praticas em troca ionica.pdf
TRANSCRIPT

Práticas em Cromatografia de íonsUma Introdução
Monografia
Metrohm Pensalab Instrumentação Analítica Ltda.Rua Minerva, 167 – PerdizesSão Paulo – SP – BrasilCEP 05007-030Fone +55 (11) 3868-6599Fax +55 (11) 3868-6575E-mail [email protected]


Práticas em Cromatografia de Íons 1
Práticas em Cromatografia de íons
Uma Introdução
2ª edição
Eng. Claudia Eith Prof. Dr. Maximilian Kolb Químico Achim Rumi Prof. Dr. Andreas Seubert Dr. Kai Henning Viehweger (Editor)
Monografia Metrohm Todos os direitos reservados a Metrohm, incluindo tradução. Impresso pela Metrohm Ltda., CH-9101 Herisau, Suíça. 8.792.5013PT – 2006-07

2 Monografia Metrohm

Práticas em Cromatografia de Íons 3
Índice
1. Os autores ............................................................................................................................................5
2. Introdução.............................................................................................................................................6
3. Seção teórica........................................................................................................................................7
3.1. A história e a importância da cromatografia de íons ................................................................... 73.2. Teoria da Cromatografia .............................................................................................................. 9
3.2.1. Divisões da cromatografia e terminologia ............................................................................ 93.2.2. Conceitos teóricos para a descrição do processo cromatográfico..................................... 12
3.3. Princípios básicos da cromatografia de íons (CI) ...................................................................... 163.3.1. Terminologia e classificação em CL................................................................................... 163.3.2. Troca iônica ........................................................................................................................ 173.3.3. Formação de par iônico ...................................................................................................... 183.3.4. Exclusão de íons ................................................................................................................ 18
3.4. Modelos de retenção em cromatografia de íons ..................................................................... 193.4.1. Modelos de retenção em cromatografia de ânions ............................................................ 193.4.2. Modelos de retenção em cromatografia de cátions ........................................................... 24
3.5. Sistemas de detecção em cromatografia de íons.................................................................... 273.5.1. Métodos de detecção eletroquímica................................................................................... 273.5.2. Métodos espectroscópicos de detecção ............................................................................ 31
3.6. Fases estacionárias em cromatografia de íons ....................................................................... 323.6.1. Visão geral das fases estacionárias comuns ..................................................................... 323.6.2. Fases estacionárias para cromatografia de ânions............................................................ 343.6.3. Fases estacionárias em cromatografia de cátions ............................................................. 353.6.4. Trocadores de cátions baseados em sílica gel .................................................................. 353.6.5. Trocadores de cátions baseados em polímeros orgânicos................................................ 353.6.6. Trocadores de cátions peliculares...................................................................................... 353.6.7. Fases estacionárias em cromatografia de exclusão iônica................................................ 363.6.8. O significado da capacidade dos trocadores de íons......................................................... 36
3.7. Eluentes em cromatografia de íons ......................................................................................... 373.7.1. Cromatografia de ânions .................................................................................................... 373.7.2. Cromatografia de cátions.................................................................................................... 40
3.7.2.1. Cromatografia de cátions de íons alcalinos, alcalino-terrosos e amônia com detecção de condutividade .......................................................................................................................................... 403.7.2.2. Cromatografia de cátions de íons metal de transição e alcalino-terrosos com detecção pela derivatização pós-coluna e fotométrica ................................................................................................... 403.7.2.3. Cromatografia de exclusão de íons............................................................................................ 42

4 Monografia Metrohm
4. Seção prática......................................................................................................................................43
4.1. Informações sobre a parte prática ............................................................................................. 434.2. Experimentos abrangendo a teoria da cromatografia de íons................................................ 47
4.2.1. Experimento 1 – Cromatografia de íons com e sem supressão ........................................ 474.2.2. Experimento 2 – Capacidade das colunas de separação .................................................. 514.2.3. Experimento 3 – Seletividade das colunas de separação.................................................. 544.2.4. Experimento 4 – Limites de calibração, detecção e determinação na cromatografia de íons...................................................................................................................................................... 60Experimento 4a – Determinação de ânions com supressão química .......................................... 614.2.5. Experimento 5 – Alteração da seletividade com o auxílio de éteres coroa (18 Crown-6) . 644.2.6. Experimento 6 – Alteração da seletividade pela utilização de agentes complexantes...... 674.2.7. Experimento 7 – Técnica de pré-concentração.................................................................. 72
4.3. Experimentos para a determinação de ânions ....................................................................... 754.3.1. Experimento 8 – Ânions em água potável.......................................................................... 754.3.2. Experimento 9 – Ânions em etanol e destilados (licores) .................................................. 794.3.3. Experimento 10 – Ânions em alface................................................................................... 854.3.4. Experimento 11 – Ácido fosfórico em refrigerantes tipo “cola”........................................... 884.3.5. Experimento 12 – Ácidos orgânicos em vinho ................................................................. 934.3.6. Experimento 13 – Contaminantes em borato – determinação de cloretos e sulfatos em soluções de bórax......................................................................................................................... 984.3.7. Experimento 14 – Determinação de Ânions em água efluente ........................................ 1044.3.8. Experimento 15 – Fluoreto em creme dental ................................................................... 1084.3.9. Experimento 16 – Ânions em açúcar refinado branco e mascavo................................... 1114.3.10. Experimento 17 – Contaminantes em peróxido de hidrogênio ...................................... 115
4.4. Experimentos para a determinação de cátions .................................................................... 1214.4.1. Experimento 18 – Metais alcalinos e metais alcalino-terrosos em água potável............. 1214.4.2. Experimento 19 – Determinação de metais de transição................................................. 125Experimento 19a – Determinação de metais de transição com um eluente contendo ácido tartárico, ácido cítrico, etilenodiamina e acetona ....................................................................... 1264.4.3. Experimento 20 – Contaminantes em sílica gel – determinação de íons cálcio e magnésio.................................................................................................................................................... 1314.4.4. Experimento 21 – Cosméticos e proteção contra corrosão: determinação de etanolaminas e metais alcalinos ....................................................................................................................... 1344.4.5. Experimento 22 – Metais alcalinos e metais alcalino-terrosos em vinho......................... 138
5. Literatura citada............................................................................................................................... 142

Práticas em Cromatografia de Íons 5
1. Os autores
Claudia Eith
Estudou Química na Fachhochschule em Aalen; realizou trabalhos práticos na área de análise de água potável e efluentes em Adelaide (Austrália). Desde 2000 desenvolve trabalhos na divisão de Pesquisa e Desenvolvimento da Metrohm Ltda.
Maximilian Kolb
Estudou Química na Technical University em Munique, com ênfase em pesquisas na área de catálise homogênea. Gerenciou o setor de qualidade da Divisão Pública de Águas em Traunstein por cinco anos. Desde 1982, é professor na Fachhochschule em Aalen; áreas de trabalho: tecnologia e controles ambientais e quimiometria.
Achim Rumi
Estudou Química no Swiss Federal Institute of Technology (ETH), Zurique, Suíça. Desde 2005 é Especialista de Produto IC na Metrohm Ltda.
Andreas Seubert
Estudou Química na Hannover University; promoção em 1990: “Análise de ultratraços em metais refratários altamente puros com separação de traços da matriz por Cromatografia de Íons”; habilitação em 1995: “Aplicações em linha da Cromatografia Líquida acoplada a Espectrometria Atômica em análise elementar”. De 1998 a 2000: Professor temporário em Química Analítica na Kassel University. Desde março de 2000 é Professor de Química Analítica na Philipps University em Marburg.
Kai Henning Viehweger
Estudou Química na Hamburg University; tese na área de análise inorgânica; promoção na área de pesquisa em sistemas ecológicos marinhos e em estuários. Desde 1996, no Setor de Marketing na Metrohm Ltda. – Gerência internacional do centro de competência em cromatografia de íons.

6 Monografia Metrohm
2. Introdução
Examinar coisas que não se revelam por si mesmas é sempre um desafio. As razões para isto variam desde uma simples curiosidade até uma real necessidade de sobrevivência. Há várias formas de se “desvendar estes mistérios”. A forma mais simples é usando os sentidos humanos: audição, tato, olfato, paladar e visão. Nos primórdios, os alquimistas gostavam de usar estes cinco sentidos. É por isso que sentimos um sabor azedo quando provamos ácidos e, o bromo tem seu nome derivado de “bromos”, fétido em Grego. A olho nu, uma solução de cromo mostra-se colorida, sendo desta forma associada ao termo Grego “chroma”, ou seja, cor. Os alquimistas também expressavam seus sentimentos com insultos tais como “you kobold” para cobalto, cuja presença causou aos nossos ancestrais grande dificuldade na produção de ferro.
Muitas vezes, componentes presentes em diversos tipos de materiais não podem ser vistos diretamente. Eles estão tão fortemente misturados que os sentidos humanos não são capazes de identificá-los. É neste momento que a análise é utilizada. É possível extrair informação precisa de uma mistura indefinida de componentes, a qual não pode ser obtida apenas pelos sentidos humanos.
Embora o organismo seja repleto deles, os sentidos humanos não podem identificá-los diretamente. Estamos falando dos íons, aqueles átomos ou moléculas carregados eletricamente que são parte integral de praticamente toda matéria viva ou morta. Os íons são responsáveis pela transferência de informação através dos nervos, pela garantia de que a digestão ocorrerá, de que a pressão sanguínea está correta e de que há oxigênio suficiente no sangue. Os íons produzem sal no mar, controlam sua sede e os constituintes iônicos são usados como alimento por todos os seres vivos – desde as bactérias até os seres humanos.
O conhecimento sobre os tipos e número de íons presentes no ambiente nos ajuda a entender as interações ecológicas e bioquímicas. Caso as concentrações iônicas em um determinado alimento sejam conhecidas, isto nos permitirá saber se o alimento é seguro para o consumo ou não.
Há diferentes formas de se determinar os íons qualitativamente (pelo tipo) e quantitativamente (pela quantidade). Cada parte da informação é importante. Um método usado para a obtenção desta informação é a cromatografia de íons. Basicamente, cromatografia significa “escrever em cores”. Em análise química clássica isto significa a separação de substâncias, de acordo com suas cores, e sua determinação, pela observação visual. Apesar de nem todos os íons serem caracterizados por cores visíveis, o termo é mantido, mas outros métodos de detecção são usados hoje em dia.
A cromatografia de íons é um membro da grande família de métodos cromatográficos. Basicamente, ela pode ser usada para determinar qualquer íon que carregue uma ou mais cargas. No passado, a cromatografia de íons ou “CI” era um método muito dispendioso, mas hoje em dia, tem um preço bem mais favorável. Por isso, ela se tornou uma ferramenta analítica poderosa e universal, sendo fácil de usar.
Esta monografia “Práticas em Cromatografia de íons” demonstrará que a IC não é apenas um assunto abstrato ou somente teórico, mas que ela pode fornecer respostas rápidas para os problemas do dia-a-dia tais como: A água potável está adequada para os bebês consumirem? Qual é a quantidade de nitrato que há no espinafre? Um determinado efluente causa poluição ambiental? Visto que, um trabalho analítico prático exato é quase impossível sem uma base teórica, esta monografia também contém informações detalhadas em uma seção teórica.
“Práticas em Cromatografia de íons” tem como objetivo, não apenas fornecer conhecimentos sobre os princípios básicos da CI, mas também uma visão geral dos princípios cromatográficos. Além disso, a cromatografia pode proporcnionar muitos feitos: satisfazer a curiosidade científica e assugurar uma sobrevivência saudável em um ambiente poluído.

Práticas em Cromatografia de Íons 7
3. Seção teórica 3.1. A história e a importância da cromatografia de íons Os primórdios da Cromatografia de Íons (CI) ou, mais precisamente da cromatografia de troca iônica, remetem-se à metade do século passado. Entre 1935 e 1950, os conhecimentos sobre trocadores iônicos e suas aplicações foram consideravelmente expandidos pelo “Projeto Manhattan”. Nos anos 50 e 60, foram desenvolvidos modelos teóricos para a compreensão do fenômeno de troca iônica e da cromatografia de íons, nos quais esta monografia se baseia. Detectores para fluxos contínuos foram usados nos anos 70; isto permitiu um salto na mudança da cromatografia de baixa-pressão para a de alta-eficiência.
Tabela 1. História dos trocadores de íons e da cromatografia de íons, a técnica analítica baseada na troca iônica
O termo “cromatografia de íons” foi criado em 1975 por Small, Stevens e Baumann com a introdução da detecção por condutividade combinada com um método químico de redução da condutividade; subseqüentemente, foi usada por um longo tempo como um nome comercial patenteado para fins de marketing. Enquanto isso, o termo abreviado “Cromatografia de íons” se estabeleceu como o termo específico para os métodos de cromatografia de formação de pares iônicos, exclusão iônica e troca iônica, incluídos sob a cromatografia líquida de alta eficiência - CLAE (HPLC, do inglês: High Performance Liquid Chromatography) [1]. Hoje, a técnica CI é aplicada na determinação de ânions enquanto os métodos de espectrometria atômica, comumente usados para a determinação de cátions, são raramente úteis para a determinação dos ânions eletronegativos do quinto ao sétimo grupo do sistema periódico.
A área de aplicação mais importante da cromatografia de ânions, hoje, é a investigação de rotina dos sistemas aquosos; isto é de vital importância na análise de água potável [2,3,4,]. A CI é também usada para a análise das espécies de elementos em complexos ou elementos aniônicos, isto é, principalmente para resolver problemas de relevância ambiental. O terceiro campo de aplicação da cromatografia de ânions é o controle de ultratraços em processamento de reagentes ultrapuros requeridos principalmente na indústria de semicondutores.
Hoje, os trocadores de íons, normalmente usados na CLAE (HPLC), consistem de partículas esféricas de polímero com um diâmetro de aproximadamente 5 a 15 μm. Vários métodos são usados para anexar os chamados grupos âncoras na superfície deste polímero; estes são usados como espaçadores entre o polímero básico e os grupos funcionais efetivos. Estes grupos funcionais normalmente consistem de íons amônio quaternário, os quais são quimicamente anexados aos grupos âncoras. O número total de grupos funcionais é conhecido como a capacidade de troca; isto é uma característica básica dos trocadores de íons.
Materiais de empacotamento, disponíveis comercialmente para cromatografia de ânions, apresentam natureza de baixa capacidade com capacidades de troca de 50 a 100 μmol por coluna de separação.
CL
CLAE

8 Monografia Metrohm
Desta forma, as principais aplicações requerem o uso de detector de condutividade, o qual é usado universalmente devido sua sensibilidade, e de eluentes que tenham a mais baixa condutividade possível (background ou sinal de fundo). Trocadores aniônicos de baixa capacidade permitem o uso de soluções aquosas de NaOH ou de tampão carbonato muito diluídas, cuja condutividade pode ser reduzida até mesmo pelo recurso da supressão química [2,4].
Em cromatografia de ânions, são usados hoje em dia principalmente os grupos funcionais do Tipo I (trimetilamônio ou TMA) e do Tipo II (dimetiletanolamônio ou DMEA). Visto que a interação mais significativa entre a fase estacionária e os ânions do analito acontece no grupo funcional, isto significa que sua estrutura tem uma influência decisiva no comportamento seletivo dos materiais de empacotamento. De acordo com o conhecimento atual, a polaridade dos grupos funcionais, a qual pode ser controlada por um número de resíduos hidroxietila (-CH2CH2OH) no nitrogênio quaternário, é de particular importância [2,4].
O termo cromatografia de íons inclui todas as separações de espécies iônicas dentro da Cromatografia com detecção em fluxo contínuo e é independente de limitações em equipamentos [5]. A CI tem sido o método escolhido, dentre os disponíveis em análise aniônica, graças à grande variedade de colunas de separação, sistemas de eluição e detectores que hoje estão mais disponíveis. A razão para isto é que existem poucos processos de separação de ânions; estes são raramente disponibilizados para fins práticos, enquanto que os métodos gravimétricos e volumétricos são limitados pela sua sensibilidade e seletividade. Mesmo com o desenvolvimento espetacular da cromatografia gasosa desde 1965 ela não apresentou grandes vantagens para os ânions, visto que estes não são voláteis. Desta forma, precisavam ser primeiramente derivatizados, mas a sensibilidade deste método não foi suficiente para atender a demanda que existe hoje em análise de traços [6]. Para a análise de cátions, existem alternativas de espectrometria atômica que possuem alta eficiência (exemplo, ICP-AES/MS), portanto, o valor da cromatografia de cátions é consideravelmente menor quando comparado com o da cromatografia de ânions. No entanto, a cromatografia de cátions tem alcançado grande importância na análise de metais alcalinos e alcalino-terrosos, na determinação de nitrogênio amoniacal (análise em água potável) e na especiação de compostos iônicos, em combinação com detectores de elementos específicos. Os trabalhos de Haddad e Weiss [2,4] oferecem uma boa visão geral das aplicações de CI em vários setores.

Práticas em Cromatografia de Íons 9
3.2. Teoria da Cromatografia
3.2.1. Divisões da cromatografia e terminologia
Cromatografia é um método físico-químico para separar misturas de substâncias. O efeito de separação é baseado na distribuição entre duas fases: uma fase é estacionária e a segunda é uma fase móvel que flui em uma determinada direção [7,8]. As técnicas cromatográficas são divididas de acordo com os estados físicos das duas fases mencionadas:
Figura 1 Divisão dos métodos cromatográficos de acordo com os estados físicos das fases estacionária e móvel.
Uma adicional diferenciação dos métodos cromatográficos pode ser feita de acordo com os processos básicos que ocorrem durante a separação, tais como adsorção ou distribuição; ou de acordo com o tipo de procedimento utilizado (cromatografia planar ou por coluna) [9].
Parâmetros de retenção Se uma mistura de substâncias for submetida à separação cromatográfica, um equilíbrio de distribuição é formado entre as fases estacionária e móvel para cada componente individual. As substâncias só podem ser separadas com sucesso quando os coeficientes de distribuição D dos componentes diferirem suficientemente uns dos outros. D é definido como a relação entre as concentrações de uma substância A na fase estacionária (índice S) e na fase móvel (índice M):
M
SA [A]
[A]D (1)
As substâncias que possuem maiores coeficientes de distribuição D serão retidas mais fortemente do que aquelas com D menores. O procedimento de separação cromatográfica é mostrado na forma de um cromatograma, no qual um sinal de um detector é registrado em função do volume (ou do tempo) de eluição da fase móvel. Isto significa que ele corresponde ao perfil de massa ou concentração em função do tempo. O sinal detectado deve ser proporcional à concentração do analito no final do processo de migração [8]. Como demonstrado na Equação 2, o tempo de retenção ou tempo de retenção bruto tR de uma substância na fase estacionária é obtido pela adição do tempo de retenção líquido tS, o qual corresponde ao tempo de retenção real no processo de migração, e o tempo de corrida na fase móvel sem qualquer interação, o tempo morto tM.
MSR ttt (2)
Devido à formação de canais, processos de difusão ou irregularidades no equilíbrio adquirido entre as fases estacionária e móvel, algumas partículas podem passar através da fase estacionária mais lentamente ou mais rapidamente do que é esperado pelo tempo de retenção líquido tS. Isto significa que um cromatograma não consiste de um número infinito de sinais discretos, mas idealmente de picos Gaussianos (vide Figura 2).
Líquido Fase Móvel
Fase
Est
acio
nária
Líquido
Líquido
Gasoso
Sólido CSL
CLL CLG
CSG

10 Monografia Metrohm
Figura 2
Cromatograma de eluição de uma separação por cromatografia de íons com indicação dos parâmetros mais importantes.
Figura 3
Distribuição Gaussiana com as quantidades mais importantes.
Como resultado de processos de difusão, os quais aumentam em importância conforme aumento do tempo de residência na fase estacionária, a largura do pico de uma substância aumenta com o aumento do tempo de retenção. Este fenômeno é característico de todos os métodos cromatográficos.
Conforme foi mencionado, sob circunstâncias ideais, um pico em um cromatograma mostra a distribuição Gaussiana. A Figura 3 mostra um exemplo da distribuição Gaussiana.
A largura na metade da altura do pico é conhecida como largura-média b0,5 e corresponde a uma variância de 2,354-vezes a distribuição. A largura da base w é definida como a distância entre os pontos de intersecção das tangentes inclinadas com o eixo y, a qual é a mesma que a variância 4-vezes da função de Gauss. Ambas as quantidades são parâmetros de medida do desempenho de uma coluna de separação cromatográfica e, com uma forma de pico ideal, podem ser usadas para calcular o número de pratos teóricos.
Analito 1
Analito 2
Sinal
área
A lturaReso lução
Form ação de Caud a
Tem po m orto
Tem po d e retenção, analito n
Tem po d e retenção líquido
Fator d e retenção
Fator d e ass imetria
Resolução
Coefici ente de sel etivi dade
Temp o o u vo lum e
Sinal
Tempo ou volume

Práticas em Cromatografia de Íons 11
Variações da forma ideal de pico podem ser descritas pelo chamado fator de assimetria T. Isto é definido pela relação entre as distâncias A e B medidas entre a linha vertical central e as inclinações da distribuição a 10% de sua altura (vide Figuras 2 e 3) e pode ser calculado por:
ABT (3)
Para picos perfeitamente Gaussianos, T = 1. Para valores de T maior que 1, o pico apresentará “cauda”, enquanto que para valores menores, uma deformação frontal. Na prática, a intenção é adquirir um fator de assimetria de T = 0,9 a 1,1.
Fator de retenção, seletividade e resolução Como o tempo de retenção bruto tR depende muito das condições cromatográficas, somente sob condições definidas que ele é característico para uma substância em particular e, portanto, pode ser usado para identificação qualitativa. Se introduzirmos uma constante adimensional, o fator de retenção k’; conseguiremos comparar diferentes sistemas cromatográficos. Esta constante fornece informação mais precisa sobre quanto tempo a mais uma substância permanece na fase estacionária do que na fase móvel [8]. O fator de retenção é matematicamente definido como o produto do coeficiente de distribuição D pela proporção entre os volumes das fases estacionária e móvel ou como a proporção entre o tempo de retenção líquido e o tempo morto. Com um cálculo envolvendo o comprimento L do caminho de migração e a velocidade linear u da fase móvel também é possível definir o fator de retenção (Equação 4).
1-Ltu
tt=
VVD=k' R
M
S
M
S (4)
Para valores pequenos de k’, uma substância elui próximo ao tempo (ou volume) morto do sistema cromatográfico; isto significa que a separação é pobre. Se o k’ for muito grande significa que, embora a separação seja boa, a um longo tempo de residência no caminho de migração e o pico torna-se mais largo. Idealmente, o fator de retenção deve estar entre 2 e 5.
Duas substâncias serão adequadamente separadas apenas se seus fatores de retenção diferirem um do outro significativamente. O coeficiente de seletividade , também conhecido como o fator de separação relativa, é uma medida da capacidade de separação de duas substâncias e é definido como segue:
(5)
Se duas substâncias não podem ser separadas, então = 1 e a coeluição ocorre. Quanto maior o valor de , melhor a separação. Entretanto, se aumenta, o tempo requerido para a separação também aumenta, sendo que na prática, os coeficientes de seletividade são ideais quando são próximos de 1.5 [10].
O coeficiente de seletividade não descreve a qualidade do processo de separação. A resolução R,entretanto, considera tanto as posições relativas dos picos, bem como suas larguras-médias b0.5 ou larguras da base w, como pode ser visto na Equação 6.
(6)
Se a diferença entre os tempos de retenção de dois picos for grande em relação à das larguras da base ou larguras-médias, então a resolução é boa. No caso de uma simetria de pico ideal, duas substâncias podem ainda ser identificadas com R = 0,5. Para separação qualitativa, R deve ser de, pelo menos, 1 (separação com 4 ); para quantificação, uma resolução de R = 1,2 a 1,5 é requerida [25]. Resoluções de R 2 (separação com 8 ) são evitadas devido ao longo tempo de análise envolvido.

12 Monografia Metrohm
3.2.2. Conceitos teóricos para a descrição do processo cromatográfico
O modelo dos estágios teóricos de separação
O modelo dos estágios teóricos de separação é derivado do processo de destilação e é usado para descrever separações cromatográficas [11]. Ele divide a fase estacionária em seções discretas, estágios ou pratos teóricos de separação, nos quais, a princípio, um equilíbrio infinitamente rápido e completamente reversível entre as fases estacionária e móvel é alcançado. A performance (eficiência) de um sistema cromatográfico é, portanto, caracterizada por um número maior possível de estágios teóricos de separação.
O número de pratos teóricos N pode ser determinado diretamente do cromatograma pela utilização da variância , a largura da base w ou a largura-média b0.5, e é calculado como se segue [12]:
2R
2
0,5
R2
R t=
bt
(2)ln8=wt
16=N (7)
Ao invés do número de pratos teóricos, a Altura Equivalente ao Prato Teórico (HETP) pode também ser usada para descrever a eficiência da separação.
(8)
Das Equações 5 a 8, pode ser visto que uma fase estacionária com um grande número de pratos teóricos pode ainda separar duas substâncias, mesmo que seus fatores de retenção não sejam tão diferentes. As equações também permitem o cálculo do número de pratos teóricos, os quais são necessários para resolver um problema de separação.
O modelo de estágios teóricos de separação pode ser usado para explicar a ocorrência de sinais Gaussianos na cromatografia se for assumido que, devido aos processos de fluxo e difusão, apenas um equilíbrio incompleto e finitamente rápido é alcançado entre as fases estacionária e móvel. Isto resulta em um processo de alargamento do pico, visto que uma zona estreita contendo a substância, no início do caminho de migração, torna-se, claramente, mais larga com o aumento do tempo de residência na fase estacionária.
O cálculo do número dos pratos teóricos, de acordo com a Equação 7, assume que a forma do pico é ideal; entretanto, isto raramente ocorre na realidade. Com picos de formas assimétricas, o cálculo deve ser realizado de acordo com o método momentâneo [13]. A Equação 9 inclui o fator de assimetria T e produz valores aproximados.
(9)
Um número de pratos efetivos n, que representa a real eficácia de separação mais precisamente do que o número de pratos teóricos N é corrigido pelo fator de retenção k’ e é obtido de:
(10)
A teoria dinâmica (teoria de Van Deemter)
A decisiva fragilidade do modelo de estágios teóricos de separação é que a destilação e a cromatografia são baseadas em dois processos físico-químicos fundamentalmente diferentes. Além disso, nenhuma hipótese foi feita sobre a influência de parâmetros importantes, que sejam experimentalmente acessíveis e que não afetam o tipo ou qualidade da fase estacionária em si [14,15]. Estes poderiam ser:
Vazão da fase móvel Diâmetro das partículas da fase estacionária Espessura dos filmes superficiais no material de empacotamento.
1,25+Tbt
41,7=N
2
0,5
R
2
k'+1k'N=n

Práticas em Cromatografia de Íons 13
Além disso, quantidades tais como os coeficientes de difusão nas fases estacionária e móvel, a temperatura ou o volume do detector na cromatografia líquida são muito importantes para a eficiência da separação.
A teoria dinâmica desenvolvida por van Deemter é, em princípio, uma extensão do modelo de estágios teóricos de separação, a qual não leva em consideração as condições limitantes não-ideais [16]. As seguintes suposições são feitas:
não há alcance espontâneo ou desimpedido de equilíbrio há transporte de massa atrasado nas fases estacionária e móvel não há vazão homogênea da fase móvel sobre a completa seção cruzada da coluna há ocorrência de difusão de espalhamento e formação de canais na fase estacionária há difusão longitudinal independente da velocidade da fase móvel e diretamente proporcional ao tempo de retenção no caminho
A relação entre os efeitos dinâmicos mencionados acima e a altura equivalente a um prato teórico é dada pela equação de van Deemter.
(11)
Os três termos A, B e C dependem, de diferentes maneiras, da vazão u da fase móvel. Os termos Ae B descrevem o completo transporte de massa através da fase estacionária; o termo C é determinado pela interferência no alcance do equilíbrio entre as fases estacionária e móvel.
O Termo A descreve a difusão circular em contra-fluxo, a qual pode ser considerada como sendo uma causa do alargamento do pico devido a um efeito multi-vias. Este termo é também conhecido como o fator de empacotamento e, é independente da vazão linear u da fase móvel, pelo menos em uma primeira aproximação. A seguinte relação se aplica ao termo A:
pd2A (12)
Na Equação (12), dP é o diâmetro médio de partículas na fase estacionária, descreve a irregularidade estatística do empacotamento; este deveria ser o mais homogêneo possível e consistir de partículas uniformes.
uCuB A HETP

14 Monografia Metrohm
O Termo B descreve a difusão longitudinal a favor ou contra a direção do fluxo da fase móvel. É de particular interesse quando colunas capilares são usadas em cromatografia gasosa (CG), uma vez que os coeficientes de difusão em gases são maiores em 4 a 5 potências de dez do que em líquidos. B é calculado como sendo o produto do coeficiente de difusão na fase móvel DM e o fator labirinto , o qual descreve a porosidade da fase estacionária.
MD2B (13)
Como a importância da difusão diminui com o aumento da vazão da fase móvel, isto significa que B é inversamente proporcional a u.
O Termo C é conhecido como o termo de transferência de massa. O atraso na transferência de massa entre as fases estacionária e móvel, geralmente, tem a maior influência sobre o alargamento do pico. A interferência no alcance do equilíbrio entre as fases estacionária e móvel aumenta com o aumento de u, e é por isso que há uma proporcionalidade direta para com a vazão linear. Atrasos na transferência de massa resultam de um coeficiente de difusão DS muito pequeno na fase estacionária em comparação com a fase móvel. O termo C pode ser consideravelmente reduzido por caminhos de difusão curtos e processos de transferência rápidos. Isto pode, principalmente, ser alcançado pela localização dos poros na superfície, para que apenas um pouco se estenda para o interior da fase estacionária. O termo C de transferência de massa é calculado como segue:
S
2
Ddp
k'+1k'16=C (14)
A representação gráfica da equação de van Deemter mostra uma curva hiperbólica, da qual o valor mínimo de vazão u para a mínima altura de prato (número máximo de pratos) pode ser determinada (Figura 4).
Termo B Termo A
Termo C
Vazão u
Figura 4 Representação dos termos individuais da teoria de van Deemter com a curva mostrando o valor ótimo de vazão.
Mesmo a teoria dinâmica é baseada em aspectos ideais. Na realidade, os três termos A, B e C só são independentes um do outro em uma primeira aproximação, havendo uma influência adicional da vazão u na difusão circular em contra-fluxo (Termo A). O termo C pode ser diferenciado pela utilização dos termos CM e CS, os quais descrevem a transferência de massa na fase móvel (CM) e da fase estacionária (CS). É por isso que a equação original de van Deemter tem sido modificada por numerosas aplicações em CLAE, CG e camada delgada [17,18].
Cromatografia líquida moderna
A cromatografia líquida (CL) deve ser considerada como o termo genérico para muitos métodos modernos de separação usando componentes no estado líquido. Ela pode ser usada para uma grande variedade de substâncias diferentes e é caracterizada por sua excelente performance analítica. A CL também inclui a cromatografia de íons (CI), que é provavelmente o mais importante método de separação usado em química analítica moderna [3].
A CLAE é um desenvolvimento subseqüentemente lógico da cromatografia líquida clássica (CL). Na CL clássica, introduzida por Tswett em 1906, colunas de vidro com diâmetro de 1 a 5 cm e

Práticas em Cromatografia de Íons 15
comprimento de até 500 cm foram usadas; estas eram preenchidas com fases de separação contendo partículas medindo de 150 a 200 μm. Mesmo as separações de substâncias em misturas simples freqüentemente duravam horas com uma eficácia média de separação. Como resultado da compreensão do processo cromatográfico, o qual foi posteriormente desenvolvido (vide Equação 11), tornou-se claro que o aumento na eficácia só poderia ser alcançado pela redução do diâmetro das partículas da fase estacionária; entretanto, isto implicou em exigências completamente novas para os equipamentos usados em cromatografia.
Desde aproximadamente 1970, uma tecnologia especial e poderosa de instrumentos tornou-se disponível, a qual é capaz de ultrapassar uma alta contra-pressão de 10 a 50 MPa que ocorre quando materiais de empacotamento com partículas de diâmetro de 3 a 10 μm e colunas de separação de 125 a 250 mm de comprimento por 4 mm de diâmetro interno são usadas.
Como resultado da miniaturização, a CLAE desenvolveu-se como um método de separação puramente analítico; em contraste, a CL clássica atualmente é utilizada praticamente apenas para fins preparativos. As vantagens da CLAE em comparação à CL clássica são principalmente:
excelente eficiência cromatográfica processo de trabalho contínuo detecção “on-line” das substâncias separadas alta sensibilidade e reprodutibilidade utilização do tempo de retenção para identificação qualitativa das substâncias menor tempo de análise
Independente de sua área de aplicação, um sistema CLAE consiste principalmente dos componentes mostrados na Figura 5: a bomba de alta eficiência com estoque da fase móvel (eluente), injetor (para introdução da amostra), coluna de separação e sistema de detecção (incluindo derivatização, aquisição e tratamento dos dados):
Eluente
Válvula de injeção
Coluna de Separação
“Loop” de Amostra
Condutividade UV – VIS Amperometria Espectrometria atômica
Reator pós-coluna/
derivatização
Amostra
Detector
Bomba CLAE
Figura 5 Unidade de CLAE ou CI montada com os componentes mais importantes.
Com exceção da coluna de separação, a bomba é o coração de qualquer sistema CLAE. Ela deve transferir (bombear) o eluente tão uniformemente e livre de pulsação quanto possível, mesmo em presença de alta contra-pressão. Isto significa que é também necessário usar um injetor com “loop” especial para a introdução da amostra. Uma válvula de seis vias é normalmente usada; ela é capaz de receber um volume definido da amostra na alça “loop” em pressão padrão e transferir para o sistema CLAE operando em alta pressão. A composição da fase móvel e o tipo de coluna de separação devem ser adaptados ao problema analítico a ser resolvido. Isto também se aplica à seleção do sistema de detecção. Hoje, a aquisição e o tratamento de todos os dados são realizados por computador. Esta montagem básica de um sistema CLAE pode ser estendida conforme necessário para resolver um problema analítico particular.
Princípios de separação em CL
A CLAE pode ser diferenciada de acordo com diferentes interações físico-químicas entre as substâncias da amostra e da fase estacionária. Embora, na realidade existam vários mecanismos diferentes responsáveis pela separação eficiente [9], uma classificação geral de acordo com os seguintes mecanismos de separação é possível:

16 Monografia Metrohm
adsorçãodistribuição exclusão por tamanho afinidade troca iônica formação de par iônico exclusão iônica
Cromatografia de adsorção é definida por reações interfaciais, nas quais substâncias líquidas ou gasosas são enriquecidas em uma fase sólida. Vários modelos estão disponíveis fornecendo uma descrição qualitativa e quantitativa dos processos de adsorção; aqui, nós apenas nos referimos à relevante literatura físico-química [19]. Duas técnicas diferentes são usadas. Em cromatografia de fase normal, a fase estacionária é geralmente a sílica gel e, portanto, consideravelmente mais polar do que a fase móvel (hidrocarbonetos). Em cromatografia de fase reversa CFR (ou RPC, em Inglês), as condições são exatamente opostas. Por razões práticas, principalmente quando se refere ao manuseio do eluente, a CFR é mais utilizada atualmente [3,9].
Na cromatografia de distribuição, a fase estacionária é um líquido, o qual é imiscível com a fase móvel. A separação é baseada nas diferentes solubilidades dos analitos nas duas fases. Em um caso ideal, é aplicada a lei de distribuição de Nernst. Este mecanismo de separação tem um importante papel, particularmente em cromatografia gasosa quando capilares recobertos com líquidos de separação são usados como fase estacionária. A cromatografia de distribuição também pode ocorrer em CLAE se a sílica gel modificada com hidrocarbonetos não-polares, ou seja, as chamadas fases com grupo octadecil (ou octil) forem usadas como material de separação.
Cromatografia por exclusão de tamanho (SEC, em Inglês) permite a separação de acordo com o tamanho molecular como resultado de efeitos de seleção de partículas. Sílica gel ou resinas de polímero orgânico, com uma estrutura de poro definida, são usadas como fase estacionária. Analitos menores podem se difundir nos poros e são retardados. Com o aumento do tamanho da molécula, torna-se menor qualquer interação com os poros, até que com um certo tamanho, as moléculas são completamente excluídas e praticamente eluem no volume morto. Este tipo de cromatografia é muito utilizado na análise de polímeros e bioanálise.
Cromatografia de afinidade permite a separação de misturas de substâncias pela seletividade ou por forças de interação específicas. Interações altamente específicas podem ser observadas entre antígenos e anticorpos (princípio chave-fechadura), bem como entre enzimas e seus substratos particulares. Na prática, enzimas ou anticorpos são quimicamente imobilizados na fase estacionária. Se houver um substrato ou antígeno correspondente na amostra, então ocorre seu retardo com extrema seletividade. É por isso que a cromatografia de bioafinidade é indispensável para o setor de análise de princípios ativos (farmacologia).
Cromatografia de troca iônica (CI) juntamente com as cromatografias por exclusão iônica e de par iônico estão descritas em detalhes na seção seguinte.
3.3. Princípios básicos da cromatografia de íons (CI)
3.3.1. Terminologia e classificação em CL
Cromatografia de troca iônica ou cromatografia de íons (CI) é uma subdivisão da CLAE. De acordo com a IUPAC, a cromatografia de troca iônica é definida como segue [7,8]:
“Na cromatografia de troca iônica, a separação é baseada nas diferenças das afinidades de troca iônica dos analitos individuais. Se íons inorgânicos são separados e podem ser detectados por detectores de condutividade ou por detecção por ultravioleta (UV) indireta, então, esta também é chamada de cromatografia de íons”.
Por várias razões, esta definição é uma escolha infeliz. A técnica de detecção deveria ser considerada separadamente do mecanismo de separação existente. Além disso, uma limitação do termo “cromatografia de íons” para íons inorgânicos é difícil de ser entendida, visto que na prática, tanto os íons orgânicos como os inorgânicos podem ser separados e identificados simultaneamente em qualquer sistema.
Uma definição antiga e mais geral é mais adequada para definir a cromatografia de íons [20]:

Práticas em Cromatografia de Íons 17
«Cromatografia de íons inclui qualquer separação cromatográfica líquida rápida de íons em colunas acopladas “on-line” com detecção e quantificação em um detector em fluxo.»
Esta definição caracteriza a cromatografia de íons independente do mecanismo de separação e do método de detecção, ao mesmo tempo em que a distingue da troca iônica clássica. Os seguintes princípios de separação aplicam-se na cromatografia de íons:
troca iônica formação de par iônico exclusão iônica
Os métodos cromatográficos são definidos pelo principal mecanismo de separação utilizado. Hoje, a cromatografia de troca iônica é simplesmente conhecida como cromatografia de íons (CI), enquanto a cromatografia de par iônico e a cromatografia por exclusão iônica são consideradas como sendo aplicações mais específicas.
3.3.2. Troca iônica
A cromatografia de troca iônica (CI) é baseada em uma reação química estequiométrica entre os íons de uma solução e uma substância sólida contendo os grupos funcionais, que podem fixar íons como resultado de forças eletrostáticas. No caso mais simples em cromatografia de cátions, são grupos de ácido sulfônico; em cromatografia de ânions, são grupos de amônio quaternário. Teoricamente, íons com mesma carga podem ser completa e reversivelmente trocados entre as duas fases. O processo de troca iônica leva a uma condição de equilíbrio. O lado em que ocorre o equilíbrio depende da afinidade dos íons participantes em relação aos grupos funcionais da fase estacionária. A Figura 6 é um diagrama esquemático mostrando os processos de troca para cátions e ânions. Os íons do analito são chamados A, os íons do eluente competindo com eles para a troca são marcados com E.
Fase Móvel
Fase EstacionáriaTroca Aniônica
A: íons do analito E: íons do eluente
Troca Catiônica
Vazão
Figura 6 Diagrama esquemático mostrando o processo de troca iônica em cromatografia de íons. Esquerda: troca catiônica. Direita: troca aniônica.
Aspectos termodinâmicos do processo de troca iônica
Trocadores de íons normalmente consistem de fases sólidas em cuja superfície são fixados os grupos iônicos. Por causa da condição de eletroneutralidade, há sempre um contra-íon de carga oposta nas proximidades do grupo funcional. O contra-íon geralmente se origina da fase móvel e é, portanto, também conhecido como íon do eluente.
Se uma amostra for adicionada contendo dois íons A- e B-, então estes brevemente substituem os íons do eluente E- e são retidos na carga fixa antes que sejam de volta trocados pelo íon do eluente. Para a cromatografia de ânions isto resulta nos seguintes equilíbrios reversíveis:
resina - N+R3 E– + A– resina - N+R3 A– + E– (15)
resina - N+R3 E– + B– resina - N+R3 B– + E– (16)
As diferentes afinidades de A- e B- para com os grupos funcionais significam que a separação é possível. A constante de equilíbrio K é também conhecida como o coeficiente de seletividade e é calculada, como se segue, para o ânion A-:
(17)

18 Monografia Metrohm
Pode-se assumir que a concentração dos íons do eluente é normalmente maior do que a dos íons do analito por várias potências de dez, então [E-] pode ser considerada como sendo uma constante nas fases estacionária e móvel. Isto significa que o coeficiente de distribuição DA (Equação 1) e o fator de retenção K’A (Equação 4) podem ser calculados. Estritamente falando, tais cálculos apenas são permissíveis se as concentrações na Equação 17 corresponderem às atividades; entretanto, este só é o caso para uma diluição infinita [19]. A princípio, as atividades dos íons na fase estacionária são inacessíveis [4]. Para os trocadores de íons de baixa capacidade mais freqüentemente usados, os quais só podem ser usados como fase móvel com eletrólitos bastante diluídos, as atividades são simplesmente desconsideradas. Tais estimativas tão grosseiras não são mais válidas para materiais de empacotamento de alta capacidade (>200 mmol/g) e eluentes concentrados; isto mostra variações claras do comportamento ideal.
3.3.3. Formação de par iônico
Com o auxílio da cromatografia de par iônico é possível separar os mesmos analitos da mesma forma que na cromatografia por exclusão iônica, mas o mecanismo de separação é completamente diferente. As fases estacionárias usadas são materiais de fase reversa como as que são usadas na cromatografia de distribuição. O chamado reagente de par iônico é adicionado aos eluentes e este consiste de surfactantes catiônicos ou aniônicos, tais como sais de tetraalquilamônio ou ácidos n-alquilsulfônicos. Juntamente com os íons do analito de carga oposta, os reagentes do par iônico formam um par de íons não carregado, o qual pode ser retardado na fase estacionária por interações hidrofóbicas. A separação é possível devido às constantes de formação dos pares iônicos e seus diferentes graus de adsorção. A Figura 7 mostra um modelo simplificado de troca iônica estática, no qual é assumido que interações com os analitos só ocorrem após a adsorção do reagente do par iônico na fase estacionária.
Figura 7 Diagrama esquemático mostrando o modelo de troca iônica estática em cromatografia de par iônico. O princípio de separação aplica-se tanto para ânions como para cátions.
3.3.4. Exclusão de íons
A cromatografia por exclusão de íons é principalmente usada para a separação de ácidos ou bases fracas [2,4]. Sua maior importância é a determinação de ácidos fracos, tais como ácidos carboxílicos, carboidratos, fenóis ou aminoácidos. A Figura 8 mostra o princípio de separação, usando um ácido carboxílico R–COOH como exemplo.
Figura 8 Exclusão de Donnan como o princípio de separação em cromatografia por exclusão de íons.
Reagente do par iônico
Íon do Eluente
Fase Móvel
Íon do Analito
Fase Estacionária
Vazão
Membrana de Donnan
Fase Móvel
R – COOH (Analito)
H+ Cl- (Eluente)
Fluxo
Fase Estacionária

Práticas em Cromatografia de Íons 19
Na cromatografia por exclusão iônica, um trocador de cátions completamente sulfonado, cujos grupos de ácidos sulfônicos são eletricamente neutralizados com prótons como contra-íons, é freqüentemente usado como material de empacotamento. Em eluentes aquosos, os grupos funcionais são hidratados. A camada de hidrato é limitada por uma membrana (imaginária) carregada negativamente (membrana de Donnan). Ela só é atravessada por moléculas não-dissociadas e não carregadas como a água. Ácidos carboxílicos orgânicos podem ser separados se ácidos minerais fortes, por exemplo, o ácido sulfúrico, forem usados como fase móvel. Devido às constantes baixas de ácido (valores pKA) dos ácidos carboxílicos, eles estão presentes em forma completamente não-dissociada em eluentes fortemente ácidos. Eles podem passar através da membrana de Donnan e serem adsorvidos na fase estacionária, enquanto os íons sulfato do ácido sulfúrico completamente dissociado são excluídos.
A Figura 9 mostra a dependência típica do volume de eluição de um ácido em seu valor pKA para separação por exclusão de íons. Adsorção sobreposta (ácidos carboxílicos de cadeia longa, H2S) e os limites da faixa de trabalho prático podem ser claramente reconhecidos. Em última instância, ácidos carboxílicos são separados por causa de seus diferentes valores de pKA.
Volume de Eluição
Forç
a do
Áci
do /
pKA
Figura 9 Dependência do volume de eluição sobre o valor pKA particular de ácidos em cromatografia por exclusão de íons.
3.4. Modelos de retenção em cromatografia de íons Em uma situação ideal, a retenção de um analito em cromatografia de íons só poderia ser determinada por sua afinidade aos grupos funcionais do trocador de íons. Esta afinidade pode ser descrita pela formulação de uma reação química, a reação de troca iônica, e pode ser explicada pelo uso da lei da ação das massas.
Os modelos de retenção descritos abaixo tentam predizer sobre o comportamento de retenção dos analitos participantes sob condições cromatográficas particulares, baseadas na lei da ação das massas. Se os modelos resultantes são aptos a explicar observações macroscópicas, então, com sua ajuda é possível, por exemplo, otimizar um sistema de eluição para um problema de separação particular.
3.4.1. Modelos de retenção em cromatografia de ânions
As seguintes observações envolvem, inicialmente, apenas a eluição por deslocamento isotônico como o mais simples mecanismo de eluição em cromatografia de íons. O exemplo real refere-se à cromatografia de ânions, mas as mesmas considerações aplicam-se similarmente à cromatografia de cátions. Só quando agentes complexantes são adicionados aos eluentes, é necessário estender o modelo de retenção; isto é descrito na seção «Modelo de retenção para eluição em presença de agentes complexantes» (capítulo 3.4.2.).
Modelo de retenção para eluentes com um ânion
Se a eletroneutralidade for assumida, então o mais simples enfoque para um modelo de retenção para deslocamento isotônico é aquele em que um único íon eluente Ey– compete com um ânion de

20 Monografia Metrohm
analito Ax– em relação aos grupos funcionais da fase estacionária [4]. A concentração dos ânions do eluente Ey– permanece constante com o tempo (eluição isocrática).
Os sítios de troca em uma coluna de separação com uma capacidade Q são ocupados por ânions do eluente Ey– no início do processo cromatográfico. Se uma amostra contendo o íon do analito Ax– for adicionada, então o seguinte equilíbrio se estabelece entre a fase estacionária (índice S) e a fase móvel (índice M):
y · AMx– + x · ES
y– y · ASx– + x · EM
y– (18)
De acordo com a lei da ação das massas, este equilíbrio pode ser descrito por uma constante de equilíbrio termodinâmico. Se as atividades dos íons participantes foram consideradas, então a seguinte constante de equilíbrio termodinâmico é obtida:
xE
yA
xE
yA
xyS
yxM
xyM
yxS
EA,yS
xM
yM
xS
][E][A][E][A
K (19)
Visto que as atividades dos íons participantes não podem ser determinadas nas fases estacionária e móvel, a atividade na fase estacionária é ignorada e assumida como 1.
Se para o ânion do analito Ax– duas quantidades, o coeficiente de distribuição DA e o fator de retenção K’A, são agora introduzidas (capítulo 3.2.1.),
M
SA [A]
[A]D com
M
SAA V
VDk' (20)
então, incluindo estas quantidades e desprezando as atividades, a Equação 19 pode ser convertida para:
x
yS
yM
y
S
MAEA, ][E
][EVVk'K (21)
Como a concentração dos íons do eluente E é normalmente maior do que a dos ânions do analito Ax–
por várias potências de dez, uma boa estimativa pode ser obtida assumindo-se que todos os grupos funcionais são ocupados por Ey–. Sob esta hipótese, a concentração não-determinável de Ey– na fase estacionária pode ser substituída pelos parâmetros mais facilmente acessíveis de capacidade de troca Q e carga do ânion do eluente y:
yQ][Ey
S (22)
Isto significa que a Equação 21 pode ser convertida para:
xyM
xy
S
M'AEA, ][E
yQ
VVkK (23)
O fator de retenção K’A do ânion do analito Ax– pode facilmente ser obtido de um cromatograma. A Equação 23 é, portanto, resolvida para esta quantidade.
yx
yM
yx
y1
EA,M
S'A ][E
yQ)(K
VV
k (24)
Esta equação é de suma importância para cromatografia de ânions, uma vez que ela fornece uma relação quantitativa entre o fator de retenção K’A e vários parâmetros, experimentalmente acessíveis tais como a concentração do eluente e a capacidade de troca. Na prática, uma versão logarítmica da Equação 24 é usada por razões de clareza.
][Elogyxlog
yQlog
yxKlog
y1k'log y
MEA,A para
M
S
VV (25)
Da Equação 25 pode ser visto que:
Aumentando a concentração do eluente [Ey–], a eluição se acelera, o maiores fatores de retenção resultam de maiores constantes de equilíbrio KA,E, maior
capacidade de troca Q e maior proporção fase-volume .
Analitos multivalentes Anx– são retardados mais fortemente do que monovalentes Ax–,

Práticas em Cromatografia de Íons 21
o pelo menos enquanto a concentração do eluente [Ey–] for relativamente baixa. Isto é também conhecido como eletro-seletividade.
Eluentes multivalentes Eny– têm uma capacidade de eluição maior do que monovalentes Ey–,o a eluição de analitos multivalentes Anx– é mais fortemente influenciada por
concentrações elevadas de íons eluentes monovalentes Ey– do que daquela de analitos monovalentes Ax–.
Como uma primeira aproximação, pode-se assumir que coeficientes de seletividade são independentes de Q quando é constante; isto resulta na seguinte proporcionalidade:
(26)
Da Equação 26 pode ser visto que se a capacidade de troca Q for aumentada, então a concentração do eluente [Ey–] deve ser aumentada proporcionalmente a fim de se obter fatores de retenção constantes. É por isso que fases de separação de baixa capacidade são, normalmente, usadas em cromatografia de íons, uma vez que altas concentrações de eletrólitos tornariam o uso do mais importante método de detecção em cromatografia de íons, detecção por condutividade, praticamente impossível.
A fim de otimizar os problemas de separação, a concentração do eluente [Ey–] é freqüentemente variada. Se todos os outros parâmetros presentes na Equação 25 permanecerem constantes, então isto pode ser simplificado para:
][ElogyxCk'log y
M1A (27)
Uma apresentação gráfica da Equação 27 origina uma linha reta com uma inclinação m = –x/y e uma interseção no eixo C1, que contém as quantidades Q, e KA,E. Se um eluente aniônico monovalente for usado, então m é também conhecido como a carga efetiva. A Figura 10 mostra o resultado da Equação 27 para várias combinações de ânions do analito e de eluente diferentemente carregados.
Figura 10 Apresentação gráfica da Equação 27 para várias combinações de ânions do analito e de eluentes diferentemente carregados [4].
A Equação 27 tem sido confirmada por numerosas publicações; entretanto, sob a hipótese de que materiais de separação de baixa capacidade e eluentes diluídos têm sido usados.
Se a capacidade de troca Q variar enquanto os outros parâmetros permanecerem constantes, então a Equação 25 pode ser simplificada para:
yQlog
yxCk'log A (28)
A representação gráfica desta equação é similar à Figura 10, mas com uma inclinação positiva. Investigações cromatográficas sobre a variação de Q foram realizadas em apenas uma ocasião, até hoje, para a separação de cátions bivalentes. Isto tem mostrado que, em contraste às hipóteses prévias, o fator de retenção e os coeficientes de seletividade não podem ser considerados como sendo independentes da capacidade de troca. Para a otimização dos problemas de separação, está claro que além da concentração do eluente [Ey–], a capacidade de troca Q é também uma quantidade importante.

22 Monografia Metrohm
As considerações acima só se aplicam para um ânion analito. Se dois ânions diferentes Ax– e Bz–
estão competindo por grupos funcionais, então o seguinte se aplica para os coeficientes de seletividade KA,B:
xzS
zxM
xzM
zxS
BA, ][A][A][B][A
K (29)
Considerando-se a Equação 20, a seletividade é primeiramente obtida
][B][A][B][A
k'k'
zS
xM
zM
xS
B
ABA,
(30)
e depois, converte-se nas Equações 31 a e b,
S
MBBA,A.B V
Vk'logz
zxKlogz1log (31a)
S
MABA,A.B V
Vk'log
zzxKlog
x1log (31b)
as quais podem ser simplificadas para analitos com mesma carga (x = z):
BA,BA, Klogz1log (32) ou
A,BA,B Klogx1log (33)
Para a seletividade entre dois ânions analitos similarmente carregados, isto significa que:
é apenas uma função dos coeficientes de seletividade KA,B e das cargas z e x, com KA,B constante, a seletividade não depende da concentração [Ey–] nem da constituição química do ânion eluente
Se A e B têm cargas diferentes, então:
A,B depende do fator de retenção de um dos dois analitos, os dois fatores de retenção K’A e K’B não são independentes um do outro
Nas Equações 31 a 33, é particularmente interessante que as seletividades de dois ânions, inicialmente, não dependem da constituição química nem da carga do ânion eluente, fazendo com que a proporção fase-volume e o coeficiente de seletividade sejam constantes. Entretanto, na prática, uma alteração em A,B pode ser alcançada pela variação de [Ey–], uma vez que dois analitos com a mesma carga podem ter, todavia, propriedades químicas diferentes, por exemplo, polarizabilidade e hidratação; isto pode resultar em diferentes afinidades para com a fase estacionária. Entretanto, estas interações não são consideradas na derivatização clássica.
Modelos de retenção para eluentes com vários ânions
As observações prévias referem-se aos sistemas de eluição com apenas um ânion eluente. Na prática, geralmente estão presentes várias espécies eluentes, por exemplo, em tampões carbonato/hidrogenocarbonato ou em ácidos polipróticos tais como ácido fosfórico, cuja dissociação e, conseqüentemente a distribuição de espécies, dependem fortemente do pH.
Mesmo em casos simples, nos quais nenhum dos ânions eluentes participantes está envolvido no equilíbrio ácido-base, a relação entre o fator de retenção K’ e a concentração do eluente [E–] não pode ser representada na forma de uma simples relação log-log, de acordo com a Equação 28. Isto só seria possível se a concentração ou o poder de eluição dos outros ânions eluentes fossem ignorados; isto então corresponderia ao modelo de retenção para eluentes monovalentes.
Na literatura, vários modelos sobre eluentes polivalentes são descritos; estes estão brevemente discutidos abaixo:
modelo do equilíbrio dominante [21] modelo da carga efetiva [22-24] modelo do eluente de múltiplas espécies [25, 26]

Práticas em Cromatografia de Íons 23
Se um eluente baseado em fosfato com H2PO4–, HPO4
2– e PO43–, (ou H2P–, HP2– e P3–) e o íon
analisado monovalente A– forem considerados, então os seguintes equilíbrios são formados:
S2M PHA M2S PHA ; x1 (34)
2S2
1M HPA 2
M21
S HPA ; x2 (35)
3S3
1M PA 3
M31
S PA ; x3 (36)
Aqui, as quantidades x1….3 correspondem às contribuições de uma dada reação na retenção, é por isso que:
x1 + x2 + x3 = 1 (37)
Ambos os modelos, do equilíbrio dominante e da carga efetiva, postulam uma carga particular para o ânion eluente, mesmo que várias espécies estejam presentes; isto significa que o modelo de retenção para eluentes aniônicos monovalentes (vide acima) pode ser usado.
O modelo do equilíbrio dominante assume que o equilíbrio na Equação 36 está completamente do lado direito, uma vez que P3– está ligado muito mais fortemente à fase estacionária do que H2P– eHP2–, como um resultado de sua maior carga. Isto significa que P3– sozinho é decisivo para a eluição sendo que a carga do ânion eluente é –3. Entretanto, na prática este modelo só alcança uma boa concordância com analitos multivalentes [4].
No modelo da carga efetiva, uma carga efetiva é calculada, levando-se em consideração o valor do pH, a partir das frações molares das espécies possíveis H2P–, HP2– e P3– [22]. Utilizando-se estes juntamente com as concentrações das espécies eluentes, uma relação análoga à Equação 27 pode ser obtida. Entretanto, um pré-requisito para este tipo de cálculo é que as seletividades das espécies eluentes não sejam muito diferentes em relação às dos íons analitos A–. O modelo de carga efetiva é principalmente adequado para uso com analitos monovalentes [4].
Na realidade, o modelo do eluente de múltiplas espécies é o mais adequado para a descrição de eluentes cujos componentes são quimicamente derivados uns dos outros. As observações seguintes são baseadas no modelo de Mongay [27], que é um desenvolvimento posterior do trabalho de Jenke e Pagenkopf [25].
As Equações 34 a 36 podem ser usadas para expressar o equilíbrio global na coluna de separação (Equação 38). Considerando-se a Equação 37, as constantes de equilíbrio KA,P podem ser definidas para o processo de troca se as atividades forem ignoradas (Equação 39).
(38)
][HP][HP]P[H][A][HP][HP]P[H][AK /3X3
S/2X2
S/1X
S2M
/3X3M
/2X2M
/1XM2S
PA, 321
321
(39)
O posterior tratamento matemático é realizado voltado para a derivação do modelo de retenção para eluentes aniônicos monovalentes. O que se segue deve ser considerado:
a (possível) dissociação do ânion analito A–
a concentração total das espécies eluentes: CP= [H3P] + [H2P–] + [HP2–] + [P3–]a extensão das interações entre as espécies do eluente e os grupos funcionais
A introdução dos fatores de retenção k’A (Equação 20) e a capacidade Q (Equação 22) supre, após posterior conversão matemática, uma expressão complicada para k’A [28]; a qual é dada aqui apenas em sua forma logarítmica e, posteriormente, simplificada:
P321
3A clog3x
2x
1x
Ck'log (40)
C3 é uma constante que, similarmente à Equação 27, contém quantidades tais como a proporção fase-volume, a capacidade e a constante de equilíbrio; CP é a soma das concentrações das espécies do eluente. Da Equação 40, pode-se deduzir que inclinações das linhas retas em um gráfico bilogarítmico devem ser sempre menores do que aquelas obtidas com o modelo de retenção simples para eluentes aniônicos monovalentes (Equação 27), uma vez que o total entre parênteses é sempre

24 Monografia Metrohm
menor que um. Está claro também que o valor do pH tem uma influência decisiva na extensão para a qual a relação log-log é influenciada.
Para as espécies do eluente que não são quimicamente derivadas umas das outras, Janoš (et al.) desenvolvou um modelo para descrever eluentes contendo tampão fosfato e perclorato adicionalmente [29]. Este modelo foi derivado de acordo com considerações similares às descritas acima, porém, um equilíbrio de troca deve ser considerado para um posterior íon eluente monovalente. Os cálculos fornecem expressões muito complicadas para o fator de retenção; eles podem ser dramaticamente simplificados para eluentes ácidos ou neutros. Se, apenas uma adicional espécie monovalente do eluente estiver presente, além do perclorato, então a Equação 41 é obtida onde x e y representam as contribuições das reações de equilíbrio correspondentes (x: tampão fosfato, y: perclorato) à retenção. Assim como em outros modelos, C é uma constante, enquanto o fator a, tão precisamente definido, deve mostrar o quanto mais fortemente o íon perclorato está ligado à fase estacionária do que as espécies de fosfato envolvidas.
(41)
Como na Equação 41, os termos entre parênteses são sempre menores que um, a inclinação da plotagem log-log é sempre menor do que seria esperado do modelo de retenção simples. Em aplicações reais, o modelo fornece boa concordância com os dados experimentais. Entretanto, a forma descrita acima não pode ser usada para sistemas alcalinos de eluição.
3.4.2. Modelos de retenção em cromatografia de cátions
A cromatografia de cátions deve ser dividida em dois grupos de modelos de retenção. Um grupo está relacionado com cátions metais alcalinos e alcalino-terrosos como analitos e só requerem um sistema de eluição baseado em deslocamento isotônico. Neste caso, a fase estacionária tem grupos ácido-carboxílicos como grupos funcionais. Na separação de íons metálicos com duas ou mais cargas, o uso de um agente complexante é essencial; sua influência na retenção é descrita abaixo.
Modelo de retenção para eluentes com um cátion
As explicações dadas na seção «Modelo de retenção para eluentes com um ânion» aplicam-se de forma análoga para cromatografia de cátions com eluição por deslocamento isotônico. Na prática, isto é relevante para a determinação de metais alcalinos e alcalino-terrosos, amônio e aminas de cadeia curta. Além de H+, cátions orgânicos tais como o ácido 2,3-diaminopropiônico (DAP), são usados como cátions eluentes em combinação com ácido clorídrico diluído. Dependendo do pH do eluente, DAP está presente nas formas iônicas (1) e (2) (Figura 11). Após supressão, a forma zwitteriônica (3) é obtida, a qual não tem condutividade própria.
Figura 11 Espécies iônicas do ácido diaminopropiônico.
Modelo de retenção para eluição na presença de agentes complexantes
Em cromatografia de cátions, eluentes contendo um agente complexante além do cátion eluente En+
são usados para separação de íons de metais pesados, alcalino-terrosos e de transição. Os agentes complexantes usados são principalmente os ácidos dicarboxílicos H2L tais como os ácidos tartárico, oxálico, cítrico e também o piridinodicarboxílico. Os analitos formam complexos de diferentes estabilidades com os ânions dos agentes complexantes HL– e L2–; suas estequiometrias também podem diferir. Como resultado do processo de complexação, a carga efetiva, ou seja, a carga do analito presente sob um período de tempo médio é reduzida. Isto ocorre de acordo com a cinética da formação do complexo e as constantes de estabilidade dos complexos, as diferenças na seletividade aumentam e a separação torna-se possível, mesmo sendo de analitos similares. Além da troca iônica, a formação de complexo é decisiva para a separação de íons metálicos com altas cargas.

Práticas em Cromatografia de Íons 25
(42)
(43)
(44)
Levando-se em consideração a influência do agente complexante na separação por cromatografia de íons, o modelo de retenção para deslocamento isotônico (vide seção «Modelo de retenção para eluentes com um ânion», capítulo 3.4.1.) é ampliado. O valor M é introduzido como quantidade influente que descreve o grau de formação de complexo do analito. A fração M dos íons analitos livres na fase móvel é dada como:
Me'Me
MeLMeLMeHLMeMe x
4x2
2x1xx
x
M (45)
sendo [Me’] a concentração total dos íons metálicos. O valor M pode ser calculado a partir das constantes de formação de complexos, da constante de dissociação dos ácidos carboxílicos e do pH do eluente. Considerando-se a formação de complexos, a seguinte equação é obtida para o coeficiente de distribuição DMe:
xx
Mx
Me MeMeR
Me'MeRD (46)
Assumindo-se que apenas os íons analitos livres Mex+ interagem com o ácido carboxílico ou com os grupos de ácido sulfônico e que c(Ez+) >> c(H+), então, obtém-se o seguinte para a Equação 23:
xx
yy
M
MeEMe, E
yQk'
K (47)
De maneira similar à Equação 25, a forma logarítmica da Equação 47 é obtida como:
][Elogyxlog
yQlog
yxKlog
y1logk'log y
MEMe,MMe (48)
Se várias espécies metálicas catiônicas estiverem juntas, isto é, Mex+ e MeHL(x–1)+ , então normalmente um único pico é obtido no cromatograma para os analitos envolvidos. O número de picos obtidos depende da cinética dos equilíbrios de complexação e de descomplexação na fase móvel. Apenas um pico é obtido se os equilíbrios dos complexos são alcançados mais rapidamente na fase móvel em comparação ao tempo de residência do complexo na fase estacionária. Por outro lado, se o processo de complexação ocorre lentamente, então, picos assimétricos ou múltiplos podem surgir. Assumindo-se que todas as espécies metálicas presentes na fase móvel podem interagir com a fase estacionária, então, obtém-se o seguinte para o fator de capacidade k’exp do analito, experimentalmente determinado:
(49)
Consideração da dependência do fator de capacidade sobre as quantidades influentes Q, [Ey+], bem como M requer que a relação apresentada na Equação 48 seja usada como base, uma vez que analitos bivalentes formam, principalmente, complexos aniônicos ou neutros com fortes agentes complexantes.
Cálculos dos valores M
De acordo com a Equação 45, o valor M é definido como a razão entre a concentração dos íons metálicos livres e a concentração total dos íons metálicos. As concentrações das espécies metálicas presentes na fase móvel podem ser calculadas a partir das constantes de formação de complexos e constantes de dissociação dos ácidos carboxílicos usados.
Se for usado ácido tartárico como agente complexante em eluentes, então, complexos MeL 1:1 neutros serão, principalmente, formados com metais pesados, alcalino-terrosos e de transição

26 Monografia Metrohm
juntamente com uma quantidade menor do complexo hidrogenotartarato MeHL+. Para eluentes contendo ácido tartárico, o seguinte é obtido para o cálculo do valor M:
LHLMeHLLLMeL2
2
M cKcK11
MeHLMeLMeMe (50)
onde cL é a concentração total de ácido tartárico e HL e L são as frações molares dos ânions ácidos HL– e L2–.Além dos complexos 1:1, alguns íons metálicos também formam complexos estáveis MeL2
2– com ácido oxálico e/ou ácido piridinodicarboxílico, sendo que M pode ser calculado como segue:
2L
2LMeLMeLLLMeL
22
2
2
M cKKcK11
MeLMeLMeMe
2
(51)
Cálculo da dissociação de ácidos
O pH e a concentração do agente complexante na fase móvel determina a concentração de ligante e, portanto, o quanto do analito está complexado. Um ácido com dois prótons se dissocia em dois estágios:
(52)
(53)
com as constantes de ácidos KS1 e KS2. As frações molares H2L, HL e L, usadas para calcular o valor M, são obtidas das leis da ação das massas dos estágios individuais de desprotonação:
211
2
SSS2
2
22
2LH
KKHKHH
LHLLHLH (54)
211
1
SSS2
S2
2HL
KKHKH
HKLHLLH
HL (55)
211
21
SSS2
SS2
2
2
LKKHKH
KKLHLLH
L (56)

Práticas em Cromatografia de Íons 27
3.5. Sistemas de detecção em cromatografia de íons Diferentes métodos são usados para a detecção de substâncias em CLAE. A seleção do detector adequado deve sempre estar em concordância com os problemas analíticos a serem resolvidos. Os requisitos de um detector podem ser sumarizados como se segue:
alta sensibilidade de medição e curto tempo de resposta; sinal de medição proporcional à concentração do analito (faixa linear larga); pequenas mudanças na linha de base; baixo ruído de fundo (background);o menor volume possível para reduzir o alargamento do pico.
Uma diferenciação geral é feita entre detectores seletivos e não-seletivos. Enquanto um detector seletivo responde diretamente a uma propriedade do analito, detectores não-seletivos reagem a uma alteração de uma das propriedades físicas do completo sistema de eluição causada pelo analito. Como os detectores usados em cromatografia de íons não diferem, a princípio, daqueles usados em CLAE “convencional”, os sistemas de detecção mais importantes serão, pelo menos, mencionados nesta seção. O detector universal e mais freqüentemente usado em CI é o detector de condutividade.
3.5.1. Métodos de detecção eletroquímica
Detecção de condutividade
Detecção de condutividade, também conhecida como detecção condutométrica, tem uma participação de mercado de aproximadamente 55% no setor de cromatografia de íons [4]. Se for considerado o número de cromatógrafos de íons vendidos, então, esta participação é provavelmente muito maior hoje em dia. Detecção de condutividade é um princípio de detecção não-seletiva; assim, ambas as determinações com detecção direta e indireta são possíveis. Uma vez que eletrólitos aquosos são freqüentemente usados como fase móvel em cromatografia de íons, o detector deve ser capaz de responder às mudanças relativamente pequenas na condutividade total do eluente causada pelos íons analisados. Pelo uso das chamadas técnicas de supressão, a condutividade de fundo de alguns eluentes pode ser drasticamente reduzida; no caso de ânions de ácido forte, é possível melhorar consideravelmente a sensibilidade. A condutividade é determinada como a recíproca da resistência R que um líquido produz entre dois eletrodos com uma área A em uma distância L.
= L / (A*R) (57)
A condutividade equivalente de uma solução pode ser determinada como:
= / c (58)
A condutividade limite e a variação da condutividade com a concentração pode ser determinada pela Equação 59. As constantes A e B são constantes empíricas das substâncias.
= – (A + B ) c (59)
A condutividade de um eletrólito é obtida pela adição das condutividades iônicas -–Ânion e
+Cation
= c (-–Ânion +
+Cation) (60)

28 Monografia Metrohm
Distância L
Área A
Figura 12 Construção de uma célula de condutividade.
De acordo com a lei de Kohlrausch, a condutividade de uma solução diluída é proporcional à soma das condutividades de todos os íons multiplicada por suas concentrações,
1000ici (61)
onde é a condutividade em S cm–1, é a condutividade limitante em S cm2 (z mol)–1 e c é a concentração em z mol L–1 (z corresponde à carga no íon). O fator 1000 surge do fato de que 1 litro é igual a 1000 cm3.
A mudança na condutividade causada pelo analito é proporcional à sua concentração no eluato,
1000cSES (62)
onde S e E correspondem ao íon analisado e íon eluente, respectivamente. Sabemos que na detecção de condutividade a alteração (variação) desta é medida, assim, na cromatografia de ânions, eluentes com altas condutividades de fundo ocasionam pequenas alterações na condutividade devido à passagem do analito. Isto significa que é vantajoso manter a condutividade de fundo o mais baixo possível, conforme o exemplo abaixo:
Figura 13 Mudanças na condutividade do eluato durante a separação de uma mistura de múltiplas substâncias por cromatografia de íons. Os cromatogramas mostram o desempenho de um eluente com condutividade alta (vermelho) e baixa (azul).
Em cromatografia de íons, a condutividade de um eluato pode ser determinada diretamente ou após a passagem por um supressor. Estas versões são conhecidas como técnicas de coluna simples e supressora. A melhor versão a ser adotada pode ser determinada realizando-se um cálculo simples.
Se a detecção por condutividade direta for usada em cromatografia de ânions, então, a sensibilidade Pico da medida depende da diferença das condutividades equivalentes entre os ânions analisados e
Kanalito 1
Keluente 1 (baixo)
Keluente 2 (alto)
Con
dutiv
idad
e

Práticas em Cromatografia de Íons 29
eluentes; tendo o cloreto como analito e o carbonato como ânion eluente, as seguintes equações são obtidas:
Pico cAnalito (-–Cl– –
-–CO32–) Pico cAnalito (76 – 72)
Pico cAnalito • 4
Se o eluente estiver adaptado aos requisitos da detecção de condutividade direta, então, a seguinte sensibilidade é obtida pela substituição do eluente carbonato pelo eluente ftalato:
Pico cAnalito (-–Cl– –
-–Ftalato) Pico cAnalito (76 – 38)
Pico cAnalito • 38
Se, por outro lado, a condutividade do eluente for quimicamente suprimida (troca dos cátions eluentes por H+), a sensibilidade dependerá da soma das condutividades equivalentes do ânion analisado e do íon H+; o seguinte então se aplica para Cl– como ânion analisado:
Pico cAnalito(-–Cl– +
+H+) Pico cAnalito (76 + 350)
Pico cAnalito • 426
A partir deste cálculo estimado pode ser observado que, para ânions, a detecção por condutividade direta é menos sensível por um fator de 10 em relação à detecção por condutividade após supressão química.
Similarmente, o seguinte cálculo simples pode ser feito para cromatografia de cátions, com Na+ como o cátion analisado e H+ como o cátion eluente. No caso de detecção de condutividade direta (NaCl/HCl) as seguintes equações são obtidas para a sensibilidade Pico da medida:
Pico cAnalito (+Na+ –
+H+) Pico cAnalito (50 – 350)
Pico cAnalito • (–300)
Se, por outro lado, a condutividade do eluente for suprimida quimicamente (troca dos ânions eluentes Cl– por OH–), o seguinte se aplica:
Pico cAnalito (+Na+ +
-–OH–) Pico cAnalito (50 + 198)
Pico cAnalito • 248
Isto significa que a sensibilidade é melhor para a detecção por condutividade direta do que para detecção por condutividade após supressão química.
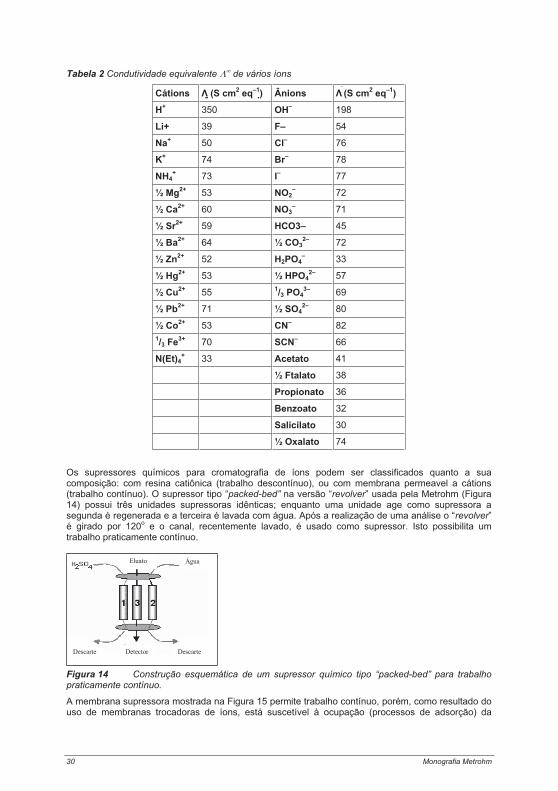
30 Monografia Metrohm
Tabela 2 Condutividade equivalente de vários íons
Cátions (S cm2 eq–1) Ânions (S cm2 eq–1)
H+ 350 OH– 198
Li+ 39 F– 54
Na+ 50 Cl– 76
K+ 74 Br– 78
NH4+ 73 I– 77
½ Mg2+ 53 NO2– 72
½ Ca2+ 60 NO3– 71
½ Sr2+ 59 HCO3– 45
½ Ba2+ 64 ½ CO32– 72
½ Zn2+ 52 H2PO4– 33
½ Hg2+ 53 ½ HPO42– 57
½ Cu2+ 55 1/3 PO43– 69
½ Pb2+ 71 ½ SO42– 80
½ Co2+ 53 CN– 821/3 Fe3+ 70 SCN– 66
N(Et)4+ 33 Acetato 41
½ Ftalato 38
Propionato 36
Benzoato 32
Salicilato 30
½ Oxalato 74
Os supressores químicos para cromatografia de íons podem ser classificados quanto a sua composição: com resina catiônica (trabalho descontínuo), ou com membrana permeavel a cátions (trabalho contínuo). O supressor tipo “packed-bed” na versão “revolver” usada pela Metrohm (Figura 14) possui três unidades supressoras idênticas; enquanto uma unidade age como supressora a segunda é regenerada e a terceira é lavada com água. Após a realização de uma análise o “revolver”é girado por 120o e o canal, recentemente lavado, é usado como supressor. Isto possibilita um trabalho praticamente contínuo.
Descarte Detector Descarte
Água Eluato
Figura 14 Construção esquemática de um supressor químico tipo “packed-bed” para trabalho praticamente contínuo.
A membrana supressora mostrada na Figura 15 permite trabalho contínuo, porém, como resultado do uso de membranas trocadoras de íons, está suscetível à ocupação (processos de adsorção) da

Práticas em Cromatografia de Íons 31
superfície da membrana; isto reduz a capacidade de supressão e finalmente faz com que o supressor pare de funcionar.
Figura 15 Construção esquemática de uma membrana supressora de trabalho contínuo.
Detecção amperométrica
Em princípio, detectores voltamétricos podem ser usados para todos os compostos com grupos funcionais que possam ser reduzidos ou oxidados. O detector amperométrico é a versão mais importante. Neste detector, um determinado potencial é aplicado entre um eletrodo de trabalho e um eletrodo de referência. Se um analito eletroquimicamente ativo, cujo potencial de meia-onda é tal que o potencial aplicado causa sua redução ou oxidação, passar entre os eletrodos então a corrente fluirá e esta representará o sinal de medida. A amperometria é muito sensível; sua taxa de conversão é de aproximadamente 10%. Com exceção de uns poucos cátions (Fe3+, Co2+), são principalmente os ânions tais como nitrito, nitrato, tiossulfato bem como haletos e pseudohaletos que podem ser determinados em análise de íons. As aplicações mais importantes consistem, entretanto, na análise de açúcares por cromatografia de ânions e em análises clínicas. Devido ao seu princípio de trabalho diferente, o detector coulométrico provê um resultado quantitativo sem, entretanto, haver qualquer aumento da sensibilidade.
Detecção potenciométrica
Na detecção potenciométrica, eletrodos íon-seletivos são usados, alguns dos quais têm uma alta seletividade. Apesar de sua necessidade de alto grau de miniaturização, os sensores devem funcionar com segurança; isto ainda causa problemas na prática. É por isso que até agora, a detecção potenciométrica, em cromatografia de íons, está limitada a poucas aplicações especiais.
3.5.2. Métodos espectroscópicos de detecção
Detecção fotométrica
Devido à sua área de aplicação extremamente ampla, a detecção fotométrica ou UV/VIS é o mais importante método de detecção usado em CLAE, já que praticamente todas as moléculas orgânicas possuem grupos cromóforos, os quais são capazes de absorver na região do visível ou do ultravioleta do espectro. Um requisito é que o eluente usado não absorva nos comprimentos de onda usados. Com detecção direta na absorção máxima de um analito, a detecção UV/VIS é praticamente seletiva. Substâncias que possuem apenas uma absorção limitada ou que não possuem absorção em uma determinada faixa de comprimento de onda podem ser determinadas indiretamente, medindo-se a absorção máxima do sistema de eluição. Na área de análise de íons inorgânicos, a detecção UV/VIS tem um papel menor. Ânions comuns tais como nitrato, brometo e iodeto absorvem radiação UV, porém outros analitos importantes tais como fluoreto, sulfato ou fosfato só podem ser medidos indiretamente [4]. Muitos cátions não absorvem, mas, particularmente metais de transição e multivalentes podem ser convertidos em uma derivatização pós-coluna, com formadores de quelatos tais como 4-(2-piridilazo)-resorcinol (PAR) ou Tiron, para formar complexos coloridos. Analitos redox-ativos tais como bromato e outros íons oxi-haletos podem ser analisados por detecção UV/VIS após sofrerem uma reação pós-coluna com um indicador eletroquimicamente ativo.

32 Monografia Metrohm
Detecção por fluorescência
Detecção por fluorescência é muito sensível, desde que os analitos possam ser estimulados a fluorescer; é principalmente o caso de compostos orgânicos com sistemas de elétrons- conjugados. Isto significa que aplicações típicas são encontradas nas áreas de análises clínicas e orgânicas. Conectada à cromatografia de íons, a detecção por fluorescência é usada em poucos casos especiais, visto que apenas determinados íons tais como Ce3+ estão diretamente acessíveis e íons não fluorescentes só podem ser detectados após derivatização. É extremamente difícil desenvolver sistemas de eluição para este método de detecção devido à sua grande susceptibilidade às interferências por contaminantes. Além disso, a taxa linear do método é relativamente pequena (freqüentemente menor que 100 vezes) devido aos seus efeitos de absorção.
Técnicas de acoplamento
As chamadas técnicas de acoplamento representam a estreita ligação, ou “link-up” de um sistema cromatográfico com um método analítico independente, geralmente a espectrometria [3]. Recentemente, estes métodos têm adquirido uma importância crescente. Embora o acoplamento de uma cromatografia gasosa com um espectrômetro de massa (GC-MS, em Inglês) esteja bem estabelecido, o acoplamento da CLAE com métodos espectrométricos gera alguns problemas técnicos. Na CLAE clássica, ou seja, a análise de compostos orgânicos, acoplamentos com um espectrômetro de massa, espectrômetro de infravermelho e espectrômetro de ressonância magnética nuclear estão disponíveis [3]. Em particular, poderosos detectores de espectrometria atômica são usados em cromatografia de íons. Como exemplo há espectrometria de massa e emissão atômica com plasma indutivamente acoplado e o resultado de sua sensibilidade e especificidade de cada elemento, este acoplamento proporciona excelentes resultados. É por isso que, apesar do alto custo, tais sistemas são usados para especiação e em análises de ultratraços de elementos.
Refratometria
Refratometria diferencial é um outro método de detecção baseado em método óptico. Este detector é também chamado de detector RI (do Inglês Refractive Index = índice de refração). Ele não é específico, pois a unidade de medida é a mudança (variação) no índice de refração do eluente puro causado pelo analito. Entretanto, a grande sensibilidade de temperatura deste parâmetro significa que o método é muito susceptível a interferências. Assegurando-se que a temperatura seja absolutamente estável, o método tem uma faixa linear de três potencias de dez. Uma vez que íons inorgânicos possuem um índice de refração extremamente baixo, eles só podem ser determinados indiretamente com o uso de eluentes aos quais foram adicionados compostos com alto índice de refração.
3.6. Fases estacionárias em cromatografia de íons A cromatografia de íons eficiente requer materiais de empacotamento, sendo estes feitos de partículas muito pequenas as mais esféricas possíveis e que tenham uma taxa de distribuição reduzida do tamanho de partícula. Partículas com diâmetros de 2 a 10 μm são usadas. Além disso, o material de empacotamento deve apresentar cinéticas de troca iônica o mais rápido possível. Com exceção das partículas, isto também determina a eficiência dos trocadores de íons.
3.6.1. Visão geral das fases estacionárias comuns
Um grande número de materiais orgânicos e inorgânicos diferentes é apropriado para o uso em cromatografia de íons. O que todos têm em comum é que suas superfícies carregam grupos funcionais que podem trocar íons. As seguintes classes de substâncias podem ser usadas [4]:
Resinas de polímeros orgânicos modificadas; Sílica gel modificada; Sais inorgânicos (por exemplo, polifosfatos); Vidros; Zeólitas;Óxidos metálicos (por exemplo, Al2O3);Derivados de celulose.
Fora estes, os sistemas com uma constituição muito complexa são também possíveis, tais como os trocadores de ânions com grupos funcionais consistindo de íons metálicos alcalinos ligados a uma

Práticas em Cromatografia de Íons 33
fase estacionária por éteres coroa. Na prática, os representantes mais importantes são baseados em resinas de polímero orgânico modificadas e sílica gel. A Figura 16 fornece uma visão geral dos materiais de separação usados na CI:
Figura 16. Fases estacionárias comumente usadas em cromatografia de íons [4].
Todas as fases estacionárias podem posteriormente ser diferenciadas de acordo com seu tipo de aplicação (cromatografia de ânions ou de cátions) ou a estrutura do grupo funcional. Os materiais de empacotamento baseados em sílica gel foram originalmente usados para cromatografia de íons. Embora tenham uma eficácia de separação muito boa e sejam mecanicamente muito estáveis, sua estabilidade química não é muito grande, tanto que podem somente ser usados em pH = 2 a 7.
A partir dos anos 80, os trocadores de íons tornaram-se disponíveis, os quais eram baseados em polímeros orgânicos e podiam ser usados em cromatografia de íons; estes eram manufaturados modificando-se as resinas adsorventes comercialmente disponíveis. Hoje, os materiais de empacotamento usados são normalmente baseados nos copolímeros do poliestireno-divinilbenzeno (PS-DVB) ou polímeros metacrilato (MMA).
Estes dois tipos básicos de copolímero diferem principalmente em sua polaridade. Os copolímeros do PS-DVB são completamente não-polares e representam fases reversas enquanto os polímeros MMA são relativamente polares. Esta situação é uma vantagem em CI visto que as fases de separação mais polares têm uma menor tendência às interações secundárias tais como a adsorção.
A maior vantagem das resinas orgânicas poliméricas é sua grande estabilidade química em toda a escala de pH. Depois que problemas iniciais são superados, sua eficiência cromatográfica é similar àquela da sílica gel. Entretanto, as fases de MMA podem ter uma estabilidade mecânica limitada, isto poderia limitar o comprimento da coluna de separação usada ou da vazão máxima possível do eluente.
Na cromatografia de íons, dois tipos de fases estacionárias são usados hoje, os quais diferem no princípio; trocadores de íons de superfície funcional e trocadores de íons peliculares. No primeiro tipo, os grupos funcionais estão localizados diretamente na superfície do polímero ou nos poros; os materiais peliculares têm partículas muito pequenas (também de superfície funcional) que estão ligadas às partículas centrais maiores [4]. A ligação pode ser mecânica ou resultar das interações hidrofóbicas ou eletrostáticas. A Figura 17 mostra o arranjo dos dois tipos de material de empacotamento usando trocadores de ânions como exemplos.
Ligaçãoeletrostática

34 Monografia Metrohm
Figura 17. Estrutura de trocadores de ânions de superfície funcional (a) e peliculares com ligação mecânica (b).
Os materiais de empacotamento do tipo pelicular têm uma maior eficiência cromatográfica, pois as vias de difusão são mantidas muito curtas devido à grande distância dos grupos funcionais para com o material de base; isto resulta em excelente transferência de massa. Entretanto, a estabilidade química destas fases de separação é consideravelmente menor do que a dos materiais de superfície funcionalizada.
3.6.2. Fases estacionárias para cromatografia de ânions
O grupo funcional usado em cromatografia de ânions é produzido normalmente pela conversão de um grupo âncora com uma amina apropriada. Isto produz íons amônio que são fixados na superfície do polímero. Os grupos funcionais baseados em nitrogênio são praticamente os únicos usados para a separação de ânions por CI. Isto é baseado principalmente em sua estabilidade química e no número quase ilimitado de substituintes possíveis no átomo do nitrogênio.
Grupos amônio são gerados na superfície do polímero pela conversão de um grupo âncora com uma amina.
Os resíduos alquilas no nitrogênio positivamente carregado podem ser enormemente variados. No caso mais simples, R=H e um íon amônio primário são formados. Entretanto, isto pode ser desprotonado em pH elevado e perder sua carga. Com este tipo de material de empacotamento a capacidade de troca depende do pH do eluente, é por isso que eles devem ser fracamente alcalinos. Se os átomos do hidrogênio forem agora substituídos sucessivamente por grupos alquilas, então, primeiramente os grupos amônio secundários e depois os terciários são formados; estes podem também ser desprotonados. Somente quando todos os resíduos R consistirem de grupos alquilas, a capacidade ou a carga, independente do pH do eluente, e um trocador de ânions quaternário fortemente básico são obtidos. Em cromatografia, uma capacidade independente de pH é normalmente o esperado para que somente as resinas completamente alquiladas sejam usadas. Para aplicações especiais tais como técnicas de pré-concentração ou análise de proteínas, materiais fracamente básicos são também usados.
Os dois grupos funcionais mais importantes em cromatografia de ânions são derivados de trimetilamina (TMA) e dimetiletanolamina (2-dimetilaminoetanol, DMEA). Praticamente todos os materiais de separação comercialmente disponíveis usam um destes dois grupos. Os grupos TMA são também conhecidos na literatura como os grupos do Tipo I, e os grupos DMEA como do Tipo II. Outros grupos funcionais, estreitamente relacionados aos mencionados acima, são mostrados na Figura 18:
Figura 18. Visão geral dos grupos funcionais mais importantes mencionados neste trabalho TMA: trimetilamina (Tipo I); DEMA: Dietanolmetilamina; EDMA: Etildimetilamina; TEA: Trietanolamina; DMEA: Dimetiletanolamina (Tipo II).
Em materiais comerciais, que são derivados geralmente do tipo I ou II, o arranjo exato dos grupos funcionais é altamente secreto [4].

Práticas em Cromatografia de Íons 35
3.6.3. Fases estacionárias em cromatografia de cátions
Em cromatografia de cátions, tanto os materiais baseados em sílica gel como os materiais baseados em polímero estão em uso. Em contraste à cromatografia de ânions com seus eluentes alcalinos usuais, as condições para a cromatografia de cátions permitem também o uso de sílica gel.
3.6.4. Trocadores de cátions baseados em sílica gel
Com os trocadores de cátions baseados em sílica gel uma diferenciação é feita entre os materiais diretamente funcionais e aqueles que são revestidos por um polímero.
Praticamente todos os trabalhos a respeito dos materiais funcionais indicam o uso de trocadores fortemente ácidos, possuindo um grupo ácido sulfônico [2,4]. Estes têm uma boa eficiência cromatográfica, mas são inadequados para a determinação simultânea de metais alcalinos e alcalino-terrosos devido às grandes diferenças em afinidade.
Com a sílica gel revestida de polímeros, as chamadas fases de Schomburg, a superfície de silicato é revestida por um “pré-polímero” que é então imobilizado por ligação cruzada (cross-linking). Pela subseqüente funcionalização, vários tipos diferentes de trocadores podem ser produzidos. A fim de se obter trocadores de cátions fracamente ácidos, o ácido polibutadienomaleico (PBDMA) é usado, proporcionando ligação cruzada (cross-linked) in-situ [30]. Em conseqüência da camada fina de polímero de aproximadamente 1 a 5 nm [31], as vias de difusão do analito são pequenas visando um elevado grau de eficiência cromatográfica
Existe um grande número de aplicações para trocadores de íons baseados em sílica gel. A separação simultânea de metais alcalinos e alcalino-terrosos é uma das mais freqüentes aplicações; a separação de íons de metal pesado e de transição também é possível. Entretanto, várias desvantagens impedem o uso universal dos trocadores de íons baseados em sílica gel:
Em pH < 2, a ligação entre a matriz de silicone e o grupo funcional torna-se muito mais fraca; isto leva a uma erosão gradual dos grupos funcionais e uma perda da capacidade;
Em pH > 7, a solubilidade da sílica gel aumenta consideravelmente; isto resulta em uma redução da estabilidade mecânica do empacotamento, quebra da partícula e um volume morto no início da coluna.
3.6.5. Trocadores de cátions baseados em polímeros orgânicos
As resinas produzidas por co-polimerização estireno-divinilbenzeno são usadas principalmente como matrizes. As limitações mencionadas para trocadores sílica gel não ocorrem aqui; elas podem ser usadas em toda a escala de pH, de 0 –14, e são também inertes ao fluoreto. Embora sua resistência de pressão seja menor do que a de sílica gel, ainda é geralmente adequada, com exceção de algumas resinas metacrilato.
A maioria dos trocadores de cátions comercialmente disponíveis usa grupos de ácido sulfônico como grupos funcionais. Estes podem ser ligados ao sistema aromático diretamente ou através de um espaçador cujo comprimento seja variável. Os trocadores de ácido sulfônico com espaçadores têm uma eficiência cromatográfica consideravelmente melhor; o comprimento do espaçador é de importância secundária. Eles não são apropriados para a determinação simultânea de metais alcalinos e alcalino-terrosos devido às grandes diferenças em afinidade.
3.6.6. Trocadores de cátions peliculares
Assim como as resinas diretamente funcionalizadas, os trocadores peliculares estão também disponíveis. Eles têm um arranjo de camada dupla não sendo possível apresentar uma partícula de substrato completamente aminada. É por isso que uma partícula substrato inteiramente sulfonada é primeiramente cercada por uma camada de partículas de látex aminadas e então por uma camada de partículas de látex sulfonadas. A fixação ocorre através das interações eletrostáticas e de van der Waals. O diâmetro relativamente grande das partículas do substrato (10 a 30 μm) resulta em uma contra-pressão comparativamente baixa. Os diâmetros pequenos (20 a 250 nm) das partículas de látex permitem vias curtas de difusão com processos de troca rápidos e, conseqüentemente, uma alta eficiência da coluna de separação. A desvantagem destes materiais peliculares é sua sensibilidade aos solventes orgânicos [32] e às fases móveis com uma força iônica elevada, visto que neste caso as partículas de látex são gradualmente removidas.
A determinação simultânea de metais alcalinos e alcalino-terrosos é possível usando-se uma versão modificada na qual a camada aminada é ligada covalentemente. Entretanto, as diferenças na

36 Monografia Metrohm
seletividade entre potássio, magnésio e cálcio são desnecessariamente grandes, proporcionando tempos de análise relativamente longos. Isto pode ser justificado pela presença de grupos ácido sulfônico fortemente ácidos que são usados.
3.6.7. Fases estacionárias em cromatografia de exclusão iônica
A escolha das fases estacionárias para este tipo de separação é muito limitada. A formação da membrana de Donnan por grupos funcionais dissociados é importante para o processo da separação. Além disso, o material de empacotamento deve prevenir o máximo possível a adsorção do analito na fase estacionária e, como os analitos são principalmente dos grupos ácido carboxílico e açúcar, uma superfície tão polar quanto possível é o esperado para uma fase estacionária em cromatografia de exclusão iônica. Praticamente nenhuma demanda é requerida nas cinéticas da reação de troca iônica, uma vez que a separação é baseada em um mecanismo de exclusão. Os polímeros de baixa ligação cruzada a base de PS/DVB são quase que os únicos usados e estes devem ser completamente sulfonados.
3.6.8. O significado da capacidade dos trocadores de íons
Além do tipo de matriz e do tipo do grupo funcional, a capacidade de troca Q é a quantidade decisiva para caracterizar trocadores de íons. Ela fornece informação sobre o número de sítios ativos disponíveis para a troca iônica.
A capacidade de troca é expressa normalmente em gramas do material seco de empacotamento, em microequivalentes (μeq/g) ou micromols (μmol/g) [2]. Outras informações também são normalmente dadas [33]. Para finalidades analíticas, trocadores de íons podem ser grosseiramente divididos de acordo com sua capacidade em:
Materiais de baixa-capacidade: Q < 100 μmol/g; Materiais de média capacidade: 100 < Q < 200 μmol/g; Materiais de alta capacidade: Q > 200 μmol/g.
As fases da separação usadas em trocadores clássicos de íons são todos tipos de alta capacidade com 3 a 5 mmol/g [4].
A definição de capacidade acima é baseada em um equilíbrio completo alcançado entre as fases estacionária e móvel, por isso, também é conhecida como capacidade estática. Em contraste, a capacidade dinâmica (eficaz) é conhecida como sendo o número dos grupos funcionais que estão realmente disponíveis durante um processo cromatográfico. É sempre menor do que a capacidade estática [2,4].
A capacidade de troca pode ser determinada de maneiras diferentes [33]:
Volumetricamente ou titrimetricamente; pela análise elementar; pela determinação dos tempos de retenção.
Todos os métodos fornecem valores diferentes para Q para o mesmo material. Os métodos volumétricos são os mais freqüentemente usados na prática. Para trocadores de ânions, uma coluna de separação empacotada ou uma quantidade definida de resina são carregadas, por exemplo, com solução de cloreto. Após a lavagem do cloreto residual, a coluna ou a resina pode ser eluída com nitrato. A quantidade de cloreto eluído, que corresponde à capacidade de troca estática sob condições de equilíbrio, pode ser titulada contra uma solução de AgNO3 e quantificada.
A dependência do pH pelos trocadores de íons pode geralmente ser desprezada. Com trocadores de cátions, a capacidade dos grupos ácido carboxílico fracamente ácidos (R-OOH) é dada apenas em valores elevados de pH (desprotonação), enquanto que grupos de ácido sulfônico fortemente ácidos (R-SO3H) sempre estão completamente desprotonados e fornecem uma capacidade independente do pH.
Em combinação com os modelos de retenção (capítulo 3.4), a importância de Q para a seleção dos sistemas de eluição e detecção torna-se óbvia. A detecção universal de condutividade era praticamente impossível de ser realizada com eluentes com uma força iônica elevada, independente de qual técnica especial fosse usada. Esta é a razão pela qual, desde a introdução da cromatografia de íons em 1975, muitas colunas de separação de baixa capacidade têm sido usadas. Até agora, trocadores de íons de alta capacidade têm sido descritos geralmente para aplicações da CI em

Práticas em Cromatografia de Íons 37
combinação com as técnicas de detecção que não são tão limitadas pelo sistema de eluição, tais como a detecção UV/VIS [2,4].
Enquanto os materiais de separação de baixa capacidade são muito apropriados para a análise de amostras com uma força iônica baixa e, conseqüentemente, para uma matriz de carga baixa, em forças iônicas mais elevadas eles alcançam rapidamente seus limites. Os efeitos da sobrecarga ocorrem por causa de seu baixo número de grupos funcionais; estes resultam em picos deformados e em uma queda drástica na eficiência. Os problemas ocorrem também se os analitos estiverem presentes em concentrações muito diferentes; isto é quase sempre o caso em análise de ultratraços.
3.7. Eluentes em cromatografia de íons Como em todas as separações por cromatografia líquida, a fase móvel é também o parâmetro mais fácil de se alterar na cromatografia de íons a fim de influenciar a separação enquanto que a coluna de separação e o sistema de detecção são, na maioria dos casos, pré-definidos.
A escolha de um sistema de eluição adequado pode ser feita usando-se uma grande variedade de critérios. Em cromatografia de ânions, os seguintes parâmetros, entre outros, devem ser considerados [4]:
Compatibilidade com o método de detecção; Natureza química e concentração do íon eluente; pH;Capacidade de tamponamento; Conteúdo de solvente orgânico (modificadores);
Na literatura [2,4,5], a discussão sobre as fases móveis ocorre principalmente sobre a adequação referentes às técnicas com ou sem supressão. Este será também o caso abaixo, mesmo que o foco do assunto deste trabalho seja a capacidade de troca Q da coluna de separação. Primeiramente, será feita uma breve explanação sobre os termos “técnica de coluna simples” (sem supressão) e “técnica de supressão”. Como na seção anterior, as observações limitam-se principalmente à cromatografia de ânions.
3.7.1. Cromatografia de ânions
Técnica da coluna simples
Na técnica de coluna simples, que foi estabelecida na cromatografia de íons por Gjerde em 1979 [33], a saída de uma coluna de separação é conectada diretamente a um detector; isto corresponde ao arranjo clássico de um equipamento da CLAE. Para distinguí-la da segunda versão principal da CI, esta técnica é também conhecida como “cromatografia de íons não suprimida”. O eluente contendo os analitos sai da coluna de separação e não é alterado quimicamente. O número de sistemas possíveis de eluição com características químicas diferentes é quase ilimitado. Exceto para a própria questão da separação, na qual somente uma compatibilidade do eluente com o detector deve ser considerada. Por exemplo, se a detecção UV direta for usada, então o eluente não deve absorver na faixa de espectro relevante. Na técnica de coluna simples, nenhuma limitação é colocada no sistema de detecção de modo que, em princípio, todos os detectores usuais de CLAE podem ser usados.
Para trocadores de ânions da baixa capacidade (Q < 100 μmol/coluna de separação), uma grande variedade de eluentes com diferentes características químicas está disponível para a técnica de coluna simples. A concentração dos eluentes corresponde normalmente a uma faixa de mmol/kg ou menor. As classes de substâncias mencionadas abaixo podem ser usadas [4], sendo que em casos especiais, também podem ser usados agentes complexantes como o EDTA ou complexos borato-manitol.
Ácidos carboxílicos aromáticos; Ácidos carboxílicos alifáticos; Ácidos sulfônicos; Hidróxidos alcalinos; Ácidos inorgânicos tais como H2SO4, HCl ou H3PO4.
A classe mais importante de compostos é a dos ácidos carboxílicos aromáticos e seus sais. Os representantes mais freqüentemente usados são mostrados na Figura 19.

38 Monografia Metrohm
Figura 19. Estruturas dos ácidos carboxílicos aromáticos mais importantes usados com técnica de coluna simples.
Esta classe de substâncias é usada tão freqüentemente porque as soluções de ácidos e seus sais têm uma elevada força de eluição combinada com uma condutividade de fundo relativamente pequena, de modo que possam ser usadas para a detecção direta de condutividade. Com ácidos polipróticos a carga e conseqüentemente a força de eluição podem ser controladas através do pH. Entretanto, isto deve ser mantido de forma muito precisa. Os ácidos carboxílicos aromáticos têm uma absorção elevada no espectro UV de modo que podem ser usados vantajosamente em detecção indireta UV/VIS.
Para ácidos sulfônicos aromáticos, tais como o ácido p-toluenossulfônico, aplicam-se, em princípio, as mesmas indicações; estes estão sempre presentes em um estado desprotonado e não possuem conseqüentemente capacidade tamponante. Sua força de eluição pode somente ser controlada através da concentração.
Os ácidos carboxílicos alifáticos, tais como o ácido oxálico e o ácido cítrico, têm uma alta condutividade de fundo, mas são “transparentes” em UV, podendo ser usados para a detecção direta UV/VIS. Isto também se aplica aos ácidos sulfônicos alifáticos, dos quais o ácido metanossulfônico é um dos mais freqüentemente usados. Compostos similares de ambas as classes, com suas cadeias carbônicas maiores, os quais estão associados a baixas condutividades equivalentes, oferecem a possibilidade de serem usados para a detecção direta da condutividade [2, 4].
Os hidróxidos alcalinos têm um uso muito limitado na técnica de coluna simples, pois os íons OH– têm a mais baixa afinidade aos grupos amônio quaternário de todos os ânions eluentes possíveis. Em conseqüência, concentrações altas do eluente são requeridas mesmo em capacidades baixas, sendo que na melhor das hipóteses, somente a detecção indireta de condutividade possa ser usada. Em contraste, a detecção UV/VIS é facilmente possível, embora os íons hidróxido basicamente “transparentes” em UV tenham uma absorção característica abaixo de 220 nm em concentrações mais altas.
Ácidos inorgânicos ou seus sais parcialmente ou totalmente dissociados apresentam alta condutividade. Isto significa que somente detectores fotométricos podem ser usados. Por exemplo, se o ácido fosfórico ou fosfatos forem usados, então, a capacidade tampão e a força de eluição podem ser controladas através do pH do eluente.
A maioria dos eluentes mencionados aqui permite também o uso de outros sistemas de detecção tais como técnicas de amperometria, fluorimetria ou de acoplamento. Se você deseja usar os sistemas de eluição mencionados acima com estes detectores, por favor, consulte uma literatura mais detalhada [2,4,5].
Técnica de supressão A técnica de supressão utilizada desde os princípios da CI [1] é caracterizada principalmente pelo uso do detector de condutividade na maioria das vezes, diferindo da técnica da “coluna simples”. Na técnica de supressão, o chamado supressor é inserido entre a coluna de separação e o detector‚ e é por isso que esta técnica também é conhecida como “cromatografia de íons suprimida” [2, 4]. Tanto os eluentes quanto os analitos são modificados quimicamente no supressor, isto é de particular importância em relação à subseqüente detecção de condutividade. O supressor tem a tarefa de reduzir a condutividade de fundo do eluente e, se possível, aumentar a detectabilidade dos analitos.
A supressão é realizada com um trocador de cátion na forma H+ fortemente ácida. Se utilizarmos um eluente NaHCO3 e a amostra contendo o íon cloreto, teremos as seguintes reações 63 e 64
R-SO3– H+ + Na+ + HCO3
– R-SO3– Na+ + H2O + CO2 (63)
R-SO3– H+ + Na+ + Cl– R-SO3
– Na+ + H+ + Cl– (64)
No caso mais simples, a unidade supressora consiste em uma coluna inserida após a coluna de separação, de onde se origina o antigo termo “técnica de dupla coluna”. O eluente bicarbonato de

Práticas em Cromatografia de Íons 39
sódio é neutralizado de acordo com a Equação 63, uma vez que os íons de sódio são substituídos por prótons, diminuindo drasticamente a condutividade de fundo do eluente. O analito Cl– não é alterado (Equação 64), mas seu contra-íon Na+ é trocado por H+, que tem uma condutividade equivalente consideravelmente maior [19]. Uma vez que o detector se sensibiliza com a soma das condutividades do analito e do contra-íon como o sinal, as duas reações que ocorrem resultam em uma sensibilidade claramente maior.
As colunas com trocadores de cátions na forma H+, originalmente usadas como supressoras, contribuíram consideravelmente para o alargamento dos picos. Além disso, elas só podiam ser operadas descontinuamente, visto que o trocador de cátion tinha que ser regenerado. É por isso que, supressores com regeneração paralela ou de membrana operando continuamente são principalmente usados hoje; nestes, o agente regenerante geralmente é o ácido sulfúrico diluído[6].
Apesar de suas vantagens, a técnica de supressão tem também algumas desvantagens. Na prática, somente os eluentes que são baseados, principalmente, em hidróxidos alcalinos ou carbonatos podem ser suprimidos com sucesso na cromatografia de ânions. Os ânions de ácidos mais fracos, por exemplo, acetato ou propionato, são quase completamente protonados após a reação de supressão, sendo que sua detectabilidade torna-se menor quando comparada com a técnica de coluna simples.
Como já vimos, a separação e quantificação de ânions podem ser realizadas com técnica de supressão ou pela detecção direta de condutividade [2, 4]. Esta última técnica é menos utilizada em cromatografia de ânions, principalmente porque em muitos casos ela é menos sensível. Um dos fatores é a condutividade de fundo: para os eluentes clássicos da técnica de coluna simples (por exemplo, 2 mmol/kg de ftalato, pH = 8) este valor é de aproximadamente 200 μS/cm e para eluentes capazes de serem suprimidos, ela é geralmente menor em pelo menos dez vezes após o processo da supressão.
A supressão química da condutividade de fundo é possível somente para alguns eluentes. Estes são soluções de [4]:
Hidróxidos alcalinos; Carbonatos alcalinos e bicarbonatos; Boratos (por exemplo, B4O7
2–);Aminoácidos.
Na prática, dos eluentes mencionados acima somente os hidróxidos alcalinos e os tampões carbonatos são de grande importância, significando que a escolha das potenciais fases móveis é limitada.
O íon hidróxido é um íon eluente extremamente fraco, de modo que mesmo com materiais de separação com baixa capacidade, o trabalho deve ser realizado em altas concentrações, acima de 50 mmol/kg. Se, grupos funcionais muito polares estiverem presentes na resina trocadora, então, a força de retenção relativa do íon OH– pode ser consideravelmente aumentada utilizando-se a seletividade do hidróxido. Com eluentes OH–, a manipulação dos tempos de retenção ou das seletividades pode somente ser realizada através da concentração.
O uso de carbonatos ou bicarbonatos alcalinos permite trabalho com sistemas de eluição consideravelmente mais flexíveis. Ambas as espécies, HCO3
– e CO32–, estão presentes como ácido
carbônico H2CO3 após a supressão e só dissociam uma fração muito pequena. E quanto a força de eluição, o bicarbonato é ligeiramente mais forte do que o hidróxido, visto que o carbonato é um eluente relativamente mais forte. Ambos os ânions são normalmente usados juntos; isto resulta em um eluente com um efeito tampão cuja força de eluição pode ser facilmente controlada através das concentrações dos dois componentes e da proporção de suas concentrações. Por causa das cargas nas espécies eluentes, a seletividade para analitos monovalentes e divalentes pode ser influenciada muito seletivamente. A proporção de concentração dos dois íons eluentes pode também ser ajustada com precisão através do pH, é por isso que a faixa de pH usada para os eluentes HCO3
–/CO32– está
entre 8 e 11. Assim como para eluentes OH–, a eluição pode ser acelerada pelo uso de fases estacionárias com grupos polares funcionais.
Ambos os sistemas de eluição podem apenas ser usados com sucesso com trocadores aniônicos de superfície funcionalizada quando a matriz (co-polímeros de metacrilato) ou o grupo funcional tiver uma polaridade elevada. Para colunas de separação baseadas em PS-DVB, picos com simetria menor e longos tempos de retenção têm sido observados para analitos fracos (nitrato, brometo) mesmo quando grupos funcionais polares são usados. Com materiais peliculares, estes efeitos não são tão marcantes, o que explica o uso excepcionalmente grande difundido da técnica de supressão [2, 4].

40 Monografia Metrohm
3.7.2. Cromatografia de cátions
3.7.2.1. Cromatografia de cátions de íons alcalinos, alcalino-terrosos e amônio com detecção de condutividade
Na separação de íons metálicos alcalinos e amônio, assim como aminas alifáticas de cadeia curta em fases sulfonadas de separação pela cromatografia de íons, os ácidos minerais tais como HCl ou HNO3 são muito usados como eluente [4]. A concentração ácida do eluente depende do tipo e da capacidade do trocador de cátion usado, mas são necessários alguns mmol/L. Cátions divalentes tais como os alcalino-terrosos não podem ser eluídos somente com ácidos minerais, pois eles têm uma afinidade distintamente maior para com a fase estacionária e, um aumento excessivo na concentração ácida tornaria a detecção muito insensível, ou seja, cátions monovalentes coeluindo e a pobre separação entre os cátions divalentes. Como uma alternativa, uma base orgânica tal como a etilenodiamina pode também ser usada para a separação de íons alcalino-terrosos. Em baixos valores de pH, ela é protonada e está presente como um cátion divalente.
A análise simultânea de cátions alcalinos e alcalino-terrosos em trocadores de cátions fortemente ácidos é realizada principalmente com eluentes que contêm o ácido clorídrico e o ácido 2,3-diaminopropiônico [2]. Variando-se o pH é possível alterar o grau de protonação dos grupos aminas e conseqüentemente da força de eluição do ácido 2,3-diaminopropiônico (veja o item “Modelo de retenção para eluentes com um cátion”, capítulo 3.4.2).
Em sistemas de cromatografia de íons sem supressão, ácidos orgânicos fracos podem também ser usados como eluentes, além dos ácidos minerais. Os ácidos orgânicos usados são, por exemplo, ácido oxálico, ácido cítrico e ácido tartárico. Os agentes complexantes tais como o ácido 2,6-piridinodicarboxílico (PDCA) e o éter coroa 18 Crown-6 são usados para influenciar seletivamente os tempos de análise de cátions individuais.
Há dois tipos diferentes de detecção de condutividade. Se a condutividade de fundo dos eluentes for elevada, por exemplo, com ácidos minerais diluídos por causa da alta condutividade dos íons H+altamente dissociados, então a detecção direta de condutividade é possível com os analitos que têm uma condutividade evidentemente mais baixa do que a do eluente. Isto significa que picos negativos são produzidos, entretanto, eles podem ser convertidos em um cromatograma normal pela reversão da polaridade do detector. Se a qualidade do detector de condutividade não permite medida sensível da condutividade em um nível elevado, então, a supressão é também usada para a cromatografia de cátions. A construção de supressores para a cromatografia de cátions é bem mais difícil do que para a cromatografia de ânions e eles também têm um tempo de vida útil menor devido a precipitação de hidróxidos de metais de transição.
3.7.2.2. Cromatografia de cátions para determinação de metais de transição e de íons alcalino-terrosos com detecção pela derivatização pós-coluna e fotométrica
Cátions monovalentes tais como H+ ou Na+ são inadequados para o uso como um eluente para a separação de íons de metais pesados e de transição porque os coeficientes de seletividade dos analitos raramente diferem em um mesmo número da carga. Entretanto, a separação pode ser realizada pela introdução de um equilíbrio secundário. Isto é feito usando-se ácidos carboxílicos formadores de complexos (vide Figura 20) tais como o ácido cítrico, o ácido oxálico e o ácido tartárico como eluente. Junto com os íons do metal, eles formam complexos neutros ou aniônicos (vide capítulo 3.4).
Figura 20. Vários ácidos carboxílicos como eluentes em cromatografia de cátions
Complexando-se cátions de metal, a densidade da carga efetiva dos analisados é reduzida. Além disso, constantes formando diferentes complexos dos íons individuais de metal aumenta a seletividade da separação. O mecanismo de eluição é um resultado da remoção isoiônica pelo contra-íon (efeito de “empurrar”) e pela formação de complexos (efeito de “puxar”) pelos ligantes formadores de complexos [4].

Práticas em Cromatografia de Íons 41
A Figura 21 mostra os equilíbrios entre os analisados M2+, o agente complexante L2– e os contra-íons E+, os quais participam do mecanismo de eluição.
Figura 21 Diagrama esquemático dos participantes no equilíbrio em um processo de troca catiônica [4].
A extensão da remoção isoiônica pelos contra-íons E+ é determinada pela afinidade do cátion à fase estacionária. Com contra-íons monovalentes, o cátion possuindo uma maior afinidade à fase estacionária tem um efeito eluente mais forte. A influência do agente complexante pode ser variada alterando-se o pH e a concentração do eluente. Além disso, a força de eluição pode ser influenciada pelo uso de diversos agentes complexantes, bem como pelo uso de um cátion divalente como contra-íon.
Enquanto os dois tipos de detecção de condutividade são apropriados para a detecção de metais alcalinos e alcalino-terrosos, íons de transição e de metal pesado só podem ser determinados pela medida direta da condutividade sem supressão. Se a técnica de supressão fosse usada, então, estes metais seriam convertidos (a maioria) em hidróxidos insolúveis pela reação de supressão. A detecção direta de condutividade só é possível quando eluentes com uma baixa condutividade de fundo são usados junto com trocadores de cátions de baixa capacidade.
É por isso que a derivatização pós-coluna, com a formação de complexos de metais fotometricamente detectáveis, é usada principalmente para a detecção de íons de transição e de metais pesados. Após sair da coluna de separação, o eluato é misturado com um reagente colorimétrico em um reator pós-coluna. Este reage com os analitos para formar complexos coloridos do metal que podem ser detectados fotometricamente. Uma variedade grande de azo-corantes pode ser usada como agentes de coloração [2,4]. Metais de transição e lantanídeos reagem com o 4-(2-piridilazo)-resorcinol (PAR) (Figura 22) para formar complexos coloridos. Lantanídeos e actinídeos podem ser detectados usando-se o ácido 2,7-bis(2-arsenofenilazo)-1,8-dihidroxinaftaleno-3,6-dissulfônico (Arsenazo III). O ácido pirocatecol-3,5-dissulfônico (Tiron) é apropriado para a derivatização pós-coluna do alumínio.
Figura 22 Estrutura do indicador de metal PAR
PAR é usado principalmente para a detecção de íons de transição e de metal pesado. Como o Fe, Co, Ni, Cu, Zn, Mn, Pb e Cd, o PAR forma complexos coloridos; estes absorvem em comprimento de ondas de 490 a 520 nm com coeficientes de absorção de até 104 L mol–1 cm–1 e podem ser detectados fotometricamente. A sensibilidade do método é baseada no fato de que os coeficientes de

42 Monografia Metrohm
extinção dos complexos de metal-PAR são grandes comparados com o coeficiente de absorção do reagente no comprimento de ondas usado.
3.7.2.3. Cromatografia de exclusão de íons
Na cromatografia por exclusão iônica, a escolha do agente de eluição, como a seleção do material de empacotamento, não permite muitas variações. A faixa se estende da água pura até ácidos minerais diluídos. Ao selecionar o sistema mais apropriado é também necessário considerar o sistema de detecção. Os métodos de detecção mais freqüentemente usados são detecção fotométrica e de condutividade. Com detecção fotométrica, o ácido sulfúrico ou perclórico diluídos são freqüentemente usados por causa de sua absoluta “transparência” em UV. Ao aplicar a detecção da condutividade, o ácido sulfúrico diluído é o eluente de escolha porque ele produz bons cromatogramas com um mínimo de equipamento, com ou sem supressor.
A concentração de ácido no eluente determina o grau de dissociação dos analitos e conseqüentemente também a sua retenção. Em geral, a retenção diminui enquanto a concentração ácida aumenta. A forma do pico é influenciada também pela concentração ácida. Para ácidos orgânicos, a água pura não é geralmente apropriada devido à sua elevada adsorção, embora produza excelentes resultados para o carbonato e borato.

Práticas em Cromatografia de Íons 43
4. Seção prática A seção prática desta monografia apresenta experimentos que fornecem uma introdução detalhada da cromatografia de íons. Inicialmente, os experimentos abrangem os conceitos da cromatografia de íons (capítulo 4.2); a segunda parte (capítulo 4.3) continua com a determinação de ânions, e a terceira e última parte (capítulo 4.4) trata da determinação de cátions orgânicos e inorgânicos.
Em princípio, os experimentos podem ser realizados em qualquer cromatógrafo de íons. Entretanto, o instrumento deve satisfazer os seguintes requisitos:
Bomba de duplo pistão e baixa pulsação, se possível, operando sem suprimento externo de nitrogênio ou hélio; Controle de fluxo de acordo com os requisitos da coluna; Válvula elétrica de injeção; Possibilidade de trocar vários “loops” de amostra e colunas de pré-concentração; Supressão eletrônica para cátions e ânions; Supressão química para ânions; Detector de condutividade estabilizado termicamente, se possível, melhor que ±0,01°C; Gabinete do instrumento isolado térmica e eletronicamente; Operação de ânions e cátions; Controle por PC é recomendado;
Todos os sistemas modulares ou compactos de CI da Metrohm satisfazem os requisitos acima. Entretanto, o CI básico 792 é especialmente projetado para treinamento e ensino, sendo assim, recomendado para realizar os experimentos descritos abaixo.
Figura 23. CI básico 792 da Metrohm: desenvolvido especialmente para treinamento e ensino.
4.1. Informações sobre a parte prática
Crescimento bacteriano
A fim de prevenir o crescimento bacteriano, o eluente, as soluções de lavagem e as de regeneração devem ser sempre preparados no momento do uso, e não devem ser usados por um período de tempo longo. Para impedir o crescimento de bactérias ou algas, pode-se adicionar 5% de metanol ou acetona ao eluente. Isto não pode ser feito se forem usados supressores de membrana, pois estes serão destruídos pelos solventes orgânicos. Entretanto, o módulo supressor da Metrohm «MSM» é 100% resistente a solventes.
Análise de cátions
Em cada análise realizada, a amostra pode ser acidificada (pH 2,5 a 3,5) com ácido nítrico (aproximadamente 100 μL de HNO3 a 2 mol/L para 100 mL de amostra). Caso contrário, a análise de cátions bivalentes poderá não apresentar resultados reprodutíveis.

44 Monografia Metrohm
Grau químico
Todos os produtos químicos usados devem ter pelo menos grau p.a. (grau analítico) ou puríssimo (extra-puro). Quaisquer padrões usados devem ser especialmente adequados para a cromatografia de íons; por exemplo, sais sódicos devem ser dissolvidos em água, não em ácido.
Fontes de contaminação
Todas as soluções, as amostras, as soluções de regeneração, a água e os eluentes usados devem estar livres de partículas, pois estas poderão bloquear as colunas de separação ou capilares no decorrer do tempo (aumento da pressão da coluna). Isto é especialmente importante no momento da preparação de eluentes, visto que estes fluem pela coluna continuamente (500 a 1000 mL por dia de operação, enquanto a amostra injetada é de aproximadamente 0,5 mL).
Degaseificação de eluentes
A fim de evitar a formação de bolhas, é recomendado que a água usada para a preparação de eluente seja degaseificada antes de serem adicionados os reagentes. Isto pode ser feito aplicando-se vácuo proveniente de trompa d’água ou bomba de vácuo por 10 minutos ou utilizando-se um banho ultrassônico.
Proteção ambiental
Uma das grandes vantagens da cromatografia de íons é que, geralmente, meios aquosos são usados. Por isso, os produtos químicos usados em cromatografia de íons são praticamente atóxicos e não poluem o ambiente. Entretanto, se ácidos, bases, solventes orgânicos ou metais pesados forem usados, deve-se tomar o cuidado de descartá-los apropriadamente após o uso.
Desligando o instrumento
Se o cromatógrafo de íons não for usado por um longo período de tempo (>1 semana), então, a coluna de separação deve ser removida e a solução metanol/água (1:4) deve ser bombeada através do cromatógrafo. Deve-se tomar o cuidado para que todos os três canais de supressores sejam lavados.
Informações de segurança
Óculos de segurança, roupa apropriada e, se necessário, luvas protetoras devem ser usadas na realização de qualquer experimento. Observe as informações de segurança especificadas para os produtos químicos.
Selecionando uma coluna
A maioria dos experimentos descritos é realizada com colunas de baixo custo – “Metrosep Anion Dual 1” para ânions e “Metrosep C2” para cátions. Estas colunas promovem a separação adequada para todos os experimentos descritos. Naturalmente, o conjunto de produtos Metrosep inclui colunas de separação que têm uma eficiência consideravelmente melhor, porém são de custo mais altos.

Práticas em Cromatografia de Íons 45
Armazenamento das colunas de separação
Coluna para ânions “Metrosep A Supp 4” (6.1006.430)
A coluna é armazenada no eluente.
Figura 24. Coluna para ânions “Metrosep A Supp 4” (6.1006.430)
Coluna para ânions “Metrosep A Supp 5” (6.1006.530)
A coluna é armazenada no eluente.
Coluna para cátions “Metrosep C2” (6.1010.210)
A coluna é armazenada no eluente num refrigerador (4 °C). É essencial que a coluna não permaneça seca.
Figura 25. Coluna de cátion “Metrosep C2” (6.1010.210).
Coluna para cátions “Nucleosil 5SA” (6.1007.000)
Para armazenagem a curto-prazo (dias), a coluna é mantida em eluente, e para longo-prazo (semanas), em metanol/água (1:4).
Coluna “Metrosep Organic Acids” (6.1005.200)
Para armazenagem a curto-prazo (dias), a coluna é mantida em eluente, para longo-prazo (semanas), em água deionizada.

46 Monografia Metrohm
Colunas de separação de outros fabricantes
Favor seguir as instruções do fabricante.
Qualidade da água
Em cromatógrafos de íons são usados, principalmente, meios aquosos. É por isso que a qualidade da água é crucial para bons resultados cromatográficos. Se a qualidade da água for insatisfatória, então, certamente os resultados serão insatisfatórios. Além disso, há o perigo de danificar o aparelho e a coluna de separação. Portanto a água deionizada usada deve atender a classificação Tipo I da ASTM, ou seja, ter uma resistência maior que 18 M e ser livre de partículas. É recomendado que a água seja filtrada em filtro de 0,45 μm.

Práticas em Cromatografia de Íons 47
4.2. Experimentos abrangendo a teoria da cromatografia de íons
4.2.1. Experimento 1 – Cromatografia de íons com e sem supressão
A condutividade equivalente é sempre obtida pela soma das condutividades equivalentes de todos os ânions – e cátions + na solução:
= + + –
Em princípio, a condutividade aumenta com o aumento da concentração dos eletrólitos ou íons. Uma relação linear entre concentração e condutividade só existe para soluções diluídas, visto que depende da concentração (lei de Kohlrausch). Os valores encontrados nas tabelas (vide seção “Detecção de Condutividade”) aplicam-se para - condutividade equivalente em soluções de concentrações infinitas.
A medida da condutividade equivalente depende, em grande parte, da temperatura ( 2% / °C). Em particular, para medidas sem supressão química, ou seja, principalmente contra uma alta condutividade de fundo, erros causados por uma mudança na temperatura são extremamente aparentes. É por isso que oscilações na temperatura dentro do detector não devem ser maiores que 0,01 °C.
Em cromatografia de íons sem supressão química, na qual o sinal de fundo é suprimido eletronicamente, o eluente deve ter uma condutividade tão baixa quanto possível. Os sais de ácidos fracos tais como ácido ftálico, ácido salicílico e ácido benzóico são freqüentemente usados aqui. Um valor medido sem supressão química depende da diferença das condutividades equivalentes do íon da amostra e dos íons do eluente.
~ ( P – E)
Picos negativos ocorrem sempre que a condutividade do íon da amostra for menor do que a dos íons eluente. Um exemplo disto é o ânion fosfato. Em pH = 5, ele está presente principalmente como H2PO4
–. Como a condutividade equivalente do H2PO4– de 33 S*cm2/mol é menor do que a
condutividade equivalente do ftalato com 38 S*cm2/mol, um pequeno pico negativo é obtido. Isto pode ser corrigido selecionando-se um pH maior, no qual o fosfato estará presente como HPO4
2– com condutividade equivalente de 57 S*cm2/mol.
Cromatografia de íons com supressão química significa que a condutividade de fundo é reduzida quimicamente. Um eluente com uma alta condutividade é convertido, em uma reação pós-coluna – reação de supressão –, em um eluente com uma baixa condutividade.
Em análise de ânions, isto é realizado com o uso de sais de ácidos fracos, ou seja, HCO3–
, CO32– e
BO33–, no eluente. Todos os cátions no eluente e na amostra são trocados por H+ no supressor.
Ácidos fracamente dissociados são formados a partir do eluente de acordo com a seguinte reação:
Na+ + HCO3– H2CO3
O ácido carbônico resultante está presente como CO2 + H2O. Em conseqüência, a condutividade residual é muito baixa. Como os contra-íons dos ânions a serem determinados são também trocados por H+ no supressor, a seguinte equação pode ser formulada:
Na+ + Cl– H+ + Cl–
Ao invés de Na+ e Cl–, que estavam originalmente presentes na amostra, a condutividade equivalente consideravelmente maior de H+ e Cl– é agora medida também contra uma baixa condutividade de fundo. Teoricamente, o sinal com supressão química esperado deverá ser dez vezes maior do que sem supressão química. Entretanto, na prática, a sensibilidade aumenta de 2 a 4 vezes.
Em contraste à função de calibração linear obtida quando operando sem supressão química, uma função de calibração quadrática será obtida quando a supressão química for usada; isto significa que uma calibração multi-pontos é necessária para o cálculo. A região de operação linear é consideravelmente menor (aproximadamente 1/20 a 1/50) do que para CI sem supressão química.
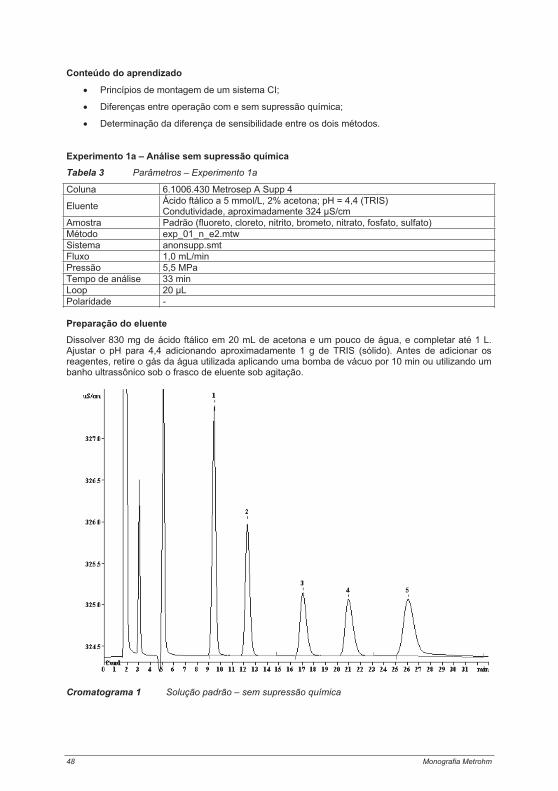
48 Monografia Metrohm
Conteúdo do aprendizado
Princípios de montagem de um sistema CI;
Diferenças entre operação com e sem supressão química;
Determinação da diferença de sensibilidade entre os dois métodos.
Experimento 1a – Análise sem supressão química
Tabela 3 Parâmetros – Experimento 1a
Coluna 6.1006.430 Metrosep A Supp 4
Eluente Ácido ftálico a 5 mmol/L, 2% acetona; pH = 4,4 (TRIS) Condutividade, aproximadamente 324 μS/cm
Amostra Padrão (fluoreto, cloreto, nitrito, brometo, nitrato, fosfato, sulfato) Método exp_01_n_e2.mtw Sistema anonsupp.smt Fluxo 1,0 mL/min Pressão 5,5 MPa Tempo de análise 33 min Loop 20 μL Polaridade -
Preparação do eluente
Dissolver 830 mg de ácido ftálico em 20 mL de acetona e um pouco de água, e completar até 1 L. Ajustar o pH para 4,4 adicionando aproximadamente 1 g de TRIS (sólido). Antes de adicionar os reagentes, retire o gás da água utilizada aplicando uma bomba de vácuo por 10 min ou utilizando um banho ultrassônico sob o frasco de eluente sob agitação.
Cromatograma 1 Solução padrão – sem supressão química

Práticas em Cromatografia de Íons 49
Tabela 4 Componentes – Experimento 1a
Experimento 1b – Análise com supressão química
Tabela 5 Parâmetros – Experimento 1b
Coluna 6.1006.430 Metrosep A Supp 4
Eluente Na2CO3 a 1,8 mmol/L / NaHCO3 a 1,7 mmol/L + acetona 2% Condutividade após supressão química aprox. 14 μS/cm
Amostra Padrão (fluoreto, cloreto, nitrito, brometo, nitrato, fosfato, sulfato) Método exp_01_s_e2.mtw Sistema asupp.smt Fluxo 1,0 mL/min Pressão 3 MPa Tempo de análise 18 min Loop 20 μL
Supressor Agentes de regeneração: H2SO4 a 50 mmol/L e água ultrapura alimentados continuamente com mudança automática dos canais de supressão, regeneração e lavagem
Polaridade +
Preparação do eluente
Dissolver 191 mg de carbonato de sódio (anidro) e 143 mg de hidrogenocarbonato de sódio em 980 mL de água ultrapura e, então, adicionar 20 mL de acetona.
Cromatograma2 Solução padrão – com supressão química
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 21 Cloreto 9,45 25 4834 61,48 32 Nitrito 12,30 25 4709 43,67 43 Brometo 17,06 25 4272 29,83 54 Nitrato 20,96 25 4174 33,44 65 Sulfato 26,06 25 2636 52,62

50 Monografia Metrohm
Tabela 6 Componentes – Experimento 1b
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Fluoreto 4,05 5 5605 66,28 2 Cloreto 5,65 5 8573 43,59 3 Nitrito 6,56 5 8013 27,43 4 Brometo 7,90 5 7963 17,70 5 Nitrato 8,83 5 7653 22,26 6 Pico do sistema 10,50 7 Fosfato 13,50 5 6477 11,66 8 Sulfato 15,62 5 7922 29,28

Práticas em Cromatografia de Íons 51
4.2.2. Experimento 2 – Capacidade das colunas de separação
Picos cortados, picos em forma de “barbatana de tubarão”, tempos de retenção alterados para os mesmos íons, picos com cauda ou deformação frontal – todos estes podem ter uma causa em comum: sobrecarga da coluna.
Cada coluna tem um número finito de sítios de troca iônica. Antes da injeção da amostra, eles estão completamente ocupados pelos íons do eluente. Quando a amostra é injetada, inicia-se a troca iônica: íons do eluente são trocados por íons da amostra e, então, íons da amostra são trocados por íons do eluente. Como as espécies de íons diferem em suas constantes de ligação, eles eluem da coluna em velocidades diferentes. O resultado obtido é a separação das substâncias da mistura e, em seguida, o cromatograma.
Este processo só funciona perfeitamente se o número dos sítios de troca for substancialmente maior do que o número dos pontos de ligação requeridos pela amostra. Por exemplo, uma coluna com capacidade de troca de 1 meq (miliequivalente) pode ligar no máximo 1 mmol de íons monovalentes.
Um segundo efeito com uma influência negativa na separação é o fato de que, em princípio, cada íon pode agir como um íon eluente. Se a coluna estiver sobrecarregada, então muitos íons eluentes são substituídos pelos íons da amostra. Isto resulta em alterações incalculáveis no equilíbrio da coluna e a separação se torna ruim.
A capacidade de uma coluna pode ser determinada pela sua ocupação completa com íons cloreto. Após a etapa de lavagem com água deionizada, os íons cloreto são eluídos com eluente carbonato e então quantificados por cromatografia de íons ou titulação argentométrica.
No experimento seguinte, a concentração de cloreto é elevada até que a coluna esteja sobrecarregada. Alguns efeitos associados com a sobrecarga da coluna são descritos abaixo:
os tempos de retenção dos íons seguintes tornam-se mais curtos; o pico do íon dominante é cortado; o pico do íon dominante forma cauda; o número de pratos teóricos (PT) torna-se menor; a proporção área/altura torna-se pior (em área constante, a altura do pico diminui); a simetria do pico torna-se deficiente.
A simetria do pico também se torna deficiente com o bloqueio dos discos de vedação “frits”, formação de elevados volumes mortos como resultado dos efeitos de absorção no material da coluna.
Conteúdo do aprendizado
Esclarecimentos sobre os seguintes parâmetros cromatográficos: tempo de retenção, resolução, área, número de pratos, simetria e proporção área/altura;
Influência de um componente dominante principal sobre os componentes com uma concentração muito menor: alterações nos parâmetros acima mencionados.

52 Monografia Metrohm
Tabela 7 Parâmetros – Experimento 2
Coluna 6.1006.430 Metrosep A Supp 4
Eluente Na2CO3 a 1,8 mmol/L / NaHCO3 a 1,7 mmol/L / + 2% acetona Condutividade após supressão química aprox. 14 μS/cm
Amostra Padrão (fluoreto, cloreto, nitrito, brometo, nitrato, fosfato, sulfato) + NaCl (aprox. 5%; m:v)
Método exp_02_s_e2.mtw Sistema asupp.smt Fluxo 1,0 mL/min Pressão 5,5 MPa Tempo de análise 20 min
Loop 20 μL
Supressor Agentes de regeneração H2SO4 a 50 mmol/L e água ultrapura alimentados continuamente com mudança automática dos canais de supressão, regeneração e lavagem
Polaridade +
Preparação do eluente
Dissolver 191 mg de carbonato de sódio (anidro) e 143 mg de hidrogenocarbonato de sódio em 980 mL de água ultrapura e, então, adicionar 20 mL de acetona.
Cromatograma 3 Solução padrão com uma concentração muito elevada de cloreto.

Práticas em Cromatografia de Íons 53
Cromatograma 4 Solução padrão com uma concentração de cloreto muito alta; parte ampliada do cromatograma 3
Observações
O cloreto de sódio sólido foi pesado e dissolvido na solução-padrão dos outros ânions. Os tempos de retenção dos picos, bem como os valores para o número de pratos e a área podem variar dependendo da quantidade do NaCl pesada. O enorme pico do cloreto interfere na avaliação dos picos seguintes. A identificação correta de picos individuais apenas é possível através da adição de padrão na amostra dos respectivos ânions, visando o aumento da altura dos picos.

54 Monografia Metrohm
4.2.3. Experimento 3 – Seletividade das colunas de separação
Se a força iônica do eluente for plotada contra o logaritmo dos tempos de retenção dos íons mono e divalentes, então uma linha reta é obtida. A inclinação desta linha é maior para os íons divalentes do que para os monovalentes. O aumento na força de eluição do eluente, portanto, tem uma maior influência no tempo de retenção dos íons divalentes do que no tempo de retenção dos íons monovalentes.
A influência pode ser observada no íon sulfato. Com o aumento da concentração do eluente, a eluição do sulfato é acelerada mais do que a dos íons monovalentes.
Em geral, os íons bivalentes são eluentes mais efetivos do que os monovalentes, uma vez que podem formar duas ligações com a fase estacionária e, conseqüentemente, realizar interações mais fortes com a mesma.
Em uma mesma concentração molar, o hidróxido de sódio tem um pH maior do que o carbonato/hidrogenocarbonato. O poder de eluição do íon OH– é menor quando comparado com a do HCO3
–, uma vez que os íons hidróxidos não interagem tão fortemente com a fase estacionária.
O tempo de retenção do íon fosfato depende fortemente do valor do pH. Em valores altos, o equilíbrio é deslocado de HPO4
2– para PO43–. Se for adicionado hidróxido de sódio a um eluente carbonato de
sódio, isto resulta em um aumento no tempo de retenção do fosfato, enquanto os tempos de retenção de todos os outros íons são diminuídos.
Conteúdo do aprendizado
Comparação dos eluentes com ânions mono e divalentes; Comparação de eluentes hidróxido de sódio e carbonato/hidrogenocarbonato; Influência do pH do eluente na retenção do fosfato.
Tabela 8 Parâmetros – Experimentos 3a a 3d
Coluna 6.1006.430 Metrosep A Supp 4 Eluente a) Na2CO3 a 0,5 mmol/L / NaHCO3 a 11 mmol/L
b) Na2CO3 a 1,8 mmol/L / NaHCO3 a 1,7 mmol/L c) Na2CO3 a 1 mmol/L / NaHCO3 a 4 mmol/L d) Na2CO3 a 4 mmol/L / NaOH a 1 mmol/L e) Na2CO3 a 1 mmol/L / NaOH a 4 mmol/L
Amostra Padrão (fluoreto, cloreto, nitrito, brometo, nitrato, fosfato, sulfato) Método exp_03_s_e2.mtw Sistema asupp.smt Fluxo 1,0 mL/min Pressão 5,5 MPa Tempo de análise a) 18 min
b) 16 min c) 20 min d) 10 min e) 25 min
Loop 20 μL Supressor Agentes de regeneração: H2SO4 a 50 mmol/L e água ultrapura alimentados
continuamente com mudança automática dos canais de supressão, regeneração e lavagem
Polaridade +

Práticas em Cromatografia de Íons 55
Experimento 3a – Eluente: Na2CO3 a 0,5 mmol/L / NaHCO3 a 11 mmol/L
Preparação do eluente
Dissolver 53,0 mg de carbonato de sódio (anidro) e 924,4 mg de hidrogenocarbonato de sódio em 1 L de água ultrapura.
Cromatograma 5 Solução padrão – Eluente: Na2CO3 a 0,5 mmol/L / NaHCO3 a 11 mmol/L.
Tabela 9 Componentes – Experimento 3ª
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Fluoreto 4,12 10 5936 128,6 2 Cloreto 6,07 10 7548 86,9 3 Brometo 9,07 10 6711 36,3 4 Nitrato 10,41 10 6318 44,8 5 Fosfato 12,19 10 5104 23,1 6 Sulfato 14,71 10 5061 61,4

56 Monografia Metrohm
Experimento 3b – Eluente: Na2CO3 a 1,8 mmol/L / NaHCO3 a 1,7 mmol/L
Preparação do eluente
Dissolver 191 mg de carbonato de sódio (anidro) e 143 mg de hidrogenocarbonato de sódio em 1 L de água ultrapura.
Cromatograma 6 Solução padrão – Eluente: Na2CO3 a 1,8 mmol/L / NaHCO3 a 1,7 mmol/L.
Tabela 10 Componentes – Experimento 3b
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Fluoreto 3,51 10 5109 134,7 2 Cloreto 4,88 10 6664 82,5 3 Brometo 6,89 10 6083 32,9 4 Fosfato 7,71 10 5500 40,35 Nitrato 10,81 10 4651 21,26 Sulfato 12,41 10 5837 56,6
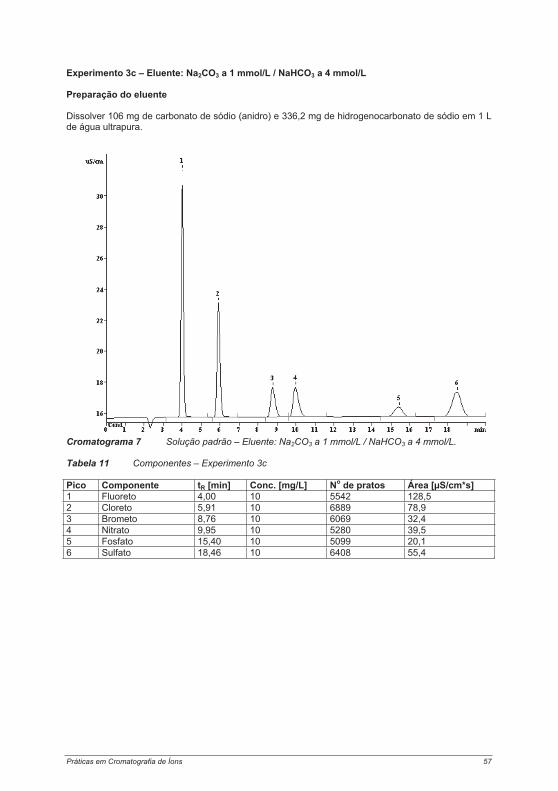
Práticas em Cromatografia de Íons 57
Experimento 3c – Eluente: Na2CO3 a 1 mmol/L / NaHCO3 a 4 mmol/L
Preparação do eluente
Dissolver 106 mg de carbonato de sódio (anidro) e 336,2 mg de hidrogenocarbonato de sódio em 1 L de água ultrapura.
Cromatograma 7 Solução padrão – Eluente: Na2CO3 a 1 mmol/L / NaHCO3 a 4 mmol/L.
Tabela 11 Componentes – Experimento 3c
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Fluoreto 4,00 10 5542 128,5 2 Cloreto 5,91 10 6889 78,9 3 Brometo 8,76 10 6069 32,4 4 Nitrato 9,95 10 5280 39,5 5 Fosfato 15,40 10 5099 20,16 Sulfato 18,46 10 6408 55,4

58 Monografia Metrohm
Experimento 3d – Eluente: Na2CO3 a 4 mmol/L / NaOH a 1 mmol/L
Preparação do Eluente
Dissolver 424 mg de carbonato de sódio (anidro) e 100 μL NaOH 30% em 1 L de água ultrapura.
Cromatograma 8 Solução padrão – Eluente: Na2CO3 a 4 mmol/L / NaOH a 1 mmol/L.
Tabela 12 Componentes – Experimento 3d
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Fluoreto 3,67 10 4662 123,5 2 Cloreto 4,65 10 6672 79,5 3 Brometo 6,04 10 7201 32,3 4 Nitrato 6,61 10 6902 41,4 5 Fosfato + Sulfato 7,94 10 + 10 4889 81,8

Práticas em Cromatografia de Íons 59
Experimento 3e – Eluente: Na2CO3 a 1 mmol/L / NaOH a 4 mmol/L
Preparação do Eluente
Dissolver 106 mg de carbonato de sódio (anidro) e 400 μL NaOH 30% em 1 L de água ultrapura.
Cromatograma 9 Solução padrão – Eluente: Na2CO3 a 1 mmol/L / NaOH a 4 mmol/L.
Tabela 13 Componentes – Experimento 3e
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Fluoreto 4,36 10 4654 127,8 2 Cloreto 5,6 10 7884 80,7 3 Brometo 7,31 10 8307 33,1 4 Nitrato 7,98 10 8204 42,8 5 Sulfato 14,29 10 7676 55,4 6 Fosfato 22,84 10 6477 21,8

60 Monografia Metrohm
4.2.4. Experimento 4 – Limites de calibração, detecção e determinação na cromatografia de íons
Os parâmetros importantes para métodos de determinação analítica são a faixa linear, limite de detecção e limite de determinação. Os métodos matemáticos para eles baseiam-se em métodos padrões, por exemplo, em DIN 32645.
Se os cromatogramas são registrados com um detector de condutividade, a área do pico é, geralmente, usada para avaliação. A área do pico é proporcional à quantidade de substância. Se a área do pico for plotada contra a concentração, então a função calibração é obtida. Ela é linear para medidas sem supressão química. Como uma primeira aproximação, ela é uma função quadrática para medidas com supressão química. Programas de avaliação calculam as funções de calibração automaticamente.
A avaliação utilizando a altura do pico é usada, preferencialmente, para picos com significativa cauda ou para picos insuficientemente separados, com grandes diferenças na relação área/altura, visto que nestes casos, a avaliação baseada na área prporciona grandes erros.
O limite de detecção é a concentração mínima de um analito que pode ser detectada com uma conhecida certeza estatística. A menor concentração teórica, que pode ser distinta de um valor do “branco”, deve ser calculada.
Há dois métodos para calcular o limite de detecção:
Método do valor branco:
A amostra “branco” deve ser uma amostra que não contém o íon a ser determinado, mas que produz um sinal na mesma posição que o íon da amostra. Medidas repetidas de uma amostra “branco” geram para uma concentração «0» (valor x) valores medidos (valores y) cujo modo (freqüência média máxima) é conhecido como o valor branco. Utilizando-se uma curva de calibração, o valor y máximo é associado com o valor de concentração no eixo x; isto é o limite de detecção.
Método da curva de calibração:
Este método é usado quando não é possível determinar o valor do branco, devido ao fato de que o íon analisado não pode ser detectado na amostra. No método da curva de calibração, múltiplas medidas são realizadas em diferentes concentrações do íon. Uma faixa de confiança é obtida a partir do desvio padrão. Desta forma, a concentração «0» corresponde a um intervalo y particular. A função calibração é usada para associar o intervalo y a um intervalo de concentração cujo valor máximo é o limite de detecção.
A relação sinal/ruído é freqüentemente usada para determinar o limite de detecção. Por exemplo, o limite de detecção é definido como sendo a concentração do analito na qual o sinal de medida é 3, 5 ou 7 vezes o ruído da linha de base.
O limite de quantificação é considerado como o menor valor que possa ser quantificado com uma determinada confiança. Este valor deve ser sempre maior que o limite de detecção e, como estimativa, pode ser dito que o limite de quantificação é maior por um fator de três.
Conteúdo do aprendizado
O que significa calibração?
Comparação de uma calibração de ponto único e de pontos múltiplos – estimativa de erros;
Determinação de ruído do sistema;
Estimativa do limite de detecção.

Práticas em Cromatografia de Íons 61
Experimento 4a – Determinação de ânions com supressão química
Tabela 14 Parâmetros – Experimento 4a
Coluna 6.1006.430 Metrosep A Supp 4
Eluente Na2CO3 a 1,8 mmol/L NaHCO3 a 1,7 mmol/L / + 2% acetona Condutividade após supressão química aprox. 14 μS/cm
Amostra Padrão (fluoreto, cloreto, nitrito, brometo, nitrato, fosfato, sulfato) Método exp_04_s_e2.mtw Sistema asupp.smt Fluxo 1,0 mL/min Pressão 5,5 MPa Tempo de análise 18 min Loop 20 μL
Supressor Agentes de regeneração: H2SO4 a 50 mmol/L e água ultrapura alimentados continuamente com mudança automática dos canais de supressão, regeneração e lavagem
Polaridade +
Preparação do eluente
Dissolver 191 mg de carbonato de sódio (anidro) e 143 mg de hidrogenocarbonato de sódio em 980 mL de água ultrapura e, então, adicionar 20 mL de acetona.
Cromatograma 10 Sobreposição de cromatogramas – Soluções padrão com concentrações diferentes – com supressão química

62 Monografia Metrohm
Tabela 15 Componentes – Experimento 4a
Conc. [mg/L] Pico Componente tR [min] Nível 1 Nível 2 Nível 3 Nível 4 1 Fluoreto 4,04 1 5 25 50 2 Cloreto 5,66 1 5 25 50 3 Nitrito 6,58 1 5 25 50 4 Brometo 7,95 1 5 25 50 5 Nitrato 8,89 1 5 25 50 6 Fosfato 13,49 1 5 25 50 7 Sulfato 15,62 1 5 25 50
Experimento 4b – Determinação de ânions sem supressão química
Tabela 16 Parâmetros – Experimento 4b
Coluna 6.1006.430 Metrosep A Supp 4
Eluente Ácido ftálico a 5 mmol/L, 2% acetona; pH = 4,4 (TRIS) Condutividade aprox. 324 μS/cm
Amostra Padrão (fluoreto, cloreto, nitrito, brometo, nitrato, fosfato, sulfato) Método exp_04_n_e2.mtw Sistema anonsupp.smt Fluxo 1,0 mL/min Pressão 5,5 MPa Tempo de análise 33 min Loop 20 μL Polaridade –
Preparação do eluente
Dissolver 830 mg de ácido ftálico em 20 mL de acetona e um pouco de água e completar para 1 L. Ajustar o pH para 4,4 adicionando aprox. 0,6 g de TRIS (sólido).
Cromatograma 11 Sobreposição de cromatogramas – Soluções padrão com concentrações diferentes – sem supressão química

Práticas em Cromatografia de Íons 63
Tabela 17 Componentes – Experimento 4b
Conc. [mg/L] Pico Componente tR [min] Nível 1 Nível 2 Nível 3 Nível 4 1 Cloreto 9,45 1 5 25 50 2 Nitrito 12,30 1 5 25 50 3 Brometo 17,06 1 5 25 50 4 Nitrato 20,96 1 5 25 50 5 Sulfato 26,06 1 5 25 50

64 Monografia Metrohm
4.2.5. Experimento 5 – Alteração da seletividade com o auxílio de éteres coroa (18 Crown-6)
Os tempos de retenção de cátions podem ser alterados pela adição de agentes complexantes aos eluentes. O agente complexante age como um ligante, o cátion analisado é incluso como o íon metálico central. Quanto mais seletivo for um ligante em relação ao íon metálico central, mais forte a influência no tempo de retenção. Em um caso ideal, os tempos de retenção dos outros cátions só serão alterados levemente.
Os agentes complexantes são usados para obter uma melhor separação de íons de metais alcalinos. A adição de éter coroa 18 Crown-6 ao eluente proporciona uma melhor separação de Na+, NH4
+ e K+.Por exemplo, o éter coroa pode ser adicionado ao eluente para melhorar a separação entre Na+ e NH4
+ quando traços de NH4+ têm que ser determinados em águas naturais. O aumento no tempo de
retenção do K+ é muito acentuado. Isto pode ser explicado pela formação do complexo K+ e o éter 18-Crown-6. O K+ se ajusta perfeitamente no “espaço central” do éter. Ele é complexado através de pares de elétrons dos átomos de oxigênio. Após a complexação, uma molécula consideravelmente maior, com a mesma carga, é separada. Isto significa que o tempo de retenção do potássio é aumentado como resultado de impedimento estérico.
Figura 26 Complexo Potássio-18 Crown-6
O nome 18 Crown-6 indica que um sistema de anel consiste de 18 átomos dos quais 6 são átomos de oxigênio. Os éteres coroa não só têm um papel importante na cromatografia de íons, como também são usados como fase de seleção de íons em eletrodos de potássio.
Conteúdo do aprendizado
Efeito de um agente complexante muito seletivo nos tempos de retenção
Esclarecimentos sobre o efeito em comparação com o Experimento 6
Tabela 18 Parâmetros – Experimentos 5a e 5b
Coluna 6.1010.210 Metrosep C2 (4 x 100 mm)
Eluente
a) ácido tartárico a 4 mmol/L / ácido dipicolínico a 0,75 mmol/L b) ácido tartárico a 4 mmol/L / ácido dipicolínico a 0,75 mmol/L + éter coroa a 0,75 mmol/L Condutividade aprox. 470 μS/cm
Amostra Padrão (lítio, sódio, amônia, potássio, cálcio, magnésio + HNO3 a 2 mmol/L) Método exp_05_c.mtw Sistema cátion.smt Fluxo 1 mL/min Pressão 8 MPa
Tempo de análise a) 15 min b) 20 min
Loop 20 μL Polaridade –

Práticas em Cromatografia de Íons 65
Experimento 5a – Eluente sem éter coroa
Preparação do Eluente
Dissolver, sob aquecimento, 600 mg de ácido tartárico e 125 mg de ácido dipicolínico em 100 mL de água ultrapura e, então, completar para 1 L com água ultrapura.
Cromatograma 12 Solução padrão – Eluente sem éter coroa.
Tabela 19 Componentes – Experimento 5a
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Lítio 2,7 1 2584 22 2 Sódio 3,5 5 3203 30 3 Amônio 3,9 5 2919 33 4 Potássio 5,3 10 2675 285 Cálcio 9,7 10 3168 26 6 Magnésio 12,5 10 2298 53

66 Monografia Metrohm
Experimento 5b – Eluente com éter coroa
Preparação do eluente
Dissolver, sob aquecimento, 600 mg ácido tartárico e 125 mg de ácido dipicolínico em 100 mL de água ultrapura, adicionar 200 mg de éter coroa e completar para 1 L com água ultrapura.
Cromatograma 13 Solução padrão – Eluente com éter coroa.
Tabela 20 Componentes – Experimento 5b
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Lítio 2,7 1 2653 22 2 Sódio 3,8 5 3108 30 3 Amônia 4,9 5 2207 32 4 Cálcio 10,7 10 2914 27 5 Magnésio 12,2 10 2378 53 6 Potássio 14,9 10 1889 26

Práticas em Cromatografia de Íons 67
4.2.6. Experimento 6 – Alteração da seletividade pela utilização de agentes complexantes
Na análise dos íons magnésio, sódio e potássio na presença dos íons zinco e cálcio, a habilidade destes em formar complexos com o ácido dipicolínico (DPA, ácido 2,6-piridinodicarboxílico) é utilizada.
Me2+
HH
OO
O OCC N
Figura 27 Complexo Me2+ ácido dipicolínico
A constante que resulta da formação de complexo
Me2+ + (DPA) [Me(DPA)]2+
é diferente para cada metal.
Dependendo do pH, os seguintes complexos podem ser formados (aumento da remoção de H+ com o aumento de pH):
condições ácidas condições fracamente ácidas condições alcalinas
Isto significa que, dependendo do pH, o complexo formado tem uma dupla carga positiva ou uma única carga positiva ou é neutro.
O critério primário de separação em uma coluna trocadora de cátion é a carga positiva dos íons a serem separados. Os complexos neutros não são retardados enquanto os complexos com três cargas positivas são ligados muito fortemente. Como resultado da constante de formação de complexo e do pH do eluente, o complexo tem uma carga média devido ao equilíbrio. Esta carga de equilíbrio determina o tempo de retenção. É por isso que íons metálicos bivalentes podem ser acelerados pela adição de ácido dipicolínico em uma determinada faixa de pH.
Não há influência nos tempos de retenção dos íons metálicos monovalentes, os quais não formam um complexo com o ácido dipicolínico.
Conteúdo do aprendizado
Influência da constante de formação de complexo no tempo de retenção – comparação de zinco e cálcio;
Comportamento de outros íons de metais de transição;
Influência do valor do pH na carga total do complexo;
Explicação sobre os efeitos em comparação ao Experimento 5.
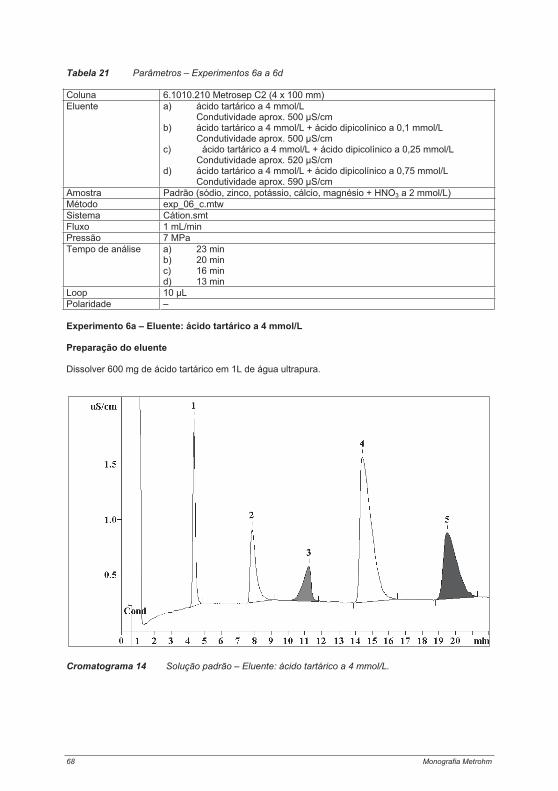
68 Monografia Metrohm
Tabela 21 Parâmetros – Experimentos 6a a 6d
Coluna 6.1010.210 Metrosep C2 (4 x 100 mm) Eluente a) ácido tartárico a 4 mmol/L
Condutividade aprox. 500 μS/cm b) ácido tartárico a 4 mmol/L + ácido dipicolínico a 0,1 mmol/L Condutividade aprox. 500 μS/cm c) ácido tartárico a 4 mmol/L + ácido dipicolínico a 0,25 mmol/L Condutividade aprox. 520 μS/cm d) ácido tartárico a 4 mmol/L + ácido dipicolínico a 0,75 mmol/L Condutividade aprox. 590 μS/cm
Amostra Padrão (sódio, zinco, potássio, cálcio, magnésio + HNO3 a 2 mmol/L) Método exp_06_c.mtw Sistema Cátion.smt Fluxo 1 mL/min Pressão 7 MPa Tempo de análise a) 23 min
b) 20 min c) 16 min d) 13 min
Loop 10 μL Polaridade –
Experimento 6a – Eluente: ácido tartárico a 4 mmol/L
Preparação do eluente
Dissolver 600 mg de ácido tartárico em 1L de água ultrapura.
Cromatograma 14 Solução padrão – Eluente: ácido tartárico a 4 mmol/L.

Práticas em Cromatografia de Íons 69
Tabela 21 Componentes – Experimento 6a
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Sódio 4,3 5 4450 17 2 Potássio 7,8 10 2330 16 3 Zinco 11,2 5 2330 114 Magnésio 14,4 10 2010 63 5 Cálcio 19,5 10 2640 34
Experimento 6b – Eluente: ácido tartárico a 4 mmol/L + ácido dipicolínico a 0,1 mmol/L
Preparação do eluente
Dissolver, sob aquecimento, 600 mg de ácido tartárico e 17 mg de ácido dipicolínico em 100 mL de água ultrapura e completar para 1L.
Cromatograma 15 Solução padrão – Eluente: ácido tartárico a 4 mmol/L + ácido dipicolínico a 0,1 mmol/L.
Tabela 23. Componentes – Experimento 6b
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Zinco 1,9 5 554 6.72 Sódio 4,4 5 4390 17 3 Potássio 7,6 10 2750 16 4 Magnésio 14,8 10 2100 64 5 Cálcio 17,9 10 2720 33

70 Monografia Metrohm
Experimento 6c – Eluente: ácido tartárico a 4 mmol/L + ácido dipicolínico a 0,25 mmol/L
Preparação do eluente
Dissolver, sob aquecimento, 600 mg de ácido tartárico e 42 mg de ácido dipicolínico em 100 mL de água ultrapura e completar para 1L.
Cromatograma 16 Solução padrão – Eluente: ácido tartárico a 4 mmol/L + ácido dipicolínico a 0,25 mmol/L.
Tabela 24. Componentes – Experimento 6c
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [ S/cm*s] – Zinco 1,1 51 Sódio 4,3 5 4450 18 2 Potássio 7,8 10 2330 17 3 Cálcio + Magnésio 14,4 10 + 10 2010 98

Práticas em Cromatografia de Íons 71
Experimento 6d – Eluente: ácido tartárico a 4 mmol/L + ácido dipicolínico a 0,75 mmol/L
Preparação do eluente
Dissolver, sob aquecimento, 600 mg de ácido tartárico e 125 mg de ácido dipicolínico em 100 mL de água ultrapura e completar para 1L.
Cromatograma 17 Solução padrão – Eluente: ácido tartárico a 4 mmol/L + ácido dipicolínico a 0,75 mmol/L.
Tabela 25 Componentes – Experimento 6d
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] – Zinco 1,1 51 Sódio 3,8 5 4540 17 2 Potássio 6,4 10 2900 17 3 Cálcio 8,5 10 2630 334 Magnésio 10,5 10 2190 67
Observações:
Todas as soluções devem ser estocadas em recipientes plásticos. Para uma correta determinação de sódio, deve-se evitar qualquer contato com vidro. O pH da solução padrão e da amostra deve estar entre 2,5 e 3,5.
Após a troca do eluente, deixar o sistema em funcionamento até que a linha de base se apresente constante.
Cromatogramas 14 e 15: o zinco é complexado pelo ácido dipicolínico e elui no pico de injeção.

72 Monografia Metrohm
4.2.7. Experimento 7 – Técnica de pré-concentração
Na cromatografia de íons, a injeção da amostra é realizada com o auxílio de um “loop” de amostra integrado com a válvula de injeção. Em condições padronizadas, o volume do “loop” de amostra é de 20 μL para ânions e 10 μL para cátions. Com um sistema CI simples, “loops” de amostra deste tamanho cobrem limites de detecção de 100 μg/L ou 100 ppb sem qualquer problema.
Aumentando o “loop” de amostra, ou seja, para 100 μL, por exemplo, os limites de detecção podem ser diminuídos dez vezes, aproximadamente. Entretanto, um volume maior de amostra produz um pico de água, “volume morto”, substancialmente maior, o que afeta a avaliação dos picos que eluem no inicio do cromatograma. Além disso, o formato dos picos assimétricos torna-se pior.
Um método simples para diminuir os limites de detecção em vários níveis de magnitude é a pré-concentração da amostra. Uma coluna de pré-concentração é montada ao invés do “loop” de amostra. Um volume elevado de amostra – 5 mL no nosso exemplo – é passado através da coluna de pré-concentração. A princípio, esta coluna é preenchida com o mesmo material que é usado na própria coluna de separação. Isto assegura que todos os ânions ou cátions da amostra a ser analisada sejam completamente retidos durante muito tempo, enquanto nenhum íon do eluente atual esteja presente. Entretanto, a pré-concentração só funciona se a amostra em si não contiver íons (analitos) em altas concentrações.
A capacidade da coluna de pré-concentração é significantemente menor do que a da coluna de separação. Para eluir os íons pré-concentrados em um volume menor possível, a extração é realizada pelo eluente em contracorrente, ou seja, a direção da vazão durante a pré-concentração é oposta àquela durante a eluição.
Figura 28 Técnica de pré-concentração: à esquerda posição «alimentar», à direita posição «injetar».
A técnica de pré-concentração permite amostras com concentrações menores que g/L, ou ppb, sejam determinadas mesmo em um sistema de cromatografia simples.
Conteúdo do aprendizado
O que é preparação da amostra?
Onde é a interface entre a cromatografia de íons e a preparação da amostra?
Qual é a vantagem da pré-concentração da amostra?
Quais são os limites do método?
Qual é o efeito de carbonato/CO2 na amostra?

Práticas em Cromatografia de Íons 73
Tabela 26 Parâmetros – Experimento 7
Coluna 6.1006.430 Metrosep A Supp 4
Eluente Na2CO3 a 1,8 mmol/L / NaHCO3 a 1,7 mmol/L / + 2% acetona Condutividade após supressão química aprox. 14 μS/cm
Amostra Padrão (fluoreto, cloreto, nitrito, brometo, nitrato, fosfato, sulfato) Método exp_07_s_e2.mtw Sistema asupp.smt Fluxo 1,0 mL/min Pressão 5,5 MPa Tempo de análise 18 min Coluna de pré-concentração 6.1006.300 Metrosep Anion Volume da amostra 5 mL em coluna de pré-concentração
Supressor Agentes de regeneração H2SO4 a 50 mmol/L e água ultrapura alimentados continuamente com mudança automática dos canais de supressão, regeneração e lavagem
Polaridade +
Preparação do eluente
Dissolver 191 mg de carbonato de sódio (anidro) e 143 mg de hidrogenocarbonato de sódio em 980 mL de água ultrapura e, então, adicionar 20 mL de acetona.
Cromatograma 18 Solução padrão (1 ppb de cada ânion) para análise de água ultrapura

74 Monografia Metrohm
Tabela 27 Componentes – Experimento 7 – Padrão
Pico Componente tR [min] Conc. [μg/L] No de pratos Área [μS/cm*s] 1 Fluoreto 3.81 1 3196 0.36 2 Acetato 4.35 n.d. 3 Cloreto 5.22 1 4919 0.32 4 Nitrito 6.01 1 5545 0.18 5 Brometo 7.23 1 5307 0.12 6 Nitrato 8.02 1 5900 0.14 7 Pico do sistema 9.88 8 Fosfato 11.79 1 10497 0.10 9 Sulfato 13.28 1 7033 0.33
Cromatograma 19 Água ultrapura
Tabela 28 Componentes – Experimento 7- Água Ultrapura
Pico Componente tR [min] Conc. [μg/L] No de pratos Área [μS/cm*s] 1 Acetato 4.37 n.d. 4945 0.139 2 Pico do sistema 9.89 3 Sulfato 13.33 n.d. 8154 0.231
Observações:
Lavar com muito cuidado e por várias vezes todos os recipientes, seringas e o sistema; utilize recipientes plásticos.

Práticas em Cromatografia de Íons 75
4.3. Experimentos para a determinação de ânions
4.3.1. Experimento 8 – Ânions em água potável
A água potável é o nosso mais importante tipo de alimento. Ela é obtida principalmente de água subterrânea e água de superfície. Águas de superfície incluem águas de lagos e reservatórios, água subterrânea enriquecida com águas de superfície e água de rios.
De acordo com a DIN 2000, a água potável deve satisfazer os seguintes requisitos:
Deve ser incolor, clara, fria e livre de odor e sabor estranho; Se possível, deve ser naturalmente livre de patógenos e substâncias consideradas de risco para a saúde; Não deve conter muitos sais, particularmente componentes sólidos, ferro, manganês, bem como substâncias orgânicas (lodo e substâncias húmicas);
Não deve causar corrosão;
A quantidade disponível deve ser suficiente para suprir todas as necessidades da população que a utiliza.

76 Monografia Metrohm
Dependendo do grau de poluição, vários métodos são usados para o tratamento da água potável.
Triagem, remove solo grosso e partículas grandes.
Filtro de areia, para filtração; processo de biodegradação também ocorre na areia e ajuda na purificação.
Filtro de carvão ativo absorve substâncias orgânicas dissolvidas, por exemplo, pesticidas.
Remoção de ferro e manganês pela oxidação de Fe(II) e Mn(II); este processo ocorreria, por outro lado, em aqueduto de água potável e resultaria em turbidez marrom ou flocos na água potável.
Desinfecção é sempre necessária quando a água não está livre de patógenos. Cloro, ozônio, dióxido de cloro e irradiação UV são usados.
Cloração preventiva antes da liberação na tubulação de água potável, a fim de impedir o crescimento de microrganismos na água que chega ao consumidor.
Figura 29 Tratamento de água potável – diagrama feito por Thomas Seilnacht, Tuttling.
Conteúdo do aprendizado
Investigação do nosso alimento “Número 1”;
Conferindo as informações fornecidas nas garrafas de água mineral.
Tabela 29 Valores limitantes para água potável (Alemanha)
Fluoreto 1,5 mg/L como F–
Nitrato 50 mg/L como NO3–
Nitrito 0,1 mg/L como NO2–
Cloreto 250 mg/L Sulfato 250 mg/L
Tabela 30 Parâmetros – Experimento 8
Coluna 6.1006.430 Metrosep A Supp 4 Eluente NaHCO3 a 1,8 mmol/L / Na2CO3 a 1,7 mmol/L + 2% acetona
Condutividade após supressão química aprox. 14 μS/cm Amostra Água potável (água de torneira, água mineral)
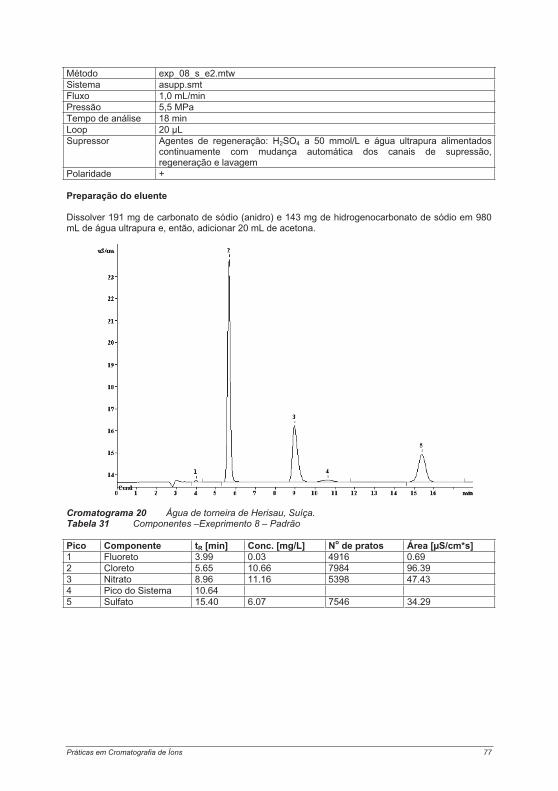
Práticas em Cromatografia de Íons 77
Método exp_08_s_e2.mtw Sistema asupp.smt Fluxo 1,0 mL/min Pressão 5,5 MPa Tempo de análise 18 min Loop 20 μL Supressor Agentes de regeneração: H2SO4 a 50 mmol/L e água ultrapura alimentados
continuamente com mudança automática dos canais de supressão, regeneração e lavagem
Polaridade +
Preparação do eluente
Dissolver 191 mg de carbonato de sódio (anidro) e 143 mg de hidrogenocarbonato de sódio em 980 mL de água ultrapura e, então, adicionar 20 mL de acetona.
Cromatograma 20 Água de torneira de Herisau, Suíça.Tabela 31 Componentes –Exeprimento 8 – Padrão
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Fluoreto 3.99 0.03 4916 0.69 2 Cloreto 5.65 10.66 7984 96.39 3 Nitrato 8.96 11.16 5398 47.43 4 Pico do Sistema 10.64 5 Sulfato 15.40 6.07 7546 34.29

78 Monografia Metrohm
Cromatograma 21 Água mineral sem dióxido de carbono.
Tabela 32 Componentes – Exeprimento 8 – Água mineral sem dióxido de carbono
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Fluoreto 3.93 0.18 1789 7.77 2 Cloreto 5.76 46.24 2456 468.9 5 Nitrato 8.75 0.23 4659 0.94 3 Pico do sistema 11.15 6 Sulfato 15.55 95.61 4400 637.1
Observações
A água mineral contendo dióxido de carbono deve ser desgaseificada antes da medição – por quê?

Práticas em Cromatografia de Íons 79
4.3.2. Experimento 9 – Ânions em etanol e destilados (licores)
A determinação de ânions em solventes orgânicos é possível, embora ocorra a presença de picos marcantes nos cromatogramas no início do sistema. Análises de rum, vodca e etanol são usadas como exemplos.
Para se obter limites de detecção menores, a técnica de pré-concentração pode ser aplicada (vide capítulo 4.2.7). Em alguns casos, entretanto, a matriz da amostra (por exemplo, etanol) pode interferir principalmente com a determinação, levando a picos com formatos irregulares que não podem ser avaliados (vide Cromatograma 26). É, então, necessário remover a matriz pela lavagem da coluna de pré-concentração com água ultrapura (eliminação da matriz). Desta forma, são obtidos bons cromatogramas (vide Cromatograma 25).
A técnica de eliminação da matriz pode ser usada para analisar solventes polares e até mesmo não-polares.
Um método alternativo para remover matéria orgânica de soluções aquosas é usar uma coluna de preparação da amostra RP (Fase Reversa)
Conteúdo do aprendizado
Análise de alimentos;
Influência da matriz na cromatografia;
Preparação da amostra;
Pré-concentração da amostra e eliminação da matriz.
Tabela 33 Parâmetros – Experimentos 9a a 9c
Coluna 6.1006.430 Metrosep A Supp 4 Eluente NaHCO3 a 1,7 mmol/L / Na2CO3 a 1,8 mmol/L + 2% acetona
Condutividade após supressão química aprox. 14 μS/cm Amostra a) Rum
b) Vodca Método exp_09_s e2.mtw Sistema asupp.smt Fluxo 1,0 mL/min Pressão 5,5 MPa Tempo de análise a) 20 min
b) 19 minLoop a) 100 μL
b) 100 μL Supressor Agentes de regeneração: H2SO4 a 50 mmol/L e água ultrapura alimentados
continuamente com mudança automática dos canais de supressão, regeneração e lavagem
Polaridade +
Preparação do eluente
Dissolver 191 mg de carbonato de sódio (anidro) e 143 mg de hidrogenocarbonato de sódio em 980 mL de água ultrapura e, então, adicionar 20 mL de acetona.

80 Monografia Metrohm
Experimento 9a – Determinação de ânions em rum
Cromatograma 22 Solução padrão para análise de rum.
Tabela 34 Componentes – Experimento 9a – Padrão
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Cloreto 5.51 5.011 5759 225.321 2 Nitrato 8.65 0.248 4834 5.110 3 Sulfato 14.91 5.005 5925 143.508 4 Oxalato 16.51 0.250 4732 5.411

Práticas em Cromatografia de Íons 81
Cromatograma 23. Análise de rum.
Tabela 35 Componentes – Experimento 9a – Rum
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 n.d. 4.15 n.d. 1535 54.68 2 n.d. 4.43 n.d. 3358 41.49 3 Cloreto 5.32 0.837 5456 35.44 4 Nitrato 8.32 0.059 5554 1.46 5 Sulfato 14.17 0.580 4624 15.90 6 Oxalato 15.76 0.233 5268 5.05

82 Monografia Metrohm
Experimento 9b – Determinação de ânions em vodca
Cromatograma 24 Análise de vodca
Tabela 36 Componentes – Experimento 9b – Vodca
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 n.d. 3.89 50.52 2 n.d. 4.50 38.89 3 Cloreto 5.39 3.04 4942 132.57 4 Nitrato 8.32 0.26 4991 5.33 5 Sulfato 14.23 2.87 3100 80.17 6 Oxalato 15.78 0.57 3530 11.95
Observações:
Os picos 1 e 2 não foram avaliados.

Práticas em Cromatografia de Íons 83
Experimento 9c – Determinação de ânions em etanol com pré-concentração e eliminação da matriz
Tabela 37 Parâmetros – Experimento 9c Coluna 6.1006.430 Metrosep A Supp 4 Eluente Na2CO3 a 1,8 mmol/L / NaHCO3 a 1,7 mmol/L + 2% acetona
Condutividade após supressão química aprox. 14 μS/cm Amostra Etanol Método exp_09_me e2.mtw Sistema asupp.smt Fluxo 1,0 mL/min Pressão 5,5 MPa Tempo de análise 16 min Loop 2 mL na coluna de pré-concentração Metrosep Ânion 6.1006.300; enxágüe
com 2 mL de água ultra-pura para eliminação de matriz Supressor Agentes de regeneração: H2SO4 a 50 mmol/L e água ultrapura alimentados
continuamente com mudança automática dos canais de supressão, regeneração e lavagem
Polaridade +
Preparação do eluente
Dissolver 191 mg de carbonato de sódio (anidro) e 143 mg de hidrogenocarbonato de sódio em 980 mL de água ultrapura e, então, adicionar 20 mL de acetona.
Cromatograma 25 Determinação de ânions em etanol com eliminação da matriz.
Tabela 38 Componentes – Experimento 9c – Etanol sem eliminação da matriz
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Cloreto 5.2 1.82 5416 1.81 2 Nitrito 6.03 0.42 2658 0.41 3 Nitrato 8.08 1.83 3887 1.17 4 Pico do sistema 9.93 5 Sulfato 13.34 2.61 5440 3.89 6 Oxalato 14.77 0.99 6025 1.11
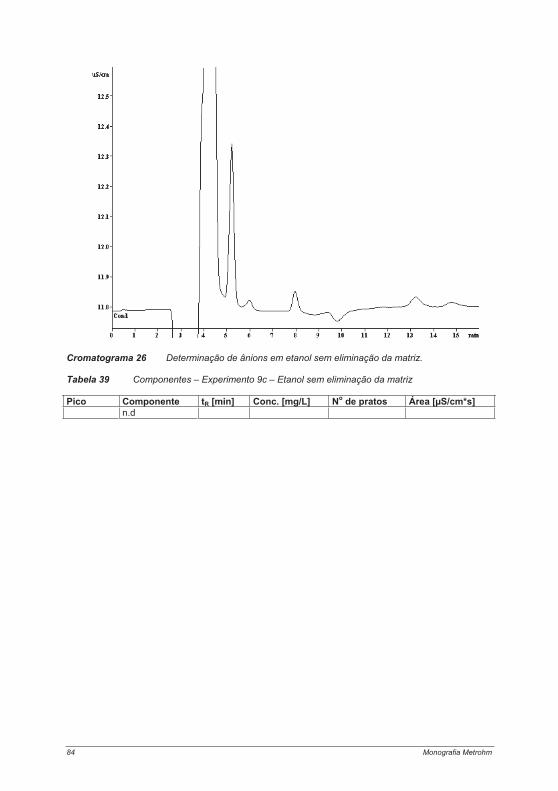
84 Monografia Metrohm
Cromatograma 26 Determinação de ânions em etanol sem eliminação da matriz.
Tabela 39 Componentes – Experimento 9c – Etanol sem eliminação da matriz
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] n.d

Práticas em Cromatografia de Íons 85
4.3.3. Experimento 10 – Ânions em alface
Os nitratos entram no corpo humano não só através da água potável, mas também pela ingestão de vegetais, saladas etc. A OMS (Organização Mundial da Saúde) recomenda que o consumo diário de nitrato não exceda 220 mg por pessoa. Na Alemanha, o consumo médio diário de nitrato é de aproximadamente 130 mg por pessoa. Aproximadamente 5% desta quantidade origina-se de carnes e lingüiças; o restante é dividido em quase meio a meio entre a água potável e os vegetais.
O nitrato pode causar efeitos adversos na saúde humana por diferentes formas. O nitrato em si é relativamente atóxico. Porém, o consumo de grandes quantidades pode levar à inflamação do trato gastrintestinal. Entretanto, sob certas condições, particularmente em presença de bactérias da região bucal humana, o nitrato é reduzido a nitrito pela enzima nitrato-redutase:
NO3– NO2
–
O nitrito é capaz de converter a hemoglobina, a coloração vermelha do sangue, em metemoglobina (Fe2+ é oxidado a Fe3+). Ao contrário da hemoglobina, a metemoglobina não pode transportar o oxigênio no sangue. Em seres humanos adultos, este dano pode ser sanado por reações metabólicas. Entretanto, o metabolismo de bebês até os 5 meses de idade ainda não é capaz de realizar tais reações com eficiência. Isto resulta em falta de oxigênio no sangue dos bebês e a pele se torna azulada. Esta cianose ou metemoglobinemia pode, às vezes, resultar em morte.
Nas proximidades ácidas do estômago, o nitrito pode reagir com várias aminas secundárias (dois átomos de H da molécula amônia são substituídos por grupos alquilas), que entram no corpo humano através de medicamentos e alimentos, para formar as nitrosaminas.
Figura 30 Formação de nitrosaminas a partir de nitrito e aminas secundárias
As nitrosaminas estão entre as substâncias mais carcinogênicas conhecidas. Elas são formadas dentro do organismo a partir de compostos não-carcinogênicos.
Conteúdo do aprendizado
Análise de alimentos;
Controle dos alimentos, particularmente, em relação ao nitrito e nitrato.
Tabela 40 Parâmetros – Experimento 10
Coluna 6.1006.430 Metrosep A Supp 4 Eluente Na2CO3 a 1,0 mmol/L / NaHCO3 a 4,0 mmol/L + 2% acetona
Condutividade após supressão química aprox. 8 μS/cm Amostra Alface Método exp_10_s_e2.mtw Sistema asupp.smt Fluxo 1,0 mL/min Pressão 5,5 MPa Tempo de análise 20 min Loop 20 μL Supressor Agentes de regeneração H2SO4 a 50 mmol/L e água ultrapura alimentados
continuamente com mudança automática dos canais de supressão, regeneração e lavagem
Polaridade +
Preparação do eluente
Dissolver 106 mg de carbonato de sódio (anidro) e 336 mg de hidrogenocarbonato de sódio em 750 mL de água ultrapura e, então, adicionar 250 mL de acetona.

86 Monografia Metrohm
Preparação da amostra
Picar a alface, espalhar, adicionar água ultrapura (proporção 1:100) e filtrar.
Cromatograma 27 Solução padrão para a análise de alface.

Práticas em Cromatografia de Íons 87
Tabela 41 Componentes – Experimento 10 – Padrão
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Cloreto 6.53 1.00 4994 7.30 2 Nitrato 8.96 10.0 4789 39.98 3 Fosfato 19.68 1.0 3328 2.29 4 Sulfato 22.40 3.0 3946 3.99 5 Oxalato 24.35 1.0 4395 5.63
Cromatograma 28 Análise de alface (diluição 1:100).
Tabela 42 Componentes – Experimento 10 – Alface
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Cloreto 6.51 172 5223 12.13 2 Nitrato 8.91 1346 4659 54.04 4 Fosfato 19.72 88 3697 2.06 5 Sulfato 22.40 351 3345 4.68 6 Oxalato 24.33 103 3926 5.93

88 Monografia Metrohm
4.3.4. Experimento 11 – Ácido fosfórico em refrigerantes tipo “cola”
A Coca-Cola é uma bebida não-alcoólica que contém dióxido de carbono e cafeína. Ela foi produzida pela primeira vez em 1885, pelo farmacêutico americano Pemberton. Entre os seus ingredientes estão os extratos de noz de cola, laranjas amargas, essência de gengibre, açúcar, ácido fosfórico (pH = 2,7), corante caramelo, dióxido de carbono e cafeína.
Muitos refrigerantes tipo “cola” estão disponíveis em supermercados (Pepsi Cola, Club Cola, River Cola, Afri Cola, Baré Cola, Dolly Cola etc.); a composição destes pode diferir da composição da Coca-Cola.
A determinação do conteúdo de ácido fosfórico em refrigerantes tipo “cola” é de especial importância, uma vez que os engarrafadores locais deste refrigerante adquirem o concentrado e o engarrafamento é controlado através do conteúdo de fosfato, requerendo uma determinação altamente precisa.
O ácido fosfórico é um ácido tribásico (pKA1= 2,161; pKA2= 7,207; pKA3= 12,325). As quantidades das diferentes espécies variam com o pH, como pode ser visto no seguinte diagrama:
Figura 31 Efeito tampão do ácido fosfórico em uma escala de pH de 6 a 8.
Uma mistura do fosfato primário e secundário é freqüentemente usada como uma solução tampão em pH de 6 a 8 (90% de H2PO4
– + 10% de HPO42– até 10% de H2PO4
– + 90% de HPO42–).
Veja também a equação de Henderson-Hasselbalch (equação de tampão):
)()(lg
HAcAcpKpH S
Conteúdo do aprendizado
Análise de bebidas;
Química do ácido fosfórico;
Influência do pH na separação cromatográfica;
Estatística: reprodutibilidade.
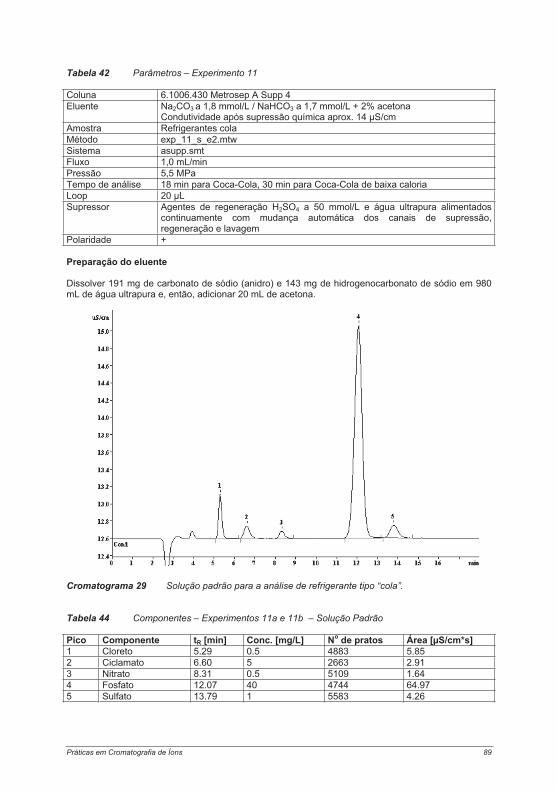
Práticas em Cromatografia de Íons 89
Tabela 42 Parâmetros – Experimento 11
Coluna 6.1006.430 Metrosep A Supp 4 Eluente Na2CO3 a 1,8 mmol/L / NaHCO3 a 1,7 mmol/L + 2% acetona
Condutividade após supressão química aprox. 14 μS/cm Amostra Refrigerantes cola Método exp_11_s_e2.mtw Sistema asupp.smt Fluxo 1,0 mL/min Pressão 5,5 MPa Tempo de análise 18 min para Coca-Cola, 30 min para Coca-Cola de baixa caloria Loop 20 μL Supressor Agentes de regeneração H2SO4 a 50 mmol/L e água ultrapura alimentados
continuamente com mudança automática dos canais de supressão, regeneração e lavagem
Polaridade +
Preparação do eluente
Dissolver 191 mg de carbonato de sódio (anidro) e 143 mg de hidrogenocarbonato de sódio em 980 mL de água ultrapura e, então, adicionar 20 mL de acetona.
Cromatograma 29 Solução padrão para a análise de refrigerante tipo “cola”.
Tabela 44 Componentes – Experimentos 11a e 11b – Solução Padrão
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Cloreto 5.29 0.5 4883 5.85 2 Ciclamato 6.60 5 2663 2.91 3 Nitrato 8.31 0.5 5109 1.64 4 Fosfato 12.07 40 4744 64.97 5 Sulfato 13.79 1 5583 4.26
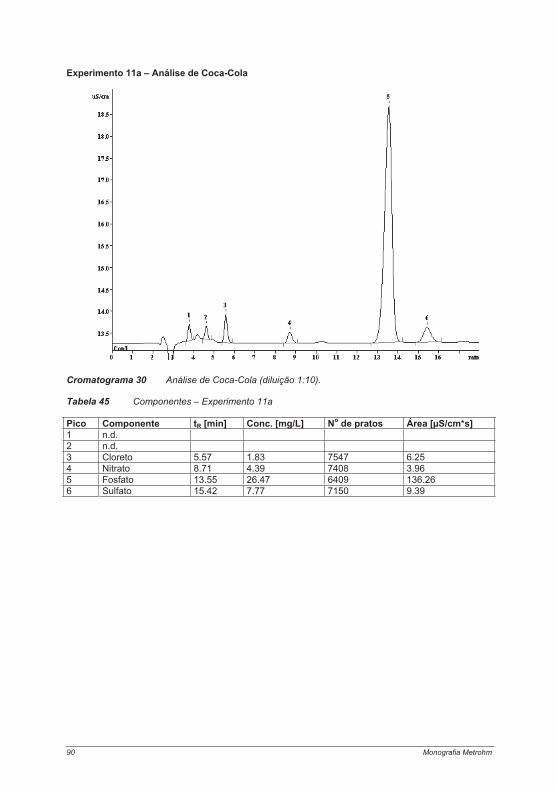
90 Monografia Metrohm
Experimento 11a – Análise de Coca-Cola
Cromatograma 30 Análise de Coca-Cola (diluição 1:10).
Tabela 45 Componentes – Experimento 11a
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 n.d. 2 n.d. 3 Cloreto 5.57 1.83 7547 6.25 4 Nitrato 8.71 4.39 7408 3.96 5 Fosfato 13.55 26.47 6409 136.26 6 Sulfato 15.42 7.77 7150 9.39

Práticas em Cromatografia de Íons 91
Experimento 11b – Análise de Coca-Cola de baixa caloria
Cromatograma 31 Análise de Coca-Cola de baixa caloria (diluição 1:10).
Tabela 46 Componentes – Experimento 11b
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 n.d. 2 n.d. 3 Cloreto 5.56 3.23 8140 1.84 4 Ciclamato 7.07 360.76 3670 46.02 5 Nitrato 8.69 2.69 4290 2.27 6 Fosfato 13.54 247.96 1850 102.58 7 Sulfato 15.41 9.33 1630 6.03
Observações:
Os picos 1 e 2 não foram avaliados.
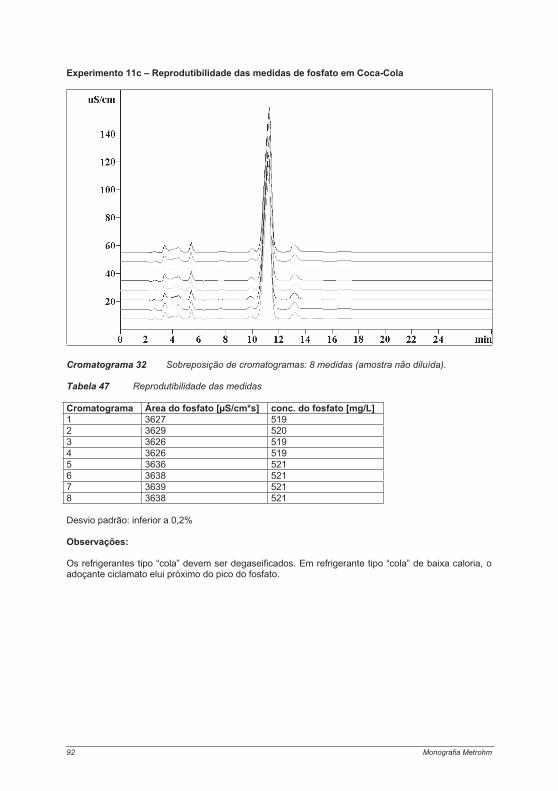
92 Monografia Metrohm
Experimento 11c – Reprodutibilidade das medidas de fosfato em Coca-Cola
Cromatograma 32 Sobreposição de cromatogramas: 8 medidas (amostra não diluída).
Tabela 47 Reprodutibilidade das medidas
Cromatograma Área do fosfato [μS/cm*s] conc. do fosfato [mg/L] 1 3627 519 2 3629 520 3 3626 519 4 3626 519 5 3636 521 6 3638 521 7 3639 521 8 3638 521
Desvio padrão: inferior a 0,2%
Observações:
Os refrigerantes tipo “cola” devem ser degaseificados. Em refrigerante tipo “cola” de baixa caloria, o adoçante ciclamato elui próximo do pico do fosfato.

Práticas em Cromatografia de Íons 93
4.3.5. Experimento 12 – Ácidos orgânicos em vinho
De acordo com a lei alemã de vinhos, o vinho é um produto que só pode ser obtido através de uma fermentação alcoólica, parcial ou completa, de uvas frescas ou amassadas, ou do suco de uvas. O suco de uvas contém de 12 a 25% de carboidratos (glicose, frutose) e 0,9 a 1,5% de ácidos; os mais importantes são o ácido L-(+) tartárico (2R, 3R) e o ácido málico, mas o ácido cítrico, o ácido cetoglutárico, o ácido succínico e o ácido lático podem também estar presentes.
Um critério importante para analisar o suco de uva é o grau Oechsle (Oe°); quanto maior for o grau, mais açúcar o suco contém. Isto indica o número de gramas pelo qual 1 L de suco a 20 °C é mais pesado do que 1 L de água destilada. Por exemplo, um suco com uma densidade de 1,115 kg/L (115 g a mais que em 1 L de água) tem 115 Oe°. A partir de graus Oechsle, um cálculo simples permite a determinação do conteúdo de açúcar e álcool. 1,7 g de açúcar produz 1 mL (0,794 g) de etanol. Em uma fração de 12 a 15% do volume de álcool a fermentação se paralisa, uma vez que as leveduras são mortas pelo álcool que elas produziram.
O aroma do vinho é constituído de 600 a 800 componentes: hidrocarbonetos, álcoois, aldeídos, cetonas, ácidos, ésteres, lactonas, éteres, fenóis, entre outros.
De interesse analítico são os (2R, 3R)-ácidos tartárico, ácido málico, ácido cítrico, ácido lático e ácido succínico. O conteúdo total de ácido (calculado como ácido tartárico) está, geralmente, entre 5,5 e 8,5 g/L. O ácido acético, ácido propiônico, ácidos graxos maiores e quantidades anormais de ácido lático ocorrem em vinhos “estragados” e são, principalmente, produzidos por microrganismos.
COOH
C OHH
C HHO
COOH
2
3
Figura 32 Ácido L-(+) tartárico, forma (2R, 3R)
A fermentação alcoólica ocorre de acordo com a seguinte equação:
C6H12O6 + 2 ADP + 2 P 2 C2H5OH + 2 CO2 + 2ATP
Abreviações: ADP = difosfato de adenosina, ATP = trifosfato de adenosina, P = fosfato
Os ácidos orgânicos fracos podem ser determinados por cromatografia de exclusão de íons (vide capítulos 3.3.4, 3.6.7 e 3.7.2.3).

Tabe
la 4
7. C
onte
údo
de á
cido
(mg/
L) e
m v
inho
s al
emãe
s (v
alor
méd
io d
e 4
ou 2
aná
lises
indi
vidua
is; a
s de
nom
inaç
ões
alem
ães
fora
m m
antid
as)
Obs
erva
ções
Ries
ling
(uva
bra
nca
alem
ã)
Mül
ler-T
hurg
au
Vinh
o típ
i-co
de
alta
qu
alid
ade
Vinh
o Ti
nto
Portu
guês
Ka
bine
ttSp
ätle
seAu
slese
Be
eren
aus-
-le
se, E
iswei
n Si
lvane
rKa
bine
ttSp
ätle
seAu
slese
Be
eren
aus-
lese
Ácid
o ox
álico
15
,7
13,0
11
,045
,816
,021
,716
,740
,915
,419
,5Ác
ido
succ
ínico
19
4,0
260,
1 20
5,8
282,
123
0,2
149,
224
3,8
215,
126
4,9
269,
2Ác
ido
fum
árico
44
,2
31,9
24
,628
,320
,836
,020
,951
,827
,040
,6Ác
ido
glut
árico
3,
7 1,
9 4,
52,
62,
37,
02,
62,
21,
96,
2Ác
ido
c- o
u t-
acon
ítico
2,
1 1,
9 +
5,5
4,9
6,4
1,4
2,5
1,6
4,5
Ácid
o gl
icólic
o
1,8
1,3
++
1,4
6,9
3,7
3,4
1,7
5,8
Ácid
o D-
+ L
- lát
ico
1789
23
64
319,
815
9,8
151,
021
4179
322
1,0
208,
120
96Ác
ido
angl
icéric
o 43
,5
62,6
50
,139
,559
,053
,049
,687
,068
,849
,9Ác
ido
trigl
icéric
o 3-
M-2
,3-D
HBS
Ácid
o m
álico
39
71
5080
38
5038
7142
3522
5128
9331
6334
1028
97Ác
ido
tartá
rico
1586
99
4 14
2814
3181
712
5112
8010
1480
588
7Ác
ido
citra
mál
ico
94,5
53
,7
63,0
18,6
46,8
48,2
31,3
33,8
24,2
41,0
Ácid
o-h
idro
xiglu
táric
o 24
,9
27,5
18
,224
,528
,420
,618
,815
,326
,922
,2Ác
ido
cítri
co
138,
9 15
0,2
241,
122
2,8
298,
628
7,3
200,
722
1,7
240,
233
5,1
Ácid
o gl
ioxí
lico
+ 1,
0 +
+4,
06,
12,
91,
81,
13,
1Ác
ido
pirú
vico
20,6
29
,8
17,8
12,4
16,8
8,6
17,6
22,7
18,6
33,0
Ácid
o-c
etog
lutá
rico
41,1
45
,7
38,6
36,8
55,7
35,6
41,0
33,7
40,3
27,5
Ácid
o gl
ucôn
ico
59,0
35
7,2
54,5
95,9
452,
133
7,2
53,9
131,
137
3,9
540
4,1
20,4
+4,
511
,2Ác
ido
glicu
rôni
co
2,1
3,4
3,8
5,8
7,9
14,5
1,7
6,0
7,6
10,2
Ácid
o ga
lact
urôn
ico
11,1
11
,7
14,1
19,7
22,0
43,4
10,0
17,3
23,2
34,3
Ácid
o L-
ascó
rbico
12
,9
9,0
13,7
10,1
9,8
9,6
12,0
10,4
9,7
8,5
Ácid
os to
tais
7977
94
91
6349
6302
6474
6744
5685
5284
5565
7333
2 Monografia Metrohm 94

Práticas em Cromatografia de Íons 95
Conteúdo do aprendizado
Análise de alimentos;
Cromatografia de exclusão de íons;
Quais íons podem ser determinados por cromatografia de exclusão de íons?
Tabela 49 Parâmetros – Experimentos 12a e 12b
Coluna 6.1005.200 Metrosep Organic Acids (7.8 x 250 mm) Eluente H2SO4 a 0,5 mmol/L / acetona (85:15) Amostra Vinho branco e vinho tinto Método exp_12_o.mtw Sistema orgacids.smt Fluxo 0,5 mL/min Pressão 3 MPa Tempo de análise 20 min Loop 20 μL Polaridade +
Preparação do eluente
Misturar H2SO4 a 0,5 mmol (850 mL) e acetona (150 mL).
Preparação da amostra
Diluir a amostra de vinho 1:100 e filtrar em membrana com poros de 0,45 μm.
Cromatograma 33 Solução padrão para a determinação de ácidos orgânicos em vinho.

96 Monografia Metrohm
Tabela 50 Componentes – Experimentos 12a e 12b – Solução Padrão
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Citrato 7,2 5 26200 1,1 2 Tartarato 7,5 20 7990 31 3 Malato 8,3 5 9210 4,5 4 Succinato 9,9 5 7140 1,2 5 Lactato 10,8 20 8830 11 6 Acetato 13,1 10 11500 1,2 Pico do sistema 15,9
Experimento 12a – Determinação de ácidos orgânicos em vinho branco
Cromatograma 33. Determinação de ácidos orgânicos em vinho branco (diluição 1:100).
Tabela 51 Componentes – Experimento 12a
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Citrato 7,2 2 Tartarato 7,5 1210 8430 19 4 Malato 8,2 7 Succinato 9,8 350 5660 0,8 8 Lactato 10,7 2190 8820 13 9 Acetato 13,0 710 5200 0,8 Pico do sistema 15,8
Observações:
Os picos 3, 5 e 6 não foram avaliados.
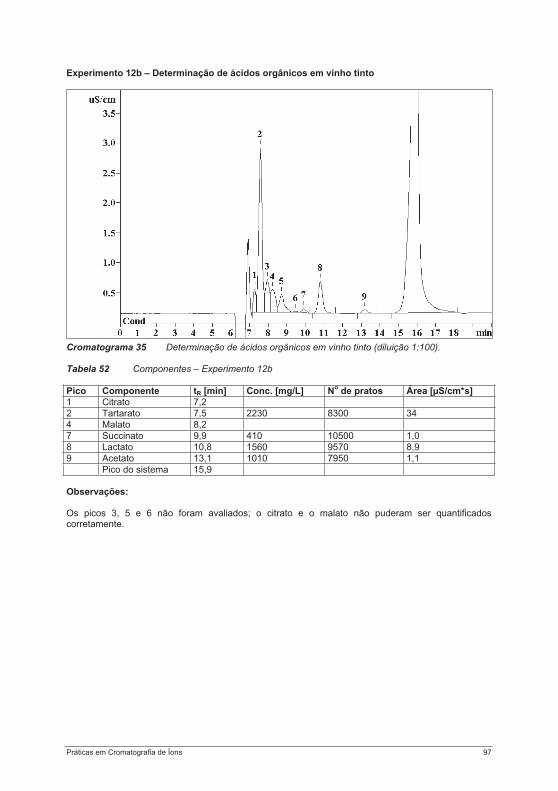
Práticas em Cromatografia de Íons 97
Experimento 12b – Determinação de ácidos orgânicos em vinho tinto
Cromatograma 35 Determinação de ácidos orgânicos em vinho tinto (diluição 1:100).
Tabela 52 Componentes – Experimento 12b
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Citrato 7,2 2 Tartarato 7,5 2230 8300 34 4 Malato 8,2 7 Succinato 9,9 410 10500 1,0 8 Lactato 10,8 1560 9570 8,9 9 Acetato 13,1 1010 7950 1,1 Pico do sistema 15,9
Observações:
Os picos 3, 5 e 6 não foram avaliados; o citrato e o malato não puderam ser quantificados corretamente.

98 Monografia Metrohm
4.3.6. Experimento 13 – Contaminantes em borato – determinação de cloretos e sulfatos em soluções de bórax
O bórax (tetraborato dissódico, Na2B4O7·10 H2O, Na2[B4O5(OH)4]·8H2O) tem a seguinte estrutura:
BHOO
O
B
B
OH
OH
O
OB OHO 2 Na 8 H2O
Figura 33 «Bórax» (tetraborato dissódico, Na2B4O7·10 H2O, Na2[B4O5(OH)4]·8H2O)
Ao se fundir, o bórax pode dissolver muitos óxidos metálicos com a formação de cores características. Estas “pérolas de bórax” são muito conhecidas em prática de química inorgânica. O bórax é também usado na produção de vidro, cerâmica vitrificada, porcelana e como um fundente em solda. A 100°C, o bórax perde 5 moléculas de água e se torna o pentaidrato “pedra bórax”. Se um metal fundente for adicionado a ele, os contaminantes de superfície – principalmente camadas de óxido – serão destruídos. Estes, por outro lado, afetariam a formação de uma liga entre a solda (uma liga de prata, cobre e estanho) e o material básico.
As soluções de tetraborato de sódio (bórax) são usadas em circuito interno de resfriamento de usinas nucleares como absorvedores de nêutrons. A pureza do material é extremamente importante, uma vez que traços de cloreto e sulfato causam corrosão na tubulação, o que deve ser evitado de todas as maneiras.
O ácido bórico H3BO3 ou B(OH)3 é um ácido monobásico muito fraco, cujo valor de pKA corresponde a 9,25, aproximadamente, ao do HCN. O ácido bórico não é um doador de H+ (definição de ácido segundo Bronsted), mas sim um aceptor de OH– (definição de ácido segundo Lewis).
B(OH)3 + HOH H+ + B(OH)4-
A cromatografia de íons sem supressão química usa um eluente ácido com pH = 4,1 para a determinação de contaminantes em borato. Neste pH, o borato está presente quase que totalmente como ácido bórico B(OH)3. Ao contrário dos ânions carregados negativamente, o ácido bórico não interage com a fase estacionária da coluna de separação. Isto significa que ele elui no volume morto.
Entretanto, com supressão química é usado o eluente carbonato/hidrogenocarbonato fracamente alcalino. Embora seja possível separar o borato como um ânion, a detecção não funciona. A causa disto pode estar no supressor, que troca todos os íons Na+ por íons H+. Em conseqüência, é formado o ácido bórico fraco.
A grande quantidade de ácido bórico promove distorções no cromatograma, tornando quase impossível a determinação dos ânions. A eliminação da matriz (vide capítulo 4.3.2) é a melhor maneira de resolver o problema.
Em usinas de energia, traços de ânions devem, freqüentemente, ser determinados em amostras de água contendo 2 a 4% de ácido bórico. Nestes casos, aplica-se a técnica de eliminação da matriz com pré-concentração da amostra (vide Cromatograma 39).
Conteúdo do aprendizado
Ácidos fortes e fracos;
Preparação da amostra;
Pré-concentração da amostra e eliminação da matriz.
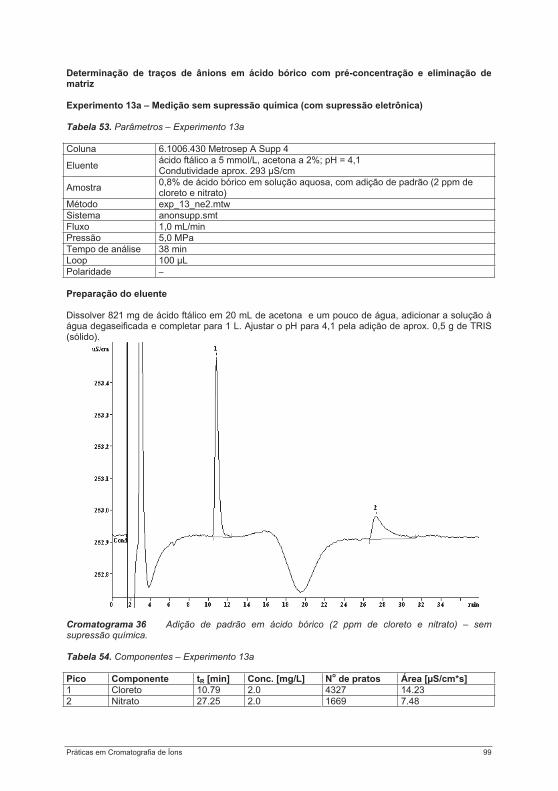
Práticas em Cromatografia de Íons 99
Determinação de traços de ânions em ácido bórico com pré-concentração e eliminação de matriz
Experimento 13a – Medição sem supressão química (com supressão eletrônica)
Tabela 53. Parâmetros – Experimento 13a
Coluna 6.1006.430 Metrosep A Supp 4
Eluente ácido ftálico a 5 mmol/L, acetona a 2%; pH = 4,1 Condutividade aprox. 293 μS/cm
Amostra 0,8% de ácido bórico em solução aquosa, com adição de padrão (2 ppm de cloreto e nitrato)
Método exp_13_ne2.mtw Sistema anonsupp.smt Fluxo 1,0 mL/min Pressão 5,0 MPa Tempo de análise 38 min Loop 100 μL Polaridade –
Preparação do eluente
Dissolver 821 mg de ácido ftálico em 20 mL de acetona e um pouco de água, adicionar a solução à água degaseificada e completar para 1 L. Ajustar o pH para 4,1 pela adição de aprox. 0,5 g de TRIS (sólido).
Cromatograma 36 Adição de padrão em ácido bórico (2 ppm de cloreto e nitrato) – sem supressão química.
Tabela 54. Componentes – Experimento 13a
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Cloreto 10.79 2.0 4327 14.23 2 Nitrato 27.25 2.0 1669 7.48

100 Monografia Metrohm
Experimento 13b – Medições com supressão química
Tabela 55 Parâmetros – Experimento 13b
Coluna 6.1006.430 Metrosep A Supp 4 Eluente Na2CO3 a 1,8 mmol/L / NaHCO3 a 1,7 mmol/L + 2% acetona
Condutividade após supressão química aprox. 12 μS/cm Amostra 0,8% de ácido bórico em solução aquosa com adição de padrão (2 ppm de
cloreto e nitrato) Método exp_13_s_e2.mtw Sistema asupp.smt Fluxo 1,0 mL/min Pressão 5,0 MPa Tempo de análise 20 min Loop Agentes de regeneração: H2SO4 a 50 mmol/L e água ultrapura alimentados
continuamente com mudança automática dos canais de supressão, regeneração e lavagem
Supressor 100 μL Polaridade +
Preparação do eluente
Dissolver 191 mg de carbonato de sódio (anidro) e 143 mg de hidrogenocarbonato de sódio em 980 mL de água ultrapura e, então, adicionar 20 mL de acetona.
Cromatograma 37 Adição de padrão em ácido bórico (2 ppm de cloreto e nitrato) – com supressão química. Tabela 56 Componentes – Experimento 13b
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Cloreto 7.95 2 2714 70.7 2 Nitrato 14.17 2 85005 57.8

Práticas em Cromatografia de Íons 101
Experimento 13c – Determinação, com supressão química, de traços de ânions em ácido bórico com pré-concentração e eliminação de matriz
Tabela 57 Parâmetros – Experimento 13c
Coluna 6.1006.430 Metrosep A Supp 4 Eluente Na2CO3 a 1,8 mmol/L / NaHCO3 a 1,7 mmol/L + 2% de acetona
Condutividade após supressão química aprox. 12 μS/cmAmostra Solução aquosa de ácido bórico a 0,2% Método exp_13_me_e2.mtw Sistema asupp.smt Fluxo 1,0 mL/min Pressão 5,5 MPa Tempo de análise 20 min Introdução da amostra
2 mL da amostra são passados pela coluna de pré-concentração onde os traços de ânions a serem determinados são retidos. Posteriormente, a coluna de pré-concentração é lavada com água ultrapura para remover o ácido bórico (eliminação da matriz) e, então, é injetada. Os traços de ânions retidos são re-eluídos, transportados para a coluna Metrosep A Supp 4 e lá separados.
Supressor Agentes de regeneração: H2SO4 a 50 mmol/L e água ultrapura alimentados continuamente com mudança automática dos canais de supressão, regeneração e lavagem
Polaridade +
Preparação do eluente
Dissolver 191 mg de carbonato de sódio (anidro) e 143 mg de hidrogenocarbonato de sódio em 980 mL de água ultrapura e, então, adicionar 20 mL de acetona.
Cromatograma 38 Determinação de traços de ânions em padrão com pré-concentração

102 Monografia Metrohm
Tabela 58 Componentes – Experimento 13c
Pico Componente tR [min] Conc. [μg/L] No de pratos Área [μS/cm*s] 1 Cloreto 5.35 1.5 4421 3.48 2 Nitrito 6.19 5 3455 2.42 3 Nitrato 8.58 1 5324 0.24 4 Pico do sistema 10.09 5 Sulfato 13.54 5 5441 3.19 6 Oxalato 15.00 5 4855 3.19
Cromatograma 39 Determinação de traços de ânions em 0,2% de ácido bórico com pré-concentração e eliminação de matriz
Tabela 59 Componentes – Experimento 13c
Pico Componente tR [min] Conc. [μg/L] No de pratos Área [μS/cm*s] 1 Cloreto 5.37 2.8 4833 5.65 2 Nitrito 6.20 4.8 4452 2.36 3 Nitrato 8.36 n.d. n.d. 4 Pico do sistema 9.94 5 Sulfato 13.51 7.0 5269 4.30 6 Oxalato 14.98 3.5 5293 2.25

Práticas em Cromatografia de Íons 103
Cromatograma 40 Sobreposição de um branco (cromatograma inferior) e solução de ácido bórico a 0,2% (cromatograma superior) com pré-concentração e eliminação de matriz idênticas.
Observações:
A sobreposição dos dois cromatogramas mostra claramente que a maioria dos ânions determinados é proveniente do ácido bórico e não da água ultrapura utilizada para diluir o ácido bórico.

104 Monografia Metrohm
4.3.7. Experimento 14 – Determinação de Ânions em água efluente
Em estações de tratamento biológico, o efluente é purificado, principalmente, com o auxílio de bactérias. As substâncias orgânicas são oxidadas com o consumo de oxigênio.
Substâncias orgânicas + O2 CO2 + H2O + massa celular
Além da oxidação de substâncias orgânicas, a amônia também pode ser oxidada para nitrato (nitrificação) em estações adequadamente projetadas.
NH4+ + 2 O2 NO3
– + H2O + 2 H+
Em processos com ausência de oxigênio, as bactérias utilizam o oxigênio do nitrato para a oxidação de substâncias orgânicas (desnitrificação).
Substâncias orgânicas + 2 NO3– 2 CO2 + 2 OH– + 2 H2O + N2
O fosfato – um nutriente para plantas, assim como NH4+ e NO3
– - pode ser precipitado, por exemplo, pela adição de soluções de sal de Fe3+. Além dos chamados parâmetros de fechamento – demanda bioquímica de oxigênio (DBO), demanda química de oxigênio (DQO), carbono orgânico total (TOC) – que são uma medida da carga orgânica do efluente ou água em geral, a análise de NH4
+, NO3– e
PO43– é também importante. Cl– e SO4
2– são normalmente apenas analisados sob circunstâncias especiais.
Em estações de tratamento comunitárias com mais de 100.000 dos chamados “equivalentes de população” (1 equivalente de população é o fluxo de esgoto por pessoa), os seguintes limites são estabelecidos na Alemanha:
DBO 15 mg/L DQO 75 mg/L NH4-N 10 mg/L*
Ntot 18 mg/L* (soma de NH4-N, NO2-N, NO3-N)PO4-Ptot 1 mg/L
* Só se aplica a uma temperatura de água efluente 12 °C, uma vez que a nitrificação é fortemente influenciada pela temperatura.
NH4-N, NO2-N, NO3-N significa que os valores se referem ao nitrogênio contido nos respectivos íons; o mesmo se aplica para PO4-P.
Conteúdo do aprendizado
Análise ambiental;
Técnicas de preparação da amostra;
Análise de mistura de substâncias com grandes diferenças em concentração – taxa da concentração dinâmica.

Práticas em Cromatografia de Íons 105
Tabela 60 Parâmetros – Experimentos 14a e 14b
Coluna 6.1006.430 Metrosep A Supp 4 Eluente NaHCO3 a 1,8 mmol/L / Na2CO3 a 1,7 mmol/L + 2% acetona
Condutividade a supressão química aprox. 13 μS/cm Amostra Água potável municipal (de Herisau, Suíça) Método exp_14_n_e2.mtw Sistema asupp.smt Fluxo 1,0 mL/min Pressão 5,5 MPa Tempo de análise 20 min Loop 20 μL Supressor Agentes de regeneração: H2SO4 a 50 mmol/L e água ultrapura alimentados
continuamente com mudança automática dos canais de supressão, regeneração e lavagem
Polaridade +
Preparação do eluente
Dissolver 191 mg de carbonato de sódio (anidro) e 143 mg de hidrogenocarbonato de sódio em 980 mL de água ultrapura e, então, adicionar 20 mL de acetona.
Preparação da amostra
A filtração das amostras é essencial. Usar membrana com poros de 0,45 μm ou menor. Mesmo as soluções aparentemente limpas podem conter partículas muito finas que danificam a coluna.
Experimento 14a – Influxo em estação de tratamento
Cromatograma 41 Cromatograma padrão para a análise do influxo em estação de tratamento.

106 Monografia Metrohm
Tabela 61 Componentes – Experimento 14a – Padrão
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Fluoreto 3.68 0.08 3545 0.9 2 Cloreto 5.24 150.97 5416 1354.2 3 Nitrito 5.99 0.49 5510 1.8 4 Brometo 7.26 0.49 5228 1.2 5 Nitrato 8.07 4.95 4949 15.9 6 Fosfato 12.01 4.99 4440 8.3 7 Sulfato 13.94 50.30 5886 249.8
Cromatograma 42 Análise do influxo em estação de tratamento.
Tabela 62 Componentes – Experimento 14a – Influxo em estação de tratamento
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Fluoreto 3.68 0.15 1454 1.77 2 n.d. 4.25 3 Cloreto 5.20 72.86 5581 1880.47 4 Nitrito 6.03 1.13 4562 0.89 5 Brometo 7.35 0.37 2898 2.16 6 Nitrato 8.23 8.40 4244 40.97 7 Pico do sistema 9.78 8 Fosfato 12.05 3.85 3381 7.01 9 Sulfato 13.90 27.21 5981 492.44

Práticas em Cromatografia de Íons 107
Experimento 14b – Efluxo em estação de tratamento
Cromatograma 43 Cromatograma padrão para a análise do efluxo em estação de tratamento.
Tabela 63 Componentes – Experimento 14b – Efluxo em estação de tratamento
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Fluoreto 3.68 0.06 3163 0.8 2 n.d. 4.23 3 Cloreto 5.19 56.54 5782 473.1 4 Nitrito 6.00 0.27 2252 0.6 5 Nitrato 8.09 27.73 3882 94.9 6 Pico do sistema 9.76 7 Fosfato 12.06 1.12 4318 1.9 8 Sulfato 13.89 23.95 5827 113.3

108 Monografia Metrohm
4.3.8. Experimento 15 – Fluoreto em creme dental
Os cremes dentais são constituídos por diferentes ingredientes:
Água - 30 a 40%;
Substâncias assépticas – são substâncias inorgânicas insolúveis que intensificam a ação de limpeza da escovação dental. Elas possuem partículas com tamanho < 15 μm. As substâncias assépticas comumente usadas são silicato de alumínio e sódio, Al2O3, CaCO3, CaHPO4·2H2O, SiO2·H2O;
Fluoreto – aumenta a resistência do esmalte dos dentes contra o ataque dos ácidos originados por placas bacterianas. Os fluoretos tornam também possível a mineralização do esmalte dos dentes (a parte inorgânica do esmalte dos dentes consiste principalmente de fosfato de cálcio, hidroxiapatita, carbonato de cálcio, fosfato de magnésio, fluoreto de cálcio e cloreto de cálcio). Isto significa que o fluoreto é importante na prevenção de cáries. Os compostos fluorados mais freqüentemente usados são o fluoreto de sódio, monofluorfosfato de sódio e fluoretos de amônio quaternária, os quais também contêm íons fluoreto livres. Em alguns países, o fluoreto é adicionado na água potável.
Figura 34 Monofluorfosfato de sódio
Surfactantes (por exemplo, lauril sulfato de sódio) – reduz a tensão da superficial e ajuda a distribuir homogeneamente o creme dental. Eles aumentam os efeitos de limpeza, particularmente em locais difíceis de se alcançar com a escova dental.
Outros ingredientes de cremes dentais são:
Umectantes (por exemplo, glicerol, sorbitol, polietilenoglicol) – melhoram a estabilidade a baixas temperaturas e impedem o ressecamento;
Ligantes e espessantes (por exemplo, carboximetilcelulose de sódio) – impedem a separação em fases líquida e sólida;
Edulcorantes (por exemplo, sacarina sódica) – melhora o sabor do creme dental;
Conservantes (por exemplo, ácido 4-hidroxibenzóico) – são usados para proteger o creme dental contra a decomposição bacteriana;
Substâncias aromáticas – são adicionadas para aumentar tanto a aceitação do creme dental como também a sensação de higiene. Estes incluem óleo de hortelã, mentol, óleo de semente de anis, óleo de eucalipto, óleos aromáticos, óleos cítricos;
A análise de creme dental por cromatografia de íons determina o fluoreto proveniente de fluoreto de sódio juntamente com o monofluorfosfato de sódio.
Observações
O citrato elui da coluna Metrosep Anion Dual 1 (3 x 150 mm) só após aprox. 90 min. A fim de checar se o citrato está presente no creme dental, o Experimento 15b usa o cartucho (6.1006.030) da pré-coluna Metrosep Anion Dual 1 ao invés da coluna de separação Metrosep Anion Dual 1.
Conteúdo do aprendizado
Controle de qualidade;
Preparação da amostra.

Práticas em Cromatografia de Íons 109
Experimento 15a – Creme dental
Tabela 64 Parâmetros – Experimento 15a
Coluna 6.1006.430 Metrosep A Supp 4 Eluente NaCO3 a 1,8 mmol/L / Na2HCO3 a 1,7 mmol/L + 2% acetona
Condutividade após supressão química aprox. 14 μS/cm Amostra Creme dental Método exp_15_s_e2.mtw Sistema asupp.smt Fluxo 1,0 mL/min Pressão 5,5 MPa Tempo de análise 37 min Loop 20 μL Supressor Agentes de regeneração: H2SO4 a 50 mmol/L e água ultrapura alimentados
continuamente com mudança automática dos canais de supressão, regeneração e lavagem
Polaridade +
Preparação do eluente
Dissolver 191 mg de carbonato de sódio (anidro) e 143 mg de hidrogenocarbonato de sódio em 980 mL de água ultrapura e, então, adicionar 20 mL de acetona.
Preparação da amostra
Dissolver 1 g de creme dental em 20 mL de água ultrapura e, então, filtrar esta solução em membrana com poros de 0,45 μm.
Cromatograma 44 Padrão para análise de creme dental

110 Monografia Metrohm
Tabela 65 Componentes – Experimento 15a - Padrão
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Fluoreto 3.91 2.50 2714 684.38 2 Cloreto 5.27 0.50 5447 85.02 3 Nitrato 8.25 0.01 4889 1.05 4 Sulfato 13.91 5.04 5456 689.44 5 Sacarina 30.63 0.25 2044 5.84
Cromatograma 45 Creme dental (diluição 1:20)
Tabela 66 Componentes – Experimento 15
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Fluoreto 3.9 76.38 2063 1022.00 2 n.d. 4.36 2.93 3 Cloreto 5.25 6.18 5066 51.25 4 Nitrato 8.22 0.70 4890 2.77 5 Sulfato 13.95 155.16 4545 1115.672 6 Sacarina 29.70 183.92 2262 116.15

Práticas em Cromatografia de Íons 111
4.3.9. Experimento 16 – Ânions em açúcar refinado branco e mascavo
Os açúcares são compostos orgânicos com vários grupos hidroxilas. Usualmente, o termo “açúcar” se refere à sacarose (dissacarídeo).
Figura 35 Estrutura da sacarose (dissacarídeo).
Na produção de açúcar a partir de beterraba, estas são inicialmente lavadas e repicadas, então, extraídas com água chamadas unidades de difusão em contra-fluxo. Os constituintes solúveis, por exemplo, açúcar, sais, ácidos, proteínas e pectinas, são dissolvidos. A maioria dos constituintes que não são açúcares é precipitada com a adição de cal viva (CaO). O dióxido de carbono (CO2) é, então, usado para precipitar o excesso de hidróxido de cálcio como CaCO3. Após a filtração, a solução de açúcar é concentrada em evaporadores multi-estágios para formar um “xarope”, é filtrado novamente e concentrado, posteriormente, até que o açúcar se separe como uma “massa branca”. O açúcar é separado em centrífugas e, após a purificação (recristalização), obtém-se o açúcar cristal branco com uma pureza de 99,95%. O líquido da centrífuga no último estágio corresponde ao xarope de cor marrom – melaço.
O açúcar mascavo é purificado com menor intensidade e pode ser colorido pela adição de melaço. Além de contaminantes orgânicos, contém íons de metais alcalinos e alcalino-terrosos, bem como ânions; também tem um sabor diferente em relação ao açúcar cristal branco.
Os contaminantes orgânicos podem ser separados com uma coluna RP (Fase Reversa) antes que os ânions sejam determinados.
Conteúdo do aprendizado
Análise de alimentos;
Controle do processo;
Controle de alimentos contendo grande quantidade de açúcar, por exemplo, o mel.
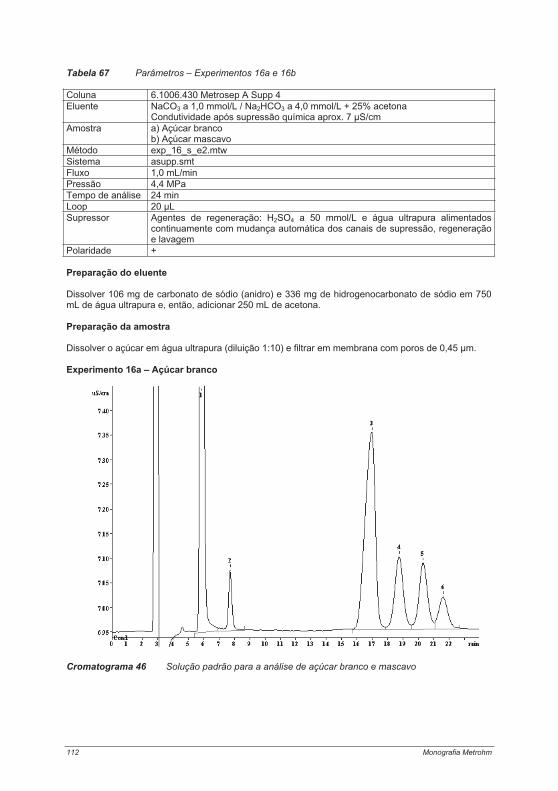
112 Monografia Metrohm
Tabela 67 Parâmetros – Experimentos 16a e 16b
Coluna 6.1006.430 Metrosep A Supp 4 Eluente NaCO3 a 1,0 mmol/L / Na2HCO3 a 4,0 mmol/L + 25% acetona
Condutividade após supressão química aprox. 7 μS/cm Amostra a) Açúcar branco
b) Açúcar mascavo Método exp_16_s_e2.mtw Sistema asupp.smt Fluxo 1,0 mL/min Pressão 4,4 MPa Tempo de análise 24 min Loop 20 μL Supressor Agentes de regeneração: H2SO4 a 50 mmol/L e água ultrapura alimentados
continuamente com mudança automática dos canais de supressão, regeneração e lavagem
Polaridade +
Preparação do eluente
Dissolver 106 mg de carbonato de sódio (anidro) e 336 mg de hidrogenocarbonato de sódio em 750 mL de água ultrapura e, então, adicionar 250 mL de acetona.
Preparação da amostra
Dissolver o açúcar em água ultrapura (diluição 1:10) e filtrar em membrana com poros de 0,45 μm.
Experimento 16a – Açúcar branco
Cromatograma 46 Solução padrão para a análise de açúcar branco e mascavo
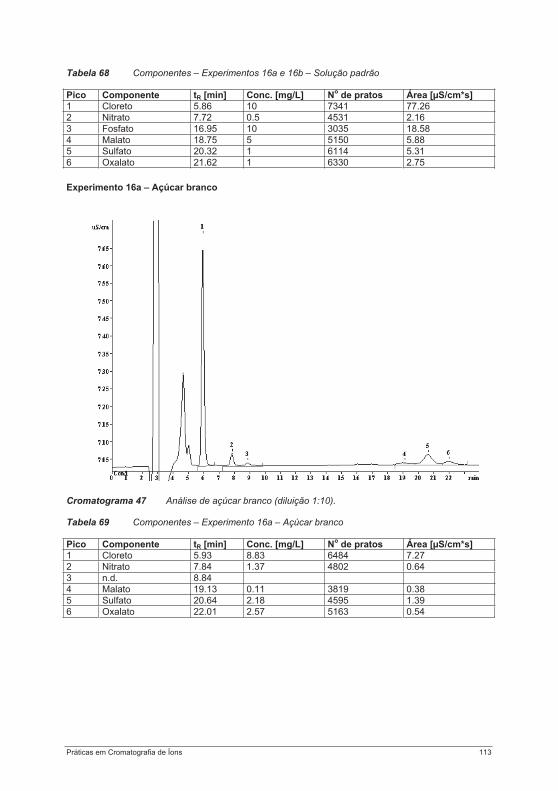
Práticas em Cromatografia de Íons 113
Tabela 68 Componentes – Experimentos 16a e 16b – Solução padrão
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Cloreto 5.86 10 7341 77.26 2 Nitrato 7.72 0.5 4531 2.16 3 Fosfato 16.95 10 3035 18.58 4 Malato 18.75 5 5150 5.88 5 Sulfato 20.32 1 6114 5.31 6 Oxalato 21.62 1 6330 2.75
Experimento 16a – Açúcar branco
Cromatograma 47 Análise de açúcar branco (diluição 1:10).
Tabela 69 Componentes – Experimento 16a – Açúcar branco
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Cloreto 5.93 8.83 6484 7.27 2 Nitrato 7.84 1.37 4802 0.64 3 n.d. 8.84 4 Malato 19.13 0.11 3819 0.38 5 Sulfato 20.64 2.18 4595 1.39 6 Oxalato 22.01 2.57 5163 0.54

114 Monografia Metrohm
Experimento 16b – Açúcar mascavo
Cromatograma 48 Análise de açúcar mascavo (diluição 1:10)
Tabela 70 Componentes – Experimento 16b – Açúcar mascavo
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Cloreto 5.81 232.60 7502 209.60 2 Nitrato 7.66 1.58 983 0.74 3 n.d. 12.75 4 Fosfato 16.61 16.49 4952 3.09 5 Malato 18.54 30.35 5389 3.56 6 Sulfato 20.38 200.92 5843 96.95 7 Oxalato 21.45 10.05 6986 2.82
Observações
Com o açúcar mascavo, os contaminantes podem eluir em tempos de retenção retardados; isto pode interferir com os cromatogramas seguintes se o tempo de corrida for muito curto. É por isso que o açúcar branco deve ser medido primeiro, e em seguida, o mascavo.

Práticas em Cromatografia de Íons 115
4.3.10. Experimento 17 – Contaminantes em peróxido de hidrogênio
O peróxido de hidrogênio puro (H2O2) se funde a 0,4°C e entra em ebulição a 150°C. O peróxido de hidrogênio puro só pode ser preparado por cristalização fracionada sob condições restritas de segurança. O H2O2 comercial está disponível como uma solução aquosa com concentrações de 35, 50, 60 ou 70%. Estas soluções são purificadas através de trocadores iônicos ou por destilação. A decomposição fortemente exotérmica de H2O2 em H2O e O2 não ocorre espontaneamente, porém metais pesados agem como catalisadores.
2 H2O2 ===> 2 H2O + O2 + 196.2 kJ
Íons de metais pesados são ligados pela adição de estabilizadores tais como polifosfatos, EDTA, estanato etc. Isto inibe a decomposição. A propriedade característica do H2O2 é seu efeito oxidante. O potencial padrão (E0) é +1,8 V em pH = 0 e +0,78 V em pH = 14.
O peróxido de hidrogênio é um dos produtos mais importantes no setor de química básica com uma produção anual de 2,7 milhões de toneladas. É produzido industrialmente pela conversão de hidroquinona com O2 atmosférico. Isto produz o correspondente antraquinona e H2O2. A antraquinona é reduzida novamente a hidroquinona por hidrogênio natural e um catalisador de paládio, retornando ao sistema.
Um dos processos que mais utiliza o H2O2 é o alvejamento de papel e celulose. Em muitos países, alvejantes clorados (Cl2 ou ClO2), com os quais formam subprodutos organo-clorados dificilmente biodegradáveis no efluente, têm sido parcial ou totalmente substituídos por outros métodos. Isto significa que o H2O2 tem se tornado muito importante.
Outras áreas de aplicação são:
Remoção de ferro e manganês de água potável;
Descontaminação de efluentes contendo cianetos (CN– CNO–), por exemplo, efluente de banhos galvânicos, pela ação do H2O2 puro ou pela mistura H2O2/H2SO4;
A combinação de H2O2 e radiação UV provê radicais OH (radicais hidroxilas), os quais são oxidantes extremamente fortes (E0 = +2.8 V). Estes são de interesse para a purificação de tipos especiais de efluente;
O desenvolvimento de um método muito promissor para a produção de óxido de propileno a partir de propeno e H2O2 (catalisador de titânio), ao invés do método da cloridrina que gera poluentes;
Entre os produtos secundários de H2O2, o perborato de sódio e o percarbonato de sódio são os mais importantes; estes são usados como alvejantes em sabão em pó;
H2O2 é usado na indústria eletrônica para limpar camadas de silício e circuitos impressos.
A determinação de contaminantes em H2O2 é de grande importância, principalmente na indústria eletrônica mencionada acima. Mesmo em menor quantidade de íons não identificados (ng/L ou ppt) diminui significantemente o rendimento das camadas como, por exemplo, o fosfato e o nitrato causam n-desarranjos cristalinos no cristal de silício.
Em concentrações elevadas, o peróxido de hidrogênio afeta tanto a coluna de separação como o equilíbrio de carbonato do eluente (vide Cromatograma 49). É por isso que a eliminação de matriz (vide capítulo 4.3.2) é aplicada na análise de traços com as seguintes vantagens: Primeiro, as soluções concentradas podem ser analisadas sem diluição; segundo, volumes maiores de amostra podem ser usados (pré-concentração).
Conteúdo do aprendizado
Controle do processo;
Preparação da amostra;
Pré-concentração da amostra e eliminação da matriz;
Seletividade de colunas diferentes.

116 Monografia Metrohm
Tabela 71 Parâmetros – Experimentos 17a e 17b
Coluna 6.1006.430 Metrosep A Supp 4 Eluente Na2CO3 a 1,0 mmol/L / NaHCO3 a 4,0 mmol/L + 20% acetona
Condutividade após supressão química aprox. 7 μS/cm Amostra Peróxido de hidrogênio 30%, ultrapuro, diluição 1:30 Método exp_17_s_e2.mtw Sistema asupp.smt Fluxo 1,0 mL/min Pressão 6,6 MPa Tempo de análise 20 min Coluna de pré-concentração
6.1006.300 Metrosep Anion
Introdução da amostra 2 mL da amostra em coluna de pré-concentração; para eliminação da matriz, lavar com 4 mL de água ultrapura
Supressor Agentes de regeneração: H2SO4 a 50 mmol/L e água ultrapura alimentados continuamente com mudança automática dos canais de supressão, regeneração e lavagem
Polaridade +
Preparação do eluente
Dissolver 106 mg de carbonato de sódio (anidro) e 336 mg de hidrogenocarbonato de sódio em 800 mL de água ultrapura e, então, adicionar 200 mL de acetona.
Introdução da amostra
Lavar com muito cuidado e por várias vezes com água ultrapura. Passar 2 mL da amostra através da coluna de pré-concentração e lavar com 4 mL de água ultrapura.
Cromatograma 49 Solução padrão para a determinação de ânions em peróxido de hidrogênio.
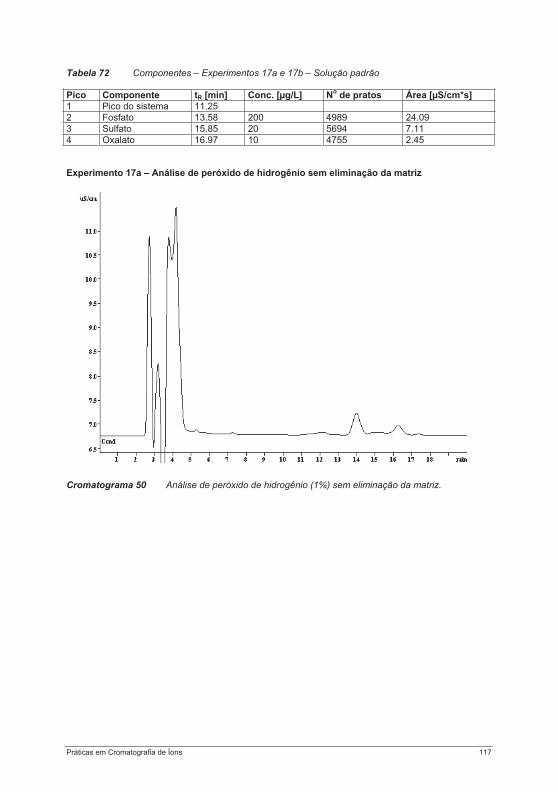
Práticas em Cromatografia de Íons 117
Tabela 72 Componentes – Experimentos 17a e 17b – Solução padrão
Pico Componente tR [min] Conc. [μg/L] No de pratos Área [μS/cm*s] 1 Pico do sistema 11.25 2 Fosfato 13.58 200 4989 24.09 3 Sulfato 15.85 20 5694 7.11 4 Oxalato 16.97 10 4755 2.45
Experimento 17a – Análise de peróxido de hidrogênio sem eliminação da matriz
Cromatograma 50 Análise de peróxido de hidrogênio (1%) sem eliminação da matriz.
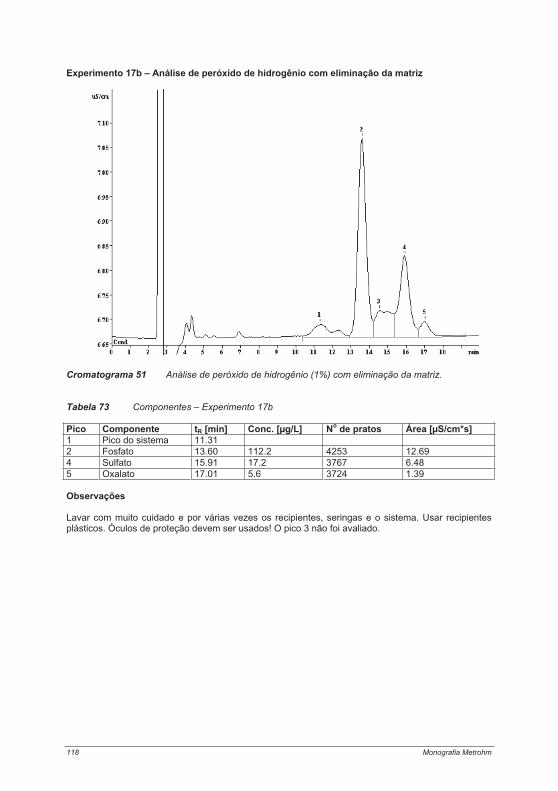
118 Monografia Metrohm
Experimento 17b – Análise de peróxido de hidrogênio com eliminação da matriz
Cromatograma 51 Análise de peróxido de hidrogênio (1%) com eliminação da matriz.
Tabela 73 Componentes – Experimento 17b
Pico Componente tR [min] Conc. [μg/L] No de pratos Área [μS/cm*s] 1 Pico do sistema 11.31 2 Fosfato 13.60 112.2 4253 12.69 4 Sulfato 15.91 17.2 3767 6.48 5 Oxalato 17.01 5.6 3724 1.39
Observações
Lavar com muito cuidado e por várias vezes os recipientes, seringas e o sistema. Usar recipientes plásticos. Óculos de proteção devem ser usados! O pico 3 não foi avaliado.

Práticas em Cromatografia de Íons 119
Experimento 17c – Análise de peróxido de hidrogênio com eliminação da matriz em uma coluna de alta eficiência
Tabela 75 Parâmetros – Experimento 17c
Coluna 6.1006.530 Metrosep A Supp 5 (4 x 250 mm) Eluente NaHCO3 a 1 mmol/L / Na2CO3 a 3,2 mmol/L
Condutividade após supressão química aprox. 15 μS/cm Amostra Peróxido de hidrogênio 30%, ultrapuro Método exp_17c_s.mtw Sistema asupp5.smt Fluxo 1,0 mL/min Pressão 11 MPa Tempo de análise 30 min Coluna de pré-concentração
6.1006.300 Metrosep Anion
Introdução da amostra 1 mL da amostra na coluna de pré-concentração; para eliminação da matriz, lavar com 1 mL de água ultrapura
Supressor Agentes de regeneração: H2SO4 a 50 mmol/L e água ultrapura alimentados continuamente com mudança automática dos canais de supressão, regeneração e lavagem
Polaridade +
Preparação do eluente
Dissolver 339 mg de carbonato de sódio (anidro) e 84 mg de hidrogenocarbonato de sódio em 1 L de água ultrapura.
Introdução da amostra
Lavar o sistema com muito cuidado com água ultrapura, então, passar 1 mL da amostra através da coluna de pré-concentração e lavar com 1 mL de água ultrapura.
Cromatograma 52 Análise de peróxido de hidrogênio (30%) com eliminação da matriz em coluna Metrosep A Supp 5.
120 Monografia Metrohm
Tabela 76 Componentes – Experimento 17c
Pico Componente tR [min] Conc. [μg/L] No de pratos Área [μS/cm*s] 3 Acetato 6,3 6,8 2850 219 4 Formiato 6,7 0,77 8740 123 6 Cloreto 8,9 0,78 17810 380 7 Pico do sistema 13,0 8 Nitrato 16,4 0,15 19100 37 10 Sulfato 23,3 0,35 16640 86 13 Oxalato 27,0 0,04 12100 7,7
Observações
Os picos 1, 2, 5, 9, 10, 11 e 12 não foram avaliados.

Práticas em Cromatografia de Íons 121
4.4. Experimentos para a determinação de cátions
4.4.1. Experimento 18 – Metais alcalinos e metais alcalino-terrosos em água potável
Além dos gases dissolvidos O2, N2 e CO2, a água na natureza também contém sais que são retirados principalmente do solo e rochas que esta água penetra. Outros tipos de ingredientes da água são os compostos orgânicos polares que se originam, por exemplo, das camadas de húmus no solo. A contaminação por efluente é também uma possível fonte de vários sais e compostos orgânicos. Os sais mais importantes são os cloretos, sulfatos e hidrogenocarbonatos de sódio, cálcio e magnésio.
Um parâmetro importante da água potável, tanto para o seu uso como um alimento, assim como para o uso em processos de lavagem ou processos industriais é a chamada dureza da água.
A dureza total da água é entendida como sendo a soma das concentrações molares (mmol/L) dos íons cálcio e magnésio. A dureza que pode ser removida pelo aquecimento é conhecida como dureza carbonato (anteriormente, dureza temporária). A dureza residual é causada pelos íons sulfato e cloreto, cujos sais de Ca e Mg não podem ser precipitados pelo aquecimento.
Nos sabões clássicos – sais sódicos de ácidos graxos – os íons cálcio e magnésio formam compostos insolúveis dos respectivos íons. Os dois são ineficazes em lavagem e são também depositados nos tecidos como «metal cinzento». Modernos agentes para lavagem contêm alquil-sulfatos que não formam compostos insolúveis de cálcio e magnésio. As zeólitas, que são também adicionadas a estes agentes, funcionam como trocadores iônicos e ligam os íons Ca2+ e Mg2+. Isto efetivamente impede a precipitação de CaCO3 insolúvel e de carbonato básico de magnésio sob aquecimento.
Normas sobre qualidade da água potável, as quais são implementações das diretivas da CE para água potável (80/778/EEC), estipulam os seguintes valores para cátions:
NH4+ 0.5 mg/L
Na+ 150 mg/L K+ 12 mg/L Mg2+ 50 mg/L Ca2+ 400 mg/L
Uma ingestão elevada de íons sódio pelos seres humanos causa pressão alta, entre outras alterações. A necessidade diária para uma pessoa é de 1 g de Na+, atualmente verificamos que o consumo, em média, é maior (de 3 a 7g). Em todas as análises realizadas, as amostras foram acidificadas com ácido nítrico (pH 2,5 a 3,5), caso contrário, alguns resultados poderão não se reproduzir, para cátions bivalentes.
Conteúdo do aprendizado
Controle do alimento “Número 1”;
Checando as informações contidas nas garrafas de água mineral;
Por que é necessário acidificar as amostras?

122 Monografia Metrohm
Tabela 76 Parâmetros – Experimento 18
Coluna 6.1010.210 Metrosep C2 (4 x 100 mm) Eluente Ácido tartárico a 4 mmol/L / ácido dipicolínico a 0,75 mmol/L
Condutividade aprox. 590 μS/cm Amostra – Água potável, acidificada a pH = 2,5 a 3,5 (+ aprox. 100 μL de HNO3 a 2 mol/L
para 100 mL da amostra) – Água mineral, acidificada a pH = 2,5 a 3,5 (+ aprox. 100 μL de HNO3 a 2 mol/L para 100 mL da amostra)
Método exp_18_c.mtw Sistema cátion.smt Fluxo 1 mL/min Pressão 7 MPa Tempo de análise 12 min Loop 10 μL Polaridade –
Preparação do eluente
Dissolver, sob aquecimento, 600 mg de ácido tartárico e 125 mg de ácido dipicolínico em 100 mL de água ultrapura e completar para 1 L com água ultrapura.
Cromatograma 53 Solução padrão para determinação de cátions em água potável

Práticas em Cromatografia de Íons 123
Tabela 77 Componentes – Experimento 18 – Padrão
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Sódio 3,8 0,5 4200 1,5 2 Potássio 6,5 0,2 3090 0,9 3 Cálcio 8,4 20 1960 66 4 Magnésio 11,0 5 2700 34
Cromatograma 54 Água de torneira (Herisau, Suíça, diluição 1:10).
Tabela 79. Componentes – Experimento 18 – Água de torneira
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Sódio 3,8 5 4840 1,4 2 Cálcio 8,7 85 2640 29 3 Magnésio 11,2 19 3520 13

124 Monografia Metrohm
Cromatograma 55 Água mineral (diluição 1:10)
Tabela 79 Componentes – Experimento 18 – Água mineral
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Sódio 3,7 4,3 4190 1,3 2 Potássio 6,4 0,8 6350 0,3 3 Cálcio 8,3 344 1520 113 4 Magnésio 10,9 62 2550 43
Observações
Todas as soluções devem ser mantidas em recipientes de plástico. Para uma correta determinação do sódio, qualquer contato com vidro deve ser evitado. Os valores do pH da solução padrão e da amostra devem estar entre 2,5 e 3,5.

Práticas em Cromatografia de Íons 125
4.4.2. Experimento 19 – Determinação de metais de transição
Agentes complexantes são adicionados à fase móvel a fim de separar íons de metais de transição. Eles reduzem a densidade da carga e melhoram a seletividade da separação, a qual é relativamente baixa para íons de metais de transição com a mesma carga. Além do equilíbrio entre a resina trocadora e o íon analito, o agente complexante que foi adicionado promoverá um equilíbrio adicional, isto é, entre o íon metálico e este complexante. Este último equilíbrio é também influenciado pelo pH da solução, se várias etapas da dissociação do agente complexante forem possíveis ou se os íons metálicos formarem hidróxi-complexos.
Os ácidos orgânicos fracos são freqüentemente usados como agentes complexantes, por exemplo, ácido tartárico, ácido cítrico, ácido oxálico, ácido 2,6-piridinodicarboxílico (ácido dipicolínico – vide capítulo 4.2.6 – Experimento 6 – Alteração da seletividade pelo uso de agentes complexantes). O número de ligantes L ao redor do íon metálico central (Me) pode variar, por exemplo, MeL, MeL2,MeL3. A carga resultante pode também diferir. Dependendo da carga do íon metálico, dos ligantes e sua quantidade, podem estar presentes complexos aniônicos, neutros ou catiônicos. Isto significa que, dependendo de suas cargas, os complexos podem ser separados em trocadores catiônicos ou trocadores aniônicos. Teoricamente é possível o uso de trocadores contendo os dois grupos de trocadores, catiônicos e aniônicos.
A separação pode ser influenciada e otimizada pela variação do pH e agentes complexantes – dois ou mais agentes complexantes diferentes podem ser adicionados à fase móvel. Em parte, as influências são contrárias e difíceis de serem previstas. Algumas informações gerais são dadas abaixo:
Estabilidades diferentes (constantes de formação do complexo) influenciam na separação.
A adição de um solvente orgânico, por exemplo, a acetona, ao eluente também influencia na separação. Os íons lipofílicos eluem mais rapidamente após a adição.
Em cromatografia de troca catiônica, a etilenodiamina tem se mostrado um bom eluente. Sob condições normais, ele está presente como um cátion carregando dois prótons.
Um aumento no pH resulta em uma redução do tempo de retenção pela redução da carga total.
Um aumento na concentração de ligante na fase móvel, além de remover o equilíbrio do complexante, também aumenta a concentração do contra-íon H+ ou Na+ e acelera a remoção do complexo dos sítios de troca negativamente carregados, ou seja, o tempo de analise é reduzido.
A detecção dos complexos pode ser realizada através da condutividade ou fotometricamente (derivatização pós-coluna)
Para detecção de condutividade, apenas a detecção sem supressão química pode ser usada, uma vez que hidróxidos insolúveis seriam formados pela reação supressora e os únicos ânions eluentes disponíveis seriam carbonato ou hidróxido.
A detecção fotométrica requer derivatização pós-coluna, na qual, um agente complexante cromóforo adicional remove o complexo com o eluente (por exemplo, ácido tartárico, ácido oxálico etc.). Este complexo recentemente formado, que absorve radiação UV ou VIS, é detectado caso a máxima absorção dos ligantes livres não ultrapasse o valor do máximo do complexo. Assim como, para a maioria dos corantes complexantes, a máxima absorção dos complexos de metais de transição individuais ocorre em regiões de diferentes comprimentos de onda, o que acarreta limites de detecção de metais individuais bem diferentes.
Conteúdo do aprendizado
Influência do eluente na seletividade;
Complexantes de íons de metais de transição;
Agentes complexantes para a detecção fotométrica;
Fotometria.

126 Monografia Metrohm
Tabela 80 Parâmetros – Experimentos 19a e 19b
Coluna 6.1007.000 Nucleosil 5SA (4 x 125 mm) Eluente a) ácido tartárico a 4 mmol/L, ácido cítrico a 0,5 mmol/L, etilenodiamina a 3
mmol/L, 5% acetona Condutividade aprox. 500 μS/cm b) ácido oxálico a 3,5 mmol/L, 5% acetona, pH = 4 (aprox. 120 μL de
etilenodiamina) Condutividade aprox. 350 μS/cm
Amostra Água de torneira Método exp_19_c.mtw Sistema nucleosil.smt Fluxo 1,5 mL/min Pressão 13 MPa Tempo de análise a) 12 min
b) 13 min Loop 100 μL Polaridade –
Experimento 19a – Determinação de metais de transição com um eluente contendo ácido tartárico, ácido cítrico, etilenodiamina e acetona
Preparação do eluente
Dissolver 600 mg de ácido tartárico e 105 mg de ácido cítrico em água ultrapura. Adicionar 200 μL de etilenodiamina e 50 mL de acetona e completar para 1L com água ultrapura.
Cromatograma 56 Solução padrão 1 para determinação de metais de transição.

Práticas em Cromatografia de Íons 127
Tabela 81 Componentes – Experimento 19a – Solução padrão 1
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Níquel 3,4 5 1020 40 2 Zinco 4,4 5 5640 36 3 Cobalto 5,6 5 4370 34 4 Ferro(II) 8,1 10 6150 30 5 Cálcio 9,8 5 6340 29 6 Magnésio 10,8 5 6390 39
Cromatograma 57 Solução padrão 2 para determinação de metais de transição
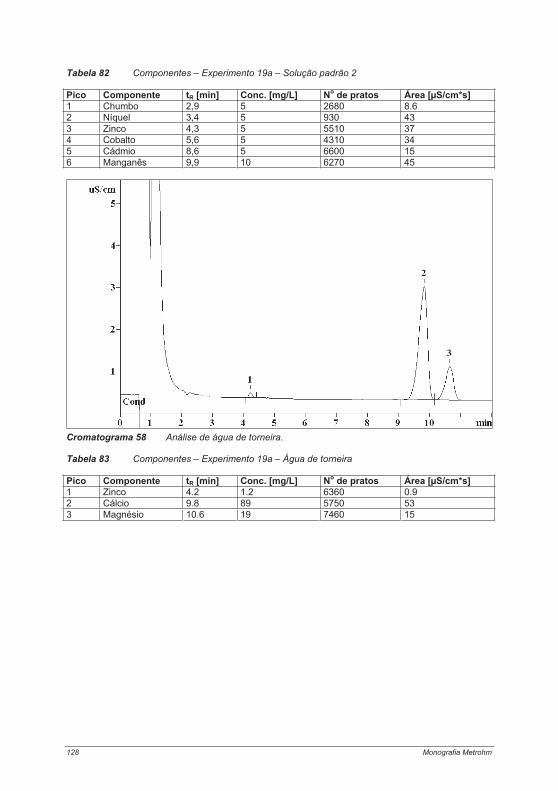
128 Monografia Metrohm
Tabela 82 Componentes – Experimento 19a – Solução padrão 2
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Chumbo 2,9 5 2680 8.6 2 Níquel 3,4 5 930 43 3 Zinco 4,3 5 5510 37 4 Cobalto 5,6 5 4310 34 5 Cádmio 8,6 5 6600 15 6 Manganês 9,9 10 6270 45
Cromatograma 58 Análise de água de torneira.
Tabela 83 Componentes – Experimento 19a – Água de torneira
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Zinco 4.2 1.2 6360 0.9 2 Cálcio 9.8 89 5750 53 3 Magnésio 10.6 19 7460 15
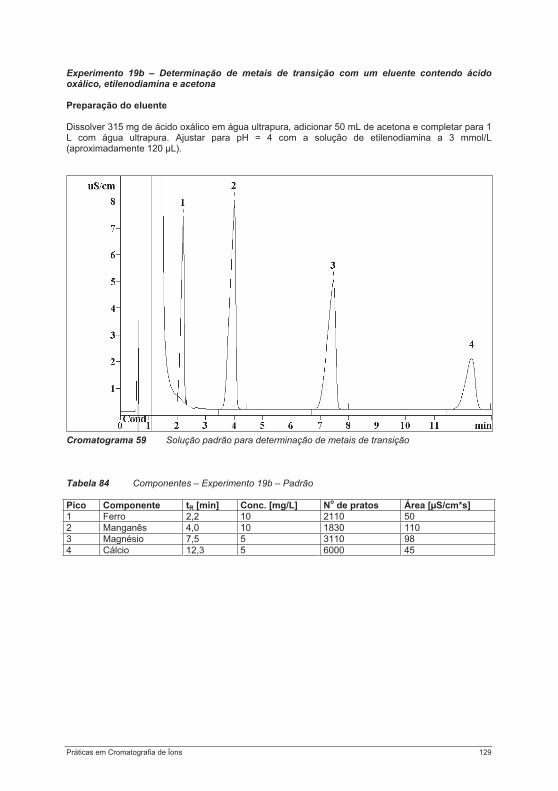
Práticas em Cromatografia de Íons 129
Experimento 19b – Determinação de metais de transição com um eluente contendo ácido oxálico, etilenodiamina e acetona
Preparação do eluente
Dissolver 315 mg de ácido oxálico em água ultrapura, adicionar 50 mL de acetona e completar para 1 L com água ultrapura. Ajustar para pH = 4 com a solução de etilenodiamina a 3 mmol/L (aproximadamente 120 μL).
Cromatograma 59 Solução padrão para determinação de metais de transição
Tabela 84 Componentes – Experimento 19b – Padrão
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Ferro 2,2 10 2110 50 2 Manganês 4,0 10 1830 110 3 Magnésio 7,5 5 3110 98 4 Cálcio 12,3 5 6000 45
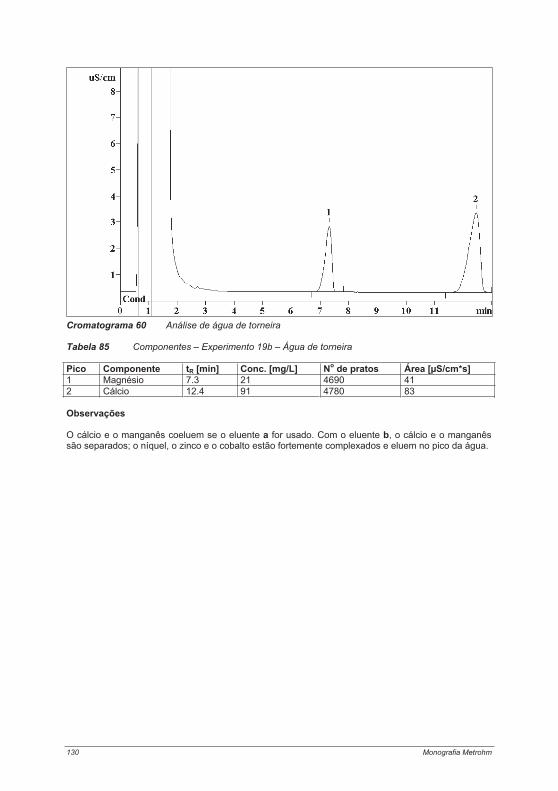
130 Monografia Metrohm
Cromatograma 60 Análise de água de torneira
Tabela 85 Componentes – Experimento 19b – Água de torneira
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Magnésio 7.3 21 4690 41 2 Cálcio 12.4 91 4780 83
Observações
O cálcio e o manganês coeluem se o eluente a for usado. Com o eluente b, o cálcio e o manganês são separados; o níquel, o zinco e o cobalto estão fortemente complexados e eluem no pico da água.

Práticas em Cromatografia de Íons 131
4.4.3. Experimento 20 – Contaminantes em sílica gel – determinação de íons cálcio e magnésio
A sílica gel é produzida, por exemplo, pela hidrólise de silicato de sódio e pela remoção quase completa da água; ela tem a seguinte estrutura:
Si
Si
SiSi
Si
O
O
O
OO
O
O
OHOH
OH
Figura 36 Estrutura da sílica gel
Além de seu uso como um agente técnico de absorção, a sílica gel é usada tanto em cromatografia a gás como em cromatografia líquida de alta pressão.
Em cromatografia gasosa, originalmente, apenas colunas empacotadas eram usadas – estas ainda são usadas atualmente, em uma menor proporção. Uma fase líquida, por exemplo, óleo de silicone, é aplicada para um arraste particulado como a sílica gel. O número de pratos teóricos de tais colunas é baixo em comparação ao das colunas capilares usadas atualmente.
Em cromatografia líquida de alta eficiência (CLAE), a sílica gel é usada como uma fase estacionária ou material de arraste para adsorção, fase reversa (RP), fase normal e cromatografia de íons. Para algumas destas aplicações, a superfície da sílica gel deve ser modificada com grupos funcionais (por exemplo, C18 ou grupos trocadores iônicos).
Em cromatografia de adsorção, a sílica gel também é usada como fase estacionária ou juntamente com Al2O3. A separação cromatográfica é baseada em ligações dipolo, dipolo-dipolo, ligações de hidrogênio e ligações -complexos. A cromatografia de adsorção é usada para a separação de substâncias não polares que são difíceis de dissolver em água.
Em cromatografia fase reversa, os grupos silanóis livres da sílica gel são hidrofobizados pelas ligações químicas de longas cadeias alquilas (por exemplo, grupos octila ou octadecila):
Figura 37 Reação de sílica gel com clorosilano (R = C8H17 to C18H37).
Nesta conversão, os grupos silano residuais permanecem; estes podem adsorver compostos polares e originar picos com caudas. Estes grupos silano residuais podem ser desativados por trimetilclorosilano.
Em cromatografia de fase normal, os resíduos polares são quimicamente ligados aos grupos silanos, por exemplo:
(CH2)n C N ou (CH2)n NH2
Uma fase móvel menos polar ou não-polar é usada com esta fase estacionária polar para separação cromatográfica. Na cromatografia de fase reversa, usada em quase 75% de todas as aplicações de HPLC, as polaridades são reversas e uma fase estacionária não-polar é usada com a fase móvel polar. Isto significa que o nome fase reversa tem uma base histórica, visto que este método foi desenvolvido posteriormente.
Em cromatografia de íons, a sílica gel, na qual foram ligados grupos de ácidos sulfônicos, é usada como um trocador catiônico para a análise de íons alcalinos.
A contaminação da sílica gel por íons metálicos é importante em cromatografia, uma vez que podem causar interações não específicas ou valores nulos. Um trocador catiônico fortemente ácido é usado para a determinação destes contaminantes; cátions monovalentes eluem no volume morto e só os

132 Monografia Metrohm
cátions bivalentes são separados. Fe(III) deve ser reduzido a Fe(II) com ácido ascórbico antes da análise.
Conteúdo do aprendizado
Controle de qualidade; Experimento adicional: adição de padrão ao extrato com Fe(III); determinação com e sem a adição de ácido ascórbico (vitamina C); Colunas de separação para CLAE baseadas em sílica gel.
Tabela 86 Parâmetros – Experimento 20
Coluna 6.1007.000 Nucleosil 5SA (4 x 125 mm) Eluente Ácido tartárico a 4 mmol/L, ácido cítrico a 0,5 mmol/L, etilenodiamina a 3
mmol/L, 5% acetona Condutividade aprox. 500 μS/cm
Amostra Sílica gel (5% solução) Método exp_20_c.mtw Sistema nucleosil.smt Fluxo 1,5 mL/min Pressão 13 MPa Tempo de análise 12 min Loop 100 μL Polaridade –
Preparação do eluente
Dissolver 600 mg de ácido tartárico e 105 mg de ácido cítrico em água ultrapura. Adicionar 200 μL de etilenodiamina e 50 mL de acetona e completar para 1 L com água ultrapura.
Cromatograma 61 Solução padrão para a determinação de cátions em sílica gel.

Práticas em Cromatografia de Íons 133
Tabela 87 Componentes – Experimento 20 – Solução padrão
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Cálcio 9,6 0,5 6990 3,1 2 Magnésio 10,5 0,1 7260 0,7
Cromatograma 62 Determinação de cátions em sílica gel.
Tabela 88 Componentes – Experimento 20 – Sílica gel
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] Ferro 8,1 n.d. – – 1 Cálcio 9,7 9,4 6850 2,9 2 Magnésio 10,6 1,4 10100 0,4

134 Monografia Metrohm
H NCH2
CH2
CH2
CH2
OH
OHHO CH2 CH2 N
CH2
CH2
CH2
CH2
OH
OHH2N CH2 CH2 OH
4.4.4. Experimento 21 – Cosméticos e proteção contra corrosão: determinação de etanolaminas e metais alcalinos
As três etanolaminas seguintes, cujos nomes sistemáticos são aminoetanol, iminodietanol e nitrilotrietanol, são produzidas pela reação de amônia com óxido de etileno.
As três substâncias são usadas para remover CO2 e H2S de misturas de gases, por exemplo, para a remoção de dióxido de carbono de gás síntese e na síntese de amônia, bem como para a remoção de sulfeto de hidrogênio em gases de refinarias.
NR3 + H2O + CO2 HNR3+ + HCO3
-T
Em temperaturas maiores, o CO2 é novamente liberado e a etanolamina NR3 retorna ao processo de absorção.
2NR3 + H2ST
(HNR3)2S
O H2S novamente liberado em elevada temperatura é oxidado para enxofre líquido em “unidades de Claus”; esta é, então, convertida para ácido sulfúrico via SO2 e SO3.
A monoetanolamina (MAK = concentração máxima permitida em ambiente de trabalho = 3 ppm) é usada em surfactantes como um éster de ácido graxo. A dietanolamina é usada em produtos de proteção de móveis e pisos, graxas de sapatos e lubrificantes.
A trietanolamina é empregada em sabões com ácidos graxos, por exemplo, com o ácido esteárico C17H35COOH. Estes são facilmente solúveis em água e em óleos minerais e são, portanto, usados como emulsificantes. Eles podem também estar contidos em preparações cosméticas (por exemplo, espuma de barbear).
A monoetanolamina é usada como inibidor de corrosão em usinas hidrelétricas, uma vez que ele mantém um pH fracamente alcalino e arrasta o excesso de íons H+.
Para serem determinados por cromatografia de íons, as etanolaminas são protonadas pela adição de ácido e, então, podem ser determinados na forma de seus derivados de amônio.
Conteúdo do aprendizado
Determinação de cátions inorgânicos e orgânicos;
Mudança na seletividade pela adição de agentes complexantes;
Determinação de aminas por cromatografia de íons.
Figura 38. Aminoetanol Figura 39. Iminodietanol Figura 40. Nitrilotrietanol
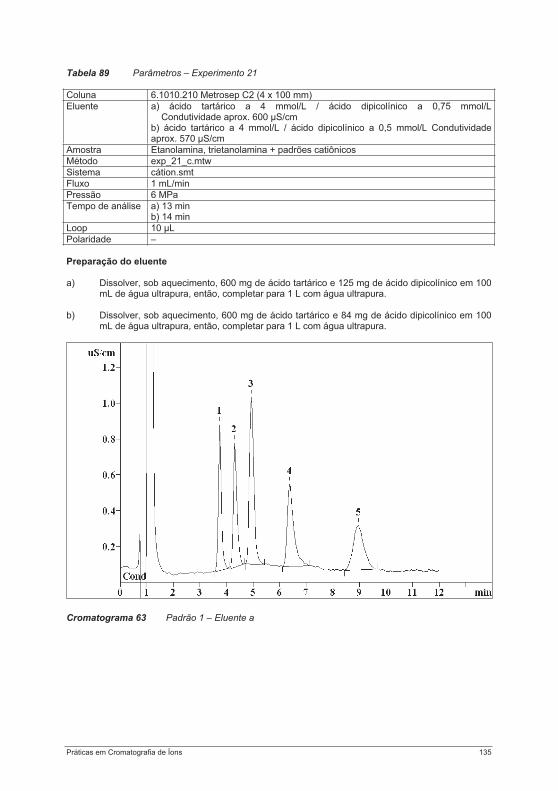
Práticas em Cromatografia de Íons 135
Tabela 89 Parâmetros – Experimento 21
Coluna 6.1010.210 Metrosep C2 (4 x 100 mm) Eluente a) ácido tartárico a 4 mmol/L / ácido dipicolínico a 0,75 mmol/L
Condutividade aprox. 600 μS/cm b) ácido tartárico a 4 mmol/L / ácido dipicolínico a 0,5 mmol/L Condutividade aprox. 570 μS/cm
Amostra Etanolamina, trietanolamina + padrões catiônicos Método exp_21_c.mtw Sistema cátion.smt Fluxo 1 mL/min Pressão 6 MPa Tempo de análise a) 13 min
b) 14 min Loop 10 μL Polaridade –
Preparação do eluente
a) Dissolver, sob aquecimento, 600 mg de ácido tartárico e 125 mg de ácido dipicolínico em 100 mL de água ultrapura, então, completar para 1 L com água ultrapura.
b) Dissolver, sob aquecimento, 600 mg de ácido tartárico e 84 mg de ácido dipicolínico em 100 mL de água ultrapura, então, completar para 1 L com água ultrapura.
Cromatograma 63 Padrão 1 – Eluente a
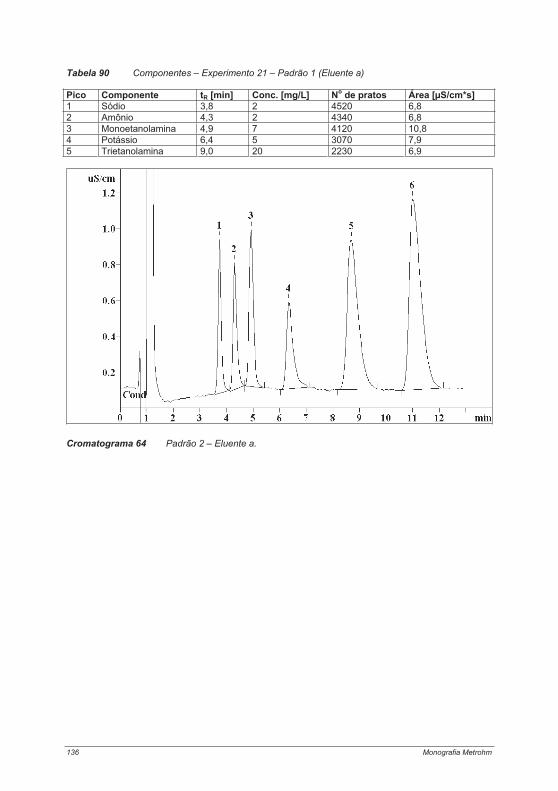
136 Monografia Metrohm
Tabela 90 Componentes – Experimento 21 – Padrão 1 (Eluente a)
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Sódio 3,8 2 4520 6,8 2 Amônio 4,3 2 4340 6,8 3 Monoetanolamina 4,9 7 4120 10,8 4 Potássio 6,4 5 3070 7,9 5 Trietanolamina 9,0 20 2230 6,9
Cromatograma 64 Padrão 2 – Eluente a.

Práticas em Cromatografia de Íons 137
Tabela 91 Componentes – Experimento 21 – Padrão 2 (eluente a)
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Sódio 3,7 2 4500 7,2 2 Amônio 4,3 2 4190 7,0 3 Monoetanolamina 4,9 7 4380 9,8 4 Potássio 6,4 5 2930 8,6 5 Cálcio + trietanolamina 8,7 5 + 20 2070 24 6 Magnésio 11,0 5 2970 32
Cromatograma 65 Padrão 2 – Eluente b.
Tabela 92 Componentes – Experimento 21 – Padrão 2 (eluente b)
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Sódio 3,9 2 4700 7,2 2 Amônio 4,5 2 4110 7,0 3 Monoetanolamina 5,1 7 4400 9,6 4 Potássio 6,6 5 2920 8,4 5 Trietanolamina 9,3 20 2400 7,3 6 Cálcio 10,8 5 3240 16 7 Magnésio 12,2 5 2820 32
Observações
Se o eluente a for usado, então o cálcio e a monoetanolamina coeluem.
Todas as soluções devem ser mantidas em recipientes plásticos para uma correta determinação do sódio, qualquer contato com vidro deve ser evitado.

138 Monografia Metrohm
4.4.5. Experimento 22 – Metais alcalinos e metais alcalino-terrosos em vinho
As substâncias minerais presentes no suco de uva são, principalmente, fosfatos de cálcio, magnésio e potássio. As concentrações destes compostos variam entre 3 e 5 g/L. O conteúdo mineral, também conhecido como “cinzas” do vinho, é menor do que o do suco, uma vez que parte dos minerais é consumida pelas leveduras durante seu metabolismo. O conteúdo mineral é também reduzido pela separação de tartaratos. Isto significa que o vinho contém só 1,8 a 2,5 g/L de “cinzas”. Importantes constituintes catiônicos são dados como óxidos, tais como: K2O (aprox. 40%), MgO (aprox. 6%), CaO (aprox. 4%) e Na2O (aprox. 2%). (vide Experimento 12)
Conteúdo do aprendizado
Análise alimentícia;
Análise do processo – checando a precipitação do tartarato.
Tabela 93 Parâmetros – Experimento 22
Coluna 6.1010.210 Metrosep C2 (4 x 100 mm) Eluente Ácido tartárico a 4 mmol/L / ácido dipicolínico a 0,75 mmol/L
Condutividade aprox. 600 μS/cm Amostra Vinho tinto (Pinot Noir, Suíça), Vinho branco (Fechy, Suíça) Método exp_22_c.mtw Sistema cátion.smt Fluxo 1 mL/min Pressão 6 MPa Tempo de análise 20 min Loop 10 μL Polaridade –
Preparação do eluente
Dissolver, sob aquecimento, 600 mg de ácido tartárico e 125 mg de ácido dipicolínico em 100 mL de água ultrapura, então, completar o volume para 1 L com água ultrapura.
Preparação da amostra
Filtrar e diluir a amostra (1:10).

Práticas em Cromatografia de Íons 139
Cromatograma 66 Solução padrão para determinação de cátions em vinho
Tabela 94 Componentes – Experimento 22 – Padrão
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Sódio 3,7 1 4360 3,5 2 Potássio 5,9 100 1480 180 3 Cálcio 8,6 5 2950 16 4 Magnésio 10,8 10 2150 66
Observações
Todas as soluções devem ser mantidas em recipientes plásticos. Para uma correta determinação do sódio, qualquer contato com vidro deve ser evitado. O pH da solução padrão e da amostra deve estar entre 2,5 e 3,5.
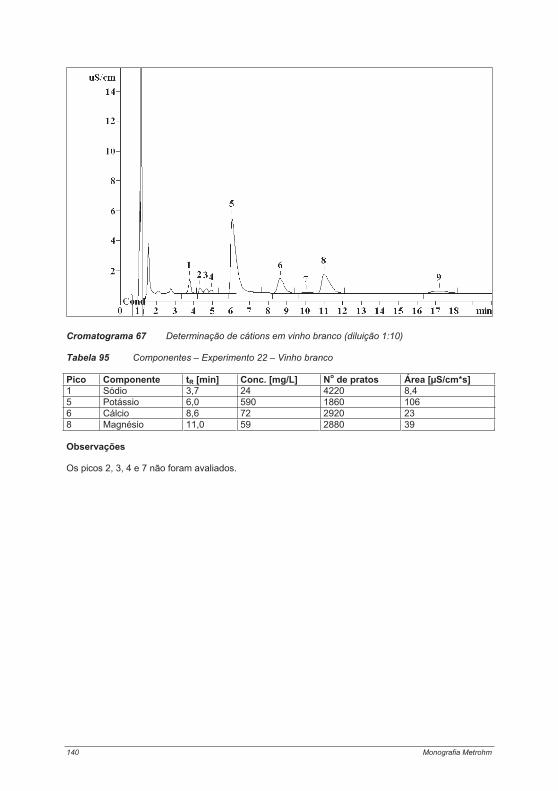
140 Monografia Metrohm
Cromatograma 67 Determinação de cátions em vinho branco (diluição 1:10)
Tabela 95 Componentes – Experimento 22 – Vinho branco
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Sódio 3,7 24 4220 8,4 5 Potássio 6,0 590 1860 106 6 Cálcio 8,6 72 2920 23 8 Magnésio 11,0 59 2880 39
Observações
Os picos 2, 3, 4 e 7 não foram avaliados.
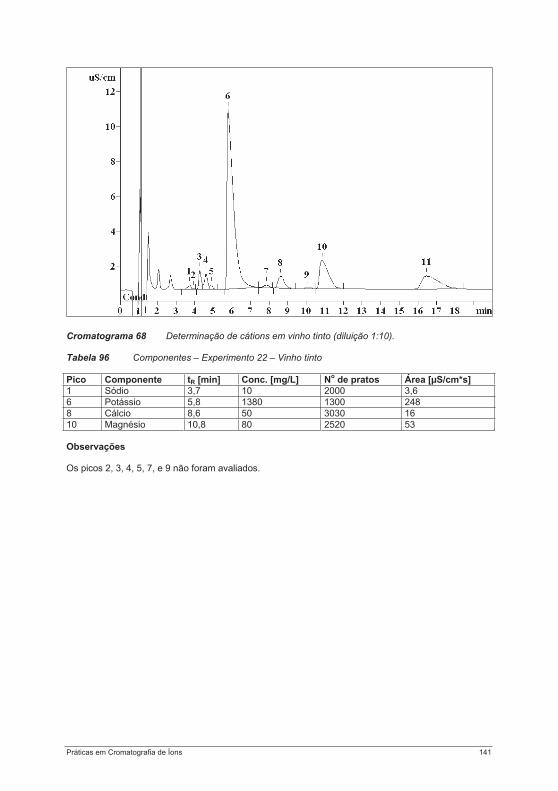
Práticas em Cromatografia de Íons 141
Cromatograma 68 Determinação de cátions em vinho tinto (diluição 1:10).
Tabela 96 Componentes – Experimento 22 – Vinho tinto
Pico Componente tR [min] Conc. [mg/L] No de pratos Área [μS/cm*s] 1 Sódio 3,7 10 2000 3,6 6 Potássio 5,8 1380 1300 248 8 Cálcio 8,6 50 3030 16 10 Magnésio 10,8 80 2520 53
Observações
Os picos 2, 3, 4, 5, 7, e 9 não foram avaliados.

142 Monografia Metrohm
5. Literatura citada [1] H. Small, T. S. Stevens, W. C. Bauman, Anal. Chem. 47 (1975) 1801. [2] J. Weiss, Ionenchromatographie, 2. Aufl. (1991), Verlag Chemie, Weinheim. [3] J. M. Mermet, M. Otto, H. M. Widmer, Analytical Chemistry, 1. Aufl. (1998), Wiley-VCH,
Weinheim, New York. [4] P. R. Haddad, P. E. Jackson, Ion Chromatography – Principles and Applications, 1st ed.
(1990), J. Chromatogr. Library Vol. 46, Elsevier Verlag, Amsterdam. [5] J. S. Fritz, D. T. Gjerde, Ion Chromatography, 3rd ed., Wiley-VCH, Weinheim, 2000. [6] G. Schwedt, Chromatographische Methoden in der Anorganischen Analytik, 1. Aufl. (1980),
Dr. Alfred Hüthig Verlag, Heidelberg. [7] International Union of Pure and Applied Chemistry (IUPAC), Pure & Appl. Chem. 65 (1993),
819.[8] H. Engelhardt, L. Rohrschneider, Deutsche chromatograpische Grundbegriffe zur IUPAC-
Nomenklatur (1998), Universität Saarbrücken. [9] G. Schwedt, Chromatographische Trennmethoden, 3. Aufl. (1994), Thieme Verlag, Stuttgart. [10] V. R. Meyer, Praxis der Hochleistungsflüssigchromatographie, 5. Aufl. (1988), Diesterweg
Verlag, Frankfurt/Main. [11] A. J. P Martin, R. L. M. Synge, Biochem. J. 35 (1941) 1358. [12] B. A. Bindlingmeyer, F. V. Warren, Anal. Chem. 56 (1984) 1583. [13] J. P. Foley, J. G. Dorsey, Anal. Chem. 55 (1983) 730. [14] E. Grushka, L. R. Snyder, J. H. Knox, J. Chrom. Sci. 13 (1975) 25. [15] E. Katz, K. L. Ogan, R. P. W. Scott, J. Chromatogr. A 270 (1983) 51. [16] J. J. van Deemter, F. J. Zuiderweg, A. J. Klinkenberg, Chem. Eng. Sci. 5 (1956) 271. [17] J. H. Knox, H. P. Scott, J. Chromatogr. A 282 (1983) 297. [18] A. Berthold, F. Chartier, J. L. Rocca, J. Chromatogr. A 469 (1989) 53. [19] G. Wedler, Lehrbuch der physikalischen Chemie, 3. Aufl. (1987), Verlag Chemie, Weinheim. [20] G. Schwedt, Fresenius Z. Anal. Chem. 320 (1985) 423. [21] M. J. van Os, J. Slania, C. L. de Ligny, W. E. Hammers, J. Agterdendos, Anal. Chim. Acta 144
(1982) 73. [22] P. R. Haddad, C. E. Cowie, J. Chromatogr. A 303 (1984) 321. [23] A. Diop, A. Jardy, M. Caude, R. Roset, Analusis 14 (1986) 67. [24] A. Jardy, M. Caude, A. Diop, C. Curvale, R. Roset, J. Chromatogr. A 439 (1988) 137. [25] D. R. Jenke, G. K. Pagenkopf, Anal. Chem. 56 (1984) 85 and 88. [26] T. B. Hoover, Sep. Sci. Tech. 17 (1982) 295. [27] P. R. Haddad, P. E. Jackson, A. L. Heckenberg, J. Chromatogr. A 346 (1985) 139. [28] P. Janoš, J. Chromatogr. A 789 (1/2) (1997) 3. [29] P. Janoš, P. Aczel, J. Chromatogr. A 749 (1996) 115. [30] P. Kolla, J. Köhler, G. Schomburg, Chromatographia 23 (1987) 465. [31] C. A. Pohl, J. R. Stillian, P. E. Jackson, J. Chrom. A 789 (1997) 29. [32] K. Dorfner (Ed.), Ion Exchangers, 4th ed. (1991), Walter de Gruyter Verlag, Berlin. [33] D. T. Gjerde, J. S. Fritz, G. Schmuckler, J. Chromatogr. A 186 (1979) 509.