scaly skin a fishy story - mm3 admin : login · scaly skin§ a fishy story workshop: common...
TRANSCRIPT

§Scaly SkinA Fishy Story
Workshop:Common Allergic Skin Disorders
ALLSA CongressDr Candice Royal

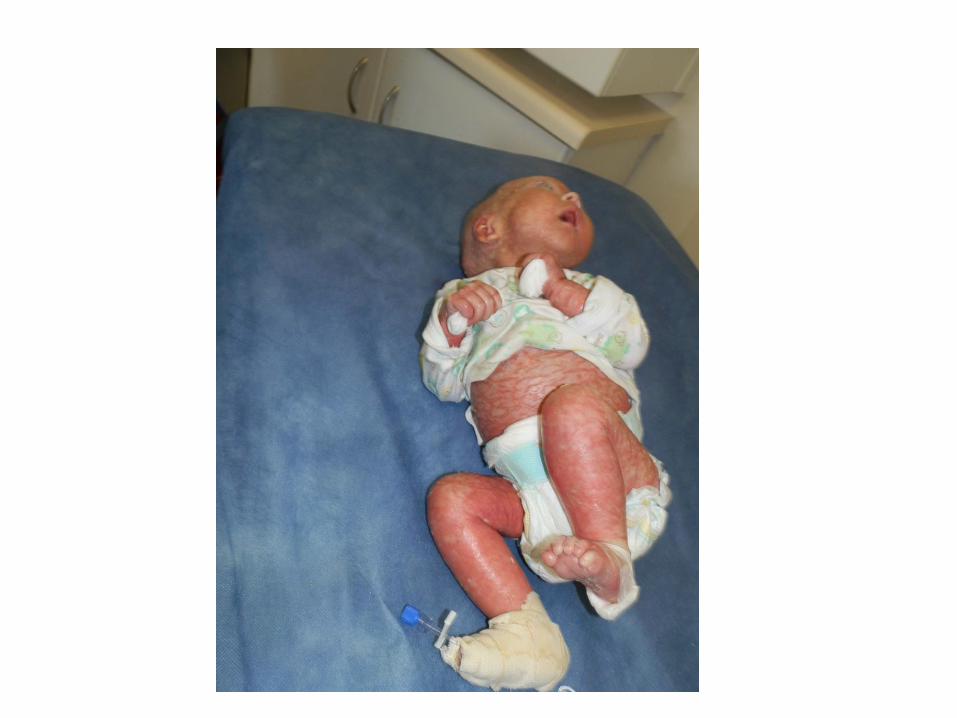
Baby S
• First-born to married non-consanguineous parents
• Uncomplicated antenatal and birth history
• Birth weight 3330g, no abnormalities detected
• Discharged after 6 hours
• Exclusively breast-fed
• Thrived during first month of life

Age One Month
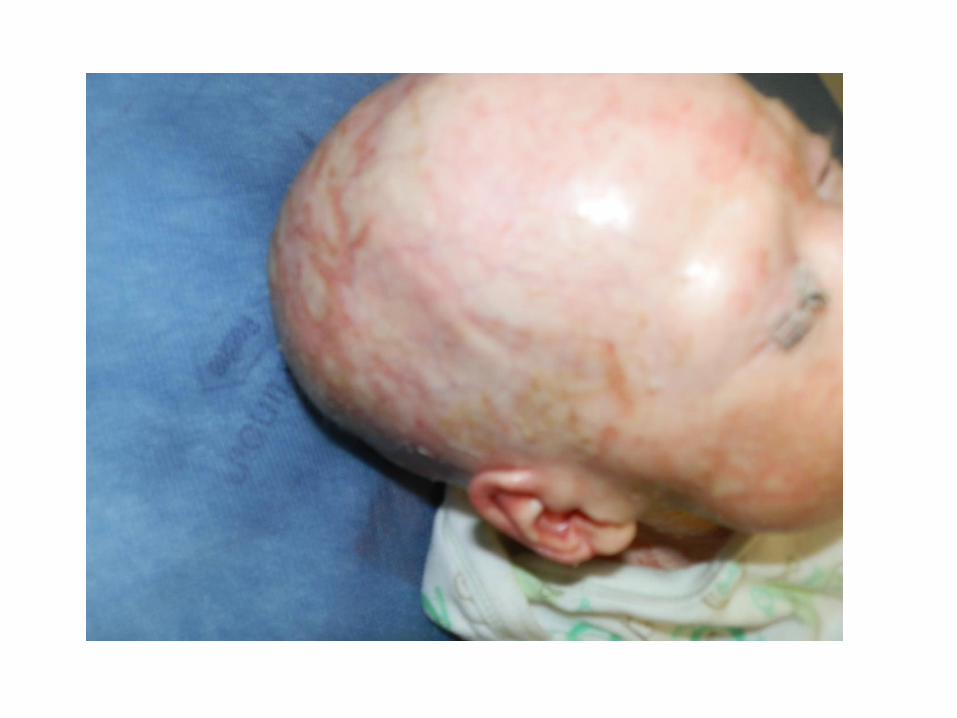
• Developed a red and scaly rash predominantly of the face and scalp
• GP visit: Seborrhoeic Dermatitis
• Rx: emollients, topical steroids
• Both parents had eczema in childhood- not severe, no other atopic disorders
• No family history of any other skin disorder

Age Three Months
• Skin condition significantly deteriorated
• No longer growing
• Referred to Red Cross Dermatology Services
• Noted to have erythroderma, extensive dry and scaly skin, hair loss
• ? Ichthyosiform disorder
• Skin biopsy

Admission to Red Cross
• Unwell, febrile and losing weight
• Missed 10 and 14 week vaccinations
• Skin worsened despite emollient wraps
• WFA < -3 Z score
• WFL < -2 Z score
• Referred to general paediatrics for further management ? Serious bacterial infection



Investigations
HIV Rapid Negative
FBC: WCC 28 (N) Hb 9.8 Plts 825
CRP: 94
Renal function and elecs: Normal
TP 51 Albumin 22

Investigations
Blood culture: No growth
Urine dipstix NAD, Micro Neg, Culture Neg
CSF: P 56 L 26 E 16640 Prot 0.29 Gluc 6.2
Skin Biopsy: “favours an ichthyosiform genodermatosis”

Differential Diagnosis
?
Serious Infection
Skin Rash
Failure to thrive

Differential Diagnosis
Inflammatory Skin Disorder
Congenital Ichthyosis
Infection
Metabolic: Zinc, CF, Biotinidasedeficiency
Primary Immune Deficiency

Inflammatory Skin Disorder
Inflammatory Skin Disorder
Infection

Inflammatory Skin Disorder
Infection
Congenital Ichthyosis
Inflammatory Skin Disorder
Infection
Congenital Ichthyosis
Metabolic: Zinc, CF, Biotinidase deficiency

Inflammatory Skin Disorder
Congenital Ichthyosis
Infection
Metabolic: Zinc, CF, Biotinidasedeficiency
Primary Immune Deficiency

Course of Admission
• Rigorous skin care
• Nutritional Support
• IV Ceftriaxone
• Extensive work-up

Course of Admission
• Continued Deterioration
• D10 developed rapidly progressive conjunctivitis- Bilateral Perforated Corneal Ulcers- Cultured Pseudomonas
• New sepsis - fever, raised WCC and CRP- Blood Culture: Acinetobacter, Enterococcus
Faecalis
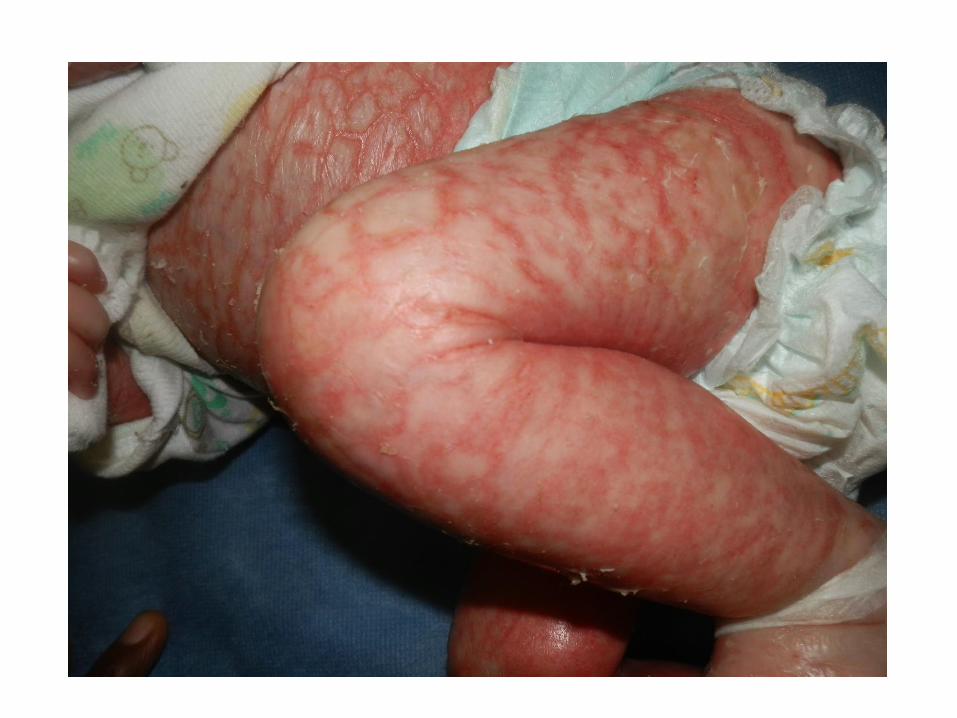

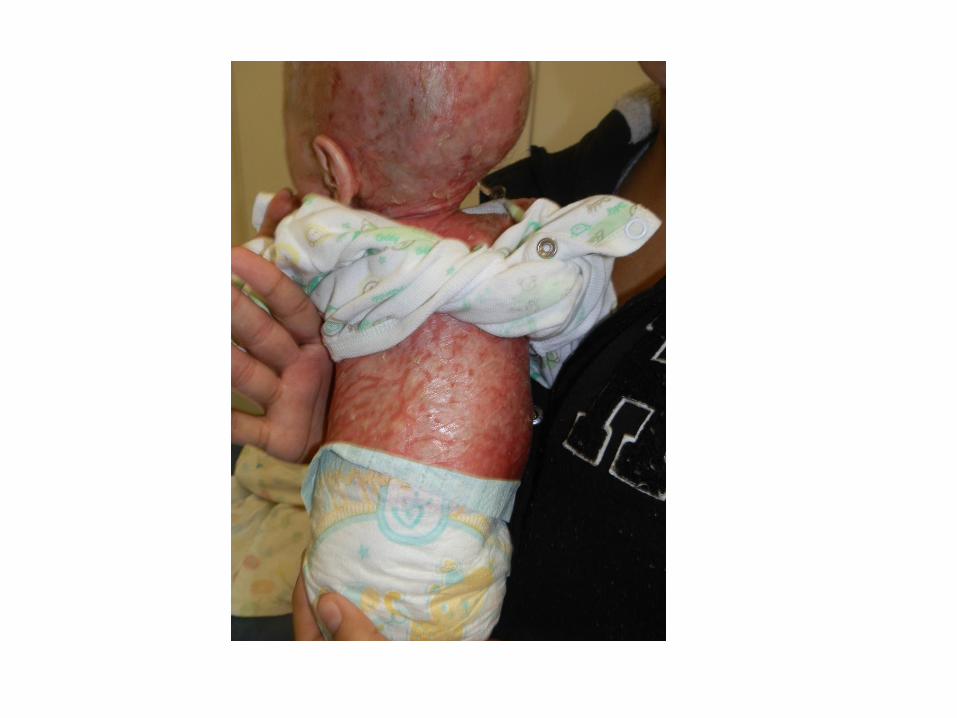

Investigations
Congenital Syphillis Excluded
Metabolic: Normal Zinc
Faecal Elastase 189ug/g, CF genetics Neg
Immune profile: Normal lymphocyte subsets, IgE 246
Immunocaps

Investigations
• Fx5:
- IgE to paediatric foods 6.38
- IgE Cow’s milk 1.00
- IgE Wheat 0.15
- IgE Peanuts 24.3
- IgE Soya 0.13
- IgE Fish <0.1
- IgE Egg White 9.62

Diagnosis

Progress
• D15 given IVIG
• Continued weight loss
• Repeated infections incl fungal sepsis
• Skin refractory to emollients, topical steroids
• Difficulties with IV access and sampling
• Feed changed to alimentum
• D34 profound hypernatremic dehydration complicated by AKI and neurologic sequelae

ICU admission
• Difficulties managing electrolytes and renal function
- Peak Na+ 197
- Peak Creatinine 267
• Ongoing episodes of sepsis
• MDT decision not for escalation of care
• Demise on D 48 of admission

Cutaneous Manifestations of PIDD
• Early diagnosis of a PID is critical
• Skin manifestations are present in 40-70%
• Skin manifestations were the presenting features in 39% of total PID
Al-Herz et al, Pediatric Dermatology, 2011.
• Although not pathognomic skin manifestations should trigger consideration of a PID

Skin Manifestation
Primary Immune Deficiency
Telangiectasia Ataxia Telangiectasia
Partial Albinism Griscelli syndrome type 2, Chediak-Higashi syndrome and Hermansky– Pudlak syndrome type 2
Eczematous skin rash Hyperimmunoglobulin-E syndromeAutosomal dominant (STAT3 mutation)Autosomal recessive (DOCK8 mutation)IPEX, Wiskott–Aldrich syndrome, Omenn syndrome , Nethertonsyndrome
Erythroderma Netherton’s syndrome, Omenn syndrome and SCID with graft vshost disease
Epidermal Dysplasia NEMO deficiency, Dyskeratosis congenita, Papillon–Lefèvre syndrome

Eczematous Rashes in PIDD
• Eczema affects between 13 % and 22 % of PIDD cohortsstudied
• Non-specific eczema in children with humoralimmunodeficiencies
• Severe eczema presenting shortly after birth especially with eosinophilia and high IgE levels is suggestive of immunodysregulation

Autosomal Dominant HIES
• Caused by STAT 3 mutation
• Characteristic facial, dental, and skeletal abnormalities
• Recurrent infections - Mucocutaneous esp cold staphylococcal abscesses - Pulmonary infections with resultant bronchiectasis and/ or
pneumatoceles
• Eczema 80-100%



Autosomal Recessive HIES
• Caused by DOCK8 or Tyk2 mutations
• Present primarily with immunodeficiency: viral infections (herpes zoster, molluscum contagiosum, varicella)
• Lack the skeletal and facial characteristics of the AD form
• Neurological sequelae
• Eczema in DOCK8 is more severe and occurs in the newborn period in 24%
• High prevalence of other atopic disorders including food allergy
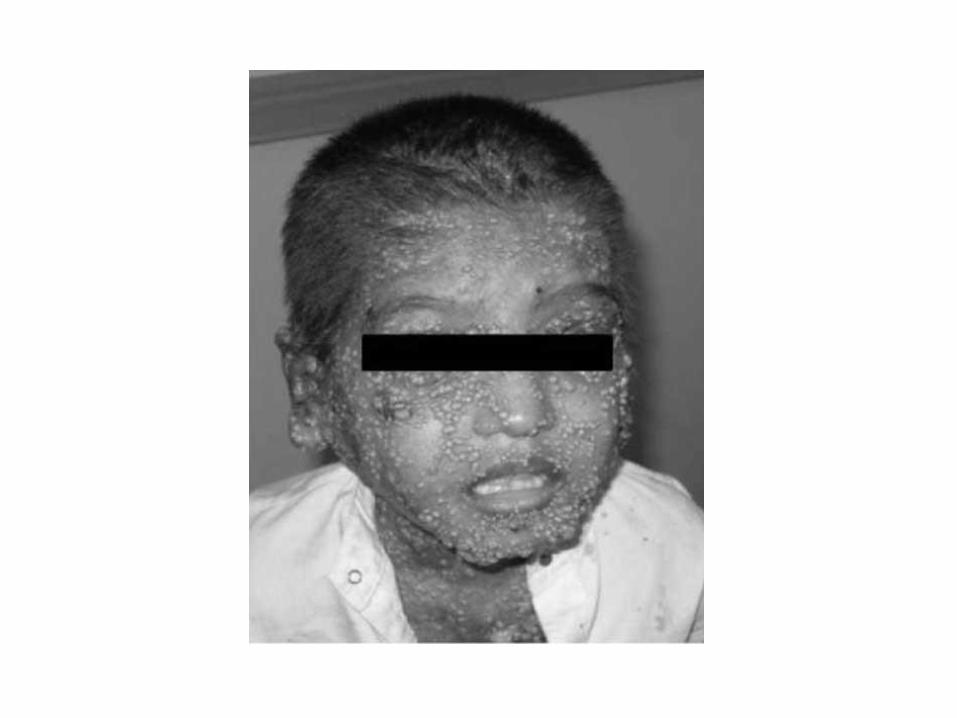

Wiskott-Aldrich Syndrome
• X-linked recessive
• Triad thrombocytopenia, eczema and immune deficiency.
• Variable expression but the immunodeficiency is usually characterized by low IgM and IgE high IgA.
• Eczema is present in 81% of patients
• Presentation is usually with petechiae or bleeding
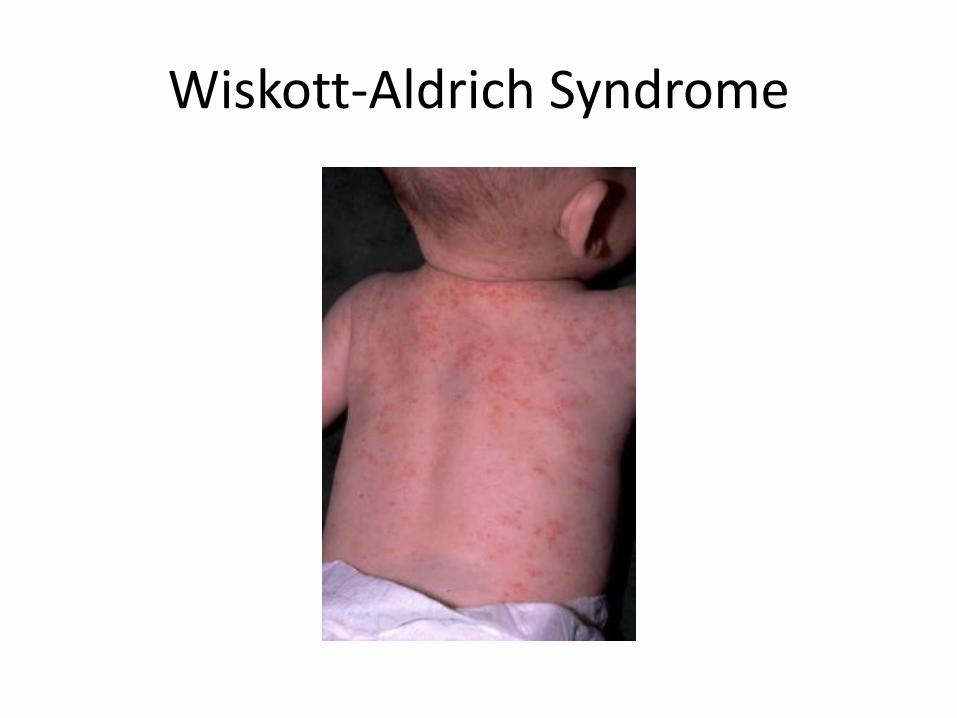
Wiskott-Aldrich Syndrome

IPEX Syndrome
• Immunodysregulation polyendocrinopathy enteropathy X-linked syndrome
• Linked to the dysfunction of the transcription factor FOXP3-regulator of the regulatory T cell lineage
• Leads to autoimmunity
• Features incl: intractable diarrhoea, endocrinopathies (type 1 diabetes, thyroiditis), eczema, infections

Erythroderma
• Involvement of >90 % of the total body surface area with erythema and/or scaling
• A survey of erythroderma in infancy found 48 % of cases to be due to primary immunodeficiency
Al-Dhalimi et al, J Derm, 2007
• Specific disorders: Omenn, Netherton’s, SCID

Omenn Syndrome
• Autosomal recessive form of SCID • Mutation in RAG1, RAG2
• Clinical features:- Exfoliative erythroderma with diffuse alopecia, - Lymphadenopathy, hepatosplenomegaly- Failure to thrive.
• Leucocytosis with eosinophilia, raised IgE, increased numbers of clonal T cells and decreased B cells with hypogammaglobulinaemia.

Omenn Syndrome

Netherton’s Syndrome
Classic Triad• Congenital ichthyosiform erythroderma
• Hair shaft defect: trichorrhexis invaginata
• Atopic manifestations

Epidemiology
• Inheritance is AR
• Incidence is estimated at 1/200,000 births
• Thought to be the cause of up to 18% cases of congenital erythroderma
• 20% Fatality rate in the first year of life

Aetiology
• Mutation on chromosome 5q32, gene SPINK5 (serine protease inhibitor, Kazal type-5)
• Encodes an inhibitor of serine proteases called LEKTI (lympho-epithelial Kazal-type 5 related inhibitor)
• Increase in trypsin-like hydrolytic activity in the stratum corneumcausing premature desquamation and a severe skin barrier defect

Clinical Features
• Presents at or shortly after birth with non-bullous ichthyosiformerythroderma, may be eczematous
• Erythroderma may be more pronounced with infection
• Pruritus is an inconsistent feature, no white dermatographism
• Palms and soles are normal
• Nails and teeth are unaffected
• Later many develop atopic dermatitis and/or ichthyosis lineariscircumflexa

Ichthyosis Linearis Circumflexa

Clinical Features
• Severe cases: Failure to thriveHypernatremic dehydration Recurrent infectionsIntestinal malabsorption
• 44 Case ReportsHypernatraemic Dehydration in 7%Wasting in 16%Recurrent Infections 30%
Smith et al, JACI, 1995.

Clinical Features
• Hair is characteristically sparse and easily broken
• Pathognomonic feature: trichorrhexis invaginata “ball and socket defect”
• Pili torti and trichorrhexis nodosa are also seen
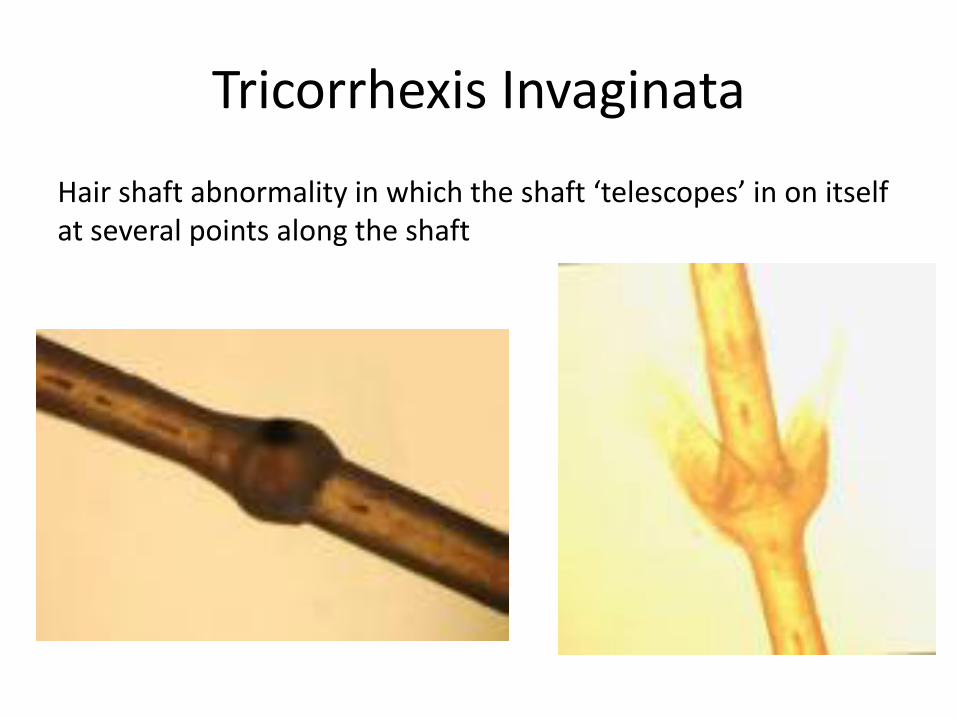
Tricorrhexis Invaginata
Hair shaft abnormality in which the shaft ‘telescopes’ in on itself at several points along the shaft

Tricorrhexis Invaginata
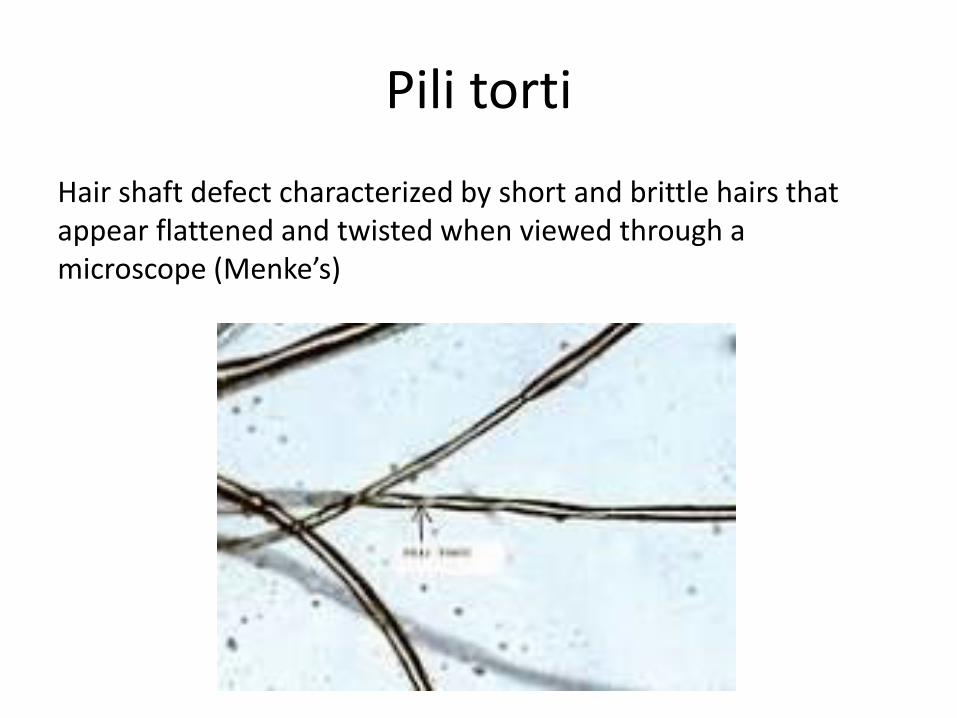
Pili torti
Hair shaft defect characterized by short and brittle hairs that appear flattened and twisted when viewed through a microscope (Menke’s)

Trichorrhexis Nodosa
Minute nodes are formed in the hair shafts, weakening the hair and causing splitting and breaking

Clinical Features
• Other features: delayed growthshort statureintermittent aminoaciduriaintellectual deficit
• 75% of patients develop atopic manifestations: asthma, atopic dermatitis, food allergies, urticaria, angioedema
• Nut and fish allergy is the most frequently reported food allergy

Diagnosis
• Suggestive: eosinophilia, elevated IgE, positive skin prick tests or Immunocaps to food and aeroallergens
• Skin biopsy specimens are generally nonspecific and largely noncontributory in the diagnosis of NS
• Immunostaining of a skin biopsy with a specific monoclonal antibody against LEKTI
• Examination of hairs for the trichorrhexis invaginata
• Molecular diagnosis through identification of the genetic mutation

Treatment
• Topical corticosteroids are largely ineffective and may be absorbed in greatly increased amounts rendering these patients particularly susceptible to AE.
• Therapy includes emollients, etretinate, PUVA, topical tacrolimus, and pimecrolimus 1% with moderate and temporary effects
• Ammonium lactate lotionSmith et al, JACI, 199
Wehr et al, J Am Acad Derm, 1988
• Systemic drugs like methotrexate and cyclosporine have been found ineffective
Braun et al. Dermatology 1997


Thank You