sin título de diapositivacampus.usal.es/~ogyp/clases teoricas 2013 2014/licenciatura...
TRANSCRIPT


Componentes de Defensa
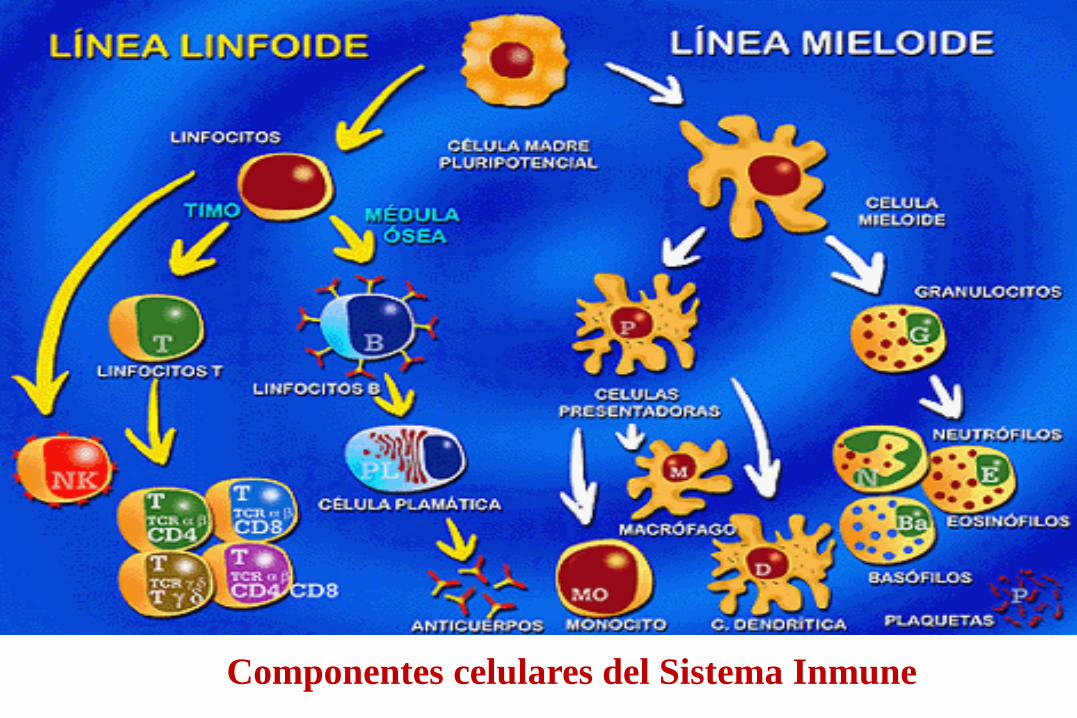
Tejidos: Piel y mucosas Órganos: 1º):Timo, 2º): ganglios linfáticos, bazo Células: Macrófagos, Monocitos, Leucocitos, Eosinófilos, Linfocitos (T-B-K-NK), Proteínas: Anticuerpos, Interleucinas, Interferones Proteína c, Complemento, etc.

Célula madre Stem cell
CFU-GM CFU-Eo CFU-Baso CFU-Mas CFU-meg CFU-E
MONOCITOS NEUTROFILOS EOSINOFILOS BASOFILOS MASTOCITOS MEGACARIOCITOS ERITROCITOS
C. D. Plasmocitoides Células dendríticas Macrófagos
L.B C. Plasmática
L.T. Célula NK
Célula NK-T Célula madre Linfoide
Precursor C.Dendríticas
Origen células sistema inmune. Células hematopoyéticas
Célula madre Mieloide
Igs
Linfocinas

Célula madre Linfoide
Pre-timocito CD3-,TCR- CD4-CD8-
T I M O Zona subcapsular Zona Cortical Zona Medular
TCR αβ + CD3 + CD4 + CD8 + Doble +
Selección clonal Positiva. No afinidad
Ag HLA propios
APOPTOSIS
TCR αβ + CD3 + CD4 + CD8 - Simple +
TCR αβ + CD3 + CD4 - CD8 + Simple +
Selección clonal Negativa. Excesiva Afinidad péptidos y
Ag HLA propios
APOPTOSIS
TCR αβ + CD3 + CD4 + CD8 -
L.T Cooperadores
TCR αβ + CD3 + CD4 - CD8 +
L.T Citotóxicos
Linfocito γδ
TCR γδ + CD3 + CD4 - CD8 -
Células epiteliales Células epiteliales y dendriticas
Diferenciación y Maduración de linfocitos T

Diferenciación y desarrollo de linfocitos B
Célula Madre linfoide
µ µ µ
IgM
IgM
IgD
IgM
Célula Pre-pre B VHDHJH
Célula Pre B VDJµ
L. B. Inmaduro
VDJµδ
L. B. Maduro
Independiente de antígeno Dependiente de antígeno
Activación Ag Cambio Isotipo
Linfocito B Memoria
Célula Plasmática
CD19-CD45 CD19-CD45 CD19-CD45 CD19-CD45
CD19-CD45 sIgM
sIgG
sIgA
sIgA
IgM
IgG
IgA
IgE
Complemento sérico
Componentes celulares del Sistema Inmune

I. ESPECIFICA I. INESPECIFICA C E L U L A R
H U M O R A L
COMPLEMENTO INTERLEUCINAS L.B
INMUNOGLOBULINAS
L.T MACROFAGOS K-NK

Inmunodeficiencias
Primarias: -Congénitas Secundarias: -Enf. Neoplásicas -Trat. Inmunosupresores -Infecciones VIH -Malnutrición -Esplenectomía -Síndrome nefrótico -Infecciones víricas y bacterianas -Otras causas

Inmunodeficiencia de la Inmadurez
•I. Inespecífica humoral: Deficiente
•I. Inespecífica Celular: Deficiente
•I. Específica Celular: LB LT
•I. Especifica Humoral:
• IgG: normal, IgA e IgM: disminuidas
• Anticuerpos: Solo maternos

Paso placentario de anticuerpos Maternos al Feto
• Antitoxinas DF y T • Aglutininas Pertusis • Antiestreptolisinas • Antiestafilolisinas • Poliomielitis • Saramp/Rubeola/parotiditis • Herpes Simple • Estreptococo B • Salmonella H
– Rh incompleto – Insohemaglutininas anti A y
B – VDRL – Antitiroideos – Antinucleares
Antiinfecciosos Otros anticuerpos
Muy elevado

Paso placentario de anticuerpos Maternos al Feto
• H. Influenzae • B. Pertusis • Shigela • Estreptococo • Estafilococo PC(-)
• Salmonella • E. Coli • Heterofilos • Wasserman • Reagians IgE
Deficiente Nulo

Clasificación de las I.D. Primarias 2011
• I.D. Combinadas T y B: 33 cuadros
• I.D. Predominantemente Ac:26 Cuadros
• Síndromes bien definidos con I.D.: 22 cuadros
• Cuadros de Disregulación inmune: 19 cuadros
• Defectos de Fagocitos: 32 Cuadros
• Defectos en Inmunidad innata: 14 cuadros
• Desórdenes autoinflamatorios: 11 cuadros
• Defectos de complemento: 24 cuadros
181 cuadros

ESID, Marzo2012 www.esid.com Clin Exp Immunol. 2012 Apr;168(1):58-9
• Predominantly antibody disorders: 56.14% (n=8,660)
• Predominantly T-Cell deficiencies : 7.64% (n=1168)
• Phagocytic disorders: 8.66% (n=1,336)
• Complement deficiencies : 4.19% (n=647)
> 15.000 casos registrados

ESID, Marzo2012 www.esid.com Clin Exp Immunol. 2012 Apr;168(1):58-9
• Other well defined PIDs: 15.16% (n=2,338)
• Autoimmune & immunedysregulation syndromes 3.75% (n=579)
• Autoinflammatory syndromes: 1.94% (n=300)
• Defects Innate Immunity: 0.97% (n=149)
• Unclassified PIDs: 1.54% (n=238)
Francia, Marzo 2011: 5,3/ 100.000

I.D. PRIMARIAS EDAD DE DIAGNOSTICO
5%
15%
40%
40% > 1 Año< 5 Años< 16 AñosAdultos

Bases del diagnóstico de las Inmunodeficiencias
1. Diagnostico Clínico
2. Diagnóstico Inmunofenotípico
3. Diagnóstico Molecular y Genético

Puntos clave en Inmuno-Deficiencias
-Por lo general cursan con infecciones de repetición
-Pueden presentar además diversas características
-Es vital la sospecha temprana y tratamiento precoz
-Existen signos clínicos relevantes en diferentes órganos
-Ante sospecha derivar a servicio de Inmunología
-Diagnostico definitivo y tratamiento por especialista


Bases del diagnóstico de I.D. Datos de sospecha (Jeffrey Modell)
1. Ocho o más infecciones óticas en un año
2. Dos o mas sinusitis serias en un año
3. Dos o mas meses de antibioticoterapia
sin resultados
4. Dos o más neumonías en un año
5. Muguet persistente en boca o Candidiasis en la piel

Bases del diagnóstico de I.D. Datos de sospecha (Jeffrey Modell)
5. Abscesos profundos o en órganos
7. Dos o mas infecciones severas como meningitis,
osteomielitis, sépsis
8. Necesidad de antibióticos intravenosos en infecciones
9. Fallo en el crecimiento ponderoestatural
10. Historia familiar de Inmunodeficiencias primarias.
Sensibilidad 63 %; Especificidad 23%

Sospecha de ID en Neonatos
1. Diarrea crónica refractaria/falta de medro
2. Candidiasis oral
3. Infecciones por gérmenes persistentes
4. Neumonitis persistente
5. Lesiones cutáneas (eritrodermia, eczema)
6. Retraso caída de cordón (mas de 30 días)

Sospecha de ID en Neonatos
7. Hepato-esplenomegalia
8. Defectos congénitos corazón (anomalías conotruncales)
9. Historia familiar de ID o fallecimientos tempranos
10.Ausencia de sombra del timo en radiografía.
11. Hallazgo en laboratorio de: -Linfopenias (< 3000 linfocitos) -Citopenias -Leucocitosis sin infección -IgM < de 20 mg/dl; IgA < 5 mg/dl -Hipocalcemia

Sospecha de ID en niño < 6 meses
1. Retraso ponderoestatural
2. Diarrea crónica refractaria y/o mala absorción
3. Muguet persistente
4. Infecciones por gérmenes persistentes
5. Neumonías antes de los 3 meses de vida
6. Infecciones que no se resuelven con antibióticos I.V.

Sospecha de ID en niño menor de 6 meses
7. Escaso efecto con Antibióticos mas de 2 meses
8. Historia familiar de ID primaria
9. Reacciones sistémicas ante vacunas con gérmenes vivos
10. Ausencia de sombra tímica en RX torax
11. Hipoplasia tejido linfoide (amigdalas, ganglios)
Por edad debe sospecharse una ID celular o combinada

Sospecha de ID en niño mayor de 6 meses
1. Ocho o mas episodios de otitis media en 1 año (sobre todo si
se cronifica o se asocia a infecciones broncopulmonares o sinusitis )
2. Dos o mas episodios de sinusitis severa en 1 año
3. Dos o mas episodios de neumonía en un año
4. Dos o mas infecciones invasivas (meningitis, celulitis, mastoiditis,
osteomielitis o sépsis)
5. Infecciones que no se resuelven con antibióticos I.V.
6. Escaso efecto con antibióticos durante mas de 2 meses

Sospecha de ID en niño mayor de 6 meses
8. Enfermedades alérgicas, autoinmunes o síndromes
asociados a inmunodeficiencias
9.Neoplasias de origen linfático
10. Historia familiar de inmunodeficiencia primaria
11. Reacciones sistemicas ante vacunas de gérmenes vivos
Por la edad la 1ª opción será una ID por déficit de AC

Sospecha de ID en niño a cualquier edad
-Déficit de Complemento: Dos o más infecciones invasivas como
meningitis o sepsis por Neisseria.
-Defecto de Fagocitosis: -Abscesos recurrentes cutáneos, pulmonares o en órganos
profundos (bazo, hígado, etc) -Caída retrasada del cordón umbilical -Dificultad cicatrización heridas -Periodontitis/estomatítis persistente -Fiebre mantenida -Infecciones que no se resuelven con antibioticos IV -Historia familiar de Inmunodeficiencias primarias

MANIFESTACION CLÍNICA SOSPECHA DIAGNÓSTICA
Def. de Anticuerpos Neumonias repetidas con buena Respuesta a antibióticos habituales
Neumonias lobares repetidas Def. de Anticuerpos
Neumonia persistente sin Respuesta a antibióticos habituales
Deficiencia de Celulas T IDCS
Defecto de Fagocitos SIDA
Neumonias con derrame pleural Defecto Fagocitos Def de anticuerpos

MANIFESTACIÓN CLÍNICA SOSPECHA DIAGÓSTICA
Infecciones Fúngicas Def. de Células T
SCID Defecto de Fagocitos
Aislamiento de S. aureus en paciente con infecciones recurrentes / grave / persistente
Defecto de Fagocitos Def. de Anticuerpos
Aislamiento de S. pneumoniae en paciente com infecciones de repetición
Def. de Anticuerpos Def. de Complemento
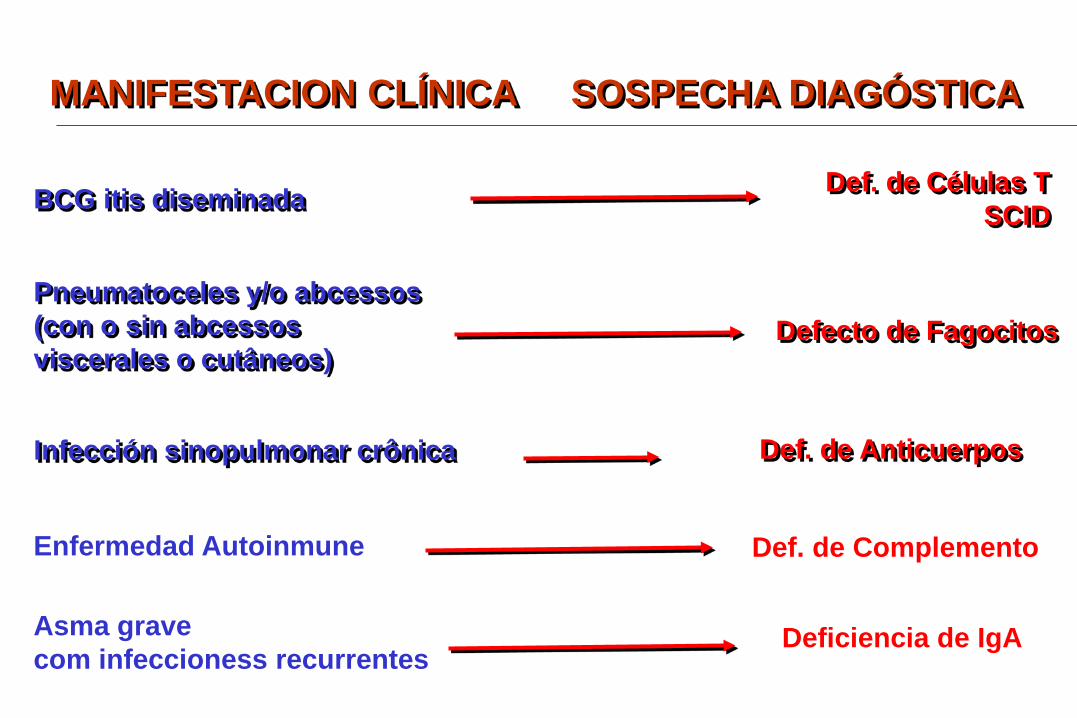
BCG itis diseminada Def. de Células T SCID
Pneumatoceles y/o abcessos (con o sin abcessos viscerales o cutâneos)
Defecto de Fagocitos
Infección sinopulmonar crônica Def. de Anticuerpos
Enfermedad Autoinmune Def. de Complemento
Asma grave com infeccioness recurrentes
Deficiencia de IgA
MANIFESTACION CLÍNICA SOSPECHA DIAGÓSTICA

SINTOMATOLOGIA CLINICA
• INFECCIONES • ALTERACIONES DIGESTIVAS • ALTERACIONES CUTANEAS • DETERIORO CRECIMIENTO Y
DESARROLLO • OTRAS MANIFESTACIONES

I.D. PRIMARIAS ENFERMEDADES ASOCIADAS
• NEOPLASIA • ENFERMADES AUTOINMUNES • ENFERMEDADES ALERGICAS

CARACTERISTICAS DE LAS INFECCIONES
• FRECUENTES • PROLONGADAS • REITERATIVAS • CON MALA EVOLUCION • RESISTENTES • GRAVES

I.D. PRIMARIAS LOCALIZACION INFECCIONES
• RESPIRATORIAS • DIGESTIVAS • CUTANEAS • GENERALIZADAS

Señales de alerta Respiratorias
- Neumatocele
- Neumonitis
- Bronquiectasias
- Rinosinusitis crónica
- Faringoamigdalitis de repetición
- Otitis de repetición
- Sinusitis de repetición

I.D. PRIMARIAS MANIFESTACIONES CLINICAS
• OTITIS • SINUSITIS • NEUMONIAS • GASTROENTERITIS • MENINGITIS • HEPATITIS • ENCEFALITIS • DERMATOMIOSITIS
• CITOMEGALOVIRUS • SARAMPION • VARICELA • CANDIDIASIS CRONICA • DIARREAS VIRICAS • GENERALIZACION
VACUNAL
D. ANTICUERPOS D. CELULARES

I.D. PRIMARIAS MANIFESTACIONES CLINICAS
• MENINGITIS • GONOCOCIAS • INFECCIONES PIOGENAS
(PIEL)
• HIPERGAMMAGLOBULINEMIA.
• INFECCIONES REPETIDAS DE PIEL Y MUCOSAS
• ABCESOS CUTANEOS • NEUMONIAS ENCAPSULADAS • ECZEMAS
• HIPERGAMMAGLOBULINEMIA
D. COMPLEMENTO D. FAGOCITOSIS

I.D. PRIMARIAS BACTERIAS INFECTANTES
• STAFILOCOCCUS • STREPTOCOCCUS • H. INFLUENZAE • SALMONELA • MENINCOCOCCUS • CAMPILOBACTER
• SALMONELLAS • PSEUDOMONAS • MYCOBACTERIAS • LISTERIA • SERRATIA
D. ANTICUERPOS D. CELULARES

I.D. PRIMARIAS VIRUS INFECTANTES
• ECHO • POLIO • ROTAVIRUS • HEPATITIS B • HEPATITIS no A no B
• VACCINIA • HERPES SIMPLE • VARICELA ZOSTER • CITOMEGALOVIRUS • ROTAVIRUS • V.S.R. • ADENOVIRUS • ENTEROVIRUS
D. ANTICUERPOS D. CELULARES

I.D. PRIMARIAS OTROS MICROORGANISMOS
• LAMBLIAS • CRIPTOSPORIDIOS • PNEUMOCISTIS CARINII
• MICOPLASMA • UREAPLASMA
• NEUMOCISTIS CARINII • TOXOPLASMA
• CANDIDA • ASPERGILLUS
D. ANTICUERPOS D. CELULARES
Protozoos: Protozoos:
Hongos Otros:

I.D. PRIMARIAS AGENTES INFECTANTES
• NEISERIAS • STREPTOCOCCUS • H. INFLUENZAE • E. COLI • BRUCELA • S. AUREUS
• S. AUREUS • STREPTOCOCCUS • E. COLI • KLEBSIELLA • SERRATIA • PSEUDOMONA • MYCOBACTERIAS • CANDIDA • ASPERGILLUS • NOCARDIA
D. COMPLEMENTO D. FAGOCITOS

Señales de alerta Digestivas
- Diarreas agudas
- Diarrea crónica con o sin malabsorción
- Enfermad inflamatoria intestinal
- Abscesos hepáticos, esplénicos
- Enteropatías

Señales de alerta dermatológicas (1/3)
- Infecciones bacterianas de piel
- Candidiasis mucocutánea
- Moluscum contagioso
- Verrugas
- Eczema
- Eritrodermia generalizada
- Alopecia
- Acné quístico
- Pioderma gangrenoso

I.D. PRIMARIAS ALTERACIONES HEMATICAS
• LINFOPENIA • NEUTROPENIA • TROMOBOCITOPENIA • ANEMIAS • ALTERACIONES MORFOLOGICAS

I.D. PRIMARIAS OTRAS MANIFESTACIONES
• ENDOCRINOLOGICAS • NEUROLOGICAS • METABOLICAS • ESQUELETICAS • CROMOSOMICAS

Estudios de Imagen en I. Deficiencias
Presencia de tejido Linfoide: Timo, Adenoides Signos pulmonares: (Variable): -I.D anticuerpos: -Bronquitis -Bronquiectasias -Atelectasias -I.D. Celulares: Neumonías -Patrón reticular (Neumocistis jrovenci) -Patrón milliar (Tb, Becegeitis) -Imágenes abigarradas y difusas (en EGC)

Estudios de Imagen en I. Deficiencias
Hallazgos digestivos: -Adenopatías abdominales (Def IgA) -Hiperplasia nodular linfoide (SVC, Def IgA, def Ac) -Lesiones hepáticas, granulomas (EGC) -Abscesos subfrénicos, perianales (EGC)
Alteraciones esqueléticas:
-Osteomielitis infecciosa (EGC) -Artritis cronica (Def. anticuerpos) -Acortamiento diafisario de extremidades (Hipoplasia C.Pelo) -Ensanchamientos metafisarios (Deficit ADA)

Coronel Bruton

P t i

Inmunoelectroforesis

Bases del diagnóstico de las Inmunodeficiencias
1. Diagnostico Clínico
2. Diagnóstico Inmunofenotípico
3. Diagnóstico Molecular y Genético

Componentes celulares del Sistema Inmune

Características de las I.D. Deficiencias de anticuerpos
* Infecciones por bacterias encapsuladas
* Localización respiratoria > digestivo > Oseo >
piel, S.N.C > Sepsis.
* No mayor incidencia de infecciones víricas
* Escasa infección por hongos.
* Comienzo > 9 meses.

Características de las I.D. Deficiencias de anticuerpos
•Otras manifestaciones: •Cuadros malabsortivos
•Poliartitris (Ureaplasma o micoplasma)
•Autoinmunidad (Citopenias, tiroiditis, conectivopatías)
•Tumores (linfomas, carcinoma gástrico)

Evaluación de los linfocitos B
Pruebas de cribado:
-Cuantificación de inmunoglobulinas IgG, A, M y E
-Síntesis de anticuerpos específicos Ag proteicos y polisacáridos
-Anticuerpos naturales (Isohemaglutininas anti A o anti B)
-En niños pequeños sin inmunizar (Batería amplia de Ags)
-Neoantígenos-Bacteriófago (si recibió Igs)
-Determinación IgA secretora

Evaluación de los linfocitos B
Pruebas especiales:
-Subclases de Inmunoglobulinas
-Cuantificación de linfocitos B (CD19, CD20)
-Cuantificación linfocitos B “vírgenes” y de “memoria”
-Células formadoras de anticuerpos
-Linfocitos T y células NK
-Biopsia ganglionar
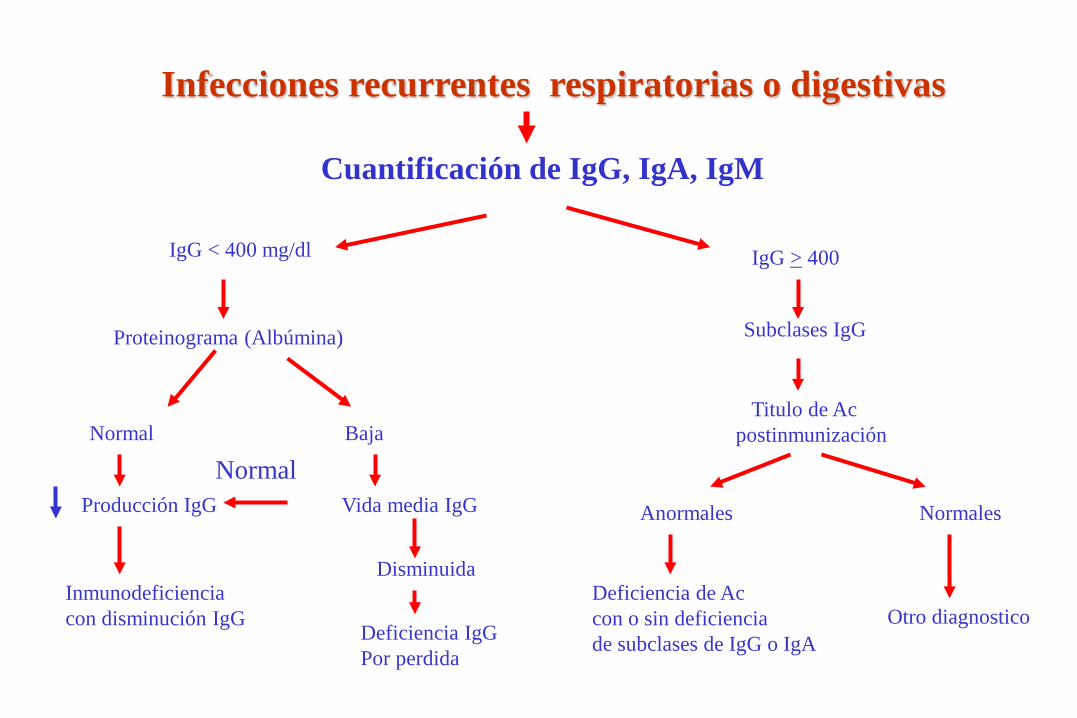
Infecciones recurrentes respiratorias o digestivas
Cuantificación de IgG, IgA, IgM
IgG < 400 mg/dl
Proteinograma (Albúmina)
Normal Baja
Producción IgG Vida media IgG
Inmunodeficiencia con disminución IgG
Disminuida
Deficiencia IgG Por perdida
IgG > 400
Subclases IgG
Titulo de Ac postinmunización
Anormales
Deficiencia de Ac con o sin deficiencia de subclases de IgG o IgA
Normales
Otro diagnostico
Normal

Evaluación de la Inmunidad mediada por anticuerpos
2. Estudio molecular y genético: * Detección del defecto molecular en linfocitos T y B * Identificación de la anomalía genética.

Diagnóstico de Agammaglobulinemia ligada al sexo
Probable: * Linfocitos B CD19+ menores de 2 % y positivas todas las siguientes circunstancias: - Infecciones bacterianas repetidas en los 5 primeros años - IgG, IgA e IgM 2 DS inferiores a lo normal - Ausencia de isohemaglutininas o - Pobre respuesta de Ac a vacunaciones - Excluidas otras causas de Hipogammaglobulinemia

Diagnóstico de Agammaglobulinemia ligada al sexo
Definitivo: * Linfocitos B CD19+ menores de 2 % y al menos una de las siguientes circunstancias: - Mutación en BTK - Ausencia de BTK mRNA en C. Mononucleadas o neutrófilos - Historia materna de angammaglobulinemuia y L. B. bajos

Shock séptico por ps. aeruginosa
Lesiones puntuales, afectación del estado general y fiebre alta 2 días antes, catalogadas de “virasis”
Ausencia de cel´s B en sangre periférica por defecto en una molécula (Btk) necesaria para su maduración

Diagnóstico de Hipogammaglobulinemia con hiper IgM ligada al sexo
Probable: * Varón con IgG sérica inferiores en 2 DS de lo normal y todas las siguientes circunstancias: - Numero y proliferación de L.T. normales. - Numero de LB normales o elevados pero anormal Ac. IgG - Una de las siguientes infecciones o complicaciones: * Infecciones bacterias recurrentes en el 1º año de vida * Infección por Neumocitis carinii en el 1º año de vida * Neutropenia - Ausencia del CD40 en Linfocitos CD4+

Diagnóstico de Hipogammaglobulinemia con hiper IgM ligada al sexo
Definitivo: * Varón con IgG sérica inferiores en 2 DS de lo normal y al menos una de las siguientes circunstancias: - Mutación en el gen CD40 - Historia materna de primos o tíos con diagnostico confirmado de hiper IgM ligado al sexo.

Otras IDP por déficit de Anticuerpos de interés
Déficit aislado de IgA Hipogamaglobulinemia transitoria:

Componentes celulares del Sistema Inmune

Inmunodeficiencias Combinadas
Grupo de enfermedades heterogéneas Cuadros muy variados: Defecto en el metabolismo de las purinas (adenosin deaminasa), maduración anómala de los linfocitos, deficiencia de la cadena gamma del receptor de IL-12 Se caracterizan por la alteración de la Inmunidad celular y humoral (LT y LB)

Características de las I.D. Combinadas
* Cuadro clínico muy grave * Predominio de infecciones víricas y fúngicas y de bacterias intracelulares. * Frecuente debut por neumonía intersticial o por infecciones cutáneas por Cándida * Comienzo muy temprano (anterior a los 6 meses) * Detección de desarrollo póndero-estatural

Evaluación de los linfocitos T
Pruebas de cribado: -Recuento de linfocitos (linfopenia en ID celulares) -Pruebas cutáneas Hipersensibilidad retardada
Pruebas especiales: -Recuento de linfocitos T y subpoblaciones -Pruebas Funcionales con PHA, Con A y PWM -Pruebas funcionales con Ac contra TCR o Ags -Producción de citocinas -Activación de moléculas TLR -Determinación de enzimas (ADA, PNP) -Biopsia


Inmunodeficiencia variable comun
Diagnóstico frecuentemente en adultos Afectación de I. Celular y Humoral Deterioro progresivo de la Inmunidad En ocasiones precedida de: -Déficit de IgA -PTI

Síndromes de Bien definidos
Síndromes de Inmunodeficiencia asociados a otros defectos mayores: -Síndrome de Wiskott-Aldrich
-Ataxia Telangiectasia
-Anomalía de Di George
-Síndrome de Duncam

Síndrome de Wiskott-Aldrich
Clínica:
- Eczema
- Infecciones de repetición
- Diátesis hemorragica

Síndrome de Wiskott-Aldrich Datos de laboratorio
* Plaquetopenia
* Disminución de IgM; IgA e IgE elevadas
* Titulo de Isohemaglutininas bajo
* Degradación inmunológica progresiva:
Disminución de CD4+
Los linfocitos carecen de microvilli

Ataxia Telangiectasia
Clínica:
-Ataxia cerebelosa
-Telangiectasias
-Infecciones de repetición

Ataxia Telangiectasia Datos de laboratorio
* 80 % de casos se asocia a Inmunodeficiencia:
- Ausencia de IgA e IgG2
* Deterioro progresivo del Sistema Inmune:
- Linfopenia
- Elevación de linfocitos T TCR-g/d
* Frecuentes roturas cromosomicas y translocaciones
* Elevación de α fetoproteina

Anomalía de Di George
Clinica:
- Tetania hipocalcémica
- Malformaciones congénitas
* Paratiroides
* Timo
* Otras
- Infecciones de I.D Celular

Anomalía de Di George Datos de laboratorio
Frecuentes delecciones de Cromosoma 22
1. Anomalía total:
* Ausencia de linfocitos T
* Normalidad de linfocitos B y células NK
* Deficiente formación de anticuerpos
2. Anomalía parcial: Linfocitos T en número normal
o con diferente grado de linfopenia.

Síndrome de Duncam
Enfermedad linfoproliferativa ligada a X
Incapacidad respuesta a virus de Epstein-Barr
Sucumben a la infección por este virus o
desarrollan Neoplasia secundaria
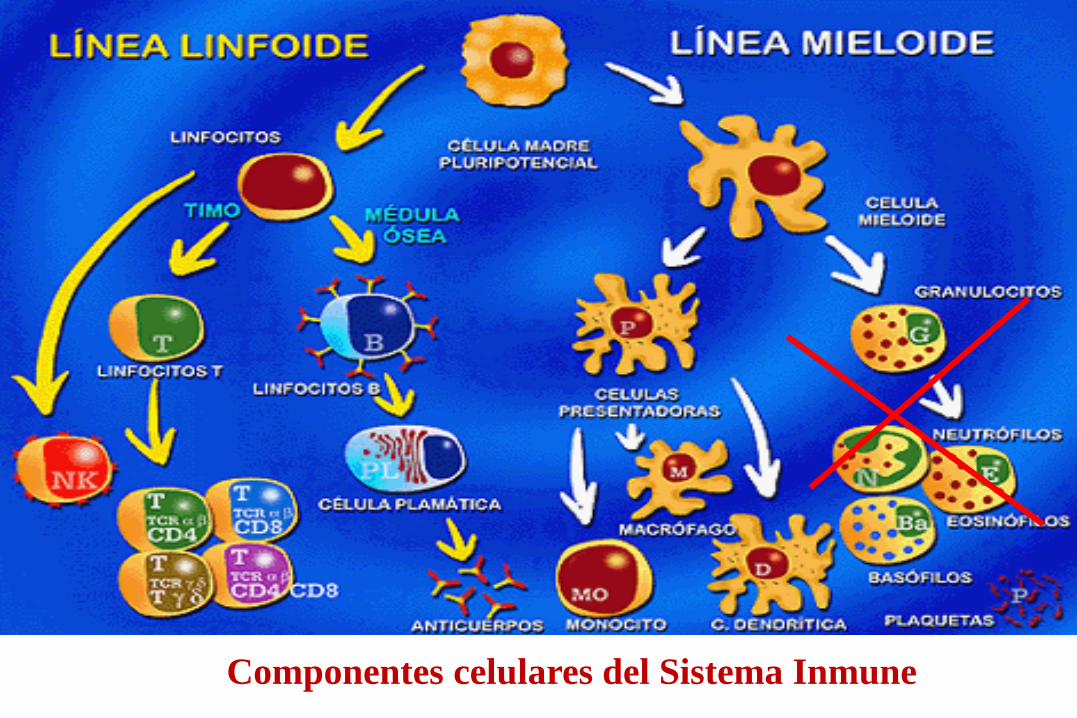
Componentes celulares del Sistema Inmune

Defectos de Fagocitos
•Defectos de la función leucocitaria
-Adherencia
-Movilidad
•Defectos de la función fagocítica bacteriana
-Defecto del metabolismo oxidativo
-Deficiencias de enzimas proteolíticos

Características de las I.D. Por defectos de Fagocitos
* Infecciones por bacterias de crecimiento
intracelular y hongos. (50 % Stafilococcus aureus)
* Comienzo en el primer año de vida.
* Abscesos, linfadenitis, granulomas y neumonías
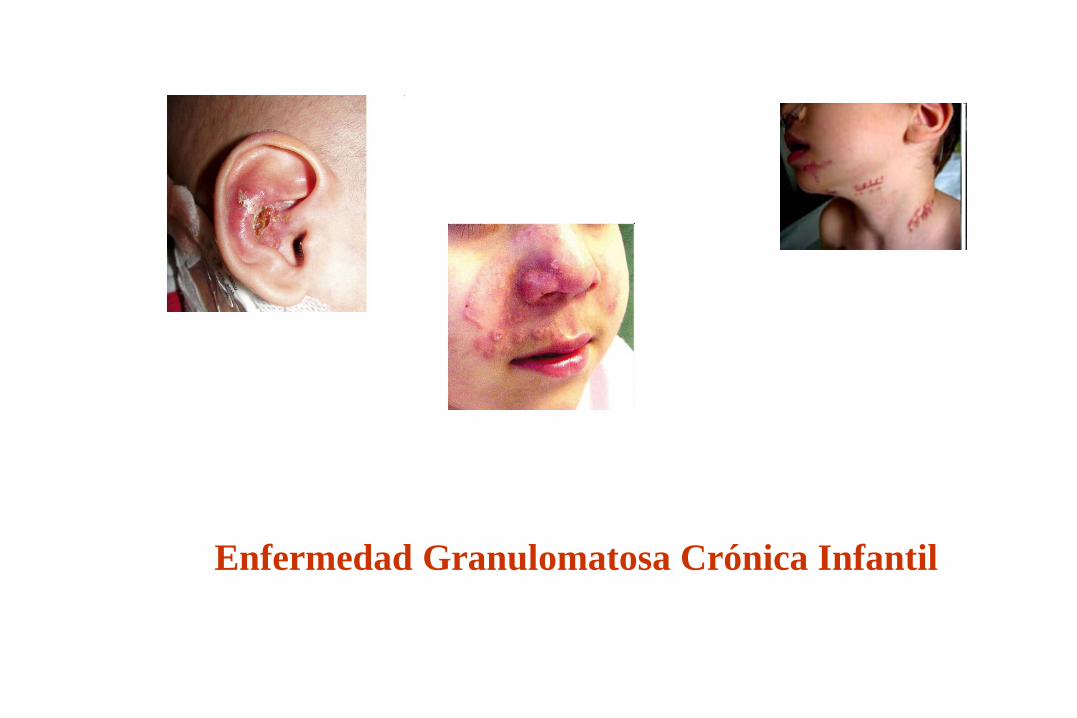
Enfermedad Granulomatosa Crónica Infantil
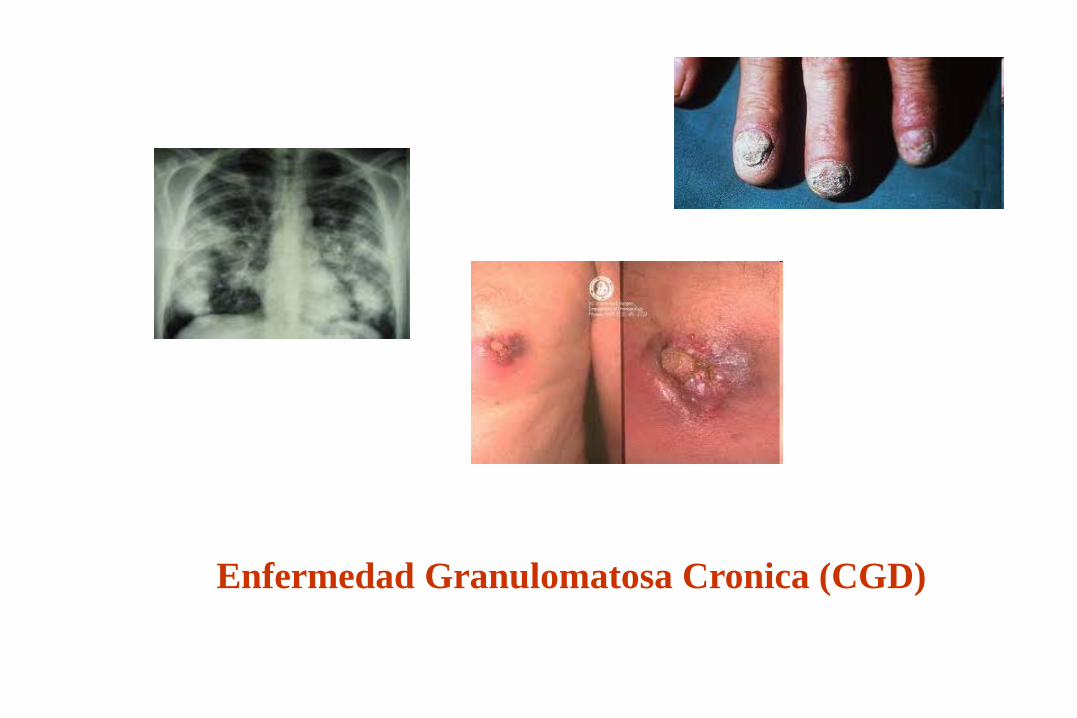
Enfermedad Granulomatosa Cronica (CGD)

Evaluación de la Fagocitosis
Pruebas de cribado: -Cuantificación celular (recuento y fórmula) -Morfología celular -Cribado de EGC (Test de NBT, citometría)
Pruebas especiales: -Valoración de la Quimotaxis (Rebuck, Boyden) -Estudios de moléculas de adhesividad (CD11, CD18,CD15) -Estudios de fagocitosis y opsonización -Capacidad bactericida -Estudio metabolismo oxidativo -Anticuerpos anti neutrofilos
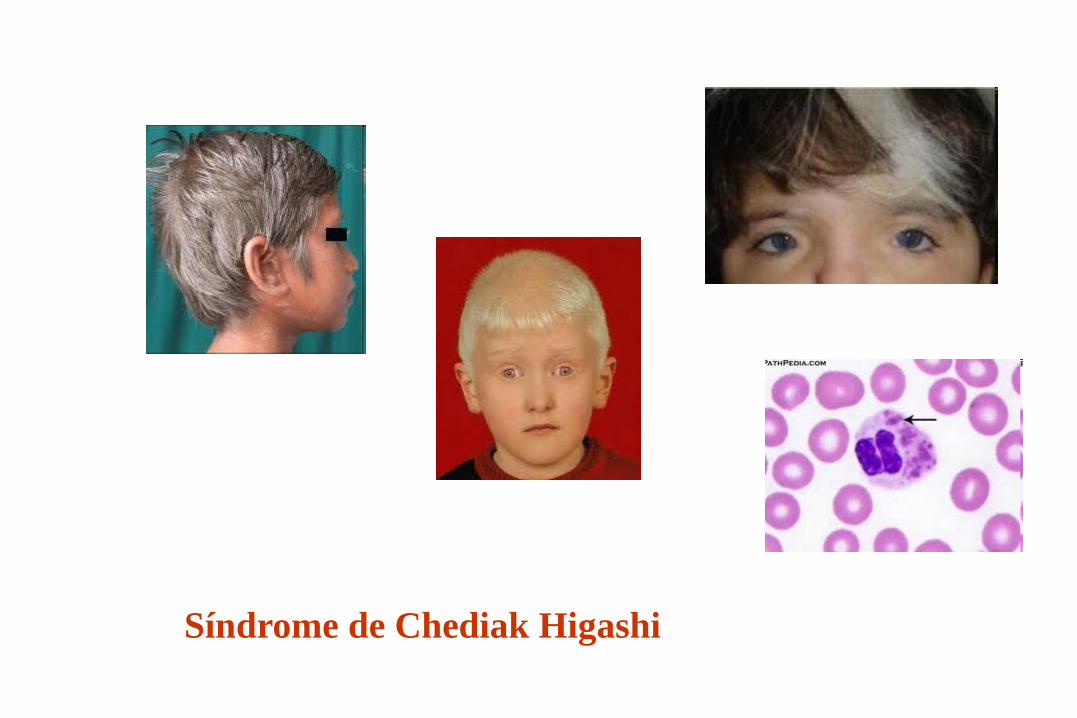
Síndrome de Chediak Higashi
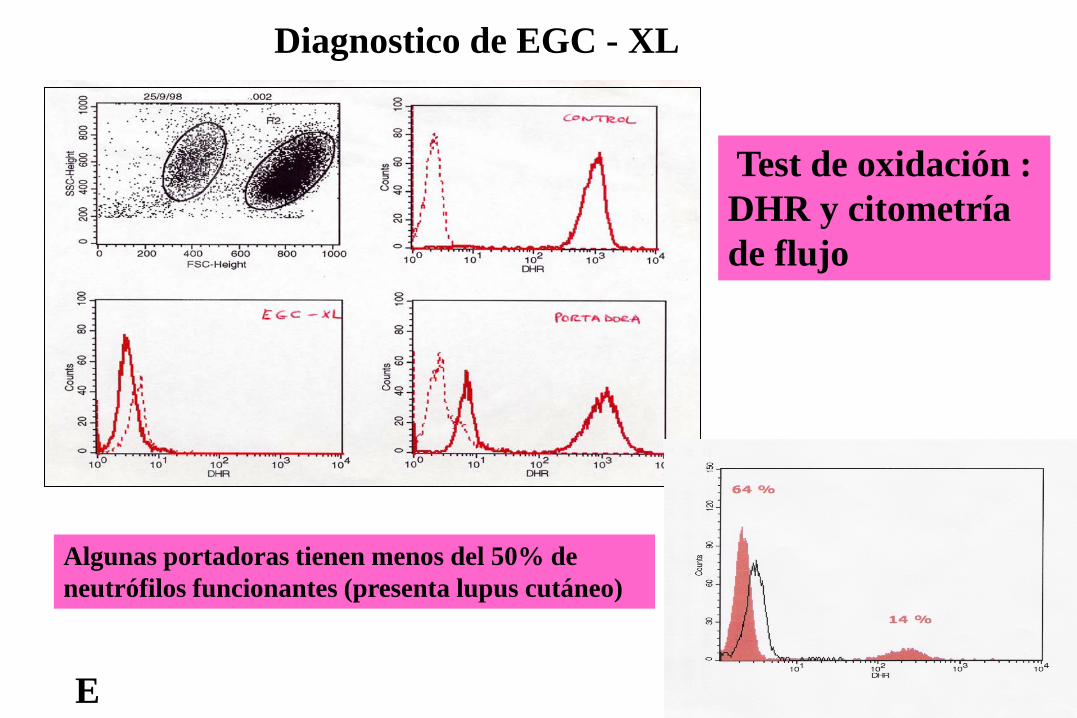
Test de oxidación : DHR y citometría de flujo
Diagnostico de EGC - XL
E
Algunas portadoras tienen menos del 50% de neutrófilos funcionantes (presenta lupus cutáneo)

Deficiencias del Complemento Características Generales
* Enfermedades autoinmunes (primeros componentes)
* Infecciones por bacterias piógenas (C3)
* Infecciones por Neisserias (Ultimos componentes)
* Edema angioneuroticos familiar (C1INH)

Deficiencia de C1 INH Edema angioneurotico
Clínica:
* Comienzo segunda década
* Angioedema
* Urticaria
* Abdominalgias
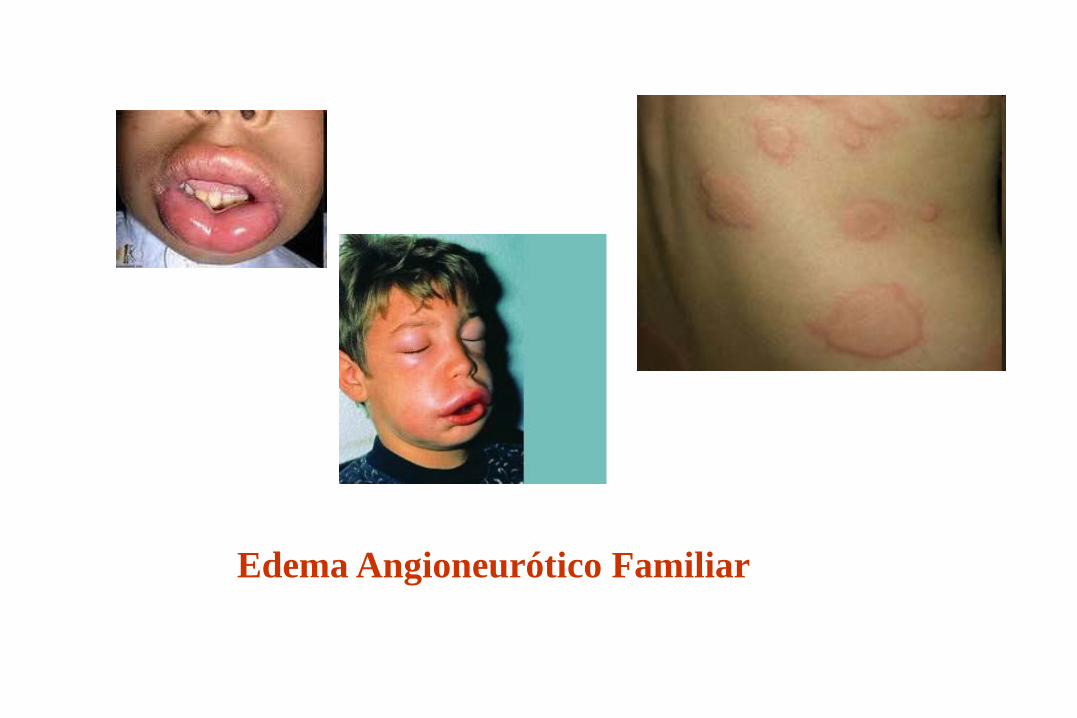
Edema Angioneurótico Familiar

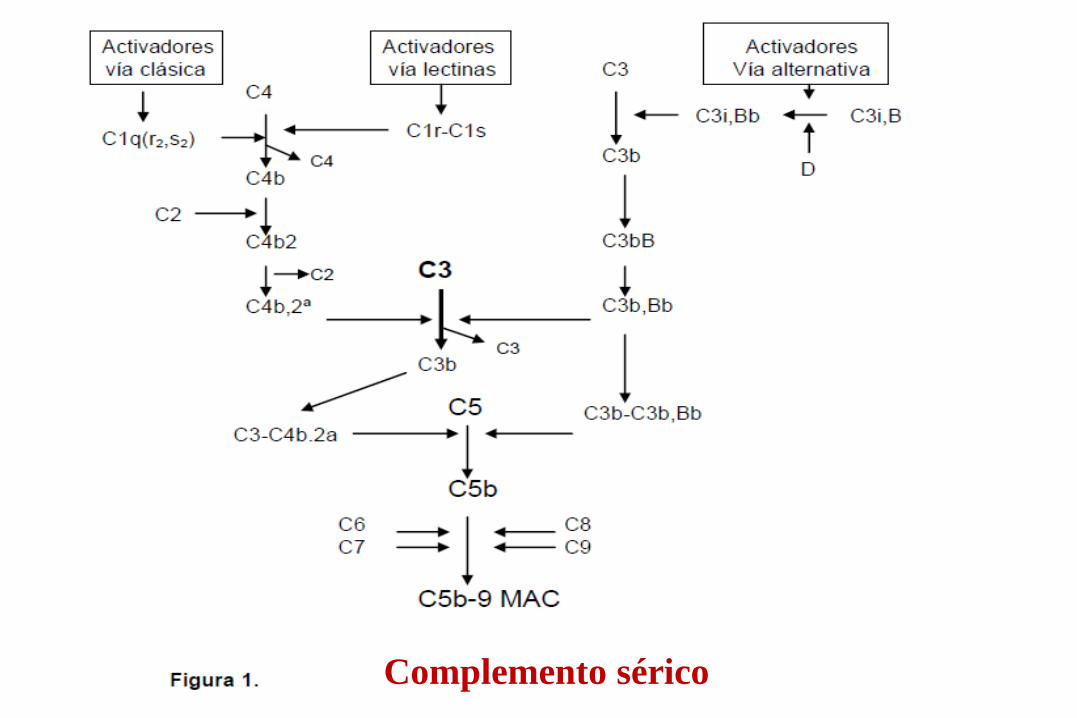
Evaluación del Complemento
Capacidad Hemolítica: -CH50 -AP50 -Vías lectinas
Cuantificación Factores complemento: -C3 y C4 -Otros Factores
Inhibidor del C1: Niveles, función, Ac contra inhibidor C1
Estudios especiales: Estudios función del complemento

Deficiencia de C1 INH Edema angioneurotico
Laboratorio:
* Deficiencia de C1 INH
* C4 Disminuido
* C3 Normal
* 20 % C1 INH no funcionante.

Bases del diagnóstico de las Inmunodeficiencias
1. Diagnostico Clínico
2. Diagnóstico Inmunofenotípico
3. Diagnóstico Molecular y Genético

Utilidad de las tecnicas de Genetica en Inmunodeficiencias
1. Confirmación diagnostica
2. Diagnóstico de Portadores
3. Diagnóstico Prenatal

Utilización del diagnóstico prenatal en Inmunodeficiencias
* IDCS LX o AR con mutación conocida ( cg del IL-R2, ZAP-70, Jak 3). * Desconomiento de mutación pero con hermano con fenotipo de IDCS LX * Agammaglobulinemia LX (Btk) * Sindrome de Hiper IgM LX (CD154) * EGC Lx y AR * Otras ID ( Cd11/CD18; HLA, Complemento)

Localización de mutantes de proteínas en linfocitos T y B Responsables de Inmunodeficiencias primarias.

Inmunodeficiencias primarias LX Defecto Molecular
Enfermedad Gen responsable
Agammaglobulinemia LX Btk IDCS LX Cadena g R-IL-2 S. De Wiskkot-Aldrich WASP S. De Hiper IgM CD40 ligando CGD-LX Gp91/phox Síndrome Proliferativo LX SAP Deficiencia de Properdina Properdina

Inmunodeficiencias primarias A.R. Defecto Molecular (I)
Enfermedad Gen responsable
•Agammaglobulinemia AR: Cadena m, l 5/14 Vpre B •IDCS por defecto recombinación: RAG1 y RAG2 •Deficiencia ADA: Adenosina desaminasa •Deficiencia PNP: Purin nucleotido fosforilasa •Deficiencia en Jak 3: Jak 3 •Deficiencia en receptor IL-7: Cadena a R-IL-7 •Deficiencia en receptor L.T: CD3g/CD3e •Deficiencia en Zap 70: Zap 70 •CGD AR: p47/phox,p67/phox,p22/phox

Inmunodeficiencias primarias A.R. Defecto Molecular (II)
Enfermedad Gen responsable Deficiencia moléculas adhesión: CD11/CD18 Síndrome de Chediak-Higashi: LYST Ataxia Telangiectasia: ATM Deficiencia de MHC-II: MHC2TA,RFXANK,RFX5 Deficiencia de MHC-I: TAP 1, TAP 2 IDCS con anomalía CD45: CD45 Síndrome linfopr. Autoinmune: Fas Susceptibilidad aumentada a micobacterias: T-INF-g, b1_IL-12; IL- 12,STAT 1 Deficiencia de C1 INH: C1 INH

Evitar Infecciones Gammmaglobulina Cuidad nutrición Transplante médula Antibioticoterapia Otras fuentes de selectiva. Células madre Evitar transfusiones Terapéutica génica No vacunas con Interferon gamma gérmenes vivos. C1 INH
Consejo genético Diagnóstico prenatal Diagnóstico precoz
Inmunodeficiencias Primarias TRATAMIENTO
MEDIDAS PROFILACTICAS
MEDIDAS GENERALES
TERAPUTICA SUSTITUTIVA
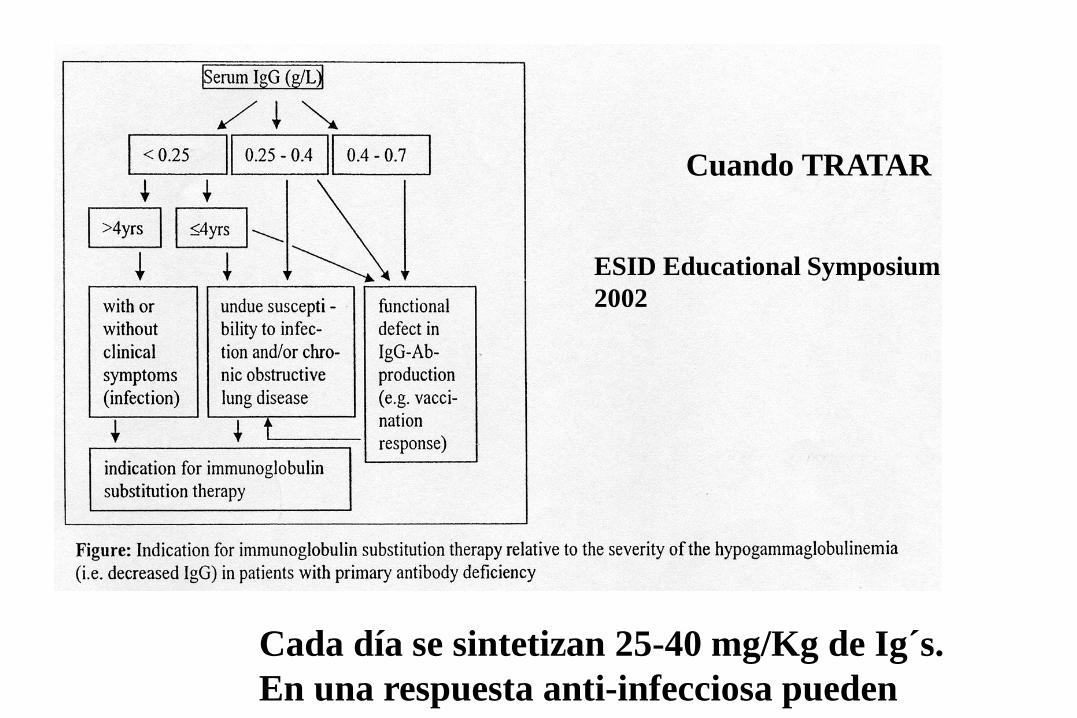
Cuando TRATAR
ESID Educational Symposium 2002
Cada día se sintetizan 25-40 mg/Kg de Ig´s. En una respuesta anti-infecciosa pueden

Indicaciones del uso de gammaglobulinas
• Indicada: • Agammaglobulinemia ligada al sexo • Agammaglobulinemia con hiper IgM • Síndrome variable común de I.D. • Niños con infecciones de repetición y
portadores de: – Defecto de cadenas pesadas – Deficiencia de subclases de de IgG – Ataxia Telangiectasia

Indicaciones del uso de gammaglobulinas
• Util::
• No aconsejada:
• Contraindicada:
I.D. Combinada Severa Déficit de ADA y PNP Deficiencias subclases de IgG. Síndrome de Di George Síndrome de Wiskot-Aldrich
• Hipogammaglobulinemia transitoria
• Déficit selectivo de IgA

Comparación Gammaglobulina IV y SC
IGIV * Intervalos más largos (3-4 semanas) * Inmediata subida de la IgG sérica * No contraindicación en pacientes hemorrágicos * Procedimiento mas habitual para los sanitarios IGSC * No precisa acceso venoso * Tasas séricas de IgG más estables y sin “valles” * Sin reacciones secundarias sistémicas * Flexibilidad de la aplicación * Mayor facilidad para uso domiciliario * Sin ausencia escolar
Ventajas

Comparación Gammaglobulina IV y SC
IGIV * Precisa acceso venoso * Inmovilización del enfermo durante 3-5 h * Dependencia hospitalaria * Posibles reacciones sistémicas graves
IGSC * Intervalos cortos (administración semanal) * Riesgo de peor cumplimiento * Necesidad de una bomba de perfusión * Más reacciones locales leves * Dificultad para infundir grandes cantidades * Menor biodisponibilidad de la IgG
Desventajas

Administración mas “fisiológica” Buena tolerancia y menor cantidad de Ig necesaria y a domicilio. Brit J Hosp Med 2007

Evitar Infecciones Gammmaglobulina Cuidad nutrición Transplante médula Antibioticoterapia Otras fuentes de selectiva. Células madre Evitar transfusiones Terapéutica génica No vacunas con Interferon gamma gérmenes vivos. C1 INH
Consejo genético Diagnóstico prenatal Diagnóstico precoz
Inmunodeficiencias Primarias TRATAMIENTO
MEDIDAS PROFILACTICAS
MEDIDAS GENERALES
TERAPUTICA SUSTITUTIVA
Evitar Infecciones Gammmaglobulina Cuidad nutrición Transplante médula Antibioticoterapia Otras fuentes de selectiva. Células madre Evitar transfusiones Terapéutica génica No vacunas con Interferon gamma gérmenes vivos. C1 INH
Consejo genético Diagnóstico prenatal Diagnóstico precoz
Inmunodeficiencias Primarias TRATAMIENTO
MEDIDAS PROFILACTICAS
MEDIDAS GENERALES
TERAPUTICA SUSTITUTIVA

Tipos de trasplante de células madre
• En función del donante: -Autólogo -Homologo
-Alogénico (Idéntico, haploidéntico, no idéntico)
-Xenogénico • Según procedencia: -Células médula ósea
-Células periféricas -Células sangre de cordón -Células madre propias corregidas
• Contraindicada: Acondicionamiento previo trasplante

Indicaciones trasplante de células madres hematopoyéticas en I.D.
• Inmunodeficiencia combinada y severa • Inmunodeficiencias combinadas:
-Síndrome de Wiskott –Aldrich -Síndrome de Di George -Síndrome de Hiper IgM -Deficiencia ligando de CD40 (CD40L) -Deficiencia de antígenos HLA (Clase I y II) -Hipoplasia cartílago pelo -Enfermedad linfoproliferativa ligada al X -Candidiasis mucocutánea crónica

Indicaciones trasplante de células madres hematopoyéticas en I.D.
• Alteraciones de polimorfonuclares: -Enfermedad Granulomatosa Crónica -Deficiencia de moleculas de adhesion leucocitaria -Síndrome de Chediak-Higashi -Sindrome de Grscelli -Sindrome de Schwachmann-Diamon
• Otras enfermedades: -Enfermedades metabólicas -Fallos medulares -Neoplasias hematopoyéticas -Tumores

Evitar Infecciones Gammmaglobulina Cuidad nutrición Transplante médula Antibioticoterapia Otras fuentes de selectiva. Células madre Evitar transfusiones Terapéutica génica No vacunas con Interferon gamma gérmenes vivos. C1 INH
Consejo genético Diagnóstico prenatal Diagnóstico precoz
Inmunodeficiencias Primarias TRATAMIENTO
MEDIDAS PROFILACTICAS
MEDIDAS GENERALES
TERAPUTICA SUSTITUTIVA


Faringitis

Amigdalitis

Sinusitis
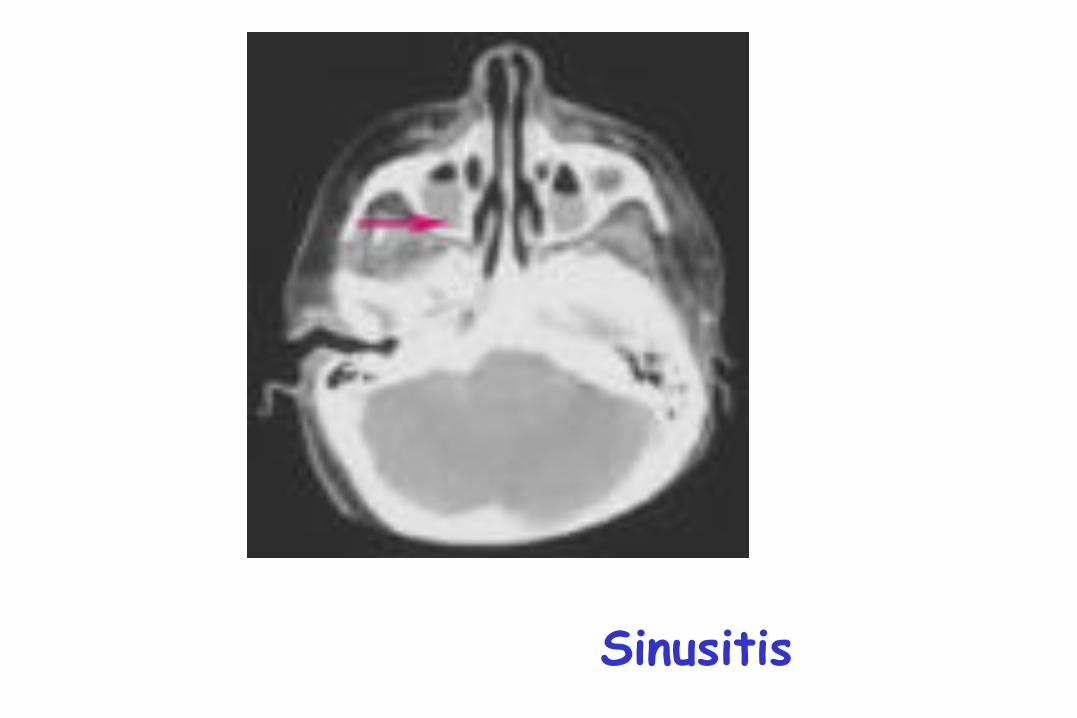
Sinusitis

Otitis
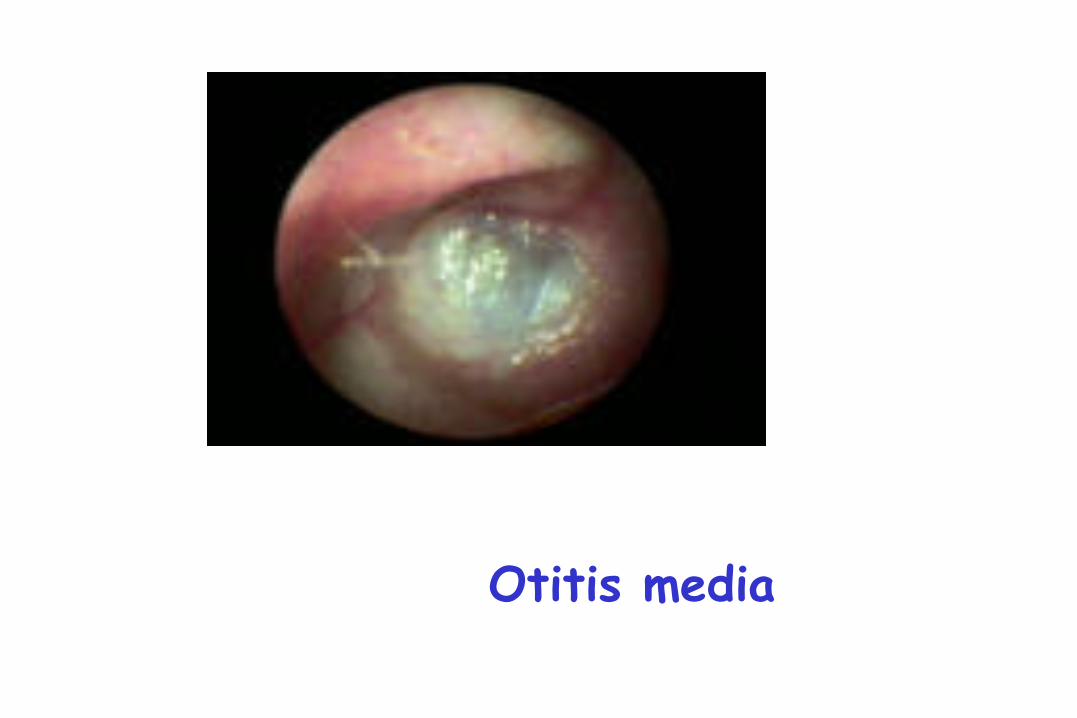
Otitis media
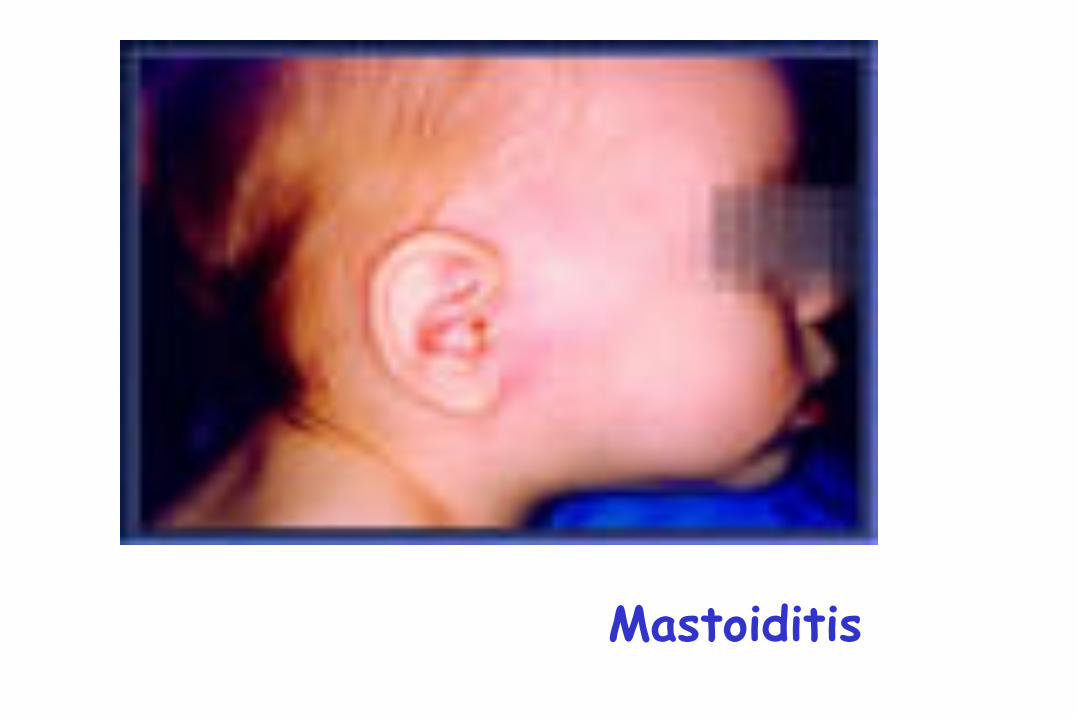
Mastoiditis

Mastoiditis

Neumonía
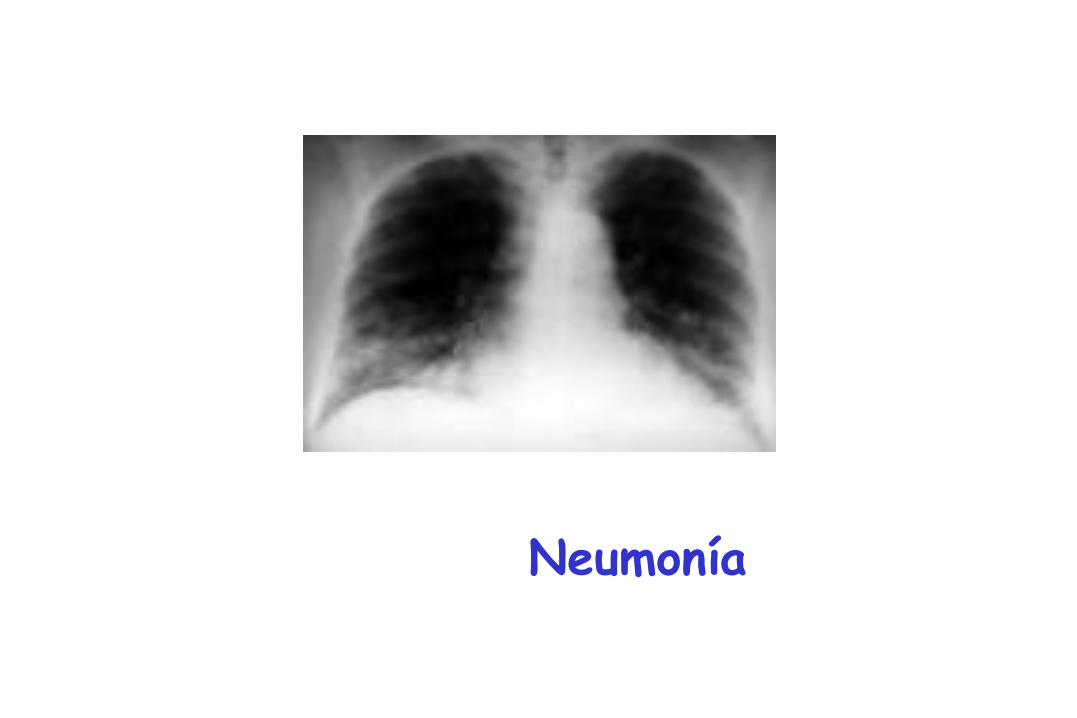
Neumonía

Neumonía

Neumonía por Pneumocystis

Pneumocystis Carynii
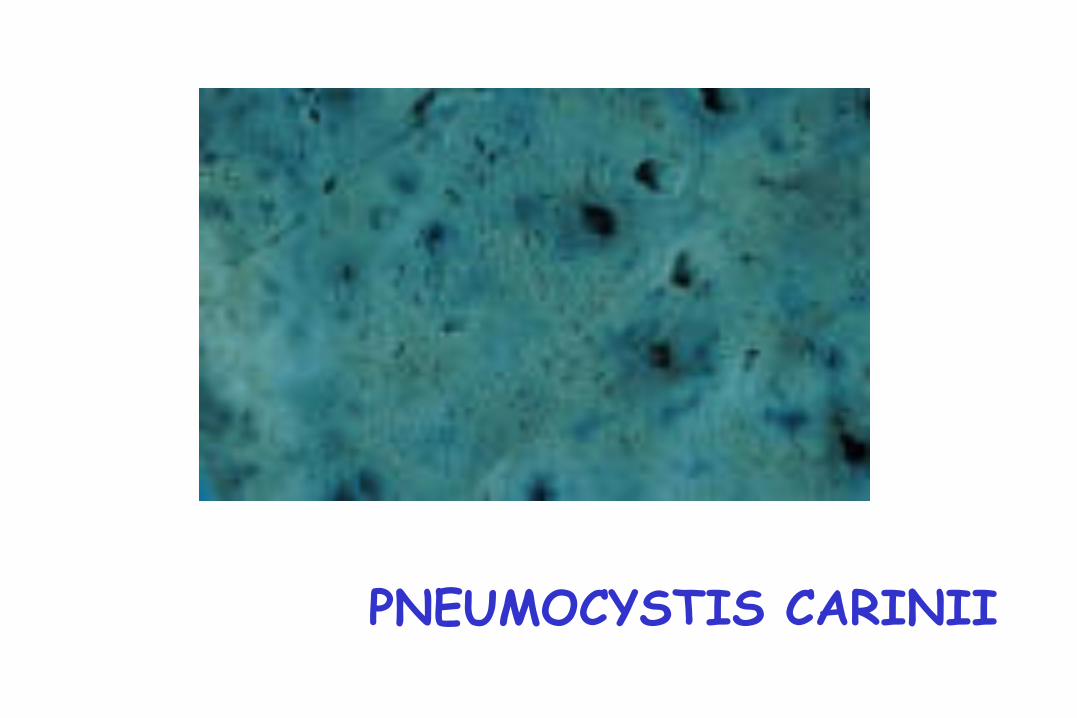
PNEUMOCYSTIS CARINII

Neumonia por cytomegalovirus

Neumonia por Legionella

Neumonía por Cándidas

Furunculosis

Gingivitis

Gingivitis
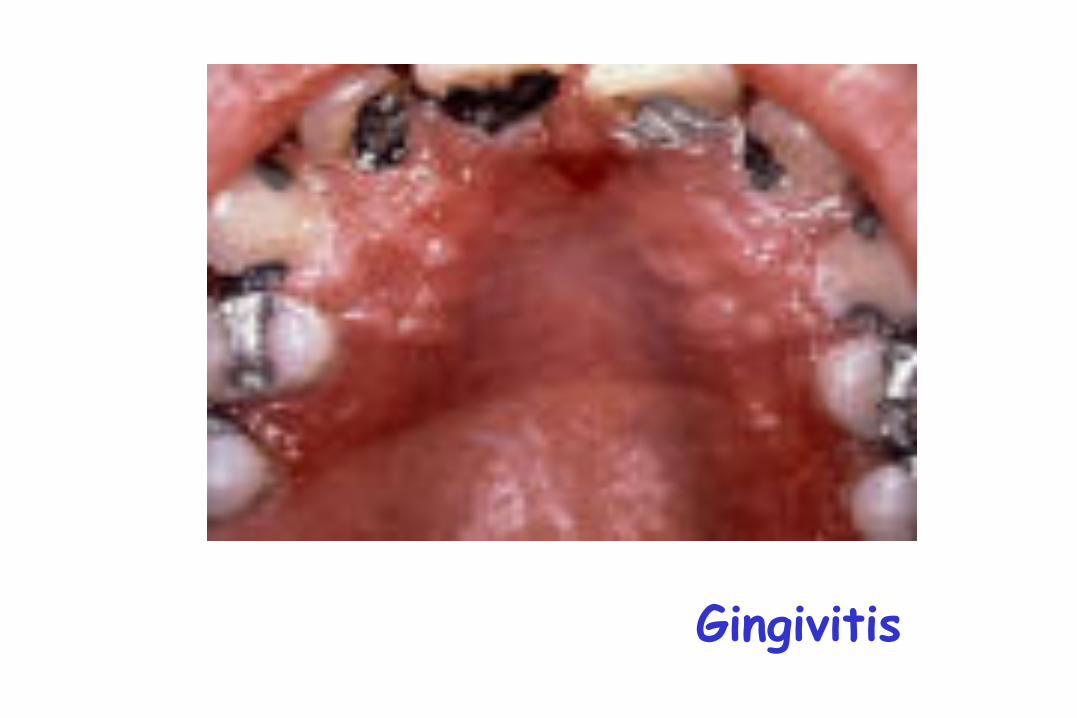
Gingivitis

Candidiasis oral-Muguet

Muguet

Candidiasis genital

Onicomicosis

Dermatitis atópica

Dermatitis

Dermatitis seborreica

Exantema
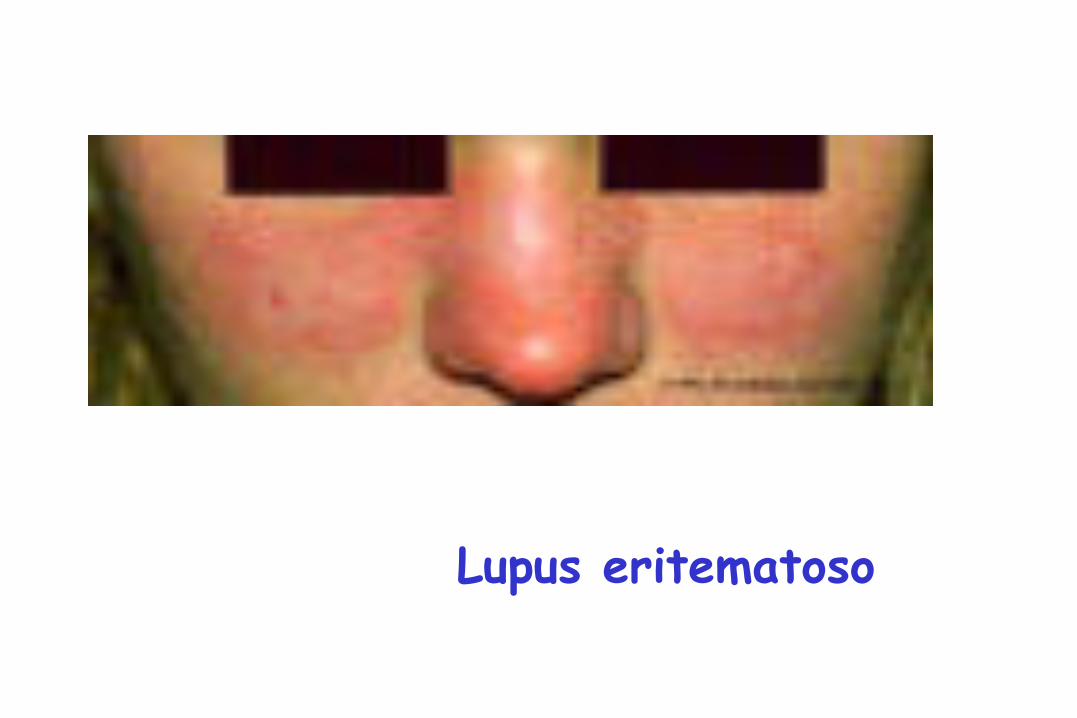
Lupus eritematoso

Síndrome de Di George

Síndrome de Di George

Osteomielitis
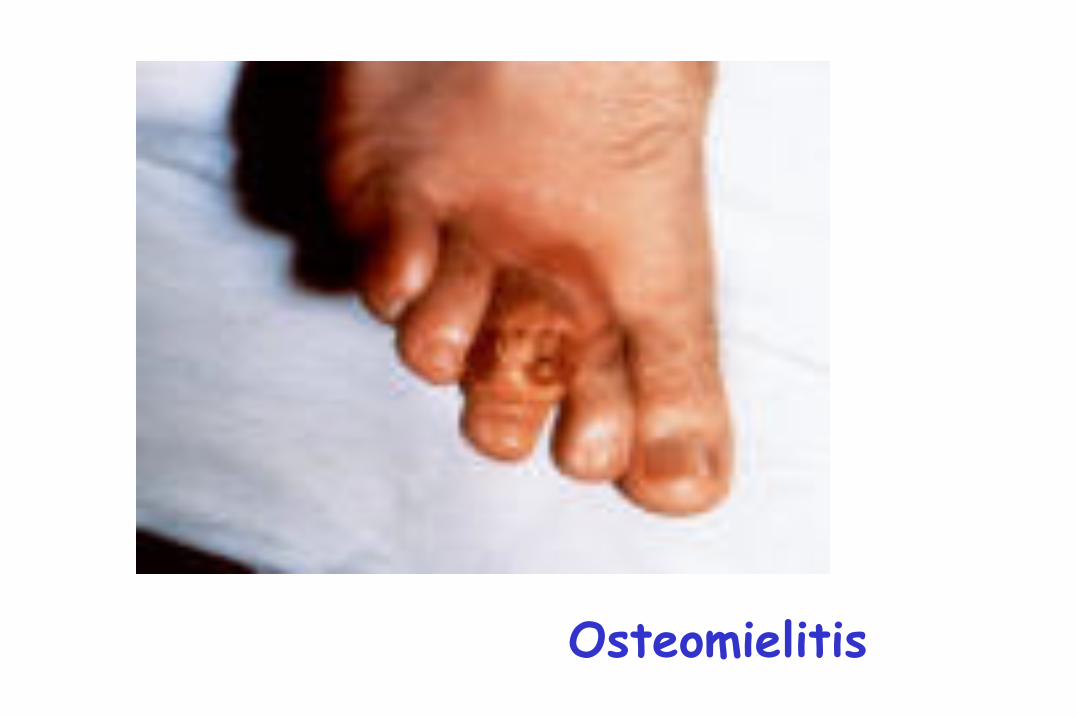
Osteomielitis

Meningitis meningocócica

Meningitis