pharmakologie der hyperlipidämie - home | meduni wien · microsoft powerpoint -...
TRANSCRIPT

Lipoproteine (=Transportform der Lipide; Lipide = Sammelbegriff, z.B. Fette, Wachse )
Kernlipid Spez. Dichte
Apolipoprotein Stoffwechsel
HDL Phospholipid, Cholesterinester
A, E, C > Aufnahme in die Leber oder > Cholesterin-Abgabe an LDL/VLDL
LDL Cholesterinester B-100 LDL-Rezeptor Aufnahme (75% in d. Leber)
IDL (VLDL-Remnants)
Cholesterinester, Triglyzerid
B-100, E, C 50% umgewandelt in LDL 50% Aufnahme in Leber
VLDL Triglyzerid aus d. Leber
C, B-100, E Triglyzerid-Hydrolyse
Chylomikrone
Aus der Nahrung: Triglyzerid,
Cholesterinester
Triglyzerid-Hydrolyse (Lipoproteinlipase) FFA + Chylomikronenremnant
Chylomikronen-Remnants
Cholesterinester, wenig Triglyzerid
B-48, C, E, A
Aufnahme in Leber
Pharmakologie der Pharmakologie der HyperlipidHyperlipidäämiemie(siehe Freissmuth, Offermanns, Böhm, Pharmakologie & Toxikologie, Kapitel 43, S 452-471 )
Autor: C. NanoffInstitut für Pharmakologie, Med.Universität Wien

LPL...Lipoproteinlipase, HL...hepatische Lipase, FFA...freieFettsäuren
Lipidtransport

Fett in der Nahrung besteht zum Großteil aus Triglyzeriden und Cholesterinestern. Mit Hilfe der Gallensäuren wird das Fett in die Darmepithelzelle aufgenommen, von wo der Weitertransport in Form von Chylomikronen erfolgt. Für die Aufnahme von Cholesterin und pflanzlichen Sterinen wird NPC1L1 (Niemann-Pick C1-like protein) benötigt. Phytosterine kommen nicht in Chylomikrone sondern werden aktiv ins Darmlumen zurückgepumpt. Chylomikrone sind Lipoproteine, bestehen daher aus einem Proteinanteil (Apolipoproteinen=„apo“) und Lipiden (Triglyzeride, Cholesterinester, Phospholipide); die Apolipoproteine bestimmen das Schicksal jedes Lipoproteinpartikels. Chylomikrone werden im Darmepithel gebildet, enthalten zunächst nur Apo-B48 und nehmen erst im Blutstrom funktionell bedeutsame Apolipoproteine auf: (i) ApoC-II aktiviert die Lipoproteinlipase (LPL) in der Endstrombahn verschiedener Gewebe (Muskulatur, Fettgewebe), Triglyzerid wird hydrolysiert und die gewonnenen Fettsäuren an die Zellen geliefert. Dadurch entstehen die „Chylomikronen-Remnants“, die sich von der Kapillarwand ablösen und sehr rasch von den Leberzellen aufgenommen werden. Für die Aufnahme wird der Remnant-Rezeptor benötigt (auch LRP = LDL-Rezeptor related protein), der (ii) apoE als Liganden erkennt.
Lipoproteine - Abbildungslegende

VLDL wird in der Leber produziert und zwar, wenn der Leber ausreichende Mengen an Triglyzerid zur Verfügung stehen. Das Apolipoprotein ist B-100. Komplett wird VLDL aber erst nach Aufnahme der Apoproteine, apoC und apoE, im Blut. Wie die Chylomikrone wird VLDL durch apoC-II-aktivierte LPL verstoffwechselt. Die entstehenden VLDL-Remnants (=IDL) werden durch LDL-Rezeptoren (Ligand = apoB-100) oder LRP (Ligand = apoE) in die Leber gebracht. Etwa die Hälfte des IDL erfährt jedoch ein anderes Schicksal. Dieses wird weiter durch LPL und HL (= hepatische Lipase) abgebaut, wobei auch apoC und apoE entfernt werden. So entsteht LDL, das aus apoB-100 und Cholesterinestern aufgebaut ist. Wegen seiner geringen Clearance ist der LDL-Spiegel höher als der von VLDL/IDL. 75% des LDL geht in die Leber, der Rest in periphere Zellen, die mit LDL-Rezeptoren ausgestattet sind. Eine Zunahme der LDL-Rezeptoren wird durch Cholesterinmangel in der Leber induziert, dies führt verlässlich zur Abnahme der LDL-Spiegel. der Effekt wird durch SREBP2, einen cholesterinempfindlichen Transkriptionsfaktor vermittelt. Ebenfalls steigernd wirken Schilddrüsenhormon und Östrogen. LDL ist ein für die Atherosklerose wichtiges Lipidpartikel; nach Oxidation („ranzig Werden“) wird es von subendothelialen Makrophagen durch „Scavenger-Rezeptoren“ wie SR-B1 endozytiert ( Entstehung von Schaumzellen atherosklerotischer Plaque). Hohe LDL-Werte sind ein Risikofaktor für Atherosklerose und Herzinfarkt.
Lipoproteine - Abbildungslegende, Fortsetzung

HDL entsteht aus Apolipoprotein A-I und Phospholipid in Leber/Darm, kann sich aber auch spontan in der Blutbahn formieren. Diese Vorstufe (= Prä-1HDL) nimmt unverestertes Cholesterin aus Gefäßwänden auf (dank Bindung von apoA-I an einen spezifischen Transporter=ABCA1). Das Prä-1HDL Partikel wächst durch Aufnahme von mehr Cholesterin. Große HDL Partikel vom Typ HDL2 enthalten hauptsächlich Apo A-I und schützen vor Atherosklerose. Durch Andocken an einen Rezeptor (SR-B1) übernimmt die Leberzelle Cholesterin aus dem Partikel; über den gleichen Rezeptor kann Cholesterin aber auch ins Partikel nachfließen. Cholesterin wird durch Lecithin-Cholesterin-Acyltransferase (LCAT) verestert; in dieser Form wird es vermittels CETP auch auf LDL-Partikel übertragen (CETP = Cholesterinester-Transferprotein). HDL sind für den „reversen Cholesterintransport“ in die Leber mitverantwortlich: nur Leberzellen können Cholesterin abbauen, aber alle Körperzellen können im Prinzip Cholesterin synthetisieren.
Lipoproteine - Abbildungslegende, Fortsetzung

SRELDL-RezeptorHMG-CoA-Reduktase
Cholesterin
SCAPInsig-1
SREBP2SREBP2
Cholesterin
Insig-1 SCAP
SREBP2nSREBP2
SRESRELDL-RezeptorHMG-CoA-Reduktase
Cholesterin
SCAPInsig-1
SREBP2
Cholesterin Cholesterin
SCAPInsig-1
SREBP2SREBP2
Cholesterin
Insig-1 SCAP
SREBP2
Cholesterin
Insig-1 SCAP
Cholesterin Cholesterin
Insig-1 SCAP
SREBP2nSREBP2
ER
Golgi-Apparat
Zellkern
Insig-1 retiniert SCAP im endoplasmatischenReticulum (ER)
SCAP…SREBP cleavageactivating protein
SREBP…Sterolregulatory elementbinding protein
Cholesterin regelt die zelluläre Aufnahme von LDL und die Cholesterin- (= seine eigene) Neusynthese. Die Cholesterinkonzentration in der Membran des endoplasmatischenReticulums (ER) bestimmt den Weitertransport von SCAP-SREBP2 zum Golgi-Apparat; Cholesterin stabilisiert die Bindung zwischen Insig-1 und SCAP und verhindert dadurch den Einbau von SCAP-SREBP2 in Vesikel, die sich vom ER abspalten und mit dem Golgi-Apparatfusionieren. An der Golgi-Membran werden Proteasen aktiv, die den Transkriptionsfaktor nSREBP2 freisetzen. Nach seiner Translokation in den Kern, steigert er die Expression von LDL-Rezeptoren und HMG-CoA-Reduktase.

Insulinresistenz: Einfluss der Hyper-insulinämie auf Blutlipide
Lebe
rzel
le
Übermäßige Aktivierung des Insulinrezeptors in der Leber führt zu typischen Veränderungen des Lipidprofils. Zunehmende Insulinspiegel fördern die Bildung des Transkriptionsfaktors SREBP1-c (verwandt mit SREBP2, das den Cholesterinhaushalt reguliert aber nicht durch Insulin induziert wird). SREBP1-c hält die Kontrolle über den Insulin-abhängigen Fett- und Kohlenhydratstoffwechsel in der Leber. SREBP1-c steigert durch Induktion die Fettsäure- und Triglyzeridsynthese, fördert damit die Bildung und Freisetzung von VLDL und kann darüber hinaus zur Entstehung von Fettleber bei Typ II Diabetes führen. Aus einer intronischen Sequenz des SREBP1-c Gens entsteht ferner eine mikroRNA die - selbst komplementär zu einem Abschnitt der ABCA1-Sequenz - zum Abbau der ABCA1 –Transporter kodierenden mRNA führt (für diese Prozesse steht jeder Zelle ein Enzymapparat – RISC Komplex - zur Verfügung). Zu wenig ABCA1 reduziert die HDL-Spiegel. Hohes VLDL und niedriges HDL bei Patienten mit Insulinresistenz. Physiologisch fördert Insulin hingegen durch einen permissiven Effekt auf die Lipoproteinlipase (LPL) den Abbau von Triglyzerid in der Blutbahn. AMPK Aktivität vermindert SREBP1c und senkt VLDL.
AMPK

Störungen im Lipoproteinstoffwechsel = Dyslipidämie Primäre Dyslipidämie: polygenetische Ätiologie (monogenetische Defekte selten) Sekundäre Dyslipidämie - Beispiele Hypothyreose ( LDL) Diabetes mellitus, Nierenerkrankungen (Hypertriglyzeridämie,
HDL, LDL oder ) Medikamente – z.B.: -Blocker, Thiazid-Diuretika
(Triglyzeride) Risikofaktoren für die Atherosklerose Messwert Ungünstig Apo B/ Apo A-I Verhältnis LDL-gebundenes Cholesterin Apo A-I HDL-gebundenes Cholesterin LDL : HDL Verhältnis Lipoprotein(a) Hypertriglyzeridämie

Gesamtcholesterin < 190 mg/dl Wünschenswert>240 mg/dl Hoch HDL-C(holesterin) < 40 mg/dl Niedrig > 60 mg/dl Hoch LDL-C < 100 mg/dl Wünschenswert>160 mg/dl Hoch Triglyzeride < 150 mg/dl Normal > 200 mg/dl Hoch
Sollwerte
Indikation für Behandlung mit Lipidsenker abhängig von Atheroskleroserisiko und LDL-Cholesterin Ohne Risikofaktor + LDL-C 160-190mg/dl Bei 2 Risikofaktoren + LDL-C > 160 mg/dl Bei koronarer Herzkrankheit + LDL-C >130 mg/dl Bei KHK + Diabetes mellitus + LDL-C > 100 mg/dl

Lipidsenker Angriffspunkte und Wirksamkeit Angriffspunkt Stoffgruppe Abfall
des LDL-C Zunahme von HDL-C
HMG-CoA-Reduktase () Statine 20-55%
10%
Enterohepatischer Kreislauf der Gallensäuren ()
Anionenaus-tauscherharze
20%
4%
PPAR Leberstoffwechsel Fibrate ? 5-15% Unklar Nikotinsäure 25% 15 – 30% Cholesterintransport () am Darmepithel (NPC1L1)
Ezetimibe 20% unbekannt
Kombinationstherapie LDL-C Abfall um mehr als 50% Partner: Statin + Anionenaustauscherharz Simvastatin + Ezetimibe
Statin + Fenofibrat Statin + Nikotinsäure

Statine = kompetitive Hemmung der HMG-CoA-Reduktase, einem Schlüsselenzym der Cholesterinbiosynthese [HMG-CoA = Hydroxy-Methyl-Glutarsäure-Koenzym A] Vertreter (gereiht nach Affinität für HMG-CoA-Reduktase) Fluva- < Prava -, Lovas- < Simva- < Atorva- < Rosuva- < Pitava
Effekt 1. Reduzierte Cholesterinsynthese in der Leber ...
LDL-Rezeptoren LDL-Aufnahme Abfall des Plasma LDL titrierbar
2. VLDL-Synthese Triglyzeride Metabolismus der Statine First-pass effect: Aufnahme in Leberzelle durch OATP, Abbau durch CYPs
und Glucuronyltransferase 5-20% der Dosis erreicht die systemische Zirkulation (Gefahr von tödlichen
Zwischenfällen durch Bioverfügbarkeit = Interaktionen! z.B. Lipobay=Cerivastatin) Kein Einfluss auf NNR- und Keimdrüsenfunktion Halbwertszeit < 12 hr Orale Gabe, einmal täglich (am besten abends)

Verträglichkeit = gut. Nebenwirkungen 1. Muskelbeschwerden 2. Statin-Myopathie Risiko der Rhabdomyolyse (Inzidenz gering <0.1%, Lebensgefahr)
Symptome wie beim grippalen Infekt Zunehmende Schmerzen, Abgeschlagenheit Serum-Creatinkinase, Myoglobinurie Akutes Nierenversagen
3. KI = Schwangerschaft. 4. Hepatotoxizität = sehr selten
höhergeringBioverfügbarkeit
< x 2< x 2CYP2C9 Inhibitor
50%
x 5
< x 2
PravaRosuvaFluvaPitava
75%Induktion
x 10Ciclosporin
x 10CYP3A4 Inhibitor
SimvaLovaAtorva
Effekte auf AUC
Pharmakokinetische Eigenschaften und Wechselwirkungen von Statinen

Gemfibrozil
CYP2C9-Inhibitoren:Nife-, NicardipinDiclofenacOmeprazolAzol-Antimykotika(Fluconazol, Voriconazol)
CYP3A4-InhibitorenAzol-AntimykotikaMakrolid-AntibiotikaHIV-Protease-InhibitorenCiclosporin
Risikozunahme durch Wechselwirkung
Wenn Kombination mit einem dieser Mittel notwendig: Statin-Dosisniedrig halten = ein Viertel der maximal empfohlenen Dosis

keine
Thiazolidindione
Fibrate
Pharmakol.Agonist
Lactatdehydrogenase
Adiponectin
Enzyme der Fettsäureoxidation
Zielgene
Ungesättigte Fettsäuren(Arachidonsäuremetabolite)
Rezeptorligand
PlacentaFoetusSkeletmuskelHerzmuskel
2 in Fettzellen und Vorstufen
1, 2
Leber
GewebePPAR-Subtyp
PPAR – nukleäre Rezeptoren Angriffspunkte für stoffwechselaktive Pharmaka

Fibrate, Liganden des PPAR (Peroxisomen-Proliferator-Aktivator-Rezeptor ) Vertreter: Fenofibrat, Bezafibrat, Gemfibrozil Aktivierung von PPAR in Leberzellen Fettsäureoxidation apoC LPL VLDL
Fenofibrat apoA HDL Abnahme der „small dense“ LDL-Partikel AG = MCI Prävention, wenn HDL-Cholesterin niedrig (und Triglyzeridspiegel hoch) Indikationen Dyslipidämie wie bei Typ II Diabetes (HDL, Triglyzeride), aber: Fenofibrat-
Kombi mit Statin bringt keinen Nutzen im Vergleich zu Statin allein Typ IIb Hyperlipidämie (LDL-C, Triglyceride) Hypertriglyzeridämie Verträglichkeit = gut Gastrointestinale Störungen (schwach leberschädlich) Relative Kontraindikation= Nieren-, Leberinsuffizienz Risiko für Entstehung von Gallensteinen KI = Schwangerschaft und Kindesalter

Nikotinsäure (Präparate: Nikotinsäure in Retardgalenik; Acipimox)aktiviert in supraphysiologischen Dosen (1-2 g/Tag) einen G-Protein-gekoppelten Rezeptor…im Fettgewebe Hemmung der Lipolyse (vorübergehend)…in Hautzellen Prostaglandinfreisetzung Flush (vorübergehend)Lipidsenkung: Triglyzeride, LDL-Cholesterin, HDLMechanismus verzögert (HDL), indirekt und im Detail unbekannt
Pharmakokinetik: Hohe Clearance (15 ml/min/kg)• Rasche Verstoffwechslung zu Nikotinamid, NAD; Konjugation mit Glycin Nikotinursäure• Aus Nierentubuluszellen wird Nikotinsäure über den URAT1-Transporter in den Harn
sezerniert - als „Gegenanion“ treibt Nikotinsäure die Rückresorption der HarnsäureAG = Kombi mit Statin (in submaximaler Dosis)
Nebenwirkungen• Bei Therapiebeginn Flushsymptomatik mit Juckreiz, Parästhesien und Kopfschmerz – Dosis
langsam steigern („einschleichen“), zur Prävention auch Gabe von COX-Hemmern oder Laropiprant (Prostaglandin D2-Rezeptorantagonist)
• Insulinresistenz• Magenschmerzen (präventiv Antazida)• Leberschäden• Sehstörung infolge Retinaödems (reversibel)
KIDiabetesUlcus(anamnese)Hyperurikämie
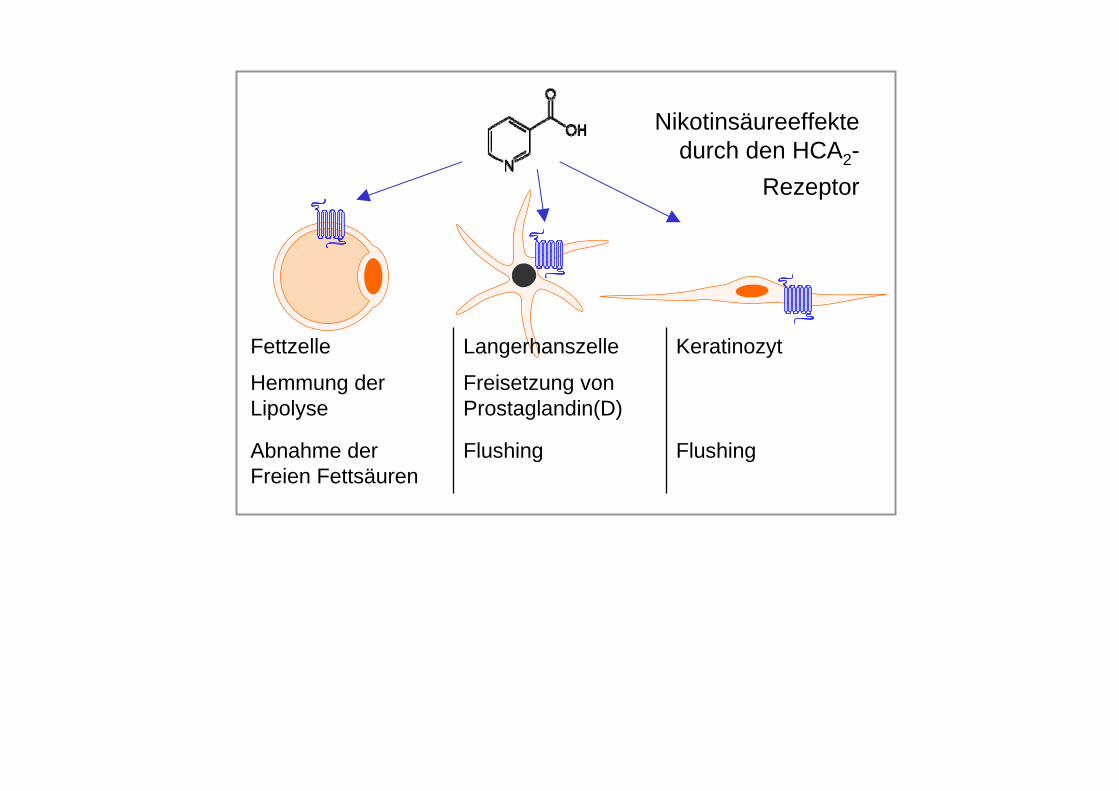
FlushingFlushingAbnahme der Freien Fettsäuren
Freisetzung von Prostaglandin(D)
Hemmung der Lipolyse
KeratinozytLangerhanszelleFettzelle
Nikotinsäureeffekte durch den HCA2-
Rezeptor

Fettresorptionshemmung Ezetimibe hemmt intestinalen Cholesterintransport Anwendung allein: Cholesterinsynthese Anwendung in Kombination mit Statinen : LDL Pharmakokinetik: Sehr lipophil hepatische Glucuronidierung aktiver Metabolit Enterohepatischer Kreislauf Halbwertszeit ~ 20h Verträglichkeit: gut, wenig Erfahrung AG = ? (verhindert nicht Atherosklerose) Anionen-Austauscherharze Kolloidale nicht resorbierbare Kunsstoffpolymere (hygroskopisch)- Cholestyramin, Colesevelam - hemmen Gallensäure- und Cholesterin-Resorption LDL-Rezeptor-Induktion und vermehrte Cholesterinsynthese. 1. LDL um 20% 2. VLDL-Spiegel anfänglich 3. HDL Nebenwirkungen Verdauungsstörungen (Obstipation, Völlegefühl, Fettstühle), Präparate schmecken schlecht Einnahmehinweise 2*tägl. vor dem Frühstück und Abendessen Mittel im Kühlschrank mit Wasser quellen lassen! Können Resorption von Pharmaka reduzieren– Einnahmezeitpunkte versetzen!