neuraminidase inhibitiors
TRANSCRIPT

7/27/2019 Neuraminidase inhibitiors
http://slidepdf.com/reader/full/neuraminidase-inhibitiors 1/21
176 VARGHESE
© 1999 Wiley-Liss, Inc.
DRUG DEVELOPMENT RESEARCH 46:176–196 (1999)
Research Overview
Development of Neuraminidase Inhibitors as Anti-Influenza Virus Drugs
Joseph N. Varghese*Biomolecular Research Institute, Parkville, Victoria, Australia
ABSTRACT Structure-based design and synthesis of potent influenza virus neuraminidase inhibitors are
now being evaluated in human trials as anti-influenza virus drugs. The first drug of this class, Relenza™(Zanamivir/GG167), is now awaiting pharmaceutical evaluation and registration in Australia, Europe, andNorth America for both treatment and prophylaxis of influenza. The target for the drug is the active site of neuraminidase, which is a pocket that has been totally conserved in both Type A and B influenza in allknown subtypes of influenza (animal and human). Mutations in residues that surround this conservedpocket allow the virus to escape binding to circulating antibodies that recognise the molecular surfacearound the active site of the wild-type virus. High-affinity neuraminidase inhibitors have been designed thatinteract only with the conserved active site residues. The design of these sialic acid analogues was based onthe crystal structure of influenza virus neuraminidase and its complex with N-acetyl neuraminic acid (sialicacid) and 2-deoxy-2,3-dehydro-N-acetyl neuraminic acid. These novel inhibitors are highly specific for in-fluenza neuraminidase, and have been shown to inhibit influenza virus replication in both cell culture andanimal models. The development of drugs against a rapidly mutating organism like influenza has to addressto the possibility of emerging drug resistance. This is examined in the light of drug resistant mutants se-lected after in vitro passaging of virus in the presence of neuraminidase inhibitors. Drug Dev. Res. 46:176–
196, 1999. © 1999 Wiley-Liss, Inc.
Key words: sialidase; Zanamivir; Relenza™
Strategy, Management and Health Policy
Venture CapitalEnabling Technology
PreclinicalResearch
Preclinical Development Toxicology, FormulationDrug Delivery,Pharmacokinetics
Clinical Development Phases I-IIIRegulatory, Quality,Manufacturing
Postmarketing Phase IV
INTRODUCTION
Influenza, a viral infection of the upper respiratorytract in humans, has plagued mankind since the dawn of history. The disease in modern times continues to affecta significant proportion of the population irrespective of age or previous infection history. These periodic epidem-ics that reinfect otherwise healthy people have devas-
tated communities worldwide. Some pandemics like the1917–1919 “Spanish flu” were responsible for the deathsof tens of millions of people throughout the world. Theorigins, spread, and severity of influenza epidemics havebeen a puzzle that has only in the last two decades beenadequately addressed. The virus is spread by aerosolsproduced by infected animals, and the continual produc-tion of new strains of the virus results in reinfection [re-viewed by Kilbourne, 1987].
There are three types of influenza virus classified
on their serological cross-reactivity with viral matrix pro-teins and soluble nucleoprotein (A, B, and C). Only type A and B are known to cause severe human disease. TypeB is only found in humans, while type A has a naturalreservoir in birds and some mammals like pigs and horses[Webster et al., 1993]. Influenza, an orthomyxovirus, is a 100 nm lipid-enveloped virus containing an eight-seg-
ment negative single-stranded genome [Lamb, 1989]. Twoof the segments code for the surfaces glycoproteins he-magglutinin HA (which binds to terminal sialic acid) andneuraminidase NA (which cleaves terminal sialic acid)which appear as spikes protruding out of the viral enve-
*Correspondence to: Joseph N. Varghese, Biomolecular Re-search Institute, 343 Royal Parade, Parkville, Victoria 3052, Austra-lia. E-mail: [email protected]

7/27/2019 Neuraminidase inhibitiors
http://slidepdf.com/reader/full/neuraminidase-inhibitiors 2/21
ANTI-VIRAL DRUGS FOR INFLUENZA 177
lope. The viral target in humans is the upper respiratorytract epithelial cells.
Replication begins with penetration of the virionthrough the mucin layer covering the epithelial surface,followed by attachment to the viral receptor by the HA.Penetration of the cell is achieved by endocytosis and
the virion core is released after the fusion of the virionand vesicle membrane, mediated by the HA. Fusion isenabled by a conformational change in the HA, madepossible by lowering the pH of the endosome by the M2ion channel protein. Following replication, the progenyvirions are released by budding off the cell membrane[Murphy and Bang, 1952; Compans and Dimmons, 1969].
The release of virons occurs 8 h postinfection andthe onset of infection is sudden, resulting in pyroxia,muscular and joint pain, and a dry cough [Murphy andWebster, 1990]. Virus shedding continues for up to a week,when a rise in virus-specific antibody clears the virusfrom the host. The vulnerability of the host succumbingto viremea during this week of rising viral titre is depen-dent on interferon induction [Ennis and Meager, 1981]48 h postinfection, which attenuates viral replication untilthe cell-mediated immune response begins to clear thevirus. The severity of the illness is thought to depend onthe level of cross protection arising from antibodies raisedfrom previous influenza infections [Fox et al., 1982]. Thecourse of the illness can be dehabilitating, and no effec-tive treatment is available at present to halt the progres-sion of the disease. Death can result for susceptiblepopulations (neonate and elderly), primarily as a resultof secondary infections [Sprenger et al., 1993].
In this article, a structural basis for the continualemergence of new influenza strains is discussed, and thereasons why current vaccines against influenza fail toprotect against all strains of influenza. The discovery inthe early 1980s of the molecular structure of influenza virus NA, the detailed atomic description of the activesite of the molecule, and the exploitation of its structuralconservation is discussed in terms of the design of po-tent NA inhibitors. The therapeutic use of these inhibi-tors as anti-viral drugs against influenza virus infectionsshall be examined and the possibility of drug resistanceto NA inhibitors shall be addressed.
Antigenic VariationThe plethora of different strains of the virus that
are responsible for continued reinfection of the virus inhumans is primarily related to mutations in the viral genesof two surface glycoproteins, HA and NA [Smith andPalase, 1989]. The current paradigm for this genetic varia-tion [Skehel, 1974; Webster and Laver, 1975] is that thesemutations arise primarily from incremental changes inthe amino acid sequences of these glycoproteins by se-lection pressure of the immune system of the infected
host. This mechanism, termed “Antigenic Drift,” accountsfor most of the strain variation within a particular sub-type of influenza.
However, infrequently a mutation arises by geneticre-assortment of viruses from different animal hosts (“An-tigenic Shift”), whereby an entirely new gene for one of
the surface glycoproteins is generated which is signifi-cantly different (~50%) in amino acid sequence of theparent virus. This is the mechanism by which new sub-types of influenza arise which are primarily responsiblefor the major pandemics that occur. Such a great changein the antigenic surface of these glycoproteins results inlittle or no cross-protection by circulating antibodies toprevious influenza infections, leading to the global spreadof severe influenza and the high mortality rate suchpandemics inflict.
Strains of influenza virus are classified by type (A,B, or C), geographic location, date of original isolation,and the subtype of the HA and NA antigens. There existnine known subtypes (N1 to N9) of NA and 13 knownsubtypes (H1 to H13) of HA from influenza A in all ani-mal populations. Two NA (N1 and N2) and three HA (H1,H2, and H3) subtypes of influenza A have occurred instrains that have infected humans since 1933, when iso-lates were first characterised [Smith et al., 1933]. Priorto 1933 there is indirect evidence of antigenic shift oc-curring in human populations [Beveridge, 1977]. The N1subtype was associated with virus isolated between 1933and 1957, after which time the N2 subtype appeared inthe Asian influenza. No major change in the structure of NA has occurred since, although the HA subtype has
changed from H2 to H3 in 1968 in the Hong Kong pan-demic, and H1N1 reappeared in 1978 as the Russianpandemic. Recently (1998) an avian strain, H5N1, causeda brief outbreak in humans in Hong Kong that was quicklycontained, as it appears that infections were only trans-mitted from avian to human and not human to human[Yeun et al., 1998; Subbaroa et al., 1998; Claas et al., 1998].Influenza B, which infects only human hosts, has onlyone subtype, but like type A undergoes continual anti-genic drift.
Current Therapeutics and Vaccines
Amantadine and rimantadine are the only class of
drugs that have been approved for therapy [Douglas,1990; Wintermeyer and Nahata, 1995]. At high concen-tration (>50µg/mL), amantadine is thought to buffer thepH of the endosome and prevent the conformationalchange of the HA necessary for fusion. Drug-resistantmutants arise where the hemagglutinin trimers arethought to be less stable than the wild type [Daniels etal., 1985]. At low concentrations (<1 µg/mL), amanta-dine blocks the activity of the viral M2 ion channel pro-tein, which plays a role in virus uncoating and

7/27/2019 Neuraminidase inhibitiors
http://slidepdf.com/reader/full/neuraminidase-inhibitiors 3/21
178 VARGHESE
glycoprotein maturation [Hay et al., 1993]. Amantadineprevents fusion by altering the ability of the virus tochange the ion balance [Pinto et al., 1992] within the en-dosome and the trans-Golgi network. However, amanta-dine rapidly gives rise to drug-resistant strains bymutations in the HA and the M2 protein that circumvent
or block the activity of the drug. Amantidine andrimantadine can cause adverse CNS complaints and othercomplications [Srange et al., 1991; Monto et al., 1995],although rimantadine can be taken at lower doses, in partrelated to lower plasma levels, and much less dependenceon renal secretion for elimination than amantidine.Rimantadine has been shown to lead to drug resistancein humans 2 days posttreatment [Hayden, 1993].
Influenza virus vaccines [Tyrrell, 1980; Murphy andWebster, 1990] prepared from killed (formalin inactivated)virus are used currently worldwide as the only prophy-lactic treatment available against the disease. Killed vac-cines contain whole virus or fractionated subunits. Thesevaccines attempt to incorporate antigens from influenza strains that are predicted to circulate in the communityduring the next season. They also confer immunity to vi-ral strains that are closely related to the strains incorpo-rated into the vaccine. However, the susceptibility of thevirus to neutralisation by antibodies raised by immuni-sation is reduced by antigenic drift. For example, com-mercial vaccines containing inactivated A/Beijing/353/89H3N2 strain circulating among humans from 1990 to 1993provided significantly less protection [Chakraverty et al.,1993] against the antigenic drift [Both et al., 1983] vari-ant A/Georgia/03/93.
A more radical vaccination treatment involving thedirect injection into muscle of plasmid DNA encoding HA,nuclear protein, and M1 influenza viral proteins has suc-cessfully been attempted in animal models [Donnelly etal., 1995]. This is thought to involve the incorporation of the plasmid into muscle cells, and the production of theviral proteins cell-mediated immune (CMI) response elic-ited offers cross-protection due to reactivity with the moreconserved epitopes of the HA and nuclear proteins, thusovercoming antigenic drift in the virus. The success of themethod arises from a CMI response to segments of con-served amino acids in these proteins. Problems arising frompossible autoimmune response to DNA, and in ensuring
the exogenous DNA is not integrated into the cell genomeor sensitive cell lines elsewhere in the host, have precludedtesting in humans at present. Furthermore, mutations inthe nuclear proteins of influenza (which have been rela-tively stable to date) arising from CMI selection pressurecould undermine the efficacy of this technique.
VIRAL NEURAMINIDASE AS AN ANTI-VIRAL TARGET
Enzyme activity on the surface of influenza viruswas first detected by Hirst [1942], who observed that red
blood cells once agglutinated by influenza virus couldnot again be agglutinated by either the eluted virus orfresh virus preparations. This activity is now attributedto NA, which is one of the two integral membrane glyco-proteins of influenza virus [for review, see Colman, 1994;Varghese, 1997]
Neuraminidase is an exoglycosidase which destroysthe hemagglutinin receptor by cleaving the α-ketosidiclinkage of terminal sialic acid (N-actylneuraminic acid(Neu5Ac)) to an adjacent sugar [Klenk et al., 1955;Gottschalk, 1957]. Viral HA binds specifically to Neu5Ac-containing receptors on the surface of susceptible cells[Rogers and Paulson, 1983]. NA, which also removes ter-minal sialic acid from a range of glycoconjugates, playsan important, but not completely understood, role in theviral replication cycle. Without NA activity, viruses[Burnett, 1947] were thought to be immobilised by mu-cosal secretions in the upper respiratory tract. By remov-ing terminal sialic acid from the sialic acid-rich mucouslayer protecting target cells [Gottschalk, 1957, 1958], NAcould facilitate penetration of the virus to the cell sur-face. It has been shown that neuraminidase-deficient vi-rus [Liu and Air, 1993] can still replicate in vivo, albeit ata much reduced rate [Liu et al., 1995]. This shows thatNA does not play an essential role in viral entry, replica-tion, assembly, or budding in mice. It has an importantrole in facilitating the spread of the infection by prevent-ing aggregation at the cell surface and possibleimmobilisation in the mucin by HA. During virus repli-cation, the freshly synthesised viral glycoproteins haveto be desialylated to prevent self-aggregation at the in-
fected host cell surface by HA binding to terminal sialicacid on these glycoproteins. Finally, on elution of prog-eny virions from infected cells, NA activity is required tofacilitate viral escape from the cell surface.
Inactivation or inhibition of NA during budding hasbeen observed to result in aggregation of virons on thecell surface [Palese et al., 1974; Palese and Compans,1976; Griffin and Compans, 1979]. Inhibition of thisglycohydrolase thus provides a means of controlling thisdisease, while the number of infected cells is low, by slow-ing the rate of viral attachment and subsequent releaseof progeny virons. This would allow the host immunesystem to eliminate the virus.
Molecular Structure of Neuraminidase
There are between 50 to 100 NA spikes per virion[Bucher and Palese, 1975], which are approximately 10%of the visible spikes projecting out of the surface of thevirion [White, 1974]. These spikes can be removed fromthe virus by treatment with detergent [Laver, 1963]. Elec-tron microscopic images of the NA spikes [Laver andValentine, 1969] reveal a mushroom-shaped moleculemade up of a box-like head of about 80 × 80 × 40 A, with

7/27/2019 Neuraminidase inhibitiors
http://slidepdf.com/reader/full/neuraminidase-inhibitiors 4/21
ANTI-VIRAL DRUGS FOR INFLUENZA 179
a narrow centrally attached stalk (15 A wide and 100 Along) which terminates in a hydrophobic knob anchoredin the viral envelope. The detergent-released spikes canbe digested by pronase to release the NA “heads,” whichretain full antigenic and enzyme activity [Drzenick et al.,1968; Laver, 1978]. The molecule was found to be a tet-
ramer of molecular weight 240,000, reducing to 200,000when treated with pronase [Blok et al., 1982].
The X-ray 3-dimensional molecular structure of NAheads was determined [Varghese et al., 1983] for two N2subtypes, A/Tokyo/3/67 and A/RI/5+/57. The structureof A/Tokyo/3/67 N2 has since been refined [Varghese andColman, 1991] to higher resolution (2.2 A), as have thestructures of two avian N9 subtypes [Baker et al., 1987;Tulip et al., 1992a] and influenza type B [Burmeister etal., 1991]. They were found to have an identical proteinfold with 60 residues (including 16 conserved cysteineresidues) being invariant. Bacterial sialidases from Sal- monella [Crennell et al., 1993] and cholera [Crennell etal., 1994] have homologous structures to influenza NA,but only a few of the residues in the active site are struc-turally invariant.
The protein fold consists of a symmetric arrange-ment of six four-stranded antiparallel β-sheets arrangedas the blades of a propeller, the propeller axis being ap-proximately parallel to but tilted away from the circular4-fold axis of the tetramer. This tilt angle varies betweenthe known subtypes.
There is a calcium-binding site connected to theactive site via conserved residues. The functional role of calcium in the structure is unknown, although calcium
has been shown to be necessary for NA activity [Bakerand Gandhi, 1976] and it is essential for the thermostablityof the molecule [Burmeister et al., 1994].
Carbohydrate Structure
Carbohydrates at four N-linked glycosylation siteswere observed in N2 NA at residues 86, 146, 200, and234 in the X-ray structure. Two N-actylglucosamines wereresolved at Asp86 and Asp234, both at the bottom sur-face of the monomer. The carbohydrate at Asp200 con-sists of at least eight sugar residues with linkagesconsistent with known mannose-rich simple N-linked car-bohydrate [Wagh and Bahl, 1981]. This oligosaccharide
emerges from the side of the monomer and covers a neighbouring subunit (see Fig. 1).The oligosaccharide site at Asn146 is the most con-
served of all neuraminidase glycosylation sites, exceptthat of the neurovirulent virus A/WSN/33 (H1N1) [Francisand Moore, 1940; Hitte and Nayak, 1982]. The absenceof this glycosylation site in A/WSN/33 has been shownto confer neurovirulence in mice [Li et al., 1993]. It hasrecently been shown [Goto and Kawaoka, 1998] that theNA of A/WSN/33 (a descendent of the virus responsible
for the 1918 pandemic, selected by serial passaging bymouse brain) binds and sequesters plasminogen. Thisleads to higher local concentrations of this protease pre-cursor and, thus, increasing cleavage of HA. This is a possible explanation for the pantropism of this strain[Kunkel et al., 1987]. The structural basis for this unusual
function of NA appears to be the presence of a carboxyl-terminus lysine and the absence of the carbohydrate at Asn146. Goto and Kawkaoka [1998] suggest that this car-bohydrate obstructs the binding of plasminogen to theterminal lysine, suppressing HA cleavage in cells otherthan its usual targets. This oligosaccaride is a complexsugar containing N-acetylgalactosamine [Ward et al.,1983] that is not found in any other of the knownoligosaccarides of influenza virus glycoproteins, and isthe only glycopeptide antigenically related to chick em-bryo “host antigen” [Ward et al., 1982]. The oligosaccha-ride appears as a spike emanating from the top of themonomer, forming a crystal contact with a neighbouringtetramer in crystals of A/Tokyo/3/67 NA. The four carbo-hydrate spikes of a tetramer form an open “barrel” struc-ture of eight carbohydrate chains with the neighbouringtetramer, with no apparent inter-carbohydrate contacts[Varghese and Colman 1991]. This oligosaccharide mayplay an important but as yet unidentified role in NA struc-ture or activity.
A Second Sialic Acid Binding Site
Hemagglutination activity has been reported for theN9 subtype of NA of influenza type A virus [Laver et al.,1984] at 4°C, which was not related to aberrant
neuraminidase activity, but was associated with a secondNeu5Ac binding site on the surface of NA away from theactive site [Webster et al., 1987]. The residues on thesurface of NA responsible for the haemabsorbing (HB)activity have been identified by monoclonal variantswhich lost the capacity to bind red blood cells [Websteret al., 1987], and the activity was successfully transferredto the N2 subtype of NA [Nuss and Air, 1991] by site-directed mutagenesis. Furthermore, it was shown thatN9 NA activity did not remove the putative sialic acid-related moiety that bound red blood cells to this HB site[Air and Laver, 1995], as the agglutination was restoredon cooling to 4°C. An HB site has also been discovered
on the NA of A/FPV/Rostock/34 H7N1 [Hausmann et al.,1995], which appears to have the same location on theNA surface as in N9 NA.
Recently, Varghese and co-workers [1997] locatedthe HB site on N9 by X-ray diffraction (Fig. 2). They haveshown that six residues on three separate loops of N9NA interact directly with the sialic acid in the secondsialic acid binding site. These are the three serine resi-dues (367, 370, and 372) in the loop containing residues367-SIASRS-372, Asn400, and Trp403 in the loop con-

7/27/2019 Neuraminidase inhibitiors
http://slidepdf.com/reader/full/neuraminidase-inhibitiors 5/21
180 VARGHESE
Fig. 1. A CPK atomic model of the upper surface of an influenza virusneuraminidase N2 tetrameric head [Varghese and Colman, 1991]. The
long spike-like structures emerging from the face of the tetramer towardsthe right are carbohydrate moieties attached to Asn146. The dark spheresrepresent those residues that have been totally conserved in all known
taining 400-NTSW-403, and Lys432 in a third loop. Thesequence variation at residues 400 and 432 in NAs withknown HB activity indicates that these residues are notessential for HB activity, and the triple serine SxxSxS loopof 367 to 372 and Trp403 are a minimal signature for HBbinding in NA.
All known strains from N1 to N9 which carry thesignature are avian (or equine), with the exception of thetwo human N2 strains, RI5+/57 and Leningrad/134/57[Varghese et al., 1997]. All avian sequences appear to carrythis signature, and in general human and swine NAs donot, indicating that the HB site has a functional role in
influenza viruses, and they cluster near the corners of the molecule wherethe active site of the enzyme is situated. It is this site to which the neuramini-
dase inhibitors are targeted. Residues that have mutated since 1933 arerepresented by lighter shaded spheres, and they surround the conservedactive site.
7/27/2019 Neuraminidase inhibitiors
http://slidepdf.com/reader/full/neuraminidase-inhibitiors 6/21
ANTI-VIRAL DRUGS FOR INFLUENZA 181
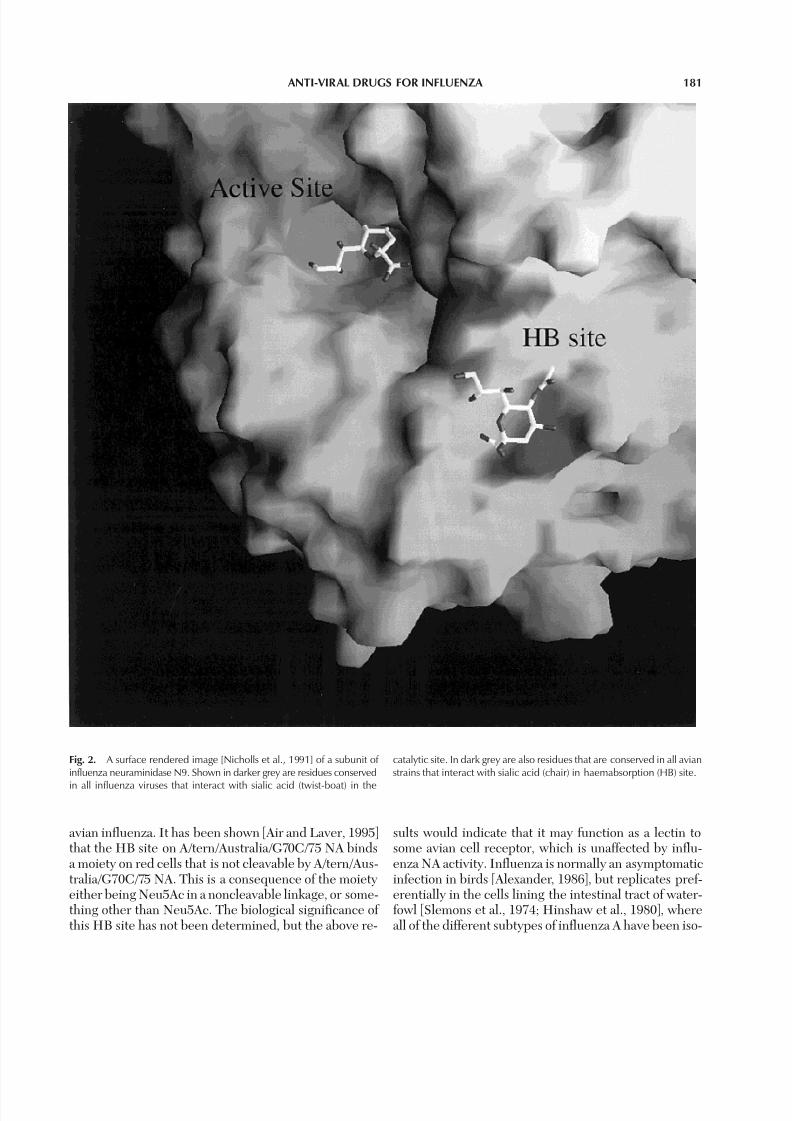
Fig. 2. A surface rendered image [Nicholls et al., 1991] of a subunit of influenza neuraminidase N9. Shown in darker grey are residues conserved
in all influenza viruses that interact with sialic acid (twist-boat) in the
catalytic site. In dark grey are also residues that are conserved in all avianstrains that interact with sialic acid (chair) in haemabsorption (HB) site.
avian influenza. It has been shown [Air and Laver, 1995]that the HB site on A/tern/Australia/G70C/75 NA bindsa moiety on red cells that is not cleavable by A/tern/Aus-tralia/G70C/75 NA. This is a consequence of the moietyeither being Neu5Ac in a noncleavable linkage, or some-thing other than Neu5Ac. The biological significance of this HB site has not been determined, but the above re-
sults would indicate that it may function as a lectin tosome avian cell receptor, which is unaffected by influ-enza NA activity. Influenza is normally an asymptomaticinfection in birds [Alexander, 1986], but replicates pref-erentially in the cells lining the intestinal tract of water-fowl [Slemons et al., 1974; Hinshaw et al., 1980], whereall of the different subtypes of influenza A have been iso-

7/27/2019 Neuraminidase inhibitiors
http://slidepdf.com/reader/full/neuraminidase-inhibitiors 7/21
182 VARGHESE
lated. This has led to the proposition that waterfowl arethe primary vectors for the global spread of the disease[Webster et al., 1992] through faecal droppings. Thisavirulent adaptation of the virus to avian species enablesit to survive and persist in a vast global reservoir.
If this HB signature relates to some as-yet unidenti-
fied role in avian influenza, then it could be inferred thatthe first human 1957 pandemic N2 strains carrying the HBsignature (RI5+/57 and Leningrad/134/57) were most prob-ably derived from a genetic reassortment event with an avianstrain [Skehel, 1974]. This HB signature has since disap-peared under antigenic drift in post-1957 human and swinestains, indicating that it serves no biological function inpathogenesis of influenza in humans and pigs.
Antigenic Variation in Neuraminidase Structures
Comparison of all known sequences of approximately390 residues of the NA globular head [Varghese and Colman,1991], indicates that only 54 (excluding 16 conserved cys-teine residues) are invariant (Fig. 1). Apart from 21 resi-dues involved in preserving the structural integrity of themolecule [Varghese and Colman, 1991], the main cluster-ing of these invariant residues is within the enzyme activesite (Fig. 1), where 17 are in the active site and 16neighbouring the active site. This is a cavity on the uppersurface of the molecule into which sialic acid has been ob-served to bind [Colman et al., 1983; Varghese et al., 1992;Burmeister et al., 1991]. Excluding the active site pocket,strain variation occurs over the entire surface of the NAheads. The active site was found to be in a pocket of totallyconserved (over all animal subtypes) residues [Colman et
al., 1983]. In this way the enzyme active site pocket is sur-rounded by highly variable surface residues that preventimmune recognition by antibody molecules of the activesite [Colman, 1994]. X-ray diffraction studies of NA–anti-body complexes have shown that the footprint of an anti-body in the complex is larger than the exposed surface of the conserved region of the active site [Colman et al., 1987,1989; Malby et al., 1994]. These structural results indicatethat antibodies are unable to exert mutational pressure onthe conserved active site because they cannot bind therewithout engaging strain variable residues as part of the bind-ing surface [Colman, 1997]. However, since antibodies bindto strain variable elements of the structure, the virus can
overcome host immune pressure by point mutations of theseresidues that do not have a catalytic or structural role[Varghese, 1988; Tulip et al., 1992b], but are able to disruptthe antigen–antibody binding interface. The rapid emer-gence of these escape mutants explains the failure to pro-duce a universal vaccine for influenza.
Enzyme Active Site
The structures of N-acetyl neuraminic acid (sialicacid Neu5Ac) and the 2-deoxy-2,3-dehydro-N-acetyl
neuraminic acid (Neu5Ac2en) inhibitor complexedwith N2 NA [Varghese et al., 1992] revealed the na-ture of the interactions of the molecules in the activesite pocket (Fig. 3). Sialic acid binds in the active sitein the α-anomer, and in a distorted half-chair confor-mation through the same face as used in its interac-
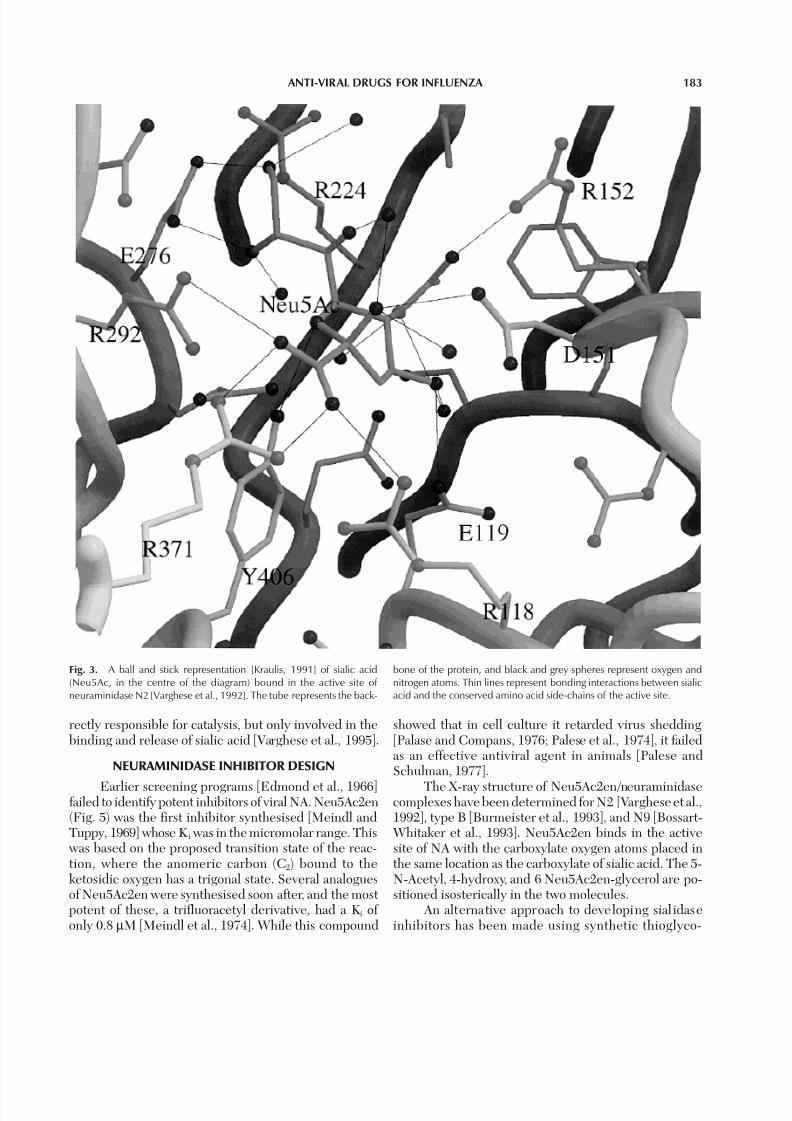
tion with HA [Weis et al., 1998]. The carboxylate groupof the sugar interacts with three guanidinium groupsof arginine residues 118, 292, and 371 and has an equa-torial conformation with respect to the sugar ring (thisgroup, axial toward the floor in the undistorted struc-ture, is probably held equatorial by interactions withthese arginine residues). The NH group of the 5-N- Acetyl side chain interacts with the floor of the activesite cavity, via a bound water molecule. The oxygen of the 5-N-Acetyl side chain is hydrogen bonded to theNε of Arg152, while the methyl group lies in a hydro-phobic pocket near Ile222 and Trp178. The last twohydroxyl groups of the 6-glycerol sidechain are hy-drogen bonded to carboxylate oxygens of Glu276 andthe 4-hydroxyl is directed to a carboxylate oxygen of Glu119, and the glycosidic oxygen O2 interacts witha carboxylate oxygen of Asp151. Similar binding of sialic acid in the active site was observed in type B[Burmeister et al., 1991].
Comparison of active sites of N2, N9, and type BNA [Varghese et al., 1995] show there are no significantdifferences between active site orientations, except forsome minor displacements of Arg224 and Glu276, wherethe major interactions with the 6-glycerol group of sialicacid occur. However, there are some differences in the
water structure in the active sites of the different sub-types. Comparison of the active sites of influenza neuraminidases and bacterial sialidases [Crennell et al.,1993, 1994] indicates that there is considerable conser-vation of the catalytic site at the carboxylate-binding end.The residues Asp151, Arg118, Glu277, Arg292, Val orIle349, Arg371, Try406, and Glu425 are conserved overall known viral and bacterial strains. The argininyl resi-dues 118, 292, and 371 position the 2-carboxylate groupand the Val(or Ile)349, Glu425 and Glu 277 are impor-tant in positioning the triarginyl cluster. Asp151 andTyr406 are presumably important in bond cleavage, butthe precise mechanism is still unclear. These eight resi-
dues (Fig. 4) are thus most likely to be conserved in allneuraminidases. Differences between viral, bacterial, andmammalian NA structures may correspond with the dif-ferent role these enzymes have in vivo. These differencesare likely to be in the interactions of the 6-glycerol, 5-N-acetyl, and 4-OH groups of silaic acid. In influenza, theturnover rate must be balanced against the requirementto maintain sufficient sialic acid at the cell surface to en-able attachment via the HA. This balance may requiresome configuration of residues in the active site not di-
7/27/2019 Neuraminidase inhibitiors
http://slidepdf.com/reader/full/neuraminidase-inhibitiors 8/21
ANTI-VIRAL DRUGS FOR INFLUENZA 183
rectly responsible for catalysis, but only involved in thebinding and release of sialic acid [Varghese et al., 1995].
NEURAMINIDASE INHIBITOR DESIGN
Earlier screening programs [Edmond et al., 1966]failed to identify potent inhibitors of viral NA. Neu5Ac2en(Fig. 5) was the first inhibitor synthesised [Meindl andTuppy, 1969] whose Ki was in the micromolar range. Thiswas based on the proposed transition state of the reac-tion, where the anomeric carbon (C2) bound to theketosidic oxygen has a trigonal state. Several analoguesof Neu5Ac2en were synthesised soon after, and the mostpotent of these, a trifluoracetyl derivative, had a Ki of only 0.8 µM [Meindl et al., 1974]. While this compound
showed that in cell culture it retarded virus shedding[Palase and Compans, 1976; Palese et al., 1974], it failedas an effective antiviral agent in animals [Palese andSchulman, 1977].
The X-ray structure of Neu5Ac2en/neuraminidasecomplexes have been determined for N2 [Varghese et al.,1992], type B [Burmeister et al., 1993], and N9 [Bossart-Whitaker et al., 1993]. Neu5Ac2en binds in the activesite of NA with the carboxylate oxygen atoms placed inthe same location as the carboxylate of sialic acid. The 5-N-Acetyl, 4-hydroxy, and 6 Neu5Ac2en-glycerol are po-sitioned isosterically in the two molecules.
An alternative approach to developing sial idaseinhibitors has been made using synthetic thioglyco-
Fig. 3. A ball and stick representation [Kraulis, 1991] of sialic acid(Neu5Ac, in the centre of the diagram) bound in the active site of neuraminidase N2 [Varghese et al., 1992]. The tube represents the back-
bone of the protein, and black and grey spheres represent oxygen andnitrogen atoms. Thin lines represent bonding interactions between sialicacid and the conserved amino acid side-chains of the active site.

7/27/2019 Neuraminidase inhibitiors
http://slidepdf.com/reader/full/neuraminidase-inhibitiors 9/21
184 VARGHESE
side-analogues of gangliosides such as Neu5Acα(2-S-6)Glcβ(1-1)Ceramide [Suzuki et al., 1990], which in-hibit different subtypes of human and animal influenza virus with a K i of up to 2.8 µM. These metabolicallystable ganglioside analogues contain a thioglycosidiclinkage to the terminal neuraminic acid that resistscleavage by the enzyme. Several flavonoid neuramini-dase inhibitors have been isolated from plant extracts[Nagai et al., 1990], one of which, 5,7,4′-trihydroxy-8-methoxyflavone was a more potent inhibitor than
Neu5Ac2en. Recently, in vivo anti-influenza virus activ-ity of a Kampo (Japanese herbal medicine) preparationhas shown promising results in inhibiting influenza vi-rus replication in mice [Nagai and Yamada, 1994], butthe mode of action of these compounds is unclear.
Enzyme Mechanism
The similar positioning of the carboxylate oxygensand ring of Neu5Ac2en and sialic acid suggests thatNeu5Ac2en is probably a transition state analogue. As
Fig. 4. A ball-and-stick representat ion [Kraul is, 1991] of Zanamivir/ GG167 (centre in light grey) in the active site of neuraminidase, show-ing “catalytic” residues (black) that are conserved over both bacte-
rial and viral neuraminidases, and “recognition” residues (grey) con-served only in influenza virus neuraminidase. The tube is a repre-sentation of the protein backbone.

7/27/2019 Neuraminidase inhibitiors
http://slidepdf.com/reader/full/neuraminidase-inhibitiors 10/21
ANTI-VIRAL DRUGS FOR INFLUENZA 185
Neu5Ac2en has a higher affinity for NA than sialic acid,
a mechanism of catalysis was proposed [Miller et al., 1978]that involves the distortion of the substrate by the forma-tion of a oxycarbonium ion intermediate, which has a simi-lar structure to Neu5Ac2en. However, the structuralresults from sialic acid/NA complexes suggest that thetighter binding of Neu5Ac2en arises more likely from therelaxation of the conformational strain arising from thetransition from chair to boat of the pyranose ring of sialicacid in the active site [Varghese et al., 1992].
Evidence for a sialyl cation transition state by iso-
topic effects [Chong et al., 1992] support the existence of a oxycarbonium ion intermediate. However, the struc-tural basis for NA activity is still unclear. It has been sug-gested [Varghese et al., 1992] that the tyrosyl oxygen of Tyr406 assisted by the sialic acid carboxylate itself couldstabilise the developing charge on the oxycarbonium ion
intermediate. The reaction is completed by the activa-tion of a water molecule by a deprotonated Asp151, andits attack on the carbonium, resulting in the formation of the α anomer of sialic acid [Friebolin et al., 1980]. How-ever, the pH activity profile of neuraminidase [Lentz etal., 1987; Chong et al., 1991; Burmeister et al., 1993] sug-gest a bell-shaped profile, indicating normal activity froma pH range of about 4.5 to 9, which would indicate thatthe role of Asp151 as the acid group in the catalysis isunclear. A nonspecific proton donor has been proposed[Taylor and von Itzstein, 1994], probably a water mol-ecule as the acid group, with a deprotonated Tyr406stabilising the oxycarbonium ion, and a proton transferredfrom water to the departing aglycon group. It has alsobeen postulated that a proton elimination at C3 leads tothe transformation of the oxycarbonium ion intoNeu5Ac2en, which is produced irreversibly at low levelsfrom sialic acid by the enzyme [Burmeister et al., 1993]. A SN1-type mechanism has been suggested [Taylor andvon Itzstein, 1994] that is facilitated by an activated wa-ter molecule that can be expelled upon inhibitor bind-ing. The catalytic mechanism could possibly proceedwithout an acid group, by the electrostatic potential of the enzyme lowering the barrier preventing the break-ing of the ketosidic bond, and the solvent protonating
the aglycon after release [Janakiraman et al., 1994].Clearly, details of the enzyme mechanism have yet to beelucidated definitively, as structural considerations canonly indicate that Tyr406 (and possibly Glu277) and thetriarginyl cluster are essential in the enzyme mechanism,and that Asp151 could be implicated in it.
Inhibitor Design Principles
All the nearest neighbour interactions with sialicacid and Neu5Ac2en and the protein are with totally con-served amino acids. Thus, an inhibitor designed to bindonly to the conserved active site residues of NA wouldinhibit NA activity across all strains of influenza. This
would enable the development of an anti-viral drug thatwould affect the spread of viral replication potentially inthree ways, i.e., transport through the protective mucosallayer, desialyation of freshly synthesised viral glycopro-teins, and elution of progeny virions from infected cells.
The development of potent inhibitors was based onthe structural information of the N2 NA conserved ac-tive site and its complex with sialic acid and Neu5Ac2en.There are no reports of de novo molecules designed tofit into the cavity, and the most useful approach was to
Fig. 5. Influenza neuraminidase inhibitors: 1) Sialic Acid, N- Acetylneuraminic ac id (Neu5Ac) ; 2) 2-deoxy-2,3-dehydro-N-acetylneuraminic acid (Neu5Ac2en); 3) 4-amino-Neu5Ac2en; 4)Zanamivir, 4-guanidino-Neu5Ac2en; 5) 5-N-acetyl-4-guanidino-6-methyl(propyl) carboxamide-4,5-dihydro-2H-pyran-2-carboxylic acid;6)5-N-acetyl-4-amino-6-diethyl carboxamide-4,5-dihydro-2H-pyran-2-car-boxylic acid; 7 ) GS4071, 4-N-acetyl-5-amino-3-(1-ethylpropoxy)-1-cyclohexene-1-carboxylic acid.

7/27/2019 Neuraminidase inhibitiors
http://slidepdf.com/reader/full/neuraminidase-inhibitiors 11/21
186 VARGHESE
consider molecules that were structurally related toNeu5Ac2en. This involved the design of molecules thatwould bind isosterically to Neu5Ac2en, but which weremodified to increase the number of favourable interac-tions with the protein.
The method of Goodford [1985] enabled the calcu-
lation of favourable binding sites for a variety of chemi-cal probes. The validity of this method was indicated byits method’s ability to identify the positions of the car-boxylate binding site of sialic acid as an energy minima for a carboxylate probe, and the successful prediction of known bound water sites in the active sites by a waterprobe. Utilising this methodology, predictions of ener-getically favourable substitutions to Neu5Ac2en wereexamined [von Itzstein et al., 1993]. A replacement of the hydroxyl at the 4-position of the pyranose ring of Neu5Ac2en by an amino group was identified by thisprocedure as an energetically favourable substitution. Aprotonated primary amine probe identified a favourablebinding site of -16 kcal mol–1 at this location, and in a pocket in the active site near two conserved glutamateresidues, Glu119 and Glu227. This suggested that thesubstitution of the 4-hydroxyl group by an amino groupwould increase the overall binding interactions by form-ing a salt link with Glu119. Furthermore, the substitu-tion of the 4-hydroxyl group with a much more bulkyguanidinyl group would lead to even tighter binding as a result of lateral interactions of the terminal nitrogens of the guanidinyl group and the carboxylate groups of Glu119 and Glu277.
This led to the design and syntheses of 3 and 4 (4-
amino-Neu5Ac2en and 4-guanidino-Neu5Ac2en in Fig.5) [von Itzstein et al., 1993] (4 is GG167, Zanamivir,Relenza™), which bound to A/Tokyo/3/67 with a K i of 50nM and 0.2 nM, respectively. These compounds werelater shown by X-ray studies of 3 and 4 complexed with A/Tokyo/3/67 NA to bind close to that predicted by thedesign studies. However, details of the interactions of theguanidinyl group of 4 with the glutamic acid groups(Glu119 and Glu277) in the floor of the active site wereslightly different. This was confirmed on a higher reso-lution X-ray study (Fig. 4) of 4 complexed with Tern N9NA [Varghese et al., 1995]. One of the primary guanidinylnitrogens of 4 is hydrogen bonded to the main chain oxy-
gen at residue 178, a carbxylate oxygen of Glu227, and a water molecule. The other primary guanidinyl nitrogeninteracts with the main chain oxygen of residues 178 and151. The secondary guanidinyl nitrogen interacts withthe carboxylate of Glu119 and Asp 151. The interactionswith Glu119 lack the hydrogen bonding geometry (pos-tulated in the design study), as the carboxylate group of Glu119 stacks parallel to the guanidinyl group, with itsinteractions being electrostatic and van der Waals in char-acter. Furthermore, theoretical energy-minimised struc-
tures of the complex using AMBER [Pearlman et al., 1990]converged to the X-ray structure only if the proteinnonhydrogen atom were kept rigid in the X-ray struc-ture [Varghese et al., 1995]. Otherwise, this resulted inactive site residues showing large distortions in their con-formation. This is an example of the difficulty in correctly
modelling even modest changes in the interactions of aninhibitor/active site complex.
Zanamivir shows potent inhibition of NA activityin all known wild strains of influenza. Furthermore, it isvery specific to influenza, as it shows weak inhibition tobacterial, para influenza, and mammalian neuraminidases[von Itzstein et al., 1993; Woods et al., 1993]. This is pos-sibly due to the specific interactions of the 4-guanidinogroup within the subpocket of the active site of influenza NA that is not conserved in other neuraminidases. Inbacterial neuraminidases [Crennell et al., 1993, 1994],this pocket is much smaller, and would prevent the bind-ing of the 4-guanidino group in this region of the activesite. This is consistent with the proposition that the in-teractions of sialic acid with the active site of NA are func-tion-specific at the C4,C5, and C6 position of sialic acid,and that modification at these positions confers specific-ity to the target enzyme [Varghese et al., 1995].
Recently, a new class of potent NA inhibitors, ex-emplified by 5 and 6 (Fig. 5) has been reported in whichhydrophobic substituents have replaced the glycerolmoiety at the 6-position [Sollis et al., 1996; Smith et al.,1996; Taylor et al., 1998]. A small conformational changein the active site of NA occurs to enable these inhibitorsto be accommodated (Fig. 6). Glu276 changes its posi-
tion to form a salt link with Arg224, and thereby createsthe necessary hydrophobic pocket for the carboxamidesubstituents. It has been proposed that the unexpectedstrong binding of these inhibitors is a result of the burialof hydrophobic surface area and salt-bridge formation inan environment of low dielectric. Taylor and co-workers[1998] have shown that there is a greater degree of dis-tortion of the active site residues in influenza B NA thaninfluenza A NA on binding with these carboxamide de-rivatives. This correlates with the decreased affinity forinfluenza B NA when compared with influenza A NA onbinding to ligand. They have also shown by moleculardynamics calculations that the tendency for salt-bridge
formation is greater in influenza A NA than influenza BNA, and this is a useful descriptor for the prediction of inhibitor potency.
A carbocyclic analogue of sialic acid (GS4071,Ro640796) [Kim et al., 1997] (7 in Fig. 5), which has a hydrophobic group attached to the 6 position via an etherlink, has also been shown to inhibit NA and virus repli-cation in vitro. This inhibitor binds in a similar mode asthe 6-carboximide derivatives of Neu5Ac2en [Vargheseet al., 1998], by forming a hydrophobic pocket created

7/27/2019 Neuraminidase inhibitiors
http://slidepdf.com/reader/full/neuraminidase-inhibitiors 12/21
ANTI-VIRAL DRUGS FOR INFLUENZA 187
by the salt link of Glu276 and Arg224. It could be in-ferred, as with the carboxamide derivatives, that this com-pound would have a differential binding to influenza Aand B neuraminidases. GS4071 has a similar low bio-availablity compared to GG167 [Li et al., 1998]; however,
an ethyl ester linked to one of the carboxylate oxygens of GS4071 improves the oral bioavailablity following rapidconversion to the active form during gastrointestinal ab-sorption. This prodrug, GS4104, when administeredorally results in high and sustained systemic absorptionin animal tests [Li et al., 1998] with a half-life of 5 h inmost tissues. Whole body autoradiography in rats hasshown a twofold higher concentration in the lungs com-pared to plasma 6 h postdose, and a 30-fold higher con-centration at 24 h postdose [Eisenberg et al., 1997].Furthermore, distribution in brain tissue was minimal.This would indicate that the GS4104 inhibitor is orallyactive and the inhibitor is currently undergoing human
clinical trials for use as an anti-influenza therapeutic.Other approaches [Luo et al., 1995] to structure-based design of inhibitors have to date produced onlymM inhibition of neuraminidase activity.
ANTIVIRAL ACTIVITY OF NEURAMINIDASEINHIBITORS
In vitro inhibition of viral replication in tissue cul-ture was demonstrated [Palese and Compans, 1976] forthe tri-fluro derivative of Neu5Ac2en, but its antiviral
activity in vivo was not demonstrated [Palese andSchulman, 1977]. As a consequence of this, efforts weredirected towards HA, which was then considered a bet-ter target for anti-influenza drugs. The interest inneuraminidase inhibitors as anti-influenza drugs has only
been revived with the success of 4-guanidino-Neu5Ac2enand its analogues in attenuating viral titres in mice whenadministered directly into the lungs [von Itzstein et al.,1993; Ryan et al., 1994].
Inhibition In Vitro
von Izstein and co-workers [1993] have shown thatthe 4-amino- and 4-guanidino-Neu5Ac2en inhibit influ-enza strains A/Singapore/1/57 and B/Victoria/102/95 inMDCK cells with IC50 values (the concentration requiredto inhibit plaque formation in MDCK cells by 50%) of 1.5 µM and 0.065µM (4-amino) and 0.014 µM and 0.005µM (4-guanidino), respectively. These IC50 values, in
particular for the 4-guanidino compound, are well belowthose found for amantadine, ribavirin, and Neu5Ac2en.Furthermore, in comparison to Neu5Ac2en the 4-guanidino-Neu5Ac2en inhibitor was 100-fold less activeagainst human lysosomal sialidase and over 1,000-foldmore active against a wide range of clinical isolates of influenza A and B, including amantidine- and riman-tadine-resistant variants [Woods et al., 1993].
The inhibition of replication of virus in MDCK cellshas been confirmed [Thomas et al., 1994] and Hayden
Fig. 6. A surface rendered image [Nicholls et al., 1991] of neuramini-dase N9 with a ball and stick representation of (a) Zanamivir [Varghese et al., 1993] and (b) a carboxamide derivative of Neu5Ac2en in the activesite [Taylor et al., 1998]. The change in conformation of Glu276 (grey
patch at the upper centre of diagram) from H-bond interactions with theglycerol group of GG167 to formation of a salt-link with Arg224 is shownby the creation of a hydrophic binding pocket in (b). Black spheres repre-sent bound water molecules.

7/27/2019 Neuraminidase inhibitiors
http://slidepdf.com/reader/full/neuraminidase-inhibitiors 13/21
188 VARGHESE
and co-workers [1994] extended the inhibition studies tohuman respiratory epithelium cells in vitro, indicatinghigh antiviral activity for strains of A(H1N1) and A(N3N2)isolates. They also found that delayed administration of the drug after viral replication was well established wasassociated with inhibition of virus replication. However,
the viral titre was higher (1.3 log10 compared to 4.0 log10
at 10 µg/ml concentration of the drug) for the delayedadministration compared with the viral titre when thedrug was present throughout the period of viral expo-sure. The clinical significance of this in the treatment of established infections has been explored in detail in clini-cal trails.
Administration and Inhibition In Vivo
The earlier work of Palese and Schulman [1977]indicated that the failure in inhibiting viral replicationafter intranasal and subcutaneous treatment by Neu5a-C2en in mice would produce similar results forNeu5Ac2en analogues. This failure of Neu5Ac2en as ananti-viral treatment in animal models can now be ascribedto the rapid excretion of the compound [Nohle et al.,1982], thereby not delivering sufficient concentration of the inhibitor to the infected tissue. It has been shown[von Itzstein et al., 1993] that when Zanamivir is admin-istrated intranasally in mice it is considerably more ef-fective than when the drug is injected intra-peritoneally.This can be attributed to the localisation of sufficientlyhigh concentrations of inhibitor in the lining of the nasaland respiratory epithelia, where influenza virus replica-tion is believed to occur.
Animal trials with ferrets challenged with influenza virus have shown that Zanamivir is effective in studiesinvolving prophylactic administration of the drug [vonItzstein et al., 1993]. When the drug is administrated in-tranasally, 50 µg/kg, twice daily, 1 day before infectionwith the virus and the succeeding 6 days, it substantiallyreduced virus titre in nasal washings and abolished feverthat usually appears 3 days after infection.
Results from a double-blind, randomised, placebo-controlled trial using this compound have been successfulboth for early treatment and prophylaxis of experimentalinoculation of 166 adult human volunteers [Hayden et al.,1996] with 105TCID50 of influenza A/Texas/91 (H1N1). For
all dose groups combined, Zanamivir was 82% effective inpreventing laboratory evidence of infection, and 95% ef-fective in preventing febrile illness. These results indicatethat NA is important for viral replication in humans.
In separate randomised, double-blind studies in 38centres in North America and 32 centres in Europe dur-ing the 1994–1995 influenza season, a total of 417 adultswith influenza-like illness of less than 2 days durationwere randomly assigned to the following groups: 6.4 mgof Zanamivir by intranasal spray plus 10 mg by inhala-
tion; 10 mg of Zanamivir plus placebo spray; or placeboby both routes [Hayden et al., 1997]. Treatments wereself-administrated for 5 days. This study showed that inadult infections of both influenza A or B, Zanamivir di-rectly administered to the respiratory tract was safe andreduced symptoms if begun early. Administration of
Zanamivir by both groups reduced from 7 to 4 days themedian time to the alleviation of major symptoms whencompared with the placebo group, if treatment com-menced within 30 h after onset of symptoms. Similar re-sults were obtained in a double-blind randomised,placebo-controlled study completed in the southernhemisphere winter of 1997, on healthy and “high risk”patients, showing Zanamivir was well tolerated and alle-viated symptoms of influenza 1.5 to 2.5 days earlier thanplacebo, and “high risk” patients had fewer complica-tions and a reduction in the use of antibiotics with thesecomplications [Silagy et al., 1998]. In a double-blind pla-cebo controlled prophylaxis trail, Zanamivir (10 mg in-haled daily) was shown to be 67% (P < 0.001) effective inpreventing laboratory-confirmed clinical influenza illness[Monto et al., 1998], and when confirmed illness was re-stricted to those with fever, the efficacy rose to 84% (P =0.001). Efficacy in prevention of total influenza infectionwas 31% (P = 0.034), indicating subclinical infection,which allows immunity to develop in subsequent infec-tions. Reduced single daily dosage of 6.4mg inhaledZanamivir has also been shown to be protective againstexperimental infections in humans [Calfee et al., 1998].
Similar results have been obtained with the orallyactive prodrug GS4104. The parent drug, GS4071, has
low bioavailability like GG167 (2–4%) compared to thehigh bioavailability of GS4104 [Li et al., 1998]. GS4104undergoes rapid enzymatic conversion to GS4101 follow-ing gastrointestinal absorption. The oral administrationof GS4104 in mice produced dose-dependent protectiveeffects against the neurotropic influenza strain A/NWS/ 33 (H1N1), where 1 mg/kg/day afforded 100% protec-tion [Mendel et al., 1998]. Increasing the dose to 10 mg/ kg/day afforded similar protection against A/Victoria/3/ 75 (H3N2) and B/HongKong/5/72. Similar results wereobtained with the ferret model [Oxford et al., 1998]. Nosignificant drug-related toxicity was observed after theadministration of oral dosages of GS4104 of up to 800
mg/kg/day for 14 days in nonclinical toxicology studieswith rats [Mendel et al., 1998].In human clinical trials in the US, following oral
treatment (75 mg or 150 mg) of GS4104 for 5 days, afterfebrile respiratory naturally acquired illness of 36 h orless, in a placebo-controlled, double-blind study of healthy non-immunized adults ages 18–65 have shownthe efficacy of the drug [Treanor et al., 1998]. The resultsshowed a 30% reduction in both 75 mg (P = 0.0001) and150 mg (P = 0.001) groups in duration of symptoms, 40%

7/27/2019 Neuraminidase inhibitiors
http://slidepdf.com/reader/full/neuraminidase-inhibitiors 14/21
ANTI-VIRAL DRUGS FOR INFLUENZA 189
reduction (both 75 mg, P = 0.0001; and 150 mg, P =0.001) in severity of symptoms, and a 50% reduction (P= 0.017) of secondary complications. Similar results wereobtained for treatment of naturally acquired influenza during the 1997–1998 influenza season in Canada, Eu-rope, and China [Aoki et al., 1998], and no serious ad-
verse events were reported. A study of the long-termprophylaxis of natural influenza [Hayden et al., 1998]showed an overall protective efficacy of 74% against in-fluenza illness when 75 mg was taken once or twice dailyduring the period of local influenza activity. Long-termadministration of once daily GS4104 was found to be safeand well tolerated.
DRUG RESISTANCE
Although the active site of influenza virus has beenconserved in all known field strains of the virus, the pos-sibility of drug resistance needs to be addressed. NA in-hibitors, if used extensively as anti-influenza drugs, willapply selection pressure on the active site residues forthe first time in its evolutionary history. Experience withinfluenza and other viruses, in particular HIV, have shown[Kimberlin et al., 1995] that drug-resistant mutants arisevery rapidly, resulting in the effectiveness of antiviraldrugs being short-lived. One attempted solution to theproblem is the use of several drugs during therapy[Madren et al., 1995], making it more difficult for the vi-rus to develop resistance.
In the case of influenza virus, to date there havebeen no reports of drug resistance to Zanamivir fromfield strains [Osterhaus et al., 1998], except in an
immunocompromised host [Gubereva et al., 1998].However, it has recently been reported [McKimm-Breschkin et al., 1994; Gubareva et al., 1995] that 4-guanidino-Neu5Ac2en-resistant mutants can arisefrom multiple serial passages of virus in MDCK cellsin the presence of the inhibitor. It has been demon-strated [McKimm-Breschkin et al., 1996] that almostall of the mutations arise in the HA receptor bindingsite and not on the NA. These altered HA variants, whichhave weaker binding to HA receptors, appear to arise asa result of increased inhibition of the NA by the drug.This is consistent with the earlier proposition that therate of desialylation of receptor is critically related to the
rate of attachment to receptor for the virus infection andelution [Varghese et al., 1995]. The decreased activity of the NA by the drug selects HA mutants with decreasedbinding to receptors.
E119G Neuraminidase Variant
A Zanamivir-resistant NA mutation has been iso-lated [Blick et al., 1995; Gubareva et al., 1995] that re-sults in a single active-site residue mutation, withapparently unaltered activity, of glutamic acid 119 to gly-
cine in Tern N9. The crystal structure of this mutant andits complex (Fig. 7) with 4-guanidino-Neu5Ac2en hasbeen determined [Blick et al., 1995]. The structures sug-gest that the decrease in inhibitor binding arises fromthe loss of stabilising interaction of the 4-guanidino groupof Zanamivir [Varghese et al., 1995] and alterations in the
solvent structure of the active site. This alteration arisesfrom a water molecule that binds near the location of oneof the carboxylate oxygens of the glutamic acid in thewild-type molecule. The location of the 4-guanidino-Neu5Ac2en drug in the complex with the mutant enzymeis isosteric compared to the drug/wild-type complex[Varghese et al., 1995], the only differences being the in-teractions with residue 119.
This variant is, however, inherently thermally un-stable, as measured both by enzyme activity and NC10monoclonal antibody reactivity [Sahasrabudhe et al.,1998]. Substrate, inhibitor, or monoclonal antibodiesstabilised the NA against all methods of denaturation.These results suggests that the instability of the variantis primarily due to the level of polypeptide chain foldingrather than the level of association of monomers and tet-ramers. Furthermore, the presence of high level of sub-strate, either cell- or virus-associated, may be sufficientto stabilise the NA during virus replication.
R276K Neuraminidase Variant
Resistance to the carboxamide inhibitor 5 (Fig. 5)has been investigated in type A influenza with the N9subtype NA, and an active site mutant, R292K, has beenisolated [McKimm-Breschkin et al., 1998]. The specific
activity of the R292K variant is only 20% of that of wild-type, and virus bearing this mutant NA shows decreasedsensitivity to all NA inhibitors [McKimm-Breschkin etal., 1998]. The R292K mutant has also been reported toarise in an avian N2 background after passaging inZanamivir [Gubareva et al., 1997].
Varghese and co-workers [1998] have shown by X-ray diffraction that the mutation of Arg292 to Lys resultsin a very local change of structure when compared to thewild-type enzyme. In the wild-type NA Arg292, togetherwith Arg118 and Arg371, engages the 2-carboxylate groupof sialic acid in the active site and is partly responsiblefor distorting the pyranose ring from a chair to a boat
conformation [Varghese et al., 1992]. The distal primaryguanidinyl amide of Arg292 interacts with Glu277 andthe tyrosyl oxygen of Tyr406 (Fig. 2), and the other twoguanidinyl nitrogens interact with Asn294. The guani-dinyl group of Arg292 is parallel with the peptide planeof mainchain atoms of residues 348–349. Two water mol-ecules in the active site cavity have H-bond interactionswith the primary amides of the Arg292 prior to the entryof substrate into the active site.
The R292K variant of influenza NA affects the bind-

7/27/2019 Neuraminidase inhibitiors
http://slidepdf.com/reader/full/neuraminidase-inhibitiors 15/21
190 VARGHESE
ing of substrate by modification of the interaction withthe substrate carboxylate. This may be one of the struc-tural correlates of the reduced enzyme activity of thevariant. Inhibitors which have replacements for the glyc-erol at the 6-position are further affected in the R292Kvariant because of structural changes in the binding sitewhich apparently raise the energy barrier for the confor-mational change in the enzyme required to accommo-date such inhibitors.
It has been shown [Varghese et al., 1998] that there
is a correlation between the effect of the Arg292 to Lyssubstitution on the structure of complexes with inhibi-tors and on the binding properties of the inhibitors. Thereis also a correlation between the degree of resistance of the variant over wild-type and the degree of structuraldissimilarity of the inhibitor to the transition state ana-logue 2. The largest structural consequences occur withthe inhibitors whose binding is most severely compro-mised, i.e., 7 and then 6 and 5. While 6 and 5 are able toform the hydrophobic pocket to accommodate the sub-
Fig. 7. A ball-and-stick representation of the complex of Zanamivir inthe active site of wild-type N9 neuraminidase and the Glu199 to Glymutation superimposed on each other. Spheres represent oxygen, nitro-gen, and carbon atoms. A black sphere (Wat) represents a solvent mol-ecule that occupies the position of the carboxylate group of Glu119 when
it mutates to a Gly. Thin lines represent H-bond interactions of theguanidinium group of Zanamivir and the enzyme. The loss of the interac-tions with Glu119 in the mutant is thought to be responsible for the re-duction of binding of Zanamivir to the mutant. The tubes represent thebackbone of the protein.
7/27/2019 Neuraminidase inhibitiors
http://slidepdf.com/reader/full/neuraminidase-inhibitiors 16/21
ANTI-VIRAL DRUGS FOR INFLUENZA 191
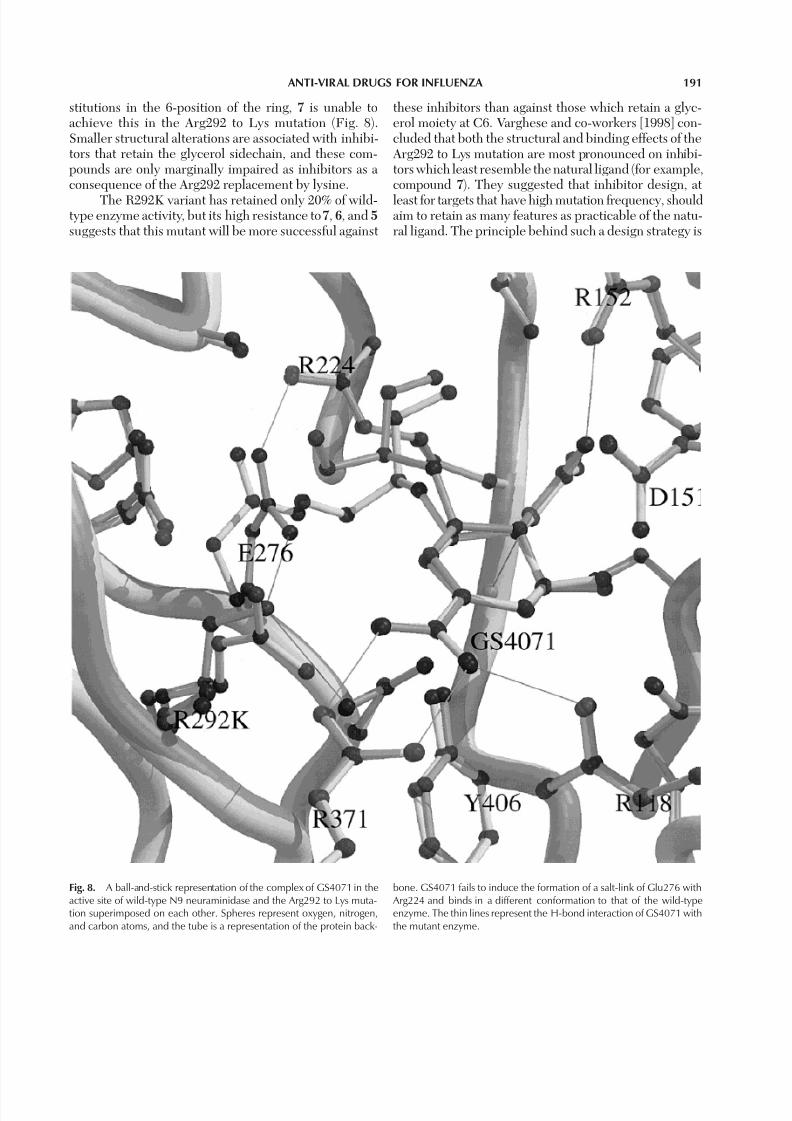
stitutions in the 6-position of the ring, 7 is unable toachieve this in the Arg292 to Lys mutation (Fig. 8).Smaller structural alterations are associated with inhibi-tors that retain the glycerol sidechain, and these com-pounds are only marginally impaired as inhibitors as a consequence of the Arg292 replacement by lysine.
The R292K variant has retained only 20% of wild-type enzyme activity, but its high resistance to 7, 6, and 5suggests that this mutant will be more successful against
these inhibitors than against those which retain a glyc-erol moiety at C6. Varghese and co-workers [1998] con-cluded that both the structural and binding effects of the Arg292 to Lys mutation are most pronounced on inhibi-tors which least resemble the natural ligand (for example,compound 7). They suggested that inhibitor design, at
least for targets that have high mutation frequency, shouldaim to retain as many features as practicable of the natu-ral ligand. The principle behind such a design strategy is
Fig. 8. A ball-and-stick representation of the complex of GS4071 in theactive site of wild-type N9 neuraminidase and the Arg292 to Lys muta-tion superimposed on each other. Spheres represent oxygen, nitrogen,and carbon atoms, and the tube is a representation of the protein back-
bone. GS4071 fails to induce the formation of a salt-link of Glu276 with Arg224 and binds in a different conformation to that of the wild-typeenzyme. The thin lines represent the H-bond interaction of GS4071 withthe mutant enzyme.

7/27/2019 Neuraminidase inhibitiors
http://slidepdf.com/reader/full/neuraminidase-inhibitiors 17/21
192 VARGHESE
simple — variants in the natural ligand binding site mustretain some binding to that ligand to preserve the pro-tein function.
CONCLUSION
It has now been 15 years since the structure of in-
fluenza virus neuraminidase was determined [Vargheseet al., 1983; Colman et al., 1983]. The development of the 4-subtituted Neu5Ac2en analogue inhibitors datesfrom 1987, when the structure of sialic acid andNeu5Ac2en complexed with neuraminidase were deter-mined to sufficient accuracy to permit modelling of po-tential inhibitors [Varghese et al., 1992]. The developmentwas a multidisciplinary collaboration of biochemists, crys-tallographers, molecular modellers, and synthetic chem-ists, and culminated in the synthesis [von Itzstein et al.,1994] and biological testing of the compounds [vonItzstein et al., 1993]. Similar results have been obtainedby the orally active Gilead carbocyclic analogue GS4104.Preliminary data on the efficacy of potent neuraminidaseinhibitors on humans indicate its effectiveness in bothprophylaxis and treatment of the disease [Hayden, 1998].Zanamivir has completed stage III clinical trials in Aus-tralia, Europe, and North America, and awaits clearanceas a pharmaceutical by regulatory authorities for use as a pharmaceutical both for prophylaxis and treatment of allinfluenza A and B infections. GS4104 is still undergoingclinical trials and offers an alternative oral therapy forinfluenza virus infections. While drug resistance of thevirus has been observed in vitro, the question of whetherthis translates into resistance in field strains (as has oc-
curred in the amantidine class of drugs) infecting humanpopulations will be a cause for concern in the long-termviability of this class of antivirals. It has recently beenreported that the Arg292 to Lys mutation in neuramini-dase has emerged in two immunocompetent patients inclinical trials of GS4104 [Hayden, 1998]. The significanceof these isolates in terms of infectivity and spread of re-sistant variants is yet to be determined. The structuralstudies on drug-resistant variants of neuraminidase havesuggested a minimalist approach [Varghese et al., 1998]to drug design against shifting targets like influenza vi-rus proteins. The further removed the drug is from itsnatural ligand the greater the potential of the organism
developing resistance.This is one of the first structure-based design com-pounds to be a useful anti-viral drug and portends wellfor this methodology to be used as an additional weaponin controlling the many pathogens that have plaguedhumanity for so long.
ACKNOWLEDGMENTS
The author would like to acknowledge PeterColman for crystallographic and drug design strategies,
Jenny McKimm-Breschkin for drug resistance, BrianSmith for enzyme mechanisms.
REFERENCES
Air GM, Laver WG. 1995. Red cells bound to influenza virus N9neuraminidase are not released by the N9 neuraminidase activity.
Virology 211:278–284. Alexander DJ. 1986. Avian diseases—historical aspects. In: Proceed-
ings of the 2nd International Symposium on Avian Influenza. Rich-mond, VA: U.S. Animal Heath Assosiation. p 4–13.
Aoki F, Osterhaus A, Rimmelzwaan, Kinnersley N, Ward P. 1998. OralGS4104 successfully reduces duration and severity of naturally ac-quired influenza. San Diego: ICAAC 24–27 Sept. (Abstr. LB-5).
Baker NJ, Gandhi SS. 1976. Effect of Ca++ on the stability of influ-enza virus neuraminidase. Arch Virol 52:7–18.
Baker AT, Varghese JN, Laver WG, Air GM, Colman PM. 1987. Thethree-dimensional structure of neuraminidase of subtype N9 froman avian influenza virus. Proteins 1:111–117.
Beveridge WI. 1977. Influenza: the last great plague. London:Heinemann.
Blick TJ, Tiong T, Sahasrabudhe A, Varghese JN, Colman PM, HartGJ, Bethell RC, McKimm-Breschkin JL. 1995. Generation and char-acterization of an influenza virus variant with decreased sensitivityto the neuraminidase specific inhibitor 4-guanidino-Neu5Ac2en.Virology 214:475–484.
Blok J, Air GM, Laver WG, Ward CW, Lilley GG, Woods EF, RoxburghCM, Inglis AS. 1982. Studies on the size, chemical composition andpartial sequence of the neuraminidase (NA) from type A influenza virus show that the N-terminal region of the NA is not processedand serves to anchor the NA in the viral membrane. Virology119:109–121.
Bossart-Whitaker P, Carson M, Babu YS, Smith CD, Laver WG, AirGM. 1993. Three dimensional structure of influenza A N9 neuramini-dase and its complex with the inhibitor 2-deoxy 2,3-dehydro-N-
acetyl neuraminic acid J Mol Biol 232:1069–1083.Both GW, Sleigh MJ, Cox NJ, Kendal AP. 1983. Antigenic drift in influ-
enza virus H3 haemagglutinin from 1968 to 1980: multiple evolu-tionary pathways and sequential amino acid changes at key antigenicsites. J Virol 48:52–60.
Bucher DJ, Palese P. 1975. The biologically active proteins of influ-enza virus neuraminidase. In: Kilbourne ED, editor. Influenza vi-rus and influenza. New York: Academic Press. p 83–123.
Burmeister WP, Ruigrok RWH, Cusack S. 1991. The 2.2Å resolutioncrystal structure of influenza B neuraminidase and its complex tosialic acid. EMBO J 11:49–56.
Burmeister WP, Henrissat B, Bosso C, Cusack S, Ruigrok RWH. 1993.Influenza B virus neuraminidase can synthesize its own inhibitor.Structure 1:19–26.
Burmeister WP, Cusack S, Ruigrok RWH. 1994. Calcium is neededfor the thermostability of influeza B virus neuraminidase. J Gen Virol75:381–388.87.
Burnett FM. 1947. Mucins and mucoids in relation to influenza virusaction. IV. Inhibition by purified mucoid of infection andhaemagglutinin with the virus strain WSE. Aust J Exp Biol Med Sci26:381–387.
Calfee DP, Peng AW, Hussey EK, Lobo M, Hayden FG. 1998. Protec-tive efficacy of reduced frequency dosing of intranasal zanamivir inexperimental human influenza. San Diego: ICAAC 24–27 Sept.(Abstr. H-68).

7/27/2019 Neuraminidase inhibitiors
http://slidepdf.com/reader/full/neuraminidase-inhibitiors 18/21
ANTI-VIRAL DRUGS FOR INFLUENZA 193
Chakraverty et al., 1993. Influenza activity — United States and world-wide, and composition of the 1993–94 influenza vaccine. JAMA269:1778–1779.
Chong AKJ, Pegg MS, von Itzstein M. 1991. Characterisation of anionisable group involved in the binding and catalysis by sialidasefrom influenza virus. Biochem Int 24:165–171.
Chong AKJ, Pegg MS, Taylor NR, von Itzstein M. 1992. Evidence fora sialyl cation transition state complex in the reaction of sialidasefrom influenza. Eur J Biochem 207:335–343.
Claas EC, Osterhaus AD, van Beek R, De Jong JC, Rimmelzwaan GF,Krauss S, Shortridge KF, Webster RG. 1998. Human influenza AH5N1 virus related to a highly pathogenic avian influenza virus.Lancet 351:472–477
Colman PM. 1989. Influenza virus neuraminidase: Enzyme and anti-gen. In: Krug RM, editor. The influenza viruses. New York: PlenumPress. p 175–218.
Colman PM. 1994. Influenza virus neuraminidase: structure, antibod-ies, and inhibitors. Protein Sci 3:1687–1696.
Colman PM. 1997. Virus versus antibody. Structure 5:591–593.
Colman PM, Laver WG. 1981. The structure of influenza virus
neuraminidase at 5A resolution. In: Structural aspects of recogni-tion and assembly in biological macromolecules. Rehovot, Israel:I.S.S. p 869–872.
Colman PM, Varghese JN, Laver WG. 1983. Structure of the cata-lytic and antigenic sites in influenza virus neuraminidase. Na-ture 303:41–44.
Colman PM, Laver WG, Varghese JN, Baker AT, Tullock PA, Air GM,Webster RG. 1987. Three-dimensional structure of a complex of antibody with influenza virus neuraminidase. Nature 326:358–363.
Colman PM, Tulip WR, Varghese JN, Tulloch PA, Baker AT, LaverWG, Air GM. 1989. 3-D structures of influenza virus neuramini-dase-antibody complexes. Proc R Soc Lond B323:511–518.
Compans RW, Dimmock NJ. 1969. An electron microscopic study of
single-cycle infection of chick embryo fibroblasts by influenza vi-rus. Virology 39:499.
Crennell SJ, Garman EF, Laver WG, Vimr ER, Taylor GL. 1993. Crys-tal structure of a bacterial sialidase (from Salmonella typhimuriumLT2) shows the same fold as an influenza virus neuraminidase. ProcNatl Acad Sci USA 90:9852–9856.
Crennell SJ, Garman E, Laver G, Vimr E, Taylor GL. 1994. Crystalstructure of Vibro Cholerae neuraminidase reveals dual lectin-likedomains in addition to the catalytic domain. Structure 2:535–544.
Daniels RS, Downie JC, Hay AJ, Knossow M, Skehel JJ, Wang ML,Wiley DC. 1985. Fusion mutants of the influenza virus haemag-glutinin glycoprotein. Cell 40:431–439.
Donnelly JJ, Friedman A, Martinez D, Montgomery DL, Shiver JW,Motzel SL, Ulmer JB, Liu MA. 1995. Preclinical efficacy of a proto-
type DNA vaccine: enhanced protection against antigenic drift ininfluenza virus. Nat Med 1:583–586.
Douglas RG Jr. 1990. Drug therapy, prophylaxis and treatment of in-fluenza. N Engl J Med 322:443–450.
Drzenick R, Frank H, Rott R. 1968. Electron microscopy of purifiedinfluenza virus neuraminidase. Virology 36:703–707.
Edmond JD, Johnston RG, Kidd D, Rylance HJ, Sommerville RG. 1966.The inhibition of neuraminidase and antiviral action. Br J PharmacolChemother 27:415–426.
Eisenberg EJ, Bidgood A, Cundy KC. 1997. Penetration of GS4071, a novel influenza neuraminidase inhibitor, into rat bronchoalveolar
lining fluid following oral administration of prodrug GS4104, Antimicrob Agents Chemother 41:1949–1952.
Ennis FA, Meager A. 1981. Immune interferon produced to high lev-els antigenic stimulation of human lymphocytes with influenza vi-rus. J Exp Med 154:1279–1289.
Fox JP, Cooney MK, Hall CE, Foy HM. 1982. Influenza virus in-
fections in Seattle families, 1975–1979. II. Pattern of infectionof invaded households and relation of age and prior antibody tooccurrence of infection by time and age. Am J Epidemiol 116:228–242.
Francis T Jr, Moore AE. 1940. A study of the neurotropic tendency instrains of the virus of epidemic influenza. J Exp Med 72:717–728.
Friebolin H, Brossmer R, Keilich G, Ziegler D. 1980. Supp M. 1H-NMR spektroskopischer nachweis der N-acetyl-a-D-neuraminsaureals primares spaltprodukt der neuraminidasen. Hoppe Seyler’s ZPhysiol Chem 361:697–702.
Goodford PJ. 1985. A computational procedure for determining ener-getically favourable binding sites on biologically important macro-molecules. J Med Chem 28:849–857.
Gottschalk A. 1957. Neuraminidase: the specific enzyme of influenza virus and vibrio cholerae. Biochem Biophys Acta 23:645–646.
Gottschalk A. 1958. Neuraminidase: its substrate and mode of action. Adv Enzymol 20:135–145.
Goto H, Kawaoka Y. 1998. A novel mechanism for the acquisition of virulence by a human influenza A virus. Proc Natl Acad Sci USA95:10224–10228.
Griffin JA, Compans RW. 1979. Effect of cytochalsin B on the matura-tion of enveloped viruses. J Exp Med 150:379–391.
Gubareva LV, Bethell R, Hart GJ, Penn CR, Webster RG. 1995. Char-acterization of mutants of influenza virus selected with 4-guanidino-Neu5Ac2en. Abstracts, 14th Annual Meeting, Am Soc Virol, Austin,Texas W44-1.
Gubareva LV, Robinson MJ, Bethell R, Webster RG. 1997. Catalyticframework mutations in the neuraminidase active site of influenza viruses that are resistant to 4-guanidino-Neu5Ac2en. J Virol 71:3385–3390.
Gubareva LV, Matrosovich MN, Brenner MK, Bethel RC, WebsterRG. 1998. Evidence for Zanamivir resistence in an immuno-compromised child infected with influenza B virus. J Infect Dis-eases 178:1257–1262.
Hausmann J, Kretzschmar E, Garten W, Klenk H-D. 1995. N1neuraminidase of influenza virus A/FPV/Rostock/34 has haemad-sorbing activity. J Gen Virol 76:1719–1728.
Hay AJ, Thompson CA, Geraghty A, Hayhurst S, Grambas S, BennettMS. 1993. The role of the M2 protein in influenza virus infection.In: Hannoun C et al., editors. Options for the control of influenza virus II. Amsterdam: Excerpta Medica. p 281–288.
Hayden FG. 1993. Update on antiviral agents and viral drug resis-tance. In: Mandell GL, Douglas RG, Bennett JE, editors. Principles
and practice of infectious disease. New York: Churchill Livingston.p 2(2):3–15.
Hayden FG. 1996. Amantidine and rimantidine – clinical aspects. In:Richmond DD, editor. Antiviral drug resistance. Chichester, UK:John Wiley. p 69–77.
Hayden FG, Rollins BS, Madren LK. 1994. Anti-influenza activity of the neuraminidase inhibitor 4-guanidino-Neu5Ac2en in cell cultureand in human respiratory epithelium. Antiviral Res 25:123–131.
Hayden FG, Treanor JJ, Betts RF, Lobo M, Esinhart JD, Hussey EK.1996. Safety and efficacy of the neuraminidase inhibitor GG167 inexperimental human influenza. JAMA 275:295–299.

7/27/2019 Neuraminidase inhibitiors
http://slidepdf.com/reader/full/neuraminidase-inhibitiors 19/21
194 VARGHESE
Hayden FG, Osterhaus ADME, Treanor JJ, Fleming DM, Aoki FY,Nicholson KG, Bohnen AM, Hirst HM, Keene O, Whiteman K.1997. Efficacy and safety of the neuramimnidase inhibitorZanamivir in the treatment of influenza virus infections. N Engl JMed 337:874–880.
Hayden FG, Atmar R, Schilling M, Johnson C, Poretz D, Parr D, HusonL, Ward P, Mills R. 1998. Safety and efficacy of oral GS4104 in
longterm prophylaxis of natural influenza. San Diego: ICAAC 24–27 Sept. (Abstr. LB-6).
Hinshaw VS, Webster RG, Turner B. 1980. The perpetuation of orthomyxoviruses and paramyxoviruses in Canadian waterfowl. CanJ Microbiol 26:622–629.
Hirst GK. 1942. Adsorption of influenza hemagglutinins and virus byred blood cells. J Exp Med 76:1740–1743.
Hitte AL, Nayak DP. 1982. Complete nucleotide sequences of theneuraminidase gene of human influenza virus A/WSN/33. J Virol41:730–734.
Janakiraman MN, White CL, Laver WG, Air MA, Luo M. 1994. Struc-ture of influenza virus neuraminidase B/Lee/40 complexed with sialicacid and a dehydro analog at 1.8A-resolution: implications for thecatalytic mechanism. Biochemstry 33:8172–8179.
Kilbourne ED. 1987. Influenza. New York: Plenum Press.Kim CU, Lew W, Williams MA, Liu H, Zhang L, Swaminathan S,
Bischofberger N, Chen MS, Mendel DB, Tai CY, Laver WG, StevensRC. 1997. Influenza neuraminidase inhibitors possessing a novelhydrophobic interaction in the enzyme active site: design, synthe-sis, and structural analysis of carbocyclic sialic acid analogues withpotent anti-influenza activity. J Am Chem Soc 119:681–690.
Kimberlin DW, Crumpacker CS, Straus SE, Biron KK, Drew WL,Hayden FG, McKinlay M, Richman DD, Whitley RJ. 1995. Antivi-ral resistance in clinical practice. Antiviral Res 26:423–438.
Klenk E, Faillard H, Lempfrid H. 1955. Uber die enzymatishe Wirkungvon Ifluenza. Z Physiol Chem 301:235–246.
Kraulis PJ. 1991. MOLSCRIPT: A program to produce both de-tailed and schematic plots of protein structures. J Appl Cryst
D50:869–873.Kunkel TA, Roberts JD, Zakaur RA. 1987. Rapid and efficient site-
specific mutagenesis without phenotypic selection. MethodsEnzymol 154:369–382.
Lamb RA. 1989. Genes and proteins of the influenza virus. In:Krug RM, editor. The influenza viruses. New York: PlenumPress. p 1–87.
Laver WG. 1963. The structure of influenza virus 3. Disruption of thevirus particle and separation of neuraminidase activity. Virology20:251–262.
Laver WG. 1978. Crystallization and peptide maps of neuraminidase“heads” from H2N2 and H3N2 influenza virus strains. Virology86:78–87.
Laver WG, Valentine RC. 1969. Morphology of the isolated hemag-
glutinin and neuraminidase subunits of influenza virus. Virology38:105–119.
Laver WG, Colman PM, Webster RG, Hinshaw VS, Air GM. 1984.Influenza virus neuraminidase with hemagglutinin activity. Virol-ogy 137:314–323.
Lentz MR, Webster RG, Air GM. 1987. Site-directed mutation of theactive site of influenza neuraminidase and implications for the cata-lytic mechanism. Biochemistry 26:5351–5358.
Li S, Schulman J, Itamura S, Palase P. 1993. Glycosylation of neuramini-dase determines the neurovirulence of influenza A/WSN/33 virus.J Virol 67:6667–6673.
Li W, Escarpe PA, Eisenberg EJ, Cundy KC, Sweet C, Jakeman KJ,Merson J, Lew W, Williams M, Zhang L, Kim CU, Bischofberger N,Chen MS, Mendel DB. 1998. Identification of GS4104 as an orallybioavailable prodrug of the influenza virus neuraminidase inhibitorGS4071. Antimicrob Agents Chemother 42:647–653.
Liu C, Air GM. 1993. Selection and characterization of a neuramini-dase-minus mutant of influenza virus and its rescue by cloned
neuraminidase genes. Virology 194:403–407.Liu C, Eichelberger MC, Compans RW, Air GM. 1995. Influenza type
A viurs neuraminidase does not play a role in viral entry, replica-tion, assembly, or budding. J Virol 69:1099–1106.
Luo M, Jedrzejas MJ, Singh S, White CL, Brouillette WJ, Air GM,Laver WG. 1995. Benzoic acid inhibitors of influenza virusneuraminidase. Acta Crystallogr D51:504–510.
Madren LK, Shipman C, Hayden FG. 1995. In vitro inhibitory effectsof combinations of anti-influenza agents. Antiviral Chem Chemother6(2):109–113.
Malby RL, Tulip WR, Harley VR, McKimm-Breschkin JL, Laver WG,Webster RG, Colman PM. 1994. The structure of a complex be-tween the NC10 antibody and influenza virus neuraminidase andcomparison with the overlapping binding site of the NC41 antibody.
Structure 2:733–746.McKimm-Breschkin JL, Marshall D, Penn CR. 1994. Phenotypic
changes observed in influenza viruses passaged in 4-amino or 4-guanidino-Neu5Ac2en in vitro. Abstracts, 9th International Con-ference on Negative Strand Viruses, Estoril, Portugal, p 260.
McKimm-Breschkin JL, Blick TJ, Sarasrabudhe AV, Tiong T, MarshallD, Hart GJ, Bethell RC, Penn CR. 1996. Generation andcharacterisation of variants of the NWS/G70C influenza virus afterin vitro passage in 4-amino-Neu5Ac2en and 4-guanidino-Neu5Ac2en. Antimicrob Agents Chemother 40:40–46.
McKimm-Breschkin JL, Sahasrabudhe A, Blick TJ, McDonald M,Colman PM, Hart GJ, Bethell RC, Varghese JN. 1998. Mutations ina conserved residue in the influenza virus neuraminidase active sitedecreases sensitivity to Neu5Ac2en derivatives. J Virol 72:2456–
2462.Meindl P, Tuppy H. 1969. 2-Deoxy-2,3-dehydrosialic acids. I. Synthe-
sis and properties of 2-deoxy-2,3-dehydro-N-acetylneuraminic ac-ids and their methyl esters. Monatsh Chem. 100:1295–1306.
Meindl P, Bodo G, Palase P, Schulman J, Tuppy H. 1974. Inhibition of neuraminidase activity by derivatives of 2-deoxy-2,3-dehydro-N-acetylneuraminic. Virology 58:457–463.
Mendel DB, Tai CY, Escarpe PA, Sidwell RW, Huffman JH, Sweet C,Jakeman KJ, Merson J, Lacy SA, Lew W, Williams MA, Zhang L,Chen MS, Bischofberger N, Kim CU. 1998. Oral administration of a prodrug of the influenza virus neuraminidase inhibitor GS4071 pro-tects mice and ferrets against influenza infection. Antimicrob AgentsChemother 42:640–646
Miller CA, Wang P, Flashner M. 1978. Mechanism of Arthro-bacter
sialophilus neuraminidase: the binding of substrates and transitionstate analogues. Biochem Biophys Res Commun 83:1479–1487.
Monto AS, Ohmir Se, Hornbuckle K, Pearce CL. 1995. Safety andefficacy of long term use of rimantadine for prophylaxis of type 1influenza in nursing homes. Antimicrob Agents Chemother 39:2224–2228.
Monto AS, Robinson DP, Herlocher L, Hinson JM, Elliott M,Keene O. 1998. Efficacy and safety of zanamivir in preventionof influenza among healthy adults. San Diego: ICAAC 24–27Sept. (Abstr. LB-7).
Murphy JS, Bang FB. 1952. Observations with the electron micro-

7/27/2019 Neuraminidase inhibitiors
http://slidepdf.com/reader/full/neuraminidase-inhibitiors 20/21
ANTI-VIRAL DRUGS FOR INFLUENZA 195
scope on cells of chick chorio-allantoic membrane infected with in-fluenza virus. J Exp Med 95:259.
Murphy BR, Webster RG. 1990. Orthomyxovirus. In: Fields BN, KnipeDM, editors. Virology. New York: Raven Press. p 1091–1152.
Nagai T, Yamada H. 1994. In vivo anti-influenza virus activity of Kampo(Japanese herbal) medicine “Sho-seiryu-to” and its mode of action.
Int J Immunopharmacol 16(8):605–613.Nagai T, Miyaichi Y, Tomimori T, Suzuki Y, Yamada H. 1990. Inhibi-tion of influenza virus sialidase and anti-influenza virus activity byplant flavonoids. Chem Pharm Bull 38(5):1329–1332.
Nicholls A, Sharp K, Honig B. 1991. Protein folding and association:insights from the interfacial and thermodynamic properties of hy-drocarbons. Proteins 11:281–296.
Nohle U, Beau J-M, Schauer R. 1982. Uptake, metabolism andexcretion of orally and intravenously administered, double-la-beled N-glycoloylneuraminic and single-labeled 2-deoxy-2,3-dehydro-N-acetylneuraminic acid in mouse and rat. Eur JBiochem 126:543–548.
Nuss JM, Air GM. 1991. Transfer of the hemagglutinin activity of in-fluenza virus neuraminidase subtype N9 into N2 neuraminidasebackground. Virology 183:496–504.
Osterhaus ADME, Tisdale M, Elliot M. 1998. Double blind randomisedtrial of zanamivir in the treatment of acute influenza — clinical andvirological efficacy results. San Diego: ICAAC 24–27 Sept. (Abstr.H-67).
Palese P, Compans RW. 1976. Inhibition of influenza virus replicationin tissue culture 2-deoxy-2,3-dehydro-N-trifluroacetylneuraminicacid. FANA: mechanism of action. J Gen Virol 33:159–163.
Palese P, Schulman JL. 1977. Inhibitors of viral neuraminidase as po-tential antiviral drugs. In: Chemoprophylaxis and virus infectionsof the upper respiratory tract, vol 1. Boca Raton, FL: CRC Press. p189–205.
Palese P, Tobita K, Ueda M, Compans RW. 1974a. Characterisation of temperature sensitive influenza virus mutants defective inneuraminidase. Virology 61:397–410.
Palese P, Schulman JL, Bodo G, Meidl P. 1974b. Inhibition of influ-enza and parainfluenza virus replication in tissue culture by 2-deoxy-2,3-dehydro–N–trifluroacetylneuraminic acid. FANA Virol59:490–498.
Pearlman DA, Case DA, Caldwell J, Seibel G, Singh UC, Weiner PK,Kollman PA. 1990. AMBER 4.0 San Franscisco, CA: University of California.
Pinto LH, Holsinger LJ, Lamb RA. 1992. Influenza virus M2 proteinhas ion channel activity. Cell 69:517–528.
Rogers GN, Paulson JC. 1983. Receptor determinants of human andanimal influenza virus isolates: difference in receptor specificityof the H3 haemagglutinin based on species of origin. Virology127:361–373.
Ryan DM, Ticehurst J, Dempsey MH, Penn CR. 1994. Inhibition of influenza virus replication in mice by GG167 (4-guanidino-2,4-dideoxy-2,3-dehydro-N-acetylneuraminic acid) is consistent withextracellular activity of viral neuraminidase. Antimicrob ChemChemother 38(10):2270–2275.
Sahasrabudhe A, Lawrence L, Epa VC, Varghese JN, Colman PM,McKimm-Breschkin JL. 1998. Substrate, inhibitor, or antibodystabilises the Glu 119 Gly mutant influenza virus neuraminidase.Virology 247:14–21.
Skehel JJ. 1974. The origin of pandemic influenza virus. Symp SocGen Microbiol 24:321–342.
Slemons RD, Johnson DC, Osborn JS, Hayes F. 1974. Type A influ-enza viruses iosloated from wild free-flying ducks in California. AvianDis 18:119–125.
Smith FI, Palese P. 1989. Variation in influenza virus genes. Epi-demiological, pathogenic, and evolutionary consequences. In:Krug RM, editor. The influenza viruses. New York: PlenumPress. p 319–359.
Smith W, Andrewes CH, Laidlaw PP. 1933. A virus obtained from in-fluenza patients. Lancet 2:66–68.
Smith PW, Sollis SL, Howes PD, Cherry PC, Cobley KN, Taylor H,Whittington AR, Scicinski J, Bethell RC, Taylor N, Skarzynski T,Cleasby A, Singh O, Wonacott A, Varghese J, Colman P. 1996. Novelinhibitors of influenza sialidases related to GG167. Structure-activ-ity, crystallographic and molecular dynamics studies with 4H-py-ran-2-carboxylic acid 6-carboxamides. Bioorg Med Chem Lett6(24):2931–2936.
Sollis SL, Smith PW, Howes PD, Cherry PC, Bethell RC. 1996. Novelinhibitors of influenza sialidase related to GG167. Synthesis of 4-amino and 4-guanidino-4H-pyran-2-carboxylic acid-6-propylamides;selective inhibitors of influenza A virus sialidase. Bioorg Med ChemLett 6(15):1805–1808.
Sprenger MJW, Beyer WEP, Kempen BM, Mulder PGH, Masurel N.1993. Risk factors for influenza mortality. In: Hannoun C et al., edi-tors. Options for the control of influenza virus II. Amsterdam:Excerpta Medica. p 15–23.
Srange KC, Little DW, Blarnik B. 1991. Adverse reactions to amanta-dine prophylaxis of influenza in a retirement home. J Am GeriatrSoc 33:700–705.
Subbarao K, Klimov A, Katz J, Regnery H, Lim W, Hall H, Perdue M,Swayne D, Bender C, Huang J, Hemphill M, Rowe T, Shaw M, XuX, Fukuda K, Cox N. 1998. Characterisation of an avian influenza A(H5N1) virus isolated from a child with fatal respiratory illness. Sci-ence 279:393–396.
Suzuki Y, Sato K, Kiso M, Hasegawa A. 1990. New ganglioside analogsthat inhibit influenza virus sialidase. Glycoconjugate J 7:349–356.
Taylor NR, von Itzstein M. 1994. Molecular modeling studies on ligandbinding to sialidase from influenza virus and the mechanism of ca-talysis. J Med Chem 37:616–624.
Taylor NR, Cleasby A, Singh O, Skarzynski T, Wanacott A, Smith PW,Sollis SL, Howes PD, Cherry PC, Bethell R, Colman P, Varghese J.1998. Dihydropyran carboxamides related to Zanamivir. A new seriesof inhibitors of influenza virus sialidases. Part 2: A crystallographicand molecular modeling study of complexes of 4-amino-4H-pyran-6-carboxamides and sialidase from influenza types A and B. J Med Chem41:798–807.
Thomas GP, Forsyth M, Penn CR, McCauley JW. 1994. Inhibition of growth of influenza virus in vitro by 4-guanidino-2,4-dideoxy-2,3-dehydro-N-acetylneuraminic acid. Antiviral Res 24:351–356.
Treanor JJ, Vrooman PS, Hayden FG, Kinnersley N, Ward P, Mills RG.
1998. Efficacy of oral GS4104 in treating acute influenza. San Di-ego: ICAAC 24–27 Sept. (Abstr. LB-4).
Tulip WG, Varghese JN, Baker AT, van Donkelaar A, Laver WG,Webster RG, Colman PM. 1992a. Refined atomic structures of N9subtype influenza virus neuraminidase and escape mutants. J MolBiol 221:487–497.
Tulip WG, Varghese JN, Laver WG, Webster RG, Colman PM. 1992b.Refined crystal structure of the influenza virus N9 neuraminidase-NC41 Fab complex. J Mol Biol 227:122–148.
Tyrrell DAJ. 1980. Influenza vaccines. Phil Trans R Soc LondB288:449–460.

7/27/2019 Neuraminidase inhibitiors
http://slidepdf.com/reader/full/neuraminidase-inhibitiors 21/21
196 VARGHESE
Varghese JN. 1997. The design of anti-influenza virus drugs from theX-ray molecular structure of influenza virus neuraminidase. In:Veerapandian P, editor. Structure based drug design: diseases, tar-gets, techniques and developments, vol 1. New York: Marcel Dekker.p 459–486.
Varghese JN, Colman PM. 1991. Three-dimensional structure of theneuraminidase of influenza virus A/Tokyo/3/67 at 2.2Å resolution. J
Mol Biol 221:473–486.Varghese JN, Laver WG, Colman PM. 1983. Structure of the influenza
virus glycoprotein antigen neuraminidase at 2.9Å resolution. Na-ture 303:35–40.
Varghese JN, Webster RG, Laver WG, Colman PM. 1988. The three-dimensional structure of an escape mutant of the neuraminidase of influenza virus A/Tokyo/3/67. J Mol Biol 200:201–203.
Varghese JN, McKimm-Breschkin JL, Caldwell JB, Kortt AA,Colman PM. 1992. The structure of the complex between influ-enza virus neuraminidase and sialic acid, the viral receptor.Proteins 14:327–332.
Varghese JN, Epa VC, Colman PM. 1995. Three-dimensional struc-ture of 4-guanidino-Neu5Ac2en and influenza virus neuraminidase.Protein Sci 4:1081–1087.
Varghese JN, Colman PM, van Donkelaar A, Blick TJ, Sahasrabudhe A, McKimm-Breschkin JL. 1997. Structural evidence for a secondsialic acid binding site in avian influenza virus neuraminidase. ProcNatl Acad Sci USA 94:11808–11812.
Varghese JN, Smith PW, Sollis SL, Blick TJ, Sahasrabudhe A, McKimm-Breschkin JL, Colman PM. 1998. Drug design against a shiftingtarget: a structural basis for resistance to inhibitors in a variant of influenza virus neuraminidase. Structure 6:735–746.
von Itzstein M, Wu W-Y, Kok GB, Pegg MS, Dyson JC, Jin B, VanPhanT, Symthe ML, White HF, Oliver SW, Colman PM, Varghese JN,Ryan DM, Woods JM, Bethell RC, Hotham VJ, Cameron JM, PennCR. 1993. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 363:418–423.
von Itzstein M, Wu W-Y, Jin B. 1994. The synthesis 2,3-didehydro-
2,4-dideoxy-4-guanidinyl-N-acetylneuraminic acid: a potent influ-enza virus inhibitor. Carbohydr Res 259:301–305.
Wagh PV, Bahl OP. 1981. Sugar residues on proteins. Crit Rev Biochem307–377.
Ward CW. 1981. Structure of influenza virus hemagglutinin. Curr TopMicrobiol Immunol 94/95:1–74.
Ward CW, Elleman TC, Azad AA. 1982. Amino acid sequence of the pronase-released heads of neuraminidase subtype N2 fromthe Asian strain A/Tokyo/3/67 of influenza virus. Biochem J207:91–95.
Ward CW, Murry JM, Roxburgh CM, Jackson DC. 1983. Chemi-cal and antigenic characterisation of the carbohydrate sidechains of an Asian (N2) influenza virus neuraminidase. Virol-
ogy 126:370–375.Webster RG, Laver WG. 1975. Antigenic variation in influenza viruses.
In: Kilbourne ED, editor. Influenza virus and influenza. New York: Academic Press. p 269–314.
Webster RG, Air GM, Metzger DW, Colman PM, Varghese JN, Baker AT, Laver WG. 1987. Antigenic structure and variation in an influ-enza N9 neuraminidase. J Virol 61:2910–2916.
Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y. 1992.Evolution and ecology of influenza A viruses. Microbiol Rev56:152–179.
Webster RG, Schafer JR, Suss J, Bean WJ Jr, Kawaoko Y. 1993. Evolu-tion and ecology of influenza viruses. In: Hannoun C et al., editors.Options for the control of influenza virus II. Amsterdam: Excerpta Medica. p 177–185.
Weis W, Brown JH, Cusack S, Paulson JC, Skehel JJ, Wiley DC. 1988.Structure of the influenza virus haemagglutinin complexed with itsreceptor, sialic acid. Nature 333:426–431.
White DO. 1974. Influenza viral proteins: identification and synthe-sis. Curr Top Microbiol Immunol 63:1–48.
Wintermeyer SM, Nahata MC. 1995. Rimantadine: a clinical perspec-tive. Ann Pharmacother 29:299–310.
Woods JM, Bethell RC, Coates JAV, Healy N, Hiscox SA, Pearson BA,Ryan DM, Ticehurst J, Tilling J, Walcott SM, Penn CR. 1993. 4-Guanidino-2-4-dideoxy-2,3-dehydro-n-acetylneuraminic acid is a highly effective inhibitor both of the Sialidase (Neuraminidase) andgrowth of a wide range of influenza A and B viruses in vitro. Antimicrob Agents Chemother 37:1473–1479.
Wright CE, Laver WG. 1978. Preliminary crystallographic data for influenza virus neuraminidase ‘heads.’ J Mol Biol 120:133–136.
Yeun KY, Chan PK, Peiris M, Tsang DN, Que TL, Shortridge KF,Cheung PT, To WK, Ho ET, Sung R, Cheng AF. 1998. Clinical fea-tures and rapid viral diagnosis of human disease associated withavian influenza A H5N1 virus. Lancet 351:467–471.