mutagenic impurities, metal residues, and extractables … · impurities training event agenda •...
TRANSCRIPT

Impurities and residues:
mutagenic impurities, metal
residues, and extractables
and leachables
MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration
and Control, “A Scientific Approach”
A Teasdale, AstraZeneca

Impurities Training Event
Agenda
• Introduction – what is an impurity?
•Impurity Qualification
•Genotoxic (Mutagenic) Impurities
•Metal Impurities
•Extractables and Leachables
2 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”

Impurities
Introduction
WHAT IS AN IMPURITY? •Impurities are substances which differ from the chemical composition of the material or compound.
WHY ARE THEY PRESENT? •Impurities in pharmaceuticals arise as a consequence of a number of factors most relating to the manufacture of the active
• Also include impurities arising from storage of the product e.g. Degradants and/or leachables.
•Impurities should be effectively controlled and where possible eliminated.
• Approach taken needs to be pragmatic and based on safety hazard.
MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach” 3

Qualification ‘The process of acquiring and
evaluating data that establishes the
biological safety of an individual
impurity or a given impurity profile at
the level(s) specified.’

Background to qualification
• Drug substance impurities must be qualified in preclinical studies
before drug is used in man.
• Risk assessment for man based on preclinical studies will include any
contribution to toxicity profile resulting from presence of impurities at
levels tested
• Decision tree and thresholds for reporting, identification and
qualification of impurities for Marketing Authorisation Applications or
New Drug Applications are contained in ICH Q3A(R) and Q3B(R)
guidance documents
• For a drug dosed at up to 2g/day, the threshold for qualification for
impurities is 0.15% or 1.0mg/day, whichever is lower
MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach” 5

ICH Q3a decision tree for qualification studies
Greater than id threshold?
Any known human relevant risks?
YES
NO
Greater than qualification threshold?
YES
Reduce to qualification thresholdc?
No action NO
NO
No further
action
NO
Any clinically relevant adverse effects?
Reduce to safe level
YES
YES
Structure identified?
NO Reduce to
identification
thresholdc
Consider need for:
1. Genotox studies (point mutations, chromosomal aberrations)
2. General tox studies (1 species, min 14 days, max 90 days)
3. Other specific tox endpoints, as appropriate
NO
No further
action
YES
Qualified NO
YES Reduce to safe level
YES
MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach” 6

Design of qualification studies
• Careful design required: objective is to identify any significant change to
safety profile of a drug resulting from the impurity
• Studies should be compared with previous toxicology studies (e.g.. dose
levels normally focused on no-effect or low effect levels) NB. taking safety
margins into account
• Species selected should enable potential impurity effects to be identified;
often species that tolerates highest doses of drug substance – the rat in
most cases
• Study duration - at least 14 days and up to 3 months (dependent on
duration of clinical dosing, duration of existing toxicology studies, whether
the impurity or degradation product was present or not in the original
toxicology tests (but at low concentrations), and evidence or suspicion of
accumulation, e.g. heavy metals).
MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach” 7
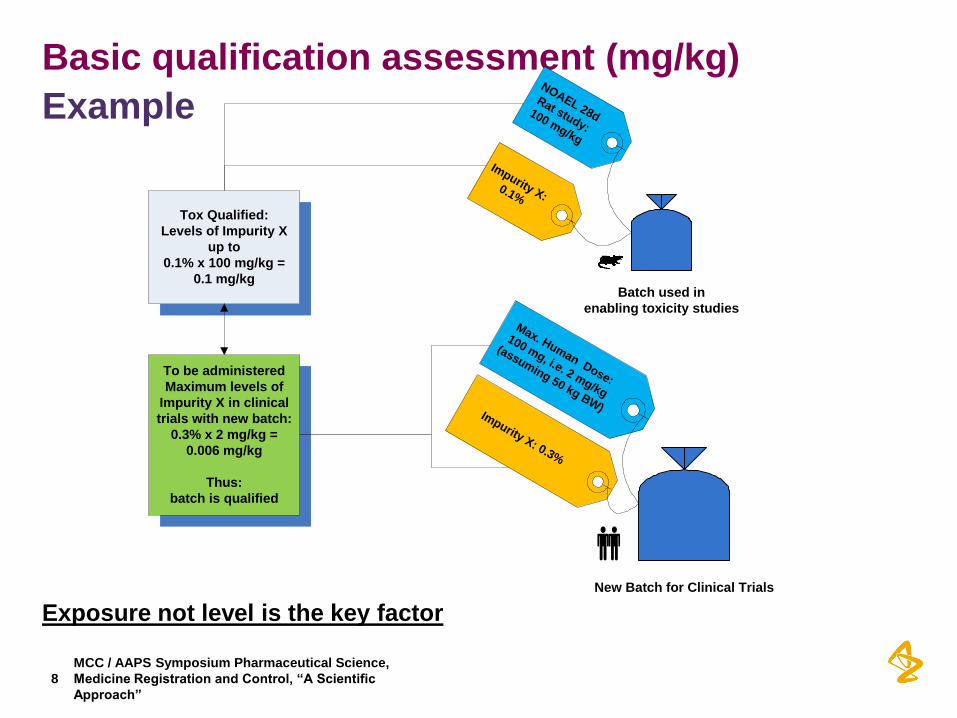
Basic qualification assessment (mg/kg)
Example
Exposure not level is the key factor
8
MCC / AAPS Symposium Pharmaceutical Science,
Medicine Registration and Control, “A Scientific
Approach”
Batch used in
enabling toxicity studies
Impurity X: 0.1%
Tox Qualified:
Levels of Impurity X
up to
0.1% x 100 mg/kg =
0.1 mg/kg
New Batch for Clinical Trials
Impurity X: 0.3%
NOAEL 28d
Rat study:
100 mg/kg
Max. Human Dose:
100 mg, i.e. 2 mg/kg
(assuming 50 kg BW)
To be administered
Maximum levels of
Impurity X in clinical
trials with new batch:
0.3% x 2 mg/kg =
0.006 mg/kg
Thus:
batch is qualified

Genotoxic (Mutagenic)
Impurities
Current and Future
Guidelines - Implications

Introduction
What is a genotoxin?
•Genotoxicity defined as: Toxic (damaging) to
DNA.
•Of specific interest are substances that bind
directly to DNA thereby causing mutations which
may or may not lead to cancer.
•This is mutagenicity
MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach” 10

Introduction – Current Guidelines
• Currently the only formal guideline pertaining to genotoxic impurities is the EMEA (now EMA) guideline.
• Published in mid-2006 this became effective 1st January 2007.
• Supplemented since by a Q&A Process. - Industry questions answered by the EMA
• FDA published a draft guideline in Dec 2008 - Will not be adopted
• Now an ICH topic – ICH M7
11 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”

Historical Perspective
The beginning
•In 2002 the EU position paper caused a great deal of concern.
•Paper was based on the belief that current controls over genotoxic residues were inadequate.
• i.e. Not addressed through the ICH quality guidelines.
•Paper structured in similar way to the current EMA guideline.
• Two sections • Quality Assessment and • Toxicological Assessment.
12 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”
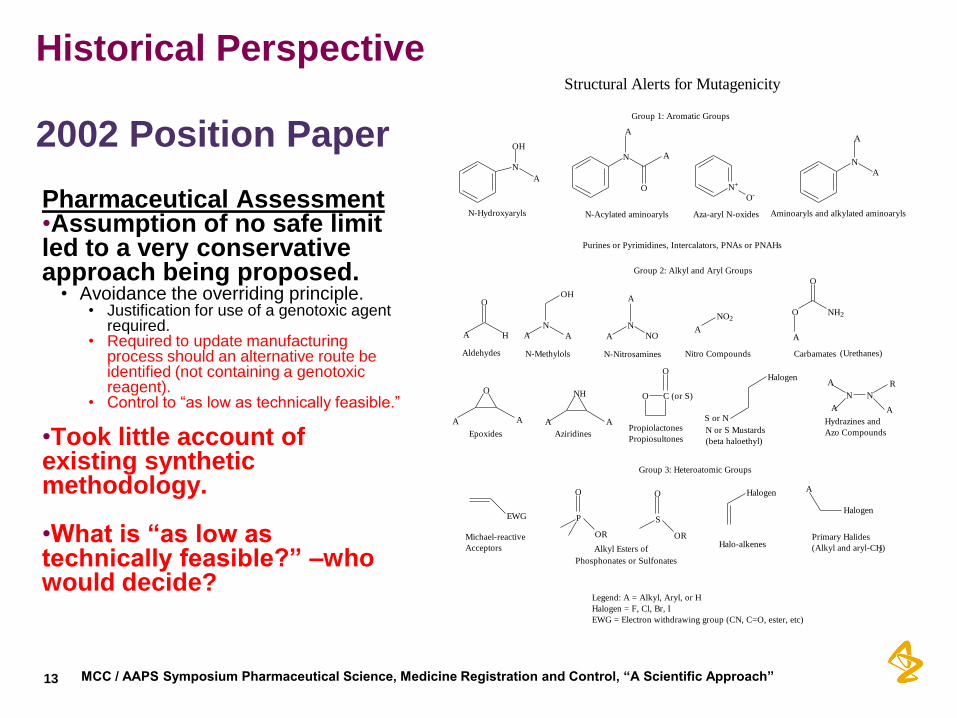
Historical Perspective
2002 Position Paper Pharmaceutical Assessment •Assumption of no safe limit led to a very conservative approach being proposed.
• Avoidance the overriding principle. • Justification for use of a genotoxic agent
required. • Required to update manufacturing
process should an alternative route be identified (not containing a genotoxic reagent).
• Control to “as low as technically feasible.”
•Took little account of existing synthetic methodology.
•What is “as low as technically feasible?” –who would decide?
13 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”
N
OH
N
O N+
O-
N
A
A
Aminoaryls and alkylated aminoarylsAza-aryl N-oxides
A
N-Hydroxyaryls
A
A
N-Acylated aminoaryls
Purines or Pyrimidines, Intercalators, PNAs or PNAHs
Group 1: Aromatic Groups
Group 2: Alkyl and Aryl Groups
O
N
OH
NO
N
O NH2
O
NO2
A H A A
A
AA
A
Aldehydes N-Methylols N-Nitrosamines Nitro Compounds Carbamates (Urethanes)
O NH O C (or S)
AA
Epoxides
A A
O
Propiolactones
PropiosultonesAziridines
Halogen
S or N
N or S Mustards
(beta haloethyl)
N N
R
A
A
A
Hydrazines and
Azo Compounds
Group 3: Heteroatomic Groups
EWG
Michael-reactive
Acceptors
P
O
OR
S
O
OR
Alkyl Esters of
Phosphonates or Sulfonates
Halogen
Halo-alkenes
Halogen
A
Primary Halides
(Alkyl and aryl-CH2)
Legend: A = Alkyl, Aryl, or H
Halogen = F, Cl, Br, I
EWG = Electron withdrawing group (CN, C=O, ester, etc)
Structural Alerts for Mutagenicity

Historical Perspective
Draft EMEA Guideline – June 2004 •Significant step forward from an industry perspective – far more conciliatory.
• E.g “as low as technically feasible replaced by “as low as reasonably practical”.
• No longer required to search for a viable alternative at a later date.
•By far the most important change was the inclusion of the acceptable risk concept –the TTC. Set at 1.5µg/day (lifetime exposure).
•Also introduced the idea of flexible limits based on factors such as duration / disease area but no guidance of values.
14
GOOD NEWS
MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”
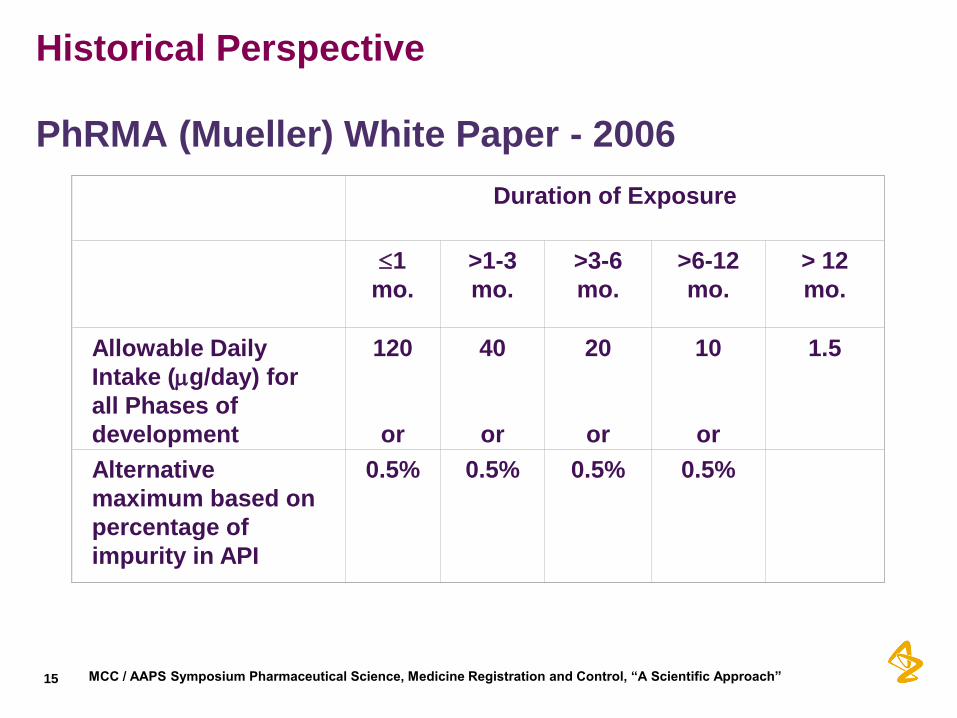
Historical Perspective
PhRMA (Mueller) White Paper - 2006
Duration of Exposure
1
mo.
>1-3
mo.
>3-6
mo.
>6-12
mo.
> 12
mo.
Allowable Daily
Intake (g/day) for
all Phases of
development
120
or
40
or
20
or
10
or
1.5
Alternative
maximum based on
percentage of
impurity in API
0.5%
0.5%
0.5%
0.5%
15 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”

ICH M7
Format / Content
• Many elements common to
EMEA and FDA guidelines •Safety and Quality sections
(different terminology)
•Specific section addressing
Marketed Products.
•New section focused on
documentation.
•Case Studies
16 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”
ICH M7 Step 2
Safety perspective
• Areas of potential significance:
• Specific focus on mutagenic impurities. • Recognised that the primary test is the
Ames test. • No requirement to specifically look to
assess other risks e.g. Clastogenicity.
• SAR evaluation. • Heightened scrutiny around the use of in
silico systems to assess potential mutagenic risk.
• May require the use of multiple systems rule based and statistically based (QSAR). • May have a big impact on small /
medium organisations – these systems are expensive and require expert knowledge
• No clear evidence to support the view that accuracy improved by use of more than 1 system.
17
SAR Evaluation -
• Think spot the difference !
• A structure activity relationship (SAR) relates
features of the molecular structure of a
chemical to a property, effect or biological
activity associated with that chemical.
• Predictions made from chemical structure for
mutagenicity (Ames test)
• Best indicator of genotoxicity.
MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”
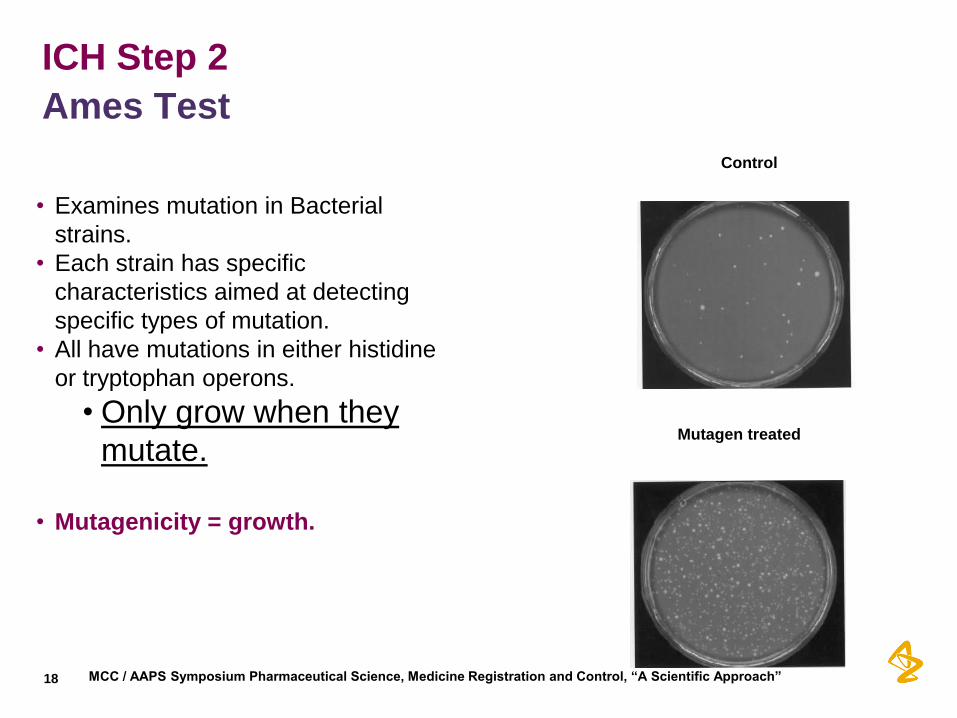
ICH Step 2
Ames Test
18
• Examines mutation in Bacterial
strains.
• Each strain has specific
characteristics aimed at detecting
specific types of mutation.
• All have mutations in either histidine
or tryptophan operons.
• Only grow when they
mutate.
• Mutagenicity = growth.
Control
Mutagen treated
MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”

ICH M7 Step 2
Safety perspective
Areas of Significance cont..
•Greater acceptance of limits based on specific safety
data. • Initiative to develop a table analogous to solvents in Q3c.
•Single set of limits irrespective of route of
administration and/or patient population.
•Greater use of compound specific risk assessment • Construct of table analogous to that use for solvents
19 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”

20
Reagent Ames
Mutagenic /
Non-mutagenic
Mutagen, but AI from linear extrapolation not appropriate not a mutagenic
carcinogen, but
sometimes treated as
one
carco data
problematic*
carcinogenic mechanism not related to
mutagenicity, has threshold,( site-specific)
or rodent specific mechanism
(nat = also prevalent in diet/endogenous
metabolism)
mutagenicity has
practical threshold
(demonstrated in
vivo)
acetaldehyde M X (nat)
Acrolein M X
Allyl bromide M X
Aniline NM X
Benzyl chloride M
Bis-chloromethyl ether M
Bromoacetic acid M
chloro-nitrobenzene M
dimethyl aminopyridine M
Dimethyl sulphate M
DMCC M
Epichlorohydrin M
Ethyl chloride M
Ethyl methane sulfonate M X
Formaldehyde NM X (nat)
gycidol M
hydrogen peroxide M
X
Hydroxylamine NM
Isopropyl chloride M?
methyl chloride M X
Methyl Iodide M X
Methyl methane sulfonate M
N-nitroso pyridine/morpholine/piperazine M
p-chloro-aniline M
X
Phenol NM X
Phenyl hydrazine M
MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”

ICH M7 Step 2
Safety perspective Areas of significance
continued..
•Modification and extension
of variable limits based on
duration to marketed
products.
21
Staged TTC limits
0
20
40
60
80
100
120
140
1 day 2-14 days 15days - 1
month
1-3 months 3-6 months 6-12 months >12 months
Duration
Perm
issib
le l
imit
(u
g/d
ay)
Muller
EMEA
MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”

ICH M7 Step 2
Quality
•Greater flexibility in terms of mechanism to prove
absence. • Options other than to simply test for presence in final API.
• Ability to more widely use chemical / process based arguments to
assess purging. (will examine this further in case studies)
•Tacit recognition of evolutionary nature of risk
assessment. • e.g. Degradants.
•Emphasis on more detail / clarity in submissions. • What will this mean in practice?
• Certainly will need to provide more detail relating to SAR evaluations
22 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”
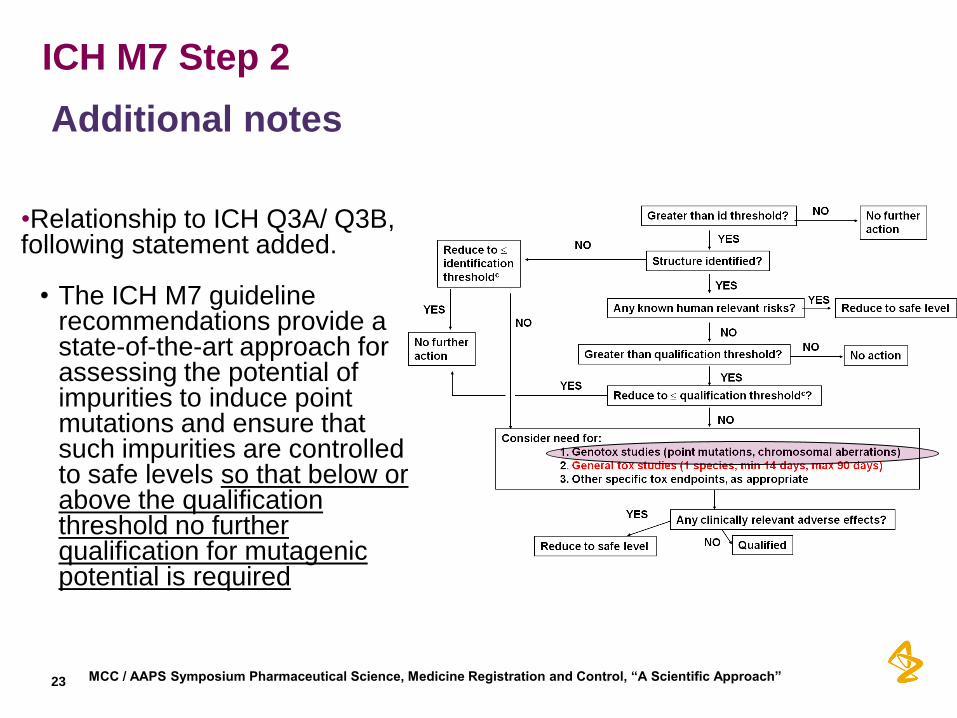
ICH M7 Step 2
Additional notes
•Relationship to ICH Q3A/ Q3B, following statement added. • The ICH M7 guideline
recommendations provide a state-of-the-art approach for assessing the potential of impurities to induce point mutations and ensure that such impurities are controlled to safe levels so that below or above the qualification threshold no further qualification for mutagenic potential is required
23 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”

ICH M7 Step 2
Conclusions
•In general the step 2 document is a positive
development.
•Addressed many previous areas of concern
from quality and safety perspective.
•Little or no impact on current strategy.
•Main areas of uncertainty
• SAR evaluation
• Documentation.
24 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”
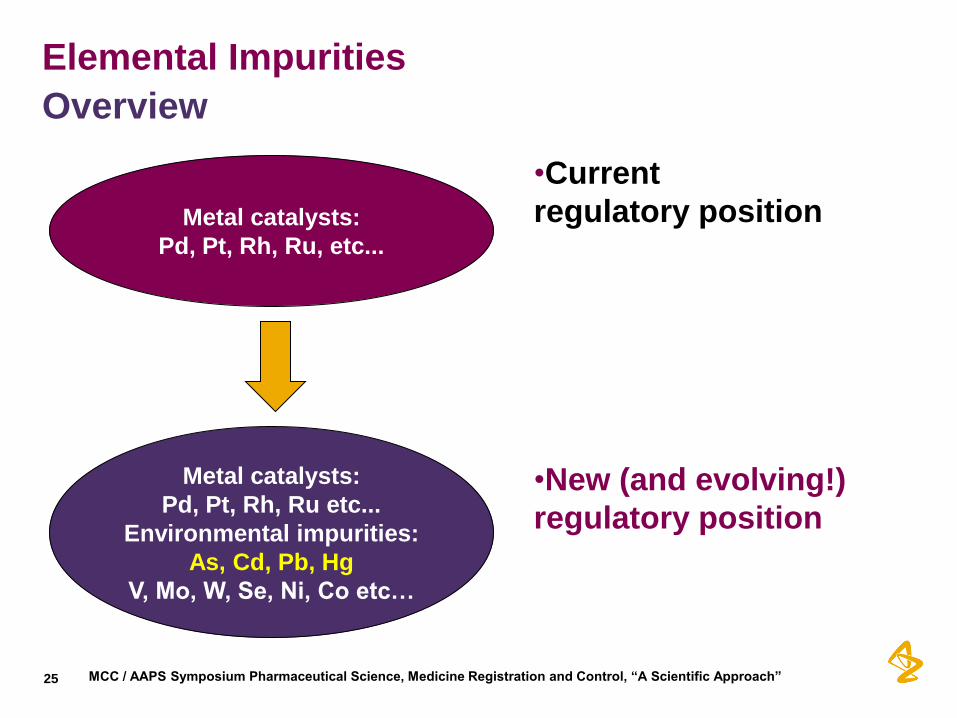
Elemental Impurities
Overview
•Current
regulatory position
•New (and evolving!)
regulatory position
Metal catalysts:
Pd, Pt, Rh, Ru, etc...
Metal catalysts:
Pd, Pt, Rh, Ru etc...
Environmental impurities:
As, Cd, Pb, Hg
V, Mo, W, Se, Ni, Co etc…
25 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”

Metal Impurities
Current regulatory guidance
•ICH Q3A Impurities in New Drug Substances (2002) ‘Inorganic impurities are normally detected and quantified using pharmacopoieal or
other appropriate procedures. Carry-over of catalysts to the new drug substance
should be evaluated during development. The need for inclusion or exclusion of
inorganic impurities in the new drug substance specification should be discussed.
Acceptance criteria should be based on pharmacopoieal standards or known
safety data.’
•EMEA guidelines for Metal catalysts (2008) - recommended specification limits for metal catalyst/reagent residues
- focus is control of API and excipients.
26 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”

Introduction
Historical approach to elemental impurity testing
•Historically risk posed by metals
addressed through ‘wet chemical test’ –
Heavy metals limit test.
•This test relies on the formation of a
sulphide precipitate of the metal in
question
•Issues • Inaccurate
• Non-specific
• Some key metals form soluble sulphide salts
27 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”

Example of a metal impurity control strategy
Routine testing
For Pt
Risks controlled by GMP: purified water, equipment compatibility
Pharmacopoieal tests
e.g. Heavy metals
USP <231>, EP 2.4.8,
JP 1.07
28 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”

New elemental/metal impurities guidelines
Changes a) Move to risk based control strategies that consider all potential sources
of metal impurities in drug products
b) decrease in some permitted daily exposure (PDE) limits c) limits for some ‘new’ elements d) updated analytical technology
USP <232/233> and ICH Q3D
Metal
impurities
in drug
Product
Excipient 2Water
Primary
packaging
materials
Excipient 4
Drug
Substance
Excipient 3Manufacturing
equipment
Excipient 1
29 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”
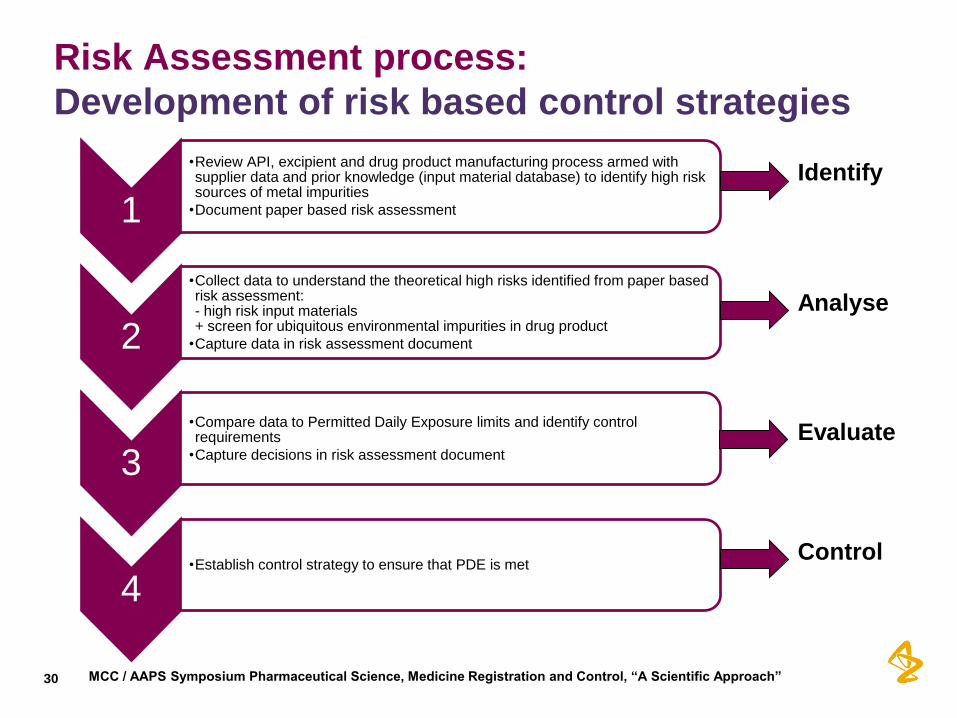
Risk Assessment process:
Development of risk based control strategies
1
•Review API, excipient and drug product manufacturing process armed with supplier data and prior knowledge (input material database) to identify high risk sources of metal impurities
•Document paper based risk assessment
2
•Collect data to understand the theoretical high risks identified from paper based risk assessment: - high risk input materials + screen for ubiquitous environmental impurities in drug product
•Capture data in risk assessment document
3
•Compare data to Permitted Daily Exposure limits and identify control requirements
•Capture decisions in risk assessment document
4 •Establish control strategy to ensure that PDE is met
Identify
Analyse
Evaluate
Control
30 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”
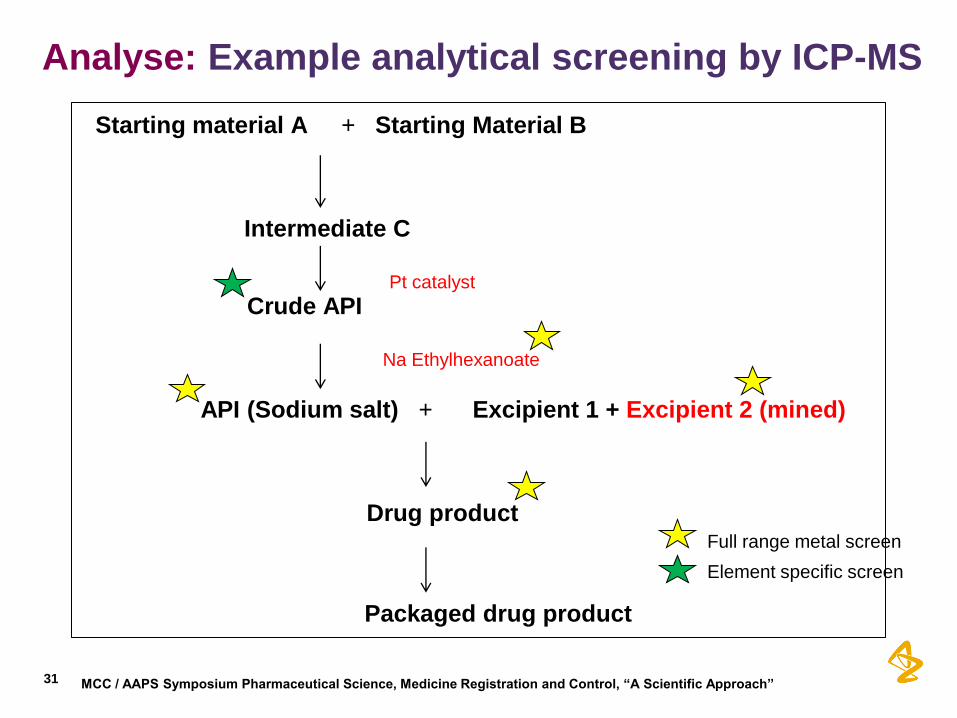
Analyse: Example analytical screening by ICP-MS
Starting material A + Starting Material B
Intermediate C Pt catalyst
Crude API Na Ethylhexanoate
API (Sodium salt) + Excipient 1 + Excipient 2 (mined)
Drug product
Packaged drug product
Full range metal screen
Element specific screen
31 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”

New Guidelines: compliance deadlines
•From 1 December 2015, USP <231> Heavy Metals test will be invalid
• By 1 December 2015, new and existing marketed products must
upgrade from USP <231> to compliance with USP <232> & <233>:
- Documented risk assessment for potential metal impurities
- Updated specification limits (for metals of concern)
- Updated analytical methodology
• By –date undefined – all marketed products must comply with the ICH
Q3D metal impurities guidance:
- Documented risk assessment for potential metal impurities
- Updated specification limits (for metals of concern)
- Updated analytical methodology
•Guidelines are not specifically applicable to clinical development
- But as metal impurities are now a regulatory hot topic we are likely to
receive questions during the clinical trial application process
32 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”

• Lack of harmonisation of global limits (EMEA, USP, ICH
timelines).
• Lack of guidance on acceptable risk based control
strategy principles.
• Many supplier/manufacturing sites aren’t analytically
equipped for the required testing
Evaluate and Control:
Challenges
33 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”

Introduction to
Extractables and Leachables

Overview
- Polymers used in the pharmaceutical industry
- Potential problems with additives
- Definitions
- Route of administration versus interactions
- Guidelines
- Dose & detection limits
- Change control & LCM
35 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”

Polymers used in Pharmaceutical applications
Polypropylene (PP) Polyethylene (PE)
Polyvinylchloride (PVC) • Polysulfone (PS)
Polyurethane (PU) Polymethylmethacrylate (PMMA)
Polycarbonate (PC) Polyamide (PA)
• Rubber (natural & synthetic) Polyvinylidendifluoride (PVDF)
Ethyl vinyl alcohol (EVOH) Polyethylene Terephthalate
(PET)
• Cyclic Olefin Polymer (COC) Silicones
Etc.
36 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”

So what’s the problem using polymers? All plastics, elastomers, film coatings etc. are complex formulations
which can contain various additives, initiators processing agents etc.,
e.g.:-
Monomers oligomers Accelerators
Antioxidants Blowing agents catalysts
Pigments cross linking agents curing agents
Fillers Lubricants plasticisers
Vulcanising agents
Some of these can degrade or react further with the formulation.
Irgafos 168 used
as an antioxidant
in PP & PE
These may end up as Leachables by migrating into the container
closure systems, may be of toxicological concern !
37 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”

What are Extractables and
Leachables?
- Extractables: chemical compounds that are forcibly removed from drug product container closure systems, packaging, or from devices under rigorous laboratory conditions (e.g. reflux) • If low levels are found then Leachables may not be needed
- Leachables: chemical compounds that migrate from drug product
container closure systems, packaging, or from devices under normal-use/stability • May use simulation studies, e.g. placebos, chopped up polymer
etc to accelerate the formulation of leachables
Definitions
38 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”

Extractables and Leachables
A B
C
D
E
F G
H
I
J K
L M N P
A
C
E
H J K
M
Extractables
Drug Product A
Leachables
Forced
Extraction
Testing Drug Product B
Leachables
Product B
A
C
D J K
M
Product A
Different products will have
unique leachables profiles
from the same packaging
system
Note: Some new Leachables may be seen outside the “design space”!
39 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”

Examples of Packaging Concerns for Common Classes of Drug Products.
Degree of Concern
Associated with the
Route of
Administration
Likelihood of Packaging Component-Dosage Form Interaction
High Medium Low
Highest Inhalation Aerosols and
Solutions;
Injections and Injectable
Suspensions
Inhalation powders
Sterile Powders and
Powders for Injection;
High Nasal Aerosols and Sprays
Ophthalmic Solutions and
Suspensions;
Transdermal Ointments
and Patches;
Low Topical Solutions and
Suspensions;
Topical and Lingual
Aerosols;
Oral Solutions and
Suspensions
Topical Powders; Oral
powders
Oral Tablets
Oral (Hard and Soft
Gelatin) Capsules
Regulatory Concern – FDA – Route v
Packaging Interaction
40 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”

Regulatory Concern – detection limits Authorities have increased scrutiny and focus on E&L compounds over
last several years
• PQRI guidelines exist for E&L in Orally Inhaled & Nasal Drug Products
(OINDPs)
•Safety Concern Threshold (SCT) <0.15 ug/day (ref TTC concept in
PGIs 1.5ug/day)
• Compounds seen at ≤ 5ug/day just need SARs evaluation
• Compounds above this threshold must be toxicology justified
• PAH’s, Nitrosamines & 2-mercaptobenzothiazole are exceptions
• PODP (Parenteral, Ophthalmic Drug Products) guidelines are in
preparation by the PQRI and the SCT is likely to be 1.5ug/day
• Solid dosage forms do not normally require E&L data & just require
materials of construction & specification
41 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”

Analytical Concern – detection & quantification – may
vary with dose!
42 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”

Monomer
Synthesis Polymer
Manufacturing
Molding
Shop
(Converter)
Packaging
Containers/
Device
Components
Change Control & LCM - Complex supply chains – Support to Operations
Bulk Chemicals
Storage Stabilizers
Catalysis
Stabilizers
Antioxidants
Processing
aids
Others
Lubricants
Colorants
(N)
End user N-1 N-3 N-4
Master batcher
N-2
Stabilizers
Antioxidants
Processing aids
Antistatic
Others
Trade Secret Protected
43

Conclusions - Impurities in Pharmaceuticals
• Safe medicines can be achieved through appropriate control
of impurities.
• Such control should be predicated on control to safe levels,
not total avoidance.
• There is a strong regulatory framework that supports the
assessment process.
• Care is though needed to ensure the approach taken in
commensurate with the risk.
• By utilising a risk based approach it is possible to ensure
actual risks are identified and mitigated.
44 MCC / AAPS Symposium Pharmaceutical Science, Medicine Registration and Control, “A Scientific Approach”