myc via an intron 1 x box in undiffer
TRANSCRIPT

Activation of protein kinase C induces nuclear translocation of RFX1 and down-regulates c-
myc via an intron 1 X box in undifferentiated leukemia HL-60 cells*
Lei Chen, Lucinda Smith, Martin R. Johnson, Kangsheng Wang, Robert B. Diasio, and Jeffrey
Bingham Smith‡
Department of Pharmacology & Toxicology and Comprehensive Cancer Center, Schools of
Medicine and Dentistry, University of Alabama at Birmingham, Birmingham, Alabama 35294
Running title: Role of RFX1 in the down-regulation of c-myc by PKC
‡To whom correspondence should be addressed: Jeffrey B. Smith, Ph.D., Department of
Pharmacology & Toxicology, Schools of Medicine and Dentistry, University of Alabama at
Birmingham, Birmingham, AL 35294-0019; Tel: (205) 934-7434; Fax: (205) 975-5841; E-mail:
Copyright 2000 by The American Society for Biochemistry and Molecular Biology, Inc.
JBC Papers in Press. Published on July 28, 2000 as Manuscript M002645200 by guest on M
arch 17, 2018http://w
ww
.jbc.org/D
ownloaded from

2
ABSTRACT
Treatment of human promyelocytic leukemia cells (HL-60) with phorbol 12-myristate 13-
acetate (PMA) is known to decrease c-myc mRNA by blocking transcription elongation at sites
near the first exon/intron border. Treatment of HL-60 cells with either PMA or bryostatin 1
(Bryo), which acutely activates protein kinase C (PKC), decreased the levels of myc mRNA and
Myc protein. The inhibition of Myc synthesis accounted for the drop in Myc protein because PMA
treatment had no effect on Myc turnover. Treatment with PMA or Bryo increased nuclear protein
binding to MIE1, a c-myc intron 1 element that defines an RFX1-binding X box. RFX1 antiserum
supershifted MIE1-protein complexes. Increased MIE1 binding was independent of protein
synthesis and abolished by a selective PKC inhibitor, which also prevented the effect of PMA on
myc mRNA and protein levels and Myc synthesis. PMA treatment increased RFX1 in the nuclear
fraction and decreased it in the cytosol without affecting total RFX1. Transfection of HL-60 cells
with myc-reporter gene constructs showed that the RFX1-binding X box was required for the
down-regulation of reporter gene expression by PMA. These findings suggest that nuclear
translocation and binding of RFX1 to the X box cause the down-regulation of myc expression
which follows acute PKC activation in undifferentiated HL-60 cells.
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

3
The c-myc protooncogene encodes nuclear proteins, which heterodimerize with Max and
regulate the expression of genes implicated in cell growth (size), metabolism, differentiation,
apoptosis, tumorigenesis, and genomic stability (1-4). Activation of the c-myc gene is a crucial
oncogenic determinant in a wide variety of human cancers (1, 3). Uncontrolled expression of the
normal Myc proteins is associated with a wide variety of animal and human tumors, with almost a
third of breast and colon carcinomas having elevated c-myc expression (1). Mammalian cells
express multiple Myc polypeptides: Myc1, Myc2, and MycS, which are produced by initiation of
translation at different codons (5). Normal cell function depends on tightly modulated Myc protein
levels. Myc proteins and myc mRNA turnover rapidly (t1/2 < 30 min) in eukaryotic cells, and
multiple redundant systems regulate myc transcription and translation (1-3). Myc and MycS
proteins are degraded by the tightly regulated ubiquitin-proteasome system (6, 7), and stabilization
of Myc has been suggested to be caused by certain cancer-associated mutations (6).
Overexpression of Myc in mammalian cells blocks differentiation, predisposes to malignant
transformation, and can initiate apoptosis (8-10).
Initiation of c-myc transcription at the two major promoters (P1 and P2) is under the
control of several protein factors and DNA elements (2). Furthermore, c-myc was the first
eukaryotic gene shown to be regulated at the level of transcription elongation (11, 12). Premature
transcription termination near the first exon/intron junction depends on initiation at the predominant
P2 promoter and explains the early phase of c-myc down-regulation following induction of
differentiation (2, 11-13). For example, in human promyelocytic leukemia HL-60 cells, induction
of differentiation along either the monocytic/macrophage pathway by phorbol 12-myristate 13-
acetate (PMA)1 and perhaps by 1,25-dihydroxyvitamin D3, or along the granulocytic pathway by
retinoic acid or DMSO, blocks c-myc transcription near the first exon/intron border (11-15).
Protein kinase C β plays a critical role in the differentiation response to PMA, retinoic acid, and
1,25-dihydroxyvitamin D3 (16-18). Cotransfection of Burkitt’s lymphoma cells with a c-myc-
reporter gene construct together with myc gene fragments suggested that the 5’ half of the first
intron contained sequences that competed for one or more putative negative regulatory factors
(19). Remarkably somatic mutations in a 20 bp c-myc intron 1 element (called MIE1 or MIF-1)
abolished nuclear protein binding to the element and were associated with c-myc activation in
Burkitt’s lymphoma cell lines (20). Burkitt’s lymphoma mutations also appeared to be clustered in
two additional protein-binding elements (MIE2 and MIE3) that were just downstream of MIE1
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

4
(21). The functional significance of the intron 1 elements in myc expression remains to be
established. Deletion of MIE1 and MIE2 had no effect on c-myc-driven reporter gene expression,
and deletion of MIE3 modestly increased reporter gene activity in transfected cells (21). Recently
five tandem repeats of MIE1 were shown to suppress the activity of the SV40 promoter in
hepatocarcinoma cell lines (22, 23).
MIE1 essentially consists of a regulatory factor X (RFX) consensus binding site (5’-
GTNRCC(0-3N)RGYAAC), which is called an X box, EP element, or MDBP site (22-25). X
boxes are key positive elements in the promoters of MHC class II (22-24) and interleukin-5
receptor α chain (28). EP elements are enhancers of genes encoded by hepatitis B virus, polyoma
virus, cytomegalovirus, and Epstein-Barr virus (29, 30). MDBP sites occur in a wide variety of
mammalian genes and bind RFX when they are methylated at CpG dinucleotides or when they
contain TpG or TpA at the analogous positions of the methylated cytosine (24, 31).
RFX proteins are the chief component of nuclear complexes previously referred to as MDBP,
MIF-1, NF-X, EF-C, or EP protein (21, 24, 27, 31). RFX family members (RFX1-5) have a
highly conserved winged-helix DNA binding domain (32). RFX proteins homo- and
heterodimerize with one another and up- or down-regulate transcription of target genes in a DNA
context-dependent manner (27, 33-35). RFX1, which appears to be ubiquitously expressed in
mammalian cells, has an N-terminal activation domain and a C-terminal repression domain that
overlaps the dimerization domain (27, 28, 31). The functional regions can neutralize one another
resulting in a nearly inactive transcription factor (34). Association of RFX1with other family
members, with non-RFX proteins such as c-Abl, or with other DNA-bound proteins apparently
determines whether it has enhancer or silencer activity, although the determinants of the activity
are not understood (33-36).
In this study, we measured the rates of synthesis and degradation of Myc proteins
following the treatment of undifferentiated HL-60 cells with PMA or Bryo. Bryo, like PMA, binds
to the zinc finger C1 domain of conventional (α, β, γ) and novel (δ, ε, η, θ, µ) isoforms of PKC,
which turns on its kinase function and concomitantly predisposes it to ubiquitinylation and
degradation by the 26S proteasome (37, 38, 39). In contrast to PMA, Bryo fails to induce
differentiation of HL-60 cells and prevents the induction of HL-60 differentiation by PMA (40).
Bryo, which is in clinical trials as an anticancer agent, apparently down-regulates PKC more
rapidly and efficiently than PMA (41). Our results indicate that a brief activation of PKC in
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

5
undifferentiated HL-60 cells decreased the Myc protein by blocking its synthesis without affecting
its turnover. Studies of HL-60 cells transfected with c-myc-luciferase reporter constructs suggest
that the RFX-binding X box of intron 1 is essential for the down-regulation of myc by PKC. We
also show, for the first time, that PMA treatment induced nuclear translocation of RFX1. Our
findings suggest that nuclear translocation and binding of RFX1 to the X box contributes to the
down-regulation of myc expression following acute activation of PKC.
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

6
MATERIALS AND METHODS
Cell culture and protein concentration. HL-60 cells were grown in RPMI 1640
containing 15% FBS, 100 units/ml penicillin G and 0.1 mg/ml streptomycin. The medium was
diluted with fresh medium three-times per week and cell density was kept below a million per ml.
The cells were collected by centrifugation and incubated in RPMI 1640 containing the additions
indicated in the figure legends. Protein concentration was measured by the BCA method (Pierce
Chemical) with bovine serum albumin as a standard.
Plasmids. pMPCAT (42), which contains a 3.2 kb HindIII/SacI fragment (nt -2238 to
+936) of human c-myc upstream of the CAT gene, was used to produce the luciferase reporter
constructs. A 2 kb fragment of the c-myc (nt –1058 to +936 relative to P1) was excised from
pMPCAT with KpnI and EcoRV and inserted into the pGL3 control vector at the KpnI and HindIII
(blunt) sites in place of the SV40 promoter. The 2 kb c-myc promoter was upstream of the SV40
enhancer and the firefly luciferase gene. The same procedure was used to subclone the myc
promoter mutants from pMPCAT∆287 and pMPCAT∆220 (21). pMP-Luc∆14, which lacked the
14 bp intron 1 X box (nt 3004-3017) was produced by overlap extension polymerase chain
reaction mutagenesis with the following forward and reverse primers, respectively: 5’-TTT TCT
CAG ATG GGG CTG GGG TGG GGG GTA and 5’-CCC AGC CCC ATC TGA GAA AAG TGT
CAA TAG. Each of the constructions was validated by sequencing, carried out on double-
stranded DNA with dye-terminator chemistry, and the products were resolved using an ABI Prism
377 automated sequencer.
Electroporation and dual-luciferase assay. HL-60 cells were collected by centrifugation
and rinsed once with antibiotic-free RPMI 1640, and 20 million cells were suspended with 0.8 ml
of this medium. Electroporation was done at room temperature at 350 V and 960 µF with a Gene
Pulser (Bio-Rad) and 15 µg of the indicated myc-reporter vector and 15 µg of pRL-TK (Promega)
to control for transfection efficiency. After electroporation the cells were placed on ice for 30 min
prior to dilution with 20 ml of RPMI 1640 containing 10% FBS without antibiotics. To assay
luciferase the cells were harvested by centrifugation, rinsed once with room temperature PBS, and
suspended with passive lysis buffer (Promega). The cells were lysed by three freeze-thaw cycles
using liquid nitrogen and a room temperature water bath. The protein concentration of the
samples was measured, and each was diluted to 3 µg/µl with lysis buffer. Luciferase activity (60
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

7
µg) was determined with the Dual-Luciferase Reporter Assay System (Promega) as recommended
by the manufacturer. Statistical analysis was done by two-tailed Student’s t test.
Western blot analyses. For Myc protein analysis HL-60 cells were lysed with hot
(>95oC) SDS lysis buffer which contained 1% (w/v) SDS, 2 mM EDTA, 2 mM EGTA, and 10
mM Tris-HCl, pH 7.4. Sample of containing 30 µg protein were fractionated by SDS-PAGE on a
10% gel (Myc proteins) or 7% gel (MIBP1 or RFX1). Proteins were electrophoretically
transferred to a PVDF membrane (Millipore Corp.), and the membrane was blocked for 1 h at
room temperature with 5% (w/v) nonfat dry milk in TBS. TBS contained (per liter): 8 g of NaCl,
0.2 g of KCl, 3 g of Tris base, and was adjusted to pH 7.4 at room temperature, and in the case of
TBST, 0.05% (w/v) Tween 20. Membranes were incubated overnight at 4oC with 2 µg/ml OP10
primary antibody in TBS containing 1% nonfat dry milk. For western analysis of MIBP1, RFX1
and NFκB, the membrane was incubated overnight at 4oC with a mouse monoclonal antibody to
NFκB (200 fold dilution, F-6, Santa Cruz Biotechnology, Inc.), rabbit antiserum to MIBP1 (250
fold dilution), rabbit antiserum to RFX1 (1000 fold dilution), or the respective preimmune serum
in TBS containing 1% nonfat dry milk. The preparation and specificity of anti-MIBP1 and anti-
RFX1 were described previously (22). Membranes were rinsed and processed with horseradish
peroxidase-conjugated goat anti-mouse IgG (Transduction Laboratories) or with horseradish
peroxidase-conjugated goat anti-rabbit IgG (BioSource) and a chemiluminescent substrate as
described (7). Films were scanned and analyzed with a model GS-670 Imaging Densitometer
using Molecular Analyst software (Bio-Rad).
Immunoprecipitations and pulse-labeling of Myc. For immunoprecipitation a sample of
the SDS lysate (usually 0.5 mg protein) was diluted ten fold with immunoprecipitation buffer
which contained (in mM): 70 NaCl, 50 NaF, 1 EDTA, 1 EGTA, 0.2 mM sodium orthovanadate,
0.2 phenylmethylsulfonyl fluoride, 1% Triton X-100, 0.5% NP-40, and 10 Tris-HCl, pH 7.4. Two
µg of the C-8 anti-Myc monoclonal (C-8, Santa Cruz Biotechnology), or 5 µl of anti-MIBP1, anti-
RFX1, or preimmune sera were added, and the solution was rotated overnight at 4oC, with 30 µl
of a 50% suspension of protein A agarose (Life Technologies) present during the last h.
Immunoprecipitates were collected by centrifugation and washed twice with each of two buffers,
which differed only NaCl concentration: the first buffer contained (in mM): 1 EDTA, 1 EGTA,
500 NaCl, 0.5% NP40, 1% Triton and 10 Tris HCl, pH 7.4; the second buffer lacked NaCl.
Finally immunoprecipitates were washed twice with 10 mM Tris-HCl, pH 7.4, extracted with 20
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

8
µl 2X SDS sample loading buffer, and incubated for 5 min in a boiling water bath. Proteins were
fractionated by SDS-PAGE (10% gel), and the dried gel was fluorographed with Kodak PPB film
to visualize [35S] labeled proteins. For pulse-labeling experiments, the cells were labeled with
[35S]Met/Cys and subjected to the treatments indicated in the figure legends. The [35S] labeled c-
Myc2 band was cut out of the gel and [35S] was quantified by liquid scintillation counting. To
determine the effects of the cell treatments on the overall translation rate, some cells were labeled
with 5 µCi [35S]Met/Cys for 10 min prior to lysis with >95oC SDS buffer and treated with or
without 20 nM PMA or Bryo. Proteins (30 µg) were fractionated by SDS-PAGE (10% gel) and
fluorographically visualized.
Pulse-chase labeling of Myc. HL-60 cells (107 cells per condition) were rinsed twice
with PBS and once with Met/Cys free culture medium. Pulse-labeling was usually done for 60
min with medium containing 10% of the usual concentration of Cys and Met, and 0.15 mCi of
[35S]Met/Cys. Labeling was stopped by addition of 10 mM each of Met and Cys. After the
indicated chase interval the cells were lysed with hot SDS buffer as described above, and Myc
was immunoprecipitated from a sample containing 0.5 mg protein as described above with the
OP10 antibody. Half-lives were determined by nonlinear regression curve fitting to a single
exponential decay equation.
c-myc northern blot analysis. Total RNA was extracted by the acidified guanidinium
thiocyanate-phenol chloroform method with Trizol as recommended by the manufacturer (Life
Technologies) and quantified by absorbance at 260 nm. RNA samples (10 µg) were size
fractionated by electrophoresis on 1% agarose gel containing (in mM): 20 MOPS, pH 7.4, 1
EDTA, 5 sodium acetate, 0.2 M formaldehyde, and 0.5 µg/ml ethidium bromide. RNA samples
contained 50% formamide. The gel was illuminated with a UV lamp and photographed to compare
the quality and quantity of the rRNA. RNA was transferred to Duralon (UV) membranes
(Stratagene) by downward capillary transfer with the Turboblotter (Schleicher and Schuell) and
cross-linked to the membrane with a Stratalinker 1800 (Stratagene). Membranes were
prehybridized with 6 ml QuikHyb (Stratagene) for 10 min at 68oC in a roller-bottle oven. 32P
labeled cDNA probe (3-5 µCi/ml, >109 cpm/µg) was mixed with 0.1ml of denatured salmon
sperm DNA (10 mg/ml) and added to the roller-bottle. After 2 h at 68oC the membrane was
washed twice at room temperature for 15 min with twice concentrated sodium chloride sodium
citrate (SSC) containing 0.1% SDS and twice at 60oC for 15 min with SSC containing 0.1% SDS.
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

9
SSC contained 8.8 g NaCl and 4.4 g sodium citrate per liter and was adjusted to pH 7.0 with HCl.
A 1.4 kbp Cla1-EcoRI fragment of pHSR-1 of human c-myc (ATCC 41010) was agarose gel
purified and labeled using [α-32P] dCTP and the Klenow fragment of DNA polymerase I (Life
Technologies). c-myc transcript was quantified by autoradiography with Konica PPB film and an
intensifying screen for <24 h at -70oC. Autoradiograms were scanned and analyzed with a model
GS-670 Imaging Densitometer using Molecular Analyst software (Bio-Rad).
Nuclear extracts. The cells (3 x 107 per condition) were rinsed and subjected to hypotonic
lysis without mechanical disruption in buffer A, which contained (in mM): 10 Hepes-Tris, pH 7.9,
10 KCl, 1.5 MgCl2, 5 sodium pyrophosphate, 1 sodium orthovanadate, 0.5 DTT, 0.5 PMSF, and 20
nM calyculin A. The cells were incubated on ice for 10 min and observed by phase-contrast
microscopy to determine that >95% lysis had occurred. A nuclear pellet was obtained by
centrifugation (16,000 x g for 10 s) and extracted with buffer B for 20 min on ice with intermittent
dispersal by pipetting. Buffer B contained (in mM): 20 Hepes-Tris, pH 7.9, 25% glycerol, 420
NaCl, 1.5 MgCl2, 0.2 EDTA, 5 sodium pyrophosphate, 1 sodium orthovanadate, 0.5 DTT, 0.5
PMSF, and 100 nM calyculin A. Particulate material was removed by centrifugation for 15 min at
16,000 x g at 4oC and the supernatant was used for EMSA. Nuclear extracts were diluted 3 fold
with buffer C which contained: 20 mM Hepes-Tris, pH 7.9, 20% glycerol, 100 mM KCl, 0.5 mM
DTT, 0.2 mM EDTA, and the following phosphatase inhibitors: 2 nM calyculin A, 1 mM Na3VO4,
and 5 mM sodium pyrophosphate. Nuclear extracts (~1 µg/µl protein) were used immediately for
EMSA and then frozen in liquid N2 and stored at -80oC.
Electrophoretic mobility shift assays (EMSA) and supershift assays. Binding reactions,
which contained nuclear extract (2 µg protein), 12 µl buffer C, 1 µl poly (dI-dC) (1 µg/µl), were
incubated for 10 min at 25oC in the absence or presence of the indicated competitor double
stranded oligonucleotide or 1 µl of rabbit antiserum to MIBP1, RFX1, or the respective
preimmune serum. [32P] labeled double stranded MIE1, (0.1 ng, ~20,000 cpm) was added and
incubation continued for 30 min. After the addition of gel loading buffer (2 µl), which contained
250 mM Tris-HCl, pH 7.4, 0.2% bromphenol blue, and 40% glycerol, the entire reaction was
loaded onto a 4% acrylamide gel which had been prerun for 1 h at 100 V at 4oC. Electrophoresis
was for 1 h at 100 V and 4oC, and the gel was dried and autoradiographed. The gel buffer
contained (in mM): 380 glycine, 50 Tris, 2 EDTA, and had a pH of 8.5.
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

10
Complementary strands of the oligonucleotides (Life Technologies, Inc.), having the
sequences indicated in Figure 5B, were mixed in annealing buffer ( 20 mM Tris-HCl, pH 7.4, 50
mM NaCl, and 1 mM EDTA), incubated at 65oC for 10 min and allowed to cool slowly (>4 h) to
room temperature. The double stranded oligonucleotides were 5' end labeled with [32P] using T4
kinase (Life Technologies, Inc.) and PAGE purified on a 20% gel which had been prerun for 1 h at
150 V.
Materials. Three antibodies which recognize c-Myc were used: OP10 (Calbiochem)
which is a monoclonal to the Myc epitope tag (amino acids 410-419); a rabbit polyclonal to c-Myc
(cat. no. 06-340, Upstate Biotechnology) and C-8 (Santa Cruz Biotechnology, Inc.) which is a
monoclonal produced by immunization with full length human c-Myc. [α-32P] dCTP (3,000
Ci/mmol) and [35S]Met/Cys (>1000 Ci/mmol, EXPRE35S35S) was from Dupont NEN. Bryo, PMA,
and Bis were dissolved in dimethylsulfoxide and added to culture medium from thousand-fold
concentrated stock solutions.
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

11
RESULTS
Activation of PKC decreases c-myc mRNA and protein levels and Myc2 synthesis.
Treatment of HL-60 cells with Bryo or PMA for 4 h markedly decreased the steady state level of
the 64 kDa Myc2 protein (Figure 1A). The decrease in Myc2 protein depended on the
concentration of Bryo or PMA. At 1 nM neither compound affected the Myc2 level, and at 10 nM
each compound almost maximally decreased Myc2 (Fig. 1A). PMA maximally decreased Myc2
by 82%, whereas Bryo maximally decreased Myc2 by 58% (Fig. 1B). Bis, a selective inhibitor of
PKC (43), completely prevented PMA or Bryo from decreasing Myc2 (Figure 2, top panel).
Treatment of the cells with Bis alone moderately increased Myc2, which may be caused by the
inhibition of residual PKC activity in cells not treated with PMA or Bryo (Fig. 2). In addition to
decreasing the Myc2 level, treatment with 20 nM PMA or Bryo for 4 h decreased the level of c-
myc mRNA (Fig. 2, middle panel). Bis prevented either PMA or Bryo from decreasing the c-myc
transcript in the cells (Fig. 2). These results indicate that the decreases in myc mRNA and protein
were produced by the activation of PKC. The 67 kDa Myc1 and 45-50 kDa MycS proteins are
much less abundant in HL-60 cells than Myc2.2 Although we quantified MycS protein levels and
synthesis rates, we present only Myc2 data because all of the treatments described had essentially
the same effects on MycS and Myc2.2
To determine whether the activation of PKC affected Myc translation, the cells were
treated with PMA or Bryo for 1 h and pulse-labeled with [35S]Met /Cys for 10 min. Pulse labeling
of Myc2 increased linearly between 10 and 30 min.2 Treatment with 20 or 200 nM PMA or Bryo
for 1 h markedly decreased Myc2 labeling (Fig. 3A). Treatment with 20 nM PMA decreased the
pulse-labeling of Myc2 to 45 ± 3% control (n = 3). Simultaneous treatment with Bis prevented
PMA from decreasing Myc2 labeling (Fig. 3B). Treatment with Bis alone slightly increased Myc2
labeling (Fig. 3B), in agreement with the effects of Bis on the Myc2 level (Fig. 2). Treatment
with PMA had no effect on general protein synthesis which was determined by the rate of
incorporation of [35S]Met /Cys into protein.2 These results show that a 1 h treatment with PMA or
Bryo decreased the rate of Myc2 synthesis in HL-60 cells. Treatment of the cells with 20 nM
PMA for 1 h decreased myc mRNA to 42 ± 3% control (n = 3) (Fig. 3C). These findings show
that a 1 h treatment with PMA produced similar decreases in the myc mRNA and Myc2 synthesis.
Treatment of the cells with 20 nM Bryo for 1 h decreased myc mRNA level and Myc2 synthesis
similarly to PMA.2
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

12
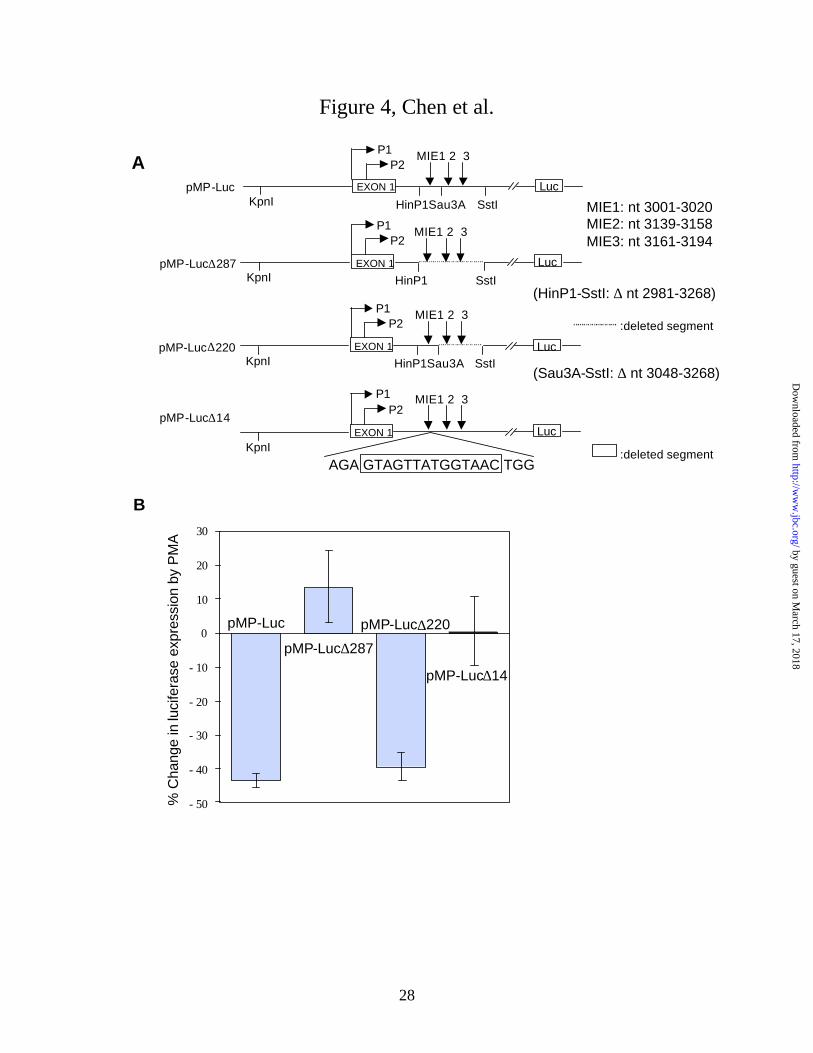
Down-regulation of myc depends on the X box of MIE1. To determine whether the
previously identified myc intron 1 elements (MIEs) were involved in the down-regulation of myc
by PMA, we transfected HL-60 cells with a c-myc-luciferase reporter vector which contained a 2
kb c-myc cDNA upstream of the SV40 enhancer and luciferase gene (Fig. 4A). The 2 kb myc
cDNA consisted of 1057 bp upstream of exon 1, exon 1 (554 bp), and the first 387 bp of intron 1.
Two myc deletion mutants, pMP-Luc∆287, which lacked all three MIEs, and pMP-Luc∆220,
which lacked MIE2 and MIE3 were used to determine whether one or more of the MIEs affected
reporter gene expression (Fig. 4A). HL-60 cells were transfected with the pMP-Luc wild type or
mutant vectors and 18 h later half of the cells were treated with 20 nM PMA for 6 h. PMA
treatment significantly decreased myc-driven luciferase expression by 44 ± 2% (p < 0.005) in
cells transfected with pMP-Luc and by 40 ± 4% (p < 0.02) in cells transfected with the deletion
mutant which lacked MIE2 and MIE3 (Fig. 4B). In cells transfected with pMP-Luc deletion mutant
which lacked all three MIEs, PMA treatment had no significant effect on luciferase expression (p =
0.19) (Fig. 4B). These findings are consistent with the hypothesis that only MIE1 is required for
the down-regulation of myc-driven luciferase expression by PMA. Next we deleted only the 14 bp
X box to determine whether it was required for the down-regulation of myc. PMA had no
significant effect on reporter gene expression in cells transfected with pMP-Luc∆14 (p = 0.44)
(Fig. 4B). Luciferase expression in the untreated cells was essentially the same for each of the
pMP-Luc constructs. These findings indicate the down-regulation of reporter gene expression by
PMA required the myc intron 1 X box.
Activation of PKC increases protein binding to MIE1 DNA. Treatment of HL-60 cells
with 20 nM PMA or Bryo for 1 h increased MIE1-binding activity in the nuclear fraction (Fig.
5A). Two MIE1-protein complexes with slightly different electrophoretic mobilities were
observed by EMSA (Fig. 5). Treatment with Bis eliminated the increases in both MIE1-protein
complexes produced by PMA or Bryo (Fig. 5A), as expected if the increases depended on the
activation of PKC. The specificity of the binding to MIE1 was determined by addition of
competitor oligonucleotides with the indicated sequences (Fig. 5B). A 10 fold excess of
unlabeled MIE1 was sufficient to completely block the MIE1-protein binding (Fig. 5C). A 100
fold excess of duplex MIE2 and MIE3 oligonucleotides relative to the 32P labeled MIE1 probe had
no effect on binding (Fig. 5C). Therefore, the MIE1-protein complexes were specific for MIE1.
A 10 fold excess of the duplex BL1+2 (Burkitt’s lymphoma mutation 1 + 2) oligonucleotide, a
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

13
mutant with two substitutions in the 3’ half of the MIE1 X box (Fig. 5B), had no effect on
[32P]MIE1 binding, and a 100 fold excess of the X box mutant only partially reduced binding (Fig.
5C). This finding confirms the role of the X box in nuclear protein binding to MIE1 as reported
previously (20, 24). These results show that a brief treatment of undifferentiated HL-60 cells with
PMA or Bryo is sufficient to increase specific MIE1-binding activity in the nuclear fraction.
Treatment of the cells with PMA for 0.5, 1, 2, 3, and 4 h increased MIE1-binding activity.2
However, a 48 h treatment of the cells with 0.1 µM PMA, which induced differentiation, as
indicated by the attachment and elongation of cells on the culture surface, had no effect on MIE1
binding.2 A 48 h treatment with 0.1 µM Bryo, which failed to induce attachment and
differentiation, also had no effect on MIE1 binding.2 These findings indicate that MIE1-binding
activity returned to the basal level between 4 and 48 h of PMA treatment , and they suggest that
there is no difference in MIE1 binding activity between undifferentiated and differentiated HL-60
cells, as observed by Erhlich and coworkers (24).
Presence of RFX1 and MIBP1 in MIE1 DNA-protein complexes. Supershift analysis of
MIE1-protein complexes was carried out with antisera to RFX1 and MIBP1 (Fig. 6). MIBP1 is a
160 kDa protein which is present in MIE1 complexes and apparently associates with RFX1 (22).
The formation of the both MIE1-protein complexes depended on the addition of the nuclear extract
as expected (Fig. 6A). A 10 fold excess of unlabeled duplex MIE1, but not the BL1+2 MIE1
mutant, abolished the supershifted complexes indicating that 32P MIE1 binding was specific (Fig.
6A). Antiserum to RFX1 supershifted both of the complexes, but the MIBP1 antiserum
supershifted only the slower mobility (“Complex 1”) (Fig. 6A). This finding suggests that both of
the MIE1-protein complexes contained RFX1, but only the slower mobility complex contained
MIBP1, as reported MIE1-protein complex from HeLa cells (22). Western blot analysis of
nuclear extracts from undifferentiated HL-60 cells confirmed that the antisera specifically
recognized proteins with the expected electrophoretic mobility of RFX1 and MIBP1, respectively
(Fig. 6B). The preimmune sera were not reactive with MIBP1 or RFX1 (Fig. 6B).
PMA treatment increases nuclear accumulation of RFX1, but not MIBP1. Treatment of
HL-60 cells with PMA for 1 h increased RFX1 protein in the nuclear extract as determined by
western blot (Fig. 6B and 7A). PMA increased nuclear fraction RFX1 2.5 ± 0.2 fold (n = 4, p =
0.001). NFκB was also increased in the nuclear fraction by PMA treatment (Fig. 7A), which is
known to induce NFκB translocation from the cytoplasm to the nucleus (41). PMA treatment
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

14
decreased RFX1 in the cytosol and had no effect on total cell RFX1 (Fig. 7A). PMA treatment had
no effect on the level of MIBP1 in the nuclear fraction (Fig. 7B). The increase in nuclear RFX1
was independent of protein synthesis. Blockade of protein synthesis with cycloheximide had no
effect on the accumulation of RFX1 in the nuclear fraction (Fig. 7B). Blockade of protein or RNA
synthesis with cycloheximide or actinomycin D, respectively, had no effect on increased MIE1-
binding activity produced by PMA (Fig. 7C). These findings indicate that the increases in nuclear
extract RFX1 and MIE1-protein binding were independent of protein synthesis.
Lack of effect of PMA treatment on Myc protein turnover. Although decreased synthesis
appeared to cause the fall in Myc protein level, pulse-chase labeling experiments were done to
determine whether activation of PKC also affected Myc turnover. HL-60 cells were labeled with
[35S]Met /Cys for 1 h in the presence or absence of 20 nM PMA. Excess unlabeled Cys and Met
were added to terminate the labeling, and after the indicated chase interval, the cells were lysed
and [35S] labeled Myc was immunoprecipitated and fractionated by SDS-PAGE. The gel was
fluorographed and [35S] labeled Myc1 plus Myc2 was quantified by scintillation counting. These
experiments indicated that [35S] labeled Myc had a half-life of 23 ± 2 min in the cells (Fig. 8).
Following the PMA treatment, [35S] labeled Myc disappeared with a half life of 22 ± 2 min
(Figure 8).
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

15
DISCUSSION
It has been known for some time that premature termination of transcription plays a
critically important role in the down-regulation of c-myc in the early phase of the response to
differentiation inducing compounds in undifferentiated HL-60 cells (11-15). During the later
phase of differentiation of HL-60 cells, however, a loss of transcriptional initiation occurs (45).
In this report we observed that acute activators of conventional and novel isoforms of PKC,
namely Bryo and PMA, rapidly and markedly decreased the steady state level of Myc protein and
mRNA in undifferentiated HL-60 cells (Fig. 1 and 2). Treatment with PMA or Bryo for 1 h
strongly inhibited the rate of Myc synthesis (Fig. 3). Because the half-life of the Myc protein was
unaffected by PMA (Fig. 8), the inhibition of Myc synthesis explained the decrease in the Myc
level. For this inference to be correct the Myc protein must have a relatively short half-life in
untreated HL-60 cells, which it does (~20 min) (Fig. 8).
Our findings indicate that PMA or Bryo markedly increased nuclear protein binding to
MIE1 as determined by EMSA (Fig. 5). Protein binding depended on the MIE1 X box because
substitution of two nucleotides in the 3’ half of the X box strongly decreased the ability of the
BL1+2 oligonucleotide to compete with MIE1 (Fig. 5C). Supershift analysis with antiserum to
RFX1 showed that it was present in both of the MIE1-protein complexes that were resolved by
EMSA (Fig. 6). Only one of the complexes contained MIBP1, which is a 160 kDa uncharacterized
MIE1 binding protein from HeLa cells (22). Increased MIE1-protein binding depended on PKC
activation, but was independent of protein synthesis (Fig. 5 and 7). Bis, a selective inhibitor of
PKC (41), which prevented the down-regulation of myc mRNA and protein by PMA or Bryo, also
abolished their effects on MIE1-binding activity (Fig. 5A).
The mechanism by which acute activation of PKC increased protein binding to MIE1
appears to be indirect and due at least in part to the nuclear translocation of RFX1 (Fig. 7). Thus
we have been unable to detect the 32P labeled RFX1 following immunoprecipitation from 32P
labeled cells.2 Although 32P labeling of MIBP1 was readily detected, PMA treatment had no effect
on the labeling.2 Nuclear translocation of RFX1 could explain the PMA- or Bryo-evoked increase
in the MIE1 complex that contained MIBP1 because this complex also contained RFX1 (Fig. 6A,
“Complex 1”). In agreement with this idea, MIBP1 and RFX1 coimmunoprecipitated from HeLa
cell nuclear extracts in the absence of MIE1 (22). No RFX nuclear localization signals have been
reported, and there appear to be no reports of a dynamic change in the subcellular localization of
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

16
an RFX family protein. One possible explanation for nuclear translocation of RFX1 is the
phosphorylation of an RFX1 associated and cotranslocated protein in response to PKC activation.
Although RFX proteins are most well known as essential transactivators of MHC class II
genes and as a cellular transactivators of pathogenic viruses such as hepatitis B virus (27, 29, 30),
RFX1 appears to be ubiquitously expressed in mammalian cells (25, 27, 28), including
undifferentiated HL-60 cells as determined by western blot analysis (Fig. 6 and 7) and northern
blot analysis (28). The present results suggest that RFX1 binding to the X box of intron 1 has
silencer activity towards myc expression and are consistent with the apparent silencer function of
tandem MIE1 repeats towards SV40 promoter activity in hepatocarcinoma cell lines (22, 23).
Apparently the interaction of RFX proteins with other DNA bound proteins determines whether it
has enhancer or silencer activity , although the determinants of the activity are unknown (36).
A recent report by Pan and Simpson (18) of the suppression of c-myc following a 2 day
treatment of HL-60 cells with 1,25-dihydroxyvitamin D3 implicated MIE1 and activation of PKC,
in agreement with our study using acute PKC activators. However, they suggested that the
homeobox HOXB4 protein was a major MIE1-binding protein and that the 1,25-dihydroxyvitamin
D3 treatment down-regulated c-myc by increasing the level of HOXB4 (18). It is not known if
HOXB4 complexes with RFX1 or other MIE1 binding proteins. The following evidence supports
the view that RFX1 plays a role in the rapid down-regulation of c-myc following acute activation
of PKC: 1) down-regulation of myc-driven reporter gene expression by PMA was abolished by
deletion of the RFX-binding X box (Fig. 4); 2) acute treatment with a PKC activator, PMA or
Bryo, increased protein binding to the X box of MIE1 (Fig. 5); 3) RFX1 was present in the MIE1-
protein complexes (Fig. 6); 4) a selective inhibitor of PKC prevented PMA or Bryo from
increasing MIE1-binding activity and decreasing myc expression (Fig. 2, 3, and 5); and 5) PMA
treatment increased RFX1 in the nuclear fraction and decreased it in the cytosol (Fig. 7). It is
noteworthy that PMA and Bryo only transiently increased MIE1-protein binding.2 We observed no
difference in MIE1-binding activity between undifferentiated and differentiated (2 day PMA
treatment) HL-60 cells as reported previously (24).2 The lack of increased MIE1-binding activity
in differentiated cells is consistent with the role of transcription termination near the first
exon/intron junction in the early response to a stimulus of differentiation (14, 45). Hence, acute
induction of nuclear translocation and binding of RFX1 to the intron 1 X box produced by PMA
correlates with the rapid onset of the blockade of transcription elongation following the addition of
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

17
PMA or other stimuli of differentiation, in contrast to the later phases of myc down-regulation
which involve a loss of transcriptional initiation (14, 45). Bryo treatment downregulated
endogenous c-myc similarly to PMA (this report), however Bryo fails to induce a differentiation
response and antagonizes differentiation produced by PMA (40). PKC activation by Bryo is
shorter in duration than that produced by PMA because Bryo more rapidly and efficiently
downregulates PKC (41). Additional studies are needed to identify critical differences in the
cellular responses to PMA and Bryo which are subsequent to the rapid downregulation of c-myc.
In conclusion, the present findings implicate nuclear translocation of RFX1 and an intron 1
X box in the early phase of the down-regulation of c-myc produced by acute PKC activators in
undifferentiated HL-60 cells. Considering the pivotal role of myc overexpression in malignant
tumors (1, 2), biochemical understanding of myc regulation by RFX1 should help to devise novel
strategies for silencing myc.
ACKNOWLEDGMENTS
We thank Drs. Maria Zajac-Kaye for the generous gift of the pMPCAT constructs and
antisera to RFX1 and MIBP1; G. R. Pettit for the bryostatin 1; and Svetlana A. Shestopal for
helpful discussions.
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

18
REFERENCES
1. Dang, C. V. (1999) Mol Cell Biol 19(1), 1-11
2. Marcu, K. B., Bossone, S. A., and Patel, A. J. (1992) Annu Rev Biochem 61, 809-60
3. Iritani, B. M., and Eisenman, R. N. (1999) Proc Natl Acad Sci U S A 96(23), 13180-5
4. Felsher, D. W., and Bishop, J. M. (1999) Proc Natl Acad Sci U S A 96(7), 3940-4
5. Hann, S. R., King, M. W., Bentley, D. L., Anderson, C. W., and Eisenman, R. N. (1988)
Cell 52(2), 185-95
6. Salghetti, S. E., Kim, S. Y., and Tansey, W. P. (1999) Embo J 18(3), 717-26
7. Chen, L., Smith, L., Accavitti-Loper, M. A., Omura, S., and Bingham Smith, J. (2000) Arch
Biochem Biophys 374(2), 306-12
8. Coppola, J. A., and Cole, M. D. (1986) Nature 320(6064), 760-3
9. Land, H., Parada, L. F., and Weinberg, R. A. (1983) Nature 304(5927), 596-602
10. Evan, G. I., Wyllie, A. H., Gilbert, C. S., Littlewood, T. D., Land, H., Brooks, M., Waters,
C. M., Penn, L. Z., and Hancock, D. C. (1992) Cell 69(1), 119-28
11. Bentley, D. L., and Groudine, M. (1986) Nature 321(6071), 702-6
12. Eick, D., and Bornkamm, G. W. (1986) Nucleic Acids Res 14(21), 8331-46
13. Spencer, C. A., LeStrange, R. C., Novak, U., Hayward, W. S., and Groudine, M. (1990)
Genes Dev 4(1), 75-88
14. Salehi, Z., Taylor, J. D., and Niedel, J. E. (1988) J Biol Chem 263(4), 1898-903
15. Simpson, R. U., Hsu, T., Wendt, M. D., and Taylor, J. M. (1989) J Biol Chem 264(33),
19710-5
16. Tonetti, D. A., Henning-Chubb, C., Yamanishi, D. T., and Huberman, E. (1994) J Biol
Chem 269(37), 23230-23235
17. Simpson, R. U., O'Connell, T. D., Pan, Q., Newhouse, J., and Somerman, M. J. (1998) J
Biol Chem 273(31), 19587-19591
18. Pan, Q., and Simpson, R. U. (1999) J Biol Chem 274(13), 8437-44
19. Chung, J., Sinn, E., Reed, R. R., and Leder, P. (1986) Proc Natl Acad Sci U S A 83(20),
7918-22
20. Zajac-Kaye, M., Gelmann, E. P., and Levens, D. (1988) Science 240(4860), 1776-80
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

19
21. Yu, B. W., Ichinose, I., Bonham, M. A., and Zajac-Kaye, M. (1993) J Biol Chem 268(26),
19586-92
22. Reinhold, W., Emens, L., Itkes, A., Blake, M., Ichinose, I., and Zajac-Kaye, M. (1995) Mol
Cell Biol 15(6), 3041-8
23. Blake, M., Niklinski, J., and Zajac-Kaye, M. (1996) J Virol 70(9), 6060-6
24. Zhang, X. Y., Supakar, P. C., Wu, K. Z., Ehrlich, K. C., and Ehrlich, M. (1990) Cancer Res
50(21), 6865-9
25. Emery, P., Strubin, M., Hofmann, K., Bucher, P., Mach, B., and Reith, W. (1996) Mol Cell
Biol 16(8), 4486-94
26. Reith, W., Barras, E., Satola, S., Kobr, M., Reinhart, D., Sanchez, C. H., and Mach, B.
(1989) Proc Natl Acad Sci U S A 86(11), 4200-4
27. Mach, B., Steimle, V., Martinez-Soria, E., and Reith, W. (1996) Annu Rev Immunol 14,
301-31
28. Iwama, A., Pan, J., Zhang, P., Reith, W., Mach, B., Tenen, D. G., and Sun, Z. (1999) Mol
Cell Biol 19(6), 3940-50
29. Ben-Levy, R., Faktor, O., Berger, I., and Shaul, Y. (1989) Mol Cell Biol 9(4), 1804-9
30. Ostapchuk, P., Scheirle, G., and Hearing, P. (1989) Mol Cell Biol 9(7), 2787-97
31. Zhang, X. Y., Jabrane-Ferrat, N., Asiedu, C. K., Samac, S., Peterlin, B. M., and Ehrlich,
M. (1993) Mol Cell Biol 13(11), 6810-8
32. Gajiwala, K. S., Chen, H., Cornille, F., Roques, B. P., Reith, W., Mach, B., and Burley, S.
K. (2000) Nature 403(6772), 916-21
33. Westerheide, S. D., and Boss, J. M. (1999) Nucleic Acids Res 27(7), 1635-41
34. Katan, Y., Agami, R., and Shaul, Y. (1997) Nucleic Acids Res 25(18), 3621-8
35. Katan-Khaykovich, Y., and Shaul, Y. (1998) J Biol Chem 273(38), 24504-12
36. Dikstein, R., Heffetz, D., Ben-Neriah, Y., and Shaul, Y. (1992) Cell 69(5), 751-7
37. Newton, A. C. (1995) J Biol Chem 270(48), 28495-8
38. Lee, H. W., Smith, L., Pettit, G. R., Vinitsky, A., and Smith, J. B. (1996) J Biol Chem 271,
20973-20976
39. Lee, H. W., Smith, L., Pettit, G. R., and Smith, J. B. (1997) Mol Pharm 51, 439-447
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

20
40. Kraft, A. S., Smith, J. B., and Berkow, R. L. (1986) Proc Natl Acad Sci U S A 83(5),
1334-8
41. Lee, H. W., Smith, L., Pettit, G. R., and Bingham Smith, J. (1996) Am J Physiol 271(1 Pt
1), C304-11
42. Avigan, M. I., Strober, B., and Levens, D. (1990) J Biol Chem 265(30), 18538-45
43. Toullec, D., Pianetti, P., Coste, H., Bellevergue, P., Grand-Perret, T., Ajakane, M., Baudet,
V., Boissin, P., Boursier, E., Loriolle, F., and et al. (1991) J Biol Chem 266(24), 15771-
81
44. Ghosh, S., and Baltimore, D. (1990) Nature 344, 678-682
45. Siebenlist, U., Bressler, P., and Kelly, K. (1988) Mol Cell Biol 8(2), 867-74
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

21
FOOTNOTES*This work was supported by grant HL44408 from the National Institutes of Health.
‡To whom correspondence should be addressed: Dr. Jeffrey B. Smith, Department of
Pharmacology & Toxicology, Volker Hall G133E, 1670 University Blvd., UAB, Birmingham, AL
35294-0019. Tel.: 205-934-7434; Fax: 205-975-5841; E-mail: [email protected]
1The abbreviations used are: AD, actinomycin D; Bis, bisindoylmaleimide; Bryo, bryostatin 1;
CAT, choramphenicol acetyltransferase; CHX, cycloheximide; DMSO, dimethylsulfoxide; DTT,
dithiothreitol; EMSA, electrophoretic gel mobility shift assay; FBS, fetal bovine serum; MIBP1,
myc intron binding protein 1; MIE, myc intron 1 element; PAGE, polyacrylamide gel
electrophoresis; PKC, protein kinase C; PMA, phorbol 12-myristate 13-acetate; RFX, regulatory
factor X; SDS, sodium dodecylsulfate; TBS, Tris buffered saline.
2Chen, L., Smith, L., and Smith, J. B., unpublished data
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

22
FIGURE LEGENDS
Figure 1. Down-regulation of Myc2 protein following the treatment of HL-60 cells with Bryo
or PMA. A. HL-60 cells (107 per condition) were incubated with the indicated concentration of
Bryo or PMA for 4 h in RPMI 1640 containing 2% FBS. The cells were lysed with >95oC SDS
buffer, and proteins (30 µg) were fractionated by SDS-PAGE for western blot analysis with the
OP10 monoclonal antibody. B. Graph of Myc2 protein level determined as indicated for A.
Values are mean ± S.E. (n = 3 experiments).
Figure 2. Effects of Bryo, PMA, and Bis on myc mRNA and Myc2 protein. HL-60 cells (107
per condition) were incubated for 30 min in RPMI 1640 containing 2% FBS and 4 µM Bis as
indicated. Then Bryo or PMA (20 nM) was added, and 4 h later the cells were lysed with >95oC
SDS buffer and proteins (30 µg) were subjected to western blot analysis (top panel). For
determination of myc mRNA, the cells (3 x107 each) were treated with Bis, Bryo, and PMA as
described for western analysis, and total RNA was extracted, size fractionated, and subjected to
northern blot analysis (middle panel). 28S rRNA was visualized by ethidium bromide staining
(bottom panel).
Figure 3. Effects of PMA, Bryo, or Bis on Myc2 synthesis in HL-60 cells. A. HL-60 cells (107
each) were incubated in RPMI 1640 containing one-tenth the normal Met and Cys and treated with
the indicated concentration of PMA or Bryo for 1 h. Then the cells were labeled with 0.15 mCi
[35S]Met/Cys for 10 min and lysed with >95oC SDS buffer. c-Myc was immunoprecipitated from
lysate proteins (0.2 mg), fractionated by SDS-PAGE (10% gel), and visualized by fluorography.
B. The cells were incubated in RPMI 1640 containing one-tenth the normal Met and Cys for 10
min with or without 4 µM Bis as indicated. PMA or Bryo (20 nM) was added for 1 h. Then the
cells were pulse-labeled for 10 min and processed to quantify [35S] labeled Myc2 as indicated for
part A. C. Northern blot analysis of myc mRNA following a 1 h treatment with PMA. HL-60 cells
(3 x 107 each) were incubated in the presence or absence of 20 nM PMA as indicated prior to
extraction of total RNA, which was size fractionated, and subjected to northern analysis. 28S
rRNA was visualized by ethidium bromide staining (bottom panel).
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

23
Figure 4. Effects of c-myc intron 1 elements on the down-regulation of reporter gene
expression by PMA treatment of transfected HL-60 cells. A. Diagram of human c-myc-firefly
luciferase construct (pMP-Luc) and deletion mutants lacking all three MIEs (pMP-Luc∆287) or
lacking MIE2 and MIE3 (pMP-Luc∆220). pMP-Luc∆14 lacked the 14 bp X box of MIE1.
Nucleotide positions are relative to the P1 transcription start site. B. Change in firefly luciferase
expression produced by a 6 h treatment of transfected HL-60 cells with 20 nM PMA. After
electroporation with the indicated vector, the cells were incubated for 18 h in 20 ml of RPMI 1640
containing 10% FBS before adding PMA to half the cells from each electroporation. The cells
were cotransfected with pRL-TK vector and the Dual-Luciferase Assay System (Promega) was
used to measure myc-driven firefly luciferase and Ranilla luciferase activity as a control for
transfection efficiency. Values are mean ± S.E. for 5-7 experiments. Percentage change in
luciferase activity produced by PMA was significantly different from pMP-Luc to pMP-Luc∆287
(p = 0.003) and to pMP-Luc∆14 (p = 0.004), but not to pMP-Luc∆220 (p = 0.419).
Figure 5. Effects of PMA, Bryo, and Bis on protein binding to 32P labeled MIE1 duplex DNA.
A. EMSA autoradiogram obtained with nuclear extracts from HL-60 cells following treatment
with 20 nM PMA or Bryo for 1 h as indicated. Some cells were treated with 4 µM Bis for 30 min
before the addition of PMA and Bryo. Nuclear extracts were prepared and 2 µg protein was
incubated with 1 µg poly dI-dC for 10 min at 25oC as described in “Methods”. Then 0.1 ng 32P
labeled duplex MIE1 was added to each reaction and the incubation continued for 30 min. Binding
reactions were size-fractionated by Tris-glycine PAGE (4% gel). The positions of the two
protein-32P-MIE1 complexes are indicated. B. Diagram of c-myc intron 1 elements (MIEs) and
nucleotide sequences of the MIEs and the BL1+2 mutant. C. Specificity of the 32P-MIE1-protein
complexes was determined by addition of 1 or 10 ng (“10X” or “100X”) of the unlabeled duplex
MIE1, MIE2, MIE3, or the BL1+2 mutant of MIE1 to the binding reaction described in A.
Unlabeled competitor was present during the 10 min incubation of the nuclear extract with poly dI-
dC before the addition of 32P labeled MIE1.
Figure 6. Supershift analysis of 32P labeled MIE1 DNA-protein complexes. A. Antiserum to
RFX1 or MIBP1 or the corresponding preimmune serum (1 µl each) was added to the binding
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

24
reaction which contained 0.1 ng 32P labeled MIE1, 2 µg nuclear extract from cells treated with 20
nM PMA for 1 h, 1 ng duplex MIE1 or the BL1+2 mutant as indicated. The indicated antiserum or
preimmune serum and the indicated duplex DNA competitor was present during the 10 min
incubation of the nuclear extract with poly dI-dC before the addition of 32P labeled MIE1. B.
Western blot analysis of MIBP1 and RFX1 in nuclear extracts from PMA-treated and untreated
HL-60 cells. The cells were treated with 20 nM PMA for 1 h and nuclear extracts were prepared
as described in “Methods”. Nuclear extract proteins (30 µg) were size fractionated by SDS-
PAGE (7% gel) and subjected to western blot analysis with the indicated serum.
Figure 7. Effect of PMA on the level of RFX1 in nuclear extracts, cytosol, and total cell
lysate. A. Western blot analysis of nuclear extract, cytosol, and total cell lysate from untreated
and PMA-treated cells. After the PMA treatment (20 nM for 1 h in RPMI 1640 containing 2%
FBS) a sample of the treated and untreated cells was extracted with >95oC SDS lysis buffer to
obtain the total cell lysate. Another sample of the cells was subjected to hypotonic lysis and the
cytosol was collected after centrifugation to remove nuclei. Nuclear extracts were prepared as
described in “Methods”. Cytosol was concentrated by with Centricon 10 concentrators, and
proteins (30 µg nuclear extract, 60 µg cytosol, or 100 µg total lysate) were size fractionated by
SDS-PAGE (7% gel) and subjected to western blot analysis with the indicated antiserum. B.
Western blot analysis of nuclear extracts (30 µg protein) from cells that were incubated with 10
µg/ml cycloheximide (“CHX”) or 2.5 µg/ml actinomycin D (“AD”) for 10 min prior to the
addition of 20 nM PMA as indicated. One h later nuclear extracts were prepared. C. EMSA was
carried out on nuclear extracts that were prepared from cells that were treated as described in B.
Figure 8. Lack of effect of PMA on the disappearance of [35S]labeled Myc in HL-60 cells.
A. HL-60 cells (107 each) were incubated with or without 20 nM PMA in medium containing a
tenth of the Met and Cys concentrations of RPMI 1640, 2% dialyzed FBS, and 0.15 mCi
[35S]Met/Cys for 1 h. Labeling was stopped by the addition of 10 mM each of Met and Cys, and
after the indicated interval, the cells were lysed with >95oC SDS buffer, and Myc was
immunoprecipitated, fractionated by SDS-PAGE, and visualized by fluorography. B. The
percentage of [35S]Myc remaining after the indicated interval was determined by liquid
scintillation counting of [35S]Myc1 plus Myc2 containing gel slices.
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

25
Figure 1, Chen et al.
100
80
60
40
20
0
Myc
2 (%
con
trol
)
B
Con
trol Bryo PMA
10-3
10-2
100
10-1
10-3
10-2
100
10-1A
Myc2
µM
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

26
Figure 2, Chen et al.
PMA+
Bis
Con
trol
PMA
Bryo
+Bis
Bis
Bry
oMyc2
myc mRNA
28S rRNA
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

27
Figure 3, Chen et al.
Contro
l200 20 200 20
PMA BryonM
S Myc235
S Myc235
Contro
l
PMA
Bis PMA+B
is
A
B
PMA - +myc mRNA
28S rRNA
C
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from
28
Figure 4, Chen et al.
% C
hang
e in
luci
fera
se e
xpre
ssio
n by
PM
A
B
- 50
- 40
- 30
- 20
- 10
0
10
20
30
pMP-Luc
pMP-Luc∆287
pMP-Luc∆220
pMP-Luc∆14
EXON 1
P1P2
HinP1Sau3A SstIpMP-Luc
pMP-Luc∆220
EXON 1
P1P2
HinP1 SstI
EXON 1
P1P2
HinP1Sau3A SstI
:deleted segment
KpnI
MIE1 2 3
KpnI
KpnI
Luc
Luc
Luc
MIE1: nt 3001-3020MIE2: nt 3139-3158MIE3: nt 3161-3194
(HinP1-SstI: ∆ nt 2981-3268)
(Sau3A-SstI: ∆ nt 3048-3268)
A
MIE1 2 3
MIE1 2 3
pMP-Luc∆287
pMP-Luc∆14EXON 1
P1P2
KpnILuc
MIE1 2 3
AGA GTAGTTATGGTAAC TGG:deleted segment
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

29
Figure 5, Chen et al.
PMA+
Bis
Con
trol
PMA
Bryo
+Bis
Bis
Bryo
P-MIE132
A C
Competitor MIE
1M
IE2
MIE
3B
L1+2
10x
100x
P-MIE132
Complex21 Complex
21
Complex21
EXON1 EXON2
P1P2
1 2 3
MIE1: 5’-AGAGTAGTTATG GTA ACTGGBL1+2: 5’-AGAGTAGTTATG ATT ACTGGMIE2: 5’-CCTTATGAATATATTCACGCMIE3: 5’-CTCCCGGCCGGTCGGACATTCCTGCTTTATTGT
MIEB
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

30
Figure 6, Chen et al.
Competitor
Nuclear extract
MIE1BL1+2
+ + + + + + + + + + + + + + + + + + + +
+ + + + +
antiserum none MIBP1 preimmuneMIBP1
RFX1 preimmuneRFX1
AComplex
21
B PMA - + - + - + - +
antiserum: MIBP1 pre-immuneMIBP1
RFX1 pre-immuneRFX1
217
126
73
MIBP1RFX1
kDa by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

31
Figure 7, Chen et al.
Contro
l
PMA
PMA+
CHX
CHX
RFX1
MIBP1
B
PMA - +Nuclear extract
Cytosol
Total lysateNuclear extract
RFX
NFκΒ
A
Contro
lPMACH
X
PMA+
AD
PMA+
CHX
AD
C
P-MIE132
Complex21
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

32
Figure 8, Chen et al.
S Myc35
Control PMA
0 20 40 60 0 20 40 60Chase(min)
A
75
-PMA
+PMA
50
25
0
100
0 20 40 60
S M
yc (
% r
emai
ning
)35
Chase (min)
B
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Jeffrey B. SmithLei Chen, Lucinda Smith, Martin R. Johnson, Kangsheng Wang, Robert B. Diasio and
down-regulates c-myc via an intron 1 X box in undifferentiated leukemia HL-60 cellsActivation of protein kinase C induces nuclear translocation of RFX1 and
published online July 28, 2000J. Biol. Chem.
10.1074/jbc.M002645200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on March 17, 2018
http://ww
w.jbc.org/
Dow
nloaded from