strategies to optimize residual renal risk in chronic
TRANSCRIPT

University of Groningen
Strategies to optimize renoprotective therapy in proteinuric renal patientsVogt, Liffert
IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite fromit. Please check the document version below.
Document VersionPublisher's PDF, also known as Version of record
Publication date:2008
Link to publication in University of Groningen/UMCG research database
Citation for published version (APA):Vogt, L. (2008). Strategies to optimize renoprotective therapy in proteinuric renal patients. s.n.
CopyrightOther than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of theauthor(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons).
The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license.More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne-amendment.
Take-down policyIf you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediatelyand investigate your claim.
Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons thenumber of authors shown on this cover page is limited to 10 maximum.
Download date: 08-01-2022

Strategies to optimize renoprotective therapy in
proteinuric renal patients
Liffert Vogt

CIP-GEGEVENS KONINKLIJKE BIBLIOTHEEK, DEN HAAG Vogt, L. Strategies to optimize renoprotective therapy in proteinuric renal patients Proefschrift Groningen. – Met lit. opg. – Met samenvatting in het Nederlands. ISBN 978-90-367-3279-6 © Copyright 2008 L. Vogt All rights are reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, mechanically, by photocopying, recording, or otherwise, without the written permission of the author. This work was supported by an educational grant from Merck & Co., Inc. and performed within the framework of the research school GUIDE (Groningen University Institute for Drug Exploration) Financial support by the Dutch Kidney Foundation is gratefully acknowledged. Financial support of Actelion Pharmaceuticals, Ltd. is highly appreciated. Further financial support was kindly provided by the University of Groningen, University Medical Center Groningen, GUIDE, Merck Sharp & Dohme BV, Boehringer Ingelheim BV, Amgen BV, AstraZeneca BV, Baxter BV, Bristol-Myers Squibb BV, Eli Lilly Nederland BV, Fresenius Medical Care Nederland BV, Genzyme Nederland BV, Itémedical BV- distributor of SpaceLabs Medical ABP monitors in the Netherlands, Novartis Pharma BV, Novo Nordisk BV, Pfizer BV, Roche BV, Sanofi-Aventis BV, Servier Nederland Farma BV Cover illustrations: Minerva defeats Ignorantia, oil on canvas by Peter Paul Rubens, Koninklijk Museum voor Schone Kunsten, Antwerp, Belgium (front). Photograph, ornament from the sanctuary of Sacromonte di Varese, Italy (back). Proofreading and language advice: B. Crompton Proofreading: C.L. Verhoeven, E. Vogt-Noordenbos Printed by: Ponsen & Looijen, Wageningen, the Netherlands

RIJKSUNIVERSITEIT GRONINGEN
Strategies to Optimize Renoprotective Therapy in Proteinuric Renal Patients
Proefschrift
ter verkrijging van het doctoraat in de Medische Wetenschappen
aan de Rijksuniversiteit Groningen op gezag van de
Rector Magnificus, dr. F. Zwarts, in het openbaar te verdedigen op
woensdag 2 april 2008 om 16.15 uur
door
Liffert Vogt
geboren op 4 juni 1975 te Groningen

Promotores: Prof. dr. G.J. Navis Prof. dr. D. de Zeeuw Copromotores: Dr. R.P.F. Dullaart Dr. A.J.J. Woittiez Beoordelingscommissie: Prof. dr. M.M. Levi Dr. M. Noris
Prof. dr. P.A.B.M. Smits

Paranimfen: Drs. J.L.P. Brouwer Mr. A.G. Jonkman


Scire est per causas cognoscere
—Aristoteles Analytica posteriora, liber I


CONTENTS Chapter 1 Introduction and scope of this thesis 11 PART I. RENOPROTECTIVE STRATEGIES: BLOOD PRESSURE OR MORE?
Chapter 2 Renoprotection: a matter of blood pressure or agent characteristics? Updated from: Journal of the American Society of Nephrology 2002;13(Suppl):S202-7
21
Chapter 3 The angiotensin II receptor antagonist telmisartan reduces urinary albumin excretion in patients with isolated systolic hypertension: results of a randomized, double-blind, placebo-controlled trial 33 Journal of Hypertension 2005;23:2055-61
PART II. OPTIMIZING RENOPROTECTION: REDUCING RESIDUAL PROTEINURIA
Introduction Renoprotection by dual blockade of renin-angiotensin system 49 Letter in: Lancet 2003;361:1170-1
Chapter 4 Altering the dosing time of trandolapril does not overcome nocturnal resistance to the antiproteinuric effect of ACE inhibition in non-diabetic kidney disease 53
Chapter 5 Independent and added effects of low sodium diet and diuretic on the antiproteinuric effect of the AT1 antagonist losartan in non-diabetic proteinuric patients 63
Journal of the American Society of Nephrology 2008; in press
Chapter 6 Individual titration for maximal blockade of the renin-angiotensin-system in proteinuric patients: a feasible strategy? 81
Journal of the American Society of Nephrology 2005;16 (Suppl):S53-7 PART III. OPTIMIZING RENOPROTECTION: INTERVENTION IN NEW PATHOPHYSIOLOGICAL PATHWAYS
Chapter 7 Selective cyclooxygenase-2 inhibition reduces proteinuria in renal patients 95
Chapter 8 Effect of the urotensin receptor antagonist palosuran on albuminuria in hypertensive patients with type 2 diabetic nephropathy: results from the PROLONG proof-of-concept study 109
PART IV. OPTIMIZING RENOPROTECTION: REDUCTION OF CARDIOVASCULAR RISK?
Chapter 9 Lipid management in the proteinuric patient: do not overlook the importance of proteinuria reduction 123
Nephrology Dialysis Transplantation 2004;19:5-8
Chapter 10 Cellular cholesterol efflux to plasma from proteinuric patients is elevated and remains unaffected by antiproteinuric treatment 131
Nephrology Dialysis Transplantation 2006;21:101-6

Chapter 11 Change in urinary albumin excretion is predictive of cardiovascular outcome in normotensive patients with type 2 diabetes mellitus and microalbuminuria 143
Diabetes Care 2007;30:3119-21 GENERAL DISCUSSION
Chapter 12 Towards optimal long-term protection of kidney and heart in the proteinuric renal patient 153 Adapted and updated from: Minerva Medica 2004;95:395-409
List of abbreviations 173 Nederlandse samenvatting (summary in Dutch) 175 Dankwoord 181 Curriculum vitae 183
List of publications 184

Chapter 1
Introduction and scope of this thesis

CHAPTER 1
12

INTRODUCTION AND SCOPE
‘ reaming of no more dialysis’ entitled the interview with G. Remuzzi in The Lancet of 1998 when the results of the REIN follow-up trial were published in the same journal (1,2). The study showed that in patients with progressive renal failure assigned to treatment with the angiotensin-converting enzyme (ACE) inhibitor ramipril, the need for dialysis could be prevented within the study follow-up of almost 4 years. Patients that initially were assigned to placebo but shifted to ramipril during the study displayed a considerable reduction of the rate of renal function decline when ramipril was started. These results elicited the hope that the dream of ‘no more dialysis’ could be fulfilled.
D
Yet, since the publication of the REIN study, the number of patients that suffer from progressive renal failure has still increased and, notably, also the number of patients depending on renal replacement therapy. The Dutch End Stage Renal Disease Registry reported in their annual report of 2006 that nowadays about 11,000 patients depend on renal replacement therapy in The Netherlands, of whom 5,500 patients are receiving dialysis treatment (3). The last decade the number of patients that depend on dialysis showed an 1.3% yearly increase. Albeit life-saving, dialysis has a tremendous negative impact on the lives of renal patients, as it impairs the quality of life and the risk of premature cardiovascular death is enormous. For example, in The Netherlands the annual mortality rate in dialysis patients amounts to 20%.
Obviously to enable further improvements in prevention of renal function loss, or even remission of renal function loss, new strategies are urgently needed. To this purpose, different approaches could be followed. On the one hand, many innovative approaches are extensively studied. Unfortunately, promising new strategies, as gene therapy and stem cell therapy, have shown limited or no success in experimental conditions, whilst clinical application will not be foreseen in the near future. An
13

CHAPTER 1
illustrative experimental study indicates, for instance, that stem cell therapy, representing a so-called regenerative strategy directed to in situ repair of the kidney, seems to act as a two-edged sword, as stem cells may contribute to repair processes as well as the persistence of triggers for renal scarring (4). On the other hand, from clinical studies, as the REIN, several common risk factors for progressive renal failure have emerged that could be subject to therapy. Such symptomatic approach comprising the identification and treatment of new determinants of progressive renal failure is challenging and is the focus of this thesis. CURRENT INSIGHTS OF PROGRESSIVE RENAL FUNCTION DECLINE
Throughout the last decades substantial progress has been made in understanding progressive renal failure. In respect of renoprotective treatment strategies, three important early hypotheses are still relevant. Firstly, the notion that high blood pressure is not merely a symptom of renal failure but also an important risk factor in the progression of renal function decline was a crucial step forward in renoprotective treatment (5,6). Since then, antihypertensive treatment is regarded as the cornerstone of therapy directed to attenuation of progressive renal function decline. Secondly, Brenner and co-workers postulated that loss of nephrons as result of renal injury can elicit a vicious circle of progressive renal function decline (7). They hypothesized that after loss of functioning nephrons, the remaining (remnant) nephrons exhibit a compensatory response aimed at preservation of glomerular filtration rate, at the expense of glomerular hypertension that eventually leads to glomerular capillary damage, glomerular protein leakage, and finally glomerulosclerosis, and further nephron loss. The fact that antihypertensive treatment exerts a beneficial effect on renal function is, according to this hypothesis, due to the reduction of glomerular pressure that results from the systemic blood pressure lowering, and/or specific reduction of glomerular pressure by post-glomerular vasodilation, as occurs during blockade of the renin-angiotensin-aldosterone system (RAAS). Thirdly, more than a decade ago, Remuzzi and co-workers came up with the concept that proteinuria is not only a symptom of renal damage but plays a key role itself in progressive renal function loss (8,9). They demonstrated that albumin that leaks into the tubular lumen exerts tubulotoxic effects leading to renal scarring, and that antiproteinuric treatment can prevent these sequelae (8). Accordingly, they postulated that proteinuria reduction is pivotal for renoprotection, as subsequently supported by the results of the previously mentioned REIN trial and several subsequent studies. Figure 1 summarizes the current understanding of progressive renal failure. In this figure, several factors are included that were identified previously to play a role in the sequelae of progressive renal failure and might be accessible for therapeutic intervention.
14

INTRODUCTION AND SCOPE
Figure 1. Current understanding of the processes involved in progressive renal failure (adapted from Remuzzi & Bertani, N Engl J Med 1998;339:1448-56)
AIMS AND SCOPE OF THE THESIS
Starting from the generally held concept that antihypertensive treatment is a prerequisite for renoprotective treatment, and that the degree of proteinuria reduction predicts long-term renal outcome, a series of intervention studies are carried out aimed to optimize the antiproteinuric response. Feasibility of improving the response to therapy by better use of currently available classes of drugs as well as new modes of intervention was studied. Finally, the impact of better proteinuria reduction on cardiovascular risk was assessed, as outlined in the following paragraphs. Part I. Renoprotective strategies: blood pressure or more? The benefit of antihypertensive therapy in chronic renal failure is reviewed in chapter 2. The evidence from the large clinical trials showing that antihypertensive treatment with agents that interfere in the RAAS exert a specific renoprotective effect as compared to other classes of antihypertensive drugs is discussed. In addition, available evidence that specific renoprotective action of an agent seems related to its proteinuria lowering effect is reviewed. In chapter 3, it is hypothesized that the angiotensin type 1 receptor (AT1) antagonist telmisartan may have renoprotective characteristics by a specific antiproteinuric action beyond its blood pressure lowering effect. In this study, a direct comparison of telmisartan with an agent of another antihypertensive class, hydrochlorothiazide, in a double-blind, randomized, placebo-controlled large scale
15

CHAPTER 1
clinical trial in patients with isolated systolic hypertension blood pressure was performed. Part II. Optimizing renoprotection: reducing residual proteinuria RAAS blockade is effective in attenuating the progression of renal function decline but can not prevent the development of end-stage renal disease in many patients. In order to fully confer the benefit of the renoprotective effects of RAAS blockers, different therapeutic approaches to optimize the response to RAAS blockade may be useful. Such optimization strategies could then be used to maximize the antiproteinuric response to RAAS blockers, as also discussed in the introduction to Part II. In chapter 4, 5 and 6, different strategies to improve the antiproteinuric effectiveness of RAAS blockade are tested. Firstly, changing the dosing time of the ACE inhibitor trandolapril on proteinuria in non-diabetic proteinuric patients was tested in chapter 4. Previously a relative therapy resistance to this long-acting ACE inhibitor was observed during the night when dosed in the morning (10). It was hypothesized that better antiproteinuric efficacy during the night by changing the dosing time may contribute to lower residual 24-h proteinuria. The effect of dosing in the evening and twice daily on the nocturnal therapy resistance and residual proteinuria was studied as compared to dosing in the morning. Secondly, in chapter 5 the modulation of sodium status during RAAS blockade-based proteinuria reduction was studied in non-diabetic proteinuric patients in a double blind randomized placebo-controlled cross-over design. Early on, it has been demonstrated that low sodium intake improved the response to an ACE inhibitor, and that co-treatment with hydrochlorothiazide restored the response to ACE inhibitor therapy that had been blunted by high sodium intake (11,12). The studies on effects of intensified measures on sodium status have not so far been undertaken. The combination of two sodium-depleting measures was assumed to further improve the response to the AT1 antagonist losartan. In chapter 6, feasibility of titration with different measures to recommended levels of residual proteinuria (< 1 g/d) was tested. It has been postulated earlier that lowest residual proteinuria will allow to stop further progression of renal function decline, or to enable remission of renal function (13). In this study, low sodium in combination with hydrochlorothiazide, dual RAAS blockade and dose-titration with an ACE-inhibitor were stepwise applied to pursue lowest levels of proteinuria. Part III. Optimizing renoprotection: intervention in new pathophysiological pathways In the following chapters, several strategies to reduce proteinuria by targeting pathways other than the RAAS were explored. In Chapter 7, it was hypothesized that cyclooxygenase-2 (COX-2) inhibitors may have antiproteinuric efficacy. COX-2 inhibitors may represent an old concept in a new fashion. Non-steroidal anti-
16

INTRODUCTION AND SCOPE
inflammatory drugs (NSAID), acting by interfering in prostaglandin synthesis, were the first effective antiproteinuric agents for symptomatic proteinuria reduction, even before ACE inhibitors were introduced (14). Their full development as renoprotective agents was hampered, among others due to high incidence of intolerance to these drugs. For instance, these agents are associated with adverse effects on the gastrointestinal and central nervous systems. The new selective COX-2 inhibitors also interfere in the prostaglandin system, albeit in a different way. In the study described in chapter 7, the antiproteinuric potency of two doses of the selective COX-2 inhibitor rofecoxib were tested and compared with the traditional NSAID indomethacin. In addition, the antiproteinuric efficacy of rofecoxib was compared with the ACE inhibitor lisinopril. In chapter 8, antiproteinuric efficacy of the newly developed drug palosuran was tested in a phase II double blind, randomized, placebo-controlled cross-over study in diabetic patients characterized by having microalbuminuria and hypertension. This drug represents a new concept of interfering in the sequelae of hyperfiltration, development of albuminuria and progression of chronic renal failure as observed in renal patients. Palosuran has shown to be an effective blocker of the urotensin system. The peptide urotensin II has been described as one of the most potent vasoconstrictive peptides in mammals, possibly even more potent than angiotensin and endothelin, and is involved in the pathophysiology of diverse nephropathies as well as in hypertension (15). Accordingly, blockade of this system was hypothesized to exert renoprotection, as measured by proteinuria reduction. Part IV. Optimizing renoprotection: reduction of cardiovascular risk? Hitherto, one could conclude that residual proteinuria predicts subsequent renal risk, but it is not completely clear whether this is also true for the residual cardiovascular risk. Considering that proteinuria is a potent cardiovascular risk factor, residual proteinuria might also be a predictor of cardiovascular risk, as for instance by data from a recent large clinical trial conducted in patients with diabetic nephropathy (RENAAL study) (16). Chapters 9 and 10 address effects of antiproteinuric intervention on proteinuria-associated cardiovascular risk factors. Firstly, the prevalence of the elevated cardiovascular risk in proteinuric patients in relation to the classical risk factors, as well as the possible benefits of the current antiproteinuric treatments in cardiovascular risk management, are discussed in chapter 9. Proteinuric patients have distinct abnormalities in their cholesterol metabolism and these abnormalities may contribute to their elevated cardiovascular risk. Secondly, chapter 10 shows the results of dose-titration with single and dual RAAS blockade on proteinuria and lipid profiles, especially HDL metabolism and HDL function, in non-diabetic proteinuric patients. Finally, in chapter 11 the predictive value of proteinuria reduction on long-term major cardiovascular
17

CHAPTER 1
complications was investigated. General discussion In conclusion, the results of the studies described in this thesis are discussed in chapter 12 and put into the perspective of the available literature, thereby identifying specific patient factors that may contribute to the residual renal risk. It is questioned whether these specific factors can be seized in order to resolve the remaining risk for progression to end-stage renal disease and the remaining cardiovascular risk in each individual patient. REFERENCES 1. Simini B. Giuseppe Remuzzi: dreaming of no more renal dialysis. Lancet 1998;352:296 2. Ruggenenti P, Perna A, Gherardi G, Gaspari F, Benini R, Remuzzi G. Renal function and requirement for dialysis in chronic
nephropathy patients on long-term ramipril: REIN follow-up trial. Lancet 1998;352:1252-6 3. Dutch End Stage Renal Disease Registry (Stichting RENINE). Http://www.renine.nl (2006) 4. Broekema M, Harmsen MC, van Luyn MJ et al. Bone marrow-derived myofibroblasts contribute to the renal interstitial
myofibroblast population and produce procollagen I after ischemia/reperfusion in rats. J Am Soc Nephrol 2007;18:165-755. Alvestrand A, Gutierrez A, Bucht H, Bergstrom J. Reduction of blood pressure retards the progression of chronic renal failure in
man. Nephrol Dial Transplant 1988;3:624-31 6. Herlitz H, Bjorck S, Nyberg G, Granerus G, Aurell M. Clinical evaluation of felodipine in patients with refractory hypertension.
Drugs 1987;34 Suppl 3:151-5 7. Brenner BM, Meyer TW, Hostetter TH. Dietary protein intake and the progressive nature of kidney disease: the role of
hemodynamically mediated glomerular injury in the pathogenesis of progressive glomerular sclerosis in aging, renal ablation, and intrinsic renal disease. N Engl J Med 1982;307:652-9
8. Abbate M, Zoja C, Rottoli D et al. Antiproteinuric therapy while preventing the abnormal protein traffic in proximal tubule abrogates proteinuria. J Am Soc Nephrol 1999;10:804-13
9. Remuzzi G, Bertani T. Pathophysiology of progressive nephropathies. N Engl J Med 1998;339:1448-56 10. Buter H, Hemmelder MH, van Paassen P, Navis GJ, de Zeeuw D, de Jong PE. Is the antiproteinuric response to inhibition of the
renin-angiotensin system less effective during the night? Neprol Dial Transplant 1997;12:S53-611. Heeg JE, de Jong PE, van der Hem GK, de Zeeuw D. Efficacy and variability of the antiproteinuric effect of ACE inhibition by
lisinopril. Kidney Int 1989;36:272-9 12. Buter H, Hemmelder MH, Navis GJ, de Jong PE, de Zeeuw D. The blunting of the antiproteinuric efficacy of ACE inhibition by
high sodium intake can be restored by hydrochlorothiazide. Nephrol Dial Transplant 1998;13:1682-5 13 Ruggenenti P, Schieppati A, Remuzzi G. Progression, remission, regression of chronic renal diseases. Lancet 2001:357:1601-8 14. Arisz L, Donker AJ, Brentjens JR, van der Hem GK. The effect of indomethacin on proteinuria and kidney function in the nephrotic
syndrome. Acta Med Scand 1976;199:121-5 15. Ashton N. Renal and vascular actions of urotensin II. Kidney Int 2006;70:624-9 16. De Zeeuw D, Remuzzi G, Parving H-H et al. Albuminuria, a therapeutic target for cardiovascular protection in type 2 diabetic
patients with nephropathy. Circulation 2004;110:921-7
18

PART I
RENOPROTECTIVE STRATEGIES: BLOOD PRESSURE OR MORE?

PART I
20

Chapter 2
Renoprotection: a matter of blood pressure or agent-characteristics? Liffert Vogt , Gerjan Navis and Dick de Zeeuw
Updated from: Journal of the American Society of Nephrology 2002; 13: S202-7

CHAPTER 2
22

RENOPROTECTION: BLOOD PRESSURE OR AGENT?
Data from recent clinical trials show that lowering of blood pressure reduces the rate of renal function loss in chronic renal disease. Much evidence supports the hypothesis that blood pressure lowering obtained by intervention in the renin-angiotensin-aldosterone system (RAAS) has an additive renoprotective effect both in diabetic and non-diabetic renal diseases. However, to dissociate between blood pressure dependent and non-blood-pressure dependent action of RAAS blockade, the relevant trials are in many cases flawed by design, resulting in blood pressure differences between the comparative antihypertensive strategies. This review discusses whether the relevant literature allows concluding that RAAS intervention has renoprotective effects in addition to its effects on blood pressure. In particular, the main evidence for a specific renoprotective action of RAAS blockade is provided by its consistent antiproteinuric action, which cannot completely be attributed to the reduction in blood pressure. Indeed, other strategies that lower proteinuria without having an antihypertensive effect, such as lowering dietary protein intake or the use of non-steroidal anti-inflammatory drugs, appear to have a renoprotective effect as well. Interestingly, a consistent finding across different intervention studies is that the more proteinuria is reduced, the better the kidney appears to be protected. Therefore, it is concluded that agent-characteristics of RAAS intervention (i.e., antiproteinuric properties) independently influence renal function loss, in addition to its blood pressure lowering effect. Future studies should further explore the renoprotective benefit of non-antihypertensive intervention measures, alone and in combination with antihypertensive strategies.
ntihypertensive therapy has always been the cornerstone of renoprotective intervention. Recent large trials indicate that particularly intervention in the renin-angiotensin-aldosterone system (RAAS) appears to be effective in retarding the decline of renal function loss in both diabetic and non-diabetic renal diseases. In non-diabetic patients, the AIPRI (1) and REIN study (2) showed that angiotensin-converting enzyme (ACE) inhibitors delay the progression of renal function loss. Lewis et al. (3) showed a renoprotective effect of ACE inhibitors in type 1 diabetic patients. Moreover, other recent studies, RENAAL (4), IDNT (5) and IRMA-2 (6), demonstrated angiotensin II type 1 receptor (AT1) antagonists to be renoprotective in type 2 diabetics.
A
The above-mentioned trials can be taken as impressive evidence for RAAS intervention being superior to other treatment strategies. However, other large studies could not confirm the renoprotective superiority of RAAS blockade over other antihypertensives. Recently, the ALLHAT study, which is the largest trial ever comparing different classes of antihypertensives, failed to demonstrate additional benefit of ACE inhibitors over conventional antihypertensives in diabetic and non-
23

CHAPTER 2
diabetic patients (7). Moreover, the UKPDS study, another large, well-conducted trial in type 2 diabetic patients did not support better renoprotection by RAAS blockade over beta-blockade (8). These data point towards the importance of blood pressure lowering as such. However, whether the renoprotective effects of RAAS intervention are due to the specific pharmacological effects of RAAS blockade, or due to their antihypertensive potency is a crucial question. The issue is still open, because in many of the above trials demonstrating superiority of RAAS blockade, the obtained blood pressure levels were lower in the patients treated with an agent that intervenes in the RAAS, as compared to the control groups. The current review therefore focuses on the particular question, that is, is renoprotection obtained by a lower blood pressure per se, or do the specific pharmacological properties of the agent exert an independent renoprotective effect? REDUCTION OF BLOOD PRESSURE
Blood pressure is an important risk factor for renal function loss. In the MRFIT study
(9), blood pressure was a strong predictor for the development of end-stage renal failure during 16 years of follow-up in middle-aged men. The study identified a strong graded relation between both systolic and diastolic blood pressure and end-stage renal disease. Several other studies pointed out that a more aggressive blood pressure control is beneficial to the course of renal function loss in renal patients.
In patients with diabetic nephropathy, the importance of aggressive blood pressure reduction for renal function preservation has been demonstrated (10,11). Early on, Parving et al. demonstrated in an observational study that the long-term, aggressive antihypertensive treatment retards the rate of renal function loss in type 1 diabetic patients.
Type 2 diabetic patients with nephropathy were studied in the ABCD study (12). In this study of 950 patients, the presence of hypertension was associated with nephropathy. Patients with hypertension were randomized to an intensive blood pressure target (diastolic blood pressure of 75 mmHg) vs. a moderate blood pressure target (diastolic blood pressure of 80-89 mmHg). After 5 years of follow-up, an equally stabilizing effect on GFR decline was reported in both intervention groups (13). Also, in the non-hypertensive patients in this study a more aggressive blood pressure control did not influence the GFR, although a lower percentage of patients progressed from normoalbuminuria to overt albuminuria (14).
In non-diabetic patients, several studies showed that blood pressure level was an important contributor to progression of chronic renal failure (15,16,17). Within the MDRD study, (non-IDDM) patients with a diverse array of renal disease were randomly assigned to either a usual or a low blood pressure goal (17). In this (sub)study, a higher mean arterial blood pressure over 3 years was associated with a faster decline in GFR.
24

RENOPROTECTION: BLOOD PRESSURE OR AGENT?
Remarkably, these relations were even stronger for patients with a greater baseline proteinuria.
Bakris et al. performed a meta-analysis of long-term clinical trials directed to blood pressure reduction in both diabetic and non-diabetic patients (18). This analysis showed a linear relationship between the obtained blood pressure and the rate of decline of renal function across the different studies. Thus, the available evidence indicates that blood pressure reduction exerts a beneficial effect on the decline of renal function. Nevertheless, interesting differences in renoprotective potency between different regimens have also been observed.
RENOPROTECTIVE ACTION DEPENDS ON ANTIHYPERTENSIVE TREATMENT MODALITY
Over the last decade, several studies found additional renoprotective benefits of RAAS blockade in comparison with conventional antihypertensive treatment. These observations were made both in diabetes (type 1 and type 2) as well as in a variety of non-diabetic renal diseases.
Early studies on the effect of the ACE inhibitor captopril in small groups of both hypertensive (19) and non-hypertensive type 1 diabetic patients (20) demonstrated that the rate of renal function loss was effectively inhibited by RAAS intervention. In particular, in type 1 diabetic patients with nephropathy, Bjorck et al. showed that RAAS intervention with enalapril does reduce the rate of renal function decline more than antihypertensive treatment with metoprolol at a similar blood pressure (21,22). Interestingly, in the patients of the enalapril group a significant reduction in proteinuria was observed. A large randomized controlled trial (Collaborative trial) performed by Lewis et al. (3) compared captopril with placebo in type 1 diabetic patients with mild proteinuria. For both groups, additional conventional drugs were used to titrate the blood pressure to a similar level. This study showed a better renal outcome in the RAAS intervention group that was still apparent after adjustment for small, but non-significant differences in blood pressure.
In type 2 diabetic patients, a beneficial effect of intervention in the RAAS was found as well. In normotensive type 2 diabetic patients, RAAS intervention by enalapril has been reported to attenuate progression of renal function loss (23). During 7-year follow-up treatment with enalapril, a risk reduction of 42% (95% CI: 15-69%) for developing nephropathy was found. Recently, two extensive randomized double-blind placebo controlled studies investigated the renoprotective effect of RAAS intervention by AT1 antagonists. In the RENAAL trial (4), less patients in the losartan treatment group in comparison to the placebo group reached the primary endpoint, defined as a composite of doubling of the baseline serum creatinin, end-stage renal disease or death. In both groups also additional conventional antihypertensive drugs were used to lower
25

CHAPTER 2
blood pressure to the target level. Although there were again small differences in blood pressure observed between the two arms in favour of the losartan arm, the benefit exceeded that attributable to blood pressure reduction after statistical adjustment. In accord with earlier findings, treatment with losartan was associated with a reduction in proteinuria. Also in the IDNT trial (5), treatment with irbesartan was associated with a lower risk of reaching the primary composite endpoint compared with the placebo group and compared with a calcium-channel antagonist group. The changes or differences in blood pressure that were achieved could not explain these differences. Again, after treatment with irbesartan proteinuria was reduced. Of note, the IRMA-2 study demonstrated that the renoprotective effect of irbesartan seems to be dose-dependent, as treatment with irbesartan 300 mg QD resulted in additional risk reduction of the development of overt diabetic nephropathy as compared to irbesartan 150 mg (6). Also in this study, the differences could not be explained by blood pressure effects.
In contrast, the type 2 diabetic patients in the UKPDS did not show that RAAS intervention is superior to conventional antihypertensive treatment, although the study did show the long-term benefit of a lower than usual blood pressure goal (24). Moreover, an additional benefit of the ACE inhibitor captopril against conventional treatment with the beta-blocker atenolol could not be confirmed in reaching the endpoint of renal failure (8). Also, a small randomized double blind parallel study comparing lisinopril and atenolol in hypertensive type 2 diabetic patients reported an identical blood pressure reduction and GFR decline after a follow-up of almost 3 years (25). In addition, the ABCD study did not show a larger benefit of ACE inhibition on renal function loss in either hypertensive and non-hypertensive type 2 diabetic patients (13,14). These contrasting findings may be due to the design of the studies comparing two single drugs, while the IDNT and RENAAL study were designed to compare the AT1 antagonist with placebo in addition to conventional antihypertensive therapy. Furthermore, beta-blockers may not be the suited comparator drug, since it is reported that beta-blockade effectively inhibits RAAS, by another mechanism than ACE inhibitors and AT1 antagonists (26).
In non-diabetic patients, the benefits of RAAS intervention have also been established in large randomized trials, although Apperloo et al., in a relative small clinical trial, could not demonstrate better renoprotection in patients treated with an ACE inhibitor as compared to beta-blockade (27). Maschio et al. in the AIPRI study (1) conducted a randomized double blind placebo controlled trial comparing benazepril against placebo. Also in this study, additive conventional antihypertensive medication was used to titrate to the blood pressure goal. After three years follow-up an overall risk reduction of 53 (95% CI: 27-50%) was found in the treatment group for reaching the primary endpoint, i.e. doubling baseline serum creatinin concentration or the need for
26

RENOPROTECTION: BLOOD PRESSURE OR AGENT?
dialysis. Notably, a significant blood pressure difference was observed in favour of the RAAS intervention arm. After adjustment for the lower blood pressure in the benazepril group, the overall risk reduction prevailed. Also, benazepril induced a significant proteinuria reduction compared to placebo. The REIN trial (2) in patients with overt proteinuria demonstrated a clear-cut beneficial effect of ramipril. In this prospective randomized double blind trial, a pre-stratification recognized two levels in proteinuria in patients assigned to ramipril or placebo plus conventional antihypertensive therapy. In patients with proteinuria of 3 grams or more ramipril safely reduced the rate of GFR decline and halved the combined risk of doubling serum creatinin or end-stage renal disease. These effects were accompanied by a substantial lowering of the urinary protein excretion rate. The reported renoprotective effect appeared to exceed what could be expected from the degree of blood pressure reduction. The notion that the renoprotective effect of an ACE inhibitor is accompanied with reduction of proteinuria was also demonstrated in the already mentioned study of Apperloo et al. (27). In a recent meta-analysis of 11 randomized trials in non-diabetic renal disease, the antihypertensive strategy with ACE inhibition was compared with placebo or conventional antihypertensive medication (28). In most reviewed studies a better blood pressure control was reached during treatment with an ACE inhibitor. After adjustment for blood pressure, this meta-analysis showed also a significant beneficial effect in favour of ACE inhibition.
In conclusion, the available data strongly suggest that RAAS intervention has a renoprotective effect that goes beyond its antihypertensive effect in different renal diseases. However, in most studies blood pressure was not similar in the tested arms, and notably lower in the RAAS intervention arm. One could therefore still state that blood pressure is the sole determinant of renoprotection, and that no extra benefit is to be expected from RAAS intervention giving a similar blood pressure control. Importantly, all of the studies showed that intervention in the RAAS led to a reduction of urinary protein excretion. This antiproteinuric effect was significantly higher than all the other treatment strategies. PROTEINURIA: MARKER FOR PROGRESSION
The above findings, adding to earlier data, have drawn attention to the role of proteinuria as a predictor of progressive renal function loss. Proteinuria is nowadays looked upon as a marker, or maybe even a causal factor, of progressive renal function loss, and not merely a consequence of renal disease. In different renal conditions, both in patients as well as in the experimental setting, proteinuria consistently determines the rate of progression of renal function loss (29,30). This may indeed point to the pathogenic role of proteinuria in progressive renal damage. The MDRD study (17)
27

CHAPTER 2
showed that baseline proteinuria was an important determinant of the renoprotective benefit in the follow-up after reduction of blood pressure. The additional benefit of a lower blood pressure goal was clearly more pronounced in patients with a higher baseline proteinuria.
In patients with diabetes, it was demonstrated that the amount of reduction in proteinuria achieved during treatment with captopril was associated with a better long-term effect on the decline of renal function loss (31). Also post-hoc data of the RENAAL study show that the more one reduces proteinuria, the lower the risk of development of end-stage renal disease (32).
Similar effects are found in non-diabetic patients. Two studies reported that the short-term antiproteinuric response to antihypertensive treatment predicted the GFR decline during follow-up (27,33). These correlations were independent of baseline proteinuria. From a therapeutic perspective, it is important to note that the residual proteinuria was correlated with the subsequent progression of renal function loss (27). Moreover, the REIN trial, with hard endpoint data, showed baseline proteinuria to be an independent and accurate predictor of disease progression and end-stage renal disease (34). In response to ramipril treatment, a stronger short-term antiproteinuric effect is a important predictor of more effective protection against end-stage renal disease in the long term (35). In this respect, the COOPERATE trial (36), comparing the combination treatment with the ACE inhibitor trandolapril and the AT1 antagonist losartan vs. both monotherapies, is of interest. In this study, blood pressure was kept practically equal in the three treatment arms, but demonstrated an almost 50% risk reduction after dual RAAS blockade as compared to monotherapy. The renoprotective effect was accompanied with better proteinuria reduction and, importantly, the individual short-term antiproteinuric response was predictive for the renal outcome.
Thus, the large clinical trials comparing RAAS intervention with conventional antihypertensive strategies or placebo show that the renoprotective effect related to RAAS intervention is associated with a better antiproteinuric effect during RAAS intervention. Considering the predictive value of antiproteinuric potency for long-term renoprotection, and the consistent antiproteinuric efficacy of RAAS blockade, it would be logical to assume, that these specific antiproteinuric effects are involved in a renoprotective action exceeding the reduction of blood pressure. ANTIPROTEINURIC PROPERTIES COUNT: ANOTHER STRATEGY?
The importance of proteinuria reduction for renoprotection is supported by the renoprotective action of several blood-pressure-neutral interventions. As to non-pharmacological intervention, data indicate that a low-protein diet lowers proteinuria and reduces the rate of renal function loss (37). In an interesting parallel to
28

RENOPROTECTION: BLOOD PRESSURE OR AGENT?
pharmacological intervention, the amount of initial proteinuria reduction induced by the diet is correlated with the degree of subsequent renal function loss.
The effect of non-antihypertensive pharmacological intervention on proteinuria is also of interest. Immunosuppressive treatment was already reported to reduce proteinuria and with that, to influence the renal prognosis (38). Before the era of RAAS blockade, several groups of investigators focused on the effect of intervention in the synthesis of prostaglandins on proteinuria. Prostaglandins are modulators of vascular tone, glomerular haemodynamics, salt and water homeostasis, and renin-secretion in the kidney. The prostaglandin cascade is activated in several renal conditions. As such, prostaglandins might be involved in the pathophysiology of progressive renal function loss.
In non-diabetic renal disease, inhibition of prostaglandins by non-steroidal-anti-inflammatory-drugs (NSAIDs) leads to renal haemodynamic changes, with a reduction in intraglomerular pressure, and reduction of proteinuria (39,40). Historically, besides corticosteroids, NSAIDs were the first drugs with a marked antiproteinuric effect (41,42). Similar antiproteinuric effects were observed in diabetic nephropathy (43). The effect on proteinuria appears related to the degree of prostaglandin E-2 (PGE-2) inhibition in the urine (44,45). Moreover, when ACE inhibitors are compared with NSAIDs, it is reported that there is equal reduction in urinary protein excretion (46,47). A combination of both treatments resulted even in a more potent antiproteinuric effect (47,48). Interestingly, Vriesendorp et al. reported on the renoprotective effect of indomethacin when used as an antiproteinuric tool in patients with proteinuria (49). Unfortunately, this is a retrospective open label, non-randomized study and prospective studies have never been carried out.
Notably, the antiproteinuric effect of NSAIDs, and also of RAAS intervention, appears to occur only under special conditions. Firstly, the antiproteinuric effect is clearly dependent on the dose of the drugs. For indomethacin, naproxen and flurbiprofen, the maximal allowed chronic dose had to be used for achieving an optimal antiproteinuric effect (44,50). Secondly, not only the dose of the NSAID, but also the specific NSAID determined the degree of response. Indomethacin was considered to be superior to respectively diclophenac and flurbiprofen (44). Thirdly, the patient has to be on a sodium-restricted diet or use a diuretic to attain the full potential of this antiproteinuric treatment (39). These findings underscore that without systemic effects on blood pressure specific agents can induce an antiproteinuric response and subsequent renoprotection. Like RAAS intervention, treatment with NSAIDs as indomethacin or naproxen may have a beneficial effect on renal function. Because of the well-known side effects of NSAIDs and the required high dose, it is not attractive to study the long-term renoprotective effect in patients with nephropathy.
29

CHAPTER 2
A NOVEL STRATEGY: COX-2 INHIBITION
Several recent data suggest that COX-2 inhibition may be a novel and promising renoprotective strategy for diabetic and non-diabetic patients with proteinuria. In animal studies, COX-2 is up-regulated in progressive renal disease models, such as ablation and diabetes (51). Moreover, COX-2 inhibition has antiproteinuric and renal protective effects in these animal models (51,52). In humans, so far no data on the antiproteinuric or renoprotective action of COX-2 inhibition are available. There are data however, showing similarity between NSAIDs and COX-2 inhibitors on the renal haemodynamics and sodium handling (53). Therefore, it may be worthwhile to investigate whether COX-2 inhibitors also share the antiproteinuric properties of the NSAIDs. The application of NSAIDs in renoprotective strategies has been limited because of the high frequency of side-effects, whereas the use of COX-2 inhibition was better tolerated and has been reported to be associated with lower incidence of gastrointestinal complications (54,55). It would allow studying the antiproteinuric effect of a drug that has no lowering effects on the systemic blood pressure. Thus, we would further elucidate the role of proteinuria in progressive renal damage, and also test a new application in proteinuric patients in whom the optimal reduction of proteinuria cannot always be obtained due to drug-induced symptomatic hypotension. It should be noted however; that the lessons drawn from the results of the APPROVe study indicate that the clinical application of COX-2 inhibitors might be problematic as the chronic use of the COX-2 inhibitor rofecoxib was associated with a higher incidence of myocardial infarction, at least in non-proteinuric patients (56).
CONCLUSION
To summarize, agents that reduce blood pressure provide renoprotection in chronic renal diseases. An additional renoprotective effect is achieved by intervention in the RAAS. ACE inhibitors and AT1 antagonists show this additional, non-pressure-related effect on decline of renal function. The characteristic potential of both agents to reduce the proteinuria appears to be the strongest predictor for renal outcome. Apparently, the antiproteinuric characteristics of the specific agent used for lowering the blood pressure determine the degree of renoprotection reached, more than reduction in blood pressure alone. Agents that do not lower blood pressure, but specifically reduce proteinuria, may exert a renoprotective effect too. We conclude that it is not only the blood pressure reduction that is important for renal protection. The particular agent (RAAS blocker) has a specific renal protective effect beyond the blood pressure lowering, and is thus the agent of choice in patients with progressive renal function loss. To obtain more effective renoprotective strategies in the future, it would be of interest to specifically explore the renoprotective action of non-antihypertensive agents.
30

RENOPROTECTION: BLOOD PRESSURE OR AGENT?
REFERENCES 1. Maschio G, Alberti D, Janin G et al. Effect of the angiotensin-converting-enzyme inhibitor benazepril on the progression of chronic
renal insufficiency. N Engl J Med 1996;334:939-45 2. The GISEN Group. Randomized placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of
terminal renal failure in proteinuric, non-diabetic nephropathy. Lancet 1997;349:1857-63 3. Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. N
Engl J Med 1993;329:1456-62 4. Brenner BM, Cooper MA, de Zeeuw D et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2
diabetes and nephropathy. N Engl J Med 2001;345:861-9 5. Lewis EJ, Hunsicker LG, Clarke WR et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with
nephropathy due to type 2 diabetes. N Engl J Med 2001;345:851-60 6. Parving H-H, Lehnert H, Brochner-Mortensen J, Gomis R, Andersen S, Arner P. The effect of irbesartan on the development of
diabetic nephropathy in patients with type 2 diabetes. N Engl J Med 2001;345:870-8 7. The ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. Major outcomes in high-risk
hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs. diuretic. JAMA 2002;288:2981-97
8. UK Prospective Diabetes Study Group. Efficacy of atenolol and captopril in reducing risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 39. BMJ 1998;317:713-20
9. Klag MJ, Whelton PK, Randall BL et al. Blood pressure and end-stage renal disease in men. N Engl J Med 1996;334:13-8 10. Parving H-H, Andersen AR, Smidt UM, Hommel E, Mathiesen ER, Svendsen PA. Effect of antihypertensive treatment on kidney
function in diabetic nephropathy. BMJ 1987;294:1443-7 11. Mogensen CE. Long-term antihypertensive treatment inhibiting progression of diabetic nephropathy. BMJ 1982;285:685-8 12. Mehler PS, Jeffers BW, Estacio R, Schrier RW. Associations of hypertension and complication in non-insulin-dependent diabetes
mellitus. Am J Hypertens 1997;10:152-61 13. Estacio RO, Jeffers BW, Gifford N, Schrier RW. Effect of blood pressure control on diabetic microvascular complications in
patients with hypertension and type 2 diabetes. Diabetes Care 2000;23(Suppl. 2):B54-B64 14. Schrier RW, Estacio RO, Esler A, Mehler P. Effects of aggressive blood pressure control in normotensive type 2 diabetic patients on
albuminuria, retinopathy and strokes. Kidney Int 2002;61:1086-97 15. Alvestrand A, Gutierrez A, Bucht H, Bergstrom J. Reduction of blood pressure retards the progression of chronic renal failure in
man. Nephrol Dial Transplant 1988;3:624-31 16. Hannedouche T, Albouze G, Chauveau P, Lacour B, Jungers P. Effects of blood pressure and antihypertensive treatment on
progression of advanced chronic renal failure. Am J Kidney Dis 1993;21(Suppl. 2):131-7 17. Peterson JC, Adler S, Burkart JM et al. Blood pressure control, proteinuria, and the progression of renal disease. Ann Intern Med
1995;123:754-62 18. Bakris GL, Williams M, Dworkin L et al. Preserving renal function in adults with hypertension and diabetes: a consensus approach.
Am J Kidney Dis 2001;36:646-61 19. Parving H-H, Hommel E, Smidt UM. Protection of kidney function and decrease in albuminuria by captopril in insulin dependent
diabetes with nephropathy. BMJ 1998;297:1086-91 20. Parving H-H, Hommel E, Damkjaer Nielsen M, Giese J. Effect of captopril on blood pressure and kidney function in normotensive
insulin dependent diabetics with nephropathy. BMJ 1989;299:533-6 21. Bjork S, Mulec H, Johnsen SA, Norden G, Aurell M. Renal protective effect of enalapril in diabetic nephropathy. BMJ
1992;304:339-43 22. Mulec H, Johnsen SA, Bjorck S. Long-term enalapril treatment in diabetic nephropathy. Kidney Int Suppl 1994;45:S141-4 23. Ravid M, Lang R, Rachmani R, Lishner M. Long-term renoprotective effect of angiotensin-converting enzyme inhibition in non-
insulin-dependent diabetes mellitus. A 7-year follow-up study. Arch Intern Med 1996;156:286-9 24. UK Prospective Diabetes Study Group. Tight blood pressure control and risk of macrovascular and microvascular complications in
type 2 diabetes: UKPDS 38. BMJ 1998;317:703-13 25. Nielsen FS, Rossing P, Gall MA, Skott P, Smidt UM, Parving H-H. Long-term effect of lisinopril and atenolol on kidney function in
hypertensive NIDDM subjects with diabetic nephropathy. Diabetes 1997;46:1182-8 26. Blumenfeld JD, Sealey JE, Mann SJ et al. Beta-adrenergic receptor blockade as a therapeutic approach for suppressing the renin-
angiotensin-aldosterone system in normotensive and hypertensive subjects. Am J Hypertens 1999;12:451-9 27. Apperloo AJ, de Zeeuw D, de Jong PE. Short-term antiproteinuric response to antihypertensive treatment predicts long-term GFR
decline in patients with non-diabetic renal disease. Kidney Int Suppl 1994;45:S174-8 28. Jafar TH, Schmid CH, Landa M et al. Angiotensin-converting-enzyme inhibitors and progression of non-diabetic renal disease. Ann
Intern Med 2001;135:73-87 29. Williams PS, Fass G, Bone JM. Renal pathology and proteinuria determine progression in untreated mild/moderate chronic renal
failure. Q J Med 1988;67:343-54 30. Remuzzi G, Bertani T. Is glomerulosclerosis a consequence of altered glomerular permeability to macromolecules? Kidney Int
1990;38:384-394 31. Rossing P, Hommel E, Smidt UM, Parving H-H. Reduction in albuminuria predicts a beneficial effect on diminishing the
progression of human diabetic nephropathy during antihypertensive treatment. Diabetologia 1994;37:511-6 32. De Zeeuw D, Remuzzi G, Parving H-H et al. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy:
lessons from RENAAL. Kidney Int 2004;65:2309-20 33. Gansevoort RT, de Zeeuw D, de Jong PE. Long-term benefits of the antiproteinuric effect of angiotensin-converting enzyme
inhibition in nondiabetic renal disease. Am J Kidney Dis 1993;22:202-6 34. Ruggenenti P, Perna A, Mosconi L, Pisoni R, Remuzzi G. Urinary protein excretion rate is the best independent predictor of ESRF
in non-diabetic proteinuric chronic nephropathies. Kidney Int 1998;53:1209-16 35. Ruggenenti P, Perna A, Remuzzi G. Retarding progression of chronic renal disease: the neglected issue of residual proteinuria.
Kidney Int 2003;63:2254-61 36. Nakao N, Yoshimura A, Morita H, Takada M, Kayano T, Ideura T. Combination treatment of angiotensin-II receptor blocker and
angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomized controlled trial. Lancet 2003;361:117-24
37. El Nahas AM, Masters-Thomas A, Brady SA et al. Selective effect of low protein diets in chronic renal diseases. BMJ 1984;289:1337-41
31

CHAPTER 2
38. Idelson BA, Smithline N, Smith GW, Harrington JT. Prognosis in steroid-treated idiopathic nephrotic syndrome in adults. Analysis of major predictive factors after ten-year follow-up. Arch Intern Med 1977;137:891-6
39. Donker AJ, Brentjens JR, van der Hem GK, Arisz L. Treatment of the nephrotic syndrome with indomethacin. Nephron 1978;22:374-81
40. Arisz L, Donker AJ, Brentjens JR, van der Hem GK. The effect of indomethacin on proteinuria and kidney function in the nephrotic syndrome. Acta Med Scand 1976;199:121-5
41. Conte J, Suc JM, Mignon-Conte M. Anti-proteinuric effect of indomethacin in glomerulopathies (nephrotic syndrome, acute glomerulonephritis and chronic glomerulonephritis). J Urol Nephrol 1967;73:850-6
42. Michielsen P, Verberckmoes R, Desmet V, Hermerijckx W. Histological course of diffuse proliferative glomerulonephritis treated with indomethacin. J Urol Nephrol 1969;75:315-8
43. Hommel E, Mathiesen E, Arnold-Larsen S, Edsberg B, Olsen UB, Parving H-H. Effects of indomethacin on kidney function in type 1 (insulin-dependent) diabetic patients with nephropathy. Diabetologia 1987;30:78-81
44. Vriesendorp R, de Zeeuw D, de Jong PE, Donker AJM, Pratt JJ, van der Hem GK. Reduction of urinary protein and prostaglandin E2 excretion in the nephrotic syndrome by nonsteroidal anti inflammatory drugs. Clin Nephrol 1986;25:105-10
45. Shehadeh IH, Demers LM, Abt AB, Schoolwerth AC. Indomethacin and the nephrotic syndrome. JAMA 1979;241:1246-66 46. Garini G, Mazzi A, Buzio C et al. Renal effects of captopril, indomethacin and nifedipine in nephrotic patients after an oral protein
load. Nephrol Dial Transplant 1996;11:628-34 47. Heeg JE, de Jong PE, Vriesendorp R, de Zeeuw D. Additive antiproteinuric effect of the NSAID indomethacin and the ACE
inhibitor lisinopril. Am J Nephrol 1990;10(Suppl. 1):S94-7 48. Perico N, Remuzzi A, Sangalli F et al. The antiproteinuric effect of angiotensin antagonism in human IgA nephropathy is
potentiated by indomethacin. J Am Soc Nephrol 1998;9:2308-17 49. Vriesendorp R, Donker AJM, de Zeeuw D, de Jong PE, van der Hem GK, Brentjens JRH. Effects of nonsteroidal anti-inflammatory
drugs on proteinuria. Am J Med 1986;81(Suppl. 2B):84-94 50. Vriesendorp R, Donker AJM, de Zeeuw D, de Jong PE, van der Hem GK. Antiproteinuric effect of naproxen and indomethacin. Am
J Nephrol 1985;5:236-42 51. Harris RC. Cyclooxygenase-2 in the kidney. Cyclooxygenase-2 in the kidney. J Am Soc Nephrol 2000;11:2387-94 52. Wang JL, Cheng HF, Shappell S, Harris RC. A selective cyclooxygenase-2 inhibitor decreases proteinuria and retards progressive
renal injury in rats. Kidney Int 2000;57:2334-42 53. Rossat J, Maillard M, Nussberger J, Brunner HR, Burnier M. Renal effects of selective cyclooxygenase-2 inhibition in normotensive
salt-depleted subjects. Clin Pharmacol Ther 1999;66:76-84 54. Goldstein JL, Correa P, Zhao WW et al. Reduced incidence of gastroduodenal ulcers with celecoxib, a novel cyclooxygenase-2
inhibitor, compared to naproxen in patients with arthritis. Am J Gastroenterol 2001;96:1019-27 55. Bombardier C, Laine L, Reicin A et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with
rheumatoid arthritis. N Engl J Med 2000;343:1520-8 56. Bresalier RS, Sandler RS, Quan H et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention
trial. N Engl J Med 2005;352:1092-102
32

Chapter 3
The angiotensin II receptor antagonist telmisartan reduces urinary albumin excretion in patients with isolated systolic hypertension: results of a randomized, double-blind, placebo-controlled trial
Liffert Vogt, Gerjan Navis, Jürgen Köster, Athanasios J. Manolis, John L. Reid and Dick de Zeeuw. For the Angiotensin II Receptor Antagonist Telmisartan in Isolated Systolic Hypertension (ARAMIS) Study Group Journal of Hypertension 2005;23:2055-61

CHAPTER 3
34

TELMISARTAN IN IN ISOLATED SYSTOLIC HYPERTENSION
Objective—To examine the effect of telmisartan or hydrochlorothiazide (HCT) on the control of urinary albumin excretion (UAE) in patients with isolated systolic hypertension (ISH) and unselected for albuminuria in a pre-planned substudy of a large, multicentre, double-blind, placebo-controlled, randomized study. Methods—The Angiotensin II Receptor Antagonist Micardis in Isolated Systolic hypertension (ARAMIS) study compared the antihypertensive efficacy after 6 weeks of once-daily fixed doses of telmisartan 20, 40 or 80 mg versus HCT 12.5 mg or placebo in patients (n = 1039, age 35-84 years) with ISH (seated blood pressure (BP) 150-179/< 90 mmHg). The prospective substudy analysed UAE using spot morning samples. Results—Urinary albumin (> 2.2-901.6 mg/L) was detected at baseline in 614/918 patients, who were included in the substudy analysis. In the telmisartan group (n = 354, all doses combined), a median reduction in UAE from baseline of 14.1% (95% confidence intervals 7.3, 21.8) was observed versus 1.1% (-13.5, 16.0) and 2.7% (-0.9, 19.9) in the HCT (n = 140) and placebo (n = 120) groups, respectively. The difference between telmisartan and HCT was significant (p = 0.017). Reductions in UAE with telmisartan were observed in patients with baseline normoalbuminuria, microalbuminuria or macroalbuminuria. Telmisartan and HCT produced comparable reductions in systolic BP in these patients. Conclusion—In patients with ISH and unselected for baseline albuminuria, telmisartan 20-80 mg after 6 weeks’ treatment afforded significantly greater lowering of UAE than HCT 12.5 mg, irrespective of the baseline UAE, and despite comparable reductions in systolic BP with both drugs.
solated systolic hypertension (ISH) is an important treatment target, recognized as a dominant risk factor for heart disease, stroke and renal failure (1,2). Microalbuminuria and macroalbuminuria are also widely accepted risk factors for cardiovascular and renal disease in patients with diabetes (3,4) and hypertension (5-7). In patients with urinary albumin excretion (UAE) below the definition of microalbuminuria (< 20 mg/L), increased cardiovascular and renal risk still exists (8), and epidemiological studies show that such individuals are at increased risk of cardiovascular death (9) and all-cause mortality (9,10). Moreover, in hypertensive patients with left ventricular hypertrophy, elevated UAE results in heightened cardiovascular risk without any thresholds for albuminuria (11). In patients with ISH, macroalbuminuria is predictive of cardiovascular mortality and cardiovascular and renal morbidity (12), but the relationship between slightly elevated UAE in these patients and cardiovascular risk has not been studied.
I
Antihypertensive treatment should ideally not only lower the risk associated with high blood pressure, but also lower UAE and thus reduce its associated risk (13). Agents targeting the renin-angiotensin-aldosterone system (RAAS) lower blood pressure and are
35

CHAPTER 3
particularly effective in reducing microalbuminuria and macroalbuminuria (14). This reduction of UAE contributes independently to improved cardiovascular and renal outcomes in many different patient groups (15-21).
Until now there has been no information on the effects of different antihypertensive agents on UAE in hypertensive patients with UAE below microalbuminuric levels. Data in the present report are derived from a pre-planned substudy of a large-scale, multicentre study—Angiotensin II Receptor Antagonist telmisartan (Micardis) in Isolated Systolic hypertension (ARAMIS) (22). The purpose of the main study was to identify telmisartan doses that are more effective than placebo and non-inferior to hydrochlorothiazide (HCT) in lowering systolic blood pressure (SBP) in patients with ISH and that are well tolerated. The primary objective of this substudy was to evaluate the effect of different once-daily telmisartan doses (20, 40 and 80 mg) on UAE in patients with ISH and with UAE of any degree (including below the threshold for microalbuminuria), compared with once-daily HCT 12.5 mg or placebo. To determine whether any effect on UAE is independent of blood pressure control, efficacies of the different telmisartan doses on the reduction in SBP and diastolic blood pressure (DBP) were compared with those of HCT and placebo. PATIENTS AND METHODS
Study population A total of 1039 patients with ISH (seated SBP/DBP 150-179/< 90 mmHg) were randomized to treatment in the ARAMIS study, which was conducted in 17 countries in Europe, Australia and South Africa (22). Participants of either sex and between 35 and 84 years of age were recruited from primary-care or specialist hypertension centres. Patients receiving antihypertensive medication immediately prior to the study could only be enrolled if it was considered that withdrawal of that treatment and the possible receipt of up to 10 weeks’ placebo would not compromise their health. Exclusion criteria comprised secondary hypertension, hepatic and/or renal dysfunction (defined as serum creatinine > 159 µmol/L (> 1.8 mg/dL)), clinically relevant hypo- or hyperkalaemia, uncorrected volume or sodium depletion, primary aldosteronism, symptomatic cardio- or cerebrovascular disease, previous percutaneous transluminal angioplasty or coronary artery bypass grafting, inadequately controlled or recently stabilized type 2 diabetes mellitus, type 1 diabetes mellitus, hypersensitivity to telmisartan or HCT, or gout. Women who were pregnant, nursing or of childbearing potential were not eligible. Patients from Australia (n = 65) were excluded from the substudy because correct collection of urine was not possible for UAE assessment. All patients gave written, informed consent.
36

TELMISARTAN IN IN ISOLATED SYSTOLIC HYPERTENSION
Study design Local institutional review boards approved the protocol for the randomized, double-blind, placebo-controlled, parallel-group substudy. After screening, eligible patients entered a single-blind run-in period before randomization. If a patient was not receiving antihypertensive treatment at the time of enrolment, placebo was administered for 2 weeks. For patients who had received antihypertensive treatment immediately prior to enrolment, the placebo washout period was extended to 4 weeks. Thereafter, patients were randomized to 6 weeks’ double-blind, once-daily treatment with telmisartan 20, 40 or 80 mg, HCT 12.5 mg or placebo. Patients were instructed not to take their trial medication on the clinic visit days, which were always in the morning at approximately the same time and within 23-26 h of the most recent study medication intake. Determination of UAE, renal function and blood pressure Patients were evaluated at baseline and after 6 weeks’ double-blind treatment. UAE was measured as the urinary albumin concentration in a morning urine sample (first micturition after getting out of bed) determined by a commercial immunoturbidimetry assay (BNII, Bade Behring Diagnostica), lower limit of quantification 2.2 mg/L and inter- and intra-assay coefficients of variation 4.4% and 4.3%, respectively. Microalbuminuria was defined as a UAE of 20-200 mg/L and macroalbuminuria as UAE > 200 mg/L (23).
Urinary creatinine was determined in spot urine sample and serum creatinine was analysed. Estimated glomerular filtration rate (eGFR) was calculated from serum creatinine according to the Cockcroft formula (24).
Seated trough blood pressure was measured three times at every clinic visit, with at least 2 min. between measurements, after the patient had been seated for 5 min. A manual cuff sphygmomanometer was used, with measurements to the nearest 2 mmHg. SBP was recorded as Korotkoff phase I and DBP as phase V. Statistics analysis For the pre-planned substudy, the analysis was performed on the per-protocol population, comprising all randomized patients in whom UAE was measured at baseline and after 6 weeks’ treatment and who met the substudy inclusion criteria. Based on our previous clinical experience, we assumed a 15% UAE reduction (log-transformed change 0.4 mg/L) to be clinically relevant. Based on data from the Prevention of Renal and Vascular Endstage Disease Intervention Trial (PREVEND IT) (25), the mean UAE is 31.3 mg/L (log 2.87) with a standard deviation (SD) of 85.5 (log 0.93) for patients with ISH. Using the log-transformed values of 2.8 mg/L (SD = 0.9), a 0.4 mg/L (15%)
37

CHAPTER 3
Figure 1. Selection of patients for substudy analysis of effects of fixed doses of telmisartan 20, 40 or 80 mg, hydrochlorothiazide (HCT) 12.5 mg or placebo on urinary albumin excretion (UAE) from the 1039 patients randomized to treatment in the ARAMIS study (22)
change could be detected with 5% error probability and 80% power using a two-sided t-test in a population of 81 patients per group. Baseline values are expressed as means and 95% confidence intervals (CI), except for UAE which is expressed as median and 95% CI of the median range. Effects of treatment on UAE, expressed as percentage and absolute changes from baseline, and UAE-to-creatinine ratios were analysed by the non-parametric Kruskal-Wallis test. In case of significance, pair-wise post-hoc comparisons between the separate groups were performed using the Mann-Wilcoxon-Whitney test. Blood pressure effects between the different treatment groups were analysed by a general linear model of covariance, adjusting for treatment and country effects with baseline as covariates. RESULTS
Of the 1039 patients included in the ARAMIS study, spot morning urine samples were available both before and after the 6-week intervention and UAE was determined in 918 patients (figure 1). There was a wide baseline distribution of UAE (> 2.2-901.6 mg/L, figure 2), with UAE detectable (i.e., above lower level of quantification of 2.2 mg/L) in 614 patients (66.9%); these patients were included in the substudy analysis. Microalbuminuria (20-200 mg/L) was present in 70 (7.6%) patients and macroalbuminuria (> 200 mg/L) in 86 (9.4%). All treatment groups were comparable with regard to the number of patients with detectable UAE and baseline characteristics (table 1).
Efficacy of different telmisartan doses
After 6 weeks’ telmisartan treatment, the median (95% CI) reductions in UAE with telmisartan 20, 40 and 80 mg were 0.8 mg/L (0.5, 1.8), 1.2 mg/L (0.6, 2.1) and 0.3 mg/L
38

TELMISARTAN IN IN ISOLATED SYSTOLIC HYPERTENSION
Table 1. Demographic and baseline values (mean (SD)) for patients with detectable urinary albumin excretion (UAE)
Telmisartan
Placebo
(n = 120)
HCT
12.mg
(n = 140)
All doses
(n = 354)
20 mg
(n = 117)
40 mg
(n = 119)
80 mg
(n = 118)
Male (%) 41.7 46.4 44.6 46.2 42.9 48.3
Age (years) 64.0 (10.5) 63.6 (11.5) 62.6 (11.3) 62.7 (12.2) 62.2 (11.2) 62.8 (10.6)
BMI (kg/m²) 27.3 (4.0) 27.5 (4.3) 28.0 (4.5) 28.0 (4.4) 27.8 (4.3) 28.1 (4.9)
SBP (mmHg) 164.3 (7.8) 162.5 (8.3) 163.7 (8.0) 162.9 (7.9) 164.5 (8.2) 163.5 (8.0)
DBP (mmHg) 83.3 (5.6) 83.5 (4.4) 83.4 (4.8) 83.7 (4.7) 83.5 (4.5) 83.1 (5.1)
Pulse (bpm) 72.3 (9.8) 71.9 (9.5) 73.5 (10.0) 73.4 (9.5) 73.4 (10.0) 73.6 (10.4)
eGFR (mL/min/1.73m2)a 95 (32) 102 (40) 106 (38) 103 (37) 105 (35) 109 (41)
UAE (mg/L)b 5.1 (4.1–6.0)
4.8 (4.0–5.6)
5.2 (4.6–5.7)
5.2 (4.2–6.3)
5.7 (4.6–6.6)
4.9 (3.9–5.5)
Type 2 diabetes (%) 7.5 10.7 11.3 11.1 12.6 10.2
Microalbuminuria (%)c 10.0 9.3 12.7 14.5 8.4 15.3
aCockcroft formula (24). bMedian (95% CI), cUAE 20-200 mg/L. BMI, body mass index.
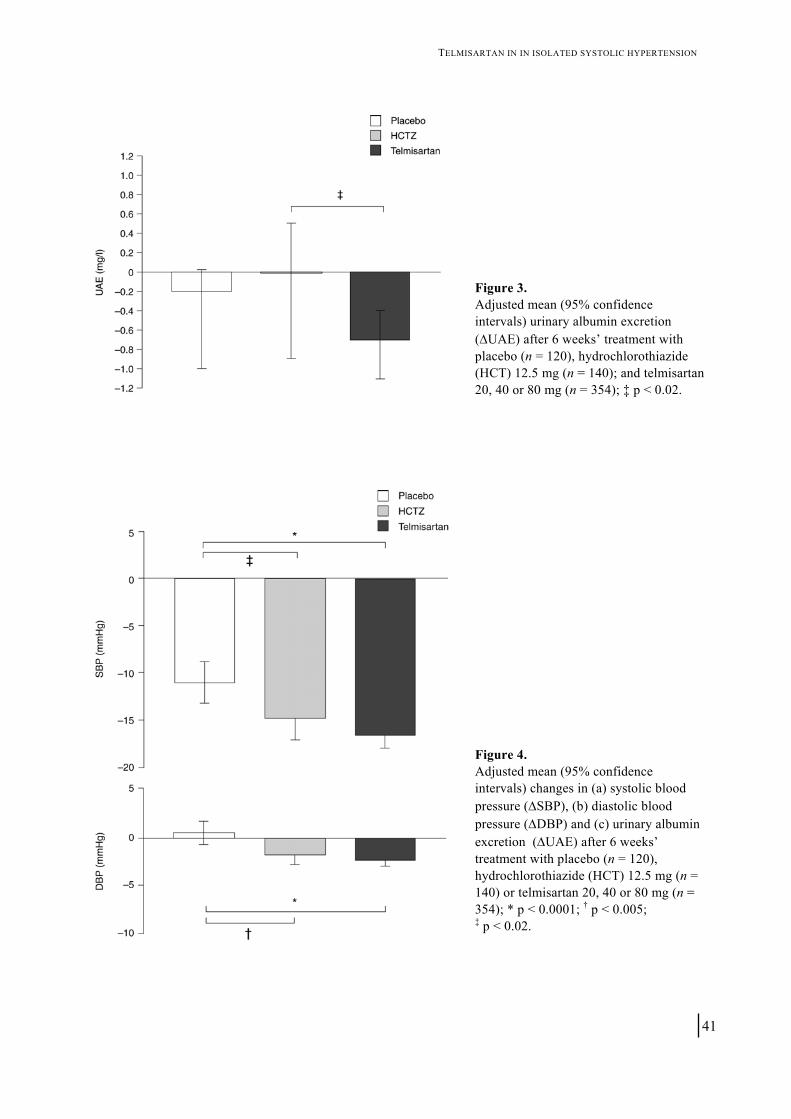
(0.0, 1.0), respectively. Mean (95% CI) reductions in seated trough SBP in the 20-, 40-and 80-mg treatment groups were 15.1 mmHg (12.8, 17.4), 17.6 mmHg (15.3, 19.9) and 16.9 mmHg (14.4, 19.5), respectively. Since no true dose–response was observed for either UAE or SBP, the values of the three telmisartan groups were combined (n = 354) for comparison with HCT and placebo. Efficacy of telmisartan compared with HCT and placebo on UAE The effects of the different treatments on absolute UAE values are shown in figure 3. Baseline UAE (median (95% CI)) in the total telmisartan group was 5.2 mg/L (4.2, 6.3). After treatment, median (95% CI) UAE was reduced significantly from baseline by 14.1% (7.3, 21.8) in the telmisartan group, where as non-significant reductions from baseline of 1.1% (-13.5, 16.0) and 2.7% (-0.9, 19.9) were observed in the HCT and placebo groups, respectively. The reduction in the telmisartan group differed significantly from that in the HCT group (p = 0.0165).
Correcting for the urinary creatinine excretion revealed the same pattern: in the telmisartan group, UAE-to-creatinine ratio was significantly reduced by 12.7% (5.4, 21.8). In the placebo groups, the median reduction was 8.0% (-7.9, 22.0) whereas there was a 1.8% (-25.3, 20.1) increase in the HCT group. Again, the difference between telmisartan and HCT was significant (p = 0.0378).
39

CHAPTER 3
Figure 2. Distribution of baseline UAE in patients with spot urine evaluation (n = 918). Division of categories is made based on doubling of the successive intervals for UAE. LoQ, lower limit of quantification (2.2 mg/L).
In the telmisartan group, when the response was analysed based on baseline UAE (> 2.2-20 mg/L (normoalbuminuria), > 20-200 mg/L (microalbuminuria) and > 200 mg/L (macroalbuminuria/overt proteinuria)), UAE reductions after 6 weeks’ telmisartan treatment of 10.2% (1.5,16.3), 55.4% (3.9, 77.0) and 50.4% (-371.7, 81.1), respectively, were detected. Antihypertensive efficacies The effects of treatment on blood pressure (adjusted mean) are depicted in figure 4. Baseline (mean (95% CI)) SBP in the total telmisartan group was 163.7 mmHg (162.8, 164.5); this was comparable to the SBP in the HCT and placebo groups of 162.5 mmHg (161.1, 163.9) and 164.3 (162.9, 165.8) mmHg, respectively. After 6 weeks, SBP was significantly reduced from baseline by 6.7% (5.4, 8.0) in the placebo group. Telmisartan treatment resulted in a larger reduction in SBP of 10.1% (11.0, 9.3). With HCT, a 9.1% (10.4, 7.7) reduction from baseline was detected. At baseline, the DBP in the total telmisartan group was 83.4 mmHg (95% CI 82.9, 83.9) and this was reduced by 2.7% (95% CI 2.0, 3.5) after 6 weeks’ treatment. In the placebo group, DBP did not change significantly from a baseline of 83.3 mmHg (82.2, 84.3), whereas a 2.0% (0.9, 3.1) reduction from a baseline value of 83.5 mmHg (82.8, 84.2) was detected in the HCT group. The differences in SBP and DBP reductions between HCT and telmisartan were not statistically significant. Safety Incidences of all-cause adverse events in the per-protocol population (n = 918) were 17.6%, 14.0% and 16.9%, respectively, for telmisartan 20, 40 and 80 mg. By
40
TELMISARTAN IN IN ISOLATED SYSTOLIC HYPERTENSION
Figure 3. Adjusted mean (95% confidence intervals) urinary albumin excretion (ΔUAE) after 6 weeks’ treatment with placebo (n = 120), hydrochlorothiazide (HCT) 12.5 mg (n = 140); and telmisartan 20, 40 or 80 mg (n = 354); ‡ p < 0.02.
Figure 4. Adjusted mean (95% confidence intervals) changes in (a) systolic blood pressure (ΔSBP), (b) diastolic blood pressure (ΔDBP) and (c) urinary albumin excretion (ΔUAE) after 6 weeks’ treatment with placebo (n = 120), hydrochlorothiazide (HCT) 12.5 mg (n = 140) or telmisartan 20, 40 or 80 mg (n = 354); * p < 0.0001; † p < 0.005; ‡ p < 0.02.
41

CHAPTER 3
Table 2. The five most frequent reported adverse events after 6 weeks’ treatment
Telmisartan
Placebo
(n = 183)
HCT
12.5 mg
(n = 185)
20 mg
(n = 180)
40 mg
(n = 187)
80 mg
(n = 183)
Headache 4 (2.2%) 4 (2.1%) 3 (1.6%) 4 (2.2%) 4 (2.2%)
Bronchitis 3 (1.6%) 2 (1.1%) 4 (2.2%) 3 (1.6%) 0 (0.0%)
Upper respiratory tract infection 5 (2.7%) 1 (0.5%) 4 (2.2%) 0 (0.0%) 1 (0.5%)
Back pain 1 (0.5%) 3 (1.6%) 3 (1.6%) 0 (0.0%) 1 (0.5%)
Influenza-like symptoms 1 (0.5%) 2 (1.1%) 2 (1.1%) 1 (0.5%) 0 (0.0%)
comparison, in the HCT and placebo groups, 19.8% and 18.7%, respectively, experienced an adverse event. The five most frequently reported events in any of the treatment groups are summarized in table 2. The total incidence of drug-related events was 3.0%, being comparable in the different treatment groups.
No significant changes from baseline in clinical laboratory parameters were detected during the study. The mean (95% CI) calculated creatinine clearance decreased by 3.6 mL/min (1.3, 5.8) after 6 weeks’ treatment with telmisartan. A reduction of 2.1 mL/min (-1.6, 5.7) was observed in the HCT group and an increase of 0.6 mL/min (-3.1, 4.3) was detected in the placebo group. DISCUSSION
This study demonstrates that, in patients with ISH and unselected for the degree of albuminuria at baseline, UAE was effectively reduced after telmisartan treatment, but not after treatment with HCT. This was despite similar reductions in SBP and DBP with the two treatments. Further analysis showed that this reduction in UAE was not only observed in patients with microalbuminuria or macroalbuminuria, but also in those with UAE within the range generally considered to be normal.
Based on the outcomes of Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack (ALLHAT) (26), Systolic Hypertension in the Elderly Program (SHEP) (27) and Systolic Hypertension in Europe trial (Syst-Eur) (28), The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure proposes the use of a diuretic as first-choice therapy for the treatment of ISH, with a long-acting calcium channel blocker as an alternative (29). However, it is noted that the choice of the initial agent is of less importance than the degree of blood pressure reduction achieved (29). The results of ARAMIS provide a different perspective, with telmisartan proving non-inferior to hydrochlorothiazide in the control of SBP (22). The results of our substudy show that telmisartan could have an additional benefit as it reduced UAE, whereas HCT did not. To our knowledge, there
42

TELMISARTAN IN IN ISOLATED SYSTOLIC HYPERTENSION
have been no previous studies comparing the effect of an angiotensin II receptor blocker with that of HCT on UAE. However, there is evidence that targeting the RAAS using an angiotensin-converting enzyme inhibitor, as well as reducing blood pressure, decreased UAE in hypertensive diabetic patients, whereas HCT was ineffective despite comparable antihypertensive efficacy (30). Another study showed that, in normotensive diabetic patients, an angiotensin-converting enzyme inhibitor, but not HCT, reduced microalbuminuria (31).
The reduction in UAE we observed in the short term among patients with varying degrees of albuminuria may confer clinically significant long-term cardiovascular benefit. Data from 40 000 individuals in PREVEND demonstrated that, after adjustment for other well-recognized risk factors, UAE is predictive of cardiovascular death in the general population (9). An individual with a UAE of 10-20 mg/L is reported to have 28% higher risk of cardiovascular death than one with a UAE of 0-10 mg/L (9). Findings of the Heart Outcomes Prevention Evaluation (HOPE) study also demonstrated that any degree of albuminuria is a risk factor for cardiovascular events (19). A similar relationship exists between UAE and the development of renal damage, patients with high-normal UAE being at higher risk of developing microalbuminuria and macroalbuminuria (32).
Whether or not a reduction in UAE, as a consequence of antihypertensive treatment—even when established within baseline ranges below the threshold for microalbuminuria—contributes to a risk reduction still needs to be established. Nevertheless, data from studies performed in both diabetic and non-diabetic microalbuminuric or macroalbuminuric patients show that a reduction in albuminuria using antihypertensive treatment favourably impacts on the incidence of end-organ damage (16-18,33). In particular, the beneficial effects of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers on cardiovascular and renal risk are predicted by their antiproteinuric activity (34). Currently, there are no data on changes in UAE from comparative studies conducted in patients with very low levels of UAE. Given that over 80% of the patients in our study had levels of UAE lower than the criteria for microalbuminuria, the significantly greater reduction in UAE with telmisartan compared with placebo is all the more striking. The effect on cardiovascular and renal risk reduction is uncertain at present, but deserves further examination.
In the interpretation of our results, we acknowledge deficiencies. Firstly, although the reduction in UAE with telmisartan compared with placebo exceeded our prespecified limit for clinical significance, it did not reach statistical significance. Furthermore, SBP was also significantly reduced by placebo, although to a lesser extent than the two active treatments. It is, therefore, not feasible to distinguish between possible favourable effects of telmisartan or the more detrimental effect of HCT.
43

CHAPTER 3
Secondly, our patient population displayed a marked variation in baseline UAE, ranging from < 2.2 mg/L to 901.6 mg/L. After treatment, the changes of UAE varied greatly in the three treatment groups. This wide distribution can be partly explained by the use of spot sampling, as opposed to 24-h collection of urine, and the collection of only one sample at baseline and one at the end of treatment. Nevertheless, correction of UAE for the creatinine excretion, which would allow for confounding factors, did not affect the overall pattern of our results. Thirdly, we did not observe a dose-response effect on UAE reduction with telmisartan. By contrast, in macroalbuminuric patients, uptitration of doses of RAAS-targeting agents may bring about additional UAE reduction (35). Since optimal blood pressure reduction is of great importance for optimizing outcome, it is interesting that no dose-response effect on SBP and DBP was detected with telmisartan. This suggests that telmisartan doses used in our study were at the plateau of the dose–response curve, both for the reduction of UAE and for blood pressure control. A previous study was unable to detect any significant linear trend in blood pressure reduction among telmisartan doses in the range 40-120 mg (36).
In conclusion, treatment of ISH with the angiotensin II receptor antagonist telmisartan reduced UAE. In contrast, hydrochlorothiazide did not affect UAE, despite bringing about a similar reduction in blood pressure. With respect to optimal risk management, the telmisartan-induced reduction in UAE could be of additional benefit, although no cause-effect relationship has been demonstrated so far. Future studies should be directed at aggressive treatment of risk factors to establish the cardiovascular and renal benefit. REFERENCES 1. Kannel WB. Blood pressure as a cardiovascular risk factor: prevention and treatment. JAMA 1996;275:1571-6 2. Klag MJ, Whelton PK, Randall BL et al. Blood pressure and end-stage renal disease in men. N Engl J Med 1996;334:13-8 3. Mogensen CE. Microalbuminuria predicts clinical proteinuria and early mortality in maturity-onset diabetes. N Engl J Med
1984;310:356-60 4. Mogensen CE. Preventing end-stage renal disease. Diabet Med 1998;15(Suppl 4):S51–S56 5. Jensen JS, Feldt-Rasmussen B, Borch-Johnsen K et al. Microalbuminuria and its relation to cardiovascular disease and risk factors.
A population-based study of 1254 hypertensive individuals. J Hum Hypertens 1997;11:727-32 6. Ruilope LM. Microalbuminuria as risk in essential hypertension. Nephrol Dial Transplant 1997;12 (Suppl 2):2-5 7. Borch-Johnsen K, Feldt-Rasmussen B, Strandgaard S, Schroll M, Jensen JS. Urinary albumin excretion. An independent predictor of
ischemic heart disease. Arterioscler Thromb Vasc Biol 1999;19:1992-7 8. Diercks GF, Hillege HL, van Boven AJ et al. Relation between albumin in the urine and electrocardiographic markers of myocardial
ischemia in patients without diabetes mellitus. Am J Cardiol 2001;88:771-4 9. Hillege HL, Fidler V, Diercks GF et al. Urinary albumin excretion predicts cardiovascular and noncardiovascular mortality in
general population. Circulation 2002;106:1777-82 10. Romundstad S, Holmen J, Kvenild K, Hallan H, Ellekjaer H. Microalbuminuria and all-cause mortality in 2,089 apparently healthy
individuals: a 4.4-year follow-up study. The Nord-Trondelag Health Study (HUNT), Norway. Am J Kidney Dis 2003;42:466-73 11. Wachtell K, Ibsen H, Olsen MH et al. Albuminuria and cardiovascular risk in hypertensive patients with left ventricular
hypertrophy: the LIFE study. Ann Intern Med 2003;139:901-6 12. De Leeuw PW, Thijs L, Birkenhager WH et al. Prognostic significance of renal function in elderly patients with isolated systolic
hypertension: results from the Syst-Eur trial. J Am Soc Nephrol 2002;13:2213-22 13. De Jong PE, Brenner BM. From secondary to primary prevention of progressive renal disease: the case for screening for
albuminuria. Kidney Int 2004;66:2109-18
44

TELMISARTAN IN IN ISOLATED SYSTOLIC HYPERTENSION
14. Navis G, de Jong PE, de Zeeuw D. A comparison of progression in diabetic and non-diabetic renal disease: similarity of progression promotors. In: Mogensen CE (editor). The Kidney and Hypertension in Diabetes Mellitus. Dordrecht: Kluwer Academic Publishers 2000 pp. 587–600
15. Maschio G, Alberti D, Janin GM et al. Effect of the angiotensin-converting-enzyme inhibitor benazepril on the progression of chronic renal insufficiency. N Engl J Med 1996;334:939-45
16. The GISEN Group (Gruppo Italiano di Studi Epidemiologici in Nefrologia). Randomized placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of terminal renal failure in proteinuric, non-diabetic nephropathy. Lancet 1997;349:1857-63
17. Brenner BM, Cooper ME, de Zeeuw D et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001;345:861-9
18. Lewis EJ, Hunsicker LG, Clarke WR et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001;345:851-60
19. Gerstein HC, Mann JF, Pogue J et al. Prevalence and determinants of microalbuminuria in high-risk diabetic and nondiabetic patients in the Heart Outcomes Prevention Evaluation Study. Diabetes Care 2000;23(Suppl. 2):B35-9
20. Heart Outcomes Prevention Evaluation Study Investigators. Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy. Lancet 2000;355:253-9
21. Lindholm LH, Ibsen H, Dahlöf B et al. Cardiovascular morbidity and mortality in patients with diabetes in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomized trial against atenolol. Lancet 2002;359:1004–10.
22. Manolis AJ, Reid JL, de Zeeuw D et al. Angiotensin II receptor antagonist telmisartan in isolated systolic hypertension (ARAMIS) study: efficacy and safety of telmisartan 30, 40 or 80 mg vs. hydrochlorothiazide 12.5 mg or placebo. J Hypertens 2004;22:1033-7
23. Bangstad HJ, Try K, Dahl-Jorgensen K, Hanssen KF. New semiquantitative dipstick test for microalbuminuria. Diabetes Care 1991;14:1094-7
24. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron 1976;16:31-41 25. Diercks GF, Janssen WM, van Boven AJ et al. Rationale, design, and baseline characteristics of a trial of prevention of cardio-
vascular and renal disease with fosinopril and pravastatin in nonhypertensive, nonhypercholesterolemic subjects with microalbumin-uria (the Prevention of REnal and Vascular ENdstage Disease Intervention Trial (PREVEND IT)). Am J Cardiol 2000;86:635-8
26. ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2002;288:2981-97
27. SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension. Final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA 1991;265:3255-64
28. Staessen JA, Fagard R, Thijs L et al. Randomized double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension. Lancet 1997;350:757-64
29. Chobanian AV, Bakris GL, Black HR et al. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 2003; 42:1206-52
30. Lacourcière Y, Nadeau A, Poirier L, Tancrede G. Comparative effects of converting enzyme inhibition and conventional therapy in hypertensive non-insulin dependent diabetics with normal renal function. Clin Invest Med 1991; 14:652-60
31. Hallab M, Gallois Y, Chatellier G, Rohmer V, Fressinaud P, Marre M. Comparison of reduction in microalbuminuria by enalapril and hydrochlorothiazide in normotensive patients with insulin dependent diabetes. BMJ 1993;306:175-82
32. Verhave JC, Gansevoort RT, Hillege HL et al. An elevated urinary albumin excretion predicts de novo development of renal function impairment in the general population. Kidney Int Suppl 2004;66:S18-S21
33. Stuveling EM, Verhave JC, Hillege HL et al. Incidence and risk factors for microalbuminuria in the non-diabetic general population. J Am Soc Nephrol 2003;14(Suppl 3):679A
34. Nakao N, Yoshimura A, Morita H, Takada M, Kayano T, Ideura T. Combination treatment of angiotensin-II receptor blocker and angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomized controlled trial. Lancet 2003;361:117-24
35. Laverman GD, Navis G, Henning RH, de Jong PE, de Zeeuw D. Dual renin-angiotensin system blockade at optimal doses for proteinuria. Kidney Int 2002;62:1020-5
36. Smith DH, Matzek KM, Kempthorne-Rawson J. Dose response and safety of telmisartan in patients with mild to moderate hypertension. J Clin Pharmacol 2000;40:1380-90
ACKNOWLEDGEMENTS Boehringer Ingelheim has financed and executed the angiotensin II receptor antagonist telmisartan in isolated systolic hypertension (ARAMIS) trial (BI Trial No. 502.254). Drs Reid and Manolis were on the Steering Committee. Dr de Zeeuw was an advisor and provided laboratory measurements in the present substudy. Drs Vogt and Navis had no financial interest or arrangement in the ARAMIS trial. Elements of this study were presented at the American Society of Nephrology 2002 Annual Meeting in Philadelphia (abstract J Am Soc Nephrol 2002; 13: 241A)
45

CHAPTER 3
46

PART II
OPTIMIZING RENOPROTECTION: REDUCING RESIDUAL PROTEINURIA

PART II
48

Introduction to chapters 4, 5, and 6
Renoprotection by dual blockade of the renin-angiotensin system
Liffert Vogt, Gozewijn D Laverman, Dick de Zeeuw and Gerjan Navis Letter in: Lancet 2003; 361: 1170-1

INTRODUCTION TO CHAPTERS 4,5 & 6
50

RENOPROTECTION BY DUAL RAAS BLOCKADE
ir—Naoyuki Nakoa and colleagues (Jan 11, p 117 (1)) show that, in patients with non-diabetic chronic renal disease, the risk of reaching end-stage renal disease or doubling of serum creatinine concentration was reduced by almost 50% after dual blockade of the renin-angiotensin aldosterone system (RAAS) compared with use of angiotensin-converting enzyme (ACE) inhibitors or angiotensin II receptor type 1 (AT1) blockers alone. The differences in renoprotection are probably due to the much larger antiproteinuric effect of dual blockade.
S
The value of proteinuria reduction with respect to long-term renoprotection has been consistently shown in clinical and experimental studies (2). The effect of proteinuria reduction holds true not only for between-group difference for regimens with different antiproteinuric efficacies—as in Nakoa and colleagues’ study—but also for within-group differences in renal outcome between individuals with a poor, compared with a good, antiproteinuric response (3). Thus, the value of proteinuria reduction is independent of the way it is obtained.
Although the COOPERATE study shows the improved benefit of combination treatment, many patients still progressed to end-stage renal disease. Nakoa and colleagues identified non-responding patients as having a smaller antiproteinuric response. Thus, to optimise renoprotective strategies, should we not aim for more effective proteinuria reduction (4)? The individual antiproteinuric dose response to ACE inhibitor or AT1 antagonist reveals that some patients need a higher dose to obtain the same antiproteinuric effect. We have shown individual dose-dependent differences in maximum reduction of proteinuria from baseline after 6 weeks of monotherapy, with a much more effective reduction of proteinuria after combined treatment (5).
Given these data, we argue that for optimum renoprotection, one should not only apply dual RAAS blockade with fixed dose-titration for proteinuria—as Nakoa and colleagues did—but specifically pursue the lowest level of proteinuria by individual dose-titration in combination with additive measures that enhance antiproteinuric efficacy, such as dietary sodium restriction, diuretic therapy, and protein restriction (4).
51

INTRODUCTION TO CHAPTERS 4,5 & 6
REFERENCES 1 Nakao N, Yoshimura A, Morita H, Takada M, Kayano T, Ideura T. Combination treatment of angiotensin-II receptor
blocker and angiotensin-converting enzyme inhibitor on non-diabetic renal disease (COOPERATE): a randomised controlled trial. Lancet 2003;361:117-24
2 Remuzzi G, Bertani T. Pathophysiology of progressive nephropathies. N Engl J Med 1998;339:1448-56 3 Apperloo AJ, de Zeeuw D, de Jong PE. Short-term antiproteinuric response to antihypertensive therapy predicts long-
term GFR decline in patients with non-diabetic renal disease. Kidney Int 1994;45(suppl 45):174-8 4 Ruggenenti P, Schieppati A, Remuzzi G. Progression, remission, regression of chronic renal disease. Lancet 2001;
357:1601-8 5 Laverman GD, Navis GJ, Henning RH, de Jong PE, de Zeeuw D. Dual renin-angiotensin system blockade at optimal
doses for proteinuria. Kidney Int 2002;62:1020-5
52

Chapter 4
Altering the dosing time of trandolapril does not increase the antiproteinuric effect of ACE inhibition in non-diabetic kidney disease Els A. van der Wouden, Liffert Vogt, Robert H. Henning, Gerjan Navis, Dick de Zeeuw, and Marc H. Hemmelder

CHAPTER 4
54

EFFECT OF DOSING TIME OF TRANDOLAPRIL ON PROTEINURIA
Background—Angiotensin-converting enzyme (ACE) inhibitors are drugs of first choice to reduce proteinuria in renal diseases, but the antiproteinuric response may vary highly. We previously observed a relative therapy resistance to blockade of the renin-angiotensin-aldosterone system (RAAS) during the night. Since higher residual proteinuria is associated with more rapid renal function loss, it is important to enhance the nocturnal antiproteinuric response. We therefore questioned whether altering the dosing time of the ACE inhibitor trandolapril could overcome the relative nocturnal therapy resistance. Methods—Fourteen non-diabetic proteinuric patients on stable RAAS inhibition, with residual proteinuria of > 1 g/d were converted to trandolapril (4 mg) in the morning. Other antihypertensive medication was continued. After 6 weeks, patients were randomized to evening (4 mg) or BID (2 x 2 mg) dosing of trandolapril in a cross-over fashion (6 weeks each). During the last study period, patients again used trandolapril (4 mg) in the morning. Patients collected 2 x 24-h urine in day- and nighttime portions every 6-week period. Proteinuria and blood pressure were measured. Results—Total residual proteinuria and blood pressure were equal during all periods. Daytime and nighttime proteinuria were also comparable during all periods. Blood pressure diurnal rhythm was similar during all periods. Evening and BID dosing did not affect total residual proteinuria, daytime or nighttime proteinuria. Sodium and protein intake were not significantly different among the different dosing regimens. Conclusions—Altering the dosing time of the ACE inhibitor, trandolapril, does not increase the antiproteinuric response. Therefore, once daily dosing of the long-acting ACE inhibitor, trandolapril, at maximum dose results in its optimal antiproteinuric effect.
ntervention in the renin-angiotensin-aldosterone system (RAAS) with ACE inhibitors or angiotensin type 1 receptor (AT1) antagonists is the preferred antiproteinuric therapy for renal disease since these drugs reduce proteinuria beyond their blood pressure lowering effect (1,2). This specific property of RAAS inhibitors results in a reduced rate of renal function loss compared to non-RAAS antihypertensive treatment (3). Yet, in a given patient population, there is a large variability in antiproteinuric therapy response to ACE inhibitors (4). This is of clinical importance because the lower the residual proteinuria (presumably < 1 g/d) the better kidney function is preserved on the long term (5,6).
I
Proteinuria usually displays a circadian rhythm with maximum urinary protein excretion during the day (7), probably involving diurnal changes in blood pressure, renal haemodynamics (8,9), glomerular permeability (9) and tubular reabsorption. We previously observed a relative therapy resistance during the night to the antiproteinuric
55

CHAPTER 4
action of a single morning dose of the ACE inhibitor trandolapril (10), despite comparable reduction of blood pressure during both daytime and nighttime. Consequently, nighttime protein excretion under ACE inhibitor therapy contributes relatively strongly to the residual proteinuria, and may thus represent an important target for optimization of the therapeutic response. As ACE inhibitors are routinely dosed in the morning, we questioned whether alternative timing of dosing may overcome nocturnal therapy resistance in non-diabetic patients with significant residual proteinuria during RAAS inhibition. METHODS
Patients This study was performed in accordance to the Declaration of Helsinki, approved by the local Medical Ethical Committee, and all patients gave their informed consent. Fourteen Caucasian, non-diabetic patients with chronic renal disease and residual proteinuria > 1 g/d on stable RAAS-inhibiting therapy were enrolled. Only patients with an age between 18 and 70 yrs, and a creatinine clearance (CLcr) ≥ 30 mL/min/1.73m2 were included. Exclusion criteria were uncontrolled hypertension (systolic blood pressure (SBP) > 180 mmHg or diastolic blood pressure (DBP) > 100 mmHg) during the run-in period, a history of myocardial infarction, unstable angina, heart failure, coronary by-pass surgery or cerebrovascular accident during the past 6 months, frequent use of non-steroidal anti-inflammatory drugs (> 2 doses weekly), use of immunosuppressants, high rate of renal function loss (decline in CLcr > 6 mL/min/year during the previous year), serum potassium ≥ 6 mmol/L, intolerance or contra-indication for the use of ACE Table 1. Baseline patient characteristics. Data are expressed as median (95% CI)
Male/female 7/7 Age (yrs) 48 (28-62) BMI (kg/m2) 29.1 (22.8-35.0) Diagnosis 3 FGS, 2 MGP, 3 IgA, 3 other, 3 no/NCBx Residual proteinuria (g/24h) 3.0 (1.3-5.7) SBP (mmHg) 126 (118-148) DBP (mmHg) 76 (67-88) CLcr (mL/min) 87.5 (38.1-99.3) Antihypertensive medication (number of users)
ACE inhibitors AT1 antagonists Dual RAAS blockade Diuretics ß-blockers Calcium antagonists
12 6 4 9 4 4
BMI, body mass index. FGS, focal glomerulosclerosis. MGP, membranous glomerulopathy. IgA, IgA nephropathy. No/NCBx, no or non-conclusive biopsy.
56

EFFECT OF DOSING TIME OF TRANDOLAPRIL ON PROTEINURIA
inhibitors and inability to understand the patient information. The characteristics of the 14 patients enrolled in this study are shown in table 1. Study protocol This prospective, randomized, open-label, cross-over study was performed on an ambulatory basis (figure 1). Patients visited the outpatient clinic with 6 weeks intervals. All RAAS inhibition was stopped and converted to trandolapril 4 mg in the morning after patients gave informed consent. We chose not to use a washout period without any RAAS inhibition, because this may result in a deterioration of renal function. More importantly, a washout period is not necessary to answer the study question whether alteration of the dosing time of an ACE inhibitor can amplify its antiproteinuric efficacy. Other antihypertensive medication was continued during the study, but the dosage of these drugs was not changed during the complete protocol. Patients were instructed to take their non-study medication on regular fixed times of the day, preferably in the morning. The study consisted of 4 6-week periods. As previous studies demonstrated that proteinuria stabilizes 4 weeks after the start of RAAS inhibition (1,2), we chose to use 6-week treatment periods to rule out the influence of carry-over effects. After an initial run-in period of 6 weeks of morning dosing of trandolapril (4 mg), the patients were randomized to periods of evening dosing (4 mg at 10.00 p.m.) and BID (2x 2 mg at 8.00 a.m. and 8.00 p.m.) in a cross-over fashion (6 weeks each). To control for time influences, all patients were treated with a morning dose of trandolapril (4 mg at 8.00 a.m.) during the final 6-week period of the study. Measurements The patients visited the outpatient clinic in the morning after an overnight fast, without taking any blood pressure affecting medication. A blood sample was drawn and trough blood pressure was measured in a semisupine position with a Dinamap apparatus for 15 min with a 1 min interval. The mean of the last 5 measurements was taken for analysis. In addition, 9 patients underwent a 24-h blood pressure measurement in each study period at 15 min intervals during the day and 30 min intervals during the night using an
Figure 1. Study design: dosing times of trandolapril. Half of the patients (n = 7) were randomly allocated to the upper arm and the other half (n = 7) to the lower arm.
57

CHAPTER 4
ambulatory blood pressure meter (Spacelabs model 90207). At the end of each 6-week period, patients collected 2 x 24 h urine in daytime and nighttime portions. The daytime period was from 8.00 a.m. until 10.00 p.m., whereas the nighttime period was from 10.00 p.m. until 8.00 a.m. Patients were instructed to adhere to a mild sodium restricted diet (< 100 mmol Na/day) throughout the study. Proteinuria was measured using the benzethonium chloride method. Serum and urinary creatinine were analyzed by Jaffé reaction. Urinary sodium was measured with an ion-selective electrode and urinary urea was measured enzymatically using urease. Statistical analysis Data are expressed as median with 95% confidence interval. Based on previous clinical experience, a 25% reduction of proteinuria, after shifting from morning to evening or BID dosing, was considered clinically relevant. For a desired power of 0.8 and a two-sided alpha of 0.05, a total of at least 10 patients was needed to detect such a reduction of proteinuria. Missing value analysis was performed for the 24-h blood pressure measurements using expectation maximization. Because of the small number of patients data from the 4 study periods were analyzed with a non-parametric Friedman’s ANOVA for paired observations with correction for multiple comparisons. Wilcoxon signed rank test was used for differences between daytime and nighttime data. A p-value < 0.05 was considered statistically significant. RESULTS
The median residual proteinuria of patients at recruitment was 3.0 (1.3-5.7) g/24h on their pre-study RAAS-inhibiting medication (table 1). The conversion to morning dosing of trandolapril 4 mg resulted in a similar residual proteinuria of 3.0 (1.9-4.2) g/24h after 6 weeks. Changing of dosing in the subsequent 6-week periods to evening (4 mg) and BID (2 x 2 mg) dosing did not influence residual proteinuria significantly (figure 2A).
Sodium and protein intake were not significantly different among the different dosing regimens, although the sodium intake was higher than prescribed. Urinary creatinine excretion remained stable throughout the study (table 2).
Subsequently, we examined whether changing of the dosing time affected the amount of residual proteinuria during daytime and nighttime (figure 2B). Both daytime and nighttime urinary protein excretion were not significantly different among the 4 dosing periods. Three patients showed a favourable response of > 25% reduction in residual proteinuria on switching from morning to evening dosing. Five patients had such a response on switching from morning to BID dosing. These numbers are too small to identify specific patient characteristics which determine a favourable response to a switch in dosing time.
58

EFFECT OF DOSING TIME OF TRANDOLAPRIL ON PROTEINURIA
Figure 2. Influence of dosing regimen periods on total (A), and day- and nighttime proteinuria (B). Data are displayed as median and 95% CI.
Both trough SBP and DBP were equal during all therapy regimen periods (table 2). Ambulatory measurement showed a significant lower SBP and DBP during the night compared to daytime (p < 0.05), without any significant differences between the dosing regimes. DISCUSSION
In this study, we found no effect of altering the dosing time of the long-acting ACE inhibitor trandolapril on residual proteinuria in non-diabetic renal patients. We observed an equal urinary protein excretion rate during daytime and nighttime in non-diabetic patients treated with a single morning dose of trandolapril. Changing from morning dosing to BID or evening dosing did not result in greater reduction of proteinuria during nighttime compared to daytime. As in untreated proteinuric patients proteinuria during daytime exceeds nocturnal proteinuria (10), our data also suggest a relative therapy
Table 2. Proteinuria, blood pressure and urinary urea, sodium and creatinine excretion during the 4 treatment periods (median and 95% CI)
Dosing time trandolapril Morning BID Evening Morning
SBP (mmHg) daytime (mmHg) nighttime (mmHg)
130 (112-149)
124 (117-137) 109 (105-119)*
128 (110-145) 121 (117-138) 107 (102-117)*
134 (107-154) 128 (120-139) 110 (105-115)*
130 (109-146) 124 (109-130) 111 (91-120)*
DBP (mmHg) daytime (mmHg) nighttime (mmHg)
79 (66-84)
81 (74-85) 69 (60-76)*
74 (64-87) 82 (76-83) 64 (55-71)*
79 (65-85) 83 (72-89) 68 (55-72)*
80 (66-85) 83 (72-86) 68 (54-78)*
Uurea (mmol/24h) 334 (263-477) 316 (267-450) 297 (249-438) 312 (253-444)
UNa+ (mmol/24h) 175 (126-248) 141 (119-191) 174 (144-216) 156 (99-203)
UCr (mmol/24h) 13.7 (12.1-18.1) 12.2 (10.7-17.6) 12.2 (11.0-17.1) 11.5 (10.8-15.8)
Uurea, urinary urea excretion. UNa+, urinary sodium excretion. UCr, urinary creatinine excretion. * p < 0.05 nighttime vs. daytime
59

CHAPTER 4
resistance to ACE inhibitors during nighttime.
What could explain the relative therapeutic resistance of nocturnal proteinuria to ACE inhibition? A possible explanation may be offered by the physiological diurnal variation in the activity of the RAAS, having its peak renin and aldosterone activity early in the morning and the lowest activity during the evening (11), and ACE-activity peaking in the afternoon (12). Optimal efficacy of RAAS blockade may be dependent on activation of the system, as shown by an augmentation of the antiproteinuric response to ACE inhibitors by diuretics and low salt diet (13). Consequently, the lower nocturnal activity of RAAS may impair its therapeutic efficacy. Such a proposition is supported by the observation that blockade of the RAAS on another level by the renin inhibitor, remikiren (10), and dual RAAS inhibition with an ACE inhibitor and an AT1 antagonist (14) were also less effective in reducing nighttime proteinuria. Therefore, augmentation of the nighttime RAAS activity and the application of non-RAAS inhibiting antiproteinuric strategies may be more effective in reducing nighttime proteinuria.
Could the choice for trandolapril in our study have influenced our results? Trandolapril is a long-acting ACE inhibitor. The effective half-life of its active metabolite, trandolaprilat, is approximately 15-23h (15,16), due to its lipophilicity and saturable binding to ACE. We chose to use trandolapril at a dose of 4 mg per day, since with this drug and dose the optimal antiproteinuric effect is achieved (17). In spite of using an optimal antiproteinuric dose of a long-acting ACE inhibitor, we previously observed nocturnal therapy resistance to its antiproteinuric effect when the drug was administered in the morning (10). Trandolapril is claimed to inhibit the RAAS for 24 h by once daily dosing, supported by effective blood pressure reduction for 24 hours (10,18). Indeed, in our study no differences in SBP or DBP were observed during different dosing regimens. It should be noted that ACE inhibition in specific compartments (e.g. kidney tissue) may not last for 24 hours and antiproteinuric responses might therefore dissociate from systemic haemodynamic responses (1). The effect of changing the dosing time of RAAS blockade at the antiproteinuric efficacy in renal patients has not been previously studied. Only preliminary data are available from a study in Japanese proteinuric patients comparing morning dosing, evening dosing, and TID dosing of 3 mg of trandolapril (19). The antiproteinuric effect of trandolapril was most prominent during evening dosing, while TID dosing resulted in variable responses. Besides the differences in study design, a difference in genetic background of the patients or the use of a smaller dose may be an explanation for this difference with our results.
We conclude that altering the dosing time of the ACE inhibitor, trandolapril, is not an effective strategy to further reduce residual proteinuria. For clinical purposes, this means that once daily dosing of the long-acting ACE inhibitor trandolapril, at maximum
60

EFFECT OF DOSING TIME OF TRANDOLAPRIL ON PROTEINURIA
dose on an arbitrary time, results in its optimal antiproteinuric effect. Further research should be directed at other interventions which could further reduce residual proteinuria by ACE inhibitors in non-diabetic renal disease. REFERENCES 1. Gansevoort RT, de Zeeuw D, De Jong PE. Dissociation between the course of the hemodynamic and antiproteinuric effects of
angiotensin I converting enzyme inhibition. Kidney Int 1993;44:579-84 2. Hemmelder MH, de Zeeuw D, Gansevoort RT, De Jong PE. Blood pressure reduction initiates the antiproteinuric effect of ACE
inhibition. Kidney Int 1996;49: 174-80 3. Jafar TH, Stark PC, Schmid CH et al. Proteinuria as a modifiable risk factor for the progression of non-diabetic renal disease.
Kidney Int 2001; 60: 1131-40 4. Heeg JE, De Jong PE, van der Hem GK, de Zeeuw D. Efficacy and variability of the antiproteinuric effect of ACE inhibition by
lisinopril. Kidney Int 1989;36:272-9 5. Peterson JC, Adler S, Burkart JM et al. Blood pressure control, proteinuria, and the progression of renal disease. The Modification of
Diet in Renal Disease Study. Ann Intern Med 1995;123:754-62 6. Ruggenenti P, Schieppati A, Remuzzi G. Progression, remission, regression of chronic renal diseases. Lancet 2001;357:1601-8 7. Koopman MG, Krediet RT, Koomen GC, Strackee J, Arisz L. Circadian rhythm of proteinuria: consequences of the use of urinary
protein:creatinine ratios. Nephrol Dial Transplant 1989;4:9-14 8. Koopman MG, Krediet RT, Zuyderhoudt FJ, De Moor EA, Arisz L. A circadian rhythm of proteinuria in patients with a nephrotic
syndrome. Clin Sci (Lond) 1985;69:395-401 9. Koopman MG, Koomen GC, van Acker BA, Arisz L. Circadian rhythm in glomerular transport of macromolecules through large
pores and shunt pathway. Kidney Int 1996;49:1242-9 10. Buter H, Hemmelder MH, van Paassen P et al. Is the antiproteinuric response to inhibition of the renin-angiotensin system less
effective during the night? Nephrol Dial Transplant 1997;12(Suppl 2):53-6 11. Hurwitz S, Cohen RJ, Williams GH. Diurnal variation of aldosterone and plasma renin activity: timing relation to melatonin and
cortisol and consistency after prolonged bed rest. J Appl Physiol 2004;96:1406-14 12. Cugini P, Lucia P. Circadian rhythm of the renin-angiotensin-aldosterone system: a summary of our research studies. Clin Ter
2004;155:287-91 13. Buter H, Hemmelder MH, Navis G, De Jong PE, de Zeeuw D. The blunting of the antiproteinuric efficacy of ACE inhibition by high
sodium intake can be restored by hydrochlorothiazide. Nephrol Dial Transplant 1998;13:1682-5 14. Laverman GD, Navis G, Henning RH, de Jong PE, de Zeeuw D. Dual renin-angiotensin system blockade at optimal doses for
proteinuria. Kidney Int 2002;62:1020-5 15. Arner P, Wade A, Engfeldt P et al. Pharmacokinetics and pharmacodynamics of trandolapril after repeated administration of 2 mg to
young and elderly patients with mild-to-moderate hypertension. J Cardiovasc Pharmacol 1994;23(Suppl 4):S44-9 16. Danielson B, Querin S, LaRochelle P et al. Pharmacokinetics and pharmacodynamics of trandolapril after repeated administration of
2 mg to patients with chronic renal failure and healthy control subjects. J Cardiovasc Pharmacol 1994;23(Suppl 4):S50-9 17. Nakao N, Yoshimura A, Morita H et al. Combination treatment of angiotensin-II receptor blocker and angiotensin-converting-
enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomised controlled trial. Lancet 2003;361:117-24 18. Mancia G, De Cesaris R, Fogari R et al. Evaluation of the antihypertensive effect of once-a-day trandolapril by 24-hour ambulatory
blood pressure monitoring. The Italian Trandolapril Study Group. Am J Cardiol 1992;70:60-6 19. Nakao N, Yoshimura A, Taira T et al. Effect of timing of administration on the antiproteinuric activity of angiotensin-converting
enzyme inhibitor. Nephrol Dial Transplant 2002;17(Suppl 12):44A ACKNOWLEDGEMENTS This study was financially supported by the scientific board of the Medical Center Leeuwarden, Leeuwarden, the Netherlands, and Abbott, Hoofddorp, the Netherlands.
61

CHAPTER 4
62

Chapter 5
Independent and combined effects of low sodium diet and diuretic on the antiproteinuric efficacy of the AT1 antagonist losartan in non-diabetic proteinuric patients Liffert Vogt, Femke Waanders, Frans Boomsma, Dick de Zeeuw and Gerjan Navis Journal of the American Society of Nephrology2008; in press

CHAPTER 5
64

LOW SODIUM DIET AND DIURETIC ON TOP OF LOSARTAN
The variability in the response to blockade of the renin-angiotensin aldosterone system (RAAS) is large. Lowest proteinuria is propagated for optimal renoprotection. Intervention in sodium status by low sodium diet (LS) or diuretics improve the responses of proteinuria and blood pressure to RAAS blockade, but the efficacy of their combination is unknown. Therefore, we examined the separate and combined effects of LS (50 mmol Na+/d) and hydrochlorothiazide (HCT, 25 mg/d) on proteinuria and blood pressure during losartan treatment (100 mg/d) in 34 non-diabetic proteinuric patients in a randomized, double-blind, placebo-controlled, cross-over study. Baseline proteinuria (3.8 ± 0.4 g/d (mean (SD) on placebo HS) displayed a stepwise significant reduction after LS (-22%), losartan (-30%), losartan + LS (-55%), losartan + HCT (-56%) and losartan + LS/ HCT (-70%; all p < 0.05). Mean arterial pressure showed a similar stepwise reduction (all p < 0.05). Particularly, individuals with an antiproteinuric response resistant to losartan monotherapy benefited to intervention in sodium status. In conclusion, LS and HCT are equally effective in reducing proteinuria and blood pressure when added to losartan and are specifically beneficial in patients resistant to RAAS blockade. Intensified intervention in sodium status by combining LS and HCT is an effective tool in maximizing the antiproteinuric efficacy of RAAS blockade.
roteinuria is, next to high blood pressure, a major risk factor for progression to end-stage renal disease in diabetic and non-diabetic nephropathies. Blockade of the renin-angiotensin aldosterone system (RAAS) with angiotensin-converting enzyme (ACE) inhibitors or angiotensin II type 1 receptor (AT1) antagonists protects against progressive renal function loss by reduction of blood pressure and proteinuria (1-3). However, individual differences in antiproteinuric response to RAAS blockade are large, ranging from zero to complete reduction in proteinuria (4). Interestingly, residual proteinuria during RAAS blockade predicts the long term renal risk for that individual patient (5,6). Therefore, enhancing the antiproteinuric efficacy is advocated to improve renoprotection (7).
P
Sodium restriction and diuretic treatment enhance the responses of proteinuria and blood pressure to RAAS blockade (8-10). Of note, intervention in sodium status might increase the top of the dose-response to RAAS blockade (9,11) and, therefore, a larger maximum response can be obtained. In an earlier study, we showed that the antiproteinuric response to ACE inhibition was almost completely annihilated by high sodium intake, with a blunted blood pressure response as well (4,9). Antiproteinuric and blood pressure responses could be restored by adding sodium restriction (9) or a diuretic (4). Presumably, diuretics and sodium restriction enhance the efficacy of RAAS
65

CHAPTER 5
blockade by similar mechanisms, that is, by their effect on volume status. Whether their combination has added effects on the antiproteinuric efficacy of RAAS blockade is unknown. It is also unknown whether patients resistant to RAAS blockade have specific benefits of sodium depletion. We therefore examined the effects of sodium restriction, hydrochlorothiazide (HCT) and their combination on proteinuria and blood pressure during losartan in non-diabetic proteinuric patients in a randomized, double-blind, placebo-controlled, cross-over study. CONCISE METHODS
Patients and protocol The protocol, which was in accordance with the Declaration of Helsinki, was approved by our local ethical committee and conducted according to the guidelines of good clinical practice. Written informed consent was obtained from each patient before inclusion. Patients were selected from our outpatient renal clinic and were enrolled between March 2004 and June 2006. All patients fulfilled the inclusion criterion of a stable proteinuria > 2 g/d and < 10 g/d. Only patients with stable renal function (i.e. creatinine clearance > 30 mL/min and < 6mL/min/yr decline) and age between 18 and 70 years were included. Patients with uncontrolled hypertension (mean arterial pressure (MAP) > 100 mmHg), serum potassium > 5.5 mmol/L, cardiovascular disease (myocardial infarction, unstable angina, percutaneous transluminal coronary angioplasty, coronary artery by-pass grafting or stroke within the last 6 months), contraindication for AT1 antagonist or diuretic use, and/or diabetes mellitus were excluded, as well as frequent users of non-steroidal anti-inflammatory drugs (NSAIDs; > 2 doses/week). Additional antihypertensive drugs except for RAAS blocking agents or diuretics were allowed for blood pressure control. These drugs were kept stable during the study.
Selected patients entered this single center, prospective, randomized, placebo-controlled, cross-over study and were consecutively treated during 6 weeks with placebo, losartan (100 mg QD; Cozaar®; Merck & Co. Inc., Whitehouse Station, NJ, USA), losartan plus HCT (100 / 25mg QD; Fortzaar®; Merck & Co. Inc., Whitehouse Station, NJ, USA) in random order. Patients were instructed to take the study medication once daily, in the morning, except on study days; on those days the study drug was not taken before data collection at the hospital (between 8:00 and 9:30 a.m.), allowing blood pressure measurement at trough. At the same time, patients were randomized to either a high sodium diet (200 mmol (~ 4.8 g) Na+/d) or a low sodium diet (50 mmol (~ 1.2 g) Na+/d) during 18 weeks (three 6-week periods). After 18 weeks, patients changed diet and the three 6-week periods were repeated.
66

LOW SODIUM DIET AND DIURETIC ON TOP OF LOSARTAN
During the whole study, patients were closely supported by a dietary consultant and were instructed to adhere to a stable protein diet (1.1 g/kg/d) throughout the study. Differences in sodium intake were achieved by replacing sodium-rich products with a low-sodium product of the same product group in order to remain isocaloric with a similar balance between protein, carbohydrate and fat. To prevent concurrent changes in dietary habits, the diets were based on the personal food habits of each subject and fitted to the individual caloric need. Every two weeks after an overnight fast, patients collected 24hr urine, blood pressure was measured and blood was sampled to control dietary compliance and to monitor renal function and blood pressure. Collected data at the end of each 6-week treatment period were used for analysis. Study measurements The primary endpoint was the 24-h proteinuria at the end of each treatment period. Secondary end points were the mean arterial pressure (MAP) and serum creatinine, urea, cholesterol, triglycerides, total protein and albumin. At the end of each period, the day previous to every visit, patients collected 24-h urine samples to determine proteinuria and urinary sodium, urea, potassium and creatinine excretion. Urinary protein was determined using the pyrogallol red-molybdate method. Serum and urinary electrolytes, creatinine and cholesterol, triglycerides, total protein and albumin levels were determined using an automated multi-analyzer (SMA-C®; Technicon, Tarrytown, NY, USA). Aldosterone was measured with a commercially available radioimmunoassay kit (Diagnostic Products Corporation, Los Angeles, CA, USA). Plasma renin activity (PRA) was measured as described previously with a radioimmunoassay that detects the amount of angiotensin I produced per hour in the presence of excess angiotensinogen (nanograms of angiotensin I produced per mL of plasma per h). This assay measures the enzymatic activity of active plasma renin in the presence of an excess of its (exogenous) substrate (37). Blood pressure was measured under constant conditions, at 1-min intervals by an automatic device (Dinamap®; GE Medical systems, Milwaukee, WI), with the patient in semi-supine position. After 15 min of measurements, the mean of the last four readings was used for further analysis. MAP was calculated as the sum of one-third systolic and two-thirds of the diastolic blood pressure. Sample size We hypothesized that patients would present a mean proteinuria (SD) of 3.5 (2) g/d at baseline (placebo on HS). Assuming a reduction of 1.0 (0.7) g/d with losartan and an extra reduction of 0.6 (0.7) g/d by the addition of HCT or LS, it was estimated that 32 patients had to complete the cross-over design sequence to give the study a 90% power
67

CHAPTER 5
to detect a statistically significant difference (α = 0.05). Based on an expected drop-out rate of 5%, we included 34 patients. Data analysis Results are expressed as mean and standard error (SD). Baseline data were obtained after 6-week placebo treatment with HS. Drug effects in all periods were evaluated using linear mixed effect models. Tukey tests were used to localize the differences. Statistical significance was assumed at the 5% level of probability. We used SPSS 12.0.1 for Windows (SPSS Inc., Chicago, Illinois, USA) for all analyses. Table 1. Compliance to diet and effects on body weight, fractional protein excretion, protein/creatinine ratio, blood pressure and creatinine clearance during treatment with placebo, losartan and losartan+HCT combined with a high sodium and low sodium diet
n = 33 Placebo Losartan Losartan + HCT
HS 200 (10) 197 (11) 193 (11) Urinary sodium excretion (mmol/d) LS 90 (10) # 92 (8) # 93 (8) #
HS 391 (16) 428 (22) ‡ 436 (24) ‡Urinary urea excretion (mmol/d)
LS 338 (23) # 372 (27) #₣ 362 (20) #
HS 91 (3) 90 (3) 89 (3) †‡
Body weight (kg) LS 89 (3) # 88 (3) #‡₣ 88 (3) #‡
HS 591 (78) * 387 (53) 253 (43) †Protein excretion (ng) / mL filtrate
LS 518 (85) 286 (47) #‡ 189 (30) #†‡
HS 2.45 (0.27) * 1.69 (0.22) ‡ 1.01 (0.15) ‡†Proteinuria-to-creatinine ratio (mg/mg)
LS 2.10 (0.36) 1.18 (0.19) #‡ 0.73 (0.12) #†‡
HS 143 (4) * 135 (3) 125 (3) †‡
Systolic blood pressure (mmHg) LS 137 (3) 128 (3) #‡ 121 (2) #†‡
HS 86 (2) * 80 (2) 75 (1) †‡
Diastolic blood pressure (mmHg) LS 83 (1) 78 (1) ‡ 74 (1) †‡
HS 89 (5) 94 (6) 86 (6) †Creatinine clearance (mL/min)
LS 82 (6) # 83 (7) # 75 (5) #†‡
HCT, hydrochlorothiazide. HS, high sodium. LS, low sodium. * p < 0.05 vs. all periods; # p < 0.05 vs. same treatment on HS (effect of LS), † p < 0.05 vs. losartan treatment on same diet (effect of HCT), ‡ p < 0.05 vs. placebo on same diet, ₣ p < 0.05 vs. losartan + HCT/ HS (comparison between addition of LS and HCT to losartan).
68

LOW SODIUM DIET AND DIURETIC ON TOP OF LOSARTAN
RESULTS
Patients characteristics and dietary compliance We included 34 patients (25 males, 9 females, all Caucasians) with a mean age (range) of 50 (23-68) years and mean body mass index (SE) of 27.5 (0.8) (kg/m2). Baseline proteinuria was 3.9 (0.4) g/d. Diagnoses were membranous glomerulopathy (7), focal segmental glomerular sclerosis (7), membranoproliferative glomerulonephritis (2), minimal change disease with secondary glomerulosclerosis (2), hypertensive nephropathy (5), IgA nephropathy (5), Alport syndrome (1) and non-conclusive diagnosis (4). One patient could not fulfil the complete protocol (due to psychological distress unrelated to the study medication) and was excluded for further analysis.
During HS mean urinary sodium excretion was 196 (9) mmol/d and during LS 92 (8) mmol/d (p < 0.001), indicating an adequate dietary compliance with achieved urinary sodium values in the physiological range (table 1). LS was accompanied by a lower body weight in all three periods. During losartan, LS reduced body weight significantly more than HCT (table 1). Urinary urea excretion was significantly lower during LS (table 1). Proteinuria and blood pressure Proteinuria showed a stepwise decrease. Baseline proteinuria (3.8 (0.4) g/d on placebo-HS) was significantly reduced by all interventions (p < 0.01 for trend, figure 1A). Percentage change of proteinuria from baseline showed the same pattern, i.e. placebo on LS induced a reduction of 22 (6)%, losartan/ HS 30 (4)%, losartan/ LS 55 (4)%, losartan + HCT/ HS 56 (4)% and losartan + HCT/ LS 70 (4)%. The shift from HS to LS significantly reduced proteinuria during all regimens. Proteinuria was similarly reduced by addition of HCT or LS to losartan, but the lowest proteinuria was achieved with both measures combined (figure 1A). To account for the confounding effect of hemodynamic changes in creatinine clearance associated with blood pressure reduction and/or RAAS blockade, protein excretion per mL filtrate was calculated. This showed similar results (table 1). The number of patients who reached target proteinuria < 1 g/d was stepwise increased by intensifying antiproteinuric treatment. Target proteinuria was obtained in 6% of patients (2/33) by LS, in 12% (4/33) by losartan/ HS, in 30% (10/33) by losartan/ LS, in 33% (11/33) by losartan + HCT/ HS and in 49% (16/33) by losartan + HCT/ LS.
Normalization for 24-h urinary creatinine excretion did not influence the results on proteinuria (protein-to-creatinine excretion values are shown in table 1), indicating that the results were not influenced by urine collection errors. The differences in urinary urea excretion did not influence the results on proteinuria when urinary urea excretion was entered as covariate in the linear mixed effect model. Therapy was equally effective in patients with primary glomerulopathies (n = 19) as in patients with secondary
69

CHAPTER 5
Figure 1. Proteinuria (A) and mean arterial pressure (B) after 6-week treatment with losartan and losartan + HCT combined with a high sodium (black bars) and low sodium diet (grey bars) compared to baseline (placebo HS). Values are expressed as mean (SE). * p < 0.05 vs. all periods; # p < 0.05 vs. same treatment on HS (effect of LS), † p < 0.05 vs. losartan treatment on same diet (effect of HCT); ‡ p < 0.05 vs. placebo on same diet.
glomerulopathies (IgA or hypertensive nephropathies; n = 10), excluding the patients with non-conclusive diagnoses (n = 4) (figure 2).
Blood pressure also displayed a stepwise decrease, as shown for MAP in figure 1B p<0.01 for the trend). Baseline blood pressure (143 (4)/86 (2) mmHg on placebo/ HS) was significantly reduced by all interventions (systolic and diastolic blood pressures are given in table 1). Blood pressure was similarly reduced by adding HCT or LS during losartan, but the lowest blood pressure was obtained by their combination. The shift from HS to LS significantly reduced MAP during all regimens.
To analyse for possible blood pressure dependency of the antiproteinuric response, we entered blood pressure (MAP, systolic or diastolic blood pressure, respectively) as covariate in our linear effect model. However, neither measure of blood pressure contributed significantly to the model. Thus, the overall effects of therapy on proteinuria cannot be explained by the effects on blood pressure. Moreover, baseline proteinuria, but not baseline degree of blood pressure, predicted outcome in terms of residual proteinuria, with a higher residual proteinuria in patients with a higher baseline proteinuria.
Comparison of concordance of blood pressure and proteinuria responses to LS and HCT when added to losartan To assess whether there are differences in mechanisms of antiproteinuric action between the two different measures of volume depletion, we also analyzed the possible concordance of blood pressure and proteinuria responses separately for the periods with
70
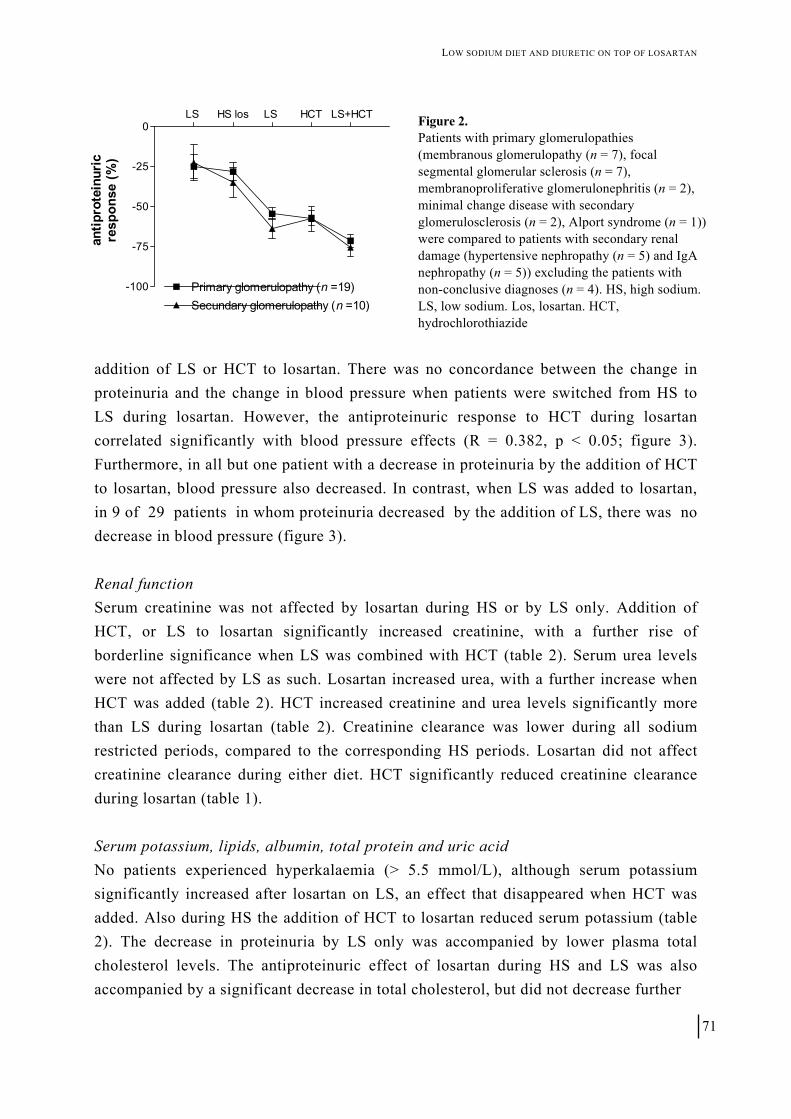
LOW SODIUM DIET AND DIURETIC ON TOP OF LOSARTAN
LS HS los LS HCT LS+HCT
-100
-75
-50
-25
0
Primary glomerulopathy (n =19)Secundary glomerulopathy (n =10)
antip
rote
inur
icre
spon
se (%
)
Figure 2. Patients with primary glomerulopathies (membranous glomerulopathy (n = 7), focal segmental glomerular sclerosis (n = 7), membranoproliferative glomerulonephritis (n = 2), minimal change disease with secondary glomerulosclerosis (n = 2), Alport syndrome (n = 1)) were compared to patients with secondary renal damage (hypertensive nephropathy (n = 5) and IgA nephropathy (n = 5)) excluding the patients with non-conclusive diagnoses (n = 4). HS, high sodium. LS, low sodium. Los, losartan. HCT, hydrochlorothiazide
addition of LS or HCT to losartan. There was no concordance between the change in proteinuria and the change in blood pressure when patients were switched from HS to LS during losartan. However, the antiproteinuric response to HCT during losartan correlated significantly with blood pressure effects (R = 0.382, p < 0.05; figure 3). Furthermore, in all but one patient with a decrease in proteinuria by the addition of HCT to losartan, blood pressure also decreased. In contrast, when LS was added to losartan, in 9 of 29 patients in whom proteinuria decreased by the addition of LS, there was no decrease in blood pressure (figure 3). Renal function Serum creatinine was not affected by losartan during HS or by LS only. Addition of HCT, or LS to losartan significantly increased creatinine, with a further rise of borderline significance when LS was combined with HCT (table 2). Serum urea levels were not affected by LS as such. Losartan increased urea, with a further increase when HCT was added (table 2). HCT increased creatinine and urea levels significantly more than LS during losartan (table 2). Creatinine clearance was lower during all sodium restricted periods, compared to the corresponding HS periods. Losartan did not affect creatinine clearance during either diet. HCT significantly reduced creatinine clearance during losartan (table 1). Serum potassium, lipids, albumin, total protein and uric acid No patients experienced hyperkalaemia (> 5.5 mmol/L), although serum potassium significantly increased after losartan on LS, an effect that disappeared when HCT was added. Also during HS the addition of HCT to losartan reduced serum potassium (table 2). The decrease in proteinuria by LS only was accompanied by lower plasma total cholesterol levels. The antiproteinuric effect of losartan during HS and LS was also accompanied by a significant decrease in total cholesterol, but did not decrease further
71
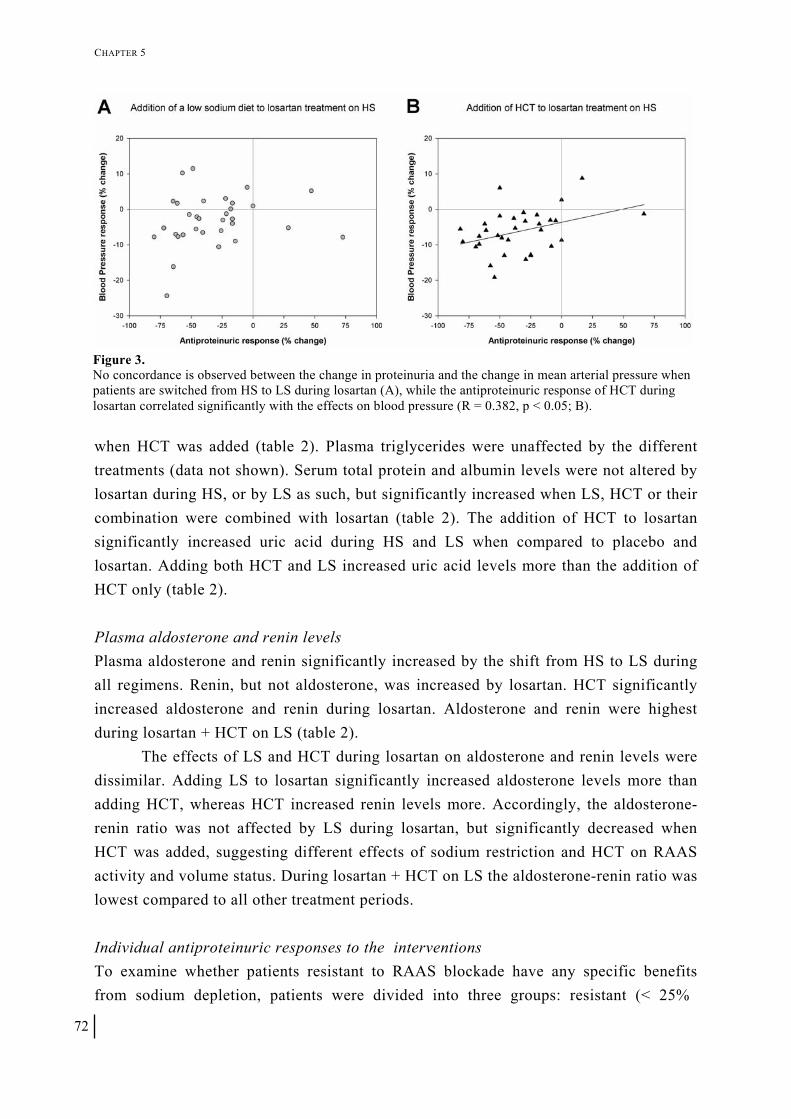
CHAPTER 5
Figure 3. No concordance is observed between the change in proteinuria and the change in mean arterial pressure when patients are switched from HS to LS during losartan (A), while the antiproteinuric response of HCT during losartan correlated significantly with the effects on blood pressure (R = 0.382, p < 0.05; B).
when HCT was added (table 2). Plasma triglycerides were unaffected by the different treatments (data not shown). Serum total protein and albumin levels were not altered by losartan during HS, or by LS as such, but significantly increased when LS, HCT or their combination were combined with losartan (table 2). The addition of HCT to losartan significantly increased uric acid during HS and LS when compared to placebo and losartan. Adding both HCT and LS increased uric acid levels more than the addition of HCT only (table 2). Plasma aldosterone and renin levels Plasma aldosterone and renin significantly increased by the shift from HS to LS during all regimens. Renin, but not aldosterone, was increased by losartan. HCT significantly increased aldosterone and renin during losartan. Aldosterone and renin were highest during losartan + HCT on LS (table 2).
The effects of LS and HCT during losartan on aldosterone and renin levels were dissimilar. Adding LS to losartan significantly increased aldosterone levels more than adding HCT, whereas HCT increased renin levels more. Accordingly, the aldosterone-renin ratio was not affected by LS during losartan, but significantly decreased when HCT was added, suggesting different effects of sodium restriction and HCT on RAAS activity and volume status. During losartan + HCT on LS the aldosterone-renin ratio was lowest compared to all other treatment periods. Individual antiproteinuric responses to the interventions To examine whether patients resistant to RAAS blockade have any specific benefits from sodium depletion, patients were divided into three groups: resistant (< 25%
72

LOW SODIUM DIET AND DIURETIC ON TOP OF LOSARTAN
Table 2. Effects of treatment with placebo, losartan and losartan + HCT during high and low sodium diet n = 33 Placebo Losartan Losartan + HCT
HS 125 (8) 121 (7) 136 (9) †‡
Serum creatinine (µmol/L) LS 126 (7) 129 (7) #₣ 143 (10) †‡
HS 7.0 (0.4) 7.6 (0.5) 9.9 (0.9) †‡ Serum urea (mmol/L)
LS 6.9 (0.5) 7.6 (0.6) ‡₣ 10.8 (1.1) †‡
HS 4.3 (0.1) 4.4 (0.1) 4.1 (0.1) †‡
Serum potassium (mmol/L) LS 4.3 (0.1) 4.5 (0.1) ‡₣ 4.0 (0.1) †‡
HS 6.1 (0.3) * 5.7 (0.2) 5.8 (0.2) Serum total cholesterol (mmol/L) LS 5.9 (0.2) 5.5 (0.2) ‡₣ 5.6 (0.3) ‡
HS 68.9 (1.4) 68.0 (1.3) 69.8 (1.2) †‡
Serum total protein (g/L) LS 69.2 (1.4) 70.1 (1.1) # 72.3 (1.1) #†‡
HS 38.7 (0.7) 38.8 (0.6) 39.7 (0.6) †
Serum albumin (g/L) LS 39.0 (0.7) 40.1 (0.6) #‡ 41.0 (0.5) #†‡
HS 0.42 (0.01) 0.39 (0.01) 0.46 (0.02) †‡ Serum uric acid (mmol/L)
LS 0.43 (0.02) 0.42 (0.02) ₣ 0.52 (0.02) #†‡
HS 93 (15) 78 (12) 113 (12) † Plasma aldosterone (pg/mL)
LS 140 (17) # 132 (13) #₣ 189 (18) #†‡
HS 4.2 (0.4) * 11.1 (2.1) ‡ 20.1 (2.8) †‡ Plasma renin (ngAI/mL/h)
LS 5.2 (0.5) 16.6 (2.5) #‡₣ 37.2 (4.4) #†‡
HS 29 (5) 15 (5) ‡ 10 (2) †‡ Aldosterone-to-renin ratio (pg/ngAIxh) LS 36 (7) 16 (5) ‡₣ 7 (1) #†‡
HCT, hydrochlorothiazide. HS, high sodium. LS, low sodium. * p < 0.05 vs. all periods; # p < 0.05 vs. same treatment on HS; † p < 0.05 vs. losartan treatment on same diet (effect of HCT); ‡ p < 0.05 vs. placebo on same diet, ₣ p < 0.05 vs. losartan + HCT/ HS (comparison between addition of LS and HCT to losartan).
reduction in proteinuria, -10 (4)%, n = 16), intermediate (25-50% reduction, -41 (3)%, n = 10) and good responders (> 50% reduction, -62 (3)%, n = 7) according to their antiproteinuric response to losartan from baseline (on placebo/ HS). Baseline characteristics (age, gender, diagnoses, BMI, creatinine clearance), dietary compliance and blood pressure response to losartan did not differ between the groups (table 3). Baseline proteinuria was similar (figure 4, upper panel). Antiproteinuric response in resistant and intermediate responders was significantly enhanced by LS or HCT, whereas in good responders antiproteinuric response did not significantly improve
73

CHAPTER 5
Table 3. Baseline characteristics according to the antiproteinuric response to losartan from baseline. n = 33 Resistant
(n = 16) Intermediate
(n = 10) Good (n = 7)
Male/Female (n) 11/5 8/2 5/2
Age (years) 54 (3) 47 (3) 47 (6)
BMI (kg/m2) 28 (1) 28 (2) 25 (1)
Creatinine clearance (at baseline, mL/min) 88 (7) 93 (8) 85 (14)
Mean urinary sodium excretion on HS (mmol/d) 200 (11) 202 (20) 177 (10)
Mean urinary sodium excretion on LS (mmol/d) 88 (7) 93 (8) 85 (14)
Blood pressure response to losartan on HS (%) 6.2 (2.3) 4.9 (1.9) 8.1 (2.6)
Patients on additional antihypertensive drugs (n) 4 / 16 2 / 10 1 / 7
BMI, body mass index. HS, high sodium. LS, low sodium. Patients were divided into three groups: resistant (< 25% reduction in proteinuria, n = 16), intermediate (25-50% reduction, n = 10) and good responders (> 50% reduction, n = 7) according to their antiproteinuric response to single losartan on HS.
further by LS or HCT (figure 4, lower panel). In all groups lowest proteinuria was obtained by adding both LS and HCT to losartan without differences between the groups (figure 2, upper panel). With this regimen only 1 patient remained resistant, 5 were intermediate and the number of good responders increased to 26 (Chi square: p < 0.001).
DISCUSSION
The objective of this study was to characterize the independent and combined effects of sodium restriction and a diuretic on the responses of proteinuria and blood pressure to AT1 antagonist therapy in non-diabetic proteinuria. Firstly, we found that sodium restriction and HCT were equally effective in reducing proteinuria and blood pressure during losartan, whereas the largest decrease in proteinuria and blood pressure was obtained by their combination. Secondly, sodium depletion by a low sodium diet or a diuretic is specifically beneficial in those patients in whom proteinuria is resistant to RAAS blockade. Thirdly, remarkably sodium restriction as such already exerted a modest, but significant antiproteinuric effect. Our data indicate that for optimal reduction of proteinuria and blood pressure by an AT1 antagonist, both diuretic and sodium restriction should be applied.
It would be of interest to know whether the added antiproteinuric efficacy of volume depletion was due to lower blood pressure. However, on analysis by the linear mixed effect model the added effects on proteinuria could not be explained by the effects on blood pressure. Yet, on univariate analysis for the different treatment periods, during HCT the effects on blood pressure correlated to the proteinuria effects. This suggests that other antihypertensives may have had a similar effect, but we have no data to support this assumption.
74

LOW SODIUM DIET AND DIURETIC ON TOP OF LOSARTAN
BL HS los LS HCT LS+HCT0
1
2
3
4
5 ResistantIntermediateGood
Prot
einu
ria
(g/d
)
-100
-75
-50
-25
0
Ant
ipro
tein
uric
res
pons
e (%
)
*
*
*
*
*
*
*
*
*
*
*
* *
#
#
#
#
##
Figure 4. Effects of a low sodium diet (LS), hydrochlorothiazide (HCT) and their combination (LS + HCT) on residual proteinuria (upper panel) and antiproteinuric response (lower panel) during losartan treatment (los). According to their antiproteinuric response to losartan from baseline (BL, on placebo/ HS) patients were divided into three groups: resistant (< 25% reduction in proteinuria, n = 16), intermediate (25-50% reduction, n = 10) and good responders (> 50% reduction, n = 7). Values are expressed as mean ± SE. # p < 0.05 vs. resistant patients on same treatment. * p < 0.05 vs. losartan on high sodium.
The reduction of proteinuria by low sodium alone has not previously been observed in patients with overt proteinuria due to primary glomerular disorders. It is in line with prior data on the effects of low sodium, in hypertensive blacks, where it was attributed to the concomitant reduction in blood pressure (12). Moreover, recent data from our own group in healthy young adults, demonstrated that sodium restriction significantly reduced urinary albumin excretion within the normal range, with only a borderline effect on blood pressure, suggesting that direct renal effects are not excluded (14). Obviously, the effect of sodium restriction alone in our overtly proteinuric patients was not sufficient to refrain from pharmacological intervention. We did not study the effects of HCT monotherapy, but in prior studies, HCT as a single measure did not influence proteinuria (14,15). It should be noted that reliable data on the latter are sparse and the aforementioned studies were corrected for urinary sodium excretion nor blood pressure (14,15).
We compared the added effects of low sodium and HCT to losartan for several reasons. Firstly, compliance with sodium restriction can be cumbersome, and it would be convenient if there was an alternative with similar efficacy. Secondly, there is some evidence that the effects of sodium restriction and HCT might not be equivalent, despite the fact that both act on sodium status. An earlier study in patients with severe renal failure demonstrated that thiazides exert their antihypertensive effect by specific
75

CHAPTER 5
vascular changes, rather than by volume depletion (16). Moreover, in uninephrectomized SHR rats, a model for hypertension and proteinuria, sodium restriction, but not diuretic therapy, diminished renal hypertrophy, while blood pressure was similar (17). In our study, addition of sodium restriction or HCT to losartan was equally effective in reducing proteinuria. On univariate analysis, the added effect of HCT on proteinuria was associated with blood pressure reduction, whereas this was not the case for the added effect of low sodium, suggesting possible different modes of action. Our data do not allow substantiating such mechanisms, and this issue requires further study.
Sodium restriction and HCT induced similar decreases in creatinine clearance during losartan, with the largest decrease during their combination, accompanied by a rise in serum urea. This decrease in GFR did not account for the decrease in proteinuria, as shown by the decrease in protein excretion per mL filtrate, indicating a specific antiproteinuric effect of losartan beyond its effects on renal function. It is well-established that during RAAS blockade changes in volume status exert distinct effects on renal function, facilitating pre-renal failure if volume depletion is severe. Here, the effect on renal function was mild, which is in line with relatively mild interventions in volume status. The clinical significance of the decrease in renal function for long term outcome cannot be derived from our data, but a decrease in GFR at the onset of treatment generally predicts a subsequent slower decline of renal function, presumably because it reflects a decrease in glomerular pressure (5,18).
Optimization of antiproteinuric treatment strategies can conceivably be obtained by combining diuretic with sodium restriction on top of RAAS blockade, although this approach might be limited by adverse events (19). Addition of HCT to losartan significantly increased uric acid, renin and aldosterone. Recent data showed that uric acid is an independent risk factor for the development of ESRD (20) and experimental studies provide evidence that uric acid may be a mediator of renal disease progression (21). Moreover, through their pro-fibrotic actions aldosterone (22-25) and (pro)renin (26-28) can also directly contribute to renal damage. Furthermore, a large meta-analysis in non-diabetic proteinuric patients showed that the risk of ESRD was adversely influenced when proteinuria reduction by RAAS blockade was accompanied by pronounced blood pressure reduction (29). This suggests a J-curve for blood pressure and long term renal outcome, with a worse outcome in subjects with pronounced blood pressure reduction. However, no adjustment for orthostasis and heart rate was made. Also, patients with lower blood pressure might be those with more severe proteinuria and hypoalbuminemia, secondary to the nephrotic syndrome, which could explain their faster progression. The assumption of a J-curve for renoprotection is supported by animal studies (30). However, for proteinuria, there is no evidence for a J-curve, and the target should be below 1 g/d (31).
76

LOW SODIUM DIET AND DIURETIC ON TOP OF LOSARTAN
Several measures can maximize the effects of RAAS blockade. These include dual blockade (32) and intervention in volume status, with sodium restriction, thiazides, loop diuretics, and aldosterone blockade. Esnault et al. showed in patients with a proteinuria comparable to ours, but with a worse renal function, that intensified furosemide therapy and avoiding excessive sodium on top of dual RAAS blockade resulted in a further decrease in proteinuria at the expense of a rise in serum creatinine (8). Our data are in line with theirs and, moreover, show the separate effects of sodium restriction and a diuretic on top of RAAS blockade by an AT1 antagonist only. Our data suggest that further reduction of proteinuria could have been obtained by further sodium depletion, possibly at the expense of a further rise in serum creatinine. The effects of more vigorous interventions in volume status on top of dual RAAS blockade deserve further investigation. This also applies to volume intervention by aldosterone blockade that may exert specific direct renoprotective effects (23,24,33,34). We consider it of particular interest that the sodium depleting measures were effective in improving therapy response in subjects with a poor response to monotherapy losartan.
We acknowledge possible weaknesses in our study. Urinary urea excretion was lower during sodium restriction. This could question the specificity of the effect of low sodium, as part of the antiproteinuric effect could be due to lower protein intake. We were unable to detect statistically significant effects of the differences in urinary urea excretion on proteinuria on multivariate analysis, but this does not fully exclude an effect. In a clinical outpatient setting, it is difficult to establish sodium restriction without any effect on protein intake, and vice versa. This was recently demonstrated in patients with stage 4 and 5 kidney disease in whom blood pressure reduction by a very-low-protein diet was independently related to urinary sodium excretion, but not to protein intake (35). In an editorial commentary it was considered likely that effects of the low-protein-diet on blood pressure were due to modification of sodium intake (36). Here, we cannot exclude an effect of altered protein intake on proteinuria, but an effect on blood pressure would not be likely. We studied short-term effects of sodium depletion on top of RAAS blockade on proteinuria. However, short-term reductions in proteinuria predict a slower decline in GFR in non-diabetic nephropathy (31). Whether combined sodium depletion is effective in slowing renal function decline has to be confirmed in long-term studies.
We conclude that sodium restriction and diuretics are equally effective in reducing proteinuria and blood pressure when added to AT1 antagonist and are specifically beneficial in those patients in whom proteinuria is resistant to RAAS blockade. The largest effect on proteinuria and blood pressure is obtained during their combination. Intensified intervention in sodium status by combining sodium restriction
77

CHAPTER 5
and a diuretic is an effective tool to maximize the antiproteinuric efficacy of RAAS blockade. REFERENCES 1. Randomised placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of terminal renal failure in
proteinuric, non-diabetic nephropathy. The GISEN Group (Gruppo Italiano di Studi Epidemiologici in Nefrologia). Lancet 1997;349:1857-63
2. Brenner BM, Cooper ME, de Zeeuw D et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001;345:861-9
3. Lewis EJ, Hunsicker LG, Clarke WR et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001;345:851-60
4. Buter H, Hemmelder MH, Navis G, de Jong PE, de Zeeuw D. The blunting of the antiproteinuric efficacy of ACE inhibition by high sodium intake can be restored by hydrochlorothiazide. Nephrol Dial Transplant 1998;13:1682-5
5. Apperloo AJ, de Zeeuw D, de Jong PE. Short-term antiproteinuric response to antihypertensive treatment predicts long-term GFR decline in patients with non-diabetic renal disease. Kidney Int 1994;Suppl 45:S174-8
6. de Zeeuw D, Ramjit D, Zhang Z et al. Renal risk and renoprotection among ethnic groups with type 2 diabetic nephropathy: a post hoc analysis of RENAAL. Kidney Int 2006;69:1675-82
7. Kopple JD. The National Kidney Foundation K/DOQI clinical practice guidelines for dietary protein intake for chronic dialysis patients. Am J Kidney Dis 2001;38:S68-S73
8. Esnault VL, Ekhlas A, Delcroix C, Moutel MG, Nguyen JM. Diuretic and enhanced sodium restriction results in improved antiproteinuric response to RAS blocking agents. J Am Soc Nephrol 2005;16:474-81
9. Heeg JE, de Jong PE, van der Hem GK, de Zeeuw D. Efficacy and variability of the antiproteinuric effect of ACE inhibition by lisinopril. Kidney Int 1989;6:272-9
10. Houlihan CA, Allen TJ, Baxter AL et al. A low-sodium diet potentiates the effects of losartan in type 2 diabetes. Diabetes Care 2002;25:663-71
11. Wapstra FH, van Goor H, Navis G, de Jong PE, de Zeeuw D. Antiproteinuric effect predicts renal protection by angiotensin-converting enzyme inhibition in rats with established adriamycin nephrosis. Clin.Sci.(Lond) 1996;90:393-401
12. Swift PA, Markandu ND, Sagnella GA, He FJ, MacGregor GA. Modest salt reduction reduces blood pressure and urine protein excretion in black hypertensives: a randomized control trial. Hypertension 2005;46:308-12
13. Krikken JA, Lely AT, Bakker SJ, Navis G. The effect of a shift in sodium intake on renal hemodynamics is determined by body mass index in healthy young men. Kidney Int 2007;71:260-5
14. Donker AJ, Brentjens JR, van der Hem GK, Arisz L. Treatment of the nephrotic syndrome with indomethacin. Nephron 1978;22:374-81
15. Vogt L, Navis G, Koster J, Manolis AJ, Reid JL, de Zeeuw D. The angiotensin II receptor antagonist telmisartan reduces urinary albumin excretion in patients with isolated systolic hypertension: results of a randomized, double-blind, placebo-controlled trial. J Hypertens 2005;23:2055-61
16. Jones B, Nanra RS. Double-blind trial of antihypertensive effect of chlorothiazide in severe renal failure. Lancet 1979;2:1258-60 17. Benstein JA, Feiner HD, Parker M, Dworkin LD. Superiority of salt restriction over diuretics in reducing renal hypertrophy and
injury in uninephrectomized SHR. Am J Physiol 1990;258:F1675-81 18. Hansen HP, Rossing P, Tarnow L, Nielsen FS, Jensen BR, Parving H-H. Increased glomerular filtration rate after withdrawal of
long-term antihypertensive treatment in diabetic nephropathy. Kidney Int 1995;47:1726-31 19. Vogt L, Navis G, de Zeeuw D. Individual titration for maximal blockade of the renin-angiotensin system in proteinuric patients: a
feasible strategy? J Am Soc Nephrol 2005;16 (Suppl 1):S53-7 20. Iseki K, Ikemiya Y, Inoue T, Iseki C, Kinjo K, Takishita S. Significance of hyperuricemia as a risk factor for developing ESRD in a
screened cohort. Am J Kidney Dis 2004;44:642-50 21. Kang DH, Nakagawa T, Feng L et al. A role for uric acid in the progression of renal disease. J Am Soc Nephrol 2002;13:2888-97 22. Aldigier JC, Kanjanbuch T, Ma LJ, Brown NJ, Fogo AB. Regression of existing glomerulosclerosis by inhibition of aldosterone. J
Am Soc Nephrol 2005;16:3306-14 23. Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic
kidney disease. Kidney Int 2006;70:2116-23 24. Chrysostomou A, Becker G. Spironolactone in addition to ACE inhibition to reduce proteinuria in patients with chronic renal
disease. N Engl J Med 2001;345:925-6 25. Miyata K, Rahman M, Shokoji T et al. Aldosterone stimulates reactive oxygen species production through activation of NADPH
oxidase in rat mesangial cells. J Am Soc Nephrol 2005;16:2906-12 26. Huang Y, Wongamorntham S, Kasting J et al. Renin increases mesangial cell transforming growth factor-beta1 and matrix proteins
through receptor-mediated, angiotensin II-independent mechanisms. Kidney Int 2006;69:105-13 27. Huang Y, Noble NA, Zhang J, Xu C, Border WA. Renin-stimulated TGF-beta1 expression is regulated by a mitogen-activated
protein kinase in mesangial cells. Kidney Int 2007;72:45-52 28. Nguyen G, Delarue F, Burckle C, Bouzhir L, Giller T, Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II
production and cellular responses to renin. J Clin Invest 2002;109:1417-27 29. Jafar TH, Stark PC, Schmid CH et al. Progression of chronic kidney disease: the role of blood pressure control, proteinuria, and
angiotensin-converting enzyme inhibition: a patient-level meta-analysis. Ann Intern Med 2003;139:244-52 30. Hamming I, Navis G, Kocks MJ, van Goor H. ACE inhibition has adverse renal effects during dietary sodium restriction in
proteinuric and healthy rats. J Pathol 2006;209:129-39 31. Ruggenenti P, Schieppati A, Remuzzi G. Progression, remission, regression of chronic renal diseases. Lancet 2001;357:1601-8
78

LOW SODIUM DIET AND DIURETIC ON TOP OF LOSARTAN
32. Nakao N, Yoshimura A, Morita H, Takada M, Kayano T, Ideura T. Combination treatment of angiotensin-II receptor blocker and angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomised controlled trial. Lancet 2003;361:117-24
33. Kramer AB, van der Meulen EF, Hamming I, van Goor H, Navis G. Effect of combining ACE inhibition with aldosterone blockade on proteinuria and renal damage in experimental nephrosis. Kidney Int 2007;71:417-24
34. Sato A, Hayashi K, Naruse M, Saruta T. Effectiveness of aldosterone blockade in patients with diabetic nephropathy. Hypertension 2003;41:64-8
35. Bellizzi V, Di Iorio BR, De Nicola L et al. Very low protein diet supplemented with ketoanalogs improves blood pressure control in chronic kidney disease. Kidney Int 2007;71:245-51
36. Weir MR. Is it the low-protein diet or simply the salt restriction? Kidney Int 2007;71:188-90 37. Derkx FH, Tan-Tjiong L, Wenting GJ, Boomsma F, Man in 't Veld AJ, Schalekamp MA. Asynchronous changes in prorenin and
renin secretion after captopril in patients with renal artery stenosis. Hypertension 1983;5:244-56 ACKNOWLEDGEMENTS We thank Corrie Nieuwenhout for her skilful assistance at the outpatient clinic. A part of this work (an interim analysis in n = 17) was presented at the congress of the American Society of Nephrology, November 2005, Philadelphia, USA (J Am Soc Nephrol 16: 2005; 37A). STATEMENT OF COMPETING FINANCIAL INTERESTS This study was supported by Merck & Co. Inc. (grant MSGP NETH-15-01).
79

CHAPTER 5
80

Chapter 6
Individual titration for maximal blockade of the renin-angiotensin system in proteinuric patients: a feasible strategy? Liffert Vogt, Gerjan Navis and Dick de Zeeuw Journal of the American Society of Nephrology 2005;16:S53-7

CHAPTER 6
82

INDIVIDUAL TITRATION FOR MAXIMAL RAAS BLOCKADE
Agents that interfere with the renin-angiotensin-aldosterone system (RAAS) reduce proteinuria and afford renal protection. Combination of different measures serving maximization of RAAS blockade is thought to improve the antiproteinuric efficacy. We studied in non-diabetic patients with residual proteinuria during previous RAAS blockade the feasibility and efficacy of such a combination strategy by individual antiproteinuric titration. Previous medication was replaced by irbesartan 300 mg combined with a diuretic. Lisinopril was added in increasing doses until a maximal dose of 40 mg/d. Titration stopped when target proteinuria (< 1 g/d) was reached or further dose-titration was not tolerated because of side effects. Residual proteinuria (median (95% CI): 3.2 g/d (1.8; 5.2)) was significantly reduced with 55.6% (73.2; 16.0) (p < 0.02) on the maximal additional tolerated dose of lisinopril. The maximal dose of lisinopril was 10 mg in 2/8, 20 mg in 2/8, 30 mg in 1/8, and 40 mg in 3/8 patients. At this dose, target proteinuria of < 1 g/d was reached in 2/8 patients. The number of patients with adverse events during dose-titration was 5/8 patients: two had cough, two had hyperkalaemia (> 5.5 mmol/L), of whom one had > 50% increase of serum creatinine, and one had dizziness. In conclusion, individual titration for maximal RAAS blockade, entailing dose-titration of an angiotensin-converting enzyme inhibitor on top of a highly dosed angiotensin II type 1 receptor antagonist with diuretic, induces further reduction of residual proteinuria. However, this occurs at expense of adverse events. To further improve renoprotective treatment strategies, it would therefore be important to explore other modes of antiproteinuric intervention in patients with residual proteinuria during RAAS blockade.
roteinuria is nowadays looked upon as an important and independent risk factor for progression of renal disease (1,2). Moreover, evidence from large clinical trials has become available, showing that reduction of proteinuria is important for long-term renoprotection (3,4). In addition, it has been noted that both residual proteinuria and the amount antiproteinuric response are also predictive for renal outcome in individual patients, indicating that residual proteinuria during therapy is a predictor of the individual renal prognosis (5,6). Accordingly, maximum reduction of proteinuria has been advocated as a treatment target for individual renal patients, in addition to control of blood pressure (7-9). For optimal renoprotection, therefore, recent data suggest that treatment target for proteinuria should be below 1 g/d, and likely near zero (7,8).
P
Intervention in the renin-angiotensin-system (RAAS) is currently the most effective strategy that combines renoprotection with proteinuria lowering. Roughly, the average antiproteinuric response of RAAS blocking agents is about 50%—both for angiotensin-converting enzyme (ACE) inhibitors and for angiotensin II type 1 (AT1)
83

CHAPTER 6
antagonists (10,11). There are several strategies to optimize the response, including dose-titration of the RAAS intervening agents (12), combining RAAS blockade with low sodium diet or a diuretic (13), and combining the different RAAS blocking strategies (14). Indeed, ACE inhibitor plus AT1 antagonist therapy renders more antiproteinuric effect and also more renoprotection (15). Although each of these measures is widely studied on group level, until now, no individual data of maximal RAAS blockade on proteinuria are available (9). Moreover, it is unknown whether it is possible, in a prospective fashion, to obtain the target level of proteinuria below 1 g/d by titrating these measures in individual patients. In the present study, our aim was to investigate the antiproteinuric potential of additional up-titration with an ACE inhibitor to maximal tolerated dose against a background of a maximal dosed AT1 antagonist combined with diuretic therapy, in a sodium restricted setting. METHODS
Patients and protocol Patients were selected from our renal outpatient clinic. All patients gave informed consent and fulfilled the inclusion criterion of a stable proteinuria > 1 g/d and < 10 g/d while they were still on their previous (non-immune suppressive) antiproteinuric treatment. Only patients with blood pressure < 140/< 90 mm Hg, creatinine clearance ≥ 30 mL/min/1.73 m2 and age between 18 and 70 years were included. Patients with cardiovascular disease or diabetes mellitus were excluded, as well as frequent users of non-steroidal anti-inflammatory drugs (NSAIDS; > 2 doses/week).
The study was designed as a practice-based clinical trial. Patients were treated according to an individual dose-titration with ACE inhibitors added to a standard treatment of an AT1 antagonist combined with diuretic therapy. Patients were instructed to take the study medication once daily and to adhere to a dietary sodium restriction of 5 to 7 g/d. Our pre-defined treatment goal was to reduce proteinuria below 1 g/d. After selection, eligible patients entered the run-in phase in which previous medication was replaced for the highest recommended daily dose irbesartan 300 mg (16), combined with the diuretic hydrochlorothiazide 12.5 mg once daily. In patients previously treated with diuretic hydrochlorothiazide or furosemide, the diuretic therapy was continued and solely irbesartan 300 mg was added. Then, patients with proteinuria > 1 g/d and serum potassium concentration < 5.5 mmol/L entered the phase of dose-titration. In this phase, lisinopril was added in increasing daily doses to a maximum of 40 mg. All periods of treatment (run-in and up-titration) lasted at least 6 weeks. The treatment protocol was discontinued when the pre-defined goal of proteinuria < 1 g/d was reached or further dose-titration was not tolerated because of side effects. Expected side effects were
84

INDIVIDUAL TITRATION FOR MAXIMAL RAAS BLOCKADE
defined as hyperkalaemia > 5.5 mmol/L, complaints of cough or dizziness, or serious increase of serum creatinine > 50%. Clinical and laboratory procedures Blood pressure was measured at every visit under similar conditions, at one-minute intervals by an automatic device (Dinamap®), with the patient in supine position. After 15 min of measurements, the mean of the last four readings was used for further analysis. Mean arterial pressure (MAP) was calculated as the sum of one-third systolic and two-thirds of the diastolic blood pressure. Urinary protein was determined using the pyrogallol red-molybdate method. At the end of each period, the day previous to every visit, patients collected 24-h urine samples to obtain proteinuria. Serum and urinary electrolytes, uric acid and creatinine were determined using an automated multi-analyzer (SMA-C®; Technicon, Tarrytown, NY, USA). Data analysis Results are expressed as median and 95% confidence intervals (95% CI). For each patient, the level of residual proteinuria after treatment with irbesartan combined with hydrochlorothiazide and at the maximal tolerated dose of lisinopril was established. The Wilcoxon signed ranks test was used to test whether paired values from standardized irbesartan 300 mg + diuretic treatment and after treatment with added maximal individual lisinopril dose differed. Differences were considered significant if the p-value was < 0.05. Based on previous data (14), we calculated that a sample size of n = 6 is needed to detect mean reduction in proteinuria of 40% with expected standard deviation of 28% and with a desired power of 80% and α = 0.05. RESULTS
Patient characteristics Twelve non-diabetic proteinuric patients were selected from our outpatient clinic. Four patients, however, did not enter the study protocol because of dizziness (n = 1) or proteinuria < 1 g/d (n = 3) during treatment with irbesartan and diuretic. The remaining 8 patients were included for up-titration with lisinopril and completed the treatment protocol. These patients, all of whom were middle-aged Caucasians, had mild to moderately impaired renal function and had biopsy proven non-diabetic nephropathy with the exception of one patient, in whom it was not possible to perform a biopsy due to anatomical abnormalities (horse shoe kidney) (table 1). During up-titration, three patients achieved the maximal daily dose of lisinopril 40 mg, whereas lisinopril 30 mg, 20 mg, and 10 mg was achieved in 1, 2 and 2 patients, respectively.
85

CHAPTER 6
Table 1. Patient characteristics at baseline (median and 95% CI or number) Male / female 5 / 3 Age (years) 51 (35; 60) Creatinine clearance (mL/min) 75 (31; 137) Systolic blood pressure (mmHg) 126 (106; 147) Diastolic blood pressure (mHg) 75 (63; 94) Diagnosis FSGS (1), HSK (1), IgA (1), MCD (1), MGP (3), NCBx (1) Previous standard medication Fos / furo (1), irb (1), lis / hct (3), los/hct (3)
FSGS, focal and segmental glomerulosclerosis. HSK, horseshoe kidney. IgA, IgA nephropathy. MCD, minimal change disease. MGP, membranous glomerulopathy. NCBx, non-conclusive hiopsy. Fos, fosinopril. Furo, furosemide. Hct, hydrochlorothiazide. Irb, irbesartan. Lis, lisinopril. Los, losartan.
Proteinuria Off-treatment proteinuria (i.e. without treatment with antihypertensive agents, especially no RAAS intervening agents, or immune suppressive medication) was 5.6 g/d (95% CI: 4.1; 8.4). Residual proteinuria was 2.9 g/d (1.7; 5.2) on treatment with the combination irbesartan, diuretic, and low sodium diet. Additional dose-titration with lisinopril to the maximal tolerated dose, showed a further proteinuria reduction of 55.6% (16.0; 73.2) to a residual proteinuria of 1.6 (0.8; 3.6) (p = 0.018 vs. irbesartan + diuretic; figure 1). Proteinuria-to-creatinine ratio was also significantly reduced (table 2). Individual up-titration with lisinopril led to further proteinuria reduction in all patients. No relation between the amount of proteinuria reduction (% change) and the dose of lisinopril could be observed (table 3).
Mean arterial blood pressure was 92 mmHg (95% CI: 77; 111). After up-titration to maximal lisinopril doses, MAP was significantly reduced to 85 mmHg (73; 109) (p = 0.043; figure 1). Body weight, serum electrolytes, albumen and lipids were not affected by lisinopril up-titration (table 2).
With regard to the pre-defined treatment goal, 2 out of 8 patients reached proteinuria of < 1 g/d after the maximal individual dose of lisinopril 10 mg and 40 mg, respectively (table 3). Six out of 8 did not reach the target proteinuria: 2/6 because they had still residual proteinuria (> 1 g/d) on the highest lisinopril dose, and 4/6 because they experienced side effects that prevented further dose increase of the ACE inhibitors. Side effects During up-titration with lisinopril, 5 out of 8 patients experienced adverse symptoms: two patients had hyperkalaemia (> 5.5 mmol/L), two patients complained of cough, and one patient experienced dizziness and had simultaneously > 50% increase of serum creatinine (table 3). One of these five patients also reached the treatment target. Notably, of those patients who experienced adverse effects, none received the maximal additional dose of lisinopril (40 mg QD), whereas in those patients in whom up-titration to the maximal lisinopril dose was possible no side effects were observed.
86

INDIVIDUAL TITRATION FOR MAXIMAL RAAS BLOCKADE
Table 2. Median (95% CI) values of the different parameters in eight non-diabetic patients after irbesartan 300 mg diuretic combination and after the maximal lisinopril dose added
Irbesartan 300 + diuretic Addition of maximum lisinopril dose
Uprot (g/d) 3.2 (1.8;5.2) 1.6 (0.8; 3.6)†
RatioUprot/Ucreat*10 1.8 (0.3; 3.3) 1.0 (0.6; 2.3)†
BPSystolic (mmHg) 126 (106; 147) 117 (108; 143)*
BPDiastolic (mmHg) 75 (63; 94) 69 (56; 91)†
CCreat (mL/min) 74 (31; 137) 65 (45; 101)*
BW (kg) 84 (71; 101) 83 (70; 101) UNa (mmol/d) 140 (76; 319) 144 (78; 244) SUA (mmol/L) 0.48 (0.44; 0.57) 0.49 (0.31; 0.57) Schol (mmol/L) 4.8 (4.8; 6.1) 5.3 (4.8; 5.9) STG (mmol/L) 3.5 (1.2; 7.8) 3.1 (1.1; 5.3) Salb (g/L) 41 (36; 45) 41 (33; 47)
SCreat (μmol/L) 129 (83;285) 151 (92; 300)†
SK (mmol/L) 4.6 (3.7; 5.5) 4.4 (3.9; 6,4)
Uprot, proteinuria. Ucreat, urinary creatinine. BP, blood pressure. CCreat, creatinine clearance. BW, body weight. UNa, urinary sodium excretion. SUA, serum uric acid. Schol, serum total cholesterol. Salb, serum albumen. Screat, serum creatinine. SK, serum potassium (* p < 0.05; † p < 0.02 vs. baseline). DISCUSSION The results of this study show that maximal RAAS blockade, entailing individual up-titration of the ACE inhibitor lisinopril to maximal tolerated dose in addition to a maximum therapy of the AT1 antagonist irbesartan combined with diuretic and dietary sodium restriction, induces a more than 50% reduction of residual proteinuria in the non-diabetic proteinuric patient. However, this benefit occurs at the expense of a number of adverse events, which clearly limit the feasibility in a considerable proportion of patients to obtain optimal antiproteinuric efficacy with the currently available therapeutic options. Indeed, in only 2 patients, the treatment target of proteinuria < 1 g/d was obtained. The high incidence of adverse events in our study seems to be at variance with the expectation based on studies in the different renal populations (14,15,17,18). Clinical studies in both diabetic and non-diabetic renal patients show that dual RAAS blockade led to further proteinuria reduction than with maximal recommended doses of monotherapy (ACE inhibitors or AT1 antagonists) could be obtained, without leading to extra adverse symptoms (14,17,18). Also, in the large trial with 263 non-diabetic patients of Nakao et al., in which the long-term benefit of dual RAAS blockade on hard renal endpoints was demonstrated, the combination therapy of ACE inhibitor and AT1 antagonist was very well tolerated and did not lead to extra side effects compared to monotherapy with trandolapril (15). To explain the high prevalence of adverse effects in
87

CHAPTER 6
Table 3. Individual blood pressure and proteinuria changes after the maximal tolerated additional lisinopril dose in eight non-diabetic proteinuric patients on irbesartan 300 mg diuretic combination treatmenta
Nr. Maximal lisinopril dose
Baseline MAP
(mmHG) % Change Baseline
Uprot (g/d) % Change Reason
1 40 mg 111 -2 5.2 -31 Maximal dose reachedc
2 40 mg 92 -10 1.8 -56 Uprot ≤ 1 g/d 3 40 mg 86 1 2.5 -16 Maximal dose reachedc
4 30 mg 92 -14 4.1 -73 Hyperkaliaemia, Screat increase > 50% 5 10 mg 99 -7 2.9 —b Hyperkaliaemia 6 10 mg 77 -6 2.3 -61 Uprot ≤ 1 g/d, cough 7 20 mg 89 -8 3.4 -53 Dizziness 8 20 mg 96 -7 4.1 -56 Cough
aReason for failure of the continuation of up-titration is given. bMissing value. cLisinopril 40 mg QD.
our study, one should take into account that most of the observed adverse symptoms (dizziness, hyperkalaemia, serious impairment of renal function) in our study tend to show a dose-dependent relation, i.e. the further maximization of RAAS blockade is obtained the more adverse effects can be expected. Our treatment schedule was characterized by two distinct measures that contribute to obtain the maximum antiproteinuric effect by a RAAS blockade-based regimen. First, in our treatment schedule, patients were individually titrated for maximal proteinuria reduction with additional lisinopril doses on top of a fixed high dose of irbesartan. Although no data are available on the dose-response of irbesartan for proteinuria reduction, it is expected that the maximal recommended dose for blood pressure reduction also renders the maximal antiproteinuric response, as indicated by most titration studies in proteinuric patients with other AT1 antagonists, as losartan and candesartan, in which a flattening of the dose-response curve is seen after treatment with higher doses (11,12,19). By contrast, increasing the ACE inhibitor dose seems not to show such recline in the dose-response for proteinuria reduction, indicating that higher doses of ACE inhibitor than needed for blood pressure control result in further proteinuria reduction (14). Therefore, we expect that individual up-titration with increasing doses lisinopril on top of high dosed AT1 antagonist leads to further maximization of blockade of the RAAS for the individual patient. As a consequence, this does not only result in improvement of the antiproteinuric effect, but also to high prevalence of side effects. In contrast, in the study of Nakao et al., the doses of both trandolapril and losartan used for proteinuria reduction were not based on observations in individual patients, but on group level for both trandolapril and losartan (15). Second, the dual RAAS blockade was given on top of diuretic therapy and dietary sodium restriction. Data from studies in hypertensive patients, but also in proteinuric
88

INDIVIDUAL TITRATION FOR MAXIMAL RAAS BLOCKADE
0
1
2
3
4
5
6
7
irb / diu max lis
Prot
einu
ria (g
/d)
60
70
80
90
100
110
120
irb / diu max lis
MAP
(mm
Hg)
-100
-75
-50
-25
0
25
irb / diu max lis
% C
hang
e in
pro
tein
uria
-100
-75
-50
-25
0
25
irb / diu max lis
% C
hang
e in
MAP
†
†
*
ns
Figure 1. Absolute and percentage change of proteinuria and MAP after individual uptitration to maximal tolerated doses with lisinopril (max lis) on background of irbesartan 300 mg + diuretic (Irb / diu) treatment in eight non-diabetic proteinuric patients. Dotted line represents the proteinuria treatment target of < 1 g/d (* p < 0.05; † p < 0.02).
patients, indicate that both low sodium diet and diuretic therapy can restore the blunted therapy response during RAAS intervening therapy (13). In our study, patients were instructed to adhere to a sodium-restricted diet which was only moderately successful, as estimated from their 24-h sodium excretion. Moreover, all patients were treated with a diuretic. Since it is known from hypertensive patients that both measures combined act synergistically on the therapy response for blood pressure during ACE inhibitor (20), it can be anticipated that the combination also leads to further improvement of the antiproteinuric response during dual RAAS blockade. Therefore, individual titration with an ACE inhibitor for the optimal antiproteinuric response on top of a treatment consisting of an AT1 antagonist, a diuretic and dietary low sodium, results in more effective blockade of the RAAS, as indicated by further optimization of proteinuria reduction and blood pressure response. However, as apparent from our data, this strategy has it limits and it may not only be so because of the occurrence of side effects.
We have previously demonstrated that individual antiproteinuric responsiveness to RAAS blockade is an important determinant of the renoprotective efficacy of intervention, and that, despite proven efficacy at group level, the renoprotective effect of RAAS blockade shows a marked between-patient heterogeneity (5). Therefore, the feasibility of individual titration for proteinuria by optimising the RAAS blockade was explored in this study. However, our present data show that this strategy to overcome individual therapy resistance does not result in abolishment of therapy resistance, which is in accord with other studies, showing that, despite efficacy at group level, poor responders still fail to catch up with good responders (21,22).
How could we approach the problem of improving antiproteinuric efficacy for
89

CHAPTER 6
individual patients without enhancing side effects? It would be interesting to explore other non-RAAS intervening modes of therapy directed to maximization of reduction of residual proteinuria. First, different studies demonstrated that intervention in the synthesis of prostaglandins by using non-steroidal-anti-inflammatory drugs (NSAIDs) has antiproteinuric properties with equal effectiveness as ACE inhibitor therapy (23,24). Because of the well-known side effects of non-selective NSAIDs, it would be of great interest to explore whether the relative new selective COX-2 inhibitors, of whom lower rates of side effects are reported, share the antiproteinuric properties of NSAIDs, as discussed previously (25). This issue is currently under investigation by our group. Until now, limited data suggest that addition of COX-2 inhibitors can overcome therapy resistance during ACE inhibitor therapy in membranous glomerulopathy by further reducing residual proteinuria (26). Second, lipid-lowering drugs, statins in particular, may exert a renoprotective effect, independently from their lipid-lowering effect. In renal patients and in hypertensive patients, long-term treatment with statins is reported to reduce proteinuria (27,28). Importantly, statin treatment added to ACE inhibitor and AT1 antagonist therapy was shown to improve proteinuria reduction (28,29). Therefore, statins could play role in renal conditions of relative resistance to intervention with RAAS blockade.
In conclusion, individual titration for maximal antiproteinuric efficacy by a RAAS blockade-based regimen results in successful reduction of residual proteinuria. However, this benefit occurs at expense of a number of adverse events. Achievement of proteinuria to lowest levels may probably only be pursued by a multi-drug approach, with intervention in other relevant pathways, such as prostaglandins or lipids. Future studies should address optimal dosing schedules and evaluate the eventual benefit in terms of renal risk. REFERENCES 1. Peterson JC, Adler S, Burkart JM et al. Blood pressure control, proteinuria, and the progression of renal disease. The Modification of
Diet in Renal Disease Study. Ann Intern Med 1995;123:754-62 2. Ruggenenti P, Perna A, Mosconi L, Pisoni R, Remuzzi G. Urinary protein excretion rate is the best independent predictor of ESRF
in non-diabetic proteinuric chronic nephropathies. ‘Gruppo Italiano di Studi Epidemiologici in Nefrologia’ (GISEN). Kidney Int 1998;53:1209-16
3. Randomized placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of terminal renal failure in proteinuric, non-diabetic nephropathy. The GISEN Group (Gruppo Italiano di Studi Epidemiologici in Nefrologia). Lancet 1997;349:1857-63
4. Brenner BM, Cooper ME, de Zeeuw D et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001;345:861-69
5. Apperloo AJ, de Zeeuw D, de Jong PE. Short-term antiproteinuric response to antihypertensive treatment predicts long-term GFR decline in patients with non-diabetic renal disease. Kidney Int 1994;Suppl 45:S174-8
6. Rossing P, Hommel E, Smidt UM, Parving H-H. Reduction in albuminuria predicts a beneficial effect on diminishing the progression of human diabetic nephropathy during antihypertensive treatment. Diabetologia 1994;37:511-6
7. Ruggenenti P, Schieppati A, Remuzzi G. Progression, remission, regression of chronic renal diseases. Lancet 2001;357:1601-8 8. de Jong PE, Navis G, de Zeeuw D. Renoprotective therapy: titration against urinary protein excretion. Lancet 1999;354:352-3 9. Vogt L, Laverman GD, de Zeeuw D, Navis G. The COOPERATE trial. Lancet 2003;361:1055-6
90

INDIVIDUAL TITRATION FOR MAXIMAL RAAS BLOCKADE
10. Gansevoort RT, de Zeeuw D, de Jong PE. Is the antiproteinuric effect of ACE inhibition mediated by interference in the renin-angiotensin system? Kidney Int 1994;45:861-7
11. Andersen S, Tarnow L, Rossing P, Hansen BV, Parving H-H. Renoprotective effects of angiotensin II receptor blockade in type 1 diabetic patients with diabetic nephropathy. Kidney Int 2000;57:601-6, 2000
12. Laverman GD, Henning RH, de Jong PE, Navis G, de Zeeuw D. Optimal antiproteinuric dose of losartan in nondiabetic patients with nephrotic range proteinuria. Am J Kidney Dis 2001;38:1381-4
13. Buter H, Hemmelder MH, Navis G, de Jong PE, de Zeeuw D. The blunting of the antiproteinuric efficacy of ACE inhibition by high sodium intake can be restored by hydrochlorothiazide. Nephrol Dial Transplant 1998;13:1682-5
14. Laverman GD, Navis G, Henning RH, de Jong PE, de Zeeuw D. Dual renin-angiotensin system blockade at optimal doses for proteinuria. Kidney Int 2002;62:1020-5
15. Nakao N, Yoshimura A, Morita H, Takada M, Kayano T, Ideura T. Combination treatment of angiotensin-II receptor blocker and angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomized controlled trial. Lancet 2003;361:117-24
16. Lewis EJ, Hunsicker LG, Clarke WR et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001;345:851-60
17. Rossing K, Jacobsen P, Pietraszek L, Parving H-H. Renoprotective effects of adding angiotensin II receptor blocker to maximal recommended doses of ACE inhibitor in diabetic nephropathy: a randomized double-blind crossover trial. Diabetes Care 2003;26:2268-74
18. Jacobsen P, Andersen S, Rossing K, Jensen BR, Parving H-H. Dual blockade of the renin-angiotensin system versus maximal recommended dose of ACE inhibition in diabetic nephropathy. Kidney Int 2003;63:1874-80
19. Andersen S, Rossing P, Juhl TR, Deinum J, Parving H-H. Optimal dose of losartan for renoprotection in diabetic nephropathy. Nephrol Dial Transplant 2002;17:1413-8
20. Wing LM, Arnolda LF, Harvey PJ et al. Low-dose diuretic and/or dietary sodium restriction when blood pressure is resistant to ACE inhibitor. Blood Press 1998;7:299-307
21. Bos H, Henning RH, De Boer E et al. Addition of AT1 blocker fails to overcome resistance to ACE inhibition in adriamycin nephrosis. Kidney Int 2002;61:473-80
22. Laverman GD, de Zeeuw D, Navis G. Between-patient differences in the renal response to renin-angiotensin system intervention: clue to optimising renoprotective therapy? J Renin Angiotensin Aldosterone Syst 2002;3:205-13
23. Heeg JE, de Jong PE, Vriesendorp R, de Zeeuw D. Additive antiproteinuric effect of the NSAID indomethacin and the ACE inhibitor lisinopril. Am J Nephrol 1990;10(Suppl 1):S94-7
24. Garini G, Mazzi A, Buzio C et al. Renal effects of captopril, indomethacin and nifedipine in nephrotic patients after an oral protein load. Nephrol Dial Transplant 1996;11:628-34
25. Vogt L, Navis G, de Zeeuw D. Renoprotection: a matter of blood pressure reduction or agent- characteristics? J Am Soc Nephrol 2002;13(Suppl 3):S202-7
26. Costanzi S, Sturniolo A, Fulignati P et al. Cyclooxygenase (COX)-2 selective inhibitors + ACE inhibitors: a new protocol in the treatment of heavy proteinuria [Abstract]. Nephrol Dial Transplant 2003;18:79A
27. Fried LF, Orchard TJ, Kasiske BL. Effect of lipid reduction on the progression of renal disease: a meta- analysis. Kidney Int 2001;59:260-9
28. Lee TM, Su SF, Tsai CH. Effect of pravastatin on proteinuria in patients with well-controlled hypertension. Hypertension 2002;40:67-73
29. Bianchi S, Bigazzi R, Caiazza A, Campese VM. A controlled, prospective study of the effects of atorvastatin on proteinuria and progression of kidney disease. Am J Kidney Dis 2003;41:565-70
ACKNOWLEDGEMENTS This study was supported by a grant from Sanofi-Synthélabo. We are greatly indebted to our colleague, Dr. G.D. Laverman, for support in collecting clinical observations.
91

CHAPTER 6
92

PART III
OPTIMIZING RENOPROTECTION: INTERVENTION IN NEW PATHOPHYSIOLOGICAL PATHWAYS

PART III
94

Chapter 7
Selective cyclooxygenase-2 (COX-2) inhibition reduces proteinuria in renal patients Liffert Vogt, Arend Jan J. Woittiez, Dick de Zeeuw and Gerjan Navis

CHAPTER 7
96

COX-2 INHIBITION REDUCES PROTEINURIA
Background—The antiproteinuric effect of antihypertensive agents interfering in the renin-angiotensin-aldosterone system (RAAS) affords renoprotection in chronic renal failure. Non-steroidal anti-inflammatory drugs (NSAID) interfering non-selectively in the prostaglandin system have strong antiproteinuric potency without reducing blood pressure. The effect of selective COX-2 inhibitors in proteinuric patients is unknown. Therefore, short-term effects of the selective COX-2 inhibitor rofecoxib on proteinuria and blood pressure as compared to NSAID and RAAS blockade were studied. Materials and methods—Sixteen stable patients (mean proteinuria 4.4 g/d; MAP 103 mmHg)) were included after a wash-out period. Hydrochlorothiazide 12.5 mg QD was given throughout. Additional blood pressure control was ensured by non-RAAS blocking antihypertensive agents. Patients received rofecoxib 25 mg QD, 50 mg QD and indomethacin 75 mg BID in randomised order for 4 weeks. Thereafter, a subset of the included patients (n = 11) received lisinopril 40 mg QD for 6 weeks preceded by a wash-out period. Results—Rofecoxib exerted a dose-dependent antiproteinuric effect. As compared to rofecoxib 25 and 50 mg, indomethacin was more effective (-18, -28% vs -49% (n = 16; p < 0.05)). As compared to rofecoxib 50 mg, lisinopril was more effective (-21 vs -51 (n = 11; p < 0.05)). No significant blood pressure changes were observed after rofecoxib and indomethacin, whereas lisinopril had a significant antihypertensive effect. Conclusions—Selective COX-2 inhibition reduces proteinuria without blood pressure reduction, pointing towards a specific renoprotective effect, and may serve as a novel non-hypotensive adjunct antiproteinuric treatment.
eduction of blood pressure and proteinuria are the cornerstones of long-term renoprotection (1). After onset of treatment the amount of residual proteinuria is the main predictor of subsequent renal function loss (2,3). Reduction of blood pressure is generally associated with reduction in proteinuria. As RAAS-blockade exerts an antiproteinuric effect on top of the reduction of blood pressure, RAAS-blockade is the therapy of choice for antiproteinuric intervention. To stop progressive renal function loss the target for proteinuria is < 1 g/d (4). However, this target is not always reached as adverse effects, such as hypotension, limit the use of maximal blockade of the RAAS for titration of proteinuria towards the target (5). Moreover, very low systolic blood pressure values (< 110 mmHg) may negatively influence long-term renal outcome (6). Therefore, exploration of non-hypotensive strategies that lower proteinuria may be fruitful to expand the therapeutic arsenal.
R
Before the era of RAAS blockade non-steroidal anti-inflammatory drugs (NSAID) have demonstrated to be highly effective to reduce proteinuria, in particular when volume excess was corrected by sodium restriction and diuretics (7). Moreover,
97

CHAPTER 7
antiproteinuric treatment with the NSAID indomethacin may retard progression of renal function loss, as demonstrated in two independent retrospective studies (8,9). Need for high doses and the related high frequency of side effects hampered further development of this drug class for renoprotection (10). In fact, NSAIDs are mostly known for their adverse effects on the kidney (11). Traditional NSAIDs exert their effects by blocking the production of prostaglandins, which are modulators of vascular tone, glomerular filtration, salt and water homeostasis and renin-secretion in the kidney (12,13,14). In particular, the degree of prostaglandin E2 (PGE2) inhibition was associated with the antiproteinuric effect (15,16). PGE2 is mainly derived from cyclooxygenase-2 (COX-2) that is up regulated and newly expressed in renal tissue in response to renal disease (14), indicating that the degree of COX-2 inhibition may account for the renoprotective effect in proteinuric patients. The analgesic effects of NSAIDs, that inhibit the two isoforms of COX, COX-1 and COX-2, are commonly attributed to inhibitory effects on COX-2, whereas their COX-1 inhibiting effects are associated with adverse effects on the gastrointestinal and central nervous system (13).
Selective cyclooxygenase-2 (COX-2) inhibitors comprise a class of drug with reduced gastrointestinal complications. In non-diabetic and diabetic experimental renal disease, selective COX-2 inhibition reduced proteinuria and retarded the progression of glomerular injury (14). In humans, however, little is known about the effects of selective COX-2 inhibition in chronic renal disease. The primary purpose of the present open-label study was to investigate the effect of the COX-2 inhibitor rofecoxib in two different doses on proteinuria, blood pressure and renal function in proteinuric patients, including a comparison with the traditional NSAID indomethacin. The secondary purpose of this study was to compare the effects of selective COX-2 inhibition with RAAS blockade, i.e. the ACE inhibitor lisinopril at maximal recommended dose. PATIENTS AND METHODS
Patients and protocol The study was approved by the local medical ethics committee and all participants provided written informed consent. Patients with stable proteinuric nephropathy of glomerular origin or overtly diabetic nephropathy were selected from our renal outpatient department. Eligibility for participation in the study was considered after a run-in period (at least 6 weeks). In this period, patients refrained from RAAS blocking agents. Hydrochlorothiazide 12.5 mg QD was started and patients were instructed to adhere to a restricted sodium intake (< 100 mmol/d) and standardized protein intake (1 g/kg bodyweight/d). Patients had to fulfill the following inclusion criteria after run-in: proteinuria ≥ 2 g/d, diastolic blood pressure < 90 mmHg, creatinine clearance ≥ 30 mL/min, and age between 18 and 70 years. During the run-in period, addition of
98

COX-2 INHIBITION REDUCES PROTEINURIA
amlodipine (maximal daily dose 10 mg) or doxasozine (maximal dose 8 mg/d) was allowed for blood pressure control. These drugs were kept stable during the rest of the study. Patients with proteinuria due to a non-primary renal disorder other than diabetic nephropathy, as well as patients with systemic diseases and recent cardiovascular events (< 6 months) were excluded. None of the participants received any immunosuppressive treatment previous (< 6 months) or during the study.
The study was performed before rofecoxib (VIOXX®, Merck & Co., Inc., Whitehouse Station, N.J., USA) was withdrawn from the market in response to the preliminary results of the APPROVe [Adenomatous Polyp Prevention on Vioxx] trial (17). Patients were treated according to a prospective open-label study protocol, consisting of two parts. In the first part (protocol A), patients were treated with rofecoxib 25 mg QD, rofecoxib 50 mg QD, and indomethacin 75 mg BID (retard formula; Indocid® Merck & Co., Inc., Whitehouse Station, N.J., USA) in random order, each preceded by a baseline period without the study medication. Indomethacin, at the dose of 75 mg BID was chosen as comparator since this dose had showed maximal antiproteinuric efficacy in previous studies as compared to other maximally dosed traditional NSAIDs (15). Each period lasted 4 weeks. In the second part (protocol B), for direct comparison with the current treatment standards with proven renoprotective action, i.e. RAAS blockade, patients were also treated for 6 weeks with lisinopril at maximal recommended dose (40 mg QD) preceded by a 6-week wash-out period directly after protocol A. The treatment periods were 6 weeks, as previous studies have shown that within this period the maximal established antiproteinuric effect of RAAS blockade can be expected (18). A pre-planned treatment period with the combination of rofecoxib on top of lisinopril could not be performed due to the withdrawal of rofecoxib. Measurements At the end of the run-in period and at the end of each study period, patients visited the hospital after an overnight fast. Blood was sampled, 24-h urine was collected, and blood pressure was measured by an automatic device (Dinamap®, GE Healthcare, Waukesha, Wisconsin, USA). Mean arterial blood pressure (MAP) was calculated as: 2/3 * diastolic blood pressure + 1/3 * systolic blood pressure. Mean value of four readings after 15 min was used for analysis. Urinary protein was determined with the pyrogallol red-molybdate method. Serum creatinine, albumin and lipids were determined using an automated multi-analyzer (MEGA®, Merck, Darmstadt, Germany). Statistical analysis Results are expressed as mean and standard error (SE). Baseline data were obtained after the first run-in. Student t-test was used for comparisons at baseline. Drug effects in all
99

CHAPTER 7
periods were evaluated by one-way ANOVA. In case of significance, post-hoc Duncan correction was used for multiple comparisons. Correlation coefficients of treatment effects within patients were calculated using Pearson correlation. A p-value < 0.05 was considered significant. RESULTS
Patient characteristics Sixteen patients (11 males and 5 females) with mean (range) age of 55 (39-70) years were included. Of these patients, 9 patients suffered from non-diabetic nephropathy (including membranous glomerulopathy (3), primary focal segmental glomerular sclerosis (2), IgA nephropathy (2) and non-conclusive diagnosis (2)), and 7 from (type 2) diabetic nephropathy. After protocol A, a subset of patients comprising 11 of 16 patients were included for protocol B. Not all patients completed the protocol B due to Table 1. Baseline characteristics at end of run-in
Mean (SE)
Protocol A (n = 16)
NDN (n = 9)
DN (n = 7)
Protocol B (n = 11)a
Age (yrs) 55 (3) 59 (3)
52 (4)
55 (3)
Male sexe (%) 65% 50%
83% 78%
Diabetes mellitus (n) 7 4
Body weight (kg) 92 (4) 88 (6)
96 (6) 93 (5)
BMI (kg/m2) 31 (1) 28 (1)
34 (2) * 30 (1)
Systolic BP (mmHg) 146 (4) 138 (5)
156 (6) * 146 (6)
Diastolic BP (mmHg) 81 (2) 82 (3)
79 (3) 84 (2)
Proteinuria (g/d) 4.4 (1.0) 5.8 (1.5)
2.7 (1.0) 5.6 (1.3)
Serum creatinine (µmol/L) 125 (17) 129 (19)
120 (32) 148 (21)
Creatinine clearance (mL/min) 93 (11) 89 (14)
98 (20) 79 (11)
Serum albumin (g/L) 35 (1) 35 (1)
36 (1) 35 (1)
Serum glucose (mmol/L) 5.7 (0.3)
14.2 (2.0) *
Serum HbA1c (%) NA
8.1 (0.5)
Serum total cholesterol (mmol/L) 7.4 (0.5) 8.3 (0.7)
6.2 (0.3) * 7.9 (0.7)
Serum triglycerides (mmol/L) 3.8 (0.5) 4.1 (0.8)
3.3 (0.6) 4.3 (0.7)
NA, not analysed. NDN, non-diabetic nephropathy. DN, diabetic nephropathy. a subset of patients from protocol A. * p < 0.05 non-diabetic vs. diabetic nephropathy
100

COX-2 INHIBITION REDUCES PROTEINURIA
preliminary withdrawal of the study drug rofecoxib (4 patients) and one patient left the study due to personal circumstances. Baseline characteristics at the end of the run-in period are given in table 1. Protocol A: Effects of rofecoxib as compared to indomethacin In table 2 and figure 1, the results in the 16 proteinuric patients are summarized. Mean (SE) off-treatment proteinuria was 4.8 (1.1) g/d. After treatment with rofecoxib 25 mg and 50 mg QD, mean proteinuria was significantly reduced by 18.6 (5.9)% and 27.7 (7.5)%, respectively (p < 0.05). The largest antiproteinuric response was observed after treatment with indomethacin 75 mg BID (48.9 (5.4)% (p < 0.05 vs. baseline; p < 0.05 vs. rofecoxib)). Both diabetic and non-diabetic proteinuric patients showed similar antiproteinuric responses after the different treatments, also after correction for 24h-urinary creatinine excretion.
MAP was 103 (2) mmHg. After rofecoxib 25 mg, MAP did not change significantly (103 (3) mmHg (0.4 (2.6)%). Dose-titration with rofecoxib to 50 mg led to a borderline significant rise of MAP to 108 (2) mmHg (6.2 (3.0)% (p = 0.076)). After indomethacin, MAP changed non-significantly to 104 (2) mmHg (0.9 (1.9)%). When analyzing diabetic and non-diabetic patients separately, a significant increase of MAP was only present in non-diabetic patients, but not in diabetic patients (figure 1). Body weight was significantly increased at the end of all active treatments. Separate analysis showed that changes in non-diabetic patients accounted for the significant differences in
Figure 1. Percentage change (mean (SE)) of proteinuria and blood pressure in 16 proteinuric patients from protocol A (table shows results of 9 non-diabetic and 7 diabetic patients separately analysed) after 4-week treatment with rofecoxib 25 mg QD, rofecoxib 50 mg QD and indomethacin 75 mg BID, respectively. * p < 0.05 vs. baseline; † p < 0.05 vs. rofecoxib 25 mg; ‡ p < 0.05 vs. rofecoxib 50 mg.
101

CHAPTER 7
Table 2. Results of protocol A summarized n = 16 Mean (SE)
Baseline 1 Rofecoxib
25 mg Baseline 2
Rofecoxib 50 mg
Baseline 3 Indomethacin
150 mg
Proteinuria (g/d) 4.8 (1.1) 3.8 (0.8)* 4.5 (1.1) 3.7 (0.8)* 5.2 (1.3) 2.8 (0.9)*†‡ Proteinuria/creatinine (mg/mg)
3.1 (0.6) 2.4 (0.5)* 2.9 (0.7) 2.3 (0.5)* 3.3 (0.8) 1.8 (0.5)*†‡
Systolic BP (mmHg) 147 (5) 147 (6) 146 (5) 155 (6) 143 (6) 150 (5) Diastolic BP (mmHg) 81 (2) 82 (3) 81 (2) 84 (3) 80 (2) 81 (2) Body weight (kg) 91 (5) 93 (5)*# 91 (5) 92 (5)# 90 (5) 92 (5)# Creatinine clearance (mL/min) 87 (12) 84 (12) 88 (13) 81 (15) 85 (13) 73 (11) * Serum creatinine (µmol/L) 133 (17) 142 (19) 134 (19) 157 (21)* 144 (25) 155 (22)* Urinary sodium (mmol/d) 160 (21) 147 (20) 160 (21) 163 (17) 173 (18) 129 (15)# Proteinuria/creatinine clearance (g/mL/min)
8.0 (2.4) 6.3 (1.7)# 7.9 (2.6) 7.0 (2.6)# 9.6 (3.9) 5.6 (2.0)*#
BP, blood pressure. *p < 0.05 vs. baseline 1; † p < 0.05 vs. rofecoxib 25 mg; ‡ p <0.05 vs. rofecoxib 50 mg; # p < 0.05 vs. baseline 3.
body weight, since body weight did not alter significantly in diabetic patients (data not shown).
Renal function expressed as mean creatinine clearance was non-significantly decreased after rofecoxib 25 mg and 50 mg. After indomethacin, creatinine clearance decreased significantly (p < 0.05). Mean serum creatinine was not significantly increased after rofecoxib 25 mg. After rofecoxib 50 mg and indomethacin serum creatinine was significantly increased (p < 0.05). To study the antiproteinuric efficacy related to changes in creatinine clearance during all treatments, the proteinuria-to-creatinine clearance ratio (i.e. fractional protein excretion) was calculated. After rofecoxib 25 mg and 50 mg, the ratio was significantly lowered as compared to baseline. The ratio was lowest after indomethacin treatment (p < 0.05 vs. baseline; p < 0.05 vs. rofecoxib).
Protocol B: Effects of rofecoxib as compared to lisinopril In table 3 and figure 2, the effects of treatment with rofecoxib 50 mg QD, indomethacin 75 mg BID and lisinopril 40 mg QD are summarized for the 11 patients that entered protocol B. The antiproteinuric response after lisinopril was greater than after rofecoxib (51.4 (9.6) vs. 21.3 (7.4)% reduction (p < 0.05)), but not different from indomethacin (44.6 (6.5)% reduction). Lisinopril led to a significant reduction in systolic and diastolic blood pressure (p < 0.05), whereas no significant effect on systolic or diastolic blood pressure was observed after rofecoxib or indomethacin. After lisinopril and indomethacin treatment, serum creatinine increased significantly, but not after rofecoxib. Moreover, creatinine clearance showed a non-significant trend to decrease in response to lisinopril and indomethacin. The fractional protein excretion measured as
102

COX-2 INHIBITION REDUCES PROTEINURIA
Table 3. Results of protocol B summarized
n = 11 Mean (SE) Baseline Rofecoxib
50 mg Indomethacin
150 mg Lisinopril
40 mg
Proteinuria (g/d) 5.8 (1.3) 4.1 (0.9)* 3.5 (1.2)* 2.8 (0.8)* Proteinuria/creatinine (mg/mg) 3.6 (0.8) 2.6 (0.5)* 2.2 (0.7)** 1.8 (0.5)* Systolic BP (mmHg) 147 (6) 151 (5) 148 (6) 128 (7)* Diastolic BP (mmHg) 82 (3) 84 (3) 81 (3) 70 (2)* Body weight (kg) 93 (5) 94 (5) 94 (5) 92 (5)‡ Creatinine clearance (mL/min) 78 (11) 77 (15) 64 (10) 64 (9) Serum creatinine (µmol/L) 151 (22) 160 (25) 177 (27)* 186 (32)*† Urinary sodium (mmol/d) 139 (15) 163 (12) 114 (19) 165 (21) Proteinuria/creatinine clearance (g/mL/min) 10.1 (3.2) 8.0 (3.3) 7.3 (2.7)* 5.7 (1.9)*
BP, blood pressure. *p < 0.05 vs. baseline; † p < 0.05 vs. rofecoxib 50 mg; ‡ p < 0.05 vs. indomethacin 150 mg
the proteinuria-to-creatinine clearance ratio was lowest after lisinopril treatment and differed significantly from rofecoxib (p < 0.05). Individual responses To investigate whether individual patients with a poor antiproteinuric responsiveness to ACE inhibitor therapy could benefit from a switch to rofecoxib, individual responses were analyzed, as illustrated in figure 3. It shows that the individual responses to lisinopril and rofecoxib were positively correlated (R = 0.61 and R = 0.67, for 25 and 50 mg rofecoxib, respectively (p < 0.05). Thus, the patients with a favorable response to rofecoxib were also the ones in whom lisinopril was effective. Side effects Besides the effects on renal function, as described above, 4 patients reported adverse events. One diabetic and one non-diabetic patient complained of both dizziness and somnolence during indomethacin treatment. One diabetic and one non-diabetic patient newly developed pitting edema at the lower extremities during rofecoxib 50 mg treatment. DISCUSSION
This study demonstrates that, in patients with proteinuric renal disease from diabetic or non-diabetic origin, proteinuria was dose-dependently reduced by rofecoxib treatment. This antiproteinuric effect was obtained without reduction of blood pressure. Treatment with the traditional NSAID indomethacin showed better antiproteinuric efficacy as compared to selective COX-2 inhibition with rofecoxib. Moreover, the ACE inhibitor lisinopril at maximal recommended dose was also more effective as compared to rofecoxib treatment. On the individual level, the antiproteinuric responses after
103
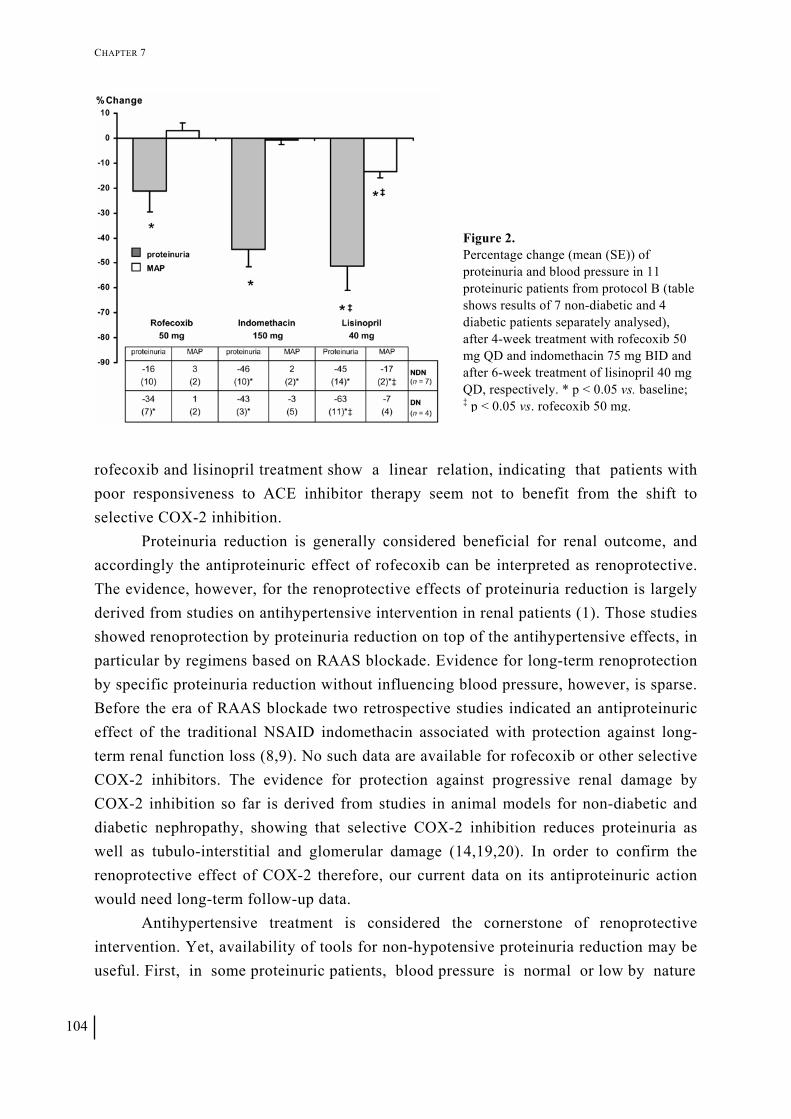
CHAPTER 7
Figure 2. Percentage change (mean (SE)) of proteinuria and blood pressure in 11 proteinuric patients from protocol B (table shows results of 7 non-diabetic and 4 diabetic patients separately analysed), after 4-week treatment with rofecoxib 50 mg QD and indomethacin 75 mg BID and after 6-week treatment of lisinopril 40 mg QD, respectively. * p < 0.05 vs. baseline; ‡ p < 0.05 vs. rofecoxib 50 mg.
rofecoxib and lisinopril treatment show a linear relation, indicating that patients with poor responsiveness to ACE inhibitor therapy seem not to benefit from the shift to selective COX-2 inhibition. Proteinuria reduction is generally considered beneficial for renal outcome, and accordingly the antiproteinuric effect of rofecoxib can be interpreted as renoprotective. The evidence, however, for the renoprotective effects of proteinuria reduction is largely derived from studies on antihypertensive intervention in renal patients (1). Those studies showed renoprotection by proteinuria reduction on top of the antihypertensive effects, in particular by regimens based on RAAS blockade. Evidence for long-term renoprotection by specific proteinuria reduction without influencing blood pressure, however, is sparse. Before the era of RAAS blockade two retrospective studies indicated an antiproteinuric effect of the traditional NSAID indomethacin associated with protection against long-term renal function loss (8,9). No such data are available for rofecoxib or other selective COX-2 inhibitors. The evidence for protection against progressive renal damage by COX-2 inhibition so far is derived from studies in animal models for non-diabetic and diabetic nephropathy, showing that selective COX-2 inhibition reduces proteinuria as well as tubulo-interstitial and glomerular damage (14,19,20). In order to confirm the renoprotective effect of COX-2 therefore, our current data on its antiproteinuric action would need long-term follow-up data.
Antihypertensive treatment is considered the cornerstone of renoprotective intervention. Yet, availability of tools for non-hypotensive proteinuria reduction may be useful. First, in some proteinuric patients, blood pressure is normal or low by nature
104

COX-2 INHIBITION REDUCES PROTEINURIA
Figure 3. Individual responses (%) to rofecoxib 25 mg QD plotted against lisinopril 40 mg QD in 11 proteinuric patients from protocol B (comprising 7 non-diabetic (closed circles) and 4 diabetic patients (open circles).
(21). Second, treatment strategies that lead to more complete blockade of the RAAS are associated with a high incidence of adverse events, such as hypotension (5). Third, a too low blood pressure may adversely affect renal outcome on the long term, as a recent meta-analysis showed a J-curved relation between systolic blood pressure and long-term renoprotection during ACE inhibition (6). Thus, aggressive blood pressure titration for proteinuria reduction apparently has its limits, indicating that non-hypotensive proteinuria reduction could be desirable under certain conditions.
We found a correlation between the individual responses to lisinopril and rofecoxib, indicating that good responding patients remain good-responders after shifting to another class of antiproteinuric drug, whereas poor-responders remain poor-responders. This phenomenon has been observed previously and points towards the presence of therapy resistance in some individual proteinuric patients that seems to be determined by intrinsic patient factors (22). Apparently, a therapy change into COX2 inhibition is not a feasible strategy to overcome resistance to RAAS-blockade based therapy. It would be of interest to know whether the combination of COX-2 inhibition and lisinopril would be effective to this purpose, as the added antiproteinuric efficacy of the combination of traditional NSAID and ACE inhibition as well as limited data on the combination of selective COX-2 inhibition and ACE inhibition in membranous glomerulopathy suggest better antiproteinuric potential (23,24,25). Due to preliminary withdrawal of rofecoxib from the market we could unfortunately not address this issue.
Rofecoxib (at least at a dose of 50 mg) leads to a reversible rise in serum creatinine, similar to that after indomethacin. Older studies attributed (part of) the antiproteinuric effect of indomethacin to the decrease in GFR (7,12). However, we found a reduction in fractional proteinuria, with both indomethacin and rofecoxib, indicating a specific antiproteinuric effect as well. A reversible decline in renal function
105

CHAPTER 7
at onset of treatment has been assumed to indicate drop in glomerular pressure that predicts a more favourable outcome in the long term (26), but it should be mentioned that the (non) selective interference in the prostaglandin system is associated with impairment of autoregulation of glomerular filtration that can facilitate acute renal failure under conditions of acute volume depletion. The long-term renal consequences and safety of COX-2 inhibition obviously would need further study.
COX-2 derived prostaglandins (as PGE2) are important in modulating sodium excretion in the medulla (13,27). As suggested by the increase in body weight, blockade of COX-2 seems to be associated with sodium and volume retention, leading also to increments of blood pressure and development of oedema—as experienced in two patients in our study after rofecoxib therapy (28,29). Intriguingly, we found that these effects may be different between diabetic and non-diabetic patients. In particular, non-diabetic patients seem to be more vulnerable to increases in blood pressure due to sodium retention than diabetic patients. A possible explanation could be a different interaction between the PG pathway and the RAAS during COX-2 inhibition in hyperglycaemic patients. COX-2 derived PGE2 showed to decrease and renal haemodynamic function to restore to normal after induction of normoglycaemia, indicating that the PG system may play a more prominent role in renal functional alteration during hyperglycaemia (30).
In non-renal patients having a history of colorectal adenoma, the APPROVe trial found a higher incidence of cardiovascular events in patients assigned to rofecoxib after chronic use of already 18 months, leading to premature closure of the trial and withdrawal of rofecoxib from the market (17). Whether the observation of elevated cardiovascular risk during rofecoxib treatment is an agent-specific effect or a class-dependent effect, including selective COX-2 inhibitors as well as traditional NSAIDs, is currently subject to debate. The associated elevated risk may not be present in the proteinuric patient, as rofecoxib and indomethacin have demonstrated to reduce proteinuria in our study. Proteinuria as such has been regarded as an important cardiovascular risk factor (31,32), whereas proteinuria reduction can be regarded as beneficial against progressive renal functional decline and consequent cardiovascular risk. It should be noted that rofecoxib may raise blood pressure, influencing the cardiovascular risk it is associated with.
In the interpretation of our results, we acknowledge several limitations. First, this study was a proof-of-concept study in which only the short-term effects on proteinuria and renal function were studied. Second, since NSAIDs and ACE inhibitors have shown that a better antiproteinuric effect was established under conditions of correction of the volume excess, patients were instructed to adhere to a sodium restricted diet (7,10). Most patients had, however, a higher salt intake than desirable which may have
106

COX-2 INHIBITION REDUCES PROTEINURIA
hampered the exploration of the antiproteinuric potential of rofecoxib, despite the standard background therapy consisting of hydrochlorothiazide in all patients. Third, due to preliminary withdrawal of rofecoxib, the pre-planned combination of RAAS blockade on top of rofecoxib on proteinuria could not be studied.
We conclude that pharmacological intervention in the prostaglandin system by selective COX-2 inhibition is effective in non-hypotensive reduction of proteinuria. Application of traditional NSAIDs for antiproteinuric treatment is limited due to high incidence of side effects and gastrointestinal complications, whereas further exploration selective COX-2 inhibition by rofecoxib is hampered by safety concerns based on an elevated cardiac risk in non-renal populations. Study of still available selective COX-2 inhibitors, that share the profile of fewer side effects, may be helpful to expand the therapeutic arsenal for proteinuria reduction. REFERENCES 1. Vogt L, Navis G, de Zeeuw D. Renoprotection: a matter of blood pressure reduction or agent-characteristics? J Am Soc Nephrol.
2002;13:S202-7 2. de Zeeuw D, Remuzzi G, Parving H-H et al. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy:
lessons from RENAAL. Kidney Int 2004;65:2309-20 3. Ruggenenti P, Perna A, Remuzzi G. Retarding progression of chronic renal disease: the neglected issue of residual proteinuria.
Kidney Int 2003;63:2254-61 4. Ruggenenti P, Schieppati A, Remuzzi G. Progression, remission, regression of chronic renal disease. Lancet 2001;357:1601-8 5. Vogt L, Navis G, de Zeeuw D. Individual titration for maximal blockade of the renin-angiotensin system in proteinuric patients: a
feasible strategy? J Am Soc Nephrol 2005;16:S53-7 6. Jafar TH, Stark PC, Schmid CH et al. Progression of chronic kidney disease: the role of blood pressure control, proteinuria, and
angiotensin-converting enzyme inhibition: a patient-level meta-analysis. Ann Intern Med 2003;139:244-52 7. Donker AJM, Brentjens JR, van der Hem GK et al. Treatment of the nephrotic syndrome with indomethacin. Nephron 1978;22:374-
81 8. Vriesendorp R, Donker AJM, de Zeeuw D et al. Effects of nonsteroidal anti-inflammatory drugs on proteinuria. Am J Med.
1986;81:84-94 9. Lagrue G, Laurent J, Belghiti D. Renal survival in membranoproliferative glomerulonephritis (MPGN): role of long-term treatment
with non-steroid anti-inflammatory drugs (NSAID). Int Urol Nephrol 1988;20:669-77 10. Heeg JE, de Jong PE, van der Hem GK et al. Efficacy and variability of the antiproteinuric effect of ACE inhibition by lisinopril.
Kidney Int 1989;36:272-9 11. Adams DH, Howie AJ, Michael J et al. Non-steroidal anti-inflammatory drugs and renal failure. Lancet 1986;1:57-60 12. Arisz L, Donker AJM, Brentjens JR et al. The effect of indomethacin on proteinuria and kidney function in the nephrotic syndrome.
Acta Med Scand 1976;199:121-5 13. Feldman M, McMahon AT. Do cyclooxygenase-2 inhibitors provide benefits similar to those of traditional nonsteroidal anti-
inflammatory drugs, with less gastrointestinal toxicity? Ann Intern Med 2000;132:134-43 14. Cheng HF, Harris RC. Cyclooxygenases, the kidney, and hypertension. Hypertension 2004;43(3):525-30 15. Vriesendorp R, de Zeeuw D, de Jong PE et al. Reduction of urinary protein and prostaglandin E2 excretion in the nephrotic
syndrome by non-steroidal anti-inflammatory drugs. Clin Nephrol 1986;25:105-10 16. Alavi N, Lianos EA, Venuto RC et al. Reduction of proteinuria by indomethacin in patients with nephrotic syndrome. Am J Kidney
Dis 1986;8:397-403 17. Bresalier RS, Sandler RS, Quan H et al; Adenomatous Polyp Prevention on Vioxx (APPROVe) Trial Investigators. Cardiovascular
events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352:1092-102 18. Gansevoort RT, de Zeeuw D, de Jong PE. Is the antiproteinuric effect of ACE inhibition mediated by interference in the renin
angiotensin system. Kidney Int 1994;45:861-7 19. Wang JL, Cheng HF, Shappell S et al. A selective cyclooxygenase-2 inhibitor decreases proteinuria and retards progressive renal
injury in rats. Kidney Int 2000;57:2334-42 20. Cheng HF, Wang CJ, Moeckel GW et al. Cyclooxygenase-2 inhibitor blocks expression of mediators of renal injury in a model of
diabetes and hypertension. Kidney Int 2002;62:929-39. 21. Maschio G, Cagnoli L, Claroni F et al. ACE inhibition reduces proteinuria in normotensive patients with IgA nephropathy: a
multicentre, randomized, placebo-controlled study. Nephrol Dial Transplant 1994;9:265-9 22. Bos H, Andersen S, Rossing P et al. The role of patient factors in therapy resistance to antiproteinuric intervention in non-diabetic
and diabetic nephropathy. Kidney Int 2000;75:S32-7. 23. Heeg JE, de Jong PE, Vriesendorp R et al. Additive antiproteinuric effect of the NSAID indomethacin and the ACE inhibitor
lisinopril. Am J Nephrol 1990;10:S94-7
107

CHAPTER 7
24. Perico N, Remuzzi A, Sangalli F et al. The antiproteinuric effect of angiotensin antagonism in human IgA nephropathy is potentiated by indomethacin. J Am Soc Nephrol 1998;9:2308-17
25. Costanzi S, Sturniolo A, Fulignati P et al. Cyclooxygenase (COX)-2 selective inhibitors + ACE inhibitors: a new protocol in the treatment of heavy proteinuria. Nephrol Dial Transplant 2003;18:79A
26. Apperloo AJ, de Zeeuw D, de Jong PE. Short-term antiproteinuric response to antihypertensive treatment predicts long-term GFR decline in patients with non-diabetic renal disease. Kidney Int 1994;45:S174-8
27. Rossat J, Maillard M, Nussberger J et al. Renal effects of selective cyclooxygenase-2 inhibition in normotensive salt-depleted subjects. Clin Pharmacol Ther 1999;66:76-84
28. Sowers JR, White WB, Pitt B et al. The Effects of cyclooxygenase-2 inhibitors and nonsteroidal anti-inflammatory therapy on 24-hour blood pressure in patients with hypertension, osteoarthritis, and type 2 diabetes mellitus. Arch Intern Med 2005;165:161-8
29. Solomon DH, Schneeweiss S, Levin R et al. Relationship between COX-2 specific inhibitors and hypertension. Hypertension 2004;44:140-5
30. Esmatjes E, Levy I, Gaya J et al. Renal excretion of prostaglandin E2 and plasma renin activity in type I diabetes mellitus: relationship to normoglycemia achieved with artificial pancreas. Diabetes Care.1987;10:428-31
31. Ordonez JD, Hiatt RA, Killebrew EJ et al. The increased risk of coronary heart disease associated with nephrotic syndrome. Kidney Int 1993;44:638-42
32. Hillege HL, Fidler V, Diercks GF et al. Urinary albumin excretion predicts cardiovascular and noncardiovascular mortality in general population. Circulation 2002;106:1777-82
ACKNOWLEDGEMENTS Part of this study was supported by Merck & Company (grant MSGP 9661). The study was conducted in the Twenteborg hospital, Almelo, The Netherlands. We thank Ms Marja van de Klok for her assistance at the outpatient clinic and Mr J. Sanderman for his skillful help in the clinical-chemical laboratory of the Twenteborg Hospital.
108

Chapter 8
Effect of the urotensin receptor antagonist palosuran on albuminuria in hypertensive patients with type 2 diabetic nephropathy: results from the PROLONG proof-of-concept study Liffert Vogt, Carlos Chiurchiu, Harbajan Chadha-Boreham, Parisa Danaietash, Samy Hadjadj, Henry Krum, Gerjan Navis, Eric Neuhart, Aneliya I. Parvanova, Piero Ruggenenti, Arend Jan Woittiez, Reuven Zimlichman, Giuseppe Remuzzi and Dick de Zeeuw. For the PROLONG Study Group

CHAPTER 8
110

EFFECTS OF UROTENSIN RECEPTOR ANTAGONIST ON ALBUMINURIA
Background—The urotensin system has been hypothesized to play an important role in the pathophysiology of diabetic nephropathy. PROLONG, a multicenter, randomized, double-blind, placebo-controlled, 2-period, crossover study, assessed the effects of the urotensin receptor antagonist palosuran on urinary albumin excretion (UAE) and blood pressure in hypertensive patients with type 2 diabetic nephropathy treated either with an angiotensin II receptor type 1 (AT1) antagonist or an angiotensin-converting enzyme (ACE) inhibitor. Methods—Patients with macroalbuminuria (24-h UAE > 0.5 and < 3.0 g) and systemic hypertension (supine systolic blood pressure > 135 and < 170 mmHg and/or diastolic > 85 and < 110 mmHg) received both palosuran 125 mg twice daily and placebo for 4 weeks. Results—Out of the 62 enrolled patients, 54 were included in the per-protocol population: male/female: 43/11, mean age: 61.6 years, (geometric) mean UAE: 1016 mg/24h, mean systolic/diastolic blood pressure: 155/ 84 mmHg, mean creatinine clearance: 84 mL/min. Palosuran did not affect UAE: the palosuran ratio (UAE at end of treatment over UAE at baseline) over placebo ratio (geometric mean) was 0.99 (95% CI: 0.85, 1.14). Similarly, palosuran did not affect placebo-subtracted systolic or diastolic casual blood pressure: -1.9 mmHg (16.5) and -0.2 mmHg (9.5), respectively. Renal function remained stable throughout the study. Conclusion—In hypertensive patients with type-2 diabetic nephropathy, treated either with an AT1 antagonist or an ACE inhibitor, combined treatment with the urotensin receptor antagonist palosuran did not affect albuminuria or blood pressure. These results suggest that inhibition of the urotensin system may not represent a new treatment strategy in this high-risk patient population.
ype 2 diabetic patients with overt nephropathy and systemic hypertension are characterized by elevated cardiovascular and renal risk. The widely accepted standard treatment strategy in these patients includes at least one inhibitor of the renin-angiotensin-aldosterone system (RAAS), an angiotensin-converting enzyme (ACE) inhibitor or an angiotensin II type 1 receptor (AT1) antagonist, as both classes of drugs have been proven to reduce cardiac and renal morbidity and mortality. In patients treated with a RAAS inhibitor, the risk reduction in morbidity and mortality is correlated with the extent of blood pressure and albuminuria decrease (1,2). However, the residual cardiovascular and renal risk in patients treated with an ACE inhibitor (3,4) or an AT1 antagonist (5,6) remains high. Despite attempts to improve RAAS blockade by combining ACE inhibitor and AT1 antagonist treatment, patients still progress to end-stage renal disease (7). Therefore, still an unmet medical need for new treatments to be combined with a RAAS inhibitor in this high-risk patient population is present.
T
The recently discovered urotensin system has been hypothesized to play an
111

CHAPTER 8
important role in the pathophysiology of cardiac and renal diseases (8). In the kidney, human urotensin II and the urotensin receptor are mostly expressed in glomeruli and capillary endothelial cells (9,10). Urotensin II, initially described as the most potent vasoconstrictor known in mammals (8,11), exerts it effects by modulating vascular tone (12-14), but these effects vary according to vascular bed. In the kidney, one in vivo rat study reported that infusion of urotensin II reduced renal plasma flow (RPF) and GFR (15,16), while in another study intra-renal infusion of urotensin II led to an increase in RPF (17). In patients with diabetic nephropathy, both urotensin II and urotensin receptor gene expression were up-regulated in kidney tissue (18), and renal dysfunction was associated with elevated plasma urotensin II levels (9,19,20).
Palosuran (ACT-058362) is an oral, selective, competitive, non-peptidic antagonist of the human urotensin receptor. Palosuran was studied both in animal and humans to assess the role of the urotensin system in the pathogenesis of diabetic nephropathy. In different rat models of renal failure, palosuran has been shown to prevent the development of renal failure by improving RPF and renal vascular resistance, GFR, and serum creatinine, and by reducing glomerular and tubulo-interstitial lesions, without affecting systemic blood pressure (21,22). In a diabetic rat model, palosuran delayed development of albuminuria and improved survival (22).
In humans, data from phase I studies in healthy adult males showed that palosuran was well tolerated and had no effects on systemic blood pressure, heart rate, or plasma urotensin II levels (23,24). Apart from an open-label pilot study in patients with type 2 diabetic nephropathy suggesting that palosuran might reduce albuminuria (25), the effects of palosuran in patients were largely unknown. Based on these preliminary preclinical and clinical data, it was hypothesized that the urotensin system might be a mediator in the pathogenesis of diabetic nephropathy, and that its inhibition would have a protective effect on the kidney. Therefore, PROLONG, a multicenter, randomized, double-blind, placebo-controlled, 2-period crossover, proof-of-concept study was designed to assess whether palosuran would reduce urinary albumin excretion (UAE) and/or systemic blood pressure in type hypertensive patients with type 2 diabetic nephropathy on stable treatment with either an ACE inhibitor or AT1 antagonist. METHODS
Patients Patients of both sex, between 30 and 75 years of age, with type 2 diabetes (with or without insulin treatment) and Hb A1c < 10%, systemic hypertension (supine systolic blood pressure > 135 and < 170 mmHg and/or diastolic > 85 and < 110 mmHg), macroalbuminuria (24-h UAE > 0.5 and < 3.0 g), and a measured creatinine clearance ≥ 30 ml/min/1.73 m2, were randomized to this 2-period crossover study.
112

EFFECTS OF UROTENSIN RECEPTOR ANTAGONIST ON ALBUMINURIA
Patients had to be on stable treatment with either an ACE inhibitor or AT1 antagonist for at least 3 months. Any treatment with diuretics, calcium channel blockers, beta-blockers, statins and non-steroidal antiinflammatory drugs, had to be stable for at least one month before screening. Finally, patients should have had no history of clinically relevant worsening of renal function in the last 6 months. Patients could not be included if they were women of childbearing potential, were treated with a combination of an AT1 antagonist and an ACE inhibitor, had clinically relevant signs of: nephrotic syndrome; significant renal artery stenosis; moderate to severe hepatic dysfunction; serum albumin < 25 g/L; serum potassium ≥ 5.5 mmol/L; or a urinary tract infection within one month before screening. Ethics committees approved the study according to national regulations. All patients gave their written informed consent. Study design PROLONG was a randomized, multicenter, double-blind, placebo-controlled, 2-period crossover study. After screening, eligible patients entered a 4-week run-in period. Thereafter, patients were randomly treated during two treatment periods of 4 weeks with palosuran (ACT-058362) 125 mg and placebo twice daily separated by a 6-week washout period. Finally, patients entered a 4-week follow-up recovery period. At the end of each study period, patients were instructed not to take their trial medication in the morning of that day. A permuted block method was used for randomization. Determination of UAE, renal function and blood pressure To reduce the known high intra-individual variability of UAE, UAE was assessed as the mean albuminuria calculated from 3 consecutive 24-h urine collections measured locally using the same center-specific measurement method throughout the study. Creatinine clearance was calculated from serum creatinine and mean 24-h urinary creatinine excretion. Trough supine systemic blood pressure was measured every 3 min during 15 min by an automated oscillometric device. Blood pressure assessment was the mean of the three last measurements.
In addition, a monocenter ancillary study has been performed in the 22 patients included at the Mario Negri institute in Ranica, Italy. Renal plasma flow and glomerular filtration rate were measured by inulin and para-aminohippurate clearance methods, respectively. Blood pressure was measured by 24-h ambulatory blood pressure monitoring (ABPM).
Statistics analysis As PROLONG was designed as a proof-of-concept study, the main analysis was performed on the per-protocol population, which included all randomized patients who
113

CHAPTER 8
Table 1. Demographic and baseline characteristics at screening
Per protocol population n = 54
Male / Female (n) 43 (80%) / 11 (20%) Age (years) 61.6 (8.4) Body weight (kg) 91.8 (16.9) BMI (kg/m2) 31.7 (5.3) Systolic/diastolic blood pressure (mmHg) 155.2 (15.8) / 84.5 (8.6) HbA1c (%) 7.49 (1.45) Creatinine clearance (ml/min) 84.4 (36.4) UAE (mg/24hour) 1016 (58) Duration of type 2 diabetes (years) 16.2 (9.5) Duration of hypertension (years) 14.4 (10.7) Duration of macroalbuminuria (years) 4.0 (4.6) Use of ACE inhibitor (n) 36 (67%) Use of AT1 antagonist (n) 18 (33%)
* Data are expressed as arithmetic mean with standard deviation (SD), except UAE expressed as geometric mean with coefficient of variation (CV%). completed both treatment periods and had complete UAE data. Based on previous clinical experience, a 30% (log-transformed: 0.357) difference in the mean change from baseline in the 24-h UAE was considered clinically relevant. Standard deviation (SD) was estimated to be 1 on the logarithmic scale. A total of 50 patients were needed to demonstrate this hypothesis, using a two-sided paired t-test with type-1 error of 10% and a power of 80%. Data are expressed as arithmetic means with standard deviation, except for the UAE and UAE over creatinine ratio, which were log-transformed prior to analysis to take into account the asymmetric distribution of UAE. The UAE and UAE over creatinine ratio are expressed as geometric mean with coefficient of variation (CV%). RESULTS
A total of 62 patients was included in the PROLONG study conducted between February 2004 and April 2005 in 11 centers from Italy (n = 24), The Netherlands (n = 10), Israel (n = 12), Australia (n = 7), France (n = 7), and Switzerland (n = 2). Of the 62 patients, 54 completed the study according to the protocol, 28 were randomized first to placebo followed by palosuran, whereas 26 received first palosuran followed by placebo. The reasons for excluding 8 patients from the per-protocol analysis were: no valid or missing UAE data (n = 4), low compliance to the treatment (n = 2) and no treatment received during the second treatment period (n = 2). Demographic and baseline characteristics at screening are summarized in table 1.
114

EFFECTS OF UROTENSIN RECEPTOR ANTAGONIST ON ALBUMINURIA
Efficacy Results are summarized in table 2. No period effect (i.e., the disease remains stable), or treatment carry over effect were observed in this crossover study. The mean UAE remained stable during the total study duration (figure 1). The intra and inter-patient variability of UAE were high but not different to those observed in other studies (30). Compared to placebo, palosuran did not significantly affect UAE: end of treatment over baseline ratio of 1.0 (95% CI: 0.93, 1.15) and 1.02 (95% CI: 0.93, 1.12), respectively, with a palosuran ratio over placebo ratio (geometric mean of log-transformed data) of 0.99 (95% CI: 0.85, 1.14). The use of the urinary albumin over creatinine ratio to correct for potential urine collection bias did not modify the results. Similarly, palosuran did not affect placebo-subtracted systolic or diastolic blood pressure: -1.9 mmHg (16.5) and -0.2 mmHg (9.5), respectively, or heart rate. In the ancillary study, data from 19 patients out of 22 confirmed the absence of clinically relevant effect on blood pressure measured by 24-hour ambulatory blood pressure monitoring. Palosuran had no effect on serum glucose levels.
Renal function measured by serum creatinine and creatinine clearance, and urinary electrolytes (sodium and potassium) remained stable after palosuran or placebo treatment. In the ancillary study, per-protocol data from 20 patients out of 22, did not show any significant difference between palosuran and placebo on RPF and GFR, measured by inulin and para-aminohippurate clearance methods, respectively. With palosuran RPF decreased from 325.0 to 298.2 ml/min/1.73m2 and GFR decreased from Table 2. Results
Per protocol population n = 54 Placebo Palosuran
Baseline End Baseline End Systolic BP (mmHg) 154.2 (17.0) 155.0 (18.0) 153.1 (17.3) 152.0 (15.9) Diastolic BP (mmHg) 83.3 (10.4) 84.3 (10.4) 82.9 (10.0) 83.7 (10.4) UAE * (mg/24h) 1047 (76) 1084 (77) 945 (94) 964 (101) UAE/creatinine * (mg/mmol) 87 (78) 90 (85) 78 (98) 82 (101) Albumin fractional clearance 37 (31) 40 (37) 34 (28) 38 (33) Serum creatinine (umol/L) 122 (43) 120 (41) 116 (39) 124 (48) Serum urea (mmol/L) 9.8 (4.9) 9.6 (4.6) 9.5 (4.7) 9.8 (5.1) Serum potassium (mmol/L) 4.3 (0.5) 4.3 (0.5) 4.2 (0.5) 4.2 (0.5) Creatinine clearance (mL/min) 79 (30) 80 (33) 83 (34) 79 (35) Urinary urea (mmol/24h) 653 (339) 661 (360) 643 (339) 638 (311) Urinary sodium (mmol/24h) 190 (67) 181 (62) 189 (69) 194 (72)
IR-hUII (pg/mL) 1.5 (1.3) 1.4 (1.3) 1.3 (0.8) 1.2 (1.0)
BP, blood pressure. UAE, urinary albumin excretion. IR-hUII, immunoreactive human urotensin II. * Data are expressed as arithmetic mean with standard deviation (SD), except UAE and UAE over creatinine ratio expressed as geometric mean with coefficient of variation (CV%).
115

CHAPTER 8
Figure 1. Effects on urinary albumin excretion (per protocol population)
69.6 to 68.6 ml/min/1.73m2, while with placebo, RPF decreased from 326.5 to 304.6 ml/min/1.73m2 and GFR decreased from 66.9 to 62.2 ml/min/1.73m2.
Plasma urotensin II levels measured at the end of palosuran treatment did not show any identifiable pattern as compared to baseline.
Safety In the 62 patients who received the study drug, the number of patients with at least one adverse event was 14 (23%) and 17 (27%) when treated with placebo and palosuran, respectively. The most frequently reported adverse events were headache (4.8 vs. 1.6%), nausea (3.2 vs. 3.2%), and first-degree atrioventricular block (3.2 vs. 1.6%) during palosuran and placebo treatment, respectively. During the study, no adverse event led to study drug discontinuation and no patient died. Two serious adverse events not considered by the investigators to be related to study drug were reported: one basal cell carcinoma and one cerebrovascular accident. DISCUSSION
PROLONG was the first placebo-controlled study assessing the effect of the urotensin receptor antagonist palosuran in diabetic nephropathy. Although previous preclinical and clinical studies suggested that blockade of the urotensin system might be beneficial in hypertensive patients with type 2 diabetic nephropathy, palosuran did not show any effect on UAE or casual blood and 24-h ambulatory blood pressure, in patients treated with either an ACE inhibitor or AT1 antagonist. Renal function, measured as creatinine clearance and serum creatinine as well as RPF and GFR in the ancillary study, remained stable.
116

EFFECTS OF UROTENSIN RECEPTOR ANTAGONIST ON ALBUMINURIA
These neutral results do not support the hypothesis that an antagonist of the urotensin receptor such as palosuran would counteract the deleterious effects of urotensin II in the pathophysiology of diabetic nephropathy. However, these findings may provide additional insight into the putative role of the urotensin system in the cardio-renal physiology and pathophysiology, still subject to debate (12-14).
Similarly to angiotensin II, human urotensin II was thought to contribute to kidney damage, by successively triggering hyperfiltration, albuminuria, glomerular and interstitial lesions. Indeed, it has been shown that the urotensin system was upregulated in patients with diabetic nephropathy (18), or various degrees of renal dysfunction (9,19,20). However, preclinical and clinical studies with intravascular infusion of urotensin II showed different results. In rats, intravenous administration of urotensin II reduced renal blood flow and GFR (15,16), but intra-renal infusion lead to an increase in renal blood flow, abolished by nitric oxide synthase inhibition (17). In humans, intravenous infusion of urotensin II in healthy volunteers had no effect on forearm blood flow in one study (26), whereas another study showed a dose-dependent decrease in forearm blood flow (27). These conflicting data suggest that urotensin II may influence vascular tone by different mechanisms, i.e. direct vasoconstriction and/or indirect vasodilatation via the release of nitric oxide or prostacyclin (17,29).
Preclinical studies with different urotensin receptor antagonists showed more consistent results suggesting that the urotensin system may be an important mediator in the development of nephropathy (16,21,22). In different rat models of diabetic and non-diabetic nephropathy, it has been shown that palosuran prevented the development of renal failure, improved RPF and GFR, delayed development of albuminuria and reduced glomerular and tubulo-interstitial lesions (21,22). In addition, a pilot study in hypertensive patients with type-2 diabetic nephropathy treated either with an ACE inhibitor or AT1 antagonist, showed that palosuran administered during 2 weeks decreased UAE by 24%, without affecting RPF and GFR, or systemic blood pressure (25). Of note, the assessment of UAE was not the primary objective of this open-label, non-comparative study, its assessment in this study (one 24-h urine collection) may explain the discrepant results compared to the PROLONG study.
Finally, the study results should be interpreted knowing the limited available data on the role of the urotensin system in the pathophysiology of nephropathy. Firstly, since all patients were on stable treatment with an inhibitor of the RAAS, the independent effects of urotensin receptor blockade could not be studied. In addition, potential interactions between the two hormonal systems have not been investigated. Therefore, one cannot rule out that the urotensin system plays a minor role when the RAAS is inhibited. Secondly, no dose-titration for albuminuria or blood pressure was performed in this study. Thus, a too low dose of palosuran might have been given in this study.
117

CHAPTER 8
Thirdly, while all proven antiproteinuric agents exert their effects within several weeks (29), one cannot rule out that the time of exposure was too short to observe an effect on albuminuria. However, at that time, the available preclinical toxicology data did not allow a longer exposure of patients to palosuran.
In summary, these results suggest that inhibition of the urotensin system does not seem to represent a new treatment strategy in hypertensive patients with type 2 diabetic nephropathy. The inhibition of the RAAS by an ACE inhibitor, an AT1 antagonist or their combination, remains the cornerstone in treatment modalities protecting cardiac and renal function in this high-risk patient population. REFERENCES 1. Zhang Z, Shahinfar S, Keane WF et al. Importance of baseline distribution of proteinuria in renal outcomes trials: lessons from the
reduction of endpoints in NIDDM with the angiotensin II antagonist losartan (RENAAL) study. J Am Soc Nephrol 2005;16:1775-80 2. The GISEN Group: Randomized placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of
terminal renal failure in proteinuric, non-diabetic nephropathy. Lancet 1997;349:1857-63 3. Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The
Collaborative Study Group. N Engl J Med 1993;329:1456-62 4. Maschio G, Alberti D, Janin G et al. Effect of the angiotensin-converting-enzyme inhibitor benazepril on the progression of chronic
renal insufficiency. The Angiotensin-Converting-Enzyme Inhibition in Progressive Renal Insufficiency Study Group. N Engl J Med 1996;11;334:939-45
5. Brenner BM, Cooper ME, de Zeeuw D et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345:861-9
6. Parving H-H, Lehnert H, Brochner-Mortensen J et al. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N Engl J Med 2001;345:870-8
7. Nakao N, Yoshimura A, Morita H et al. Combination treatment of angiotensin-II receptor blocker and angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomised controlled trial. Lancet 2003;361:117-24
8. Ames RS, Sarau HM, Chambers JK et al. Human urotensin-II is a potent vasoconstrictor and agonist for the orphan receptor GPR14. Nature 1999;40:282-6
9. Matsushita M, Shichiri M, Imai T et al. Co-expression of urotensin II and its receptor (GPR14) in human cardiovascular and renal tissues. J Hypertens 2001;19:2185-90
10. Shenouda A, Douglas SA, Ohlstein EH, Giaid A. Localization of urotensin-II immunoreactivity in normal human kidneys and renal carcinoma. J Histochem Cytochem 2002;50:885-9
11. Douglas SA, Ohlstein EH. Human urotensin-II, the most potent mammalian vasoconstrictor identified to date, as a therapeutic target for the management of cardiovascular disease. Trends Cardiovasc Med 2000;10:229-37
12. Douglas SA. Human urotensin-II as a novel cardiovascular target: 'heart' of the matter or simply a fishy 'tail'? Curr Opin Pharmacol 2003; 3:159-67
13. Maguire JJ, Davenport AP. Is urotensin-II the new endothelin? Br J Pharmacol 2002;137:579-88 14. Krum H, Gilbert RE. Urotensin II: a new player in vascular and myocardial disease? Clin Sci (Lond) 2003;104:65-7 15. Gardiner SM, March JE, Kemp PA, Davenport AP, Bennett T. Depressor and regionally-selective vasodilator effects of human and
rat urotensin II in conscious rats. Br J Pharmacol 2001;132:1625-9 16. Song W, Abdel-Razik AE, Lu W, Ao Z, Johns DG, Douglas SA, Balment RJ, Ashton N. Urotensin II and renal function in the rat.
Kidney Int 2006;69:1360-8 17. Zhang AY, Chen YF, Zhang DX, Yi FX, Qi J, Andrade-Gordon P, de Garavilla L, Li PL, Zou AP. Urotensin II is a nitric oxide-
dependent vasodilator and natriuretic peptide in the rat kidney. Am J Physiol Renal Physiol 2003; 285:F792-8 18. Langham RG, Kelly DJ, Gow RM, Zhang Y, Dowling JK, Thomson NM, Gilbert RE. Increased expression of urotensin II and
urotensin II receptor in human diabetic nephropathy. Am J Kidney Dis 2004; 44:826-31 19. Totsune K, Takahashi K, Arihara Z et al. Role of urotensin II in patients on dialysis. Lancet 2001; 358:810-1 20. Totsune K, Takahashi K, Arihara Z et al. Increased plasma urotensin II levels in patients with diabetes mellitus. Clin Sci 2003;
104:1-5 21. Clozel M, Binkert C, Birker-Robaczewska M, Boukhadra C, Ding SS, Fischli W et al. Pharmacology of the urotensin-II receptor
antagonist palosuran (ACT-058362; 1-[2-(4-benzyl-4-hydroxy-piperidin-1-yl)-ethyl]-3-(2-methyl-quinolin-4-yl)-urea sulfate salt): first demonstration of a pathophysiological role of the urotensin System. J Pharmacol Exp Ther 2004;311:204-12
22. Clozel M, Hess P, Qiu C, Ding SS, Rey M. The urotensin-II receptor antagonist palosuran improves pancreatic and renal function in diabetic rats. J Pharmacol Exp Ther 2006;316:1115-21.
23. Sidharta PN, van Giersbergen PLM, Schaarschmidt D et al. Pharmacokinetics and pharmacodynamics of the urotensin-II receptor antagonist palosuran in healthy human subjects. J Clin Pharmacol 2004;44:1192
24. Sidharta PN, van Giersbergen PLM, Schaarschmidt D, et al. Multiple-dose tolerability, safety, pharmacokinetics, and pharmacodynamics of the urotensin-II receptor antagonist palosuran in healthy human subjects. Br J Clin Pharmacol 2005;60:677-8
118

EFFECTS OF UROTENSIN RECEPTOR ANTAGONIST ON ALBUMINURIA
25. Sidharta PN, Wagner FD, Bohnemeier H, Jungnik A, Halabi A, Krahenbuhl S et al. Pharmacodynamics and pharmacokinetics of the urotensin II receptor antagonist palosuran in macroalbuminuric, diabetic patients. Clin Pharmacol Ther 2006;80:246-56
26. Bohm F, Pernow J. Urotensin II evokes potent vasoconstriction in humans in vivo. Br J Pharmacol 2002;135:25-7 27. Affolter JT, Newby DE, Wilkinson IB, Winter MJ, Balment RJ, Webb DJ. No effect on central or peripheral blood pressure of
systemic urotensin II infusion in humans. Br J Clin Pharmacol 2002;54:617-21 28. Bottrill FE, Douglas SA, Hiley CR, White R. Human urotensin-II is an endothelium-dependent vasodilator in rat small arteries. Br J
Pharmacol 2000;130:1865-70. 29. Buter H, Navis G, Dullaart RP, de Zeeuw D, de Jong PE. Time course of the antiproteinuric and renal haemodynamic responses to
losartan in microalbuminuric IDDM. Nephrol Dial Transplant 2001;16:771-5 30. Chau NP, Bouhanick B, Mestivier D, Taki M, Marre M. Normal and abnormal day-to-day variability of urinary albumin excretion in
control and diabetic subjects. Diabetes Metab 2000;26:36-41
CONTRIBUTORS Principal Investigators: Prof. Dick de Zeeuw, University Medical Center Groningen, Groningen, The Netherlands, and Prof. Giuseppe Remuzzi, Mario Negri Institute, Ranica, Italy. Investigational centers: Italy: Dr. Norberto Perico, Dr. Aneliya I. Parvanova, Dr. Carlos Chiurchiu, Mario Negri Institute Ranica; Dr. Piergiorgio Messa, Prof. Attilio Elli, Ospedale Maggiore Milano – Netherlands: Dr. Arend Jan Woittiez, Dr. Liffert Vogt, Almelo Hospital; Prof. Gerjan J. Navis, Dr. Liffert Vogt, University Medical Center Groningen – Israel: Prof. Reuven Zimlichman, Dr. Ninel Wolfson, The E.Wolfson Medical Center, Holon; Prof. Naftali Stern, Dr. Yonit Marcus, Tel Aviv Sourasky Medical Center; Prof. Michael Bursztyn, Hadassah University Hospital Mt. Scopus Jerusalem – Australia: Prof. Henry Krum, Drs. Topliss, Pranckunas, Mukherjee, Kemp, Johnston, Jandeleit Dam, Aw, The Alfred Hospital Melbourne – France: Dr. Samy Hadjadj, Dr. David Gendre, Clinical Research Center, Poitiers University Hospital; Dr. Fabrice Bonnet, Dr. Faiza Lamine, Hôpital Louis Pradel Lyon – Switzerland: Prof. Michel Burnier, Dr. Pascal Maier, Centre Hospitalier Universitaire Vaudois Lausanne. CONFLICTS OF INTEREST Actelion Pharmaceuticals Ltd has sponsored the PROLONG study. The investigators had the opportunity to interpret the data and write the related publication independently from Actelion. ACKNOWLEDGEMENTS We would like to acknowledge the work done by the investigators and study nurses at the different centers. A special thank you to the study team in Ranica, who performed the ancillary study in the 22 patients included at their center.
119

CHAPTER 8
120

PART IV
OPTIMIZING RENOPROTECTION: REDUCTION OF CARDIOVASCULAR RISK?

PART IV
122

Chapter 9
Lipid management in the proteinuric patient: do not overlook the importance of proteinuria reduction
Liffert Vogt, Gozewijn D Laverman, Robin PF Dullaart and Gerjan Navis
Nephrology Dialysis and Transplantation 2004; 19: 5-8

CHAPTER 9
124

LIPID MANAGEMENT IN THE PROTEINURIC PATIENT
he markedly elevated cardiovascular risk observed in renal patients is increasingly recognised as an important treatment target (1). Among the renal populations, proteinuric patients are at particularly high risk, as apparent from the observation of an almost 6-fold increased incidence for myocardial infarction in such patients (2). Moreover, proteinuria has been shown to be an independent risk factor for cardiovascular morbidity and mortality (3,4). Most likely, proteinuria-associated lipid abnormalities play a main role in the high cardiovascular risk in proteinuric patients, and thus provide an important treatment target.
T
Several studies underlined the efficacy of statins, not only to improve the lipid profile, but also to reduce cardiovascular morbidity and mortality in hyperlipidemic and hypertensive populations (5,6), and recent post-hoc data from the CARE study showed that statin treatment reduces cardiovascular morbidity in subjects with chronic renal insufficiency (7). Most likely therefore, statins will be a cornerstone in cardiovascular prevention for the years to come in non-renal and renal populations.
For overtly proteinuric patients, however, solid data on cardiovascular risk management are still lacking, in spite of the obvious need for aggressive risk management in this high-risk population. Rational principles for cardiovascular risk management in this population nevertheless can be formulated, based on the available evidence. In this respect, lipid management is an important target. Importantly, proteinuria reduction exerts a clear-cut lipid lowering effect—irrespective of the way (class of drug, dietary intervention, or both) it is achieved. Here, we will briefly review the distinct effects of proteinuria reduction and antihyperlipidemic treatment on lipid status in proteinuric patients, and discuss their implications for lipid management and overall risk management in proteinuric patients. PLASMA LIPOPROTEINS IN PROTEINURIA
Lipid abnormalities are among the hallmarks of the nephrotic syndrome. Experimental data have causally linked hyperlipidemia to urinary protein loss (8). The typical
125

CHAPTER 9
abnormalities include elevated concentrations of plasma total cholesterol, due to an increase in low-density lipoprotein (LDL) and very-low-density lipoprotein (VLDL) cholesterol, and of plasma triglycerides (9). Consequently, the plasma level of apolipoprotein (Apo) B, the major apolipoprotein of LDL and VLDL is elevated. High-density lipoprotein (HDL) cholesterol and Apo A-I, the quantitatively most abundant apolipoprotein associated with this lipoprotein, is found to be decreased in many studies, albeit not uniformly so (9,10). As a result, proteinuria is associated with an unfavourable high total cholesterol/HDL cholesterol ratio. Moreover, the highly atherogenic lipoprotein(a) (Lp[a]) is elevated in proteinuric patients (11). In many patients, proteinuria as such is not the only factor that exerts an unfavourable effect on lipid profile. Other factors, such as renal function impairment, insulin resistance and diabetes frequently coincide with proteinuria, and may therefore also contribute to dyslipidemia. A recent study in 150 non-diabetic patients showed that the lipid profile—i.e. plasma total cholesterol, total cholesterol/HDL cholesterol ratio—is not only correlated with proteinuria, but also with the severity of renal function impairment (12). EFFECTS OF SYMPTOMATIC ANTIPROTEINURIC TREATMENT ON PLASMA LIPOPROTEINS
In patients and experimental animals, reduction of proteinuria results in a proportional decrease in plasma total cholesterol and triglycerides (13,14), irrespective of the type of intervention. The quantitative relationship between proteinuria reduction and decrease in total cholesterol is illustrated in figure 1, summarising mean proteinuria and total cholesterol before and after different antiproteinuric regimens in clinical studies (15). Apparently, the reduction in total cholesterol and triglycerides does not depend on the class of drug, but is related to the efficacy of proteinuria reduction—as it is obtained with angiotensin-converting enzyme (ACE) inhibitors, angiotensin II type 1 receptor (AT1) antagonists, non-steroidal inflammatory drugs (NSAID) or a combination of these drugs (16-18). Moreover, further reduction of proteinuria by supportive measures such as protein restriction, sodium restriction, or co-treatment with diuretic also results in a further decrease of cholesterol. Accordingly, for a single drug regimen, the dose-response for lipid reduction closely follows the dose-response for antiproteinuric efficacy, as shown in experimental animals and in man (13,14). Also, when the top of the dose-response for proteinuria is shifted towards greater maximal efficacy by sodium restriction, lipid reduction is more effective (13). Roughly, for a proteinuria reduction of 2-3 g/d a decrease in total cholesterol of approximately 1 mmol/L may be expected.
The reduction in total cholesterol is largely explained by a drop in LDL cholesterol, and accompanied by a fall in triglycerides (14). Moreover, proteinuria reduction leads to a decrease in Lp[a] (17,19). Thus, the lipoprotein profile appears to be altered favourably by reduction of proteinuria. However, antiproteinuric treatment has
126

LIPID MANAGEMENT IN THE PROTEINURIC PATIENT
Figure 1. Overview of the effects of different antiproteinuric treatment schedules on plasma total cholesterol as a function of proteinuria (mean values before and during intervention (figure and data adapted from refs (14,15)). Different regimens are indicated by 1: ACE inhibitors; 2: low protein diet; 3: AT1 antagonist; 4: NSAID
also been reported to be associated with a reduction in HDL cholesterol in several independent studies (14,17). The mechanism and the clinical significance of this unexpected finding are still uncertain. Theoretically, effects on reverse cholesterol transport could lead to a drop in HDL. However, data on cholesterol metabolism during antiproteinuric treatment are scarce. Our group has shown that the drop in HDL cholesterol during RAS blockade was not associated with a decrease in lecithin: cholesterol acetyltransferase activity (19), the enzyme responsible for cholesterol esterification in HDL—one of the first steps in the reverse cholesterol transport pathway. Thus, an explanation for the drop in HDL is lacking so far. The effects of antiproteinuric treatment on other aspects of reverse cholesterol transport, and/or oxidative properties of HDL are currently unknown, and deserve further exploration. EFFECTS OF LIPID LOWERING DRUGS IN PROTEINURIC PATIENTS
Like in other populations, in proteinuric patients different classes of lipid-lowering agents reduce elevated plasma lipid levels (20). Statins are particularly effective, as illustrated by double-blind data in 56 non-diabetic proteinuric patients, showing a reduction of 47% in LDL cholesterol after 9 months simvastatin therapy (21). However, it should be noted that statins do not modify Lp[a] levels in non-proteinuric or in proteinuric subjects (21,22).
For appropriate risk management, target levels of plasma lipoproteins have been defined. Guidelines of the National Kidney Foundation and the American Diabetes Association recommend strict control of LDL cholesterol (< 100 mg/dL or < 2.58 mmol/L) in patients with end-stage renal disease and diabetes (23,24). No target lipid levels have been recommended for proteinuric subjects specifically. However, given
127

CHAPTER 9
their high overall risk it is highly desirable that strict lipid control should be pursued in this population.
Achievement of target lipid levels with monotherapy, however, can be problematic. A large-scale analysis in general practices reported that with statins the recommended target level was reached in only 40% of the patients (25). The same appears to apply to proteinuric patients (21,26), indicating that despite statin treatment, lipid control in these patients is often suboptimal. RENOPROTECTIVE EFFECTS OF STATINS IN PROTEINURIC CONDITIONS
Interestingly, long-term therapy with statins may have a renoprotective effect that may partly be independent from the lipid-lowering effects. Animal studies support such a renoprotective effect. Moreover, statin treatment was shown to restore the response to ACE inhibition in renal conditions resistant to intervention with RAS blockade (27). In man, long-term treatment with statins was reported to reduce proteinuria in renal patients and in hypertension, although not uniformly so (20,28). The renoprotective potential of statins is furthermore supported by post-hoc data from the CARE study, showing reduction of rate of renal function loss with statins in subjects with previous myocardial infarction and chronic renal insufficiency (29). Statins added to ACE inhibitor or AT1 antagonist therapy can result in further proteinuria reduction, as recently shown in man (28,30). As RAAS blockade is an established first-choice therapy in proteinuric conditions, this added efficacy is relevant for the clinical application of statins in proteinuric patients. Although the renoprotective properties of statins in man still await prospective confirmation, the odds seem to be in its favour. CONCLUSION
Proteinuric patients are at a high risk for progressive renal function loss as well as cardiovascular morbidity and mortality. Overall risk management in these patients requires appropriate control of blood pressure, proteinuria, lipid profile, and intervention in other cardiovascular and renal risk factors, such as smoking and obesity. Obviously, these risk factors are closely linked and require an integrated approach.
RAAS blockade, as first-choice treatment in proteinuric patients, together with statins for specific lipid control provides a rational combined approach for overall risk reduction, with added efficacy and with complementary properties regarding the specific effects on HDL cholesterol and Lp[a]. Achievement of the intermediate targets, i.e. optimal blood pressure control, maximal proteinuria reduction and lipids within target levels, should be actively pursued by appropriate dosing and supportive measures. Future studies should address optimal dosing schedules for this setting and evaluate the eventual benefit in terms of cardiovascular and renal risk.
128

LIPID MANAGEMENT IN THE PROTEINURIC PATIENT
REFERENCES 1. Baigent C, Burbury K, Wheeler D. Premature cardiovascular disease in chronic renal failure. Lancet 2000;356:147-52 2. Ordonez JD, Hiatt RA, Killebrew EJ, Fireman BH. The increased risk of coronary heart disease associated with nephrotic syndrome.
Kidney Int 1993;44:38-42 3. Roodnat JI, Mulder PG, Rischen-Vos J et al. Proteinuria after renal transplantation affects not only graft survival but also patient
survival. Transplantation 2001;72:438-44 4. Hillege HL, Fidler V, Diercks GF et al. Urinary albumin excretion predicts cardiovascular and noncardiovascular mortality in
general population. Circulation 2002;106:1777-82 5. Shepherd J, Cobbe SM, Ford I et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West
of Scotland Coronary Prevention Study Group. N Engl J Med 1995;333:1301-7 6. Sever PS, Dahlof B, Poulter NR et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have
average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial—Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet 2003;361:1149-58
7. Tonelli M, Moye L, Sacks FM, Kiberd B, Curhan G. Pravastatin for secondary prevention of cardiovascular events in persons with mild chronic renal insufficiency. Ann Intern Med 2003;138:98-104
8. Thomas ME, Morrison AR, Schreiner GF. Metabolic effects of fatty acid-bearing albumin on a proximal tubule cell line. Am J Physiol 1995:268:F1177-84
9. Keane WF, Kasiske BL. Hyperlipidemia in the nephrotic syndrome. N Engl J Med 1990;323:603-4 10. Joven J, Villabona C, Vilella E et al. Abnormalities of lipoprotein metabolism in patients with the nephrotic syndrome. N Engl J
Med 1990;323:579-84 11. Wanner C, Rader D, Bartens W et al. Elevated plasma lipoprotein(a) in patients with the nephrotic syndrome. Ann Intern Med
1993;119:263-9 12. Ozsoy RC, Kastelein JJ, Arisz L, Koopman MG. Atorvastatin and the dyslipidemia of early renal failure. Atherosclerosis
2003;166:187-94 13. Wapstra FH, van Goor H, Navis G, de Jong PE, de Zeeuw D. Antiproteinuric effect predicts renal protection by angiotensin-
converting enzyme inhibition in rats with established adriamycin nephrosis. Clin Sci (Lond) 1996;90:393-401 14. Ruggenenti P, Mise N, Pisoni R et al. Diverse effects of increasing lisinopril doses on lipid abnormalities in chronic nephropathies.
Circulation 2003;107:586-92 15. Navis G, Buter H, de Jong PE, Dullaart RP, de Zeeuw D. Effect of antiproteinuric treatment on the lipid profile in nondiabetic renal
disease. Contrib Nephrol 1997;120:88-96 16. Gansevoort RT, de Zeeuw D, de Jong PE. Additive antiproteinuric effect of ACE inhibition and a low-protein diet in human renal
disease. Nephrol Dial Transplant 1995;10:497-504 17. Gansevoort RT, Heeg JE, Dikkeschei FD et al. Symptomatic antiproteinuric treatment decreases serum lipoprotein (a) concentration
in patients with glomerular proteinuria. Nephrol Dial Transplant 1994;9:244-50 18. Buter H, van Tol A, Navis GJ et al. Angiotensin II receptor antagonist treatment lowers plasma total and very low + low density
lipoprotein cholesterol in Type 1 diabetic patients with albuminuria without affecting plasma cholesterol esterification and cholesteryl ester transfer. Diabet Med 2000;17:550-52
19. Dullaart RP, Gansevoort RT, Dikkeschei BD et al. Role of elevated lecithin: cholesterol acyltransferase and cholest-eryl ester transfer protein activities in abnormal lipoproteins from proteinuric patients. Kidney Int 1993;44:91-7
20. Fried LF, Orchard TJ, Kasiske BL. Effect of lipid reduction on the progression of renal disease: a meta-analysis. Kidney Int 2001;59:260-9
21. Olbricht CJ, Wanner C, Thiery J, Basten A. Simvastatin in nephrotic syndrome. Simvastatin in Nephrotic Syndrome Study Group. Kidney Int 1999;71(Suppl):S113-6
22. Kostner GM, Gavish D, Leopold B et al. HMG CoA reductase inhibitors lower LDL cholesterol without reducing Lp(a) levels. Circulation 1989;80:1313-19
23. National Kidney Foundation. K/DOQI Clinical practice guidelines for managing dyslipidemias in chronic kidney disease. Am J Kidney Dis 2003;41:S1-92
24. Haffner SM. Management of dyslipidemia in adults with diabetes. Diabetes Care 2003;26 (Suppl 1): S83-6 25. Pearson TA, Laurora I, Chu H, Kafonek S. The lipid treatment assessment project (L-TAP): a multicenter survey to evaluate the
percentages of dyslipidemic patients receiving lipid- lowering therapy and achieving low-density lipoprotein cholesterol goals. Arch Intern Med 2000;160:459-67
26. Keane WF, St Peter JV, Kasiske BL. Is the aggressive management of hyperlipidemia in nephrotic syndrome mandatory? Kidney Int 1992;38(Suppl):S134-41
27. Zoja C, Corna D, Rottoli D et al. Effect of combining ACE inhibitor and statin in severe experimental nephropathy. Kidney Int 2002;61:1635-45
28. Lee TM, Su SF, Tsai CH. Effect of pravastatin on proteinuria in patients with well-controlled hypertension. Hypertension 2002;40:67-73
29. Tonelli M, Moye L, Sacks FM, Cole T, Curhan GC. Effect of pravastatin on loss of renal function in people with moderate chronic renal insufficiency and cardiovascular disease. J Am Soc Nephrol 2003;14:1605-13
30. Bianchi S, Bigazzi R, Caiazza A, Campese VM. A controlled, prospective study of the effects of atorvastatin on proteinuria and progression of kidney disease. Am J Kidney Dis 2003;41:565-70
129

CHAPTER 9
130

Chapter 10
Cellular cholesterol efflux to plasma from proteinuric patients is elevated and remains unaffected by antiproteinuric treatment Liffert Vogt, Gozewijn D. Laverman, Arie van Tol, Albert K. Groen, Gerjan Navis and Robin P.F. Dullaart Nephrology Dialysis Transplantation 2006; 21:101-6

CHAPTER 10
132

CHOLESTEROL EFFLUX IN PROTEINURIC PATIENTS
Background—Lipid derangements are assumed to contribute to the elevated cardiovascular risk in proteinuric patients. The impact of proteinuria on reverse cholesterol transport (RCT) is unknown. The first step in RCT, cellular cholesterol efflux to plasma, may be altered in proteinuria, consequent to changes in pre-β HDL formation and plasma phospholipid transfer protein (PLTP) activity. Methods—In 6 non-diabetic male patients with nephrotic-range proteinuria and 12 matched healthy men plasma (apo)lipoproteins, pre-ß HDL formation, PLTP activity as well as the ability of plasma to promote cholesterol efflux out of cultured human skin fibroblasts were determined. These variables were also measured in response to antiproteinuric treatment, consisting of single and dual RAAS blockade by losartan and lisinopril. Results—Plasma total cholesterol (p < 0.05), triglycerides (p < 0.05), apolipoprotein (apo) A-I (p < 0.001), apo B (p < 0.001), PLTP activity (p < 0.005) and pre-ß HDL formation (p < 0.001) were higher in proteinuric patients. Cellular cholesterol efflux to plasma from proteinuric patients was 41% higher than to plasma from healthy subjects (p < 0.001). Reduction of proteinuria from 5.0 to 1.4 g/d by dual RAAS blockade was associated with a 23 % reduction in plasma apo B levels (p < 0.05). Pre-β HDL formation and plasma PLTP activity did not significantly change. Combined antiproteinuric treatment did not reduce the elevated cellular cholesterol efflux. Conclusion—Cellular cholesterol efflux to plasma from patients with nephrotic-range proteinuria is enhanced, in conjunction with elevated pre-ß HDL formation and plasma PLTP activity. These changes may attenuate the cardiovascular risk assocated with proteinuria-associated hyperlipidemia. Antiproteinuric therapy lowers plasma apo B, but does not affect cell-derived cholesterol efflux, suggesting that this therapy beneficially affects cardiovascular risk in proteinuric patients.
n proteinuric patients, plasma total cholesterol, very low density lipoprotein (VLDL), low density lipoprotein (LDL) cholesterol and triglycerides (TG) are usually elevated (1,2). These proteinuria-associated abnormalities in apolipoprotein (apo) B-containing lipoproteins may well contribute to the elevated cardiovascular risk in proteinuric patients (3). The impact of proteinuria on HDL metabolism, relevant as this may be to cardiovascular risk, is incompletely understood.
I
HDL plays an important role in the reverse cholesterol transport (RCT) pathway (4), which provides transport of excess cellular cholesterol from peripheral cells to the liver for metabolism and excretion in the bile. Thus, the RCT pathway plays a protective role in cardiovascular risk. Among other factors, cholesteryl ester transfer protein (CETP) and phospholipid transfer protein (PLTP) are involved in HDL remodelling and metabolism. CETP transfers cholesteryl esters from HDL to apo B-containing lipoproteins, whereas PLTP transfers phospholipids between lipoproteins and is able to
133

CHAPTER 10
convert HDL in smaller and larger HDL particles (5). During this process small apo A-I containing pre-β HDL particles are generated (6). These particles are initial acceptors of cell-derived cholesterol and are likely to be important for removal of cholesterol from the vessel wall.
The effect of nephrotic syndrome on HDL metabolism in humans, and more specifically on these processes of the RCT pathway, is not fully understood. Plasma HDL cholesterol and its most abundant apolipoprotein, apo A-I, were reported to be low, unaltered or even elevated (1,2,7) in proteinuric patients. Plasma CETP levels and cholesteryl ester transfer rates are elevated in nephrotic patients (2,8), contributing to a low cholesterol content in HDL. Plasma PLTP activity is increased in hypertriglyceridemic subjects (5), but no data are available with respect to plasma PLTP activity and pre-β HDL in proteinuric patients. The effect of proteinuria on the ability of plasma to promote cellular cholesterol efflux, representing a functional measure to evaluate the effectiveness of early steps in the RCT pathway with respect to the constellation of extracellular cholesterol acceptors, is also unknown. Antiproteinuric treatment results in reduction of plasma apo B-containing lipoproteins, irrespective of the mode of antiproteinuric intervention (3,8-10). HDL cholesterol may also drop in response to proteinuria reduction (9,10), but it is unknown whether cell-derived cholesterol efflux is affected by antiproteinuric treatment.
In view of high plasma triglycerides in proteinuric patients, we hypothesized that proteinuria is accompanied by changes in plasma PLTP activity and pre-β HDL, which may affect the ability of plasma to stimulate cellular cholesterol efflux. This hypothesis was tested in untreated proteinuric patients. Moreover, we investigated the effect of antiproteinuric treatment on these components of RCT. PATIENTS AND METHODS
Patients and protocol The study was approved by the local medical ethics committee and all participants provided written informed consent. Patients with proteinuric nephropathies were selected from our renal outpatient department and eligibility for participation in the study was considered after a wash-out period from all antihypertensive medication (at least 6 weeks) and all lipid-lowering agents (at least 8 weeks). All patients had to fulfil the following inclusion criteria after wash-out: proteinuria ≥ 2 g/d after wash-out, diastolic blood pressure between 80 and 110 mmHg, creatinine clearance ≥ 30 mL/min/1.73 m2, and age between 18 and 70 years. Patients with nephrotic syndrome consequent to a non-primary renal disorder, as well as patients with systemic diseases, cardiovascular disorders or diabetes mellitus were excluded. Only men participated to avoid effects of sex-related differences in HDL cholesterol. For baseline comparisons,
134

CHOLESTEROL EFFLUX IN PROTEINURIC PATIENTS
each male patient was matched with 2 male healthy subjects with respect to race (Caucasian), age (within 5 yrs) and body mass index (BMI, calculated as weight divided by height squared; within 2 kg/m2). Six patients were identified for study participation and completed the protocol. All patients had proteinuria of biopsy proven glomerular non-diabetic origin (focal segmental glomerular sclerosis (n = 3), membraneous glomerulopathy (n = 2), and IgA nephropathy (n = 1). None of the participants received any immunosuppressive treatment.
Patients were treated according to a prospective open-label study protocol designed to obtain individualized maximal antiproteinuric response. In short, patients were treated with losartan (subsequently 50, 100 and 150 once daily) and lisinopril (subsequently 10, 20 and 40 once daily) in random order, each preceeded by a baseline period without medication. After these single drug periods, all patients received combined treatment, using the optimal individual antiproteinuric doses for each drug (usually lisinopril 40 mg and losartan 100 mg), in order to obtain the maximal antiproteinuric effect. Each treatment period lasted 6 weeks. At the end of the initial baseline period and at the end of the dual RAAS blockade period, patients visited the hospital after an overnight fast. Data obtained from these visits were used for this study. Blood pressure was measured by an automatic device (Dinamap®). Mean arterial pressure (MAP) was calculated as: 2/3 * diastolic blood pressure + 1/3 * systolic blood pressure. The values shown represent the mean value of three readings after 15 min of supine rest. Laboratory measurements Urinary protein was determined with the pyrogallol red-molybdate method (mean of two 24-h urine collections). Serum creatinine and albumin were determined using an automated multi-analyzer (MEGA®, Merck, Darmstadt, Germany). Venous blood was collected in tubes containing 1.5 mg/mL ethylenediaminetetraacetic acid and was directly placed on ice. Plasma was obtained within 30 min by centrifugation at 3000 rpm for 15 min at 4 °C and was kept frozen at -20 °C until analysis. Plasma total cholesterol and triglycerides were measured enzymatically. HDL cholesterol was assayed by a homogeneous method using a commercially available assay system (Abbott Inc., Cat. № 30-3064/R3, Abbott Park, Ill, USA). VLDL + LDL cholesterol was calculated as the difference between plasma total cholesterol and HDL cholesterol. Apo A-I and B were assayed by immunoturbidimetry using commercially available kits (Serapak, Bayer, Leverkusen, Germany, Cat. № 682, and 6822, respectively). Plasma PLTP activity was assayed in a liposome vesicles-HDL system using a previously described method, which is not affected by the phospholipid transfer
135

CHAPTER 10
promoting action of CETP (11). Plasma PLTP activity is linearly related to the amount of plasma used in the incubations and is not influenced by the endogenous lipoproteins in plasma. Plasma PLTP activity is expressed in arbitrary units (AU), corresponding to the percentages of the activities in normal human pool plasma. Plasma pre-β HDL formation was measured by crossed immuno-electrophoresis using frozen plasma essentially as described (12). In brief, plasma samples were thawed while kept on ice with addition of proteolysis inhibitors. Subsequently, iodoacetate, an LCAT inhibitor was added and the samples were incubated at 37 ˚C for 24 h to measure the formation of pre-beta HDL. At the end of the procedure, the gel was stained with Coomassie brilliant blue R250 and subsequently dried. Areas under the pre-β HDL and α HDL peaks were scanned and calculated using Scion software. The pre-β HDL area was calculated as the percentage of the sum of the pre-β HDL and the α HDL areas. Pre-β HDL was expressed in apo A-I concentration (g/L).
Cholesterol efflux to plasma was determined using human fibroblasts, as described (12). Fibroblasts were obtained from normolipidemic control persons by explant culture from a 3 mm punch biopsy at a 1 mm skin thickness. The cells were then cultered in DMEM and subsequently were loaded with [3H]-cholesterol and unlabelled cholesterol during 24 h. Unlabelled cholesterol was added to induce ATP-binding cassette transporter-AI (ABCA1) expression in fibroblasts. Subsequently, the cells were extensively washed and the efflux assay was started by adding plasma diluted to 1% in efflux medium. At this concentration the plasma dose-response curve for cholesterol efflux is in the linear range. An incubation time of 4 h at 37 0C was chosen to minimize analytical errors. [3H]-cholesterol was quantified by liquid scintillation counting after collection of the medium. Total cellular [3H]-cholesterol was determined after extraction of the cells with 2-propanol and the percentage efflux was calculated. All values were corrected for radioactivity appearing in the culture medium in the absence of plasma. To be able to normalize between series of experiments and to correct for between-day variation, efflux to 50 μg protein/mL HDL (Calbiochem, San Diego, CA, USA) was determined in triplicate. Statistical analysis Results are expressed as mean and range. Data from patients before, at baseline, and after the three treatment periods were compared with data from healthy subjects by one-way ANOVA. Where appropriate, one-way ANOVA with post-hoc Duncan correction for multiple comparisons or paired t-tests were used to evaluate the effect of antiproteinuric treatment in these patients. In proteinuric patients, relationships between variables were assessed by Spearman’s rank correlation analysis using four data sets, obtained at baseline and after each treatment period. Z-transformation was applied to
136

CHOLESTEROL EFFLUX IN PROTEINURIC PATIENTS
Table 1. Proteinuria, mean arterial pressure, serum albumin and creatinine clearance (mean (range)) at baseline and after losartan 100 mg, lisinopril 40 mg and combined treatment with losartan and lisinopril in 6 non-diabetic proteinuric patients.
Baseline Losartan 100 mg Lisinopril 40 mg Losartan + Lisinopril
Uprot (g/d) 5.0 (2.2-8.2) 3.0 (1.1-6.5)* 1.8 (0.0-6.0)* 1.4 (0.0-5.6)*$ MAP (mmHg) 105 (90-127) 90 (71-97)* 85 (73-102)* 78 (68-89)*$# SeAlb (g/L) 38 (36-42) 41 (35-45)* 40 (34-43) 40 (35-46)* Clcreat (mL/min) 78 (40-92) 81 (57-107) 73 (37-99) 67 (40-82)
Uprot, proteinuria. MAP, mean arterial pressure. SeAlb, serum albumin. Clcreat, creatinine clearance. * p < 0.05 vs. baseline; $ p < 0.05 vs. losartan; # p<0.05 vs. lisinopril
correct for multiple comparisons. A p-value < 0.05 was considered significant. RESULTS
Age was 47 (39-54) years in proteinuric patients and 51 (45-56) years in control subjects (NS). BMI was also not different between the groups (24.5 (21.9-27.0) kg/m2 and 24.4 (22.9-25.8) kg/m2 for proteinuric patients and control subjects, respectively (NS). Renal data and blood pressure from the proteinuric patients are given in table 1. In table 2, further clinical characteristics, plasma lipids, lipoproteins, apolipoproteins, pre-ß HDL formation and PLTP activity levels of the proteinuric and matched healthy control subjects are given. Plasma total cholesterol, triglycerides and apo B levels were higher in proteinuric patients. The difference in plasma VLDL + LDL between patients and healthy subjects did not reach statistical significance. HDL cholesterol was similar in patients and healthy subjects, whereas plasma apo A-I was higher in patients. Plasma PLTP activity was elevated in the patient group, coinciding with an increase in pre-β HDL formation. As shown in figure 1, cholesterol efflux out of fibroblasts amounted to 18.3 (17.7-18.7) %/4 h to plasma from proteinuric patients vs. 13.0 (12.2- 13.8) %/4 h to plasma from healthy subjects (p < 0.001), resulting in a 41% higher efflux using patient plasma.
In patients, proteinuria was lowered in response to losartan and lisinopril treatment (table 1). Dual RAAS blockade, consisting of the combination of the individual optimal antiproteinuric doses of losartan and lisinopril, resulted in the maximal reduction in proteinuria which amounted to 73 (50-97) % compared to baseline. Proteinuria reduction was accompanied by a rise in serum albumin. MAP fell during each treatment period with the lowest values being recorded during dual RAAS blockade. The changes in creatinine clearance did not reach statistical significance.
Table 2 shows that plasma apo B levels dropped in response to antiproteinuric treatment with lisinopril and dual RAAS blockade, whereas the reductions in plasma total cholesterol and VLDL + LDL cholesterol were significant after lisinopril treatment. Plasma apo A-I was lowered after lisinopril and combined treatment, but HDL
137

CHAPTER 10
Table 2. Lipids profiles, apo(lipoprotein)s, pre-ß HDL formation and PLTP activity (mean (range)) of male non-diabetic proteinuric and healthy subjects.
Male proteinuric patients (n = 6)
Male control
subjects (n = 12) Baseline Losartan
100 mg Lisinopril
40 mg Losartan + lisinopril
Total C (mmol/L) 4.65 (4.27-5.03)
6.00 # (4.60-7.36)
5.78 # (4.72-6.85)
4.89 * (4.22-5.57)
5.10 (4.56-5.63)
TG (mmol/L) 1.05 (0.68-1.43)
1.88 # (1.02-2.73)
1.72 (0.86-2.58)
1.69 (0.93-2.47)
1.72 # (1.22-2.21)
VLDL + LDL-C (mmol/L) 3.07 (2.70-3.45)
4.09 (2.48-5.41)
3.97 # (2.86-5.07)
3.18 * (2.54-3.82)
3.34 (2.73-3.94)
HDL-C (mmol/L) 1.11 (0.96-1.25)
1.05 (0.80-1.31)
1.05 (0.70-1.39)
0.95 (0.57-1.33)
0.99 (0.60-1.37)
Plasma Apo A-I (g/L) 1.14 (1.03-1.25)
1.46 $ (1.29-1.64)
1.41 # (1.23-1.58)
1.28 * (1.06-1.49)
1.31 * (1.05-1.56)
Plasma Apo B (g/L) 0.77 (0.69-0.85)
1.45 $ (1.07-1.82)
1.32 $ (0.98-1.65)
1.18 $* (0.98-1.37)
1.12 $*
(0.90-1.34)
PLTP activity (AU) 89.8 (81.6-98.1)
112.1 ‡ (96.2-127.9)
119.3 ‡ (96.6-142.0)
117.9 $ (103.1-132.7)
117.5 $ (104.3-130.7)
Pre-β HDL formation (apo A-I, g/L)
0.19 (0.15-0.22)
0.36 $ (0.24-0.47) 0.25
(0.15-0.35)
TC, plasma total cholesterol. TG, plasma triglycerides. C, cholesterol. VLDL + LDL, very-low + low-density lipoproteins. HDL, high-densitiy lipoproteins. Apo A-I, apolipoprotein A-I. Apo B, apolipoprotein B. PLTP, plasma phospholipid transfer protein. $ p < 0.001, ‡ p < 0.005, # p < 0.05 vs. control subjects; * p < 0.05 vs. baseline. cholesterol did not change. Plasma triglycerides and PLTP activity were unaltered after proteinuria lowering, and PLTP activity remained higher compared to control subjects at each treatment period. In the proteinuric patients, there was no relationship of plasma PLTP activity with the degree of proteinuria as measured during the four observation periods (averaged r = -0.17, p = 0.56). In contrast, plasma apo B was positively correlated with proteinuria (averaged r = 0.52, p < 0.05). Neither plasma PTLP activity nor apo B was correlated with creatinine clearance (averaged r = 0.39, p = 0.17; and averaged r = 0.29, p = 0.33, respectively).
Pre-β HDL formation and cellular efflux were only measured at baseline and during dual RAAS blockade. The changes in pre-β HDL formation were not significant. Figure 1 shows that the ability of patient plasma to induce cholesterol efflux out of fibroblasts was not affected by dual RAAS blockade and remained elevated compared to plasma from healthy subjects. DISCUSSION This study shows for the first time that plasma pre-ß HDL formation is enhanced and that the plasma PLTP activity level is higher in proteinuric patients compared to healthy subjects. Another novel finding is that plasma from proteinuric patients has a 41% higher ability to stimulate cholesterol efflux out of human fibroblasts than plasma
138
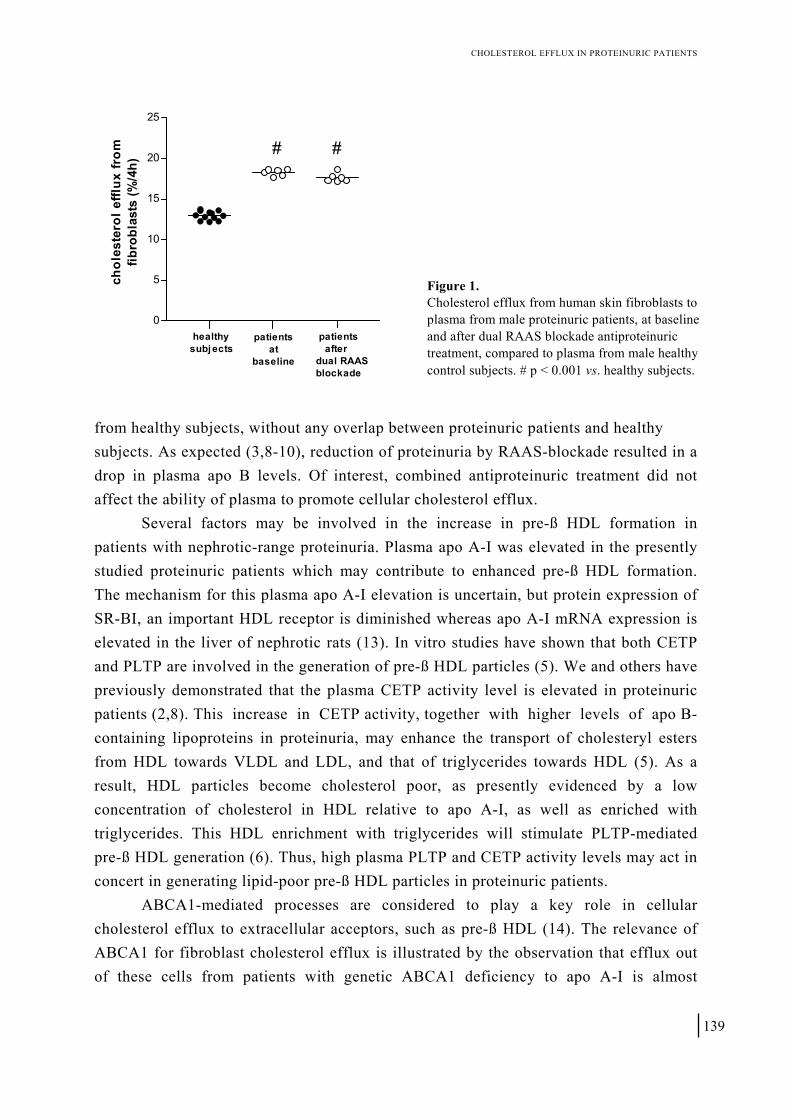
CHOLESTEROL EFFLUX IN PROTEINURIC PATIENTS
0
5
10
15
20
25
healthysubjects
patientsat
baseline
patients afterdual RAASblockade
# #
chol
este
rol e
fflux
from
fibro
blas
ts (%
/4h)
Figure 1. Cholesterol efflux from human skin fibroblasts to plasma from male proteinuric patients, at baseline and after dual RAAS blockade antiproteinuric treatment, compared to plasma from male healthy control subjects. # p < 0.001 vs. healthy subjects.
from healthy subjects, without any overlap between proteinuric patients and healthy subjects. As expected (3,8-10), reduction of proteinuria by RAAS-blockade resulted in a drop in plasma apo B levels. Of interest, combined antiproteinuric treatment did not affect the ability of plasma to promote cellular cholesterol efflux.
Several factors may be involved in the increase in pre-ß HDL formation in patients with nephrotic-range proteinuria. Plasma apo A-I was elevated in the presently studied proteinuric patients which may contribute to enhanced pre-ß HDL formation. The mechanism for this plasma apo A-I elevation is uncertain, but protein expression of SR-BI, an important HDL receptor is diminished whereas apo A-I mRNA expression is elevated in the liver of nephrotic rats (13). In vitro studies have shown that both CETP and PLTP are involved in the generation of pre-ß HDL particles (5). We and others have previously demonstrated that the plasma CETP activity level is elevated in proteinuric patients (2,8). This increase in CETP activity, together with higher levels of apo B-containing lipoproteins in proteinuria, may enhance the transport of cholesteryl esters from HDL towards VLDL and LDL, and that of triglycerides towards HDL (5). As a result, HDL particles become cholesterol poor, as presently evidenced by a low concentration of cholesterol in HDL relative to apo A-I, as well as enriched with triglycerides. This HDL enrichment with triglycerides will stimulate PLTP-mediated pre-ß HDL generation (6). Thus, high plasma PLTP and CETP activity levels may act in concert in generating lipid-poor pre-ß HDL particles in proteinuric patients. ABCA1-mediated processes are considered to play a key role in cellular cholesterol efflux to extracellular acceptors, such as pre-ß HDL (14). The relevance of ABCA1 for fibroblast cholesterol efflux is illustrated by the observation that efflux out of these cells from patients with genetic ABCA1 deficiency to apo A-I is almost
139

CHAPTER 10
abrogated (15). In our study, we used normal human skin fibroblasts which, after cholesterol loading, abundantly express ABCA1 (12). Lipid poor acceptors including pre-ß HDL particles are considered to be the initial acceptors of cholesterol from these cells (16). Besides, PLTP could also directly promote cholesterol efflux out of fibroblasts (17). Thus, it is likely that enhanced pre-β HDL formation and elevated PLTP activity are involved in the higher cholesterol efflux out of fibroblasts to proteinuric plasma, although effects of other HDL-related variables remain possible. Moreover, it should be realized that we did not document whether the in situ capacity of periheral cells to transport cholesterol to the extracellular space is abnormal in proteinuric patients. We propose that an enhanced ability of plasma to stimulate cellular cholesterol efflux can be envisaged to favorably modulate the increased cardiovascular risk in proteinuric patients, which is in part attributable to high levels of apo B-containing lipoproteins (3). Since it has been previously suggested that plasma apo AI and / or HDL cholesterol can decrease after rigorous antiproteinuric treatment (9,10), we tested whether this intervention would decrease the ability of plasma to stimulate cellular cholesterol removal. In our study, plasma apo A-I decreased in response to lisinopril and dual RAAS blockade, but the changes in HDL cholesterol and pre-β HDL formation were not significant. Plasma PLTP activity did not change after antiproteinuric treatment and remained elevated compared to control subjects. Importantly, cellular cholesterol efflux from fibroblasts was also unaltered after combined antiproteinuric treatment. This suggests that even after pronounced antiproteinuric therapy this early step in the reverse cholesterol transport process remains unaffected, as far as the ability of plasma to stimulate cellular cholesterol efflux is concerned. However, since no data are available concerning effects of lisinopril or losartan on plasma PLTP activity, pre-β HDL and cell-derived cholesterol transport to plasma in subjects without proteinuria, we cannot completely exclude that effects of proteinuria reduction on these variables were masked by opposite effects of these medications per se. PLTP is a multifacetted lipid transfer protein that, besides its role in stimulating cellular cholesterol efflux via direct and indirect mechanisms in vitro, has atherogenic properties as well. Of interest, in vivo studies in mice transgenic for human PLTP show increased atherosclerosis, which is attributable to decreased HDL levels and a stimulatory effect of PLTP on hepatic VLDL secretion (18). Limited data available so far in humans suggest that plasma PLTP activity may be elevated in patients with cardiovascular disease (19). To which extent high PLTP activity contributes to the cardiovascular risk in proteinuric patients is unknown at present. Finally, in the interpretation of the present findings in terms of cardiovascular risk, it should be realized that HDL also has antiinflammatory and antioxidative properties that were not
140

CHOLESTEROL EFFLUX IN PROTEINURIC PATIENTS
evaluated in our study (20). In conclusion, albeit in a small number of patients, the present study demonstrates that the ability of plasma from proteinuric patients to stimulate cholesterol efflux out of human skin fibroblasts is strongly elevated. This abnormality is probably due to enhanced pre-ß HDL formation and PLTP activity. Thus, with respect to plasma-related acceptors of cell-derived cholesterol, this early step in RCT appears to be favorably altered in proteinuric patients, which may alleviate the cardiovascular consequences of their hyperlipidemia. Remarkably, the elevated cellular cholesterol efflux to plasma persists despite pharmacological reduction of proteinuria—along with a drop in plasma apo B levels. These effects lend support to the possibility that antiproteinuric treatment beneficially affects cardiovascular risk in proteinuric patients. REFERENCES 1. Joven J, Villabona C, Vilella E et al. Abnormalities of lipoprotein metabolism in patients with the nephrotic syndrome. N Engl J
Med 1990;323:579-84 2. Deighan CJ, Caslake MJ, McConnell M et al. The atherogenic lipoprotein phenotype: small dense LDL and lipoprotein remnants in
nephrotic range proteinuria. Atherosclerosis 2001;157:211-20 3. Vogt L, Laverman GD, Dullaart RPF, Navis GJ. Lipid management in the proteinuric patient: do not overlook the importance of
proteinuria reduction. Nephrol Dial Transplant 2004;19:5-8 4. Fielding CJ, Fielding PE. Molecular physiology of reverse cholesterol transport. J Lipid Res 1995;36:211-28 5. Borggreve SE, De Vries R, Dullaart RPF. Alterations in high-density lipoprotein metabolism and reverse cholesterol transport in
insulin resistance and type 2 diabetes mellitus: role of lipolytic enzymes, lecithin:cholesterol acyltransferase and lipid transfer proteins. Eur J Clin Invest 2003;33:1051-69
6. Rye KA, Jauhiainen M, Barter PJ, Ehnholm C. Triglyceride-enrichment of high density lipoproteins enhances their remodelling by phospholipid transfer protein. J Lipid Res 1998;39:613-22
7. Muls E, Rosseneu M, Daneels R et al. Lipoprotein distribution and composition in the human nephrotic syndrome. Atherosclerosis 1985;54:225-37
8. Dullaart RPF, Gansevoort RT, Dikkeschei LD et al. Role of elevated lecithin: cholesterol acyltransferase and choles-teryl ester transfer protein activities in abnormal lipoproteins from proteinuric patients. Kidney Int 1993;44:91-7
9. Ruggenenti P, Mise N, Pisoni R et al. Diverse effects of increasing lisinopril doses on lipid abnormalities in chronic nephropathies. Circulation 2003;107:586-92
10. Gansevoort RT, Heeg JE, Dikkeschei LD et al. Symptomatic antiproteinuric treatment decreases serum lipoprotein (a) concentration in patients with glomerular proteinuria. Nephrol Dial Transplant 1994;9:244-250
11. Speijer H, Groener JE, Van Ramshorst E, Van Tol A. Different locations of cholesteryl ester transfer protein and phospholipid transfer protein activities in plasma. Atherosclerosis 1991;90:159-68
12. De Vries R, Kerstens MN, Sluiter WJ, Groen AK, Van Tol A, Dullaart RPF. Cellular cholesterol efflux to plasma from moderately hypercholesterolaemic type 1 diabetic patients is enhanced, and is unaffected by simvastatin treatment. Diabetologia 2005;48:1105-13
13. Liang K, Vaziri ND. Down-regulation of hepatic high-density lipoprotein receptor, SR-B1, in nephrotic syndrome. Kidney Int 1999;56:621-6
14. Yancey PG, Bortnick AE, Kellner-Weibel G et al. Importance of different pathways of cellular cholesterol efflux. Arterioscler Thromb Vasc Biol 2003;23:712-9
15. Hovingh GK, Van Wijland MJ, Brownlie A et al. The role of the ABCA1 transporter and cholesterol efflux in familial hypoalphalipoproteinemia. J Lipid Res 2003;44:1251-5
16. Favari E, Lee M, Calabresi L et al. Depletion of pre-β-high density lipoprotein by human chymase impairs ATP-binding cassette transporter A1-but not scavenger receptor class B type I-mediated lipid efflux to high density lipoprotein. J Biol Chem 2004;279:9930-6
17. Oram JF, Wolfbauer G, Vaughan AM et al. Phospholipid transfer protein interacts with and stabilizes ATP-binding cassette transporter A1 and enhances cholesterol efflux from cells. J Biol Chem 2003;278:52379-85
18. Van Haperen R, Van Tol A, Van Gent T et al. Increased risk of atherosclerosis by elevated plasma levels of phospholipid transfer protein. J Biol Chem 2002;277:48938-43
19. Schlitt A, Bickel C, Thumma P et al. High plasma phospholipid transfer protein levels as a risk factor for coronary artery disease. Arterioscler Thromb Vasc Biol 2003;23:1857-62
20. Nofer JR, Kehrel B, Fobker M et al. HDL and arteriosclerosis: beyond reverse cholesterol transport. Atherosclerosis 2002;161:1-16 ACKNOWLEDGEMENTS G.D. Laverman was appointed by a grant of the Dutch Kidney Foundation (program grant 5023). The expert technical assistance of H. Elias, T. van Gent and L.M. Scheek is highly appreciated.
141

CHAPTER 10
142

Chapter 11
Change in urinary albumin excretion is predictive of cardiovascular outcome in normotensive patients with type 2 diabetes mellitus and microalbuminuria Adrienne A. Zandbergen, Liffert Vogt, Dick de Zeeuw, Steven W.J. Lamberts, Rob J.T.H. Ouwendijk, Marinus G.A. Baggen and Aart H. Bootsma Diabetes Care 2007;30:3119-21

CHAPTER 11
144

ALBUMINURIA PREDICTIVE OF CARDIOVASCULAR RISK
icroalbuminuria is associated with cardiovascular complications and all-cause mortality in patients with diabetes mellitus (1-3). Inhibitors of the renin-angiotensin aldosterone system (RAAS) protect renal and cardiac function in these patients, at least partly independent of the associated blood pressure reduction (4-9). Recently, a few studies showed that reduction in albuminuria in hypertensive diabetic patients reduces the risk of subsequent cardiovascular events (10-12). The question remains whether this risk reduction is not fully explained by the reduction of a too high blood pressure. No data are available of normotensive diabetic patients. Therefore, we investigated whether sustained change in albuminuria independently predicts cardiovascular outcome in patients with type 2 diabetes mellitus and microalbuminuria, without hypertension.
M
RESEARCH DESIGN AND METHODS
The present study is a prospective follow-up study of 67 normotensive patients (baseline blood pressure ≤ 140/90 mmHg without antihypertensive treatment) with type 2 diabetes and microalbuminuria (urinary albumin excretion 20-200 mg/L), who participated in a previously published, larger, randomized, double-blind, placebo-controlled trial investigating short-term effects of the angiotensin-receptor antagonist losartan on microalbuminuria (7). Exclusion criteria included a history of macrovascular complications and a baseline serum creatinine level > 150 μmol/L. After the original 20-week study period, a cohort of 67 patients from that study was prospectively followed during 4.7 ± 0.1 (± SEM) years. They were recruited on the base of their address and received standard medical care. Data were collected on current and past health, medication use, blood pressure, renal function and albuminuria, annually assessed in morning spot-urines. The endpoint was a composite of death, cardiovascular disease, cerebrovascular events and peripheral artery disease. The paired Student’s t-test was used for comparisons within similar variables. Regarding the rate of change in albuminuria from baseline over year, three groups were discerned: one with reduction of albuminuria of 30% or more, one with stable
145

CHAPTER 11
albuminuria (change less than 30%), and one with rapid progression of albuminuria of at least 30%. The correlation between rate of change in albuminuria at one year and cumulative event free survival was analyzed with the Kaplan-Meier method and log-rank test, as well as multivariate Cox regression. The 95% CI of the Hazard ratio was calculated as the exponent of the regression coefficient. P-values < 0.05 defined statistical significance. We used SPSS 12.0.1 (SPSS, Inc., Chicago, Illinois) for all analyses. RESULTS
Baseline characteristics of the three groups, including blood pressure and albuminuria, did not differ significantly, excepting age, which we corrected for. Albuminuria reduced from 69.1 mg/L at baseline to 39.4 mg/L after one year (mean difference -29.7 mg/L [95% CI, -39.7 to -19.8 mg/L; p < 0.0001]), and returned to 62.0 mg/L at the end of follow-up in the group with albuminuria reduction. In patients with rapid progression of albuminuria, mean levels were 84.3 mg/l at baseline, 223.3 mg/l after one year (mean difference 139.0 mg/L [95% CI, 46.3 to 209.7 mg/L; p < 0.01]) and 354.1 mg/L at the end of follow-up. Albuminuria levels did not change significantly in the group with stable albuminuria. Importantly, the course of blood pressure was similar in the three groups, without significant changes in neither systolic nor diastolic blood pressures (figure 1). During follow-up, 14 (21%) patients reached the composite endpoint. A significant difference in event free survival was observed between the three groups (p = 0.02). Patients with rapid progression of albuminuria were at highest risk to reach the endpoint, whereas patients with reduction in albuminuria of 30% or more were at lowest risk. After adjustment for sex, age, baseline systolic blood pressure, total cholesterol over HDL-cholesterol ratio and current smoking in a multivariate Cox regression model, change of albuminuria remained an independent, significant predictor (HR 5.1; 95% CI, 1.5-18.1; p = 0.01).
Relevant medication during follow-up was used similar in the three groups. Following the original study protocol, 63 (95%) patients received a RAAS inhibitor because of microalbuminuria. At the end of follow-up, 62 (93%) patients still used RAAS inhibition. Moreover, neither statin nor aspirin use differed between groups.
DISCUSSION This study demonstrates that normotensive patients with type 2 diabetes and microalbuminuria run a marked risk for cardiovascular complications. This risk depends on the rate of one year change in urinary albumin excretion. Patients with rapid progression of albuminuria were at highest risk, whereas patients with regression of
146

ALBUMINURIA PREDICTIVE OF CARDIOVASCULAR RISK
147
0
50
100
150
200
250
300
350
400
UAE dif ference < 30% ns
UAE reduction > 30% p < 0.0001
UAE progression > 30% p < 0.01
baseline 1 yr end of study
urin
ary
albu
min
exc
reti
on(m
g/l)
010
UAE dif ference < 30% ns
UAE reduction > 30% p < 0.0001
UAE progression > 30% p < 0.01
end of studybaseline 1 yr
130
135
140
145
syst
olic
blo
od p
ress
ure
(mm
Hg)
010
UAE difference < 30% ns
UAE reduction > 30% p < 0.0001
UAE progression > 30% p < 0.01
baseline 1 yr end of study
75
80
85
dias
tolic
blo
od p
ress
ure
(mm
Hg)
a
a
Figure 1. Mean urinary albumin excretion, systolic and diastolic blood pressure at baseline, after 1 year, and at the end of study in type 2 diabetic patients. Patients were divided into three groups based on albuminuria reduction after 1 year of treatment (stable albuminuria (< 30%; ns vs. baseline), reduction of albuminuria (> 30%; p < 0.0001 vs. baseline), rapid albuminuria progression (> 30%; p < 0.01 vs. baseline)). UAE, urinary albumin excretion.

CHAPTER 11
albuminuria have lowest risk. This association persisted after adjustment for the classical cardiovascular risk factors and baseline albuminuria. Besides reducing blood pressure, RAAS inhibitors are effective in preserving renal and cardiac function in diabetic patients (4-9). Moreover, they reduce albuminuria up to 40%, significantly more than other classes of antihypertensive drugs (4). Since albuminuria is strongly associated with cardiovascular outcome, changes in albuminuria during treatment might reflect changes in cardiovascular disease risk (13,14). Few studies recently showed that reduction of albuminuria in hypertensive diabetic patients reduces the risk of subsequent cardiovascular events (10-12,15). However, these studies investigated hypertensive patients, thereby leaving the possibility of blood pressure lowering being the explanation for the cardiovascular risk reduction, and albuminuria changes just being an innocent bystander. To our knowledge, our study is the first one demonstrating that, even with no appreciable changes or even rises in blood pressure, change in albuminuria differentiates the cardiovascular outcome in type 2 diabetic patients without hypertension.
An important limitation of this study is the small sample size. Nonetheless, the association we observed between changes in albuminuria and cardiovascular outcome were statistically significant in multivariate analysis. The strength of our study comprises the fact that we studied type 2 diabetic patients with microalbuminuria, but without hypertension at baseline, in a prospective design. Clearly, this study needs further follow-up in larger cohorts. In summary, sustained reduction in albuminuria reflected cardiovascular risk reduction in type 2 diabetic patients without hypertension. Hence, albuminuria during treatment seems to reveal therapeutic responsiveness independent of blood pressure changes, and therefore is useful as modifiable treatment goal. These observations advocate more aggressive approach of albuminuria in addition to more aggressive cardioprotective treatment in normotensive diabetic patients with remaining raised levels of albuminuria. REFERENCES 1. Gerstein HC, Mann JF, Yi Q et al. Albuminuria and risk of cardiovascular events, death, and heart failure in diabetic and nondiabetic
individuals. JAMA 2001;286:421-6 2. Arnlov J, Evans JC, Meigs JB et al. Low-grade albuminuria and incidence of cardiovascular disease events in nonhypertensive and
nondiabetic individuals: the Framingham Heart Study. Circulation 2005;112:969-75 3. Yuyun MF, Adler AI, Wareham NJ. What is the evidence that microalbuminuria is a predictor of cardiovascular disease events?
Curr Opin Nephron Hypertens 2005;14:271-6 4. Ruilope LM, Segura J. Losartan and other angiotensin II antagonists for nephropathy in type 2 diabetes mellitus: a review of the
clinical trial evidence. Clin Ther 2003;25:3044-64 5. Brenner BM, Cooper ME, de Zeeuw D et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2
diabetes and nephropathy. N Eng J Med 2001;345:861-9
148

ALBUMINURIA PREDICTIVE OF CARDIOVASCULAR RISK
6. Parving H-H, Lehnert H, Brochner-Mortensen J, Gomis R, Andersen S, Arner P. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N Eng J Med 2001;345:870-8
7. Zandbergen AA, Baggen MG, Lamberts SW, Bootsma AH, de Zeeuw D, Ouwendijk RJ. Effect of losartan on microalbuminuria in normotensive patients with type 2 diabetes mellitus. A randomized clinical trial. Ann Intern Med 2003;139:90-6
8. Dahlöf B, Devereux RB, Kjeldsen SE et al. Cardiovascular morbidity and mortality in the losartan intervention for endpoint reduction in hypertensive study (LIFE): a randomized trial against atenolol. Lancet 2002;359:995-1003
9. Heart Outcomes Prevention Evaluation Study Investigators. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med 2000;342:145-53
10. Ibsen H, Olsen MH, Wachtell K et al. Reduction in albuminuria translates to reduction in cardiovascular events in hypertensive patients. Losartan intervention for endpoint reduction in hypertension study. Hypertension 2005;45:198-202
11. De Zeeuw D, Remuzzi G, Parving H-H et al. Albuminuria, a therapeutic target for cardiovascular protection in type 2 diabetic patients with nephropathy. Circulation 2004;110:921-7
12. Yuyun MF, Dinneen SF, Edwards OM, Wood E, Wareham NJ. Absolute level and rate of change of albuminuria over 1 year independently predict mortality and cardiovascular events in patients with diabetic nephropathy. Diabet Med 2003;20:277-82
13. De Zeeuw D. Albuminuria, not only a cardiovascular / renal risk marker, but also a target for treatment? Kidney Int 2005;68:1899-1901
14. Burnier M, Zanchi A. Blockade of the renin-angiotensin-aldosterone system: a key therapeutic strategy to reduce renal and cardiovascular events in patients with diabetes. J Hypertens 2006;24:11-25
15. Ibsen H, Olsen MH, H Wachtell K et al. Does albuminuria predict cardiovascular outcomes on treatment with losartan versus atenolol in patients with diabetes, hypertension, and left ventricular hypertrophy? The LIFE study. Diabetes Care 2006;29:595-600
149

CHAPTER 11
150

GENERAL DISCUSSION

GENERAL DISCUSSION
152

Chapter 12
Towards optimal long-term protection of kidney and heart in the proteinuric renal patient Adapted and updated from: Liffert Vogt, Menno J.A. Kocks, Gozewijn D. Laverman and Gerjan Navis Minerva Medica 2004;95:395-409

CHAPTER 12
154

GENERAL DISCUSSION
Data of numerous clinical trials show that lowering of blood pressure is pre-requisite for reducing the rate of renal function loss in chronic renal disease. There is impressive evidence supporting that blood pressure lowering obtained by intervention in the renin-angiotensin-aldosterone system (RAAS) has an additive renoprotective effect over reduction of blood pressure alone, both in diabetic and non-diabetic renal diseases. The main evidence for renoprotective action of RAAS blockade is provided by its consistent antiproteinuric action, which cannot completely be attributed to the reduction in blood pressure. Proteinuria reduction during therapy is the single most important factor predicting the renal prognosis, independent from the class of drugs used. As a consequence, residual proteinuria predicts residual renal risk. Moreover, as proteinuria has been linked to an elevated cardiovascular risk, residual proteinuria predicts residual cardiovascular risk as well. In this thesis, different strategies directed to better reduction of residual proteinuria have been tested for the sake of better long-term protection. Despite effectiveness of these strategies on group level, not in all patients optimal levels of residual proteinuria could be achieved, as individual factors seem to determine the therapy response. In this chapter, the putative factors for therapy resistance are discussed. Experimental data from studies in rats suggest a specific involvement of intrarenal factors in therapy resistance. These factors comprise, among others, pre-existing renal damage and renal angiotensin-converting enzyme (ACE) activity. Identification of such factors in individual renal patients provides insight in mechanisms by which renoprotective strategies fail to overcome therapy resistance. This prompts for a dual approach to improve renoprotection, namely unravelling these specific intrarenal mechanisms on the one hand, and development of better strategies for early detection of renal risk on the other hand.
PROTEINURIA: INDEPENDENT RISK FACTOR FOR RENAL PROGNOSIS DURING THERAPY
Proteinuria is looked upon as an independent risk factor of progressive renal function loss and not merely a consequence of renal disease, as pointed out firstly by Remuzzi et al. (as also discussed in detail in chapter 1) (1). In different renal conditions, in the experimental as well as in the clinical setting, proteinuria consistently determines the rate of progression of renal function loss (1,2). Taken together with experimental data, demonstrating a tubulotoxic effect of leaked proteins (3), this may indeed point to the pathogenic role of proteinuria in progressive renal damage.
The relation between proteinuria and renal function decline appears to be three-fold. Firstly, the MDRD study (4) showed that baseline proteinuria was an important determinant of the renoprotective benefit in the follow-up after reduction of blood pressure. The additional benefit of a lower blood pressure goal was clearly more pronounced in patients with a higher baseline proteinuria. Secondly, from early studies,
155

CHAPTER 12
since the introduction of ACE inhibitors, it has been noted that not only baseline proteinuria, but also the extent to which proteinuria is lowered during antihypertensive treatment is of prognostic value, in diabetic as well as non-diabetic renal patients. For example, in patients with diabetes, it was demonstrated that the extent of reduction in proteinuria achieved during treatment with captopril was associated with a better long-term effect on the decline of renal function loss (5). Also, in non-diabetic patients, the short-term antiproteinuric response during enalapril treatment predicted the GFR decline during follow-up. The antiproteinuric response could be a marker of the accessibility to intervention, as such, but may point also towards the specific role of proteinuria. The latter possibility is supported by yet another consistent finding, that is: thirdly, the residual proteinuria during treatment is an important predictor of the subsequent rate of renal function loss. This phenomenon, which was firstly reported by Apperloo et al., appears to be independent of the class of drug, as the residual proteinuria during treatment was correlated with the subsequent progression of renal function loss, no matter what drug was used, be it ACE inhibitor or beta-blocker (6).
The above mentioned findings have now been confirmed by the results from large randomised clinical trials with ACE inhibitors as well as AT1 antagonists. The RENAAL study showed that the more one reduces proteinuria the better the renoprotection is achieved, defined as risk reduction for reaching end-stage renal disease. Moreover, not only the efficacy of reduction, but also the achieved level of residual proteinuria during therapy predicts renal outcome (7). Again, the observed benefit is independent from class of drugs used. The REIN trial showed baseline proteinuria to be an independent and accurate predictor of disease progression and end-stage renal disease (8). In response to treatment, a stronger short-term antiproteinuric effect was a predictor of more effective protection against end-stage renal disease in the long term (9). In addition, residual proteinuria during treatment predicted renal outcome independently. Finally, the COOPERATE trial confirmed the importance of proteinuria reduction as well, as the study revealed again that the initial antiproteinuric response was an independent predictor for the renal outcome (10).
The observation that residual proteinuria is a main prognostic factor has considerable implications for therapy, as measuring residual proteinuria during therapy can serve as short-term indicator of long-term renoprotective efficacy. This allows titration of therapy, especially using RAAS blockade, to obtain optimal renoprotection by reducing proteinuria as far as possible. Indeed, maximum reduction of proteinuria to lowest levels as a treatment target for each individual renal patient has been recommended (11-13). It has to be noted, however, that prospective studies titrating for proteinuria have not yet been performed, and prospective evidence that titrating for proteinuria will improve renoprotection is still lacking. In this respect, it may be
156

GENERAL DISCUSSION
relevant that experimental data indicate that residual proteinuria may reflect a poor renal prognosis as such; however, considering the evidence for an independent nephrotoxic effect of leaked proteins into urine, the next step in optimizing renoprotective strategies is obviously to titrate for proteinuria. Also, the COOPERATE trial indicates that individual response to therapy should be considered (10), since this study demonstrated that, despite much progress can be made by optimizing RAAS blockade on group level, large differences between patients exist in their antiproteinuric response and that these differences contribute to the renal prognosis. Thus, optimization of renoprotection will require an individual approach, aimed at reduction of blood pressure and, in addition, proteinuria. This approach awaits confirmation in a prospective fashion, but some inference can already be made from the results of a number of small studies, among the studies as described in this thesis. OPTIMIZING RENOPROTECTION BY TITRATING TO THE ANTIPROTEINURIC RESPONSE
Based on the data from the MDRD and REIN study, it has been advocated that the treatment target for proteinuria should be below 1 g/d, and likely near zero, for each renal patient to ensure optimal renoprotection (4,12). Different strategies, as also studied in this thesis, are available in order to optimize the antiproteinuric response during blockade of the RAAS (table 1).
Firstly, the effect of changing the time of dosing of RAAS blockade has been studied. It has been noted previously that urinary protein excretion displays a circadian rhythm with maximum excretion during the day (14). RAAS blockade by using a long-acting ACE inhibitor effectively reduced daytime proteinuria, whereas nocturnal proteinuria was less affected (15). By contrast, blood pressure reduction was for the complete 24 h reduced. These observations indicate the presence of relative nocturnal resistance to RAAS blockade as far as its antiproteinuric action is concerned. As daytime proteinuria can more successfully be reduced, residual proteinuria consequently is to a greater extent determined by urinary protein excretion during the night. In chapter Table 1. Strategies for optimizing the antiproteinuric response during RAAS blockade
• correction of volume excess: - restriction of dietary sodium to 50 mmol/d - combined treatment with diuretic - combined treatment with both above measures
• dose-titration with ACE inhibitor or AT1 antagonist to proteinuria further than needed for blood pressure control • combined treatment of both ACE inhibitor and AT1 antagonist • combined treatment with statin • intervention in prostaglandins: - combined treatment with NSAID
- combined treatment with selective COX-2 inhibitor Not effective strategies: • change of dosing time of ACE inhibitor from morning to evening or twice daily • combined treatment with urotensin II antagonist
157

CHAPTER 12
4, the switch of dosing time of the long-acting ACE inhibitor from morning to evening as well as the effect of two half doses twice daily were studied. Unfortunately, no benefit of such a change in dosing time could be observed.
Secondly, it has long since been shown that the therapeutic effects of ACE inhibitors depend on sodium status (16,17). The antiproteinuric response of ACE inhibition, for example, was almost annihilated by an increase of sodium intake from 50 mmol/d to 200 mmol/d in patients with proteinuria in the nephrotic range (16), whereas the blood pressure response was blunted to a lesser extent. Notably, a small study in non-diabetic proteinuric patients showed that this blunted antiproteinuric effect can be restored by the addition of diuretic therapy with hydrochlorothiazide (18). Combined effects of diuretic and low sodium intake had not been tested so far. In addition, whether these measures directed to a lower sodium status positively influence the response to AT1 antagonist treatment as they do to ACE inhibitor therapy has limitedly been studied (19). This issue was studied in chapter 5, in which the effects of these two measures inferring in the sodium status on proteinuria during AT1 antagonist therapy (losartan) were investigated. It was shown that low sodium intake reduced proteinuria as a single measure. Moreover, when added to losartan treatment, low sodium intake led to better proteinuria reduction than achieved by losartan monotherapy. Losartan in combination with hydrochlorothiazide had an equal effect on proteinuria as losartan combined with low sodium intake. Importantly, lowest proteinuria in this study was obtained by combining low sodium intake and hydrochlorothiazide on top of losartan treatment, emphasizing the high efficacy of intensified intervention in the sodium status in maximizing the antiproteinuric response to RAAS blockade.
Thirdly, different studies in diabetic and non-diabetic proteinuric patients demonstrate that the dose-response curves of RAAS blocking agents for blood pressure and proteinuria may not always be similar, as apparent from the IRMA-2 study (20,21). In this trial, irbesartan 150 mg QD showed less protection against progression to overt diabetic nephropathy than irbesartan 300 mg, accompanied with a less effective proteinuria reduction, but equal efficacy on blood pressure reduction. However, not all titration studies by using different AT1 antagonists, as candesartan, losartan or telmisartan, show that doses higher than the maximal recommended doses for blood pressure reduction will lead to better proteinuria reduction (22-26). By contrast, increasing the ACE inhibitor dose seems not to show such flattening in the dose-response, indicating that higher doses of ACE inhibitor than needed for blood pressure control result in further proteinuria reduction (27).
Fourthly, as mentioned earlier, the COOPERATE trial demonstrated better renoprotection by dual RAAS blockade with both an ACE inhibitor and an AT1 antagonist (10). Smaller clinical studies in diabetic as well as non-diabetic renal patients
158

GENERAL DISCUSSION
Figure 1. Number of non-diabetic proteinuric patients (n = 12) that reached proteinuria target (< 1 g/d; light-grey boxes), still had residual proteinuria (> 1 g/d; white boxes), or experienced side effects (dark-grey boxes) during a RAAS intervening titration protocol consisting of replacement of previous RAAS blocking therapy into irbesartan (Irb) 300mg/ diuretic, and subsequently dose-titration with lisinopril (Lis) to a maximal dose of 40 mg QD (adapted from: Vogt L et al. J Am Soc Nephrol 2005;16:S53-7).
showed that dual RAAS blockade led to further proteinuria reduction than with maximal recommended doses of monotherapy could be obtained (27-29), as also was observed in our study described in chapter 10. Finally, combination strategies with drugs not interfering in the RAAS, as prostaglandin synthesis inhibitors (i.e. NSAIDs) or statins, may also render higher antiproteinuric efficacy. These strategies will be discussed in more detail further on in this chapter.
Despite the demonstrated efficacy of these measures on group level, limited data are available on the feasibility to specifically pursue the lowest level of proteinuria by individual dose-titration with these measures. The small practice-based clinical study, described in chapter 6, in non-diabetic patients with residual proteinuria during previous RAAS intervening therapy indicates that individual titration for maximal RAAS blockade has its limits. In this study, individual dose-titration of an ACE inhibitor on top of high dosed AT1 antagonist therapy with diuretic in order to lower proteinuria below 1 g/d induced, as expected, further reduction of the residual proteinuria on group level. However, in regard with the individual patient, the treatment target was reached in a low number of patients as compared with the increasing amount of patients that experienced adverse events for each titration step (figure 1) (30). These findings indicate that attempts to intensify the RAAS blockade based regimen to overcome individual therapy resistance may not be the solution in all patients. Again, despite increasing
159

CHAPTER 12
efficacy at group level, measures for optimization will hardly be of benefit to poor responders. This prompts for specific focus on the individual determinants of therapy response, in order to design specific measures that improve therapy response in these high risk subjects.
INDIVIDUAL PATIENT FACTORS
As the individual differences in responsiveness to therapy are large, the individual effect of various measures that could in principle lead to an improved therapy response have been studied. The effect of dose on the antiproteinuric effect was studied in diabetic and non-diabetic patients by Bos et al. (31). Increasing the dose from 10 to 20 mg enalapril and from 50 to 100 mg losartan, respectively, improved the response at group level, but patients with a poor response to lower doses failed to catch up to good-responders at a higher dose. Moreover, in this study the shift from ACE inhibition to AT1 antagonist therapy was studied in the same patients, showing that patients with a poor response to the ACE inhibitor were also the poor responders to AT1 antagonist therapy (figure 2). It would be of interest whether combined blockade would be of benefit in these non-responders. Data from the COOPERATE trial, however, suggest that this is not the case, as this study showed that a poor therapy response during previous treatment with ACE inhibitors was independently predictive for the eventual renal outcome (10). However, to specifically answer this question, the appropriate design would be to stratify subjects as being poor vs. good responders to ACE inhibitors. This has been done in an experimental study in adriamycin-nephrotic rats, i.e. a model of proteinuria-related renal damage, which showed that addition of AT1 antagonist therapy or higher doses of an ACE inhibitor could not resolve resistance for single ACE inhibitor therapy (32).Other important measures to improve therapy resistance are sodium restriction and/or diuretic
Figure 2. Antiproteinuric effect of the ACE inhibitor enal-april 10 mg QD and the AT1 antagonist losartan 100 mg QD administered to the same patients. Close correlation is observed between the effect of both interventions in the same patient, be it in a non-diabetic (NDRD, triangles) or in diabetic (IDDM) proteinuric patient (adapted from Bos H et al. Kidney Int 2000;75(Suppl):S32-37).
160

GENERAL DISCUSSION
Figure 3. A. Individual data on residual proteinuria during ACE inhibitor monotherapy (ACEi) and high sodium intake (x-axis), and during ACEi combined with hydrochlorothiazde (HCT) during high sodium intake (HS), or combined with low sodium intake (LS) (y-axis). The regression lines for both combined therapies (continuous line: HCT; broken line: LS) and line of identity are given (adapted from Buter H et al. Nephrol Dial Transplant 1998;13:1682-5). B. Individual data on residual proteinuria during AT1 antagonist losartan monotherapy (AT1A) and high sodium intake (x-axis), and during AT1A during low sodium intake (LS), or during LS combined with hydrochlorothiazide (HCT) (y-axis). The regression lines for both combined therapies and line of identity are given (AT1A + HS vs. AT1A + LS (r = 0.86, p < 0.001; solid line); AT1A + HS vs. AT1A + LS + HCT (r = 0.40, p < 0.01; broken line)). Data from chapter 5 (Vogt L et al. J Am Soc Nephrol 2008 [in press])
therapy. The individual effects of these measures are shown in figure 3. Indeed, addition of a diuretic or dietary sodium restriction on top of ACE inhibition or AT1 antagonist therapy led to a better antiproteinuric response at group level. However, in subjects in whom the response was already relative poor during ACE inhibition and liberal sodium intake, also the residual proteinuria during sodium restriction and/or diuretic therapy remained highest (18). It supports the presence of individual factors as main determinants of therapy response. The factors involved in the individual resistance to antiproteinuric therapy are poorly understood, but several possibilities should be explored. Firstly, the underlying renal disorder may be relevant, as suggested by the observation that type 2 diabetic patients with overt nephropathy tend to have a worse response compared to non-diabetic patients (33). Secondly, the severity of the renal damage that is already present at onset of therapy may be of importance. A retrospective analysis of renal transplant recipients showed that the antiproteinuric effect of fosinopril in proteinuric renal transplant patients was inversely related to the degree of pre-existing histologically proven
161

CHAPTER 12
interstitial damage of the renal graft (34). Thirdly, genetic factors may be involved in the response to therapy as well (35). The insertion/deletion (I/D) gene polymorphism of the ACE gene strongly affects the plasma and cellular levels of ACE, with the D allele associated with increased levels. DD homozygosity displays higher ACE activity levels and is considered to be a renal risk factor in both diabetic and non-diabetic nephropathy, as this genotype is associated with a high rate of renal function loss, despite renoprotective therapy (36). Whereas the antiproteinuric and renal hemodynamic response to ACE inhibition at first glance does not appear to be modified by ACE genotype (37), a retrospective analysis suggests that the blunting of therapy response by high sodium may be particularly prominent in DD homozygote subjects, pointing towards gene-environment interaction between sodium status and ACE genotype (38). This was confirmed by prospective data, showing that high sodium intake appears to unmask the unfavourable phenotype in the ACE DD genotype with respect to the blood pressure and renal responsiveness to angiotensin I infusion in both healthy (non-diabetic) volunteers and uncomplicated diabetic subjects (39,40). This gene-environment interaction is of particular clinical interest, as sodium intake is accessible to therapeutic intervention, whereas genetic make-up is not. In terms of mechanisms underlying this gene-environmental interaction, the effect of high sodium intake on ACE activity would be of interest. RENAL MECHANISMS UNDERLYING RESISTANCE TO RAAS BLOCKADE
Recent experimental data could shed more light on several of the determinants of therapy resistance, i.e. the severity of pre-existing renal damage at onset of therapy, the role of the renal ACE activity, and the role of sodium status, particularly in relation to ACE activity. The role of pre-existent renal damage was prospectively studied in adriamycin-nephrotic rats. In this study, it was found that the efficacy of ACE inhibition in preventing development of focal glomerulosclerosis and macrophage influx was strongly set by prevalent damage before treatment, despite the fact that the extent of renal damage was still very mild and in fact did not exceed the stage of pre-fibrotic changes (41). A post hoc analysis of the REIN in renal patients support these findings from animal studies, as in this sub-study baseline GFR predicted renal outcome during ramipril treatment after 2.6 years follow-up (42). None of the patients within the highest tertile of baseline GFR reached the end-point of end-stage renal disease, whereas, importantly, in patients within the lowest tertile the risk of end-stage renal disease was lowered with 22% compared to those patients treated with conventional antihypertensives.
Also, the extent of inhibition of ACE activity specifically in renal tissue has been proposed to be an important determinant for the eventual renoprotection. In this respect,
162

GENERAL DISCUSSION
a recent study observed that both proteinuria and focal glomerulosclerosis strongly correlate with the amount of intrarenal ACE activity (43). However, plasma ACE activity was negatively associated with renal damage in this study. These findings indicate, firstly, the importance of intrarenal ACE as opposed to plasma ACE for the development of renal damage, and, secondly, that it is unlikely that the higher renal ACE in the animals with more damage is due to less effective dosing, as this would presumably have been reflected in plasma ACE as well. During AT1 antagonist therapy a higher intrarenal ACE activity correlated with a poor antiproteinuric and renoprotective efficacy. This suggests that intrarenal ACE activity may be the permissive or promoting factor in the processes by which proteinuria eventually leads to renal structural damage. It also suggests that intrarenal ACE activity during ACE inhibition (at least within the context of these well-established high doses) is not primarily governed by the pharmacologic properties of the drug. In line with this hypothesis, it would be of interest whether a circadian pattern of intrarenal ACE activity exists; such pattern is known to be present in relation to plasma ACE activity, with high activity during the night. If so, higher intrarenal ACE activity at night may serve as a logical explanation for the relative therapy resistance during the night despite treatment with a long-acting ACE inhibitor or changing the time of dosing of this drug. Moreover, the role of sodium intake on intrarenal ACE activity is very interesting, as recently studied by our group (44). Firstly, we found in normal Wistar rats that rats on a high sodium diet had a higher renal ACE activity than rats on a low sodium diet, whereas no difference in plasma ACE activity was observed (figure 4). When these healthy animals were treated with an ACE inhibitor, however, renal ACE was lowered equally, during high as well as low sodium diet. Subsequently, in diseased adriamycin rats, the
Figure 4. Effects of sodium intake on plasma and renal ACE activity in healthy male Wistar rats. During low sodium (LS; 0.05% NaCl) and high sodium intake (HS; 2.0% NaCl), plasma ACE activity (pACE) is not similar, but renal ACE activity (rACE) is increased in the animals on HS diet.
163

CHAPTER 12
Figure 5. Individual cross-over data on the modulating effect of dietary sodium shift on proteinuria (Uprot) and renal ACE (rACE) during ACE inhibition (ACEi) in two parallel treated groups of adriamycin rats. 5A: the shift from low sodium (LS; 0.05% NaCl) to high sodium diet (HS; 2.0% NaCl) resulted in an increase of both Uprot and rACE; 5B: the switch from HS to LS resulted in a reduction of Uprot and rACE activity—with a less pronounced or in some rats no reduction of rACE. The change of rACE was of borderline significance (figure adapted from data presented earlier in: Kocks MJA et al. J Am Soc Nephrol 2002;13:339A).
modulating effects of sodium intake on renal ACE activity and antiproteinuric response during ACE inhibition were studied. In this cross-over study with two paralleled treated groups of nephrotic rats, the shift from low to high sodium during ACE inhibition in one group led to a rise in proteinuria, paralleled by a rise in residual renal ACE activity. When shifting sodium-intake the other way round in the other group, a pronounced fall in proteinuria occurred, whilst renal ACE activity showed a less pronounced fall that, moreover, failed to reach statistical significance (figure 5). This supports the assumption that sodium-induced effects on renal ACE activity are relevant to therapy response. However, proteinuria as such could also lead to elevation of renal ACE (43,45). It cannot be dissected by these data whether the changes in renal ACE activity are primarily mediated by the change in sodium status or also by the change in proteinuria. This issue requires further study. Nevertheless, the experimental data suggest that sodium-induced rise in renal ACE activity may contribute to the vicious circle of
164

GENERAL DISCUSSION
proteinuria-induced rise in ACE activity with consequent renal damage. Therefore, probably not only proteinuria, but also the regulation of renal ACE activity may be important with respect to individual factors that determine responsiveness to RAAS blocking therapy (46). This may also explain the observations made in the human situation, as the dose-response for proteinuria may flatten during AT1 antagonist therapy, but not during ACE inhibition (27). Clearly, further studies are needed, though this may be somehow troublesome, since organ-specific ACE activity is difficult to measure in man.
As the human representation of difference in regulation of ACE activity, the impact of the ACE I/D gene polymorphism and its interaction with the sodium status for prognosis and therapy response are relevant. Subjects with the deletion (DD) genotype do not only have a higher plasma ACE activity (47) and higher mRNA (48), but have also been reported to have higher levels of renal ACE on immunohistochemical localization than patients with the ID or II genotype (49). In addition, as noted above, the ACE DD genotype is associated with a worse prognosis in diabetic and non-diabetic renal disease, although not invariably so (50). Related to sodium status, we already pointed out, that high sodium intake is a modifiable patient factor eliciting therapy resistance in man and experimental animals. Moreover, high sodium intake unmasks the unfavourable phenotype in the ACE DD genotype, both with respect to therapy response and to responsiveness to angiotensin I (but not angiotensin II) in non-diabetic and diabetic subjects (39,40). A possible explanation might be that high sodium intake stimulates tissue ACE activity, a notion also supported by human angiotensin I and II infusion data by Boddi et al. (51) and by our observations in animal studies. As plasma ACE is unaltered during high sodium, this would be expected to be an effect at tissue (e.g. renal) level.
Considering the impact of renal ACE on renal damage, it may be of interest that a homologue of ACE, ACE-2, was recently discovered (52). ACE-2 appears to counteract effects of ACE by cleaving angiotensin I into vaso-inactive angiotensin(1-9) and cleaving angiotensin II into the vasodilative angiotensin(1-7). Moreover, in the kidney it co-localises with ACE, a finding consistent with an alleged counteracting function (53,54). Future functional studies will have to elucidate the role of ACE-2 in the development and prevention of progressive renal damage.
In summary, for a large part, specific patient factors are involved in the individual therapy response to renoprotective intervention. These will have to be taken into account when pursuing optimization of intervention. The extent of renal damage at time of onset of treatment seems to be an important factor in the eventual outcome, indicating that renoprotective intervention directed to reduction of proteinuria should be started in the early stage of the renal disease. Moreover, the notion that intrarenal ACE
165

CHAPTER 12
activity depends on sodium state and is involved in the antiproteinuric response, stresses the importance of dietary sodium restriction, and suggests that dose-titrating with an ACE inhibitor might be more fruitful, as indicated by the specific action of ACE inhibitors on ACE. Finally, we found that rigorous application of different measures to optimize the effects of a RAAS blockade-based regimen in clinical practice may be troublesome, since the limits of this maximising approach are likely to be encountered in terms of tolerability (30). Therefore, alternative approaches should be explored to circumvent therapy resistance. OTHER MODES OF PHARMACOLOGIC INTERVENTION TO OVERCOME THERAPY RESISTANCE
Several non-RAAS intervening modes of therapy directed to optimization of renoprotection, such as blockade of the prostaglandin system or lipid-lowering drugs, would be relevant to explore, as was done in studies described in chapter 7 and 8. Different studies demonstrated that intervention in the prostaglandin synthesis by using NSAIDs has antiproteinuric properties with equal effectiveness as ACE inhibition (55,56). The mechanism by which blockade of the prostaglandin system exerts an antiproteinuric effect has been related to alteration of the intraglomerular pressure as reflected by a drop in FF and GFR. Indeed, treatment with indomethacin seems to slow down the progression of renal function decline on the long-term. Because of the poor tolerability of non-selective NSAIDs, it would be interesting to explore whether the relatively new selective COX-2 inhibitors, of whom lower rates of side effects are reported, share the antiproteinuric properties of NSAIDs. Unfortunately, the use of selective COX-2 inhibitors as an analgesic as well as a drug applied for broader therapeutical purposes became part of a violent debate, since the COX-2 inhibitor rofecoxib was withdrawn from the market by Merck & Co. in response to the interim results of the APPROVe study (57). In this randomized placebo-controlled study, a higher frequency of cardiovascular morbidity was reported in patients with a history of colorectal adenoma after 18 months of treatment with rofecoxib as compared to placebo. Yet, in regard to the results of the study as described in chapter 7, renal patients may benefit from selective COX-2 inhibitor therapy as these drugs may have renoprotective effects. Rofecoxib dose-dependently reduces proteinuria, albeit less effectively than the comparator drugs (i.e. the non-selective NSAID indomethacin and the ACE inhibitor lisinopril at optimal antiproteinuric doses). The combined therapy of rofecoxib and lisinopril could not be studied due to premature withdrawal of rofecoxib, but may be particularly of interest as individual patients with limited antiproteinuric therapy response to lisinopril did not benefit to change of treatment into single rofecoxib. So far, limited data suggest such addition of COX-2 inhibitors to RAAS blockade can overcome therapy resistance—at least at group level—during ACE inhibition in membranous
166

GENERAL DISCUSSION
glomerulopathy by further reducing residual proteinuria (58). In this respect, it is noteworthy that the RAAS and prostaglandin system are closely related in the maintenance of renal function. Intriguing experimental data have recently been published showing that during low sodium in combination with ACE inhibitor therapy, the usual vasodilative response by prostaglandins, counteracting angiotensin II-mediated vasoconstrictory effects in the kidney, seems to be shifted towards the production of vasoconstrictive prostaglandins (59). Moreover, COX-2 derived prostaglandins have been proposed to contribute to high-renin states, as reviewed earlier (60). The role of such prostaglandins and its pharmacological inhibition in relation to renoprotection is interesting to explore, especially during combined therapy of ACE inhibition and NSAIDs, compared to single therapy.
Another vasoactive system that has been hypothesized to play an important role in the pathophysiology of chronic renal failure and recently became accessible for pharmacological intervention comprises the urotensin system. The main effector peptide of this system urotensin II, initially described as the potent vasoconstrictor known in mammals (61), exerts its effects by modulating vascular tone, including the tone of the renal vasculature. Urotensin induced renal hemodynamic changes were hypothesized to contribute to the sequelae of renal function decline (i.e. elevated intraglomerular pressure, hyperfiltration, leakage of proteins into the urine and renal scarring). In different experimental models for renal failure, the newly developed urotensin II receptor antagonist palosuran had beneficial effects on renal function with delayed development of albuminuria. Interestingly, limited evidence shows that palosuran may reduce residual proteinuria when added to a regimen of RAAS blockade, whereas the well-conducted trial in diabetic nephropathy, as described in chapter 8, could not confirm such an added effect on residual proteinuria in patients already treated with RAAS blockade.
Not only prostaglandins and urotensin II may interfere with the therapy response during RAAS blockade, but also lipid lowering therapy. One experimental study reported that the antiproteinuric response and renoprotection was enhanced by the addition of statin therapy to ACE inhibition (62). Indeed, lipid-lowering drugs, statins in particular, may exert a renoprotective effect, independently from their lipid-lowering effect, as in renal patients and in hypertensive patients, long-term treatment with statins is reported to reduce proteinuria (63,64). In addition, it has been noted that elevated plasma levels of cholesterol are related to limited therapy response to RAAS blockade (65). Two prospective trials showed that statin treatment added to ACE inhibition and AT1 antagonist therapy improved proteinuria reduction in human as well (64,66). Finally, respecting progression of renal function loss, it should be noted that single statin treatment has been demonstrated to modestly reduce the rate of GFR decline,
167

CHAPTER 12
although no data on the risk of development of renal endpoints are available thus far (63,67). PROTEINURIA AND PROTEINURIA REDUCTION: CONSEQUENCES FOR CARDIOVASCULAR
RISK?
The question whether the different optimization strategies directed to proteinuria reduction and eventual renoprotection should be pursued for optimal cardiovascular risk reduction is still open. Recent epidemiologic studies in diverse populations have demonstrated that renal function as such is an important cardiovascular risk factor (68). Consequently, patients with chronic renal failure are characterized by an elevated cardiovascular risk. Proteinuric patients are considered to be at high risk for myocardial infarction and death indicating that proteinuria is a risk factor for cardiovascular outcome (69,70), as also demonstrated in the general population (71). Therefore, different risk factors, classical as well as renal, within the proteinuric patient seem contribute to the elevated cardiovascular risk, among others: blood pressure, presence of diabetes, renal function (GFR), proteinuria and lipid derangements. In this thesis, two studies indicate that the effect of antiproteinuric treatment in terms of cardiovascular risk seem to be related to the effects on lipid derangements as well as the amount of residual proteinuria.
Firstly, in proteinuric patients, plasma total cholesterol, very-low-density lipoprotein (VLDL), low-density lipoprotein (LDL) cholesterol and triglycerides (TG) are usually elevated (72). It should be noted that these abnormalities can be restored by reduction of proteinuria, no matter what antiproteinuric drug class is used. As outlined in chapter 9, proteinuria reduction is a main independent measure in the management of lipid abnormalities. Numerous studies have demonstrated the beneficial effect of proteinuria reduction on (V)LDL, as ApoB and (V)LDL cholesterol which are normally elevated in proteinuric conditions are reduced when proteinuria is reduced. As a rule of thumb, one could expect an 1 mmol/L reduction of plasma total cholesterol in response to a 2-3 g/d reduction of proteinuria. Moreover, proteinuria reduction has been linked to reductions in plasma levels of the highly atherogenic Lp[a] (73). These effects can be interpreted as cardioprotective in addition of the renoprotective effects of proteinuria. However, it has been noted that high-density lipoprotein (HDL), also regarded as an important factor in relation to cardiovascular risk, may negatively be influenced by pronounced proteinuria reduction (74). HDL plays an important role in the reverse cholesterol transport (RCT) pathway (75), which provides transport of excess cellular cholesterol from peripheral cells to the liver for metabolism and excretion in the bile. In chapter 10, HDL and components of the RCT have thoroughly been studied in proteinuric patients as well as the influence of proteinuria reduction. This study showed
168

GENERAL DISCUSSION
that cellular cholesterol efflux to plasma from proteinuric patients is enhanced, in conjunction with elevated pre-ß HDL formation and plasma PLTP activity. These changes could attenuate the increased cardiovascular risk associated with proteinuria-associated hyperlipidaemia. Moreover, antiproteinuric therapy lowered plasma apoB, but did not affect cell-derived cholesterol efflux, suggesting that proteinuria reduction beneficially affects cardiovascular risk by correction of the proteinuria-associated lipid derangements. Secondly, the effect of proteinuria reduction on cardiovascular risk is not well-established yet. In chapter 11, the effect of residual proteinuria on the development of cardiovascular events in diabetic nephropathy without the presence of hypertension was studied. It was demonstrated that residual proteinuria after 1 year from baseline predicted the occurrence of macrovascular events after a mean follow-up duration of 4.7 years. Detailed analysis of the study indicated that this effect was related to specific antiproteinuric effects as blood pressure was not reduced or even elevated after 1 year of therapy. In summary, optimization of proteinuria reduction is important for optimal cardiovascular risk reduction as residual proteinuria appears to be predictive for the development of major cardiovascular events on the one hand, and low residual proteinuria is related to a better lipid profile on the other hand. CONCLUSION
Reduction of blood pressure and proteinuria are pre-requisites for renoprotection. Blockade of the RAAS, by ACE inhibitors or AT1 antagonists, appears to be particularly effective in this respect. Important advances have been made in the efficacy of renoprotection. Nevertheless, therapy resistance is an important problem to tackle. Current evidence indicates that therapy resistance seems to be determined by individual factors with a role for specific intrarenal factors, such as pre-existing renal damage and renal ACE activity (the latter being modulated by genetic factors, sodium status, and renal damage as such). Further elucidation of the interplay between these factors may help to design additional strategies for renoprotection, and circumvention of therapy resistance. Such strategies will attenuate renal function decline and may afford additional cardiovascular risk reduction as well. So far, RAAS blockade remains the select strategy to reduce proteinuria.
169

CHAPTER 12
REFERENCES 1. Remuzzi G, Bertani T. Is glomerulosclerosis a consequence of altered glomerular permeability to macromolecules? Kidney Int
1990;38:384-94 2. Williams PS, Fass G, Bone JM. Renal pathology and proteinuria determine progression in untreated mild/moderate chronic renal
failure. Q J Med 1988;67:343-54 3. Abbate M, Zoja C, Corna D, Capitanio M, Bertani T, Remuzzi G. In progressive nephropathies, overload of tubular cells with
filtered proteins translates glomerular permeability dysfunction into cellular signals of interstitial inflammation. J Am Soc Nephrol 1998;9:1213-24
4. Peterson JC, Adler S, Burkart JM et al. Blood pressure control, proteinuria, and the progression of renal disease. Ann Intern Med 1995;123:754-62
5. Rossing P, Hommel E, Smidt UM, Parving H-H. Reduction in albuminuria predicts a beneficial effect on diminishing the progression of human diabetic nephropathy during antihypertensive treatment. Diabetologia 1994;37:511-6
6. Apperloo AJ, de Zeeuw D, de Jong PE. Short-term antiproteinuric response to antihypertensive therapy predicts long-term GFR decline in patients with non-diabetic renal disease. Kidney Int 1994;45(Suppl):174-8
7. De Zeeuw D, Remuzzi G, Parving H-H et al. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: Lessons from RENAAL. Kidney Int 2004;65:2309-20
8. Ruggenenti P, Perna A, Mosconi L, Pisoni R, Remuzzi G. Urinary protein excretion rate is the best independent predictor of ESRF in non-diabetic proteinuric chronic nephropathies. Kidney Int 1998;53:1209-16
9. Ruggenenti P, Perna A, Remuzzi G. Retarding progression of chronic renal disease: the neglected issue of residual proteinuria. Kidney Int 2003;63:2254-61
10. Nakao N, Yoshimura A, Morita H, Takada M, Kayano T, Ideura T. Combination treatment of angiotensin-II receptor blocker and angiotensin-converting enzyme inhibitor on non-diabetic renal disease (COOPERATE): a randomised controlled trial. Lancet 2003;361:117-24
11. De Jong PE, Navis G, de Zeeuw D. Renoprotective therapy: titration against urinary protein excretion. Lancet 1999;354:352-3 12. Ruggenenti P, Schieppati A, Remuzzi G. Progression, remission, regression of chronic renal disease. Lancet 2001;357:1601-8 13. Vogt L, Laverman GD, de Zeeuw D, Navis G. The COOPERATE trial. Lancet 2003;361:1055-6 14. Koopman MG, Krediet RT, Koomen GC, Strackee J, Arisz L. Circadian rhythm of proteinuria: consequences of the use of urinary
protein:creatinine ratios. Nephrol Dial Transplant 1989;4:9-14 15. Buter H, Hemmelder MH, van Paassen P et al. Is the antiproteinuric response to inhibition of the renin-angiotensin system less
effective during the night? Nephrol Dial Transplant 1997;12(Suppl. 2):53-6 16. Heeg JE, de Jong PE, van der Hem GK, de Zeeuw D. Efficacy and variability of the antiproteinuric effect of ACE inhibition by
lisinopril. Kidney Int 1989;36:272-9 17. Navis G, de Jong PE, Donker AJ, van der Hem GK, de Zeeuw D. Moderate sodium restriction in hypertensive subjects: renal effects
of ACE-inhibition. Kidney Int 1987;31:815-9 18. Buter H, Hemmelder MH, Navis G, de Jong PE, de Zeeuw D. The blunting of the antiproteinuric efficacy of ACE in-hibition during
high sodium intake can be restored by hydrochlorothiazide. Nephrol Dial Transplant 1998;13:1682-5 19. Esnault VL, Ekhlas A, Delcroix C, Moutel MG, Nguyen JM. Diuretic and enhanced sodium restriction results in improved
antiproteinuric response to RAS blocking agents. J Am Soc Nephrol 2005;16:474-81 20. Parving H-H, Lehnert H, Bröchner-Mortensen J, Gomis R, Andersen S, Arner P. The effect of irbesartan on the development of
diabetic nephropathy in patients with type 2 diabetes. N Engl J Med 2001;345:870-8 21. Laverman GD, Andersen S, Rossing P, Navis G, de Zeeuw D, Parving H-H. Renoprotection with and without blood pressure
reduction. Kidney Int 2005;94(Suppl):S54-9 22. Laverman GD, Henning RH, de Jong PE, Navis G, de Zeeuw D. Optimal dose of losartan in non-diabetic patients with nephrotic
range proteinuria. Am J Kidney Dis 2001;38:1381-4 23. Andersen S, Tarnow L, Rossing P, Hansen BV, Parving H-H. Renoprotective effects of angiotensin II receptor blockade in type 1
diabetic patients with diabetic nephropathy. Kidney Int 2000;57:601-6 24. Andersen S, Rossing P, Juhl TR, Deinum J, Parving H-H. Optimal dose of losartan for renoprotection in diabetic nephropathy.
Nephrol Dial Transplant 2002;17:1413-8 25. Schmieder RE, Klingbeil AU, Fleischmann EH, Veelken R, Delles C. Additional antiproteinuric effect of ultrahigh dose
candesartan: a double-blind, randomized, prospective study. J Am Soc Nephrol 2005;16:3038-45 26. Rossing K, Schjoedt KJ, Jensen BR, Boomsma F, Parving H-H. Enhanced renoprotective effects of ultrahigh doses of irbesartan in
patients with type 2 diabetes and microalbuminuria. Kidney Int. 2005;68:1190-8 27. Laverman GD, Navis G, Henning RH, de Jong PE, de Zeeuw D. Dual renin-angiotensin system blockade at optimal doses for
proteinuria. Kidney Int 2002;62:1020-5 28. Rossing K, Jacobsen P, Pietraszek L, Parving H-H. Renoprotective effects of adding angiotensin II receptor blocker to maximal
recommended doses of ACE inhibitor in diabetic nephropathy: a randomised double-blind crossover trial. Diabetes Care 2003;26:2268-74
29. Jacobsen P, Andersen S, Rossing K, Jensen BR, Parving H-H. Dual blockade of the renin-angiotensin system versus maximal recommended dose of ACE inhibition in diabetic nephropathy. Kidney Int 2003;63:1874-80
30. Vogt L, Navis G, de Zeeuw D. Individual titration for maximal blockade of the renin-angiotensin system in proteinuric patients: a feasible strategy? J Am Soc Nephrol 2005;16(Suppl 1):S53-7
31. Bos H, Andersen S, Rossing P et al. The role of patient factors in therapy resistance to antiproteinuric intervention in non-diabetic and diabetic nephropathy. Kidney Int 2000;75(Suppl):32-7
32. Bos H, Henning RH, De Boer E et al. Addition of AT1 blocker fails to overcome resistance to ACE inhibition in adriamycin nephrosis. Kidney Int 2002;61:473-80
33. Ruggenenti P, Laverman GD, Chiurchiu C, Remuzzi G. Ineffectiveness of high dose ACE inhibitor therapy in overt nephropathy of type 2 diabetes. In: Laverman GD. Optimisation of renoprotective therapy with RAS blockade. Thesis. Groningen: University of Groningen 2004 pp. 113-26
34. Lufft V, Kliem V, Hamkens A et al. Antiproteinuric efficacy of fosinopril after renal transplantation is detemined by the extent of vascular and tubulointerstitial damage. Clin Transplant 1998;12:409-15
35. Marshall A. Laying the foundations for personalized medicines. Nat Biotechnol 1997;15:954-7 36. Van Essen GG, Rensma PL, de Zeeuw D et al. Association between angiotensin-converting-enzyme gene polymorphism and failure
of renoprotective therapy. Lancet 1996;347:94-5
170

GENERAL DISCUSSION
37. Van der Kleij FGH, Navis G, Gansevoort RT et al. ACE polymorphism does not determine short-term renal response to ACE inhibition in proteinuric patients. Nephrol Dial Transplant 1997;12(Suppl):S42-6
38. Van der Kleij FGH, Schmidt A, Navis G et al. ACE I/D polymorphism and short-term renal response to ACE inhibition; role of sodium status. Kidney Int 1998;53(Suppl):S23-6
39. Van der Kleij FGH, de Jong PE, Henning RH, de Zeeuw D, Navis G. Enhanced responses of blood pressure, renal function and aldosterone to angiotensin I in DD genotype are blunted by low sodium intake. J Am Soc Nephrol 2002;13:1025-33
40. Luik PT, Hoogenberg K, Kersten MN et al. The influence of the ACE (I/D) polymorphism on systemic and renal vascular responses to angiotensins in normotensive, normoalbuminuric type I diabetes mellitus. Diabetologia 2003;46:1131-9
41. Kramer AB, Laverman GD, van Goor H, Navis G. Inter-individual differences in anti-proteinuric response to ACEi in established adriamycin nephrotic rats are predicted by pretreatment renal damage. J Pathol 3003;201:160-7
42. Ruggenenti P, Perna A, Remuzzi G. ACE-inhibitors to prevent end-stage renal disease: when to start and why possibly never stop: a post hoc analysis of the REIN trial results. J Am Soc Nephrol 2001;12:2832-7
43. Bos H, Laverman GD, Henning RH et al. Involvement of renal ACE activity in proteinuria-associated renal damage in untreated and treated adriamycin nephrotic rats. J Renin Angiotensin Aldosterone Syst 2003;4:106-12
44. Kocks MJA, van Goor H, de Zeeuw D, Navis G. High residual renal ACE during ACE inhibition and high sodium intake: cause or consequence of therapy resistance to ACE inhibition? J Am Soc Nephrol 2002;13:339A
45. Largo R, Gomez-Garre D, Soto K et al. Angiotensin-converting enzyme is upregulated in the proximal tubules of rats with intense proteinuria. Hypertension 1999;33:732-9
46. Laverman GD, van Goor H, Henning RH, de Jong PE, de Zeeuw D, Navis G. Residual renal ACE activity during ACE inhibition predicts resistance to renoprotection in nephrotic rats. J Am Soc Nephrol 2001;12:818A
47. Rigat B, Hubert C, Alhenc-Gelas F, Cambien F, Corvol P, Soubrier F. An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J Clin Invest 1990;86:1343-6
48. Mizuiri S, Hemmi H, Kumanomidou H et al. Angiotensin-converting enzyme (ACE) I/D genotype and renal ACE gene expression. Kidney Int 2001;60:1124-30
49. Mizuiri S, Yoshikawa H, Tanegashima M et al. Renal ACE immunohistochemical localization in NIDDM patients with nephropathy. Am J Kidney Dis 1998;31:301-7
50. Boonstra AH, de Zeeuw D, de Jong PE, Navis G. Role of genetic variability in the renin-angiotensin system in diabetic and non-diabetic renal disease. Seminars in Nephrology 2001;21:580-92
51. Boddi M, Poggesi L, Coppo M et al. Human vascular renin-angiotensin system and its functional changes in relation to different sodium intakes. Hypertension 1998;31:836-42
52. Donoghue M, Hsieh F, Baronas E et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res 2000;87:E1-9
53. Hamming I, Lely AT, Navis G, van Goor H. Renal ACE2 expression parallels renal ACE expression: comparison of different rat strains and experimental condition. J Am Soc Nephrol 2003;14:412A
54. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 2004 Jun;203(2):631-7
55. Heeg JE, de Jong PE, Vriesendorp R, de Zeeuw D. Additive antiproteinuric effect of the NSAID indomethacin and the ACE inhibitor lisinopril. Am J Nephrol 1990;10:94-7
56. Garini G, Mazzi A, Buzio C, Mutti A, Allegri L, Savazzi G, Borghetti A. Renal effects of captopril, indomethacin and nifedipine in nephrotic patients after an oral protein load. Nephrol Dial Transplant 1996;11:628-34
57. Bresalier RS, Sandler RS, Quan H et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med 2005;352:1092-102
58. Costanzi S, Sturniolo A, Fulignati P et al. Cyclooxygenase (COX)-2 selective inhibitors + ACE inhibitors: a new protocol in the treatment of heavy proteinuria. Nephrol Dial Transplant 2003;18:79A
59. Kocks MJ, Gschwend S, De Zeeuw D, Navis GJ, Buikema H. Low Sodium Modifies the Vascular Effects of ACE Inhibitor Therapy in Healthy Rats. J Pharmacol Exp Ther 2004; 10:1183-9
60. Harris RC, Breyer MD. Physiological regulation of cyclooxygenase-2 in the kidney. Am J Physiol Renal Physiol 2001;281:F1-11 61. Ashton N. Renal and vascular actions of urotensin II. Kidney Int 2006;70:624-962. Zoja C, Corna D, Rottoli D et al. Effect of combining ACE inhibitor and statin in severe experimental nephropathy. Kidney Int
2002;61:1635-45 63. Fried LF, Orchard TJ, Kasiske BL. Effects of lipid reduction on the progression of renal disease: a meta-analysis. Kidney Int
2001;59:260-9 64. Bianchi S, Bigazzi R, Caiazza A, Campese VM. A controlled, prospective study of the effects of atorvastatin on proteinuria and
progression of kidney disease. Am J Kidney Dis 2003;41:565-70 65. Bos H, Henning RH, De Jong PE, De Zeeuw D, Navis G. Do severe systemic sequelae of proteinuria modulate the antiproteinuric
response to chronic ACE inhibition? Nephrol Dial Transplant 2002;17:793-7 66. Lee TM, Su SF, Tsai CH. Effect of pravastatin on proteinuria in patients with well-controlled hypertension. Hypertension
2002;40:67-73 67. Ozsoy RC, van der Steeg WA, Kastelein JJP, Arisz L, Koopman MG. Dylipidaemia as predictor of progressive renal failure and the
impact of treatment with atorvastatin. Nephrol Dial Transplant 2007;22:1578-86 68. Weiner DE, Tighiouart H, Amin MG et al. Chronic kidney disease as a risk factor for cardiovascular disease and all-cause mortality:
a pooled analysis of community-based studies. J Am Soc Nephrol 2004;15:1307-15 69. Ordonez JD, Hiatt RA, Killebrew EJ, Fireman BH. The increased risk of coronary heart disease associated with nephrotic syndrome.
Kidney Int 1993;44:638-42 70. Mahmoodi BK, Ten Kate MK, Waanders F, Veeger NJ, Brouwer JLP, Vogt L, Navis G, van der Meer J. High absolute risks and
predictors of venous and arterial thromboembolic events in patients with nephrotic syndrome. Results from a large retrospective cohort study. Circulation 2008;117:224-30.
71. Hillege HL, Fidler V, Diercks GF et al. Urinary albumin excretion predicts cardiovascular and noncardiovascular mortality in general population. Circulation 2002;106:1777-82
72. Vogt L, Laverman GD, Dullaart RPF, Navis G. Lipid management in the proteinuric patient: do not overlook the importance of proteinuria reduction. Nephrol Dial Transpl 2004;19:5-8
73. Gansevoort RT, Heeg JE, Dikkeschei FD, de Zeeuw D, de Jong PE, Dullaart RP. Symptomatic antiproteinuric treatment decreases serum lipoprotein (a) concentration in patients with glomerular proteinuria. Nephrol Dial Transplant. 1994;9:244-50
171

CHAPTER 12
74. Ruggenenti P, Mise N, Pisoni R et al. Diverse effects of increasing lisinopril doses on lipid abnormalities in chronic nephropathies. Circulation 2003;107:586-92
75. Fielding CJ, Fielding PE. Molecular physiology of reverse cholesterol transport. J Lipid Res 1995;36:211-28
172

173
LIST OF ABBREVIATIONS ABCD Appropriate Blood pressure Control in Diabetes ABPM ambulatory blood pressure monitoring ACE angiotensin-converting enzyme AIPRI ACE Inhibition in Progressive Renal Insufficiency Apo apolipoprotein APPROVe Adenomatous Polyp PRevention On Vioxx trial ARAMIS Angiotensin II Receptor Antagonist Micardis in Isolated Systolic hypertension
trial AT1 angiotensin II type 1 receptor BMI body mass index BID bis in die CARE Cholesterol And Recurrent Events trial CETP cholesteryl ester transfer protein COOPERATE Combination treatment of angiotensin-II receptor blocker and angiotensin-
converting-enzyme inhibitor in non-diabetic renal disease study COX cyclooxygenase DBP diastolic blood pressure FF filtration fraction GFR glomerular filtration rate HCT hydrochlorothiazide HDL high-density lipoprotein HOPE Heart Outcome Prevention Evaluation study HS high sodium diet IDNT Irbesartan in Diabetic Nephropathy Trial IRMA Irbesartan Microalbuminuria Type 2 Diabetes in Hypertensive Patients trial LCAT lecithin-cholesterol acyltransferase LDL low-density lipoprotein LS low sodium diet MAP mean arterial pressure MDRD Modification of Diet in Renal Disease study MFRIT Multiple Risk Factor Intervention Trial NSAID non-steroidal anti-inflammatory drug PG prostaglandin PLTP phospholipid transfer protein PREVEND (IT) Prevention of REnal and Vascular ENdstage Disease (Intervention Trial) study QD quaque die RAAS renin-angiotensin-aldosterone system REIN Ramipril Efficacy In Nephropathy trial RENAAL Reduction of Endpoints in NIDDM with the Angiotensin II Antagonist Losartan
trial RCT reverse cholesterol transport SBP systolic blood pressure TID ter in die UKPDS United Kingdom Prospective Diabetes Study UAE urinary albumin excretion Uprot urinary protein excretion VLDL very-low-density lipoprotein

174

NEDERLANDSE SAMENVATTING
NEDERLANDSE SAMENVATTING ‘ romen van een wereld zonder dialyse’, luidt het citaat waarmee het eerste hoofdstuk van dit proefschrift begint naar aanleiding van de resultaten van de REIN studie. Uit deze studie, een groot opgezet geneesmiddelenonderzoek met patiënten die lijden aan een chronische nierziekte, bleek dat toen deze patiënten behandeld werden met een bepaalde klasse van bloeddrukverlagende geneesmiddelen, de zogenaamde ACE-remmer, gedurende 4 jaar observatie in geen van de patiënten de nierfunctie zo ernstig achteruitging dat dialyse noodzakelijk was. Het zijn hoopvolle resultaten, omdat de aanwezigheid van een chronische nierziekte—ongeacht de onderliggende oorzaak—een geleidelijke achteruitgang van de nierfunctie betekent, die uiteindelijk kan resulteren in eindstadiumnierfalen en, als geen nierfunctievervangende therapie wordt toegepast, overlijden. Volgens cijfers van de Nierstichting waren in 2006 11.000 patiënten afhankelijk van een vorm van nierfunctievervangende therapie, waarvan 5.500 dialyseafhankelijk waren. Dit aantal groeit nog steeds. Dialyse betekent een levensreddende therapie, maar vormt eveneens een ernstige aanslag op het leven van de patiënt, doordat de kwaliteit van leven er flink op achteruitgaat en bovendien het risico op voortijdige dood aan hart- en vaatziekten sterk verhoogd is. De jaarlijkse mortaliteit wordt geschat op 20%. Er bestaat dus een onmiskenbaar belang om de voortschrijding van nierfunctieachteruitgang tot eindstadiumnierfalen een halt toe te roepen. Genees-middelstudies met patiënten die lijden aan een chronische nierziekte, zoals de REIN trial, laten zien dat dit wellicht mogelijk is.
D
Nadere bestudering van de geneesmiddelenstudies die tot dusver verricht zijn in patiënten met een chronische nierziekte wijst op twee belangrijke factoren die de mate van nierfunctieachteruitgang voorspellen. Ten eerste laten deze studies zien dat hoge bloeddruk niet alleen een uiting van de nierziekte is maar tevens de belangrijkste risicofactor voor nierfunctieachteruitgang. Het nierfunctiebeschermende effect van
175

bloeddrukverlagende geneesmiddelen valt hiermee te verklaren. Ofwel: hoe lager de bloeddruk, hoe meer nierbescherming. Ten tweede laten recente studies met bloeddruk-verlagende geneesmiddelen zien dat het uitmaakt welk geneesmiddel gekozen wordt ter behandeling van de chronische nierziekte. Het blijkt dat een specifieke groep van bloeddrukverlagende geneesmiddelen eveneens de eiwituitscheiding door de nier in de urine (proteïnurie) verlagen. Proteïnurie is, net als hoge bloeddruk, een uiting van het bestaan van een nierziekte, maar proteïnurie draagt bovendien bij aan de voortschrijding van nierfunctieachteruitgang. Het proteïnurieverlagende effect van een geneesmiddel in samenhang met goede bloeddrukregulatie bepaalt in sterke mate de effectiviteit van de behandeling op lange termijn. Ofwel: hoe lager de proteïnurie, hoe meer nierbe-scherming en wellicht een lager risico op de ontwikkeling van hart- en vaatziekten. DOEL VAN HET ONDERZOEK
De vraag op welke wijze het doel van een zo laag mogelijke proteïnurie, als maat voor de te verwachten nierfunctiebescherming op lange termijn, in patiënten met chronische nierinsufficiëntie kan worden behaald staat centraal. Hiertoe zijn verschillende strategieën onderzocht. De mogelijkheden om de proteïnurieverlaging zo optimaal mogelijk te vergroten door beter gebruik te maken van de huidige toegepaste geneesmiddelen zijn onderzocht, alsmede de effecten van nieuw ontwikkelde genees-middelen op de proteïnurie. Als laatste is het effect van effectievere proteïnurieverlaging op het cholesterolmetabolisme en het risico op de ontwikkeling van hart- en vaatziekten onderzocht. Deel I. Nierfunctiebescherming: meer dan bloeddrukverlaging alleen? In het overzichtsartikel van hoofdstuk 2 worden de meeste geneesmiddelstudies opgesomd die verricht zijn in patiënten met chronische nierziekte. Het blijkt dat bloeddrukverlagende geneesmiddelen die aangrijpen op het renine-angiotensine-aldosteronsysteem (RAAS) een specifiek voordeel hebben boven andere bloeddruk-verlagers. Behandeling met deze zogenaamde RAAS-blokkers brengt een lager risico op de ontwikkeling van eindstadiumnierfalen met zich mee. Het specifieke beschermende effect van RAAS-blokkers lijkt bij nadere bestudering van de onderzoeksdata van deze geneesmiddelstudies telkens gerelateerd te zijn aan een proteïnurieverlagend effect. RAAS-blokkers worden onderscheiden in ACE-remmers en AT1-receptorantagonisten. Hoofdstuk 3 laat zien dat de bloeddrukverlager telmisartan, een AT1-receptorantagonist, na 6 weken net zo effectief de bloeddruk verlaagt als hydrochloorthiazide, een niet-RAAS-blokker, in patiënten met een verhoogde systolische bloeddruk. In deze patiënten werd de proteïnurie echter alleen door telmisartan verlaagd, wijzend op een specifiek
176

NEDERLANDSE SAMENVATTING
bloeddrukonafhankelijk effect van RAAS-blokkers, dat belangrijk is voor de bescherming van de nier. Deel II. Optimaliseren van nierfunctiebescherming: optimaliseren van proteïnurie-reductie RAAS-blokkers mogen dan zeer effectief zijn in het afremmen van de snelheid van nierfunctieachteruitgang, toch ontwikkelen nog steeds veel patiënten uiteindelijk eindstadiumnierfalen. Drie verschillende studies zijn verricht in patiënten met een niet-diabetische nierziekte om de effectiviteit van RAAS-blokkers, gemeten aan hun proteïnurieverlagende effect, te vergroten. In hoofdstuk 4 wordt nagegaan of door het veranderen van het tijdstip van doseren van de lang werkende ACE-remmer trandolapril van ’s ochtends naar ’s avonds de proteïnurie lager wordt. Een eerdere studie liet name-lijk zien dat bij ‘s ochtends doseren van trandolapril de nachtelijke eiwituitscheiding in de urine veel minder verlaagd werd dan overdag. Onze studie laat zien dat een verandering van doseertijdstip van trandolapril de proteïnurie niet beïnvloedt. Hoofdstuk 5 laat zien dat de beïnvloeding van de water- en zouthuishouding belangrijk is voor de proteïnurieverlagende effectiviteit van de AT1-antagonist losartan. Wanneer de patiënten tijdens losartantherapie zich aan een zoutbeperkt dieet hielden, bleek het proteïnurieverlagende effect te worden vergroot. In plaats van een zoutbeperking bleek de toevoeging van de plastablet hydrochloorthiazide aan losartan even effectief te zijn. De combinatie van zoutbeperking en hydrochloorthiazide leidde tot de beste proteïnurieverlaging tijdens losartantherapie. Bovendien bleek dat tijdens de placebo-behandeling (de periode zonder losartan) een zoutbeperking de proteïnurie verlaagde. Deze bevinding wijst op de mogelijkheid dat een zoutbeperking op zichzelf een nierfunctiebeschermend effect kan uitoefenen. In hoofdstuk 6 zijn verschillende strategieën stap voor stap gecombineerd die het proteïnurieverlagende effect van RAAS-blokkade kunnen vergroten met als doel de proteïnurie te verlagen tot onder het niveau van 1 g/d. Eerder onderzoek heeft uitgewezen dat bij deze proteïnuriewaarde een stabilisatie van de nierfunctie te verwachten valt. Om het proteïnuriedoel te bereiken kregen patiënten een zoutbeperkt dieet voorgeschreven en werden behandeld met de AT1-antagonist irbesartan in hoge dosering. Vervolgens werd de ACE-remmer lisinopril in opklimmende doseringen toegevoegd. Indien bij een patiënt het proteïnuriedoel gedurende de stapsgewijze behandeling was bereikt of een bijwerking was opgetreden, verliet de patiënt het studieprotocol. Het bleek slechts bij een aantal patiënten mogelijk met deze titratiestrategie het behandelingsdoel te bereiken; dit voordeel ging echter ten koste van het optreden van bijwerkingen in een even zo groot aantal patiënten. Verbetering van de proteïnurieverlaging door maximaal gebruik te maken van RAAS-blokkade is hiermee een effectieve, maar niet voor alle patiënten haalbare strategie.
177

Deel III. Optimaliseren van nierfunctiebescherming: ingrijpen in andere regelsystemen In dit deel zijn de proteïnurieverlagende effecten van twee strategieën die niet op het RAAS aangrijpen onderzocht. Ten eerste is in hoofdstuk 7 het effect van twee doseringen van de COX-2-remmer rofecoxib vergeleken met indomethacine en de ACE-remmer lisinopril. COX-2-remmers omvatten een nieuwe klasse binnen de prostaglandinesyntheseremmers, die normaal gesproken als pijnstiller worden toegepast. De oudere generatie van deze remmers, zoals indomethacine, heeft reeds in de jaren ’70 uitgewezen effectief te zijn in de behandeling van proteïnurie. In verband met het veelvuldig optreden van bijwerkingen tijdens gebruik van deze oudere prostaglandine-syntheseremmers, zoals maag- en darmbloedingen en klachten van het centrale zenuwstelsel, is nader onderzoek naar de mogelijk nierfunctiebeschermende effecten nooit voldoende uitgevoerd. Van de COX-2-remmer rofecoxib (VIOXX®) is gebleken dat minder frequent deze bijwerkingen optreden. Het was tot dusver niet bekend of behandeling met rofecoxib zou leiden tot proteïnurieverlaging. Onze studie demonstreert dat rofecoxib dosisafhankelijk de proteïnurie verlaagt—weliswaar minder effectief dan indomethacine of lisinopril. Een opvallende bevinding is dat de bloeddruk tijdens de behandeling met rofecoxib niet verlaagd was. De COX-2-remmers zouden dienten-gevolge vooral toegepast kunnen worden in patiënten die gekenmerkt worden door een lage bloeddruk, waarbij verder verlaging als gevolg van behandeling met RAAS-blokkers niet wenselijk is. Recentelijk is rofecoxib echter van de markt gehaald omdat bij langdurig gebruik ervan gebleken is, dat er een hoger risico op het ontwikkelen van een hartaanval of herseninfarct bestaat. De toepassing van rofecoxib in proteïnurie-patiënten lijkt daarmee van de baan. In hoofdstuk 8 is het effect van de urotensine-II-receptorantagonist palosuran op proteïnurie in patiënten met een nierziekte veroorzaakt door diabetes mellitus type II onderzocht. Deze patiënten werden reeds behandeld met RAAS-blokkers en hadden ondanks deze therapie toch proteïnurie. In eerder onderzoek is aangetoond dat het urotensinesysteem een pathofysiologische rol speelt in verschillende nierziekten en ook in het bestaan van hoge bloeddruk. Blokkade van het systeem met palosuran bleek in onze studie geen effect te hebben op de proteïnurie. Deel IV. Optimaliseren van nierfunctiebescherming: verlaging van het risico op hart- en vaatziekte? In de voorgaande hoofdstukken is ingegaan op het beschermende effect proteïnuriever-laging op de nierfunctie. In het opiniestuk van hoofdstuk 9 wordt besproken dat proteïnurie eveneens gepaard gaat met een verhoogd risico op hart- en vaatziekten. Een andere belangrijke risicofactor voor hart- en vaatziekte is een veranderd vetspectrum in het bloed, zoals een verhoogd cholesterolgehalte. Het is bekend dat proteïnurie geassocieerd is met een veranderd vetspectrum. Het precieze mechanisme hierachter is
178

NEDERLANDSE SAMENVATTING
niet helemaal duidelijk, maar deze bevinding zou kunnen verklaren waarom proteïnurie bijdraagt aan het verhoogde risico op hart- en vaatziekten. Hoofdstuk 10 laat zien dat proteïnurieverlaging als het gevolg van behandeling met RAAS-blokkers leidt tot een verbetering van het vetspectrum—een effect dat het verhoogde risico op hart- en vaatziekten gunstig zou kunnen beïnvloeden. Hoofdstuk 11 demonstreert dat de hoeveelheid proteïnurie op zichzelf ook bepalend is voor het risico op hart- en vaatziekten. In patiënten met een nierziekte tengevolge van diabetes mellitus type II bleek dat de patiënten bij wie de proteïnurie het succesvolst verlaagd was, gekenmerkt werden door een lager risico op de ontwikkeling van een hart- of vaataandoening of overlijden op de lange termijn. Optimaliseren van de proteïnurieverlaging is dus niet alleen voordelig voor de nier, maar lijkt ook een belangrijk onafhankelijk therapiedoel ter bescherming van hart en vaten. AANBEVELINGEN
Voor optimale nierfunctiebescherming, gemeten aan de proteïnurie, in patiënten met een chronische nierziekte dienen verschillende strategieën te worden toegepast. Hiertoe is de toepassing van een specifieke groep bloeddrukverlagende geneesmiddelen, de RAAS-blokkers, in het bijzonder geschikt. Het proteïnurieverlagende effect van deze geneesmiddelen kan worden vergroot door een zoutbeperkt dieet en een plastablet toe te voegen. Hogere doseringen van RAAS-blokkers evenals het combineren van de ACE-remmer en de AT1-receptorantagonist kunnen de proteïnurieverlaging ook verbeteren. De toepassing van COX-2-remmers is controversieel maar zou kunnen worden gebruikt in bijzondere situaties, zoals het bestaan van een (laag)normale bloeddruk. Optimale proteïnurieverlaging is niet alleen voordelig voor de nier, maar is ook een behandelings-strategie die leidt tot verbetering van het vetspectrum in het bloed en samenhangt met een lager risico op de ontwikkeling van hart- en vaatziekten.
179

180

DANKWOORD Dit proefschrift omvat de resultaten van drieënhalf jaar onderzoek dat ik verricht heb op de afdelingen nefrologie van het Universitair Medisch Centrum Groningen, Medisch Centrum Leeuwarden en het Twenteborgziekenhuis te Almelo. In tegenstelling tot de wijze waarop de buitenwereld soms tegen wetenschappers aankijkt, verloopt het doen van onderzoek niet in volledige eenzaamheid. Een woord van dank is daarom ten zeerste op zijn plaats aan allen die een bijdrage hebben geleverd aan de totstandkoming van dit proefschrift. Een aantal van hen wil ik bij naam noemen.
Allereerst zou ik de deelnemende patiënten aan de onderzoeken binnen de verschillende ziekenhuizen van harte willen bedanken. Hun bereidheid om vele malen de polikliniek te bezoeken, nauwkeurig 24-uursurines te verzamelen, zich strikt aan verschillende diëten te houden en bovendien extra of nieuwe medicijnen te gebruiken heeft een schat aan informatie opgeleverd, waarop dit proefschrift is gebaseerd.
Graag bedank ik mijn promotoren Gerjan Navis en Dick de Zeeuw. Twee persoonlijkheden die mij met grote deskundigheid, creativiteit en enthousiasme hebben begeleid. De mate van vrijheid die ik mocht genieten om mijn weg in het onderzoek te ontdekken heb ik uiterst gewaardeerd. Het heeft ondermeer geleid tot de betrokkenheid van mijn co-promotoren Robin Dullaart en Arend Jan Woittiez bij het project. Dankzij Robin heb ik me kunnen verdiepen in het cholesterolmetabolisme van de proteïnuriepatiënt, waardoor dit onderwerp een belangrijke plaats inneemt in mijn proefschrift. Veel dank ook aan Arend Jan. Door zijn enthousiaste begeleiding ben ik met veel plezier in Almelo aan de slag gegaan; onze samenwerking toont overtuigend dat onderzoekspoli’s niet alleen een academische aangelegenheid zijn. Tenslotte wil ik Hauw The bedanken, die me al als tweedejaarsstudent geneeskunde geïnspireerd heeft tot het doen van wetenschappelijk onderzoek.
Dit proefschrift is het resultaat van nog een aantal samenwerkingsverbanden. De wekelijkse autoritjes naar Leeuwarden met Els van der Wouden hebben geleid tot een heuse onderzoekspoli onder enthousiaste begeleiding van Marc Hemmelder. De kennismaking met Adrienne Zandbergen, werkzaam in het Erasmus Medisch Centrum te Rotterdam, heeft al in twee publicaties geresulteerd, waarvan een opgenomen is in dit proefschrift.
De leden van de beoordelingscommissie, prof. dr. M. Levi, dr. M. Noris en prof. dr. P. Smits, wil ik hartelijk danken voor hun bereidheid mijn proefschrift te beoordelen.
181

In het bijzonder dank ik dr. Noris. Na afronding van mijn klinisch geneesmiddelonderzoek, heb ik met erg veel plezier een leerzame tijd doorgebracht in het laboratorium van dr. Noris. Marina, é stato un grande piacere lavorare con te e il tuo gruppo. Lo considero un onore che tu abbia accettato di partecipare alla mia commissione di laurea.
Op de polikliniek waar alle patiënten gezien worden sta je er als onderzoeker niet alleen voor, en gelukkig maar. Dankzij de steun van Corrie van Nieuwenhout konden de complexe protocollen, die leidden tot spitsuren op de proteïnuriepoli van het UMCG, draaiende gehouden worden. Marja van de Klok is uiterst behulpzaam geweest in het opzetten van een onderzoekspoli in het Twenteborgziekenhuis.
Alle mensen werkzaam binnen het Kidney Center wil ik bedanken voor de prettige sfeer en samenwerking, die essentieel zijn geweest voor de voortgang van het onderzoek. Enkelen van hen wil ik bij naam noemen: Paul de Jong, het afdelingshoofd, Rieta Graler en Winie de Jonge, die het secretariaat bemanden, en natuurlijk mijn directe collega’s. Met mijn kamergenoten Menno Kocks en Titia Lely heb ik meer dan een kamer gedeeld; ik koester de herinneringen aan onze enthousiaste brainstormsessies en bevlogen discussies. Met Goos Laverman, Peter Luik, Erik Stuveling, Jorden Veeneman, Jacobien Verhave en Aiko de Vries, de zogenaamde oude garde, en Eelke Bos, Martin de Borst, Auke Brantsma, Jacoline Brinkman, Inge Hamming, Andrea Kramer, Jan Krikken, Wynand Melenhorst, Leendert Oterdoom, Mieneke Rook, Mirjan van Timmeren en Folkert Visser, de jonge garde, heb ik altijd goed samengewerkt met als resultaat geslaagde stuk voor stuk memorabele congresbezoeken aan de VS. Als laatste wil ik Femke Waanders bedanken voor de voortvarende wijze waarop zij de lopende proteïnuriestudie heeft opgepakt. Femke, veel succes met jouw promotie straks en je verdere carrière.
Zonder de steun van familie en vrienden is promoveren niet mogelijk. Jan-Leendert Brouwer en Erwin Jonkman, veel dank dat jullie mijn paranimfen zijn. Dank ook aan Elsbeth ten Kate, Hanneke Klaaijssen, Ward Libourel, Hester van Meer, wijlen Richard Nahuis, Patrick Oderkerk en Bram Rutgers. Als onze existentiële vragen van het hoe en waarom opnieuw niet beantwoord waren, dan was er altijd nog het café-, disco- of buitenlandbezoek om dat leed te verzachten. Als laatste wil ik de belangrijkste mensen, het thuisfront, bedanken. Berber, Betty en Frank. En Igor. Liffert Vogt Amsterdam, februari 2008
182

CURRICULUM VITAE Liffert Vogt was born on June 4, 1975, in Groningen, The Netherlands. After the comprehensive school, he obtained his certificate of Lyceum (VWO-beta) in 1993 at the Ichthus College in Drachten. After the summer of that year, he started with his study of medicine at the University of Groningen. On January 27, 2000, he received his graduate medical degree. For one year he worked as a resident in internal medicine at the Isala Clinics of Zwolle, where after he returned to the University of Groningen to become coordinator of the Junior Scientific Master Class—a scientific training program for talented medical students. From April 2002 until 2005, he was PhD trainee. In this period, he performed different clinical trials in the outpatient nephrology clinics of the University Medical Center Groningen (UMCG), the Medical Center Leeuwarden (MCL), and the Twenteborg Hospital Almelo. A final research year, in 2006, was dedicated to experimental research in the laboratories for molecular medicine of the Mario Negri Institute in Bergamo, Italy. On October 1, 2006, his specialty training in internal medicine has started in the Academic Medical Center (AMC) of Amsterdam.
183

LIST OF PUBLICATIONS Mahmoodi BK, Ten Kate MK, Waanders F, Veeger NJ, Brouwer JLP, Vogt L, Navis G, van der Meer J. High absolute risks and predictors of venous and arterial thromboembolic events in patients with nephrotic syndrome. Results from a large retrospective cohort study. Circulation 2008;117:224-30 Vogt L, Waanders F, Boomsma F, de Zeeuw D, Navis G. Independent and combined effects of low sodium diet and diuretic on the antiproteinuric efficacy of the AT1 antagonist losartan in non-diabetic proteinuric patients. J Am Soc of Nephrol 2008; in press Zandbergen AA, Vogt L, de Zeeuw D, Lamberts SW, Ouwendijk RJ, Baggen MG, Bootsma AH. Change in albuminuria is predictive of cardiovascular outcome in normotensive patients with type 2 diabetes and microalbuminuria. Diabetes Care 2007;30:3119-21 Vogt L, Laverman GD, van Tol A, Groen AK, Navis G, Dullaart RP. Cellular cholesterol efflux to plasma from proteinuric patients is elevated and remains unaffected by antiproteinuric treatment. Nephrol Dial Transplant 2006;21:101-6 Vogt L, Navis G, Köster J, Manolis AJ, Reid JL, de Zeeuw D; Angiotensin II Receptor Antagonist Telmisartan Micardis in Isolated Systolic Hypertension (ARAMIS) Study Group. The angiotensin II receptor antagonist telmisartan reduces urinary albumin excretion in patients with isolated systolic hypertension: results of a randomized, double-blind, placebo-controlled trial. J Hypertens 2005;23:2055-61 Vogt L, Navis G, de Zeeuw D. Individual titration for maximal blockade of the renin-angiotensin system in proteinuric patients: a feasible strategy? J Am Soc Nephrol 2005;16 (Suppl 1):S53-7 Vogt L, Kocks MJA, Laverman GD, Navis G. Renoprotection by blockade of the renin-angiotensin-aldosterone system in diabetic and non-diabetic chronic kidney disease. Specific involvement of intra-renal angiotensin-converting enzyme activity in therapy resistance? Minerva Med 2004;95:395-409 Vogt L, Laverman GD, Dullaart RP, Navis G. Lipid management in the proteinuric patient: do not overlook the importance of proteinuria reduction. Nephrol Dial Transplant 2004;19:5-8 Vogt L, Laverman GD, de Zeeuw D, Navis G. The COOPERATE trial. Lancet 2003;361:1055-6 Vogt L, Navis G, de Zeeuw D. Renoprotection: a matter of blood pressure reduction or agent-characteristics? J Am Soc Nephrol 2002;13 (Suppl 3):S202-7
184