nÖroblastoma hastalarinda kromozom … · 9q22, 13q14, 14q11, 14q24 ve 15q22 kromozom...
TRANSCRIPT

T.C. ÇUKUROVA ÜNİVERSİTESİ
SAĞLIK BİLİMLERİ ENSTİTÜSÜ TIBBİ BİYOLOJİ ANABİLİM DALI
NÖROBLASTOMA HASTALARINDA KROMOZOM
DÜZENSİZLİKLERİ, MYCN ve AURKA GEN DEĞİŞİKLİKLERİNİN
İNCELENMESİ
Biyolog Nihal İNANDIKLIOĞLU
YÜKSEK LİSANS TEZİ
DANIŞMANI
Prof. Dr. Osman DEMİRHAN
ADANA – 2010

T.C.
ÇUKUROVA ÜNİVERSİTESİ SAĞLIK BİLİMLERİ ENSTİTÜSÜ TIBBİ BİYOLOJİ ANABİLİM DALI
NÖROBLASTOMA HASTALARINDA KROMOZOM
DÜZENSİZLİKLERİ, MYCN ve AURKA GEN DEĞİŞİKLİKLERİNİN İNCELENMESİ
Biyolog Nihal İNANDIKLIOĞLU
YÜKSEK LİSANS TEZİ
DANIŞMANI
Prof. Dr. Osman DEMİRHAN
Bu tez, Çukurova üniversitesi Bilimsel Araştırma Projeleri Birimi
tarafından TF.2009YL10 nolu proje olarak desteklenmiştir.
Tez No:..............
ADANA – 2010

i
KABUL ve ONAY FORMU
Çukurova Üniversitesi Sağlık Bilimleri Enstitüsü
Tıbbi Biyoloji Anabilim Dalı Yüksek Lisans Programı çerçevesinde yürütülmüş olan NÖROBLASTOMA HASTALARINDA KROMOZOM DÜZENSİZLİKLERİ MYCN ve AURKA GEN DEĞİŞİKLİKLERİNİN İNCELENMESİ adlı çalışma, aşağıdaki jüri tarafından Yüksek Lisans tezi olarak kabul edilmiştir.
Tez Savunma Tarihi: 20/01/2010
İmza
Jüri Başkanı Prof. Dr. Osman DEMİRHAN
Çukurova Üniversitesi
İmza
Prof. Dr. Davut ALPTEKİN Çukurova Üniversitesi
İmza
Prof. Dr. Atila TANYELİ Çukurova Üniversitesi
Yukarıdaki tez, Yönetim Kurulunun ........................tarih ve ..........................
sayılı kararı ile kabul edilmiştir.
Prof. Dr. Halil KASAP
Enstitü Müdürü

ii
TEŞEKKÜR
Lisans üstü eğitimim boyunca bana yol gösteren ve her konuda yardımını
esirgemeyen, sorunların çözümlenmesinde çok önemli katkılar sağlayan ve beni
destekleyen saygıdeğer hocam ve tez danışmanım sayın Prof. Dr. Osman
DEMİRHAN'a saygı ve şükranlarımı arz ediyor ve kendileriyle çalışmanın benim
için büyük bir şans olduğunu belirtmek istiyorum.
Tez izleme jürisinde yer alan ve bu vesileyle de hem çalışmanın
şekillenmesinde hem de karşılaşılan sorunların giderilmesindeki katkılarından
dolayı saygıdeğer hocam sayın Prof. Dr. Atilla TANYELİ’ne ve aynı zamanda
Anabilim Dalı başkanımız sayın Prof.Dr.Davut Alptekin’e, çalışmalarımızı her
zaman destekleyen ve güler yüzünü esirgemeyen sayın Dr. Sema YILMAZ
başta olmak üzere Çukurova Üniversitesi Pediatrik Onkoloji Anabilim Dalı’nın
tüm çalışanlarına ve Anabilim Dalımızdaki değerli hocalarım sayın Prof. Dr.Halil
KASAP, Prof. Dr. Mülkiye KASAP, Doç. Dr. Ümit LÜLEYAP, Doç. Dr.Ayfer
PAZARBAŞI ve Öğr. Gör. Dr. Ali İrfan GÜZEL’e de saygı ve şükranlarımı arz
ediyorum.
Çalışmada istatistiksel analizlerdeki yardımlarından dolayı değerli
arkadaşım sayın Çağla Sarıtürk’e, laboratuar çalışmalarındaki yardım ve
desteklerinden dolayı sayın Araş. Gör. M. Bertan YILMAZ, Araş. Gör. Dr. İlker
GÜNEY, Erdal TUNÇ, Müzeyyen İZMİRLİ ve Sabriye KOCATÜRK SEL’e ve
bölümümüzde çalışanları sayın Ömer DEMİR, Nadir BUDAK ve Mustafa
BAŞDİNÇ’e de teşekkür ediyorum.
Ayrıca, bu tez çalışmasını Bilimsel Araştırma Projeleri Birimi kanalıyla
TF.2009YL10 nolu proje olarak destekleyen Çukurova Üniversitesi
Rektörlüğüne ve lisansüstü eğitimi koordine eden Sağlık Bilimleri Enstitüsü
Müdürlüğüne de teşekkür ediyorum.
Her zaman manevi destekçim olan değerli aileme sonsuz sevgilerimi
iletiyor ve onları kucaklıyorum.

iii
İÇİNDEKİLER
KABUL ve ONAY TEŞEKKÜR İÇİNDEKİLER ŞEKİLLER DİZİNİ ÇİZELGELER DİZİNİ SİMGELER ve KISALTMALAR DİZİNİ ÖZET ABSTRACT 1. GİRİŞ 2. GENEL BİLGİ
2.1. Epidemiyolojisi 2.2. Etyolojisi 2.3. Genetik
2.3.1. Allelik Fazlalık ve Onkogen Aktivasyonu 2.3.1.1. Ploidi 2.3.1.2. MYCN Amplifikasyonu 2.3.1.3. 17q’da Dengesiz Fazlalık 2.3.1.4. Diğer Onkogenler
2.3.2. Allelik Kayıplar ve Tümör Supresör Genler 2.3.2.1. 1p Delesyonu 2.3.2.2. 11q Delesyonu 2.3.2.3. Diğer Bölgeler 2.3.2.4. Spesifik Tümör Supresör Genler
2.3.3. Bazı Genlerin Ekspresyonundaki Anormal Değişimler 2.3.3.1. Nörotrofin Reseptörlerinin Ekspresyonları 2.3.3.2. Diğer Önemli Genlerin Ekspresyonları
2.4. Klinik Bulgular 2.4.1. Paraneoplastik Sendromlar
2.5. Evreleme 2.6. Tanı
2.6.1. Tümör Belirleyiciler 2.6.2. Radyolojik Tanı Yöntemleri 2.6.3. Sintigrafik Tanı Yöntemleri 2.6.4. Patolojik Değerlendirme
2.7. AURKA Geni 3. GEREÇ ve YÖNTEM
3.1. Araç ve Gereçler 3.1.1.Kimyasal Malzemeler 3.1.2. Aygıtlar
3.2. Örneklerin Sağlanması İçin Kullanılan Yöntemler 3.2.1. Sitogenetik Çalışmalar
3.2.1.1. Kan Kültürünün Yapılması 3.2.1.2. Preparatların GTG (G-bands by Tripsin using Giemsa)
Bantlama Yöntemiyle Boyanması 3.2.2. Moleküler Sitogenetik Çalışmalar
3.2.2.1. Fluorescence In Situ Hybridization (FISH) Yöntemi 3.2.2.2. FISH Değerlendirmesi
i ii iii v vi vii x xi 13 14 14 16 17 17 17 18 21 22 22 22 23 23 23 24 24 26 28 31 31 33 33 35 36 36 38 40 40 40 41 42 42 42 43
44 44 46

iv
3.2.3. İstatistiksel Analiz 4. BULGULAR
4.1. Sitogenetik Bulgular 4.1.1. Kan Kültürlerinden Elde Edilen Sitogenetik Bulgular
4.1.1.1. Kan Dokularında Saptanan Sitogenetik Bulgular 4.2. Moleküler Sitogenetik Bulgular
4.2.1. Parafin Dokulardan Elde Edilen Moleküler Sitogenetik Bulgular 5. TARTIŞMA
5.1. Sitogenetik 5.2. Moleküler sitogenetik
6. SONUÇLAR ve ÖNERİLER 7. KAYNAKLAR EKLER
EK-1 EK-2
ÖZGEÇMİŞ
46 47 49 49 49 66 66
68 68 73 75 77 83 83 83 84

v
ŞEKİLLER DİZİNİ
Şekil 2.1. Şekil 2.2. Şekil 2.3. Şekil 2.4. Şekil 2.5. Şekil 2.6. Şekil 2.7. Şekil 4.1. Şekil 4.2. Şekil 4.3.
N-myc’in diğer proteinlerle interaksiyonu. TrkA tirozin kinaz reseptörünün yolağı Sağ sürrenal Nöroblastom tümörü Arka mediastende Nöroblastom tümörü Nöroblastomda periorbital infiltrasyona bağlı “rakun gözü” görünümü Katekolamin metabolizması. Nöroblastomun Patolojik Sınıflandırması (International Neuroblastoma Pathology Committe-1999). Hasta grubunda gözlenen kromozom anomalilerinden bazılarının karyotip görüntüleri. Kontrol grubunda saptanan kromozom anomalilerinden bazıları. Hastaların AURKA (a) ve MYCN (b,c,d) genlerine ait FISH görüntüleri
22 25 29 30 31 34 37 57 60 67

vi
ÇİZELGELER DİZİNİ
Çizelge 2.1. Çizelge 2.2. Çizelge 2.3. Çizelge 2.4. Çizelge 2.5 Çizelge 2.6. Çizelge 2.7. Çizelge 4.1. Çizelge 4.2. Çizelge 4.3. Çizelge 4.4. Çizelge 4.5. Çizelge 4.6. Çizelge 4.7.
Nöroblastoma ile birlikteliği olan hastalıklar. Nöroblastom evreleri N-Myc amplifikasyonu ilişkisi. Nöroblastomun genetik/klinik grupları ile ilişkisi. Nöroblastom tümör oluşumunda kanıtlanmış veya içerdiği tahmin edilmiş genler ve kromozomal bölgeler. Uluslararası Nöroblastom Evreleme Sistemi (INSS). Nöroblastomda prognostik faktörler. Uluslararası Nöroblastom Patolojik Sınıflaması (INPC). Cinsiyetin, yaş, LDH, NSE ve VMA değerleri ile karşılaştırılması. Evrelerin; yaş, LDH, NSE ve VMA değerleri ile karşılaştırılması. Hasta grubunun cinsiyet, tümör yeri, evre ve histoloji parametrelerinin dağılımı Hasta ve kontrol grubunun lenfosit hücrelerinde gözlenen yapısal ve sayısal kromozom düzensizlikleri Hasta grubunda saptanan yapısal anomalilerin her kromozom için bölgelere göre dağılımı Hasta ve kontrol grubunda bulunan hasarlı bölgelerin sayısı, hücre başına düşen hasarlı bölge sayısı, nisbi oranı ve önemlilik sırası 9 Hastanın klinik ve biyolojij verileri
15 19 27 28 32 35 38 47 48 48 53 61 63 67

vii
SİMGELER ve KISALTMALAR DİZİNİ
ace AURKA BCL2 BCLX BDNF bHLH-LZ BT CGH chtb chrb del DI Dmin DNA EDTA EtBr FISH GTG H HCl hsr HVA K KCl LDH LOH M
Asentrik kromozom Aurora kinaz A B-cell CLL/ Lymphoma 2 geni B-cell CLL/ Lymphoma X geni Beyin kaynaklı nörotropik faktör Helik-sloop-heliks/lösin fermuar motifi Bilgisayarlı tomografi Karşılaştırmalı genomik hibridizasyon Tek kromatit kırığı Kromozom kırığı Delesyon DNA indeksi Double minutes Deoxiribo nükleik asit Etilen diamine tetra asetik asit Etidyum bromit (Ethidium Bromide) Fluoresan in suti hibridizasyon Giemsa ve tripsinle G bantlama Hasta Hidroklorik asit Homojen boyanan bölgeler Homovanilik asit Kontrol Potasyum klorid Laktik dehidrogenaz Heterozigosite kaybı Molar

viii
MAO mar MAX MDR1 MIBG mg ml MRG mRNA µg µl NAG NB NF1 NGF ng NMYC NSE NT-3 NT-4 OMS PCR RASSF1 RNA RT-PCR SDS ser STK6 STK15
Monoamin oksidaz Marker Faktör X ile ilişkili MYC geni Çoklu ilaç direnç geni Metaiyodobenzilguanidin Miligram (1/1000 gr) Mililitre Manyetik rezonans görüntüleme Mesajcı RNA Mikrogram (1/1000 mg) Mikrolitre (1/1000 ml) Nöroblastom amplifikasyon geni Nöroblastom Nörofibromatozis tip 1 Nevre growth factor Nanogram (1/1000 µg) Nöroblastoma miyelomatosis onkogeni Nöron spesifik enolaz Nörotropin-3 Nörotropin-4 Opsomyoklonus sendromu Polimeraz zincir reaksiyonu (Polymerase Chain Reaction) Ras ile ilişkili domain ailesinin birinci üyesi Ribonükleik asit Rivörs transkriptaz-PCR (Revers Transcriptase-PCR) Sodium dodesil sulfat Serin Serin/treonin protein kinaz 15 fare homoloğu Serin/treonin protein kinaz 15

ix
t TE TNF TP53 TP73 U
Translokasyon Tris-EDTA Tümör nekroz faktör (Tumor Necrosis Factor)
Tümör protein p53 Tümör protein p73 Ünite (Unit)

x
ÖZET
NÖROBLASTOMA HASTALARINDA KROMOZOMAL DÜZENSİZLİKLERİ, MYCN ve AURKA GEN DEĞİŞİKLİKLERİNİN İNCELENMESİ
Nöroblastoma (NB), sempatik sinir sisteminin öncül hücrelerinden kaynaklanan bir embriyonel tümördür. NB hastalığının oluşumunda, birçok kromozom düzensizliği ve MYCN geninin etkili olduğu bilinmektedir. Ayrıca, AURKA geninin MYCN geni üzerinde etkili olduğu da ileri sürülmektedir. Bu nedenle, çalışmamızda NBnin etiyolojisinde etkili olduğu düşünülen bu genetik değişikliklerinin incelenmesi amaçlandı. Çalışmamız, Çukurova Üniversitesi, Tıp Fakültesi, Pediatrik Onkoloji Polikliniğine NB ön tanısı ile başvuran 25 hasta ile bu hastalara yaş ve cinsiyet açısından uygun olan sağlıklı ve ailesinde kanser öyküsü olmayan 25 kontrol grubu sitogenetik açıdan incelendi. Hasta grubundan, 9 hastanın (%36) parafin dokusunda MYCN ve AURKA gen değişiklikleri FISH tekniği ile araştırıldı. Hasta ve kontrol grubunun kan dokusundaki kromozom düzensizlikleri standart sitogenetik prosedürler uygulanarak incelendi. Çalışmamızda, 25 hastanın 21’nün (%84) kan dokusunda kromozom düzensizlikleri bulundu. Hasta grubunda, hücrelerin %18,4’ünde ve kontrol grubunda hücrelerin %2,6’sında kromozom düzensizliği tespit edildi. Hasta ve kontrol grubu arasındaki bu fark istatistiksel olarak anlamlı bulundu (p<0,0001). Hasta grubunda bu düzensizliklerin %72’si yapısal ve %28’i sayısal olduğu rapor edildi. Bu yapısal düzensizliklerden; 1q21, 1q32, 2p24, 2q21, 2q31, 4q31, 9q11, 9q22, 13q14, 14q11, 14q24 ve 15q22 kromozomal bölgelerinin NBda önemli kritik bölgeler olduğu kaydedildi. Diğer çalışmalarda da bu bölgelerin çoğunun NB etiyolojisinde yer alan onkogen ve protoonkogenlerinin bulunduğu sıcak noktalar olduğu bildirilmiştir. Parafin dokuları çalışılan 9 hastanın, 6’sında (%66,7) MYCN ve AURKA gen amplifikasyonları ve bunların sadece evre 4 hastalarına (%85.7) ait olduğu bulundu. Elde edilen bu oranların literatür bilgileri ile uyumlu olduğu saptandı. Sonuç olarak çalışmamızda; kromozomal düzensizliklerinin NB etiyolojisinde önemli olduğu; 1q21, 1q32, 2p24, 2q21, 2q31, 4q31, 9q11, 9q22, 13q14, 14q11, 14q24 ve 15q22 kromozom bölgelerinin hastalığın tanısında ve prognozunda önemli ve hastalıkla ilişkili genlerin araştırılmasında hedef noktalar olabileceği ileri sürülebilir. Bununla birlikte, MYCN ve AURKA gen amplifikasyon artışı ile hastalığın ileri evreleri (3,4) arasında paralel bir ilişki bulunduğundan, bu amplifikasyon değerleri hastalığın tanısı ve prognozunda kriter olarak alınabilir. Anahtar kelimeler: Kromozom, Gen, Kromozom Düzensizlikleri, AURKA, MYCN

xi
ABSTRACT
AN OBSERVATION OF CHROMOSOMAL ABNORMALITIES AND MYCN and
AURKA GENE CHANGES IN THE NEUROBLASTOMA PATIENTS Neuroblastoma is an embryonal tumor stemming from the precursor cells of the sympathetic nervous system. Numerous gene abnormalities and the MYCN gene are known to be the causative effects in the formation of the NB disease. Furthermore, the AURKA gene is claimed to have an influence on the MYCN gene. For this reason, these genetic changes considered to play a role in the etiology of NB were aimed to be investigated in the present study.
In our study, 25 patients applying to University of Çukurova, Faculty of Medicine, Pediatric Oncology with a neuroblastoma pre-diagnosis were included. Also, a control group consisting of 25 individuals who were the same age and gender as the target group and without a family history of cancer were included in the study and investigated in cytogenetic terms. The MYCN and AURKA gene changes in the paraffin tissue of 9 of the patient group (36%) were determined through the FISH technique. The chromosomal abnormalities in the blood tissues of both the patient group and the control group were investigated through the utilization of standard cytogenetic procedures. In the present study, chromosomal abnormalities in the blood tissues were identified in 21 (%84) patients of the 25 patient group. Chromosomal abnormalities were observed in 18,4% of the cells of the patient group and 2,6% of the control group. The difference between the patient and the control group was considered to be statistically significant (p<0,0001). It was reported that 72% of these abnormalities were structural while 28% of them quantitative. Of these abnormalities, 1q21, 1q32, 2p24, 2q21, 2q31, 4q31, 9q11, 9q22, 13q14, 14q11, 14q24 and 15q22 were identified to be critical regions in the formation of NB. These areas were also reported to be the hot regions where oncogenes and proto-oncogenes are present and are involved in the etiology of NB. In 6 (66,7%) of the 9 patients whose paraffin tissue was studied, MYCN, and AURKA gene amplifications were identified in 85.7% of only the grade 4 patients. The percentages obtained in the present study were found to be consistent with the related literature. In conclusion, based on the finding of the present study, we can maintain that chromosomal abnormalities are important in the etiology of NB and that 1q21, 1q32, 2p24, 2q21, 2q31, 4q31, 9q11, 9q22, 13q14, 14q11, 14q24 and 15q22 are the regions of utmost importance in the diagnosis and prognosis of the disease and could be the target regions in the investigation of the genes associated with this disease. Moreover, since there is a parallel connection between the increase in MYCN and AURKA gene amplification and the advanced stages (3, 4) of the disease, these

xii
amplifications values could be regarded as important criteria in the diagnosis and prognosis of the disease. Key words: Chromosome, Gene, Chromosomal abnormalities, AURKA, MYCN

13
1. GİRİŞ
Nöroblastoma (NB) çocukluk çağının; Ewing sarkomu, non-Hodgkin
lenfoma, rabdomyosarkom ve primitif nöro-ektodermal tümör gibi küçük, mavi,
yuvarlak hücreli tümörlerinden biridir. Sempato-adrenal sistemi oluşturan nöral
krest hücrelerinden köken alır ve yerleşim yeride normal sempatik sinir
sisteminin bulunduğu adrenal kromafin hücreleri veya spinal sempatik ganglion
hücreleridir. Periferal nöroblastik tümörler; nöroblastom, ganglionöroblastom ve
ganglionörom’u bulundururlar ve çocukluk çağının en sık görülen nöroblastik
ekstrakranial solid tümörlerini oluştururlar1,2.
Diğer tümörlerle karşılaştırıldığında; primer tümörün, metastatik hastalığın
ve paraneoplastik sendromların farklı klinik prezentasyonu olabilir. Spontan
regresyonlar, benign hastalığa farklılaşma, ileri yaştaki çocuklarda oldukça malign
seyretmesi, nöroblastomun farklı davranışlarına örnek olarak verilebilir. En sık
görülen ekstrakraniyal solid tümördür. Tüm çocukluk çağı malignitelerinin %8-10
kadarını oluşturur2.
NB tümör dokusundaki bir çok genetik değişikliğin prognoz üzerine etkisi
gösterilmiştir. Örneğin yaklaşık triploidi iyi prognozla ilişkiliyken, MYCN onkogen
amplifikasyonu, 17q dengesiz fazlalığı, 1p veya 11q allelik kaybı daha agresif
tümör özellikleri ve kötü prognozla ilişkilidir. Biyolojik ve genetik değişiklikler,
tedaviye yanıt ve prognozun önemli göstergesi olup, tanı sırasında tümörün
karakterini bize belirlemektedir. Tümörün biyolojik özellikleri ile ilgili bilgiler
arttıkça tedavisi ile ilgili gelişmeler de hızlanmaktadır. Yüksek riskli hastalarda
bile yoğun tedavinin azaltılması, hatta düşük riskli hastalarda tedavi edilmemesi
tartışılmaktadır2,3,4.

14
2. GENEL BİLGİ
2.1. Epidemiyolojisi
NB tüm çocukluk çağı kanserlerinin yaklaşık %8-10 kadarını oluşturur.
Onbeş yaş altında yıllık insidansı milyonda 7-12 iken, süt çocuklarında milyonda
25-51 arasındadır. Canlı doğumlara bakıldığında, NB sıklığı yenidoğan ve bir
yaş altı süt çocuklarında milyonda 64 olarak bildirilir. İnsidans, 1-4 yaş için
milyonda 19.6, 5-9 yas için milyonda 2.9 ve 10-14 yas için milyonda 0.7’dir.
Erkeklerin kızlara oranı 1,2:1 ‘dir. Tanı anında ortalama yaş 18 aydır. Tanı
sırasında olguların %36’sı 1 yaş altında, %89’u 5 yaş altında ve %98’i 10 yaş
altındadır1-6.
NB insidansı, Japonya, Avrupa ve Amerika kıtalarındaki bazı
merkezlerde yapılan tarama çalışmalarında her yıl 7000 canlı doğumda 1 olarak
bildirilir. Tarama çalışmaları öncesinde bu oran her yıl 10000 canlı doğumda 1
olarak saptanmıştır. Japonya’da tarama çalışması başlamadan önce insidans
milyonda 7-9 iken, tarama çalışması sonrasında milyonda 19’a çıkmıştır. Diğer
merkezlerde de bir yaşın altındaki bebeklerde yapılan tarama çalışmaları
sonrasında insidans milyonda 26 olarak yükselmiş, bir yaş üstündekiler de
hastalığın görülme sıklığında değişiklik olmamıştır7-9.
Doğrulanmış bazı çalışmalar, seçici çevresel faktörler (prenatal dönemde
alkole, fenobarbital ve hidantoine maruziyet gibi) ile nöroblastomdaki yüksek
risk arasında bir ilişki olduğu ileri sürülmektedir10-12. Ebeveyn mesleği
(elektronik, tarım, iş nedeniyle böcek zehiri, elektromanyetik alan, boya,
radyasyona maruz kalanlar), anne yaşı, annenin kullandığı ilaç (amfetamin,
diüretik, trankilizan, kas gevsetici, fenitoin vs...) ve hormonlar, alkol ve sigara,
düşük öyküsü, tekrarlayan sezeryan doğum ile bebeğin düşük doğum ağırlığı,
preterm doğum, tonsillektomi ve/veya adenoidektomi öyküsü nöroblastom ile
ilişkilendirilmiştir. Annenin hamilelikte aldığı vitaminlerin ve 6 aydan çok anne
sütü vermenin de riski azalttığı bildirilmiştir13-16.

15
Bir grup hastada, otozomal dominant geçişli ailevi NB gösterilmiş olup bu
olgulardaki genetik predispozisyon germinal mutasyona bağlıdır. Tüm NB
olgularının %22’sinin bu germinal mutasyon sonucu ortaya çıktığı
bildirilmektedir. Ailevi NB olguları diğer herediter kanserlerde olduğu gibi daha
erken yaşta (ortanca 9 ay) tanı almakta ve çoklu primer tümörle (olguların
%20’sinde bilateral adrenal tümör veya multifokal tümör var) çıktığı
bildirilmektedir. Bu olgularda yapılan genetik analizler ile kalıtsal NB
predispozisyon lokusunun (HNB1) 16. kromozom kısa kolunda (16p12-13)
olduğu gösterilmiş ve sporadik NB %13’ünde saptanmıştır. Ancak, bulgular
birden fazla predispozisyon geni olduğunu işaret etmektedir17-18.
İlginç olan, hirschsprung hastalığı ve/veya santral hipoventilasyon
sendromu gibi nor-adrenerjik hücrelerin patolojisi ile ortaya çıkan hastalıklar ve
nörofibromatozis Tip 1 (NF1) ile NB birlikte görülme sıklığının fazla olmasıdır.
Ayrıca, NB tümör dokusunda NF1 genindeki homozigot inaktivasyonlar
gösterilmiştir. Bu bulgular, hirschsprung hastalığı ve santral hipoventilasyon
sendromu ve/veya NF1 hastalığını ortaya çıkaran genlerin (RET, EDNRB,
EDN3, GDNF, ECE1, ZFHX1B, BDNF ve PHOX2B) NB gelişiminden de
sorumlu olabileceğini işaret etmektedir19-21. Beckwith-Wiedemann sendromu,
hirschsprung hastalıgı, ailede feokromasitom tanısı, fetal hidantoin sendromu,
fetal alkol sendromu ve nesidioblastozis gibi bazı hastalıklarla birlikteligi söz
konusudur. NB eşlik edebilecek durumlar Çizelge 2.1’de gösterilmektedir.
Çizelge 2.1. Nöroblastoma ile birlikteliği olan hastalıklar
Nörofibromatozis
Hirschsprung hastalığı ile aganglionik kolon
Ailede feokromasitom tanısı
Fetal hidantoin sendromu
Fetal alkol sendromu
Nesidioblastozis

16
2.2. Etiyolojisi
NB’un etyolojisi tam olarak bilinmemektedir. NB, sempatik sinir sisteminin
öncül hücrelerinden kaynaklanan bir embriyonel tümördür. Doğumdan sonra
yıllar süren bir zamanda sempatik sinir sistemi yeniden düzenlenir. Bu sürecin
nöroblastomun baslangıç yaşı ile örtüşmesi, nöroblastik tümörlerin gelişiminde
sempatik farklılaşmada rol alan temel mekanizmalarda bozukluk olduğunu
düşündürmektedir. Tümör dokusunda embriyonik veya nöral katlantı dönemi ile
ilişkili MASH1, HES1, c-kit, Notch, dHAND ve hASH1 gibi genlerin saptanması
embriyonun normal gelişim ve farklılaşması ile tümör oluşumu arasında bağ
olduğu düşüncesini desteklemektedir. Nöral katlantı hücrelerinden gelişen
sempatik sinir sistemi hücreleri nöronlar (ganglion hücreleri), kromafin hücreler
ve küçük floresan ile boyanan (SIF- “small intensely fluorescent”) hücrelerden
oluşur. Nöronlar, paravertebral sempatik zincir ganglionlarını, paraaortik
trunkusu ve pelvik ganglionu oluşturur. Adrenal bez medullasında da bulunurlar
ancak, gelişim süreci içinde kaybolurlar. SIF hücreleri, sempatik ganglionlar
içinde tek veya kümeler halinde bulunurlar ve fonksiyonları henüz
bilinmemektedir. Kromafin hücreler ise paraganglionları ve adrenal medullayı
oluşturur. Paraganglia diğer sempatik yapılarla birlikte oluşur ancak, doğumdan
sonraki 2-3 yıl içinde gerilemeye başlar ve kaybolur. Bu döneme kadar merkezi
sinir sistemi dışında en önemli katekolamin kaynağıdır. Bundan sonra, adrenal
medulla önem kazanır. Tümör hücre kaynağının özellikle adrenal
nöroblastomlarda, gelişmekte olan adrenal medullada geçici olarak bulunan
nöroblast toplulukları olduğu varsayılır. Dolayısı ile sempatik sinir sisteminde
hücre büyümesi, farklılaşması ve apopitozu sırasında olacak değisikliklerin
malign degisiklige neden oldugu düşünülmektedir2,19,22.
Periferik nöroblastik tümörlerin gelişim sürecinde gerileme, ilerleme ve
maturasyon evreleri yer alır. Tümörün sınırlı ve metastazların yalnızca cilt,
karaciğer ve kemik iliğinde olduğu bebeklerde hastalıktan ölüm %10-15’ten
fazla değildir. Eğer, ilerleyici safha yaşamı tehlikeye sokacak sonuçlara neden
olmaz ise veya kemoterapi veya radyoterapi ile durdurulabilir ise apopitoz ile
tümör lezyonları kendiliğinden geriler ve kaybolur. Bazen tümörde gerileme tam
olmayabilir veya maturasyon bulguları izlenir2,19,23. Üç aydan küçük bebeklerde

17
tanımlanan “in situ” NBda genetik olarak anormal olan klonun yaşla birlikte
gerilediği veya matür olduğu düşünülür. Onaltı yaş altında adrenal tümörlerden
40-50 kat fazla görülür ki bu sıklık tarama ile saptanan NBlardan fazladır24.
Nöroblastik tümörlerde, maturasyonun son aşaması ganglionöromdur.
Histogenetik ve hücre kültür çalısmalarında tümör dısından gelen ve dokuyu
istila eden schwann hücrelerinin rolü olduğu düşünülür. Kemoterapinin matür
olmayan nöroblastik hücrelerde apopitoza neden olduğu, hatta alkilleyici
ajanların maturasyonu uyardığı düşünülür. Evre 4 hastaların çoğunda ve düşük
evreli hastalığın bir kısmında ilerleme bulguları izlenir19,25.
2.3. Genetik
Normal hücrelerin malign dönüşümü iki grup kanser geni aracılığı ile olur.
Onkogenler, hücresel büyümeyi baskın olarak artırırken tümör baskılayıcı
genler, normal büyüme ve farklılaşmayı kontrol eder. Yapılarının bozulması,
allel kaybı, fazla kopya sayısı veya genin fazla ifade edilmesi tümör gelisimi ile
ilişkilidir. Aynı zamanda, klinik gidişin ön belirleyicileri olarak kullanılırlar. Tümör
gelişimi iki nokta hipotezi ile açıklanır. Doğustan bir kromozomda olan allel
kaybının NB gelişimine zemin hazırladığı ve aynı genin diğer allelinde nokta
mutasyon olduğunda kanserin olustuğu düşünülmektedir2.
2.3.1. Allelik Fazlalık ve Onkogen Aktivasyonu 2.3.1.1. Ploidi
NB hücresindeki DNA miktarı ya artmış (yaklaşık triploid) ya da normal
(diploid) olarak iki grupta toplanır. Tümörün DNA indeksi (DI) akım sitometrisi ile
kolayca belirlenebilir. DI, teşhiste hastalığı yayılmış ve özellikle Uluslar arası
Nöroblastoma Evreleme Sistemince (INSS) 4S evreli, 2 yaşından genç hastalar
için güçlü bir prognostik biyomarkerdır. Hiperdiplioid tümör (DI>1), diploid
tümöre (DI=1) göre daha iyi prognoz gösterir. Bazı araştırıcılar, tümörün ploidi
evresinin NB klinik davranışında heterogenitede önemli bir rol oynadığı
hipotezini açıklamışlardır26-28. Hastalarda, hiperdiplöidi iyi gidişle ilişkili iken
diploidili hastalarda kemoterapotik ajanlara direnç söz konusudur. Hiperdiploidik

18
hastalar, diploid hastalarda daha az yoğun kemoterapotik ajanlarla tedavi
edilirler.
2.3.1.2. MYCN Amplifikasyonu
Bazı NB hastaları, sitogenetik açıdan ekstra küçük kromozom parçaları
dms (double minutes) veya aynı kromozom içinde bir bölgenin yinelenmesi hsr
(homojen boyanan bölgeler) ile karakterize edilir. 1983 yılında, Schwab ve
arkadaşları Nb hücre hatlarının bir panelinde amplifiye edilmiş yeni bir myc
akraba onkoge MYCN tanımladılar29.
MYCN geni, 2. kromozomun kısa kolunda p24 bölgesinde bulunur.
Ancak, hücre hatlarında MYCN amplifikasyonu dms veya hsr şeklinde
haritalanır. Kromozom 2p24 bölgesinden (MYCN’inde içinde olduğu) büyük bir
bölge amplifiye olmaktadır. Tahminen bu durum, hücrelere bazı seçici avantajlar
sağlamaktadır. Amplifikasyon mekanizması tam olarak bilinmemektedir ancak,
MYCN lokusu ekstra kromozomal sirküler element formu veya dm olarak kopya
edilmektedir. DMs, mitoz boyunca dengesiz ayrılmasıyla birikmekte ancak, bazı
durumlarda amplifiye DNA hsr formu olarak kromozomal bir lokusa
yapışabilmektedir. Diğer genler ise bir takım olaylarda MYCN ile birlikte
amplifiye olabilir fakat, MYCN bu lokustan sürekli olarak amplifiye olan tek
gendir30-33.
MYCN amplifikasyonu Southern blot, FISH, kantitatif PCR,
immunositokimya ve komparatif genomik hibridizasyon (CGH) gibi çeşitli
moleküler teknikler ile tanımlanabilir. Birçok laboratuar, FISH tekniğini tercih
etmektedir. Çünkü, MYCN sinyalinin morfolojik isbatı kaliteli kontrol durumu
sağlar ve düşük seviye amplifikasyonun ve/veya intratümoral heterogenitenin
tanımlanmasına izin vermektedir34,35.
MYCN amplifikasyonu, çoğunlukla ileri evre hastalık, hızlı tümör
büyümesi, yüksek rölaps riski ve kötü prognozla anlamlı olarak ilişkilendirilir.
ABD’de 3000 nöroblastom tanısı konmuş hastada yapılan bir çalışmada MYCN
amplifikasyonunun toplam prevelansı %22 olarak saptanmıştır (Çizelge 2.2).
Bugün, prognostik önemi nedeniyle dünyada Amerika, Avrupa, Japonya ve
ülkemizde tanı alan her olguda rutin olarak bakılmakta tedavi planı buna göre
çizilmektedir36,37.

19
Çizelge 2.2. Nöroblastom evreleri ile MYCN amplifikasyonu ilişkisi37
Teşhiste Evre MYCN amplifikasyonu 3 yıllık yaşam
Benign ganglionöroma 0/64 (0%) 100%
Düşük evreler (1,2) 31/772 (4%) 90%
Evre 4S 15/190 (8%) 80%
İleri evreler(3,4) 612/1974 (31%) 30%
Toplam 658/3000 (22%) 50%
Genellikle, MYCN kopya sayısı ile ekpresyonu arasında bir korelasyon
vardır. Amplifikasyonlu tümörler genelde amplifikasyonsuz tümörlerden daha
yüksek seviyede MYCN ekpresyonu yapar ve bu nöroblastom alt türü yüksek
derecede malignant olur. Ancak, n-myc amplifikasyonu olmayan tümörlerde N-
Myc mRNA’sının veya MYCN proteininin aşırı ekspresyonunun olup
olmadığının prognostik öneme sahip olduğu tartışılmaktadır. Gen
amplifikasyonundan yoksun bazı Nb hücre hatlarında MYCN mRNA’sı veya
MYCN proteini yüksek seviyede ekprese edilir. Bu değişiklikler, MYCN
transkripsiyon otoregülasyon kaybı yerine normal protein parçalayıcı yolaklara
bağlı olabildiği bildirilmektedir38,39.
Bir çalışmada; MYCN ekspresyonu ile yaşama olasılığı arasında tersine
bir ilişki olduğu açıklanmış, oysa diğer çalışmalar ya bir ilişki bulunmadığını ya
da ilişkiyi daha büyük çocuklarla sınırlandırılmıştır40-42. MYCN geni, 3 ekzon
içerir ve GC dizilerince zengindir. Ekzon 1 kodlanmaz ve 5’ UTR bölgesi 2
potansiyel TATA kutusu içerir. Bunlardan ilki majör promotordur31.
MYCN, sinir sisteminde ve seçilmiş organlarda eksprese edilen bir
protoonkogendir. MYCN ürünü N-myc kısa ömürlü nüklear bir fosfo proteindir.
Bütün Myc ailesi proteinleri gibi N-terminal ucunda bir transaktivasyon domaini
(Myc box) ve C terminal ucunda temel bir heliks-loop-heliks/lösin fermuar motifi
içerir (bHLH-LZ). bHLH-LZ bölgesi, diğer Max ve Mad gibi diğer bHLH-LZ
proteinleri ile interaksiyon yaparak DNA’ya bağlanmalarında aracılık eder
(Şekil2.1). N-Myc’in transkripsiyonu aktive etmesi için önce Max ile dimerize
olmalı43.

20
Şekil 2.1. N-myc’in diğer proteinlerle etkileşimi
Myc protein ailesinin karakteri olan amino terminal domaininden yoksun
olan Max her hücrede eksprese edilen uzun yarı ömürlü bir nüklear proteindir.
Sabit evrede hücre Go evresindeyken, Max ekspresyonu yüksektir ve Max/Max
homodimer formasyonunu destekler ve transkripsiyonu baskılar. Ancak, MYCN
ürününün artmasıyla, hücre döngüsüne girilmesiyle veya genomik
amplifikasyonun bir sonucu olarak N-myc ile Max heterodimerizasyonu oluşur.
Bu durum, henüz tanımlanmamış büyümeyi uyaran genleri transkripsiyonel
aktivasyona yönlendirir. Ayrıca, Max; Mad ve Mxi1 gibi diğer nüklear proteinler
ile heterodimerizasyon oluşturarak Max’a bağlamış N-myc ile yarışarak
transkripsiyonu baskılar44.
MYCN, klasik dominant onkogen olarak fonksiyon gösterir ve ekspresyon
baskısı normal hücreleri düzenleyebilir ve genellikle onkogenik Ras ile ilişki
içindedir. N-Myc’in fazla ekspresyonu embriyonik fibrobastları yaşlanmadan
kurtarabilir. NB hücre hatlarına antisense-RNA ile N-Myc eklenmesi -fazla
ekspresyonu- poliferasyonu azaltabilir ve/veya farklılaşmayı teşvik edebilir45.
MYCN amplifikasyonu, yüksek malignansi gösteren NB hastalarını
teşhis etmesi çok net olmasına rağmen, N-myc proteininin amplifiye olmayan
tümörlerdeki kesin rolü halen tartışmalıdır. Bazı gen amplifikasyonsuz NB hücre
hatlarında N-myc proteini yüksek seviyede eksprese edilir. Bunun nedeni; N-

21
myc’in transkripsiyonal otoregülasyon kaybından ziyade normal protein
parçalanma yolaklarındaki değişikliklerden dolayı olabilir. Primer tümörlerde,
MYCN ekspresyonu hastalığın bütün evrelerinde bulunabilir ve klinik fenotip ile
ilişkisi önerilmez. Ancak, MYCN amplifikasyonu olmayan tümörlerin bir alt
grubunda bazı çalışmalar, MYCN ekspresyonunu hayatta kalma ihtimali ile ters
ilişkili olduğunu açıklamıştır41,46.
MYCN lokusunda, amplifikasyonun asıl hedefi olan 130 kb bir çekirdek
domain bulunur. Bugüne kadar, bu çekirdek domain içinde MYCN’in yanında
başka bir gen tanımlanmamıştır. Ancak, RNA helikaz geni olan DDX1, MYCN’in
5’ ucunda 400 kb bir alan içinde haritalanır ve MYCN amplifikasyonu yaklaşık
%40-%50 NB hastasında MYCN ile birlikte amplifikasyon gösterir. Bununla
birlikte, DDX1 amplifikasyonu MYCN amplifikasyonu bulunmadığı durumlarda
tanımlanmamıştır. MYCN amplifikasyonlu tümörlerinin malignant davranışı
DDX1 ile veya son zamanlarda keşfedilmiş NAG (Nöroblastom amlifikasyon
geni) gibi diğer 2p24 genleri ile birlikte amplifikasyonunu etkileyebilme imkanı
kalmaktadır47,48.
2.3.1.3. 17q’da Dengesiz Fazlalık
Bir diğer saptanmış spesifik karyotipik anomali, kromozom 17’nin uzun
kolundaki dengesiz artış (17q). Allelotipleme ve CGH çalışmalarında, bu
anomalinin NB yarısından fazlasında ortaya çıktığı ileri sürülmektedir49.
17q’daki dengesiz artış, kendiliğinden meydana gelebildiği gibi 1. ve 17.
kromozomlar arasında sık oluşan dengesiz bir translokasyon sonucu da ortaya
çıkmaktadır. 17q’de değişik kırıklar oluşmaktadır ancak, 17q22-qter‘deki bir
bölgenin tercihen fazlalığı bir veya birden çok genin seçici bir avantaj sağladığı
yani dozaj etkisinin olduğu öne sürülmektedir50. Bu gen veya genlerin, seçici
avantajdan sorumlu olduğu bilinmemektedir ancak, survivin (apoptozis inhibitör
proteini) ve NME1’in (NM23) (nükleosid trifosfatın sentezinde görev alır) aşırı
ekspresyonun genomik materyaldeki bu fazlalıktan kaynaklandığı ileri
sürülmektedir51. 17q dengesiz artışın prognostik önemi açıkça agresif tümör ve
kötü prognozla ilişkilendirilmektedir.

22
2.3.1.4. Diğer Onkogenler
NBda diğer insan onkogenlerinin sürekli mutasyona uğraması, fazla
eksprese olması veya amplifiye olması gösterilmemiştir. İlk başta NRAS, NB
hücre hatlarında taşınan bir gen olarak tanımlandı bu gen, RAS ve MYCN ile
işbirliği yaparak embriyonik fibroblastların dönüşümünü sağlamaktadır. Ayrıca,
farelerin nöroektodermini hedefleyen HRAS’ın aşırı ekpresyonu ganglionöroma
ve zaman zaman NB neden olmaktadır. Ancak, ras mutasyonlarının aktive ettiği
primer NB tümörleri nadiren gözlenir. MYCN ile MYCL veya MDM2’nin beraber
amplifikasyonu birkaç NB hücre hattında gözlenmiştir. En son CGH çalışmaları,
bazı tümörlerde 2p13-14, 2p23, 3q24-26, 4q33-35 ve 6p-11-22’de yeni
amplifikasyon bölgeleri tanımladı. Ancak, bu gözlemlerin biyolojik önemi henüz
tanımlanmamıştır52-54.
2.3.2. Allelik Kayıplar ve Tümör Supresör Genler 2.3.2.1. 1p Delesyonu
İlk kez, 1977 yılında Brodeur ve arkadaşları primer tümör ve hücre
hattının sitogenetik analizlerinde 1p delesyonun NB genetiğindeki önemini
açıkladılar55. Devam eden sitogenetik çalışmalar, 1p delesyonunu ve diğer 1p
yeniden düzenlenmelerinin yüksek oranda sıklığını teyit ettiler. Heterozigosite
kaybı çalışmaları (LOH), NB tümör supresör genin 1p36 bölgesinde lokalize
olduğunu gösterdi56. Tanı sırasında, primer tümör dokusunda %25-35 oranında
1p delesyonu rapor edildi57,58. İleri evre hastalarda, kromozom 1 delesyonu çok
yaygın bulunur ve 1p allelik kayıp MYCN amplifikasyonu ile birlikteliği fazladır.
1p LOH’un bağımsız prognostik önemi tartışmalara yol açmış fakat bugünki
bulgular 1p36’daki allelik kaybın lokalize tümörlü hastalarda hastalığın rölaps
riskinde bir artışı önceden haber verdiği ileri sürülmektedir59,60. 1p’nin uç kısmı,
NB çabuk ilerleyerek büyümesinde öneme sahip gen veya genler içermektedir.
Sitogenetik çalışmalar, hem terminal delesyonlar hem de dengesiz yeniden
düzenlenmeler için 1p’de 1p22’den 1p36’ya kadar geniş bir aralığın kırıldığını
göstermektedir61. 1p36 bölgesindeki delesyon Nöroblastom tümör supresör
genin kaybına neden olmaktadır. Varlığı kötü prognoz ile ilişkilidir20. Bazı genler,
1p36 NB süpresör geni için olası adaylar olarak analiz edilmiştir. Nöroblastom

23
araştırmalarında p73’ün 1p36’da haritalanmasının keşfi ile büyük bir ilgi
oluşturdu. p73’ün, bir p53 homoloğu olup apoptozis uyarır ve hücre
büyümesinin inhibe eder. Bunlar, TP53’ün homoloğu TP73; CDK2’nin homoloğu
CDCL2L1 (p58); HKR3, DAN, PAX7, ID3 ve E2F2 transkripsiyon faktörleri,
TCEB3 (Elongin A) transkripsiyon uzama faktörü ve tümör nekrosis faktör
reseptör ailesinin iki üyesi olan TNFR2 ve DR3’ü içerir. Ancak, HKR3 ve DR3
dışındaki bu genlerden her biri mevcut konsensus bölgesi dışında lokalizedir ve
hiçbir adayın delesyon olmayan allelinde hiç mutasyon bulunmamıştır62-64.
2.3.2.2. 11q Delesyonu
Kromozom 11q düzensizlikleri büyük olasılıkla, 11q14-q23 bandlarını
etkiler ve NBda kötü porgnozu gösterir. 1p allelic kaybı pimer tümörlerin
tanısında %35-%45 ‘inde bulunur65-67. İlginç olanı, 11q delesyonu MYCN
amplifikasyonlu tümörlerde bulunmaz ancak hala ileri evre, ileri yaş ve kötü
patoloji gibi diğer yüksek risk özellikleri ile birlikteliğ fazladır. Bu nedenle, 11q
delesyonu önemli bir prognostik göstergedir. Çünkü, çoğunlukla MYCN
amplifikasyonsuz ve/veya 1p delesyonsuz tümörlerin alt türünde oluşmakta ve
yeni veriler güçlü bir şekilde hastalık rölapsının bağımsız öngörü değeri
olduğunu önermektedir68.
2.3.2.3. Diğer Bölgeler
Diğer kromozomların bazı bölgelerde de allelik kayıpları bildirilmiştir. 14 q
delesyonu NBda genel bir anomalidir. 1989’da Suzuki ve arkadaşları, 12 NB
hastasının 6’ında tanımlamışlardır69. Devam eden çalışmalar, 14q32’de allelic
kayıplar olduğunu göstermiştir70. 3p, 4p, 5q, 9p ve 18q kromozom kollarında
allelik ve/veya LOH dengesizlikleri bildirilmiş ama 1p LOH’tan düşük frekansta
olduğu rapor edilmiştir71-74. Polimeraz zicir reaksiyonu tabanlı verilerle bu
çalışmaları doğruladı54.
2.3.2.4. Spesifik Tümör Supresör Genler
TP53 geni, p53 proteini kodlar ve genellikle insan kanserlerinde
mutasyona uğrayan genlerden biridir. Ancak, mutasyonlar nadiren primer NBda

24
bulunur. En son kanıt, TP53 geni rölaps hastalardan çoğaltılan hücre hatlarında
mutasyona uğrayabildiğidir. Ancak bu durum, eğer p53 anomalileri primer
tümörlerde anlamlıysa net değildir. CDKN2A (INK4A/p16) geni, yetişkin
kanserlerinin çoğunda mutasyona ya da delesyona uğrar. P16 hücre
kontrolünde kritik bir rol oynar. Primer NBlardaki bazı çalışmalar, P16
inaktivasyonuna ait veya CDKN2B (KIP1/p27) ve CDKN2C (INK4C/p18) genleri
ile ilgili kanıt bulamamışlardır16.
Klonlanmış NB spesifik tümör supresör genler olmamasına rağmen,
bilinen tümör süpresör genler içindeki mutasyonlar için hala tutarlı bir kanıt
yoktur. TP53, insan kanserinde en sık mutasyona uğrayan gendir. Ancak,
sadece nadir olarak NBda mutasyonla veya delesyonla inaktive olur. NBda, p53
proteininin sitoplazmik lokalizasyon aberasyonu gözlenmiş ve G1/S kontrol
noktasını bozduğu bulunmuştur. Bununla birlikte, NB hücrelerinin DNA hasarı
nükleusta wild-type p53’ün normal translokasyonuna ve p21 uyarılmasına
neden olmaktadır. CDKN2, p16’yı kodlar ve diğer hücre döngüsü kontrol
proteini genellikle insan kanserlerinde aktif değildir. Ama, mutasyon ve delesyon
NBda hiç bulunmamıştır16.
2.3.3. Bazı Genlerin Ekspresyonundaki Anormal Değişimler 2.3.3.1. Nörotrofin Reseptörlerinin Ekspresyonları
Sempatik sisteme ait nöroblastların nasıl NB hücrelerine malign dönüşüm
gösterdiği hala tam olarak bilinmemektedir. Ancak, nöroblastların malign fenotipi
kısmen farklılaşmayı düzenleyen sinyallere verilen yanıtın gerektiği gibi
olmamasına bağlanır. Nöroblastların, normal farklılaşmasını uyaran nörotropin
reseptör yolaklarının bu malign dönüşümde rol oynadığı düşünülmektedir.
Nöronal farklılaşmayı düzenleyen nörotropin ailesinde birçok faktör ve reseptör
yer almaktadır. Bunlar arasında, “nerve growth factor” NGF, beyin kaynaklı
nörotropik faktör (BDNF), nörotropin-3 (NT-3) ve nörotropin-4 (NT-4) sayılabilir.
NGF nöral hücre farklılasmasından sorumlu bir nörotropin ailesinin üyesidir.
NGF tek başına hastalık evresi veya prognoz ile iliskili degildir. Ancak NGF,
BDNF ve NT-3 ve NT-4 ligandlarının nörotrofin reseptörleri olan TrkA, TrkB ve
TrkC’nin NB gelişiminde önemi olduğu düşünülmektedir75,76.

25
TrkA aktivasyonu, hücrelerin faklılaşmsını uyarırken, angiogenezi inhibe
eder ve tümörün gerilemesine neden olur. Bu aktivitenin baskılanması, hücreyi
apopitozise yönlendirir. TrkA/NGF yolağı, nöroblastların ganglion hücresine
farklışlaşmasını veya apopitozis yoluyla tümörün kendiliğinden gerilemesini
kontrol eder. Tümör dokusunda TrkA’nın yüksek ifadesi iyi klinik özellikler
(küçük yaş, düşük evre), MYCN amplifikasyonunun yokluğu ile ilişkilidir ve iyi
prognoza işaret eder. TRK-B’nin tümör hücresinin yaşaması, invazyonu ve
metastaz yapmasını kolaylaştırdığı düsünülmektedir. TrkB yüksek ekspresyonu
ise agresif tümör ve MYCN amplifikasyonu ile güçlü olarak ilişkilidir.
TrkB/BDNF yolağı hücrenin ilaç direncini belirler. TRK-C’nin fonksiyonu ise
henüz kesin değildir. TrkC, TrkA reseptörü ile aynı özelliklere sahiptir, ek bir
prognostik önemi yoktur. İyi klinik özelliklere sahip tümörlerde eksprese olduğu
bildirilmiştir75,76.
Şekil 2.2. TrkA tirozin kinaz reseptörünün sinyal yolağı

26
2.3.3.2. Diğer Önemli Genlerin Ekspreyonları
Çoğu NB hastası başlangıçta kemoterapiye iyi cevap verir. Fakat
sonuçta, ya tedavide ya da durduktan kısa süre sonra ilerler. Ayrıca, NB hücre
hatlarının eski hallerine dönerek tümörün gelişmesi standart kemoterapötik
ajanlara karşı özellikle dirençlerinin arttığını gösterir. Böylece, ilaçlara karşı
direnç kazanmak NB tedavisinin başarızıslığının önemli bir nedenidir.
Kromozom 7q21.1 altbandında bulunan PGY1 geni (genel adıyla MDR1) hep
eksprese edilen P-glikoproteinini kodlar. Kanser hücrelerinde, transkripsiyonal
aktivasyon boyunca artan P-glikoprotein ekspresyonu, artan mRNA stabilitesi
veya genomik amplifikasyon gibi doğal ürün ilaçlara direnç kazanımı için
sorumlu kabul edilmiştir. Çoklu ilaç direnci (MDR), gen ekspresyonu MYCN
kopya sayısı fazla olmasa da büyük çocuklarda kötü klinik gidiş ile ilişkilidir.
MDR ile ilişkili proteinler, MYCN kopya sayısındaki artış ile orantılıdır ve
olumsuz gidişten sorumlu önemli bir belirleyicidir77,78.
BCL2 ve BCLX gibi apoptoz-süpresör genler, erken nöral ontojenide
yüksek düzeyde eksprese edilir. BCL2, çoğu NB hücre hatlarında ve primer
tümörlerde aşırı ifade edilir. BCL2 geni, kötü histoloji ve tümörigenezis ile ilişkili
olabilir. Bcl-2 proteini, ganglionöromlarda nadiren saptanır. Tümör regresyonu
apoptozis inhibitör yokluğuna bağlı olabilir. Ayrıca, Bcl-2 proteininin yüksek
düzeyleri tümörün ilaca direncinde önemli rol oynar79.
Hücrelerdeki CD95 reseptörü, CD95 ligandı ve kaspaz sisteminin ilaca
bağlı apopitoza aracılık ettiği; NB hücrelerinin de bu sisteme ve dolayısı ile
sisplatin ve doksorubisine dirençli olduğu bildirilmiştir. Tümör nekrozis faktör
(TNF) reseptör ailesine dahil olan p75, CD95/Fas ve retinoik asit reseptör ailesi
üyelerinin apopitozun uyarılmasına aracılık ettikleri düşünülmektedir. Ayrıca,
CD95’in ifade artışı apoptoz indükleyen kemoterapinin esas bir bileşeni
olduğunu gösterir79. Yaklaşık hastaların %50’sinin teşhiste hastalığının yayılmış
olmasına rağmen, NB invazyon ve metastaz biyolojisi hakkında çok az şey
bilinir. Hücre yüzey glikoproteini CD44’ün, tümör hücre adezyonu etkilediği
kabul edilir ve çeşitli insan ve fare kanserlerinde aşırı ekprese olduğu
gösterilmiştir. CD44 kromozomda 11p13’te bulunur. Bir tümör adezyon
molekülü olan CD44’ün ekspresyonunun MYCN amplifikasyonu ile ters ilişkili

27
olarak düşük evreli hastalarda bulunduğu olumlu prognostik işaret olduğu
bildirilmektedir. CD44 ifadesinde kayıp genellikle, artmış MYCN kopya sayısı ile
birliktedir ve kötü prognosa işarettir79.
Kromozom uçalarının sağlamlığı telomerazla korunur. Telomeraz aktivite
artışı, çoğu kanser hücresinde saptanabilir ve malignant dönüşüm için bir ön
koşul olarak görülmektedir. NBların büyük bir kısmında telomeraz salınımı
saptanmış ancak, normal adrenal doku veya gangliyonöromlarda
saptanmamıştır. NB hücrelerinde, hücre farklılaşması ve apoptoz ile ilşkili
genlerin ekspresyonu arttığında telomeraz aktivitesi düşük bulunmuştur. Ancak,
hücre döngüsü ile ilişkili genler ve transkripsiyon faktörlerin aşırı ekpresyonu
yüksek telomeraz aktivitesi ve kötü prognostik belirleyici olarak bildirilmiştir19,79.
Çizelge 2.3. Nöroblastomun genetik/klinik grupları ile ilişkisi
Bulgular Tip 1 Tip 2A Tip2B
MYCN Normal Normal Amplifiye
DNA ploidi Hiperdiploid/
Yaklaşık triploid
Yaklaşık diploid/
Yaklaşık tetraploid
Yaklaşık diploid/
Yaklaşık tetraploid
+17q Nadir Sık Sık
3p, 11q LOH Nadir Sık Nadir
1pLOH Nadir Çok sık değil Sık
Trk A
ekspresyonu
Yüksek Düşük veya yok Düşük veya yok
TrkB salınımı Düşük veya yok Yüksek
TrkC salınımı Yüksek Düşük veya yok Düşük veya yok
Yaş Genellikle < 1 yaş Genellikle > 1 yaş Genellikle 1-5 yaş
Evre Genellikle 1,2,4S Genellikle 3,4 Genellikle 3,4
3 yıllık yaşam %95 ≈ %50 ≈ %25

28
Çizelge 2.4. Nöroblastom tümör oluşumunda kanıtlanmış veya içerdiği tahmin edilmiş genler ve
kromozom bölgeleri (Referans; 79)
Kromozom lokusu Genin adı Genin tipi/fonksiyonu
1p36.2-p36.3 Tümör süpresör
1p13 NGF Nörotropin; NTRK1 ligandı
1q23-q31 NTRK1 (TRK-A) Reseptör tirozin kinaz
2p12-p13 MAD MYCN’i düzenleyebilir
2p24.1 MYCN Proto-onkogen
2p24 DDX1 RNA-helikaz / onkogen
3p Tümör süpresör
4p Tümör süpresör
7q21 PGY1 (MDR1) Çoklu ilaç direnci
9q22.1 NTRK2 (TRK-B) Reseptör tirozin kinaz
11p13 CD44 İntegrin/metastazı baskılar
11p13 BDNF Nörotropin; NTRK1 ligandı
11q23 Tümör süpresör 12p13 NTF3 (NT-3) Nörotropin; NTRK3 ligandı 14q23 MAX MYCN’i düzenler 14q23-qter Tümör süpresör
15q24-q25 NTRK3 (TRK-C) Reseptör tirozin kinaz 16p13.1 MRP Çoklu ilaç direnci 17q22 NME1 Nükleosid kinaz / metastazı baskılar 17q23-qter Onkogen
18q21.1 DCC Tümör süpresör 18q21.3 BCL2 Apoptozu baskılar
19 NTF4 (NT-4) Nörotropin; NTRK2 ligandı
2.4. Klinik Bulgular
NB, sempatik sinir sisteminin herhangi bir yerinden gelişebilir. Bu
doğrultuda tümörün yeri çok değişkendir ve yaşa göre farklılık gösterir. Çoğu
primer tümör hastaların %65’inde karın bölgesi yerleşimlidir. Adrenal tümör
sıklığı, bebeklerde %25, büyük çocuklarda %40’tır. NB, lenfatik ve hematojen

29
yolla yayılım gösterir. Yerleşik tümörlü hastalarda bölgesel lenf bezi tutulumu
%35’tir. Yayılım en sık kemik iliği, kemik, karaciğer ve ciltte görülür. Nadiren
akciğer ve beyin parankiminde metastaz yapabilir2,19. Klinik bulgular, primer tümörün yerine ve metastaza göre ortaya çıkar. En
sık abdominalde asemptomatik ele gelen kitle yakınmasıyla başvurulur (Şekil
2.3). Karında şişkinlik ve karın ağrısı bulguları olabilir. Fiziki incelemede,
hareketsiz ve sert kitle fark edilir. Eğer primer tümör Zuckerkandl organından
köken alıyorsa basıya bağlı mesane ve anal sifinkter disfonksiyonu ortaya
çıkabilir. Bebeklerde kitlesel karaciğer tutulumu sıktır (Evre 4S) ve çok büyümüş
karaciğer solunum sıkıntısına neden olur. Nadiren, renal arter basısına bağlı
olarak hipertansiyon gelişebilir2,19.
Şekil 2.3. Sağ sürrenal nöroblastom tümörü
Primer torasik yerleşimli tümörler, genellikle travma veya infeksiyon
nedeniyle çekilen akciğer grafilerinde tesadüfen saptanır. Toraks üst kısmı yada
servikal yerleşimli tümör “Horner sendromu”nu geliştirebilir. Büyük torasik
yerleşimli tümörler, superior vena kava sendromu yapabilir2,19 (Şekil 2.4).

30
Şekil 2.4. Arka mediastende Nöroblastom tümörü
Parasipinal yerleşimli tümörler, toraks, abdomen ve pelvik bölgede
paraspinal sempatik zincirden gelişen, vertebraların nöral foraminalarından
spinal kanal içerisine uzanıp sinir kökleri ya da medulla spinalise bası yapabilen
tümörlerdir. Buna bağlı olarak sırt ağrısı, subakut veya akut parapleji, mesane
ya da anal sifinkter fonksiyon kaybıyla karşılaşılır2,19.
Farklı bulgular ve semptomlar metastatik hastalık ile ilişkilendirilir.
Propitozis ve periorbital ekimoz (rakun gözü) periorbital kemiklerin metastatik
infiltrasyonuna bağlıdır (Şekil 2.5). Yaygın kemik veya kemik iliği metastazı
sonucu kemik ağrıları ve buna bağlı topallama görülür. Kemik iliği tutulumuna
bağlı anemi, kanama ya da enfeksiyon olabilir. Cilt tutulumu, evre 4S
bebeklerde, cilt altında ağrısız, mavimsi subkutan nodüller olarak ortaya çıkar.
Metastaza bağlı ateş, solunum yetmezliği, letarji, kilo kaybı ortaya çıkabilir.
Nadiren nöroblastom ergenler veya erginlerde de görülebilir. Primer tümör
yerleşimi çocuklardaki gibi olup, daha sinsidir. Bu hastalar, kemoterapiye daha
az duyarlıdır. Biyolojik olarak genetik Tip 2A’nın özelliklerini taşırlar. MYCN
amplifikasyonu yoktur genellikle, yapısal değişiklere sahip diploid karyotip
vardır2,19.

31
Şekil 2.5. Nöroblastomda periorbital infiltrasyona bağlı “rakun gözü” görünümü
2.4.1. Paraneoplastik Sendromlar
Opsomyoklonus sendromu (OMS) yeni tanı almış NB olgularının %2-
4’ünde görülen, hızlı göz hareketleri (opsoklonus), ataksi ve myoklonik
sıçramalar ile karakterize edilen bir sendromdur. Tümöre karşı gelişen
antikorların serebellar ve nöral hücrelerle çapraz reaksiyon vermesi sorumlu
tutulmaktadır. İyi biyolojik özelliklere sahip tümörlerle birliktelik gösterdiğinden
bu hastaların tümör prognozu iyidir. Tümörün kaybolmasına rağmen hastaların
%70-80’ninde uzun süreli motor gelişmede gecikme, dil sorunları ve davranışsal
bozukluklar gibi nörolojik fonksiyon kayıpları görülebilir. OMS tanısı alan
çocukların %30-50’sinde NB ortaya çıkabilmektedir. Bu nedenle, OMS tanısı
alan her hastaya I-MIBG sintigrafisi çekilmeli ve BT ile tüm vücut
taranmalıdır2,19,80.
2.5. Evreleme
Dünya genelinde, ilk olarak 1986’da geliştirilen ve daha sonra 1993’te
tekrar gözden geçirilip düzenlenen Uluslar Arası Nöroblastoma Evreleme
Sistemi (INSS) uygulanmaktadır (Çizelge 2.5). INSS kriterine göre, NB teşhisi
ya tümör dokusunun karakteristik histopatolojik gelişimi ile ya da kemik iliğindeki
tümör hücrelerinin biyopsisi/aspirasyonu ve idrardaki kateşolamin seviyesi ile
konulabilir. Evreleme için özel gereksinimler bilateral kemik iliği aspirasyonları
ve biyopsileri, vücudun tomografi hesapları, kemik taraması ve
metaiodobenzilguanadine (mIBG) sintiografisini içerir81.
Evre 1’de, lokalize bir tümör vardır ve cerrahi operasyonla alınır.
Mikroskobik tümör artığı sınırlıdır. Evre 2’de, lokalize tümörler göze batan kalıntı

32
hastalıkla (2A) veya lokalize tümör ipsilateral lenf nodu tutulumuna (2B) göre
değerlendirilir. Evre 3’de, tümörler orta hattı aşan invazyon gösterir. Evre,3
tümörlerin çoğu, karın bölgesinde ortaya çıkar. Çünkü, ya infilitrasyon ile ya da
lenf nodu tutulumuyla orta hattı aşan tümörler toraksta yaygın değildir. Uzak lenf
nodu, kemik, kemik iliği, karaciğer ve/veya diğer organlar içeren yaygın
hastalıklı hastalar evre 4 olarak karakterize edilir. Evre 4S’de, iyi prognoza
sahip farklı bir gruptur. Bu olguları, 1 yaşından küçük evre 4 gibi seyreden
olgulardan ayırmak önemlidir. Bu ayırımda, tümör dokusundan yapılan genetik
analizler önemlidir. Evre 4S’lerin çoğunda DI hiperdiploiddir ve olguların ancak
%10’nunda MYCN amplifikasyon vardır. Evre 4’de DI daha sık diploiddir ve
MYCN amplifikasyonu daha sıktır (1/3 olguda). Ayrıca, kemik iliğinin yoğun
tutulumu (>%10) evre 4 olarak kabul edilmelidir81. Genellikle, Zuckerkandl
ganglionu veya pelvisteki sempatik ganglionlardan köken alan tümörler orta hat
tümörü olarak kabul edilir. Orta hat tümörü, total çıkarıldıysa ve lenf nodu
tutulumu yoksa evre 1 olarak kabul edilir. Multifokal primer tümörde (örneğin
bilateral sürrenal tümör), evreleme en büyük tümöre göre yapılmalıdır19.
Çizelge 2.5. Uluslararası nöroblastom evreleme sistemi (INSS)
EVRE TANIM
1 Tümör köken aldığı organa sınırlı, makroskobik tam rezeksiyon.
Mikroskobik tümör artığı olabilir veya olmayabilir. İpsilateral veya
kontrlateral lenf nodu tutulumu yok.
2a Unilateral tümör, tam olmayan makroskobik rezeksiyon. İpsilateral veya
kontrlateral lenf nodu tutulumu yok.
2b Unilateral tümör, makroskobik tam veya tam olmayan makroskobik
rezeksiyon. İpsilateral bölgesel lenf nodu tutulumu var, kontrlateral lenf
nodu tutulumu yok.
3 Orta hattı aşan tümör ± bölgesel lenf nodu tutulumu
Unilateral tümör + kontrlateral lenf nodu tutulumu
Orta hat tümör + bilateral lenf nodu tutulumu
4 Yaygın hastalık, uzak metastazlar (kemik iliği, kemik, uzak lenf nodu,
karaciğer ve/veya diğer organlar)

33
4S Hastanın yaşı < 365 gün; Evre 1 ve 2 gibi lokalize primer tümör var;
sadece karaciğer, cilt ve/veya kemik iliği tutulumu (kemik iliğinde tümör
hücrelerinin oranı < %10 olmalı).
2.6.Tanı
NB tanısında, INSS kriterlerine göre tümörün mikroskopta
değerlendirilmesi esas olup, idrarda artmış katekolamin düzeyi, sintigrafik ve
radyolojik bulgular da tanı değerlendirmesini kuvvetlendirir2,13,14.
2.6.1. Tümör Belirleyiciler
NBda yaşın prognostik önemi 1971 yılında rapor edilmiş olup halen
önemini korumaktadır. Hastalığın bütün evreleri için tümörün yerinin ötesinde, 1
yaşından küçük bebekler aynı evreye sahip daha büyük çocuklardan önemli
derecede daha iyi pronoza sahiptir. Çoğu malignensiler gibi, hastalığın evresi
NB’da en önemli prognostik faktördür ve çeşitli retrospektif analizler INSS
evreleme sisteminin klinik anlamını doğrulamışlardır82,83. NBda, klinikte en çok
değerlendirilen ve duyarlı olan belirleyiciler vanil mandelik asit (VMA) ve
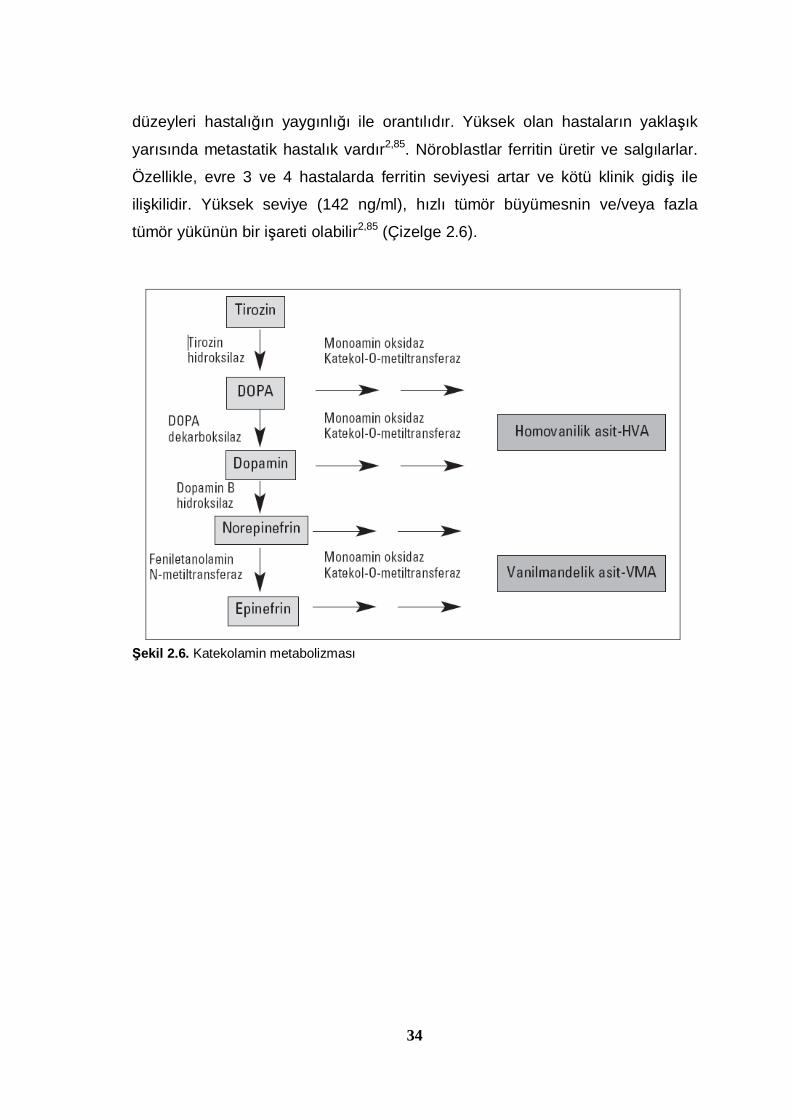
homovanilik asittir (HVA). Katekolamin sentezinin şeması Şekil 2.6’da
gösterilmektedir. Vanil mandilik asit epinefrin, norepinefrin, HVA ise dopamin
metabolizmasındaki son üründür. Nöroblastomda monoamin oksidaz (MAO) ve
katokol-O-metiltransferaz enzimleri vardır. Fakat eniletanolamin-N-
metiltransferaz enzimi olmadığından VMA ve HVA değerleri artar. VMA ve
HVA’nın spot idrar örneğinde araştırılması (mg/kreatinin) tanıda ve tedaviye
yanıtın izlenmesinde çok önemlidir. Sonuçlar, idrar kreatinin konsantrasyonuna
göre değerlendirilirse, ölçüm için 24 saatlik idrar toplanmasına
gerekmeyebilir19,84. Laktik dehidrogenaz (LDH), NB tanısında spesifik olmamasına rağmen
serum düzeyi (>1500 U/ml) prognostik marker olarak kullanılır. Hızlı hücre
döngüsü veya fazla tümör yükünü işaret eder. LDH’nin yüksek değeri büyük
tümörlerde ve yüksek riskli hastalarda görülür2,85. Nöron spesifik enolaz proteini
(NSE), nöroblastom hücreleri tarafından salgılanır. Yüksek NSE değerli (>100
ng/ml) ve ileri evre tümörlü hastalar büyük ölçüde klinik açıdan kötüdür. Serum
34
düzeyleri hastalığın yaygınlığı ile orantılıdır. Yüksek olan hastaların yaklaşık
yarısında metastatik hastalık vardır2,85. Nöroblastlar ferritin üretir ve salgılarlar.
Özellikle, evre 3 ve 4 hastalarda ferritin seviyesi artar ve kötü klinik gidiş ile
ilişkilidir. Yüksek seviye (142 ng/ml), hızlı tümör büyümesnin ve/veya fazla
tümör yükünün bir işareti olabilir2,85 (Çizelge 2.6).
Şekil 2.6. Katekolamin metabolizması

35
Çizelge 2.6. Nöroblastomda prognostik faktörler
Prognostik Faktörler Olumlu Olumsuz
KLİNİK FAKTÖRLER
EVRE 1, 2, 4S 3, 4
YAŞ <365 gün >365 gün
TÜMÖR MARKERLARI
Ferritin Düşük Yüksek
LDH (laktat dehidrogenaz)
Düşük Yüksek
NSE (nöron-spesifik enolaz)
Düşük Yüksek
PATOLOJİ Olumlu Olumsuz
BİYOLOJİK FAKTÖRLER
MYCN onkogeni Normal kopya Amplifikasyon
DNA index >1.0 (hiperdiploid)
1.0 (diploid)
Kromozom 1p Normal Delesyon
Kromozom 17q Normal Artış
TrkA ekspresyonu Yüksek Düşük
TrkC ekspresyonu Yüksek Düşük
TrkB ekspresyonu ---- Yüksek / FL
CD44 ekspresyonu Yüksek Düşük
MRP ekspresyonu (çokluilaç ilişkili protein)
Düşük Yüksek
Damarlanma Düşük Yüksek
FL= full-lenght transcript
2.6.2. Radyolojik Tanı Yöntemleri
NB tanısında ve evrelemesinde görüntüleme yöntemleri oldukça
önemlidir. Bazı hastalarda akciğer grafisi, bacak grafileri ve ultrasanografi ile
veya paraneoplastik sendrom araştırılırken NB tanısı konulabilir86.
Ultrasonografi, primer bölge (batın, paravertebral) değerlendirilmesinde
kullanılır. Deri, yumuşak doku tutulumlarında lezyon büyüklüğünün
belirlenmesinde kullanılabilir (opsiyonel). Ayrıca, karaciğer metastazlarının

36
değerlendirilmesi ve fontanel açıksa beyin değerlendirilmesinde kullanılır87.
Radyografi, akciğer grafisi ve kemik grafilerinin değerlendirilmesinde kullanılır.
Başlangıçta çekilmeli, tutulum söz konusu ise bu lezyonların takibi de
yapılmalı87. Bilgisayarlı tomografi (BT) ve manyetik rezonans görütüleme (MRG)
tümörün üç boyutlu tanımlanmasını sağlar. Primer bölge değerlendirilmesi,
özellikle paravertebral kitlelerde spinal MRG gerekir. Kranial MRG; evre 4
olgularında intrakraniyal/orbital tutulumu göstermek amaçı ile çekilmelidir87.
2.6.3. Sintigrafik Yöntemler
NB evrelemesinde sintigrafik yöntemlerin önemli ve yeri vardır.
Metaiyodobenzilguanidin (MIBG), katekolamin öncüllerinin analoğu olması
nedeniyle 131I veya 123I ile işaretlenerek yeni veya rölaps hastalığın
tanımlanmasında duyarlı ve özgül bir yöntem olarak kullanılır. Kemik, kemik iliği
ve lenf nodu tutulumunu gösterir. I123-MIBG, I131 MIBG’ e göre daha duyarlı
bir yöntem olduğundan küçük çocuklarda I123- MIBG tercih edilir. Büyük
çocuklarda I131 -MIBG ile sintigrafik görüntüleme yapılabilir. NB olgularının
%85’i I123-MIBG tutar. Bu nedenle tanı ve izlemde çok önemlidir2,87,88. 99mTc-
dimerkaptofosfonat (MDP) ile kemik sintigrafisi yeni tanı alan hastalarda kortikal
kemik tutulumunu göstermede kullanılır89.
2.6.4. Patolojik Değerlendirme
NB çocukluk çağının; ewing sarkomu, non-Hodgkin Lenfoma,
rabdomyosarkom ve primitif nöro-ektodermal tümör gibi küçük, mavi, yuvarlak
hücreli tümörlerinden biridir. Nöral krest hücrelerinden köken alır ve yerleşim
yeride normal sempatik sinir sisteminin bulunduğu adrenal kromafin hücreleri
veya spinal sempatik ganglion hücreleridir90,91. Periferal nöroblastik tümörler;
nöroblastom, ganglionöroblastom ve ganglionörom’u bulundururlar ve çocukluk
çağının en sık görülen nöroblastik ekstrakranial solid tümörlerini oluştururlar92.
Bunlar ortak bir neoplazmın matürasyonları farklı 3 alt grubudurlar.
Nöroblastom bu yelpazenin en ilkel kısmını oluşturur. Küçük, homojen, yuvarlak
hücreler arasında nöropil tipiktir. Eosinofilik nöropil çevresindeki nöroblastlar
Homer-Wright pseudorozetlerini oluşturur. Ganglionöroblastom histolojik

37
bulguları nöroblastom ve ganglionöromun bulgularını birlikte içeren heterojen bir
tümördür. Primitif nöroblastlar ve matür ganglion hücrelerini içerir. Diffuz veya
fokal olabilir, fokal ganglionöroblastom daha agresiftir ve nöroblastik komponent
MYCN amplifikasyonu taşıyabilir. Ganglionörom tamamen diferansiye beniyn
tümördür. Matür ganglion hücreleri, nöropil ve Schwann hücrelerinin karışımı ile
karakterizedir2,92,93(Şekil 2.7).
Şekil 2.7. Nöroblastomun Patolojik Sınıflandırması (İnternational Nueroblastoma
Pathology Committe-1999)
Nöroblastom’da histolojik bulgular prognostik öneme sahiptir. Shimada
ve arkadaşlarının geliştirdiği prognostik sınıflama hasta yaşı, Schwannian
stromanın varlığı-yokluğu, farklılaşma derecesi ve mitoz oranları kullanılarak
histopatoloji iyi veya kötü olarak belirlenmiştir. 1999’dan bu yana Shimada
sistemine dayanarak geliştirilen “Uluslararası Nöroblastom Patolojik Sınıflaması
(INPC)” kullanmaktadır93(Çizelge 2.7).

38
Çizelge 2.7. Uluslararası nöroblastom patolojik sınıflaması (INPC)
Yaş Farklılaşma Durumu MKI Prognostik
Kategori
< 18 ay AD Herhangi biri Kötü histoloji
< 18 ay KD veya D Düşük veya
orta
İyi histoloji
< 18 ay Herhangi biri Yüksek Kötü histoloji
18 ay- 5 yaş UD veya PD Herhangi Kötü histoloji
18 ay- 5 yaş D Düşük İyi histoloji
18 ay- 5 yaş D Orta veya
yüksek
Kötü histoloji
> 5 yaş Herhangi Herhangi biri Kötü histoloji
Ganglionöroblastom
Nodüler
İçindeki nöroblastik
komponente ve yaşa
göre
İyi veya kötü
histoloji
İntermiks Schwannian
stromadan
Zengin
İyi histoloji
Ganglionörom Schwannian stroma
dominant
İyi histoloji
AD: Farklılaşmamış/andiferansiye, D: Farklılaşmamış/ diferansiye KD: Kötü farklılaşmış/ kötü
diferansiye M: Mitoz karyoreksis indeksi
2.7. AURKA Geni
AURKA (Aurora Kinase A/ STK6/STK15) mitotik sentrozomal proteini
kodlayan bir gendir. Tümör gelişiminde ana görevi, mitoz boyunca kromozom
ayrılmasının kontrolünde bulunmaktır94,95. Fluoresan in situ hibridizasyon (FISH)
ile AURKA geninin 20q13.2-q13.3 ve 1q41-q42 kromozom bantlarında 2 sinyal
ile ifade edildiği gösterilmiştir. Somatik hücre hibrid panel analizinde,
fonksiyonal genin kromozom 20’de ve psödogenin kromozom 1’de olduğunu
gösterilmiştir96. AURKA mRNA’sı, proteini ve katalitik aktivitesi hücre
döngüsünü düzenler, hücre döngüsünün G2 ve M fazında seviyesi artar. Aurora

39
A proteini, interfaz hücrelerinin sentrozomlarında ve profazdan telofaza kadar
mitotik iğ ipliğinde lokalizedir. AURKA aktivitesi, geç G2’de ve profazda en üst
düzeydedir94.
AURKA, memeli hücrelerinde sentrozom duplikasyonunun uyarılması,
düzensizliklerin dağılımı ve anöploidi ile ilişkilendirilir. Sentrozomlar, replike olan
kromozomların kardeş hücrelere eşit dağılımı garantilenmiş hücre bölünmesi
boyunca bipolar iğ ipliklerinin kurulumu yoluyla genomik kararlığı korumak için
gereklidir. Kontrolsüz duplikasyon ve sentrozomların dağılımı, kromozom
ayrılma düzensizliklerine dahil edilir. Çoğu kanser hücre tipinde anöploidiye yol
açtığı görülür. AURKA amplifikasyonu meme, ovaryum, kolon, prostat, sevikal
ve nöroblastom kanser hücre hatlarında bulunmuştur. Ayrıca, AURKA
mRNA’sının fazla salınımı gen amplifikasyonunun delili olmayan tümör
hücrelerde keşfedilmiştir97.
AURKA geni, birçok tümörde fazla ifade edilir. Memeli hücrelerinde,
AURKA’nın normalin dışında fazla salınımı sentrozom amplifikasyonunu,
kromozom kararsızlığı, onkogenik transformasyonu ve p53 fonksiyon kaybı
mutasyonuna neden olur. AURKA, p53’ü ser315’ten fosforile eder, MDM2
tarafından ubikutinasyonuna ve proteolizisine yol açar. P53, inaktif AURKA veya
ubikutinasyon- MDM2 defekti- varlığında parçalanmaz. Susturulmuş AURKA,
ser315’te p53’ün daha az fosforilasyonuna ve kararlığına neden olur, hücre
döngüsü G2/M fazında tutulur98.

40
3. GEREÇ ve YÖNTEM
3.1. Araç ve Gereçler
3.1.1. Kimyasal Malzemeler
• RPMI 1640 Besiyeri (Biological Industries)
• Heparin (Panpharma)
• Kolşisin (Seromed)
• Penisilin- Streptomisin (Sigma)
• Fetal Calf Serum (Gibco)
• L-Glutamin (Biochrom)
• Phytohemmaglutinin (Biochrom)
• Sodyum Klorid (NaCl) (Merck)
• Potasyum Klorid (KCl) (Sigma)
• di-Sodyum Hidrojen Fosfat Dihidrat ( Na2HPO4. 2H2O)(Merck)
• Potasyum Dihidrojen Fosfat (KH2PO4)(Merck)
• Tripsin (Difco Bacto)
• Glasiyel Asetik Asit (Merck)
• Metanol (Merck)
• Etanol (Merck)
• Xylol (Merck)
• Aseton (Merck)
• İmmersiyon yağı (Merck)
• Giemsa (Merck)
• Entellan (Merck)
• Serumfizyolojik (Eczacıbaşı-Baxter)
• Hidroklorik Asit (HCl) (Merck)
• 20xSSC (Abbott Moleculer Inc.)
• Sodyum Tiyosiyanit (NaSCN) (Riedel-de Haen Ag Seelze Hannover)
• Formaldehit (Merck)

41
• PBS
• Probe (Poseidon)
• Proteaz (Sigma)
• Tween 20 (American Bioanalytical)
• DAPI (Abbott)
• Rubber Cement (Marabu)
3.1.2. Aygıtlar
• Etüv (Heraeus, Dedeoğlu)
• Santrifüj (Nuve CN180)
• Steril Kabin (Kojair KR-125 Safety)
• Derin Dondurucular (Siemens, Bosch)
• Buzdolabı (Arçelik)
• Mikroskop (Olympus BX51 ve Olympus CH40)
• Cytovision Görüntüleme Sistemi (Applied Imaging)
• Steril Kültür Tüpü (Greiner)
• Hassas Terazi (Sartorius)
• pH Metre (İnolab)
• Steril Plastik Pipet
• Steril Disposable Farklı Büyüklükte Enjektör (2 ml, 5 ml, 10 ml, 20 ml)
• Otomatik Pipetler (Gilson, Biohit ve Socorex)
• Mezür
• Şale (Yatay- dikey)
• Lam (Rodajlı, Pozitif şarjlı)
• Lamel (24x32)
• Thermobrite (Stat Spin)
• Su Banyosu (Gen-Probe)
• Fırın (Nüve FN 500)

42
3.2. Örneklerin Sağlanması İçin Kullanılan Yöntemler
Çalışma, Şubat 2009 Ocak 2010 tarihleri arasında Çukurova Üniversitesi,
Tıp Fakültesi, Tıbbi Biyoloji Anabilim Dalı’nda gerçekleştirildi. Aynı fakültenin
Pediatrik Onkoloji Kliniğine NB ön tanısı ile başvuran 25 hasta ile bu hastalara
yaş ve cinsiyet açısından uygun olan sağlıklı ve ailesinde kanser öyküsü
olmayan Pediatri Polikliniğine gelen 25 çocuktan (kontrol grubu) etik onay
alındıktan sonra, çalışmamızın amacı doğrultusunda kan kültürü için heparinize
enjektöre 2 ml kan alındı. Kan kültüründe ve kromozom eldesinde standart
sitogenetik yöntemlerden yararlanıldı. Kromozomlarda görülen sayısal ve
yapısal bozukluklar ve kromozomlar üzerindeki dağılımı Uluslararası İnsan
Sitogenetik İsimlendirme Sistemine (ISCN) göre isimlendirilerek kaydedildi.
Araştırma ve kontrol grublarında elde edilen yapısal ve sayısal anomaliler kendi
aralarında karşılaştırıldı.
Hastalardan cerrahi operasyon sonucu alınan ve Patoloji Kliniğinde
parafinlenen dokulardan pozitif sarjı lamlara 3 µ kalınlığında kesitler alınarak
uygun şartlarda Tıbbi Biyoloji Anabilim Dalı laboratuvarına getirildi. Alınan doku
kesitlerinde FISH tekniği kullanılarak her iki gendeki değişimler kaydedildi.
3.2.1. Sitogenetik Çalışmalar
Pediatrik Onkoloji Polikliniğine, NB ön tanısı ile başvuran hastalardan
(25) ve kontrol grubundan (25) alınan kan dokularına 72 saatlik kültür yötemi
uygulandı99,100.
3.2.1.1. Kan Kültürünün Yapılması
Kromozom analizi için hasta ve kontrol grubundan steril şartlarda
heparinle yıkanmış enjektöre 2 ml kan alındı. Her birey için, içerisinde 3’er ml
normal (RPMI-1640, R0883) (EK-1) besiyerinin bulunduğu steril kültür tüplerine
her olgunun kanından 3’er damla ilave edilerek ekim yapıldı. İsim karışıklığını
önlemek amacıyla tüplerin üzerine önceden bireyin ismi yazıldı. Ekim işlemi
yapıldıktan sonra kültür tüpleri hafifçe karıştırıldı. Daha sonra, tüpler 37°C’de 72
saat süreyle inkübatörde tutuldu. Kültüre alınmış tüplerin her birine 70 saat
sonra final konsantrasyonu 10 µg/ml olacak şekilde 30 µl kolşisin (EK-2) eklendi

43
ve iki saat daha kültüre devam edildi. Süre sonunda, tüpler hafifçe
karıştırıldıktan sonra 1200 rpm’de 10 dakika santrifüj edildi. Daha sonra, tüplerin
üstünde kalan süpernatan pastör pipeti ile atıldı. Dipte kalan çökleti, tüpün alt
kısmına parmakla hafifçe vurarak karıştırıldı. Tüplere 6’şar ml hipotonik
solüsyonu (0.075 M KCl) (EK-2) ilave edildi ve 13 dakika etüvde bekletildi. Süre
sonunda, her tüpe 6 damla soğuk fiksatif solüsyonu (EK-2) eklenerek
karıştırıldı. Tüpler 1200rpm’de 10 dakika santrifüj edildi. Süpernatan atıldıktan
sonra çökelti tekrar hafifçe karıştırıldı ve üzerine taze hazırlanmış 6 ml fiksatif
solüsyonu ilave edildi. 1200 rpm’de 10 dakika santrifüj edildi. Fiksatif ile yıkama
3 kez tekrarlandıktan sonra, çökeltinin miktarına göre çok az fiksatif bırakılıp
pipetajlanarak hücre süspansiyonu elde edildi. Daha önceden, temizlendikten
sonra asetonda bekletilen ve yaymadan 24 saat önce saf sudan geçirilerek
buzdolabında soğutulmuş olan lamlar üzerine 30°‘lik bir eğimle hücre
süspansiyonu damlatılarak yayma yapıldı. Lamlara hastanın ismi yazılarak
havada kurutuldu. Yayılan preparatlar yaşlandırmak amacıyla 3 gün 37°C’de
inkübatörde bekletildi. Yaşlanan preparatlar mikroskopta incelenmek üzere
aşağıda açıklandığı gibi Giemsa-Trypsin-Giemsa (GTG) bantlama yöntemiyle
boyandı.
3.2.1.2. Preparatların GTG (G-bands by Trypsin using Giemsa)
Bantlama Yöntemiyle Boyanması
Analiz için 3 gün oda ısısında bekletilen preparatlar GTG bantlama
tekniği uygulanarak hazır hale getirildi116.
Bunu için sırasıyla şu işlemler yapıldı:
• 1,5 ml’lik tripsin stok solüsyonu (EK-2) eritilerek bir şale içerisinde 75 ml
serum fizyolojik içerisine eklendi ve pH’sı 7.5-7.8’e ayarlandı.
• İkinci bir şaleye 75 ml fosfat tamponu (EK-2) ilave edildi.
• Üçüncü bir şaleyede fosfat tamponu içerisinde %7-10’luk olacak şekilde
giemsa boya çözeltisi (EK-2) hazırlandı.
• Metafaz kalitesine göre sıralanan preparatlar, mikroskop altında
metafazların açık ve koyuluğuna göre değişik saniyelerde (35-40 saniye)
tripsinle muamele edilidi.

44
• Preparatlar daha sonra fosfat tamponundan geçirildi. Hafifçe
çalkalanarak yıkandı.
• Daha sonra Giemsa boya solüsyonu ile 3-5 dakika boyandı.
• Akan musluk suyundan geçirilen preparatlar havada kurutuldu.
• Bantları boyanmış metafaz kromozomları mikroskop altında sayısal ve
yapısal olarak incelendi.
• Hazırlanan preparatlar 100x objektifte incelendi.
• Her hasta için 50 metafaz plağı incelendi. Ancak, mitotik indeksi düşük
olan hasta preparatlarında mevcut olan metafaz plakları incelendi.
• Preparatlar inceleme sonrası, Xylol’den geçirilerek immersiyon yağından
temizlendikten sonra entellan ve lamel ile kapatılarak devamlı hale
getirildi.
3.2.2. Moleküler Sitogenetik Çalışmalar 3.2.2.1. Fluorescence In Situ Hybridization (FISH) Yöntemi
Pozitif sarjlı lam üzerine alınan 3 mikron kalınlığındaki kesitler 56°C’lik
etüvde 16 saat bekletilerek deparafinize edildi.
• 2 kez 10’ar dakika süre ile %100 xylene içeren şalede oda sıcaklığında
bekletildi.
• 2 kez 5’er dakika süre ile %100 etanol içeren şalede oda sıcaklığında
bekletildi.
• 45-50 derecelik hotplate üzerinde 2-5 dk. kurutuldu.
• 20 dk. süre ile 0.2 N HCL içeren şalede oda sıcaklığında bekletildi.
• 3 dk. süre ile distile su içeren şalede oda sıcaklığında bekletildi.
• 3 dk. süre ile yıkama tamponu içeren salede oda sıcaklıgında bekletildi.
• 30 dk. süre ile 80 derecelik su banyosunda önceden bu ısıya getirilmiş
uygulama öncesi solüsyonu içeren şalede inkübe edildi.
• Lamlar 1 dakika süre ile distile su içeren şalede oda sıcaklığında
bekletildi.
• 2 kez 5’er dakika süre ile yıkama tamponu içeren şalede oda
sıcaklığında bekletildi.

45
• 10 dk. süre ile 37 derecelik etüvde önceden bu ısıya getirilmiş proteaz
solüsyonu içeren şalede bekletildi.
• 2 kez 5’er dakika süre ile yıkama tamponu içeren şalede oda
sıcaklığında bekletildi.
• 45-50 derecelik hotplate üzerinde 2-5 dk. kurutuldu.
• 10 dk. süre ile %10 tamponlanmış formalin içeren şalede oda
sıcaklığında bekletildi.
• 2 kez 5’er dakika süre ile yıkama tamponu içeren şalede oda
sıcaklığında bekletildi.
• 45-50 derecelik hotplate üzerinde 2-5 dk. kurutuldu.
• Bundan sonraki aşamalar karanlıkta gerçekleştirildi.
• Önceden oda ısısına getirlimiş prob tüpü vortekslendi, mikrosantrifüjde
spin atıldı ve tekrar vortekslendi.
• Herbir lamın hedef alanına 10 μl prob (ON AURKA (20q13)/20q11,
Repeat Free FISH Probe, Poseidon; ON MYCN (2p24)/LAF (2q11),
Repeat Free FISH Probe, Poseidon) damlatıldı ve hemen uygun boyutta
bir lamelle hava kabarcığı kalmayacak şekilde kapatıldı. Lamellerin etrafı
rubber cement ile çevrelendi.
• Lamlar Thermobrite cihazına yerleştirilip kapağı kapatıldı ve önceden
programlanan prosedür başlatıldı. (denatürasyon 73°C-5 dk.,
Hibridizasyon 37°C -18 saat)
• Bir gece hibridizasyondan sonra lamların üzerinden rubber cement
ayrıldı ve oda ısısındaki yıkama solüsyonu içerisinde çalkalanarak
lamellerin ayrılması sağlandı.
• 2 dk. süre ile su banyosunda önceden 73°C a getirilmis olan yıkama
solüsyonu içerisinde bekletildi.
• Lamlar oda ısısında distile su içeren saleye batırılıp çıkarıldıktan sonra
karanlıkta kurumaya bırakıldı.
• Herbir hibridizasyon alanına 10 μl. 4’,6-diamidino-2-phenylinodole (DAPI
II Counterstain, Abbott Laboratories) damlatılarak uygun boyutta lamelle
kapatıldı.
• Lamlar analiz öncesi en az 30 dakika buzdolabında bekletildi.

46
3.2.2.2. FISH Değerlendirmesi
ON AURKA (20q13)/20q11 ve ON MYCN (2p24)/LAF (2q11) probları için
her bir preparatta 100 tümör hücresi sayıldı ve bu 100 hücre içinde pozitif
hücrelerin yüzdeleri hesaplandı. ON MYCN (2p24)/LAF (2q11) probe için
hücrede iki kırmızı iki yeşil sinyal varlığı normal, %10 üzeri iki yeşil ve 5 kırmızı
ve üzerindeki sinyal varlığı amplifikasyon kabul edildi. ON AURKA
(20q13)/20q11 probe için hücrede iki kırmızı iki yeşil sinyal varlığı normal, %10
üzeri iki yeşil ve 3 kırmızı ve üzerindeki sinyal varlığı amplifikasyon kabul edildi.
3.2.3. İstatistiksel Analiz
Verilerin istatistiksel analizinde SPSS 15.0 paket programı kullanıldı.
Kesikli değişkenler sayı ve yüzde olarak, sürekli değişkenlerse ortalama ve
standart sapma (gerekli yerlerde ortanca, maksimum ve minimum) olarak
özetlendi. Sürekli değişkenlerin karşılaştırılmasında nonparametrik istatistikler
kullanılmıştır. Tüm testlerde anlamlılık düzeyi p<0.05 alındı.

47
4. BULGULAR
Çalışmamızda, 11’i kız (%44) ve 14’ü erkek (%66) olmak üzere toplam
25 hasta çocuğun ve sağlıklı, ailesinde kanser öyküsü bulunmayan 10’u kız
(%40) ve 15’i erkek (%60) olmak üzere toplam 25 çocuktan oluşan kontrol
grubunun analizleri yapıldı. Hastaların yaş aralığı; 1 ay-108 ay arasında olup,
genel yaş ortalaması 33.4 ay, kontrol grubunun yaş aralığı 5 ay-144 ay arasında
olup, genel yaş ortalaması 57,6 ay olarak belirlendi.
Hastaların nöroblastomaya yakalandıkları yaş ile cinsiyetleri
karşılaştırıldığında; erkeklerde hastalığın daha ileri yaşta (42ay), kız
çocuklarında daha erken yaşta (18ay) ortaya çıktığı Çizelge 4.1.’de
gösterilmiştir. Bu fark, İstatistiksel olarak anlamlı bulundu (p=0.020).
Çizelge 4.1. Cinsiyetin, yaş, LDH, NSE ve VMA değerleri ile karşılaştırılması.
Hasta grubunda yer alan 25 hastanın 24’ünün klinik bilgilerine ulaşıldı.
Hastaların p=0,044 LDH, p=0,049 NSE değerleri evrelere göre (evre 2b, evre 4,
evre 4S) farklılık göstermektedir. Evre 4’te LDH ve NSE değerleri daha yüksek
seyretmiştir (Çizelge 4.2). Bu değerler, istatistiksel olarak anlamlı bulundu
(p=0.044 ve p=0.049).
Erkek Kız Total p Ort±SS Medyan (min-
max) Ort±SS Medyan (min-
max) Ort±SS Medyan
(min-max)
Yaş (ay) (n=25)
42.0±30.5 39.0(6.0-108.0) 18.1±21.8 12.0(1.0-72.0) 33.4±29.6 24.0(1.0-108.0)
0.020
LDH (n=20)
2015.7±2097.2
1242.0(473.0-7930.0)
900.0±392.9 721.0(603.0-1627.0)
1625.2±1767.7 887.5(473.0-7930.0)
0.115
NSE (n=22)
225.7±117.2
261.0(44.0-370.0)
115.3±128.1 60.0(20.0-370.0) 190.6±128.9 181.5(20.0-370.0)
0.066
VMA (n=6)
58.0±80.4 23.0(1.0-150.0) 145.7±189.2 75.0(2.0-360.0) 101.8±138.6 49.0(1.0-360.0)
0.700

48
Çizelge 4.2. Evrelerin; yaş, LDH, NSE ve VMA değerleri ile karşılaştırılması.
Klinik verilerine ulaşılan 24 hastanın, 20’sinin tümör yeri sürrenal, 2’sinin
parasipinal, 1’nin mediasten ve 1’nin kranial olduğu saptandı. Hastalarımızın
21’nin evre 4, 2’sinin evre 2b ve 1’nin evre 4S; 15’i yüksek histolojiye sahipken
5’i kötü histolo-yüksek, 2’si iyi histo-yüksek ve 2’sinin düşük histolojiye sahip
olduğu Çizelge 4.3’te gösterildi.
Çizelge 4.3. Hasta grubunun cinsiyet, tümör yeri, evre ve risk parametrelerinin dağılımı
Hasta sayısı %
CİNSİYET
Erkek 16 64.0
Kız 9 36.0
TÜMÖR YERİ
Sürrenal 20 80.0
Mediasten 1 4.0
Kranial 1 4.0
Paraspinal 2 8.0
EVRE
2b 2 8.0
Evre 4 21 84.0
Evre 4S 1 4.0
RİSK
Düşük 2 8.3
Yüksek 15 62.5
Orta (iyi-kötü) 7 29.2
Evre 2b Evre 4 Evre 4S P
Ort±SS Medyan (min-max)
Ort±SS Medyan (min-max)
Ort±SS Medyan (min-max)
YAŞ (ay) (n=24)
55.5±74.2 55.5(3.0-
108.0)
33.2±26.2 30.0(1.0-
108.0)
10.0±- 10.0(10.0-
10.0)
0.641
LDH (n=20) 545.5±102.
5
545.5(473.0
-618.0)
1808.2±1
863.3
1242.0(603.0
-7930.0)
673.0±- 673.0(673.0-
673.0)
0.044
NSE (n=22) 35.5±12.0 35.5(27.0-
44.0)
215.7±12
0.4
188.0(20.0-
370.0)
24.0±- 24.0(24.0-
24.0)
0.049
VMA (n=6) 1.0±-- 1.0(1.0-1.0) 133.8±16
4.4
86.5(2.0-
360.0)
75.0±- 75.0(75.0-
75.0)
0.343

49
4.1. Sitogenetik Bulgular
4.1.1. Kan Kültürlerinden Elde Edilen Sitogenetik Bulgular
Hasta grubunda yer alan 25 hastanın 23’ünde (%92) ve kontrol grubunda
yer alan sağlıklı ve ailesinde kanser öyküsü olmayan 25 (%100) çocuğun kan
kültürlerinden kromozom analiz sonuçları elde edildi. Hasta grubundan 2 kişi
yetersiz ve kalitesiz mitoz nedeniyle değerlendirmeye alınamadı. Her iki grup için
kan kültürleri normal besiyerinde (RPMI-1640) yapıldı.
4.1.1.1. Kan Dokularında Saptanan Sitogenetik Bulgular
Çalışmamızda; 50 metafaz plağı incelenerek önceden ya da sonradan
kazanılmış kromozomal bozuklukları belirlendi. Hasta grubu için toplam 1150
metafaz hücresi incelendi. Bu hücrelerin, 212’sinin (%18,4) sayısal ve/veya
yapısal kromozom düzensizlikleri taşıdıkları gözlendi. Hasta grubunda bu
düzensizliklerin %72’sinin yapısal ve %28’inin sayısal olduğu rapor edildi.
Kontrol grubunda ise toplam 1250 metafaz hücresi incelendi. Bu hücrelerin,
33’ünde (%2,64) kromozom düzensizlikleri tespit edildi. Hasta ve kontrol grubu
arasındaki bu fark istatistiksel olarak anlamlı bulundu (p<0,0001). Mevcut olan
kromozom anomalileri Çizelge 4.4’de gösterildi.
Hasta grubunda, toplam 21 kişinin (%84) değişik sayısal ve/veya yapısal
kromozom düzensizlikleri taşıdığı saptandı. Karyotipi normal olan (46,XX ve
46,XY) iki hastanın olduğu kaydedildi (H19 ve H20). Bu düzensizliklerin bazıları
Şekil 4 ve Şekil 4’de gösterilmektedir. Hastalarımızın 18’inde (%72), 1, 2, 3, 4, 5,
7, 8, 9, 10, 11, 12, 13, 14, 15, 17, 18, 19, 20 ve X kromozomlarının belirli
bölgelerini içeren delesyonlar yüksek oranda tespit edildi. Bu delesyonlar
hastalaın bir yada birkaç hüçresindeki dağılımı; birinci hastada H1’de
del(1)(q21.3→qter)(1/50) ve del(8)(q12→qter)(1/50), H3’de
del(1)(q21.3→qter(1/50)), del(2)(p24→pter)(1/50), del(3)(p22→pter)(1/50),
del(4)(p12→pter)(1/50), del(7)(q32.1→qter)(1/50), del(9)(p11→pter)(1/50),
del(9)(q22→qter)(1/50), del(13)(q22.1→qter)(1/50), del(14)(q13→qter)(1/50),
del(X)(q21→qter)(1/50), H4’de del(8)(q11→qter)(1/50), del(10)(q11→qter)(1/50)
ve aynı metafazda del(4)(q31→qter)(1/50) ile del(7)(q32→qter)(1/50) birlikte,
H5’te del(1)(q32→qter)(1/50) ve del(12)(q11→qter)(1/50), H6’da

50
del(3)(p23→pter)(1/50), del(14)(q11.2→pter)(1/50), H7’de
del(20)(p12→pter)(1/50), H8’de del(4)(q22→q25)(1/50), H9’da del(8)(q22-
qter)(1/50), del(13)(p12→pter)(1/50), del(13)(q14→pter)(1/50) ve aynı metafazda
del(6)(p23→pter)(1/50) ile del(8)(p11→pter)(1/50) birlikte, H10’da
del(2)(p23→pter)(1/50) ve del(11)(q13→qter)(1/50), H12’de
del(1)(p32→pter)(1/50), del(1)(p22→pter)(1/50), del(2)(q12→q21)(1/50),
del(5)(q31→qter)(1/50) ve del(15)(q22→qter)(1/50), H13’te
del(2)(p24→pter)(1/50), del(15)(q22→qter)(1/50), del(9)(q22→qter)(1/50) ve
del(14)(q11.2-q13)(1/50), H14’te del(3)(p25→pter)(1/50) ve
del(17)(q25→qter)(1/50), H16’da del(1)(q32→qter)(1/50) ve
del(2)(p24→pter)(1/50), H17’de del(2)(p23→pter)(1/50), H18’de
del(9)(q11.1→qter)(1/50), del(2)(q31→qter)(1/50), del(3)(q11→qter)(1/50),
del(7)(q11.1→qter)(1/50), del(9)(q13→qter)(1/50), del(10)(q24→qter)(1/50),
del(12)(p11→qter)(2/50), del(18)(q21→qter)(1/50), del(19)(q13.3→qter)(1/50) ve
aynı metafazda del(13)(q14.3→p13)(1/50) ile del(15)(q25→qter)(1/50),
del(1)(q32 →qter)(1/50) ile del(3)(cen→qter)(1/50) birlikte, H21’de
del(15)(q22→qter)(1/50), H24’de del(3)(q28→qter)(1/50) ve
del(19)(q13→qter)(1/50) ve H25’te del(2)(p13→pter)(1/50),
del(9)(q32→qter)(1/50) ve del(14)( pter→q11.1)(1/50) düzensizlikleri tespit edildi
(Çizelge 4.4) (Şekil 4.1).
Hastalarımızın 13’ünde (%52) 1, 2, 3, 5, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16,
17, 18, 20, 21, 22 ve X kromozomlarının belirli bölgelerini içeren translokasyonlar
ve buna bağlı olarak derivatif kromozom düzensizlikleri yüksek oranda tespit
edildi. Bu translokasyonlar hastalara göre;H1’de t(7;14)(p22;q24)(1/50), H3’de
t(20;22)(p12→pter;q13)(1/50) ve t(14;16)(q24;p13)(1/50), H4’de
t(8;18)(p11;p11)(1/50), t(3;5)(p21;p11)(1/50) ve t(10;17)(p14;p13)(1/50), H9’de
t(1;?)(p16;?)(1/50), H13’de rbt(13;22)(p11;p11)(1/50), H16’de
t(9;14)(q34;q24.3→qter)(1/50), H23’de aynı metafazda t(5;15)(p12-pter;p11)(1/50)
ile t(2;22)(p11-pter;p12)(1/50) birlikte ve H25’de t(10;14)(q26;q13)(1/50)
düzensizlikleri tespit edildi (Çizelge 4.4)(Şekil 4.1).
Translokasyonlara bağlı olarak oluşan derivatif düzensizlikler hastalara
göre; H3’de der(21)t(21;21)(q22;q22)(1/50), H9’de der(1)(?)(1/50), aynı metafazda

51
der(21)t(?)(1/50), der(14)t(14;?)(q11-pter;?)(1/50) ve der(7)t(7;?)(p11;?)(1/50) ve
aynı metafazda der(7)t(2;7)(q21;p13)(1/50) ile der(4)t(4;?)(q35;?)(1/50), H12’de
der(18)t(13;18)(q11;p11.3)(1/50), H13’de der(4)t(4;7)(p16;q21)(1/50) ve
der(12)t(12;17)(p13;q12)(1/50), H14’de der(X)t(8;X)(p23;p22)(1/50), H17’de
der(8)t(8;22)(p23;q11)(1/50), H18’de der(3)t(3;12)(p26;q15→qter)(1/50) ve
der(11)t(11;18)(q25;pter→q21)(1/50) ve H21’de der(15)t(15;22)(q11;q11)(1/50)
tespit edildi (Çizelge 4.4) (Şekil 4.1).
Hastalarımızın 4’ünde (%16) 1, 3, 7 ve 9 kromozomlarını içeren frajilitelerin
olduğu ve bunların hastaların birer metafazında olmak üzere dağılımları; H1’de
fra(9)(q22), H3’de fra(12)(q24), H7’de fra(11)(q23), H9’de fra(2)(q21), fra(2)(q31)
ve fra(7)(q36) tespit edildi (Çizelge 4.4) (Şekil 4.1).
Hastalarımızın 8’inde (%32); 1, 2, 3, 4, 5, 8, 9, 11, 12, 19 ve X
kromozomlarının belirli bölgelerini içeren kromatit kırıkları (chtb), hastalarımızın
5’inde (%20) 4, 8 ve 9 kromozomlarının belirli bölgelerini içeren kromozom
kırıkları yüksek oranda kaydedildi. Bunların hastalara göre dağılımı; H3’de
chtb(5)(q33)(1/50), H6’da chtb(3)(q21)(1/50), H9’da chtb(19)(q13)(1/50),
chbr(2)(q31)(1/50) ve chtb(1)(q21)(q32)(1/50), H10’da chtb(X)(q22)(1/50),
chtb(3)(p13)(1/50) ve chtb(2)(q21)(1/50), H14’de chtb(2)(p11)(1/50) ve
chtb(11)(q23)(1/50), H16’da chtb(2)(q32.2)(1/50), H17’de chtb(4)(q31)(1/50) ve
H21’de chtb(12)(q14.1)(2/50) kromatit kırıkları tespit edildi. H3’te
chrb(9)(q11.1)(1/50); H4’te chrb(4)(q31)(1/50), H9’da chbr(2)(q31)(1/50),
H11’de chrb(8)(q22)(1/50) ve H12’de chrb(8)(q23)(1/50) kromozom kırıkları
tespit edildi (Çizelge 4.4) (Şekil 4.1).
Ayrıca, hastalarımızın 3’ünde (%12) 2, 5, 12 ve 19 kromozomlarının
belirli bölgelerini içeren duplikasyonlar; H3’te dup(12)(q24→qter)(1/50) ve
dup(2p)(1/50), H4’te dup(19)(p13.2)(1/50), H24’te ?dup(5)(p12→p14)(1/50).
Hastalarımızın 2’inde (%8) 5, 9 ve 15 kromozomlarının belirli bölgelerini içeren
gapler; H5’te gap(5)(q31)(1/50), H15’te gap(15)(q15)(1/50); hastalarımızın
3’ünde (%12) 3, 9 ve 16 kromozomlarının belirli bölgelerini içeren inversiyonlar;
H4’te inv(13)(p13;q14)(1/50), H11’de inv(9)(p11;q12)(1/50), H14’te
inv(16)(p11.2-pter;q24)(1/50) ve hastalarımızın 1’inde (%4) insersiyon; H21’de
ins(3;8)(q11; q11-q24)(1/50) tespit edildi (Çizelge 4.4) (Şekil 4.1).

52
Yapısal kromozom düzensizliklerinin yanı sıra otozomal ve gonozomal
kromozom anöploidileri gözlendi. Üç hastada (%12) gonozomal kromozom
anöploidileri (X, XYY) gözlendi. Bu hastaların 2’sinde (%8) H18 ve H21’de
47,XYY ve 1’inde (%4) (H12) 45,X kromozom kuruluşuna rastlandı. On iki
hastada (%48); (H3, H4, H6, H7, H8, H9, H12, H13, H17,H23, H24, H25) azalış
ve artışlar şeklinde sayısal otozomal kromozom anöploidilerine rastlandı.
Özellikle; 4, 6, 7, 8, 9, 10, 12, 13, 14, 15, 16, 18, 19, 20, 21, 22
kromozomlarında azalış; 1, 7, 10, 13, 15, 16, 19, 20, 21, 22 kromozomlarında
artış şeklinde anöploidiler gözlendi. Ayrıca, asentrik (H1, H9, H12, H18), marker
(H16), double minutes (H3, H21) ve p+ (H6, H9, H12) kromozomlar da
gözlenen diğer anormal kromozomal bulgular olarak kaydedildi (Çizelge 4.4).
Sayısal ve yapısal düzensizlikler dışında 9qh+ (H10, H24, H25), Yqh+
(H4) ve 15p+ (H21) gibi kromozomal polimorfik yapılar da gözlendi (Çizelge 4.4)
Kontrol grubunda 25 kişinin (%100) normal karyotipe sahip olduğu
gözlendi (46,XX/46,XY). Kontrol grubundan onunda (%40) gaplere; bunların
hastalara göre dağılımı; K1’de 1/50 oranında, K4’te 1/50, K6’da 3/50, K7’de
2/50 oranında gap(1)(q32) ve K9’da 1/50 oranında gap(1)(q36); K7’de 1/50
oranında gap(3)(q27), K1 ve K3’te 1/50 oranında gap(4)(q31), K2 ve K22’de
1/50 oranında gap(4)(q27), K16 ve K19’da 1/50 oranında gap(7)(q22), K1’de
1/50 oranında gap(5)(q32) ve gap(10)(q24) ve K2’de 1/50 oranında gap(5)(q31)
tespit edildi. Dokuzunda (%36) frajil bölgelere ve ikisinde (%8) kromatit
kırıklarına rastlandı. Bu düzensizliklerin hastalara göre dağılımı; frajil bölgeler
K2’de fra (X)(q27)(1/50), K4’de fra(X)(q26)(1/50), K6’da fra(3)(p25)(1/50) ve
fra(7)(q33)(1/50), K10’da fra(12)(p13)(1/50), K11’de fra(7)(q36)(1/50) ve
far(15)(q24)(1/50), K14’te fra(4)(q27)(1/50), K17’de fra(X)(q27)(1/50), K19’da
fra(1)(q42)(1/50), K23’te fra(5)(q31)(1/50) ve K25’de fra(1)(q32)(1/50) ve
fra(1)(q42)(1/50) tespit edildi. Kromatit kırıkları da K15’te chtb(12)(q13)(1/50)
ve K25’te chtb(X)(p22)(1/50) saptandı. Ayrıca, K5’te 5/50, K9’da 2/50, K15 ve
K18’de 3/50 ve K25’de 1/50 oranında 9qh+ heteromorfizmlerine rastlandı
(Çizelge 4.4)(Şekil 4.2).

53
Çizelge 4.4. Hasta ve kontrol grubunun lenfosit hücrelerinde gözlenen yapısal ve sayısal
kromozom düzensizlikleri
Yapısal ve Sayısal Kromozom Düzensizlikleri (Anomali Saptanan Hücre Sayısı/Total Hücre Sayısı) Has. No.
Kont. No.
H1 46,XY,del(1)(q21.3→qter) (1/50)
46,XY,t(7;14)(p22;q24) (1/50)
47,XY,+ace (1/50)
46,XY,del(8)(q12→qter) (1/50)
46,XY,fra(9)(q22) (1/50)
K1 46,XY,gap(1)(q32) (1/50)
46,XY,gap(5)(q32) (1/50)
46,XY,gap(4)(q31) (1/50)
46,XY,gap(10)(q24) (1/50)
H2 Üreme olmadı. K2 46,XY,gap(5)(q31) (1/50)
46,XY,fra (1)(q32)(1/50)
46,XY,gap(4)(q27) (1/50)
H3 46,XX,del(1)(q21.3→qter) (1/50)
46,XX,del(7)(q32.1→qter) (1/50)
46,XX,del(13)(q22.1→qter) (1/50)
45,XX,chrb(9)(q11.1),-15 (1/50)
45,XX,del(9)(p11→pter),-21 (1/50)
46,XX,del(2)(p24→pter) (1/50)
46,XX,t(20;22)(p12→pter;q13),del(X)
(q21→qter) (1/50)
46,XX,dup(12)(q24→qter),fra(12)(q24) (1/50)
46,XX,dup(2p) (1/50)
46,XX,del(4)(p12→pter) (1/50)
46,XX,+1,-14,del(9)(q22→qter) (1/50)
45,XX,-8 (1/50)
46,XX,del(3)(p22→pter) (1/50)
45,XX,-10 (1/50)
51,XX,der(21)t(21;21)(q22;q22),+5dmin(1/50)
46,XX,chtb(5)(q33) (1/50)
46,XX,del(14)(q13→qter) (1/50)
46,XX,t(14;16)(q24;p13) (1/50)
K3 46,XX,gap(4)(q31) (1/50)
H4 46,XY,Yq+ (50/50)
46,XY,inv(13)(p13;q14),Yq+ (1/50)
46,XY,del(4)(q31→qter),del(7)(q32→qter),Yq+
(1/50)
46,XY,t(8;18)(p11;p11),Yq+ (1/50)
K4 46,XX,gap(1)(q32) (1/50)
46,XX,far(X)(q26) (1/50)

54
45,XY,t(3;5)(p21;p11),-8 Yq+ (1/50)
46,XY,t(10;17)(p14;p13),Yq+ (1/50)
46,XY,del(10)(q11→qter),Yq+ (1/50)
46,XY,chrb(4)(q31),Yq+ (1/50)
46,XY,dup(19)(p13.2),Yq+ (1/50)
46,XY,del(8)(q11→qter), Yq+ (1/50)
H5 46,XX,hsr(2)(q13→q21)(25/50)
46,XX,del(1)(q32→qter) (1/50)
46,XX,del(12)(q11→qter) (1/50)
46,XX,gap(5)(q31) (1/50)
K5 46,XY,9qh+ (5/50)
H6 43,XY,del(3)(p23→pter),-4,-8,-18 (1/50)
47,XY,+21 (1/50)
46,XY,chtb(3)(q21) (1/50)
46,XY,8p+,del(14)(q11.2→pter)(1/50)
K6 46,XY,gap(1)(q32) (3/50)
46,XY,fra(3)(p25) (1/50)
46,XY,fra(7)(q33) (1/50)
H7 46,XY,hsr(2)(q11→q21) (20/50)
45,XY, -16,del(20)(p12→pter) (1/50)
46,XY, fra(11)(q23) (1/50)
45,XY,-20 (1/50)
45,XY,-19 (1/50)
K7 46,XX,gap (1)(q32) (2/50)
46,XY,gap(3)(q27) (1/50)
H8 46,XY,del(4)(q22→q25) (1/50)
46,XY,?10q/8q (1/50)
44,XY,-18,-21 (1/50)
43, XY, -15,-18,-19 (1/50)
45,XY,-21 (1/50)
K8 46,XX
H9 46,XY,fra(2)(q21),del(13)(q14→pter) (1/50)
Endoreduplikasyon (1/50)
46,XY,del(13)(p12→pter),
chtb(19)(q13),fra(2)(q31),der(21)t(?),
der(14)t(14;?)(q11pter;?),
der(7)t(7;?)(p11;?),t(1;?)(p16;?) (1/50)
46,XY,der(7)t(2;7)(q21;p13),
chbr(2)(q31),der(4)t(4;?)(q35;?),-19,-22x2(1/50)
46,XY,del(8)(q22-qter) (1/50)
55,XY,der(1)(?),-4,+10x2,+13,+20x2,
+21,+22x2,+ace (1/50)
53,XY,-6,+1,+7,+16x2,+19x2,+21,+22 (1/50)
46,XY,chtb(1)(q21)(q32),
del(6)(p23→pter),fra(7)(q36), del(8)(p11→pter),
K9 46,XY,gap(1)(q36) (1/50)
46,XY,9qh+ (2/50)

55
10p+,+ace (1/50)
H10 46,XX,9qh+ (50/50)
46,XX,hsr(2)(q11→q21),9qh+ (25/50)
46,XX,hsr(2)(q11→q21),chtb(3)(p13),9qh+
(1/50)
46,XX,del(2)(p23→pter),9qh+ (1/50)
46,XX,chtb(X)(q22),9qh+ (1/50)
46,XX,chtb(2)(q21),9qh+ (1/50)
46,XX,del(11)(q13→qter),9qh+ (1/50)
K10 46,XY,fra(12)(p13) (1/50)
H11 46,XX,9qh+ (50/50)
46,XX,hsr(2)(q11→q21),9qh+ (8/50)
46,XX,chrb(8)(q22),9qh+ (1/50)
46,XX,inv(9)(p11;q12),9qh+ (1/50)
45,XX,hsr(2)(q11→q21),9qh+,-17 (1/50)
K11 46,XY,fra(15)(q24) (1/50)
46,XY,fra(7)(q36) (1/50)
H12 48,XX,+ace(1)(q21→qter),+16 (1/50)
46,XX, del(2)(q12→q21) (1/50)
46,XX,del(5)(q31→qter) (1/50)
46,XX,del(15)(q22→qter) (1/50)
45,X,-8,-9,-X,+21 (1/50)
47,XX,?+(p) (1/50) 46,XX,der(18)t(13;18)(q11;p11.3) (1/50)
45,XX,del(1)(p32→pter),-19 (1/50)
46,XX,del(1)(p22→pter) (1/50)
46,XX,chrb(8)(q23) (1/50)
45,XX,-19 (1/50)
K12 46,XY
H13 46,XX,del(15)(q22→qter) (1/50)
45,XX,rbt(13;22)(p11;p11) (1/50)
45,XX,del(9)(q22→qter),-21 (1/50)
46,XX,del(2)(p24→pter) (1/50)
46,XX,der(4) t(4;7)(p16;q21) (1/50)
46,XX,der(12)t(12;17)(p13;q12) (1/50)
45,XX,-8,del(14)(q11.2-q13) (1/50)
45,XX,-21 (2/50)
K13 46,XX
H14 46,XY,del(17)(q25→qter) (1/50)
45,XY,der(X) t(8;X)(p23;p22) (1/50)
46,XY,del(3)(p25→pter) (1/50)
46,XY,inv(16)(p11.2-pter;q24) (1/50)
46,XY,chtb(2)(p11) (1/50)
K14 46,XY,fra(4)(q27) (1/50)

56
46,XY,chtb(11)(q23) (1/50)
H15 46,XY,hsr(2)(q11→q21),gap(9)(p13)(1/50)
46,XY,gap(15)(q15) (1/50)
K15 46,XY,9qh+ (3/50)
46,XY,chtb(12)(q13) (1/50) H16 47,XX,+mar (1/50)
46,XX,del(1)(q32→qter),
t(9;14)(q34;q24.3→qter) (1/50)
46,XX,hsr(2)(q11-q21) (1/50)
46,XX,chtb(2)(q32.2) (1/50)
46,XX,del(2)(p24→pter) (1/50)
K16 46,XX,gap(7)(q22) (1/50)
H17 46,XX,del(2)(p23→pter) (1/50)
45,XX,der(8)t(8;22)(p23;q11),-22 (1/50)
46,XX,chtb(4)(q31) (1/50)
K17 46,XY,fra(X)(q27) (1/50)
H18 46,XY,del(9)(q11.1→qter) (2/50)
46,XY,der(3)t(3;12)(p26;q15→qter)(1/50)
46,XY,del(12)(p11→qter)(2/50)
46,XY,del(10)(q24→qter) (1/50)
46,XY,del(13)(q14.3→p13),
del(15)(q25→qter)(1/50)
45,XY,der(11)t(11;18)(q25;pter→q21),
del(18)(q21→qter) (1/50)
46,XY,del(19)(q13.3→qter) (1/50)
47,XYY (1/50)
46,XY,del(3)(q11→qter) (1/50)
46,XY,del(1)(q32→qter),del(3)(cen→qter)(1/50)
46,XY,del(9)(q13→qter) (1/50)
46,XY,del(7)(q11.1→qter) (1/50)
47,XY,+ace, del(2)(q31→qter) (1/50)
K18 46,XY,9qh+ (3/50)
H19 46,XX (50/50) K19 46,XX,fra(1)(q42) (1/50)
46,XX,gap(7)(q22) (1/50)
H20 46,XY (50/50) K20 46,XX
H21 46,XY,15p+ (50/50)
46,XY,der(15)t(15;22)(q11;q11)(1/50)
47,XYY,15p+ (1/50)
46,XY, 15p+, dmin (10<) (1/50)
46,XY,del(15)(q22→qter) (1/50)
46,XY,chtb(12)(q14.1),15p+ (2/50)
45,XY,ins(3;8)(q11; q11-q24),15p+ (1/50)
K21 46,XY
H22 Üreme olmadı K22 46,XY,gap(4)(q27) (1/50)

57
H23 45,XX,-22 (1/50)
44,XX,-13,-22 (1/50)
44,XX,-7,-22 (1/50)
45,XX,-13 (1/50)
44,XX,-13,-16 (1/50)
46,XX,t(2;22)(p11-pter;p12), t(5;15)(p12-
pter;p11) (1/50)
K23 46,XX,fra(5)(q31) (1/50)
H24 46,XX,9qh+ (50/50)
46,XX,del(3)(q28→qter), ?dup(5)(p12→p14),
9qh+ (1/50)
45,XX,del(19)(q13→qter),9qh+,-12 (1/50)
K24 46,XX
H25 46,XY,9qh+ (25/50)
46,XY,9qh+,t(10;14)(q26;q13) (1/50)
47,XY,+15 (1/50)
46,XY,del(9)(q32→qter) (1/50)
46,XY,del(2)(p13→pter),9qh+,+21,-13 (1/50)
46,XY,9qh+,del(14)(q11.1→pter) (1/50)
K25 46,XX,fra(1)(q42) (1/50)
46,XX,fra(1)(q32) (1/50)
46,XX,chtb(X)(p22) (1/50)
46,XX,9qh+ (1/50)
Şekil 4.1. Hasta grubunda gözlenen kromozom düzensizliklerinden bazılarının kısmi karyotip
görüntüleri

58
Şekil 4.1. (Devamı)

59
Şekil 4.1. (Devamı)

60
Şekil 4.1. (Devamı)
Şekil 4.2. Kontrol grubunda saptanan kromozom anomalilerinden bazıları

61
Çizelge 4.5. Hasta grubunda saptanan yapısal düzensizliklerin her kromozom için bölgelere göre
dağılımı
Kromozom İdiyogramı

62
Çizelge 4.5. (Devamı) Kromozom bölgelerinde gözlenen yapısal düzensizliklerin yoğunluğu
(delesyon, translokasyon, kromatit kırığı, kromozom kırığı, hsr bölgesi, duplikasyon, frajil bölge,
gap bölgesi, asenstrik bölge, insersiyon ve inversiyon).
Hasta ve kontrol grubunda saptanan düzensizliklerin bölgele sayısının
kromozom bölgelerine göre dağılımı, populasyondaki sıklığı (hücre başına
düşen anomali sayısı=saptanan anomali sayısı/incelenen hücre sayısı), nisbi
oranı (saptanan anomali sayısı/toplam anomali sayısı) ve önemlilik derecesi

63
Çizelge 4.6’te gösterildi. Buna göre; hasta grubunda düzensizliklerin en yoğun
olduğu kromozom bölgeleri önemlilik sırasına göre; 1q21, 2q21, 14q11.2, 1q32,
2p24, 2q31, 4q31, 9q11, 9q22, 13q14, 14q24 ve 15q22 şeklinde sıralandığı
görülmektedir. Kontrol grubunda ise; 1q32 şeklindedir. Hasta grubunda; 212
hücrede, 102 kromozomal bölgede 151 tane yapısal kromozom düzensizliği
olduğu saptandı. Kontrol grubunda 20 kromozomal bölgede 33 yapısal
kromozomal düzensizliği bulundu (Çizelge 4.6.).
Çizelge 4.6. Hasta ve kontrol grubunda bulunan hasarlı bölgelerin sayısı, hücre başına düşen
hasarlı bölge sayısı, nisbi oranı ve önemlilik sırası
Krom. Böl.
Hasta Grubu (25) Kontrol Grubu (25) Krom. Hasar sayısı
Hücre Başına Düşen Hasar Sıklığı (%)
Nisbi Oran*
Sıra Krom. Hasar Sayısı
Hücre Başına Düşen Hasar Sıklığı (%)
Nisbi Oran*
Sıra
1p16 1 0,087 0,66 4
1p22 1 0,087 0,66 4
1p32 2 0,174 1,32 3
1q21 4 0,348 2,64 1
1q32 3 0,261 1,98 2 8 0,64 24,24 1
2p11 2 0,174 1,32 3
2p23 2 0,174 1,32 3
2p24 3 0,261 1,98 2
2q12 1 0,087 0,66 4
2q21 4 0,348 2,64 1
2q31 3 0,261 1,98 2
2q32.2 1 0,087 0,66 4
3p13 2 0,174 1,32 3
3p21 1 0,087 0,66 4
3p22 1 0,087 0,66 4
3p23 1 0,087 0,66 4
3p25 1 0,087 0,66 4 1 0,08 3,03 3
3p26 1 0,087 0,66 4
3q11 2 0,174 1,32 3
3q21 1 0,087 0,66 4
64
3q28 1 0,087 0,66 4
3cen 1 0,087 0,66 4
4p12 1 0,087 0,66 4
4p16 1 0,087 0,66 4
4q22 1 0,087 0,66 4
4q25 1 0,087 0,66 4
4q31 3 0,261 1,98 2 2 0,16 6,06 2
4q35 1 0,087 0,66 4
5p11 1 0,087 0,66 4
5p12 2 0,174 1,32 3
5p14 1 0,087 0,66 4
5q31 2 0,174 1,32 3 2 0,16 6,06 2
5q33 1 0,087 0,66 4
6p23 1 0,087 0,66 4
7p11 1 0,087 0,66 4
7p13 1 0,087 0,66 4
7p22 1 0,087 0,66 4
7q11 1 0,087 0,66 4
7q21 1 0,087 0,66 4
7q32 2 0,174 1,32 3
7q36 1 0,087 0,66 4 1 0,08 3,03 3
8p11 2 0,174 1,32 3
8p23 2 0,174 1,32 3
8q11 2 0,174 1,32 3
8q12 1 0,087 0,66 4
8q22 2 0,174 1,32 3
8q23 1 0,087 0,66 4
8q24 1 0,087 0,66 4
9p11 2 0,174 1,32 3
9p13 1 0,087 0,66 4
9q11 3 0,261 1,98 2
9q12 1 0,087 0,66 4
9q13 1 0,087 0,66 4
9q22 3 0,261 1,98 2
9q32 1 0,087 0,66 4
9q34 1 0,087 0,66 4
10p14 1 0,087 0,66 4
65
10q11 1 0,087 0,66 4
10q24 1 0,087 0,66 4 1 0,08 3,03 3
11q13 1 0,087 0,66 4
11q23 1 0,087 0,66 4
11q25 1 0,087 0,66 4
12p11 1 0,087 0,66 4
12p13 1 0,087 0,66 4 1 0,08 3,03 3
12q11 1 0,087 0,66 4
12q14 2 0,174 1,32 3
12q15 1 0,087 0,66 4
12q24 2 0,174 1,32 3
13p11 1 0,087 0,66 4
13p13 2 0,174 1,32 3
13q11 1 0,087 0,66 4
13q12 1 0,087 0,66 4
13q14 3 0,261 1,98 2
13q22 1 0,087 0,66 4
14q11.2 4 0,348 2,64 1
14q13 2 0,174 1,32 3
14q24 3 0,261 1,98 2
15p11 1 0,087 0,66 4
15q11 1 0,087 0,66 4
15q15 1 0,087 0,66 4
15q22 3 0,261 1,98 2
15q25 1 0,087 0,66 4
16p11.2 1 0,087 0,66 4
16p13 1 0,087 0,66 4
17p13 1 0,087 0,66 4
17q12 1 0,087 0,66 4
17q25 1 0,087 0,66 4
18p11 1 0,087 0,66 4
18p11.3 1 0,087 0,66 4
18q21 2 0,174 1,32 3
19p13.2 1 0,087 0,66 4
19q13.1 2 0,174 1,32 3
19q13.3 1 0,087 0,66 4
20p12 2 0,174 1,32 3

66
21q22 2 0,174 1,32 3
22p11 1 0,087 0,66 4
22p12 1 0,087 0,66 4
22q11 2 0,174 1,32 3
22q13 1 0,087 0,66 4
Xp22 1 0,087 0,66 4 1 0,08 3,03 3
Xq21 1 0,087 0,66 4
Xq22 1 0,087 0,66 4
Diğer 16
Toplam 151 13,13 33 2,64
4.2. Moleküler Sitogenetik Bulgular
Çalışamamızın moeküler sitogenetik kısmı 9 hasta üzerinde yapıldı.
Hastaların parafin dokularından alınan kesitlere FISH yöntemi uygulanarak
NMYC ve AURKA genlerinin amplifikasyon değerleri; hastaların yaşı, hastalığın
evresi, tümörün yeri ve patolojisi ile karşılaştırldı. İstatistiksel olarak
karşılaştırılan değerler anlamsız bulundu.
4.2.1. Parafin Dokulardan Elde Edilen Moleküler Sitogenetik Bulgular
İncelenen 9 olgunun, 2 tanesi (%22.22) kız çocuk ve 7 tanesi (%77.78)
erkek çocuktu. Hastaların ortalama yaşları 43 ay idi. Olguların 9’unda da
(%100) tümörün yeri sürrenal bölgedeydi. Tümör lokalizasyonu ile MYCN
amplifikasyonu, AURKA amplifikasyonu ve patterni, hastalığın gidişi arasında
istatistiksel olarak anlamlı bir fark saptanmadı. Ayrıca, klinik izlemlerine ulaşılan
9 hastanın 2’sinin evre 2b ve 7’sinin evre 4 oldukları saptandı. Olguların
dokuzunun MYCN ve AURKA amplifikasyonu değerlendirildi. 9 olgunun 6’sında
(%66,7) AURKA amlifikasyonu ve MYCN amlifikasyonu gözlendi (Şekil 4.3). 6
hastada AURKA’nın ortalama amplifikasyon değeri %19, MYCN
amlifikasyonunun ortalama değeri %23 olarak hesaplanmıştır. Evre, cinsiyet ve
riske göre AURKA, MYCN değerleri karşılaştırılmıştır ancak hasta sayısı düşük
olduğu için p değeri istatistiksel olarak anlamlı çıkmamıştır. Hasta verileri
Çizelge 4.7’ de özetlenmiş, FISH görüntüleri Şekil 4.3’te gösterilmiştir.

67
Şekil 4.3 Hastaların AURKA (a) ve MYCN (b,c,d) genlerine ait FISH görüntüleri
Çizelge 4.7. 9 Hastanın klinik ve biyolojij verileri
Hasta
no
Cinsiyet Yaş
(ay)
Evre Tümör
yeri
Risk
grubu
VMA LDH NSE MYCN
amp.
AURKA
amp.
H4 Erkek 108 2b Sürrenal Orta-
iyi
1 473 44 %8 %7
H7 Erkek 42 4 Sürrenal Orta-
kötü
- 4381 370 %32 %25
H8 Erkek 6 4 Sürrenal Yüksek - 967 327 %36 %25
H9 Erkek 48 4 Sürrenal Orta-
kötü
23 1242 316 %25 %22
H10 Kız 3 2b Sürrenal Düşük - 618 27 %8 %5
H14 Erkek 6 4 Sürrenal Yüksek - 680 106 %12 %15
H15 Erkek 54 4 Sürrenal Yüksek 150 - 185 %18 %12
H17 Kız 72 4 Sürrenal Yüksek - - - %14 %15
H20 Erkek 48 4 Sürrenal Yüksek - 781 117 %4 %8

68
5. TARTIŞMA
Nöroblastom köken aldığı yere, yaşa ve evreye göre değişebilen, metastaz
yapması, histolojisi ve paraneoplastik sendromlar nedeniyle diğer çocukluk çağı
kanserlerinden farklıdır. Nöroblastik tümörler, en sık 1-4 yaş arasındaki çocuklarda
görülür ve ortalama yaş 18 ay olarak bildirilir3,101. Hastalarımızın bir kısmı, genel
yaş ortalamsa ile aynı aralıkta olmasına rağmen, ortalama yaş 33.4 ay olup bu
yaş ortalaması, genel ortalamadan 15.4 ay daha büyüktür. Başvuru yaşındaki
farklılık ailelerin sosyo-ekonomik düzeyi ve yaşadığı yerde hekime ulaşma olanağı
gibi nedenler ile ilişkili olabilir.
Evrelere göre hastalarımızın ortalama tanı yaşları; evre 4S, 2b ve 4 için
sırasıyla; 10, 55.5 ve 33.2 ay civarında olup evre 2b, literatüre göre daha
yüksektir. NBda, yaşın önemli bir yaşam belirleyici faktör olduğu bilinmektedir.
Tanı anında bir yaşından küçük çocuklarda hastalık yaygın olsa bile daha iyi
seyretmektedir102,103. İleri evrelerde, yaşın yüksek olmasının sebebi yine geç
başvuru yaşı olabilir. NB, erkeklerde kızlara göre biraz daha sık görülür ve bu oran
1.2 civarındadır3,101. Hastalarımızda ise erkek/kız oranı 1.27 olup bu oran,
literatürlerdeki oranlar ile uyumlu çıkmaktadır. Literatürde; LDH ve NSE
değerlerinin yüksekliği ile ileri evreler ilişkilendirilmiştir2,85. Nitekim, evre 4’te yer
alan hastalarımızın LDH ve NSE değerleri de yüksek seyretmiş ve bu değerler
istatistiksel olarak anlamlı bulunmuştur (p=0.044 ve p=0.049) (Çizelge 4.2).
5.1. Sitogenetik
Kromozomların sıcak bölgeleri olarak ifade edilen proto-onkogen ve
onkogenlerin bulunduğu kritik lokuslarda oluşan yapısal düzensizlikler, bu genleri
uyarılmak ve/veya baskılamak suretiyle kanserleşmeye neden olduğu
bilinmektedir. Bu nedenle, kromozom hasarları kanserlerin oluşumunda ve
prognozunda önemli göstergeler olarak kabul edilmektedir. Bu güne kadar, NBlı
hastalar üzerinde yapılan sitogenetik, moleküler sitogenetik ve moleküler genetik
çalışmalarında; NBdan sorumlu olabilecek belirli kromozomal düzensizlikleri ve

69
gen/genler bildirilmiştir16,17. Bu hastalarda, çeşitli kromozomal düzensizlikleri
saptanmış; spesifik NMYC geni amplifikasyonu, kromozom 1p delesyonu, 11q
delesyonu, 17q artışı ve DNA miktarı ile hastalığın prognozu ilişkilendirilmiştir16,17.
Çalışmamızda; lenfosit hücrelerinde NBya neden olabilecek çok önemli
kromozom düzensizlikleri (yapısal ve sayısal) rapor edildi. Bu kromozom
düzensizlikleri bakımından, hasta (%18,43) ve kontrol (%2,64) grubu arasındaki
farkın istatistiksel olarak anlamlı olduğu bulundu (p<0,0001). Bu da, NBlı
hastalarda kromozomal hasarların önemli ölçüde arttığını yani, genetik
dengesizlilik/hasarın hastalığın etiyolojisinin bir göstergesi olduğunu
göstermektedir. Nitekim diğer bir çalışmada; NB hastalarında heterojen kromozom
düzensizliklerinin rastlandığı bildirildi5. (Çizelge 4.4).
Hastalarımızda; bulduğumuz yapısal kromozom düzensizliklerin %72’sinin
1, 2, 3, 4, 5, 7, 8, 9, 10, 11, 12, 13, 14, 15, 17, 18, 19, 20 ve X kromozomlarındaki
kayıplardır (delesyon) (Çizelge 4.4 ve 4.5)(Şekil 4.1). Özellikle, 1.
kromozomundaki p32[2], q21[4] ve q32[3] bölgelerinin sıklığı ve nisbi oranları
dikkate değerde bulundu (Çizelge 4.6 ve 4.4). Birinci koromozomunun kısa
kolunun birçok tümör süpressör geni ihtiva ettiği ve solid tümörlerin %54’ünde
bulunduğu özellikle, 1p32-p36 bölgesinde heterozigozite kayıplarının olduğu
doğrulanmıştır104. NBların %86’sinda, 1p de LOH kayıplarının olduğu rapor
edilmiştir105. Birinci kromozomun kısa kolunda TP73, DR3, HRK3 ve AML2 gibi
aday tümör süpresör genlerin bulunduğu bildirilmiştir106. Bununla birlikte,
kromozomun p36 bölgesinde, TP73 tümör süpressör geni haritalanmış ve bu
genin baskılanması NBda etkili olduğu bildirilmiştir64. Çalışmamız ve diğer
çalışmalarda; 1. kromozomun p22 ve p32 bölgelerinin NB kanser öyküsünde
önemli olduğu ortaya çıkmaktadır. Kromozomun q23-q31 bölgesinde bulunan
NTRK1 (TRK-A) nörotrofin reseptör geninin aktivasyonu ile hücre farklılaşması
uyarılmakta, anjiyogenez baskılanarak tümörün gerilemesi sağlanırken hücre
apoptoza yönlendirilmektedir75,76. Tüm bu bilgiler ışığında; 1. kromozomun tümör
oluşumunda önemli genleri ihtiva ettiği; özellikle q23, q31, p32 ve p36 bölgelerinin
NB gelişiminde kritik bölgeler olduğu söylenebilir. Ayrıca, diğer çalışmalarda
rastlanmayan ancak, hastalarımızda sık rastladığımız 1p22 ve 1q21.3 bölgelerinin
de NB oluşumunda önemli olabileceğini söyleyebiliriz.

70
Hastalarımızda; 2. kromozomda yüksek sıklıkta hasar gören p11[2], p23[2],
p24[3], q21[4] ve q31[3] bölgelerinden özellikle p24[3], q21[4] ve q31[3]
bölgelerinde delesyonlar, frajilite, kırık ve translokasyonların sıklığı ve nisbi oranı
yüksek oranda bulundu (Çizelge 4.6 ve 4.4). Delesyonlar; del(2)(p23-pter) ve
del(2)(p24-pter) bölgelerde sık olup, kromozomun kısa kolunda kayıplar şeklinde
gözlenmektedir (Çizelge 4.4, 4.5 ve 4.6)(Şekil 4.1). Bununla birlikte, q21
bölgesinin yüksek oranda kırıldığı ve delesyona uğradığı görülmektedir. İkinci
koromozomun p24.1 bölgesinde hastalıkla direkt ilişkilili olan MYCN proto-
onkogeni, p24 bölgesinde ise DDX1 onkogeni yer almaktadır. MYCN
amplifikasyonu kanser prognozunun en önemli göstergelerinden birisidir. Nitekim,
hastalarımızda p24 bölgesinin fazla oranda hasar gözlenmiş olması da ve bu
bölgenin NBnın etiyolojisinde sıcak/hassas bir bölge olduğunu doğrulamaktadır.
Literatür bilgilerinde DDX1 onkogenin, yüksek malignant yapıda bazı MYCN
amplifiye tümörlerde payı olduğu düşünülmektedir79. Hastalarımızda, bulduğumuz
q21 ve q31 bölgelerinin de NBnın etiyolojisinde önemli bölgeler olduğunu
söyleyebiliriz. İkinci kromozomun sıcak bölgeleri olarak ifade edilen proto-onkogen
ve onkogenlerin bulunduğu kritik lokuslarda yapısal düzensizlikler sonucu bu
genler uyarılmak ve/veya baskılanmak suretiyle kanserleşmeye neden olduğunu
söyleye biliriz.
Üçüncü kromozomun, çeşitli kanser türlerinin etiyolojisinde etkin genler
taşıdığı bilinmektedir. Nitekim, çalışmamızda da hasarların sıklığı ve nisbi oranı
yüksek kromozomlardan biri 3. kromozomdur (Çizelge 4.4, 4.5 ve 4.6)(Şekil 4.1).
Bu kromozomun özelikle, kısa kolunda yüksek sıklıkta hasarlara rastlandı. Bunlar
arasında; p13[2], p21[2] ve q11[2] bölgeleri en hassas noktalar olarak rapor edildi
(Çizelge 4.4, 4.5 ve 4.6). Bu hasarlar; del(3)(p22-pter), del(3)(p23-pter),
del(3)(p25-pter), del(3)(q11-qter), del(3)(q28-qter), del(3)(cen-qter),
t(3;5)(p21;p11), der(3)t(3;12)(p26;q15-qter), chtb(3)(p13), chtb(3)(q21),
ins(3;8)(q11;q11-q24) şeklinde kaydedildi (Çizelge 4.4, 4.5 ve 4.6)(Şekil 4.1).
Üçüncü kromozomun kısa kol delesyonları, NB hastalarının %15’inde
tanımlanmaktadır71. Yapılan çalışmalarda; NB hastalarında 3p14.2 bölgesinde
yerleşik FHIT geninde allel kayıplarının bulunduğu bildirilmiştir71. Diğer bazı
çalışmalarda; 3p21.3’te yer alan RASSF1 geninin erken ve geç evre NBlarda

71
inaktif olduğu gösterildi107. Bulgularımzda da kromozom 3’teki p13[2], p21[2] ve
q11[2] bölgelerindeki gen/genlerin NBnın gelişiminde rol aldığını doğrulamaktadır.
Hastalarımızda, 4. kromozomda; del(4)(p12→pter), del(4)(q31→qter),
del(4)(q22→q25)], der(4)t(4;7)(p16;q11), der(4)t(4;?)(q35;?) ve chbr(4)(q31),
chtb(4)(q31) şeklinde hasarlar rapor edildi (Çizelge 4.4, 4.5 ve 4.6)(Şekil 4.1).
Özellikle, 4q31 bölgesinin sıklığı ve nisbi oranı yüksek bulundu (Çizelge 4.6).
Schleiermacher ve arkadaşları (2007) da NBlı hastalarda, 4p bölge kayıplarını
evre 2a ve 2b tümörlerde %22 oranında bulmuşlardır108. 4p bölgesinde, hastalıkla
ilişkili tümör supresör genler tanımlanmıştır16. Hastalarımızda, kısa kolun değişik
bölgelerinde hasar görülmekle birlikte en sık hasarın uzun kolun q31 bölgesinde
görülmesi bu bölgenin de NB etiyolojisinde etkin olduğunu göstermektedir. Bu
bölgeyle ilgili fazla bilgi olmasa da bazı çalışmalarda; 4q33-35 bölgesinin
amplifikasyonu, NB hastalarında nadir görüldüğü bildirilmiştir79.
Çalışmamızda; sıklıkla hasarların tespit edildiği bir başka kromozom 9.
kromozomdur. Bu kromozomdaki hasarları; del(9)(p11→pter), del(9)(q22→qter),
del(9)(q13→qter), del(9)(q32→qter), chbr(9)(q11.1), inv(9)(p11;q12), gap(9)(p13),
fra(9)(q22) ve t(9;14)(q34;q24.3-qter) şeklindedir (Çizelge 4.4, 4.5 ve 4.6)(Şekil
4.1). Bu kromozomdaki hasarlar, en fazla q11 ve q22 bölgelerinde saptandı. Daha
önce, 9q22.1 bölgesinde NTRK2 (TRK-B) reseptör tirozin kinaz geni tanımlandı76.
TRK-B’nin, tümör hücresinin yaşamını, invazyonunu ve metastazını kolaylaştırdığı
düşünülmektedir. TRK-B, yüksek oranda salgılanması agresif tümör ve N-Mcy
amplifikasyonu ile ilişkilendirilmiştir75,76. 9p21 bölgesinde yerleşik olan p16 genin
biallelik inaktivasyonunun NBda tümör oluşumunda rol alabileceği ileri
sürülmektedir109. 9p delesyonları, NBlarda az sıklıkta görülen bir durum olarak
rapor edildi79. Tüm bu bilgiler, 9q11 ve 9q22 bölgelerinin NBda etkin ve hassas
noktalar olduğunu göstermektedir.
Çalışmamızda; yapısal kromozom düzensizliklerinin sıklığı ve nisbi oranları
yüksek bulunan diğer bir kromozom 13. kromozomdur. Hasta grubunda özellikle,
delesyonları [del(13)(q22.1→qter), del(13)(q14→qter), del(13)(q12→qter),
del(13)(q14.3→qter)] dikkat çekiciydi (Çizelge 4.4, 4.5 ve 4.6)(Şekil 4.1). Bu
yapısal hasarlar, en fazla q14’te gözlendi. Bu kromozomun q14’te yer alan RB1
geninin homozigot kaybı retinoblastoma hastalarında gözlendi110. Retinoblastoma

72
ve osteosarkoma gibi çocukluk çağı kanserlerinde 13q kromozom kayıplarına
rastlandığı belirtilmiştir110. Bizim çalışmamıza göre, 13. kromozomun q14
bölgesinin NB için de önemli bir bölge olduğunu söyleyebiliriz.
Hastalarımızda 14. kromozom, sıklığı ve nisbi oranı yüksek olan
kromozomlardan biridir. Bu kromozomda bulunan düzensizlikleri; delesyonlar
[del(14)(q13→qter), del(14)(q11.2→pter), del(14)(q11.2→q13),
del(14)(q11.1→pter)], translokasyonlar [t(9;14)(q34;q24.3-qter), t(7;14)(p22;q24),
t(14;16)(q24;p13)] ve derivatif düzensizlik [der(14;?)t(14;?)(q11-qter;?)] şeklinde
gözlendi (Çizelge 4.4 ve 4.5)(Şekil 4.1). Bu kromozomun uzun kolundaki q11.2[4],
q24[3] ve q13[2] bölgelerinin sıklığı ve nisbi oranı en yüksek olan bölgelerdir
(Çizelge 4.6). Ondördüncü kromozomun q23 bölgesinde, MAX geninin yerleşik
olduğu ve bu genin MYCN’in düzenlenmesinde görev aldığı bildirildi44. Hücre, Go
evresindeyken Max ekspresyonunun yüksek olduğu homodimer halde bulunarak
transkripsiyonu baskıladığı ifade edilmiştir44. Yapılan çalışmalarda; 14q23-qter
bölgesinde tümör supresör genler olduğu gösterilmiştir79. Hastalarımızda
bulduğumuz q24 bölgesi, tümör süpressör gen bölgesi içerisinde yer almaktadır.
Ancak, q11.2[4] ve q13[2] bölgeleri çalışmamızda yeni tanımlanmış bölgeler olup,
bu bölgeler de NBda yeni aday bölgeler olabilir. Tüm bu bilgilerden, tümör
supresör genlerin protoonkogenlerin ve önemli genlerin ifade olduğu bu
bölgelerde oluşan yapısal hasarlar, ilgili genleri uyarmak ve/veya baskılamak
suretiyle kanserleşmeye neden olduğunu söylenebiliriz.
Son olarak hastalarımızda, 15. kromozomun q22 bölgesinin sıklığı ve nisbi
oranının yüksek olduğu bulundu (Çizelge 4.4 ve 4.6). Onbeşinci kromozomun
q24-q25 bölgesinde tirozin kinaz reseptörü olan NTRK3 (TRK-C) geni bulunduğu
bildirldi44. TRK-C’nin fonksiyonu ise henüz kesin bilinmemektedir. TrkC, TrkA
reseptörü ile aynı özelliklere sahiptir, ek bir prognostik önemi yoktur. İyi klinik
özelliklere sahip tümörlerde eksprese olduğu bildirilmiştir75,76.
5.2. Moleküler Sitogenetik
MYCN amplifikasyonu, kanserin en önemli prognoz göstergelerinden biri
olduğu birçok çalışma ile kanıtlanmıştır5,16,27,31,32,41. MYCN geni, 2p24.1
bölgesinde yerleşik bir proto-onkogendir31,32. Bu transkripsiyon faktörü, hücre

73
siklusun ilerlemesini etkiler ve apopitozise duyarlılığı arttırır. Bu genin
amplifikasyonu, sitogenetik olarak 2 şekilde gösterilmektedir; d-mins (double
minutes kromatin cisimcikleri) ekstra küçük kromozom parçaları ve HSR (homojen
boyanan bölgeler); aynı kromozom içinde bir bölgenin yinelenmesi şeklinde
gözlenmektedir16,30-33.
Tümör hücresinde MYCN amplifikasyonu, Southern blot, FISH, kantitatif
PCR ve immunohistokimyal tekniklerle gösterilebilir. Yapılan çalışmalarda; FISH’in
daha hızlı bir teknik olduğu ve aynı tümör dokusunda hücreler arasında MYCN
amplifikasyonunun heterojenliğini gösterebildiği için daha avantajlı olduğunu rapor
edilmiştir68. NBda, MYCN amplifikasyon oranının %20-25 aralığında bulunduğu ve
hastalığın prognozu üzerine amplifikasyonunun önemi olduğu pek çok çalışma ile
bildirilmiştir16,27,31,32,41. Amplifikasyon görülen tümörlerde, hasta yaşı ve tümör
evresi klinik gidişte önemli belirteçlerdir. Amplifikasyon bulunan, 1 yasından küçük
ve evre 4 dışı tümörler diğerlerinden daha iyi prognoza sahiptirler19,111.
Amplifikasyon bulunan lokalize NBlı hastalarda, klinik gidişin daha iyi olduğu rapor
edilmiştir111. Genel olarak, MYCN amplifikasyonu ile ileri evre, kötü prognoz,
tedaviye kötü cevap ve yüksek relaps ilişkili olduğu bilinmektedir. İleri evre
hastaların %30-50’sinde, MYCN amplifikasyonu bulunurken erken evreli
hastalarda bu oran %5-10 arasında bildirilmiştir2,68. Çalışmamızda ise 9 NBlı
hastanın 6’sında MYCN ve AURKA amplifikasyonu saptandı (Şekil 4.3), MYCN ve
AURKA amplifikasyonu hastalarda görülme oranı %66.7 olarak bulundu. Aynı
zamanda, MYCN amplifikasyonu saptanan hastaların hepsinin ileri evre (Evre 4)
oldukları rapor edildi. Bununla birlikte, çalışmamızda MYCN amplifikasyon oranı
(%23), literatürde evre 4 için bildirilen oranla (%20-25) paralellik göstermektedir.
Ayrıca, MYCN amplifikasyonu ile hastaların cinsiyetleri, yaşları, LDH, NSE ve
tümör yerleri karşılaştırıldığında fark anlamlı bulunmamıştır.
Kopya sayısı arttıkça, gen ifadesi de artmaktadır. Kopya sayısının 10 ve
üzerinde olması prognostik öneme sahiptir20. Yapılan çalışmalarda; düşük
seviyedeki amplifikasyonunun yüksek amplifikasyon seviyesinden daha iyi
prognoza sahip olduğunu ifade edilmiştir36,39,102. Çalışmamızda; MYCN ve
AURKA amplifikasyonun görüldüğü 1 hastanın öldüğü diğer hastaların evre 4’te
olduğu saptandı.

74
AURKA geni tarafından kodlanan Aurora A proteini, hücre bölünmesinde
G2/M kontrol noktasında düzenleyici, mitotik iğ ipliği oluşumu ve kromozomların
doğru ayrılması için gerekli bir serin-tireonin kinazdir. Aurora A, MYCN ile
etkileşime girer ve MYCN’i proteolitik parçalanmadan korur111,112. Aurora A
proteini, kromozom ayrılması sırasında kutuplardaki iğ ipliklerinde mikrotübül
kararlılığını ve/veya oluşmasını sağladığı gibi interfazda sentromerde ve mitozda
kutuplardaki iğ ipliklerinde bulunmuştur. Bu genin, tümör gelişiminde ve
ilerleyişinde rolü olabileceği belirtilmiştir113. Kanser hücrelerindeki düzensiz hücre
bölünmesi mitoza ve mitozdaki işlemlere bağlıdır. Kanser uygulamalarında,
başarısı kanıtlanmış etkili bir strateji, mitotik iğ ipliği fonksiyonunu engellemek
olmuştur. Son yıllarda, tubulin ve mitotik iğ ipliklerini birleştiren hedef proteinlerde
büyük ilerlemeler elde edilmiştir. Aurora ailesi gibi mitotik kinazlar tam olarak
kromozom ve sentromer ayrılmasını sağlamadaki hayati rollerinden dolayı önemli
kabul edilmektedir. Aurora A’nın mRNA’da ve/veya protein düzeyinde fazla
ekspresyonu meme, mide, ovaryum, pankreas ve hepatoselüler karsinomalar gibi
insan tümörlerinde %26-%94 aralığında fazla ifade edildiği bildirilmiştir114,115. Tüm
bu bulgular ışığında; MYCN ve AURKA gen amplifikasyon artışı ile hastalığın ileri
evreleri arasında paralel bir ilişki bulunduğu, bu amplifikasyon değerlerinin
hastalığın tanısı ve prognozunda kriter olarak alınabileceğini söyleyebiliriz.

75
6. SONUÇLAR ve ÖNERİLER
Çukurova Üniversitesi, Tıp Fakültesi, Pediatrik Onkoloji Polikliniğinde NB ön
tanısı konmuş hastalar ile yapmış olduğumuz sitogenetik ve moleküler genetik
çalışmalarında şu sonuçlar elde edilmiştir;
1. Rapor ettiğimiz kromozom düzensizlikleri bakımından hasta (%18,43)
ve kontrol (%2,64) grubları arasındaki fark istatistiksel olarak anlamlı
bulundu (p<0,0001). NBda, önemli oranda kromozom hasarı ortaya
çıkmaktadır.
2. Hastalarda, hücrelerin %78.3’ü yapısal ve %21.7’si sayısal
kromozom düzensizlikleri taşıdı bulundu. Bu da NBnın öyküsünde
yapısal kromozom düzensizliklerinin önemli olduğunu
göstermektedir.
3. Yapısal düzensizliklerin hücre başına sıklığı %13.1 oranında yüksek
değerde bulundu. NBda hüçrelerin önemli kısmı genetik hasara
uğramaktadır.
4. Yapısal kromozomal düzensizlikleri arasında; 1q21, 1q32, 2p24,
2p21, 2q31, 4q31, 9q11, 9q22, 13q14, 14q11.2, 14q24 ve 15q22
bölgelerinin NBda en sık rastlanan sıcak noktalar olduğu ve NBda
tümör oluşumunda etkili gen veya genleri taşıdığını söyleyebiliriz.
5. Bu nedenle bu bölgeler, NBnin tanısında ve prognozunda etkili gen
veya genlerin belirlenmesinde moleküler genetik çalışmalarda aday
kritik noktalar olarak alınabilir.
6. Hastalarımızın %66.7’sinin kanser dokularında AURKA ve MYCN
gen amplifikasyonları gözlendi.
7. Evre 4 hastalarında, AURKA’nın ortalama amplifikasyon değeri %19
ve MYCN’in %23 olarak hesaplandı.

76
8. Sonuç olarak, NB’li hastalarda farklı kromozom düzensizliklerinin,
MYCN ve AURKA gen amplifikasyon değerlerinin hastalığın tanısı ve
prognozunda kriter olarak alınabileceğini söyleyebiliriz.

77
7. KAYNAKLAR
1. Goodman M, Gurney JG, Smith MA, Olshan AF. Sympathetic nervous system tumors. Cancer Incidence and Survival amongChildren and Adolescents: United States SEER Program, SEER pediatric monograph. 1975-1995.65-72
2. Brodeur GM, Maris JM. Neuroblastoma In: Pizzo PA, Poplack DG. Principles and practice of pediatric oncology 5th ed. Lippincott Williams& Wilkins 2006:934-969.
3. Izbicki T, Mazur J, Izbicka E. Epidemiology of neuroblastoma: analysis of a single institution. Anticancer Res. 2003 ;23(2C):1933-8
4. Alexander F. Neuroblastoma. Urol Clin North Am. 2000;27(3):383-92, 5. Castleberry RP. Neuroblastoma. Eur J Cancer. 1997;33(9):1430-7. 6. Kutluk T. First national pediatric cancer registry in Turkey: A Turkish Pediatric
Oncology Group (TPOG) study. Pediatr Blood Cancer. 2004;43(4):452 7. Yamamoto K, Ohta S, Ito E, Hayashi Y, Asami T, Mabuchi O, Higashigawa M,
Tanimura M. Marginal decrease in mortality and marked increase in incidence as a result of neuroblastoma screening at 6 months of age: cohort study in seven prefectures in Japan. J Clin Oncol. 2002;20(5):1209-14.
8. Schilling FH, Spix C, Berthold F, Erttmann R, Sander J, Treuner J, Michaelis J. Children may not benefit from neuroblastoma screeningat 1 year of age. Updated results of the population based controlled trial in Germany. Cancer Lett. 2003;197(1-2):19-28.
9. Brodeur GM, Look AT, Shimada H, Hamilton VM, Maris JM, Hann HW, Leclerc JM, Bernstein M, Brisson LC, Brossard J, Lemieux B, Tuchman M, Woods WG. Biological aspects of neuroblastomas identified by mass screening in Quebec. Med Pediatr Oncol. 2001;36(1):157-9.
10. Allen RW, Ogden B, Bentley FL, Jung AL. Fetal hydantoin syndrome, neuroblastoma, and hemorrhagic disease in a neonate. Journal of the American Medical Association 1996,244,1464-1465.
11. Kinney H, Faix R, Brazy J. The fetal alcohol syndrome and neuroblastoma. Pediatrics 1996, 66, 130-132.
12. Schwartzbaum JA. Influence of the mother’s prenatal drug consumption on risk of neuroblastoma in the child. American Journal of Epidemiology 1996, 135, 1358-1367.
13. Haase GM, Perez C, Atkinson JB. Current aspects of biology, risk assessment, and treatment of neuroblastoma. Semin Surg Oncol. 1999;16(2):91-104.
14. Cheung NV, Cohn SL. Neuroblastoma. Springer-Verlag Berlin Heidelberg 2005. 15. Menegaux F, Olshan AF, Reitnauer PJ, Blatt J, Cohn SL. Positive association
between congenital anomalies and risk of neuroblastoma. Pediatr Blood Cancer. 2005;45(5):649-55.
16. Maris JM, Matthay KK. Molecular biology of neuroblastoma. J Clin Oncol. 1999;17(7):2264-79.
17. Tonini GP, Longo L, Coco S, Perri P. Familial neuroblastoma: a complex heritable disease. Cancer Lett. 2003;197(1-2):41-5.
18. Hiyama E, Yokoyama T, Hiyama K, Yamaoka H, Matsuura Y, Nishimura S, Ueda K. Multifocal neuroblastoma: biologic behavior and surgical aspects. Cancer. 2000;88(8):1955-63
19. Aksoylar S. Nöroblastom. Pediatrik onkoloji. Klinik gelişim 2007;20:62-74. 20. Demirkaya M, Sevinir B. Nöroblastom. Güncel Pediatri 2006;3: 128-132,. 21. Landuzzi L, Strippoli P, Giovanni CD, Nicoletti G, Rossi I, Tonelli R, Frabetti F,
Nanni P, Bagnara GP, Lollini PL. Production of stem cell factor and ezpression of C-kit in human rhabdomtosarcoma cells: Lack of autocrine growth modulation. International Journal of Cancer 1998;78: 441-445,.

78
22. Pahlman S, Stockhausen MT, Fredlund E, Axelson H. Notch signaling in neuroblastoma. Semin Cancer Biol. 2004;14(5):365-73.
23. Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, Roald B. Terminology and morphologic criteria of neuroblastic tumors: recommendations by the International Neuroblastoma Pathology Committee. Cancer. 1999;86(2):349-63.
24. Shimada H. In Situ Neuroblastoma: An Important Concept Related to the Natural History of Neural Crest Tumors. Pediatr Dev Pathol. 2005 ;8(3):305-6.
25. Berthold F, Hero B. Neuroblastoma: current drug therapy recommendations as part of the total treatment approach. Drugs. 2000;59(6):1261-77.
26. Look AT, Hayes FA,Nitschke R, et al. Cellular DNA content as a predictor of response to chemoterapy in infants with unresectable neuroblastoma. N Eng J Med 1984;311:231-235.
27. Look AT, Hayes FA, Shuster JJ, et al. Clinical relevance of tumor cell ploidy and N-myc gene amplification in childhood neuroblastoma. A Pediatric Oncology Group Study. J Clin Oncol 1991;9:581-591.
28. Kaneko Y, Knudson AG. Mechanism and relevance of ploidy in neuroblastoma. Genes Chromosomes Cancer 2000;29:89-95.
29. Schwab M, Alitalo K, Klempnauer KH, et al. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 1983;305:245-248.
30. Schwab M, Varmu HE, Bishop JM,et al. Chromosome localization in normal human cells and neuroblastomas of a gene relatedto c-myc. Nature 1984;308:288-291.
31. Corvi, R., Amler, L. C., Savelyeva, L., Gehring, M. & Schwab, M. MYCN is retained in single copy at chromosome 2 band p23-24 during amplification in human neuroblastoma cells. Proc. Natl Acad. Sci. USA 1994; 91:5523–5527.
32. Schneider, S. S. et al. Isolation and structural analysis of a 1.2-megabase N-myc amplicon from a human neuroblastoma. Mol. Cell. Biol. 1992;12: 5563–5570.
33. Reiter, J. L. & Brodeur, G. M. MYCN is the only highly expressed gene from the core amplified domain in human neuroblastomas. Genes Chromosom Cancer 1998;23: 134–140.
34. Mathew P, Valentine MB, Bowman LC et al. Detection of MYCN gene amplification in neuroblastoma by fluorescence in situ hybridization: a Pediatric Oncology Group study Neoplasia 2001;3:105-109.
35. Ambros PF, Ambros IM, Kerbl R, et al. Intratumoral heterogeneity of 1p deletion and MYCN amplification in neuroblastomas. Med Pediatr Oncol 2001;36:1-4.
36. Seeger RC, Broudeur GM, Sather H, et al. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N Engl J Med 1985;313:1111-1116.
37. Brodeur GM. Clinical and biological aspects of neuroblastoma. In: Vogelstein B, Kinzler KW,eds. The genetic basis of human cancer, 2nd ed. New York McGraw-Hill, 2002;751-772.
38. Nakagawara A, Arima M, Azar CG, et al. Inverse relationship between trk expression and N-myc amplification in human neuroblastomas. Cancer Res 1992;52:1364-1368.
39. Wada RK, Seeger RC, Brodeur GM, et al. Human neuroblastoma cell lines that Express N-myc without gene amplification. Cancer 1993;72:3346-3354.
40. Chan, H. S. et al. MYCN protein expression as a predictor of neuroblastoma prognosis. Clin Cancer Res 1997;3:1699–1706.
41. Bordow, S. B., Norris, M. D., Haber, P. S., Marshall, G. M. & Haber, M. Prognostic significance of MYCN oncogene expression in childhood neuroblastoma. J. Clin. Oncol. 1998;16:3286–3294.
42. Cohn, S. L. et al. MYCN expression is not prognostic of adverse outcome in advanced-stage neuroblastoma with nonamplified MYCN. J. Clin. Oncol. 2000;18:3604–3613.
43. Wenzel A, Cziepluch C, Hamann U, et al: The N-Myc oncoprotein is associated in vivo with the phosphoprotein Max (P20/22) in human neuroblastoma cells. EMBO J 1991;10:3703-3712.
44. Grandori C, Eisenman RN: Myc target genes. Trends Biochem Sci 1997;22:177-181.

79
45. Schmidt ML, Salwen HR, Manohar CF, et al: The biological effects of antisense N-myc expression in human neuroblastoma. Cell Growth Differ 1994;5:171-178,.
46. Sivak LE, Tai KF, Smith RS, et al: Autoregulation of the human N-myc oncogene is disrupted in amplified but not single-copy neuroblastoma cell lines. Oncogene 1997;15:1937-1946.
47. George RE, Squire JA. Structure of the MYCN amplicon. In: Brodeur GM, Sawada T, Tsuchida Y, et al, eds. Neuroblastoma. Amsterdam: Elsevier, 2000:85–100.
48. Wimmer K, Zhu XX, Lamb BJ, et al. Co-amplification of a novel gene, NAG, with the N-myc gene in neuroblastoma. Oncogene 1999;18:233–8.
49. Bown, N. et al. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. N. Engl. J. Med. 1999;340:1954–1961.
50. Lastowska, M. et al. Breakpoint position on 17q identifies the most aggressive neuroblastoma tumors. Genes Chromosom. Cancer 2002;34:428–436.
51. Islam, A. et al. High expression of Survivin, mapped to 17q25, is significantly associated with poor prognostic factors and promotes cell survival in human neuroblastoma. Oncogene 2000;19:617–623.
52. Sweetser DA, Kapur RP, Froelick GJ, et al: Oncogenesis and altered differentiation induced by activated Ras in neuroblasts of transgenic mice. Oncogene 1997;15:2783-2794.
53. Corvi R, Savelyeva L, Breit S, et al: Non-syntenic amplification of MDM2 and MYCN in human neuroblastoma. Oncogene 1995;10:1081- 1086.
54. Lastowska M, Nacheva E, McGuckin A, et al: Comparative genomic hybridization study of primary neuroblastoma tumors: United Kingdom Children’s Cancer Study Group. Genes Chromosom Cancer 1997;18:162-169,.
55. Brodeur GM, Sekhon GS, Goldstein MN. Chromosomal aberrations in human neuroblastomas. Cancer 1977;40:2256–63.
56. Caron H, Peter M, Van Sluis P, Spelemen F, de Kraker J, Laureys G, Michon J, Brugieres L, Votue PA, Westerveld A, Slater R, Delattre O, Versteeg R: Evidence for two tumor suppressor loci on chromosomal bands 1p35-36 involved in neuroblastoma: one probably imprinted, another associated with N-myc amplication. Hum Mol Genet 1995;4:535-539.
57. White PS, Maris JM, Beltinger C, et al. A region of consistent deletion in neuroblastoma mapswithin 1p36.2-3. Proc Natl Acad Sci USA1995;92:5520-5524.
58. Martinsson T, Sjoberg RM, Hedborg F, Kogner P. Deletion of chromosome 1p loci and microsatellite instability in neuroblastomas analyzed with short tandem repeat polymorphisms. Cancer Res1995;55:5681-5686.
59. Maris JM, Weiss MJ, Guo C, et al. Loss of heterozygosity at 1p36 independently predicts for disease progression but not decreased overall survival probability in neuroblastoma patients: a Children’s Cancer Group study; J Clin Oncol. 2000;18:1888;1899.
60. Spitz R, Hero B, Westermann F, et al. Fluorescence in situ hybridization analyses of chrosome band 1p36 neuroblastoma detect two classes of alterations. Genes Chromosomes Cancer 2002;34:299-305.
61. Bown N. Neuroblastoma tumor genetics: clinical and biological aspects. J Clin Pathol 2001;54:891-910.
62. White PS, Maris JM, Sulman EP, et al: Molecular analysis of the region of distal 1p commonly deleted in neuroblastoma. Eur J Cancer 1997;33:1957-1961.
63. Kaghad M, Bonnet H, Yang A, et al: Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 1997;90:809-819.
64. M. Romani, G.P. Tonini, B. Banelli, et al. Biological and clinical role of p73 in neuroblastoma. Cancer Letters 2003;197:111–117.
65. J.M. Maris, C. Guo, P.S. White, M.D. Hogarty, P.M. Thompson, D.O. Stram, et al., Allelic deletion at chromosome bands 11q14-23 is common in neuroblastoma, Med. Pediatr. Oncol. 2001;36:24–27.
66. Guo C, White PS, Weiss MJ, et al. Allelic deletion at 11q23 is common in MYCN single copy neuroblastomas. Oncogene 1999;18:4948-4957.

80
67. Spitz R, Hero B, Ernestus K, Berthold F, et al. Deletions in chromosomes arms 3p and 11q are new prognostic markers in localized and 4s neuroblastoma. Clin Cancer Res. 2003;9:52-58.
68. Spitz R, Hero B, Ernestus K, Berthold F, et al. FISH analyses for alternations chrosomes 1,2,3, and 11 define high risk groups in neuroblastoma. Med Pediatr Oncol 2003;41:30-35.
69. Suzuki T, Yokota J, Mugishima H, et al: Frequent loss of heterozygosity on chromosome 14q in neuroblastoma. Cancer Res 1989;49:1095-1098.
70. Takita J, Hayashi Y, Kohno T, et al: Allelotype of neuroblastoma. Oncogene 1995;11:1829-1834,.
71. Ejeskar K, Aburatani H, Abrahamsson J, et al: Loss of heterozygosity of 3p markers in neuroblastoma tumours implicate a tumour-suppressor locus distal to the FHIT gene. Br J Cancer 1998;77:1787-1791.
72. Caron H, van Sluis P, Buschman R, et al: Allelic loss of the short arm of chromosome 4 in neuroblastoma suggests a novel tumour suppressor gene locus. Hum Genet 1996;97:834-837.
73. Takita J, Hayashi Y, Kohno T, et al: Deletion map of chromosome 9 and p16 (CDKN2A) gene alterations in neuroblastoma. Cancer Res 1997;57:907-912.
74. Marshall B, Isidro G, Martins AG, et al: Loss of heterozygosity at chromosome 9p21 in primary neuroblastomas: Evidence for two deleted regions. Cancer Genet Cytogenet 1997;96:134-139.
75. Yano H,Chao MV. Neurotrophin receptor structure and interactions. Pharm Acta Helv 2000;74:253-260.
76. Patapoutian A, Reichardt LF. Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol 2001;11:272-280.
77. Keshelava N, Seeger RC, Reynolds CP: Drug resistance in human neuroblastoma cell lines correlates with clinical therapy. Eur J Cancer 1997;33:2002-2006.
78. Bradshaw DM,Arceci RJ: Clinical relevance of transmembrane drug efflux as a mechanism of multidrug resistance. J Clin Oncol 1998;16:3674-3690,.
79. Maris J.M. and Matthay K.K. Molecular Biology of Neuroblastoma. American Society of Clinical Oncology. J Clin Oncol 1999;17:2264-2279.
80. Weistein JW, Katzeinstein HM, Cohan SL et al. Advences in the diagnosis and treatment of neuroblastoma.The Oncologist 2003;8:278-292.
81. Brodeur GM,Pritchard J, Berthold F. et al.Revisions of the international criteria for neuroblastoma diagnosis, staging and response to treatment. J Clin Oncol 1993;11:1466-1477.
82. Castleberry RP, Shuster JJ, Smith El. The Pediatric Oncology Group experience with the international staging system criteria for neuroblastoma. J Clin Oncol 1994;12:2378-2381.
83. Haase GM, Atkinson JB, Stram DO et al. Surgical management and outcome of locoregional neuroblastoma: comparison of the Childrens Cancer Group and the international staging systems. J Pediatr Surg 1995;30:289-294.
84. Monsaingeon M, Perel Y, Simonnet G, Corcuff JB. Comparative values of catecholamines and metabolites for the diagnosis of neuroblastoma. Eur J Pediatr. 2003;162(6):397-402.
85. Titilope A. Isholab, Dai H. Neuroblastoma Chung Surgical Oncology. 2007;16, 149–156.
86. Weinstein JL, Katzenstein HM, Cohn SL. Advances in the diagnosis and treatment of neuroblastoma. Oncologist. 2003;8(3):278-92.
87. Türkiye Pediatrik Onkoloji Grubu (TPOG) Nöroblastom 2009 Tedavi Protokolü. 2009. 88. Frappaz D, Bonneu A, Chauvot P, Edeline V, Giammarile F, Siles S, Wioland M,
Gomez F. Metaiodobenzylguanidine assessment of metastatic neuroblastoma: observer dependency and chemosensitivity evaluation. The SFOP Group. Med Pediatr Oncol. 2000;34(4):237-41.
89. Hiorns MP, Owens CM. Radiology of neuroblastoma in children. Eur Radiol. 2001;11(10):2071-81.

81
90. Joshi VV. Peripheral neuroblastic tumors: pathologic classification based on recommendations of international neuroblastoma pathology committee (Modification of shimada classification). Pediatr Dev Pathol. 2000;3(2):184-99.
91. Qualman SJ, Bowen J, Fitzgibbons PL, Cohn SL, Shimada H; Cancer Committee, College of American Pathologists. Protocol for the examination of specimens from patients with neuroblastoma and related neuroblastic tumors. Arch Pathol Lab Med. 2005;129(7):874-83.
92. Shimada H, Umehara S, Monobe Y, Hachitanda Y, Nakagawa A, Goto S, Gerbing RB, Stram DO, Lukens JN, Matthay KK. International Neuroblastoma Pathology Classification for Prognostic Evaluation of Patients with Peripheral Neuroblastic Tumors: A Report from the Children’s Cancer Group. Cancer 92: 2451–61, 2001.
93. Shimada H, Ambros IM, Dehner LP, Hata JI, Joshi VV, Roald B, Stram DO, Gerbing RB, Lukens JN, Matthay KK, Castleberry RP. The International Neuroblastoma Pathology Classification (the Shimada System). Cancer 86: 364–72, 1999.
94. Bischoff JR, Plowman GD. The Aurora /IpI 1p kinase family: regulators of chrosome segregation and cytokinesis. Trends Cell Biol. 1999;9:454-459.
95. Kimura M, Kotani S, Hattori T, Sumi N, Yoshioka T, Todokoro T, Okano Y.Cell cycle-dependent expression and spindle pole localization of a novel human protein kinase Aik, related to Aurora of Drosophila and yeast IpI1. J. Biol. Chem. 1997;272:13766-13771.
96. Kimura M, Matsuma Y, Eki T, Yoshioka T, Okumura K, Hanako F, Okano Y. Assigment of STK6 to human chromosome 20q13.2-q13.3 ve a pseudugene STK6P to 1q41-q42. Cytogenet. Cell Genet. 1997;79:201-203.
97. Zhou H, Kuang J, Zhong L, Kuo W, Gray JW, Sahin A, Brinkley BR, Sen S. Tumour amplified kinase STK15/BTAK induces centrosome amplification. Nature Genet. 20: 189-193, 1998.
98. Katayama H, Sasai K, Kawai H, Yuan Z-M, Bondaruk J, Suzuki F, Fujii S, Arlinghaus RB, Czerniak BA, Sen S. Phosphorylation by Aurora kinase A induces Mdm2- mediated destabilization and inhibition of p53. Nature Genet. 36: 55-62, 2004.
99. Dracopoli NC,Haines JL,Korf BR, Moir DT, Morton CC, Seidman CE, Seidman JG, Smith DR. Current Protocols in Human Genetics. United State: Willey&Sons,1994.
100. Rooney DE, Czepulkowski BH. Prenatal Diagnosis and Tissue Culture. In: Human Cytogenetics, 1993 Volume I: 55-89.
101. Izbicki T, Mazur J, Izbicka E. Epidemiology and etiology of neuroblastoma: an overview. Anticancer Res. 2003;23:755-60.
102. DuBois SG, Kalika Y, Lukens JN, Brodeur GM, Seeger RC, Atkinson JB, Haase GM, Black CT, Perez C, Shimada H, Gerbing R, Stram DO, Matthay KK. Metastatic sites in stage IV and IVS neuroblastoma correlate with age, tumor biology, and survival. J Pediatr Hematol Oncol. 1999;21(3):181-9.
103. London WB, Castleberry RP, Matthay KK, Look AT, Seeger RC, Shimada H, Thorner P, Brodeur G, Maris JM, Reynolds CP, Cohn SL. Evidence for an age cutoff greater than 365 days for neuroblastoma risk group stratification in the Children's Oncology Group. J Clin Oncol. 2005;23(27):6459-65.
104. Ragnarsson G, Eiriksdottir G, Johannsdottir JT, Jonasson JG, Egilsson V, Ingvarsson S. Loss of heterozygosity at Chrosome 1p in different Solid Tumours: Association with Surrvival. Br J Cancer 1999;79:1467-1474.
105. Caron H, Speiker N, Godfried M, Veenstra M, van Sluis P, de Kraker J, Voute P, Versteeg R. Chromosome Bands 1p35-3 Contain Two Distinct Neuroblastoma Tumour Suppressor Loci, One of Which is Imprinted. Genes Chrosomes Cancer 2001;30:18-174.
106. Godfried M.B., Veenstra M., Valent A., Sluis P.V., Voute P.A., Versteeg R., Caron H.N. Lack of interstitial chromosome 1p deletions in clinically-detected neuroblastoma. European Journal of Cancer 2002;38:1513–1519.
107. Astuti D., Agathanggelou A, Honorio S, Dallol A., Martinsson T, Kogner P, Cummins C, Neumann HPH, Voutilainen R, Dahia P, Eng C, Maher ER and Latif F. RASSF1A promoter region CpG island hypermethylation in phaeochromocytomas and neuroblastoma tumours. Oncogene 2001;20:7573-7577.

82
108. Schleiermacher G., Michon J, Huon I, d’Enghien C.D, Klijanienko J, Brisse H, Ribeiro A, Mosseri V, Rubie H, MunzerC, Thomas C,Valteau-Couanet D, Auvrignon A, Plantaz D, Delattre O and Couturier J. Chromosomal CGH identifies patients with a higher risk of relapse in neuroblastoma without MYCN amplification. British Journal of Cancer 2007;97:238–246.
109. Brodeur G.M. Neuroblastoma: Biological Insights into a Clinical Enigma. Nature Rewievs Cancer. 2003:3;203-216.
110. Mertens F, Mandahl N, Mitelman F, Heim S. Cytogenetic analysis in the examination of solid tumors in children. Pediatr Hematol Oncol. 1994;4:361-77.
111. Spitz R, Hero B, Skowron M, Ernestus K, Berthold F. MYCN-status in neuroblastoma: characteristics of tumours showing amplification, gain, and nonamplification. European Journal of Cancer 2004;40:2753–2759,
112. John M.M. Unholy Matrimony. Aurora A and N-Myc as Malignant Partners in Neuroblastoma. Cancer Cell, 2009;15: 5-6
113. http://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?val=NM_003600.2 Erişim Tarihi:10.02.2009
114. Tobias O, Sebastian H, Markus B, et al. Stabilization of N-Myc Is a Critical Function of Aurora A in Human Neuroblastoma.Cancer Cell. 2009;15:67-78.
115. Marta M, Jorge B, Adrián M.E, et al. Aurora kinases as prognostic biomarkers in ovarian carcinoma.Human Pathology. 2008.
116. Köhler A. Chromosome Staining. In: Wegner RD. Ed. Diagnostic Cytogenetics, Berlin:Springer-Verlag; 1999:56-60.

83
EKLER
EK-1. Sitogenetik Çalışmalarda Kullanılan Besiyerleri a. Normal Kültür Besiyeri
RPMI-1640, R0883 100 ml
Fetal calf serum 25 ml
Fitohemaglutinin 1,5 ml
Penisilin/ Streptomisin 1,0 ml
L-Glutamin 1,5 ml
EK-2. Sitogenetik Çalışmalarda Kullanılan Solüsyonlar
a. Kolşisin (Seromed Cat. No. L 6210) 10 µl / ml
b. Hipotonik solüsyonu (0,075 KCl)
5,592 gr KCl (Potasyum klorür) tartılıp 1000 ml’ye distile su ile
tamamlandı.
c. Fiksatif solüsyonu
3 birim metanol ile 1 birim glasiyel asetik asit karıştırılarak her
uygulama öncesi taze olarak hazırlandı.
d. Tripsin solüsyonu (Stok solüsyonu, 30 mg / ml) e. Serum fizyolojik f. Fosfat tamponu
İki ayrı solüsyon halinde hazırlanır ve eşit oranda karıştırılır.
Solüsyon 1. 9,073 g KH2PO4 tartılıp 1000 ml bidistile suda çözülür.
Solüsyon 2. 11,87 g Na2HPO4.2H2O 1000 ml bidistile suda çözülür.
g. Giemsa boya solüsyonu
Fosfat tamponu içerisinde %7-10 olacak şekilde hazırlanır.

84
ÖZGEÇMİŞ
1983 yılında Yozgat’ın Yerköy ilçesinde doğdu. İlköğretimini Yerköy
Mehmet Akif Ersoy İlkokulu’nda ve Yerköy Şehit Şaban Karadoğan
Ortaokulu’nda tamamladı. Lise eğitimini Yozgat Erdoğan Akdağ Anadolu
Öğretmen Lisesinde tamamladı. 2006 yılında Ankara Üniversitesi Fen Fakültesi
Biyoloji Bölümününden mezun oldu. 2007 yılında Çukurova Üniversitesi, Sağlık
Bilimleri Enstitüsü, Tıbbi Biyoloji Anabilim Dalı Yüksek Lisans programını
kazandı.