neuropharmex4
DESCRIPTION
Notes from neuropharmacologyTRANSCRIPT

4/8/09 7:39 PM
← Pharmokinetics – speed of drugs through a system thus alleviating
symptoms. Slow (oral & topical). Moderate (intraparenteral – abdominal
cavity & mucosal absorption & intravenous) Fast (intraventricular – into the
ventricles & Intrathecal –injected into the space)
←← Timothy Leary, Ph.D. gave us the laws of effect. The dose, setting
(the stress or environment during study), and set (genetic makeup, eating
and sleeping habits, pH, ect any physiological variables), in order to adjust
the Effect.
←← Neuro pharmacology
← Determination of drug absorption -
← pH and pKa interact to determine the site of absorption.
← Only non-ionized ligands cross lipid barriers redily
← pKa = equilibrium constant
← pKa = pH at which a ligand is nonionized.
← PCP pka = 9.4
← The stomach has a pH of 1.4 and the ph of duodenum = 8.4 (pH) thus
it is more active in the duodenum and enters the blood stream = 7.4 (pH)
←← Plasma protein binding
← Only unbound drug diffuses from the vascular compartment
← Plasma binding protein (eg serum albumin) and free drugs bind to the
protein. “Nonspecific” binding (ie protein indiscriminately binds multiple
ligands.) The binding event doesn’t have any change of the activity of the
proteins – worthless activity for the desired effects of the drugs taken.
Plasma drug binding is almost 99% absorbed by plasma binding proteins so
only a few % end up entering your system for functionality. Look at the
formula!!
←← Fluid “compartments” determine distribution, concentration.

← If a drug is taken orally will enter the blood plasma/circulatory system
(determined from the pKa). The circulatory system needs to enter the blood
brain barrier in order to become active. Remember that most of the drug is
bound to plasma proteins and wasted! There is no conductors/tickets only
the statistical ability. Since most proteins are highly charged and large so
they can pass to the circulatory system. The liver, kidneys, heart, skin, ect
are all possible enterance sites. These might have fatty tissues (adipose)
where the drugs can trapped. Circulatory system passes through the liver
for metabolizing or the kidneys for excretion. Only a very small fraction of
the drugs are actually reaching the brain. Remember Effects and Side-
effects.
←← The BBB (Blood-Brain Barrier)
← Peripheral blood vessels have fairly loose junctions between the cells
(porous). Some substances can pass through these gap junctions. If the gap
junctions were too large you would loose blood plasma and die so these
junctions must be small. The CNS blood vessel have tight junctions. There
are multiple membranes to cross and glial cells (astrocytes) have two more
sets of membranes (total 6 membranes to pass through vs only two in the
peripheral blood vessels. The drugs must be able to be passed to and from
the brain.
←← Metabolism of Drugs – transformation of drugs from one state to
another
← Hydrolysis
← Oxidation
← Conjugation
← all via enzyme activity
←← Metabolism + Excretion = Drug Clearance
← Log plasma concentration (ng/dl) vs time (hr) graph has a negative
slope. The clearance time (lifespan of drug in system) from the highest
concentration to zero. The time to reach the halfway point between the
highest concentration to zero is the half-life of the drug. *notice the half-
time is the is going from one value (not the highest value) to half of that
value*

←← Attaining steady state concentration
← Concentration vs Time (multiples of elimination half times)
← Time to Steady State
← - attained after approximately 4 half-times
← - Time to steady state independence of dosage
← Steady State Concentrations
← - proportional to dose/dosage interval
← - Proportional to F/CL
← Fluctuations
← - Proportional to dosage interval/half time
← - Blunted by slow absorption
←← Developmental regulation of drug clearance
← Graph Clearance vs. Age
← Before 0-1 you steadily go to the highest clearance until puberty and
then there is a downward slope. Be careful to give drugs to very young and
very old because they aren’t able to clear drugs out of their system as
quickly.
←← Dose-response relationships
← Efficacy – look at graphs in book
← Kd is the concentration of 50% of the full effect (Bmax)
← Log of drug concentrations can give you a sigmoid curve.
← For each drug you can determin the potency by looking at the
concentration of drug… if it doesn’t take as much to reach the Kd then it is
more potent. The more efficacy of the drug would be the sigmoid
← Potency is how much to use to get the same effect (maximal effect)
← Efficacy is how well the drug works
←← Agonist vs Partial Agonist
← A full agonist is a drug that can fully activate a mechanism (complete
efficacy)
← A partial agonist can only partly activate a mechanism (partial efficacy)
← The potency of the partial agonist may be more potent (require less
drug) than a full agonist. *remember potency and efficacy are different!

←← Therapeutic index: LD50 vs. ED50
← The graph is the % of individuals responding vs. dose (mg)
← LD = lethal dose; LD50 is the dose to kill 50% of the population at that
dose
← ED = effective dose; ED50 is similar to LD50.
← The ED50 is much lower than the LD50. The therapeutic index is the
LD50 divided by ED50. There are over lapping information. What might be a
ED99 might be LD2. The approval of a drug depends on the therapeutic
index of at least 10.
←← Issues with repeated drug treatment
← Graph Intensity of effect vs. tolerance.
← Initial response may be higher than the second time, this is called
Desensitization (habituation). Recovery may also happen when the user
stops taking a drug for a period of time, and the intensity of effect may rise
to normal (initial response). The recovery can be hazardous because a
patient may take a higher dose thinking that
←← 1/20/09 – was 15 minutes late
← Peripheral vs. CNS
← Evoked mEPPs (mV) *NMJ & Spontaneous mESPs (mV) *CNS
←← Presynaptic Ca2+ channels are anchored to membrane binding
proteins (SNARE = Soluble NSF Attachment REceptor)
←← enhanced [Ca] 1-3x10^-4)
← VDCC and SNAREs are attached to the presynaptic membrane are
close together. The other SNAREs are attached to the vesicle and they
become closer together until the vesicle fuses to the presynaptic membrane.
Similar to the actin and myosin sliding but this causes the fusion.
←← EM shows vesicular docking
← -transmission EM of CA1 neuron synaptic transmitter release
← - Cs+2 mediated interactions of SNARE proteins resulting in vesicular
fusion

←← Vesicle associated membrane proteins (VAMPs)
← There are many different types – **synapsin, synaptobrevin, &
synaptotagmin*primary ones but there are dozens of others.
←← Vesicular fusion, and after
← NSF = N-ethymaldeimide sensitive fusion protein (ATP-ase)
← Each VAMPs have different SNAREs . The voltage dependant Ca2+
channels allow Ca into the cell activating the ATPase functionality and the
coupling of SNAREs and VAMPs is possible. Fusion of the vesicle and the
presynaptic memebrane is complete.
←← BoTOx block of presynaptic vesicular release
← Then botox is added there is no fusion of the VAMPs and SNAREs by
changing their shape. Changing the shape will cause a loss in binding to the
presynaptic membrane thus there will not be a release of NT. An increase in
the concentration of botox, they epsp will decrease incrementally until some
little excitation won’t by affective postsynaptically.
←← Presynaptic exocytoxins block fusion
← Toxin
← Clostridial Toxins
← - botulinum toxins (botox)
← - tetanus toxin
←← EM of fusion
← Scanning EM of freeze fractured presynaptic membrane unstimulated
and after depolarization (pits and omega profiles present)
←← Endocytosis
← Clathrin attached to microtubles interact with synaptotagmin for
endocytosis. Still triggered by ATPases
←← Evidence for and models of membrane recycling
← Classical model – medium time
← Kiss and run model – short time

← Bulk endocytosis model – longer time
←← Vesicular Membrane Cycling
← NTs are being pumped into the vesicle via aid of VAMPs (filled with one
quanta). The transportation and docking occur near a Ca2+ channel with
the aid of ATP. Priming and fusion (exocytosis) due to the depolarization and
sliding of the SNARE and VAMP interaction and Ca2+ concentration increase.
ATP binds the Cathrin to the VAMPs and endocytosis happens. Protons are
being pumped into the vesicle bringing the pH of the vesicle to the lower.
The endoplasmic reticulum may be included in the loop or not. The acidicy
of the interior of the vesicle aids in the NTs to be pumped back into the
vesicle and loop begins over.
←← Depolariaztion, CA2+ influx, vesicle fusion, release, Na+ influx
← 1. Ca2+ influx via presynaptic voltage dependant Ca+2 channels
← 2. Membrane proteins and VAMPS fuses vesicle to membrane
← 3. Release NT binds to gates postsynaptic receptors
←← Rapid NT clearance from the synapse
← 500 microseconds to have the clearance of the whole event.
←← 1/22/09
Post synaptic information
Receptor-effector coupling – ligands bind to receptors and then the effector
on the intracellular domain. Most receptors are bound to the lipid bilayer but
not always true. Some channels are ion channels (ionotropic) and some do
not (mechanotropic).
Stochastic nature of equilibrium binding
- ligands (L) and receptors (R) vary in number in different areas. Some
molecules of the ligand come close enough to the receptor. Over a
certain amount of time there should be a number of ligands binding to the
receptors. As time continues, free ligand molecules (F). Once they
associates there is a value Ka – the rate of free ligands binding to
receptors to form a ligand/receptor complex (or # of bound complexes
(B)).

- [L] = [F] + [LR]
- [LR] = [B]
- The whole reaction is a two way reaction until equilibrium is reached. A
ratio is expressed. To know the dissociation constant (Kd) you must know
the Kd = [F]*[R]/[B]. You use this information to determine the affinity of
drugs. The affinity is measured by using 1/Kd. Bmax = [R] + [B].
Defining classes of ligands
- agonist: binds to and activates effector site
full agonist; maximally activates effector
partial agonist; only partially activates effector
inverse agonist; reverses effector mechanism
← - antagonist: does not activate effector site
competitive; binds to effector site
noncompetitive; binds to site distant from effector site
suicide; binds irreversibly
← - modulator: alters affinity or efficacy
affinity; binding “attraction” of ligand and of receptor
efficacy; maximal effect obtainable
←← Multiple competitive types I, II, III
← Non-competitive antagonism – there are multiple sites on receptor
← All sites open: receptor inactive
← Agonist site occupied: receptor active
← Agonist site open: receptor inactive
← Antagonist site occupied: receptor inactive
←← Plasticity 1: experience-dependent shifts in dose-response curves
← Plasticity 2: antagonists shift dose-response curves
The increase in competitive antagonist will shift the agonist curve to
the right which looks like tolerance.
If a noncompetitive antagonist will reduce the maximal affect
achieved. It doesn’t change the ability of the agonist to bind but
the number of receptors useful.
←← Common pharmacological analyses

← Dose-response curves
The efficacy and potency of ligands
← Inhibitor plots (antagonist)
Track efficacy, potency of antagonists
← Hill plots
Calculate # of binding sites/receptors
← Time-dependent (kinetic) plots
Association (binding)
Dissociation (unbinding)
Clearance (half-life and half-time)
← Scatchard plots
Calculate kinetic variables
← Saturation curves
Calculate binding capacity
←← Hill coefficient = binding sites/receptor
← The number of binding sites of the receptor determined by the slope of
the effect (nA)/log [ACh] (nM). A higher hill coefficient will determine the
amount of NT needed to activate a receptor.
←← Saturation curves – specific vs. nonspecific binding
← Y axis – amount bound (fmol/mg protein)
← X axis – [Ligand] (nM)
← Specific = total – nonspecific
←← Scatchard plot: ratio of B to F ligand at equilibrium
← Y axis = log [B]/log [F]
← X axis = log [F]
← The slope = 1/Kd
← X-Intercept = Bmax
←← 1/27/09 – 5 min late
← 3 major G=protein families: Gs, Gi, Gq
← Gs – increases AC activity
← Gi – decreases AC activity
← - increase PLC activity

← Gq – increase PLC activity
← G-protein = guanosine-phosphate-binding protein
←← Both alpha and Beta-gamma subunits can be active messengers. But
the alpha subunit is the diffusible subunit and the Beta-gamma subunit is
membrane bound so the activity of the subunit is limited (sorta).
←← Life-cycle of G-protein effector coupling
← The alpha and beta-gamma subunits are bound together and the alpha
is bound to GDP. The ligand receptor complex forms when the ligand binds
to the receptor. Once the complex forms the affinity for the G-protein and it
interacts with the ligand receptor complex. The GTPase adds or converts the
GDP into a GTP. The GTP is bound to the alpha subunit/beta-gamma subunit
is now has higher energy (unstable). The alpha-GTP dissociates from the G-
protein complex and can act as a source of phosphates (adding energy to
rxns). The beta-gamma subunit now can also act as a secondary messenger.
Again a GTPase converts the GTP into GDP again, either by transferring the
phosphate to another molecule. The alpha-GDP and beta-gamma subunits of
the Gprotein can now associate once more and the ligand is release from the
receptor.
←← Small G-proteins
← Look at GAPS and GEFS in book. Pg. 80
←← Hydrolyzable and non-activating forms of GTP
← GTP can by hydrolyzed
← GTPgammaS – toxic effect because the GTP will not be able to be
removed from the active alpha subunit of G proteins.
←← Alpha subunits and toxins
← Cholera toxin *diarrhea (attacks alpha-s subunits)
← Pertussis toxin *whooping cough (attacks the alpha-I and alpha-o)
←← Enzyme terminology
←← Multiple pathways, diverging and converging

←← 1/29/09
← Second messenger
← A messenger is something that carries message.
← Messenger cascades. The book says that any the second messenger is
the molecule that transmits the “signal” after the g-protein.
←← Enzymes transiently disrupt homeostasis
← - cyclases (eg AC) vs. phosphodiesterases (PDEs)
← - synthases (eg NOS, etc.) vs. proteases, lipases (egPLC)
← - kinases *adding phosphate groups (eg. PKA, PKC, CAM-KII) vs.
phophatases *removes phosphate groups (PP1, calcineurin)
←← Cyclases produce cyclic nucleotides
← ATP is converted into camp via adenylyl cyclase (aka AC & is
membrane bound with two catalytic sites.) **important in Gs and Gi
gproteins.
← Guanylyl cyclase has only one catalytic domain but has the ability to
be unbound.
←← Gs, calmodulin (CAM) and forskolin activate AC
← G-protein alpha subunits activate or inhibit cyclases
← CAM is endogenous calcium binding protein(CBP) that can bind 4 Ca
ions; forskolin is a plant-derived organic toxin; many other pathways also
converge on AC.
← High levels of intercellular Ca will activate the adenylyl cyclase. Gi and
Go decrease the amount activity of AC
←← cAMP-dependent protein kinase (PKA)← Binding 2 molecules of cAMP to each of the 2 regulatory subunits of PKA releases the
2 catalytic subunits. See pg 92 in text. ** there needs to be 4 cAMP in order to fully release
the active subunits.
←

← 7 classes of phosphodiesterases (PDEs) ** just know that there are
seven and the major difference between them is the affinity of their
substrates are – either cAMP or cGMP creating 5’cAMP and 5’cGMP
respectively.
←← NO synthase (NOS) produces NO
← L-arginine is converted into L-citrulline + nitric oxide (NO)via NOS + a
cofactor of Ca. NOS activity is also regulated by CAM.
←← NO is a retrograde neurotransmitter
← NO activates presynaptic guanalyly cyclase which increases the cGMP
production which increases the vesicle binding to the presynaptic
membrane; increased presynaptic cGMP increases vesicular transport rate.
←← Phosphotidylinositol (PI) metabolism
← The main component of the pathway is the PIP2. Li can interfere with
the metabolism of PI. It is a cyclic pathway
←← PLC is activated by Gp or Go alpha subunits
← PIP2 is cleaved into 2 fragments by PLC:
DAG (membrane bound)
IP3 (diffusible)
← PIP2 is the substrate and the PLC acts as the enzyme to form DAG and
IP3
←← PKC is activated by free Ca and by DAG. The enzyme has a restrictive
and catalytic sites. When in the inactive form Ca and DAG form the active
form of the PKC. Since binding of DAG to the catalytic subunit is required for
activation, most forms of active PKC are membrane bound.
←← IP3 activates and intracellular IP3 receptor. The IP3 receptor
(ionotropic receptor) releases Ca sequestered within the smooth ER (an
intracellular Ca store). The Ca is sequested via a ATP pump (ATPase) and
during the reaction the Ca is used & stored inside the smooth ER.
←

← IP3 and ryanodine receptors activate Ca channels – both release Ca
from intracellular stores. Both muscles and neurons have IP3 and ryanodine
receptors. IP3 receptors are activated by IP3 but Ryanodine receptors cause
Ca to be released by ryanodine and Ca (positive feedback).
Thapsigargin – blocks the ATP pump into the smooth ER store.
Calsequestrin - (Ca binding protein) thus it gets stuck inside the
smooth ER.
←← CAM, calcineurin, and CAM-KII
← Autophosphoylation of CAM-KII happens immediately after the active
state is reached by the binding of high levels of CA/CAM and is locked in the
active state. Calcineurin has binding sites for Ca and CaM. Binding of Ca has
little affect of activity but binding of both, CaM and Ca will greatly increase
the activity. ** remembrer CAM needs Ca to be bound to it to be active**
←← Intracellular stores are one of the many intracellular free Ca sources.
Channels are sources of Ca while pumps and exhcnagers & CBP are sinks.
← Voltage Dependant Ca Channels. ATPases pump Ca outside the cell
and Na+/Ca2+ exchangers are also used where 3Na+ are pumped inside the
cell and one Ca2+ is pumped out. ** most Ca pumped outside would be
through the ATPase pump but it costs energy but the Na+/Ca2+ will
eventually cause the cell to depolarize. Ca is very important in regulation***
←← 2/3/09
← Review for test at 6pm today
←← Cyclase pathways – Adenylyl Cyclase is stimulated by Gs and inhibited
by Gi. ATP is the substrate to make cyclic AMP. Ion channels can be opened
or other processes such as increasing Ca ion
←← CAMP and Phosphodiesterases (PDEs) use cyclic nucleotides (cAMP or
cGMP) 5’cAMP or 5’cGMP.
←← IP3 receptor and products.
← Ca+2 binding proteins (CaBPs)

o Calmodulin (CaM)
o PKC
o Calsequestrin
o Calcineurin
o Calbindin
o Parvalbumin (found in inhibitory interneurons)
← Organic Ca2+ chelators
o BAPTA
o EGTA
←← Dentritic spine apparatus – limits the amount of Ca allowed localized
by the synapse.
←← PKC and CAM-KII
← PKC is activated by Ca+2 and DAG
← CAM-KII is activated by Ca+2/CAM and autophosphorylates itself in
order to remain active even when [Ca] lower.
←← Serine/threonine phosphateases
PP1
o Catalytic + target subunits/inhibitors
o Regulation
PI1
o Inhibitors
Okadaic acid & DARPP-32
PP2A
o Heterotrimer (catalytic + 2 regulatory)
o Regulation
Ceramide, phosphorylation, methylation
o Inhibitors
Okadaic acid
PP2B (calcineurin)
o Heterodimer (catalytic + CAM binding site, CAM-mimic)
o Regulatation
CA2+ / CAM

o Inhibitors
Cyclosporin, cyclophilin, pyrethroids (insect) FK506 Fepb
PP2C
o Catalytic subunit
Phosphatase Inhibitors
Okadaic acid inhibits PP1 and PP-2A
Deltamethrin inhibits calcineurin
Cyclosporin inhibits calcineurin
PHosphatase regulation: DARPP-32 (dopamine and cAMP regulated
phosphoprotein)
Phosphorylated DARPP-32 inhibits PP1, therefore provides positive feedback
for AC activation.
←
←←←←

4/8/09 7:39 PM
← Ion Channels: Depolarizing
← Ionophores: non-gated channels
← Toxins
Monensin
o Source – Bacteria
o Mechanism of Action
Macro – antibiotic
Micro – Na+ ionophore
Amphotericin
o Source Strepococcus
o Mechanism of Action
Macro – antifungal and antiprotozoal
Micro – NA+/K+ ionophore
←← ATPases (pumps) can change membrane polarization
Na+/K+-ATPase
o For every 3 Na+ pumped out, 2 K+ ions enter the cell;
causing a hyperpolarization.
←← Pump toxins disrupt function
← Toxins
Digitalis
o Blocks NA+/K+ ATPase by inhibiting dephosphorylation
Ouabain (wabain)
o Blocks NA+/K+ ATPase
Thrapsigargin
o Blocks Ca2+ATPase in ER
Depolarizing channels
Voltage-gated
o Na+ channels
o Ca+2 channels
←← Gating Models

*Regional conformational change – a selective portion of the protein
structure changes
General conformational change – entire structure changes
*Blocking particle
← * most likely to be the cause
←← Three kinetic steps
Open inactivation closed
The inactivation gate (msec) will be much slower than the activation
gate (nsec)
←← Voltage-clamp single-channel recording
The voltage would rise during the Voltage command step
The current opens during the command step and it shuts off but the
voltage still remains depolarized.
←← Active, inactive and closed
In the inactive state there is no current but the voltage still remains
depolarized.
←← Cahnnel structure conservation
← Na and K channels end up being made up of a single protein. There is
an open central space. There are inactive regions and active regions within
the protein.
←← Visualizing Na channels
← 4 subunits with 6 transmembrane segments each
← Na channel types NaV1.x don’t need to know
←← Local anesthetics block Na+ channels
Cocaine
Lidocaine widely used because of less cardiac and liver toxicity
←← TTX block Na+ channel conductance
Fugu fish (sushi)
←

← Additional Na+ channel blockers
← Toxin
Saxitoxin (STX) red tide

Tetrodotoxin (TTX)
QX-314 works from the inside, used in recording electrode to
selectively turn off neurons individually
←← Cardiostimulants prevent inactivation of Na+ currents
Aconitine – monks hood
Veratridine – rhizomes of sabadilla lily
← * both are considered neurotoxins but may be used for very low heart
rates
←← Intracellular Ca2+ channels
IP3 (neurons and muscles)
o IP3 is the agonsist
o Heparin (blood thinning agent) and caffeine are antagonists
Ryanodine (skeletal muscle, cardiac muscle, neuronal)
o Ryanodine, caffeine, and heparin are all agonists
o Dantrolene is the antagonists
←← Ca2+ channel subunit structure
← 4 subunits with 6 transmembrane domains each
←← Two major types of Ca2+ channels
LVA (low-voltage activated)
o Rapidly inactivating
HVA (high-voltage activated)
o Inactivating
o Noninactivating
←← 2/12/09
← Voltage-dependent Ca2+ channel (VDCC) types
← L
Found in neuronal (CaV1.2-1.4) and muscle (CaV1.1) cells
HVA & noninactivating
Blocked by inorganic ions (cobalt, cadmium)
← T (transient)

Cardiac, Nuronal (CaV3.1-3.3)
LVA and Fast Inactivating
← N
Neuronal only (CaV2.2)
HVA and Slow Inactivating
← P/Q (purkinje) and originally thought to be presynaptic but also in
postsynaptic
Neuronal only (CaV2.1)
HVA and Slow Inactivating (but not as slow as N type)
← R (found originally in retina)
Neuronal Only (CaV2.3)
HVA and Fast Inactivating
←← Organic Ca2+ channel toxin sources
Spiders, snakes, tree roots, and microorganisms
←← Ca2+ channel Ligands I
← Blocks L types
Dihydropyridine
o Nifedipine – widely used for hypertension to regulate heart
rate and cardiac output
o Nimodipine – Brain specific
Arylalkylamine
o Verapomil
Benzothiazepine
o Diltiazem
Alkaloids
o Phloretin (plumb tree roots)
← Blocks T type
Benzimidazole
o Flunarazine
o Mibefradil
← Blocks N type
Conotoxin peptides
o Ω-CTX-GVIA (conotoxin gene 6 A)

← Blocks N, P/Q
Conotoxin peptides
o Ω-CTX-MVIIC
←← Ca2+ channel ligands II
← Enhances L type
BAY-K-8644
← Enhances ALL types
Maitotoxin
← Blocks P/Q (funnel spider toxin)
Omega-Aga-Ia
← Blocks L type
Phloretin (fruit tree root alkaloid
Blocks ALL types
Inorganic ions (Mg2+, Cd2+, Cs2+, Ni2+, Co2+)
← ** barium flows through Ca2+ channels better than Ca2+
←← Ca2+ homeostasis in neurons
← Ca2+ Influx
VDCCs
NRs (NMDA receptors)
Na+/Ca2+ exhanger
← Ca2+ stores
CaBPs
ER
Mitochondria
← Ca2+ release
IP3Rs
RyRs
Ca2+ dependent
Ca2+ release
← Ca2+ Efflux
ATP-ases

NA+/Ca2+ exchanger
←← Ca2+ imaging with fluorescent indicators
Fura 2 most commonly used
←← Voltage-Gated Channels: Hyperpolarizing
Chloride (Cl-) channels
Potassium (K+) channels (K channels can flow inward if the cell is
above than the resting potential or outward if the cell is below the
resting potential voltage -65mV.
←← Chloride Channel Toxins
Chlorotoxin
Flufenamate (chlorea use)
Tamoxifen (breast cancer treatment)
←← K+ Channel toxin sources – organic and synthetic
←← 4 major classes of K+ channels
Voltage-Dependent K+ channels (VDKC)
o Ia (first observed),
o Id (delayed),
o Ik,
o Im (muscurine sensitive)
Rectifier (making right returning to right potential) K+ channels
o Ik,
o anomalous rectifier (activated by below resting potential),
o Ih(queer) – hyperpolarization activated
o tandem pore – form two channels per molecule
Metabotropic-Gated K+ channels
o Im (metabotropic regulated)
o ATPsensitive (requires ATP)
o Iahp (After hyperpolarization potential)
Ca2+-dependent K+ channels (CDKC)
o Ic (calcium)

o Im
o Iahp
←← 2/17/09
← VDKC structure (Ia, Id, Ik)
4 separate subunits with 6 transmembrane segments each
←←← K+ currents and action potential firing
← Ia and Id are the first to be activated and try to keep the action
potential back to resting. As the voltate rises up then the delayed rectifier Ik
and brings the resting potential. Then Ca dependant K channels are
activated . Then the Ic, Im, Iahp to lower the after hyperoplarization
potential. Then the slowIAHP, and ISahp. These determine ability for a neuon
to fire again. If we reduce the capacity for the Ic, Im, Iahp, I ahp, & Isahp.
←← VDKCs (see notes)
← Ia
LVA and Fast inactiating
Blockers – DTX (dendrotoxin)
Id
LVA
Some rectifier K chaneels structures are add
Anomalous: 4 separate subunits with 2 transmembarne segments each
Tandem Pore: 4 transmembrane segments eac, dimerize for 2 pores VIA
volatile anesthetics
Ik
Activation – HVA
Inactivating – SI
Blockers – Cs+
Tandem pore
Activation – voltage independant

Inactivating – SI
Activators
o Halothane
o Isoflurane
Blockers – none known
← Anomolous rectifier (inward rectiviers)
Activation – HA (hyperpolarization activated)
Inactivating – SI
Blockers – CS+
← Ih
Activation – HA (hyperpolarization activated)
Inactivating – SI
Blockers – Cs+
←← Metabotropic-Gated channels
← Im
Channel proteins KCNA1-5
Ca2+ sensitivity- High
Neurotransmitter sensitivity – Muscarinic Ach agonist
Activation - LVA
Inactivation - NI
Activators - retigabine
Blockers – linopirdine, muscarinic agonist
← Iatp
Channel proteins – Kir6.x
Ca2+ sensitivity - insensitive
Neurotransmitter sensitivity - ATP
Activation - None
Inactivation - NI
Activators – ninoxidil (blocks conversion of testosterone to dht)
Blockers – ATP
← Iahp
Channel proteins – 2 or more families
Ca2+ sensitivity – high
Neurotransmitter sensitivity – NE, DA, 5HT, Glu
Activation

Inactivation
Activators
Blockers
←← CAM and SK-channels
CAM remains constitutively bound to SK channels, acting as their
Ca2+ sensor
Apamin (bee venom) binding to SK channel
←← CDKCs
← Im
Channel proteins
Ca2+ sensitivity - low
Neurotransmitter sensitivity - NO
Activation
Inactivation - SI
Activators - retigabine
Blockers – linopirdine
← Ic
Channel proteins - BK
Ca2+ sensitivity - >1uM
Neurotransmitter sensitivity - Yes
Activation
Inactivation - SI
Activators - Niflumate
Blockers – charybdotoxin & TEA (uM)
← Iahp
Channel proteins - SK
Ca2+ sensitivity – 200-600nM (CAM)
Neurotransmitter sensitivity - No
Activation
Inactivation - NI
Activators
Blockers – apamin, dequalinium, neurotransmitters

← IsAHP
Channel proteins - ???
Ca2+ sensitivity
Neurotransmitter sensitivity - no
Activation
Inactivation - NI
Activators
Blockers – neurotransmitters – via PKA PKC and CAMKII
metabotropic pathways
←← Comparing CDKCs – The last three are the AHP portion of the neuronal
firing
←← 2/19/09
← Glutamate receptors
← Ligand gated Rs: non-NMDA-rs
← Ionotropic neurotransmission – cell neighboring another cell that fires
can also have an excitatory post synaptic potentials. Glutamate is the
neurotransmitter that brings about EPSPs (Glu for short). Inhibitory (IPSPs)
are GABA modulated. EPSP depolarize and IPSP hyperpolarize. They are not
separate from each other but sum. The sum of the two identical units of
stimulation from both EPSP and IPSP is still an inhibition not an excitation!
You need a larger amount of excitation than inhibition (they are not linear
function).
←← Excitatory amino acids
← Glu
← AMPA
← KA
← NMDA
←← Defining GluRs by regional ligand binding
← AMPA the most abundant receptor type in nervous systems
← Defining GluRs by effector mechanisms
AMPA is part of the receptors for Sodium (Na+) channels
← Protein Homology (genes also)

← AMPARs
Glu R1-R4
← KARs
Glur5- glur7
KA-1 & KA-2
← Defining GluRs by gene expression
←← GluRs subunits form tetramers
← AMPARs typically contain at least one GluR2, at least one other
subtype, thus act as Na+ channels (hill coefficient = 2)
←← GluR biology: unique GluR2 subunits. Amino terminals on exterior and
carboxyl group in the cytosol. There are 4 membrane spaning and a subunit
Q/R site where you can either have a Q = Gln or R = Asp. That area
determines the specificity for Na+ if not then they will also allow Ca2+ ions
through also. The flip-flop site where alternative splicing determines
polyamine sensitivity. The sensitivity will increase the amount of Na+ influx
under the right conditions.
←← Agonist
AMPA – best activator of receptors (but exogenous)
Glu (endogenous)
← Antagonist (all competitive)
NBQX (blocks AMPA receptors)
CNZX (block KA receptors
← AMPAkines (inc. affinity of AMPARs for Glu)
Aniracetam (enhances Glu binding to receptors)
←← KAR ligands
← Agonist
KA (best activator or receptors
Glu
← Antagonist (all competitive)
CNQX
←← Receptor desensitization

← Receptor – AMPA ; cyclothiazide removes desensitization
← Receptor – KA ; concavilin A (conA) removes desensitization epilepsy
possibilities
←← Iontropic effects of Glu
← AMPA receptors and NMDA receptors. An EPSP (voltage) and EPSC
(current (mA)) create an action potential.
←← Glu EPSPs are typically mediated by multiple receptor types.
←← Glu: multicomponent EPSCs See notes
←← Sulfur-containing EAA transmitters
← Homocysteic acid – produced an anoxious state drownding and
taurine does not cross the blood brain barrier but in diseased states then it
stimulates the inhibitory function.
←← 2/24/09
← Glutamate
← Defining NMDARs by gene expression
← Ionotropic NMDA, AMPA, Kainate (functional classes)
← NMDA gene families (NR1, NR2a-d, NR3a &b)
← Typical NR = 2 NR1 + 2 NR2 subunits
←← NR subunit expression varies regionally
← NR1 are found everywhere in the brain
← NR2a – d are regionally distributed
← Ex: RN2a are found in hypocampus & Nr2c – cerebellum
←← Unique protein structure of NRs – NR= 2 NR1, 2 NR2 (dual-heterodimer
= tetramer) The subunits NR2 determine the flow and glutamate binds to
the NR2s. The NR1 have binding sites for glycene (Gly). The receptors have
a co-agonist… it requires all sites activated (2 gly and 2 glu) *unique
←← Functional analysis of NRs (see notes)
←

← Uses/abuses of NMDAR ligands
Therapuetic
o Dissociative anesthesia
o Cough suppression
o Learning and /or memory enhancement
o Stroke/drowning/ischemia neuroprotection
Non-therapuetic
o Dissociative hallucinogen
←← PCP
Antagonist – blocks open channel
o PCP (phencyclidine; angel dust)
o MK-801 (dizocilpine)
o Angeldustin (endogenous)
o Ketamine (veterinary practices)
o Dextromethorphan (robitusin)
o Memantine (memory treatment in Alzheimer’s)
← Mg++
Antagonist – blocks open channel
o Mg++
o Memantine
← EtOH
Antagonist – blocks channel opening
o EtOH (ethanol)
← Polyamine
Antagonist – decrease (ANT) Glu binding affinity
o Ifenprodil
o Cadaverine (endogenous)
Agonist – increase (AGO) Glu binding affinity
o Spermine
← Heavy Metal
Agonists – prolongs open channel
o Zn++
← HPO4-
Agonist – prolongs open channel
o HPO4-

←← “Use-dependent” Mg++ block 1
← When magnesium isn’t present there is a continuous opening but with
normal [Mg++] it will “flicker”
←← NR noncompetitive antagonist impair learning
← PCP people cannot learn very well
←← Regulation of NRs by phosphorylation
← NMDAR is attached to PSD-95 connected to other proteins and mGluR
is bound to the homer, which can cause phosphorylation of the NMDA
receptor and enhances the current flow. Review Hammond…
←← Excitotoxicity via NRs
← NMDA induced apoptosis via Ca++ influx… APV, MK-801, Kyn block
apoptosis
←← NR2 Glu-binding site ligands
Agonists
o NMDA (N-methyl-Daspartate)
o Glu (glutamate) (endogenous)
Antagonist (competitive)
o APV (AP5 = D-amino-phosphovaleric acid) higher affinity
than glutamate but it doesn’t cross the BBB
o CPP (crosses the BBB)
←← NR2 antagonist impair learning
← - adding APV to mice will greatly impair their learning
←← NR1 Gly-binding site ligands
Agonists
o Ser (serine *endogenous via glial cells)
o Gly (glycine *endogenous)
Partial agonists
o DCS (D-cycloserine)
Antagonist (competitive)

o Kynurenate (kyn *endogenous)
←← Gly or Ser as a coagonist of NRs
←← NR roles in plasticity
← Antagonists block learning, and agonists generate seizures
←← 2/26/09
← Inhibitory amino acid receptors – GlyRs, GABAARs, GABACRs
←← 5pm Friday study review.
←← Pentameric receptors (five sets of subunits make up their structure)
← GABA-A – contain alpha and beta subunits (and gama)
← GABA-C – contains rho subunits
←← Structural conservation between inhibitory Rs
←← GlyR anchoring to the cytoskeleton, “clustering”
← They are attached to gepherin to form the clusters and attracted to
microtubules and neurofilaments and other gephrin. If there is a knockout of
the gepherin the GlyRs are not clustered and inhibition is affected.
←← GlyR activated IPSPs (hyperpolarization of the membrane). Activation
of the receptors cause and influx of Cl- ions.
←← GlyR activated IPSCs has a hill coefficient of 2
←← GlyR ligands (strychnine sensitive)
← Agonists (competitive)
Gly
Taurine
← Partial agonists (competitive)
B-aminobutyrate (BABA)
← Antagonists (competitive)
Strychnine (muscle spasms and breathing issues)

← Antagonists (noncompetitive)
Picrotoxin
Picrotoxinin
←← Regional heterogeneity of GABAAR alpha subunits
GABAAR mediated Cl- currents are ligand- and voltage-dependent
GABAAR desensitization – the decrease in reactivity to prolonged stimulation
Nipecotate suppresses but prolongs IPSPs
Uses/abuses of GABAAR ligands
Therapeutic
Sedatives
Hypnotics (sleeping pills)
Anxiolytics
Seizure prevention, reduction (AEDs) Anti-Epileptic Drugs
← Recreational
Date rape
Sedative-hypnotics
Anxiolytics
Agonist Antagonist Allosteric
Modulator
Ligand Competitive Noncompetitive
GABA bicuculline picrotoxin barbiturates
muscimol GABAzine picrotoxinin BDZs
gavozadol -cyphat neurosteroids
isoguvacine flucybene EtOH
propybicyphat

← Related issues:
Development of tolerance
Development of dependence/addiction
Overdose
Side-effects
←← Allosteric modulators effect GABAAR IPSPs
←← Barbiturates enhance Cl- conductance of channel
← Ligands
Agonists (competitive)
o Phenovarbital (luminal) and pentobarbital (nembutal)
←← Benzodiazepines (BDZs) enhance the affinity of GABARs for GABA.
More is likely to bind and stay bound.
← Ligands
Agonist (competitive) – bloth classes of omega receptors
o Chlordiazepoxide
o Diazepam (valium)
o Carbamazephine
Agonist (partial)
o Bretazenil
Antagonists (competitive)
o Flumazenil
Agonists (inverse)
o Carboline
← Omega 1 receptors are associated with hypnotic affects
Zolpidem (ambien) – non-benzodiazepine hypnotic
(imidazopyridine)
Eszopiclone (lunesta) – non-benzodiazepine hypnotic (cyclopyrrone)
← Omega 2 receptors are associated with anxylitic
←← Diazepam liver conversion to nordazepam oxazepam and two
products after breakdown are still BDZs
←

← Neruosteroid Ligands
Agonist (competitive)
o Pregnenolone – decrease anziety and increase GABA affinity
Antagonists (competitive)
o Allopregnanolone – increases anxiety – reduce GABA affinity
←← GABACR are unique (and rare) – found in the retina horizontal cells.
They have long durations and high current.
←← Only common in retinal horizontal neurons
← Pentameric but unique subunit expression : Rho
← Mostly insensitive to most GABAa and GABAb antagonists
← Insensitive to modulators (nosensitivity to BDX/
Barbiturates.neurosteriods/ethanol
← Very slow channel gating (activation inactivation occurs in secs not ms
← Little or no desensitization
← Cl= channel blocked by pirotoxin/picrotoxinin
←← GABACR Ligands
← Agonists (competitive)
GABA
← Agonists (partial)
CACA (cis-4-aminocrotonate)
← Antagonists (competitive)
TPMPA
← Antagonists (non-competitive)
Picrotoxinpicrotoxinin
←3/3/09
nAChR gating via conformational change. They are pentameric and use a
sliding mechanism to change the chape of the channel pore.
Was the 1st receptor purified, sequenced, cloned, imaged via torpedo fish
studies.
There is conserved subunit structure among receptors

The developmental reguation and locale of nAChRs in a child there will be a
gamma and in adults is epsilan. The will have 2 alpha, 1 beta,
nAChR alpha 7 receptors and smoking. An all alpha 7 monomer where all
the 5 subunits are identical.
nAChR mediated Na+ Currents and there is a linear I’V curve
Desensitization by phosphorylation = matabotropic receptor cascades
Uses/abuses of nAChR ligands
Clinical
o Treatment of motor dysfunction
o Treatment of cognitive impairments, ADD
o Treatment of memory impairments
o Weaning from tobacco addiction
o Regulation of autonomic functions
o Induction of surgical paralysis
Non-clinical
o Recreational smoking, chewing, snuff
o Insecticides, neurotoxins
o Paralytic hunting, assassination poisons
←← Short- and long-acting analogs of ACh
← ACh (short) methacholine (medium) carbachol (long)

← Expression ← Neuronal ← Muscle
← Name ← CNS (alpha-
bungarotoxin
insensitive)
← CNS
(alpha-
bungarotoxi
n sensitive)
← Autonomi
c Gangila
← NMJ
← Subunits ← ← (alpha
7) petamere
← ←
← Agonists
(competitive)
← ACh
← Carbachol
← Nicotine
← arecoline ←
← anatoxin ← ← anatoxin
← *epibatidine ← ← epibatidine
← lobeline ← anabas
in e
← DMPP ←
← Antagonist
s (competitive)
← coronaridine ← alpha-
bungarotoxi
n
← Hexameth
onium
← Decameth
onium
← k-
bungarotosin
← Curare
← Depolarizi
ng blocker
← ← Trimethop
han
← Succinylc
holine
← Antagonist
s
(noncompetitive)
← PCP (phencyclidine)
← TEA (tetraethylammonium)
← Histrionocotoxin
← Lophotoxin
← nereistoxin← ← buprop
rion
(wellbutrin)
←
← Antagonist
(suicide)
← bromoACh
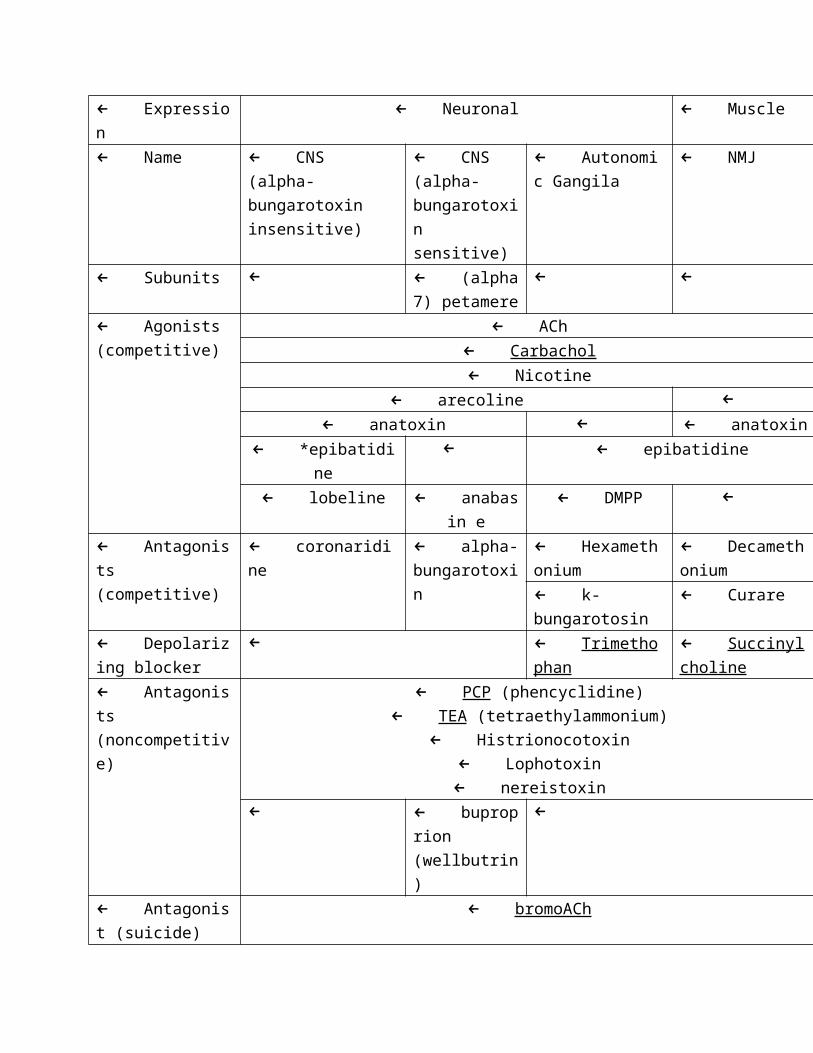
← * tree frog poison
**
Voltage and ligand gated channels and the neurotransmitters associated.
Know the agonsit and antagonist. Know the endogenous ligands. Exogenous
drugs need to know. Know which drug goes to which channel. Does it apply
to voltage gated, ligand gatied, Ia, Id, … what does the drug interact with!!!
The complicated receptors of the NMDA then the GABAa receptors and look
over the sturcutre and the modulatory sites. The NMDA subunits, NR1 and
NR2- both important. NMDA receptor in neuroplasticity. HINT HINT HINT.
Know the difference between gly and glu mediated subunits. Know the I/V
curves and what they mean for the different receptors and currents.

4/8/09 7:39 PM
← 3/10/09
← GluRs – Metabotropic glutamate receptors: mGluRs
←← L-glutamine
↓ (via glutaminase)
← L-glutamate (mitochondrial) α-ketoglutarate (mitochondrial)
dicarboxylate carrier outside of mitochondria alpha-ketoglutarate
(cytoplasmic) L-glutamate via AA (cytoplasmic) L-glutamine via
glutamine synthase (GS) or GABA via (glutamate decarboxylase aka GAD) in
inhibitory neurons because GABA is an inhibitory neurotransmitter
←← Na+- dependent: Excitatory Amino Acid Carrier-1 (EAAC-1)
← Na+- dependent: Glu Transporter-1 (GLT-1)
← Na+- dependent: Glu Aspartate Transporter (GLAST)
← H+ and Mg++ dependent Vesicular ATP-ase Glutamate Transporter
(VGlu-T)
← Na+ and G+ dependent Glutamine Transporter (Gln-T)
←← Early structural model of mGluRs
← Current structural model of mGluRs – Homodimers
← Regional expression varies: mGluR1, 5, 6 – due to experience!!
Hippocampus and cerebellum – mGluR1
← GluR5 in the amygdala
←← Gene expression and metabotropic function
← They are numbered mGluR1-8 and all homodimers
← Class I
mGluR1 and mGluR5
functionally they increase IP3 and Ca2+
Class II
mGluR2 and mGluR3
functionally decrease cAMP
← Class III
mGluR4 and mGluR6-8
functionally decrease cAMP
←

← Defining GluRs by their effectors
←← mGluR functional attributes
Involved in:
o perception of pain
o mood/affect, especially anxiety
o learning and/or memory
o blood flow / headache
Modulation of the activity of:
o Voltage-dependent ion channels
o Transmitter release
o Ca++-dependent ion channels (K+)
o Ligand-gated ion channels
←← Group I effector mechanisms
← Glu mgluR1/mGluR5 Gq PLC & PIP2 IP3 Ip3-R or DAG
PKC phosphorylation of ion channels
← Agonist (competitive)
Glu endogenous
Ibotenate
DHPG (3,5-dihydroxyphenylglycine)
← Antagonists (competitive)
CPG (carboxyphenylglycine; especially 4-CPG)
←← Group I agonist can increase excitability
There is no AHP after DHPG is added; spike-frequency
accommodation blocked and easily excited
But both pre and postsynaptic effects are observed
Ca++ hypothesis disproven
←← Group I agonist can
cause headaches
reduce transmitter release by reducing the Ca++ influx
← Group II, III effector mechanism
← Glu Group II, III Gi AC decrease cAMP decrease PKA
decrease

← Group II
agonists
o Glu
o NAAG (N-acetylaspartylglutamate)
Antagonist
o EthGlu (ethylGlu)
← Group III
Agonist
o Glu
o AP4
Antagonists
o MAP4
←← 3/12/09
← GABAbRsII
← GABA biosynthesis and metabolism
← Glu
← ↓ (GAD)
← GABA
← ↓ (GABA-T) *blocked by
GABAculine
← Succinate Semialdehyde
← ↓ (SSDH) *blocked by Valproate
← Succinate
←← Valproate strongly facilitates CNS inhibition
It facilitates the GAD enzyme and prevents the breakdown of GABA. This
increases the transfer from Glu Gaba. This causes inhibition!
←
← GABA Transporters (GAT) Pharmacology
VGAT (vesicular GABA transporter): GABA
o Nipecotate
o Vigabatrin
GAT1 (neuronal, glial): GABA
o Nipecotate
GAT2 (pia, arachnoid): GABA, Beta-alanine

o Nipecotate
GAT3 (neuronal, glial): GABA, Beta-alanine
o Nipecotate
←
← GABAergic synapse
Package gaba into vesicles ant release it onto Gaba receptors that
generate ipsps. Presynaptically there was an action potential.
GABABR are found posynaptically and presynaptically. There are
differences in their interaction tho. The gaba can be transported
back presynaptically and it can be transported into mitochondria or
broken down they can also be broken down in glial cells.
←←← Two subunits: coexpression
GABA B receptors are obligate heterodimers: one GABA b1 and one
GABAb2 subunits are needed. One binds the g-protein and one binds to
the GABA.
Gaba b1 and GABA b2 subunits exist and coexpressed in the same cells and
regions and rate.
If you only express Gaba b1 then they are stuck in the endoplasmic membrane
and doesn’t make it to the plasma bembrane. If only gabab2 are expressed then
the GABA doesn’t interact with the receptor.
←
← GABAbR-effector coupling
GABA binds to one of the two components of the dimmer and
activating the g protein on the other part of the dimmer.
The Gi/o family of proteins;
o The α subunit ↓ the AC
o The βγ subunit ↓ VDCCs (blocking depolarization) and ↑ the
activity of VDKC (decreases the resting potential).
PTX (pertussis toxin blocks the Gi/o Proteins).
←← GABA BR ligands
AGOo GABA (endogenous)
o Baclofen (exogenous antispasmatic drug)

ANTo Phaclofen
o 2-OHsaclofen
←
← Effects of divalent cations on GABA BR ligands binding Some enhance GABA binding
o Mn2+ = NI2+ >Mg2+ >Ca2+
o Physiological [Mg2+] or [Ca2+
Some inhibit GABA binding
o Hg 2+ > Pb2+
Phosophorylation of GABABRs enhances βγ coupling to GIRKs; enhanced
dissociation of βγ from Rs. The phosphorylation of the dimmer causes the G
protein to dissociate.
←← GABABR block of VDCC via Go βγ subunits
Antibody for βγ -Go will block the affect of baclofen
←
← Presynaptic GABABR decrease postsynaptic GABA IPSPs GABA will inhibits it’s own release with autoreceptor effect. If you block the
presynaptic GABABR Gi paired pulse suppression/depression.
←
← GABABR activates slow IPSC Slow outward K+ current: GIRK (more than ½ seconds) found postsynaptically in
neurons
3/24/09
Cholinergic receptors mAChRs
Diffuse central cholinergic systems – GABA is released from these systems
after activation from the
LTN = laterodorsal tegmental n.
NDB = n. diagonal band
MS = medical septum
H = Habenula

NBM – N Basilis of Meynert
ACh is involved blood pressure regulation and
Biosynthesis: ChAT (choline acetyl transferase) positive cholinergic neurons
Degradation: Multiple isoforms of AChE (Aceylcholinesterase) **it is not taken
up by glial cells or reuptaken. It is degraded in the synapse.
ACh biosynthesis and metabolism
Acetyl CoA and Choline are the substrates and ChAT is the enzyme which
catalyzes the ACh and CoA (products). ACh in the synapse is broken down
by AChE into Acetate and Choline
AChE ligands I: OrganophosphatesAChE ligands I: Organophosphates – the ACh is left inside the synapse and
the AChE isn’t able to do its job.
Ligand Duration Use (dose dependant)
Long Irreversible Insecticide WMD
Sarin [GB] X X
DDT X X
Diazinon X X
Malathion X X
AChE Ligands II: Clinical AChE drugs
Inhibitor Duration
Short Long
Donepezil (Aricept) X
Physostigmine X
Regenerator of AChE
activity
Duration
Short Long
Obidoxime X

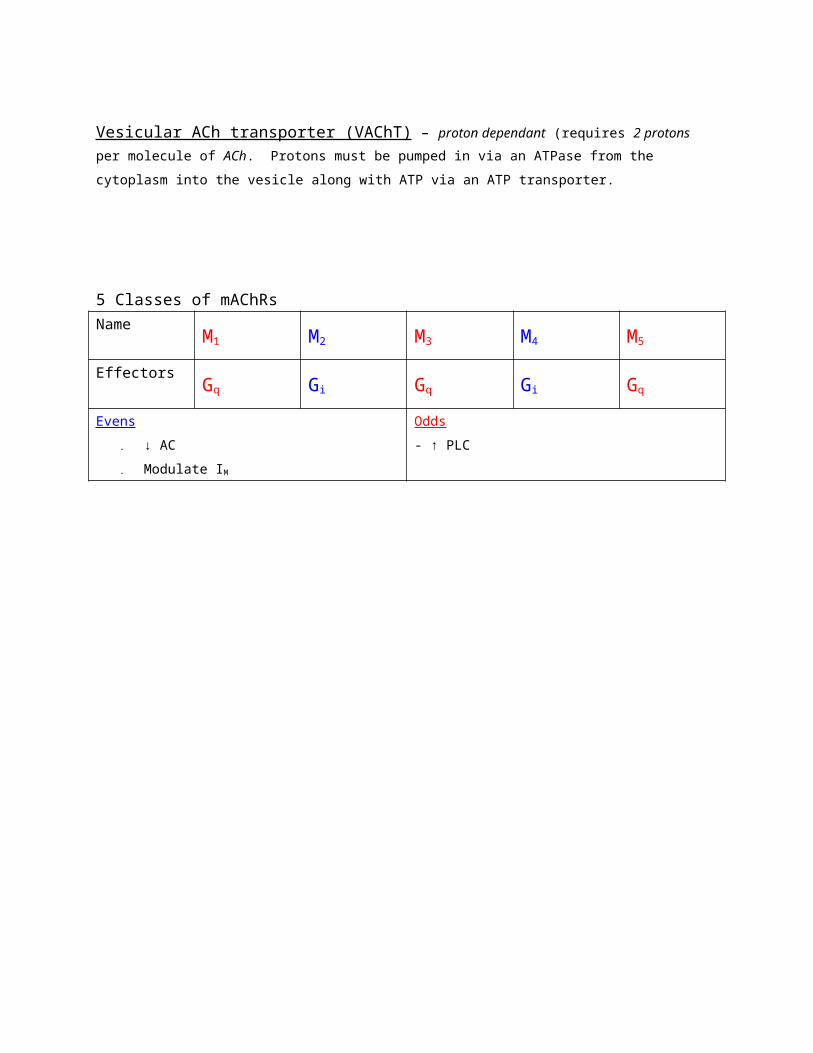
Vesicular ACh transporter (VAChT) – proton dependant (requires 2 protons per
molecule of ACh. Protons must be pumped in via an ATPase from the cytoplasm into the
vesicle along with ATP via an ATP transporter.
5 Classes of mAChRs
Name M1 M2 M3 M4 M5
Effectors Gq Gi Gq Gi Gq
Evens
- ↓ AC
- Modulate IM
Odds
- ↑ PLC

Uses/abuses of mAChR ligands
- Clinicalo reduce secretions (atropine)
o prevent motion sickness (scopolamine)
o treat Alzheimer’s (Donepezil)
o treat autonomic disorders (M3 ANT)
o dilate pupils for ophthalmic exams (M3 ANT)
o treat severe diarrhea (M3 ANT)
o slow heart rate (M2 AGO)
o treat mushroom/anti-AChE poisoning (atropine)
- Non-clinicalo dilate pupils for beauty (atropine)
*Newly discovered –allosteric modulation
- Brucine: enhance muscarinic agonist binding
mAChR Agonists
LIGAND Receptors
M1 M2 M3 M4 M5
Acetylcholine
(ACh)
X X X X X
Carbachol X X X X X
Muscarine X X X X X
Oxotremorine X X X X X
Pilocarpin E X
mAChR Antagonists
LIGAND Receptors
M1 M2 M3 M4 M5
Atropine E X X X X X
Scopolamine X X X X X
Pirenzepine X
Gallamine X
4-DAMP X
Tropicamide X

Smooth muscle contraction via m3 mAChRs via IP3R mediated
Ca2+ nM muscarine
Extremely slow IPSPs via m2AChRs
Onset 30-90 s after mAChR ligand binding; amplitude 2-3 x normal AHP;
GIRKs: Gi βϒ subunits
After-depolarization; Ca2+ spikes (m1,3 mAChRs)
Activating T-Type Ca2+ channels
Slow depolarization: Gq βϒ subunits
Block of HVA VDCCs by M1, 3 Rs
Mediated by Gq α subunits
Im block by muscarinic agonists
Gq α subunits
Block of sAHP by m1, 5 AChRs
Anti-AChEs raise synaptic ACh concentration
Anti – AchEs block “normal” slow AHPs via mAChRs (metrifonate
Effects of chronic anti AChE treatments linger in vitro
3/26/09
Central DA minergic pathways
MFB – medial forbrain bundle (path from the NS and VTA to the rest of brain)
SN substantia nigra – basal ganglia
VTA = ventral tegmental area – the rest of the brain
Catecholamine biosynthesis

Tyr (tyrosine crosses BBB) TH (tyrosine hydroxylase) DOPA AAAKC
(alpha amino acid decarboxylase) or DOPA decarboxylase (DDC)
Dopamine (in the VTA and SN this is the end) but others continue using
dopamine Beta hydroxylase norepinephrine (adonergic) in adrenal
cortex it is converted into Epi (epinephrine) via PNMT
TH: The rate limiting enzyme
- it has several amino acids that can be phosphorylated. Cam-KII, PKA, PKC, ERK, and cdc-
like-K. PHosphorylating increases activity and phosphotases decrease activity.
LDOPA when used to treat Parkinson’s it is used with Carbidopa (blocks TH)
so that tyr isn’t converted. Carbidopa doesn’t cross BBB. LDOPA is
transported into the brain slowly.
Transmitters vs false transmitters
L-DOPA is converted into DA via AAADC. False neurotransmitter (alpha-
methyl-DA) is created by the same enzyme. DbetaH converts it into alpha
methyl NE
DA degradation
DA
can be converted into 3-methoxy-tyramine via COMT. MAO converts 3-
methoxy-tyramine into HVA (homovanillate). DA can also be converted into
DOPAC via MAO + aldehyde deOH. COMT converts DOPAC into HVA.
Uses and abuses of DAminergic ligands
- appetite suppression
- antidepressants
- antipsychotics (including treatment of schizophrenia)
- fatigue suppression
- enhance attention
- treating motor dysfunctions (parkinsons)
- pressor effects
Recreational
- fatigue suppression

- enhanced attention
- mania mimicry
- mild hallucinogenic effects
Biogenic amine hypotheses of affect/psychosis
As you age your biogenic amine are reduced… may lead to depression. All
three seratonin, dopamine, norepinephrine are involved in mood, emotion.
And cognitive function
VTA frontal ctx – hypo (negative symptoms)
VTA Nucleus accumbens – hyper (positive symptoms)
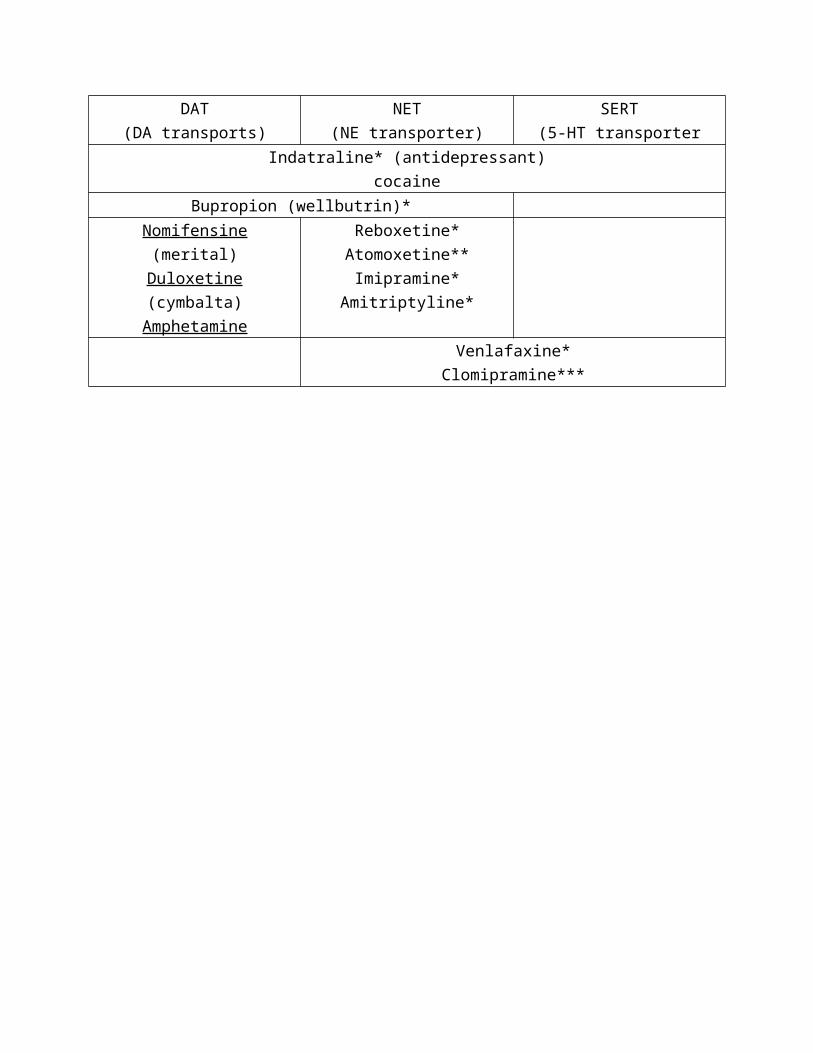
Transporters; vesicular DAT (VDAT)
12 transmembrane segments and proton-dependent transport. Only
requires one proton (ATPase) and the ATP site is outside the vesicle so no
ATP import is required.
Vesicular Transport inhibitors (block filling of all biogenic amines)
- Reserpine – most widely used
Synaptic DAT (DA back into the cell)
12 transmembrane protein. Na/CL-dependent transport; Oubain indirectly
inhibits it, by blocking Na+/K+-ATPases.
Reuptake inhibitors

DAT
(DA transports)
NET
(NE transporter)
SERT
(5-HT transporter
Indatraline* (antidepressant)
cocaine
Bupropion (wellbutrin)*
Nomifensine (merital)
Duloxetine (cymbalta)
Amphetamine
Reboxetine*
Atomoxetine**
Imipramine*
Amitriptyline*
Venlafaxine*
Clomipramine***

*antidepressants
** ADHD
*** anxiolytics
Amphetamine and analogs
Amphetamine, detroamphetamine, methamphetamine
Analogs – methylphenidate (Ritalin), cocaine, phenmetrazine, MDMA
ADD works to block the reuptake of DA in the prefrontal ctx (inhibiting
forbrain).
Amphetamines increase synaptic DA
Block reuptake in 2 ways
1. compete for DAT
2. upregulate MAP-K phosphorylate DAT internalize DAT
DA-specific neurotoxins
MPTP glial MAO-B MPP+ (shuts down kreb cycle in mitochondria)
6-OH-DA
1. Both MPP and 6OH DA are substrates for DAT
2. Both are then taken into mitochondria
3. Both block oxidative metabolism
4. Both kill DA neurons selectively and dose-dependently
5. Both produce behavioral effects mimicking parkinsonism
DARs are diverse
7 transmembrane segments; mono-, di-, and heterodi-meric (s. SST
(somatostatin), etc)
DAR types
Name D1 D2 D3 D4 D5
Synaptic
location
Post- Pre- and
Post-
Post- Post- Post-
Alpha
subunit
effector
Gs Gi Gs
Beta
gamma
subunits
effects
Decrease
VDCC,
VDKC,
CDKC
The ctx has D1-D5
Hypothalamus has D3 + D5
Corpus striatum has D1 + D2
DA agonists
LigandDA receptor type
Other catecholamine
receptors
D1 D2 D3 D4 D5 Alpha beta
Dopamine X X X X X X
Apomorphine X X X X X
Dihydrexidine XX X
bromocriptin
e
X X X
Praminpexole X XX
Ropinirole X XX

DAR antagonists
Ligand DA receptor type
D1 D2 D3 D4 D5
Spiperone XXX XXX XX XX XX
Ecopipam X
Amisulpride XXX X
Chlorpromazine* XX X X
Trifluoperazine* XX X X
Haloperidol* XX X X
Resperidone* XX X X

out patient drugs
Neuroleptics vs. tricyclic antidepressants vs. Li+
Tardive dyskinesia – intermittent inability to move. The receptors increases
and then they can’t create more. They are taken off neuroleptic drugs they
return to the drugs after symptoms of origional problem comes back.
3/31/09
Catecholamines – Norepinephrine (NE)
Adreneric projections in the CNS
LC = Locus coeruleus - cerebellum
A1-A9 = reticular adrenergic nuclei – regulatory on the hypothalamus
Methods to alter transmission**- Alter synthesis
o facilitate, block, false transmitters
- Block vesicular storage
- Block release == Ca2+ blockers, etc.
- Direct receptor effects
o agonists, antagonists, suicide antagonists
- Block reuptake
- Block degradation
Uses/abuses of NE ligands
- Clinicalo Cardiac regulation (rhythm, rate, volume)
o Antihypertensive
o Bronchial dilation (asthma relief)
o Anti-depressant
o Anti-ADHD/ADD
o Anti-OCD
o Decongestant
- Recreationalo Hallucinogenic

o Amphetamine-like effects
Release blockers indirectly effect VNETs (vesicular norepinephrine transporters)
- block Mg2+-ATPase proton pumps specific to NE neurons
- do not cross BBB; anti-hypertensive (guan-), block arrhythmias
(bretylim)
- Sympathetic
- It is proton dependant
Antidepressants, ADHD, anziolytics, etc (SEE ABOVE CHART)
DAT
(DA transports)
NET
(NE transporter)
SERT
(5-HT transporter
Degradation inhibitors (-Is)
Enzyme MAO-I COMT-I
A-specific
(NE, 5-HT)
A- or B-
(nonspecific)
B- specific
(DA, HA)
Inhibitor Reversible Irreversible
Befloxaton
e
Harmaline Iproniazid Deprenyl tocapone

MAO-Is are still prescribed as antidepressants; deprenyl in phase III for
Alzheimer’s
Classes of adrenoceptors (NE, EPI)
- alpha
o α1: smooth muscle contraction - Gq
o α2: smooth muscle contraction – Gi (inhibiting neurotransmitter
release)
- beta: cardiac muscle contraction - Gs
Alpha 1 ( α 1) Adrenoceptors
Nameα 1A α 1B
α 1D
Expressed in:
(don’t need to know)
CNS, heart, liver,
urogential smooth
muscle
Resistance vessels,
kidney, spleen
CNS, aorta, lung,
bladder
Non-selective AGO DA
NE
EPI
Phenylephrine
Ephedrine, Pseudoephedrine
Selective AGO Tetrahydrozoline
Selective ANT
(don’t need to know)
Niguldipine
5-methylurapadil
Cycloazosin
Spiperone
Non-selective ANT Phentolamine
Prazosin
Suicide ANT Phenoxybenzamine
G-protein Gq - ↑ IP3/DAG

Decongestants, eye drops (α 1 AGO)
Ephedrine
Pseudoephedrine
Phenylephrine (nyquil)
Tetrahydrozoline (eyedrops)
Alpha 2 ( α 2) Adrenoceptors
Name α2A α2B α2C
Expressed in: CNS, Lung, vascular
muscle
Thalamus, liver,
vascular, spleen
CNS, lung
Function Autoreceptor;
Vasoconstriction,
sedation, analgesia,
anesthesia
Vasoconstriction Undeterimined
Non-selective AGO NE
EPI
Clonidine
Selective partial
AGO
LSD-25
Nonselective ANT Phentolamine
Prazosin
Yohimbine
α -subunit Gi
β-ϒ subunits Inhibit VDCCs, activate CDKCs

Antihypertensive vasodilators (alpha ANT)
Phenylethylamine hallucinogen relatives of NE- LSD
- cathine
- ephedrine
- yohimbine
- mescaline
- elimicin
Beta (β) Adrenoceptors
Name β 1 β 2 β3
Expressed in: Neuronal cardiac,
kidney
Neuronal, cardiac,
bronchial
Neuronal, cardiac,
adipose
Non-Selective AGO NE
EPI
Isoproterenol
Ephedrine
Selective AGO Xamoterol Albuterol
Salmeterol
Selective ANT Atenolol Butozamine
Non-Selective ANT Propranolol
G-protein Gs - ↑ cAMP

Sympathetic effects on cardiovascular system
- Increases VDCC (HVA) and increases VDKC (IK) and increases VDCC
(LVA) resulting in each heart beat is going be faster and stronger. Bigger
volume of blood pumped harder each time. (acute effects)
- The higher blood pressure sustained causes issues also.
Antihypertensives (β1 ANT)Atenolol – used to lower blood pressure.
**notice the suffix -olol
Brochodilators (β2 AGO)Albuterol
Salmeterol
**notice the suffix –terol
4/2/09
Indolaminergic transmitters – Serotonin (5-HT)
Peripheral 5HT
- first purified from blood plasma (serotonin): 1948 and first purified
from the brain in 1957
- total quantity of the body < 10 mg (5% of that in brain)
- Intestines 0 enterochromaffin cells enhance motility of 5HT4
- Blood vessels – platelets; vasoconstriction: 5HT1D
- Also a mitogen: enhances cell proliferation in muscle, liver but not in
the bone
5HT pathways: Raphe n. which are widely distributed from the pons down
the brainstem and project throughout the rest of the brain. The largest are
the dorsal and median Raphe n. They are pyramidal neurons and they have
afterhyperpolarization (modulated by apamine). These neurons are
important in the sleep/wake cycle. They are almost completely silent when
sleeping and active during waking.
Hertogeneous 5HT fiber systems

- Doral Raphe n. has diffuse projections
- Median Raphe n. has specific projections
Serotonin (5HT) synthesis and metabolism
Compound Enzyme Inhibitors
L-tryptophan
↓
Tryptophan
hydroxylase
(TrpH)
Fenfluramine
L-tryptamine (Tra)
↓
α aromatic amino
acid decarboxylase
(DDC or AAADC)
Carbidopa
5hydroxytryptamine (5HT)
↓Monoamine oxidase
(MAO-A)
MAO-A inhibitors
5-hydroxyindoleacetaldehyde
↓Aldehyde
dehydrogenase (AD)
5-hydroxyindoleacetate (5-HIAA)

MAO-A doesn’t have A’s in abr. (NE, 5HT)
MAO-B has the A’s (DA, HA)
SERTs - ATPase pumps protons into the vesicle
- SERTs pump 5HT and Na+ & Cl- into the vesicle and 5HT and K+ outside the vesicle
SSRIs (antidepressants and anti OCD)
DAT NET SERT
Indatraline
cocaine
Bupropion (wellbutrin)
Venlafazine (Effexor)
Clomipramine (anafranil)
See other chart See other chart Fluoxetine (frozac)
Sertraline (Zoloft)
Citalopram (Celeza)
Paroxetine (Paxil)
Fluvoxamine (Luvox)

5HT1Rs
5-HT1A 5-HT1B 5-HT1D 5-HT1e 5-HT1f
Tissue Neuronal; smooth
muscle
Rodent neuronal
autoreceptor;
smooth muscle
constriction
Human
neuronal
autoreceptor;
smooth
muscle
constriction
Widely
distributed
Ubiquitous
agonist
5-HT
quipazine
Selective
full
agonists
Buspirone
DMT
(dimetheyltryptamine)
Ergotamine (mold)
sumatriptan
Selective
antagonist
s
Spiperone isamoltane
Antagonist pindolol
G-Protein G i/o

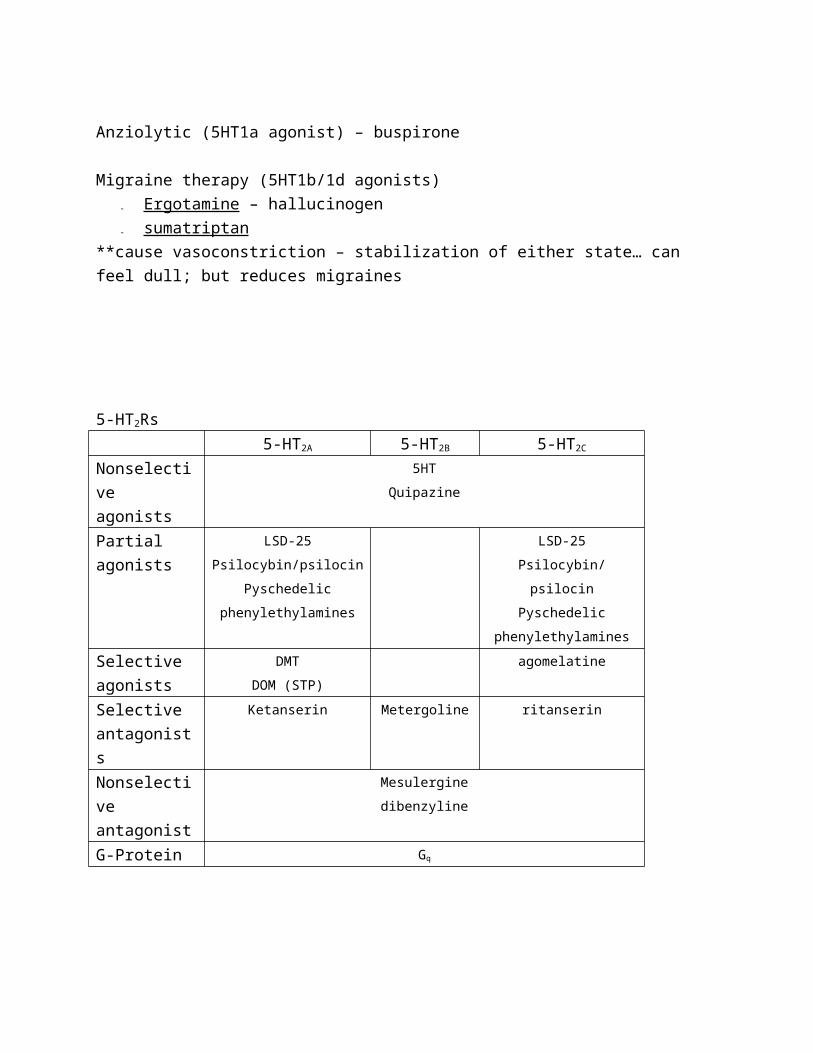
Anziolytic (5HT1a agonist) – buspirone
Migraine therapy (5HT1b/1d agonists)
- Ergotamine – hallucinogen
- sumatriptan
**cause vasoconstriction – stabilization of either state… can feel dull; but
reduces migraines
5-HT2Rs
5-HT2A 5-HT2B 5-HT2C
Nonselectiv
e agonists
5HT
Quipazine
Partial
agonists
LSD-25
Psilocybin/psilocin
Pyschedelic
phenylethylamines
LSD-25
Psilocybin/psilocin
Pyschedelic
phenylethylamines
Selective
agonists
DMT
DOM (STP)
agomelatine
Selective
antagonists
Ketanserin Metergoline ritanserin
Nonselectiv
e antagonist
Mesulergine
dibenzyline
G-Protein Gq

Psychedelic effects mediated by 5HT2ARs
- competitive antagonists do not block psychedelic effects
Indolaminergic psychedelics (50HT2a agonists)
- DMT
- Psilocybin
- LSD-25
Hallucinogens bind promiscuously
- alpha 2 receptors – assess using tritrated alpha 1 and alpha 2
antagonists
- 5ht receptors assessed using titrated 5ht1 and 5ht2 antagonis
- D2 receptors assessed using tritiated D1 and D2 antagonist
- LSD receptors assessed using titrated LSD
Clinical use of 5-HT2 ligands
- anti-schizophrenic (5HT2a antagonists)
- satiety stimulants (5HT2c agonists)
5HT3Rs
5HT3Rs
Nonselective agonists 5HT
Quipazine
Selective agonists CBG
Selective antagonists Ondansetron (keeps you from
vomiting)
Zacopride
Mechansism Petameric cation channel (weak
selectivity for Na+)
Antiemetics (5HT3 antagonists) – used extensively to alleviate nausea in
chemotherapy
- ondansetron
- zacopride
- also used in IBS treatment
Nootropics (5HT3 antagonists) – the problems is they don’t cross the BBB
and would cost $100/dose/day to take enough drugs to cause the nootropic
effects.
5HT4-like receptros
5HT4 5HT6 5HT7
Nonselective
agonists
5HT quipazine
Selective AGO EMDT COAT
Selective ANT amoxipine
Nonselective
antagonists
clozapine
G-protein Gs
5HT5Rs
5-ht5a 5ht5b
Nonselective
agonists
5HT
LSD

Melatonin
Serotonin 5HTNacetylase Nacetyl-5HT 5-OH-indole-OMT melotonin
(pineal gland) *just know that it is serotonin synthesizes melatonin in pineal
5HT specific neurotoxin
- PCPA: highly specific for 5HT neurons similar in mechanism to MPP+
REVIEW PERIOD
Synthesis and metabolism of the neurotransmitters
mGluR
Get the information off the website
- Three classes
o Class 1: mglu r1 mGluR5
o Class II: mGluR2 m GluR3
o Class II: mGlu
o Class I increase IP3 and Ca+
o Class II and III decrease cAMP
o Involved in perception of pain, affect and mood, learning and
memory, modulating activity of voltage dependent ion channels,
modulating
Glutamate metabolism (website information)
L-glutamine glutaminase lGlutamate (mitochondrial) alpha …
Don’t need to know the cofactors/substrates or inhibitors
** maybe aminooxyacetic acid as an inhibitor because it’s useful at different
sites
GABA Transporter Blockers: Nipecotate blocks all VGAT, GAT1, GAT2, GAT3
Vigabatrin blocks VGAT
ACHC
Dopamine

- reuptake inhibitors (prescribed as antidepressants)
o buproprion
o nomifensine
o indatreline
o mazindol
o amphetamine
- amphetamines and analogs
o amphetamines
o dextroamphetamine
o methamphetamine
o methylphendate
o Cocaine
o Phenmetrazine
- DA specific neurotoxins
o MPTP
o 6-OH-DA
Norepinephrine
- major source of NE in the brain – locus coeruleus
- peripheral release blockers
o quanathedine, guanadrel, bretylium
o CNS Ganglionic synapse NMJ
o Sympathetic NS
- Cental release blockers (cross BBB)
o Guanathedine, bretylium
- NE classes of receptors
o Alpha 1 accitvates Gq which increases PLC
o Alpha 2 – gi decresase cAMP
o Beta1 and beta 2 Gs increase cAMP
- phynylethylamine hallucinogenic relative
o cathrine, ephedrine, amphetamine, yohimbine, LSD-25,
Mescaline, MMDA
- NET blockers
o Atomexetine
o Desipramine

o Nortriptyline
o Amitryptyline
o Indatrelin
- Degradation Inhibitors
o MAO-A specific (NE, 5HT)
Specific receptors and antagonists – Know the non-specific
Serotonin – synthesis and metabolism
- produced in the bidbrain and the pontine nuclei, major source : raphe
nuclei
- SERTs
o VSERTs- H dependent (ATPAse)
o SERT (blocked b SSRIs) = Na and Cl dependent
o Both block by fenfluramine
- 5HT1 – Gi
- 5HT2 – Gq
SSRIs
Antidepressants
Seratonin Hallucinogens
- DMT
- Psilboc
←← 4/7/09
← HistamineHistamine (last of the biogenic amines)←← Peripheral HA
← - Inflamation (in mast cells): H1Rs orgasm
allergy
congestion
← - Digestion: H2Rs gastrin

Parietal cells create gastric acid
← - Immune Response: H4Rs basophils
neutrophils
bone marrow
←
←← HAminergic neurons: transport, synthesis, degradationHAminergic neurons: transport, synthesis, degradation
L-amino acid transporter takes HA into the cell (AAAT)
Histidine is converted by histidine decarboxylase (HD) into histamine and
transported into vesicles via Vesicular MonoAmine Transporter (VMAT).
Histamine methyltransferase (HAMT) breaks down histamine into
telemethylhistamine
←
← CNS HA : Hypothalamic tuberomamillary nuclei* (TMN) project to the
rest of the body.← *(right above the mamilary bodies)
←← Tuberomamillary neurons (TMN
← - large soma, dendrites
← - long thin poorly myelinated punctate axons (slow, large number of
synapes
← - adjacent to VLH hypocretin (hypothalamus cretine neurons)
HA: Role in waking and sleep. The firing rate is high when you are awake and drops during SWS and almost turns
off during REM.
Antihistamines make you lethargic
Modafinil – increases HA release, increases alertness

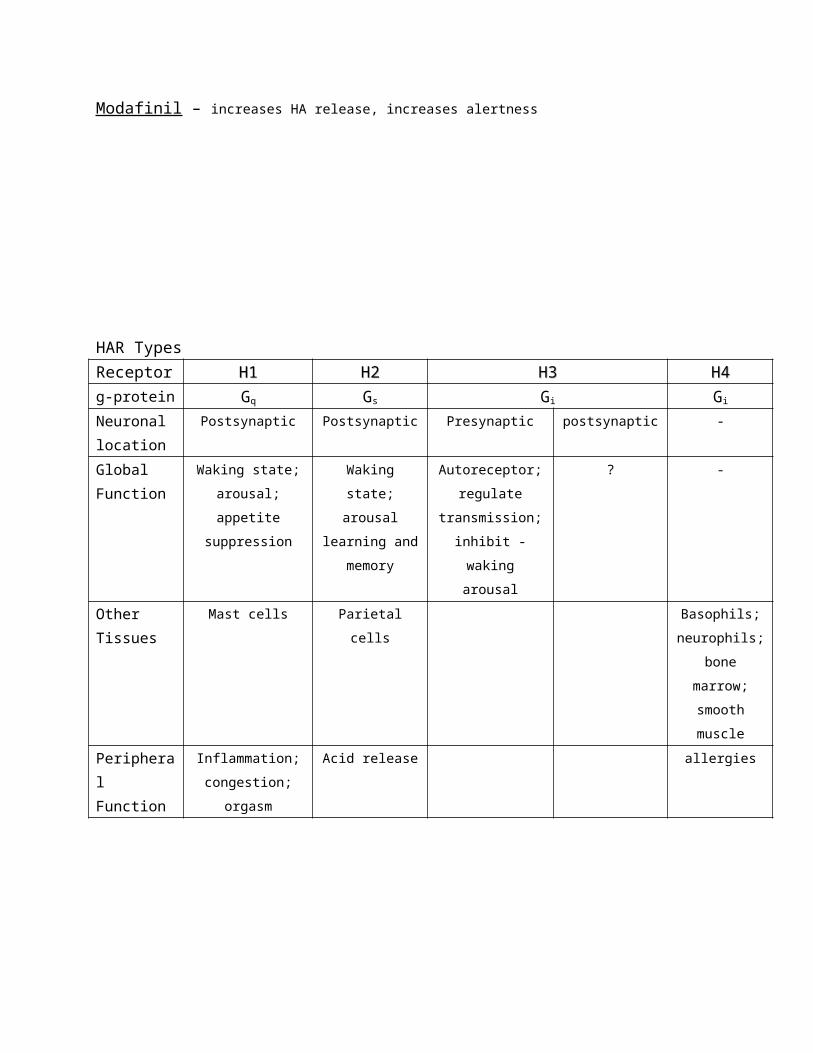
HAR Types
Receptor H1H1 H2H2 H3H3 H4H4
g-protein Gq Gs Gi Gi
Neuronal
location
Postsynaptic Postsynaptic Presynaptic postsynaptic -
Global
Function
Waking state;
arousal; appetite
suppression
Waking state;
arousal
learning and
memory
Autoreceptor;
regulate
transmission;
inhibit -waking
arousal
? -
Other
Tissues
Mast cells Parietal cells Basophils;
neurophils;
bone
marrow;
smooth
muscle
Peripheral
Function
Inflammation;
congestion;
orgasm
Acid release allergies

H3 is the only autoreceptor. - If you block the receptor you increase firing rate of the postsynaptic neuron.
- H3 presynaptic block of Voltage Dependent Ca Channels.
- An AGO will decrease the inward current through VDCCs.
- Autoreceptors act like this
Uses/potentials of HA ligands
- Peripheralo Allergy treatments (peripheral H1 ANT)
o Anti-congestive (peripheral H1 ANT)
o Anti-inflammatory (peripheral H1 ANT)
o Antacids (peripheral H2 ANT) *not really antiacids but go the problem of the
acid.
- Centralo Anti-emetics/anti-vertigo (H1 ANT)
o Hibernation, sleep (H1 ANT)
o Stimulants (H3 ANT)
o Alzheimer’s therapies (H3 ANT)
o Nootropics (H3 ANT)
o ADD therapies (H3 ANT)
o Anti-obesity treatments (H3 ANT)
HA Agonists
Competitive
AgonistHAR
H1H1 H2H2 H3H3 H4H4
Histamine (HA) X X X XDimaprit XProxyfan (partial) XImetit X X
Classic antihistamines: Competitive ANT
Competitive ANT HARH1 H2 H3 H4
Cetirizine (zyrtec) XClorpheniramine *** XDimenhydrinate (Dramamine) XDiphenhydramine (Benadryl) XDoxylamine (Unisom) XFexofenadine (Allegra) XLoratadine (Claritin) XMeclizine (Antivert) XPromethazine (phenergan) Xallergy meds
anti vestibular
Sleeping pills
Other HAR antagonistsOther HAR antagonists, inverse agonists
Competitive ANT HARH1 H2 H3 H4
Cimetidine (tagamet) XClobenpropit XInverse AGO ***Increases Neurotransmitter Release… similar to the
antagonist.
Thioperamide X
H2 agonists increase CA1 excitability
- If the H2 agonists is administered there is no afterhyperpolarization
H3 antagonists: nootropic and dietary benefits…. Every learning task the H3
has shown to enhance.
Exam… know the receptors, antagonist, non-competitive. Know the
mechanism of action. Know the clinical uses… digestion, asthma, and heart

rate information. Know the biosynthesis and the degradation of the
neurotransmitters and their enzyme and rate limiting…. Know the transports-
vesicular and cell membrane transporters. Know the drugs that blocks NETS
SERTS ECT.

4/8/09 7:39 PM
← 4/14/09
← Transcription factors etc.
← Steroid Hormones
←← Ganadal steroids and neural plasticity. –
← HRT hypotheses: for heart disease prevention: disproven;
← for osteoporosis (bone loss), “hot-flash” prevention: supported;
*possible breast cancer increase due to hormonal treatment
← for cognitive support (esp. in Alzheimer’s): uncertain *women have
higher rates of Alzheimers after menopause
←← Gonadal steroid biosynthesis
← - Cholesterol
← ↓
← - Progesterone
← ↓
← - Testosterone (testes in males, ovaries in females)
← ↓
← - Estradiol
←← Biosynthesis
← Testosterone →estradiol (E2) ⇔estrone (E1)
← ↓ (alpha-reductase) ↓
← DHT estriol (E3)
←← Hormonal regulation of estrus
← P. Pitutiary
← ↓ (GnRH) *gonadotropal releasing hormones
← Anterior Pituitary
← ↓ (FSH & LH) – the same time there is an increase in temp.
← Ovaries
← ↓ (Progesterone and Estradiol)
←← zGonadotropin hormone Rs (metabotropic and cytokine)
←

7- tansmembrane segments cytokine
receptors and homodimer- singl transmembrane segment
← Hormon
e
← GnRH ← FSH ← LH ← Prolacti
n
← Release
d by
← Posterior
pituitary
← Anterior
pituitary
← Anterior
pituitary
← Anterior
pituitary
← Recepto
rs expressed in
← Anterior
pituitary
← Egg
follicle
← Egg follicle,
testes
← Mammar
y glands
← Stimulat
es
← Producti
on of FSH, LH
← Maturati
on
← Corpus
luteum/spermatogenesis
← lactation
← Signalin
g
← Gq ← Gs← ← JAK/STAT
pathway

← JAK - Janus kinase
← STAT- Signal transduction and activator of transcription
Steroid receptors: - Intracellular, not membrane bound
- Hormones have receptors in the cytoplasm and then its translocated into the steroid
receptor complex to the nucleus. Binding of complex to DNA regulatory site.
Transcription then translation
1. Ligand binding
1.5 HSP (heat-shock protein) release
2. Translocation via nuclear pore complex
3. Dimerization and DNA binding
4. Gene transcription
5. Also includes RNA transport, insertion in ER, translocation into protein, and possible
post-translation protein modification.
Steroid Hormone Receptors

Receptor Gonadal Steroids Adrenal Steroids
Estrogen Androgen Progesterone Glucocorticoid Mineralocorticor
d
Endogenous
Agonists
Estradiol
Estriol
Estrone
Testosterone
DHT
Progesterone Cortisol
Corticosterone
aldosterone
Antagonists Tamoxifen Flutamide Mifepristone Spironolactone
Agonist
Effects
Regulate
hormone
secretion
Regulate
cholesterol
metabolism
Increase lipid
protein
synthesis
Breast tissue
stimulation
Maintain
neural
plasticity
Maintenance
of sex drive
Enhance bone
Ca2+ retention
Increase lipid
protein
synthesis
Maintain
neural
plasticity
Enhance bone
Ca2+ retention
Stabilize uterine
proliferation
Stabilize lipid
metabolism
Increase hepatic
gluconeogenesis
Increase protein
catabolism,
glycogenolysis
Increase BP
Increase HR
Immunosuppression
Na+ retention
/K_excretion by
kidney distal tubules
ERs: Estrogen receptors
Dimeric receptors: ERα ERβ
Most are heterodimers, widely distributed throughout, including the CNS
Agonists: Birth Control Pills/HRT
Formulation Generic Brand name
Estrogen only Estradiol Premarin
Combination monophasic Ethinyl estradiol Desogen
Progestin only Norethindrone Microanaar

Anti-estrogens
ER-antagonists – used in treatment of breast cancers to block proliferation of
estrogen-sensitive tumor cells
Tamoxifen
Clomiphene
Or aromatase inhibitor – anastrozole
Anti-progestins
Progesterone Rs antagonists are used as arbortifactants to disrupt
maintenance of uterine endometrial lining to block fertilized egg
implantation. Mifespristone (RU486) morning after pill prevents egg
implantation
Metabolites of Androgens
Active Inactive
DHT (another one too) Don’t need to know
Testosterone
Developmental regulation of testosterone
Plasma testosterone in the trimester during neonatal and then decreases
and then increases during first year of life. It goes way down and then
during puberty it increases until late adulthood.
Cellular effects of androgens
Pituitary (LH) Testes (testosterone) target cells
Target cells
- AR-A
o GsRH regulation
o Bone and muscle growth and anabolic effects
- AR-B
o Sermatogenisis
o Sexual differation and virgor
Anabolic steroids: androgens

o Exogenous anabolic steroids can increase skeletal (bone) and skeletal muscle
mass.
o They also increase blood pressure and cardiac muscle mass, which can lead to
rhythm disturbances and sudden cardiac arrest
o Excess anabolic steroid use may increase aggression, rage, linked to arrest for
violent, disruptive, or sexually violent behavior
o Anabolic steroids cause scalp hair loss
o Finally, anabolic steroids can trigger prostate and brain cancers
- testosterone
- DHT
- Oxandrolone
- Oxymetholone
Anti- Androgens
- competitive ANT
o flutamide
- alpha-recutase inhibitor
o finasteride
used for: treatment of sex offenders, prostate cancer, male-pattern baldness (vs minoxidil
rogaine K-ATPactivator)
Biosynthesis of adrenal steroids
Cholesterol progesterone cortisol
↓
corticosterone (smaller amt) aldosterone
Steroid synthesis inhibitors
Inhibitor Mechanism Macroscopic effects
aminoglutethimide
trilostane
ketocon

H-HPA axis and stress hormone regulation
Hippocampus (CA2 and CA3 pyramidal cells) release glu hypothalamus (paraventricular
N) posterior pituitary (neural lobe) release CRF onto the anterior pitutaty
(corticotropes) ACTH is released onto the adrenal cortex (fasiculata cells) releasing
corticosteroids which circulate causing the following:
- GREs (glucocorticoid response elements )
- Immunse suppression
- Increase HR
- Increase BP
- Increase catabolism
- OR feedback onto the hippocampus to shut the cycle down
Dual cleavage of POMC to yield ACTH
- two peptidases are cleaved via endopeptidase and caroxypeptidase
Adrenal (endogenous) steroids
Gona glomerulosa – aldosterone
Zona glaciculata – ACTH and cortisol
adrenal steroid secretion rates
Type Steroid Rate/day
Corticosteroid Cortisol 10-20 mg
Corticosterone 2-4

Clinical (exogenous) corticosteroids
- betamethasone
- dexamethasone
- hydrocortisone (synthetic cortisol)
- prednisone
Plasma protein binding affects corticosteroids
- over 9/10ths get to the target… the rest is bound to the plasma binding protein.
Major clinical corticosteroid uses
Corticosteroids and mineralocorticoid Rs
Cortisone Cortisol Aldosterone
↓X ↓binds ↓binds
mineralocorticoid receptor
4/21/09
CNS depressants: EtOH and other anesthetics
DOSE: for governemtn purposes, 1 “serving” of ethanol ~1ox. Of 80proof (40% EtOH by vol.)
Structure of common alcohols
Products of carbohydrate fermentation. We can only take in ethanol.
Differential distribution and clearance of EtOH
When taking in ethanol oralsly it goes through the gus portal vein vena cava, arteries,
peripheral veins
EtOH degradation in CNS
EtOH is broken down by alcohol dehydrogenase (AD) and NAD and H+ NADH

Acetaldehyde is carried into the mitochondria and aldehyde dehydrogenase (ALD) turns it
into acetic acid. Acetic acid turns into H20 and CO2
EtOH can alter DA metabolism, producing “opioids”
DA will take an alternative pathway and condencse into tetrahydropapaveroline (HTP). THP
will create opioids. This is due to aldehyde dehydrogenase (normally used in DA
degredation) being used to degrade EtOH
Acute effects of EtOH
- Initial , low dose (1-2 oz. EtOH/75kg)
o Stimulatnt; social lubricant” due to lowering of cognitive inhibitions; slight
motor impairment
- Laster, high does (>4 ox EtOH/75 kg)
o Depressant; obliteration of cognitive inhibitionas; servere cognitive and motor
impairments; aggression; emetic reflex; sedation
- Next day (after high dose)
o Hangover: mixture of sympathetic a tivation and dysfunction (nincreased BP,
dry mouth, etc)
Headache, irritiability common after
Chronic Effects of EtOH
Effect Probable mechanism
Sedation
Increased seizure threshold
Anterograde amnesia
BDP independent enhancement of GABA-Ar
function
Euphoria
Memory impairment
Analgesia
Sedation
Delta

Alcoholism
- tolerance tends to develop rapidly
- dependence tends to develop more slowly
- well defined genetics links (dependence prone)
- gradual development of reverse tolerance (impaired metabolism)(
- multiple motor effects, some of which habituate
- Korsakoffs, other vitamin deficiency effects
- Social issues, socieoeconomic outcasting
- Societal role in definitions of “problem dringking, binge drinking partying
- Recidivism high, long-term prognosis poor
Rating depth of anesthesia
Volatile (taken in through the lungs) Anesthetics
Nitrous oxide
Diethyl ether
Chloroform – liver toxicity
Isoflurane – commonly used
Sevolurane –
Instead of giving an injection (which goes to the liver via metabolism) it goes through the
lungs then other systems then the liver.
Old “membrane fluidity” theories – they thought the channels changed shape of all the
proteins
Tandem-pore K+ channels
Name Tamdem poore
Ca 2 sensitivity Insensitive
Ligand seneitivie ?
Activation -60mV/-40mV
Inactiavition Slow (s)
Actiavatiors Isoflurane
Sevoflurane
Blockers Unknown

Hyperbaric Oxygen as an anesthetic, toxin
Dissociative anesthetics – feeling of being detached from the physical self
- ketamine
- phencyclidine (PCP)
- dextromethorphan
Sigma Opiate effects
Name Sigma
Classical 1 2
Ligands Haloperidol
Detamine
PCP
Pentazocine
Haloperidol Haloperidol
Mechanism Unknown Unknown unknown

Neuroleptics (used to treat schizophrenia) Underlined D2 blocker
Weighing the risks, benefits of anesthesia
- benefits
o allows complex, painful surgical technique performance
o allows development of procedures (eg killing the patient during heart surgery)
that would otherwise be impossible
o gives surgeons time to be careful in procedures
- Risks
o Some patients fail to recovers
o Constant monitoring required, particularly to avai anoxia \\
o Illicit use of anesthetics increases life-threatening risks
- Balance
o Valuable ligands, absolutely required by modern medicine
o Life-threatining nature of these ligns requreiesnd
4/23/09
NSAIDs, peptides and alkaloids – opiates, opioids, and other analgesics
Next Thursday is the review for the final exam.
Analgesics (“pain-killers”)
- Analgesia = relief of pain
o Surgical
o Acute
o Chronic
o Phantom
- Analgesic formulations
o Short-acting
o Intermediate
o Long-acting
1897 Bayer Ad
NSAIDs (non-steroidal anti-inflammatory drugs)
Cyclooxygenase inhibitors (COX-inhibitors)
- salicylates:

o Aspirin (bayer) **aids in platelet breakup
- Traditional NSAIDs:
o Acetaminophen (Tylenol)
o Ibuprofen (Advil)
o Naproxen (Alleve)
COX-1 – required for normal gastric function
COX-2 – active in inflammatory actions, cardiac/cardiovascular function
- Celebrex (celecoxib) *joint pain, arthritis
Uses/abuses of opiate peptides
Clinical uses
- analgesia
- cough suppressant
- sedation
- prevention of withdrawal
Recreational uses
- analgesia
- Sedation
- Prevention of withdrawal
- Other psychotropic effects
Conserved sequences among cloned opiate Rs.

Receptor
Name
μ Δ Κ Nociceptin
Protein MOP1 MOP2 DOP1 DOP2 KOP1 KOP2 KOP3 NOP
End.
AGO
Endophins Enkephalins
Deltorphins
Dynophins Nociceptin
AGO
affects
Analgesia
Euphoria
Intestinal inhibition
Respiratory-depression
Analgesia
Intestinal inhibition
Analgesia
Dysphoria (unpleasant)
Supraspinal pain
Spinal/peripheral
analgesia
ANT
(specific)
Cyprodime Naltriben
ANT Naloxone
Naltrexone
Diprenophine
G protein Gi/Go

Stucture of endogenous ligands
POMC (proopiomelanocortinin) – precursor to the opiates
Exogenous “organic” opium alkaloids – from poppies
- Morphine
- Codeine
- Narcotine
- Papaverine
- Thebaine
Modified opiates- diacetylmorphine (heroin)
- hydromorphone (dilaudid)
- oxycodone (percodan, oxycontin)
- hydrocodone (vicodin)
Synthetic Opiates (piperidine analgesics)
- Buprenophrine (Buprenex); partial AGO, high affinity, long Kd
- Methadone (Dolophine)
- Propoxyphen (Darvon)
- Meperidine (Demerol)
- Fentanyl – regulated because
Opiate antagonists – **OD treatments**
- naloxone
- naltrexone
- diprenorphine
Side effects: instantaneous onset of severe withdrawal
Addiction/dependence- Physical
o Development of tolerance
o Other kinases upregulated to compensate for PKA suppression
o Rate of development differs by cellular phenotype
- Psychological

o Need for hedonic effects (which decline)
o Need to avoid withdrawal symptoms (increases)
o Sedation soothes anxieties re. addiction
Legality and illegality of opiate use- Schedule 1 (heroin)
o High abuse potential
o No clinical utility
- Schedule 2 (morphine, oxycodone, fentanyl)
o High abuse potential
o Clinical utility
o Prescription dispensing only, closely monitored
- Schedule 3 (hydrocodone, propoxyphene)
o Lower abuse potential
o Clinical utility
o Prescription only
Withdrawal- Early symptoms
o Watery eyes, runny noes
o Yawning, no appetite
o Anxiety, irritability
o Restlessness, insomnia
o Rapid increase of AC activity
- Longer duration, more severe symptoms
o Tremors
o Hallucinations
o Panic
o Chills and sweating
o Cramps
o Nausea, wretching
**High rate of recidivism
Methadone – agonist but does not have pleasurable effects like heroin

Overdose- Initial symptoms
o Slow, shallow breathing
o Cognitive impairment
o Constricted pupils
o Severe drowsiness
o Cold, clammy skin
- Life Threatening Symptoms
o Vomiting
o Loss of consciousness
o Convulsions
o Coma
o Severe respiratory depression anoxia
4/28/09
Drugs of abuse – illicit or illegal ligands
Major categories of controlled substances- Stimulants
o cocaine
o amphetamines
o designer drugs (ecstasy)
- Depressants
o Barbiturates/BDZs
o Opiates
o PCP/ketamine
- Hallucinogens
o Marijuana/hashish
o Peyote
o LSD
o PCP/ketamine
o Psilocybin
o Designer drugs
- Anabolic steroids
o Androgens

Sources of illicit drugs
- synthesis
o diversion from regulated sources
o extraction/modify/purify
- Nature
Drug Enforcement- Federal (sets the standards others follow) Under FBI
o DEA (justice): concentrates on trafficking (international and interstate), diversion,
elimination of foreign growers
o AFT (Treasury): concentrates on domestic production, growers, theoretical “revenue”
sources
- State
o DPS: involves all levels, including low-level dealers and users
- County
o Sheriff: all levels, including low-level dealers and users
- Local
o Police: all levels, including low-level dealers and users
o
Federal Drug Laws- Formal scheduling – The controlled substances Act (CSA;1970) places all substances which
were in some manner previously regulated under existing federal law into one of the five
schedules
1) Potential for abuse
2) Medical uses
3) Safety or dependence liability

Federal Drug Schedules
- Schedule I (heroin, LSD, marijuana, methagualone)
o High potential for abuse
o No currenly accepted medical use
o Lack of accepted safety for use
- Schedule II (morphine, cocaine, methadone, hydrcodone) **tightly regulated
prescriptions
o High potential for abuse
o Currently accepted medical use
o May lead to severe psychological or physical dependence
- Schedule III (anabolic steroids, codeine/hydrocodone, most barbiturates)
o Potential for abuse
o Currently accepted medical use
o Moderate or low physical dependence
- Schedule IV (ex BDZ and anxiolytics)
o Low potential for abuse
o Currently accepted medical use
o Limited physical or psychological dependence
- Schedule V (cough syrup with codeine
o Low potential for abuse
o Currently accepted medical use
o Limited physical or psychological dependence
Variability in enforcement I
- stimulants
o caffeine – none
o nicotine – no sales to minors, smoking bans
o cocaine – schedule II
o amphetamines – schedule II
o designer drugs – schedule I
- depressants
o EtOH – no sales to minors; driving prohibitions
o Opiates – schedule I, II, III, IV, or V
o Barbiturates – Schedule II, III, or IV
o BDZs – schedule IV

o Methaqualone – schedule I
- Hallucinogens
o Designer drugs – Schedule I
o LSD – Schedule I
o Pyote – Schedule I
o Psilocybin – Schedule I
o Marijuana – Schedule I
o PCP – Schedule I
o Ketamine – Schedule III
- Anabolic steroids – Schedule III
Stimulants
- health risks
o sudden arrythmias, etc. leading to cardiac failure risk of acute psychosis with
high doses/chronic use perceived subjective superiority in performance.
DOES NOT match objective measure
- Physical dependence
o Strong risk
o Once developed, physical withdrawal begins within hours after last does
o Withdrawal less severe than opiates
- Psychological dependence
o Stong risk
o Recidivism high for former users (as high or higher than for opiates)
Hallucinogens
- Health Risks
o Multiple reports of mental health problems (can unmask borderline or
developing problems)
o Bad trips, flashbacks
o Substantial risk of obtaining high dose/erroneous substance
- Physical dependence
o None substantiatesd
- Psychological dependence
o Can develop for piperidine dirivitives (PCP, ketamine, etc.) unlikely for others
- Footnote:

o Original medical model to manage hallucinogenic use suggested
recreational/spiritual clinics, with controlled set, setting and dose of known
compounds
o Cf. early works of Richard Alpert, Timothy Leary, Sashatchewan psychiatric
model, et al. treatment of alcoholism, etc.
Opiates
- Health risks
o Overdoes – potentially fatal coma, respiratory depression, emesis
o HIV and other serology – related risks of drug injection
o Malnutrition possible due to severe appetite suppression (coupled with GI
motility reduction)
o Issues related to contaminants/cuts/dose and purity
o Withdrawal related to categories below
- Physical dependence
o Strong risk
o Once developed, physical withdrawal begins within hours after last dose
- Psychological dependence
o Strong risk
o Recidivism high for former users
Other depressants (ETOH, BDZs, barbiturates)
- Health Risks
o Overdose – potentially fatal coma, respiratory depression, emesis (vomiting)
o Issues related to contaminants/cuts/dose and purity
- Physical dependence
o Strong risk
o Once developed, physical withdrawal begins within hours after last dose
o Withdrawal less severe than for opiates
o Psychological dependence
o Strong risk
o Recidivism high for former users (as high or higher than for opiates)
Cannabis (dronabinol is synthetic)
- Health risks
o Smoking related respiratory problems (can be more severe than those for
tobacco, need not be)
o Mild immunosuppression (stabilizes with chronic use)

o Problems associated with adulterants, herbicides, etc
- Physical dependence
o None substantiates
- Psychological dependence
o Varies tremendously by individual
- Potential clinical uses
o Antiemetic/nausea suppressant (chemotherapy)
o Relief of ocular pressure in glaucoma
o Anxiolytic
Cannaboid receptors
Receptor CB1 CB2
Tissue expressing CNS (hippocampus; ctx;
basal ganglia; cblm) rarer in
periphery ()lung; vascular
tissue; testis; uterus
Immune system cells
Endogenous AGO Anandamide
Selective AGO JWH-015
Non-selective AGO THC
Selective ANT SR 144528
Beta gamma subunints
G-Protein alpha subuning Gi

Incomplete list of other hallucinogenic drugs
Ergot derivatieves
- LSD (synthetic also from morning glories, baby wood rose seeds)
- DMT
- DET
Other “natural” hallucinogens:
- psilocybin – from psilocybe mushrooms
- mescaline (from peyote cacti)
- datura (mixure of atropine, harmaline, cscopolamine from Datura stramonium =
Jimson weed)
Hallucinogenic amphetamines:
- DMO= STP= dimethoxyamphetamine
- MDA
Anabolic steroids
- Health risks
o Immunosuppressant
o Cardiac hypertrophy, leading to cardiac failure
o Acceleration of male balding
o Induction of prostate cancer and other tumors
o Risk of acute psychosis with high doses/chronic use
- Physical dependence
o Not well studied; likely
- Psychological dependence
o Considerable risk
******Final Review******
voltage gated channel blockers
know drugs from last lectures
know the pharmakenetics
concentrate on the most recent drugs and the ones that have been recurrent. Know the
mechanisms from notes. I/V curves may come again.

Neuropharm Final Review
Steroid Hormones
Estrogens
- estradiol
- estriol
- estorone
Androgens
- testosterone
- DHT
Progesterone
- progesterone
Glucocorticoid
Know the estradiol metabolism!!
Gonadal steroid biosynthesis
Biosynthesis of adrenal steroids
Steroid hormones
- birth control pills
o ethinyl estraidol
o norethindrone
- Anti-Estrogens
Know the HHPA Axis
A

4/8/09 7:39 PM
←