payamkarisani cs ph.d. student (team lead) eugene ... · biomedical informatics and biostatistics,...
TRANSCRIPT

Presenter: Payam Karisani
Team members:Payam Karisani, CS Ph.D. Student (Team lead) Eugene Agichtein, Associate Professor/Advisor
Intelligent Information Access Laboratory (IR Lab)Computer Science & Informatics, Emory University
Kenong Su, Yanting Huang: Bioinformatics Ph.D. students, Zhaohui (Steve) Qin, Associate ProfessorBiomedical Informatics and Biostatistics, Emory University

� Overview
� Architecture
� Design details
� Experiments and results
� Conclusions
Content

� bioCADDIE dataset for the text retrieval challenge:� Almost 800k biomedical dataset descriptions
� Crawled from 20 different web domains� Document fields:
� DOCNO, TITLE, REPOSITORY, METADATA� Training set: 6 queries (relevancy scale 0-2)� Test set: 15 queries (relevancy scale 0-2)� Evaluation metric: NDCG (inferred)
Recap of the BioCADDIE Challenge

� Query intent and corpus characteristics:� Queries are transactional (in contrast to informational queries in ad-hoc
retrieval)� Documents often do not explicitly contain relevant keywords
� Query and document mismatch:� A higher degree of query-document mismatch comparing to ad-hoc
retrieval� Training data:
� Relatively small of number of training queries (in ad-hoc retrieval usually 50 queries are provided)
Task Challenging for “Classical” IR

� Query-document term mismatch:ØDocument enrichment (with meta-data)ØAutomated query expansion
� Small amount of training data:� Simple probabilistic IR models (BM25), with automated tuning� Training set expansion (noisy labeling)� Learning-to-Rank with additional features
Emory University Approach

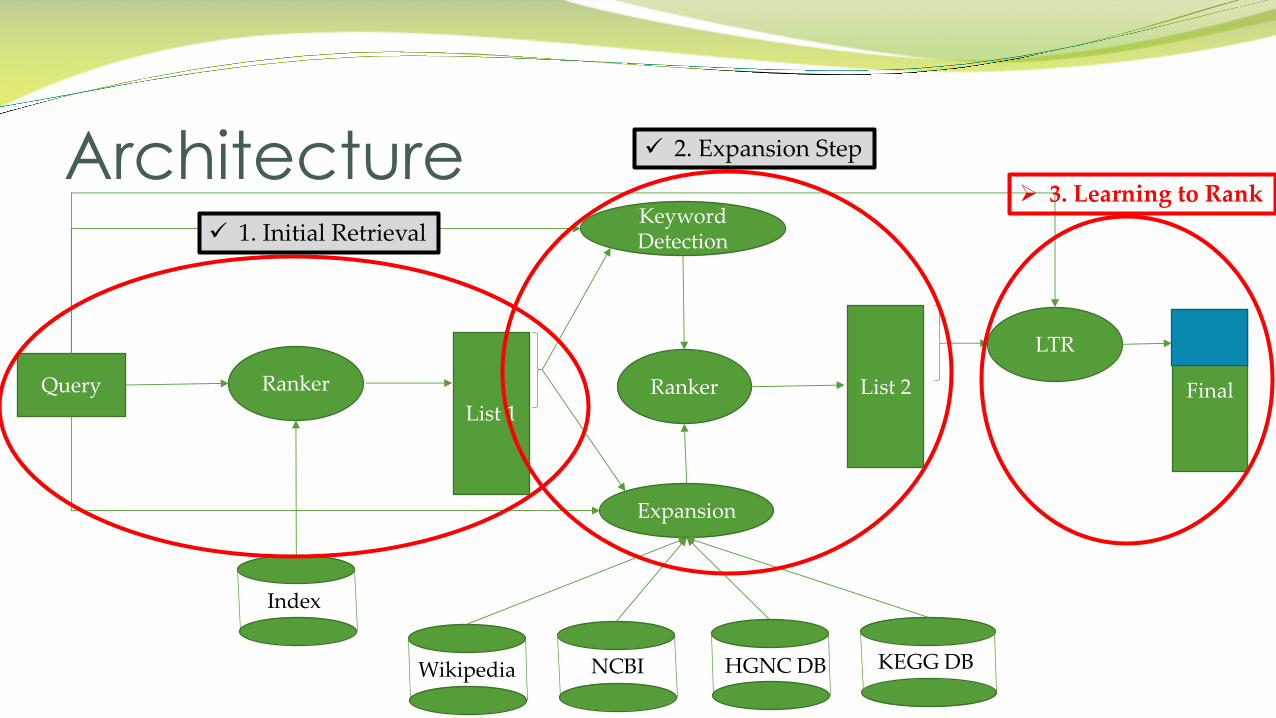
Architecture
Query Ranker
Index
List 1Ranker List 2
LTR
Final
Keyword Detection
Expansion
KEGG DBHGNC DBNCBIWikipedia
1. Initial Retrieval
2. Expansion Step
3. Learning to Rank

� Some details:� All the connections are function calls.
� Except for calling the LTR Module which is an operating system call.� To retrieve from Wikipedia and NCBI we used Google vertical search.
� Do not use Google in practice!� Used offline search in HGNC and search API for KEGG databases.
� Tools:� Apache Lucene was used for indexing and retrieval� RankLib was used for LTR step� trec_eval was used for performance evaluation
Design

� Searchable indexed fields:� Title: dataset name, as provided� Dataset description: as provided
� With simple preprocessing to remove labels� Metadata: Manually collected information about the dataset source
� Intuition: The description of the the database contains additional descriptive information about all of the contained datasets
Indexing Phase

� Grabbed from: ncbi.nlm.nih.govThe National Center for Biotechnology Information (NCBI) is part of the United States National Library of Medicine (NLM), a branch of the National Institutes of Health. The NCBI is located in Bethesda, Maryland and was founded in 1988 through legislation sponsored by Senator Claude Pepper. The NCBI houses a series of databases relevant to biotechnology and biomedicine and an important resource for bioinformatics tools and services. Major databases include GenBank for DNA sequences and PubMed, a bibliographic database for the biomedical literature. Other databases include the NCBI Epigenomics database. All these databases are available online through the Entrez search engine. NCBI is directed by David Lipman, one of the original authors of the BLAST sequence alignment program and a widely respected figure in bioinformatics. He also leads an intramural research program, including groups led by Stephen Altschul(another BLAST co-author), David Landsman, Eugene Koonin (a prolific author on comparative genomics), John Wilbur, Teresa Przytycka, and Zhiyong Lu. NCBI is listed in the Registry of Research Data Repositories re3data.org.[1] GenBank Main article: GenBank NCBI has had responsibility for making available the GenBank DNA sequence database since 1992.[2] GenBank coordinates with individual laboratories and other sequence databases such as those of the European Molecular Biology Laboratory (EMBL) and the DNA Data Bank of Japan (DDBJ).[3] Since 1992, NCBI has grown to provide other databases in addition to GenBank. NCBI provides Gene, Online Mendelian Inheritance in Man, the Molecular Modeling Database (3D protein structures), dbSNP (a database of single-nucleotide polymorphisms), the Reference Sequence Collection, a map of the human genome, and a taxonomy browser, and coordinates with the National Cancer Institute to provide the Cancer Genome Anatomy Project. The NCBI assigns a unique identifier (taxonomy ID number) to each species of organism.[4] The NCBI has software tools that are available by WWW browsing or by FTP. For example, BLAST is a sequence similarity searching program. BLAST can do sequence comparisons against the GenBank DNA database in less than 15 seconds. PubMed PubMed is a database developed by NCBI National Library of Medicine (NLM), it works as a part of the NCBI Entrez retrieval system. It was primarily designed to provide the access to references and abstracts from biomedical and life sciences journals. PubMed provides links that allow access to the full-text journal articles of participating publishers.[5] MEDLINE database is the primary data source for PubMed, which includes the fields of medicine, dentistry, nursing, health care system, veterinary and the preclinical sciences.[6] PubMed Central (PMC) was launched in February 2000, it is a free archive and serves as a digital counterpart to NLM’s extensive print journal collection. PMC provides permanent access to all of its content and is managed by NLM.[7] NCBI Bookshelf The NCBI Bookshelf is a collection of freely accessible, downloadable, on-line versions of selected biomedical books. The Bookshelf covers a wide range of topics including molecular biology, biochemistry, cell biology, genetics, microbiology, disease states from a molecular and cellular point of view, research methods, and virology. Some of the books are online versions of previously published books, while others, such as Coffee Break, are written and edited by NCBI staff. The Bookshelf is a complement to the Entrez PubMed repository of peer-reviewed publication abstracts in that Bookshelf contents provide established perspectives on evolving areas of study and a context in which many disparate individual pieces of reported research can be organized.[citation needed] Basic Local Alignment Search Tool (BLAST) BLAST is an algorithm used for calculating sequence similarity between biological sequences such as nucleotide sequences of DNA and amino acid sequences of proteins.[8] BLAST is a powerful tool for finding sequences similar to the query sequence within the same organism or in different organisms. It searches the query sequence on NCBI databases and servers and post the results back to the person's browser in chosen format. Input sequences to the BLAST are mostly in FASTA or Genbank format while output could be delivered in variety of formats such as HTML, XML formatting and plain text. HTML is the default output format for NCBI's web-page. Results for NCBI-BLAST are presented in graphical format with all the hits found, a table with sequence identifiers for the hits having scoring related data, along with the alignments for the sequence of interest and the hits received with analogous BLAST scores for these[9] Entrez The Entrez Global Query Cross-Database Search System is used at NCBI for all the major databases such as Nucleotide and Protein Sequences, Protein Structures, PubMed, Taxonomy, Complete Genomes, OMIM, and several others.[10] Entrez is both indexing and retrieval system having data from various sources for biomedical research. NCBI distributed the first version of Entrez in 1991, composed of nucleotide sequences from PDB and GenBank, protein sequences from SWISS-PROT, translated GenBank, PIR, PRF and PDB and associated abstracts and citations from PubMed. Entrez is specially designed to integrate the data from several different sources, databases and formats into a uniform information model and retrieval system which can efficiently retrieve that relevant references, sequences and structures.[11] Gene Gene has been implemented at NCBI to characterize and organize the information about genes. It serves as a major node in the nexus of genomic map, expression, sequence, protein function, structure and homology data. A unique GeneID is assigned to each gene record that can be followed through revision cycles. Gene records for known or predicted genes are established here and are demarcated by map positions or nucleotide sequence. Gene has several advantages over its predecessor, LocusLink, including, better integration with other databases in NCBI, broader taxonomic scope, and enhanced options for query and retrieval provided by Entrez system.[12] Protein Proteindatabase is an important protein resource at NCBI. It maintains the text record for individual protein sequences, derived from many different resources such as NCBI Reference Sequence (RefSeq) project, GenbBank, PDB and UniProtKB/SWISS-Prot. Protein records are present in different formats including FASTA and XML and are linked to other NCBI resources. Protein provides the relevant data to the users such as genes, DNA/RNA sequences, biological pathways, expression and variation data and literature. It also provides the pre-determined sets of similar and identical proteins for each sequence as computed by the BLAST. The Structure database of NCBI contains 3D coordinate sets for experimentally-determined structures in PDB that are imported by NCBI. The Conserved Domain database (CDD) of protein contains sequence profiles that characterize highly conserved domains within protein sequences. It also has records from external resources like SMART and Pfam. There is another database in protein known as Protein Clusters database which contains sets of proteins sequences that are clustered according to the maximum alignments between the individual sequences as calculated by BLAST.[13] Pubchem BioAssay database PubChem BioAssay database of NCBI is a public resource for biological tests of small molecules and siRNA reagents. The major purpose of PubChem repository is to provide easy and free of cost access to all deposited data, and to provide intuitive data analysis tools. It is structured as a set of relational databases organized on Microsoft SQL servers. PubChem’s BioAssay data is searchable and accessible by Entrez information retrieval system. PubChem database provides programmatic and Web-based tools for users to search, review, and download a publications, bioactivity data for a compound, a BioAssay record, a molecular target.[14]
Site-level Metadata Example

� Query No 3: Search for data on BRCA gene mutations and the estrogen signaling pathway in women with stage I breast cancer
� Examples showed the occurrences of the general words such as gene, pathway, and cancer in the NCBI description caused the improvement.
Example: Query-Metadata Match
Before METADATA After METADATA
NDCG 0.3709 0.431
P@10 0.2 0.3

� Base method: BM25: � A probabilistic model which tries to rank the documents based on the
estimated probability of relevance: P(R=1|q, d)� BM25 has two parameters to train:
� K1: to calibrate the term frequency (0 ≤K1)� b: to calibrate the document length normalization (0≤b≤1)
� To detect the most informative document section:� Lucene multiple field search to match over all the three searchable fields
Baseline IR Retrieval Model

1. Query expansion with Blind Relevance Feedback:� Assumes the top K retrieved documents are relevant and tries to extract the
relevant terms from these documents to relocate the query
Query Expansion & Reformulation (1 of 3)

2. Query Expansion with external resources� 4 external resources were used to extract expansion terms:
� NCBI and Wikipedia: Accessed through Google and the first relevant pages were retrieved
� KEGG was accessed through a search API� HGNC was accessed offline
� The terms with the highest frequency conditioned on appearing in the top documents were selected (why?)
Query Expansion & Reformulation (2 of 3)

� 3. Automated Query term weighting� bioCADDIE queries are verbose: 15.8 terms on average (web search query is ~3
terms per query)� Idea: weight query terms by “importance”� Weighted Information Gain was used:
Query Expansion & Reformulation (3 of 3)

Query Expansion ExamplesQuery
NoOriginal Query Terms and Automatically Expanded Terms NDCG before
modificationNDCG after modification
1
Find protein sequencing data related to bacterial+ chemotaxis+
across all databases+ [citat cell bacteria gradient direct responsdevelop system primari organ] <nifh ncbi thaw permafrost
alaskan 5s harbor 23 bigsdb campylobact>
0.111 0.291(+162%)
3
Search for all data types related to gene TP53INP1+ in relation to p53+ activation across all databases+ [cell protein express cancer tumor induc function apoptosi human dna] <ptm mmtv ncbi ra
sequenc muscl ebi salivari restrict express>
0.342 0.710(+107%)
10Search for data of all types related to energy metabolism+ in obese+ M. musculus+ [fat studi gene profil cell] <fat obstrut
massag apneic simpl n apnea sleep mechan therapy>0.373 0.436
(+16%)
15Find data on the NF-kB+ signaling pathway in MG
(Myasthenia+ gravis+) patients [activ cell 2 rna gene] <nfkbizstat3 thymoma dlbcl protein myc ncbi abc oci sequenc>
0.603 0.524(-13%)
Architecture
Query Ranker
Index
List 1Ranker List 2
LTR
Final
Keyword Detection
Expansion
KEGG DBHGNC DBNCBIWikipedia
ü 1. Initial Retrieval
ü 2. Expansion Step
Ø 3. Learning to Rank

� LTR is a family of machine learning methods for ranking results� LTR models find an optimal way of combining features extracted from
query-document pairs� Example: SVM-rank a variation of SVM which tries to find a way to sort
documents by classifying document pairs� We used MART: Combines boosting with regression trees as ranking
model� LTR main steps:
� Design features to represent query-document match� Represent top K results as feature vectors for the LTR model� Train the model to optimize feature weights to re-rank the results
Learning to Rank (LTR)

� 8 feature groups were extracted1. BM25 scores2. Shared unigram TF in the fields3. Shared unigram IDF in the fields4. Shared unigram TF-IDF in the fields5. Shared unigrams the concatenated fields6. Shared bigrams in the fields7. The position of the first shared term8. The web domain scale
LTR FeaturesGroup No Feature Name
1 BM251 BM25Title1 BM25Text1 BM25Meta2 1GramTFTitle2 1GramTFText2 1GramTFMeta3 1GramIDFTitle3 1GramIDFText3 1GramIDFMeta4 1GramTFIDFTitle4 1GramTFIDFText4 1GramTFIDFMeta5 1GramTFWhole5 1GramIDFWhole5 1GramTFIDFWhole6 2GramsTitle6 2GramsText6 2GramsMeta6 2GramsWhole7 DistanceFromStart8 DomainWeight
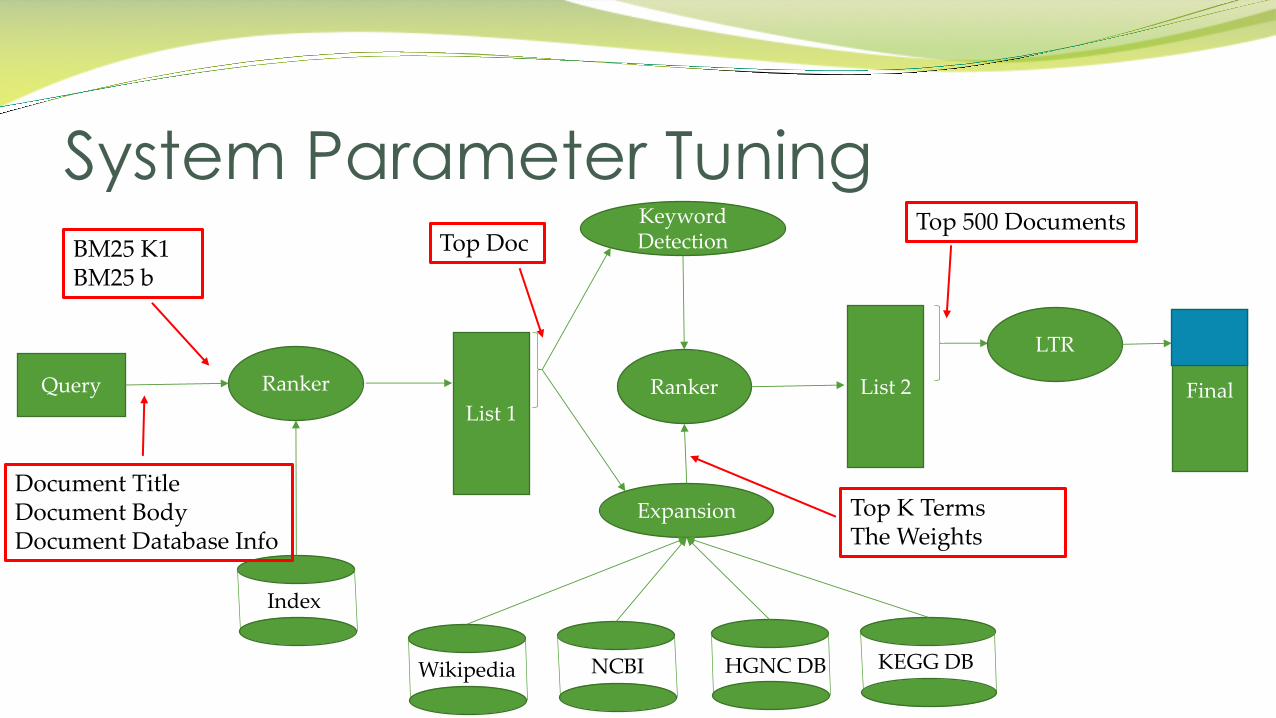
System Parameter Tuning
Query Ranker
Index
List 1Ranker List 2
LTR
Final
Keyword Detection
Expansion
KEGG DBHGNC DBNCBIWikipedia
Document TitleDocument BodyDocument Database Info
BM25 K1BM25 b
Top Doc
Top K TermsThe Weights
Top 500 Documents

� 4-fold cross validation was carried out over all the 21 queries (6 train + 15 test)
� Tuned parameters for the initial retrieval:
Parameter Optimization: Baseline retrieval
Parameter Description Range Best Value
TITLE weight Weight of TITLE in the retrieval 0.1, 0.3, 0.5, 0.7 0.1TEXT weight Weight of METADATA in the retrieval 0.1, 0.3, 0.5, 0.7 0.3METADATA weight Weight of DATASET_INFO in the retrieval 0.1, 0.3, 0.5, 0.7 0.5BM25 k1 K1 parameter in BM25 0.6, 1, 1.4, 1.8 1.8BM25 b b parameter in BM25 0.3, 0.5, 0.7, 0.9 0.7

Parameter optimization: Query Expansion
Parameter Description Range Best Value
Top datasets Top datasets selected for WIG model, BRF, and external expansion
5, 10, 30 5
Internal terms Number of terms added to the query by BRF 5, 10, 30 5Weights for internal terms
Weight of the terms selected by BRF 0.1, 0.3, 0.5 0.1
External terms Number of terms added to the query using external resources
5, 10, 30 10
Weights for external terms
Weight of the terms added using external resources
0.1, 0.3, 0.5 0.5
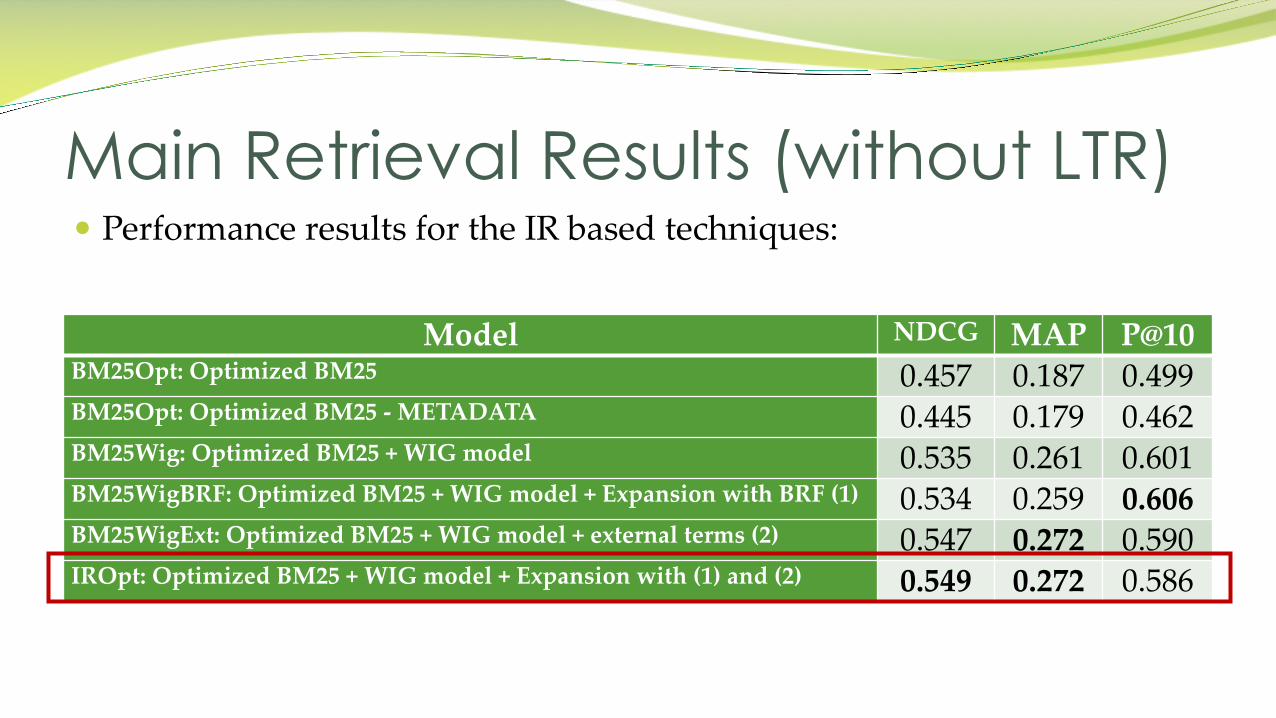
� Performance results for the IR based techniques:
Main Retrieval Results (without LTR)
Model NDCG MAP P@10BM25Opt: Optimized BM25 0.457 0.187 0.499BM25Opt: Optimized BM25 - METADATA 0.445 0.179 0.462BM25Wig: Optimized BM25 + WIG model 0.535 0.261 0.601BM25WigBRF: Optimized BM25 + WIG model + Expansion with BRF (1) 0.534 0.259 0.606BM25WigExt: Optimized BM25 + WIG model + external terms (2) 0.547 0.272 0.590IROpt: Optimized BM25 + WIG model + Expansion with (1) and (2) 0.549 0.272 0.586
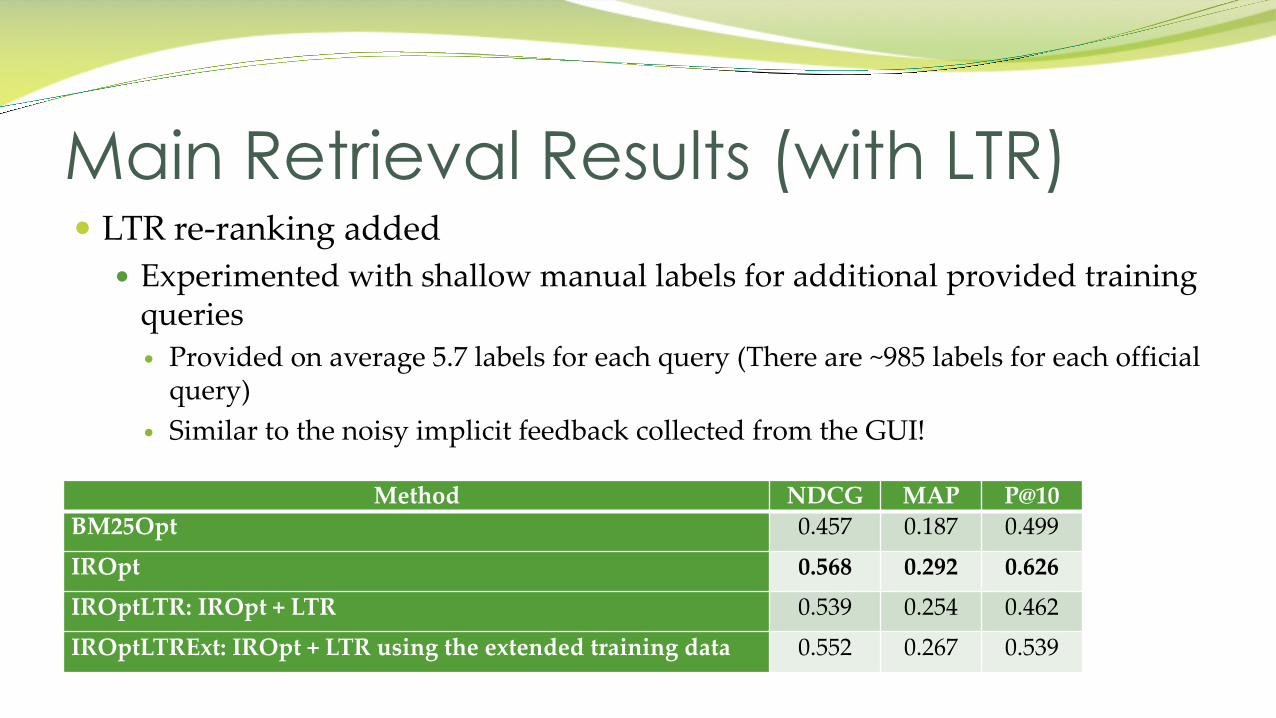
� LTR re-ranking added� Experimented with shallow manual labels for additional provided training
queries� Provided on average 5.7 labels for each query (There are ~985 labels for each official
query)� Similar to the noisy implicit feedback collected from the GUI!
Main Retrieval Results (with LTR)
Method NDCG MAP P@10BM25Opt 0.457 0.187 0.499
IROpt 0.568 0.292 0.626IROptLTR: IROpt + LTR 0.539 0.254 0.462
IROptLTRExt: IROpt + LTR using the extended training data 0.552 0.267 0.539

� Feature ablation for LTR framework
Contribution of LTR Feature groupsRank Category NDCG after
omission1 (group 1) BM25 scores 0.538
2 (group 3) unigram IDF in the dataset fields 0.544
3 (group 5) unigram in the whole (concatenated) dataset fields 0.548
4 (group 7) DistanceFromStart 0.550
5 (group 2) unigram TF in the dataset fields 0.550
6 (group 8) DomainWeight 0.5537 (group 6) shared bigrams 0.557
8 (group 4) unigram TF-IDF in the dataset fields 0.558

� We tried multiple retrieval models: VS model, and language model based methods, did not improve over baseline+query expansion
� Experimented with topic distributions for each dataset and query pair, to use as features in the LTR framework: no improvement seen
� Experimented with multiple LTR models: RankNet and Coordinate Ascent, no significant differences
Lessons learned

� Implicit feedback to re-train LTR models� Can use as “noisy labels” for training: clicks, dwell time on visited results à relevance labels
� Augment feature sets with behavior features� Revisit term generalizations (with topic models or word embeddings) with
more training labels
� Dynamic query expansion:� Learn to automatically decide whether to expand a query based on initial
retrieved results.
Potential Future Work

� Enriching the dataset descriptions with available (meta-)information on the web is helpful
� Default parameter settings should be re-optimized for dataset� Keyword detection critical� Query expansion using text based resources shown helpful� LTR prone to overfitting on small training sets, improves with more data
� Implicit feedback can be potentially helpful!� Used as “noisy labels” for training: clicks, dwell time on visited results� Could enable more sophisticated LTR and text representation methods
Conclusions

� Development partially supported by subcontract from the BioCADDIEproject
� More details: in Database article, in revision.
Thank you!