metabolic disease - goshintensivecare.files.wordpress.com · • metabolic pathways of energy...
TRANSCRIPT

Copyright 2009-10 Great Ormond Street Hospital. All rights reserved. 1
Metabolic Disease Sophie Skellett, December 2005 Updated by: Shruti Agrawal August 2009 Available guidelines:
1. Arrangements for specimens to be taken in an emergency when a metabolic disease is suspected 2. Recommendations for storage of postmortem biopsies
Fundamental Knowledge: List of topics relevant to PIC that will have been covered in membership examinations.
They will not be repeated here.
• Metabolic pathways of energy production
• Hormonal control of metabolism
• Regulation of plasma glucose
• Response to trauma
• Physiological alterations in starvation, obesity, exercise and the stress response; blood sugar
• Metabolic tests and investigations of inborn errors of metabolism
Information for Year 1 ITU Training (basic):
Year 1 ITU curriculum • Signs and symptoms that require ITU care: encephalopathy & seizures, lactic acidosis,
hyperammonaemia, neuromuscular weakness, hepatitis.
• Basics of management & investigation of these patients.
Curriculum Notes for Year 1: Commoner IEM’s – more likely to see on ICU
1. Organic acidaemias
a. Methylmalonic acidaemia (MMA) 1 per 48,000 b. Propionic acidaemia (PA) 1 per 100,000 c. Multiple carboxylase deficiency (MCD) d. Isovaleric acidaemia (IVA) 1 per 50,000 e. Biotinidase deficiency 1 per 140,000
2. Aminoacidopathies a. Maple Syrup Urine Disease (MSUD) 1 per 180,000
3. Urea Cycle Defects
a. Ornithine tanscarbamylase deficiency (OTC) 1 per 80,000
b. Arginosuccinic aciduria (ASA) 4. Disorders carbohydrate metabolism
a. Galactosaemia 1 per 40,000
Disclaimer:
The Great Ormond Street Paediatric Intensive Care Training Programme was developed in 2004 by the clinicians of that Institution, primarily for use within Great Ormond Street Hospital and the Children’s Acute Transport Service (CATS). The written information (known as Modules) only forms a part of the training programme. The modules are provided for teaching purposes only and are not designed to be any form of standard reference or textbook. The views expressed in the modules do not necessarily represent the views of all the clinicians at Great Ormond Street Hospital and CATS. The authors have made considerable efforts to ensure the information contained in the modules is accurate and up to date. The modules are updated annually. Users of these modules are strongly recommended to confirm that the information contained within them, especially drug doses, is correct by way of independent sources. The authors accept no responsibility for any inaccuracies, information perceived as misleading, or the success of any treatment regimen detailed in the modules. The text, pictures, images, graphics and other items provided in the modules are copyrighted by “Great Ormond Street Hospital” or as appropriate, by the other owners of the items.

Copyright 2009-10 Great Ormond Street Hospital. All rights reserved. 2
Differential diagnosis collapsed neonate
Collapsed, shocked neonate, cardiopulmonary
Sepsis
Inborn error of
metabolism
Primary lung
abnormalities – CDH / CCAM and PPHN
Mainly left
sided congenital cardiac lesions
Five times more common than sepsis
Prognosis in IEM’s often depends upon prompt diagnosis and some are associated with an increased risk of infection, so always consider the possibility of an IEM and investigate accordingly.
History: Previous miscarriage/ stillbirth, particularly late fetal loss. Maternal health in pregnancy
- Prolonged hyperemesis - Liver disease (AFLP)
- HELLP (haemolysis, elevated liver enzymes, low platelets)
Associated with Fatty acid oxidation (FAO)defects
Fetal movements – very active, rhythmic movements may be seizures.
Family History – consanguinity, SID (particularly late > 6/12 old), IEM.
Initial Investigations
Blood FBC LFT’s Clotting Screen Blood Glucose Blood gas Ammonia Lactate
Acylcarnitine (profile and free carnitine) Amino acids
Urine Ketones (dipstix)
Organic acids (including orotic acid) Reducing substances (galactosaemia)
(Amino acids not usu done as reflect tubular leak rather than pathology)

Copyright 2009-10 Great Ormond Street Hospital. All rights reserved. 3
Emergency Management
• Stop feeds Feeds stopped only for 48 -72 hours (to avoid catabolism). Results TMS (Tandem Mass Spectroscopy) should be back, liaise with metabolic team.
• Promote anabolism 10% Dextrose plus electrolytes as maintenance
5% Dextrose if lactic acidosis suspected Use insulin if hyperglycemic – don’t stop dextrose
• Correct electrolyte imbalance
• Elimination of toxic metabolites (see table below)
Toxic metabolite Treatment
Ammonia Sodium benzoate Ammonia Sodium phenylbutyrate Organic acids Carnitine IVA Glycine Ammonia Organic acids Leucine (MSUD)
Dialysis PD if < 2.5 kg CVVH if haemodynamically unstable
Hyperammonaemia
Important : Urgent action is needed to bring down ammonia levels as both the level and duration of elevated ammonia levels is related to the amount of irreversible neurological damage. Therefore measure ammonia in any lethargic, encephalopthic or comatose newborn where cause is unclear.
Level:
a) Normally, levels are higher in neonates, especially preterms b) Levels persistently > 100 micromol/L will need investigation and treatment. c) Levels > 300 micromols/L failing to respond to sodium benzoate or
phenylbutyrate need urgent dialysis. d) Levels > 1000 micromols/L caused
by i. Urea cycle defects ii. Hepatic failure iii. Organic acidaemias
e) Sepsis and hypoxic insults can raise ammonia but levels not usually very high < 200 micromols/L.
Presenting symptoms:
a) Encephalopathy (levels above 150 micromols/L), decorticate / decerebrate posturing.
b) Signs of raised ICP (caused by cerebral oedema). c) Lethargy d) Tachypnoea (central in origin and often respiratory alkalosis) e) Apnoea f) Fits, tremors, hypotonia, hypertonia g) Hypothermia h) Vomiting i) Cardiovascular instability j) Poor feeding

Copyright 2009-10 Great Ormond Street Hospital. All rights reserved. 4
Causes
1. Urea cycle defects – OTC, ASA, CPS, Citrullinaemia (Most likely dx with very high
ammonia levels)
2. Organic acidaemias - GA1, IVA, MMA, 3KT, MCD, PA (Likely if associated
ketoacidosis and presentation within 24 hours of birth) 3. Amino acid defects 4. Transient hyperammonaemia of the newborn (THAN) – shunting of blood away from hepatocytes via ductus venosus or PDA. Good prognosis if ammonia level quickly controlled. 5. Fatty acid oxidation defects in severe decompensation (older infant / child). 6. Liver failure.
Metabolic Acidosis
Common. Also caused by a variety of pathologies (sepsis, CHF, hypoxia). IEM more likely if large persistent base deficit and acidosis resistant to resuscitation measures.
Investigations ABG
Lactate Ketones (dipstix) Glucose Anion gap
(Na + K) – ( HCO3 + Cl) Acylcarnitines
Urine organic acids
Ketosis
• Abnormal in neonates – highly suggestive IEM.
• Ketosis present
o Diabetes Mellitus o Organic acidaemias o Ketolysis defects o Respiratory chain defects o Gluconeogenesis defects o Adrenal insufficiency
• Note absence ketones despite low glucose in fat oxidation defects.
Anion Gap:
• Diagnosis of the metabolic acidosis is aided by working out the anion gap. An increased gap > 16 is seen in many inborn errors, but a normal AG is limited to diarrhoea or GI losses and renal tubular acidosis. A normal / low gap may be due to hyperchloraemia.
AG = (Na+ + K
+) – (Bicarbonate + Cl
- )
Normal = 8 - 16
Management: ABC APPROACH
1. Adequately treat any component of shock 2. Stop protein and fat intake whilst diagnosis is unclear, start intravenous fluids with
glucose 8 to 10 mg/kg/min and rehydrate to help aid excretion of toxic metabolites. 3. For severe acidosis, haemodialysis or CVVH may be necessary.

Copyright 2009-10 Great Ormond Street Hospital. All rights reserved. 5
4. Sodium bicarbonate or THAM administration as necessary. If an IEM is suspected full correction and/ or an ongoing infusion is often required.
5. Treat hypoglycaemia and increase glucose deliver as necessary. Lactic Acidoses • Usually due to tissue hypoxia not an inborn error of metabolism and in this case will not be associated with ketosis. • In many severe cases diagnosis not found. • Management limited. • Prognosis guarded – as this usually represents a block in a fundamental energy pathway. • 5% Dextrose used as higher concentrations may exacerbate the lactic acidosis.
Investigations
ABG Lactate Glucose Ketones (dipstix) Ammonia Urate Urine organic acids LFT’s Acylcarnitnes
Cholesterol and Triglycerides
Causes 1. Respiratory chain disorders: Muscle biopsy, MRI brain
shows brain stem and basal ganglia changes, raised CSF lactate
2. Pyruvate disorders Fibroblast enzymology 3. Organic Acidaemias Urine organic acids 4. Gluconeogenesis defects Liver enzymology 5. Hepatic glycogen storage disorders Liver or rbc enzymology 6. Fat oxidation defects Acylcarnitines
7. Biotinidase deficiency Blood biotinidase level
Encephalopathy – no acidosis or hyperammonaemia
1. Non ketotic hyperglycinaemia – intractable fits in neonate. Diagnosis performed
by measuring simultaneous plasma and CSF glycine and the ratio is raised > 0.09. Fits respond to sodium benzoate but still poor neurological outcome.
2. Sulphite oxidase and Molybdenum co-factor deficiency – presents like HIE (low uric acid levels), sulphite dipstix on fresh urine (positive).
3. Biotinidase deficiency – responds to 10 mg biotin daily and should be started in neonates with intractable seizures whilst awaiting assay results.
4. Pyridoxine dependency – give trial pyridoxine.
5. Peroxisomal disorders may present with encephalopathy and fits – diagnosis made on VLCFA’s.
6. Galactosaemia (rarely may present with symptoms of CNS toxicity from accumulating galactose

Copyright 2009-10 Great Ormond Street Hospital. All rights reserved. 6
Hypoglycaemia
Definition: At present, a glucose level ≤ 2.6 mmol/L. This is based on EEG changes in children
at different blood glucose levels and may be shortly revised upwards to compensate in part for
the fact that BM stix can be 0.4 – 0.6 mmol/L in error. Likely acceptable level will be at least
3.0 mmol/L in children in future and may be higher for neonates.
Investigations (whilst hypoglycaemic)
Blood Glucose
Acylcarnitines Cortisol Free fatty acids GH U+E’s Insulin and C-peptide Lactate 3- Hydroxy butyrate
Urine Ketones (dipstix)
Organic acids
Hypoglycaemia – no ketones
1. Hyperinsulinism Need high glucose requirement > 10 mg/kg/min Measure Insulin and C-peptide levels in blood when blood sugar low. 2. Fat Oxidation defect Hepatomegaly, high lactate and LFT’s. Test blood acylcarnitines and urinary organic acids. 3. Liver failure Signs of severe liver disease, LFT’s, Ammonia & clotting.
Hypoglycaemia – with ketones
1. Sepsis 2. Adrenal insufficiency 3. Hypopituitarism 4. Respiratory chain disorders 5. Gluconeogenesis defects
6. Ketolysis defects
Treatment:
1. Definitive treatments dependent upon cause and are varied. However hypoglycaemia must be
corrected immediately to prevent neurological damage,
2. For neonates : 2mls/kg of 10% Dextrose bolus. Recheck level in 15 minutes.
3. APLS and EPLS recommendations for children:
i. 5mls/kg 10% Dextrose bolus
ii. Recheck blood sugar regularly
4. Start neonates on 6mg/kg/min glucose = 3.6 mls/kg/hr 10% Dextrose.
5. Note: Glucose mg/kg/min = ml/hr x % dextrose
6 x weight (kg)

Copyright 2009-10 Great Ormond Street Hospital. All rights reserved. 7
6. Increase glucose delivered intravenously according to response. Some infants with hyperinsulinism may need as much as 20 mg/kg/min glucose.
7. If glucose required > 12 mg/kg/min then consider glucagons infusion. CONTACT
METABOLIC TEAM.
8. Ammonia levels should always be checked in hyperinsulinism to diagnose or exclude HIHA
(hyperinsulinism, hperammonaemia syndrome) which is diazoxide responsive.
9. Oral diazoxide inhibits pancreatic secretion of insulin, stimulates glucose release from the
liver, and stimulates catecholamine release, which elevates blood glucose levels. 5-20 mg/kg/d
PO divided q8h.
10. Some patients may require sub total pancreatectomy.
Hepatitis
Presenting symptoms:
a) Jaundice - Conjugated (usually),
- Unconjugated - Mixed with predominantly unconjugated progressing to conjugated as liver dysfunction continues ( galactosaemia).
b) Vomiting c) Abdominal distention d) Lethargy, fits, tremors (hypoglycaemia) e) Encephalopathy (hyperammonaemia)
Causes:
1. Galactosaemia 2. Hereditary tyrosinaemia 3. Neonatal haemochromatosis 4. Alpha 1 anti-trypsin deficiency 5. Rare: Niemann Pick and GSD type 4, can have jaundice that resolves in few
months, then clinically normal for months to years before developing neurological disease.
6. Zellwegers – recognised by dysmorphic features and severe hypotonia accompanying the jaundice and liver dysfunction
Investigations: a) FBC and film b) U + E’s c) LFT’s
d) Bilirubin level and conjugated and unconjugated fractions e) Ammonia f) Blood glucose g) Plasma amino acids
h) Alpha 1 anti-trypsin level i) Urine succinylacetone j) Urine for reducing sugars k) G 1 PUT rbc enzyme level l) Very long chain fatty acids
m) Serum ferritin and Iron levels

Copyright 2009-10 Great Ormond Street Hospital. All rights reserved. 8
Inborn Errors of Metabolism Associated With Neonatal Liver Disease and Laboratory Studies Useful in Diagnosis
Disorder Laboratory Studies
Galactosemia Urine reducing substances; RBC galactose-1-phosphate uridyl transferase
Hereditary tyrosinemia Plasma quantitative amino acids; urine succinylacetone
1-Antitrypsin deficiency Quantitative serum 1-antitrypsin; protease inhibitor typing
Neonatal hemochromatosis
Serum ferritin; liver biopsy
Zellweger syndrome Plasma very long-chain fatty acids
Niemann-Pick disease type C
Skin biopsy for fibroblast culture; studies of cholesterol esterification and accumulation
GSD type IV (brancher deficiency)
Liver biopsy for histology and biochemical analysis or skin biopsy with assay of branching enzyme in cultured fibroblasts

Copyright 2009-10 Great Ormond Street Hospital. All rights reserved. 9
Peri-operative management of patients with inborn errors of
metabolism
1. Urea cycle defects and Organic acidaemias, MSUD– Catabolic stress causes breakdown of endogenous protein which can trigger a decompensation. So the aim perioperatively is to ensure
a. No intercurrent infections that may also precipitate a crisis.
b. Do FBC, clotting, U+E’s , LFT’s preoperatively.
c. Adequate iv fluids with glucose to maintain good hydration and prevent catabolism.
d. Monitor ABG, blood glucose
e. Look out for signs of sepsis. 2. Glycogen storage disease 1 a. Treat patients as if they have a full stomach due to hepatomegaly b. Maintain glucose containing infusion c. Monitor serum ph and glucose d. Obtain preoperative bleeding time
3. Glycogen storage 2 (Pompe)
a. ECG preop
b. Care with myocardial depressant drugs 4. Galactosaemia a. Monitor fluids, electrolytes and glucose 5. Homocystinuria a. Beware thrombosis, keep well hydrated and use TEDs 6. Hurler’s (mucopolysaccharidosis) a. Difficult airway management due to deposition of MPS in airway, short neck, poor joint mobility including TMJ b. Restrictive lung disease c. Cardiomyopathy – anaesthetic drugs d. Some other MPS (4,6,7) also associated with atlanto-axial instability. Fibreoptic intubation may be needed. 7. Gaucher disease a. May have pancytopenia, check FBC and correct b. Fracture easily – be aware when handling c. Difficult intubation – retroflexed heads and trismus d. Increased risk of aspiration, secretions++ 8. Lesch – Nyhan a. Drugs used important, use BDZ or barbiturates. Not suxamethonium and more sensitive to narcotics b. Aspiration – increased risk
9. Porphyrias a. Check that do not use trigger drugs for e.g., AIP such as barbiturates, diazepam, local anaesthetics, steroids. Caution for same drugs in VP and HCP types also.
Please see end for Websites & References.

Copyright 2009-10 Great Ormond Street Hospital. All rights reserved. 10
Information for Year 2 ITU Training (advanced):
Year 2 ITU curriculum • Clinical management of patients requiring ITU care: encephalopathy &
seizures, lactic acidosis, hyperammonaemia, neuromuscular weakness, hepatitis.
• Prognostication.
• Peri-operative management of patients with inborn errors of metabolism
Curriculum Notes for Year 2:
Timing and mode presentation depend on site IEM:
Problem synthesizing complex molecules – usually have dysmorphic features at birth
• Zellweger Peroxisomal biogenesis disorder
• Smith Lemli Opitz Block in cholesterol synthesis
• CGD 1a Block in glycosylation
Problem breaking down complex molecules – usually present later as molecules accumulate
• Hurler Failure to break down mucopolysaccharides
• Tay Sachs Failure to break down gangliosides
• Fabry disease Failure to break down glycolipids]
Intoxication – usually presents neonatally and often baby well at first then becomes poor feeder, lethargic and encephalopathic.
• Urea cycle defects Block in ammonia clearance
• Organic acidaemias Block in amino acid breakdown
• Galactosaemia Block in galactose metabolism
Problem in energy generation – can present almost immediately if severe but many different forms and age ranges of presentation
• Congenital lactic acidosis Respiratory chain disorders Pyruvate metabolism disorders (PDH, PCD)
• Energy supply Fat oxidation defects
Glycogen storage disorders Gluconeogenesis defect
Inborn errors of metabolism that acutely present to ITU: The table represents a list of the
different types of IEM. Some are more likely than others to be seen on ICU - these are those that can or usually present acutely with catastrophic decompensation (the relative likelihood is marked by a star rating) HA = Hyperammonaemia
Note: Organic acids are distinguished from amino acids in that they do not contain nitrogen.
Category of Disorder
Disorder Likelihood of acute ITU presentation
Age presentation

Copyright 2009-10 Great Ormond Street Hospital. All rights reserved. 11
Disorders of carbohydrate Metabolism
Glycogen Storage Disorders All Unlikely but: Type 1 may present with hypoglycaemic fits and lactic acidosis
Type 5 + 7 may present with ARF due to rhabdomyolysis
Infancy
Late childhood /adolescence
Galactosaemia *** Hypoglycaemic fits, haemolytic anaemia Unconj jaundice, hepatitis, E. Coli sepsis
0 – 1/12
Hereditary Fructose Intolerance
**Hypoglycaemic fits Weaning 4/12
Primary Lactic Acidaemias
Defects of Pyruvate Dehydrogenase complex
*** however some present less acutely. Metabolic acidosis, Hyperammonaemia (HA), Fits, Hypotonia, Respiratory distress, Apnea, mild dysmorphism
Birth / Infancy or chilhood – very variable depending on subunit affected
Abnormalities of gluconeogenesis a) Pyruvate carboxylase
Deficiency
*** Lethargy, coma, lactic acidosis, HA, apnoea, fits, hypoglycaemia
Usually within 24 hrs
b) Fructose 1,6,
diphosphatase deficiency c) Glucose 6 phosphatase
deficiency (Von Gierke, Type 1 GSD )
Lactic acidosis, hypoglycaemia
Hypoglycaemic fits Lactic acidosis
Newborn
Infancy
Defects in mitochondrial respiratory chain a) Disorders of respiratory
chain complexes b) Cytochrome oxidase
deficiency
**
Fits, stroke, episodes lactic acidosis, respiratory distress, apnoea
Heterogenous group Neonate to childhood (most infancy)
Aminoacidopathies Urea Cycle Defects a) Carbamoyl Phosphate
synthetase (CPS)
b) Ornithine transcarbamylase deficiency (OTC)
c) Citrullinaemia
d) Arginosuccinic acidaemia
(ASA)
****
Encephalopathy Fits Cardiovascular
Collapse Hyperammonaemia
Birth / Infancy
Neonatal
Commonly neonatal / can present later
Phenylketonuria (PKU)
Unlikely Screened for in UK Can present with fits
Early Infancy

Copyright 2009-10 Great Ormond Street Hospital. All rights reserved. 12
Hereditary Tyrosinaemia * sometimes rapidly fatal in infancy – liver failure HA / jaundice, renal failure, hypoglycaemia, bleeding Usually more chronic
Neonatal / Infancy
Maple Syrup Urine Disease (MSUD)
*** ketoacidosis Hypoglycaemia Fits, coma Hypertonia “burnt sugar urine odour”
3 – 10 days Early neonatal
Homocystinuria Unlikely – unless stroke or thromboembolic event, but this is rarely presentation
Childhood
Nonketotic hyperglycinaemia ** neonatal form , encephalopathy, fits, hypotonia
Neonatal – more common Later in infancy – less acute presentation
Sulphite Oxidase deficiency and Molybdenum co-factor deficiency
*** intractable fits Hypotonia, myoclonus Mild lactic acidosis
After first few days of life
Organic Acidaemias
Propionic acidaemia (PA)
*** Ketoacidosis Hypoglycaemia Neutropenia Hyperammonaemia Encephalopathy
Newborn form commonest 0 – 3 days Also later form, more insidious presentation
Methylmalonic acidaemia (MMA)
*** Ketoacidosis Hypoglycaemia Neutropenia Hyperammonaemia
Newborn form commonest 0 – 3 days Also later form,
Encephalopathy more insidious presentation +/- fits
Isovaleric acidaemia (IVA)
*** Ketoacidosis Hypoglycaemia Neutropenia Hyperammonaemia Encephalopathy “Sweaty foot odour”
Newborn form commonest 0 – 3 days
Also later form, more insidious presentation +/- fits
Glutaric acidaemia 1 + 2 (GA1 AND GA2)
** type 2 2 - Catastrophic neonatal form – hypoglycaemia Encephalopathy - HA Metabolic acidosis Hypotonia Infancy form of type 1 can present with subdurals Type 2 can have sweaty feet odour
Neonatal type2 – within 24 hrs
Later infancy or childhood – type 1 or some type2
Combined carboxylase deficiency
a) holocarboxylase
b) Biotinidase deficiency
*** Ketoacidosis, hypoglycaemia, coma, HA, tachypnoea
Intractable fits, acidosis, fungal infections
Within hours of birth
1st
week to 6 months
Copyright 2009-10 Great Ormond Street Hospital. All rights reserved. 13
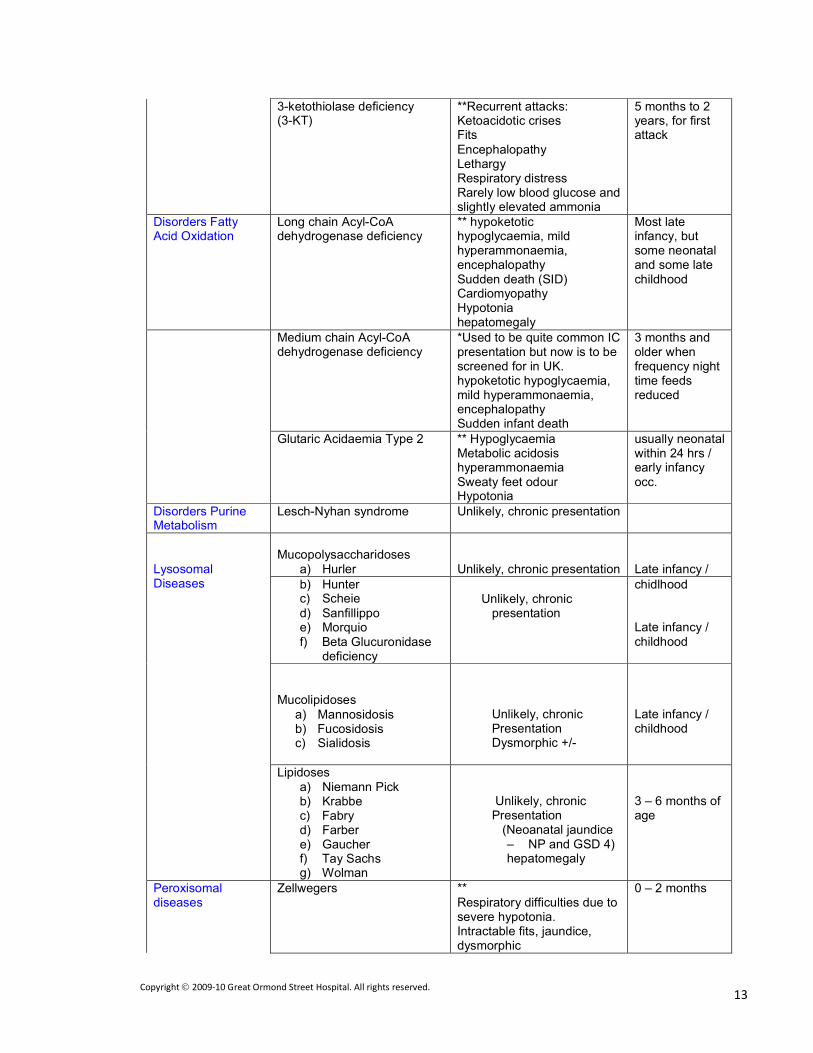
3-ketothiolase deficiency (3-KT)
**Recurrent attacks: Ketoacidotic crises Fits
Encephalopathy Lethargy Respiratory distress Rarely low blood glucose and slightly elevated ammonia
5 months to 2 years, for first attack
Disorders Fatty Acid Oxidation
Long chain Acyl-CoA dehydrogenase deficiency
** hypoketotic hypoglycaemia, mild hyperammonaemia, encephalopathy Sudden death (SID) Cardiomyopathy Hypotonia hepatomegaly
Most late infancy, but some neonatal and some late childhood
Medium chain Acyl-CoA dehydrogenase deficiency
*Used to be quite common IC presentation but now is to be screened for in UK. hypoketotic hypoglycaemia, mild hyperammonaemia, encephalopathy Sudden infant death
3 months and older when frequency night time feeds reduced
Glutaric Acidaemia Type 2 ** Hypoglycaemia Metabolic acidosis hyperammonaemia Sweaty feet odour Hypotonia
usually neonatal within 24 hrs / early infancy occ.
Disorders Purine Metabolism
Lesch-Nyhan syndrome Unlikely, chronic presentation
Lysosomal Diseases
Mucopolysaccharidoses
a) Hurler
Unlikely, chronic presentation
Late infancy /
b) Hunter c) Scheie
d) Sanfillippo e) Morquio f) Beta Glucuronidase
deficiency
Unlikely, chronic
presentation
chidlhood
Late infancy / childhood
Mucolipidoses
a) Mannosidosis b) Fucosidosis c) Sialidosis
Unlikely, chronic Presentation Dysmorphic +/-
Late infancy / childhood
Lipidoses a) Niemann Pick b) Krabbe c) Fabry d) Farber e) Gaucher f) Tay Sachs g) Wolman
Unlikely, chronic Presentation
(Neoanatal jaundice – NP and GSD 4) hepatomegaly
3 – 6 months of age
Peroxisomal diseases
Zellwegers ** Respiratory difficulties due to severe hypotonia. Intractable fits, jaundice, dysmorphic
0 – 2 months

Copyright 2009-10 Great Ormond Street Hospital. All rights reserved. 14
Neonatal Adrenoleukodystrophy
* Depends on severity hypotonia and fits, dysmorphic
neonatal
Hyperpipecolic acidaemia Depends on severity hypotonia and fits
infancy
Primary Hyperoxaluria Unlikely childhood X linked ALD unlikely Childhood Refsum’s unlikely Up to 20 yrs old

Copyright 2009-10 Great Ormond Street Hospital. All rights reserved. 15
Eyes
Corneal clouding MPS
Cystinosis
Tyrosinaemiaa II
I-cell disease Cherry red spot Tay Sachs
Niemann Pick GM 1 Pigmentary Retinopathy Respiratory chain disorders Fat Oxidation Defects Peroxisomal disorders
Congenital disorder glycosylation (CGD) Cataracts Galactosaemia
Peroxisomal disorders Respiratory chain disorder
Organomegaly
• Storage disorders presenting neonatally = I-cell disease (mucolipidoses type2)
and infantile sialic acid storage disease.
Cardiomyopathy – associated with:
• Respiratory chain disorder
• Fat oxidation defects
• Organic acidaemias
• CDG
• Storage disorder
Hypertrophic cardiomyopathy most common CMP associated with IEM.
Hydropic neonates, who often have CHF – some can be caused by lysosomal disorders, so check urine for mucopolysaccharides and oligosaccharides and send blood for vacuolated lymphocytes.
Notes: Significant hyperammonaemia usually seen in urea cycle defects, levels > 1000 micromols/L, and
organic acidaemias (these will be commonest)

Copyright 2009-10 Great Ormond Street Hospital. All rights reserved. 16

Copyright 2009-10 Great Ormond Street Hospital. All rights reserved. 17
2. Organic acidaemias will be accompanied by a usually severe metabolic acidosis and sometimes hypoglycaemia, these are less common features of a urea cycle defect. Acidosis is also a feature of mitochondrial disorders. 3. The mechanism of neurotoxicity of ammonia is not fully understood but the metabolism of pyruvate, lactate and glucose is altered and there are changes in NMDA and GABA receptors. Whatever the cause of hyperammonaemia the clinical encephalopathic picture is similar.
4. Liver dysfunction will result in hyperammonaemia but levels are usually considerably lower (rarely above 500 micromols/L) Investigations: a) Ammonia level b) Liver function tests
c) Clotting studies d) Plasma amino acids – interpret with Metabolic specialist. Results with tandem mass spectrometry should be available in 48 -72 hours or sooner. e) Urinary organic acids– interpret with Metabolic specialist. Results with tandem mass spectrometry should be available in 48 -72 hours or sooner. f) Blood lactate g) Blood gas
h) U+E’s – patients often dehydrated. (low urea often in urea cycle defects) i) FBC – BM depression causing thrombocytopenia and neutropenia in organic acidaemias PA, IVA, MMA. Treatment: ABC APPROACH
Note that ammonia causes an unstable circulation and may be refractory to conventional treatment, in which case dialysis may be urgently necessary.
1. Stop protein intake for 48 to 72 hours, start intravenous glucose to deliver 8 – 10 mg/kg/min glucose (if blood sugar high may require insulin).
2. Haemodialysis to remove ammonia > 600 micromols/L , most effective. If neonate too
unstable try CVVHD or PD. 3. Suspected urea cycle defect (little/no acidosis) an infusion of arginine can be given 6ml/kg over 90 minutes. In patients with citrullinaemia and arginosuccinic aciduria this will drop the plasma ammonia rapidly. Arginine reacts with ammonia so helping excretion. 4. Suspected organic acidaemia, vitamin B12 Img should be given IM, in case patient turns out to have a B12 responsive form of methylmalonic acidaemia. 5. Biotin 10 mg should be given orally/ NG because some patients with multiple carboxylase deficiency are biotin-responsive. 6. If acidotic pH < 7.22 or BD > -14 give bicarbonate and monitor ABG 7. Rehydrate, consider insulin to prevent catabolism. 8. Carnitine 100 mg/kg/day intravenous or oral. Buffers toxic acyl-CoA compounds within the mitochondria. Enhances elimination of toxic organic acids as carnitine esters. 9. Sodium phenylbutyrate is excreted in the urine and assists in clearing nitrogenous
waste – given in any condition with HA. 10. Sodium benzoate combines with glycine to form hippurate which is excreted in the urine – given in any condition with HA. Metabolic Acidosis
Presenting symptoms;
a) Tachycardia b) Poor feeding, persistent vomiting c) Apnoea d) Tachypnoea, Kussmaul breathing e) Vomiting f) Encephalopathy, coma, lethargy, fits. g) Unusual body odours

Copyright 2009-10 Great Ormond Street Hospital. All rights reserved. 18
1. MSUD – burnt sugar/ maple syrup. 2. IVA, GA2 – sweaty feet h) Hypotonia, hypertonia
i) Bleeding – associated with some conditions causing severe metabolic acidosisb
Burton, B. K. Pediatrics1998;102:e69
Causes: 1. Organic acidaemias – MMA, PA, IVA, ASA 2. Amino acid disorders - MSUD 3. Defects in pyruvate metabolism 4. Respiratory chain defects 5. Primary lactic acidosis
Notes:
1. Work out the anion gap:
i. If high measure lactate to work out contribution of lactate or other unmeasured acids to acidosis.
ii. If normal – unlikely IEM
iii. If low measure chloride and look at resuscitation fluids given.
2. Defects in respiratory chain or pyruvate metabolism lead to primary lactic acidosis as the cause of the metabolic acidosis – features will be unrelated to feeding, urinary organic acids will be normal. 3. In defects in respiratory chain or pyruvate metabolism plasma pyruvate will be raised. The lactate/pyruvate ratio should then be measured, If normal ratio < 25 this suggests defect in pyruvate dehydrogenase or in gluconeogenesis. But if the ratio is > 25 this suggests pyruvate carboxylase deficiency,or a respiratory chain defect or a mitochondrial myopathy) 4. Plasma lactate often raised in organic acidaemias as a result of secondary interference with co-enzyme A metabolism

Copyright 2009-10 Great Ormond Street Hospital. All rights reserved. 19
5. Often encephalopathy is a result of severe acidosis and hyperammonaemia (e.g.,organic acidaemias –see below)
6. L/P ratio can also will distinguish metabolic from ischaemic causes and can differentiate some metabolic causes (see above)
Investigations: a) ABG b) Lactate, Plasma pyruvate, Lactate / Pyruvate ratio c) Urinary organic acids and ketones, plasma amino acids d) Blood glucose e) FBC and clotting studies, U + E’s f) LFT’S and ammonia
Management
a) Treat shock
b) Stop fat and protein intake whilst diagnosis is unclear. Start dextrose infusion with at least 5mg/kg/min glucose and electrolytes.
c) Sodium bicarbonate or THAM infusion to correct acidosis. d) Thiamine in suspected MSUD e) Vitamin B12 IM if organic acidaemia suspected (B12 responsive MMA)
f) Biotin 10 mg should be given orally/ NG because some patients with multiple carboxylase deficiency are biotin-responsive.
Encephalopathy
Major Inborn Errors of Metabolism Presenting in the Neonate as an Acute Encephalopathy
Disorders Characteristic Laboratory Findings
Organic acidemias (includes MMA, PA, IVA, MCD and many less common conditions)
Metabolic acidosis with increased anion gap; elevated plasma and urine ketones; variably elevated plasma ammonia and lactate; abnormal urine organic acids

Copyright 2009-10 Great Ormond Street Hospital. All rights reserved. 20
Urea cycle defects Variable respiratory alkalosis; no metabolic acidosis; markedly elevated plasma ammonia; elevated orotic
acid in OTC; abnormal plasma amino acids
Maple syrup urine disease Metabolic acidosis with increased anion gap; elevated plasma and urine ketones; positive ferric chloride test;
abnormal plasma amino acids
Nonketotic hyperglycinemia No acid-base or electrolyte abnormalities; normal ammonia; abnormal plasma amino acids
Molybdenum co-factor deficiency No acid-base or electrolyte abnormalities; normal ammonia; normal amino and organic acids; low serum
uric acid; elevated sulfites in urine
Abbreviations: MMA, methylmalonic acidemia; PA, propionic acidemia; IVA, isovaleric acidemia; MCD, multiple carboxylase deficiency; OTC, ornithine transcarbamylase deficiency.
Hypoglycaemia
Presenting symptoms:
a) Lethargy, poor feeding, vomiting b) Tachycardia, hypertension c) Cyanosis, respiratory distress d) Fits, jitteriness, sweating e) Hypothermia
f) Symptoms due to associated abnormalities of conditions – hyperammonaemia, metabolic acidosis, accumulated toxic metabolites (GSD)
Causes:
1. Glycogen storage disorders 2. Galactosaemia 3. Hereditary Fructose Intolerance 4. Defects in gluconeogenesis 5. Fatty acid oxidation defects 6. Maple syrup Urine Disease, Leucine sensitivity 7. Organic acidaemias - PA, MMA 8. Tyrosinaemia
9. Hyperinsulinism a. Primary some transient some genetic
b. Beckwith Wiedemann (large for dates, hypotonia, visceromegaly, creases earlobes, omphalocele)
c. Adenoma d. UAC placement – transient. e. Beta cell hyperplasia focal or generalised.
10. Adrenal insufficiency
Disorder Blood Glucose
Insulin level Ketones -Beta hydroxybutyrate - Acetoactetate
Other points
Hyperinsulinism Low High Low Low IGFBP 1 Low free fatty acids
GH and cortisol deficiency
Low Low Low
Defective glycogenolysis 1a, 1b, 3 and 6
Low Low High
Fatty acid oxidation defects
Low Low Low/ none Low carnitine

Copyright 2009-10 Great Ormond Street Hospital. All rights reserved. 21
Notes on hypoglycaemia: 1. Can be seen in any sick neonate particularly if septic or small for dates. 2. Normally a healthy neonate can maintain blood glucose for six hours post feed. 3. Persistent servere hypoglycaemia most commonly seen in disorders of
carbohydrate metabolism or fatty acid oxidation defects. 4. In FA oxidation defects ketone production is impaired despite hypoglycaemia so
the hypoglycaemia is nonketotic. 5. When associated with HA, acidosis and raised liver transaminases – a Reye’s
like picture consider FA oxidation defects 6. LCAD – often have associated cardiomyopathy.
7. MCAD and LCAD may present with cardiac arrhythmias or unexplained cardiac arrest.
8. Accumulation of fatty acyl CoA’s in FA oxidation defect patients causes a secondary carnitine deficiency because the carnitine is used to excrete acylcarnitines in the urine. Hence evaluation of plasma carnitine, acylcarnitine levels are useful in diagnosis.
9. MCAD is now to be routinely screened for in the newborn. 10. Low glucose plus ketonuria - not hyperinsulinism. 11. IGFBP 1. This is insulin like growth factor-binding protein-1. Normally insulin
suppresses secretion of IGFBP-1; hence IGFBP 1 is normally elevated in the fasting or hypoglycemic child unless hyperinsulinism is present.
Investigations – done when hypoglycaemic:
a) Blood glucose
b) Insulin level c) Plasma ammonia if altered level of consciousness d) IGFBP 1 level e) Plasma amino acids f) Urinary organic acids
g) Urine reducing substances, ketones. h) Growth hormone i) Cortisol and ACTH levels j) Thyroid function tests k) Glucagon level l) Acylcarnitine, carnitine profiles m) Free fatty acids n) Plasma beta-hydroxybutyrate o) Blood gas p) FBC, Clotting profile q) Lactate, pyruvate, alanine r) LFT’s, U+E’s (often dehydrated)
s) C-peptide, which should be released with insulin and may help differentaite exogenous sources.

Copyright 2009-10 Great Ormond Street Hospital. All rights reserved. 22
Summary of investigation and emergency management of a neonate with suspected IEM
Investigations:
Full blood count with differential and film, clotting screen.
Urea and Electrolytes, liver function tests, Blood Glucose
Blood gases, lactate (+/- pyruvate)
Plasma ammonia (altered level of consciousness)
Urine reducing substances, Urine ketones if acidosis or hypoglycemia present
Plasma and urine amino acids, quantitative, Urine organic acids
Urinalysis – odour, pH
Emergency Management
1. ABC resuscitation 2. Stop all oral intake protein and fat 48 – 72 hours until stable and diagnosis clearer. 3. Provide calories in the form of an intravenous glucose drip aim 6 – 10
mg/kg/min glucose delivery, +/- insulin. 4. Treat hypoglycaemia and monitor regularly. 5. Treat acidosis with continuous sodium bicarbonate infusion and THAM
monitoring serum sodium. Monitor ABG regularly. 6. Raised ammonia - sodium phenylbutyrate/ sodium benzoate. 7. Removal metabolites and ammonia > 600 micromols/L (urgent) with dialysis or
CVVH/CVVHD or PD, also institute for severe intractable high anion gap acidosis.
8. Suspect UCD ( high ammonia, normal glucose, no acidosis) – arginine infusion and or sodium phenylbutyrate/ sodium benzoate.
9. Suspect organic acidaemia ( acidosis, high ammonia, hypoglycaemia, neutropenia, thrombocytopenia) – carnitine infusion 100 mg/kg/day and give B12 (1 mg daily IM) and biotin (5 mg daily PO or IV)
10. Suspect MSUD – give thiamine 50 mg daily PO. 11. Suspect FA oxidation defect – give carnitine. 12. Intractable seizures – pyridoxine, biotin.
13. Take all relevant samples for investigation of diagnosis, particularly important if patient is dying (see protocol)
Other Investigations
1. Very long chain fatty acids Peroxisomal disorders 2. 7-dehydrocholesterol Smith Lemli Opitz 3. CDG Transferrin isoelectric focusing 4. CSF lactate Respiratory chain disorders 5. Neuroimaging Respiratory chain disorders, MSUD, Purine disorders 6. Liver/ spleen Ultrasound Bright liver with fatty infiltration – peroxisomal, CDG 1a 7. X-rays Skeletal dysplasia, calcific stippling (perox) 8. ECG Conduction disorder – FAO, RCD
9. Muscle biopsy No ketosis
For suspected RCD
Hypoglycaemia Hepatomegaly Liver dysfuncyion Hyperammonaemia Raised CK (VLCAD, LCHAD) US liver – fatty

Copyright 2009-10 Great Ormond Street Hospital. All rights reserved. 23
Hepatic glycogen storage I, III, VI, IX and 0 Hypoglycaemia Hepatomegaly Raised cholesterol and TG’s – I, VI, IX Raised urate – I Raised CK – III
3. Given Reading for ICTPICM Curriculum
1. Levy PA. Inborn errors of metabolism: Part : Overview. Pediatr Rev 2009; 30: 131-7I
2. Burton B. Inborn errors of metabolism in Infancy: A guide to diagnosis. Pediatrics. 1998;102(e69)
3. Pandor A, Eastham J, Chilcott J. Clinical Effectiveness and cost-effectiveness of neonatal screening for inborn errors of metabolism using tandem mass spectrometry: a systematic review. Health Technol Assess 2004;8(12)
4. Summar M< Tuchman M. Proceedings of a consensus Conference for the management of patients with urea cycle disorders. Journal Pediatrics, 2001; 138(1).
5. Kosenko E, Kaminski Y, Lopata O, et al: Blocking NMDA receptors prevent the oxidative stress induced by acute ammonia intoxication. Free Radic Biol Med 1999 Jun; 26(11-12): 1369-74[Medline]. *******************
6. Riordan SM, Williams R: Treatment of hepatic encephalopathy. N Engl J Med 1997 Aug 14; 337(7): 473-9[Medline].
7.. Klose D, Kolker S, Hoffmann G. Incidence and short term outcome of children with symptomatic presentation of organic acid and fatty acid oxidation disorders in Ferriero D. Germany. Pediatrics 2002:110(6): 1204-1211
8. Nicolaides, P, Leonard, J, Surtees, R (1998). Neurological outcome of methylmalonic acidaemia. Arch. Dis. Child. 78: 508-512
9.. Ferriero D. Medical Progress: Neonatal Brain Injury.N Engl J Med 2004; 351:1985- 1995, Nov 4, 20041.
10. Wilcken B., Wiley V., Hammond J., Carpenter K. Screening Newborns for Inborn Errors of Metabolism by Tandem Mass Spectrometry. N Engl J Med 2003; 348:2304- 2312, Jun 5, 2003.
11. Champion M, Fox G. inborn Errors of Metabolism – a guide for Neonatologists. Orphan Europe 2003.
12. Handbook of Pediatric Intensive Care. Rogers and Helfaer. Williams and Wilkins 1998.H
13. Handbook of Paediatric Intensive Care. Pearson G. Saunders 2002.
5. Additional Useful Information & Internet sites
1. www.savebabies.org. – Primarily an information site for parents but has good section
named disease descriptions which details many IEMs. USA site.
2. www.eMedicine.com – huge numbers of papers on discovery of, up-to-date news about and symptomatic and dietary treatments of IEMs. USA site. 3. www.agsdus.org – All about glycogen storage diseases
4. www.oaanews.org – the organic acidaemia support association. Useful disease specific information and decompensation protocols. USA site.
5. www.ssiem.org.uk – UK site but to read any articles you need membership.