103. Moreover, it was found that
TSLP is highly expressed in keratinocytes from AD patients with acute and chronic
lesions. Additionally it is associated with the activation and migration of DCs within
the dermis103. Therefore, TSLP was suspected to be one of the initiating factors for
the development of AD.
Yoo et al. (2005) reported that keratinocyte specific overexpression of TSLP elicited
skin disease with all the characteristic features of human AD, such as edema
hyperkeratosisa, dermal mononuclear cell infiltrate49. Mice lacking T cells, but
overexpressing keratinocyte-specific TSLP still develop skin inflammation, indicating
that T cells are not required for disease progression49. Other studies with different
AD models using TSLPR-/- mice show that TSLP is necessary to induce AD i.e.
TSLP-/- mice failed to develop AD115,130.
30

Introduction
1.6 OBJECTIVES
Over the years, TNF-α have been well characterized as crucial proinflammatory
cytokine with its roles in both host defense and inflammatory diseases80.
Consequently, anti-TNF therapies are an approved treatment for autoimmune
diseases, including rheumatoid arthritis and psoriasis77 with eczema development
as the most common side effect90,91. However the role of endogenous TNF-α in
acute skin irritation and in AD development is not well understood. In this thesis, the
role of endogenous TNF in skin irritation but also in an AD model was analyzed in
TNF-α deficient mice.
Within this thesis the following questions were addressed
1) Can the clinical observations be replicated in a murine disease model? And if so,
what are the mechanisms?
2) Is irritation responsible for TSLP induction outside of a typically allergic condition,
and what are the associated mechanisms?
3) Is TSLP is the factor responsible for the exaggerated dermatitis in the absence of
TNF?
3) Are TNF-/- mice inherently prone to increased TSLP production or does it require
the micromilieu of the AD?
5) Does TNF deficiency lead to enhanced TSLP production through an indirect
mechanism by affecting the micromilieu and whether and to what extent are MCs
crucial elements in this cascade?
To answer these questions will open a novel view on the inflammatory processes
operating in the initiation and development of AD.
31

Material and methods
2. MATERIAL AND METHODS
2.1 MATERIALS
Details about antibodies, instruments, chemicals, buffers, solutions, reagents,
labwares and software used are listed below:
Table 1: List of reagents
Reagent Supplier Catalog Number
α-monothioglycerol Sigma-Aldrich M-6145 Agarose Biozym 840004 Albumin from chicken egg white (OVA) Sigma-Aldrich A5503-10G Anti IgE BD Pharmingen™ 553413 Antibody diluent (Dako REALTM) DAKO Diagnostika S0809 Aqua Braun 2351744 Avidin/Biotin Blocking Kit Vector Laboratories,
Inc. SP-2001
Bovine serum albumin (BSA) PAA K45-001 Calcitriol Sigma-Aldrich D1530 Croton oil Sigma-Aldrich C6719 DermaLife K Medium Complete Kit Lifeline Cell
Technology LL-0007
Dispase BD Biosciences 354235 Desoxyribonucleic acid (DNA) Molecuar Weight XIII – 50 base pair (bp) ladder
Roche 11721925001
DNA Molecular Weight XIV – 100 bp ladder Roche 11721933001 En Vision+ System-HRP(AEC) Dako K-4005 Ethanol J.T. Baker 8025 Ethidium Bromide Solution Invitrogen 15585-011 Fetal Bovine Serum (FBS) PAA NC9862466 IgE BD Pharmingen™ 554118 IMDM medium PAA E-15-819 Hydrogen peroxide (H2O2) Sigma-Aldrich 216763 Histamine Sigma-Alrich H7125 Human TSLP ELISA kit eBioscience 88-7497-88 LightCycler® FastStart DNA Master SYBR Green I
Roche 12239264001
LSAB2 System-HRP Dako K0675
32

Material and methods
Mouse TSLP Duo Set R&D Systems® DY555 Nafamostat mesylate Sigma-Aldrich N-0289 Nucleo Spin® RNA II Macherey-Nagel 740955.250 PBS GE Healthcare H15-002 Penicillin/Streptomycin Biochrom A 2212 Peroxidase block Dako S2001
Phorbol 12-myristate 13- acetate(PMA) Sigma-Aldrich P 8139 Proteinase K Macherey-Nagel 740506
Recombinant Mouse Mast Cell Protease-6/Mcpt6
R&D Systems® 3736-SE-010
rh Skin beta Tryptase Promega G7061 Retinoic Acid Sigma-Aldrich R4643 rhIL-1β Immunotools 11340015 rhTNF-α Immunotools 11343013 rm IL-4 Peprotech 11340043 rmIL-1β Miltenyi 130-094-053 rmTNF-α Miltenyi 130-094-085 rm IL-25 eBioscience 14-8175-62 rm IL-3 Immunotools 12340033 rm IL-33 eBioscience 14-8332-62 rm IL-4 R&D 404-ML-010 Sodium dodecyl sulphate(SDS) Sigma-Aldrich L3371 TAE buffer (50x) Genaxxon M3087.1000 Tetramethylbenzidine Sigma-Aldrich T5525 TLR3 ligand InvivoGen tlrl-pic Transcriptor High Fidelity cDNA Synthesis Kit
Roche 05081963001
Trypsin / EDTA Solution Gibco® BD R-001-100 Trypsin inhibitor from Glycine max (soybean)
Sigma-Aldrich 9035/81/8
Tween 20 Sigma-Aldrich P1379-500ML Xylol Roth 9713.3
33

Material and methods
Table 2: List of antibodies and antagonist
Antibody Supplier Catalog Number
Anti-mouse TSLP R&D Systems® AF555 Biotin-sp-conjugated affinipure F(ab’)2 fragment rabbit anti goat IgG(H+L)
Jackson immunoresearch
305-066-003
Fluorescein iso thiocyanate (FITC) conjugated αm CD117 (c-kit), Clone 2B8
eBiosciences 11-1171-82
Purified NA/LE Rat Anti-Mouse CD117 BD Pharmingen™ 553867 Purified NA/LE Rat IgG2b, κ Isotype Control
BD Pharmingen™ 556968
PE conjugated αm FceRI α, clone: MAR-1 eBiosciences 12-5898-81 Mouse IgG2a R&D Systems® MAB003 Mouse mast cell protease-6/Mcpt6 antibody
R&D Systems® AF3736
Mouse TSLP Antibody R&D Systems® MAB555 Mouse IgG2A Antibody R&D Systems® MAB003 Rabbit anti-human IL-1α antibody Abcam ab9614 Rabbit anti-mouse IL-1α antibody Abcam ab9724 Rabbit IgG Abcam ab27478 rmIL-1Ra Immunotools. 12344870 rhIL-1Ra Immunotools. 11344874
Table 3: List of materials
Material Supplier Catalog Number
Biosphere® Filter Tips 0.5-20 µL 2-100 µL 100-1000 µL
Sarstedt 70.1116.210 70.760.212 70.762.211
Cell strainer, 40 µm BD FalconTM 352340 Cell strainer ,100 µm BD FalconTM 352360 Culture flask T 75 T 175
Cellstar®, Greiner-Bio 658175 660175
Conical tube ,15 mL BD FalconTM 352096 Conical tube ,50 mL BD FalconTM 352070 Descosept AF Dr Schumacher GmbH sc 311001
34

Material and methods
LightCycler® Capillaries Roche 04929292001 Micro tube, 0.5 mL Sarstedt 72.699 Micro tube, 1.5 mL Sarstedt 72.690.001 Micro tube, 2 mL Sarstedt 72.691 Precellys Steel Kit 2.8 mm Peqlab 91-PCS-
MK28 Quality Tips without filter 10 µL 200 µL 1000 µL
Sarstedt 70.1130 70.760.002 70.762
Serological Pipet 5 mL 10 mL 25 mL
BD FalconTM 357543 357551 357525
96-well cell culture plate Cellstar®, Greiner-Bio 655185 Petri dish Greiner-Bio 632181
Table 4: List of instruments
Instrument Type Supplier
Cell counter CASY® - TTC-2FC-1142 Innovatis AG, Reutlingen
Centrifuge Megafuge 1.0R Thermo Scientific, Schwerte
CO2-Incubater HERAcell® Thermo Scientific, Schwerte
Electrophoresis System Sub-Cell® GT Bio Rad, München Gel Imager Gene Genius Syngene, Cambridge
Inverted Reflected-Light Microscope Zeiss Axiovert 10 Zeiss, Jena
Light Cycler Roche,Penzberg
Flow Cytometer MACS Quant Miltenyi Biotec, Bergisch Gladbach
Microplate reader Dynatech MRX Dynex Technoloies, Chantilly
Multipipette Multipipette® plus Eppendorf, Hamburg
Pipette Eppendorf Reference® / Research®
Eppendorf, Hamburg
Pipettor Pipetus standard Hirschmann Laborgeräte,
35

Material and methods
Instrument Type Supplier Eberstadt
PCR machine Px2 Thermal Cycler Thermo Electron Corporation
Power Supply POWER PAC 300 BioRad, ‚München
Spectrophotometer Nano Drop 1000 Thermo Scientific, Schwerte
Tabletop centrifuge with refrigeration Centrifuge 5417C Eppendorf, Hamburg
Tabletop Centrifuge Centrifuge 5417R Eppendorf, Hamburg
Thermomixer Thermomixer comfort Eppendorf, Hamburg
Tissue homogenizer Precellys 24 Bertin Technologies, Montigny-le-Bretonneux
Waterbath MA6 Lauda, Lauda-Königshofen
Vortexer REAX 2000 Heidolph, Schwabach
2.2 METHODS
2.2.1 Animal experiments
2.2.1.1 Breeding of B6;129S-Tnftm1Gkl/J (TNF-/-) mice
TNF-/- mice were provided by Professor Max Löhning from DRFZ, Berlin. To
generate these mice, targeting vector was constructed by replacing TNF gene with
MC1neopA cassette (Stratagene) the 438 bp Narl-BglII fragment containing 40 bp
of the 5' UTR, all the coding region, including the ATG translation initiation codon, of
the first exon and part of the first intron of the mTNF-α gene131. These mice were
bred and maintained under pathogen free conditions in animal facility. All
experiments were performed according to German animal protection law.
36

Material and methods
2.2.1.2 Genotyping of TNF-/- mice
Genomic DNA was isolated from 5 mm2 tail biopsies of TNF-/- mice by using the
nucleospin tissue kit, according to manufacturer’s protocol. PCR was performed to
identify the genotype of mice. TNF-α gene primer sequences were obtained from
the ‘The Jackson laboratory’ site (strain stock no.: 003008) and were synthesized
from TIB MOLBIOL, Berlin, Germany and are specified below:
Primer Sequence: Primer Sequence Primer type (short name)
oIMR4182 5’-tagccaggagggagaacaga-3’ Common (GC)
oIMR4183 5’-agtgcctcttctgccagttc-3’ Wild type Reverse (GW)
oIMR7297 5’-cgttggctacccgtgatatt-3’ Mutant Reverse (GM)
Reaction component:
Regents Volume (µl) Final concentration
10x GenTherm buffer 1.2 1x
50 mM MgCl2 0.48 2 mM
10 mM deoxyNTPs 0.24 200 nM
10 μM forward primer (GC) 1.2 1 μM
10 μM reverse primer (GW) 1.2 1 μM
10 μM reverse primer (GM) 1.2 1 μM
50 U/μl DNA polymerase 0.075 0.03 U/μl
DNA 2
dH2O (makeup the volume up to 14µl)
The following PCR program was used:
94 °C - 3 min
94 °C - 30 sec
62 °C - 1 min 35 cycles
72 °C - 1 min
72 °C - 2 min
37

Material and methods
4 °C - onhold
2 μl of 10x DNA loading dye were added to each PCR products and separated on a
2 % agarose gel. Gels were photographed with a UV light photometer and bands
were further analysed to determine the genotype of the mice.
Expected band: Mice Band size
TNF-/- homozygous 318 bp
TNF-/- heterozygous 183 bp and 318 bp
Wildtype (wt) 183 bp
2.2.1.3 In vivo skin irritation model
Figure 8: Experimental scheme of skin irritation model with different irritants treatment in vivo.
10 week old female C57BL/6 (wt) and TNF-/- mice were gently dry shaved at three
different regions and exposed 30 times either to croton oil, 1% SDS or tape
stripping using cotton swabs or cello tape. The fourth skin region was shaved 30
times with a help of wet shaver (Fig. 8). The groups of mice were sacrificed after 4
38

Material and methods
and 18 hr, and blood was collected for serum. 5 mm2 skin biopsies were collected
for immunochemistry and mRNA isolation.
2.2.1.4 Mouse model of AD
Figure 9: Experimental scheme of mouse model of AD with different antibodies/cytokines treatment in vivo
To induce AD, an adapted protocol from Dahten et al 2008 was used132. Briefly, 10
weeks old female wt and TNF-/- mice were sensitized by three subsequent
intraperitoneal injections (i.p) with 100 μl of 10 μg ovalbumin (OVA) adsorbed to 1.5
mg Al(OH)3 (alum) on days 1, 14 and 21 (black arrows in Fig. 9). On day 21, the
belly of the mice was shaved by wet shaving, further tape stripped and 100 µg OVA
allergen was applied epicutaneously by the patch test method for one week period.
Each mouse had a total of three one week allergen exposures at the same site on
the skin with a two week intervals in between without any allergen.
To better understand the intrinsic role of TNF in skin inflammation, different
mediators and specific antibodies were applied intradermal (i.d) to the mouse skin
one day before the patch, half a day before the patch renewal and in the middle of
the patch-free week (blue arrows in Fig. 9). The dose and timing schedule of the
antibody application was based on data from the literature i.e. anti-TSLP 20
µg/mice i.d.133, anti-c-Kit 40 µg/mice i.d134.
On day 71, mice were anesthetised by isoflurane and sacrificed by cervical
dislocation. Blood was collected for further analysis of immunoglobulin and
cytokines levels in the serum. Photographs of the patch area were taken for the
39

Material and methods
assessment of the symptom score. 5 mm2 skin biopsies from lesional skin were
taken for immunohistochemistry in O.C.T compound and frozen slowly into liquid
nitrogen or in formalin for paraffin embedding. Rest of the skin was frozen into liquid
nitrogen for mRNA isolation. All frozen samples were stored at -80 °C until further
analysis.
2.2.1.5 Assessment of AD symptoms
AD severity was evaluated by using a skin score which nearly resemble to a score
which is widely used in clinical practice. The SCORAD (scoring of atopic dermatitis)
considers different clinical features to determine the severity of AD in humans135. In
our model such typical features used to evaluate the severity were papulation,
erythema, excoriation/crusting, dryness and extension of the lesions. Each
parameter was evaluated independently in a blinded manner by six individuals in a
randomized order. Severity for each parameter was rated as following: 0, no
symptom; 1, mild symptoms; 2, intermediate symptoms; and 3, severe symptoms.
The score from all the six individuals for each of these factors were then summed
up together and the total skin score was taken as AD severity with maximal skin
score considered as 15 and minimal 5.
Functional skin barrier assessment AD severity was further evaluated at a functional level by measuring TEWL in the
skin. This method measures the barrier dysfunction which is developing in
eczematous skin. During the measurement, the probe was placed on the belly of
the mice to measure TEWL. The vapor gradient density was measured indirectly by
two pairs of sensors i.e. sensors of temperature and relative humidity inside the
hollow cylinder of defined volume and analyzed by a microprocessor. The
measurement of TEWL is based on diffusion principle in an open chamber TEWL
machine136.
Blood samples Blood samples were taken on days 0 and 35 from the vena facialis with a micro
lancet by punction. On day 71, complete blood was withdrawn from retro orbital
40

Material and methods
venous sinus located behind the eyes. The blood was collected into special serum
separator tubes and centrifuged at 14,000 rpm for 10 min. Serum was further stored
at -80 °C until further analysis.
2.2.2 Cell culture methods
2.2.2.1 In vitro culturing in mouse and human
2.2.2.1.1 Isolation, culturing and treatment of primary Keratinocytes
Figure 10: Example of murine keratinocyte culture. Keratinocytes were isolated from murine skin and cultured. A) Shows first growing colonies of freshly isolated
keratinocytes and B) shows the confluent cells ready for passage. (385 x magnification).
Mouse keratinocytes: Mice were anesthetised by isoflurane and sacrificed by cervical dislocation. The
skin was gently shaved and the primary keratinocytes were isolated according to a
published protocol with few adaptations137. Cells were cultured in DermaLife® K
serum-free keratinocyte culture medium supplemented with essential factors, 30 µM
calcium chloride and penicillin/streptomycin (KC medium)(Lifeline Cell Technology,
Walkersville, MD, USA).
After the KCs reached 70-80% confluency, cells were passaged using trypsin-
Ethylenediaminetetraacetic acid (EDTA) (PAA Laboratories, Cölbe, Germany). Cells
were counted and cell viability was checked by CASY® Cell Counter (CASY) or by
trypane blue using haemocytometer. After the 2nd passage, 7.5x103 cells per well
were seeded in a 96-well plate to grow for 96 hr in hydrocortisone hemisuccinate 41

Material and methods
free KC medium. Cells were stimulated with 10 μg/ml TLR3-ligand, 20 ng/ml rmIL-
1β, 20 ng/ml rmTNF-α, 20 ng/ml rmIL-4, 10 ng/ml rmIL-25, 50 ng/ml rmIL-33 or 50
ng/ml PMA for 24 hr. Supernatants were collected and measured by a mouse TSLP
enzyme linked immunosorbent assay (ELISA) Kit. (R&D Systems, Minneapolis, MN,
USA).
Human keratinocytes: Human KCs were isolated from foreskin and processed as previously described138.
The skin was obtained after circumcisions, with informed consent of the patients
and approval by the university Ethics committee. All the experiments were
conducted according to the Declaration of Helsinki Principles. After the 2nd passage,
7.5x103 cells/well were seeded in a 96-well plate in KC medium and grown to 70-
80% confluence. After reaching confluence, the medium was changed to
hydrocortisone hemisuccinate free KC medium for 24 hr, and cells were stimulated
with 10 μg/ml TLR3-ligand, 20 ng/ml rhIL-1β, 50 ng/ml rhTNF-α and 20 ng/ml rhIL-4
for 24 hr. Supernatants were collected and measured by a human TSLP ELISA Kit.
(R&D Systems, Minneapolis, MN, USA).
2.2.2.2 Ex vivo culture and stimulations Mice were anesthetized and sacrificed by cervical dislocation. Skin of the mice was
gently shaved and 5 mm2 of biopsy punches were taken from the dissected skin.
The initial protocol was adopted from as previously described139. After the skin
biopsies were treated by 1% SDS, croton oil by the aid of a cotton swab and
physical scratching by a scalpel for 30 times each, biopsies were incubated in 150
μl of KCs medium without hydrocortisone hemisuccinate for 8 hr. Skin biopsies
were also stimulated with 20 ng/ml rmIL-1β, 20 ng/ml rmTNF-α and 20 ng/ml rmIL-4
for 8 hr. After stimulation, supernatants were collected and TSLP was quantified by
ELISA.
Inhibition experiments (mouse): 5 mm2 skin biopsies were immersed in to 1% SDS for 5 min followed by 5 times
washing. After washing, biopsies were stimulated with 200 ng/ml of rmIL-1Ra, 25
ng/ml of neutralizing αm IL-1α antibody and 25 ng/ml rabbit-IgG in to 150 µl of KCs 42

Material and methods
medium without hydrocortisone for 3 hr. After 3 hr, supernatants were collected and
TSLP was quantified by ELISA.
Inhibition experiments (human): Epidermal sheet from foreskin were isolated with overnight treatment with dispase
II. 5 mm2 of small pieces of epidermal sheet were cut carefully and immersed in 1%
SDS for 3 min, followed with 5 times extensive washing with KCs medium and
treated with 200 ng/ml of rhIL-1Ra, 1 µg/ml of neutralizing anti-human (αh) IL-1α-
antibody (and its respective concentration of rabbit-IgG as control) for 3 hr in KCs
medium without hydrocortisone. After stimulation, supernatant was collected and
TSLP ELISA was performed.
2.2.3 TSLP enzyme linked immunosorbent assay (ELISA)
Figure 11: Scheme of sandwich based - enzyme linked immunosorbent assay (ELISA) (adapted from Epitomics - an Abcam Company).
ELISA is an enzyme immunoassay used to measure the unknown level of antigens
in serum or supernatant. In this study we have used sandwich based ELISA to
quantify the level of protein. Here, first the primary antibodies were coated on the
surface of the plate and the target protein from serum or supernatant were
incubated for specific binding. The detection antibodies were incubated over the
surface of bound specific antigen. In the next step, the plates were incubated with
Horseradish peroxidase (HRP) linked biotinylated antibodies, which can convert a
chromogenic substrate. The enzymatic reaction leads to the color change which
was measured by spectrophotometer. The concentration of protein in the samples
was calculated by the means of standard curve. All the steps were performed at
room temperature and in dark from HRP-linked antibody.
43

Material and methods
Mouse and human TSLP ELISA: In vitro, ex vivo, or in vivo experiments were performed and cell free supernatant or
serum from mice and human epidermal sheet were obtained and measured for
mouse and human TSLP levels. Analysis was performed based on TSLP ELISA kit
from R&D system (mouse) and ebiosciences (human) according to manufacturer’s
instructions.
2.2.4 RNA isolation
Frozen skin samples from mice were homogenized by pre-chilled precellys
homogenisation (PEQLAB, Germany) in 500 μl RA1 buffer (NucleoSpin® RNA
isolation kit) along with 5 μl β-mercaptoethanol (β-Me) at 5500 rpm for 2*30 sec with
5 sec pause. Homogenized samples were transferred to NucleoSpin filter and
centrifuged at 11,000 g for 2 min at room temperature. Supernatant was taken out
carefully without disturbing the pellet and 500 μl of RNase-free water was added
along with 10% proteinase K and mixed well for tissue digestion. The lysate was
incubated for 15 min at 55 °C. After 15 min, lysate was spun down at 10,000 g for 3
min. Further, RNA isolation was performed according to manufacturer’s instruction
along with DNase digestion step for 15 min at room temperature. RNA was eluted
with 60 μl of RNase-free water. Using NanoDrop UV-Vis spectrophotometer, RNA
concentration was measured at 260 nm. Later, quality of RNA was checked by 2%
agarose gel. The eluted samples were stored at -80 °C for further analysis.
2.2.5 Reverse transcription
Total RNA was reverse transcribed into single stranded cDNA with TaqMan®
reverse transcription reagent according to manufacturer instructions. The kit
contains a recombinant Moloney Murine Leukemia Virus Reverse Transcriptase,
random hexamers and oligo d(T). 1 µg of total RNA was used for reverse
transcribtion in to cDNA in thermo cycler with following protocol.
44

Material and methods
Steps Temperature (°C) Time (min)
Incubation 25 10
Reverse transcription (RT) 48 40
RT inactivation 95 5
All cDNA samples were stored at -20 °C.
2.2.6 Real-time polymerase chain reaction
After RNA was reverse transcribed into cDNA with TaqMan reverse transcription kit
(Applied Biosystems, Darmstadt, Germany), fluorescence based real time
quantitative polymerase chain reaction (qPCR) was performed for the quantification
of gene expression in skin samples. qPCR was performed with LightCycler®
FastStart DNA Master SYBR Green I (Roche) according to the experimental
protocol below. The cDNA was pre-diluted 1:3 and the primers used were designed
by Primer3 software and are listed below. The formation of PCR product is
measured by increased level of fluorescence caused by specific binding of SYBR
green fluorescence dye to double-stranded DNA (SYBR green- Double-
Stranded DNA (dsDNA)). To ignore the non-specific binding by SYBR green, PCR
buffer also contains a reference dye to normalize the specific binding. The cycle
number of crossing point (CP) or the threshold cycle value (CT) is the number of
cycle at which significant increase of the normalized florescence is first measured.
Depending on CT values of a gene and the efficiency of primers, the relative
expression of a gene was calculated. The expression level of target gene was
normalized to the expression level of housekeeping gene i.e hypoxanthine-guanine
phosphoribosyltransferase (HPRT) using the 2-ΔΔCT method140.
45

Material and methods
Reagent Volume/sample (µl) Final concentration
10X FastStart DNA Master SYBR
Green I
0.50 1X
25mM MgCl2 0.80 3-5 mM
10µM Forward Primer 0.25 100-500 nM
10µM Forward Primer 0.25 100-500 nM
RNase-free H2O (makeup the volume up
to 3µl)
cDNA 2 (1:3 diluted stock)
Primer Sequence:
Gene
Primers
Sequence
Size
Product size
mHPRT
forward
reverse
5’-cgtcgtgattagcgatgatg-3’
5’-aatccagcaggtcagcaaag-3’
20
20
221
mDef B2 forward
reverse
5’-cactccagctgttggaagttt-3’
5’-gcaacaggggttcttctctg-3’
20
20
148
mDef B3 forward
reverse
5’-ctccacctgcagcttttagc-3’
5’-ggaactccacaactgccaat-3’
20
20
118
mIL-1α forward
reverse
5’-gctgaaggagttgccagaaa-3’
5’-cccgactttgttctttggtg-3’
20
20
146
mIL-1β forward
reverse
5’-tgaaatgccaccttttgaca-3’
5’-cttctccacagccacaatga-3’
20
20
190
mIL-4 forward
reverse
5’-gactctttcgggcttttcg-3’
5’-tgatgctctttaggctttcca-3’
19
21
105
mIL10
forward
reverse
5’-tttaagggttacttgggttgc-3’
5’-agggtcttcagcttctcacc-3’
21
20
137
mIL-33 forward
reverse
5’-atgggaagaagctgatggtg-3’
5’-ccgaggactttttgtgaagg-3’
20
20
150
mIFN-γ forward
reverse
5’-aactattttaactcaagtggcatagat-3’
5’-tgctgttgctgaagaaggtag-3’
27
21
217
mTslp forward
reverse
5’-agagaagccctcaatgacca-3’
5’-ggacttcttgtgccatttcc-3’
20
20
82
46

Material and methods
2.2.7 Isolation and culture of bone marrow cells and generation of bone marrow-derived mast cells (BMcMCs)
To isolate and culture bone marrow-derived mast cells, initial protocol was adopted
by Mrabet-Dahbi et al. 2009141. 10 week old wt and TNF-/- mice were sacrificed by
cervical dislocation. Skin was dissected and legs were separated from the hip to
foot. Foot was removed by cutting off the skin and ligaments. Muscles and tissue
from the leg was completely removed and tibia was separated from femur bone
without breaking the bones. Both bones were cleaned by Softasept®N and placed
into the falcon with washing medium (IMDM medium + 10% of
penicillin/streptomycin). Under sterile condition, bones were cut from both the side
and flushed with 10 ml of syringe filled with washing medium into a petri dish. After
all the bones were flushed out, single cell suspension was made by pipetting the
cells up and down. Cells were centrifuged at 1200 rpm for 10 min at 4 ºC. The
medium was discarded and cells were resuspended in to 20 ml of culture medium
with 10 ng/ml of IL-3 for mast cell differentiation. On day 5 and 8 cells were
centrifuged at 1200 rpm for 10 min at 4 ºC and further resuspended in to fresh
medium along with 10 ng/ml IL-3 and placed in to a new flask. On day 15, the
medium was changed and cells were moved in to a big flask with 40 ml of culture
medium. On day 19, 20 ml of medium was changed by centrifuging the cells at 1200
rpm for 10 min at 4 ºC. The cells were resuspended in to 20 ml of fresh medium and
put back in to culture flask along with 10 ng/ml IL-3. Medium was changed twice a
week till the cells were 4 weeks old. Cells were then checked for mast cell surface
receptors markers, IgE-receptor142 and c-kit143, by flow cytometry.
47

Material and methods
2.2.8 Flow cytometry
Figure 12: Exemplary flow cytometry images of BMcMcs for their characteristic markers c-kit and IgE receptor. Cells were visualized with anti-c-kit-FITC and anti- Fc epsilon receptor I (FcεRI)-PE. Around 34% of cells were
double positive and can be regarded as bone marrow-derived mast cells.
After 4 weeks of culture, mast cells were counted by CASY cell counter. 5 * 105
cells per sample were taken and centrifuged at 2400 rpm for 10 min at 4ºC.
Supernatant was discarded and cell pellet was washed once with MACS buffer at
2400 rpm for 10 min at 4 ºC. Cells were blocked with 1:500 dilution of Fragment
crystallizable of Ig (Fc) block (FcγREC 2.4 g; 5.1 MG/ML, DRFZ) in fluorescence
activated cell sorter (FACS) buffer for 15 min at 4 ºC. After 15 min, cells were
washed by MACS buffer at 2400 rpm for 10 min at 4 ºC. Cells were further stained
by anti-CD117 FITC (c-kit) and anti-FcεRI (IgE) PE antibody 1:50 dilution in 100 μL
cell suspension for 30 min at 4 °C in dark. After 30 min, cells were centrifuged at
2400 rpm for 10 min at 4 °C. Cells were resuspended in to MACS buffer and fixed
by 1 % PFA in PBS for 15 min at 4 °C and then pelleted down at 2400 rpm for 10
min at 4 °C. Cells were resuspended afterwards with 500 µl of MACS buffer and
filtered to remove cell debris. Finally, cells were analyzed by flow cytometry within
24 hr.
48
Material and methods
2.2.9 Stimulation of BMcMCs
For ex vivo and in vitro experiments with mast cell supernatant, 2* 106 BMcMcs
were counted and washed with medium at 2400 rpm for 10 min at 4 °C. Cells were
resuspended and sensitized overnight with 1 µg/ml IgE144. Next day cells were spun
down and resuspended in 2 ml medium and rested for 1 hr. After 1 hr, 1* 106 cells
were seeded per well in a 24 well cell culture plate. One well was stimulated with 1
µg/ml of anti IgE145 for 30 min and other well served as an unstimulated control.
After 30 min, cells were transferred into a 1.5 ml tube and spun down at 2400 rpm
for 10 min at 4 °C. Supernatants were frozen in -80 ° C for further experiments.
2.2.10 Histology and immunohistochemistry
The frozen 5 mm2 of skin biopsies from patch area of mice were cut into 5 μM cross
sections by cryotome at -23 °C to -24 °C. The sections were directly transferred on
microscopic slides and dried on a hot plate for 15 min and stored at -80°C. The formalin treated 5 mm2 of skin biopsies were embedded in to paraffin blocks
and cut into 5 μM cross sections by microtone at room temperature. The sections
were directly transferred on microscopic slides and dried on a hot plate for 15 min.
After 15 min, sides were stored at room temperature.
Figure 13: Example of TSLP and MC staining in the skin of the mice. Cell infiltrates of A) TSLP B) MC respectively. Positive immunohistochemical stained cells were counted at 100X
magnification by using Axiovision software.
49

Material and methods
2.2.10.1 TSLP staining For TSLP staining, 5 mm2 skin sections were deparaffinized by following steps:
Steps Time (min)
Xylol 10
96% EtOH 5
96% EtOH 5
70% EtOH 5
H2O 5
Skin sections were blocked with 1% BSA for 20 min at room temperature and
incubated with H2O2 (Dako, Germany) for 10 min at room temperature to block
endogenous peroxidase activity. Skin sections were then washed 3 times in 1X PBS
with 0.05% Tween 20 (1X PBST) and blocked with the avidin/biotin blocking kit
(Dako, Germany) for 15 min each. Slides were washed as mentioned above and
sections were incubated with goat αm TSLP (clone no.: AF555, R&D Systems) for 1
hr followed by 3 times wash with 1X PBST. Samples were later incubated with
biotinylated SP conjugated affiniPure rabbit anti-goat IgG (Jackson
ImmunoResearch Laboratories) for 30 min at room temperature. Negative controls
were run in parallel omitting either the primary or the secondary antibody. After
washing the slides, sections were developed with the AEC substrate kit (Dako,
Germany) and counter-stained with hematoxylin (Sigma-Aldrich, Germany) to
staining the nuclei as blue and eosinophilic structures in red.
50

Material and methods
Figure 14: TSLP positive cells in the skin lesions upon acute irritation with croton oil for 18 hr.
2.2.10.2 Mast cell staining To stain the mast cells granules containing heparin and histamine (metachromatic),
skin section were rehydrated by 1X TBS for 3 min and stained with 0.1% toluidine
blue in 0.5N hydrochloric acid (HCl) for 1 hr followed by brief washing with tape
water.
Figure 15: Mast cell positive cells in the OVA induced AD skin lesions.
51

Material and methods
2.3 STATISTICAL ANALYSIS
Normally distributed data are depicted as mean ± SEM and non-normally distributed
data are shown as median ± range. Experiments with only two groups were
analyzed using t-test (paired or unpaired) or Wilcoxon matched paired test, when
groups were not normally distributed; for more than 2 groups, depending on the
data distribution, 1-way analysis of variance (ANOVA) was used, followed by
Bonferroni multiple comparisons test or Kruskal-Wallis test. Statistical analyses
were performed with GraphPad Prism version 5 (GraphPad Software, USA). P
value less than 0.05 was considered as statistically significant.
52

Results
3. RESULTS
3.1 SKIN IRRITATION LEADS TO TSLP PRODUCTION
3.1.1 Physical or chemical irritation of the skin leads to production of TSLP in vivo
As many studies have shown that a genetic manipulation of the skin can lead to
elicit TSLP expression in keratinocytes upon skin barrier disruption51,125,128,146, we
speculated whether acute skin irritation would be sufficient to initiate TSLP
production in the skin upon the treatment with physical or chemical irritants in vivo.
For this purpose, mice were subjected to 4 different irritation protocols. A defined
area of the belly and the back were treated for 4 and 18 hrs. The types of irritation
included shaving and tape stripping as mild physical injuries, 1% SDS and croton oil
were used as chemical stimuli. As shown in Fig. 16A, the TSLP gene expression
was induced in all settings, in particular by pure croton oil, which contains phorbol
esters147, followed by wet shaving, SDS and tape stripping. The induction of TSLP
mRNA expression was rapid, as it was mostly induced after 4 hr, but differences
compared to the control were still visible after 18 hr (Fig.16A). This mild irritation on
a reduced skin area was already sufficient to initiate a systemic TSLP protein
response, so that irritated (but not control) mice displayed substantial amount of
TSLP in the serum (Fig. 16B).
Figure 16: Physical and chemical irritation of the skin promotes TSLP production in vivo.
53

Results
A) TSLP mRNA expression in the irritated skin after 4 hr (left panel) and 18 hr (right panel) post-irritation,
respectively. Data are shown as mean ± SEM. B) TSLP protein levels in serum after 4 and 18 hr post-irritation,
each mouse is indicated by a single dot, the bar corresponds to the median+range; n = 7-12 mice/group (*P ≤
0.05, **P ≤ 0.01, ***P ≤ 0.001).
Likewise, TSLP protein levels in skin biopsies were detectable in the irritated skin
by immunohistochemistry (Fig. 17). Overall, even mild skin irritation lead to rapid
production of TSLP protein, which can be detected systemically as it enters to the
circulation quickly, thereby becoming systemically measurable.
Figure 17: Physical and chemical irritation of the skin promotes TSLP production in vivo. TSLP staining by immunohistochemistry in skin section after irritation with tape stripping, shaving, SDS or
croton oil after 18 hrs.
54
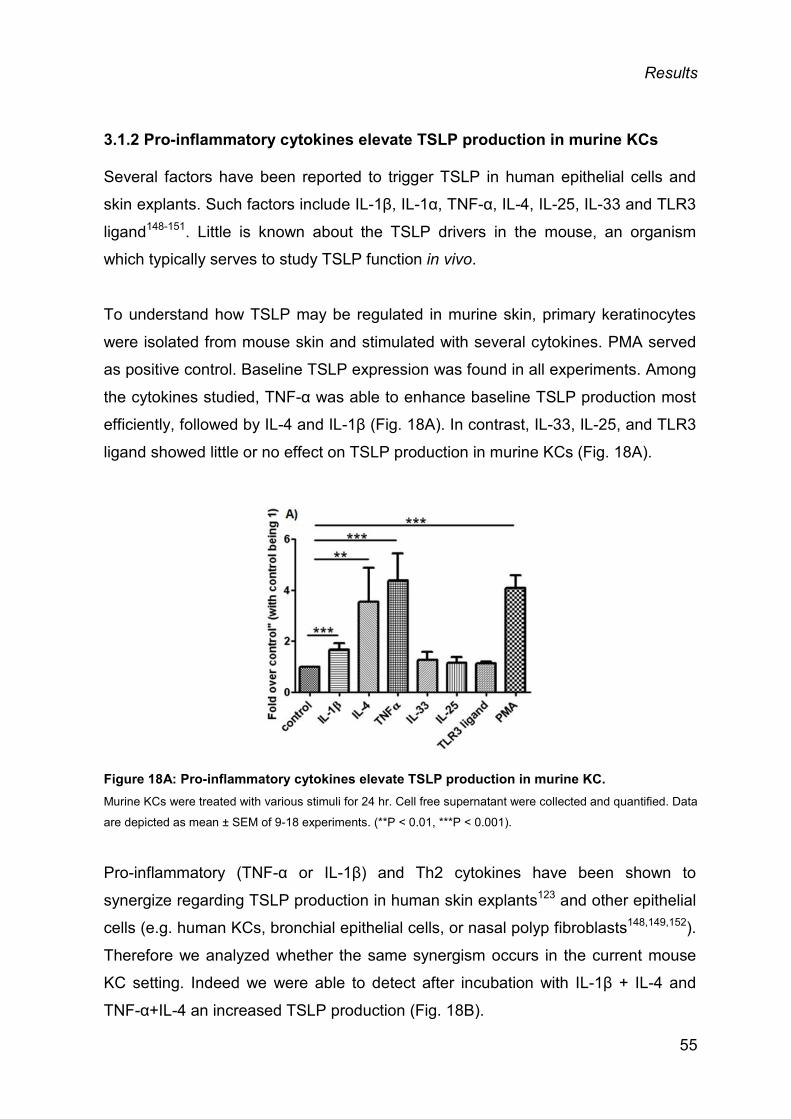
Results
3.1.2 Pro-inflammatory cytokines elevate TSLP production in murine KCs
Several factors have been reported to trigger TSLP in human epithelial cells and
skin explants. Such factors include IL-1β, IL-1α, TNF-α, IL-4, IL-25, IL-33 and TLR3
ligand148-151. Little is known about the TSLP drivers in the mouse, an organism
which typically serves to study TSLP function in vivo.
To understand how TSLP may be regulated in murine skin, primary keratinocytes
were isolated from mouse skin and stimulated with several cytokines. PMA served
as positive control. Baseline TSLP expression was found in all experiments. Among
the cytokines studied, TNF-α was able to enhance baseline TSLP production most
efficiently, followed by IL-4 and IL-1β (Fig. 18A). In contrast, IL-33, IL-25, and TLR3
ligand showed little or no effect on TSLP production in murine KCs (Fig. 18A).
Figure 18A: Pro-inflammatory cytokines elevate TSLP production in murine KC. Murine KCs were treated with various stimuli for 24 hr. Cell free supernatant were collected and quantified. Data
are depicted as mean ± SEM of 9-18 experiments. (**P < 0.01, ***P < 0.001).
Pro-inflammatory (TNF-α or IL-1β) and Th2 cytokines have been shown to
synergize regarding TSLP production in human skin explants123 and other epithelial
cells (e.g. human KCs, bronchial epithelial cells, or nasal polyp fibroblasts148,149,152).
Therefore we analyzed whether the same synergism occurs in the current mouse
KC setting. Indeed we were able to detect after incubation with IL-1β + IL-4 and
TNF-α+IL-4 an increased TSLP production (Fig. 18B).
55

Results
Figure 18B: Pro-inflammatory cytokines elevate TSLP production in murine KC. Murine KCs were treated with various combinations of stimuli for 24 hr. Cell free supernatant were collected and
quantified. Data are depicted as mean ± SEM of 9-18 experiments. (*P < 0.05, ***P < 0.001).
The above finding suggests that murine KC differ from human KC in somehow. To
confirm our results we directly compared the data from murine and human KCs. We
found profound differences between the two species regarding TSLP production
(Fig. 18C). As TNF-α failed to induce TSLP expression in human KCs compared to
mouse, while IL-1β was more effective in human KCs; TLR3 ligation was the most
active in human KCs149,153. Taken together, we conclude that overlapping
components of TSLP regulation, but at the same time important differences of the
regulation of TSLP were apparent between murine and human.
Figure 18C: Comparative analysis between murine and human TSLP production.
56

Results
KCs from mouse and human were isolated and stimulated with 10 μg/ml TLR3 ligand, 20 ng/ml IL-1β, 20 ng/ml
TNF-α or 20 ng/ml IL-4. TSLP levels were measured in the cell-free supernatants after 24 hr. The mean ± SEM
from 3-12 experiments is depicted (*P < 0.05, **P < 0.01, ***P < 0.001).
3.1.3 Skin biopsies from mouse and human produce TSLP ex vivo
By using an ex vivo strategy as previously reported for human skin139 and recently
re-established by us for murine skin154, we analyzed next, whether skin biopsies
derived from mice can be stimulated to produce TSLP ex vivo. Upon stimulation,
1% SDS was able to induce a significant amount of TSLP (Fig. 19A), while
scratching induced only a slight TSLP protein expression ex vivo, not reaching
statistical significance. Croton oil was not used for the stimulations due to its
viscous nature and ability to induce a rapid cell death when applied ex vivo (data
not shown). IL-1β, TNF-α and IL-4 induced again TSLP protein as determined in
previous in vitro experiments with mouse KCs. IL-1β was even more a potent TSLP
inducer ex vivo compared to KCs. Similar to the KCs data, IL-33 failed to enhance
TSLP production (Fig. 18A; Fig. 19B).
Figure 19: Chemical irritants and cytokines induce TSLP production ex vivo. Skin biopsies were stimulated for 8 hr with irritants or proinflammatory cytokines ex vivo. TSLP levels were
measured in the supernatants by ELISA, A) after the application of irritants, b) after stimulation with cytokines.
The mean ± SEM from 3-12 experiments is depicted (*P < 0.05, **P < 0.01, ***P < 0.001).
57

Results
3.1.4 IL-1 contributes to SDS-mediated TSLP induction
Irritation experiments revealed that TSLP mRNA and protein production was rapidly
induced upon SDS treatment (Fig. 16A; Fig. 19A). SDS is a known detergent which
can disrupt the skin barrier. This is followed by cell lysis with the release of IL-1α,155-
157 making it a key factor to explain SDS-mediated TSLP induction. To analyze this
hypothesis in more detail we first performed a kinetic study where we found that the
maximal TSLP induction was achieved after 3 hr (data not shown). We used this
system to study the influence of IL-1 by two different approaches. To counteract IL-
1 signalling, in the first approach IL-1Ra was employed158, whereas in the second
approach an IL-1α antibody was used to neutralize IL-1α by a function in SDS
treated skin biopsies in mice. Both approaches resulted in a significant reduction of
SDS-induced TSLP protein production (approximately 20%) (Fig. 20A).
Next, we investigated the influence of endogenous IL-1 on SDS-mediated TSLP
induction in human epidermal sheets considering that IL-1 had much greater effect
on TSLP induction in human compared to mouse KCs (Fig. 18C). By performing a
kinetic study on human epidermal sheets (epidermal sheet was used because total
human skin was too thick for stimulation), SDS elicited TSLP responses in human
epidermal sheets were likewise in murine skin the maximal TSLP induction was
observed after 3 hrs. In human skin, SDS-mediated TSLP induction was reduced
up to ≈50% compared to controls in both settings with the IL-1Ra and the anti-IL-1α
antibody (Fig. 20B).
58

Results
Figure 20: SDS-mediated TSLP induction is IL-1 dependent ex vivo. A) Skin biopsies from mouse were stimulated ex vivo by 1% SDS for 5 min in the presence or absence of
200ng/ml of mIL-1Ra or 25 µg/ml of αmIL-1α-Ab (rabbit IgG served as control). TSLP was quantitated in the
supernatants after 3 hr. The mean ± SEM from 8-10 experiments is depicted (**P < 0.01). B) Epidermal sheets
from human skin were isolated and stimulated ex vivo by 1% SDS for 3 min in the presence or absence of 200
ng/ml hIL-1Ra or 1 µg/ml αhIL-1α-Ab (rabbit IgG served as control). TSLP was quantitated in the supernatants
after 3 hr. The mean ± SEM from 4-5 experiments is depicted (*P < 0.05, **P < 0.01).
3.2 AGGRAVATED AD IN TNF-/- MICE
As the role of TNF-α in AD is not well understood and somewhat controversial, we
investigated this interaction in more detail. AD was induced in TNF-/- mice by
allergen dependent dermatitis. These mice developed a strong dermatitis in
comparison with their wildtype (wt) counterparts, which displayed a mild dermatitis.
The difference between the two groups of mice was highly significant (Fig. 21B).
Conversely, the skin of TNF-/- mice was normal and healthy as wt mice at the
baseline with comparable dermal and epidermal thickness, T cell, MC numbers and
KCs (data not shown, Fig. 24A), suggesting that the development and maintenance
of skin structure does not require TNF-α.
59

Results
Figure 21: TNF-/- mice exhibit increased AD severity. A) Representative illustration of skin lesions with wt and TNF-/- mice AD. C) Quantification of the symptom score
based on different criteria: erythema, extension, dryness, excoriation, crusting (Score 0-3). Each dot represents
single mouse; Median from 3 experiments is depicted (***P < 0.001)
3.3 ROLE OF TSLP IN AD AGGRAVATION UPON TNF DEFICIENCY
3.3.1 Increased TSLP levels in lesional skin of TNF-/- mice and correlation with AD severity
In search of potential AD promoting factors involved under TNF deficiency, we
analysed the local and systemic immune response in these mice. Lower levels of
IgE and IgG1 were observed in the serum of TNF-/- mice. Similarly, neither CD4+
nor CD8+ T cells were changed in lesional skin of the TNF-/- mice (Appendix Fig.
31) compared to wt mice and did not correlate with the symptom score of AD.
Similarly, key Th 1 and 2 cell cytokines, like IL-4, IL-10, IFN-γ and IL-17 were either
equally expressed or slightly enhanced (IFN-γ) in the TNF-/- mice (Appendix Fig.
32). Accordingly, enhanced AD in TNF-/- mice was unlikely due to enhanced T cell
infiltration.
Additionally, other factors like IL-1β (slightly increased), IL-1α, IL-13, IL-33, β-
defensins and skin barrier genes were either comparable or slightly increased in the
60

Results
skin of TNF-/- mice and did not correlate with the AD symptom score (Appendix Fig.
33).
Furthermore, we observed a significant increase in of TSLP expression in the TNF-/-
mice (Fig. 22A and 22B). Moreover this finding correlated with the severity of AD as
indicated by linear regression analysis (Fig. 22C). Since TSLP is an important
regulator of AD manifestation, this result implies that TNF can counter regulate AD
development by diminishing TSLP production in vivo, which supports the concept of
TSLP as an excellent candidate to explain AD aggravation under TNF deficiency.
Figure 22: Significant increase of TSLP in TNF-/- mice and its correlation to severity. Allergen triggered dermatitis was induced in to wt and TNF-/- mice. On day 71, mice were sacrificed and lesional
skin was further analysed for A) TSLP mRNA level B) TSLP protein level C) correlation of TSLP mRNA with the
clinical severity of the dermatitis. Median of n = 14 mice/group. (***P < 0.001).
61

Results
3.3.2 Anti-TSLP protect TNF-/- regarding AD onset
To further investigate, whether TSLP is responsible for AD development under TNF
deficiency, we used neutralizing antibodies in the AD model to interfere with TSLP
function. Therefore mice were injected with anti-TSLP antibodies on days 41, 45, 62
and 66 (Fig. 9). Upon anti-TSLP application, the onset of AD was diminished,
compared to the wt controls. TNF-/- treatment with appropriate isotype controls
resulted in the development of a strong dermatitis as observed earlier (Fig. 23A).
The clinical score of the AD was significantly reduced in TNF-/- mice when treated
with anti-TSLP compared to its isotype control (Fig. 23B). The TEWL measurement
revealed similar results (Fig. 23C). TNF-/- mice treated with anti TSLP still displayed
TSLP mRNA in lesional skin, (Appendix Fig. 34A) which was not unexpected.
These data clearly indicate that anti TSLP treatment results in an altered TSLP
function but not its expression. Moreover, TNF-/- mice treated with anti TSLP
showed a decrease of MCs numbers in lesional skin compared to an isotype control
group (Appendix Fig. 34B).
Figure 23: Protection of AD in TNF-/- mice upon anti TSLP treatment. A) AD suppressed in TNF-/- mice by intradermal doses of anti-TSLP antibodies B) symptom score C) TEWL
which represents a characteristic of dermatitis. Median of n = 6-9 mice/group. (**P < 0.01).
62

Results
3.4. ENDOGENOUS TNF-α DOES NOT CONTRIBUTE TO TSLP PRODUCTION
TNF is a potent inducer of TSLP in vitro, as shown by our data from murine skin
(Fig. 18A-C, Fig. 19B). As the role of TNF-α is poorly defined in vivo and as it is not
clear if TNF-/- mice are inherently prone to produce increased levels of TSLP, we
studied next the impact of endogenous TNF-α on TSLP expression by analysing
TNF-/- compared to wt mice.
As mentioned above, we confirmed that the untreated skin of TNF-/- mice was
comparable to their wt counterparts (data not shown). Purified KCs from TNF-/- and
wt were not morphologically distinguishable (Fig. 24A). KCs from both strains show
a comparable growth and survival rate (data not shown). This suggests that TNF-α
does not have a direct influence on KC differentiation.
Surprisingly the in vitro data shows that a comparable amount of TSLP was
produced by TNF-/- KCs compared to wt KCs upon stimulation with IL-1β (Fig. 24B),
exogenous TNF-α and IL-4 (data not shown).
Similar results were obtained ex vivo; skin biopsies from both strains show
comparable levels of TSLP production when treated with physico-chemical irritants
(Fig. 24C) and proinflammatory cytokines (Fig. 24D) replicating the in vitro results.
Finally, to verify that endogenous TNF-α was not required for increased TSLP
production in mice, we performed an in vivo study with acute skin irritation. This
data reveal that TSLP production was unaffected under TNF deficiency at the
mRNA (Fig.24E) and protein levels (Fig. 24F).
63

Results
Figure 24: Endogenous TNF is not required for TSLP production in murine skin. In vitro: Primary KCs were isolated from wt and TNF-/- mice and compared for TSLP levels; A) Representative
illustration of primary KCs from wt and TNF-/- mice. B) TSLP levels in cell free supernatant stimulated with
20ng/ml IL-1β after 24 hr. Ex vivo: Comparison of skin biopsies from wt and TNF-/- mice regarding TSLP
production induced by C) physical or chemical irritation and D) cytokine mediators after 8 hr. In vivo: wt and
TNF-/- mice were subjected to different irritants E) TSLP mRNA levels in skin. F) TSLP protein levels in serum
after 18 hr. Median of n = 8 mice/group. Mean ± SEM is depicted from 4-9 experiments.
64

Results
3.5 MAST CELLS CONTRIBUTE TO TSLP PRODUCTION
3.5.1 MCs are increased in lesional skin of TNF-/- mice and correlate with AD and TSLP
Our data suggest that increased levels of TSLP under TNF deficiency were not the
result of an inherently prone TNF-/- mice to over express TSLP, but may requires an
inflammatory micromilieu as present in AD. As MCs play an important role in AD, we
performed an analysis of MC in lesional skin. Indeed, MCs numbers were
significantly increased in TNF-/- mice compared to wt mice (Fig. 25).
Figure 25: MCs are increased in lesional skin of TNF-/- mice A) Representative illustration of MCs numbers in the lesional skin of TNF-deficient mice. B) MCs numbers in the
lesional skin of TNF-deficient mice as compared to wt. Median of n ≤ 14 mice/group. (***P < 0.001).
Moreover, we determined a significant correlation of the severity of dermatitis (Fig
26A) as well as the TSLP expression level in lesional skin (Fig. 26B) by linear
regression analysis.
Stem cell factor (SCF) and its receptor c-kit are well known markers for the growth
and survival of MCs 159. Therefore, we measured the mRNA expression of SCF and
c-kit in lesional skin. SCF was slightly increased in the TNF-/- mice compared to wt
and c-kit expression remained unchanged (data not shown).
65

Results
Figure 26: MC numbers in the lesional skin correlates with the severity of AD and with TSLP mRNA expression. Correlation of MCs with A) symptom score and B) TSLP mRNA expression respectively. Median of n ≤ 14
mice/group. (***P < 0.001).
3.5.2 Anti c-Kit is protective for AD development in TNF-/- mice
Since, MCs numbers were increased in the AD model and correlated with the skin
score as well as with TSLP expression, further experiments were performed to
understand the role of MCs in the AD model by application of c-kit neutralizing
antibodies to interfere with MC increase. TNF-/- mice treated with anti c-kit
antibodies show a milder appearance of AD under TNF deficiency (Fig. 27A).
Accordingly, the skin score was reduced in TNF-/- mice, when treated with anti-c-Kit
antibody compared to its isotype control, though this data did not reach statistical
significance (Fig. 27B). The data from TEWL measurements indicated similar
results (Fig. 27C).
66

Results
Figure 27: Anti c-Kit antibody treatment alleviates AD symptoms in TNF-/- mice. A) AD model depicting treatment with anti c-Kit ab. B) AD slightly suppressed in TNF-/- mice by intradermal
administration of anti-c-Kit ab C) AD skin score D) TEWL, which represents a characteristic of dermatitis.
Median of n = 5-12mice/group. (**P < 0.01).
3.5.3 MCs do not produce a relevant amount of TSLP
We investigated next how MCs contribute to TSLP production. Published studies
suggest that mast cells can produce and respond to TSLP160. To confirm MCs as a
source of TSLP under TNF deficiency, BMcMCs were sensitized with IgE and then
stimulated with anti IgE in the presence or absence of either IL-1β or TNF-α or with
their combinations (Fig. 28A). TSLP was measured at the protein level. The data
clearly indicate that BMcMCs are not able to produce TSLP upon stimulation (Fig.
28B) either when driven from wt or TNF-/- mice. This data confirm that MCs are
unlikely to contribute significantly to an increased TSLP under TNF deficiency in an
inflammatory micromilieu.
67

Results
Figure 28: MCs are not producing relevant amounts of TSLP. A) BMcMCs from wt and TNF-/- mouse were sensitized overnight with IgE and stimulated of anti IgE in the
presence or absence of either IL-1β or TNF-α or with their combinations for 24 hr. B) TSLP protein levels in cell
free supernatant. The mean ± SEM from 3 experiments is depicted.
3.5.4 MCs as instructors of TSLP production by KCs
Based on literature and our data, it is clear that MCs are not potent producers of
TSLP133. We hypothesized that MCs may be able to instruct KCs to produce TSLP
under TNF deficiency as KCs are the most potent TSLP producers. This hypothesis
might also provide a substantial link between increased TSLP levels and MC
numbers which we observed in murine AD skin lesions from TNF-/- mice.
3.5.4.1 Resting MCs supernatant enhanced TSLP levels ex vivo
To validate our hypothesis, we performed an ex vivo experiment where we
stimulated skin biopsies with supernatants from stimulated or unstimulated (resting
MCs) BMcMCs with anti IgE for 30 min. Supernatants from stimulated BMcMCs
were not able to enhance the TSLP levels significantly, moreover higher amounts of
the supernatants (2%) resulted even in a slight inhibition of TSLP production (Fig.
29A). On the other hand, supernatants (1%) from unstimulated MCs promoted a
significant increase of TSLP production in murine skin biopsies (Fig. 29B), indicating
that MCs can instruct KCs to produce TSLP.
68
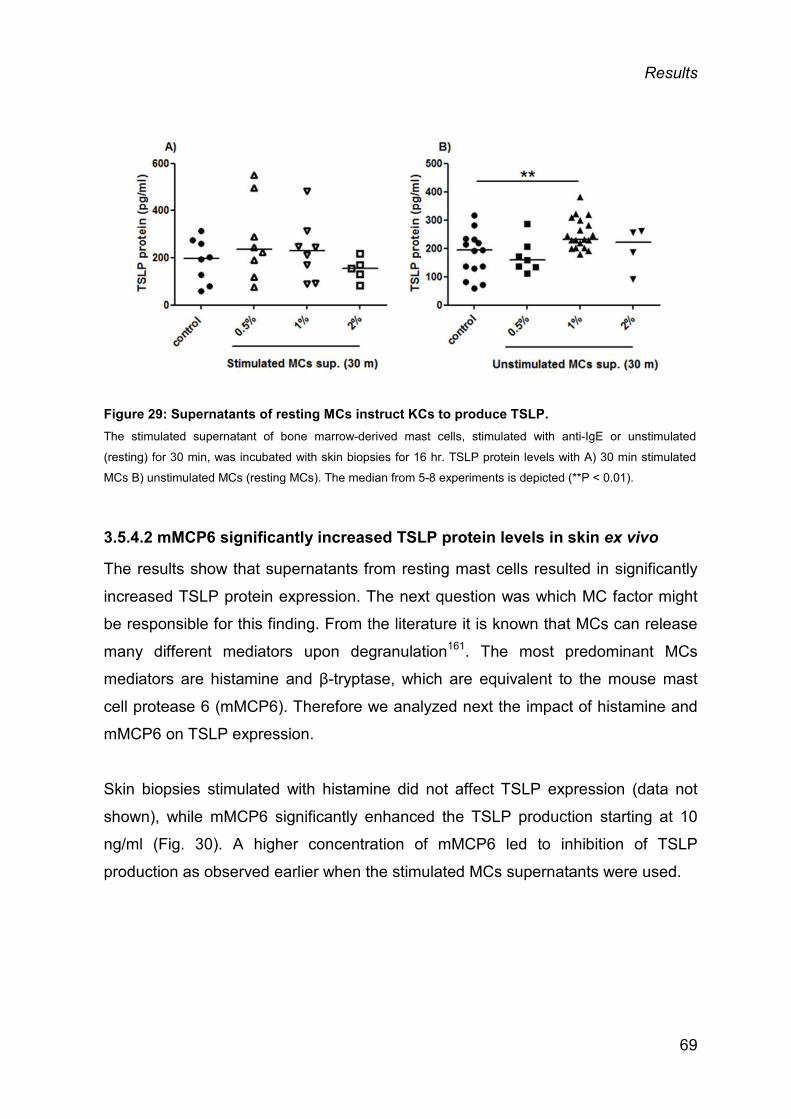
Results
Figure 29: Supernatants of resting MCs instruct KCs to produce TSLP. The stimulated supernatant of bone marrow-derived mast cells, stimulated with anti-IgE or unstimulated
(resting) for 30 min, was incubated with skin biopsies for 16 hr. TSLP protein levels with A) 30 min stimulated
MCs B) unstimulated MCs (resting MCs). The median from 5-8 experiments is depicted (**P < 0.01).
3.5.4.2 mMCP6 significantly increased TSLP protein levels in skin ex vivo
The results show that supernatants from resting mast cells resulted in significantly
increased TSLP protein expression. The next question was which MC factor might
be responsible for this finding. From the literature it is known that MCs can release
many different mediators upon degranulation161. The most predominant MCs
mediators are histamine and β-tryptase, which are equivalent to the mouse mast
cell protease 6 (mMCP6). Therefore we analyzed next the impact of histamine and
mMCP6 on TSLP expression.
Skin biopsies stimulated with histamine did not affect TSLP expression (data not
shown), while mMCP6 significantly enhanced the TSLP production starting at 10
ng/ml (Fig. 30). A higher concentration of mMCP6 led to inhibition of TSLP
production as observed earlier when the stimulated MCs supernatants were used.
69

Results
Figure 30: Mouse mast cell protease 6 (mMCP6) promote skin derived TSLP production ex
vivo. A) Stimulation of skin biopsies with mMCP6 for 16 hr, significant increase of TSLP protein levels stimulated with
concentration of 10 ng/ml. The median from 5-8 experiments is depicted (**P < 0.01).
70

Discussion
4. DISCUSSION
TNF-α is a well-known pro-inflammatory cytokine which plays a crucial role in
inflammatory diseases80. The role of endogenous TNF-α in skin inflammation and
particularly AD is not well understood. Based on the literature, an adverse
interaction between TNF and AD was described and suggests that in the absence
of TNF, AD seem to be enhanced87-89. On the other hand, TSLP which is directly
produced by keratinocytes is considered to be the initiator of the disease.
Previously, it has been shown that TSLP overexpression in mouse skin promotes
the development of a spontaneous dermatitis resembling characteristics of human
AD49,103. The role of TSLP in the development of allergic disease is well understood,
however the influence of endogenous TNF-α on TSLP production or its activation is
not yet clear.
In this thesis the irritation-induced TSLP production and its role in AD progression
was investigated. We also tried to better understand the role of endogenous TNF-α
in relation to TSLP production under irritative stimuli but also in an environment
which is present in AD. Another important component of AD pathogenesis are MCs.
They have been shown to be increased in lesional skin from AD patients but also in
lesional skin from AD mice162. To explore the role of mast cells as a trigger for TSLP
production by keratinocytes a mast cell depleting antibody was used in vivo and
mast cell supernatants but also mast cell mediators were analyzed in more detail ex
vivo.
4.1 SKIN IRRITATION LEADS TO RAPID INDUCTION OF TSLP, INDEPENDENT FROM TNF-α, BUT PARTIALLY DEPENDS ON IL-1
A disrupted barrier makes the skin more susceptible to the environment as shown
by Mogbekeloluwa O Danso et al. (2014)163. This allows allergens or irritants to
enter through the skin and to induce a Th2 response which in turn activates
keratinocytes to produce TSLP to activate dDCs thereby promoting the
inflammatory process. Many genetically modified mouse models have been used to
71

Discussion
show an essential role for TSLP in allergic diseases60,129. As our understanding
about functional aspects of TSLP in different pathophysiological conditions is slowly
increasing, the regulatory role of endogenous TSLP has been hardly well-defined.
Moreover, there are a lot of discrepancies between in vitro and in vivo studies164.
Various genetic studies with transgenic mice show that gene manipulation of
different genes in the skin was followed by an increase of the TSLP
expression51,125,146,165-167. The manipulation of different unrelated genes such as
Notch and lymphoepithelial Kazal-type-related inhibitor (LEKTI) display similar
outcomes. Therefore we hypothesized that skin perturbation caused by either
genetic manipulation or by environmental factors can initiate a specific cascade that
leads to TSLP production, even outside of an allergic scenario.
To further investigate our hypothesis and to understand whether barrier disruption
would be sufficient for the initiation of TSLP production in mouse skin, a range of
irritants were used. The data show that TSLP induction was an elicited as a
common consequence in inflamed skin when skin homeostasis was deviated. This
observation was made and confirmed by the variety of TSLP inducers, ranging from
physical trauma i.e. taking punch biopsy of the skin to mild physical irritation (wet
shaving, tape stripping) and chemical insults (croton oil, SDS). These data was
supported by a previous finding with the detergent SDS. SDS is used in different
models as irritants and can alter the stratum corneum due to its action on surface
tension. Therefore it has been used in different patch tests models and animal
assays as it is able to enhance the penetration of other substances168. Moreover
SDS can cause to a large extend alterations of the skin barrier function169. On the
other hand, croton oil has been used to induce ear edema in a mouse model170,171.
One important component of croton oil is the phenol ester 12-O-
tetradecanoylphorbol-13-acetate (TPA). Furstenberger et al. (1994) have shown
that topical application of croton oil can initiate local inflammation accompanied by
edema formation, polymorphonuclear leukocyte infiltration and epidermal
hyperproliferation, as a consequence of the production of inflammatory mediators,
such as prostaglandin E2, leukotrienes, histamine, serotonin and IL-1172. The non-
72

Discussion
specific skin inflammation elicited by SDS and croton oil resemble early phase
events during AD development in certain mouse models173,174.
In accordance with the literature, our data show a link between TSLP expression in
skin even in the absence of any particular allergic scenario. 2010, Angelova-Fischer
et al. show that injury of the stratum corneum by tape stripping and 2% SLS leads to
an increase of TSLP in human epidermis117. 2007, Allakhverdi et al. observed that a
trauma driven by a punch biopsy was sufficient to induce TSLP expression in
human skin122. These findings are in line with our data, derived from the acute
mouse skin model. Using this model we show that the increase of TSLP mRNA in
the skin but also protein in the skin and the serum was more pronounced after
physical irritation although the highest increase of TSLP expression was determined
after croton oil application. These data were further confirmed in our ex vivo cell
culture settings. Additional, evidence for the susceptibility of the TSLP gene to
various insults are known from physical irritation, UV irradiation, malignancy,
xerosis, and even in the absence of intestinal microbiota117,175-180. In support to
previous observations, our acute skin mouse model not only revealed that the
different types of stressors can lead to TSLP induction, but we could also for the
first time that TSLP induced after mild skin irritation can be measured in the serum
within in a short period of time i.e only after 4 hrs and increases over the time (18
hrs).
In search of a mechanism by which the gentle irritation and other distress can
induce TSLP in murine skin, we first looked for factors which can induce TSLP in
murine skin. As mentioned above several factors have been described to induce
TSLP in human keratinocytes. These include e.g. TNF-α, IL-1, IL-4, IL-25, IL-33 and
TLR3-ligand123,148,149,151, although their exact effects in murine keratinocytes are not
well known in detail yet. Our in vitro and ex vivo findings indicate that primary
keratinocytes and skin explants are capable to produce TSLP upon treatment with
different stimuli in a similar manner as in humans in spite of some unexpected
results. These findings are in accordance with a report from Takai et al. (2012)164.
These authors demonstrate that the gene expression of TSLP was induced in
bronchial epithelial cells (NHBE) after the exposure with different proinflammatory 73

Discussion
cytokines such as IL-1β, TNF-α and TLR2, TLR8, and TLR9 ligands. They also
indicated that the effects of these cytokines in both species are derived from the
NFκB pathway. In contrast, our data from the skin epithelial cells show that there is
considerable species dependence. TNF-α for example was able to induce TSLP to
a greater extend in mouse keratinocytes compared to human keratinocytes. By
contrast, IL-1β was more active in human keratinocytes compared to mouse. In
addition, TLR3 ligand was significantly stronger effective in human than in mouse
keratinocytes as described previously149,153.
In vitro studies with human primary keratinocytes show that cytokines like TNF-α in
combination with Th2 cytokines such as IL-4 and IL-13 can synergistically increase
the poly I:C (TLR-3 ligand) induced TSLP149. 2007, Bogiatzi et al. also found similar
results from human skin explants stimulated with different combinations of pro-
inflammatory cytokines such as TNF-α or IL-1α, in combination with Th2 cytokines
such as IL-4 and IL-13123. These results were further confirmed by many other
groups using different cell types. For example, Allakhverdi et al. (2007) show in
human airway epithelial cells, that a combination of TNF-α and IL-1α augmented
TSLP expression122. In accordance with these findings, we were able to confirm this
data in our in vitro and ex vivo settings. TSLP expression was increased in mouse
keratinocytes and in skin explants upon treatment with TNF-α and IL-1β in
combination with IL-4.
Finally, we addressed the mechanisms by which the acute skin irritation and other
physical stress can elicit TSLP in murine skin. TNF-α was the primary candidate to
be investigated as this cytokine is well-known to be induced upon skin irritation82,156
and to increase TSLP in the skin123. This was also observed in our in vitro and ex
vivo results making TNF-α to a probable intermediary in the cascade to mount
TSLP production. However, surprisingly we found that endogenous TNF-α was
expendable for TSLP production under all different settings i.e in vitro, in vivo and
ex vivo.
Taken together, it is obvious that exogenous TNF-α can induce TSLP in murine skin
under different settings, but endogenously it is not able to reach threshold levels
which may be required for its effect during skin damage. Although the impact of 74

Discussion
TNF-α regarding TSLP production in human compared to mouse skin was lower in
vitro, we investigated the role of endogenous TNF-α upon skin perturbation in
human epidermal sheets. As expected, endogenous TNF-α induced lesser TSLP
protein in the human in comparison to the mouse system emphasizing on its
dispensability in both the murine and the human skin.
Since TNF-α was found to be not required for TSLP production in the skin, an
alternative mechanism was needed to be identified. Upon trauma cells may die and
defined defense and repair processes in the respective host are activated. Such
processes or pathways include in part the activation of the ancient IL-1 family181,182.
Previous studies have shown that cytokines from the IL-1 family are involved in
SDS-mediated skin irritation156. Physical disruption of the skin can induce the
release of IL-1α. The dispensability of TNF-α and based on the literature, IL-1
seemed to be a reasonable candidate to explain the present findings. By using IL-
1Ra and IL-1α neutralizing antibodies, IL-1 was certainly shown to participate in
TSLP production in the skin when triggered by SDS. Around 20% of TSLP
production was reduced when treated with IL-1Ra or by the according neutralizing
antibodies. This indicates that IL-1 is involved in SDS mediated TSLP production in
damaged skin. As the reduction of TSLP reached only 20% we believe that other
factors may be also be involved and are likely to play an additional role in the TSLP
production upon SDS treatment. However, the other IL-1 family member IL-33183 is
not likely to be a suitable candidate as it was not able to induce TSLP in skin
constituents. Other new members of the IL-1 family whose specific contribution to
inflammatory skin conditions has only begun to be cleared182. So it is still not clear
whether these mediators such as high mobility group box chromosomal Protein 1
(HMGB1)184 might serve as a driving force for TSLP production in skin.
Nevertheless, IL-1 had a better impact on the irritation-induced TSLP production in
human skin, which was in the range of around 50%. The higher responsiveness of
human KCs towards IL-1 compared to murine KCs suggest that IL-1 is more
important in human skin compared to mouse skin. Therefore TSLP regulation in
different species will not be fully the same185. The apparent species variation in the
actions of TSLP provides another example of significant differences between the
human and murine immune system185. This is further supported by our observation 75

Discussion
that murine KCs were basically resistant to TLR3-mediated TSLP induction
compared to human KCs, although murine skin was susceptible to TLR3-ligands in
other settings186.
In summary it can be proposed based on the findings from this thesis that TSLP
acts as an executer of the innate alarm system aiming to protect the host defense
and restoring barrier function. That TSLP is critical to healing and barrier restoration
in mucosal tissues187-189, providing anti-tumor and antimicrobial effects in the skin
has already been demonstrated175,176,190 and that is moreover able to induce
extramedullary hematopoiesis191. Therefore it seems that the role of TSLP outside
of the allergic scenario will require a thorough investigation in the future.
4.2 TNF-/- MICE DEVELOP AGGRAVATED AD AND DISPLAY INCREASED TSLP EXPRESSION AND MCs NUMBERS CORRELATING WITH DISEASE SEVERITY
AD is a chronic inflammatory skin disease with a complex pathogenesis. It is
caused by a combination of an epidermal barrier dysfunction and an immune
dysregulation163. A variety of factors contribute to the pathogenesis of AD, including
environmental or genetic factors. Immunological factors as well as pharmacological
abnormalities play a role in disease progression192.
TNF-α is known for its pleiotropic functions in host defense but also for its role on
the elicitation of inflammatory diseases during immune dysregulation79,80. Anti-TNF
therapies are approved and effective for autoimmune disorders such as rheumatoid
arthritis, inflammatory bowel disease and psoriasis with relatively few side-effects77.
The two monoclonal antibodies adalimumab and infliximab and the soluble receptor
etanercept are the most commonly prescribed therapeutics in autoimmune diseases
interfering with the TNF function193,194. A direct role of TNF-α in AD initiation and
development is not well understood. A detailed analysis of the literature revealed
more evidence for negative than a positive association. 1992, Takahashi et al, has
76

Discussion
shown that the PBMCs from AD patients had decreased levels of TNF-α compared
to healthy controls86. Nomura et al. (2003) also observed lower levels of TNF-α, IL-
1β and IFN-γ in the skin from AD patients when compared to skin samples derived
from psoriasis89. Similarly dermal inflammatory DCs which produce TNF-α were
found to be reduced in the skin of AD vs psoriasis patients195. The most striking
evidence came from epidemiological studies from different clinics including our
institution, which reported the onset of AD like symptoms as a side effect of anti
TNF therapy90,91,196-198. These findings point towards a rather protective role of TNF
in the context of AD, but the experimental evidence was missing. To support
previous findings and clinical observations we used a murine model. To delineate
the underlying mechanisms, we applied an OVA-allergen AD mouse model in TNF-/-
mice to mimic the human AD scenario. Indeed, TNF-α protected against AD
development as reported in literature as worsening of the severity of AD-like
lesions occurred in TNF-/- mice. The measurement of the severity in the AD model
was adopted from the SCORAD score a tool to assess clinical severity in human
AD199. The majority of AD in human is characterized by increased levels of IgE
(extrinsic AD) and the presence of mostly Th2 cells in lesional skin32. Woodward,
A.L et al (2001) have shown that T cells play a critical role in skin inflammation200.
The key role of effector T cells and their major relevance in AD pathology was
further supported by the fact that under T cell immunodeficiency, elevated IgE levels
with eczematous skin lesions were observed32. Other studies have shown that
epidermal CD8+ cells are involved in the pathogenesis of AD201. In search of
mechanism behind AD their accumulation under TNF absence and also to
understand the clinical observations, various factors were analyzed. The analysis of
the humoral immune response from sera of mice from the AD model revealed a
decrease of the specific IgE and IgG1 concentrations in the TNF-/- mice whereas no
change was observed in infiltrating CD4+ and CD8+ T cells.
AD patients have shown to have higher frequencies of Th1, Th2, Treg and Th22
subsets as reported by Turner et al. 2012202. In addition these cells show a higher
expression of IL-5, IL-13, IL-1β IL-4, IFN-γ, IL-12, Gm-CSF, IL-10, TGF-β and IL-
688. The analysis of different proinflammatory cytokines and Th subsets; Th1, Th2,
77

Discussion
Treg, Th17 (e.g. IL-4, IL-10, IL-17, IFN-γ IL-1β, IL-1α, IL-13, IL-33 and β-defensins)
revealed no impact or slight increase on their expression under TNF deficiency
indicating that T cells were most likely not responsible for AD aggravation under
TNF deficiency.
Barrier dysfunctioning is well linked to AD pathogenesis. Aioi, A. et al. (2001) have
shown a crucial role of skin barrier function in AD manifestation using NC/Nga mice,
a spontaneous mouse AD model displaying skin barrier abnormalities like increased
TEWL and an abnormal skin conductivity under conventional conditions, but not
under specific pathogen-free conditions203. Similarly, Gupta et al. (2007) have
shown that the pathogenesis of AD is at least in part related to barrier
dysfunction204. In line with these findings, we investigated the expression of several
skin barrier genes in these mice. The expression level of transglutamase, involucrin,
loricrin and filaggrin were either comparable or slightly increased in TNF-/- mice and
did not correlate with AD symptom score (data not shown).
TSLP is a well-known key initiator of allergic diseases including AD and asthma126.
TSLP has been shown to be increased in lesional skin from AD patients but not in
nickel induced contact dermatitis103. TSLP is an IL-7 type of cytokine belonging to
the IL-2 family, which can act on DCs and can promote Th2 cell differentiation and
recruitment. TSLP can also directly act on naïve CD4+ cells to promote proliferation
in response to antigen114. Ziegler et al. (2013) demonstrated that epithelial cell-
derived TSLP can activate T cells, DCs and mast cells205. The overexpression of
TSLP in murine skin leads to the development of a spontaneous dermatitis49,103. In
agreement with the literature, TSLP was significantly overexpressed in AD skin and
also correlated with disease severity. TNF-α is known to regulate TSLP expression.
2011, Brandt and Sivaprasad provided evidence that human skin explants produce
higher levels of TSLP, when treated with a combination of pro-inflammatory
cytokines i.e. TNF-α, IL-1α and Th2 cytokines i.e IL-4 and IL-13, but not alone44.
Similar results were obtained from keratinocyte cultures149,206.
To understand why TSLP was enhanced in TNF-/- mice although TNF-α is a well-
recognized inducer of TSLP164 different factors and cell types were evaluated in 78

Discussion
TNF-/- mice. Another presumably important component in AD pathogenesis is the
mast cell. Various studies have shown that mast cells are commonly increased in
human but also the AD mouse model162. Therefore we analyzed the mast cell
numbers in lesional skin from the TNF-/- mice.
Mast cells, but not T cells as mentioned earlier were increased in AD lesions of
TNF-/- mice. Its number not only correlated with disease severity but also with
mRNA levels of lesional TSLP in the skin. In agreement with this finding Yong-Jun
Liu et al. (2006) also described that mast cells activated by IgE receptor cross-
linking express high levels of TSLP, which may support allergic inflammation121.
Interestingly, Na-Ra Han et al. in 2014 have shown as well that numbers of mast
cells in different organs of wildtype mice were significantly higher compared with
TSLP-deficient mice as well as TSLP was capable of inducing the proliferation and
differentiation of mast cells207.
4.3 ENHANCED TSLP LEADS TO AD MANIFESTATION
Th2 cytokines, IgE, mast cells, eosinophils and an increased expression of TSLP
are the common characteristics of AD pathogenesis127. Genetic screening of atopic
populations has shown an association between TSLP gene polymorphisms and AD,
clearly suggesting that TSLP plays an important role in atopic diseases208. Since
our AD model show an exaggerated TSLP expression under TNF deficiency, we
wondered if TSLP was responsible for AD development in TNF-/- mice. Saenz SA.
et al. (2008) reported that TNF-α is well-known as a positive regulator of TSLP in
vitro151. These findings are somewhat contradictory with our data were we found a
significant increase of TSLP levels under TNF deficiency. The administration of anti-
TSLP into the skin shows a clear-cut improvement of the symptom score in TNF-/-
mice, compared to the mice which were treated with its isotype control. This finding
is clearly indicating that TSLP plays a key role in the development of AD under TNF
absence. Another critical hallmark of barrier dysfunction is TEWL. An improvement
of the symptom score in AD skin is associated with a decrease in TEWL204. The
measurement of TEWL to backup the data from the symptom score show indeed a 79

Discussion
decrease of TEWL upon treatment with neutralizing TSLP antibodies which fail to
do so in the presence of an isotype control. In agreement with our data,
He and Geha (2010) have demonstrated that TSLPR-/- mice fail to develop allergic
skin inflammation in an AD mouse model upon repeated EC sensitization with OVA
after tape-stripping. They also show the blockade of TSLP by a neutralizing
antibody that inhibits the development of allergic skin inflammation, suggesting that
TSLP can amplify allergic skin inflammation during the effector phase by acting
directly on skin infiltrating T cells to induce Th2 cytokine secretion60.
The expression level of TSLP in TNF-/- mice after neutralization with anti TSLP
antibody was similar to the TSLP expression as observed after application of the
isotype control. These results indicate that the functional role of TSLP has been
altered with neutralizing antibody instead of inhibiting its production by the cells.
Ziegler and Artis (2010) hypothesize that TSLP can activate mast cells to further
produce increased levels of different cytokines such as IL-13127. In accordance with
these findings, we observed that the knockout mice treated with anti TSLP
displayed a decrease in MCs numbers in lesional skin compared to the isotype
control. This was most likely a result of an altered functional activity of TSLP after
the application of anti-TSLP.
Next the question arose, whether TNF-/- mice are prone to induce increased levels
of TSLP in general (e.g. upon trauma and skin irritation) or whether they require a
specific micromilieu as present in the AD skin. As pointed out earlier endogenous
TNF-α was not required for TSLP production, and its absence did not promote
TSLP over-expression upon acute skin irritation. These findings clearly indicate that
the milieu of AD was most likely necessary for the more pronounced production of
TSLP in lesional skin of TNF-/- mice.
80

Discussion
4.4 MCs SEEM TO PLAY A ROLE BETWEEN TNF-DEFICIENCY AND TSLP
Based on previous findings we hypothesize that TSLP is the trigger of AD under
TNF deficiency through an indirect micromilieiu mechanism as TNF can directly
induce TSLP as described earlier209. The increase of the number of mast cells in
lesional skin of TNF-/- mice and its significant correlation with the severity clearly
indicate that MCs are a hot candidate to explain the relationship between TNF-
deficiency and TSLP. Based on previous literature we assumed that MCs are
upstream of TSLP and downstream of TNF in this scenario. MCs have been linked
with TSLP in different contexts such as they are known to produce TSLP
themselves103,210, are responsive to TSLP122, and can enhance TSLP production by
epithelial cells133. Based on this literature, we aimed to clarify whether an
experimental manipulation of MC density or abrogation of MC function is facilitating
amelioration of AD.
Intradermal administration of cKit neutralizing antibodies to TNF-/- mice, resulted in
an improvement of the symptom score of the TNF-/- mice, compared to their isotype
control. Similar results were obtained with measurements by TEWL although these
data did not reach statistical significance due to a high variation within the groups.
The decrease in the symptom score of AD as well as in TEWL observed in mice
treated with cKit antibodies indicate that mast cells are involved in the process of
AD aggravation under TNF deficiency. Recently, Tomoaki Ando et al (2014) also
have demonstrated that mast cells but not the B or T cells are crucial for the onset
of spontaneous dermatitis in Plcb3-/- mice211.
Our next question was whether mast cells might contribute to TSLP production.
MCs are known to produce TSLP themselves103,210,212. Additional data from Yong-
Jun Liu (2006) suggest that mast cells which were activated by IgE receptor cross-
linking express high levels of TSLP121. In accordance to the above findings, we
activated BMcMcs with IgE cross linking in combination with other cytokines.
BMcMcs from TNF-/- mouse as well as wt mice failed to produce TSLP, indicating
that under TNF-/- deficiency MCs are not likely to be the major source of an
81

Discussion
increased TSLP production in an AD environment. Thus, we speculated that as
MCs are not able to produce TSLP, but rather may act as inducers of TSLP
production since they are also known to regulate epithelial cells regarding TSLP
expression in allergic rhinitis133.
Next our aim was to investigate the role of mast cells as triggers of TSLP production
by instructing keratinocytes as they are the best known TSLP producers127. For this
purpose mast cell supernatants and mast cell mediators were studied regarding the
onset of TSLP production. Different experimental settings with skin biopsies and
MCs were used to pin down the molecular cascade which promotes TSLP
production and consecutively support AD aggravation in a TNF deficient
environment.
Stimulation with anti-IgE led to crosslinking of the FcεRI and hence MCs
degranulate which progressively leads to the release of MC mediators like
histamine and proteases213,214. To investigate the role of MCs mediators that may
enhance TSLP expression by acting on keratinocytes in the skin, skin biopsies were
stimulated with the supernatants of either IgE cross linked stimulated or resting MCs
ex vivo. Stimulated mast cells were not able to enhance TSLP levels. This
implicates that high concentrations of MC mediators in the supernatants may even
inhibit TSLP production compared to their non-stimulated counterparts. This was
also observed by Okayama et al., 2009. These authors have shown that TSLP can
be degraded by MC-derived proteases210. Moreover, MCs release a plethora of
other mediators upon stimulation215-218 which can all have an impact on TSLP
production in the skin. Based on that, it is clear that mast cell supernatants contain
highly concentrated MC-derived proteases which can further promote TSLP
degradation.
Nevertheless, the supernatants from resting MCs contain only mediators released
spontaneously (e.g. through so called piecemeal degranulation219 (Dvorak and
Kissell, 1991)), granula-associated mediators are at lower concentrations in these
supernatants, and they are also virtually devoid of activation-induced mediators like
LTC4 and PGD2220. Interestingly, the supernatants from resting mast cells 82

Discussion
significantly increased TSLP levels in skin biopsies. These data allow to presume
that some of the MCs mediators instruct KCs to produce enhanced TSLP under
TNF deficiency.
In order to identify which factor of the mast cell supernatant may be responsible for
the increased TSLP expression by keratinocytes, the two most abundant mast cell
mediators, histamine and tryptase, were evaluated for their ability to enhance TSLP
expression.
Histamine failed to increase TSLP expression at protein levels. A dose response of
histamine was performed, but no single concentration elicited significant TSLP
expression. Histamine is believed to be a major player in the crosstalk between
mast cells and keratinocytes221,222. For example, human keratinocytes and
organotypic skin models revealed that histamine is able to down regulate the
expression of differentiation-associated proteins like filaggrin, keratin and loricrin, as
well as tight junction and desmosomal junction proteins223. These data suggest that
mast cell activation and histamine release contribute to skin barrier defects which
have been implicated in the initiation of AD223. In addition, histamine is one of the
major pruritogenic factors. Itch is a hallmark of AD, whereas histamine 1 receptor
(H1R) antagonist cannot ameliorate the itch in lesional skin of AD patients224. In
contrast it has been shown in a model of chronic allergic dermatitis in NC/Nga mice,
that the combined treatment of a H1R and histamine 4 receptor (H4R) antagonist
displayed anti-pruritogenic and anti-inflammatory effects225. Based on this evidence,
it is clear that, one of the most abundant mast cell mediators does not appear to
play a role for increased TSLP levels in AD.
The next major factor is the protease β-tryptase which is expressed in mast cells226.
Mast cell proteases constitute between 30-50% of the total mast cell protein
content218. Therefore this mast cell mediator was analyzed next as a possible
trigger of TSLP. The mouse analogue of human β-tryptase is mMCP6, which was
indeed resulting in an increased TSLP production.
83

Discussion
Based on our data it is difficult to answer the question whether tryptase is the main
component from the MC supernatant that triggers TSLP in skin. The application of
anti mMCP6 neutralizing antibody needs to be used to neutralize the induction of
TSLP by either resting mast cell supernatants or by mMCP6. Mast cells from
mMCP6-/- mice would be also required to confirm these data. But in support,
Thakurdas et al. (2007), showed that mMCP6 is not essential for migration,
retention and overall maturation of MC-committed progenitors in connective tissues
in mMCP6-/- mice227. These mice however had a reduced ability to combat K.
pneumonia infections, suggesting a critical immune protective role of mMCP6 in
bacterial infections227. Interestingly, it was reported that tryptase-like enzymes in the
stratum corneum were highly unregulated in lesional AD skin228. In addition, trends
of elevated tryptase levels in MCs from AD patients were observed229. Although,
mast cell tryptase serum levels were investigated for their suitability as a serum
marker for AD, two studies have shown no correlation between mast cell tryptase
serum levels and the severity of AD230,231. However, tryptase has been more and
more implicated with AD-mediated itch232.
Taken together, TNF-/- mice have increased mast cell numbers in lesional skin
which correlate with clinical severity and TSLP mRNA levels. Anti- cKit improved the
development of AD and reduced TEWL in TNF-/- mice compared to the controls.
BMcMcs data indicate that MCs are not producing TSLP as this is rather produced
by keratinocytes. In search of MCs mediators which enhance TSLP levels in TNF-/-
mice, we identified that supernatants from resting MCs increased TSLP production
in skin biopsies. Histamine was not able to modulate TSLP production whereas
mMCP6 was significantly able to induce TSLP production by keratinocytes in skin
biopsies, indicating that tryptase might be the relevant factor involved in instructing
KCs to produce TSLP under TNF deficiency.
4.5 CONCLUSION AND OUTLOOK
The epidermis is a rigid layer of the skin protecting an organism against external
insults. It is also the anatomical structure to provide the skin barrier. An alteration of 84

Discussion
the skin barrier initiates inflammatory processes which may lead to skin diseases. In
this thesis it has been investigated how TSLP is regulated either after exposure to
external irritants or in AD; a chronic inflammatory skin disease.
The data show that TSLP was rapidly induced in keratinocytes upon irritant
exposure. Moreover skin perturbation of different kinds led to TSLP production
starting from injury to chemical exposure. These data suggest that TSLP is one of
the alarm signals in the skin upon exposure to any trauma. The mechanistic
analysis revealed that though exogenous TNF-α was capable of inducing TSLP in
vitro or ex vivo, endogenous TNF-α failed to do so. IL-1 a well known responder
upon irritation, was partially involved in SDS mediated TSLP production. As the
cascade of TSLP regulation and its role in irritation is still not very clear, further
extensive work is required to pin point the different factors involved. To this end,
skin biopsies and keratinocytes will be treated with different inhibitors or neutralizing
antibodies or agonists and their antagonists.
To better understand the role of TNF-α in AD, an allergen dependent mouse
dermatitis model was used. It showed an increased AD severity in TNF-/- compared
to wt mice. Further analysis of these mice including the skin and serum revealed no
major alterations of single cell types or factors except the TSLP expression locally
and systemically and increased mast cell numbers in the skin which correlated with
the clinical severity. As TNF-/- mice expressed more TSLP, it was important to
understand the role of TSLP for the progression of AD. To achieve this goal, anti-
TSLP was administered to neutralize the TSLP mediated effects in eczema. Such
treated mice showed a pronounced improvement of the AD, including a reduction of
the TEWL. These findings indicate that TSLP is most likely a key cytokine in severe
AD development under TNF deficiency. These data should be confirmed using TNF-
/-TSLP-/- double knockout mice to prove the role of TSLP in this scenario.
We observed an increase of the mast cell frequency which correlates with the
symptom score and TSLP expression. The application of an anti cKit to the TNF-/-
mice showed a reduction of eczema severity, indicating that mast cells are involved
in AD in this model. 85

Discussion
As previous data pointed out the role of mast cells in AD progression, the question
arose whether mast cells can directly induce TSLP as they are known to produce
TSLP210 or whether they can instruct keratinocytes to produce TSLP during AD
progression as they are also known to instruct epithelial cells to produce TSLP133. In
our hand data from BMcMcs have suggested that mast cells are not able to produce
TSLP upon activation with anti IgE in combination with different proinflammatory
cytokines. These results suggest that mast cells can instruct KCs to produce TSLP
in TNF-/- mice during AD development. To confirm this hypothesis, we stimulated
the skin biopsies with mast cell supernatants from stimulated and resting mast cells.
Supernatants from stimulated mast cells show no effect whereas the supernatants
from resting mast cells were capable to stimulate the cells from skin biopsies to
produce TSLP at significant levels. Next we tried to investigate which mast cell
mediator is responsible for the instruction of KCs to produce TSLP. Surprisingly
‘histamine’ the most abundant mast cell mediator was not able to induce TSLP
production. By contrast, we detected a significant increase of TSLP expression
upon treatment with the mast cell protease “mMCP6”. In the future it needs to be
confirmed whether tryptase is the main component of the MC supernatant triggering
TSLP production in the skin. Such conformation would be possible if mMCP6-/- mice
will be used in further experiments. Finally to confirm that mast cells are playing a
role by instructing KCs to produce TSLP in enhancement of AD under TNF
deficiency we need to perform the AD patch-model in mMCP6-/-TNF-/-TSLPR-/- triple
knockout mice.
Irritation and inflammation of the skin is a complex process including the interplay of
resident skin cells like KC and MC. Both TNF-α and TSLP contribute to this
interplay in this scenario with a significant extend. Mouse skin models can help to
better understand this complex network by applying knockout mice models or
targeting key effector mediator in appropriate disease models.
86

References
REFERENCES
1 Proksch E, Brandner JM, Jensen JM. The skin: an indispensable barrier. Exp Dermatol 2008; 17: 1063-72. 2 Oyoshi MK, Murphy GF, Geha RS. Filaggrin-deficient mice exhibit TH17-dominated skin inflammation and permissiveness to epicutaneous sensitization with protein antigen. J Allergy Clin Immunol 2009; 124: 485-93, 93 e1. 3 Hanel KH, Cornelissen C, Luscher B et al. Cytokines and the skin barrier. International journal of molecular sciences 2013; 14: 6720-45. 4 Fuchs E. Skin stem cells: rising to the surface. The Journal of cell biology 2008; 180: 273-84. 5 Kirschner N, Brandner JM. Barriers and more: functions of tight junction proteins in the skin. Ann N Y Acad Sci 2012; 1257: 158-66. 6 Hildenbrand M, Rhiemeier V, Hartenstein B et al. Impaired skin regeneration and remodeling after cutaneous injury and chemically induced hyperplasia in taps-transgenic mice. J Invest Dermatol 2010; 130: 1922-30. 7 Madison KC. Barrier function of the skin: "la raison d'etre" of the epidermis. J Invest Dermatol 2003; 121: 231-41. 8 Richardson M. Understanding the structure and function of the skin. Nursing times 2003; 99: 46-8. 9 Janetta Bensouilah PB. Aromadermatology: Aromatherapy in the Treatment and Care of Common Skin Conditions. 2006. 10 Janetta Bensouilah, Buck P. Aromadermatology: Aromatherapy in the Treatment and Care of Common Skin Conditions. Aromadermatology 2006. 11 Kanitakis J. Anatomy, histology and immunohistochemistry of normal human skin. European journal of dermatology : EJD 2002; 12: 390-9; quiz 400-1. 12 Bruce Alberts AJ, Julian Lewis, Martin Raff, Keith Roberts, and Peter Walter. Molecular Biology of the Cell. Garland Science 2002; 4. 13 Flour M. The pathophysiology of vulnerable skin 2009. 14 Nestle FO, Di Meglio P, Qin JZ et al. Skin immune sentinels in health and disease. Nature reviews. Immunology 2009; 9: 679-91. 15 Angelova-Fischer I, Dapic I, Hoek AK et al. Skin Barrier Integrity and Natural Moisturising Factor Levels After Cumulative Dermal Exposure to Alkaline Agents in Atopic Dermatitis. Acta dermato-venereologica 2014. 16 Vinay Kumar AKA, Jon C. Aster. Robbins Basic Pathology. Elsevier Health Sciences 2012. 17 Le M, Schalkwijk J, Siegenthaler G et al. Changes in keratinocyte differentiation following mild irritation by sodium dodecyl sulphate. Archives of dermatological research 1996; 288: 684-90. 18 Varani J, Perone P, Spahlinger DM et al. Human skin in organ culture and human skin cells (keratinocytes and fibroblasts) in monolayer culture for assessment of chemically induced skin damage. Toxicologic pathology 2007; 35: 693-701. 19 Willis CM, Stephens CJ, Wilkinson JD. Differential patterns of epidermal leukocyte infiltration in patch test reactions to structurally unrelated chemical irritants. J Invest Dermatol 1993; 101: 364-70. 20 Jacobs JJ, Lehe CL, Hasegawa H et al. Skin irritants and contact sensitizers induce Langerhans cell migration and maturation at irritant concentration. Exp Dermatol 2006; 15: 432-40. 21 Escobar-Chavez JJ, Merino-Sanjuan V, Lopez-Cervantes M et al. The tape-stripping technique as a method for drug quantification in skin. Journal of pharmacy & pharmaceutical sciences : a publication of the Canadian Society for Pharmaceutical Sciences, Societe canadienne des sciences pharmaceutiques 2008; 11: 104-30.
87

References
22 Nickoloff BJ, Naidu Y. Perturbation of epidermal barrier function correlates with initiation of cytokine cascade in human skin. Journal of the American Academy of Dermatology 1994; 30: 535-46. 23 Wilmer JL, Burleson FG, Kayama F et al. Cytokine induction in human epidermal keratinocytes exposed to contact irritants and its relation to chemical-induced inflammation in mouse skin. J Invest Dermatol 1994; 102: 915-22. 24 Usatine RP, Riojas M. Diagnosis and management of contact dermatitis. American family physician 2010; 82: 249-55. 25 Coenraads PJ, Goncalo M. Skin diseases with high public health impact. Contact dermatitis. European journal of dermatology : EJD 2007; 17: 564-5. 26 Nosbaum A, Vocanson M, Rozieres A et al. Allergic and irritant contact dermatitis. European journal of dermatology : EJD 2009; 19: 325-32. 27 Slodownik D, Lee A, Nixon R. Irritant contact dermatitis: a review. The Australasian journal of dermatology 2008; 49: 1-9; quiz 10-1. 28 Dhingra N, Gulati N, Guttman-Yassky E. Mechanisms of contact sensitization offer insights into the role of barrier defects vs. intrinsic immune abnormalities as drivers of atopic dermatitis. J Invest Dermatol 2013; 133: 2311-4. 29 Vocanson M, Hennino A, Chavagnac C et al. Contribution of CD4(+ )and CD8(+) T-cells in contact hypersensitivity and allergic contact dermatitis. Expert Rev Clin Immunol 2005; 1: 75-86. 30 Khuntia A, Baldwin J. Contact Dermatitis. 2004. 31 Bieber T. Atopic dermatitis. The New England journal of medicine 2008; 358: 1483-94. 32 Leung DY, Boguniewicz M, Howell MD et al. New insights into atopic dermatitis. The Journal of clinical investigation 2004; 113: 651-7. 33 Worm M, Forschner K, Lee HH et al. Frequency of atopic dermatitis and relevance of food allergy in adults in Germany. Acta dermato-venereologica 2006; 86: 119-22. 34 Wuthrich B, Cozzio A, Roll A et al. Atopic eczema: genetics or environment? Annals of agricultural and environmental medicine : AAEM 2007; 14: 195-201. 35 Maintz L, Novak N. Getting more and more complex: the pathophysiology of atopic eczema. European journal of dermatology : EJD 2007; 17: 267-83. 36 Oyoshi MK, He R, Kumar L et al. Cellular and molecular mechanisms in atopic dermatitis. Advances in immunology 2009; 102: 135-226. 37 Bieber T. Atopic dermatitis. Annals of dermatology 2010; 22: 125-37. 38 Ong PY. New insights in the pathogenesis of atopic dermatitis. Pediatric research 2014; 75: 171-5. 39 Kuo IH, Yoshida T, De Benedetto A et al. The cutaneous innate immune response in patients with atopic dermatitis. J Allergy Clin Immunol 2013; 131: 266-78. 40 Chan LS. Atopic dermatitis in 2008. Current directions in autoimmunity 2008; 10: 76-118. 41 Bohme M, Soderhall C, Kull I et al. Filaggrin mutations increase the risk for persistent dry skin and eczema independent of sensitization. J Allergy Clin Immunol 2012; 129: 1153-5. 42 Howell MD, Kim BE, Gao P et al. Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol 2007; 120: 150-5. 43 Thyssen JP, Linneberg A, Carlsen BC et al. A possible association between a dysfunctional skin barrier (filaggrin null-mutation status) and diabetes: a cross-sectional study. BMJ open 2011; 1: e000062. 44 Brandt EB, Sivaprasad U. Th2 Cytokines and Atopic Dermatitis. Journal of clinical & cellular immunology 2011; 2. 45 Darsow U, Forer I, Ring J. Allergen-specific immunotherapy in atopic eczema. Current allergy and asthma reports 2011; 11: 277-83.
88

References
46 Jin H, He R, Oyoshi M et al. Animal models of atopic dermatitis. J Invest Dermatol 2009; 129: 31-40. 47 Gittler JK, Shemer A, Suarez-Farinas M et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J Allergy Clin Immunol 2012; 130: 1344-54. 48 Nakajima S, Kitoh A, Egawa G et al. IL-17A as an inducer for Th2 immune responses in murine atopic dermatitis models. J Invest Dermatol 2014; 134: 2122-30. 49 Yoo J, Omori M, Gyarmati D et al. Spontaneous atopic dermatitis in mice expressing an inducible thymic stromal lymphopoietin transgene specifically in the skin. J Exp Med 2005; 202: 541-9. 50 Li M, Hener P, Zhang Z et al. Topical vitamin D3 and low-calcemic analogs induce thymic stromal lymphopoietin in mouse keratinocytes and trigger an atopic dermatitis. Proc Natl Acad Sci U S A 2006; 103: 11736-41. 51 Briot A, Deraison C, Lacroix M et al. Kallikrein 5 induces atopic dermatitis-like lesions through PAR2-mediated thymic stromal lymphopoietin expression in Netherton syndrome. J Exp Med 2009; 206: 1135-47. 52 Gutermuth J, Ollert M, Ring J et al. Mouse models of atopic eczema critically evaluated. International archives of allergy and immunology 2004; 135: 262-76. 53 Spergel JM, Mizoguchi E, Oettgen H et al. Roles of TH1 and TH2 cytokines in a murine model of allergic dermatitis. The Journal of clinical investigation 1999; 103: 1103-11. 54 Niebuhr M, Werfel T. Innate immunity, allergy and atopic dermatitis. Current opinion in allergy and clinical immunology 2010; 10: 463-8. 55 Eckert RL, Rorke EA. Molecular biology of keratinocyte differentiation. Environmental health perspectives 1989; 80: 109-16. 56 Barker JN, Mitra RS, Griffiths CE et al. Keratinocytes as initiators of inflammation. Lancet 1991; 337: 211-4. 57 Palmer CN, Irvine AD, Terron-Kwiatkowski A et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nature genetics 2006; 38: 441-6. 58 De Benedetto A, Rafaels NM, McGirt LY et al. Tight junction defects in patients with atopic dermatitis. J Allergy Clin Immunol 2011; 127: 773-86 e1-7. 59 Gittler JK, Krueger JG, Guttman-Yassky E. Atopic dermatitis results in intrinsic barrier and immune abnormalities: implications for contact dermatitis. J Allergy Clin Immunol 2013; 131: 300-13. 60 He R, Geha RS. Thymic stromal lymphopoietin. Ann N Y Acad Sci 2010; 1183: 13-24. 61 Hoefakker S, Caubo M, van 't Erve EH et al. In vivo cytokine profiles in allergic and irritant contact dermatitis. Contact dermatitis 1995; 33: 258-66. 62 Wood LC, Elias PM, Calhoun C et al. Barrier disruption stimulates interleukin-1 alpha expression and release from a pre-formed pool in murine epidermis. J Invest Dermatol 1996; 106: 397-403. 63 Smith HR, Basketter DA, McFadden JP. Irritant dermatitis, irritancy and its role in allergic contact dermatitis. Clinical and experimental dermatology 2002; 27: 138-46. 64 Grangsjo A, Leijon-Kuligowski A, Torma H et al. Different pathways in irritant contact eczema? Early differences in the epidermal elemental content and expression of cytokines after application of 2 different irritants. Contact dermatitis 1996; 35: 355-60. 65 de Jongh CM, Lutter R, Verberk MM et al. Differential cytokine expression in skin after single and repeated irritation by sodium lauryl sulphate. Exp Dermatol 2007; 16: 1032-40. 66 McKenzie RC, Sauder DN. The role of keratinocyte cytokines in inflammation and immunity. J Invest Dermatol 1990; 95: 105S-7S. 67 Lee HY, Stieger M, Yawalkar N et al. Cytokines and chemokines in irritant contact dermatitis. Mediators of inflammation 2013; 2013: 916497. 68 Leung DY, Bieber T. Atopic dermatitis. Lancet 2003; 361: 151-60.
89

References
69 Kasraie S, Werfel T. Role of macrophages in the pathogenesis of atopic dermatitis. Mediators of inflammation 2013; 2013: 942375. 70 Irvine AD, McLean WH. Breaking the (un)sound barrier: filaggrin is a major gene for atopic dermatitis. J Invest Dermatol 2006; 126: 1200-2. 71 Akdis CA, Akdis M, Bieber T et al. Diagnosis and treatment of atopic dermatitis in children and adults: European Academy of Allergology and Clinical Immunology/American Academy of Allergy, Asthma and Immunology/PRACTALL Consensus Report. J Allergy Clin Immunol 2006; 118: 152-69. 72 Ong PY, Leung DY. Atopic dermatitis. Clinical allergy and immunology 2002; 16: 355-79. 73 de Koning HD, Kamsteeg M, Rodijk-Olthuis D et al. Epidermal expression of host response genes upon skin barrier disruption in normal skin and uninvolved skin of psoriasis and atopic dermatitis patients. J Invest Dermatol 2011; 131: 263-6. 74 Jessup HK, Brewer AW, Omori M et al. Intradermal administration of thymic stromal lymphopoietin induces a T cell- and eosinophil-dependent systemic Th2 inflammatory response. J Immunol 2008; 181: 4311-9. 75 Nedospasov SA, Hirt B, Shakhov AN et al. The genes for tumor necrosis factor (TNF-alpha) and lymphotoxin (TNF-beta) are tandemly arranged on chromosome 17 of the mouse. Nucleic acids research 1986; 14: 7713-25. 76 Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell death and differentiation 2003; 10: 45-65. 77 Palladino MA, Bahjat FR, Theodorakis EA et al. Anti-TNF-alpha therapies: the next generation. Nature reviews. Drug discovery 2003; 2: 736-46. 78 Bradley JR. TNF-mediated inflammatory disease. The Journal of pathology 2008; 214: 149-60. 79 Eigler A, Sinha B, Hartmann G et al. Taming TNF: strategies to restrain this proinflammatory cytokine. Immunology today 1997; 18: 487-92. 80 Kruglov AA, Kuchmiy A, Grivennikov SI et al. Physiological functions of tumor necrosis factor and the consequences of its pathologic overexpression or blockade: mouse models. Cytokine & growth factor reviews 2008; 19: 231-44. 81 Taylor PC, Feldmann M. Anti-TNF biologic agents: still the therapy of choice for rheumatoid arthritis. Nature reviews. Rheumatology 2009; 5: 578-82. 82 Lewis RW, McCall JC, Botham PA et al. Investigation of TNF-alpha release as a measure of skin irritancy. Toxicol In Vitro 1993; 7: 393-5. 83 Liao F, Rabin RL, Smith CS et al. CC-chemokine receptor 6 is expressed on diverse memory subsets of T cells and determines responsiveness to macrophage inflammatory protein 3 alpha. J Immunol 1999; 162: 186-94. 84 Corsini E, Terzoli A, Bruccoleri A et al. Induction of tumor necrosis factor-alpha in vivo by a skin irritant, tributyltin, through activation of transcription factors: its pharmacological modulation by anti-inflammatory drugs. J Invest Dermatol 1997; 108: 892-6. 85 Piguet PF, Grau GE, Hauser C et al. Tumor necrosis factor is a critical mediator in hapten induced irritant and contact hypersensitivity reactions. J Exp Med 1991; 173: 673-9. 86 Takahashi T, Sasaki Y, Hama K et al. Production of IL-4, IL-2, IFN-gamma, and TNF-alpha by peripheral blood mononuclear cells of patients with atopic dermatitis. J Dermatol Sci 1992; 3: 172-80. 87 Poulsen LK, Bindslev-Jensen C, Diamant M et al. Biomolecular regulation of the IgE immune response III. Cytokine profiles in atopic dermatitis, inhalant allergy and non-allergic donors. Cytokine 1996; 8: 651-7. 88 Jeong CW, Ahn KS, Rho NK et al. Differential in vivo cytokine mRNA expression in lesional skin of intrinsic vs. extrinsic atopic dermatitis patients using semiquantitative RT-PCR. Clin Exp Allergy 2003; 33: 1717-24.
90

References
89 Nomura I, Goleva E, Howell MD et al. Cytokine milieu of atopic dermatitis, as compared to psoriasis, skin prevents induction of innate immune response genes. J Immunol 2003; 171: 3262-9. 90 Lee HH, Song IH, Friedrich M et al. Cutaneous side-effects in patients with rheumatic diseases during application of tumour necrosis factor-alpha antagonists. The British journal of dermatology 2007; 156: 486-91. 91 Esmailzadeh A, Yousefi P, Farhi D et al. Predictive factors of eczema-like eruptions among patients without cutaneous psoriasis receiving infliximab: a cohort study of 92 patients. Dermatology 2009; 219: 263-7. 92 Rashmi Sharma CLS. TNF-alpha inhibitors: Current indications. Indian Journal of Critical Care Medicine 2007; 11: 139-48 93 Chen X, Baumel M, Mannel DN et al. Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J Immunol 2007; 179: 154-61. 94 Kassiotis G, Kollias G. Uncoupling the proinflammatory from the immunosuppressive properties of tumor necrosis factor (TNF) at the p55 TNF receptor level: implications for pathogenesis and therapy of autoimmune demyelination. J Exp Med 2001; 193: 427-34. 95 Noti M, Corazza N, Mueller C et al. TNF suppresses acute intestinal inflammation by inducing local glucocorticoid synthesis. J Exp Med 2010; 207: 1057-66. 96 Friend SL, Hosier S, Nelson A et al. A thymic stromal cell line supports in vitro development of surface IgM+ B cells and produces a novel growth factor affecting B and T lineage cells. Experimental hematology 1994; 22: 321-8. 97 Sims JE, Williams DE, Morrissey PJ et al. Molecular cloning and biological characterization of a novel murine lymphoid growth factor. J Exp Med 2000; 192: 671-80. 98 Pandey A, Ozaki K, Baumann H et al. Cloning of a receptor subunit required for signaling by thymic stromal lymphopoietin. Nat Immunol 2000; 1: 59-64. 99 Park LS, Martin U, Garka K et al. Cloning of the murine thymic stromal lymphopoietin (TSLP) receptor: Formation of a functional heteromeric complex requires interleukin 7 receptor. J Exp Med 2000; 192: 659-70. 100 Quentmeier H, Drexler HG, Fleckenstein D et al. Cloning of human thymic stromal lymphopoietin (TSLP) and signaling mechanisms leading to proliferation. Leukemia 2001; 15: 1286-92. 101 Reche PA, Soumelis V, Gorman DM et al. Human thymic stromal lymphopoietin preferentially stimulates myeloid cells. J Immunol 2001; 167: 336-43. 102 Rimoldi M, Chieppa M, Salucci V et al. Intestinal immune homeostasis is regulated by the crosstalk between epithelial cells and dendritic cells. Nat Immunol 2005; 6: 507-14. 103 Soumelis V, Reche PA, Kanzler H et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol 2002; 3: 673-80. 104 Watanabe N, Wang YH, Lee HK et al. Hassall's corpuscles instruct dendritic cells to induce CD4+CD25+ regulatory T cells in human thymus. Nature 2005; 436: 1181-5. 105 Isaksen DE, Baumann H, Trobridge PA et al. Requirement for stat5 in thymic stromal lymphopoietin-mediated signal transduction. J Immunol 1999; 163: 5971-7. 106 Arima K, Watanabe N, Hanabuchi S et al. Distinct signal codes generate dendritic cell functional plasticity. Science signaling 2010; 3: ra4. 107 Wohlmann A, Sebastian K, Borowski A et al. Signal transduction by the atopy-associated human thymic stromal lymphopoietin (TSLP) receptor depends on Janus kinase function. Biological chemistry 2010; 391: 181-6. 108 Zhong J, Kim MS, Chaerkady R et al. TSLP signaling network revealed by SILAC-based phosphoproteomics. Molecular & cellular proteomics : MCP 2012; 11: M112 017764. 109 Taylor BC, Zaph C, Troy AE et al. TSLP regulates intestinal immunity and inflammation in mouse models of helminth infection and colitis. J Exp Med 2009; 206: 655-67.
91

References
110 Olkhanud PB, Rochman Y, Bodogai M et al. Thymic stromal lymphopoietin is a key mediator of breast cancer progression. J Immunol 2011; 186: 5656-62. 111 Roan F, Bell BD, Stoklasek TA et al. The multiple facets of thymic stromal lymphopoietin (TSLP) during allergic inflammation and beyond. Journal of leukocyte biology 2012; 91: 877-86. 112 West EE, Kashyap M, Leonard WJ. TSLP: A Key Regulator of Asthma Pathogenesis. Drug discovery today. Disease mechanisms 2012; 9. 113 Zhou B, Comeau MR, De Smedt T et al. Thymic stromal lymphopoietin as a key initiator of allergic airway inflammation in mice. Nat Immunol 2005; 6: 1047-53. 114 Al-Shami A, Spolski R, Kelly J et al. A role for TSLP in the development of inflammation in an asthma model. J Exp Med 2005; 202: 829-39. 115 He R, Oyoshi MK, Garibyan L et al. TSLP acts on infiltrating effector T cells to drive allergic skin inflammation. Proc Natl Acad Sci U S A 2008; 105: 11875-80. 116 Elias PM. Stratum corneum defensive functions: an integrated view. J Invest Dermatol 2005; 125: 183-200. 117 Angelova-Fischer I, Fernandez IM, Donnadieu MH et al. Injury to the stratum corneum induces in vivo expression of human thymic stromal lymphopoietin in the epidermis. J Invest Dermatol 2010; 130: 2505-7. 118 Elias PM, Steinhoff M. "Outside-to-inside" (and now back to "outside") pathogenic mechanisms in atopic dermatitis. J Invest Dermatol 2008; 128: 1067-70. 119 Elias PM, Wood LC, Feingold KR. Epidermal pathogenesis of inflammatory dermatoses. American journal of contact dermatitis : official journal of the American Contact Dermatitis Society 1999; 10: 119-26. 120 Lee HC, Headley MB, Iseki M et al. Cutting edge: Inhibition of NF-kappaB-mediated TSLP expression by retinoid X receptor. J Immunol 2008; 181: 5189-93. 121 Liu YJ. Thymic stromal lymphopoietin: master switch for allergic inflammation. J Exp Med 2006; 203: 269-73. 122 Allakhverdi Z, Comeau MR, Jessup HK et al. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J Exp Med 2007; 204: 253-8. 123 Bogiatzi SI, Fernandez I, Bichet JC et al. Cutting Edge: Proinflammatory and Th2 cytokines synergize to induce thymic stromal lymphopoietin production by human skin keratinocytes. J Immunol 2007; 178: 3373-7. 124 Liu YJ. Thymic stromal lymphopoietin and OX40 ligand pathway in the initiation of dendritic cell-mediated allergic inflammation. J Allergy Clin Immunol 2007; 120: 238-44; quiz 45-6. 125 Demehri S, Liu Z, Lee J et al. Notch-deficient skin induces a lethal systemic B-lymphoproliferative disorder by secreting TSLP, a sentinel for epidermal integrity. PLoS Biol 2008; 6: e123. 126 Demehri S, Morimoto M, Holtzman MJ et al. Skin-derived TSLP triggers progression from epidermal-barrier defects to asthma. PLoS Biol 2009; 7: e1000067. 127 Ziegler SF, Artis D. Sensing the outside world: TSLP regulates barrier immunity. Nat Immunol 2010; 11: 289-93. 128 Zhang Z, Hener P, Frossard N et al. Thymic stromal lymphopoietin overproduced by keratinocytes in mouse skin aggravates experimental asthma. Proc Natl Acad Sci U S A 2009; 106: 1536-41. 129 Miazgowicz MM, Headley MB, Larson RP et al. Thymic stromal lymphopoietin and the pathophysiology of atopic disease. Expert Rev Clin Immunol 2009; 5: 547-56. 130 Li M, Hener P, Zhang Z et al. Induction of thymic stromal lymphopoietin expression in keratinocytes is necessary for generating an atopic dermatitis upon application of the active vitamin D3 analogue MC903 on mouse skin. J Invest Dermatol 2009; 129: 498-502.
92

References
131 Pasparakis M, Alexopoulou L, Episkopou V et al. Immune and inflammatory responses in TNF alpha-deficient mice: a critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J Exp Med 1996; 184: 1397-411. 132 Dahten A, Koch C, Ernst D et al. Systemic PPARgamma ligation inhibits allergic immune response in the skin. J Invest Dermatol 2008; 128: 2211-8. 133 Miyata M, Hatsushika K, Ando T et al. Mast cell regulation of epithelial TSLP expression plays an important role in the development of allergic rhinitis. European journal of immunology 2008; 38: 1487-92. 134 Brandt EB, Strait RT, Hershko D et al. Mast cells are required for experimental oral allergen-induced diarrhea. The Journal of clinical investigation 2003; 112: 1666-77. 135 Jahnz-Rozyk K, Targowski T, Paluchowska E et al. Serum thymus and activation-regulated chemokine, macrophage-derived chemokine and eotaxin as markers of severity of atopic dermatitis. Allergy 2005; 60: 685-8. 136 Steiner M, Aikman-Green S, Prescott GJ et al. Side-by-side comparison of an open-chamber (TM 300) and a closed-chamber (Vapometer) transepidermal water loss meter. Skin research and technology : official journal of International Society for Bioengineering and the Skin 2011; 17: 366-72. 137 Redvers RP, Kaur P. Serial cultivation of primary adult murine keratinocytes. Methods Mol Biol 2005; 289: 15-22. 138 Artuc M, Steckelings UM, Grutzkau A et al. A long-term coculture model for the study of mast cell-keratinocyte interactions. J Invest Dermatol 2002; 119: 411-5. 139 Byrne AM, Goleva E, Chouiali F et al. Induction of GITRL expression in human keratinocytes by Th2 cytokines and TNF-alpha: implications for atopic dermatitis. Clin Exp Allergy 2012; 42: 550-9. 140 Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nature protocols 2008; 3: 1101-8. 141 Mrabet-Dahbi S, Metz M, Dudeck A et al. Murine mast cells secrete a unique profile of cytokines and prostaglandins in response to distinct TLR2 ligands. Exp Dermatol 2009; 18: 437-44. 142 Nakahata T, Spicer SS, Cantey JR et al. Clonal assay of mouse mast cell colonies in methylcellulose culture. Blood 1982; 60: 352-61. 143 Ogawa M, Matsuzaki Y, Nishikawa S et al. Expression and function of c-kit in hemopoietic progenitor cells. J Exp Med 1991; 174: 63-71. 144 MacNeil AJ, Yang YJ, Lin TJ. MAPK kinase 3 specifically regulates Fc epsilonRI-mediated IL-4 production by mast cells. J Immunol 2011; 187: 3374-82. 145 Tanaka A, Matsuda H. IgE crosslinkage of Fcepsilon receptor I induces both production and activation of matrix metalloproteinase-9 in mast cells. Cellular immunology 2004; 228: 66-75. 146 Weber S, Niessen MT, Prox J et al. The disintegrin/metalloproteinase Adam10 is essential for epidermal integrity and Notch-mediated signaling. Development 2011; 138: 495-505. 147 Bertolini TM, Giorgione J, Harvey DF et al. Protein kinase C translocation by modified phorbol esters with functionalized lipophilic regions. J Org Chem 2003; 68: 5028-36. 148 Kato A, Favoreto S, Jr., Avila PC et al. TLR3- and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J Immunol 2007; 179: 1080-7. 149 Kinoshita H, Takai T, Le TA et al. Cytokine milieu modulates release of thymic stromal lymphopoietin from human keratinocytes stimulated with double-stranded RNA. J Allergy Clin Immunol 2009; 123: 179-86. 150 Lee HC, Ziegler SF. Inducible expression of the proallergic cytokine thymic stromal lymphopoietin in airway epithelial cells is controlled by NFkappaB. Proc Natl Acad Sci U S A 2007; 104: 914-9.
93

References
151 Saenz SA, Taylor BC, Artis D. Welcome to the neighborhood: epithelial cell-derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol Rev 2008; 226: 172-90. 152 Nonaka M, Fukumoto A, Ogihara N et al. Synergistic induction of thymic stromal lymphopoietin by tumor necrosis factor alpha and Th2 cytokine in nasal polyp fibroblasts. Am J Rhinol Allergy 2010; 24: e14-8. 153 Xie Y, Takai T, Chen X et al. Long TSLP transcript expression and release of TSLP induced by TLR ligands and cytokines in human keratinocytes. J Dermatol Sci 2012; 66: 233-7. 154 Mudnakudu Nagaraju KK, Babina M, Worm M. Opposing effects on immune function and skin barrier regulation by the proteasome inhibitor bortezomib in an allergen-induced eczema model. Exp Dermatol 2013; 22: 742-7. 155 Cohen C, Dossou G, Rougier A et al. Measurement of inflammatory mediators produced by human keratinocytes in vitro: A predictive assessment of cutaneous irritation. Toxicol In Vitro 1991; 5: 407-10. 156 Spiekstra SW, Toebak MJ, Sampat-Sardjoepersad S et al. Induction of cytokine (interleukin-1alpha and tumor necrosis factor-alpha) and chemokine (CCL20, CCL27, and CXCL8) alarm signals after allergen and irritant exposure. Exp Dermatol 2005; 14: 109-16. 157 Warner RR, Boissy YL, Lilly NA et al. Water disrupts stratum corneum lipid lamellae: damage is similar to surfactants. J Invest Dermatol 1999; 113: 960-6. 158 Pessolano LG, Jr., Sullivan CP, Seidl SE et al. Trafficking of endogenous smooth muscle cell cholesterol: a role for serum amyloid A and interleukin-1beta. Arterioscler Thromb Vasc Biol 2012; 32: 2741-50. 159 Welker P, Grabbe J, Gibbs B et al. Nerve growth factor-beta induces mast-cell marker expression during in vitro culture of human umbilical cord blood cells. Immunology 2000; 99: 418-26. 160 Wang YH, Liu YJ. OX40-OX40L interactions: a promising therapeutic target for allergic diseases? The Journal of clinical investigation 2007; 117: 3655-7. 161 Urb M, Sheppard DC. The role of mast cells in the defence against pathogens. PLoS pathogens 2012; 8: e1002619. 162 Kawakami T, Ando T, Kimura M et al. Mast cells in atopic dermatitis. Current opinion in immunology 2009; 21: 666-78. 163 Danso MO, van Drongelen V, Mulder A et al. TNF-alpha and Th2 cytokines induce atopic dermatitis-like features on epidermal differentiation proteins and stratum corneum lipids in human skin equivalents. J Invest Dermatol 2014; 134: 1941-50. 164 Takai T. TSLP expression: cellular sources, triggers, and regulatory mechanisms. Allergol Int 2012; 61: 3-17. 165 Murthy A, Shao YW, Narala SR et al. Notch activation by the metalloproteinase ADAM17 regulates myeloproliferation and atopic barrier immunity by suppressing epithelial cytokine synthesis. Immunity 2012; 36: 105-19. 166 Tsai M, Chen CC, Mukai K et al. Thymic stromal lymphopoietin contributes to myeloid hyperplasia and increased immunoglobulins, but not epidermal hyperplasia, in RabGEF1-deficient mice. Am J Pathol 2010; 177: 2411-20. 167 Wang Z, Zhang LJ, Guha G et al. Selective ablation of Ctip2/Bcl11b in epidermal keratinocytes triggers atopic dermatitis-like skin inflammatory responses in adult mice. PLoS One 2012; 7: e51262. 168 Lee CH, Maibach HI. The sodium lauryl sulfate model: an overview. Contact dermatitis 1995; 33: 1-7. 169 Dugard PH, Scheuplein RJ. Effects of ionic surfactants on the permeability of human epidermis: an electrometric study. J Invest Dermatol 1973; 60: 263-9. 170 Boukhatem MN, Kameli A, Ferhat MA et al. Rose geranium essential oil as a source of new and safe anti-inflammatory drugs. The Libyan journal of medicine 2013; 8: 22520.
94

References
171 Rauh V, Arunajadai S, Horton M et al. Seven-year neurodevelopmental scores and prenatal exposure to chlorpyrifos, a common agricultural pesticide. Environmental health perspectives 2011; 119: 1196-201. 172 Furstenberger G, Csuk-Glanzer BI, Marks F et al. Phorbol ester-induced leukotriene biosynthesis and tumor promotion in mouse epidermis. Carcinogenesis 1994; 15: 2823-7. 173 Flint MS, Miller DB, Tinkle SS. Restraint-induced modulation of allergic and irritant contact dermatitis in male and female B6.129 mice. Brain, behavior, and immunity 2000; 14: 256-69. 174 Terada M, Tsutsui H, Imai Y et al. Contribution of IL-18 to atopic-dermatitis-like skin inflammation induced by Staphylococcus aureus product in mice. Proc Natl Acad Sci U S A 2006; 103: 8816-21. 175 Demehri S, Turkoz A, Manivasagam S et al. Elevated epidermal thymic stromal lymphopoietin levels establish an antitumor environment in the skin. Cancer Cell 2012; 22: 494-505. 176 Di Piazza M, Nowell CS, Koch U et al. Loss of cutaneous TSLP-dependent immune responses skews the balance of inflammation from tumor protective to tumor promoting. Cancer Cell 2012; 22: 479-93. 177 Jang Y, Jeong SH, Park YH et al. UVB induces HIF-1alpha-dependent TSLP expression via the JNK and ERK pathways. J Invest Dermatol 2013; 133: 2601-8. 178 Oyoshi MK, Larson RP, Ziegler SF et al. Mechanical injury polarizes skin dendritic cells to elicit a T(H)2 response by inducing cutaneous thymic stromal lymphopoietin expression. J Allergy Clin Immunol 2010; 126: 976-84, 84 e1-5. 179 Sano Y, Masuda K, Tamagawa-Mineoka R et al. Thymic stromal lymphopoietin expression is increased in the horny layer of patients with atopic dermatitis. Clin Exp Immunol 2013; 171: 330-7. 180 Yockey LJ, Demehri S, Turkoz M et al. The absence of a microbiota enhances TSLP expression in mice with defective skin barrier but does not affect the severity of their allergic inflammation. J Invest Dermatol 2013; 133: 2714-21. 181 Carta S, Lavieri R, Rubartelli A. Different Members of the IL-1 Family Come Out in Different Ways: DAMPs vs. Cytokines? Front Immunol 2013; 4: 123. 182 van de Veerdonk FL, Netea MG. New Insights in the Immunobiology of IL-1 Family Members. Front Immunol 2013; 4: 167. 183 Cevikbas F, Steinhoff M. IL-33: a novel danger signal system in atopic dermatitis. J Invest Dermatol 2012; 132: 1326-9. 184 Keyel PA. How is inflammation initiated? Individual influences of IL-1, IL-18 and HMGB1. Cytokine 2014. 185 Leonard WJ. TSLP: finally in the limelight. Nat Immunol 2002; 3: 605-7. 186 Lin Q, Wang L, Lin Y et al. Toll-like receptor 3 ligand polyinosinic:polycytidylic acid promotes wound healing in human and murine skin. J Invest Dermatol 2012; 132: 2085-92. 187 Kamekura R, Kojima T, Koizumi J et al. Thymic stromal lymphopoietin enhances tight-junction barrier function of human nasal epithelial cells. Cell Tissue Res 2009; 338: 283-93. 188 Nishiura H, Kido M, Aoki N et al. Increased susceptibility to autoimmune gastritis in thymic stromal lymphopoietin receptor-deficient mice. J Immunol 2012; 188: 190-7. 189 Reardon C, Lechmann M, Brustle A et al. Thymic stromal lymphopoetin-induced expression of the endogenous inhibitory enzyme SLPI mediates recovery from colonic inflammation. Immunity 2011; 35: 223-35. 190 Sonesson A, Kasetty G, Olin AI et al. Thymic stromal lymphopoietin exerts antimicrobial activities. Exp Dermatol 2011; 20: 1004-10. 191 Siracusa MC, Saenz SA, Wojno ED et al. Thymic stromal lymphopoietin-mediated extramedullary hematopoiesis promotes allergic inflammation. Immunity 2013; 39: 1158-70. 192 Leung DY. Atopic dermatitis: new insights and opportunities for therapeutic intervention. J Allergy Clin Immunol 2000; 105: 860-76.
95

References
193 Prodanovich S, Ricotti C, Glick BP et al. Etanercept: an evolving role in psoriasis and psoriatic arthritis. American journal of clinical dermatology 2010; 11 Suppl 1: 3-9. 194 Youdim A, Vasiliauskas EA, Targan SR et al. A pilot study of adalimumab in infliximab-allergic patients. Inflammatory bowel diseases 2004; 10: 333-8. 195 Guttman-Yassky E, Lowes MA, Fuentes-Duculan J et al. Major differences in inflammatory dendritic cells and their products distinguish atopic dermatitis from psoriasis. J Allergy Clin Immunol 2007; 119: 1210-7. 196 Chan JL, Davis-Reed L, Kimball AB. Counter-regulatory balance: atopic dermatitis in patients undergoing infliximab infusion therapy. Journal of drugs in dermatology : JDD 2004; 3: 315-8. 197 Lai-Cheong J, Warren R, Bucknall R et al. Etanercept-induced dermatitis in a patient with rheumatoid arthritis. Journal of the European Academy of Dermatology and Venereology : JEADV 2006; 20: 614-5. 198 Mangge H, Gindl S, Kenzian H et al. Atopic dermatitis as a side effect of anti-tumor necrosis factor-alpha therapy. The Journal of rheumatology 2003; 30: 2506-7. 199 Choi YJ, Lee HJ, Lee do H et al. Therapeutic effects and immunomodulation of suanbo mineral water therapy in a murine model of atopic dermatitis. Annals of dermatology 2013; 25: 462-70. 200 Woodward AL, Spergel JM, Alenius H et al. An obligate role for T-cell receptor alphabeta+ T cells but not T-cell receptor gammadelta+ T cells, B cells, or CD40/CD40L interactions in a mouse model of atopic dermatitis. J Allergy Clin Immunol 2001; 107: 359-66. 201 Hijnen D, Knol EF, Gent YY et al. CD8(+) T cells in the lesional skin of atopic dermatitis and psoriasis patients are an important source of IFN-gamma, IL-13, IL-17, and IL-22. J Invest Dermatol 2013; 133: 973-9. 202 Turner MJ, Travers JB, Kaplan MH. T helper cell subsets in the development of atopic dermatitis. Journal of drugs in dermatology : JDD 2012; 11: 1174-8. 203 Aioi A, Tonogaito H, Suto H et al. Impairment of skin barrier function in NC/Nga Tnd mice as a possible model for atopic dermatitis. The British journal of dermatology 2001; 144: 12-8. 204 Gupta J, Grube E, Ericksen MB et al. Intrinsically defective skin barrier function in children with atopic dermatitis correlates with disease severity. J Allergy Clin Immunol 2008; 121: 725-30 e2. 205 Ziegler SF, Roan F, Bell BD et al. The biology of thymic stromal lymphopoietin (TSLP). Advances in pharmacology 2013; 66: 129-55. 206 Le TA, Takai T, Vu AT et al. Flagellin induces the expression of thymic stromal lymphopoietin in human keratinocytes via toll-like receptor 5. International archives of allergy and immunology 2011; 155: 31-7. 207 Han NR, Oh HA, Nam SY et al. TSLP Induces Mast Cell Development and Aggravates Allergic Reactions through the Activation of MDM2 and STAT6. J Invest Dermatol 2014; 134: 2521-30. 208 Gao PS, Rafaels NM, Mu D et al. Genetic variants in thymic stromal lymphopoietin are associated with atopic dermatitis and eczema herpeticum. J Allergy Clin Immunol 2010; 125: 1403-7 e4. 209 He R, Kim HY, Yoon J et al. Exaggerated IL-17 response to epicutaneous sensitization mediates airway inflammation in the absence of IL-4 and IL-13. J Allergy Clin Immunol 2009; 124: 761-70 e1. 210 Okayama Y, Okumura S, Sagara H et al. FcepsilonRI-mediated thymic stromal lymphopoietin production by interleukin-4-primed human mast cells. The European respiratory journal 2009; 34: 425-35. 211 Ando T, Xiao W, Gao P et al. Critical role for mast cell Stat5 activity in skin inflammation. Cell reports 2014; 6: 366-76.
96

References
212 Sebastian K, Borowski A, Kuepper M et al. Signal transduction around thymic stromal lymphopoietin (TSLP) in atopic asthma. Cell communication and signaling : CCS 2008; 6: 5. 213 Kuehn HS, Radinger M, Gilfillan AM. Measuring mast cell mediator release. Current protocols in immunology / edited by John E. Coligan ... [et al.] 2010; Chapter 7: Unit7 38. 214 Reuter S, Stassen M, Taube C. Mast cells in allergic asthma and beyond. Yonsei medical journal 2010; 51: 797-807. 215 Boyce JA. Eicosanoid mediators of mast cells: receptors, regulation of synthesis, and pathobiologic implications. Chemical immunology and allergy 2005; 87: 59-79. 216 Gilfillan AM, Beaven MA. Regulation of mast cell responses in health and disease. Critical reviews in immunology 2011; 31: 475-529. 217 Katsanos GS, Anogeianaki A, Orso C et al. Mast cells and chemokines. Journal of biological regulators and homeostatic agents 2008; 22: 145-51. 218 McNeil HP, Adachi R, Stevens RL. Mast cell-restricted tryptases: structure and function in inflammation and pathogen defense. The Journal of biological chemistry 2007; 282: 20785-9. 219 Dvorak AM, Kissell S. Granule changes of human skin mast cells characteristic of piecemeal degranulation and associated with recovery during wound healing in situ. Journal of leukocyte biology 1991; 49: 197-210. 220 Xue L, Barrow A, Pettipher R. Interaction between prostaglandin D and chemoattractant receptor-homologous molecule expressed on Th2 cells mediates cytokine production by Th2 lymphocytes in response to activated mast cells. Clin Exp Immunol 2009; 156: 126-33. 221 Kanda N, Watanabe S. Histamine enhances the production of nerve growth factor in human keratinocytes. J Invest Dermatol 2003; 121: 570-7. 222 Kohda F, Koga T, Uchi H et al. Histamine-induced IL-6 and IL-8 production are differentially modulated by IFN-gamma and IL-4 in human keratinocytes. J Dermatol Sci 2002; 28: 34-41. 223 Gschwandtner M, Mildner M, Mlitz V et al. Histamine suppresses epidermal keratinocyte differentiation and impairs skin barrier function in a human skin model. Allergy 2013; 68: 37-47. 224 Wahlgren CF, Hagermark O, Bergstrom R. The antipruritic effect of a sedative and a non-sedative antihistamine in atopic dermatitis. The British journal of dermatology 1990; 122: 545-51. 225 Ohsawa Y, Hirasawa N. The antagonism of histamine H1 and H4 receptors ameliorates chronic allergic dermatitis via anti-pruritic and anti-inflammatory effects in NC/Nga mice. Allergy 2012; 67: 1014-22. 226 Schwartz LB. Tryptase: a mast cell serine protease. Methods in enzymology 1994; 244: 88-100. 227 Thakurdas SM, Melicoff E, Sansores-Garcia L et al. The mast cell-restricted tryptase mMCP-6 has a critical immunoprotective role in bacterial infections. The Journal of biological chemistry 2007; 282: 20809-15. 228 Voegeli R, Rawlings AV, Breternitz M et al. Increased stratum corneum serine protease activity in acute eczematous atopic skin. The British journal of dermatology 2009; 161: 70-7. 229 Ribbing C, Engblom C, Lappalainen J et al. Mast cells generated from patients with atopic eczema have enhanced levels of granule mediators and an impaired Dectin-1 expression. Allergy 2011; 66: 110-9. 230 Amon U, Memmel U, Stoll R et al. Comparison of severity scoring of atopic dermatitis values and serum levels of eosinophil cationic protein and mast cell tryptase for routine evaluation of atopic dermatitis. Acta dermato-venereologica 2000; 80: 284-6. 231 Gerdes S, Kurrat W, Mrowietz U. Serum mast cell tryptase is not a useful marker for disease severity in psoriasis or atopic dermatitis. The British journal of dermatology 2009; 160: 736-40.
97

References
232 Tsujii K, Andoh T, Ui H et al. Involvement of Tryptase and Proteinase-Activated Receptor-2 in Spontaneous Itch-Associated Response in Mice With Atopy-like Dermatitis. Journal of pharmacological sciences 2009; 109: 388-95.
98

Appendix
APPENDIX
Figure 31: CD4+ and CD8+ cells in skin lesions of OVA-sensitized mice. A) CD4+ and B) CD8+ T cells were stained and quantified in the dermis of lesional skin of mice. Numbers of
cells per mm2 are shown as the median for each group (n = 5-7). One representative photograph of each group
is shown.
Figure 32: T cell subset cytokine expression in lesional skin. Expression levels of T cell cytokines at the mRNA level in the lesional skin of wildtype and TNF-/- mice were
analyzed by quantitative PCR. The relative expression of A) IL-10, B) IL-4 and INF-γ was measured in
comparison to the housekeeping gene HPRT and is shown as the median for each group (n ≥ 11).
99

Appendix
Figure 33: Proinflammatory cytokine and β-defensins gene expression in lesional skin. Expression levels of cytokines at the mRNA level in the lesional skin of wildtype and TNF-/- mice were analyzed
by quantitative PCR. The relative expression of A) IL-1α, B) IL-1β, C) IL-13, D) IL-33, E) Def B2 and F) Def B3
was measured in comparison to the housekeeping gene HPRT and is shown as the median for each group (n ≥
11).
Figure 34: TSLP and Mast cell levels in mice upon anti TSLP antibody treatment.
A) TSLP mRNA level in wt and TNF-/- mice after intradermal doses of anti-TSLP antibodies B) MCs numbers in
the lesional skin of wt and TNF-/- mice. Median of n ≥4 mice/group.
100

Acknowledgements
ACKNOWLEDGEMENTS
First of all, I would like to express my deepest gratitude to my advisor Prof. Dr. Margitta Worm for giving me the opportunity to do my PhD thesis in her lab at the
Department of Dermatology. Many thanks for her support and supervision and for
her kind advice and help during these years. My special thanks to Dr. Magda Babina for introducing me to the area of
immunomodulation and for her constant help, invaluable suggestions to my work
and during writing publication.
Many thanks to Prof. Dr. Andreas Radbruch for accepting me as PhD student and
for his scientific advice.
I wish to thank Prof. Max Löhning for providing me TNF-/- mice breeding pairs for
my thesis work.
Many thanks to Dana Hoser, Norman Tanner, Barbara Bleher, Tarek Hazzan,
Enya Longmuss and Laura Fleischmann for all their supporting work in my PhD
thesis.
I would like to thanks Dennis Ernst for his great help in the lab and for his
wonderful support during the mouse studies.
I am grateful to all my close friends Dr. Omera Bashir, Dr. Kamalika Mukherjee, Dr. Brinda Selvaraj, Dr. Maria Nassiri and Rajagopal Murgan for helping me in
my hardest situations and for encouraging me. Thank you for your productive
discussions, for proofreading and supporting me with my manuscript(s) and thesis. I
am glad to have you all in my life.
Many thanks to the people from the working group who supported me with
constructive advice, assistance, encouraging words and the wonderful working
atmosphere. Above alI, I would like to express my sincere thanks to all the lab
mates, Juliane Lindner, Dr. Christin Weise, Dr. Kiran Kumar, Dr. Gennadiy Drozdenko, Dr. Sabine Dölle, Kristina Heins, Marcel Wittenberg, Sandra Treptow, Davender , Dr. Guido Heine, Dr. Bernhard Ay for their cooperation and
support during my stay. I also thank previous lab colleagues, Dr. Björn Hartmann
and Dr. Kerstin Geldmeyer-Hilt for their valuable suggestions.
101

Acknowledgements
A special thanks goes to Sven Guhl and Metin Artuc for giving me useful technical
and scientific advice during the whole course of my PhD thesis, especially for
human keratinocytes and mast cell culturing.
I would like to thank and treasure the good times I spent with all my friends in Berlin.
Thank you for making my stay in Berlin a pleasant one. Lastly, I would like to show gratitude to my family for all their support and their belief
in me throughout my life. I feel myself lucky to have you all. Finally, I would like to
thank my husband and partner Krishna Kumar for his unconditional love, patience
and support during this period of time.
102

Declaration
SELBSTÄNDIGKEITSERKLÄRUNG / DECLARATION
Hiermit versichere ich, Vandana Kumari, die vorliegende Dissertation selbständig
erarbeitet und verfasst zu haben. Es wurden keine weiteren Quellen und Hilfsmittel
als die hier angegebenen verwendet.
I hereby declare that I, Vandana Kumari, have worked and wrote this dissertation
independently and did not use other than the listed support. This thesis does not
exist neither in the same or similar form nor is it submitted to another examination
procedure.
103